local fetal lung renin-angiotensin system as a target to treat...
TRANSCRIPT

INTRODUCTIONCongenital diaphragmatic hernia
(CDH) is a severe developmental anom-aly, with a mean incidence of 1:2500 Q1live births, in which etiology remainspoorly understood (1,2). This congenitalanomaly is characterized by a diaphrag-matic defect that allows intra-thoracicherniation of abdominal organs, andconsequently, maldevelopment of thealveoli and pulmonary vessels. For
many years, this malformation wasthought to be a surgical emergence,solely related to a diaphragmatic defect,and potentially curable by surgical clo-sure of this defect after birth, which al-lowed lung expansion. However, during90 years, CDH pathophysiology pro-gressed for a physiological emergence(3,4). It is now clear that lung hypoplasiaand consecutive persistent pulmonaryhypertension (PH) associated with this
disorder are the key determinants ofmortality (1–4). Despite the improve-ments in understanding CDH patho-physiology and advances in neonatalcare, such as the use of extra corporealmembrane oxygenation and inhaled ni-tric oxide, the mortality (50%) and mor-bidity rate in CDH newborns remainsexceedingly high (1–4). In humans, CDHcan be accurately diagnosed at secondtrimester during routine ultrasound ex-amination. Therefore it is amenable toantenatal therapies. Hence, antenataltherapies that promote fetal lung growthremain an appealing approach to im-prove survival of CDH fetuses. How-ever, fetal surgical interventions, such asfetal tracheal occlusion, are invasive,technically demanding, limited by thematernal and fetal risks, and their effi-cacy is still not determined, with contro-versial results in survival and morbidity
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 1
Local Fetal Lung Renin-Angiotensin System as a Target toTreat Congenital Diaphragmatic Hernia
Cristina Nogueira-Silva,1,2,3 Emanuel Carvalho-Dias,1,2,4 Paulina Piairo,1,2 Susana Nunes,5
Maria J Baptista,1,2,6 Rute S Moura,1,2 and Jorge Correia-Pinto1,2,7
1Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Braga, Portugal; 2ICVS/3B’s - PTQ5 Government Associate Laboratory, Braga/Guimarães, Portugal; 3Department of Obstetrics and Gynecology, Hospital de Braga,Braga, Portugal; Departments of 4Urology, 5Pediatrics, and 6Pediatric Cardiology, Hospital de São João, Porto, Portugal; and7Department of Pediatric Surgery, Hospital de Braga, Braga, Portugal
Antenatal stimulation of lung growth is a reasonable approach to treat congenital diaphragmatic hernia (CDH), a diseasecharacterized by pulmonary hypoplasia and hypertension. Several evidences from the literature demonstrated a possible in-volvement of renin-angiotensin system (RAS) during fetal lung development. Thus, the expression pattern of renin, angiotensin-con-verting enzyme, angiotensinogen, type 1 (AT1) and type 2 (AT2) receptors of angiotensin II (ANGII) was assessed by immunohisto-chemistry throughout gestation, whereas the function of RAS in the fetal lung was evaluated using fetal rat lung explants. Thesewere morphometrically analyzed and intracellular pathway alterations assessed by Western blot. In nitrofen-induced CDH model,pregnant rats were treated with saline or PD-123319. In pups, lung growth, protein/DNA ratio, radial saccular count, epithelial dif-ferentiation and lung maturation, vascular morphometry, right ventricular hypertrophy and overload molecular markers, gasom-etry and survival time were evaluated. Results demonstrated that all RAS components were constitutively expressed in the lungduring gestation and that ANGII had a stimulatory effect on lung branching, mediated by AT1 receptor, through p44/42 and Aktphosphorylation. This stimulatory effect on lung growth was mimicked by AT2-antagonist (PD-123319) treatment. In vivo antenatalPD-123319 treatment increased lung growth, ameliorated indirect parameters of pulmonary hypertension, improved lung functionand survival time in nonventilated CDH pups, without maternal or fetal deleterious effects. Therefore, this study demonstrated alocal and physiologically active RAS during lung morphogenesis. Moreover, selective inhibition of AT2 receptor is presented as aputative antenatal therapy for CDH.Online address: http://www.molmed.orgdoi: 10.2119/molmed.2011.00210
Address correspondence to Jorge Correia-Pinto, Life and Health Sciences Research Insti-
tute (ICVS), School of Health Sciences, University of Minho, Campus de Gualtar; 4710-057
Braga, Portugal. Phone: +351 253 604 910; Fax: +351 253 604 847; E-mail:
Submitted June 18, 2011; Accepted for publication November 17, 2011; Epub
(www.molmed.org) ahead of print November 18, 2011.

rates (5,6). Therefore, less invasive ap-proaches such as antenatal pharmaco-logical treatment to stimulate lunggrowth before birth and to treat PH arealso under investigation (3,7–10).
The renin–angiotensin system (RAS) isa classical endocrine system regulatingblood pressure, electrolyte and fluid ho-meostasis, involving several key compo-nents, namely angiotensinogen (the he-patic-derived precursor), two criticalenzymes, renin (secreted from the juxta-glomerular apparatus of the kidney) andangiotensin-converting enzyme (ACE,pulmonary-bound metalloproteinase),whose sequential actions produce an-giotensin I and the physiologically ac-tive, angiotensin II (ANGII), respectively(11). ANGII operates on two G protein-coupled receptors, the ANGII type 1(AT1) and type 2 (AT2) receptors (11).During the last two decades, in additionto this classic endocrine system, evidencehas demonstrated the presence of a localRAS with autocrine/paracrine actions inseveral developing organs, such as fetalkidney, heart, vasculature and adrenaldevelopment (12).
Regarding lung morphogenesis, thereis some evidence that lung expressesACE as well as AT1 and AT2 receptorsduring fetal development (13–15). How-ever, RAS components expression pat-tern, as well as effects during lung mor-phogenesis, is largely unknown.
In this study, we assessed the expres-sion pattern and function of RAS duringfetal lung development. Moreover, weassessed the role of RAS as a putativetarget for treatment of fetal lung hy-poplasia in CDH.
MATERIALS AND METHODSAnimal experiments were performed
according to the Portuguese law for ani-mal welfare (16; Diário da República, Por-taria 1005/92). Animals were housed inan accredited mouse house and treatedas specified by the recommendations ofthe Helsinki convention (17) and theGuide for the Care and Use of LaboratoryAnimals, published by the US NationalInstitutes of Health (18). Q6
Experimental Design and AnimalModel
In vitro studies were carried out to as-sess expression and function of RAS infetal lung, whereas in vivo studies wereperformed to explore RAS as a target totreat fetal lung hypoplasia using the nitrofen-induced CDH rat model (19).
Sprague Dawley female rats (225 g;Charles River; Barcelona, Spain) weremaintained in appropriate cages undercontrolled conditions and fed with com-mercial solid food. The rats were matedand checked daily for vaginal plug. Theday of plugging was defined as gesta-tional d 0.5 for time-dating purposes. According to the nitrofen-induced CDHrat model (19), at 9.5 d postconception(dpc), randomly selected pregnant ratswere exposed to 100 mg nitrofen (2,4-dichlorophenyl-p-nitrophenylether). Atdifferent time points, fetuses were har-vested by cesarean section. After fetal de-capitation, a toraco-laparotomy was per-formed under a binocular surgicalmicroscope (Leica, Wild M651.MSD,Heerbrugg, Switzerland) to inspect thediaphragm and harvest the organs. Fe-tuses were assigned to three experimen-tal groups: (i) Control group (C), fetusesnot exposed to nitrofen; (ii) Nitrofengroup (N), fetuses exposed to nitrofenwithout CDH; (iii) CDH group, fetusesexposed to nitrofen with CDH.
In Vitro StudiesNormal fetuses removed by cesarean
section at 13.5, 15.5, 17.5, 19.5 and 21.5dpc were killed by decapitation, andlungs dissected and processed for im-munohistochemistry (IHC). Lungs of fe-tuses with 13.5 dpc were also dissectedto perform fetal lung explants culturesand posterior western blot analysis.
IHCIHC was performed on formalin-fixed
and paraffin-embedded lungs, as previ-ously described by Nogueira-Silva et al.(7). Renin antibody (sc-27320; Santa CruzBiotechnology, Santa Cruz, CA, USA) wasused in a 1:25 dilution, ACE antibody (sc-20791; Santa Cruz Biotechnology Inc.) in a
1:75 dilution, angiotensinogen antibody(Abbiotec LLC, San Diego, CA, USA) in a1:100 dilution, AT1 receptor antibody (sc-1173; Santa Cruz Biotechnology Inc.) in a1:50 dilution and AT2 receptor antibody(sc-9040; Santa Cruz Biotechnology Inc.)in a 1:25 dilution. Incubation with the Ul-traVision detection system anti-polyvalenthorseradish peroxidase (Lab Vision Cor-poration, Fremont, CA, USA) or with thegoat ImmunoCruz™ Staining System(Santa Cruz Biotechnology Inc.) was car-ried out according to the manufacturer’sinstructions.
Fetal Lung Explant CulturesRecombinant ANGII (Sigma, St Louis,
MO, USA), AT1-antagonist (ZD-7155;Tocris Cookson, Bristol, UK) and AT2-an-tagonist (PD-123319; kindly supplied byPfizer, Groton, CT, USA) were addeddaily to lung explants to achieve increas-ing concentrations of ANGII (from 10–9 to10–4 mol/L) or ZD-7155 or PD-123319 at10–5 mol/L. These doses were selected ac-cording to the literature (20,21). This set ofexperiments created the following groups:control (n = 30), ANGII 10–9 (n = 10),ANGII 10–8 (n = 11), ANGII 10–7 (n = 11),ANGII 10–6 (n = 12), ANGII 10–5 (n = 12),ANGII 10-4 (n = 12), ZD-7155 (n = 12), andPD-123319 (n = 15). Furthermore, explantswere treated with ANGII at 10–9 mol/L inthe presence of ZD-7155 or PD-123319 at10–5 mol/L, creating the additionalgroups: ANG 10–9 + ZD-7155 (n = 12) andANG 10–9 + PD-123319 (n = 15). Cultureswere photographed daily and branchingmorphogenesis was assessed according toNogueira-Silva et al. (22).
