management of optic nerve gliomasbjo.bmj.com/content/bjophthalmol/64/8/545.full.pdf · the...
TRANSCRIPT

British Journal of Ophthalmology, 1980, 64, 545-552
Management of optic nerve gliomasJ. E. WRIGHT, W. I. McDONALD, AND N. B. CALL*From the Orbital Clinic, Moorfields Eye Hospital, City Road, London EC] V 2PD
SUMMARY Seventeen patients thought to have orbital optic nerve gliomas when first seen havebeen reviewed after up to 12 years. Enlargement of the optic canal was present in 15 of the 16patients examined, but this finding was unreliable as an indicator of the posterior extent of thetumour. Nine patients had a stable course with little change over a period of up to 8 years; therewas optic atrophy in all and neurofibromatosis was relatively common (7/9). Eight patients showedprogressive enlargement of the tumour; 6 had swollen discs, and the incidence of neurofibromatosiswas relatively low (3/8). The optic nerve was excised in 7 of the latter group. Biopsies of the opticnerve taken from the region of maximal enlargement were difficult to interpret and unhelpful inplanning management. Radical surgery should be reserved for the minority of patients in whomthere is progressively enlarging tumour without evidence of chiasmal involvement.
Gliomas of the optic nerve are relatively uncommonlesions, and their natural history and managementare the subject of considerable debate. In 1969 Hoytand Baghdassarian' suggested that these tumoursbehaved as hamartomas and concluded that themanagement of patients with them should beconservative. We have seen 17 patients with opticnerve glioma in the past 12 years, and it has becomeclear that an appreciable proportion of them havehad a progressive course. As a result our attitudetowards treatment has changed. In this paper werecord our experience of these patients and outlineour present approach to their management.
Patients and methods
The diagnosis of optic nerve glioma was made in17 patients out of more than a thousand with orbitaldisease seen at the Orbital Clinic at Moorfields EyeHospital between 1968 and 1980. The diagnosis wasinitially made clinically on the basis of poor visionin association with proptosis and optic atrophy orswelling of the optic disc, and radiographic evidenceof enlargement of the optic foramen. In 1 patientthe optic foramen was normal; 1 child was too youngto co-operate for optic foramen views; and in 1patient there was no proptosis during the period ofobservation. Enlargement of the optic nerve was,
Correspondence to Mr John E. Wright, Moorfields EyeHospital, City Road, London ECIV 2PD.
*Present address: 324 Tenth Avenue, Suite 185, Salt LakeCity, UT 84103, USA.
however, demonstrated in these 3 patients. Furtherdetails are given in the section on 'Results'. Inpatients seen later in the series enlargement of theoptic nerve on CT scans was required for thediagnosis. The clinical diagnosis was confirmedhistologically in 7 cases.
In all patients a detailed clinical history wasobtained and a ftull ocular and neurological exami-nation was carried out. If the patient was old enoughto co-operate, the visual fields were charted; insome patients pattern visual evoked potentials(VEP) were recorded. The neurologist, in additionto assessing the state of the central nervous system,checked for stigmata of Von Recklinghausen'sneurofibromatosis, using the criteria of Crowe andSchull,2 and interviewed the family to see if therewas evidence of neurofibromatosis in the relatives.Radiographic examination of the patients includedroutine skull films together with optic canal viewsin each case. In many patients axial hypocycloidaltomograms were obtained.
Patients with a slowly progressive or static lesion(as monitored by visual acuity, field charts, anddegree of proptosis) were seen at least yearly by theophthalmologist and the neurologist. Patients witha more progressive lesion were usually seen every4 weeks.
Initially a biopsy of the optic nerve was performedin 4 patients in whom there was evidence of recenttumour enlargement. This was done to differentiateoptic nerve gliomas from optic nerve meningiomas.However, without complete excision of the opticnerve the histological interpretation of the excised
545
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from
J. E. Wright, W. L. McDonald, and N. B. Call
tissue proved extremely difficult. Biopsies were nottherefore performed on the later cases.
If the clinical course indicated progressiveenlargement of the tumour, the patient was referredto our neurosurgical colleague so that the lesioncould be approached transcranially and the wholeof the optic nerve from the globe to chiasm explored.In most of these cases the optic nerve from theglobe to the chiasm was excised. If the chiasm wasinvolved, surgical resection was not performed, buta shunt was inserted if hydrocephalus was present.
