hematology& renal disease
TRANSCRIPT

Hematology& Renal disease
Dr MANAL BESSA,MD Hematology
Alexandria University

Outlines Anemia of CKDErythrocytosis in renal disease.Hemostasis and thrombosis in renal diseases,TTP and HUS.

How is anemia related to chronic kidney disease (CKD)?
Anemia commonly occurs in people with CKD.
Anemia might begin to develop in the early
stages of CKD, when someone has 20 to 50
percent of normal kidney function.
Anemia tends to worsen as CKD progresses.
Most people who have ESRD have anemia.

Erythropoiesis, role of the kidneys
www.tarleton.edu/Departments/anatomy/erythro.jpg

Erythropoiesis Failure in Renal Disease
Adapted from http://www.anemia.org/professionals/monograph/images/diabetes_kidney.gif

What are the implications of anemia in CKD patients?
FatigueDiminished quality of lifeImpaired cognitive functionCardiovascular effects:
Leading cause of death in adult population.Anemia correction has shown to decrease LV mass index.

Increased Mortality
North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) database from 1992 to 2001:
Children with an Hgb level < 9.9 g/dL versus those with an Hgb level > 9.9 g/dL showed an elevated risk for mortality: adjusted RR 1.52 (95% CI, 1.03 to 2.26; P < 0.05).

Etiology
Decreased renal synthesis of erythropoietin. Blood loss due to increased bleeding tendency due to uremia-induced platelet dysfunction.Iron deficiency: may be absolute, often due to poor dietary intake or sometimes occult bleeding, or functional ida Other causes for anemia in CKD (eg, parathyroid hormone, inflammatory cytokines) that suppress erythopoeisis and deficiencies of folate or vitamin B12
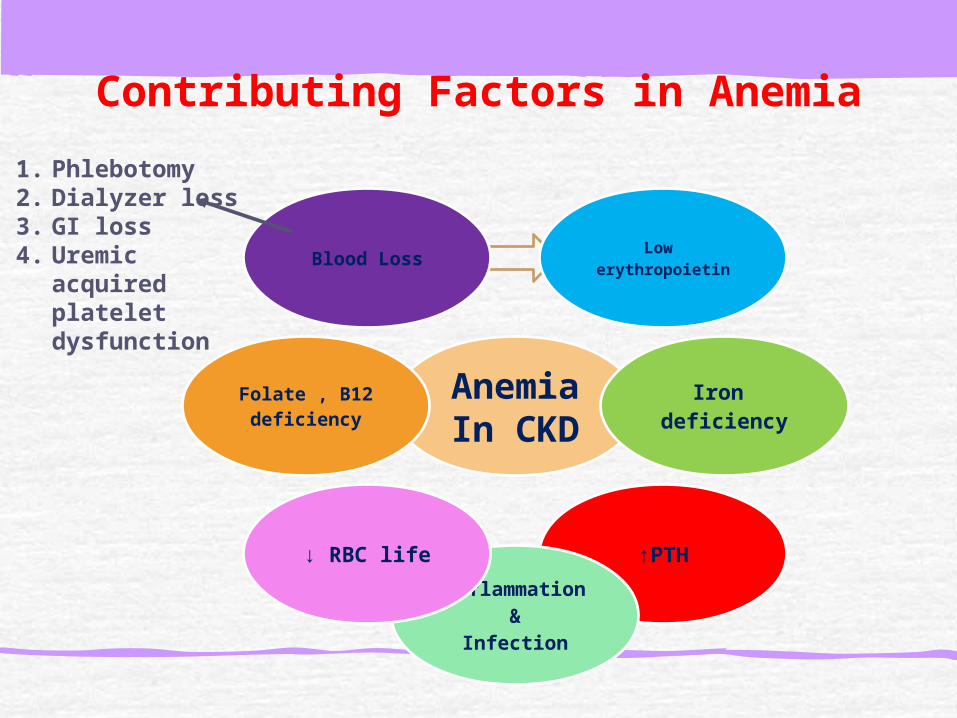
Contributing Factors in Anemia
AnemiaIn CKD
Low erythropoietin
Iron deficiency
↑PTH
Inflammation&
Infection
↓ RBC life
Folate , B12deficiency
Blood Loss
1. Phlebotomy2. Dialyzer loss3. GI loss4. Uremic
acquired platelet dysfunction

Other Potential Contributing Factors
Vit E deficiency Malnutrition ↔ surrogate marker of inflammation or caused by uremia. Medications.Increased inhibitory molecules: lead, aluminum toxicityBone marrow suppression.

