ライフサイクルモデル図の見方 - jiho.co.jp · 第1部...
TRANSCRIPT


25
第1部 CSVの基礎と関連ガイドライン解説
2
厚労省「コンピュータ化システム適正管理ガイドライン」の解説
2 ライフサイクルモデル図の見方
新ガイドラインの「ライフサイクルモデル」を図2.7に示した。これは1枚の図にコンピュータ化システムの開発,検証,運用管理および廃棄までのすべての流れを図式化したものであり,ガイドラインの主な項目(章)に対応した取り組みが記述されている。
6.運用管理業務
要求仕様(URS)
機能仕様(FS)
設計仕様(DS)
プログラムの作成プログラムテスト
据付時適格性評価(IQ)
運転時適格性評価(OQ)
性能適格性評価(PQ)
検証
検証
システムテスト
3.コンピュータ化システム管理規定
4.開発計画書
6.運用管理基準書
保守点検セキュリティ管理バックアップ及びリストア変更の管理逸脱(システムトラブル)の管理教育訓練
8.システムの廃棄
4.開発業務 5.検証業務
9.文書及び記録の管理
5.検証業務
検証
5.バリデーション計画書
検証
システムアセスメント
検証
6.3 標準操作手順書
()
7.自己点検
手順等に関する文書
システム毎に作成する文書
受入試験
図2.7 コンピュータ化システムのライフサイクルモデル(厚生労働省「医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン」より)
図は大きく2つのブロックに分かれている。上部ブロックには「手順等に関する文書」とあり「3. コンピュータ化システム管理規定」と「6.運用管理基準書」の2つのタイトルが書かれている。ちなみに,タイトルの前にある数値の「3」や「6」は新ガイドラインの章番号である。 上部ブロックの中央の点線の下には「システム毎に作成する文書」とある。これ以降が新たにシステムを導入する場合のCSVの取り組みとなる。 また,下のブロックの「4.開発業務」における初めの作業は「要求仕様書」の作成となる。図ではその前に「システムアセスメント」と書かれているが,システムアセスメントの結果を踏まえて開発業務に取り組むという主旨である。図のレイアウトの関係で「システムアセスメント」の位置がこの位置になっているのかもしれないが,正しくは「バリデーション計画書」の前になければならない。 要求仕様書の次に機能仕様,設計仕様に関する文書の作成となっているが,これはカテゴリ5製品の場合を想定している。カテゴリ3には機能仕様書,設計仕様書はなく,カテゴリ4の場合は設計仕様書の代わりに「構成定義書」となる。
解説ポイント
26
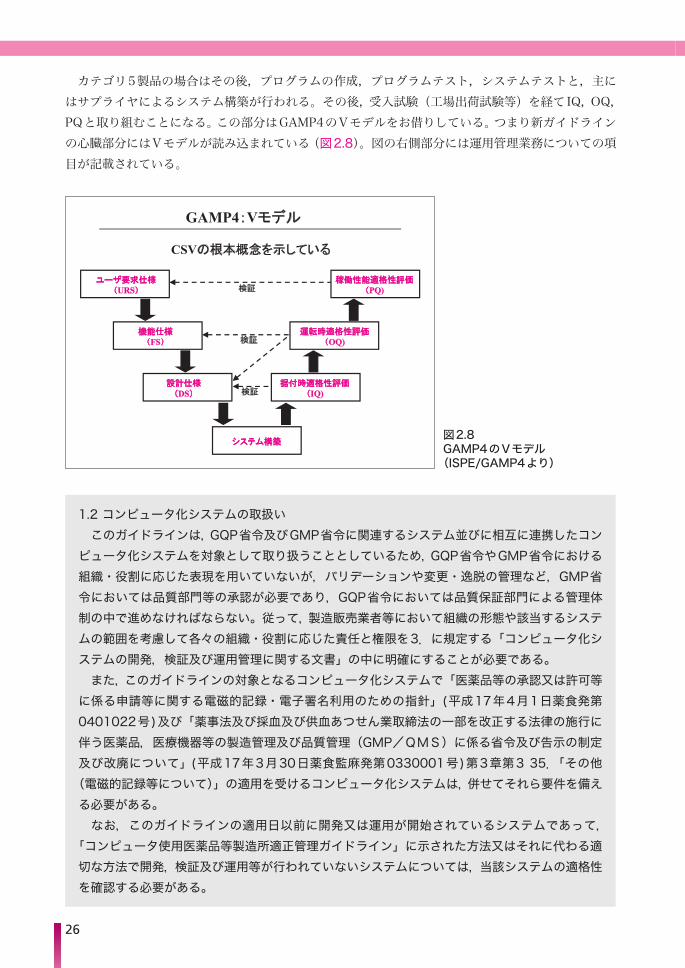
カテゴリ5製品の場合はその後,プログラムの作成,プログラムテスト,システムテストと,主にはサプライヤによるシステム構築が行われる。その後,受入試験(工場出荷試験等)を経て IQ,OQ,PQと取り組むことになる。この部分はGAMP4のVモデルをお借りしている。つまり新ガイドラインの心臓部分にはVモデルが読み込まれている(図2.8)。図の右側部分には運用管理業務についての項目が記載されている。
GAMP4:Vモデル
CSVの根本概念を示している
ユユーーザザ要要求求仕仕様様((URS))
シシスステテムム構構築築
機機能能仕仕様様((FS))
設設計計仕仕様様((DS))
据据付付時時適適格格性性評評価価((IQ)
運運転転時時適適格格性性評評価価((OQ)
稼稼働働性性能能適適格格性性評評価価((PQ)検証
検証
検証
図2.8 GAMP4のVモデル(ISPE/GAMP4より)