Western Blot AnalysisAfter 4 d in culture, lung explants
treated with ANGII at 10–9 mol/L, ZD-7155 or PD-123319 were processed forwestern blot analysis of nonphospory-lated and phosporylated forms of mitogen-activated protein kinases (MAPKs)[p44/42, p38, JNK (mAbs; Cell SignalingTechnology, Danvers, MA, USA)] andphosphatidylinositol-3-kinase (PI3K/Akt)(Cell Signaling Technology) according toNogueira-Silva et al. (22). For loading
2 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A

control, blots were probed with β-tubulinmAb (Abcam plc, Cambridge, UK) (n = 3).Quantitative analysis was performedwith Quantity One 4.6.5 1-D AnalysisSoftware (Bio-Rad Laboratories, Her-cules, CA, USA).
In Vivo StudiesPregnant rats, control or exposed to ni-
trofen, were anesthetized at 12.5 dpcwith a mix of ketamine (75 mg/kg) andmedetomidine (0.5 mg/kg) for subcuta-neous implantation of an osmotic micro-pump filled either with saline or PD-123319 (randomly), on mid scapularregion (Alzet osmotic pump Model2ML1; Durect Corporation, Cupertino,CA, USA). Saline or PD-123319 solutionwas infused with a rate of 10 μL/h(20 mg/kg/d for PD-123319). For the re-versal of the sedative effect, atipamezole(0.25 mg/kg) was used and butorfanole(1 mg/kg) was administered immedi-ately after the surgery. Fetuses were as-signed to six experimental groups: Con-trol rats treated with saline (C + S);Control rats treated with PD-123319 (C +PD); Nitrofen rats treated with saline (N + S); Nitrofen rats treated with PD-123319 (N + PD); CDH rats treated withsaline (CDH + S); and CDH rats treatedwith PD-123319 (CDH + PD).
Pregnant rats were randomly assignedfor cesarean section at 21.5 dpc or forspontaneous delivery at term.
The pregnant rats killed at 21.5 dpcwere laparotomized, and after longitudi-nal hysterotomy, each fetus (C + S, n =14; C + PD, n = 15; N + S, n = 15; N + PD,n = 12; CDH + S, n = 15; CDH + PD, n =24) was extracted, weighed and decapi-tated for organ harvesting (lungs, heartand kidneys). Organs were also weighedindependently, organs-to-body weightratios were assessed and lungs were ei-ther snap frozen in liquid nitrogen forbiochemical analyses or fixed in 4%paraformaldehyde (PAF) for histologicalanalyses. In this set of experiments, thelevel of lung hypoplasia was calculatedaccording to Baptista et al. (9).
The pups that were delivered sponta-neously at term were placed immediately
after birth in a light-heated box and ran-domly killed 5 min after birth or allowedto survive, without any care, supportstrategies or ventilatory support. Thosekilled by decapitation at min 5 (C + S, n =15; C + PD, n = 9; N + S, n = 11; N + PD,n = 6; CDH + S, n = 12; CDH + PD, n =14) were used for blood collection (neckbleeding) and gasometric evaluation (i-Stat1 analyser; Abbott, Chicago, IL, USA).These were then dissected for identifica-tion of CDH. Moreover, hearts were dis-sected for right ventricular hypertrophyevaluation and myocardial samples ofright ventricular free wall were harvestedand processed for q-PCR studies. The re-maining fetuses were allowed to survive(C + S, n = 10; C + PD, n = 9; N + S, n =14; N + PD, n = 21; CDH + S, n = 14;CDH + PD, n = 28), evaluated by two in-dependent–blind observers (MJ Baptistaand S Nunes) and scored at 1, 3, 5 andsubsequently at each 5 min, using anAPGAR-like score (Table 1, adapted fromDauger et al. [23]). The moment of deathwas registered and defined by the mo-ment in which the two observers attrib-uted an APGAR 0 (marked cyanosis,apnea, no spontaneous movements, noresponse to stimulus). All experimentswere recorded in video to clarify anydoubt. These pups were opened post-mortem for diaphragm inspection.
Lungs, heart and kidneys from allpregnant rats were weighed for maternalorgan-to-body weight ratio analysis.
Biochemical Studies for Protein/DNARatio Assessment
Total lungs (left and right) wereprocessed to determine protein and DNAcontents. Protein content was assessed
by Bradford method (24). DNA was ex-tracted using the DNeasy Blood & TissueKit (Qiagen, Hilden, Germany).
Histological StudiesThe trachea was cannulated and the
lungs were fixed with PAF under a con-stant pressure of 20 cmH2O. Lungs wereembedded in paraffin and 4-μm sectionswere used to determine radial saccularcount [RSC; using hematoxilin-eosinstain (H&E)], epithelial differentiation[IHC for clara cell secretory protein(CCSP) and prosurfactant protein C (SP-C) and determination of glycogen-content using periodic acid-Schiff stain(PAS)] and medial arterial thickness(using Weigert stain).
RSC was estimated according toEmery and Mithal adapted method (25),at 100× magnification (Olympus BX61microscope; Olympus, Tokyo, Japan), in5 animals per group, 6 slides per animal(200 μm apart each other), and 10 seg-ments per slide, by a blinded examiner(E Carvalho-Dias).
Regarding epithelial differentiation,IHC was performed as previously de-scribed by Nogueira-Silva et al. (7),briefly described above. CCSP antibody[07-623; Upstate (Millipore), Billerica,MA, USA] was used in a 1:800 dilutionand SP-C antibody (AB3428; ChemiconInternational, Temecula, CA, USA) in a1:400 dilution. These slides and PASstained slides were observed and pho-tographed (Olympus BX61 microscope).The percentage of CCSP, SP-C and PASstained cells per microscopic field wasscored in a single-blinded fashion (100×and 400× magnification, respectively) in6 independent peripheral and 4 central
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 3
Table 1. APGAR-like score.a
Sign Score 0 Score 1 Score 2
Skin color Marked cyanosis Mild cyanosis/ pale PinkBreathing Apnea Irregular or weak breathing Regular breathingSpontaneous motor No movements Weak movements Vigorous movementsactivity
Reactivity to stimulus No response Gasping movements Active movements
aThis score is obtained by assigning a value (0, 1 or 2) to 4 characteristics: skin color, breathing,spontaneous motor activity and reactivity to stimulus. The scores vary between 0 and 8.

areas per section (6 slides per animal,200 μm apart each other, and 5 animalsper each experimental group). Scoringwas as follows: 1, 0–20% cells/field; 2,20–40% cells/field; 3, 40–60% cells/field;4, 60–80% cells/field; 5, 80–100%.
Weigert resorcin fuchsin solutionstains elastic fibbers, and it was used formorphometric assessment of pulmonaryarteries (26). Pulmonary arteries weredistinguished from pulmonary veins onthe basis of the structure and position.Arteries that were approximately round[that is, the longest external diameter(ED, distance between the external elasticlaminae), did not exceed the minimal EDby more than 50%] and had both clearlyvisible external and internal elastic lami-nae were analyzed. As the structuralchanges and pharmacological effects onpulmonary arteries might be vessel sizedependent, we selected for further analy-sis arteries subcategorized into 3 sizes:ED less than 30 μm, ED 30 μm to 50 μm,and ED greater than 50 μm. Then, usingAxionVision Rel. 4.3 (Carl Zeiss, Göttin-gen, Germany), internal area (IA, definedby internal elastic lamina), external area(EA, defined by external elastic lamina)and total area (TA, defined by edge ofthe vascular sheath) were measured. Thepercentage of medial (MA) and adventi-tial arterial area (AA) were calculated ac-cording to the following formulas: MA(%) = [(EA – IA)/EA] × 100; AA (%) =[(TA – EA)/EA] × 100. At least 10 arteriesfor each section were evaluated, 6 sec-tions per animal (200 μm apart eachother), and 3 animals per each experi-mental group (at 400× magnification).
Right Ventricular HypertrophyEvaluation
Hearts (C + S, n = 8; CDH + S, n = 6;CDH + PD, n = 8) were used for rightventricle (RV) and left ventricle (LV) dis-section (LV contains septum). RV and LVwere weighed separately. The ratioRV/LV was determined and used as anindex of right ventricular hypertrophy.
Other hearts (C + S, n = 7; CDH + S, n = 6; CDH + PD, n = 6) were dissected,transversally cut and orientated accord-
ing to short-axis view of the heart, beforefixed in PAF and embedded in paraffin.Four micrometer sections were stainedwith H&E and photographed at 40×magnification (Olympus BX61 micro-scope). The right ventricular wall thick-ness was determined in the short-axisview of the heart, in the maximum dis-tance between the right side of the inter-ventricular septum to the right ventricu-lar free wall, using AxionVision Rel. 4.3(Carl Zeiss) (5 measurements per animal,20 μm apart each other).
q-PCR StudiesRight ventricular mRNA expression of
angiotensinogen, B-type natriuretic pep-tide (BNP) and endothelin-1 (ET-1),genes previously defined as ventricularoverload markers, was evaluated accord-ing to Baptista et al. (27).
Statistical AnalysisData are presented as mean ± SEM.
Statistical analysis was performed usingthe statistical software SigmaStat (ver-sion 3.5; Systat Software Inc., Chicago,IL, USA). Multiple group comparisonswere made by analysis of variance. Formorphometric explants studies, Westernblot analysis, biochemical studies, histo-logical studies, right ventricular hyper-trophy evaluation and q-PCR studies,one-way ANOVA on ranks was used.Two-way ANOVA on ranks was used fororgan-to-body weight ratio analysis andgasometric studies. Student-Newman-Keuls test was used for posttest analysis.Statistical significance was confirmed atP < 0.05.
All supplementary materials are availableonline at www.molmed.org.
RESULTS
RAS Components Expression Patternduring Fetal Lung Development
The IHC studies revealed that all RAScomponents, renin, ACE, angiotensino-gen, AT1 and AT2 receptors, were ex-pressed throughout all studied gesta-tional ages in the fetal lung (Figure 1).