Investigations
This will involve :ruling out other causes of anemia, assessment of kidney function, assessment of any cardiovascular and other complications of anemia or CKD.

Investigations
FBCHbWCCPlateletsMCVRC Htc
B12 level
Folate level
Iron studies
Iron
Ferritin
Transferrin
Transferritin
saturation (TSAT)
CRP
Inflammatory
marker

ManagementManagement of anemia should be considered in people with anemia of CKD when the hemoglobin level is less than or equal to 11 g/dL (or 10 g/dL if under 2 years of age).In people with anemia of CKD, treatment should aim to maintain stable hemoglobin levels between 10 and 12 g/dL for adults and children aged over 2 years and between 9.5 and 11.5 g/dL in children aged under 2 years

Treatment with erythropoiesis-stimulating agents ESAs should be offered to patients with anemia of CKD who are likely to benefit in terms of quality of life and physical function.
The time taken for erythropoetin treatment to be effective will depend on individual patient factors, such as degree of anemia, degree of kidney disease and presence of other adverse factors - eg, iron deficiency.
Other factors which contribute to the anemia of CKD (eg, iron or folate deficiency) should be corrected before treatment and monitored during therapy.
Aluminium toxicity, concurrent infection or other inflammatory disease may impair the response to erythropoietin

People receiving ESAs maintenance therapy should be given iron supplements (often requires intravenous iron) to keep their Serum ferritin between 200 and 500 μg/L and either:
The transferrin saturation level above 20% (unless ferritin is >800 μg/L); orPercentage hypochromic red cells less than 6% (unless ferritin is >800 μg/L).

Clinically relevant hyperparathyroidism should be treated in order to improve anemia management in patients with anemia of CKD.Where possible, blood transfusions should be avoided in patients in whom kidney transplant is a treatment option

erythropoiesis-stimulating agents ESA
Epoetin :recombinant human erythropoietin , used for the anemia associated with erythropoietin deficiency in CKD. 2 types alfa and beta of similar clinical efficacy.
Darbepoetin:Is a hyperglycosylated derivative of epoetin which has a longer half-life and may be administered less frequently than epoetin.
Methoxy polyethylene glycol-epoetin beta:Is a continuous erythropoietin receptor activator. It is licensed for the treatment of anemia associated with CKD in patients who are symptomatic. It has a longer duration of action than epoetin.

Potential side-effects of ESAs include:
increase in blood pressure or aggravation of hypertension, headache, increase in platelet count, influenza-like symptoms (may be reduced if intravenous injection is given over five minutes), thromboembolic events, pure red cell aplasia,hyperkalaemia and skin reactions.

Monitoring
In anemia of CKD, hemoglobin should be monitored:
Every 2-4 weeks in the induction phase of ESAs therapy.Every 1-3 months in the maintenance phase of ESAs therapy.More actively after dose adjustment of ESAs.


Erythrocytosis Tumor associated erythrocytosis: due to ectopic EPO production as renal cell carcinoma with good response to successful treatment of the tumor.Renal diseases:
Renal cystHydronephrosisNephrotic syndromeDiffuse renal parynchymal diseaseBartter syndrome
Renal transplant recipient: 4-17% of patient develop erythrocytosis due to increase EPO production by the naïve kidney.