Results
Seventeen patients with an orbital optic nerveglioma diagnosed by the criteria defined abovewere included in the study. The details of their ageand sex distribution are shown in Table 1. Ten ofthe 17 patients had Von Recklinghausen's neuro-fibromatosis. In 9 of these 10 cafe-au-lait spotswere the only stigmata of the disease. The exceptionwas a patient who presented at 38 years of agehaving had poor vision in one eye since at least theage of 11; she had multiple cutaneous molluscafibrosa.The presenting symptoms were particularly
interesting (Table 2). Fourteen patients had aproptosed eye which was noticed by parents. Eightof these 14 patients had no complaint other thanproptosis; only 4 patients had complained of poorvision in the affected eye, while 2 had a convergentsquint in addition to proptosis. Of the remaining 3patients 2 were noted to have a squint and diminishedvision when examined in a school clinic. Onepatient, the oldest in the series, had noticed a pro-
Table 1 Orbital optic nerve glioma, clinical data
Total patients 17Sex Male 4 Female 13Involved orbit Right 9 Left 8Age at presentation Mean 20-4 years
Median 5 5 yearsRange 2-38 years
Age at onset of symptoms Mean 4 5 years of ageRange 0-5-18 years of age
Table 2 Initial complaint
Proptosis 8Proptosis and poor visual acuity 4Proptosis and squint 2Squint and poor visual acuity 2Poor vision 1
Table 3 Clinical presentation
Present Absent
Proptosis 15 2Poor vision 17 -
Relative afferent pupillary defect 17Optic disc swelling 6Optic atrophy IIEnlarged optic canal 15 1Neurofibromatosis 10 7
Table 4 Visual acuity on presenttation
Visual acuity Number of patients
6/18 16/24 46/36 4
C.F. IPL 3NPL 4
PL=perception of light. NPL=no perception of light.
gressive deterioration in the vision of the affectedeye but did not notice that the eye was very slightlyproptosed.
All 17 patients had decreased visual acuity atthe time of presentation (Table 3), ranging from6/18 to no perception of light (Table 4), with arelative afferent pupillary defect on the affectedside. Eleven patients had a pale atrophic opticdisc; 1 of these patients had opticociliary shuntvessels. Four patients had a swollen optic disc, and1 patient had a mass extending forwards into theglobe and involving the whole of the optic disc.One patient with 6/24 vision in the affected eye hadnormal discs on presentation, but optic atrophywas noted shortly thereafter.
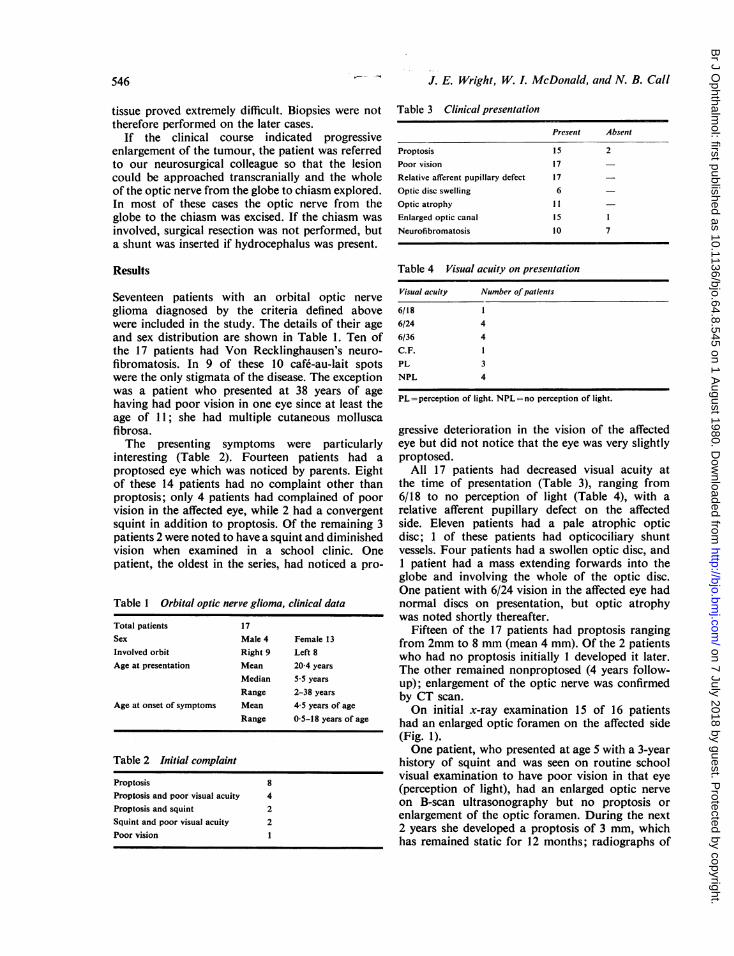
Fifteen of the 17 patients had proptosis rangingfrom 2mm to 8 mm (mean 4 mm). Of the 2 patientswho had no proptosis initially I developed it later.The other remained nonproptosed (4 years follow-up); enlargement of the optic nerve was confirmedby CT scan.On initial x-ray examination 15 of 16 patients
had an enlarged optic foramen on the affected side(Fig. 1).One patient, who presented at age 5 with a 3-year
history of squint and was seen on routine schoolvisual examination to have poor vision in that eye(perception of light), had an enlarged optic nerveon B-scan ultrasonography but no proptosis orenlargement of the optic foramen. During the next2 years she developed a proptosis of 3 mm, whichhas remained static for 12 months; radiographs of
546
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from
Management of optic nerve gliomas
Fig. 1 Oblique radiograph ofskiull showing rounded enlarge-ment of the optic canal typicalof optic nerve glioma.