Renin was expressed in bronchiolar andalso in alveolar epithelium throughoutgestation (Figure 1A) since 13.5 dpc and appears to be maximal in the mostimmature buds (see Supplemental Fig-ures 1A-D). Endothelial ACE expressionstarted in larger proximal vessels, earlyin the gestation, and spreads distally toinvolve progressively smaller vessels(Figure 1B). Interestingly, ACE proteinwas also detectable in epithelial cells since13.5 dpc (see Supplemental Figure 1E).Regarding angiotensinogen, protein ex-pression was clearly observed in epithe-lial cells since 15.5 dpc (Figure 1C). Mes-enchymal tissue also displayed scatteredangiotensinogen positive cells, first inendothelial cells and in the later gesta-tional ages (19.5 and 21.5 dpc) clearly invascular smooth muscle cells (Figure 1C).AT1 receptor protein was first mainly ex-pressed by undifferentiated mes-enchyme at 15.5 dpc and, throughoutthe gestation, it was predominantly ex-pressed by vascular smooth muscle cellsand scattered in the mesenchyme (Fig-ure 1D). AT2 receptor was expressed inbronchial epithelial cells and since 19.5dpc muscle cells of large blood vesselsalso expressed it (Figure 1E). Moreover,the epithelial AT2 expression was alsodemonstrated at 13.5 dpc (see Supple-mental Figure 1F).
Role of ANGII in Fetal LungDevelopment
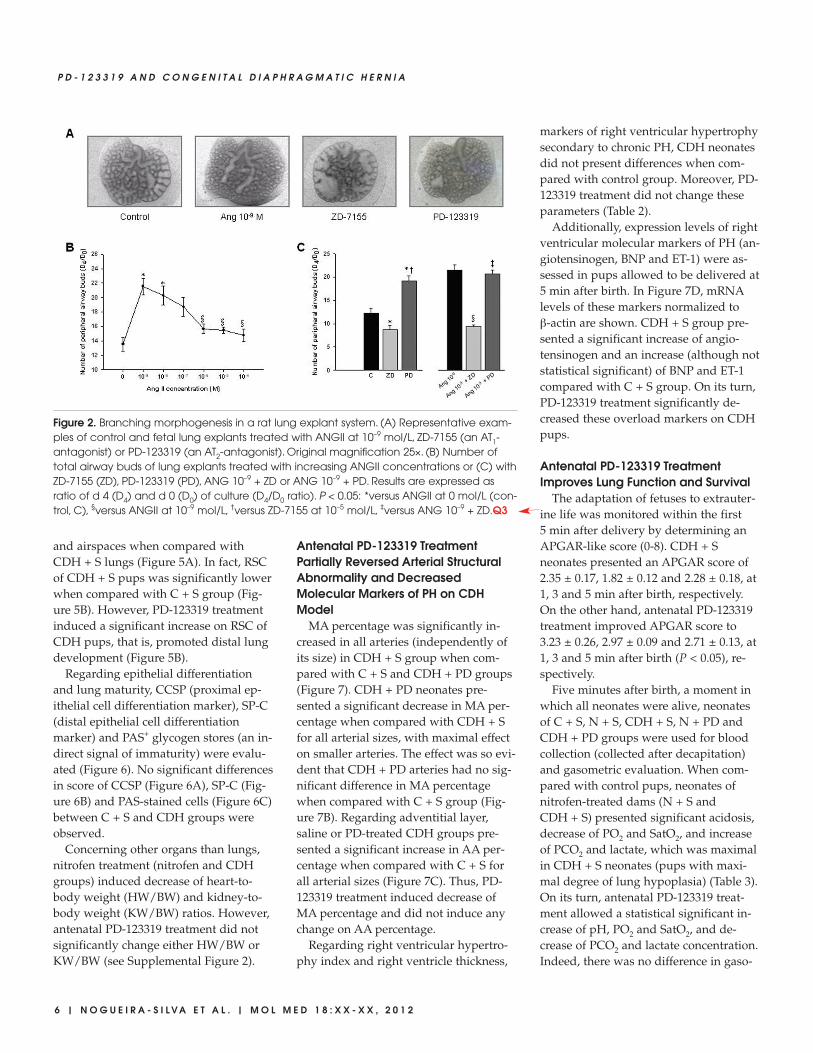
The presence of renin, ACE, angio -tensinogen and AT1 and AT2 protein on lung during fetal developmentprompted us to evaluate the role ofANGII, the physiologically active pep-tide of RAS, on lung morphogenesis. Forthis purpose, fetal lung explants weretreated with increasing concentrations ofrecombinant ANGII (Figure 2A). ANGIIsignificantly increased the number of pe-ripheral airway buds, mainly with con-centration of 10–9 mol/L, the minimalconcentration studied (Figure 2). On theother hand, treatment of lung explantswith an AT1- antagonist (ZD-7155) signif-icantly inhibited, whereas AT2-antago-nist (PD-123319) treatment significantly
4 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A

enhanced lung branching in a similarway to the dose of ANGII-inducingmaximal effect (Figure 2C). Moreover,the stimulatory effect on lung branchinginduced by ANGII 10–9 mol/L was com-pletely abolished by AT1-antagonisttreatment, and the simultaneous lungtreatment with ANG 10–9 mol/L and PD-123319 did not accomplish additionalstimulatory effect on explants growthwhen compared with ANG 10–9 mol/Ltreatment (Figure 2C). Thus, these re-
sults demonstrated that ANGII had astimulatory effect on lung branching,mediated by AT1 receptor. Interestingly,this stimulatory effect was mimicked bytreatment with AT2-antagonist alone.
To clarify the intracellular signalingpathways that mediate ANGII actions onlung growth, lung explants treated withANGII at 10–9 mol/L (selected due to itsmaximal effect in explant growth), ZD-7155 or PD-123319 were evaluatedfor MAPK and Akt pathways activation
(Figure 3A). AT1 receptor blockage in-duced a significant decrease of p-38 andJNK phosphorylation when comparedwith control explants (Figures 3B, D, re-spectively). On the other hand, the in-crease on lung branching, induced byANGII at 10–9 mol/L and AT2 receptorantagonist, significantly stimulatedp44/42 and Akt phosphorylation (Fig-ures 3C, E, respectively).
In Vivo Antenatal PD-123319Treatment Improves Fetal Lung Growth
Left, right and total lung-to-bodyweight ratio (LW/BW) were analyzedfor different experimental groups. Ac-cording to the nitrofen-induced CDHexperimental model, pups of nitrofen-treated dams presented left and rightlung hypoplasia, which was maximal inCDH + S group. Maternal PD-123319treatment induced significant growth ofboth left and right lungs in control, nitrofen and CDH groups. In fact,LW/BW was significantly higher in thecontrol, nitrofen and also CDH ratstreated with PD-123319 when comparedwith the respective saline-treatedgroups (Figure 4A). Indeed, PD-123319treatment stimulated partial recovery oflung hypoplasia in CDH neonates, in-ducing an increase of 11.4% in total lungweight (Figure 4B). Considering theseresults and to assess the potential of PD-123319 as a useful treatment for severelung hypoplasia associated with CDH,we pursued our study focused in com-paring C + S, CDH + S and CDH + PDgroups.
Biochemical analysis of lung proteinand DNA content demonstrated thatthere was no significant difference inthe protein/DNA ratio between C + S,CDH + S and CDH + PD groups (C + S0.024 ± 0.005; CDH + S 0.030 ± 0.004;CDH + PD 0.026 ± 0.002).
The histological analysis of lung archi-tecture showed that CDH + S lungs ap-peared to have a thickened septal andsaccular walls and an increased amountof interstitial tissue when compared withC + S. However, CDH + PD had a signif-icantly greater development of saccules
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 5
Figure 1. Protein expression pattern of RAS components during fetal lung development(from 15.5 until 21.5 dpc). (A) Renin was predominantly expressed in epithelium. (B) ACEexpression. (C) Angiotensinogen expression in epithelial, endothelial (arrow) and vascularsmooth muscle cells (arrowhead). (D) AT1 receptor immunostaining. (E) AT2 receptor im-munostaining. Original magnification 200×.
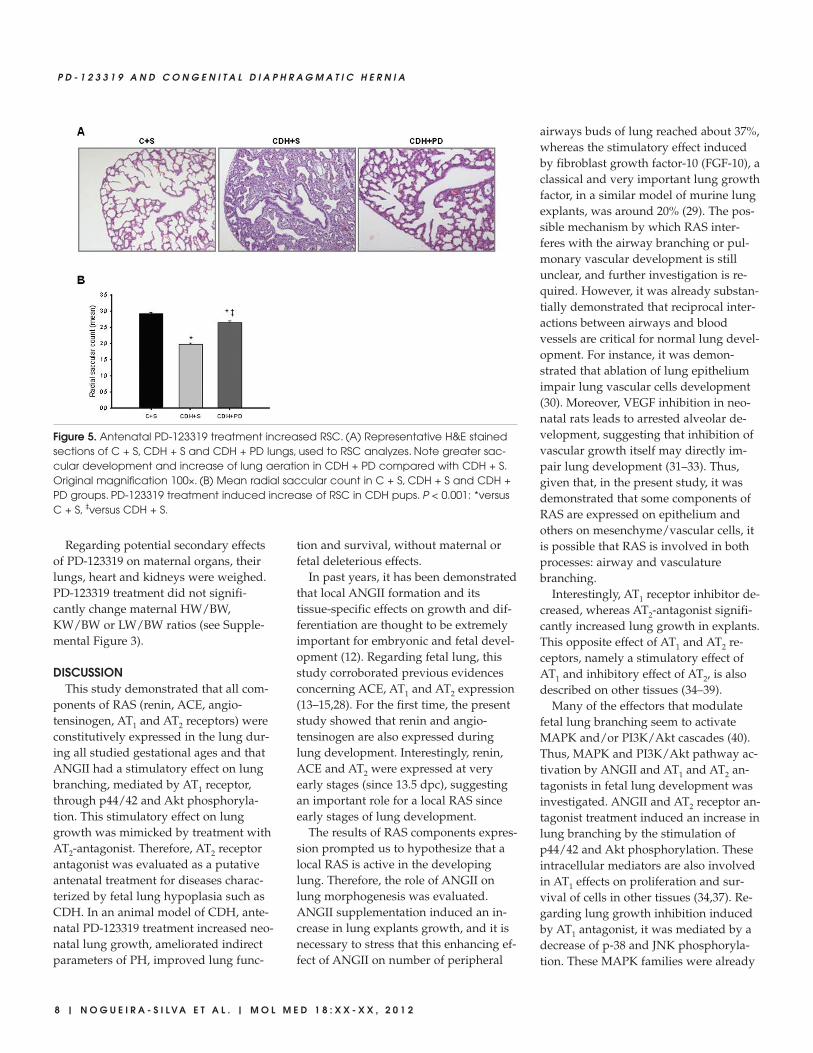
and airspaces when compared withCDH + S lungs (Figure 5A). In fact, RSCof CDH + S pups was significantly lowerwhen compared with C + S group (Fig-ure 5B). However, PD-123319 treatmentinduced a significant increase on RSC ofCDH pups, that is, promoted distal lungdevelopment (Figure 5B).
