genetic association of catalase and antigen processing genes with
TRANSCRIPT

GENETIC ASSOCIATION OF CATALASE AND ANTIGEN PROCESSING GENESWITH VITILIGO SUSCEPTIBILITY
By
COURTNEY BRADLEY CASP
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOLOF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OFDOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2003

Copyright 2003
by
Courtney Bradley Casp

This dissertation is dedicated to my husband Justin, who encouraged me every step of theway.

iv
ACKNOWLEDGMENTS
I first would like to thank my mentor, Dr. Wayne McCormack, for his guidance and
encouragement in the completion of my studies. I thank him for patiently and
knowledgeably answering the hundreds of questions I have asked him over the course of
my dissertation work. He is an inspiring educator, both in the classroom and in the lab.
Next, I would like to thank the members of my dissertation committee, Dr. Mike
Clare-Salzler, Dr. Sally Litherland, and Dr. Peggy Wallace, for their time, energy and
guidance. I would like to extend special thanks to Dr. Litherland, who patiently taught
me monocyte culture techniques, and whose lab and office were always open to me.
I am indebted to many members of the McCormack lab, past and present, most
notably Deb Fisher and Bryan Riggeal, who were always around to lend both an ear and a
hand. Special thanks also go out to the Clare-Sazler and Litherland lab members who
have shared both their expertise and their lab space with me. I also wish to thank Kim
Blenman, Linda Archer and Dr. Rita Hurst, who have provided me with scientific as well
as much needed personal support. I am forever indebted to these friends and co-workers
who have helped me to accomplish my goals.
On a personal note, I would like to thank my entire family for their unwavering
encouragement. Specifically, I thank my parents, my mother Connie Bradley, my father
Tim Bradley, and my stepmother June Bradley, who have fostered dedication and
encouraged me to always “do the right thing.” I also thank my father-in-law and mother-
in-law, Mark and Marcy Casp, who have given me encouragement and support as well as

v
a laptop for which to write this document. I thank my husband who has stood behind me
and cheered every step of the way. I thank him for his optimism and love, which have
kept me going even on my darkest days. Finally, I thank my beautiful daughter Ashlyn,
who, when I look at her innocent face, reminds me to keep everything in life in
perspective.

vi
TABLE OF CONTENTSPage
ACKNOWLEDGMENTS ................................................................................................. iv
LIST OF TABLES............................................................................................................. ix
LIST OF FIGURES ........................................................................................................... xi
ABSTRACT...................................................................................................................... xii
CHAPTER
1 INTRODUCTION ........................................................................................................1
Skin Structure and Physiology......................................................................................1Vitiligo Pathogenesis ....................................................................................................4
Autoimmune Hypothesis .......................................................................................5Cytotoxic hypothesis .............................................................................................8
Treatment....................................................................................................................10Genetics of Vitiligo.....................................................................................................12Vitiligo Candidate Genes............................................................................................15
Immune System Genes ........................................................................................15Low-molecular-weight polypeptide 2 (LMP2), low-molecular-weight
polypeptide 7 (LMP7) and multicatalytic-endopepidase-complex-like 1(MECL1).................................................................................................15
Transporter associated with antigen processing 1 and 2 (TAP1 and TAP2)16CD28 and CTLA4 (CD152) .........................................................................17CD4 ..............................................................................................................17IL-12p40.......................................................................................................18IL-1β.............................................................................................................18Autoimmune polyendocrinopathy syndrome type-1 (APS-1)/autoimmune
regulator (AIRE) .....................................................................................19Melanocyte Biochemistry and Oxidative Stress..................................................19
Catalase (CAT).............................................................................................19GTP cyclohydrolase 1 (GCH1) ....................................................................20
2 CASE/CONTROL AND FAMILY-BASED ASSOCIATION STUDIES.................21
Introduction.................................................................................................................21Materials and Methods ...............................................................................................22

vii
Subjects................................................................................................................22Blood Processing .................................................................................................22DNA Extraction...................................................................................................24Primers.................................................................................................................24Microsatellite Markers.........................................................................................25RFLP and AFLP Markers....................................................................................25Statistical Analysis ..............................................................................................28
Results.........................................................................................................................30Immune System Genes ........................................................................................30Melanocyte-specific Genes .................................................................................36
Discussion...................................................................................................................40
3 ANTIGEN PROCESSING AND PRESENTATION GENES...................................46
Introduction.................................................................................................................46Materials and Methods ...............................................................................................47
Blood Collection and Processing.........................................................................47LMP7 Sequencing ...............................................................................................48
DNA preparation and PCR amplification ....................................................48Direct sequencing of PCR products .............................................................48
Results.........................................................................................................................50Case/Control Association Studies .......................................................................50
Allele and genotype frequencies ..................................................................50Family-based association .............................................................................56
LMP7 Sequencing ...............................................................................................58Discussion...................................................................................................................58
4 CATALASE ...............................................................................................................66
Introduction.................................................................................................................66Materials and Methods ...............................................................................................67
Blood Collection and Processing.........................................................................67Catalase Sequencing............................................................................................67
DNA preparation and PCR amplification ....................................................67Direct sequencing of PCR products .............................................................69
Results.........................................................................................................................70Catalase Gene Polymorphisms ............................................................................70Association of the T/C Exon 9 (BstX I) CAT Marker with Vitiligo ...................72Family-Based Association...................................................................................72Catalase Sequencing............................................................................................74
Discussion...................................................................................................................74
5 CANDIDATE VITILIGO SUSCEPTIBILITY GENE EXPRESSION STUDIES....79
Introduction.................................................................................................................79Materials and Methods ...............................................................................................80
Monocyte Isolation and Culture ..........................................................................80

viii
Semi-Quantitative RT-PCR.................................................................................81Flow Cytometry of β2-microglobulin .................................................................83Catalase Enzyme Assay.......................................................................................83H2O2 Treatment of Monocytes ............................................................................84
Results.........................................................................................................................84Catalase Assay.....................................................................................................84β2-Microglobulin Expression..............................................................................85H2O2 Treatment ...................................................................................................89RNA Expression Studies .....................................................................................89
Discussion...................................................................................................................91
6 DISCUSSION: OXIDATIVE STRESS AND THE IMMUNE SYSTEM INVITILIGO PATHOGENESIS ..................................................................................101
Oxidative Stress ........................................................................................................101Reactive Oxygen Species and Autoimmunity ..........................................................103
Antioxidant Levels in Autoimmune Disease.....................................................103Transfection of Antioxidant Genes ...................................................................105Antioxidants and Reactive Oxygen Species in Autoimmunity .........................106
Antigen Processing, Oxidative Stress and Vitiligo...................................................108Vitiligo and Oxidative Stress.............................................................................108Vitiligo and Catalase .........................................................................................108Vitiligo and MHC class II genes .......................................................................110
Future Directions ......................................................................................................111
LIST OF REFERENCES.................................................................................................113
BIOGRAPHICAL SKETCH ...........................................................................................128

ix
LIST OF TABLES
Table page
2-1. Vitiligo patient and unaffected relative samples collected for case/control andfamily-based analyses. .............................................................................................23
2-2. Primer sequences......................................................................................................26
2-3. Microsatellite primer conditions. .............................................................................27
2-4. RFLP/SSCP PCR conditions....................................................................................27
2-5. Case/control association analysis for CD28 by microsatellite (CAA 3' UTR) ........34
2-6. Case/control association analysis for CTLA4 by microsatellite (AT 3' UTR) ........34
2-7. Case/control association analysis of CTLA4 by RFLP (Bst E II +49 A/G)............35
2-8. Case/control association analysis of CTLA4 by RFLP (Hae III intron 1 C/T)........35
2-9. Case/control association analysis of APS-1 by SSCP (C/T exon 5) ........................37
2-10. Case/control association analysis of APS-1 by RFLP (Hae III exon 10 T/C) .........37
2-11. Case/control association analysis of IL-1β by RFLP (Ava I –511 C/T) ..................38
2-12. Case/control association analysis of CD4 by Microsatellite (TTTTC repeat 5' UTR)38
2-13. Case/control association analysis of IL-12p40 by RFLP (Taq 1 C/A 3' UTR)........39
2-14. Case/control association analysis of GCH1 by RFLP (Bsa A1 C/T exon 6) ...........39
3-1. LMP7 primers for sequencing across the gene ........................................................49
3-2. Primers used for LMP/TAP and MECL1 genotyping...............................................51
3-3. Linkage disequilibrium analysis of Caucasian vitiligo patients (age of onset 0-29years) and control subjects. ......................................................................................53
3-4. Allele frequencies of LMP/TAP and MECL1 candidate genes in Caucasian vitiligopatients (age of onset 0-29 years) and control subjects............................................54

x
3-5. Genotype frequencies of LMP/TAP and MECL1 candidate genes in Caucasianvitiligo patients and (age of onset 0-29 years) control subjects. ..............................55
3-6. Family based association (transmission disequilibrium test) results for LMP/TAPand MECL1 candidate genes and vitiligo susceptibility ..........................................57
4-1. Catalase primers for sequencing across the gene .....................................................68
4-2. Sequences of primers used for CAT genotyping .....................................................71
4-3. Distribution of alleles and genotypes for the T/C SNP in CAT exon 9 in vitiligopatient and control populations ................................................................................73
4-4. Carriage rates and heterozygosity of the T/C SNP in CAT exon 9 in vitiligo patientscompared to controls ................................................................................................73
4-5. Catalase gene single nucleotide polymorphisms (SNPs). ........................................75
5-1. RT-PCR primer pairs and PCR conditions for LMP2, LMP7, TAP1, TAP2 andcatalase .....................................................................................................................82
5-2. Genotypes of patients treated with 500 U IFN-γ, and used in mRNA, catalase and,β2-microglobulin expression studies with corresponding catalase enzyme activityand mean β2-microglobulin fluorescence. ...............................................................90

xi
LIST OF FIGURES
Figure page
1-1. Melanin biosynthesis pathway ...................................................................................3
2-1. CD28 allele frequency..............................................................................................32
2-2. CTLA4 allele frequency...........................................................................................33
5-1. Monocyte catalase levels in patients and controls ...................................................86
5-2. Erythrocyte catalase levels in vitiligo patients and normal controls. .......................87
5-3. β2-microglobulin expression on patient and control monocytes.. ...........................88
5-4. Messenger RNA expression of LMP2, LMP7, TAP1 and TAP2 in patient andcontrol monocytes after overnight treatment with 1000 U IFN-γ. ...........................92
5-5. Messenger RNA expression of LMP2, LMP7, TAP1 and TAP2 in patient andcontrol monocytes after overnight treatment with 500 U IFN-γ. .............................93
5-6. Catalase/18S RNA ratios with and without overnight treatment with 500U IFN-γ.94

xii
Abstract of Dissertation Presented to the Graduate Schoolof the University of Florida in Partial Fulfillment of theRequirements for the Degree of Doctor of Philosophy
GENETIC ASSOCIATION OF CATALASE AND ANTIGEN PROCESSING GENESWITH VITILIGO SUSCEPTIBILITY
By
Courtney Bradley Casp
December 2003
Chair: Wayne T. McCormackMajor Department: Pathology, Immunology, and Laboratory Medicine
Vitiligo is a common dermatological disorder of the epidermis and hair follicles,
manifested clinically as expanding hypopigmented lesions of the skin. Vitiligo
pathogenesis is believed to be due to autoimmune destruction of the melanocyte, or
autotoxicity in the melanocyte. Vitiligo often appears in multiple family members,
suggesting a genetic component to vitiligo pathogenesis. The goal of this study was to
test the hypothesis that vitiligo pathogenesis is caused in part by genetic susceptibility to
both autoimmune and autotoxic events in the epidermis due to genetic differences in
genes involved in the regulation of the immune response, melanin production and
oxidative stress.
Through the use of case/control and family based association studies, two
susceptibility genes for vitiligo were identified. Susceptibility to vitiligo was
demonstrated for a gene(s) in or near the LMP/TAP region of the MHC class II genomic

xiii
region. The LMP/TAP gene products are responsible for processing and transport of
antigenic peptides for presentation to the immune system via MHC class I molecules.
Messenger RNA expression studies of LMP2, LMP7, TAP1 and TAP2 genes did not
show alterations in expression of mRNA for any of these genes in monocytes derived
from vitiligo patients and normal controls. However, expression studies of MHC class I
revealed decreased expression of MHC class I on monocytes from vitiligo patients,
suggesting some alterations in the MCH class I presentation pathway. The second
vitiligo susceptibility gene, catalase, is an important antioxidant, responsible for breaking
down hydrogen peroxide. Messenger RNA expression studies of catalase found no
alteration in catalase mRNA expression between patients and controls. Enzyme
expression studies of catalase, however, revealed decreased expression of catalase in the
monocytes of vitiligo patients, a result not seen in erythrocytes from these same patients.
These results demonstrate a possible role for genes involved in immune system
regulation, as well as for genes involved in regulating oxidative stress in vitiligo
susceptibility. Thus, the etiology of vitiligo may rely on both autotoxic events in the
melanocyte, allowing for increased oxidative stress in the epidermis and inappropriate
autoimmune presentation of self-peptides to the immune system.

1
CHAPTER 1INTRODUCTION
Vitiligo is a common dermatological disorder of the epidermis and hair follicles,
affecting both genders and ~1% of the population in all ethnic groups worldwide.
(Nordlund and Ortonne, 1998). Vitiligo is defined clinically by expanding areas of
hypopigmentation on the skin surface due to the destruction or inactivation of epidermal
melanocytes (Badri et al., 1993; Majumder et al., 1993; Tobin et al., 2000). Vitiligo
pathology is limited to the depigmentation of the epidermis, but it is often associated with
other autoimmune disorders such as alopecia areata and Hashimoto’s thyroiditis (Badri et
al., 1993; Kemp et al., 1999). Depigmentation can occur anywhere on the body including
the face. The striking appearance of depigmented areas flanked by normal tissue can
cause social stigmatism and physiological distress in affected individuals. Treatment for
vitiligo is often both expensive and time and labor intensive for the patient, with
insurance companies often denying coverage because it is often considered to be purely a
cosmetic disorder. This study’s goal was to determine potential genetic susceptibility of
a variety of candidate genes to vitiligo, and further characterize the expression of these
genes in vitiligo patients.
Skin Structure and Physiology
Human skin is made up of two main layers, the epidermis, which is described as a
stratified squamous epithelium mainly consisting of keratinocytes, and the dermis, an
underlying layer of vascularized connective tissue (Nordlund and Ortonne, 1998).
Melanocytes reside at the junction of the dermis and the epidermis and produce the

2
protein melanin that provides pigmentation for the skin and hair. The production of
melanin begins with the amino acid tyrosine, and ends with the production of the red-
yellow pheomelanin, and the more common brown-black eumelanin. The production of
these two pigments occurs through two different pathways, both of which require the
rate-limiting enzyme tyrosinase (Figure 1-1). Melanin is produced in melanosomes,
specialized organelles that are translocated from the center of the melanocyte cytoplasm
to the tip of its dendrites. The dendrites are then involved in the transfer of the
melanosomes to the keratinocytes.
Keratinocytes also develop at this dermal epidermal junction, where they are in a
constant state of mitosis. As new keratinocytes are made, the older ones are forced
toward the surface of the epithelium carrying their cargo of melanin. On the way to the
epidermal surface, melanosomes are degraded in the keratinocyte’s lysosomes, and the
melanin becomes finely dispersed. It is thought that the degree of dispersion of the
melanosome helps to dictate skin tone, with darker-skinned individuals seeming to have
less degradation of the melanosomes. Darker-skinned individuals also produce more
melanin, as well as melanin of a darker color than fair-skinned individuals. When the
keratinocytes reach the epithelial surface they compact and form a protective layer of
keratin before they are shed off (Nordlund and Ortonne, 1998). This keratin layer
protects the skin from injury, and the melanin cargo it carries helps to protect the skin
from the damage of ultraviolet rays. Melanin production is stimulated with excessive
exposure to ultraviolet rays, which is commonly known as tanning.
The hypopigmented lesions in vitiligo patients are a result of the destruction and/or
inactivation of the melanin-producing melanocytes. Keratinocytes still make their

3
Figure 1-1. Melanin biosynthesis pathway

4
migration to the surface of the epithelium, albeit without their cargo of pigment. This
results in patches of skin that look milky-white because they are devoid of pigment.
There are two main types of vitiligo, segmental and non-segmental, and classification
relies on the distribution of hypopigmented lesions. In segmental vitiligo the areas of
depigmentation are random and often occur on just one location on the body. Segmental
vitiligo is also sometimes marked with the loss of melanocytes from the hair follicles and
a loss of hair pigment. Segmental vitiligo is less likely to repigment than non-segmental
vitiligo; however it is also less likely to spread to other areas of the body. Non-segmental
vitiligo usually manifests in a strikingly symmetrical pattern and often does not affect the
hair follicles in the areas of depigmentation. Non-segmental vitiligo often gets
progressively worse, spreading to more areas of the body over time. Some patients with
non-segmental vitiligo almost completely depigment over the years. With non-segmental
vitiligo there is sometimes spontaneous repigmentation, or partial repigmentation with
treatment; however, the patient rarely fully returns to pre-disease pigmentation.
Segmental and non-segmental vitiligo present very differently clinically and may have
different etiologies (Bos, 1997; Nordlund and Ortonne, 1998).
Vitiligo Pathogenesis
The average age of onset in vitiligo is 20-23 years of age (Nordlund and Ortonne,
1998). Because patients are not usually born with the disease, it is thought that an
initiating event, such as illness, stress, UV exposure or injury, may trigger
depigmentation. It has been shown that the numbers of melanocytes in depigmented
lesions are vastly reduced or absent; however, the mechanisms of this apparent
destruction have been widely debated. The two theories with the most evidence are that
1) the destruction of the melanocyte is due to the buildup of toxic by-products in the

5
melanin biosynthesis pathway, and 2) the destruction is autoimmune in origin. These
theories are not mutually exclusive, and in fact, the actual onset of disease might involve
a combination of autotoxic as well as autoimmune events.
Autoimmune Hypothesis
The autoimmune theory of vitiligo etiology is the most popular as well as the most
substantiated. First serum anti-melanocytic antibodies are found in many, but not all,
vitiligo patients, and are not found in healthy pigmented individuals. These antibodies
are directed against tyrosinase, tyrosinase-related protein 1 (TRP1), tyrosinase-related
protein 2 (TRP2), and melanin-concentrating hormone receptor 1 (MCHR1) (Cui et al.,
1993; Song et al., 1994; Kemp et al., 1999; Kemp et al., 2002). However, in vitro work
with these autoantibodies has suggested that the presence of the anti-melanocytic
autoantibodies may just be a marker of active vitiligo and may have little or no active role
in initiating or maintaining the disease (Bos, 1997).
The first evidence of cell-mediated immunity playing a role in the pathogenesis of
vitiligo came from the observation of invading lymphocytes in studies of “inflammatory
vitiligo,” a disease characterized by a raised red rim surrounding the depigmented lesion
(van den Wijngaard et al., 2001). Lesional skin of non-inflammatory vitiligo patients has
more recently been shown to contain significantly higher levels of CD3+, CD4+, and
CD8+ T cells than found in control skin and in skin outside lesions in the same patients
(Badri et al., 1993; Le Poole et al., 1996). Melanocytes also have been found in rare
circumstances to both take up antigen through phagocytosis, as well as to present antigen
via MHC class II molecules (Le Poole et al., 1993a, 1993b). It has been shown that 2/3
of perilesional melanocytes in vitiligious skin express antigen on MHC class II (al Badri
et al., 1993).

6
Most immunohistochemical studies suggest that melanocytes are almost completely
destroyed, as opposed to being inactive or dormant, and this loss is accompanied by
dermal and epidermal infiltrates in the active lesion, which include an increase in CD4+
and CD8+ T cells (Hann et al., 1992; Le Poole et al., 1993a; Badri et al., 1993; Le Poole
et al., 1996). More recent work suggests that not all melanocytes are destroyed in the
lesion; however, those remaining have been rendered dysfunctional (Tobin et al., 2000).
Further evidence of an abnormal immune response is a reversal of the CD4+/CD8+ ratios
as well as a decrease in CD45RA cells in vitiligo patient peripheral blood (Mozzanica et
al., 1990; Abdel-Naser et al., 1992). Disruptions in Langerhans cells in vitiligo have also
been reported, with decreased numbers seen in active vitiligo with a return of normal
numbers in stable vitiligo (Kao and Yu, 1990). It is important to note that variation in
published data on peripheral cell imbalances may be due to a variety of factors such as
differences in study populations, differences in vitiligo disease presentation and/or
progression, and prior immune-suppressive therapy (Ongenae et al., 2003).
A subclass of lymphocytes contains an inducible carbohydrate moiety known as
cutaneous lymphocyte antigen (CLA) on their surface (Fuhlbrigge et al., 1997). This
carbohydrate moiety targets these lymphocytes to the dermis by an interaction with its
ligands E and P selectins and through the dispersion of a chemokine known as CTACK
(Picker et al., 1993; Robert and Kupper, 1999; Campbell et al., 1999; Morales et al.,
1999). Both E and P selectins, as well as CTACK, are upregulated upon local skin
inflammation, injury and UV exposure. In certain skin disorders, such as atopic
dermatitis and psoriasis, an increased percentage of CLA+ lymphocytes is observed in
peripheral blood and skin as compared to controls. Our own studies have shown

7
encouraging, but not definitive signs that this increase in CLA+ T cells (especially CD4+)
is seen in vitiligo patients’ peripheral blood (unpublished data). Cytotoxic T
lymphocytes that are specific for Mel-A, a melanocyte surface marker, and have a CLA
carbohydrate moiety have been shown to be at far higher levels in vitiligo patients than in
controls. While Mel-A+ CTLs were also found in control subjects, these cells were not
CLA+. This absence of CLA prevents these from migrating to the skin to attack the
melanocytes, thus preventing the induction of vitiligo (Ogg et al., 1998).
Further evidence of immune events mediating vitiligo is seen in the altered
expression of several cytokines in patients. The expression of ICAM-1 in vitiligo
patients has been shown to be six-fold higher than in controls (al Badri et al., 1993).
Soluble IL-2 receptor levels are expressed at high concentrations in vitiligo patients.
(Honda et al., 1997; Yeo et al., 1999; Caixia et al., 1999, Tu et al., 1999). Peripheral
blood of patients also has increased concentrations of pro-inflammatory cytokines IL-6
and IL-8 as well as a decreased production of GM-CSF, TNF-α, and INF-γ (Yu et al.,
1997).
The association of vitiligo with other known autoimmune disorders such as
Addison’s disease, Hashimoto’s thyroiditis, pernicious anemia and alopecia areata also
supports the autoimmune theory of disease. In addition, it has been observed that
successful treatment for melanoma sometimes results in spontaneous vitiligo, suggesting
that a successful anti-melanoma immune response might attack normal healthy
melanocytes, due to the sharing of many of the same surface antigens (Overwijk et al.,
1999; Bronte et al., 2000). These findings suggest that a T cell mediated attack on the

8
melanocyte can produce areas of sustained depigmentation, a scenario that has also been
suggested in the etiology of vitiligo.
The most useful animal model of vitiligo pathogenesis is the Smyth-line chicken.
The Smyth-line chicken expresses many of the major features of human vitiligo including
cutaneous depigmentation that is variable in appearance and most often occurs during
adolescence. These chickens also suffer with alopecia and autoimmune thyroiditis and
blindness. Pathogenesis in these animals also supports an autoimmune etiology as
lymphocytic infiltration is seen in the depigmented areas, melanocyte specific
autoantibodies are present, and steroid treatment can halt and sometimes improve
pigmentation (Lamoreaux and Boissy, 2000).
Cytotoxic hypothesis
The cytotoxic hypothesis is based on the premise that the loss of melanocytes in
vitiligo patients is a direct result of an inherent defect in the melanocyte, most probably
resulting in the buildup of toxic intermediates or metabolites from the melanin
biosynthesis pathway. There has been increasing evidence for a role for oxidative stress
in the involvement of vitiligo pathogenesis, based primarily on reported defects in several
key enzymes in the melanin biosynthesis pathway. Vitiligo patients show an increased
epidermal de novo synthesis and recycling of 6(R)-L-erythro-5,6,7,8-tetrahydrobiopterin
(6BH4). It is the accumulation of these oxidized pterins (6- and 7-biopterin) that results
in the fluorescence of vitiligo skin under a Woods UV lamp, which is used for the
definitive diagnosis of vitiligo in a patient (Schallreuter et al., 2001).
Vitiligo patients also show very low levels of 4a-OH-tetrahydrobiopterin
dehydratase (DH) activity which is involved in the recycling pathway of 6BH4. Due to
decreased DH activity, there is an increased buildup of the 7-isomer form of 6BH4,

9
(7BH4), which functions as an effective competitor of 6BH4. A buildup of 7BH4 will
affect phenylalanine hydroxylase (PAH) activity. PAH activity is often decreased in
vitiligo patients, which causes a buildup of epidermal L-phenylalanine levels.
All of these abnormal biochemical events in the vitiligo epidermis may contribute
to increased levels of hydrogen peroxide (H2O2). High concentrations of H2O2 can
deactivate catalase, an enzyme normally involved in breaking down H2O2 and other free
oxygen radicals. Low catalase activities have previously been reported in the epidermis
of vitiligo patients, whether this defect is due to an increase in H2O2 from the defective
6BH4 pathway or due to a separate problem in the catalase enzyme is unknown. What
has been shown is that treatment with pseudocatalase, a bis-manganese III-EDTA-
(HCO3)2 synthetic catalase substitute, has promoted repigmentation in vitiligo patients,
along with restoring DH enzyme activity and a return to normal 7BH4 levels in the
epidermis (Schallreuter et al., 2001).
Abnormal antioxidant activity in peripheral blood mononuclear cells (PBMCs) of
vitiligo patients has also been observed, with increased superoxide dismutase activity,
and reduced catalase, glutathione and vitamin E levels. This variability in antioxidant
levels was seen exclusively in subjects with active disease. These changes in
antioxidants could be responsible for the generation of intracellular reactive oxygen
species (ROS) in vitiligo patients, and may be due in part to an observed mitochondrial
impairment (Dell’Anna et al., 2001).
Vitiligo melanocytes are also found to have an abnormal dilation of the rough
endoplasmic reticulum (RER), seen on transmission electron microscope examination
(Boissy et al., 1991; Im et al., 1994). Further observation of this phenomenon is seen in

10
PIG3V cells, an immortalized vitiligo cell line. In the PIG3V cells, this abnormal
dilation is thought to be caused by a retention of a variety of proteins in the RER (Le
Poole et al., 2000).
Treatment
There is no known “cure” for vitiligo, and current treatment methods function
mainly by stopping current progression of the disease and aiding in the repigmentation of
hypopigmented lesions. Current treatments do not prevent the reappearance of future
depigmented lesions. Treatment options are often expensive and labor-intensive on the
part of the patient, and are most often not covered by patients’ insurance, as vitiligo is
considered a mainly a cosmetic disorder. Up until the late 1990’s, the options for
treatment of vitiligo were limited mainly to psoralens with UVA radiation (PUVA)
therapy and the use of topical corticosteroids on the skin. Current treatment options have
expanded recently and now include a variety of choices such as epidermal melanocyte
autografts, topical treatment with pseudocatalase, and narrow-band UVB irradiation
(Njoo and Westerhof, 2001)
PUVA is considered by many to be the “standard” treatment for vitiligo. Psoralens
are compounds found in many plants used to treat a variety of dermatological disorders,
such as psoriasis, dermatitis, and vitiligo. Psoralens, in conjunction with sun exposure,
have been used to treat skin disorders for centuries, and can be traced back to the ancient
Egyptians (McClelland et al., 1997). Upon exposure to UVA radiation, psoralens can
bind directly to macromolecules such as proteins, lipids, and DNA (Taylor and Gasparro,
1992). Once excited by UVA radiation, psoralens can interact with pyrimidines in DNA
through covalent bonding, forming interchain crosslinks (Song and Tapley, 1979). The
relative frequency of the DNA crosslinking depends on the wavelength of UVA exposure