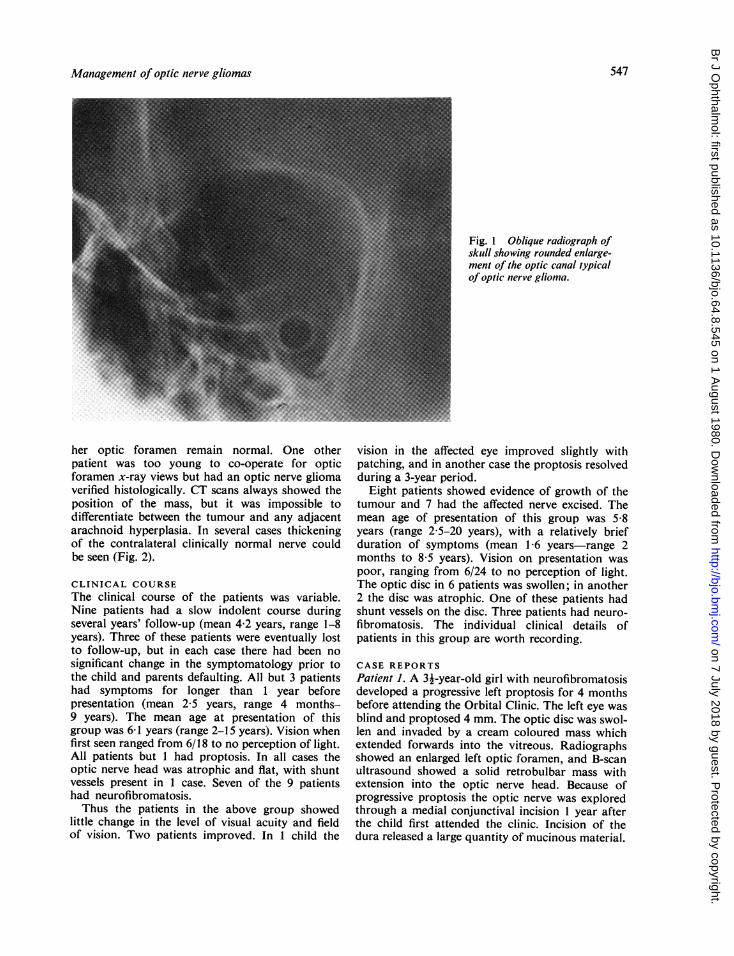
her optic foramen remain normal. One otherpatient was too young to co-operate for opticforamen x-ray views but had an optic nerve gliomaverified histologically. CT scans always showed theposition of the mass, but it was impossible todifferentiate between the tumour and any adjacentarachnoid hyperplasia. In several cases thickeningof the contralateral clinically normal nerve couldbe seen (Fig. 2).
CLINICAL COURSEThe clinical course of the patients was variable.Nine patients had a slow indolent course duringseveral years' follow-up (mean 4-2 years, range 1-8years). Three of these patients were eventually lostto follow-up, but in each case there had been nosignificant change in the symptomatology prior tothe child and parents defaulting. All but 3 patientshad symptoms for longer than 1 year beforepresentation (mean 2-5 years, range 4 months-9 years). The mean age at presentation of thisgroup was 6-1 years (range 2-15 years). Vision whenfirst seen ranged from 6/18 to no perception of light.All patients but 1 had proptosis. In all cases theoptic nerve head was atrophic and flat, with shuntvessels present in 1 case. Seven of the 9 patientshad neurofibromatosis.Thus the patients in the above group showed
little change in the level of visual acuity and fieldof vision. Two patients improved. In 1 child the
vision in the affected eye improved slightly withpatching, and in another case the proptosis resolvedduring a 3-year period.
Eight patients showed evidence of growth of thetumour and 7 had the affected nerve excised. Themean age of presentation of this group was 5-8years (range 2 5-20 years), with a relatively briefduration of symptoms (mean 1 6 years-range 2months to 8-5 years). Vision on presentation waspoor, ranging from 6/24 to no perception of light.The optic disc in 6 patients was swollen; in another2 the disc was atrophic. One of these patients hadshunt vessels on the disc. Three patients had neuro-fibromatosis. The individual clinical details ofpatients in this group are worth recording.