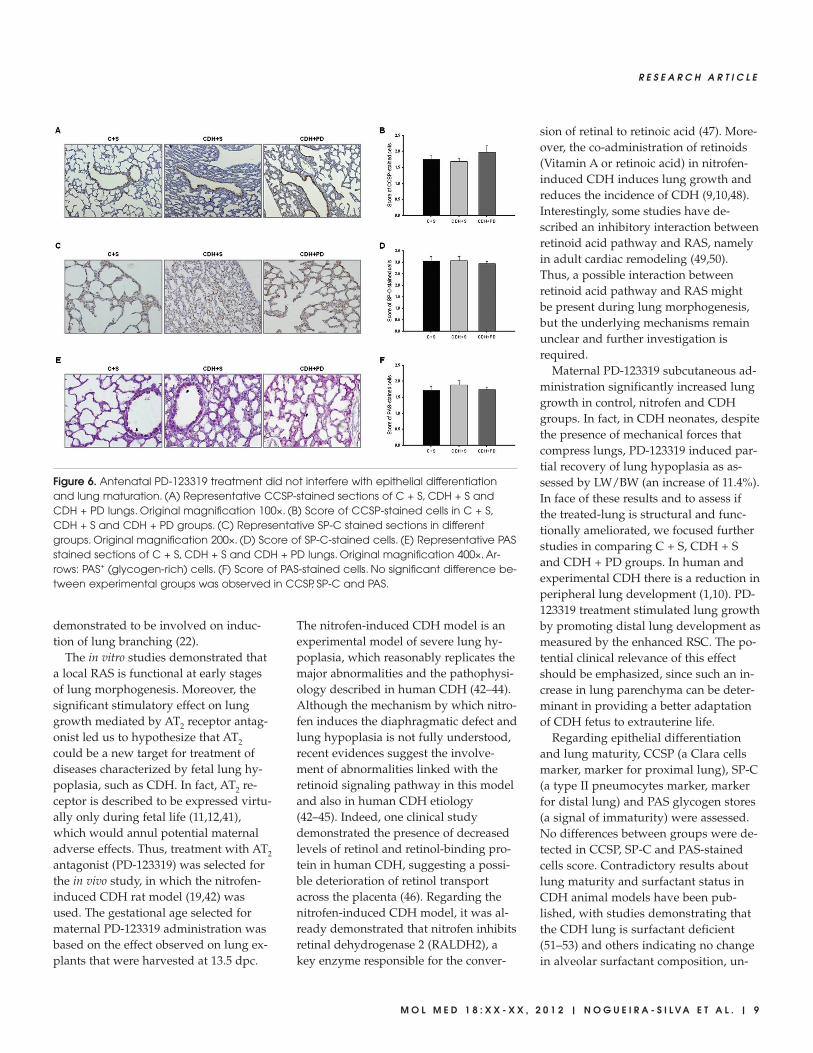
Regarding epithelial differentiationand lung maturity, CCSP (proximal ep-ithelial cell differentiation marker), SP-C(distal epithelial cell differentiationmarker) and PAS+ glycogen stores (an in-direct signal of immaturity) were evalu-ated (Figure 6). No significant differencesin score of CCSP (Figure 6A), SP-C (Fig-ure 6B) and PAS-stained cells (Figure 6C)between C + S and CDH groups wereobserved.
Concerning other organs than lungs,nitrofen treatment (nitrofen and CDHgroups) induced decrease of heart-to-body weight (HW/BW) and kidney-to-body weight (KW/BW) ratios. However,antenatal PD-123319 treatment did notsignificantly change either HW/BW orKW/BW (see Supplemental Figure 2).
Antenatal PD-123319 TreatmentPartially Reversed Arterial StructuralAbnormality and DecreasedMolecular Markers of PH on CDHModel
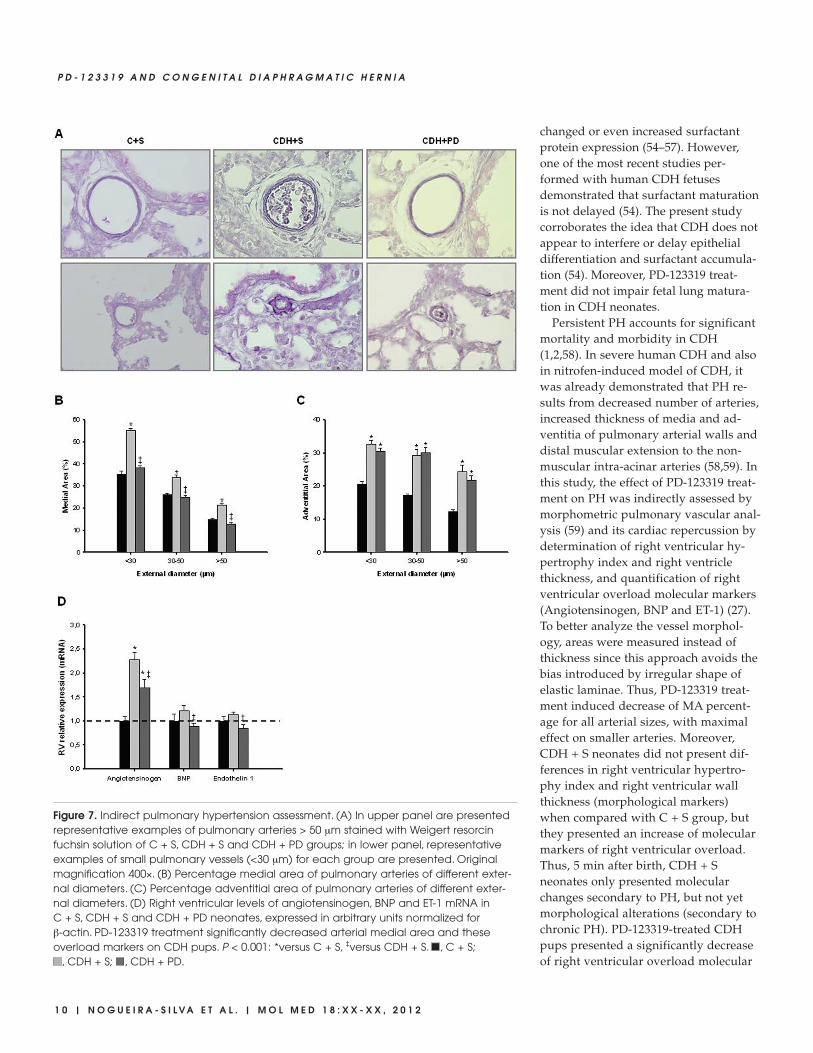
MA percentage was significantly in-creased in all arteries (independently ofits size) in CDH + S group when com-pared with C + S and CDH + PD groups(Figure 7). CDH + PD neonates pre-sented a significant decrease in MA per-centage when compared with CDH + Sfor all arterial sizes, with maximal effecton smaller arteries. The effect was so evi-dent that CDH + PD arteries had no sig-nificant difference in MA percentagewhen compared with C + S group (Fig-ure 7B). Regarding adventitial layer,saline or PD-treated CDH groups pre-sented a significant increase in AA per-centage when compared with C + S forall arterial sizes (Figure 7C). Thus, PD-123319 treatment induced decrease ofMA percentage and did not induce anychange on AA percentage.
Regarding right ventricular hypertro-phy index and right ventricle thickness,
markers of right ventricular hypertrophysecondary to chronic PH, CDH neonatesdid not present differences when com-pared with control group. Moreover, PD-123319 treatment did not change theseparameters (Table 2).
Additionally, expression levels of rightventricular molecular markers of PH (an-giotensinogen, BNP and ET-1) were as-sessed in pups allowed to be delivered at5 min after birth. In Figure 7D, mRNAlevels of these markers normalized toβ-actin are shown. CDH + S group pre-sented a significant increase of angio -tensinogen and an increase (although notstatistical significant) of BNP and ET-1compared with C + S group. On its turn,PD-123319 treatment significantly de-creased these overload markers on CDHpups.
Antenatal PD-123319 TreatmentImproves Lung Function and Survival
The adaptation of fetuses to extrauter-ine life was monitored within the first5 min after delivery by determining anAPGAR-like score (0-8). CDH + Sneonates presented an APGAR score of2.35 ± 0.17, 1.82 ± 0.12 and 2.28 ± 0.18, at1, 3 and 5 min after birth, respectively.On the other hand, antenatal PD-123319treatment improved APGAR score to3.23 ± 0.26, 2.97 ± 0.09 and 2.71 ± 0.13, at1, 3 and 5 min after birth (P < 0.05), re-spectively.