11
(Taylor and Gasparro, 1992). It is still unclear how this biochemical interaction of
psoralen with DNA functions to alleviate symptoms in the dermatological disorders it is
used to treat. One theory is that the DNA adducts formed function to hamper DNA
synthesis and cell replication, processes which may be upregulated in hypoproliferative
disorders such as psoriasis (McNeely and Goa, 1998).
PUVA-induced repigmentation in vitiligo patients is thought to be due to the
recognition of the DNA adducts as damage, which might trigger activation of genes
involved in pigmentation or melanocyte biochemistry, since DNA repair is closely
associated with transcription. A second theory hypothesizes that PUVA treatment creates
reactive oxygen species that then stimulate melanogenesis by activating melanocyte
migration from the hair follicle to the epidermis (McNeely and Goa, 1998). PUVA
therapy often requires 100-300 exposures, and treatment does not guarantee
repigmentation. One study showed 56% of vitiligo patients achieved 75%
repigmentation, with a lack of response occurring in 22% of patients (Morrison, 2000;
McNeely and Goa, 1998).
The most promising new treatment for vitiligo is the topical application of UVB-
activated pseudocatalase, a bis-manganese EDTA bicarbonate complex. Pseudocatalase
functions to mimic the human enzyme catalase, which removes H2O2 and other free
radicals from the epidermis. It has been shown that vitiligo patients have decreased
catalase expression, along with a subsequent increased expression of H2O2 concentrations
in their epidermis. Treatment of vitiligo patients with topical applications of the
pseudocatalase cream, along with frequent exposure to UVB radiation, given at a much
lower exposure than normal therapeutic doses, has allowed for the halt of depigmentation

12
in 95% of patients, and repigmentation of 60-65% of individuals. Treatment with
pseudocatalase also restores epidermal DH activities and decreases the accumulation of
7BH4 (Schallreuter et al., 1999a, 1999b, 2002).
Genetics of Vitiligo
About 20% of vitiligo patients have at least one first-degree relative also affected,
and the relative risk of vitiligo for first-degree relatives of vitiligo patients is increased by
at least 7- to 10-fold (Bhatia et al., 1992; Nath et al., 1994). Vitiligo susceptibility does
not follow a simple Mendelian inheritance pattern, and much of the available data suggest
that vitiligo is a complex hereditary disease influenced by a set of recessive alleles
occurring at several unlinked autosomal loci that collectively confer the vitiligo
phenotype (Majumder et al., 1993; Nath et al., 1994; Alkhateeb et al., 2002). This type
of inheritance is common among other autoimmune diseases. For example, in type 1
diabetes, the risk of an individual developing diabetes with a first degree relative affected
is only between 1-9% (Redondo et al., 2001), where it is likely that a combination of
genetics and environmental factors result in disease pathogenesis. Vitiligo may therefore
be considered a polygenic disease, with alleles at multiple loci possibly contributing to
increased susceptibility to and/or direct pathogenesis of vitiligo. HLA associations have
been reported between specific alleles of complement, class I and class II MHC genes
with vitiligo in various ethnic and racial subpopulations, but no common HLA association
has been observed. Studies have reported HLA associations in early onset vs. late onset
vitiligo cases, and it is possible that there are different etiologies, and different genetic
factors in early vs. late onset vitiligo, as well as in segmental vs. non-segmental vitiligo
(Finco et al., 1991; Orecchia et al., 1992; Arcos-Burgos et al., 2002). Further
complicating matters, it is probable that genetics alone do not dictate disease onset, as

13
most vitiligo patients are not born with this disorder, but develop it later in life. As with
other autoimmune diseases, environmental factors such as toxins, pollutants, viruses, UV
exposure, and stress could play a major role in the onset of vitiligo in those that are
genetically predisposed.
In order to determine potential genes involved in an inheritable disorder there are
several techniques that can be employed. Functional cloning relies on first identifying
the abnormal protein involved in the disease process, and then using its sequence to
determine the cDNA sequence, and finally the gene involved in the disease. A large
limitation to functional cloning is that the protein(s) involved in the disease process is
(are) not often known. In contrast, positional cloning allows for determination of the
gene’s location without prior knowledge of the protein involved in the disease
pathogenesis. The two main positional cloning techniques employed in disease gene
screening in recent years are linkage mapping and the candidate gene approach.
Linkage mapping scans the entire genomes of family members and individuals
afflicted with the disorder of interest, using regularly spaced genetic markers in the DNA
whose positions have already been determined. Regions where affected individuals
shared alleles more frequently than expected by chance alone are then identified, and
nearby genes are subjected to further analysis. This type of screening is extremely labor
intensive and requires large families with both affected and non-affected members.
However, it does allow for the unbiased screening of diseases where investigators have
no prior knowledge of the pathobiology of the disorder.
The candidate gene approach allows for researchers to investigate susceptibility of
a gene to a disorder by focusing on a limited number of genes selected for their potential

14
involvement in the biology or pathophysiology of the disorder. Candidate gene studies
do not rely on large multi-generation families, but can be performed using unrelated
groups of patients (cases) and controls, or through small families. Candidate gene studies
also have the advantage of being better suited for determining susceptibility genes in
more complex disorders where the relative risk associated with any one gene is relatively
small (Kwon and Goate, 2000).
Once a candidate gene is chosen based on its potential involvement of the disease
of interest, a polymorphism that will allow for suitable genotyping must be identified. It
is important to note that these polymorphisms within the gene might cause a mutation
that results in a functional change in the protein or more likely, may have no functional
relevance to protein function or stability at all. In candidate gene analyses,
polymorphisms function solely as genetic markers within the gene. If a polymorphism
within a gene is inherited at a rate that exceeds the levels determined by random chance,
this gene can then be called a potential susceptibility gene. In this case, the
polymorphism genotyped can either be the actual mutation conferring the susceptibility
or it could merely be linked to the mutation of interest due to its close physical proximity.
There are a variety of polymorphic markers within the genome that are useful for
genotyping. Single nucleotide polymorphisms, or SNPs, are heritable individual single
nucleic acid changes in the genomic sequence. Sometimes SNPs allow for the creation or
deletion of a sequence that is recognizable by a particular restriction endonuclease. This
type of polymorphism is known as a restriction fragment length polymorphism, or RFLP,
and can be genotyped using the two variable sized fragments produced through the
endonuclease activities of the restriction enzyme. Amplified fragment length

15
polymorphisms, or AFLPs, are variants of RFLPs. AFLPs are created through designing
polymerase chain reaction (PCR) primers that change one nucleotide forcing the
amplification of a sequence that creates a restriction site involving a known SNP. Repeat
regions in genomic DNA are also very useful in genotyping. The human genome
contains a large number of these normal inheritable variances in small sequence repeats.
These repeat regions can be a long sequence repeated many times (14 to 100 base pairs),
known as a variable number of tandem repeats (VNTR), or minisatellites, or a region of
multiple repeats of just a few nucleotides (2-5 base pairs), called short tandem repeats
(STR) or microsatellites (Griffiths et al., 1999).
Vitiligo Candidate Genes
Determining genes that confer genetic susceptibility to vitiligo using the candidate
gene approach requires pre-knowledge about the disease process and pathogenesis.
Because it has been hypothesized that vitiligo pathogenesis could be a direct result of
autoimmunity and/or autotoxicity due to biochemical defects in the melanocytes, genes
that regulate the immune system, melanocyte biochemistry and development and
oxidative stress are suitable choices for candidate genes.
Immune System Genes
Low-molecular-weight polypeptide 2 (LMP2), low-molecular-weight polypeptide 7(LMP7) and multicatalytic-endopepidase-complex-like 1 (MECL1)
The proteasome is a large protease composed of several subunits, which plays a
crucial role in protein degradation in the cell. Three subunits crucial for protein
proteolysis known as X, Y, and Z are constitutively expressed, but can be replaced by the
three IFN-γ inducible subunits low –molecular-weight polypeptide 2 (LMP2), low–
molecular-weight polypeptide 7 (LMP7) and multicatalytic-endopepidase-complex like 1

16
(MECL1). The induction of these three subunits allows for the creation of the
“immunoproteasome.” Whereas these induced subunits are very similar in sequence to
the constitutive subunits, functionally they cleave proteins into different peptides. The
IFN-γ inducible subunits generate peptides that are more compatible with the binding
groove of the MHC class I molecule (Driscoll et al., 1993; Tanaka et al., 1998; Pamer et
al., 1998).
The immunoproteasome genes were selected as candidate genes because several
autoimmune diseases have been shown to have significant association to genes in this
region. LMP2 and LMP7 also make good candidate genes since they are located within
the MHC class II region of the genome, a region that historically has been linked with
susceptibility to many other autoimmune diseases (Wong and Wen, 2003).
Transporter associated with antigen processing 1 and 2 (TAP1 and TAP2)
Along with the immunoproteasome genes, TAP1 and TAP2 are involved in the
MHC class I antigen-processing and presentation pathway. To reach the MHC class I
molecule for binding, the proteasome-cleaved peptide must cross into the endoplasmic
reticulum from the cytoplasm. The TAP complex allows for this transfer of peptides into
the ER. TAP is an IFN-γ inducible heterodimer made up of two subunits, TAP1 and
TAP2, which both must be present for peptide binding and translocation. The TAP
complex preferentially binds peptides of certain lengths and of certain amino acid
composition, favoring acidic, aromatic, hydrophobic and charged residues (Harding et al.,
1997). These TAP/MHC class I-preferred peptides are produced at a higher rate with the
IFN-γ induction of LMP2, LMP7, and MECL1 (Harding et al., 1997; Rechsteiner et al.,
2000).

17
TAP1 and TAP2 were also chosen as candidate genes due to their reported
associations with other autoimmune diseases such as, celiac disease (Djilali-Saiah et al.,
1994), Sjogren’s syndrome (Kumagai et al., 1997), and multiple sclerosis (Moins-
Teisserenc et al., 1995), as well as due to their location within the MHC class II genomic
region.
CD28 and CTLA4 (CD152)
In order for a naive T cell to become activated, two separate signals are required.
The first signal is the interaction of the T cell receptor on the surface of the T cell with
the peptide/MHC complex located on the surface of an antigen presenting cell (APC).
Secondly, CD28, a surface molecule on the T cell, must come into contact with its ligand,
CD80/CD86, on the surface of an APC. A T cell receiving stimulation with both signals
will be driven to activation and proliferation. Once activated, CTLA4 functions to
regulate this process by shutting down T cell activation and proliferation. CTLA4 is also
found on the surface of T cells, and is upregulated after activation. CTLA4 out-competes
CD28 for the binding of its ligand, CD80/86 as CTLA4 binds CD80/86 with an affinity
about 100 x greater than CD28. CTLA4 binding to the ligand delivers a negative signal
to the T cell, limiting its activation and proliferative response. CTLA4 polymorphisms
have been linked to various autoimmune diseases such as insulin-dependant diabetes
mellitus (IDDM), Hashimoto’s thyroiditis, multiple sclerosis, and celiac disease
(Einarsdottir et al., 2003; Udea et al., 2003; Kantarci et al., 2003).
CD4
CD4 is a single chain molecule composed of four immunoglobulin-like domains,
whose cytoplasmic domain interacts with a tyrosine kinase known as Lyk, allowing for
CD4 to participate in signal transduction. CD4 functions as a co-receptor on T cells,

18
interacting with the peptide:MHC class II complex on APCs and acting synergistically
with the T cell receptor (TCR) in signaling and activation of the T cell. CD4 was chosen
as a candidate gene because improper binding of CD4 to the peptide:MHC complex or
the TCR , or improper signaling through the CD4 receptor might allow for inappropriate
activation of T cells. Studies have also shown epidermis-infiltrating T cells exhibit an
increased CD8/CD4 ratio within perilesional skin in vitiligo patients (Le Poole et al.,
1996).
IL-12p40
IL-12 is a very important cytokine that is important in the induction of the Th1
subset that produces inflammatory cytokines. The potent pro-inflammatory activity of
IL-12 requires tight control, which is exerted at various levels. Primary control is exerted
on IL-12 production by APCs, a major factor driving the response towards the Th1 or
Th2 phenotype. A disturbed Th1/Th2 balance, and/or aberrant control of Th1 cytokines,
such as IL-12, has been hypothesized as playing a role in induction of T cell mediated
autoimmunity. IL-12p40 has also been shown to be associated with type 1 diabetes
(Davoodi-Semiromi et al., 2002).
IL-1β
IL-1 is a proinflammatory cytokine secreted mainly by macrophages. IL-1 from
activated macrophages stimulates cytokine and cytokine receptor production by T-cells as
well as stimulating B-cell proliferation. Two forms of IL-1, IL1-α and IL-1β, have the
same activities but different structures. IL-1α is primarily membrane-associated whereas
IL-1β is secreted. Excess IL-1 may induce inflammatory and autoimmune diseases in
such organs as the joints, lungs, gastrointestinal track, central nervous system, and blood

19
vessels. Treatment with IL-1 receptor antagonist has been used in animal models of
rheumatoid arthritis (Schwab et al., 1991), Inflammatory bowel disease (Cominelli et al.,
1992), and allergic asthma (Selig et al., 1992), with much success. IL-1 may be a
mediator of damage to the pancreatic islets in insulin-dependent diabetes mellitus
(IDDM), and IL-1Ra has been shown to block the IL-1-induced decrease in insulin
secretion by pancreatic beta cells in vitro (Sandler et al., 1991; Eizirik et al., 1991).
Because IL-1β has been linked to these autoimmune diseases, and due to its potent pro-
inflammatory nature, it makes a good candidate gene for vitiligo.
Autoimmune polyendocrinopathy syndrome type-1 (APS-1)/autoimmune regulator(AIRE)
The AIRE gene is responsible for autoimmune polyendocrinopathy-candidiasis-
ectodermal dystrophy syndrome (APECED). The most common features of APECED
are parathyroid gland failure, chronic susceptibility to candida yeast infection, and
Addison's disease. Other manifestations, and when the symptoms emerge, are variable.
Other autoimmune diseases associated with APECED may include alopecia, vitiligo,
ovarian failure, testicular atrophy, hypothyroidism, gastric parietal cell atrophy, hepatitis,
intestinal malabsorption, and insulin-dependent diabetes mellitus. Because vitiligo is one
of the autoimmune diseases associated with APECED, it was chosen as a candidate gene.
Melanocyte Biochemistry and Oxidative Stress
Catalase (CAT)
Catalase is a homotetrameric, heme-containing, peroxisomal enzyme that catalyzes
the conversion of hydrogen peroxide (H2O2) to water and oxygen. Catalase plays an
important role in the prevention of cell damage from highly reactive oxygen-derived free
radicals. Catalase was chosen as a candidate gene because of reports of low epidermal

20
catalase activity in both lesional and non-lesional skin of vitiligo patients. This decrease
of catalase expression is not observed in patient erythrocytes, and is also observed with a
concomitant increase in epidermal H2O2 (Schallreuter et al., 1991,1999a 1999b).
GTP cyclohydrolase 1 (GCH1)
GCH1 is the first and rate-limiting step in the synthesis of 6-tetrahydrobiopterin,
which serves as a cofactor for the hydroxylation of the amino acid L-phenylalanine to L-
tyrosine. L-tyrosine is needed as a substrate for tyrosinase to initiate melanogenesis in
the melanocyte.

21
CHAPTER 2CASE/CONTROL AND FAMILY-BASED ASSOCIATION STUDIES
Introduction
The importance of genetic factors in vitiligo susceptibility has been suggested by
reports of significant familial aggregation (Bhatia et al., 1992; Nath et al., 1994). Vitiligo
susceptibility does not follow a simple Mendelian inheritance pattern, and is considered a
complex hereditary disease influenced by a set of recessive alleles occurring at several
unlinked autosomal loci. Therefore vitiligo is considered a polygenic disease, with
alleles at multiple loci possibly contributing to increased susceptibility to and/or direct
pathogenesis of vitiligo. As previously described, two principal hypotheses concerning
the etiology of vitiligo include (1) the self-destruct model, which suggests that
biochemical and/or structural defects inherent to patient melanocytes contribute to the
initiation and/or progression of melanocyte cytolysis, and (2) the autoimmune model,
which suggests that melanocyte death occurs through inappropriate immune system
destruction of pigment cells. To evaluate genetic factors that may play a role in vitiligo
pathogenesis, a candidate gene approach was designed. Genes involved in both immune
system regulation and melanocyte regulation and biochemistry were chosen based on
their potential involvement in vitiligo etiology. Case/control and family-based analyses
were performed on genotypic data garnered from vitiligo patients, their family members,
and non-affected controls.

22
Materials and Methods
Subjects
An internet website was set up at both the University of Florida and at the National
Vitiligo Foundation. Interested patients contacted the research group via email to request
further information, or to participate in the research study. Kits were mailed out to
interested patients and family members containing three 10 mL EDTA blood collection
tubes, IRB consent forms (project numbers 130-1998 and 416-1998), personal
information surveys requesting information such as race, age of onset, family history
and/or personal presence of other autoimmune diseases, and pattern of depigmentation.
Patients and family members were collected and genotyped regardless of race and/or
ethnicity, however, due to insufficient numbers of subjects in other racial groups the
case/control and family-based analyses were performed on Caucasian subjects only
(Table 2-1). Patients requested a blood draw from their local family physician and
returned the blood kits via priority mail.
Blood Processing
Whole blood was centrifuged at 3000 rpm (2000 x g) for ten minutes in vacutainer
tubes in a clinical centrifuge. The buffy coat, the layer between the red blood cells and
plasma layer that contains white blood cells, was removed and transferred with a sterile
transfer pipette into a 50 mL conical tube. Red blood cells were then lysed to isolate
white blood cells with 6-8 volumes of red blood cell lysis solution (RBC) (139 mM
ammonium chloride, 17 mM Tris-HCl, pH 7.65), followed by a ten-minute incubation at
37°C. Samples were then centrifuged at 2000 rpm (900 x g) for seven minutes, the
supernatant discarded and the pellet washed with PBS (38 mM KCl; 21 mM KH2PO4;
1.96 M NaCl; 137 mM Na2HPO4). The pellet was then resuspended in 5 mL of high TE

23
Table 2-1. Vitiligo patient and unaffected relative samples collected for case/control andfamily-based analyses.
Vitiligo Patients Unaffected Relatives
Gender Race Gender Race
Female 255 Caucasian 315 Female 223 Caucasian 323
Male 152 Hispanic 34 Male 183 Hispanic 32AfricanAmerican 11
AfricanAmerican 8
Indian (Asian) 12 Indian (Asian) 11
Other Asian 9 Other Asian 6
Mixed 16 Mixed 14
Total 407 Not Reported 10 Total 406 Not Reported 12

24
(100 mM Tris-HCl, pH 8.0; 40 mM EDTA, pH 8.0) and 5 mL of DNA lysis solution (100
mM Tris-HCl, pH 8.0; 40 mM EDTA, pH 8.0; 400 mL dH2O; 0.22% SDS).
DNA Extraction
Lysed genomic DNA samples were mixed with 10 mL TE-saturated phenol at pH
6.6 (Fisher Scientific) and centrifuged at 2000 rpm (900 x g) for 5 minutes. The aqueous
layer which contains the DNA was transferred via sterile transfer pipette to a 50 mL
conical tube, to which 5 mL phenol (Amersham) and 5 mL chloroform (Fisher Scientific)
were added. The samples were then mixed gently, and centrifuged at 2000 rpm (900 x g)
for 5 minutes, and the aqueous layer was once again removed and transferred to a clean
50 mL conical tube. A final extraction was conducted using 5 mL chloroform. The DNA
was precipitated with the addition of ¼ the volume of 7.5 M ammonium acetate and an
equal volume of isopropanol. The precipitated DNA was rinsed with 70-100% ethanol,
using a Pasture pipette hook, and transferred into a 1.5 mL tube containing 400 µL TE
and placed on a rotator for one to two days. The DNA was quantitated using a
spectrophotometer at 260 nm, diluted to 20 ng/µL, and aliquoted 2 µL/well into a 96-well
PCR plate (DOT scientific). Control samples were prepared from Caucasian subjects
with no history of autoimmunity, and were generously provided by Dr. Jin Xiong She
(formerly at the University of Florida, now at Medical College of Georgia, Center for
Biotechnology and Genomic Medicine).
Primers
PCR primer sequences were designed using OLIGO software, or were obtained
from the literature (Table 2-2). Primers (Gibco-Life Technologies) were reconstituted
with 100 µL of 10 mM Tris-HCl and diluted to a working concentration of 20 pmol/µL

25
with 10 mM Tris-HCl. PCR conditions for each primer pair were determined
empirically, and candidate genes were genotyped using RFLP, AFLP and microsatellite
markers.
Microsatellite Markers
Forward PCR primers were labeled with 10 µCi γ-32P-ATP (6000 µCi/mol) with 1
µL T4 polynucleotide kinase (New England Biolabs), 3µL 10X kinase buffer (New
England Biolabs), 5 µL ddH2O, with a one hour incubation at 37°C and ten minutes at
65°C. Each genomic DNA sample was amplified using 10 µL of a PCR master mix
containing 710 µL ddH2O, 120 µL of 10X dNTP, 12 µL unlabeled forward primer, 10 µL
labeled forward primer, 12 µL reverse primer, and 7.2 µL Taq polymerase to each well of
the 96-well PCR plate. After thirty PCR cycles, 20 µL of stop buffer (92% formamide; 2
µM EDTA, 0.03% xylene cyanol, 0.03% bromophenol blue) was added to each of the 96
wells. Samples were then heat denatured at 97°C for 15 minutes and separated by
electrophoresis on a denaturing 6% acrylamide gel, using 0.5% TBE buffer. The gels
were vacuum dried, and exposed to Fuji X-ray film for 2-4 days at -80°C. PCR
conditions, product size and number of alleles for each marker can be found in Table 2-3.
RFLP and AFLP Markers
PCR amplification of RFLP and AFLP markers was completed using 10 µL of a
master mix containing 710 µL ddH2O, 120 µL of 10X dNTP (2 µL each dNTP, 92 µL 10
mM Tris-HCl, pH 7.5), 12 µL forward primer, 12 µL reverse primer, and 7.2 µL Taq
polymerase to each well of the 96-well PCR plate. Cycle lengths were determined
empirically. Following amplification, samples were then digested overnight with the

26
Table 2-2. Primer sequences
Gene Forwardprimer
Sequence ReversePrimer
Sequence Reference
CD28 CD28-10 GAG AAT CGC TTG AAC CTG GC CD28-15 TAG ACA AAT AAT CCT TCA CAG TA Marron et al.,2000
CTLA4 CTLA4-1 GCC AGT GAT GCT AAA GGT TG CTLA4-2 ACA CAA AAA CAT ACG TGG CTC Marron et al.,1997
CTLA4 CTLA4-4 CTG CTG AAA CAA ATG AAA CCC CTLA4-5 AAG GCT CAG CTG AAC CTG GT Marron et al.,1997
CTLA4 CTLA4-22 CCCTGGCATTGTTGTAGAGTG CTLA4-24 CACTATTTTTGAGTTGATGCAG Marron et al.,2000
IL-12p40 IL-12-1 ATTTGGAGGAAAAGTGGAAGA IL-12-2 AATTTCATGTCCTTAGCCATA Davoodi-Semiromi etal., 2002
IL-1β IL-1β-1 CAT CTG GCA TTG ATC TGG TT IL-1β-2 TTT AGG AAT CTT CCC ACT TAC di Giovine et al.,1992
APS-1 APS-1-1 TATGTGCTTGGGAACAGTCTT APS-1-2 ATCAGCCCCATCTCCCCG Genbankrs878081
APS-1 APS-1-3 GCGGGAGAGGAGGTAAGAG APS-1-4 AGGACCCACACACAGTAGG Genbankrs1800521
GCH1 GCH1-1 GGG TTG AGC CCT CTA CTT TC GCH1-2 TCG GCA CTA CAC CAC TTT TAT T Genbank rs841
SNP ID reference numbers are from the National Center for Biotechnology Information Single Nucleotide Polymorphism database(http://www.ncbi.nlm.nih.gov/SNP).