CASE REPORTSPatient 1. A 31-year-old girl with neurofibromatosisdeveloped a progressive left proptosis for 4 monthsbefore attending the Orbital Clinic. The left eye wasblind and proptosed 4 mm. The optic disc was swol-len and invaded by a cream coloured mass whichextended forwards into the vitreous. Radiographsshowed an enlarged left optic foramen, and B-scanultrasound showed a solid retrobulbar mass withextension into the optic nerve head. Because ofprogressive proptosis the optic nerve was exploredthrough a medial conjunctival incision 1 year afterthe child first attended the clinic. Incision of thedura released a large quantity of mucinous material.
547
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from

J. E. Wright, W. I. McDonald, and N. B. Call
The biopsy showed an intact dural sheath with anunderlying area of meningeal hyperplasia. CTscans showed enlargement of the whole of theoptic nerve from the globe to the chiasm. The childwas referred to our neurosurgical colleague and theaffected optic nerve removed from behind theglobe to the chiasm, which was not involved,through a frontal craniotomy. Histologically thetumour was a grade 1 astrocytoma. No tumour waspresent at the cut surface of the intracranial portionof the nerve. Intractable glaucoma developed, andthe globe was enucleated 5 months later. Ninemonths after the craniotomy the child's prosthesisbecame displaced by an enlarging orbital mass;biopsy of this lesion showed recurrence of theglioma. The mass continued to enlarge and the orbitwas eventually partially exenterated. She is alive withno sign of a recurrence of the tumour 2 years afterher last operation.
Patient 2. An 8-year-old girl with a 4-monthhistory of progressive left proptosis was referred tothis clinic. The affected eye was proptosed 3 mmand convergent 40°. Vision was reduced to 6/36, theleft optic disc appeared hypoplastic, with swellingof the nerve head in the upper nasal quadrant. Therewere no stigmata of neurofibromatosis. Radio-graphs showed enlargement of the left optic canal.The pattern VEP showed a normal response fromthe right eye. The optic nerve was explored througha medial transconjunctival approach; incision ofthe dural sheath released mucinous material. Aportion of the sheath together with the underlyingtissue was excised and was reported as showingmeningothelial cells, fibrous tissue, and a few welldifferentiated glial cells. During the next 3 years thevisual acuity in the left eye further decreased to 6/60.The proptosis increased to 10 mm. CT scans showedan ill-defined expansion of the left optic nerve, butthe intracranial scan was normal, as was an airencephalogram. The intracranial and intracanalicu-lar portion of the optic nerve were removed througha frontal craniotomy. Histopathological examina-tion showed a normal optic nerve with an extensivecollar of meningothelial cells and intermingledpsammoma bodies. No evidence of glial hyperplasiaor astrocytic glioma was seen throughout the intra-canalicular or intracranial portions of the opticnerve. She was diagnosed as having a primary opticnerve meningioma. Three months later a largetumour mass continuous with the optic nerve wasexcised through an anterior orbitotomy, and theoptic nerve was divided flush with the globe.Histological examination showed a typical grade 1astrocytic glioma of the optic nerve with extensivemeningothelial cell proliferation and numerouspsammoma bodies. The child is alive and well with
Fig. 2 Axial CT scan showing left optic nerve glioma(arrowed). The right optic nerve is diffusely enlarged;the right visual.field and acuity were normal.
no sign of a recurrence after 2 years'.Patient 3. A girl presented at the age of 51 years
with a 4-month history of a left divergent squint anda 2-month history of proptosis. Her blind left eyewas proptosed 3 mm, divergent 25', and the opticdisc was swollen. The right eye was normal in allrespects with an unaided vision of 6/6; the VEPwas normal from this side. There were no signs ofneurofibromatosis. Radiographs showed an en-larged left optic foramen. Eight months later thedivergent squint was surgically corrected and theoptic nerve explored. A biopsy of the nerve showedhyperplastic meningeal tissue. Four months laterthe VEP showed evidence of involvement of thenasal fibres from the right eye, though a field defectcould not be detected by perimetry. Twelve monthslater she experienced severe left hemicranial head-aches with nausea and vomiting, which afterseveral months subsided spontaneously. Threeyears after her initial presentation perimetry showeda right superior temporal quadrantic field defect.By this time the left eye was proptosed 9 mm. CTscans showed a large left optic nerve mass withinvolvement of the chiasm. An air encephalogramshowed a moderate degree of symetrical hydro-cephalus, with the third ventricle displaced upwardsand almost obliterated by a large suprasellar mass.A left frontal craniotomy was performed, duringwhich it was found that the tumour involved bothoptic nerves together with the chiasm. Posteriorlythe tumour surrounded the left internal carotid andanterior cerebral arteries. Portions of the tumourwere removed for histological examination. Exami-nation showed the tumour to have the features of agrade Iastrocytoma. 5500 rads of radiotherapy
548
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from

Management of optic nerve gliomas
were given to the chiasm and the left orbit. Subse-quently the shunt for hydrocephalus was removed.She remains alive and well without progression ofthe proptosis or visual field deficit for the past 4years.