End-stage kidney insufficiency are listed among the causes
of an acquired bleeding tendency.
Hemorrhagic symptoms span from minor events such as
easy bruising to severe and life-threatening complications
such as upper gastrointestinal, pericardial, and intracranial
bleeding.
The coagulation phase of hemostasis, as explored by aPTT
and PT, is normal in uremic patient outside of the period of
heparin administration during dialysis aimed to prevent
filter clotting.
The most common lab abnormality is prolongation of the
skin bleeding time, used as surrogate ex vivo measurements
of defective platelet interactions with the vessel wall and
the delayed formation of the primary hemostatic plug.
Hemostasis defects in uremia


How I treat bleeding in patients with chronic renal disease
The adoption of maintenance dialysis was the first step that improved the bleeding tendency in uremia, although not fully satisfactorily.

How I treat bleeding in patients with chronic renal disease
The management of spontaneous bleeding and its prevention at the time of invasive procedures or surgery was further improved in the 1980s by the demonstration that some drugs shortened the prolonged bleeding time (ie, desmopressin with a short-lasting effect and conjugated estrogens with a more prolonged effect).
Desmopressin: a synthetic derivative of the antidiuretic hormone that increases plasma VWF levels& shorten the bleeding time in uremia.
A conjugated estrogens, which quench the production of nitric oxide (a vasodilator and platelet function inhibitor) .

How I treat bleeding in patients with chronic renal disease
Treatment of anemia: A pioneer observation was made in 1982 by Livio et al,
who demonstrated in uremic patients that the bleeding time is prolonged in proportion to the degree of anemia, and that this test is shortened or corrected when the hematocrit is increased to at least 30% byRBC transfusion.
The improvement of anemia improves platelet adhesion because more RBCs in the circulation push more platelets and leucocytes from the axial center of flowing blood toward the periphery, thereby enhancing cell contacts with the vessel wall and the formation of the primary hemostatic plug

A step forward was the finding in 1987
that the rEPO for the management of
anemia caused a marked and sustained
shortening of the bleeding time. This
positive effect could be obtained after
the attainment of hematocrit values of
approximately 30%.

How I treat bleeding in patients with chronic renal disease

Thrombotic complications in renal disease
The risk for premature atherothrombotic manifestations of cardiovascular diseases in chronic kidney disease, even in its early clinical stages, increases by 25%-30% and this accounting for approximately 40% of all deaths.Despite increasing awareness about very high risk for incident cardiovascular events, there is a paucity of clinical trials evaluating whether the adoption of prophylactic and therapeutic measures

Thrombosis risk factors shared by cardiovascular and chronic renal diseases

Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is a rare blood disorder characterized by thrombosis in small blood vessels of the body , resulting in a low platelet count. In its full-blown form, the disease consists of the pentad of microangiopathic hemolytic anemia, thrombocytopenic purpura, neurologic abnormalities, fever, and renal disease.

History,,,
In 1924, Dr.Eli Moschcowitz described a 16- year old girl with abrupt onset of petechiae, pallor, followed by paralysis, coma, and death.Autopsy showed ‘hyaline’ thrombi occluding terminal arterioles and capillaries.

Clinical Presentation
Approximately 1000 new cases occur each yearCommon in middle aged group , median age-40Female : male (2:1).Acute onset and fulminant course Mortality rate >90% in pre-pheresis era.Relapse rates, 10-40% ranging from months to years have been reported.

Signs and symptoms
Neurologic manifestations include alteration in mental status, seizures, hemiplegia, paraesthesias, visual disturbance, and aphasia.Fatigue may accompany the anemiaAcute renal insufficiency that may be associated with anuria and may require acute dialysis Severe bleeding from thrombocytopenia is unusual, although petechiae are common.Fever occurs in approximately 50% of patientsPatients may notice dark urine from hemoglobinuria.

Pathogenesis
Congenital and acute acquired TTP are due to a deficiency of von Willebrand factor (VWF) cleaving protein, also known as ADAMTS13, (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13– von Willebrand factor cleaving protein)
In the absence of ADAMTS13, ultra large multimers of VWF (ULVWF) released from endothelium are not cleaved appropriately, and cause spontaneous platelet aggregates in conditions of high shear, such as in the microvasculature of the brain, heart and kidneys.