27
Table 2-3. Microsatellite primer conditions.
Gene PrimerPair
Marker Size Anneal°C
MarkerType
Numberof alleles
CD28 10/15 CAA 3' UTR 166 58 tri repeat 7
CTLA4 1/2 AT 3' UTR 110 58 di repeat 33+
CD4 1/2 TTTTC 5' UTR 170 58 Penta-repeat 6
Table 2-4. RFLP/SSCP PCR conditions.
Gene PrimerPair
Marker Size Anneal °C Markertype
RFLPenzyme
CTLA4 4/5 BstE II +49 A/G 152 58 RFLP BstE II
CTLA4 22/24 Hae III C/Tintron 1
257 58 RFLP Hae III
IL-12p40
1/2 Taq I C/A 3'UTR
1046 58 RFLP Taq I
IL-1β 1/2 Ava I C/Tpromoter -511
308 58 RFLP Ava I
APS-1 1/2 C/T 715 exon 5 203 60 SSCP
APS-1 3/4 Hae III 1324exon 10 T/C
150 62 RFLP Hae III
GCH1 1/2 Bsa A1 SNP841exon 6
250 54 RFLP Bsa A1

28
restriction enzyme appropriate for each RFLP/AFLP, according to the protocol of the
supplier (New England Biolabs). Restriction enzyme concentrations for complete
digestion were determined empirically for each marker. Digestion products were then
separated by agarose gel electrophoresis and visualized using ethidium bromide staining
and a 312 nm UV-transilluminator. Samples were genotyped as homozygous for the
restriction site (cut), homozygous for no restriction site (uncut) or heterozygous. PCR
conditions, and product size for each marker can be found in Table 2-4.
Statistical Analysis
Blood samples were collected from vitiligo patients and their family members
from all ethnic groups, however only the Caucasian ethnic group was large enough to
run a valid case/control analysis on the genotypic data. Patients were also divided into
two groups, segmental or non-segmental, based on the appearance/diagnosis of their
lesion types. Only patients with non-segmental lesions were included in the
case/control and TDT analysis. Allele and genotype frequencies were calculated for
each genetic marker for the patient and control data sets. These allele and genotype
frequencies were then compared between the affected and non-affected populations. If
the particular gene or allele had no association with a particular disease, the frequency
of that allele or genotype in the patient population should be very similar to the
frequency of that allele or genotype in the control population. An allele or gene that
confers susceptibility or resistance to a particular disease would hypothetically differ in
frequency between the affected and non-affected populations. To compare the
frequency of an allele or genotype between the case and control populations, a chi-

29
square (χ2) analysis is performed. Chi-square analysis is represented by the following
equation:
χ2 = Σ[(Expected Value – Obtained Value)2]/Expected Value
To further validate statistical significance determined by the case/control analysis,
the transmission disequilibrium test (TDT), a family-based association test, was
performed. The TDT examines whether alleles of the marker of interest are transmitted
at a frequency greater than 50% from an informative parent to an affected child. In the
TDT, an informative parent is defined as a parent who is heterozygous for the allele of
interest. Chi-square analysis is then performed to determine the statistical significance
of the TDT. The equation for the TDT in a 2 allele situation (i.e. RFLP, AFLP) is as
follows:
TDT = (b-c)2 / (b+c)
Where b = the number of times allele 1 is transmitted from a heterozygous parent to an
affected child and c = the number of times allele 2 is transmitted from a heterozygous
parent to an affected child (Haines and Pericak-Vance, 1998)
For a microsatellite marker where many possible alleles can be transmitted, the
TDT is then represented by the following equation:
Tkhet = [(k-1)/k] Σi (ni. – n.i)2/ ni. + n.i – 2nii
Where k = the number of marker alleles
ni. = the total number of times allele i is transmitted to affected offspring
n.i = the total number of times allele i is not transmitted to affected offspring
nii = ni. * n.i (Haines and Pericak-Vance, 1998; Spelman and Ewens, 1996)

30
Bonferroni’s correction is a statistical method of adjusting and correcting for
multiple numbers of chi-square tests. Bonferroni’s correction is represented by dividing
the test-wise significance level by the number of tests, and is represented by the
following equation:
αβ = α / k
Where α = the testwise significance level (0.05)
K = the total number of tests performed (Shaffer, 1995).
Bonferroni’s correction was used on both RFLP/AFLP and microsatellite data.
It has been suggested that there may be a difference between the etiologies of early
onset and late onset vitiligo (Arcos-Burgos et al., 2002). To investigate this
phenomenon, case/control analyses were also conducted separately on individuals
whose age of diagnosis was less than 30 years of age.
Results
Immune System Genes
Genetic association of CD28 with vitiligo was tested using a trinucleotide repeat.
Allele and genotype frequencies for the patient and control groups are seen in Figure 2-
1. Case/control analysis of this data revealed significance for allele 6, with a p value of
0.0036, and a corrected p value of 0.011 (Table 2-5). This allele is seen about 2 times
as frequently in the control population, hence it can be said to have a protective nature.
Family-based TDT analysis does not support association for this marker (Tkhet = 12.0,
p=0.101).
To determine potential susceptibility of the gene CTLA4, three polymorphisms were
used, two RFLP markers and one microsatellite marker. The microsatellite marker is an
AT dinucleotide repeat located in the 3' untranslated region. This marker has a huge

31
population variance, with at least 33 identified alleles. Figure 2-2 shows the allele and
genotype distribution for CTLA4. We found suggestion of association of single allele 8
(p=0.0140, corrected p=n.s.), which was seen more often in controls than in the patient
population and the grouped alleles 24-33, which were seen more often in patients than
in controls (p=0.0337, corrected p=n.s.) (Table 2-6) These alleles were grouped
together due to the high level of polymorphism associated with this marker. Alleles
with larger number of repeats were difficult to accurately identify, and thus alleles with
more than 24 repeats were grouped into one large group for analysis purposes. One
genotype 18/19 was also found to be possibly associated with vitiligo (p= 0.0362,
corrected p= n.s.), with this genotype seen more often in the control population than the
patient population. Neither the putative significant alleles nor the significant genotype
p values hold up after Bonferroni’s correction, which corrects for multiple chi-square
tests. The TDT also does not support association for this marker (Tkhet= 12.3, p=0.197)
(Table 2-5). The two RFLP markers CTLA 4/5 (Bst EII position +49 A/G) and CTLA4
22/24 (Hae III intron 1 C/T) were also genotyped and used in analysis. Neither marker
showed significant association with vitiligo through case/control analysis using all
patients as well as in patients with early onset vitiligo, however TDT analysis shows
significant association of both of these markers with vitiligo with p values of 0.006 and
0.043 respectively (Table 2-7 and 2-8).
Two markers were used to analyze the candidate gene APS-1, including one
RFLP and one SSCP. Analysis of the SSCP marker, APS-1 1/2, revealed no association
of this marker with vitiligo using case/control or TDT analyses. The RFLP marker,
APS-1 3/4, also showed no association with vitiligo through case/control for the total

32
Figure 2-1. CD28 allele frequency. P=vitiligo patient alleles, C=control alleles.Asterisks represent significant differences in allele frequency betweenpatient and controls.

33
Figure 2-2. CTLA4 allele frequency. The black bars are patient alleles, the checkeredbars are control alleles Asterisks represent significant differences in allelefrequency between patient and controls.

34
Table 2-5. Case/control association analysis for CD28 by microsatellite (CAA 3' UTR)
Number of AllelesGenotyped
Genotype orAllele
% Patient % Control p value Corrected pvalue
RelativeRisk (RR)
TDT
Patient Control N p value
398 366 Allele 6 5.0 10.7 0.0036 0.011 0.47 43 n.s.
Table 2-6. Case/control association analysis for CTLA4 by microsatellite (AT 3' UTR)
Number of AllelesGenotyped
Genotype orAllele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
TDT
Patient Control N p value
438 302 Allele 8 43.2 52.3 0.0140 n.s. 0.82 97 n.s.
Alleles 24-33 20.3 14.2 0.0337 n.s. 1.42
Genotype18/19
0 2 .0362 n.s. 0

35
Table 2-7. Case/control association analysis of CTLA4 by RFLP (Bst E II +49 A/G)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
Relative Risk(RR)
N p value
Total 432 368 1
2
42.1
57.9
39.4
60.6
n.s. n.s. 59 0.006
Age of onsetless than 30
176 368
Table 2-8. Case/control association analysis of CTLA4 by RFLP (Hae III intron 1 C/T)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p value
Total 404 334 1
2
43.3
56.7
41.6
58.4
n.s. n.s. 48 0.043
Age of onsetless than 30
180 334

36
vitiligo patient population, however, TDT analysis suggests association between this
marker and vitiligo (p=0.035). Grouping the patients with early onset (before 30 years
reveals allele 1 and genotype 1,1 to be possibly associated with vitiligo with p values of
0.0171 and 0.0174, respectively. Using Bonferroni’s correction, these p values remain
barely significant (p=0.05) (Tables 2-9 and 2-10).
A RFLP marker in the promoter region of IL-1β was genotyped. There was no
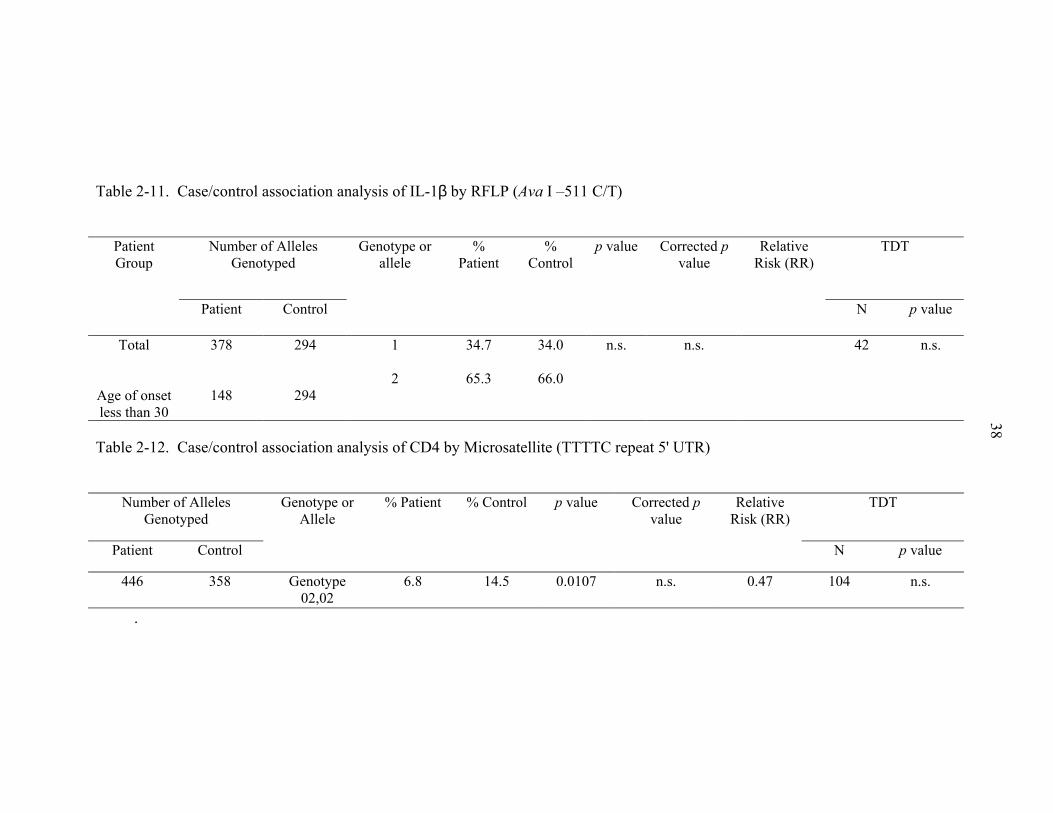
association with vitiligo suggested by either case/control or TDT analysis (Table 2-11).
The CD4 gene was genotyped using a pentanucleotide repeat (TTTTC) in the 5' UTR of
the gene. Case/control analysis found a significant p value for this microsatellite
marker (Table 2-7). Case/Control association analysis of CTLA4 by RFLP (Bst E II
+49 A/G) genotype 02,02 (p=0.0107). Bonferroni’s correction allowed the p value to
remain just within the limits of significance (Table 2-12). Family-based analysis does
not support association of this gene with vitiligo. IL-12p40 was genotyped using a C/A
SNP in the 3' UTR. Case/control analysis revealed no association of vitiligo with any
allele or genotype for the total patient population, however segregating the population
into early onset vitiligo patients found that genotype 2,2 had a significant p value
(p=0.0291, corrected p=n.s.). TDT analysis revealed a potential association of IL-12
with vitiligo with a p value of 0.0396, however the number of informative families in
this analysis was very low (n=34) (Table 2-13).
Melanocyte-specific Genes
A RFLP maker for GCH1 in exon 6 was evaluated by case/control and TDT
analysis for potential association with vitiligo. No association was found in either the
case/control or family-based association analyses (Table 2-14).

37
Table 2-9. Case/control association analysis of APS-1 by SSCP (C/T exon 5)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p value
Total 320 344 1
2
19.4
80.6
22.1
77.9
n.s. n.s. 15 n.s.
Age of onsetless than 30
72 344
Table 2-10. Case/control association analysis of APS-1 by RFLP (Hae III exon 10 T/C)
Number of AllelesGenotyped
TDTPatient Group
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p valueTotal 536 362 44 0.035
Age of onsetless than 30
174 362 Allele 1 71.8 61.3 0.0171 0.05 1.17
Age of onsetless than 30
Genotype 11 50.6 35.4 0.0174 0.05 1.87
38
Table 2-11. Case/control association analysis of IL-1β by RFLP (Ava I –511 C/T)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p value
Total 378 294 1
2
34.7
65.3
34.0
66.0
n.s. n.s. 42 n.s.
Age of onsetless than 30
148 294
Table 2-12. Case/control association analysis of CD4 by Microsatellite (TTTTC repeat 5' UTR)
Number of AllelesGenotyped
TDT
Patient Control
Genotype orAllele
% Patient % Control p value Corrected pvalue
RelativeRisk (RR)
N p value
446 358 Genotype02,02
6.8 14.5 0.0107 n.s. 0.47 104 n.s.
.

39
Table 2-13. Case/control association analysis of IL-12p40 by RFLP (Taq 1 C/A 3' UTR)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p value
Total 388 352 34 0.0396
Age of onsetless than 30
162 352 Genotype 2,2 6.2 1.7 0.0291 0.087 3.63
Table 2-14. Case/control association analysis of GCH1 by RFLP (Bsa A1 C/T exon 6)
Number of AllelesGenotyped
TDTPatientGroup
Patient Control
Genotype orallele
%Patient
%Control
p value Corrected pvalue
RelativeRisk (RR)
N p value
Total 436 284 46 n.s.
Age of onsetless than 30
174 284

40
Discussion
The candidate genes CTLA4/CD28, APS-1, IL1β, CD4 and IL-12 were analyzed
based on their roles in modulating the immune response. GCH1 was analyzed due to its
role in melanocyte biochemistry. The genetic markers chosen for both IL1β and GCH1
showed no association with vitiligo. All the other immune related genes reported here
showed at least one marker with a significant (p≤0.05) genotype or allele in either the
case/control or the TDT association studies. Although the p values for several
individual allele(s) and/or genotype(s) appeared to be significant, after application of
Bonferroni’s correction for multiple Chi square analyses, many of there p values were
not significant. These data suggest that there may be a weak or spurious association of
several of these immune response genes and the human autoimmune disease vitiligo.
Analysis of a microsatellite marker within the CD28 gene showed possible
association of a protective allele with vitiligo using case/control analysis. However,
family-based studies did not confirm this association. Three CTLA4 markers were
analyzed, including one microsatellite and two RFLP polymorphisms. The three
CTLA4 polymorphisms revealed no significant alleles or genotypes, however both
RFLP markers showed possible association with vitiligo using family-based association
studies.
Because of the important regulatory role of CTLA4 in T cell activation, it has
been considered a likely candidate for involvement in autoimmune diseases. A likely
insulin-dependent diabetes mellitus (IDDM) susceptibility locus, IDDM12, was
discovered in the CTLA4/CD28 genomic region. Using the same CTLA4 and CD28
polymorphic markers employed in this study, researchers discovered highly significant

41
association of all three CTLA4 markers with IDDM, while the CD28 marker suggested
no association (Marron et al., 1997). Association of the CTLA4 gene has also been
suggested with other autoimmune disorders, such as Graves’ disease, autoimmune
hypothyroidism (Einarsdottir et al., 2003; Udea et al., 2003), multiple sclerosis
(Kantarci et al., 2003), and rheumatoid arthritis (Rodriguez et al., 2002).
A study of 74 British vitiligo patients revealed no primary association of the
CTLA4 dinucleotide repeat with vitiligo. However, this same study demonstrated an
association of the AT dinucleotide polymorphism with vitiligo patients who also
suffered from one or more other autoimmune disorders (Kemp et al., 1999). Similar
data stratification of our patients into those who suffer from vitiligo alone, or those who
suffer from multiple autoimmune disorders does not replicate the enhanced significance
of the CTLA4 polymorphism as seen by Kemp et al. (Kristensen, 2000). While the
report by Kemp and coworkers does not support our findings of CTLA4 association
with vitiligo, it does lend evidence to an association of vitiligo with a general immune
system dysfunction. It is also important to keep in mind that there may well be several
divergent etiologies, and CTLA4 may be associated with vitiligo that is primarily
autoimmune in origin. A functional explanation for autoimmune association of the
dinucleotide repeat has been suggested. The CTLA4 AT dinucleotide repeat varies in
number from 1 repeat to greater than 33 repeats. Increased numbers of dinucleotide
repeats have been shown to affect RNA stability, and in particular AT rich regions in
UTRs have been documented to decrease stability (Kemp et al., 1999).
APS-1 or AIRE is the gene responsible for autoimmune polyendocrinopathy-
candidiasis-ectodermal dystrophy syndrome (APECED), otherwise known as

42
autoimmune polyglandular syndrome (APS). It is an autosomal recessive autoimmune
disease, caused by mutations in the AIRE (autoimmune regulator) gene. The most
common features of APECED are parathyroid gland failure, chronic susceptibility to
candida yeast infection, and Addison's disease. Other autoimmune diseases associated
with APECED include alopecia, vitiligo, ovarian failure, testicular atrophy,
hypothyroidism, gastric parietal cell atrophy, hepatitis, intestinal malabsorption, and
insulin-dependent diabetes mellitus. The AIRE protein is predominantly expressed in
thymic epithelial cells (TECs) but also in some monocyte-derived cells of the thymus,
in a subset of cells in lymph nodes, and in the spleen and in fetal liver. Due to this
expression pattern, it is thought that the AIRE protein may be involved in the
maintenance of thymic tolerance, and perhaps mutations in this gene may be
responsible for incomplete negative selection of self-antigens, resulting in the eventual
development of multiple autoimmune disorders (Pitkanen et al., 2003).
A weak association of APS-1 with vitiligo was shown in the case/control study
with the RFLP marker in exon 10 in patients with early-onset vitiligo (Table 2-10). The
family-based TDT reaffirms these results with a weakly significant p value of 0.035.
Since the APS-1 RFLP p values are not strong, and because the other SSCP APS-1
marker showed no association with vitiligo, it is not likely that a defect in the AIRE
gene is responsible for vitiligo susceptibility. It is possible that some of the vitiligo
patients used in this study are also APS patients who at the time of blood donation had
not yet been diagnosed, or who had not yet developed other autoimmune problems.
This explanation could be a reason for the significant p values found in the population
of vitiligo patients who developed the disorder before age 30.

43
CD4 is a co-receptor molecule found on CD4+ T cells, which functions by binding
the peptide:MHC class II complex on the APC and through associating with the TCR on
the T cell. CD4 is a single chained molecule composed of four immunoglobulin-like
domains. Signaling through CD4 occurs when its cytoplasmic domain interacts with a
tyrosine kinase known as Lyk. CD4 and the TCR signaling cascades function
synergistically to allow for T cell activation after interaction of the TCR and its specific
peptide located in the binding grove of the MHC class II molecule on an APC.
Theoretically any malfunction of the CD4 receptor could potentially allow for improper
activation and proliferation of CD4+ T cells. We found a weak association of the
microsatellite TTTTC repeat, located in the 5' UTR of the CD4 gene, with vitiligo.
Since this result was not supported by family-based data, thus it is unlikely that CD4 is
a true susceptibility gene for vitiligo.
Interleukin-12 is a pro-inflammatory cytokine secreted from macrophages or
dendritic cells, which plays an important role in the protection against intracellular
pathogens as well as the developmental commitment of T helper 1 cells. IL-12 is
comprised of two disulfide-linked subunits IL-12p40 and IL-12p35, which together
make up the active form of the molecule designated IL-12p70. IL-12 exerts its
biological effects through binding to specific IL-12 receptors (IL-12Rs). IL-12
receptors also play a critical role in determining Th1/Th2 balance. The IL-12Rβ2
subunit is not expressed on resting T cells, but is upregulated when the TCR comes into
contact with antigen (Chang et al., 2000). Because of its pro-inflammatory role, genes
of the IL-12 pathway are therefore good candidates for mediating susceptibility or
resistance to a range of immune disorders.

44
IL-12 polymorphisms have been associated with several autoimmune disorders
such as diabetes (Davoodi-Semiromi et al., 2002), atopic dermatitis, and psoriasis
vulgaris (Tsunemi et al., 2002). In this study the 3' UTR SNP in the IL-12 p40 gene
showed weak association with vitiligo in patients whose age of onset was less than 30
years (p= 0.0291). This association was lost when Bonferroni’s correction was applied
(p=0.087). The family-based TDT study also showed weak association of this
polymorphism with vitiligo with a p value of 0.0396. A physiological effect of this 3'
UTR polymorphism has recently been described. Seegers et al. (2002) observed an
increasing IL-12p70 secretion by monocytes in vitro in those without this
polymorphism, heterozygotes, and homozygotes with the 3' UTR polymorphism
respectively. This increase in IL-12 could contribute to autoimmunity, as it has been
hypothesized that disturbed Th1/Th2 balance, and or aberrant control of Th1 cytokines,
such as IL-12, may play an important role in induction of T cell mediated
autoimmunity.
While many of these immune system candidate genes show statistically
significant p values (p≤0.05) in the case/control and/or family-based analysis, which
would hypothetically identify them as vitiligo susceptibility genes, most of these
associations are statistically weak. It is important to note that vitiligo is most likely a
disease with a multifactorial etiology. Therefore susceptibility genes that are
uncommon in a population, or genes which are only one of many such genes
responsible for the vitiligo phenotype may not show strong associations in case/control
analyses. It is with this in mind that we dismiss these genes possessing weak genetic
association with vitiligo, CTLA4/CD28, APS-1, CD4, and IL-12p40, as being

45
improbable susceptibility genes for vitiligo. In the next chapter, we describe analyses
of a different set of candidate genes, involved in antigen processing and presentation,
using a similar approach.

46
CHAPTER 3ANTIGEN PROCESSING AND PRESENTATION GENES
Introduction
Many human autoimmune diseases have been associated with polymorphisms in
genes located in the MHC class II genomic region of the human HLA locus. This highly
polymorphic region includes several genes involved in the processing and presentation of
antigen to the immune system, including low molecular weight polypeptides -2 and -7
(LMP2 and LMP7) and transporter-associated with antigen processing proteins -1 and -2
(TAP1 and TAP2). These four genes are associated with several autoimmune diseases,
such as type 1 diabetes, juvenile rheumatoid arthritis, ankylosing spondylitis, celiac
disease, Sjögren’s syndrome, and multiple sclerosis (Deng et al., 1995; Prahalad et al.,
2001; Fraile et al., 1998; Djilali-Saiah et al., 1994; Kumagai et al., 1997; Moins-
Teisserance et al., 1995). LMP2 and LMP7 encode IFN-γ inducible subunits of the
immunoproteasome, which replace constitutively expressed subunits of the cytoplasmic
proteasome upon immune system upregulation. The immunoproteasome functions by
degrading ubiquitin-tagged cytoplasmic proteins into peptides that are especially suited
for presentation by MHC class I molecules (Tanaka et al., 1998; Pamer et al., 1998). An
additional IFN-γ inducible subunit, multicatalytic endopeptidase complex-like-1
(MECL1), is part of the immunoproteasome complex, but is encoded on chromosome 16
outside the MHC region. TAP1 and TAP2 encode subunits of an IFN-γ inducible
transporter heterodimer. TAP1 and TAP2 function by binding peptides in the cytoplasm

47
and transporting them into the endoplasmic reticulum where they can be loaded into
nascent MHC class I molecules (Pamer et al., 1998).
As reported for other autoimmune diseases, genes in the HLA region may play a
role in vitiligo pathogenesis. Many HLA associations have been reported between
specific alleles of complement, class I and class II MHC genes with vitiligo, however,
many of these associations are solely found in isolated population groups (Friedman et
al., 1999). A recent segregation and linkage disequilibrium analyses of microsatellite
markers spanning the entire HLA region suggested a major HLA genetic factor
segregating as a dominant factor in patients with early onset of vitiligo (Arcos-Burgos et
al., 2002).
Because of the reported associations of these genes with other autoimmune
diseases, and due to the location of these genes inside the MHC class II genomic region,
we chose to genotype polymorphisms in LMP2, LMP7, TAP1, TAP2 and MECL1.
Because family based association for the SNP in intron 6 of LMP7 was so significant, we
also chose to sequence across LMP7 to search for mutations in this gene. We report here
case-control and family-based genetic association studies for the TAP1, TAP2, LMP7,
LMP2 and MECL1 genes in human vitiligo patients. The LMP/TAP gene region was
found to be significantly associated with vitiligo in Caucasian patients, suggesting a
possible role for MHC class I antigen processing and/or presentation in the anti-
melanocyte autoimmune response involved in vitiligo pathogenesis (Casp et al., 2003).
Materials and Methods
Blood Collection and Processing
Blood collection and processing, DNA extraction and genotyping were performed
as described in chapter 2.

48
LMP7 Sequencing
DNA preparation and PCR amplification
Six sets of PCR primers were designed within flanking introns to amplify all 6
exons (Table 3-1). Two DNA pooled sample sets were created, one containing mixed
DNA from 50 selected vitiligo patients, the other containing a mixture of DNA from 50
unaffected controls. PCR was carried out in a 50-µL volume containing 50 mM KCl, 10
mM Tris-HCl, pH 8.3, 1.5 mM MgCl2; 60 µM of each dNTP, pooled patient and control
genomic DNA, 2 pmol of each primer and 1.5 U Taq DNA polymerase. Samples were
subjected to 35 cycles of 30 s at 94°C for denaturing, 30 s at optimum annealing
temperature, and 30 s at 72°C for extension.
Direct sequencing of PCR products
After PCR amplification, products (50 µL) were electrophoresed on a 2.0% agarose gel.
The DNA fragments were excised from the gel and transferred into 1.5-mL Eppendorf
tubes. The tubes were frozen at –20°C for 5–10 min. The gel slices were smashed while
frozen and then incubated in a 50°–60°C water bath for 10–15 min. The tubes were
briefly vortexed and the PCR products eluted out of the gels by a short centrifugation.
The supernatant was centrifuged once more to remove any remaining agarose. An
aliquot of products (30µL) was then used directly for cycle sequencing using the ABI
Prism BigDye terminator (Applied Biosystems) according to the manufacturer’s
instructions. The sequencing reactions were precipitated in 3 vol. 100% ethanol and 0.1
vol. 3 M sodium acetate, pH 5.2. Pellets were washed with 250 µL of 70% ethanol, dried
in a vacuum dryer and then dissolved in 20 µL of template suppression buffer (Perkin
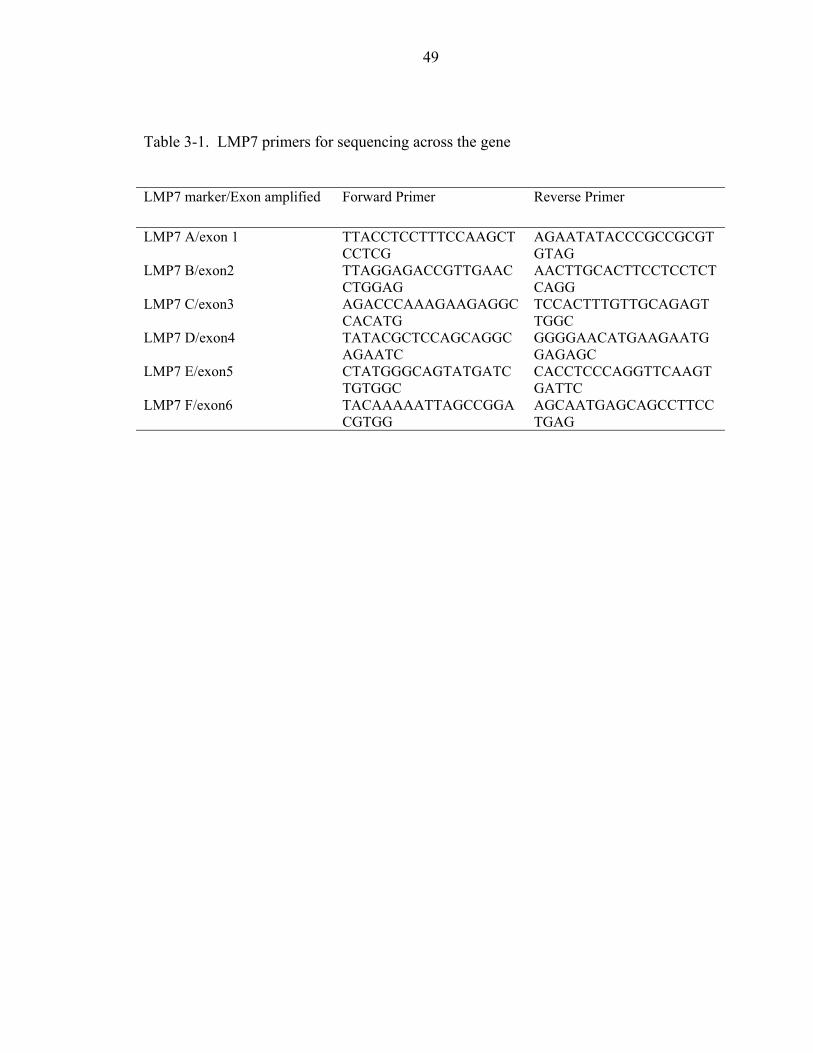
49
Table 3-1. LMP7 primers for sequencing across the gene
LMP7 marker/Exon amplified Forward Primer Reverse Primer
LMP7 A/exon 1 TTACCTCCTTTCCAAGCTCCTCG
AGAATATACCCGCCGCGTGTAG
LMP7 B/exon2 TTAGGAGACCGTTGAACCTGGAG
AACTTGCACTTCCTCCTCTCAGG
LMP7 C/exon3 AGACCCAAAGAAGAGGCCACATG
TCCACTTTGTTGCAGAGTTGGC
LMP7 D/exon4 TATACGCTCCAGCAGGCAGAATC
GGGGAACATGAAGAATGGAGAGC
LMP7 E/exon5 CTATGGGCAGTATGATCTGTGGC
CACCTCCCAGGTTCAAGTGATTC
LMP7 F/exon6 TACAAAAATTAGCCGGACGTGG
AGCAATGAGCAGCCTTCCTGAG