Patient 4. A girl was seen at the age of 24 yearswith a 2-month history of proptosis and poor visionin the left eye. Vision in the affected eye was reducedto 6/36; the eye was proptosed 3 mm, and the opticdisc was swollen. No signs of neurofibromatosiswere seen. X-rays showed an enlarged left opticforamen and CT scans enlargement of the intra-orbital part of the left optic nerve. A biopsy of theoptic nerve through a medial conjunctival ap-proach was performed and was reported as showingastrocytic proliferation with occasional Rosenthalfibres. It was not possible to tell whether the biopsywas of the nerve itself or of an extension of tumourtissue into the sheath; meningothelial cells were
not present. During the ensuing 6 months theproptosis increased 6 mm, and the child was trans-ferred to the care of our neurosurgical colleague.An air encephalogram showed no evidence of intra-cranial extension. The whole of the optic nervefrom the globe to the chiasm was removed througha transfrontal craniotomy. Histologically the tumourwas an astrocytoma grade 1. At operation it wasthought that the tumour did not involve the chiasm,but microscopically the cut end of the intracranialportion of the nerve showed gliomatous tissue.Apart from postoperative ptosis the child is well 6months later.
Patient 5. A girl aged 74 years was brought to theclinic having noticed poor vision and progressiveprotrusion of the left eye for the previous 2 months.Her left eye was proptosed 4 mm, the vision was
reduced to 6/36, and the optic disc was swollen.Radiographs showed enlargement of the left opticcanal and CT scans enlargement of the optic nerveto the orbital apex. VEPs were obtained from theright eye and were reported as being within normallimits. The patient was referred to our neurosurgicalcolleague for removal of the optic nerve because ofthe rapidity of the growth of the tumour. The nervewas totally excised, but there was histologicalevidence of glioma at the cut end of the intracranialpart of the optic nerve. Histologically the tumourwas a grade 1 astrocytoma. The child is well withno signs of a recurrence of the tumour 9 monthsafter surgery.
Patient 6. The father of a 6-year-old girl hadnoticed that his daughter's left eye had becomeprogressively prominent for 6 months prior toconsulting an ophthalmologist. The eye was blindand proptosed 8 mm. Ocular movements were full.However, the left optic disc was swollen, with
associated peripapillary haemorrhages and exudates.A VEP from the right eye and the visual fields werenormal. The child was referred to our neurosurgicalcolleague, who explored the chiasm but found thatthe glioma extended into the chiasm. Surgery wasconfined to obtaining a biopsy, which confirmed thediagnosis of glioma, astrocytoma grade 1. Duringthe ensuing 15 months the amount of proptosis hasincreased slightly, and it is probable that a portionof the orbital part of the glioma will have to beresected in order to preserve the globe.
Patient 7. A 9-year-old girl was referred to anophthalmologist. Her parents had noticed a slightright divergent squint since she was 6 months old.At a routine school examination it was found thatthe vision in her right eye was poor. At her initialexamination the right visual acuity was 6/24, withan associated slight right divergent squint. She wasinitially treated with partial occlusion of the lefteye, but after 18 months it was noticed that thevision in her right eye had deteriorated, and shecould only count fingers in the upper half of thevisual field. At the same time she was found to havea pale right optic disc, and the eye was proptosed3 mm. She was subsequently referred to the OrbitalClinic, where x-rays of her skull showed enlargementof the right optic canal. There was no evidence ofneurofibromatosis. During the next 5 years visionin her right eye deteriorated to the appreciation ofhand movements at 0 25 metre and the proptosisincreased to 6 mm. The visual field in the left eyewas examined at regular intervals and the VEPswere obtained. Four years after her initial presen-tation the VEP showed evidence of a temporal fielddefect. Twelve months later there was definiteevidence of a central temporal half field defect inthe right eye by conventional perimetry. CT scansshowed enlargement of the right optic nerve withevidence of chiasmal involvement. The child wastransferred to Maida Vale Hospital for a pneumo-encephalogram, which revealed a suprasellar massand hydrocephalus. The patient died after theprocedure. Permission for a necropsy was declined.