What is Von Willebrand factor (vWF)?
large glycoprotein encoded by a gene on chromosome 12p13.3synthesized by vascular endothelial cells and megakaryocytesSize: 270-20,000 kDa
www.vwf.group.shef.ac.uk/ .pictures html

Von Willebrand factor (vWF) VWF is stored in Weibel-Pallade bodies of endothelial cells
and the α-granules of both megakaryocytes and platelets
VWF multimers (UL-vWF), can bind better to the extracellular matrix than regular multimers and form higher strength bonds with platelet GPIb-IX-V than plasma vWF
UL-vWF are rapidly degraded into smaller forms , do not bind platelets spontaneously by ADAMTS13

ADAMTS13Degrades ultralarge vWf multimers, generating smaller form and decreasing their activity Directly cleaves the peptide bond between Tyr1605 and Met1606 of the VWF A2 domain
http://hematology.wustl.edu/faculty/sadler/vwf.gif
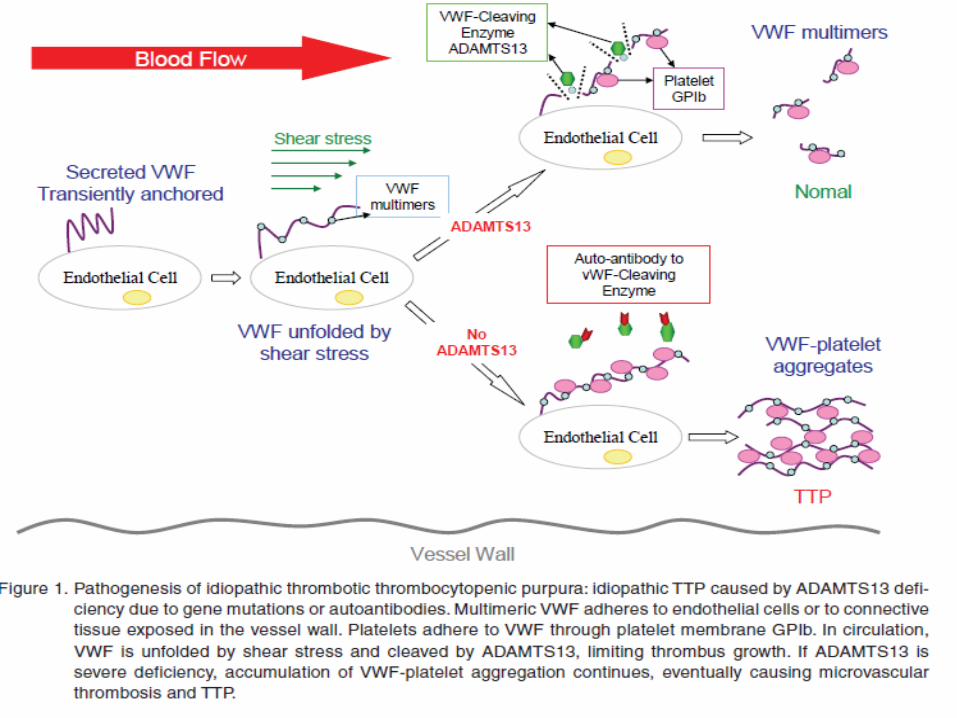

Congenital TTP is due to an inherited deficiency of ADAMTS13, But acquired immune TTP is due to the reduction of ADAMTS13 by autoantibodies directed against ADAMTS13.Other clinical forms of thrombotic microangiopathy (TMA) occur in the absence of severe deficiency.

Subgroups of TTP
Congenital TTPa rare disorder
It has a varied phenotype and can present at any age.
Neonates typically have severe neonatal jaundice. Blood film examination may show schistocytes.
The diagnosis of congenital TTP should be considered in children and adults with unexplained thrombocytopenia.
The diagnosis of congenital TTP is confirmed by ADAMTS13 activity <5%, absence of antibody and confirmation of homozygous or compound heterozygous defects of the ADAMTS13 gene

Acute idiopathic TTPthe most common form of TTP.It is an autoimmune disease characterized by antibodies, usually IgG, directed againstADAMTS13.
HIV-associated TTPmay be the initial presenting feature of HIV disease or in those with low CD4 counts following non- compliance with antiviral treatmentRemission is dependent upon improving the immune status of the patient.
Pregnancy-associated TTPlate onset adult congenital TTP or acute idiopathic TTP. Differentiating TTP from the more common pregnancy-related TMAs, such as pre-eclampsia, HELLP syndrome and HUS is difficult, especially if TTP presents post-partum .