50
Elmer). DNA sequencing was carried out using an ABI 310 automated sequencer
(Applied Biosystems).
Results
Case/Control Association Studies
Allele and genotype frequencies
The single nucleotide polymorphisms (SNPs) used in this study as genetic markers
for the LMP/TAP genes and MECL1 are listed in Table 3-2. Case/control analyses of a
patient population consisting of 230 unrelated Caucasian vitiligo patients and 188
unrelated Caucasian controls revealed significant differences in allele and/or genotype
frequencies suggesting association with the candidate genes TAP1 and TAP2 (data not
shown). However, when population heterogeneity between the case and control sets was
tested by using Wright’s F statistic according to the unbiased methods (θ value) of Weir
& Cockerham, a significant difference between the case and control populations was
indicated (Weir and Cockerham, 1984). As suggested by Arcos-Burgos et al. (2003), we
then stratified the vitiligo patient population by age of onset, as their segregation and
linkage disequilibrium analyses of the HLA region revealed a dominant genetic factor
segregating in vitiligo patients developing this disorder before age 30.
Using these new guidelines there was no significant difference in population
structure between a case population consisting of 100 Caucasian patients with an age of
vitiligo onset of 0-29 years and the control set as defined by subdivision analysis. Using
age of onset as a distinguishing factor, the θ value was not significantly different than
zero (θ = 0.0050, 95% confidence interval -0.0010 - 0.0116). Exact tests for Hardy-

51
Table 3-2. Primers used for LMP/TAP and MECL1 genotyping.
PCR Primers Annealing Restriction Fragment sizes (bp)
Gene Polymorphism Name Sequences (5′ to 3′) Temp. Enzyme Cut Uncut SNP ID
LMP2 G/A exon 3 (R60H) LMP2-2 GTGAACCGAGTGTTTGACAAGC 58°C Hha I 252 212,40 rs17587
LMP2-1 GCCAGCAAGAGCCGAAACAAGTAP1 C/T intron 7 TAP1-15 GTGCTCTCACGTTCCAAGGA 55°C Msp I 183 161,22 rs735883
TAP1-16 AGGAGTAGAGATAGAAGAACCb
TAP1 G/A exon 10 (D637G) TAP1-10 CTCATCTTGGCCCTTTGCTC 60°C Acc I 165 136,29 rs1800453TAP1-11 CACCTGTAACTGGCTGTTTG
LMP7 A/C exon 2 (Q49K) LMP7-Z TCGCTTTACCCCGGGGACTGa 63°C Pst I 212 194,18 rs2071543LMP7-BR AACTTGCACTTCCTCCTCTCAGG
LMP7 G/T intron 6 LMP7-7 TTGATTGGCTTCCCGGTACTG 58°C Hha I 583,180 428,180,155 ref. 1LMP7-4 TCTACTACGTGGATGAACATGG
TAP2 G/A exon 5 (V379I) TAP2-3 GAACGTGCCTTGTACCTGCGCc 57°C BstU I 212 192,20 rs1800454TAP2-4 ACCCCCAAGTGCAGCAC
TAP2 A/G exon 11 (T665A) TAP2-5 GGTGATTGCTCACAGGCTGCCGd 61°C Msp I 225 205,20 rs241447TAP2-6 CACAGCTCTAGGGAAACTC
MECL1 T/C exon 4 (L107L) MECL1-1 TCGACTTGGGTTGCAGGCTTAC 65°C Mlu I 973,131 535,438,131 rs20549MECL1-2 ATCTGAAGTAACCGCTGCGAC
aUnderlined nucleotide in primer LMP7-Z was changed from the germline A to a C to create the Pst I RFLP.bUnderlined nucleotide in primer TAP1-16 was changed from the germline G to a C to create the Msp I RFLP.cUnderlined nucleotide in primer TAP2-3 was changed from the germline T to a G to create the Bst UI RFLP.dUnderlined nucleotide in primer TAP2-5 was changed from the germline T to a C to create the Msp I RFLP
Underlined nucleotides were altered from germline sequence to create restriction sites at the single nucleotide polymorphism (SNP).SNP ID reference numbers are from the National Center for Biotechnology Information Single Nucleotide Polymorphism database(http://www.ncbi.nlm.nih.gov/SNP

52
Weinberg equilibrium and linkage disequilibrium are shown in Table 3-3. Deviation
from Hardy-Weinberg equilibrium was observed for the TAP2 A/G exon 5 genetic
marker in both the patient and control populations. After Bonferroni’s correction for
multiple comparisons, significant linkage disequilibrium was observed between LMP2-
TAP1, LMP2-LMP7 and TAP1-LMP7 genetic markers in patients, and between LMP2-
TAP1, LMP2-LMP7, LMP2-TAP2, TAP1-LMP7, LMP7-LMP7, and LMP7-TAP2
genetic markers in controls (Table 3-3).
The allele and genotype frequencies for the seven LMP/TAP and one MECL1
genetic markers genotyped for 100 Caucasian vitiligo patients with early age of onset (0-
29 years) and 188 unrelated Caucasian control subjects are shown in Table 3-4 and 3-5.
No significant differences were observed for the allele counts and frequencies of six of
the eight markers examined (Table 3-4). The G/T SNP in intron 6 of the LMP7 gene
displayed an excess of the G allele in the patient vs. control groups (p=0.040). The G/A
SNP in exon 10 of the TAP1 gene (D637G) exhibited a significant excess of the G allele
in the patient vs. control groups (p=0.0034).
Table 3-5 lists genotype counts and percentages for 100 Caucasian vitiligo patients
with early age of onset and 188 unrelated controls, as well as the p values determined by
χ2 analysis of 3×2 contingency tables. No significant differences were observed for
seven of the eight markers genotyped. The G/A SNP in exon 10 of the TAP1 gene
(D637G) exhibited a significant excess (p=0.0094) of the GG genotype allele in vitiligo
patients (71.4%) compared to controls (53.4%). Furthermore, comparison of the
individual allele carriage rates for this marker confirmed that the A allele carriage rate is
significantly lower in patients than in controls (28.6% vs. 46.6%, pc=0.0073), and that

53
Table 3-3. Linkage disequilibrium analysis of Caucasian vitiligo patients (age of onset 0-29 years) and control subjects.
LMP2 TAP1 TAP1 LMP7 LMP7 TAP2 TAP2 MECL1G/A exon 3 C/T intron 7 G/A exon 10 A/C exon 2 G/T intron 6 A/G exon 5 A/G exon 11 T/C exon 4
CasesLMP2 G/A exon 3 0.067TAP1 C/T intron 7 <10-5 a 0.220TAP1 G/A exon 10 0.068 0.183 0.458LMP7 A/C exon 2 0.347 0.509 0.369 1.000LMP7 G/T intron 6 <10-5 a <10-5 a 0.330 1.000 0.670TAP2 A/G exon 5 0.091 0.260 0.168 0.295 0.618 0.011
TAP2 A/G exon 11 0.812 0.863 0.335 1.000 0.811 0.185 0.625MECL1 T/C exon 4 0.603 0.622 0.647 0.330 0.160 0.640 0.503 0.521
ControlsLMP2 G/A exon 3 1.000TAP1 C/T intron 7 <10-5 a 0.237TAP1 G/A exon 10 0.044 0.027 0.154LMP7 A/C exon 2 0.248 <10-5 a 0.271 0.709LMP7 G/T intron 6 <10-5 a <10-5 a 0.268 0.0003 a 0.537TAP2 A/G exon 5 0.001 a 0.002 0.837 0.229 0.0003 a 0.010
TAP2 A/G exon 11 0.856 0.590 0.170 0.836 0.343 0.204 1.000MECL1 T/C exon 4 0.615 0.621 0.279 0.300 0.867 0.029 0.881 0.298
a p<0.05 after Bonferroni’s correction for multiple tests.P values for exact tests of Hardy-Weinberg equilibrium are shown in the diagonal (bold). Below are p values for the linkagedisequilibrium pairwise tests

54
Table 3-4. Allele frequencies of LMP/TAP and MECL1 candidate genes in Caucasian vitiligo patients (age of onset 0-29 years) andcontrol subjects.
Vitiligo Patients ControlsGene Polymorphism Allele Count Percentage Count Percentage p value
LMP2 G/A exon 3 (R60H) G 62 32.6 95 26.2 0.11A 128 67.4 267 73.8
TAP1 C/T intron 7 C 80 44.4 121 38.8 0.22T 100 55.6 191 61.2
TAP1 G/A exon 10 (D637G) A 29 14.8 89 25.6 0.0034G 167 85.2 259 74.4
LMP7 A/C exon 2 (Q49K) A 165 87.8 296 88.1 0.91C 23 12.2 40 11.9
LMP7 G/T intron 6 G 95 52.2 142 42.8 0.040T 87 47.8 190 57.2
TAP2 A/G exon 5 (V379I) A 40 21.7 74 21.0 0.85G 144 78.3 278 79.0
TAP2 A/G exon 11 (T665A) A 139 71.6 257 69.8 0.65G 55 28.4 111 30.2
MECL1 T/C exon 4 (L107L) T 146 80.2 259 80.9 0.84C 36 19.8 61 19.1

55
Table 3-5. Genotype frequencies of LMP/TAP and MECL1 candidate genes in Caucasian vitiligo patients and (age of onset 0-29years) control subjects.
Vitiligo Patients ControlsGene Polymorphism Genotype Count Percentage Count Percentage p value
LMP2 G/A exon 3 (R60H) GG 6 6.3 12 6.6 0.095GA 50 52.6 71 39.2AA 39 41.1 98 54.1
TAP1 C/T intron 7 CC 21 23.3 27 17.3 0.47CT 38 42.2 67 43.0TT 31 34.5 62 39.7
TAP1 G/A exon 10 (D637G) AA 1 1.0 8 4.6 0.0094AG 27 27.6 73 42.0GG 70 71.4 93 53.4
LMP7 A/C exon 2 (Q49K) AA 73 77.7 131 78.0 0.98CA 19 20.2 34 20.2CC 2 2.1 3 1.8
LMP7 G/T intron 6 GG 26 28.6 28 16.9 0.077GT 43 47.3 86 51.8TT 22 24.2 52 31.3
TAP2 A/G exon 5 (V379I) AA 9 9.8 14 8.0 0.84AG 22 23.9 46 26.1GG 61 66.3 116 65.9
TAP2 A/G exon 11 (T665A) AA 51 52.6 90 48.9 0.83AG 37 38.1 77 41.9GG 9 9.3 17 9.2
MECL1 T/C exon 4 (L107L) TT 60 65.9 107 66.9 0.98TC 26 28.6 45 28.1CC 5 5.5 8 5.0

56
the overall G carriage rate is higher in patients than in controls, although not statistically
significant (99.0% vs. 95.4%, pc=0.22). Taken together, these results suggest a possible
association between the TAP1 gene in the LMP/TAP gene cluster and vitiligo
susceptibility.
Family-based association
Further evidence for genetic association between LMP/TAP genes and vitiligo
susceptibility was sought using the transmission disequilibrium test (TDT), a family-
based (intrafamilial) study design that considers heterozygous (informative) parents and
evaluates the frequency with which alleles are transmitted to affected offspring (Speilman
et al., 1996). A χ2 test was used to evaluate the deviation of the rates of transmission and
non-transmission from the random expectation (Table 3-6). For the TAP1 G/A exon 10
(D637G) marker, only 35 informative (heterozygous) parents could be identified.
Although the number of informative parents is relatively low, analysis of this marker
suggests unequal transmission of alleles to affected children, with the G allele being
transmitted more frequently (69%) than expected due to random chance (p=0.028), which
is consistent with the allele and genotype frequencies described above.
TDT analysis of the other genes in the LMP/TAP cluster revealed additional
evidence for biased transmission of LMP/TAP alleles in vitiligo patients, with significant
differences detected (p≤0.05) for LMP2, TAP1 and LMP7 genetic markers. Similar to
the results for the TAP1 G/A exon 10 marker, the LMP2 G/A exon 3 (R60H) and TAP1
C/T intron 7 markers displayed allele transmission frequencies of 68%, and TDT p values
of 0.023 and 0.011, respectively. The most highly significant TDT result (p<0.00006)
was observed for the LMP7 G/T intron 6 marker, for which there were 45 informative

57
Table 3-6. Family based association (transmission disequilibrium test) results for LMP/TAP and MECL1 candidate genes and vitiligosusceptibility
Gene MarkerNumber of
Informative Parents TransmittedNot
Transmitted%
Transmitted p valueLMP2 G/A exon 3 (R60H) 38 26 12 68 0.023TAP1 C/T intron 7 50 34 16 68 0.011TAP1 G/A exon 10 (D637G) 35 24 11 69 0.028LMP7 A/C exon 2 (Q49K) 26 16 10 62 0.24LMP7 G/T intron 6 45 36 9 80 0.000057TAP2 A/G exon 5 (V379I) 22 12 10 55 0.67TAP2 A/G exon 11 (T665A) 56 31 25 55 0.42
MECL1 T/C exon 4 (L107L) 47 24 23 51 0.88

58
parents available. The G allele was transmitted from unaffected parents to affected
children much more often (80%) than expected due to random chance
LMP7 Sequencing
50 Caucasian patients and 50 Caucasian controls were pooled from the overall
patient and control groups without regard to LMP7 genotype. Pooled groups were
sequenced with the hope of discovering new polymorphisms and/or mutations in the
LMP7 gene based on preliminary results from case/control and TDT analysis, which
initially revealed highly significant p values for both the case/control and family-based
studies. As the number of patients and controls used in the analyses increased, and as
Bonferroni’s correction was applied, significant p values for LMP7 remained only in the
family based analyses. While LMP7 was sequenced completely across all 6 exons, there
was too much background from the 50 pooled samples to discern individual
polymorphisms (especially those with low patient numbers) with confidence. Even
known SNPs used as RFLP markers in the case/control and family based analyses were
not apparent within the sequenced data due to background variation (data not shown).
Discussion
Through the use of a candidate gene approach we have found evidence for genetic
association between vitiligo and the LMP/TAP region. Case/control analyses reveal
association of vitiligo and the gene encoding transporter associated with antigen
processing-1 (TAP1), whereas a family-based association method (TDT) revealed biased
transmission of specific alleles from heterozygous parents to affected offspring for the
TAP1 gene, as well as for the LMP2 and LMP7 genes. No association with vitiligo was
found for the MECL1 gene, which encodes a third immunoproteasome subunit not
located in the MHC class II region (Casp et al., 2003).

59
The LMP/TAP genes play important roles in the regulation of class I antigen
processing and presentation, as their proteins form IFN-γ inducible subunits of the
immunoproteasome and TAP transporter respectively. The proteasome is a large
protease made up of several subunits, which plays a crucial role in protein degradation in
the cell. Proteasomes reside in the cytosol in both free-floating forms and bound to
ribosomes on the endoplasmic reticulum, and in the nucleus. In most cases the
proteasomes degrade intracellular proteins that have been marked for degradation by the
attachment of an ubiquitin moiety. The complete 26S proteasome is made up of two
major components, the 20S core protease subunit, and the 19S ATP-dependant cap
(Bochtler et al., 1999). The structural prototype of the proteasome is of four stacked
heptameric rings in an α7β7β7α7 cylindrical structure. The catalytic sites of the
proteases are located in the β subunits on the interior of the cylindrical structure, and
involve an N-terminal threonine. Other conserved resides in the vicinity of the
proteasome active site include Asp17, Ser129 and Ser169 (Bochtler et al., 1999). The
20S proteasome is made up of at least 14 different subunits (Brooks et al., 2000). Three
of these subunits, known as X, Y, and Z, are constitutively expressed, but can be replaced
by the three IFN-γ inducible subunits LMP2, LMP7, and MECL1. When these inducible
subunits are present, the proteasome is known as the immunoproteasome. It was due to
their inducibility by IFN-γ that these subunits were first believed to play a role in antigen
processing. These inducible subunits have many amino acid similarities to the
constitutive subunits, however they cleave proteins into peptides differently. The IFN-γ
inducible subunits generate peptides with basic carboxyl termini, and hydrophobic
residues, while inhibiting cleavage after acidic residues. These residues are those that are

60
the most compatible with the binding groove of the MHC class I molecule (Tanaka et al.,
1998; Pamer et al., 1998; Driscoll et al., 1993).
Another subunit that is inducible by IFN-γ is known as proteasome activator 28
(PA28) or 11 S REG. PA28 has three subunits, α, β, and γ, which form a ring-shaped
heptameter (Rechsteiner et al., 2000). Evolutionarily, subunits α and β seem to have
been derived from gene duplication of the Ki antigen which is found in lower eukaryotes
(Fruh et al., 1999). Interestingly, anti-Ki antibodies are often found in conjunction with
systemic lupus erythematosus, another known autoimmune disease (Tanaka et al., 1998).
PA28 functions as an activator of the 20S proteasome, by associating with one or both
ends of the proteasome to create a football shaped structure as seen by electron
microscopy (Tanaka et al., 1998). The association of PA28 with the 20S proteasome
greatly enhances the proteasome’s peptidase activities (Rechsteiner et al., 2000).
The proteasome plays an integral role in antigen processing and presentation
through the MHC class I pathway. The MHC class I pathway is mainly responsible for
processing and presenting intracellular pathogens such as viruses and intracellular
bacteria. Processing by either the constitutive or the IFN-γ induced proteasome subunits
influence which peptides are generated and presented by MHC class I molecules
(Schwarz et al., 2000; Sijts et al., 2000; Morel et al., 2000; Maksymowych et al., 1998).
To reach the MHC class I molecule for binding, the proteasome-cleaved peptide must
cross into the endoplasmic reticulum from the cytoplasm. To do this, the peptide must
associate with a molecule known as transporter associated with antigen processing (TAP)
which will transport the peptide across the endoplasmic reticulum. TAP is an IFN-γ
inducible heterodimer that consists of two subunits, TAP1 and TAP2, both of which must

61
be present for peptide binding and translocation. Peptide binding is ATP independent,
but translocation across the ER does require ATP (Pamer et al., 1998). The TAP complex
preferentially binds peptides of certain lengths and of certain amino acid composition,
favoring basic, aromatic, hydrophobic and charged residues (Harding et al., 1997). As
mentioned before, these TAP/MHC class I-preferred peptides are produced at a higher
rate with the IFN-γ induction of the immunoproteasome (Rechsteiner et al., 2000;
Harding et al., 1997).
Without antigen in its binding groove, MHC class I molecules are sequestered in
the ER. Newly synthesized MHC class I α chain associates with a chaperone protein
known as calnexin. Association with calnexin retains the MHC in a partially folded state.
With the addition of the β-2 microglobulin chain, calnexin is released, and the MHC now
binds calreticulin, another chaperone protein, and tapasin, a protein that serves as a bridge
between the TAP complex and the MHC. The MHC will remain in this state until a
peptide that fits its specificity comes into the ER by way of the TAP transporter (Pamer et
al., 1998). MHC class I molecules are each capable of binding a repertoire of peptides
with lengths of 8-10 amino acids, characteristic anchor residues, and basic or
hydrophobic C-termini (Reichsteiner et al., 2000; Rock and Goldberg 1999).
With the majority of the proteins degraded by the proteasome being self-proteins,
the generation of altered forms of antigen by the immunoproteasome and the preferential
transport of MHC class I-compatible antigens across the ER membrane by TAP are
extremely important in influencing the expression of foreign peptide over self peptide.
Changes in function and/or expression patterns of TAP or LMP proteins could potentially
influence the peptide repertoire expressed to circulating lymphocytes, and allow for the

62
induction of inappropriate immune events to cryptic epitopes of self-peptides for which
the immune system has not been made tolerant. TAP1 amino acid polymorphisms,
including D637G (in this report) and I333V, have been reported to influence the
permissiveness of transport of specific peptides across the endoplasmic reticulum in some
models(Quadri and Singal, 1998), and the TAP2 polymorphism T665A (in this report)
has been suggested to influence the antibody response to measles virus vaccine (Hayney
et al., 1997). Two functional studies of human TAP polymorphisms, however, revealed
no significant influence of human TAP1 or TAP2 alleles on peptide binding and
translocation. (Obst et al., 1995; Daniel et al., 1997). Fewer functional studies have been
performed on LMP polymorphisms, however, it has been reported that the LMP2 R60H
and LMP7 Q49K polymorphisms (in this report) affect age-dependent TNF-α apoptosis
and response to interferon in patients with chronic hepatitis C, respectively (Mishto et al.,
2002; Sugimoto et al., 2002).
Immunoproteasomes might also play an integral role in maintaining peripheral
tolerance. Immunoproteasomes are normally expressed by cells in the thymus and by
mature dendritic cells under conditions in which most T cell activation occurs, whereas
elsewhere in the periphery constitutive proteasomes are expressed in the absence of
inflammation. Because non-professional antigen presenting cells in non-inflamed tissue
should only present antigens processed by the constitutive proteasome in their MHC class
I molecules, T cells specific for these peptides may become anergized upon presentation,
due to the lack of costimulation. T cells that become inadvertently activated by self-
peptide at a site of inflammation may “avoid” antigen from the same self-peptide in non-
inflamed sites due to differing epitopes produced by the constitutive proteasome

63
(Groettrup et al., 2001). In vitiligo patients, biochemical defects in the melanin pathway
that promote oxidative damage could be a trigger for excessive inflammation in the skin.
Inappropriate expression or function of either the constitutive proteasome or
immunoproteasome might influence antigen processing and presentation, leading to a
break in peripheral tolerance to melanocyte self-antigens.
Two other recent reports have also implicated the MHC class II region in vitiligo
susceptibility. Using a whole genome scan of a large family cluster with both vitiligo and
Hashimoto thyroiditis, a general autoimmune susceptibility locus (AIS1) was mapped to
human chromosome 1. Evidence was also reported for a Hashimoto disease
susceptibility locus within a chromosome 6 region spanning both the MHC and AIDT1, a
non-MHC locus associated with susceptibility to both Hashimoto thyroiditis and Graves’
disease (Alkhateeb et al., 2002). A linkage disequilibrium analysis of 56 multi-
generation Columbian families with vitiligo using microsatellite markers spanning the
entire human MHC region revealed a major genetic factor within the MHC at 6p21.3-
21.4 with a dominant mode of inheritance in vitiligo patients with an early age of onset
and a recessive mode of inheritance influenced by environmental effects in vitiligo
patients with an age of onset after 30 years of age. Comparisons of a variety of
inheritance models suggested that the most parsimonious genetic model was that of a
major dominant gene plus environmental effects, although multifactorial models could
not be rejected (Arcos-Burgos et al., 2002).
In recent years there have been many reports of association of LMP and TAP genes
with human autoimmune diseases. LMP2 and LMP7 have been reported to be associated
with insulin-dependent diabetes mellitus (Deng et al., 1995); LMP7 is associated with

64
ankylosing spondylitis (Fraile et al., 1998) and with juvenile rheumatoid arthritis
(Prahalad et al. 2001); TAP2 is associated with Sjögren’s syndrome (Kumagai et al.,
1997); and with multiple sclerosis (Moins-Teisserenc et al., 1995). Other HLA
associations have been sporadically reported for vitiligo patients of various ethnic groups,
e.g. DR4 in Caucasian Americans (Foley et al., 1983), A2 in German and Slovak patients
and Dw7 in Slovak patients (Buc et al.; 1996, Schallreuter et al., 1993), Cw6 and DR6 in
Dutch patients (Venneker et al., 1993), and B21, Cw6 and DR53 in Kuwaiti patients (al-
Fouzan et al., 1995). Unlike most autoimmune diseases, there seems to be no common
HLA association with vitiligo. Insufficient numbers of vitiligo patients from other ethnic
and racial populations were available for comparison to Caucasians in this study. Several
studies have reported HLA associations in early onset vs. late onset vitiligo cases (Arcos-
Burgos et al., 2002; Orecchia et al., 1992; Finco et al., 1991). Classification of vitiligo
patients by age of onset was therefore important to detect valid genetic association with
the LMP/TAP gene cluster. We detected no association between the LMP/TAP or
MECL1 genes and late-onset vitiligo (data not shown).
We interpret this association study in the LMP/TAP region with caution, given the
complications caused by a high occurrence of linkage disequilibrium in the region, for
which there are variable and inconsistent reports for markers spanning the entire region
from DM and DPB, through the LMP/TAP cluster, and extending to the DRB1, DQA1
and DQB1 genes (Djilali-Saiah et al., 1996; Van Endert et al., 1992 Carrington et al.,
1994). Similarly, after Bonferroni’s correction, significant disequilibrium values were
observed among some but not all possible pairwise LMP/TAP comparisons within the
vitiligo patient and control populations (Table 3-3). The LMP/TAP gene cluster also

65
features a well-characterized recombination hotspot within intron 2 of the TAP2 gene,
and additional recombination hotspots are located throughout the human MHC locus
(Cullen et al., 1997, 2002; Jefferys et al., 2000). Until functional data are found to
confirm associations with autoimmune disease suggested by genetic studies, definitive
conclusions about genetic association in this region will remain controversial, as
associations with autoimmune disease may be due to linkage disequilibrium with other
genes in the MHC class II region. Although most autoimmune diseases have been linked
to various genes within MHC class II region, it is important to note that to our
knowledge, causative mutations in MHC class II regions have yet to be found to explain
potential associations to disease pathology. It has been suggested that multiple MHC
genes may be contributing to disease pathogenesis, and that statistical analyses may be
averaging the effects of several genes to a centralized gene (Fu et al., 1998).
Nevertheless, our data are consistent with genetic association of the LMP/TAP region
with vitiligo susceptibility

66
CHAPTER 4CATALASE
Introduction
Catalase is a homotetrameric, heme-containing, peroxisomal enzyme that catalyses
the conversion of hydrogen peroxide (H2O2) to water and oxygen. Catalase is one of
several redundant enzymes whose main function is to prevent cell damage from highly
reactive oxygen-derived free radicals. A reduction of catalase enzyme activity (EC
1.11.1.6) has been reported in the entire epidermis of vitiligo patients (Schallreuter et al.,
1991, 1999). Defects in several enzymes involved in melanin synthesis have also been
observed in vitiligo patients, lending evidence for the autotoxic theory of vitiligo
pathogenesis. The sum of these enzymatic defects result in the buildup and accumulation
of H2O2 in the epidermis of vitiligo patients. High concentrations of H2O2 can result in
the deactivation of catalase. It is therefore unknown if the decreased concentration of
catalase found in vitiligenous epidermis is a direct result of the accumulation of H2O2 due
to previously reported biochemical defects, or whether there is a separate problem with
the catalase enzyme. Topical treatment of vitiligo patents with pseudocatalase, which
mimics the activity catalase, promotes repigmentation due to the apparent correction of
some of the biochemical defects that caused the accumulation of hydrogen peroxide in
the epidermis (Schallreuter et al., 2001).
The observed catalase deficiency in vitiligo patient skin is the basis for treatment of
vitiligo with pseudocatalase, a bis-manganese III-EDTA-(HCO3 )2 complex that can
degrade H2O2 to O2 and H2O after UVB photoactivation, thus mimicking the action of