Patient 8. A 20-year-old female student hadnoticed blurring of right vision for 18 months withprominence of the eye for 12 months before con-sulting an ophthalmic surgeon. During the ensuingyear vision in the affected eye deteriorated, and thedegree of proptosis increased. A neurosurgeon fromanother centre performed a transfrontal approachto the right orbit but was unable to find any abnor-mality. After an ineffective 6-week course of syste-mic steroids the patient was referred to this hospital.Her right eye was proptosed 5 mm and displaceddownwards 3 mm; upward movement of the globewas selectively restricted. The optic disc was pale
549
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from

J. E. Wright, W. I. McDonald, and N. B. Call
and flat, though a year previously definite swellinghad been noted. There were numerous opticociliaryshunt vessels. A CT scan showed gross enlargementof the optic nerve immediately behind the globe.Subsequently the orbital portion of the optic nervecontaining a large grade 1 astrocytoma was excisedthrough a lateral orbitotomy approach. During a4-year follow-up there has been no recurrence ofthe tumour.
Discussion
The outstanding finding of the present investigationis that our patients fell into 2 fairly distinct groupswith differing clinical course. Nine patients had anindolent course with little change in the size andextent of the tumour. In the remaining 8 patientsthe tumours progressively enlarged.The age distribution of the patients at presentation
was similar in the 2 groups, but the progressivegroup had a shorter history and a relatively rapidincrease in proptosis during the period before andsoon after the initial consultation. Other evidenceof rapid tumour enlargement was found in theocular fundus. In 6 patients the optic disc was swol-len when the patient was first seen. This was incontrast to the more stable group of patients inwhom none had evidence of optic disc swelling,though there was optic atrophy in all cases. Ofparticular interest was patient 7, in whom there wasevidence of a quiescent glioma for 9 years beforethe eye became proptosed. Five years later she haddeveloped a field defect in the contralateral eye andthe proptosis had increased considerably. This casewas exceptional, but it does illustrate that enlarge-ment of a glioma of the optic nerve can occur inpatients who have been clinically quiescent forsome years.There was a significant difference in the incidence
of neurofibromatosis in the 2 groups. The overallincidence was rather higher (10/17, 63%) than inprevious reports,13-8 as low as 10%i6 and as high as500%.8 Only 3 of 8 patients in the progressive grouphad neurofibromatosis compared with 7 out of 9with quiescent tumours. This observation accordswith the views of Martin and Cushing9 and Lloyd8and Klug,10 who reported that patients with opticnerve glioma in association with neurofibromatosishad a more benign course than patients without thisassociation. The explanation for the wide divergencein figures for the overall incidence of neurofibro-matosis is uncertain, but 3 factors are likely tocontribute: (1) the relatively small number ofpatients in most series; (2) the difficulty of recog-nising the minimal clinical signs of neurofibroma-tosis; and (3) the delay in the development of the
stigmata of the disease in some children.The recognition that there exist 2 groups of
patients with orbital optic nerve glioma has import-ant implications for management. Many surgeonshave adopted a conservative policy following Hoytand Baghdassarians's conclusion that gliomas ofthe optic pathways are indolent and slow growingand that for practical purposes they can be regardedas harmartomas, most of them requiring no activeintervention. This view has been supported by anumber of authors subsequently.11 14 There ishowever good neuropathological evidence that atleast some optic nerve gliomas are true tumourswith a potential for local invasion.'5 This observa-tion, together with our experience that nearly halfour patients showed clear evidence of progression,has led us to the conclusion that the potential forenlargement of optic nerve glioma in childhood isnot infrequently realised, and that radical surgeryshould be considered for patients in which it is.
It is clear that if the chiasm is already involvedwhen the patient presents a radical cure is notpossible. In these circumstances surgery should beconfined to the treatment of complications such asdisfiguring proptosis or hydrocephalus, as Hoytand Baghdassarian suggest.' The first step in theassessment of the patient must therefore be to try todetermine whether or not the tumour is confinedto the orbit. We have used 3 approaches to thisproblem.
(1) Radiological examination of the optic canals.There was evidence of enlargement in 15 out of 16of our patients. In several of these patients, however,surgical exploration showed that the glioma did notextend into the enlarged canal. As others havenoted,2 the widened canal could be associated withdural thickening or arachnoid hyperplasia. Thesize of the optic canal thus gives little indication ofthe posterior extent of an optic nerve glioma.
(2) CT scanning. The advent of high resolutionCT scanning in both axial and coronal planes hasenabled tumours arising from the optic nerves tobe recognised much more readily than was possiblepreviously. Again, however, it is not possible todistinguish between gliomatous tissue and meningealhyperplasia. Moreover, enlargement of the chiasmby tumour may still be difficult to detect even withthe very high resolution scanners now available.Pneumoencephalography is more reliable, but asAnderson and Spencer16 have reported, and ourcases 4 and 5 confirm, the absence of macroscopicenlargement of the chiasm does not exclude involve-ment by tumour.