Drug-associated TTP
Drugs appear to be responsible for <15% of all TTP cases include:Quinine Ticlodipine and clopidogrel.SimvastatinTrimethoprimPeglyated interferonOestrogen -containing hormonal preparations combined oral contraceptive pill (COCP) and hormone replacement therapy .Some chemotherapy agents, such as gemcitabine, bleomycin and mitomycin–C can cause HUS but not TTP.

Medications associated with precipitation of
TTP include quinine and oestrogen-containing
medications, which should be avoided to
prevent relapse in patients with a previous
episode of TTP.
Women with previous TTP should be offered
non-oestrogen containing contraception

Transplant-associated microangiopathy
Transplant-associated microangiopathy (TAM) is a MAHA and thrombocytopenia that occurs after BMT.
It may reflect endothelial toxicity associated with chemotherapy, infections, immunosuppressives, such as ciclosporin A (CSA), and GVHD.
TAM has important differences from de novo TTP, namely, absence of ADAMTS13 deficiency; rare neurological symptoms, poor response to PEX and lack of evidence of systemic microthrombi formation

Malignancy-associated thrombotic microangiopathy
Thrombotic microangiopathy occurs in association with avariety of malignancies, esp adenocarcinomas, either at an early stage of cancer or associated with disseminated disease. ADAMTS13 activity is not significantly reduced in these patients .
Pancreatitis-associated TTP
MAHA has recently been reported in association with acute pancreatitis, sometimes a number of days after resolution of pancreatitis. ADAMTS13 activity was only moderately reduced and did not correlate with the severity of TTP or pancreatitis. All patients were successfully treated with PEX and corticosteroids

Diagnosis of TTP
TTP was originally characterized by a pentad of thrombocytopenia, MAHA, fluctuating neurological signs, renal impairment and fever, often with insidious onset.However, TTP can present without the full pentad; Up to 35% of patients do not have neurological signs at presentation and renal abnormalities and fever are not prominent features. The revised diagnostic criteria state that TTP must be considered in the presence of thrombocytopenia and MAHA alone.

Diagnosis
Primary diagnostic criteriaThrombocytopenia ( often below <20,000)Microangiopathic hemolytic anemia
Negative Coomb’s test.Fragmented red cells (schistocytes) on peripheral smearLDH elevation is the hallmark of RBC destruction and tissue injury related to ischemia.
Presence of above criteria is sufficient to establish presumptive diagnosis & begin PE.


The clotting screen (PT, aPTT and fibrinogen) is usually normal.A virology screen pre-treatment is necessary to exclude HIV and other viral-associated TTP, and as a baseline prior to plasma exposure.Troponin T levels are raised in 50% of acute idiopathic TTP cases, highlighting that cardiac involvement is common. ADAMTS13 assays

Clinical differentiation of HUS and TTP can be problematic.
Differentiation is often based on the presence of CNS involvement in TTP and the more severe renal involvement in HUS.
In HUS, an antecedent history of diarrheal illness is more often present.
In children, the distinction between HUS and TTP may be of more importance, as general supportive measures (with dialysis as needed) are the standard therapy for HUS, versus plasma exchange for TTP. However, in adult with HUS plasma exchange is performed so the differentiation has less therapeutic implications at present.

ADAMTS13 assays
Blood must be taken prior to treatment to assess baseline ADAMTS13 activity. Severely reduced ADAMTS13 activity (<5%) ± the presence of an inhibitor or IgG antibodies, confirms the diagnosis.
Decreased ADAMTS13 activity (<40% but >5%) has been reported in a wide variety of non-TTP conditions such as uraemia, inflammatory states, post-operatively and during pregnancy.
The specificity of severe ADAMTS13 deficiency (<5%) in distinguishing acute TTP from HUS is 90%.