Five minutes after birth, a moment inwhich all neonates were alive, neonatesof C + S, N + S, CDH + S, N + PD andCDH + PD groups were used for bloodcollection (collected after decapitation)and gasometric evaluation. When com-pared with control pups, neonates of nitrofen-treated dams (N + S and CDH + S) presented significant acidosis,decrease of PO2 and SatO2, and increaseof PCO2 and lactate, which was maximalin CDH + S neonates (pups with maxi-mal degree of lung hypoplasia) (Table 3).On its turn, antenatal PD-123319 treat-ment allowed a statistical significant in-crease of pH, PO2 and SatO2, and de-crease of PCO2 and lactate concentration.Indeed, there was no difference in gaso-
6 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A
Figure 2. Branching morphogenesis in a rat lung explant system. (A) Representative exam-ples of control and fetal lung explants treated with ANGII at 10–9 mol/L, ZD-7155 (an AT1- antagonist) or PD-123319 (an AT2-antagonist). Original magnification 25×. (B) Number oftotal airway buds of lung explants treated with increasing ANGII concentrations or (C) withZD-7155 (ZD), PD-123319 (PD), ANG 10–9 + ZD or ANG 10–9 + PD. Results are expressed asratio of d 4 (D4) and d 0 (D0) of culture (D4/D0 ratio). P < 0.05: *versus ANGII at 0 mol/L (con-trol, C), §versus ANGII at 10–9 mol/L, †versus ZD-7155 at 10–5 mol/L, ‡versus ANG 10–9 + ZD.Q3

metric parameters between control, nitro-fen, and CDH groups treated with PD-123319 (Table 3).
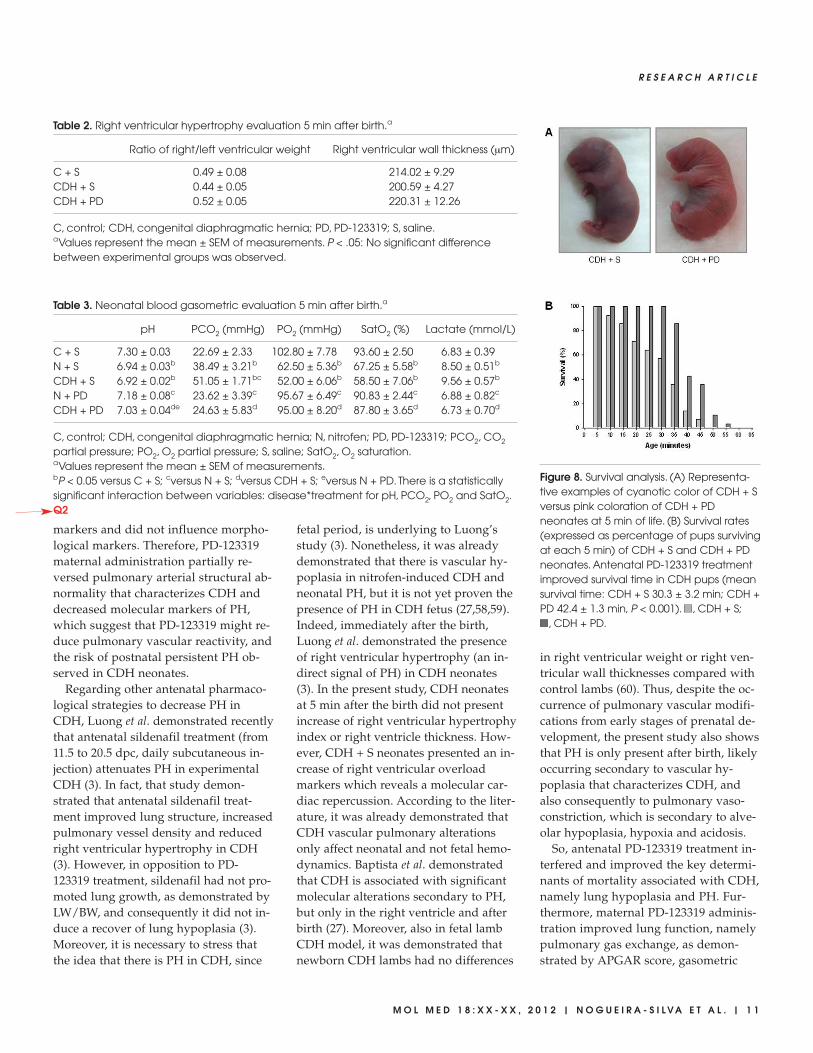
Concerning neonatal survival, the aver-age survival time was significantly longerin PD-123319-treated pups than in CDH +S pups (30.3 ± 3.2 min for CDH + S versus42.4 ± 1.3 min for CDH + PD, P < 0.001).As a result, all CDH + PD pups, but only57% of the CDH + S pups, survived forup to 30 min. At 45 min after birth, only7% of CDH + S pups were alive, as op-posed to over a third of CDH + PD pups.In fact, PD-123319 treatment increasedsurvival rates of CDH neonates for alltime points evaluated (Figure 8).
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 7
Figure 3. MAPK and Akt kinase activities in control (C) lung explants and treated withANGII at 10–9 mol/L (Ang 10–9), ZD-7155 (ZD) or PD-123319 (PD). (A) Western blot analysisof p38, p44/42, JNK1/2 and Akt and to diphosphorylated forms of p38 (dp-p38), p44/42 (dp-p44/42), SAPK/JNK (dp-JNK1/2) and Akt (dp-Akt). Control loading was performedusing β-tubulin (55 kDa). p38 corresponds to 38 kDa. p44/42 correspond to 44 and 42kDa, respectively. JNK1 and 2 correspond to 46 and 54 kDa, respectively. Akt corre-sponds to 56 kDa. Semi-quantitative analysis for dp-p38 (B), dp-p44/42 (C), dp-JNK1/2(D), and dp-Akt (E). Results are presented as arbitrary units normalized for β-tubulin. P <0.05: *versus ANGII at 0 mol/L (control, C), §versus ANGII at 10–9 mol/L, †versus ZD-7155 at10–5 mol/L.
Figure 4. In vivo antenatal PD-123319treatment effects on lung growth. (A) Ratioof left, right and total lung-to-body weightin control (C), nitrofen (N) and CDH groupstreated with saline (C + S, N + S, CDH + S)or PD-123319 (C + PD, N + PD, CDH + PD).Antenatal administration of PD-123319 en-hanced lung growth in all studied groups.(B) Effect of PD-123319 treatment on left,right and total lung hypoplasia. Prenataladministration of PD-123319 amelioratedboth left and right lung hypoplasia. Resultsare expressed as %. P < 0.001: *versus C + S,†versus N + S, ‡versus CDH + S, llversus C +PD, §versus N + PD. For left lung, there is astatistically significant interaction betweenvariables: disease*treatment (P = 0.009).
Regarding potential secondary effectsof PD-123319 on maternal organs, theirlungs, heart and kidneys were weighed.PD-123319 treatment did not signifi-cantly change maternal HW/BW,KW/BW or LW/BW ratios (see Supple-mental Figure 3).
DISCUSSIONThis study demonstrated that all com-
ponents of RAS (renin, ACE, angio -tensinogen, AT1 and AT2 receptors) wereconstitutively expressed in the lung dur-ing all studied gestational ages and thatANGII had a stimulatory effect on lungbranching, mediated by AT1 receptor,through p44/42 and Akt phosphoryla-tion. This stimulatory effect on lunggrowth was mimicked by treatment withAT2-antagonist. Therefore, AT2 receptorantagonist was evaluated as a putativeantenatal treatment for diseases charac-terized by fetal lung hypoplasia such asCDH. In an animal model of CDH, ante-natal PD-123319 treatment increased neo-natal lung growth, ameliorated indirectparameters of PH, improved lung func-
tion and survival, without maternal orfetal deleterious effects.
In past years, it has been demonstratedthat local ANGII formation and its tissue-specific effects on growth and dif-ferentiation are thought to be extremelyimportant for embryonic and fetal devel-opment (12). Regarding fetal lung, thisstudy corroborated previous evidencesconcerning ACE, AT1 and AT2 expression(13–15,28). For the first time, the presentstudy showed that renin and angio -tensinogen are also expressed duringlung development. Interestingly, renin,ACE and AT2 were expressed at veryearly stages (since 13.5 dpc), suggestingan important role for a local RAS sinceearly stages of lung development.
The results of RAS components expres-sion prompted us to hypothesize that alocal RAS is active in the developinglung. Therefore, the role of ANGII onlung morphogenesis was evaluated.ANGII supplementation induced an in-crease in lung explants growth, and it isnecessary to stress that this enhancing ef-fect of ANGII on number of peripheral
airways buds of lung reached about 37%,whereas the stimulatory effect inducedby fibroblast growth factor-10 (FGF-10), aclassical and very important lung growthfactor, in a similar model of murine lungexplants, was around 20% (29). The pos-sible mechanism by which RAS inter-feres with the airway branching or pul-monary vascular development is stillunclear, and further investigation is re-quired. However, it was already substan-tially demonstrated that reciprocal inter-actions between airways and bloodvessels are critical for normal lung devel-opment. For instance, it was demon-strated that ablation of lung epitheliumimpair lung vascular cells development(30). Moreover, VEGF inhibition in neo-natal rats leads to arrested alveolar de-velopment, suggesting that inhibition ofvascular growth itself may directly im-pair lung development (31–33). Thus,given that, in the present study, it wasdemonstrated that some components ofRAS are expressed on epithelium andothers on mesenchyme/vascular cells, itis possible that RAS is involved in bothprocesses: airway and vasculaturebranching.
Interestingly, AT1 receptor inhibitor de-creased, whereas AT2-antagonist signifi-cantly increased lung growth in explants.This opposite effect of AT1 and AT2 re-ceptors, namely a stimulatory effect ofAT1 and inhibitory effect of AT2, is alsodescribed on other tissues (34–39).
Many of the effectors that modulatefetal lung branching seem to activateMAPK and/or PI3K/Akt cascades (40).Thus, MAPK and PI3K/Akt pathway ac-tivation by ANGII and AT1 and AT2 an-tagonists in fetal lung development wasinvestigated. ANGII and AT2 receptor an-tagonist treatment induced an increase inlung branching by the stimulation ofp44/42 and Akt phosphorylation. Theseintracellular mediators are also involvedin AT1 effects on proliferation and sur-vival of cells in other tissues (34,37). Re-garding lung growth inhibition inducedby AT1 antagonist, it was mediated by adecrease of p-38 and JNK phosphoryla-tion. These MAPK families were already
8 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A
Figure 5. Antenatal PD-123319 treatment increased RSC. (A) Representative H&E stainedsections of C + S, CDH + S and CDH + PD lungs, used to RSC analyzes. Note greater sac-cular development and increase of lung aeration in CDH + PD compared with CDH + S.Original magnification 100×. (B) Mean radial saccular count in C + S, CDH + S and CDH +PD groups. PD-123319 treatment induced increase of RSC in CDH pups. P < 0.001: *versusC + S, ‡versus CDH + S.
demonstrated to be involved on induc-tion of lung branching (22).