(3) Visual field examinationi. The most sensitivemethods for detecting involvement of the nervefibres from the other eye are perimetry and VEP
550
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from

Management of optic nerve gliomas
examination. Halliday et aL." have shown that itis possible to detect abnormalities in the patternVEP before a field defect is demonstrable clinically.In 2 of our patients (patients 6 and 7) there waselectrophysiological evidence of a temporal fielddefect 6 and 12 months before it could be demon-strated by conventional perimetry. However, eventhis sensitive technique failed to show chiasmalinvolvement in 2 of the cases (4 and 5), in whom theface of the optic nerve severed at its junction withthe chiasm showed histological evidence of tumour.These observations indicate that abnormal tissuemay be present in the visual pathways without theclinician being aware of its true extent.
Having demonstrated the presence of an opticnerve tumour and obtained some idea of its extent,the ophthalmologist must then decide whether it isappropriate to biopsy the tumour in order to estab-lish the diagnosis. Our initial policy was not tobiopsy small, apparently indolent lesions. However,when there was evidence of recent tumour enlarge-ment and there was severe visual impairment, weobtained tissue from the nerve in the region ofmaximal swelling in an attempt to distinguishbetween optic nerve meningioma, which has areputation in children for aggressive growth andlocal invasion18 and therefore requires early radicalexcision, and glioma. In the event, as others havefound,6 it was difficult to interpret the biopsy find-ings. In 4 of the 5 biopsies there was arachnoidhyperplasia compatible with either glioma orprimary optic nerve meningioma. In the fifthbiopsy it was impossible to tell whether the tissuecame from the nerve itself or from an extension ofthe tumour into the sheath. Because we have notfound biopsy helpful in planning management wehave abandoned it as a routine, and we restrictsurgical intervention to total excision of tumoursshowing evidence of progressive enlargement or tothe symptomatic relief of complications.
Finally there is the question of surgical approachto the expanding tumour. In our own material, asin that of others, enlargement was produced bymeningeal hyperplasia and the production ofmucoid material containing tumour astrocytes aswell as by glial proliferation. Since it is impossibleclinically or radiologically to distinguish the con-tribution of each to any particular tumour, we havemanaged all patients in the same way. If there isno evidence of chiasmal involvement, the patientis referred to our neurosurgical colleague, whoexplores the chiasm through a frontal approach. Ifinspection confirms the absence of chiasmal swelling,the affected optic nerve is sectioned at its junctionwith the chiasm. The optic canal is unroofed andthe nerve excised to the posterior surface of the
globe. A number of authors have stated that partialremoval ofthese lesions is all that is needed and thatthe chances of enlargement of the residual tumourare extremely low.1 19 The importance of completeexcision of the nerve, however, is emphasised bythe course of case 1, in which a partial removal wasfollowed by rapid expansion of the orbital stump.
It is inevitable that in some patients tumourwill be present at the chiasm despite the absence ofswelling or of preoperative evidence of damage tofibres from the other eye. This was the case in 2patients (4 and 5) of our 4 patients in the progressivegroup (cases 1, 2, 4, and 5), who are known to havehad no macroscopic evidence of enlargement ofthe chiasm. The state of the chiasm in case 8 is notknown. Anderson and Spencer16 have suggested thatin such patients, as in those with preoperativeevidence of chiasmal enlargement, the abnormaltissue may be present in the chiasm from the start,rather than having extended there from a primaryorbital tumour. Our observations shed no light onthis suggestion. However, given that there is une-quivocal clinical and radiological evidence oftumour enlargement in some patients, it is reason-able to suppose that the chiasm might becomesecondarily involved by tumour tissue extendingalong the nerve in the same way that tumours ofsimilar histological type, such as the cerebellarastrocytomas of childhood, extend in white matterelsewhere in the central nervous system.'5 Cases 1and 2 of the 4 patients in whom there was nomacroscopic evidence of chiasmal involvement hadno tumour tissue at the cut surface of the intra-cranial optic nerve. In these 2 patients total excisionof the tumour will have removed the possibility offurther involvement of the visual pathways fromthe orbit. They are being closely followed up todetermine whether tumour subsequently developsin the chiasm. If it does, the case for an attempt atradical surgical cure will be greatly weakened.The correct management of the child with optic
nerve glioma remains a difficult problem. Ourexperience of the past 12 years has led us to movefrom a conservative to a more radical position,though as a result of our present review we thinkit likely that only a minority of patients shouldhave surgery. We now believe that parents shouldbe told that their child has a tumour of the opticnerve and that the appropriate method of treatmentwill be determined by the progress of the condition.In about half the patients there will be no deteriora-tion, and management will be conservative. In theremainder progressive deterioration will suggestthat the tumour is extending, and for these patientswe will advise radical surgery provided there is noradiological, clinical, or VEP evidence of chiasmal
551
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from

J. E. Wright, W. L McDonald, and N. B. Call
involvement. When the chiasm is involved, we willadvise against surgery unless orbital or intracranialcomplications develop, which should then bemanaged on their merits.