1.2 コンピュータ化システムの取扱い このガイドラインは,GQP省令及びGMP省令に関連するシステム並びに相互に連携したコンピュータ化システムを対象として取り扱うこととしているため,GQP省令やGMP省令における組織・役割に応じた表現を用いていないが,バリデーションや変更・逸脱の管理など,GMP省令においては品質部門等の承認が必要であり,GQP省令においては品質保証部門による管理体制の中で進めなければならない。従って,製造販売業者等において組織の形態や該当するシステムの範囲を考慮して各々の組織・役割に応じた責任と権限を3.に規定する「コンピュータ化システムの開発,検証及び運用管理に関する文書」の中に明確にすることが必要である。 また,このガイドラインの対象となるコンピュータ化システムで「医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針」(平成17年4月1日薬食発第0401022号)及び「薬事法及び採血及び供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品,医療機器等の製造管理及び品質管理(GMP/QMS)に係る省令及び告示の制定及び改廃について」 (平成17年3月30日薬食監麻発第0330001号)第3章第3 35.「その他
(電磁的記録等について)」の適用を受けるコンピュータ化システムは,併せてそれら要件を備える必要がある。 なお,このガイドラインの適用日以前に開発又は運用が開始されているシステムであって,
「コンピュータ使用医薬品等製造所適正管理ガイドライン」に示された方法又はそれに代わる適切な方法で開発,検証及び運用等が行われていないシステムについては,当該システムの適格性を確認する必要がある。
図2.8 GAMP4のVモデル(ISPE/GAMP4より)

27
第1部 CSVの基礎と関連ガイドライン解説
2
厚労省「コンピュータ化システム適正管理ガイドライン」の解説
3 CSV体制の構築
CSVの取り組みはGQPやGMP規制適合の一環である。コンピュータ化システムという特殊性はあるが,そのような理由から既存のGQPやGMPとまったく遊離した組織で運用することがないように求めている。 製造部門で使用する装置や機器,あるいは試験室の各種分析計などは元々GQPやGMPへの対応の中で取り組まれてきている。これらに変更や逸脱があればGQPやGMPの取り組みの中で変更管理や逸脱管理が行われることになるが,一方ではCSVにも変更管理や逸脱管理の要件がある。遊離した体制だと両方の取り組みが動き出すことになり,ダブってしまうこともある。実際の担当者は異なってもかまわないが,相互に連携した取り組みや体制が必要である。
4 電磁的記録・電子署名への対応
新ガイドラインではコンピュータ化システムを使用して電磁的記録を生成・保存したり,電子署名を運用する場合には「電磁的記録・電子署名」への対応を求めている。これはFDAのPart 11への対応であるが,国内でも平成17年4月に「厚労省ER/ES指針」が局長通知されている。このため,新ガイドラインでは電磁的記録・電子署名に関する要件は含まれていない。 新ガイドライン1.2項ではこれらの適用を受けるコンピュータ化システムは「厚労省ER/ES指針」に従って対応することを求めた。
5 適用日以前に開発または運用が開始されているシステム
“なお書き”部分は,ドラフトができ上がり当局のレビューが完了した段階ではなかった一文である。ドラフト版としてパブコメを出す直前の委員会において,当局メンバーより“PIC/Sガイドラインとの整合性を取ってほしい”という要求があった(その時点でCSV検討会の委員には当局がPIC/S加盟申請を行うことは知らされていなかった)。 PIC/Sガイダンスには簡単なAnnex11以外にも「GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED “GXP” ENVIRONMENTS」(PI 011-03)があるため,こちらもドラフト版との整合性を検討した。その結果,項目レベルでの齟齬はPI 011-03の16章「Retrospective validation」と21章「Electronic records and electronic signatures」だけであった(図2.9)。21章のER/ESは新ガイドラインには含まないということであったので,結局16章の「Retrospective validation」のみが問題であった。これはいわゆる「回顧的バリデーション」である。
解説ポイント