67
endogenous catalase (Schallreuter et al., 1995, 1999a, 1999b). Pseudocatalase is applied
topically to the entire epidermis twice daily accompanied by total body low dose narrow-
band UVB exposure 2-3 times per week. Clinical results are very promising, with a halt
in the progression of active vitiligo in 95% of the patients treated, and repigmentation in
60-65% of treated patients independent of disease duration. The biochemical effects of
pseudocatalase also include a dramatic decrease in epidermal H2O2 levels and restoration
of the normal 6BH4 recycling process (Schallreuter et al., 1995, 1999b, 2001).
In these experiments, case/control and family based association studies, and gene
sequencing were used to determine if the cause of decreased patient epidermal catalase
levels is due to a defect in the catalase gene itself. Results from these association studies
revealed preliminary evidence for genetic association between the catalase gene and
vitiligo susceptibility, which further supports a role for deregulation of the skin's ability
to handle oxidative stress in the pathogenesis of vitiligo (Casp et al., 2002).
Materials and Methods
Blood Collection and Processing
Blood collection and processing, DNA extraction and genotyping were performed
as described in chapter 2.
Catalase Sequencing
DNA preparation and PCR amplification
Thirteen sets of PCR primers were designed within flanking catalase (CAT) gene
introns to amplify individual exons (Table 4-1).
In order to determine the maximum DNA pool size that would allow detection of
rare mutations, DNA sample pools from vitiligo patents were created with each pool
having an incrementally increasing number of patients who had been genotyped for a

68
Table 4-1. Catalase primers for sequencing across the gene
CATmarker/Exon
amplified
Forward Primer Reverse Primer
CATx1/exon 1 TGAAGGATGCTGATAACCG CCATGAGCCCTCAATCTG
CATx2/exon 2 AAGTATTGACCAGCACAGC AACCTGAGGAAATAACCATC
CATx3/exon 3 CAGAAGGCTGGTGCTAAC AACTTCCTATGTGTCTCC
CATx4/ exon 4 AGTTCTTGGAAGTGGATTAG TTTGCCATGTTGCCCAGG
CATx5/exon 5 GCTAGTTGTCTATGCTGAG CTTTACCTTACACTACAGAC
CATx6/exon 6 TCATTAAGGGACTTTCTGG ATAATGAGATTGGGATACGC
CATx7/exon 7 GCAGTGTTACTCATAATCCT GTAAGCACTCATTCACAGC
CATx8/exon 8 ATTGAGTATGTGTATGTGGC GTGAATCCCACAAGGTAAC
CATx9/exon 9 GAAGTTTACAGCCCATTCC CAAGTAACATCTGAGGTGG
CATx10/exon 10 TAGCAGATGGCAGCGTTC GATACATCAGACAGTTGGG
CATx11/exon 11 AAAGTGAAGGACACAACCC CAAACAGCTAAGGACGATG
CATx12/exon 12 ACTCTGAGGCTGGCATTG ACAGTGGCAGGTAATGGC
CATx13/ exon 13 TTCACTGGCAAAACACATAC AAGAGTCTGGTAGCAGTTTA

69
known catalase polymorphism. This strategy allowed for the visualization of an
increased amplitude of the polymorphic SNP peak on the sequence chromatograph. This
experiment proved that in a small pool (ten patients) it would be possible to observe a
SNP or mutation present in only one of the ten patients sequenced in that pool.
Thirteen pools were then created with ten patients each, regardless of past catalase
genotype. PCR was carried out in a 10-µL volume containing 10x PCR buffer (50 mM
KCl, 10 mM Tris-HCl, pH 8.3, 1.5 mM MgCl2); 60 µM of each dNTP, pooled patent and
control genomic DNA, 2 pmol of each primer and 1.5 U Taq DNA polymerase. Samples
were subjected to 35 cycles of 30 s at 94°C for denaturing, 30 s at optimum annealing
temperature, and 30 s at 72°C for extension.
Direct sequencing of PCR products
After PCR amplification, products (10 µL) were electrophoresed on a 2.0% agarose
gel. Bands were excised from the gel and transferred into 1.5-mL Eppendorf tubes. DNA
was eluted from the agarose gel using Quantum Prep Freeze and Squeeze DNA gel
extraction spin columns (Bio-Rad). An aliquot of products (30µL) was then used directly
for cycle sequencing using the ABI Prism BigDye terminator (Applied Biosystems)
according to the manufacturer’s instructions. The sequencing reactions were then
precipitated in 3 vol. 100% ethanol and 0.1 vol. 3 M sodium acetate, pH 5.2. The pellets
were washed with 250 µL of 70% ethanol, and spun down in a dye-terminator removal
column (Qiagen), dried in a vacuum dryer and then dissolved in 20 µL of template
suppression buffer (Perkin Elmer). DNA sequencing was carried out using an ABI 310
automated sequencer (Applied Biosystems).

70
Results
Catalase Gene Polymorphisms
Three catalase gene single nucleotide polymorphisms (SNP) were genotyped in this
study, two of which were found to be uninformative (Table 4-2). A T/C substitution in
the CAT 5'-untranslated region, previously characterized in Hungarian acatalasemic and
hypocatalasemic subjects was genotyped by AFLP analysis (Goth et al., 1998). A Pst I
(CTGCAG) RFLP was created by changing one nucleotide in the reverse PCR primer
(Table 4-2). This polymorphism was found to be uninformative for our Caucasian patient
and control populations, as nearly all subjects were homozygous for the T allele (data not
shown). A T/C silent substitution in exon 10 (Leu-419), genotyped by RFLP analysis
using the restriction endonuclease BstN I (CCWGG), was also shown to be
uninformative. In this polymorphism, the T allele is cleaved by BstN I; whereas, the C
allele remains uncut. Nearly all subjects genotyped were homozygous for the T allele,
which differs from the allele frequencies of 0.78 T and 0.22 C reported in the NCBI SNP
database (data not shown).
The informative CAT genetic marker was a T/C silent substitution in CAT exon 9 (Asp-
389), which was genotyped using RFLP analysis of amplified genomic fragments with
the restriction endonuclease BstX I (CCANNNNNNTGG) (Forsberg et al., 1999). With
this polymorphism, the T allele is cleaved by BstX I, whereas the C allele remains uncut.
We observed allele frequencies of 0.82 C and 0.18 T in our control population (Table 4-
3), which were similar to those reported by Forsberg et al.(1999) for only 58 Caucasians
(0.87 C and 0.13 T).

71
Table 4-2. Sequences of primers used for CAT genotyping
PCR Primer Sequences (5′ to 3′)
PolymorphismRestriction
Enzyme Forward Primer Reverse PrimerAnnealTemp.
NCBISNP IDb
T/C 5′UTR Pst I CAT-1 GCCAATCAGAAGGCAGTCC CAT-2 GCGTGCGGTTTGCTCTGCa 60°C rs1049982T/C exon 9 BstX I CAT-5 GCCGCCTTTTTGCCTATCCT CAT-6 TCCCGCCCATCTGCTCCAC 64°C rs769217
T/C exon 10 BstN I CAT-7 CCTAAGTGCATCTGGGTGGT CAT-8 TACATCAGACAGTTGGGGCA 68°C rs704724aUnderlined nucleotide in primer CAT-2 was changed from the germline G to a C to create the Pst I RFLP.bNational Center for Biotechnology Information Single Nucleotide Polymorphism database: http://www.ncbi.nlm.nih.gov/SNP

72
Association of the T/C Exon 9 (BstX I) CAT Marker with Vitiligo
The observed allele and genotype frequencies for the T/C SNP in exon 9 in control
and patient populations are shown in Table 4-3. The allele frequencies for this CAT
genetic marker did not differ significantly between the control and patient populations
(p=0.54). When the observed control and patient genotype frequencies were compared
with expected values using a 3x2 contingency table in a standard χ2-test (Table 4-3), they
were found to be significantly different (p=0.0024), suggesting possible association of the
T/C exon 9 (BstX I) CAT marker with vitiligo. The observed genotype frequencies of the
control population did not significantly differ from those predicted by the Hardy-
Weinberg equation (p=0.26). However, the genotype frequencies observed in the vitiligo
patient group did not appear to be in Hardy-Weinberg equilibrium (p=0.016), with an
apparent excess of heterozygotes and deficiency of each homozygote class in the patient
population. Comparison of individual allele carriage rates and the percentage of
heterozygotes vs. homozygotes (Table 4-4) revealed that the overall C allele carriage rate
and the frequency of heterozygotes were significantly higher in patients than in control
subjects.
Family-Based Association
Further suggestive evidence for genetic association between the CAT gene and
vitiligo susceptibility was obtained using TDT, a family-based study design that
considers heterozygous parents and evaluates the frequency with which alleles are
transmitted to affected offspring. A χ2-test was used to evaluate the deviation of the rates
of transmission and non-transmission of the C and T alleles of the T/C exon 9 (BstX I)
CAT marker from the random expectation of 50:50. Forty-three informative

73
Table 4-3. Distribution of alleles and genotypes for the T/C SNP in CAT exon 9 in vitiligo patient and control populations
Observed allelefrequencies
Observed genotype counts (%) Expected genotype countsb
N C T C/C C/T T/T C/C C/T T/T p valueControls 177 0.82 0.18 122 (68.9) 46 (26.0) 9 (5.1) 119 52 6 0.26Patients 235 0.80 0.20 144 (61.3) 89 (37.9) 2 (0.8) 151 75 9 0.016p value 0.0024a
a controls vs. patients using the χ2 test with 3×2 contingency tableb observed vs. expected according to Hardy-Weinberg equilibrium
Table 4-4. Carriage rates and heterozygosity of the T/C SNP in CAT exon 9 in vitiligo patients compared to controls
Heterozygotes vs. homozygotesC carriage rate
(%)T carriage rate
(%)C/T (%) C/C + T/T (%)
Controls 94.9 31.1 26.0 74.0Patients 99.1 38.7 37.9 62.1p value 0.0083 n.s.a 0.011
Corrected pb 0.025 0.033a n.s., not significantb using Bonferroni’s correction

74
(heterozygous) parents were identified among all of the patients and family members
genotyped. The C allele was transmitted more frequently to affected offspring than
expected as a result of random chance (χ2=3.93, p=0.047).
Catalase Sequencing
The catalase gene was sequenced across 13 exons using 12 pools of five patients
each. Using this method, we were able to observe polymorphisms used in our genotyping
study. Although several new single nucleotide polymorphisms were identified none
resulted in amino acid changes that could affect enzyme activity or exert an obvious
effect on gene expression. No new interesting SNPs were found in exon 9, where the T/C
SNP was genotyped (Table 4-5).
Discussion
Both family based and case/control association studies suggest a role for catalase,
an enzyme responsible for the degradation of hydrogen peroxide to water and oxygen, in
vitiligo pathogenesis. Because catalase is one of many redundant antioxidant enzymes,
the absence of catalase enzyme activity in blood and tissues predisposes patients to oral
infections by peroxide-generating bacteria such as streptococci and pneumococci, but
most forms of hypocatalasemia and acatalasemia are asymptomatic (Eaton et al., 1995).
Vitiligo-like depigmentation is not a reported phenotypic feature of acatalasemia or
hypocatalasemia. However, a possible role for catalase in vitiligo susceptibility was
suggested by reports of low epidermal catalase activity in both lesional and non-lesional
skin of vitiligo patients, with concomitant increases in epidermal H2O2 (Schallreuter et
al., 1991, 1999a, 1999b). Whereas epidermal levels of catalase were decreased in vitiligo
patients, erythrocyte levels of catalase were reported to be comparable in 10 vitiligo

75
Table 4-5. Catalase gene single nucleotide polymorphisms (SNPs).
SNP Position Location
A/T 36535 15 base pairs 5' of mRNAstart site
G/A 36466 5' UTR
G/A 36303 Intron 1
C/T 36296 Intron 1
T/C 36238 Intron 1
C/T 26328 Intron 1
T/C 18934 Intron 7
G/T 6919 Intron 11
T/G 6915 Intron 11

76
patients and 20 control subjects suggesting that the catalase deficiency in vitiligo patients
may be limited to the skin and otherwise asymptomatic (Maresca et al., 1997).
The elevated levels of H2O2 observed in the epidermis of vitiligo patients, can
depress catalase activity by irreversibly inactivating the heme active site of catalase
(Aronoff et al., 1965). As discussed in detail by Schallreuter et al. (1999) several
biochemical defects have been reported for vitiligo patients can contribute to the
accumulation of epidermal H2O2, including defects in the de novo biosynthesis and/or
recycling of the essential cofactor (6R)-L-erythro-5,6,7,8-tetrahydrobiopterin (6BH4)
(Schallreuter et al., 1994), increases in epidermal monoamine oxidase A (EC 1.4.3.4)
activity (Schallreuter et al., 1996), and reduced glutathione peroxidase (EC 1.11.1.9)
activity (Beasley et al., 1999). The oxidative burst associated with NADPH oxidase
activities of inflammatory cells observed to infiltrate the peri-lesional skin of some
patients with active vitiligo is another potential source of H2O2 in patient skin (Ortonne et
al., 1993).
Other possible mechanisms of catalase deficiency in the skin of vitiligo patients
include tissue-specific differences in gene expression or enzyme structure/function in
patient melanocytes and/or keratinocytes. Qualitative comparison of catalase mRNA
levels in cultured melanocytes from lesional or non-lesional epidermis of vitiligo patients
or healthy controls showed no differences in expression between these groups
(Schallreuter et al., 1999b). Genetic association of the catalase gene with vitiligo
susceptibility is suggested by both by case-control and family based association studies,
suggesting that low catalase enzyme activity may be due to mutations within the catalase
gene that influence catalase activity in skin cells. Our association data suggest it is the

77
heterozygote genotype for T/C SNP in CAT exon 9 that confers susceptibility to vitiligo
pathogenesis. A possible explanation for this observation might be a dominant negative
effect of another mutation(s) linked to the C allele, resulting in a quantitative deficiency
of catalase enzyme activity. As observed for other multimeric proteins, mutations that
interfere with subunit assembly, interaction and/or function can lead to assembly of
mutant multimers that interfere with the function of the normal allele (Nussbaum et al.,
2001). According to this model, such a CAT mutation in the heterozygous state would
result in the expression of catalase enzyme tetramers with varying numbers of normal and
mutant subunits. Assuming equal levels of protein expression from both alleles and
depending on the nature of the CAT mutation(s), mixed molecules with possible reduced
enzyme activity could represent up to 80% of the tetramers. Interestingly, a quantitative
deficiency in catalase has also recently been suggested to play a role in diabetes
pathogenesis, as Hungarian patients with a demonstrated catalase deficiency exhibited a
higher frequency of diabetes than unaffected first-degree relatives and the general
Hungarian population (Goth et al., 2000). No CAT genetic analyses or gene expression
studies have been reported for diabetes thus far.
Sequence analysis of the entire catalase coding region was completed to screen for
the presence of any potential SNPs that may serve as functional mutations in the catalase
gene. SNPs in exon 9 were of particular interest, however no functional SNPs/potential
mutations were observed in the region. These results can be explained in several
different ways. Firstly, the mutation may not be in the sequenced exonic regions,
mutations in introns can affect splice sites, allowing for variation in translated proteins.
SNPs we have found “uninteresting” could still cause variations in the translated proteins,

78
amino acid substitutions anywhere in the coding region, even those that are conserved
could impact protein secondary structure, which could potentially affect the binding of
the two catalase subunits.
Given the lack of association between acatalasemia and vitiligo, and the localized
decrease in enzyme activity, it is possible that the effects of CAT mutations contributing
to vitiligo susceptibility may be tissue-specific, resulting in changes in gene expression or
enzyme structure/function in patient melanocytes and/or keratinocytes. Such mutations
might lead to a quantitative deficiency of catalase activity in the epidermis that is a
contributing causative factor for the accumulation of H2O2, rather than simply a
consequence of H2O2 accumulation from other sources. Overall, these results suggest
that the catalase gene may be a susceptibility gene in some vitiligo patients, and further
support the epidermal oxidative stress model for vitiligo pathogenesis. Further gene
expression studies could help to define a causative role of this enzyme in the etiology of
vitiligo, and chapter 5 discusses our approach and results.

79
CHAPTER 5CANDIDATE VITILIGO SUSCEPTIBILITY GENE EXPRESSION STUDIES
Introduction
Genetic association evidence points towards a possible involvement of catalase and
genes in the LMP/TAP region of the MHC class II region with vitiligo pathogenesis.
Gene expression and/or function studies are needed to provide further evidence for a role
of any of these genes in vitiligo pathogenesis. As our genetic studies have revealed,
vitiligo pathogenesis may include involvement of both biochemical defects in
melanogenesis as well as an inappropriate immune involvement. As a result of this
finding, we have chosen to look at gene expression in antigen presenting cells, which
serve as a bridge between intracellular events and the immune system. Because vitiligo
is a dermatological disorder, the most appropriate cell for this study should be the dermal
Langerhans cell. However, because this cell type is extremely difficult to isolate in
numbers needed for expression study analysis, we have chosen to look at gene expression
in monocytes isolated from peripheral blood. We are hypothesizing that any defect seen
in dermal APCs maybe seen in all other APCs as well.
Changes in expression of mRNA from catalase or any of the antigen processing
genes would bolster evidence of that gene’s involvement in vitiligo pathogenesis. Thus,
gene expression studies were performed on catalase, as well as on genes involved in
antigen processing and presentation, including MHC class I and the IFN-γ inducible
genes of the MHC class II region, TAP1, TAP2, LMP2 and LMP7. RNA expression
studies of the LMP and TAP genes should reflect an IFN-γ inducible upregulation of the

80
message. MHC class I expression is also IFN-γ inducible, and is easily observable by
surface β2-microglobulin (β2m) expression using flow cytometry. Catalase can be
quantitated by measuring enzyme activity as well as through semiquantitative RT-PCR of
catalase RNA. It is unknown if catalase expression is IFN-γ inducible, however, we
observed mRNA and catalase enzyme expression with and without the addition of IFN-γ.
Variability in expression of mRNA or the enzyme activity of any of these genes might
implicate that gene further in the pathogenesis of vitiligo.
Materials and Methods
Monocyte Isolation and Culture
PBMCs were obtained from whole blood from vitiligo patients who had contacted
the research group after reading about the study through a website set up at both the
University of Florida and at the National Vitiligo Foundation. Vitiligo patients and
controls donated 35 mL of blood on their first visit and 50 mL of blood on any
subsequent visit. PBMCs used for isolating monocytes used in RNA assays were isolated
by centrifugation (50 x g for 30 min at 25°C) on Ficoll gradients, washed with 1x PBS,
and resuspended in RPMI-1640 plus endotoxin-free 10% FCS. PBMCs were counted,
and their viability was assessed by trypan blue exclusion. Cells were plated at 1x107 cells
per well in a 6-well plate. Cells were allowed to adhere to the plate for 2 hours, and then
washed vigorously with 1xPBS to remove non-adherent cells. Cells for flow cytometry
and catalase enzyme activity assays were subjected to a negative selection rosetting
protocol to isolate purified monocytes using RosetteSep monocyte enrichment antibody
cocktail (StemCell Technologies). After antibody treatment to whole blood, monocytes
were isolated by centrifugation (500 x g for 20 minutes at 25°C), washed with

81
1xPBS+2% FBS, and RBCs were subjected to lysis using RBC lysis solution (139 mM
ammonium chloride; 17 mM Tris-HCl, pH 7.65). Purified monocytes were then cultured
in 5 mL polypropylene tubes (Falcon) at 2x106 cells per tube. All cells, both adherence-
purified and rosetted, were plated in duplicate, +/- the addition of either 1000 U or 500 U
human IFN-γ and incubated 24 hours at 37°C in 5% CO2.
Semi-Quantitative RT-PCR
RNA was extracted from 6-well plates using RNAqueous-4PCR Kit protocol
(Ambion), and purity of the RNA was assessed spectrophotometrically at 260 and 280
nm. LMP2, LMP7, TAP1 TAP2 and CAT RNA expression in monocytes cultured
overnight was monitored using semi-quantitative RT-PCR using the RetroSCRIPT kit
(Ambion). 250 ng of RNA from each sample were used to synthesize the first-stand
complementary DNA (cDNA), using random decamer primers and M-MLV (Ambion).
The primer sequences used to amplify cDNAs are shown in Table 5-1. Gene specific and
18S alternate primers/competimers (Ambion) were used in this experiment.
Competimers for 18S amplification chemically modify 18S primers, compete with the
18S primers and reduce the efficiency of the 18S amplification so that the 18S primers do
not become limited. Five µL aliquots of the RT reaction were used to amplify gene
specific and 18S fragments, respectively, with 10X dNTP (2 µL each dNTP, 92 µL 10
mM Tris-HCl, pH 7.5), 10X PCR buffer (50 mM KCl, 10 mM Tris-HCl, pH 8.3, 1.5 mM
MgCl2), 2.5 µM gene-specific primers (forward and reverse) or 18S primers/competimers
(8:2; 2 µL) and 1.25 U Taq polymerase. Amplification of gene specific and 18S cDNAs
was performed for 35 cycles, each cycle consisting of 1 min denaturation at 95°C, 30 s
annealing at 58°C, and 30 s extension at 72°C. cDNA was electrophoresed on a 2.0%
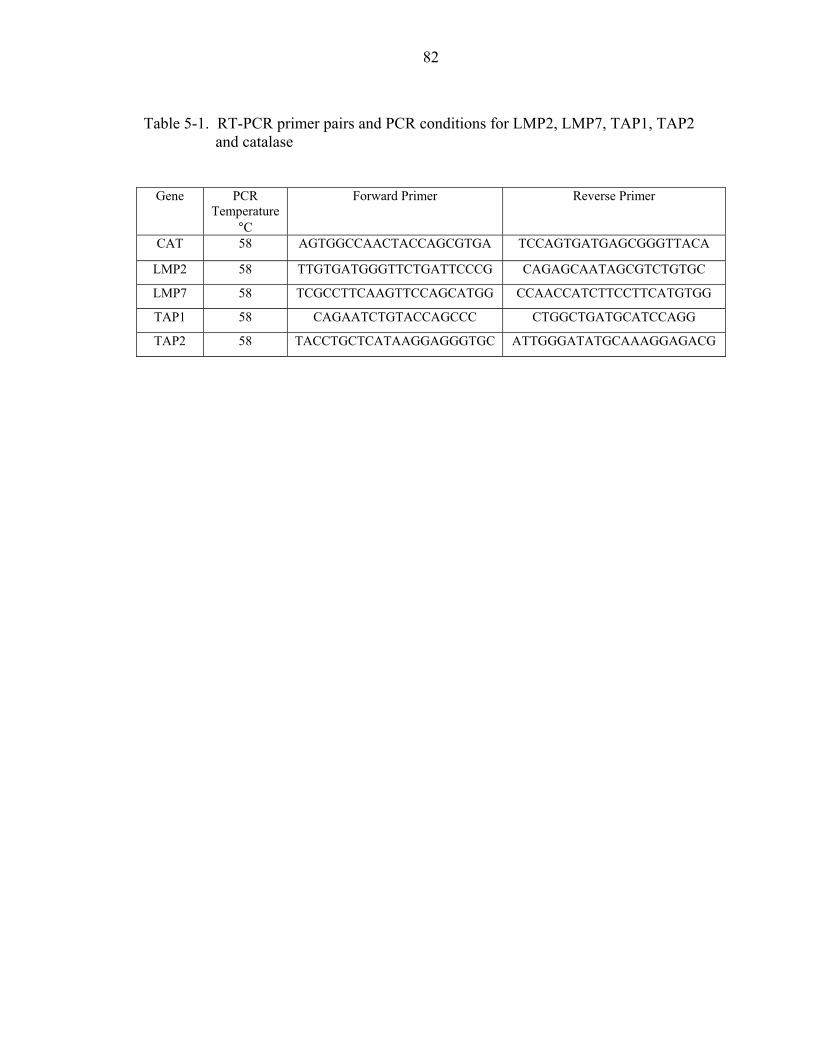
82
Table 5-1. RT-PCR primer pairs and PCR conditions for LMP2, LMP7, TAP1, TAP2and catalase
Gene PCRTemperature
°C
Forward Primer Reverse Primer
CAT 58 AGTGGCCAACTACCAGCGTGA TCCAGTGATGAGCGGGTTACA
LMP2 58 TTGTGATGGGTTCTGATTCCCG CAGAGCAATAGCGTCTGTGC
LMP7 58 TCGCCTTCAAGTTCCAGCATGG CCAACCATCTTCCTTCATGTGG
TAP1 58 CAGAATCTGTACCAGCCC CTGGCTGATGCATCCAGG
TAP2 58 TACCTGCTCATAAGGAGGGTGC ATTGGGATATGCAAAGGAGACG

83
agarose gel, and visualized using ethidium bromide staining and a 312 nm UV-
transilluminator
Flow Cytometry of β2-microglobulin
Monocytes purified by rosetting were incubated in FACS buffer (1% RIA grade
BSA, 0.1% sodium azide in 1x PBS, pH 7.4) with endotoxin-free lyophilized mouse
serum (Sigma Chemical Co.) for 20 minutes. Anti-CD14 PE (monocytes), FITC labeled
isotype control, and FITC labeled anti-β2-microglobulin (MHC class I) antibodies were
added to aliquots of cells and incubated for an additional 20 minutes. Cells were then
fixed with 4% formaldehyde for 20 minutes. Fixed cells were washed twice in FACS
buffer, and then resuspended in 200 µL FACS buffer. Flow cytometric analysis was
performed using a FACSscan or FACSCalibur flow cytometer (Becton Dickinson
Immunocytometry Systems), collecting between 5,000 to 10,000 ungated events.
Catalase Enzyme Assay
One mL of whole blood was taken from each patient and control and immediately
frozen at -70°C, to use as an erythrocyte control in the catalase enzyme activity assay.
Monocytes purified by rosetting were pelleted and washed twice in 1x PBS. The cells
were resuspended in 40 µL of sample diluent (surfactant in phosphate buffer) provided in
the Catalase-520 kit (OxisResearch) and frozen at -70°C. Samples of whole blood and
monocytes were thawed and the whole blood samples were diluted 1000-fold in sample
diluent. 30 µL of each sample were placed in a clean 1.5 mL tube, and 500 µL substrate
(10 mM H2O2) was added and incubated for exactly one minute. After 1 minute, 500 µL
of stop reagent (sodium azide) was added, and each tube was capped and inverted. 20 µL
of each reaction was added to cuvettes, and 2 mL of HRP/Chromogen reagent was added,