The in vitro studies demonstrated thata local RAS is functional at early stagesof lung morphogenesis. Moreover, thesignificant stimulatory effect on lunggrowth mediated by AT2 receptor antag-onist led us to hypothesize that AT2
could be a new target for treatment ofdiseases characterized by fetal lung hy-poplasia, such as CDH. In fact, AT2 re-ceptor is described to be expressed virtu-ally only during fetal life (11,12,41),which would annul potential maternaladverse effects. Thus, treatment with AT2
antagonist (PD-123319) was selected forthe in vivo study, in which the nitrofen-induced CDH rat model (19,42) wasused. The gestational age selected formaternal PD-123319 administration wasbased on the effect observed on lung ex-plants that were harvested at 13.5 dpc.
The nitrofen-induced CDH model is anexperimental model of severe lung hy-poplasia, which reasonably replicates themajor abnormalities and the pathophysi-ology described in human CDH (42–44).Although the mechanism by which nitro-fen induces the diaphragmatic defect andlung hypoplasia is not fully understood,recent evidences suggest the involve-ment of abnormalities linked with theretinoid signaling pathway in this modeland also in human CDH etiology(42–45). Indeed, one clinical studydemonstrated the presence of decreasedlevels of retinol and retinol-binding pro-tein in human CDH, suggesting a possi-ble deterioration of retinol transportacross the placenta (46). Regarding thenitrofen-induced CDH model, it was al-ready demonstrated that nitrofen inhibitsretinal dehydrogenase 2 (RALDH2), akey enzyme responsible for the conver-
sion of retinal to retinoic acid (47). More-over, the co-administration of retinoids(Vitamin A or retinoic acid) in nitrofen-induced CDH induces lung growth andreduces the incidence of CDH (9,10,48).Interestingly, some studies have de-scribed an inhibitory interaction betweenretinoid acid pathway and RAS, namelyin adult cardiac remodeling (49,50).Thus, a possible interaction betweenretinoid acid pathway and RAS might be present during lung morphogenesis,but the underlying mechanisms remainunclear and further investigation is required.
Maternal PD-123319 subcutaneous ad-ministration significantly increased lunggrowth in control, nitrofen and CDHgroups. In fact, in CDH neonates, despitethe presence of mechanical forces thatcompress lungs, PD-123319 induced par-tial recovery of lung hypoplasia as as-sessed by LW/BW (an increase of 11.4%).In face of these results and to assess ifthe treated-lung is structural and func-tionally ameliorated, we focused furtherstudies in comparing C + S, CDH + Sand CDH + PD groups. In human andexperimental CDH there is a reduction inperipheral lung development (1,10). PD-123319 treatment stimulated lung growthby promoting distal lung development asmeasured by the enhanced RSC. The po-tential clinical relevance of this effectshould be emphasized, since such an in-crease in lung parenchyma can be deter-minant in providing a better adaptationof CDH fetus to extrauterine life.
Regarding epithelial differentiationand lung maturity, CCSP (a Clara cellsmarker, marker for proximal lung), SP-C(a type II pneumocytes marker, markerfor distal lung) and PAS glycogen stores(a signal of immaturity) were assessed.No differences between groups were de-tected in CCSP, SP-C and PAS-stainedcells score. Contradictory results aboutlung maturity and surfactant status inCDH animal models have been pub-lished, with studies demonstrating thatthe CDH lung is surfactant deficient(51–53) and others indicating no changein alveolar surfactant composition, un-
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 9
Figure 6. Antenatal PD-123319 treatment did not interfere with epithelial differentiationand lung maturation. (A) Representative CCSP-stained sections of C + S, CDH + S andCDH + PD lungs. Original magnification 100×. (B) Score of CCSP-stained cells in C + S,CDH + S and CDH + PD groups. (C) Representative SP-C stained sections in differentgroups. Original magnification 200×. (D) Score of SP-C-stained cells. (E) Representative PASstained sections of C + S, CDH + S and CDH + PD lungs. Original magnification 400×. Ar-rows: PAS+ (glycogen-rich) cells. (F) Score of PAS-stained cells. No significant difference be-tween experimental groups was observed in CCSP, SP-C and PAS.
changed or even increased surfactantprotein expression (54–57). However,one of the most recent studies per-formed with human CDH fetusesdemonstrated that surfactant maturationis not delayed (54). The present studycorroborates the idea that CDH does notappear to interfere or delay epithelialdifferentiation and surfactant accumula-tion (54). Moreover, PD-123319 treat-ment did not impair fetal lung matura-tion in CDH neonates.
Persistent PH accounts for significantmortality and morbidity in CDH(1,2,58). In severe human CDH and alsoin nitrofen-induced model of CDH, itwas already demonstrated that PH re-sults from decreased number of arteries,increased thickness of media and ad-ventitia of pulmonary arterial walls anddistal muscular extension to the non-muscular intra-acinar arteries (58,59). Inthis study, the effect of PD-123319 treat-ment on PH was indirectly assessed bymorphometric pulmonary vascular anal-ysis (59) and its cardiac repercussion bydetermination of right ventricular hy-pertrophy index and right ventriclethickness, and quantification of rightventricular overload molecular markers(Angiotensinogen, BNP and ET-1) (27).To better analyze the vessel morphol-ogy, areas were measured instead ofthickness since this approach avoids thebias introduced by irregular shape ofelastic laminae. Thus, PD-123319 treat-ment induced decrease of MA percent-age for all arterial sizes, with maximaleffect on smaller arteries. Moreover,CDH + S neonates did not present dif-ferences in right ventricular hypertro-phy index and right ventricular wallthickness (morphological markers)when compared with C + S group, butthey presented an increase of molecularmarkers of right ventricular overload.Thus, 5 min after birth, CDH + Sneonates only presented molecularchanges secondary to PH, but not yetmorphological alterations (secondary tochronic PH). PD-123319-treated CDHpups presented a significantly decreaseof right ventricular overload molecular
1 0 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A
Figure 7. Indirect pulmonary hypertension assessment. (A) In upper panel are presentedrepresentative examples of pulmonary arteries > 50 μm stained with Weigert resorcinfuchsin solution of C + S, CDH + S and CDH + PD groups; in lower panel, representativeexamples of small pulmonary vessels (<30 μm) for each group are presented. Originalmagnification 400×. (B) Percentage medial area of pulmonary arteries of different exter-nal diameters. (C) Percentage adventitial area of pulmonary arteries of different exter-nal diameters. (D) Right ventricular levels of angiotensinogen, BNP and ET-1 mRNA in C + S, CDH + S and CDH + PD neonates, expressed in arbitrary units normalized for β-actin. PD-123319 treatment significantly decreased arterial medial area and theseoverload markers on CDH pups. P < 0.001: *versus C + S, ‡versus CDH + S. , C + S;
, CDH + S; , CDH + PD.
markers and did not influence morpho-logical markers. Therefore, PD-123319maternal administration partially re-versed pulmonary arterial structural ab-normality that characterizes CDH anddecreased molecular markers of PH,which suggest that PD-123319 might re-duce pulmonary vascular reactivity, andthe risk of postnatal persistent PH ob-served in CDH neonates.
Regarding other antenatal pharmaco-logical strategies to decrease PH inCDH, Luong et al. demonstrated recentlythat antenatal sildenafil treatment (from11.5 to 20.5 dpc, daily subcutaneous in-jection) attenuates PH in experimentalCDH (3). In fact, that study demon-strated that antenatal sildenafil treat-ment improved lung structure, increasedpulmonary vessel density and reducedright ventricular hypertrophy in CDH(3). However, in opposition to PD-123319 treatment, sildenafil had not pro-moted lung growth, as demonstrated byLW/BW, and consequently it did not in-duce a recover of lung hypoplasia (3).Moreover, it is necessary to stress thatthe idea that there is PH in CDH, since
fetal period, is underlying to Luong’sstudy (3). Nonetheless, it was alreadydemonstrated that there is vascular hy-poplasia in nitrofen-induced CDH andneonatal PH, but it is not yet proven thepresence of PH in CDH fetus (27,58,59).Indeed, immediately after the birth,Luong et al. demonstrated the presenceof right ventricular hypertrophy (an in-direct signal of PH) in CDH neonates(3). In the present study, CDH neonatesat 5 min after the birth did not presentincrease of right ventricular hypertrophyindex or right ventricle thickness. How-ever, CDH + S neonates presented an in-crease of right ventricular overloadmarkers which reveals a molecular car-diac repercussion. According to the liter-ature, it was already demonstrated thatCDH vascular pulmonary alterationsonly affect neonatal and not fetal hemo-dynamics. Baptista et al. demonstratedthat CDH is associated with significantmolecular alterations secondary to PH,but only in the right ventricle and afterbirth (27). Moreover, also in fetal lambCDH model, it was demonstrated thatnewborn CDH lambs had no differences
in right ventricular weight or right ven-tricular wall thicknesses compared withcontrol lambs (60). Thus, despite the oc-currence of pulmonary vascular modifi-cations from early stages of prenatal de-velopment, the present study also showsthat PH is only present after birth, likelyoccurring secondary to vascular hy-poplasia that characterizes CDH, andalso consequently to pulmonary vaso-constriction, which is secondary to alve-olar hypoplasia, hypoxia and acidosis.
So, antenatal PD-123319 treatment in-terfered and improved the key determi-nants of mortality associated with CDH,namely lung hypoplasia and PH. Fur-thermore, maternal PD-123319 adminis-tration improved lung function, namelypulmonary gas exchange, as demon-strated by APGAR score, gasometric
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 1 1
Figure 8. Survival analysis. (A) Representa-tive examples of cyanotic color of CDH + Sversus pink coloration of CDH + PDneonates at 5 min of life. (B) Survival rates(expressed as percentage of pups survivingat each 5 min) of CDH + S and CDH + PDneonates. Antenatal PD-123319 treatmentimproved survival time in CDH pups (meansurvival time: CDH + S 30.3 ± 3.2 min; CDH +PD 42.4 ± 1.3 min, P < 0.001). , CDH + S;
, CDH + PD.