解説ポイント
解説ポイント

50
(1) ソフトウェアカテゴリ分類 (2) 製品品質に対するリスクアセスメント (3) 供給者アセスメント
19 システムアセスメントでCSV方針を決定する
システムアセスメントは旧ガイドラインにはなかったが,近代CSVでは不可欠な取り組みである。新ガイドラインにおいてもCSV戦略を決定する重要な位置づけであり,カテゴリ分類に関する「別紙2」においても,システムアセスメントの結果により取り組みが増減されることになっている。 新ガイドラインにおけるシステムアセスメントは3つの活動を総称している。ソフトウェアカテゴリ分類,製品品質に対するリスクアセスメント,そして供給者アセスメントである。 ソフトウェアカテゴリ分類についてはすでに繰り返し説明しているが,標準システムあるいは特注システムなどのソフトウェアカテゴリで,まずは基本的なCSVの活動内容が決定される。 また,製品品質に対するリスクアセスメント(一般的には「品質リスクアセスメント」とよんでいる)により,通常は実施しなければならない項目であっても,アセスメントの結果,リスクが低い場合には省略できる。また,その逆でアセスメントの結果,リスクが高い場合には実施しなければならないなど,品質リスクアセスメントの結果がCSV活動に影響を与えることになる。
20 リスクアセスメントの考え方
新ガイドラインでは製品品質に対する“リスクアセスメント”を求めているが,アセスメントはその名のとおり“評価”であり,本来であれば“リスクマネジメント”でなければならない。 評価するだけではリスクはなくならないし,低減もされない。リスク評価を行って,リスクが高いと評価された機能があれば,それに対して対策を取り(これを「リスクコントロール」とよんでいる),受容できるリスクレベルまでリスク低減策を行うのが“リスクマネジメント”である。ちなみに ICH Q9は「Quality Risk Management」である。しかし,ここでは “リスクアセスメント”を“リスクマネジメント”と読み替えることで話を進めよう。 開発や導入を予定しているシステムが,医薬品の製品品質に対してどの程度の影響を与える可能性があるかを評価するのが品質リスクアセスメントである。一般的に品質リスクアセスメントは,「初期リスクアセスメント」と「機能リスクアセスメント」の2段階で実施することが多い。GAMP5ではシステムのライフサイクルにおいてリスクアセスメントを7回としているが,リスクアセスメントばかりやっているわけにもいかない。 「初期リスクアセスメント」は導入するシステムが決定し,当該システムのCSV戦略(CSV活動のレベル)を決定する段階で実施する。このため,機能仕様等の打合せを行う前であり,“ざっくりとした評価”となる。 図2.16に簡単な初期リスクアセスメントの例を示す。例では品質リスクだけではなく,ビジネスの観点からのリスク(ビジネスリスク)アセスメントも行っている。このため初期リスクアセスメントでは,GMPの観点から見たリスクと,ビジネス面から見たリスクの合計としている。下段ではこれ
解説ポイント
解説ポイント

51
第1部 CSVの基礎と関連ガイドライン解説
2
厚労省「コンピュータ化システム適正管理ガイドライン」の解説
らの合計値が9以上であればリスク深刻度は「高」となり,CSV実施レベルは“厳密な取り組みとする”となっている。初期リスクアセスメントはこの程度の評価でよいと考える。
観点 深刻度 レベル 評価基準
GMP
高 6 製品の品質,有効性,安全性,GxP規制に直接の影響が生じる場合
中 3 製品の品質,有効性,安全性,GxP規制に間接的な影響が生じる可能性がある
場合
低 1 製品の品質,有効性,安全性,GxP規制に影響が生じることがない場合
ビジネス
高 6 通常業務に著しい支障が生じるか,または対外的に重大な問題が生じる場合
中 3 通常業務に支障が生じるか,または対外的に問題が生じる場合
低 1 通常業務に支障が生じることがなく,また,対外的にも問題が生じることがない場合
[初期リスクアセスメント結果(リスク深刻度)] =[GMPの観点から見たリスク]+[ビジネス面から見たリスク]