ADAMTS13 assays
Do not wait for result before starting treatment in suspected TTP

Differential DiagnosisDisseminated intravascular coagulation.Sepsis: cytomegalovirus, rocky mountain spotted fever, meningococcemia.Preeclampsia/eclampsia, HELLP.Disseminated malignancy.Hemolytic-uremic syndromeEvans syndromeMalignant hypertension.

Differential Diagnosis


TTP management The diagnosis of TTP should be treated as a medicalemergency (1A).The initial diagnosis of TTP should be made on clinical history, examination and routine laboratory parameters of the patient, including blood film review (1A).In view of the high risk of preventable, early deaths in TTP, treatment with PEX should be initiated as soon as possible, preferably within 4–8 h, regardless of the time of day at presentation, if a patient presents with a MAHA and thrombocytopenia in the absence of any other identifiable clinical cause.

TTP Treatment
Plasma exchange:Untreated TTP has 80-90% mortality.Removes ULvWF multimers, autoantibody and replaces metalloproteinase.FFP as the replacement fluid is most widely used and cost effective.
Cryosupernatant plasmaTheoretically superior to FFP in refractory diseaseRemoval of cryoprecipitate from donor plasma results in removal of vWF ( only 18%), with no change in metalloproteinase concentration.
Solvent-detergent plasma Lacks high molecular weight forms of VWFInactivates lipid-enveloped viruses.

The duration of PEX and the number of procedures required to achieve remission is highly variable.
An optimal regimen has not been determined.
In the Canadian apheresis trial, 1·5 plasma volume (PV) exchange was performed on the first 3 d followed by 1·0 PV exchange thereafter.
intensive exchange, such as twice daily PEX, may be required in resistant cases especially if there is new symptomatology.
Daily exchanges should continue for a minimum of 2 d after complete remission, defined as normal platelet count (>150 X 109/l) then stop. Tapering (reducing frequency and/or volume of PEX) has not been shown to reduce relapse rates.

Further treatments in acquired TTP
Steroids are widely used in combination with PEX in the initial treatment of acute immune TTP. Higher dose pulsed steroids have shown to be associated with an improved patient outcome and usually have minimal side effects. or high dose oral prednisolone (e.g. 1 mg/kg/d) should be considered
Rituximab. Prospective studies have shown that rituximab is effective and safe in immune TTP, when patients failed to respond to daily PEX and methylprednisolone and in relapsed acute idiopathic TTP

Cyclosporine may be considered as second line therapy in patients with acute or chronic relapsing acquired TTP.
Other therapies: other drugs previously used for refractory and remitting cases, such as vincristine and cyclophosphamide, whose use is associated with severe side effects, and efficacy has been documented in small numbers of patients
Splenectomy : The mortality of open splenectomy in acute TTP was reported to be approximately 40%, rarely be considered in the non-acute period of immune-mediated TTP but has limited proven benefit.

Antiplatelet agents
The clinical efficacy of antiplatelet agents in TTP is unproven but they are relatively safe .
Low dose aspirin (75 mg OD) may be given during platelet recovery (platelet count >50 X 109/l).

Supportive therapy
Red cell transfusion should be administered according to clinical need especially if there is cardiac involvement.Folate supplementation is required during active hemolysis .Platelet transfusions are contra-indicated in TTP unless there is life-threatening hemorrhage.Thromboprophylaxis with LMWH is recommended once platelet count has reached >50 X 109/l.

Hemolytic uremic syndrome HUS
HUS is a disorder belonging to the category of thrombotic microangiopathies .
It is characterized by non-immune (Coombs negative) hemolytic anemia, low platelet count, and renal impairment .