Table 2. Right ventricular hypertrophy evaluation 5 min after birth.a
Ratio of right/left ventricular weight Right ventricular wall thickness (μm)
C + S 0.49 ± 0.08 214.02 ± 9.29CDH + S 0.44 ± 0.05 200.59 ± 4.27CDH + PD 0.52 ± 0.05 220.31 ± 12.26
C, control; CDH, congenital diaphragmatic hernia; PD, PD-123319; S, saline.aValues represent the mean ± SEM of measurements. P < .05: No significant differencebetween experimental groups was observed.
Table 3. Neonatal blood gasometric evaluation 5 min after birth.a
pH PCO2 (mmHg) PO2 (mmHg) SatO2 (%) Lactate (mmol/L)
C + S 7.30 ± 0.03 22.69 ± 2.33 102.80 ± 7.78 93.60 ± 2.50 6.83 ± 0.39N + S 6.94 ± 0.03b 38.49 ± 3.21b 62.50 ± 5.36b 67.25 ± 5.58b 8.50 ± 0.51b
CDH + S 6.92 ± 0.02b 51.05 ± 1.71bc 52.00 ± 6.06b 58.50 ± 7.06b 9.56 ± 0.57b
N + PD 7.18 ± 0.08c 23.62 ± 3.39c 95.67 ± 6.49c 90.83 ± 2.44c 6.88 ± 0.82c
CDH + PD 7.03 ± 0.04de 24.63 ± 5.83d 95.00 ± 8.20d 87.80 ± 3.65d 6.73 ± 0.70d
C, control; CDH, congenital diaphragmatic hernia; N, nitrofen; PD, PD-123319; PCO2, CO2
partial pressure; PO2, O2 partial pressure; S, saline; SatO2, O2 saturation. aValues represent the mean ± SEM of measurements.bP < 0.05 versus C + S; cversus N + S; dversus CDH + S; eversus N + PD. There is a statisticallysignificant interaction between variables: disease*treatment for pH, PCO2, PO2 and SatO2.Q2

and survival evaluation. Regarding gas-ometry it is necessary to stress that dueto low fetal blood volume, all bloodpossibly collected by decapitation wasused for gasometric evaluation. So, gas-ometry evaluated a mix of arterial andvenous blood. Nonetheless, antenatalPD-123319 treatment allowed a statisti-cal significant improvement of acidosis,hypercapnia, hypoxia and lactate con-centration that characterizes CDH fe-tuses. The results of these direct indica-tors of ventilation/perfusion matchingquality suggest an obvious improve-ment of pulmonary gas exchange andperipheral O2 delivery. Furthermore,this enhancement on lung function hadimportant consequences on neonatalsurvival, namely PD-123319 treatmentinduced significantly longer averagesurvival time. However, it is necessaryto stress that the survival evaluationwas performed without neonatal care orventilatory support. This fact might bethe explanation for the death of allneonates, despite the increase on lungfunction and survival time induced byantenatal PD-123319 treatment.
Regarding potential fetal adverse ef-fects, the nitrofen-exposed pups pre-sented decrease of HW/BW andKW/BW ratios as previously docu-mented (10,61–63). On the other hand,PD-123319 beneficial effect seemed lung-specific, since HW/BW and KW/BW ra-tios of the pups were not altered. Con-cerning potential maternal secondaryeffects induced by PD-123319, no differ-ences on heart, kidneys and lungs wereobserved. The absence of maternal dele-terious effects could be due to the fact ofAT2 receptor expression is dramaticallydecreased after birth, being restricted toa few organs (11,12,41). Indeed, an in-crease of AT2 receptor expression duringadult life has been only observed underpathological conditions (41).
CONCLUSIONIn conclusion, this study demonstrated
the existence of a functional local RAS infetal lung. Moreover, it establishes AT2
receptor antagonist (PD-123319) as a pu-
tative antenatal therapy for pathologiescharacterized by fetal lung hypoplasia,such as CDH.
ACKNOWLEDGMENTSThis project was funded by Fundação
para a Ciência e a Tecnologia(PTDC/SAU-OBD/108051/2008) and bySecção de Neonatologia da SociedadePortuguesa de Pediatria (Grant ZERU2008). P Piairo was supported by Fun-dação para a Ciência e a Tecnologia (ref-erence SFRH/BD/33410/2008). RS Moura was supported by Fundaçãopara a Ciência e a Tecnologia (referenceSFRH/BPD/15408/2005). PD-123319was kindly supplied by Medical Divisionof Pfizer Inc, Groton, Connecticut, USA.
We would like to thank to Luís Mar-tins for histological technical supportand help on animal euthanasia and toNuno M Pires for Weigert staining andvascular morphometric analysis support.
DISCLOSUREThe authors declare that they have no
competing interests as defined by Molecu-lar Medicine, or other interests that mightbe perceived to influence the results anddiscussion reported in this paper. Q4
REFERENCES1. van den Hout L, et al. (2009) Can we improve
outcome of congenital diaphragmatic hernia? Pe-diatr. Surg. Int. 25:733–43.
2. Keller RL, et al. (2010) Congenital diaphragmatichernia: endothelin-1, pulmonary hypertension,and disease severity. Am. J. Respir. Crit. Care Med.182:555–61.
3. Luong C, et al. (2011) Antenatal sildenafil treat-ment attenuates pulmonary hypertension in ex-perimental congenital diaphragmatic hernia. Cir-culation. 123:2120–31.
4. Puri P, Wester T. (1997) Historical aspects of con-genital diaphragmatic hernia. Pediatr. Surg. Int.12:95–100.
5. Harrison MR, et al. (2003) A randomized trial offetal endoscopic tracheal occlusion for severefetal congenital diaphragmatic hernia. N. Engl. J.Med. 349:1916–24.
6. Jani JC, et al. (2009) Severe diaphragmatic herniatreated by fetal endoscopic tracheal occlusion.Ultrasound Obstet. Gynecol. 34:304–10.
7. Nogueira-Silva C, Moura RS, Esteves N, Gon-zaga S, Correia-Pinto J. (2008) Intrinsic catch-upgrowth of hypoplastic fetal lungs is mediated byinterleukin-6. Pediatr. Pulmonol. 43:680–9.
8. Santos M, et al. (2006) Ghrelin expression inhuman and rat fetal lungs and the effect of ghre-lin administration in nitrofen-induced congenitaldiaphragmatic hernia. Pediatr. Res. 59:531–7.
9. Baptista MJ, et al. (2005) Antenatal vitamin A ad-ministration attenuates lung hypoplasia by inter-fering with early instead of late determinants oflung underdevelopment in congenital diaphrag-matic hernia. J. Pediatr. Surg. 40:658–65.
10. Thébaud B, et al. (1999) Vitamin A decreases theincidence and severity of nitrofen-induced con-genital diaphragmatic hernia in rats. Am. J. Phys-iol. 277:L423–9.
11. Lavoie JL, Sigmund CD. (2003) Minireview: over-view of the renin-angiotensin system—an en-docrine and paracrine system. Endocrinology.144:2179–83.
12. Paul M, Poyan Mehr A, Kreutz R. (2006) Physiol-ogy of local renin-angiotensin systems. Physiol.Rev. 86:747–803.
13. Goyal R, Leitzke A, Goyal D, Gheorghe CP, LongoLD. (2011) Antenatal maternal hypoxic stress:adaptations in fetal lung renin-angiotensin sys-tem. Reprod. Sci. 18:180–9.
14. Morrell NW, Grieshaber SS, Danilov SM, MajackRA, Stenmark KR. (1996) Developmental regula-tion of angiotensin converting enzyme and an-giotensin type 1 receptor in the rat pulmonarycirculation. Am. J. Respir. Cell Mol. Biol. 14:526–37.
15. Shanmugam S, Corvol P, Gasc JM. (1996) An-giotensin II type 2 receptor mRNA expression inthe developing cardiopulmonary system of therat. Hypertension. 28:91–7.
16. Diário da República, Portaria 1005/92). Q617. World Medical Association [WMA]. (1964) WMA
declaration of Helsinki - ethical principles formedical research involving humans. Lastamended 2008 Oct. [updated c2012; cited 2012Feb 3]. Available at: http://www.wma.net/en/30publications/ 10policies/b3/index.html Q6
18. Guide for the Care and Use of Laboratory Animals.US National Institutes of Health. NIH publica-tion no. 85-23, revised 1996. Q6
19. Tenbrinck R, et al. (1990) Experimentally inducedcongenital diaphragmatic hernia in rats. J. Pedi-atr. Surg. 25:426–9.
20. Eskildsen-Helmond YE, Mulvany MJ. (2003)Pressure-induced activation of extracellular signal-regulated kinase 1/2 in small arteries. Hy-pertension. 41:891–7.
21. Levy BI, et al. (1996) Chronic blockade of AT2-subtype receptors prevents the effect of an-giotensin II on the rat vascular structure. J. Clin.Invest. 98:418–25.
22. Nogueira-Silva C, Santos M, Baptista MJ, MouraRS, Correia-Pinto J. (2006) IL-6 is constitutivelyexpressed during lung morphogenesis and en-hances fetal lung explant branching. Pediatr. Res.60:530–6.
23. Dauger S, et al. (2001) MASH-1/RET pathway in-volvement in development of brain stem controlof respiratory frequency in newborn mice. Phys-iol. Genomics. 7:149–57.
1 2 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A

24. Bradford MM. (1976) A rapid and sensitivemethod for the quantitation of microgram quan-tities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–54.
25. Cooney TP, Thurlbeck WM. (1982) The radialalveolar count method of Emery and Mithal: areappraisal 2 - intrauterine and early postnatallung growth. Thorax. 37:580–3.