16
リスク深刻度
レベル 評価基準 CSV実施レベル
高 9以上GxP,ビジネスの観点のいずれかが高い深刻度で,
もう一方が中程度以上の深刻度の場合CSVの実施は厳密な取り組みとする。
中6以上9未満
GxP,ビジネスの観点のいずれかが高い深刻度で,
もう一方が低い深刻度の場合,あるいはいずれも中程度の深刻度の場合
CSVの実施はカテゴリ等を優先した中
程度の取り組みとする。
低 6未満GxP,ビジネスの観点のいずれかが低い深刻度で,
もう一方が中程度以下の深刻度の場合の場合CSVの実施は簡易な取り組みとする。
一方,「機能リスクアセスメント」は機能仕様書がまとまった段階で実施する。機能仕様の打合せの結果,開発が決定された機能の中で製品品質に不都合を起こすリスクはどの機能か(リスクシナリオ),そのリスクが発生する頻度はどの程度か(発生頻度),また,そのリスクはどの程度,品質に影響を与えるか(影響度)を評価する。リスク要因を洗い出し,洗い出されたリスクシナリオの「発生頻度×影響度」がリスクの大きさになる。図2.17に機能リスクアセスメントの例を示す。
入荷登録 入荷登録 異なる原料を受け入れる3 1 3
現物ラベル発行 現物ラベル ラベルの貼り間違い 3 2 1
ロット採番 ロット採番 ロット番号のダブり 3 1 1
受け入れ試験依頼 依頼忘れ 1 1 2
返品登録 返品 返品・廃棄品のラインへの混入 3 1 2
試験計画、指図作成 試験指図 試験項目の混同、抜け 3 1 1
サンプリング指図作成 サンプリング サンプルを採った梱包の劣化 2 1 1
サンプリング実績登録 異なるロットあるいは品目にデータを登録する 3 1 1
サンプリングラベル印刷 サンプリングラベル 異なるサンプルにラベルを貼る 3 2 1
試験成績登録 試験成績 試験結果の改ざん、不適データ削除 3 1 1
日程計画登録 指図作成 ロット逆転(日時とロットが逆進行) 1 1 2
製造指図の作成 製造方法、処方の間違い 3 1 2
製造指図の修正 不正修正が検知できない 3 1 1
製造指図書の印刷 製造指図書印刷 指図データと印刷結果の不一致 3 1 1
製造指図の承認(確定) 指図承認(確定) 承認ミス、 3 1 1
承認済み指図の改ざん 1 1 1
製造指図の送信 指図データの送信ミス 3 1 3
在庫の引当て(出庫指示) 出庫指示 使用期限の切れた品目を引当て 3 1 2
先入先出し違反 1 1 1
出庫(受払い)指示書作成 出庫指示書印刷 異なる原料を出庫 3 1 3
不適品の出庫 3 1 3
試験2品の出庫 2 1 3
出庫実績登録 出庫確認 異なるラベルを貼付する(倉庫出庫時にラベルを貼るケース) 3 1 1
現場在庫実績更新 実態とデータの誤差(数量) 1 2 2
実態とデータの誤差(品質情報) 3 1 1
秤量指示書作成 秤量指示 秤取換算(指示)を誤る 3 1 1
秤取量を間違える 3 1 1
秤量原料の確定 異なる原料を秤量する 3 1 1
秤取ラベルの貼り間違い 3 1 1
影響度 頻度 検知度
出庫
秤量
入荷
品管
指図
起こって欲しくない事象機能ブロック機能業務ブロック
図2.16 簡単な初期リスクアセスメントの例
図2.17 機能リスクアセスメントの例(FMEA)

86
3.1 はじめに
CSVの国際的なガイドとなっていたGAMP4が2008年2月に改定され,「GAMP5 A Risk-Based Approach to Compliant GxP Computerized Systems」 が発行された。 GAMP5はFDA(米国食品医薬品局)が2002年8月に宣言した21世紀のcGMPの取り組みである「リスクベースアプローチ」を受け,さらには国際的な標準化・規格団体である「ASTMインターナショナル」が発行したE2500-07「医薬品及びバイオ医薬品を製造するシステム及び機器の利用,設計,ベリフィケーションための標準ガイド」の強い影響を受ける形で,「バリデーションからベリフィケーション」という柔軟で合理的な提案と,SME(Subject Matter Expert:特定分野の専門家)やGEP(Good Engineering Practice:良好なエンジニアリングの実施)など数多くの新たな考え方が盛り込まれている。 GAMP5は350ページを超える膨大な内容となっており,本稿では要点や付属資料の全般的な概要紹介となる。詳細内容についてはGAMP5本誌でご確認願いたい。