History,,,,,HUS is the most common cause of acute renal failure in children and is increasingly recognized in adult.
Gasser et al first described HUS in 1955.
In 1988, Wardle described HUS and TTP as distinct entities, but in 1987, Remuzzi suggested that these 2 conditions are various expressions of the same entity.
With the discovery of VWF–cleaving metalloprotease ADAMTS-13 , HUS and TTP are clearly different diseases, despite their clinical similarities

Pathophysiology
• Damage to endothelial cells is the primary event in the pathogenesis of HUS.
• The cardinal lesion is composed of arteriolar and capillary microthrombi (thrombotic microangiopathy [TMA]) and red blood cell (RBC) fragmentation.

Classification
2 main categories
Typical HUS (Shiga-like toxin –associated HUS [Stx-HUS])
is the classic, primary or epidemic form of HUS.
Stx-HUS is largely a disease of children younger than 2-3 years and often results in diarrhea (denoted D+HUS).
One fourth of patients present without diarrhea (denoted D-HUS).
Acute renal failure occurs in 55-70% of patients, but they have a favorable prognosis, and as many as 70-85% of patients recover renal function.

Recent concepts in the pathogenesis of HUS
In North America and Western Europe, 70% of cases Stx–associated HUS are secondary to E. coli serotype O157:H7. Other E coli serotypes are O111:H8, O103:H2, O121, O145, O26, O113 and O104:H4.In Asia and Africa, it is often associated with Stx-producing Shigella dysenteriae serotype. After ingestion, Stx-E coli , it closely adheres to the epithelial cells of the gut mucosa. Stx is transported from the intestine to the kidney via polymorphonuclear neutrophils (PMNs). Susequently , Stx bind to glomerular endothelial cells by means of the terminal digalactose moiety of the glycolipid cell-surface receptor globotriaosylceramide Gb3. Stx favors leukocyte-dependent inflammation by altering endothelial cell-adhesion properties and metabolism, ultimately resulting in microvascular thrombosis.

Atypical HUS (Non–Stx-associated HUS [non–Stx-HUS])
can be sporadic or familial. infection by Stx-producing bacteria is not the cause, may occur without a gastrointestinal prodrome (D-HUS).Poor outcome, as many as 50% may progress to end-stage renal disease (ESRD) or irreversible brain damage. Up to 25% of patients die during the acute phase.The familial form is associated with genetic abnormalities of the complement regulatory proteins.

Sporadic non–Stx-associated HUS
Various triggers have been identified: Non-enteric infections, Streptococcus pneumoniae infection accounts for 40% of all causes of non–Stx-HUS and 4.7% of all causes of HUS. viruses, drugs, malignancies, transplantation, pregnancy,other underlying medical conditions (rare) (eg, antiphospholipid syndrome, SLE).

Familial non–Stx–associated HUS
It accounts for < 3% of all cases of HUS. Both autosomal dominant and autosomal recessive forms of inheritance are observed. Some data suggest genetic abnormalities in the complement regulatory proteins, including C3, factor H, factor B, factor I, and CD46 (membrane cofactor protein, MCP). Autosomal recessive HUS often occurs early in childhood with poor prognosis , recurrences are frequent, and the mortality rate is 60-70%. Autosomal dominant HUS often occurs in adults, who have a poor prognosis. The risk of death or ESRD is 50-90%.

Mutation of the gene of CFH encoding for complement factor H for 30% of gene mutations detected in a HUS and resulting in over activation of complement system,
Upon exposure to an agent that activates complement, C3b is formed in higher-than-normal amounts, and its deposition on vascular endothelial cells cannot be fully prevented as a result of loss of polyanion binding capability of mutated HF1.
This results in the formation of membrane attack complex and the recruitment of inflammatory cells, all events that cause damage and retraction of endothelial cells, adhesion and aggregation of platelets, increased local tissue factor with factor VII binding and activation, and the formation of thrombin and of fibrin polymers.