26. Pires NM, et al. (2007) Activation of nuclear re-ceptor Nur77 by 6-mercaptopurine protectsagainst neointima formation. Circulation.115:493–500.
27. Baptista MJ, Nogueira-Silva C, Areias JC, Cor-reia-Pinto J. (2008) Perinatal profile of ventricularoverload markers in congenital diaphragmatichernia. J. Pediatr. Surg. 43:627–33.
28. Shanmugam S, Monnot C, Corvol P, Gasc JM.(1994) Distribution of type 1 angiotensin II recep-tor subtype messenger RNAs in the rat fetus. Hy-pertension. 23:137–41.
29. Acosta JM, et al. (2001) Novel mechanisms inmurine nitrofen-induced pulmonary hypoplasia:FGF-10 rescue in culture. Am. J. Physiol. Lung CellMol. Physiol. 281:L250–7.
30. Sarah A, Gebb B, Shannon JM. (2000) Tissue in-teractions mediate early events in pulmonaryvasculogenesis. Dev. Dyn. 217:159–69.
31. Thébaud B, et al. (2005) Vascular endothelialgrowth factor gene therapy increases survival,promotes lung angiogenesis, and prevents alveo-lar damage in hyperoxia-induced lung injury: ev-idence that angiogenesis participates in alveolar-ization. Circulation. 112:2477–86.
32. Jakkula M, et al. (2000) Inhibition of angiogenesisdecreases alveolarization in the developing ratlung. Am. J. Physiol. Lung Cell Mol. Physiol.279:L600–7.
33. Healy AM, Morgenthau L, Zhu X, Farber HW,Cardoso WV. (2000) VEGF is deposited in thesubepithelial matrix at the leading edge ofbranching airways and stimulates neovascular-ization in the murine embryonic lung. Dev. Dyn.21:341–52.
34. Yosypiv IV, El-Dahr SS. (2005) Role of the renin-angiotensin system in the development of theureteric bud and renal collecting system. Pediatr.Nephrol. 20:1219–29.
35. Stoll M, et al. (1995) The angiotensin AT2-receptormediates inhibition of cell proliferation in coro-nary endothelial cells. J. Clin. Invest. 95:651–7.
36. Nakajima M, et al. (1995) The angiotensin II type 2(AT2) receptor antagonizes the growth effects of theAT1 receptor: gain-of-function study using genetransfer. Proc. Natl. Acad. Sci. U. S. A. 92:10663–7.
37. Inagami T, Senbonmatsu T. (2001) Dual effects ofangiotensin II type 2 receptor on cardiovascularhypertrophy. Trends Cardiovasc. Med. 11:324–8.
38. Gyurko R, Kimura B, Kurian P, Crews FT,Phillips MI. (1992) Angiotensin II receptor sub-types play opposite roles in regulating phos-phatidylinositol hydrolysis in rat skin slices.Biochem. Biophys. Res. Commun. 186:285–92.
39. Maric C, Aldred GP, Harris PJ, Alcorn D. (1998)
Angiotensin II inhibits growth of cultured em-bryonic renomedullary interstitial cells throughthe AT2 receptor. Kidney Int. 53:92–9.
40. Kling DE, et al. (2002) Pre- and postnatal lung de-velopment, maturation, and plasticity: MEK-1/2inhibition reduces branching morphogenesis andcauses mesenchymal cell apoptosis in fetal ratlungs. Am. J. Physiol. Lung Cell Mol. Physiol.282:L370–8.
41. Kaschina E, Unger T. (2003) AngiotensinAT1/AT2 receptors: regulation, signalling andfunction. Blood Press. 12:70–88.
42. Kling DE, Schnitzer JJ. (2007) Vitamin A defi-ciency (VAD), teratogenic, and surgical models ofcongenital diaphragmatic hernia (CDH). Am. J.Med. Genet. C Semin. Med. Genet. 145C:139–57.
43. Montedonico S, Nakazawa N, Puri P. (2008) Con-genital diaphragmatic hernia and retinoids:searching for an etiology. Pediatr. Surg. Int.24:755–61.
44. Gallot D, et al. (2005) Congenital diaphragmatichernia: a retinoid-signaling pathway disruptionduring lung development? Birth Defects Res. AClin. Mol. Teratol. 73:523–31.
45. Greer JJ, Babiuk RP, Thebaud B. (2003) Etiologyof congenital diaphragmatic hernia: the retinoidhypothesis. Pediatr. Res. 53:726–30.
46. Major D, et al. (1998) Retinol status of newborninfants with congenital diaphragmatic hernia.Pediatr. Surg. Int. 13:547–9.
47. Mey J, Babiuk RP, Clugston R, Zhang W, Greer JJ.(2003) Retinal dehydrogenase-2 is inhibited bycompounds that induce congenital diaphrag-matic hernias in rodents. Am. J. Pathol. 162:673–9.
48. Babiuk RP, Thebaud B, Greer JJ. (2004) Reductionsin the incidence of nitrofen-induced diaphrag-matic hernia by vitamin A and retinoic acid. Am. J.Physiol. Lung Cell Mol. Physiol. 286:L970–3.
49. Guleria RS, Choudhary R, Tanaka T, Baker KM,Pan J. (2011) Retinoic acid receptor-mediated sig-naling protects cardiomyocytes from hyperglyce-mia induced apoptosis: role of the renin-an-giotensin system. J. Cell Physiol. 226:1292–307.
50. Choudhary R, et al. (2008) All-trans retinoic acidprevents development of cardiac remodeling inaortic banded rats by inhibiting the renin-an-giotensin system. Am. J. Physiol. Heart Circ. Phys-iol. 294:H633–44.
51. Thébaud B, et al. (2001) Restoring effects of vita-min A on surfactant synthesis in nitrofen-inducedcongenital diaphragmatic hernia in rats. Am. J.Respir. Crit. Care Med. 164:1083–9.
52. Keijzer R, Liu J, Deimling J, Tibboel D, Post M.(2000) Dual-hit hypothesis explains pulmonaryhypoplasia in the nitrofen model of congenital di-aphragmatic hernia. Am. J. Pathol. 156:1299–306.
53. Asabe K, Tsuji K, Handa N, Kajiwara M, Suita S.(1998) Expression of clara cell 10-kDa protein(CC10) in congenital diaphragmatic hernia. Pedi-atr. Surg. Int. 14:36–9.
54. Boucherat O, et al. (2007) Surfactant maturationis not delayed in human fetuses with diaphrag-matic hernia. PLoS Med. 4:e237.
55. Van Tuyl M, et al. (2003) Pulmonary surfactantprotein A, B, and C mRNA and protein expres-sion in the nitrofen-induced congenital diaphrag-matic hernia rat model. Pediatr. Res. 54:641–52.
56. Chapin CJ, et al. (2005) Congenital diaphragmatichernia, tracheal occlusion, thyroid transcriptionfactor-1, and fetal pulmonary epithelial matura-tion. Am. J. Physiol. Lung Cell Mol. Physiol.289:L44–52.
57. Santos M, et al. (2007) Pulmonary epithelial celldifferentiation in the nitrofen-induced congenitaldiaphragmatic hernia. J. Pediatr. Surg. 42:1231–7.
58. Mohseni-Bod H, Bohn D. (2007) Pulmonary hy-pertension in congenital diaphragmatic hernia.Semin. Pediatr. Surg. 16:126–33.
59. Kanai M, et al. (2001) Fetal tracheal occlusion inthe rat model of nitrofen-induced congenital di-aphragmatic hernia: tracheal occlusion reversesthe arterial structural abnormality. J. Pediatr.Surg. 36:839–45.
60. Karamanoukian HL, et al. (1995) Pathophysiol-ogy of congenital diaphragmatic hernia. XI:Anatomic and biochemical characterization ofthe heart in the fetal lamb CDH model. J. Pediatr.Surg. 30:925–8.
61. González-Reyes S, Martínez L, Tovar JA. (2005)Effects of prenatal vitamins A, E, and C on thehypoplastic hearts of fetal rats with diaphrag-matic hernia. J. Pediatr. Surg. 40:1269–74.
62. Montedonico S, Nakazawa N, Shinkai T, Banni-gan J, Puri P. (2007) Kidney development in thenitrofen-induced pulmonary hypoplasia andcongenital diaphragmatic hernia in rats. J. Pedi-atr. Surg. 42:239–43.
63. Correia-Pinto J, et al. (2003) Fetal heart develop-ment in the nitrofen-induced CDH rat model:the role of mechanical and nonmechanical fac-tors. J. Pediatr. Surg. 38:1444–51.
R E S E A R C H A R T I C L E
M O L M E D 1 8 : X X - X X , 2 0 1 2 | N O G U E I R A - S I L V A E T A L . | 1 3

QueriesQ1. Correct this is twenty-five hundred?Q2: What does the asterisk indicate?Q3: Please provide a new panel B
with clear superscript nos.Q4: Please confirm that this disclo-
sure statement is correct unless youhave something to declare. If you dohave something to declare, please pro-vide that information.
Q5: Is this an abbreviated format of alonger name: ICVS/3B’s - PT?
Q6: The three items mentioned in theparagraph on p. 2 have been includedin the reference list. Please do the fol-lowing:
a. For new refs. 16 and 18, give fullstandard bibliographic information. Forref. 18, is the item at the following web-site what was used?http://www.nap.edu/openbook.php?record_id=5140
Or was it a separate NIH publication?
b. For new ref. 17, review what wehave done; access the URL and changethe date to the date you do so.
c. Check the renumbering of the ref-erences throughout the paper includingthe reference list.
QA: Please italicize any gene namesand gene symbols, but not gene de-scriptions. Please review the entirepaper for gene symbols and names.Also, indicate any symbols and wordsthat are currently italicized as genes,but that are not genes. We should un-italicize those items.
QB: Your supplemental file(s) havebeen formatted in the accompanyingdocument. Please proof it carefully toensure that everything is correct. Also,avoid making any changes to the sup-plemental document unless there is anactual error in information.
1 4 | N O G U E I R A - S I L V A E T A L . | M O L M E D 1 8 : X X - X X , 2 0 1 2
P D - 1 2 3 3 1 9 A N D C O N G E N I T A L D I A P H R A G M A T I C H E R N I A