84
and incubated for 10 minutes. In this assay, the rate of dismutation of hydrogen peroxide
to water and molecular oxygen is proportional to catalase concentration. The sample is
treated with a known quantity of H2O2, and then quenched with sodium azide. The
amount of H2O2 remaining in the reaction mixture is determined through the oxidative
coupling of 4-aminophenazone and 3,5-dichloro-2-hydroxybenzenesulfonic acid in the
presence of H2O2 and catalyzed by horseradish peroxidase. Absorbance is then read at
520 nm, and catalase concentration of each unknown was determined by fitting diluted
standards to a second order polynomial regression.
H2O2 Treatment of Monocytes
Monocytes isolated by rosetting were exposed to 100 µM H2O2 for 24 hours to
simulate oxidative stress, and to determine catalase and β2-microglobulin expression in
both vitiligo patients and controls. After 24 hours, cells were pelleted and washed twice
with 1x PBS before the catalase assay, or flow cytometry for β2-microglobulin
expression was performed.
Results
Catalase Assay
Catalase enzyme concentrations were determined in whole blood and purified
monocytes from 10 patients and 10 controls. Whole blood samples were frozen
immediately after the blood draw, and purified monocytes were cultured for 24 hours +/-
500 U IFN-γ. Catalase levels were determined using a colorimetric assay, which detects
the degradation of H2O2 by endogenous catalase present in the sample, measured as
absorbance at 520nm. Unknown sample concentrations were determined by applying the
absorbance to a standard curve of known catalase sample concentrations. Catalase

85
concentrations were found to be significantly lower in vitiligo patient monocytes (p=
0.0252), as determined by Student’s t test (Figure 5-1). Concentrations did not seem to
vary with or without the addition of 500 U IFN-γ to the monocyte cultures. As seen in
previous studies (Maresca et al., 1997), catalase concentrations did not vary significantly
in erythrocytes of the same vitiligo patients and controls (Figure 5-2). These results show
a decreased catalase enzyme level in vitiligo patients in monocytes, which is not seen in
patient whole blood.
β2-Microglobulin Expression
Flow cytometry was employed to determine expression of MHC class I on the
surface of monocytes using β2-microglobulin as a marker. PE-labeled anti-CD14
antibodies were used to label monocytes. Purified monocytes were treated +/- 500 U IFN-
γ overnight. Because the percentage of cells expressing β2-microglobulin was not
significantly different throughout patient and control populations, mean fluorescence
(MF), or the amount of β2-microglobulin on the cell surface of each monocyte, was
compared between patient and control populations. As expected, MHC class I
expression, as measured by β2-microglobulin expression, was upregulated in response to
IFN-γ in both patient and control purified monocytes. β2-microglobulin expression was
also shown to be significantly higher on control monocytes after IFN-γ treatment than in
vitiligo patients (p=0.0496 using a Student’s t test) (Figure 5-3). Expression of β2-
microglobulin on untreated monocytes also approached significance between the patient
and control populations (p=0.0512). This data suggest that vitiligo patients have a lower
expression of MHC class I on their monocytes than normal controls. It is important to
note that there is one control individual whose β2-microglobulin expression is much

86
Figure 5-1. Monocyte catalase levels in patients and controls. Patient monocytes havesignificantly less catalase activity than controls after treatment with 500 UIFN-γ (p= 0.0252 by Student’s t test). Asterisks represent significantdifferences in allele frequency between patient and controls.
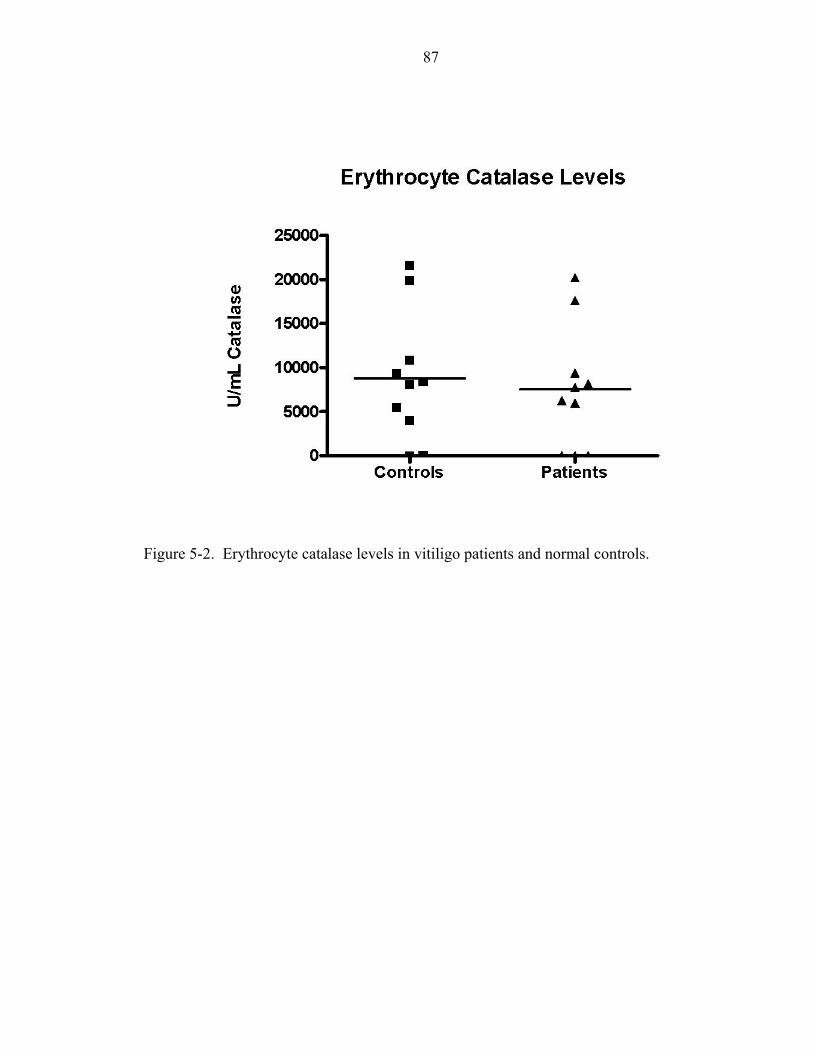
87
Figure 5-2. Erythrocyte catalase levels in vitiligo patients and normal controls.

88
Figure 5-3. β2-microglobulin expression on patient and control monocytes. There is asignificant difference between patient and control β2- microglobulin inmonocytes treated with 500 U IFN-γ (p=0.0496 Student’s t test). Asterisksrepresent significant differences in allele frequency between patient andcontrols.

89
higher than other pool drops the difference between β2-microglobulin on the surface of
patients and control controls and patients sampled. Removal of this individual from the
data removes the treated monocytes from significance. As variance in human
populations can be expected, it can be concluded that there is a trend suggesting vitiligo
patients may have lower class I expression on their monocytes. Further experiments with
a larger sample size, however, need to be run to confirm these results.
H2O2 Treatment
Purified monocytes were treated overnight with or without 500 U IFN-γ and with
100 µM H2O2. We found dramatic reduction in the number of H2O2-treated monocytes
by flow cytometry. This reduction in numbers of monocytes is most likely a result of the
death of these cells due to H2O2 toxicity. Therefore it is difficult to draw any conclusions
from these cells due to the dramatic reduction in cell number. The catalase enzyme assay
was performed as described above on these cells. No significant difference was seen
between patient and control cells treated with H2O2. Many of these patient and control
samples did not have any catalase activity, most likely due to the large loss of monocytes
as seen in the flow cytometry analysis.
RNA Expression Studies
Adherence-purified monocytes were treated overnight with or without either 1000
U or 500 U IFN-γ. RNA was harvested and used in RT-PCR to create cDNAs of several
antigen processing and presentation genes, LMP2, LMP7, TAP1, TAP2 and the
antioxidant enzyme catalase. Genotypes of the patients used in the 500 U IFN-γ
experiments, and their corresponding catalase activities and β2-microglobulin expression
on monocytes can be seen in Table 5-2. In the first set of experiments, 1000 U of IFN-γ

90
Table 5-2. Genotypes of patients treated with 500 U IFN-γ, and used in mRNA, catalaseand, β2-microglobulin expression studies with corresponding catalase enzymeactivity and mean β2-microglobulin fluorescence.
PatientNumber
CAT 5/6 CatalaseenzymeactivityU/mL
TAP1 10/11 LMP7 4/7 Mean β2-microglobulinfluorescence
243-1 CT 36.9 GG TT 360.4
358-1 CT 50.8 GG TG 744.3
513-1 CC 4.9 GG GG 337.3
517-1 CT 16.3 AG TG 372.6
518-1 CC 10.1 GG TG 381.2
521-1 CC 85.1 GG TG 882.0
523-1 CC 85.5 GG TG 320.2
524-1 CT 0 AA GG 288.0
527-1 CT 29.7 GG TT 289.7
532-1 CC 1.0 AG TT 250.2
Allelepercentagesof these 10
patients
75% T25% C
20% A80% G
55%T45%G
Allelepercentages
of totalpatients
80% T20% C
14.8% A85.2% G
47.8%T52.2%G

91
was used to treat the monocytes. Messenger RNA expression was evaluated for LMP2,
LMP7, TAP1 and TAP2 using 10 patients and 8 controls. In these preliminary
experiments the fold increase in RNA expression was determined after treatment with
1000 U of IFN-γ. There were no significant changes in induction of mRNA by IFN-γ for
LMP2, LMP7, TAP1 or TAP2 between the patient and control populations (Figure 5-4).
In the second set of experiments 500 U of IFN-γ was used to stimulate the
monocytes. 500 U was chosen to determine whether a lower dose of IFN-γ might allow
for more subtle differences in RNA expression between patients and controls to be
uncovered. Messenger RNA expression was evaluated for LMP2, LMP7, TAP1, TAP2
and CAT using 10 patients and 10 controls. The amount of RNA extracted from the
monocytes in this experiment was far lower than in the first experiment. As with the
expression studies done with 1000 U of IFN-γ, those done with 500 U showed no
difference in expression between vitiligo patients and controls (Figure 5-5). Likewise, no
differences in expression of catalase mRNA were seen between patients and controls
(Figure 5-6). This result demonstrates that the decreased expression of catalase enzyme
seen in both monocytes and in the epidermis of vitiligo patients might be caused by a
decrease in catalase enzyme function or protein expression rather than a decrease in
mRNA expression.
Discussion
Gene expression studies were performed on LMP2, LMP7, TAP1, TAP2, β2-
microglobulin, and catalase using a variety of techniques. Semi-quantitative RT-PCR
found no differences in expression of any of the antigen processing and presentation
genes and catalase between patients and controls. Enzyme expression studies of catalase

92
Figure 5-4. Messenger RNA expression of LMP2, LMP7, TAP1 and TAP2 in patientand control monocytes after overnight treatment with 1000 U IFN-γ.

93
Figure 5-5. Messenger RNA expression of LMP2, LMP7, TAP1 and TAP2 in patientand control monocytes after overnight treatment with 500 U IFN-γ.

94
Figure 5-6. Catalase/18S RNA ratios with and without overnight treatment with 500UIFN-γ.

95
using vitiligo patients and controls whole blood and monocytes showed no changes in
catalase expression in whole blood, however catalase expression in monocytes from
vitiligo patients is significantly reduced. This reduced expression of catalase does not
appear to correlate with any genotype of the T/C SNP found in exon 9 of the catalase
gene (Table 5-2). Flow cytometry using β2-microglobulin as a marker for MHC class I
expression on the surface of patient and control monocytes revealed vitiligo patients may
express a lower concentration of β2-microglobulin on the surface of their monocytes after
IFN-γ treatment than normal controls. This decreased expression of β2-microglobulin
was also not correlated with any particular genotype from either the G/T SNP in intron 6
of LMP7 or in the G/A SNP in exon 10 of TAP1 (Table 5-2).
Our data shows vitiligo patients may express lower levels of β2-microglobulin on
the surface of their monocytes than normal controls, suggesting a lower MHC class I
expression. Results showed a significantly decreased expression of β2-microglobulin on
vitiligo patients’ monocytes after treatment with IFN-γ, a trend that approached
significance even without IFN-γ treatment. The removal of one control individual results
in the loss of statistical significance. Due to this effect, it is only fair to say that there is a
trend towards decreased β2-microglobulin expression in vitiligo patients, a result that
needs to be repeated with an increased patient and control population for confirmation.
Decreased expression of MHC class I is often seen in tumor lines, and is thought to
be a mechanism for the tumor cell to avoid detection by the host’s immune system (Chen
et al., 1996; Cabrera et al., 2003; Miyagi et al., 2003). Concomitant decreases in LMP2,
LMP7, and TAP1 have also been found in various tumor lines, and associated with poor
prognosis (Cromme et al., 1994; Matsui et al., 2002; Dissemond et al., 2003). On the

96
other end of the immunological spectra, there is evidence of hyper-expression of MHC
class I molecules in other inflammatory autoimmune disorders such as IDDM and
autoimmune thyroid disease, presumably due to the local increased expression of
inflammatory cytokines such as IFN-γ (Vives-Pi et al., 1997). It may seem
counterintuitive that we observe decreased expression of β2-microglobulin on the surface
of monocytes from vitiligo patients. However, decreased expression of class I has also
been reported for other inflammatory autoimmune diseases such as diabetes (Faustman,
et al., 1991), lupus erythematosus, Sjögren’s syndrome, rheumatoid arthritis, Graves’
diseases, Hashimoto’s thyroiditis, (Fu et al., 1998) and multiple sclerosis (Li et al.,
1995a). Fu et al. (1998) argue that this decrease in MHC class I expression may
contribute to autoimmunity, as T-cell avidity and selection may be determined by the
affinity and density of complexes MHC class I:self peptide, and T-cell avidity is thought
to be critical for both positive and negative T-cell selection
Another possible explanation for this phenomenon is described by Chan et al. in
their study of lupus in the β2-microglobulin (β2m)-deficient MRL-Faslpr (MRL/lpr)
mouse. In these mice, lupus skin lesions are accelerated, whereas nephritis is
ameliorated. The authors suggest that immunoregulation differs in different organs, in
this case between the skin and the kidneys. Because the skin, like other barrier sites, is
designed to provide a rapid inflammatory response against invaders, it might also be
prone to inflammatory diseases such as lupus, atopic dermatitis, psoriasis, and vitiligo.
Chan et al. suggest that these barrier sites may be armed with specialized regulatory
mechanisms to downregulate inflammation. In this case, the regulatory cell is a β2-
microglobulin dependent regulatory cell, which may operate in the skin, but not the

97
kidneys of (β2m)-deficient animals. This suggests that skin has a natural
proinflammatory tendency, presumably not found in the nonbarrier organs (i.e. kidney),
and may be unopposed in the absence of β2-microglobulin. If this model is correct, it
would make genetic analysis of autoimmune diseases much more complex, as genetic
defects could confer pathogenesis seen only in particular target organs (Chan et al.,
2001).
Although many of the genes located in the MHC class II locus have been
associated with autoimmune disease through genetic studies, there have been far fewer
studies showing altered expression of either the message or the protein associated with
autoimmunity. Several of these studies demonstrate a deficiency of antigen processing
and presentation genes. TAP1 and LMP2 have been found to be associated with both
autoimmunity and with a decreased surface expression of MHC class I. As mentioned
previously, many tumor cell lines are deficient in expression of LMP and TAP, as well as
MHC class I (Cromme et al., 1994; Matsui et al., 2002; Dissemond et al., 2003). A
recent report has demonstrated that tumor cells that do not express MHC class I were
deficient in several proteasome subunits, including LMP2, MECL-1, PA28α and PA28β.
This suggests that impaired expression of these subunits might be involved in loss of
MHC class I expression (Miyagi et al., 2003).
Expression studies in the NOD mouse revealed decreased expression of TAP1 and
LMP2 as a result of a mutation in its shared bidirectional promoter (Yan et al., 1997). In
human lymphocytes, varying decreases in expression of mRNA for all 4 antigen
processing and presentation genes we studied, LMP2, LMP7, TAP1 and TAP2 were
reported in patients with IDDM, Sjögren’s syndrome, Graves’ disease, and Hashimoto’s

98
thyroiditis (Fu et al. 1998). Other studies have reported increases in expression of TAP1
and LMP2 mRNA and protein in juvenile dermatomyositis and Grave’s disease,
respectively (Tezak et al., 2002; Kohn et al., 2000). Increased TAP1 expression has been
reported for islet cells and thyrocytes in patients with newly diagnosed type I diabetes
and autoimmune thyroiditis (Sospedra et al., 1997; Vives-Pi et al., 1996; Hanafusa et al.,
1983). LMP7 is also found to be hyperexpressed on thyrocytes (Vives-Pi et al., 1997).
Although we did not observe any alterations in mRNA expression in any of the
genes we studied in the MHC class II region, we did observe a decrease in expression of
MHC class I on the surface of monocytes of vitiligo patients. Preliminary Western blots
performed on protein extracts from patient and control monocytes did not appear to show
a difference in expression between LMP7 and TAP1 in patients and controls, however the
sample size was far too low to draw any meaningful conclusions (data not shown).
Theoretically there may be a defect in the function of any of these proteins LMP2,
LMP7, TAP1 or TAP2 that does not affect mRNA quantity or stability, as demonstrated
by a recent study on monocytes exposed to tobacco extracts. In this study, cells exposed
to tobacco extracts in culture have reduced membrane HLA class I, as well as TAP1
protein expression (Fine et al., 2003). This reduction of TAP1 is found only at the
protein level, with no concomitant decrease in TAP1 mRNA levels. A decrease in
protein expression/function of any of these genes in the MHC class II region could be
what is driving genetic association with vitiligo, and may also be contributing to the
decreased expression of MHC class I seen on vitiligo patients. It is also possible is a
gene in the MHC class II region, other than the LMP and TAP genes, is the gene that is
responsible for conferring genetic susceptibility to vitiligo, as there are many genes in

99
this region, some of which we do not yet know their function. Factoring in the high
probability of linkage disequilibrium with other genes in the region, it is possible that the
TAP and LMP genes are not major contributing factors in the pathogenesis of vitiligo.
Nevertheless, further studies on protein expression of these genes are needed to
completely rule these genes out.
Decreased concentrations of catalase along with increased concentrations of H2O2
have been found in the epidermis of vitiligo patients (Schallreuter et al., 1991, 1999a,
1999b). This decrease is not found systemically, as catalase levels in RBCs of vitiligo
patients are not significantly different than in controls (Maresca et al., 1997). A recent
report looked at catalase concentrations in vitiligo patient PBMCs and reported a
decreased catalase expression in these cells. This study also reiterated there were no
differences in expression of catalase in RBCs of vitiligo patients versus controls
(Dell’Anna et al., 2001). Our study looked at expression of active catalase in purified
vitiligo patient monocytes, and revealed a significantly decreased expression of catalase
in the monocytes of vitiligo patients. We observed no significant difference in the
catalase activity of patient and control RBCs. We also looked at catalase mRNA
expression in these same monocytes through semi-quantitative RT-PCR. We found no
significantly different expression of catalase mRNA in vitiligo patients compared with
normal controls.
Taken together these results suggest that the decrease in vitiligo patient monocyte
catalase enzyme activity is not a result of changes in mRNA expression or quantity.
These results support those discovered by Schallreuter et al. (1999a) who found mRNA
expression extracted from keratinocytes and melanocytes from both patients and controls

100
did not show any differences in expression, while catalase activity was decreased in the
epidermis of vitiligo patients. The question remains as to whether the deficiency of
catalase enzyme activity in vitiligo patient skin is due to defects in the catalase enzyme
itself, or is due substrate inhibition caused by a localized accumulation of H2O2 from
other sources. Nevertheless, we have observed functional changes in catalase enzyme
activity in circulating monocytes of vitiligo patients, which helps to support the genetic
association found through our case/control and family-based analyses.

101
CHAPTER 6DISCUSSION: OXIDATIVE STRESS AND THE IMMUNE SYSTEM IN VITILIGO
PATHOGENESIS
This chapter discusses the interplay between the regulation of oxidative stress and
the immune system in vitiligo pathogenesis, and in other autoimmune diseases. Vitiligo
pathogenesis is likely an extremely complex event involving both genetic susceptibility
as well as environmental triggers. The two major theories of vitiligo pathogenesis
include an autoimmune etiology for the disease and an autotoxicity in the melanocyte.
There is ample evidence supporting a role for both autotoxic and autoimmune
pathogenesis mechanisms in some patients. Although these two theories are often
presented as mutually exclusive entities, it is likely that vitiligo pathogenesis may involve
both autotoxic and autoimmune events, for which there is variability within a patient
population.
Oxidative Stress
Reactive oxygen species (ROS) are produced as byproducts of melanogenesis in
melanocytes, and controlled in the epidermis by several redundant antioxidant enzymes
such as catalase and glutathione peroxidase, both of which are decreased in the epidermis
of vitiligo patients (Schallreuter et al., 1999a). Oxidative stress plays a very important
protective role in the immune system, as phagocytic cells generate reactive oxygen
intermediates such as O2- (superoxide), H2O2 (hydrogen peroxide), and NO (nitric oxide),
which are toxic to many pathogens. While ROS are damaging to pathogens, they are also

102
damaging to host cells as well. Current research is beginning to demonstrate that ROS
and oxidative stress may in fact play a critical role in initiating and/or exacerbating the
effects of autoimmunity. Our research has shown genetic association of a gene(s) in the
MHC class II region and the catalase gene with vitiligo, as well as decreased expression
of MHC class I and catalase activity in vitiligo patient monocytes. Given the role of
oxidative stress in both melanogenesis and in the immune system, we hypothesize that
biochemical defects in the melanin biosynthesis pathway, as well as possible defects in
patient antioxidant enzymes, are responsible for the generation of reactive oxygen species
in the epidermis of vitiligo patients. We suggest that it may be the build-up of ROS along
with possible immune system defects that allows for the inappropriate autoimmune
response against normal melanocytes.
Oxidative Stress and Inflammation
There is a tenuous balance between helpful and harmful oxidative stress in the
immune system. The phagocytes as part of the innate immune system are very important
in providing the first line of defense against invading pathogens. Phagocytes such as
macrophages ingest and destroy opsonized extracellular pathogens, secrete pro-
inflammatory cytokines, and generate a variety of ROS through a process known as a
respiratory burst. Under ideal situations, the ingestion of the opsonized pathogens and
the secretion of toxic oxygen species by these phagocytic cells help to kill and/or contain
the spread of these pathogens. The secreted pro-inflammatory chemokines and cytokines
attract cells of the adaptive immune system to help rid the host of the invading pathogen.
Low levels of intracellular ROS can be beneficial, as they are able to activate
transcription factors, such as NFκB and AP-1, which are involved in regulating the

103
production of inflammatory mediators, such as the cytokines TNF-α and IL-1, and a
number of matrix metalloproteinases (Dai et al., 2003).
In autoimmune disorders the immune system inappropriately targets host cells for
destruction, often creating a chronic or remitting/relapsing inflammatory milieu. The
effects of chronic inflammation can be devastating on the host, eventually causing
damage and/or destruction of the of the target organ. In this inflammatory environment,
ROS can accumulate with a toxic effect on the surrounding cells. Buildup of ROS can
cause direct damage to proteins, lipid peroxidation and DNA base modification, leading
to tissue damage (Dai et al., 2003). Current research studies are beginning to evaluate the
role of these ROS in autoimmunity. In many cases it is unclear what is causing this
aberrant inflammatory response in autoimmunity, begging the question as to whether
these ROS are a result of the chronic inflammation and autoimmunity, or part of the
cause of the autoimmune response.
Reactive Oxygen Species and Autoimmunity
Antioxidant Levels in Autoimmune Disease
Antioxidant enzymes such as catalase, superoxide dismutase, glutathione and
glutathione peroxidase usually control the buildup of reactive oxygen intermediates in the
host. Although these antioxidants are often redundant, decreases in any of these enzymes
could leave the host at an increased risk for oxidative stress. Recent studies have begun
to investigate the levels of these antioxidant enzymes in inflammatory autoimmune
disorders such as IDDM, arthritis, SLE, multiple sclerosis, and vitiligo (Lortz et al., 2003;
Gotia et al., 2001; Cimen et al., 2000; Taysi et al., 2002; Calabrese et al., 2002; Yildirim
et al., 2003; Casp et al., 2002). Studies of islet cells in IDDM have shown that beta cells
in general show very low activity levels of the antioxidant enzymes catalase, glutathione

104
peroxidase, and superoxide dismutase. This weak antioxidative defense status leaves
these cells with poor resistance against oxidative stress (Lortz et al., 2003). Studies of
antioxidant expression in islets of mice show a 30-40% reduction of SOD from levels
found in the liver of the animal, a 15% reduction of glutathione peroxidase and an
undetectable level of catalase (Lenzen et al., 1996). Studies of SLE patient sera revealed
elevated levels of superoxide dismutase activity, while the activities of glutathione
peroxidase and catalase were lower in patients SLE compared with healthy controls
(Taysi et al., 2002). Decreased levels of catalase were found in the blood of children
with juvenile rheumatoid arthritis (Gotia et al., 2001). Adult patients with rheumatoid
arthritis (RA) were found to have increased levels of SOD and normal levels of both
catalase and glutathione peroxidase (Cimen et al., 2000). In multiple sclerosis patients, a
significant decrease in reduced glutathione and significant increases in oxidized
glutathione were observed (Calabrese et al., 2002).
Significantly increased levels of erythrocyte SOD, as well as a marked reduction of
erythrocyte glutathione peroxidase and glutathione, were observed in patients with
generalized vitiligo (Yildirim et al., 2003). Catalase levels are reduced in the epidermis
of vitiligo patients as well as in PBMCs (Schallreuter et al., 1991, 1999a, 1999b;
Dell’Anna et al., 2001). We have also demonstrated a decreased expression of the
catalase enzyme in monocytes of vitiligo patients (Chapter 5). Abnormal levels of
antioxidants in these disorders could leave these individuals at a far higher risk of
autotoxic damage to cells as a result of increased oxidative stress. The chronic
inflammatory states of these diseases could potentially exacerbate the oxidative damage
caused by the abnormal expression of these antioxidant enzymes. It is unknown whether

105
the decreased expression of these enzymes is a result of a genetic defect or the result of
the chronic inflammation and increased oxidative stress in these autoimmune disorders.
However, our genetic data suggests a genetic flaw in the catalase gene may be in part
responsible for the decreased expression of the catalase enzyme seen in vitiligo patients.
Transfection of Antioxidant Genes
Decreased expression of any of these antioxidant genes could leave the host at an
increased risk of oxidative stress. Reduced expression of these enzymes could also be a
causative agent, or could exacerbate progression of autoimmune disease. In rheumatoid
arthritis (RA), the generation of reactive oxygen species within the inflamed joint has
been suggested to play a significant pathologic role. Given the potential role of ROS in
RA, Dai et al. (2003) hypothesized that the transfer of antioxidants into joints could
suppress inflammatory processes in a rat model of antigen-induced arthritis (AIA). Ex
vivo gene transfer of SOD and catalase produced about a six- to seven-fold increase in
SOD activity and a two- to three-fold increase in catalase activity compared with control
animals. Furthermore, rats treated with cells over-expressing SOD, catalase or a
combination of both enzymes showed significant suppression of knee joint swelling,
decreased infiltration of inflammatory cells within the synovial membrane, and reduced
matrix metalloproteinase activity in knee joints.
Transfection of antioxidant genes into islet cells may be a future direction in
diabetes research. Because islet cells are naturally so low in antioxidant activity,
transfection of antioxidant genes into islets may allow for these cells to better handle
oxidative stress. Lortz et al. (2003) used insulin-producing RINm5F tissue culture cells
and transfected them with SOD and catalase. Overexpression of the hydrogen peroxide-
inactivating enzyme catalase provided protection of insulin-producing cells in situations

106
of increased hydrogen peroxide generation, and a combination of an overexpression of
catalase and SOD caused an even greater protection of these cells against H2O2 damage.
This concept of increasing antioxidant levels in islet cells through gene therapy to
ameliorate oxidative damage is being investigated in conjunction with islet cell
transplantation. Because islet cells express such low levels of antioxidants, and because
both islet isolation and transplantation processes generate oxygen radicals, damage from
oxidative stress may pose a major obstacle to islet replacement therapy. Islet grafts that
overexpress SOD have been shown to function about 50% longer than control grafts
(Bertera et al., 2003). Similarly, in rat beta cell lines, adenoviral-induced overexpression
of glutathione peroxidase enhances the resistance of the cells to both ROS and reactive
nitrogen species cytotoxicity (Moriscot et al., 2003). In the future, gene therapy with
antioxidant genes may be therapeutic in controlling beta cell autoimmunity in IDDM, and
preventing further cell loss after transplantation.
Antioxidants and Reactive Oxygen Species in Autoimmunity
New research has begun to reveal more evidence for the roles of decreased
antioxidant levels and/or increased ROS in autoimmunity and/or chronic inflammatory
disorders. The production of new or altered antigenic epitopes by ROS could provide a
mechanism for the presentation of cryptic epitopes to the immune system, allowing for a
breach of peripheral immunity. Duthoit and coworkers (2001) observed that during in
vitro thyroid-hormone synthesis, hydrogen peroxide cleaves thyroglobulin (Tg) into
peptides found to contain the immunodominant region of Tg that is recognized by anti-Tg
autoantibodies from patients with autoimmune thyroid disorders. Although the normal
synthesis of thyroid hormone produces locally high levels of H2O2, the addition of extra
H2O2, and other ROS with local inflammation or immune response could be enough

107
oxidative stress to inappropriately cleave Tg into peptides with cryptic epitopes that
might allow for a breach in peripheral tolerance. Theoretically, as H2O2 levels rise both
inside the thyrocyte as a result of hormone synthesis, and external to the cell as a result of
inflammation, so does the cleavage of Tg, allowing for a buildup of these altered peptides
inside the cell. High concentrations of H2O2 also could cause cell necrosis, allowing for a
large-scale release of these cryptic epitopes, prompting APCs in the surrounding
inflammatory environment to phagocytize and present these cryptic peptides (Dulhoit et
al., 2001).
Modification of proteins by oxidative stress may also be involved in autoantibody
production in type 1 diabetes. Trigwell et al. (2001) have demonstrated that oxidation
reactions catalyzed by copper, iron or hydrogen peroxide can alter glutamic acid
decarboxylase (GAD) in the islets of rats. Oxidative modification produced high
molecular weight, covalently linked aggregates containing GAD. Sera from patients with
IDDM reacted with this modified GAD, whereas sera from normal controls did not. This
study again implicates oxidative stress as a potential mechanism for target organ
induction of autoimmunity.
Antioxidants could also play a role in the induction of autoimmunity or chronic
inflammation. Research studying patients with primary sclerosing cholangitis (PSC), a
chronic cholestatic liver disease of unknown etiology, has revealed autoantibodies against
catalase. These autoantibodies, found in 60% of patients, could implicate the loss of
catalase, and a role for oxidative stress in PSC pathology (Orth et al., 1998). Catalase
autoantibodies have also been found in patients with inflammatory bowel disease (IBD)
(Roozendaal et al., 1998).