We are grateful to Professor Lindsay Symon, who performedthe craniotomies on patients 1-6; to Dr Glyn Lloyd, whoperformed the radiological examinations; to Dr MartinHalliday, who performed the VEP studies; and to ProfessorAlec Garner, Professor Leo Duchen, and Dr Robin Barnard,who carried out the histological examinations of the tumours.We appreciate also the help of Dr T. A. Feasby and Dr I.
McG. Donaldson, who carried out preliminary analyses ofthe data, and Mrs S. J. Cole for her help with the manu-script.
References
1 Hoyt WF, Baghdassarian SA. Br J Ophthalmol 1969;53: 793-98.
2 Crowe FW, Schull WJ. Diagnostic importance ofcaf8-au-lait spot in neurofibromatosis. Arch Intern Med1953; 91: 758-65.
3 Reece AB. Tumors of the Eye. 2nd ed. New York:Harper and Row, 1963: 163-71.
4 Chutorian AM, Shwartz JF, Evans RA, Carter S.Optic gliomas in children. Neurology 1964; 14: 83-95.
5 Chang CH, Wood EH. The value of radiation therapyfor gliomas of the anterior visual pathways. In: Brock-hurst RJ, Boruohoff FA, Hutchinson BT, Lessell S, eds.Controversy in Ophthalmology. Philadelphia: Saunders,1977: 878-86.
6 Yanoff M, Davis RL, Zimmerman LE. Juvenile Pilocyticastrocytoma ('glioma') of the optic nerve. Clinicopatho-logical study of sixty-three cases. In: Jacobiec FA, ed.Ocular and Adnexal Tumors. Birmingham: Aesculapius,1978: 685-707.
7 Davis FA. Primary tumors of the optic nerve, a pheno-menon of Recklinghausen's disease: a clinical and patho-logical study with a report of 5 cases and a review of theliterature. Arch Ophthalmol 1940; 23: 735-821; 957-1022.
8 Lloyd LA. Gliomas of the optic nerve and chiasm inchildhood. Trans Am Ophthalmol Soc 1973; 71: 488-535.
9 Martin P, Cushing H. Primary gliomas of the chiasmand optic nerves in their intracranial portion. ArchOphthalmol 1973; 52: 209-41.
10 Klug GL. Gliomas of the optic nerve and chiasm inchildren. Aust N Z J Surg 1977; 47: 596-600.
11 Glaser JS, Hoyt WF, Corbett J. Visual morbidity withchiasmal glioma. Arch Ophthalmol 1971; 85: 3-12.
12 Spencer WH. Primary neoplasms of the optic nerve andits sheath; clinical features and current concepts ofpathogenetic mechanisms. Trans Am Ophthalmol Soc1972; 70: 490-528.
13 Wong IG, Lubow M. Management of optic glioma ofchildhood, a review of 42 cases. Neuro-Ophthalmology 6.St Louis: Mosby, 1972: 51-60.
14 Glaser JS, Gliomas of the anterior visual pathways inchildhood; rationale for conservative management. In:Brockhurst RJ, Boruohoff FA, Hutchinson BT, LessellS, eds. Controversy in Ophthalmology. Philadelphia:Saunders, 1977: 897-906.
15 Russell DS, Rubinstein LJ. Pathology of Tumours of theNervous System. 4th edn. London: Arnold, 1977.
16 Anderson DR, Spencer WH. Ultra structural and histo-chemical observations of optic nerve gliomas. ArchOphthalmol 1970; 83: 324-35.
17 Halliday AM, Halliday E, Kriss A, McDonald WI,Mushin J. The pattern evoked potential in compressionof the interior visual pathways. Brain 1976; 99: 357-74.
18 Walsh FB. Meningiomas, primary within the orbit,and optic canal. In: Smith JL, ed. Neuro-OphthalmologySymposium of the University of Miami and the BascomPalmer Eye Institute. St Louis: C. V. Mosby, 1970: 5:240-66.
19 Hudson AC. Primary tumours of the optic nerve.R Lond Ophthalmol Hosp Rep 1912; 18: 317-439.
552
on 7 July 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.64.8.545 on 1 August 1980. D
ownloaded from