注)GAMP5は ISPEに著作権があり,無断での複写・転写が禁じられている。本書についてはあらかじめ ISPE日本本部に「外部発表申請書」を提出し承認済みとなっているが,利用についてはご注意願いたい。
3.2 GAMP4からGAMP5への移行
ASTM E2500の影響
2002年8月にFDAは,21世紀の新cGMPを“リスクベースアプローチに移行する”と宣言し,その後,医薬品製造の近代化を提案した。その代表がPAT(Process Analytical Technology:工程分析技術)の提案である。医薬品の品質は,バリデーションと最終のラボでの出荷判定試験によって担保されている。しかし,PATは医薬品製造プロセスの中にオンライン分析計を設置し,リアルタイムでの
GAMP5の概要
3
1

87
第1部 CSVの基礎と関連ガイドライン解説
3
GGG
GG
のGG3.1 はじめに
CSVの国際的なガイドとなっていたGAMP4が2008年2月に改定され,「GAMP5 A Risk-Based Approach to Compliant GxP Computerized Systems」 が発行された。 GAMP5はFDA(米国食品医薬品局)が2002年8月に宣言した21世紀のcGMPの取り組みである「リスクベースアプローチ」を受け,さらには国際的な標準化・規格団体である「ASTMインターナショナル」が発行したE2500-07「医薬品及びバイオ医薬品を製造するシステム及び機器の利用,設計,ベリフィケーションための標準ガイド」の強い影響を受ける形で,「バリデーションからベリフィケーション」という柔軟で合理的な提案と,SME(Subject Matter Expert:特定分野の専門家)やGEP(Good Engineering Practice:良好なエンジニアリングの実施)など数多くの新たな考え方が盛り込まれている。 GAMP5は350ページを超える膨大な内容となっており,本稿では要点や付属資料の全般的な概要紹介となる。詳細内容についてはGAMP5本誌でご確認願いたい。
注)GAMP5は ISPEに著作権があり,無断での複写・転写が禁じられている。本書についてはあらかじめ ISPE日本本部に「外部発表申請書」を提出し承認済みとなっているが,利用についてはご注意願いたい。
3.2 GAMP4からGAMP5への移行
ASTM E2500の影響
2002年8月にFDAは,21世紀の新cGMPを“リスクベースアプローチに移行する”と宣言し,その後,医薬品製造の近代化を提案した。その代表がPAT(Process Analytical Technology:工程分析技術)の提案である。医薬品の品質は,バリデーションと最終のラボでの出荷判定試験によって担保されている。しかし,PATは医薬品製造プロセスの中にオンライン分析計を設置し,リアルタイムでの
GAMP5の概要
3
1
フィードバック制御を行って,製造工程の中で品質を担保していくという考え方である。 すでに石油や化学産業界では1970年代から温度,圧力,流量等の各種センサーやオンライン分析計をプロセスに設置し,これらのデータによりフィードバック制御を行い,製造プロセスの中で品質を確立し,そのまま製品として出荷している。例えば石油コンビナートにおいては,石油精製工場から隣接する他社の石油化学工場へ,エチレンプラントの原料となるナフサがパイプラインで直結されている。その間にあるのは取引用の流量計のみであり,ラボでの試験結果により出荷するようなことは行っていない。 医薬品製造プロセスにPATを導入し,リアルタイムで品質をコントロールするアプローチは考え方としてはすばらしいが,問題はどのように実装するかである。