108
These studies delineate further a role for oxidative stress, and the importance of
regulation of oxidative stress in the pathology of autoimmunity. It is obvious that a
delicate balance exists between generation and regulation of oxidative stress and the
immune system. Disrupting this balance in any way might not only increase the
complications of ROS toxicity, but may also leave the individual at a higher risk for the
development of autoimmunity or other chronic inflammatory disorders.
Antigen Processing, Oxidative Stress and Vitiligo
Vitiligo and Oxidative Stress
We believe that vitiligo pathogenesis is a multifactorial process, beginning with a
dysregulation of oxidative stress, and ending with an organ-specific autoimmune
response in the epidermis. ROS are generated through the normal production of melanin
in the melanocytes. Vitiligo patients have been shown to have an increased hydrogen
peroxide concentration in their epidermis, for which there are many different possible
sources. As reviewed by Schallreuter et al. (1999), defective de novo synthesis of 6BH4
allows accumulation of 7BH4 , which causes an inhibition of PAH resulting in a
concomitant upregulation of H2O2. Vitiligo patients also have an increase in expression
of epidermal monoamine oxidase A (MAO-A), which also contributes to H2O2. Patients
also may have an age-dependent reduction in glutathione peroxidase activities, which
also increase the H2O2 burden. Cellular immune infiltrates could potentially allow for
even more H2O2 production through phagocytic generation of ROS (Schallreuter et al.,
1999b).
Vitiligo and Catalase
We have found a genetic susceptibility of the catalase gene with vitiligo. This
susceptibility is seen with a SNP marker in exon 9 of the catalase gene. The heterozygote

109
genotype is found more frequently in patients than in controls (Chapter 4). We
hypothesize that such a heterozygous catalase gene mutation results in the expression of
catalase enzyme tetramers with varying numbers of normal and mutant subunits, leading
to decreased enzyme activity in the skin. It is possible that with this defect, enzyme
activity could be decreased up to 80% (Casp et al., 2002). We have also observed a
functional decrease in catalase enzyme activity in monocytes from catalase patients,
which is not mirrored in erythrocytes from these same patients (Chapter 5). A decrease
in catalase RNA is also not observed in these cells. This decrease in catalase activity
could be a result of either the hypothesized heterozygous mutation, or due to catalase
inactivation by H2O2, or a combination of both. Nevertheless, an increase in ROS in the
epidermis is the end result, causing toxicity to the surrounding melanocytes.
Morphological changes in melanocyte rough endoplasmic reticulum, as seen by
electron microscopy, are suggestive of retention of peptides in the RER (Boissy et al.,
1991; Im et al., 1994). Pigmentation is a multistep process critically dependent on the
functional integrity of tyrosinase, the rate-limiting enzyme in melanin synthesis. If
tyrosinase is damaged or misfolded it is often retained in the endoplasmic reticulum.
These malformed proteins are ubiquitin-tagged and targeted for degradation via the
proteasome (Halaban et al., 2002). If oxidative stress causes necrosis of these cells
retaining misfolded tyrosinase, a large amount of the protein can then be released into the
inflammatory microenvironment to potentially be picked up by APCs. It is also possible
that ROS as seen with thyroglobulin and GAD, are modifying these retained proteins,
allowing the presentation of cryptic epitopes by either APCs or by the melanocytes
themselves, which have been shown to phagocytize and present antigen themselves in

110
times of stress through MHC class II (Le Poole et al., 1993a; Le Poole et al., 1993b).
Recruitment and involvement of other cells of the immune system can function to
increase oxidative stress in the area due to respiratory bursts by phagocytes. Secretion of
chemokines and cytokines into the local environment can increase inflammation, and
potentially target cells for further destruction by necrosis and apoptosis. Dead and dying
cells may accumulate in the epidermis, drawing the attention of circulating monocytes.
This may further permit an increased presentation of self-peptides to cells of the immune
system.
Vitiligo and MHC class II genes
We have also demonstrated genetic association of vitiligo with a gene(s) in the
MHC class II region. Although RNA expression studies revealed no changes in
expression of either of the LMP or TAP genes (Chapter 5), there still may be a functional
difference in the expression of these proteins. It is tempting to hypothesize that a
mutation in one of the LMP genes is allowing for the altered cleavage of a self-peptide,
or that one of the TAP genes is allowing for the inappropriate transport of a self-peptide
across the endoplasmic reticulum. We have discovered evidence for some type of
immune system dysfunction in vitiligo patients, as we have found decreased levels of
MHC class I on monocytes of vitiligo patients. Perhaps this defect allows for the escape
of self-reactive T cells from the thymus during positive and negative selection.
Regardless, the genetic association studies, as well as the decreased class I expression
found in vitiligo patients points to some type of involvement of the immune system in
vitiligo pathogenesis. This defect, accompanied by concomitant increases in epidermal
oxidative stress, may allow for the onset of vitiligo pathogenesis in susceptible
individuals.

111
Future Directions
RNA analysis of both the MHC class II genes and catalase revealed no altered
expression at the mRNA level (Chaper 5). Because MHC class II genes are very tightly
linked, the gene that is truly conferring susceptibility to vitiligo may not be any of the
genes used in our genotyping studies. Further case/control studies could be performed on
other genes in the region, especially as new sequence data is now available with the
completion of the human genome project. Flow cytometry studies of β2-microglobulin
expression need to be performed with an greater number of vitiligo patients to determine
whether MHC class I expression is truly lower on patient monocytes. Further studies
need to be performed to rule out variations in protein expression of the MHC class II
genes and catalase as playing a role in vitiligo pathogenesis. As patient samples were
limited by IRB protocol, we were unable to perform Western analysis on these proteins
of interest. Especially with catalase, protein expression studies may help answer the
question of whether decreased enzyme expression is due to a defect in the catalase gene
or due to inactivation of the enzyme by hydrogen peroxide.
Animal studies could be designed to further elucidate the role of catalase in vitiligo.
The Smyth line chicken (SL) is the most appropriate animal model of vitiligo
pathogenesis. The development of vitiligo in SL chickens is believed to depend on two
interacting components, an inherent melanocyte defect as well as an autoimmune reaction
to melanocytes, characterized by lymphocytic infiltration into the feather (Erf et al.,
2001). Studies of catalase, other antioxidant enzymes, and hydrogen peroxide in feathers
and monocytes of these animals might further implicate a role for oxidative stress in
vitiligo pathogenesis. If catalase were found to be deficient in these animals, transgenic

112
SL chickens could be created to hyper-express catalase to determine if this phenotype
would “rescue” these animals from vitiligo.
To our knowledge, there have been no knockout catalase mice created, although
several groups are in the process of designing such animals. Knockouts of other
antioxidant enzymes such as MnSOD have been created, however, knocking out MnSOD
in these animals is a fatal phenotype (Li et al., 1995). Because acatalasemia in humans is
a mostly benign phenotype, it is unlikely that knocking out the catalase gene in mice
would be a fatal disorder. Studies have shown injecting mice with recombinant vaccinia
virus encoding murine tyrosinase related protein-1 (TRP1), a melanocyte autoantibody
often found in sera of vitiligo patients, causes spontaneous vitiligo-like lesions in mice
(Overwijk et al., 1999). Studies of rVV encoding mTRP1 in catalase-knockout mice,
might reveal if the absence of catalase would serve to increase hypopigmentation in these
animals. Likewise, performing these experiments in catalase-overproducing murine
transgenics, which do currently exist, may determine if increased catalase levels could
rescue this phenotype.
It would also be interesting to perform transfection assays of catalase into cell lines
of vitiligo patient melanocytes, to see if catalase expression could be upregulated. If
catalase enzyme expression can be upregulated, and if upregulation of the enzyme means
amelioration of vitiligo symptoms, as suggested by the efficacy of the topical
pseudocatalase cream, there may be a role in the future for gene therapy treatment of
vitiligo patients with the catalase gene.

113
LIST OF REFERENCES
Abdel-Naser, M. B., W. D. Ludwig, H. Gollnick, and C. E. Orfanos. 1992. Nonsegmentalvitiligo: decrease of the CD45RA+ T-cell subset and evidence for peripheral T-cellactivation. Int. J. Dermatol. 31:321-326.
al Badri, A. M., A. K. Foulis, P. M. Todd, J. J. Gariouch, J. E. Gudgeon, D. G. Stewart, J.A. Gracie, and R. B. Goudie. 1993. Abnormal expression of MHC class II andICAM-1 by melanocytes in vitiligo. J. Pathol. 169:203-206.
al Fouzan, A., M. al Arbash, F. Fouad, S. A. Kaaba, M. A. Mousa, and S. A. al Harbi.1995. Study of HLA class I/IL and T lymphocyte subsets in Kuwaiti vitiligopatients. Eur. J. Immunogenet. 22:209-213.
Alkhateeb, A., G. L. Stetler, W. Old, J. Talbert, C. Uhlhorn, M. Taylor, A. Fox, C. Miller,D. G. Dills, E. C. Ridgway, D. C. Bennett, P. R. Fain, and R. A. Spritz. 2002.Mapping of an autoimmunity susceptibility locus (AIS1) to chromosome 1p31.3-p32.2. Hum. Mol. Genet. 11:661-667.
Arcos-Burgos, M., E. Parodi, M. Salgar, E. Bedoya, J. Builes, D. Jaramillo, G. Ceballos,A. Uribe, N. Rivera, D. Rivera, I. Fonseca, M. Camargo, and G. Palacio. 2002.Vitiligo: complex segregation and linkage disequilibrium analyses with respect tomicrosatellite loci spanning the HLA. Hum. Genet. 110:334-342.
Aronoff, S. 1965. Catalase: kinetics of photooxidation. Science 150:72-73.
Badri, A. M., P. M. Todd, J. J. Garioch, J. E. Gudgeon, D. G. Stewart, and R. B. Goudie.1993. An immunohistological study of cutaneous lymphocytes in vitiligo. J.Pathol. 170:149-155.
Beazley, W. D., D. Gaze, A. Panske, E. Panzig, and K. U. Schallreuter. 1999. Serumselenium levels and blood glutathione peroxidase activities in vitiligo. Br. J.Dermatol. 141:301-303.
Bertera, S., M. L. Crawford, A. M. Alexander, G. D. Papworth, S. C. Watkins, P. D.Robbins, and M. Trucco. 2003. Gene transfer of manganese superoxide dismutaseextends islet graft function in a mouse model of autoimmune diabetes. Diabetes52:387-393.
Bhatia, P. S., L. Mohan, O. N. Pandey, K. K. Singh, S. K. Arora, and R. D. Mukhija.1992. Genetic nature of vitiligo. J. Dermatol. Sci. 4:180-184.

114
Bochtler, M., L. Ditzel, M. Groll, C. Hartmann, and R. Huber. 1999. The proteasome.Annu. Rev. Biophys. Biomol. Struct. 28:295-317.
Boissy, R. E., Y. Y. Liu, E. E. Medrano, and J. J. Nordlund. 1991. Structural aberrationof the rough endoplasmic reticulum and melanosome compartmentalization inlong-term cultures of melanocytes from vitiligo patients. J. Invest. Dermatol.97:395-404.
Bos, J. D. 1997. Skin Immune System 2nd edn CRC Press, Boca Raton.
Bronte, V., E. Apolloni, R. Ronca, P. Zamboni, W. W. Overwijk, D. R. Surman, N. P.Restifo, and P. Zanovello. 2000. Genetic vaccination with "self" tyrosinase-relatedprotein 2 causes melanoma eradication but not vitiligo. Cancer Res. 60:253-258.
Brooks, P., G. Fuertes, R. Z. Murray, S. Bose, E. Knecht, M. C. Rechsteiner, K. B.Hendil, K. Tanaka, J. Dyson, and J. Rivett. 2000. Subcellular localization ofproteasomes and their regulatory complexes in mammalian cells. Biochem. J. 346Pt 1:155-161.
Buc, M., B. Busova, E. Hegyi, and K. Kolibasova. 1996. Vitiligo is associated with HLA-A2 and HLA-Dw7 in the Slovak populations. Folia Biol.(Praha) 42:23-25.
Cabrera, C. M., P. Jimenez, T. Cabrera, C. Esparza, F. Ruiz-Cabello, and F. Garrido.2003. Total loss of MHC class I in colorectal tumors can be explained by twomolecular pathways: beta2-microglobulin inactivation in MSI-positive tumors andLMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 61:211-219.
Caixia, T., F. Hongwen, and L. Xiran. 1999. Levels of soluble interleukin-2 receptor inthe sera and skin tissue fluids of patients with vitiligo. J. Dermatol. Sci. 21:59-62.
Calabrese, V., G. Scapagnini, A. Ravagna, R. Bella, R. Foresti, T. E. Bates, A. M.Giuffrida Stella, and G. Pennisi. 2002. Nitric oxide synthase is present in thecerebrospinal fluid of patients with active multiple sclerosis and is associated withincreases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and withchanges in glutathione levels. J. Neurosci. Res. 70:580-587.
Campbell, J. J., G. Haraldsen, J. Pan, J. Rottman, S. Qin, P. Ponath, D. P. Andrew, R.Warnke, N. Ruffing, N. Kassam, L. Wu, and E. C. Butcher. 1999. The chemokinereceptor CCR4 in vascular recognition by cutaneous but not intestinal memory Tcells. Nature 400:776-780.
Carrington, M., J. C. Stephens, W. Klitz, A. B. Begovich, H. A. Erlich, and D. Mann.1994. Major histocompatibility complex class II haplotypes and linkagedisequilibrium values observed in the CEPH families. Hum. Immunol. 41:234-240.
Casp, C. B., J. X. She, and W. T. McCormack. 2002. Genetic association of the catalasegene (CAT) with vitiligo susceptibility. Pigment Cell Res. 15:62-66.

115
Casp, C. B., J. X. She, and W. T. McCormack. 2003 Genes of the LMP/TAP cluster areassociated with the human autoimmune disease vitiligo. Genes Immun. 4:492-499.
Chan, O. T., V. Paliwal, J. M. McNiff, S. H. Park, A. Bendelac, and M. J. Shlomchik.2001. Deficiency in beta(2)-microglobulin, but not CD1, accelerates spontaneouslupus skin disease while inhibiting nephritis in MRL-Fas(lpr) nice: an example ofdisease regulation at the organ level. J. Immunol. 167:2985-2990.
Chang, J. T., B. M. Segal, and E. M. Shevach. 2000. Role of costimulation in theinduction of the IL-12/IL-12 receptor pathway and the development ofautoimmunity. J. Immunol. 164:100-106.
Chen, H. L., D. Gabrilovich, A. Virmani, I. Ratnani, K. R. Girgis, S. Nadaf-Rahrov, M.Fernandez-Vina, and D. P. Carbone. 1996. Structural and functional analysis ofbeta2 microglobulin abnormalities in human lung and breast cancer. Int. J. Cancer67:756-763.
Cimen, M. Y., O. B. Cimen, M. Kacmaz, H. S. Ozturk, R. Yorgancioglu, and I. Durak.2000. Oxidant/antioxidant status of the erythrocytes from patients with rheumatoidarthritis. Clin. Rheumatol. 19:275-277.
Cominelli, F., C. C. Nast, A. Duchini, and M. Lee. 1992. Recombinant interleukin-1receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology 103:65-71.
Cromme, F. V., J. Airey, M. T. Heemels, H. L. Ploegh, P. J. Keating, P. L. Stern, C. J.Meijer, and J. M. Walboomers. 1994. Loss of transporter protein, encoded by theTAP-1 gene, is highly correlated with loss of HLA expression in cervicalcarcinomas. J. Exp. Med. 179:335-340.
Cui, J., Y. Arita, and J. C. Bystryn. 1993. Cytolytic antibodies to melanocytes in vitiligo.J. Invest. Dermatol. 100:812-815.
Cullen, M., J. Noble, H. Erlich, K. Thorpe, S. Beck, W. Klitz, J. Trowsdale, and M.Carrington. 1997. Characterization of recombination in the HLA class II region.Am. J. Hum. Genet. 60:397-407.
Cullen, M., S. P. Perfetto, W. Klitz, G. Nelson, and M. Carrington. 2002. High-resolutionpatterns of meiotic recombination across the human major histocompatibilitycomplex. Am. J. Hum. Genet. 71:759-776.
Dai, L., A. Claxson, S. L. Marklund, R. Feakins, N. Yousaf, Y. Chernajovsky, and P. G.Winyard. 2003. Amelioration of antigen-induced arthritis in rats by transfer ofextracellular superoxide dismutase and catalase genes. Gene Ther. 10:550-558.
Daniel S., S.Caillat-Zucman, J.Hammer, J.F. Bach, and P.M. van Endert. 1997. Absenceof functional relevance of human transporter associated with antigen processingpolymorphism for peptide selection. J. Immunol. 159: 2350-2357.

116
Davoodi-Semiromi, A., J. J. Yang, and J. X. She. 2002. IL-12p40 is associated with type1 diabetes in Caucasian-American families. Diabetes 51:2334-2336.
Dell'Anna, M. L., V. Maresca, S. Briganti, E. Camera, M. Falchi, and M. Picardo. 2001.Mitochondrial impairment in peripheral blood mononuclear cells during the activephase of vitiligo. J. Invest. Dermatol. 117:908-913.
Deng, G. Y., A. Muir, N. K. Maclaren, and J. X. She. 1995. Association of LMP2 andLMP7 genes within the major histocompatibility complex with insulin-dependentdiabetes mellitus: population and family studies. Am. J. Hum. Genet. 56:528-534.
di Giovine, F. S., E. Takhsh, A. I. Blakemore, and G. W. Duff. 1992. Single basepolymorphism at -511 in the human interleukin-1 beta gene (IL1 beta). Hum. Mol.Genet. 1:450.
Dissemond, J., P. Goette, J. Moers, A. Lindeke, M. Goos, S. Ferrone, and S. N. Wagner.2003. Immunoproteasome subunits LMP2 and LMP7 downregulation in primarymalignant melanoma lesions: association with lack of spontaneous regression.Melanoma Res. 13:371-377.
Djilali-Saiah, I., S. Caillat-Zucman, J. Schmitz, M. L. Chaves-Vieira, and J. F. Bach.1994. Polymorphism of antigen processing (TAP, LMP) and HLA class II genes inceliac disease. Hum. Immunol. 40:8-16.
Djilali-Saiah, I., V. Benini, S. Daniel, R. Assan, J. F. Bach, and S. Caillat-Zucman. 1996.Linkage disequilibrium between HLA class II (DR, DQ, DP) and antigenprocessing (LMP, TAP, DM) genes of the major histocompatibility complex.Tissue Antigens 48:87-92.
Driscoll, J., M. G. Brown, D. Finley, and J. J. Monaco. 1993. MHC-linked LMP geneproducts specifically alter peptidase activities of the proteasome. Nature 365:262-264.
Duthoit, C., V. Estienne, A. Giraud, J. M. Durand-Gorde, A. K. Rasmussen, U. Feldt-Rasmussen, P. Carayon, and J. Ruf. 2001. Hydrogen peroxide-induced productionof a 40 kDa immunoreactive thyroglobulin fragment in human thyroid cells: theonset of thyroid autoimmunity? Biochem. J. 360:557-562.
Eaton J.W.and Ma M. 1995. Acatalasemia. In The Metabolic Bases of Inherited Disease.Scriver C.R., ed. McGraw-Hill, New York, pp. 2371-2380.
Einarsdottir, E., I. Soderstrom, A. Lofgren-Burstrom, S. Haraldsson, S. Nilsson-Ardnor,C. Penha-Goncalves, L. Lind, G. Holmgren, M. Holmberg, K. Asplund, and D.Holmberg. 2003. The CTLA4 region as a general autoimmunity factor: an extendedpedigree provides evidence for synergy with the HLA locus in the etiology of type1 diabetes mellitus, Hashimoto's thyroiditis and Graves' disease. Eur. J. Hum.Genet. 11:81-84.

117
Eizirik, D. L., D. E. Tracey, K. Bendtzen, and S. Sandler. 1991. An interleukin-1 receptorantagonist protein protects insulin-producing beta cells against suppressive effectsof interleukin-1 beta. Diabetologia 34:445-448.
Erf, G. F., T. K. Bersi, X. Wang, G. P. Sreekumar, and J. R. Smyth, Jr. 2001. Herpesvirusconnection in the expression of autoimmune vitiligo in Smyth line chickens.Pigment Cell Res. 14:40-46.
Faustman, D., X. P. Li, H. Y. Lin, Y. E. Fu, G. Eisenbarth, J. Avruch, and J. Guo. 1991.Linkage of faulty major histocompatibility complex class I to autoimmune diabetes.Science 254:1756-1761.
Finco, O., M. Cuccia, M. Martinetti, G. Ruberto, G. Orecchia, and G. Rabbiosi. 1991.Age of onset in vitiligo: relationship with HLA supratypes. Clin. Genet. 39:48-54.
Fine, C. I., C. D. Han, X. Sun, Y. Liu, and J. A. McCutcheon. 2002. Tobacco reducesmembrane HLA class I that is restored by transfection with transporter associatedwith antigen processing 1 cDNA. J. Immunol. 169:6012-6019.
Foley, L. M., N. J. Lowe, E. Misheloff, and J. L. Tiwari. 1983. Association of HLA-DR4with vitiligo. J. Am. Acad. Dermatol. 8:39-40.
Forsberg, L., U. de Faire, and R. Morgenstern. 1999. Low yield of polymorphisms fromEST blast searching: analysis of genes related to oxidative stress and verification ofthe P197L polymorphism in GPX1. Hum. Mutat. 13:294-300.
Fraile, A., A. Nieto, J. Vinasco, Y. Beraun, J. Martin, and L. Mataran. 1998. Associationof large molecular weight proteasome 7 gene polymorphism with ankylosingspondylitis. Arthritis Rheum. 41:560-562.
Friedmann A. 1999. HLA and Dermatological Diseases. In: HLA in Health and Disease.Lechler R.and Warrens A., ed. Academic Press, San Diego, pp. 365-386.
Fruh, K. and Y. Yang. 1999. Antigen presentation by MHC class I and its regulation byinterferon gamma. Curr. Opin. Immunol. 11:76-81.
Fu, Y., D. M. Nathan, F. Li, X. Li, and D. L. Faustman. 1993. Defective majorhistocompatibility complex class I expression on lymphoid cells in autoimmunity. JClin Invest 91:2301-2307.
Fu, Y., G. Yan, L. Shi, and D. Faustman. 1998. Antigen processing and autoimmunity.Evaluation of mRNA abundance and function of HLA-linked genes. Ann. N. Y.Acad. Sci. 842:138-155.
Fuhlbrigge, R. C., J. D. Kieffer, D. Armerding, and T. S. Kupper. 1997. Cutaneouslymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing Tcells. Nature 389:978-981.

118
Goth, L. 1998. Genetic heterogeneity of the 5' uncoding region of the catalase gene inHungarian acatalasemic and hypocatalasemic subjects. Clin. Chim. Acta 271:73-78.
Goth, L. and J. W. Eaton. 2000. Hereditary catalase deficiencies and increased risk ofdiabetes. Lancet 356:1820-1821.
Gotia, S., I. Popovici, and B. Hermeziu. 2001. Antioxidant enzymes levels in childrenwith juvenile rheumatoid arthritis. Rev. Med. Chir. Soc. Med. Nat. Iasi 105:499-503.
Griffiths, A.J.F., J. H. Miller, D.T. Suzuki, R.C. Lewontin, and W.M. Gelbart. 1999.Introduction to Genetic Analysis. 7th edn. W.H. Freeman and Company, NewYork.
Groettrup, M., S. Khan, K. Schwarz, and G. Schmidtke. 2001. Interferon-gammainducible exchanges of 20S proteasome active site subunits: why? Biochimie83:367-372.
Halaban, R., S. Svedine, E. Cheng, Y. Smicun, R. Aron, and D. N. Hebert. 2000.Endoplasmic reticulum retention is a common defect associated with tyrosinase-negative albinism. Proc. Natl. Acad. Sci. U.S.A 97:5889-5894.
Hanafusa, T., R. Pujol-Borrell, L. Chiovato, R. C. Russell, D. Doniach, and G. F.Bottazzo. 1983. Aberrant expression of HLA-DR antigen on thyrocytes in Graves'disease: relevance for autoimmunity. Lancet 2:1111-1115.
Hann, S. K., Y. K. Park, K. G. Lee, E. H. Choi, and S. Im. 1992. Epidermal changes inactive vitiligo. J. Dermatol. 19:217-222.
Harding C.V.1997. MHC Molecules and Antigen Processing. R.G. Landes Company,Austin.
Hayney MS, G.A. Poland, P. Dimanlig, D.J. Schaid, R.M. Jacobson, and J.J. Lipsky.1997. Polymorphisms of the TAP2 gene may influence antibody response to livemeasles vaccine virus. Vaccine 15: 3-6.
Honda, Y., Y. Okubo, and M. Koga. 1997. Relationship between levels ofsolubleinterleukin-2 receptors and the types and activity of vitiligo. J. Dermatol.24:561-563.
Im, S., S. K. Hann, H. I. Kim, N. S. Kim, and Y. K. Park. 1994. Biologic characteristicsof cultured human vitiligo melanocytes. Int. J. Dermatol. 33:556-562.
Jeffreys, A. J., A. Ritchie, and R. Neumann. 2000. High resolution analysis of haplotypediversity and meiotic crossover in the human TAP2 recombination hotspot. Hum.Mol. Genet. 9:725-733.