CLINICAL MANIFESTATION
Abdominal pain, Diarrhea, Fever, Vomiting
Hematological manifestations pallor, Thrombocytopenia, Bruising, bleeding from nose or mouth, Petechiae, Bloody diarrhea.
Renal manifestation: 50% develop ARF, Hematuria, Oliguria or anuria, Proteinuria , Hypertention, Generalized edema , Metabolic acidosis,
CNS Manifestation less frequent
Other manifestation, Hyperbilirubinemia, Spleenomegaly , Pancreatic insufficiency which can lead to diabetes mellitus, Myocarditis, cardiomyopathy, Death

Treatment for Stx-HUS
There is no treatment of proven value, and care during the acute phase of the illness is still merely supportive.
Antibiotics should be avoided unless in cases with sepsis and bacteremia.
Most treatments, including plasma therapy, intravenous IgG, fibrinolytic agents, antiplatelet drugs, corticosteroids, and antioxidants, have been shown to be ineffective in controlled clinical trials in the acute phase of the disease .
Control of BP, electrolytes, careful transfusion.
Avoid antomotility drugs,
Hemodialysis for Fluid overload resistant to diuretic therapy
, hyperkalemia, symptoms of uremia and intractable acidosis

How do I treat incident patients with aHUS?
Plasma therapy
Plasma therapy in the form of plasma exchange or plasma infusion has been the cornerstone of aHUS therapy since the 1980s and was essentially the only therapy available until recently.
The effectiveness of plasma therapy is presumed to be related to its ability to deliver normal levels of CFH, CFI, CFB, and C3 and, when plasma is exchanged by apheresis, to remove mutant CFH, CFI, CFB,C3 and anti-CFH Abs.

Despite this, the majority of patients need long term renal replacement therapy within 2 years of presentation.
Plasma therapy is at least 70% effective in achieving hematologic remission.
Renal remission appears to be less certain and may relate to the particular plasma regimen used or the time lapse from disease onset to the start of therapy.

It has been recommended that plasma therapy be started within 24 hours of presumed diagnosis of aHUS.Plasma infusions or exchange should be performed daily until the platelet count, LDH, and hemoglobin levels are substantially improved or even normalized.Persistence of hemolysis or lack of improvement in thrombocytopenia after 3-5 days of plasma therapy should be considered non-response to therapy, and is an indication to stop plasma exchange and begin eculizumab.Secondary forms of HUS may also be less responsive to plasma therapy.

Hematology 2012

With the finding that excessive activation of the alternative pathway of complement underlies the pathogenesis of aHUS in most patients, it became clear that complement inhibition would be a logical therapy.

Eculizumab therapy
The activation of the terminal complement pathway is essential for the development of the endothelial lesion that characterizes aHUS.Eculizumab (trade name Soliris; Alexion Pharmaceuticals) is a recombinant, humanized, monoclonal Ig that targets C5 and blocks the cleavage of C5 to C5a and C5b, preventing the generation of the proinflammatory peptide C5a and the membrane attack complex C5b-9.Eculizumab, previously approved for PNH, and recently in September of 2011 for the treatment of aHUS after successful trials in adults and adolescents. Initially given weekly as induction therapy and then transitions to every other week.

As has been proposed plasma exchange would be a reasonable first therapy, with eculizumab introduced for non-response to plasma.The major concern with eculizumab treatment is the risk for infection with encapsulated bacterial organisms, particularly Neisseria meningitis, as a result of terminal complement blockade. Therefore, patients must receive meningococcal vaccination before being treated with eculizumab.

Liver transplantation
Because CFH, CFI, CFB, and C3 are synthesized in the liver, liver transplantation remains an appropriate option for some patients to provide a source of normal protein. Acceptable outcomes with no reported recurrences of aHUS when simultaneous liver and renal transplant.

Renal transplantation
Several patients with aHUS will progress to ESRD because of inadequate response to therapy or even delay in diagnosis. aHUS patients who need renal transplant require particular attention because of the high rate of recurrent disease without specific therapy in all patients except those with a mutation in MCP.

Approach for renal transplant
A complete genetic investigation before transplantation be performed for known genes associated with aHUS, as well as a serologic assessment for relevant autoantibodies.
Renal transplant under the cover of eculizumab, and 1 or 2 sessions of preoperative plasma therapy should be considered.
In addition to other routine pre-transplantation vaccination recommendations, meningococcal vaccine should be given to reduce the infectious risk when eculizumab therapy is used.