119
Kantarci, O. H., D. D. Hebrink, S. J. Achenbach, E. J. Atkinson, A. Waliszewska, G.Buckle, C. T. McMurray, M. de Andrade, D. A. Hafler, and B. G. Weinshenker.2003. CTLA4 is associated with susceptibility to multiple sclerosis. J.Neuroimmunol. 134:133-141.
Kao, C. H. and H. S. Yu. 1990. Depletion and repopulation of Langerhans cells innonsegmental type vitiligo. J. Dermatol. 17:287-296.
Kemp, E. H., R. A. Ajjan, E. A. Waterman, D. J. Gawkrodger, M. J. Cork, P. F. Watson,and A. P. Weetman. 1999. Analysis of a microsatellite polymorphism of thecytotoxic T-lymphocyte antigen-4 gene in patients with vitiligo. Br. J. Dermatol.140:73-78.
Kemp, E. H., E. A. Waterman, B. E. Hawes, K. O'Neill, R. V. S. R. Gottumukkala, D. J.Gawkrodger, A. P. Weetman, and P. F. Watson. 2002. The melanin-concentratinghormone receptor 1, a novel target of autoantibody responses in vitiligo. J. Clin.Invest. 109:923-930.
Kohn, L. D., G. Napolitano, D. S. Singer, M. Molteni, R. Scorza, N. Shimojo, Y. Kohno,E. Mozes, M. Nakazato, L. Ulianich, H. K. Chung, H. Matoba, B. Saunier, K.Suzuki, F. Schuppert, and M. Saji. 2000. Graves' disease: a host defensemechanism gone awry. Int. Rev. Immunol. 19:633-664.
Kristensen D.L. The genetics of human vitiligo susceptibility; a case/control associationstudy of tyrosine metabolism and immune response candidate genes. 2000. MastersThesis, University of Florida.
Kumagai, S., S. Kanagawa, A. Morinobu, M. Takada, K. Nakamura, S. Sugai, E. Maruya,and H. Saji. 1997. Association of a new allele of the TAP2 gene, TAP2*Bky2(Val577), with susceptibility to Sjogren's syndrome. Arthritis Rheum. 40:1685-1692.
Kwon, J. M. and A. M. Goate. 2000. The candidate gene approach. Alcohol Res. Health24:164-168.
Lamoreaux L. and Boissy R.E.. 2000. PUVA therapy. In: Vitiligo. Hann S.K.andNordlund J.J., ed. Blackwell Science, Oxford, pp. 281-297.
Le Poole, I. C., R. M. van den Wijngaard, W. Westerhof, R. P. Dutrieux, and P. K. Das.1993a. Presence or absence of melanocytes in vitiligo lesions: animmunohistochemical investigation. J. Invest. Dermatol. 100:816-822.
Le Poole, I. C., P. K. Das, R. M. van den Wijngaard, J. D. Bos, and W. Westerhof.1993b. Review of the etiopathomechanism of vitiligo: a convergence theory. Exp.Dermatol. 2:145-153.

120
Le Poole, I. C., R. M. van den Wijngaard, W. Westerhof, and P. K. Das. 1996. Presenceof T cells and macrophages in inflammatory vitiligo skin parallels melanocytedisappearance. Am. J. Pathol. 148:1219-1228.
Le Poole, I. C., R. E. Boissy, R. Sarangarajan, J. Chen, J. J. Forristal, P. Sheth, W.Westerhof, G. Babcock, P. K. Das, and C. B. Saelinger. 2000. PIG3V, animmortalized human vitiligo melanocyte cell line, expresses dilated endoplasmicreticulum. In Vitro Cell Dev. Biol. Anim. 36:309-319.
Lenzen, S., J. Drinkgern, and M. Tiedge. 1996. Low antioxidant enzyme gene expressionin pancreatic islets compared with various other mouse tissues. Free Radic. Biol.Med. 20:463-466.
Li, F., M. J. Linan, M. C. Stein, and D. L. Faustman. 1995. Reduced expression ofpeptide-loaded HLA class I molecules on multiple sclerosis lymphocytes. Ann.Neurol. 38:147-154.
Li, Y., T. T. Huang, E. J. Carlson, S. Melov, P. C. Ursell, J. L. Olson, L. J. Noble, M. P.Yoshimura, C. Berger, P. H. Chan, and . 1995. Dilated cardiomyopathy andneonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat.Genet. 11:376-381.
Lortz, S. and M. Tiedge. 2003. Sequential inactivation of reactive oxygen species bycombined overexpression of SOD isoforms and catalase in insulin-producing cells.Free Radic. Biol. Med. 34:683-688.
Majumder, P. P., J. J. Nordlund, and S. K. Nath. 1993. Pattern of familial aggregation ofvitiligo. Arch. Dermatol. 129:994-998.
Maksymowych, W. P., T. Ikawa, A. Yamaguchi, M. Ikeda, D. McDonald, L. Laouar, R.Lahesmaa, N. Tamura, A. Khuong, D. T. Yu, and K. P. Kane. 1998. Invasion bySalmonella typhimurium induces increased expression of the LMP, MECL, andPA28 proteasome genes and changes in the peptide repertoire of HLA-B27. Infect.Immun. 66:4624-4632.
Maresca, V., M. Roccella, F. Roccella, E. Camera, G. Del Porto, S. Passi, P.Grammatico, and M. Picardo. 1997. Increased sensitivity to peroxidative agents asa possible pathogenic factor of melanocyte damage in vitiligo. J. Invest Dermatol.109:310-313.
Marron, M. P., L. J. Raffel, H. J. Garchon, C. O. Jacob, M. Serrano-Rios, M. T. MartinezLarrad, W. P. Teng, Y. Park, Z. X. Zhang, D. R. Goldstein, Y. W. Tao, G.Beaurain, J. F. Bach, H. S. Huang, D. F. Luo, A. Zeidler, J. I. Rotter, M. C. Yang,T. Modilevsky, N. K. Maclaren, and J. X. She. 1997. Insulin-dependent diabetesmellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnicgroups. Hum. Mol. Genet. 6:1275-1282.

121
Matsui, M., S. Machida, T. Itani-Yohda, and T. Akatsuka. 2002. Downregulation of theproteasome subunits, transporter, and antigen presentation in hepatocellularcarcinoma, and their restoration by interferon-gamma. J. Gastroenterol. Hepatol.17:897-907.
McClelland, P. B., P. Morgan, E. E. Leach, and J. Shelk. 1997. Psoralenphotochemotherapy. Dermatol. Nurs. 9:403-415.
McNeely, W. and K. L. Goa. 1998. 5-Methoxypsoralen. A review of its effects inpsoriasis and vitiligo. Drugs 56:667-690.
Mishto M., M. Bonafe,S. Salvioli, F.Olivieri, and C. Franceschi. 2002. Age dependentimpact of LMP polymorphisms on TNFα-induced apoptosis in human peripheralblood mononuclear cells. Exp. Gerontol. 37:301-308.
Miyagi, T., T. Tatsumi, T. Takehara, T. Kanto, N. Kuzushita, Y. Sugimoto, M. Jinushi,A. Kasahara, Y. Sasaki, M. Hori, and N. Hayashi. 2003. Impaired expression ofproteasome subunits and human leukocyte antigens class I in human colon cancercells. J. Gastroenterol. Hepatol. 18:32-40.
Moins-Teisserenc, H., G. Semana, M. Alizadeh, P. Loiseau, V. Bobrynina, I. Deschamps,G. Edan, B. Birebent, B. Genetet, O. Sabouraud, and . 1995. TAP2 genepolymorphism contributes to genetic susceptibility to multiple sclerosis. Hum.Immunol. 42:195-202.
Morales, J., B. Homey, A. P. Vicari, S. Hudak, E. Oldham, J. Hedrick, R. Orozco, N. G.Copeland, N. A. Jenkins, L. M. McEvoy, and A. Zlotnik. 1999. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells.Proc. Natl. Acad. Sci. U.S.A 96:14470-14475.
Morel, S., F. Levy, O. Burlet-Schiltz, F. Brasseur, M. Probst-Kepper, A. L. Peitrequin, B.Monsarrat, R. Van Velthoven, J. C. Cerottini, T. Boon, J. E. Gairin, and B. J. Vanden Eynde. 2000. Processing of some antigens by the standard proteasome but notby the immunoproteasome results in poor presentation by dendritic cells. Immunity.12:107-117.
Moriscot, C., M. J. Richard, M. C. Favrot, and P. Y. Benhamou. 2003. Protection ofinsulin-secreting INS-1 cells against oxidative stress through adenoviral-mediatedglutathione peroxidase overexpression. Diabetes Metab. 29:145-151.
Morrison W.L. 2000. PUVA therapy. In: Vitiligo. Hann S.K.and Nordlund J.J., ed.Blackwell Science, Oxford, pp. 168-172.
Mozzanica, N., U. Frigerio, A. F. Finzi, A. Cattaneo, M. Negri, F. Scaglione, F.Fraschini, and S. Foppa. 1990. T cell subpopulations in vitiligo: a chronobiologicstudy. J. Am. Acad. Dermatol. 22:223-230.

122
Nath, S. K., P. P. Majumder, and J. J. Nordlund. 1994. Genetic epidemiology of vitiligo:multilocus recessivity cross-validated. Am. J. Hum. Genet. 55:981-990.
Njoo M.D., and Westerhof W. 2001. Vitiligo. Pathogenesis and Treatment. Am. J. Clin.Dermatol. 2:167-181.
Nordlund J.J.and Ortonne J.P. 1998. Vitiligo vulgaris. In: The Pigmentary System.Physiology and Pathophysiology. Nordlund J.J., Oxford University Press, NewYork, pp. 513-551.
Nussbaum R.L. 2001. Thompson and Thompson Genetics in Medicine, 6th edn. W.B.Saunders, Philadelphia.
Obst R, E.A., Armandola, M. Nijenhuis, F. Momburg , and G.J. Hammerling. 1995. TAPpolymorphism does not influence transport of peptide variants in mice and humans.Eur. J. Immunol 25:2170-2176.
Ogg, G. S., D. P. Rod, P. Romero, J. L. Chen, and V. Cerundolo. 1998. High frequencyof skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmunevitiligo. J. Exp. Med. 188:1203-1208.
Ongenae, K., N. Van Geel, and J. M. Naeyaert. 2003. Evidence for an autoimmunepathogenesis of vitiligo. Pigment Cell Res. 16:90-100.
Orecchia, G., L. Perfetti, P. Malagoli, F. Borghini, and Y. Kipervarg. 1992. Vitiligo isassociated with a significant increase in HLA-A30, Cw6 and DQw3 and a decreasein C4AQ0 in northern Italian patients. Dermatology 185:123-127.
Orth, T., R. Kellner, O. Diekmann, J. Faust, K. H. Meyer zum Buschenfelde, and W. J.Mayet. 1998. Identification and characterization of autoantibodies against catalaseand alpha-enolase in patients with primary sclerosing cholangitis. Clin Exp.Immunol. 112:507-515.
Ortonne, J. P. and S. K. Bose. 1993. Vitiligo: where do we stand? Pigment Cell Res.6:61-72.
Overwijk, W. W., D. S. Lee, D. R. Surman, K. R. Irvine, C. E. Touloukian, C. C. Chan,M. W. Carroll, B. Moss, S. A. Rosenberg, and N. P. Restifo. 1999. Vaccinationwith a recombinant vaccinia virus encoding a "self" antigen induces autoimmunevitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes.Proc. Natl. Acad. Sci. U.S.A 96:2982-2987.
Overwijk, W. W., M. R. Theoret, S. E. Finkelstein, D. R. Surman, L. A. de Jong, F. A.Vyth-Dreese, T. A. Dellemijn, P. A. Antony, P. J. Spiess, D. C. Palmer, D. M.Heimann, C. A. Klebanoff, Z. Yu, L. N. Hwang, L. Feigenbaum, A. M. Kruisbeek,S. A. Rosenberg, and N. P. Restifo. 2003. Tumor regression and autoimmunityafter reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp.Med. 198:569-580.

123
Pamer, E. and P. Cresswell. 1998. Mechanisms of MHC class I--restricted antigenprocessing. Annu. Rev. Immunol. 16:323-358.
Picker, L. J., J. R. Treer, B. Ferguson-Darnell, P. A. Collins, P. R. Bergstresser, and L.W. Terstappen. 1993. Control of lymphocyte recirculation in man. II. Differentialregulation of the cutaneous lymphocyte-associated antigen, a tissue-selectivehoming receptor for skin-homing T cells. J. Immunol. 150:1122-1136.
Pitkanen, J. and P. Peterson. 2003. Autoimmune regulator: from loss of function toautoimmunity. Genes Immun. 4:12-21.
Prahalad, S., D. J. Kingsbury, T. A. Griffin, B. L. Cooper, D. N. Glass, W. P.Maksymowych, and R. A. Colbert. 2001. Polymorphism in the MHC-encodedLMP7 gene: association with JRA without functional significance forimmunoproteasome assembly. J. Rheumatol. 28:2320-2325.
Quadri SA and D.P. Singal. 1998 Peptide transport in human lymphoblastoid and tumorcells: effect of transporter associated with antigen presentation (TAP)polymorphism Immunol. Lett. 61: 25-31.
Rechsteiner, M., C. Realini, and V. Ustrell. 2000. The proteasome activator 11 S REG(PA28) and class I antigen presentation. Biochem. J. 345 Pt 1:1-15.
Redondo M.J., P.R. Fain, and G.S.Eisenbarth. 2001 Genetics of type 1A diabetes. RecentPro. Hormone Res. 56:69-89.
Robert, C. and T. S. Kupper. 1999. Inflammatory skin diseases, T cells, and immunesurveillance. N. Engl. J. Med. 341:1817-1828.
Rock, K. L. and A. L. Goldberg. 1999. Degradation of cell proteins and the generation ofMHC class I-presented peptides. Annu. Rev. Immunol. 17:739-779.
Rodriguez, M. R., A. Nunez-Roldan, F. Aguilar, A. Valenzuela, A. Garcia, and M. F.Gonzalez-Escribano. 2002. Association of the CTLA4 3' untranslated regionpolymorphism with the susceptibility to rheumatoid arthritis. Hum. Immunol.63:76-81.
Roozendaal, C., M. H. Zhao, G. Horst, C. M. Lockwood, J. H. Kleibeuker, P. C.Limburg, G. F. Nelis, and C. G. Kallenberg. 1998. Catalase and alpha-enolase: twonovel granulocyte autoantigens in inflammatory bowel disease (IBD). Clin Exp.Immunol. 112:10-16.
Sandler, S., D. L. Eizirik, C. Svensson, E. Strandell, M. Welsh, and N. Welsh. 1991.Biochemical and molecular actions of interleukin-1 on pancreatic beta-cells.Autoimmunity 10:241-253.
Shaffer, J. P. 1995. Multiple Hypothesis Testing. Ann. Rev. Psych. 46: 561-584.

124
Schallreuter, K. U., J. M. Wood, and J. Berger. 1991. Low catalase levels in theepidermis of patients with vitiligo. J. Invest. Dermatol. 97:1081-1085.
Schallreuter, K. U., C. Levenig, P. Kuhnl, C. Loliger, M. Hohl-Tehari, and J. Berger.1993. Histocompatibility antigens in vitiligo: Hamburg study on 102 patients fromnorthern Germany. Dermatology 187:186-192.
Schallreuter, K. U., J. M. Wood, I. Ziegler, K. R. Lemke, M. R. Pittelkow, N. J. Lindsey,and M. Gutlich. 1994. Defective tetrahydrobiopterin and catecholaminebiosynthesis in the depigmentation disorder vitiligo. Biochim. Biophys. Acta1226:181-192.
Schallreuter, K. U., J. M. Wood, K. R. Lemke, and C. Levenig. 1995. Treatment ofvitiligo with a topical application of pseudocatalase and calcium in combinationwith short-term UVB exposure: a case study on 33 patients. Dermatology 190:223-229.
Schallreuter, K. U., J. M. Wood, M. R. Pittelkow, G. Buttner, N. Swanson, C. Korner,and C. Ehrke. 1996. Increased monoamine oxidase A activity in the epidermis ofpatients with vitiligo. Arch. Dermatol. Res. 288:14-18.
Schallreuter, K. U. 1999a. Successful treatment of oxidative stress in vitiligo. SkinPharmacol. Appl. Skin Physiol. 12:132-138.
Schallreuter, K. U., J. Moore, J. M. Wood, W. D. Beazley, D. C. Gaze, D. J. Tobin, H. S.Marshall, A. Panske, E. Panzig, and N. A. Hibberts. 1999b. In vivo and in vitroevidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patientswith vitiligo and its successful removal by a UVB-activated pseudocatalase. JInvestig. Dermatol. Symp. Proc. 4:91-96.
Schallreuter, K. U., J. Moore, J. M. Wood, W. D. Beazley, E. M. Peters, L. K. Marles, S.C. Behrens-Williams, R. Dummer, N. Blau, and B. Thony. 2001. EpidermalH(2)O(2) accumulation alters tetrahydrobiopterin (6BH4) recycling in vitiligo:identification of a general mechanism in regulation of all 6BH4-dependentprocesses? J. Invest. Dermatol. 116:167-174.
Schallreuter, K. U., J. Moore, S. Behrens-Williams, A. Panske, and M. Harari. 2002.Rapid initiation of repigmentation in vitiligo with Dead Sea climatotherapy incombination with pseudocatalase (PC-KUS). Int. J. Dermatol. 41:482-487.
Schwab, J. H., S. K. Anderle, R. R. Brown, F. G. Dalldorf, and R. C. Thompson. 1991.Pro- and anti-inflammatory roles of interleukin-1 in recurrence of bacterial cellwall-induced arthritis in rats. Infect. Immun. 59:4436-4442.

125
Schwarz, K., B. M. van Den, S. Kostka, R. Kraft, A. Soza, G. Schmidtke, P. M. Kloetzel,and M. Groettrup. 2000. Overexpression of the proteasome subunits LMP2, LMP7,and MECL-1, but not PA28 alpha/beta, enhances the presentation of animmunodominant lymphocytic choriomeningitis virus T cell epitope. J. Immunol.165:768-778.
Seegers, D., A. Zwiers, W. Strober, A. S. Pena, and G. Bouma. 2002. A TaqIpolymorphism in the 3'UTR of the IL-12 p40 gene correlates with increased IL-12secretion. Genes Immun. 3:419-423.
Selig, W. and J. Tocker. 1992. Effect of interleukin-1 receptor antagonist on antigen-induced pulmonary responses in guinea pigs. Eur. J. Pharmacol. 213:331-336.
Sijts, A. J., S. Standera, R. E. Toes, T. Ruppert, N. J. Beekman, P. A. van Veelen, F. A.Ossendorp, C. J. Melief, and P. M. Kloetzel. 2000. MHC class I antigen processingof an adenovirus CTL epitope is linked to the levels of immunoproteasomes ininfected cells. J. Immunol. 164:4500-4506.
Song, P. S. and K. J. Tapley, Jr. 1979. Photochemistry and photobiology of psoralens.Photochem. Photobiol. 29:1177-1197.
Song, Y. H., E. Connor, Y. Li, B. Zorovich, P. Balducci, and N. Maclaren. 1994. The roleof tyrosinase in autoimmune vitiligo. Lancet 344:1049-1052.
Sugimoto Y., N. Kuzushita, T. Takehara, T. Kanto, T. Tatsumi, T. Miyagi, M. Jinushi, K.Ohkawa, M. Horimoto, A. Kasahara, M. Hori, Y. Sasaki, and N. Hayashi .2002. Asingle nucleotide polymorphism of the low molecular mass polypeptide 7 geneinfluences the interferon response in patients with chronic hepatitis C. J. ViralHepat. 9:377-384.
Spielman, R. S. and W. J. Ewens. 1996. The TDT and other family-based tests forlinkage disequilibrium and association. Am. J. Hum. Genet. 59:983-989.
Tanaka, K. 1998. Molecular biology of the proteasome. Biochem. Biophys. Res.Commun. 247:537-541.
Taylor, A. and F. P. Gasparro. 1992. Extracorporeal photochemotherapy for cutaneous T-cell lymphoma and other diseases. Semin. Hematol. 29:132-141.
Taysi, S., M. Gul, R. A. Sari, F. Akcay, and N. Bakan. 2002. Serum oxidant/antioxidantstatus of patients with systemic lupus erythematosus. Clin. Chem. Lab. Med.40:684-688.
Tezak, Z., E. P. Hoffman, J. L. Lutz, T. O. Fedczyna, D. Stephan, E. G. Bremer, I.Krasnoselska-Riz, A. Kumar, and L. M. Pachman. 2002. Gene expression profilingin DQA1*0501+ children with untreated dermatomyositis: a novel model ofpathogenesis. J. Immunol. 168:4154-4163.

126
Tobin, D. J., N. N. Swanson, M. R. Pittelkow, E. M. Peters, and K. U. Schallreuter. 2000.Melanocytes are not absent in lesional skin of long duration vitiligo. J. Pathol.191:407-416.
Trigwell, S. M., P. M. Radford, S. R. Page, A. C. Loweth, R. F. James, N. G. Morgan,and I. Todd. 2001. Islet glutamic acid decarboxylase modified by reactive oxygenspecies is recognized by antibodies from patients with type 1 diabetes mellitus.Clin. Exp. Immunol. 126:242-249.
Tsunemi, Y., H. Saeki, K. Nakamura, T. Sekiya, K. Hirai, H. Fujita, N. Asano, M.Kishimoto, Y. Tanida, T. Kakinuma, H. Mitsui, Y. Tada, M. Wakugawa, H. Torii,M. Komine, A. Asahina, and K. Tamaki. 2002. Interleukin-12 p40 gene (IL12B) 3'-untranslated region polymorphism is associated with susceptibility to atopicdermatitis and psoriasis vulgaris. J. Dermatol. Sci. 30:161-166.
Tu, C. X., H. W. Fu, and X. R. Lin. 1999. Levels of soluble interleukin-2 receptor in thesera and skin tissue fluids of patients with vitiligo. J. Derm. Sci. 21:59-62.
Ueda, H., J. M. Howson, L. Esposito, J. Heward, H. Snook, G. Chamberlain, D. B.Rainbow, K. M. Hunter, A. N. Smith, G. Di Genova, M. H. Herr, I. Dahlman, F.Payne, D. Smyth, C. Lowe, R. C. Twells, S. Howlett, B. Healy, S. Nutland, H. E.Rance, V. Everett, L. J. Smink, A. C. Lam, H. J. Cordell, N. M. Walker, C. Bordin,J. Hulme, C. Motzo, F. Cucca, J. F. Hess, M. L. Metzker, J. Rogers, S. Gregory, A.Allahabadia, R. Nithiyananthan, E. Tuomilehto-Wolf, J. Tuomilehto, P. Bingley, K.M. Gillespie, D. E. Undlien, K. S. Ronningen, C. Guja, C. Ionescu-Tirgoviste, D.A. Savage, A. P. Maxwell, D. J. Carson, C. C. Patterson, J. A. Franklyn, D. G.Clayton, L. B. Peterson, L. S. Wicker, J. A. Todd, and S. C. Gough. 2003.Association of the T-cell regulatory gene CTLA4 with susceptibility toautoimmune disease. Nature 423:506-511.
van den Wijngaard, R., A. Wankowicz-Kalinska, S. Pals, J. Weening, and P. Das. 2001.Autoimmune melanocyte destruction in vitiligo. Lab. Invest. 81:1061-1067.
van Endert, P. M., M. T. Lopez, S. D. Patel, J. J. Monaco, and H. O. McDevitt. 1992.Genomic polymorphism, recombination, and linkage disequilibrium in humanmajor histocompatibility complex-encoded antigen-processing genes. Proc. Natl.Acad. Sci. U.S.A 89:11594-11597.
Venneker, G. T., L. P. de Waal, W. Westerhof, J. D'Amaro, G. M. Schreuder, and S. S.Asghar. 1993. HLA associations in vitiligo patients in the Dutch population. Dis.Markers 11:187-190.
Vives-Pi, M., M. P. Armengol, L. Alcalde, M. Costa, N. Somoza, F. Vargas, D.Jaraquemada, and R. Pujol-Borrell. 1996. Expression of transporter associated withantigen processing-1 in the endocrine cells of human pancreatic islets: effect ofcytokines and evidence of hyperexpression in IDDM. Diabetes 45:779-788.

127
Vives-Pi, M., F. Vargas, R. F. James, J. Trowsdale, M. Costa, M. Sospedra, N. Somoza,G. Obiols, R. Tampe, and R. Pujol-Borrell. 1997. Proteasome subunits, low-molecular-mass polypeptides 2 and 7 are hyperexpressed by target cells inautoimmune thyroid disease but not in insulin-dependent diabetes mellitus:implications for autoimmunity. Tissue Antigens 50:153-163.
Weir BS and Cockerham CC. 1984. Estimating F-statistics for the analysis of populationstructure. Evolution 38:1358-1370.
Wong FS, and L. Wen 2003. The study of HLA class II and autoimmune diabetes. Curr.Mol. Med. 3:1-15.
Yan, G., Y. Fu, and D. L. Faustman. 1997. Reduced expression of Tap1 and Lmp2antigen-processing genes in the nonobese diabetic (NOD) mouse due to a mutationin their shared bidirectional promoter. J. Immunol. 159:3068-3080.
Yeo, U. C., Y. S. Yang, K. B. Park, H. T. Sung, S. Y. Jung, E. S. Lee, and M. H. Shin.1999. Serum concentration of the soluble interleukin-2 receptor in vitiligo patients.J. Dermatol. Sci. 19:182-188.
Yildirim, M., V. Baysal, H. S. Inaloz, D. Kesici, and N. Delibas. 2003. The role ofoxidants and antioxidants in generalized vitiligo. J. Dermatol. 30:104-108.
Yu, H. S., K. L. Chang, C. L. Yu, H. F. Li, M. T. Wu, C. S. Wu, and C. S. Wu. 1997.Alterations in IL-6, IL-8, GM-CSF, TNF-alpha, and IFN-gamma release byperipheral mononuclear cells in patients with active vitiligo. J. Invest. Dermatol.108:527-529.

128
BIOGRAPHICAL SKETCH
Courtney Bradley Casp was born in Silver Spring, Maryland, to Connie and Tim
Bradley. She graduated from Walter Johnson High School in Bethesda, Maryland, in
1994. She then received her Bachelor of Science degree from the University of
Richmond in 1998, majoring in biology. She proceeded to the University of Florida to
pursue a degree in biomedical science, with a concentration in immunology. There she
did her dissertation research in the laboratory of Dr. Wayne McCormack. On August 18,
2001, she married Mr. Justin Casp in Rockville, Maryland. On May 23, 2003, she
celebrated the birth of her first daughter, Ashlyn Nicole Casp. She finished her degree in
December 2003, and is currently residing in Gainesville, Florida.