FDAはPATの実装を進めるために,医薬品業界とは無縁のASTM(American Society for Testing and Materials:米国試験材料協会 ) に検討依頼を行った。ASTMは,1898年に米国内の工業標準化と研究業務を目的として設立された世界最大の民間・非営利の国際標準化・規格設定機関であり,今日では国際的な団体となり,多くの工業規格である「ASTM規格」を設定・発行している。 2001年,ASTM規格が国際化したことを反映し「ASTMインターナショナル」と改称した。現在では120カ国以上から総計3万2,000名の製造業者,使用者,消費者,政府,学会代表者等が会員となり,製造,調達,規定等に関する活動について検討を行っている。ASTMが発行する規格は,米国内はもちろんのこと多くの国で利用され,日本工業規格(JIS)も,石油関連規格の多くが「ASTM規格」に準拠して定められている(図3.1)。
ASTMインターナショナル
規制当局
専門家団体
基準設定組織
ASTMインターナショナル
■2001年に「ASTMインターナショナルと改 」,120ヵ国以上から3万2,000名の製造業者,使用者,消費者,政府,学会代表者等が会員となり,製造,調達,規定等に関する活動について検討
■同協会の発行する規格は,米国の国内だけでなく多くの国で利用日本工業規格(JIS)も,石油関連規格の多くが「ASTM 規格」に準拠して定められている
■FDAは医薬品製造近代化の一環として「Pharmaceutical cGMPs for the 21st century - a risk-based approach (21世紀の医薬品cGMP-リスクベースアプローチ)の一部としてPAT(Process Analytical Technology:工程分析技術)を提唱。 FDAはPAT推進のために「ASTM」に協力要請 ⇒E55委委員員会会設設立立
■ ASTM は1898年,米国内の工業標準化と研究業務を目的として設立された世界最大の民間・非営利の国際標準化・規格設定機関。工業規格のASTM規格を設定・発行
■■E2500-07 医医薬薬品品およびびババイイオオ医医薬薬品品をを製製造造すするるシシスステテムム・機機器器のの利利用用,,設設計計,,ベベリリフフィィケケーーシショョンンたためめのの標標準準ガガイイドド
■E2537-08 医薬品およびバイオ医薬品の製造に連続的品質ベリフィケーション適用のための標準ガイド
E55委委員員会会
E55委委員員会会のの提提言言
■医薬品産業を直接支援し,コスト面で効果的な標準用語,ガイド,実践,試験法および規格に焦点を当て,科学およびリスクに基づくアプローチに基づいて品質の良い医薬品の開発及び製造をできるようにすることを目的とする
称
FDAの依頼を受け,ASTMでは早々にPATの実装を進めるために「E55委員会」を立ち上げた。その後,ASTMは2007年7月にE2500-07「医薬品およびバイオ医薬品を製造するシステム・機器の利用,設計,ベリフィケーションのための標準ガイド」を発行した(E2500-07の07は発行された2007年の意味を表す。以下07を省略し,「E2500」と記載する)。これは,ASTMが従来から取り組んできた石油や化学分野における取り組みから見た,医薬品産業への提案である。
図3.1 「ASTMインターナショナル」の概要

172
覧表を別紙で管理する方法もあります。
GMPですでにバリデーション体制(製造工程や設備のバリデーション)を持っている場合,CSVも既存のバリデーション体制で行うべきでしょうか?
Q2.14
A2.14 GMPですでに運用しているバリデーション体制でCSV対応が可能であれば,それでかまいません。しかし,一般的にはCSVの実施に必要なスキルは,GMPでの取り組みとは異なる部分もありますので,同じ組織や体制でCSVを実施できるとは限りません。 まずCSV管理規定の組織体制を検討する段階で,既存のバリデーション体制でCSVの運用が可能かを検討する必要があります。 また,厚労省ガイドラインでもCSV体制が既存のGMP体制とまったく遊離して活動することは好ましくないと指摘しているように,双方が連携しながら取り組むことが必要です。特に製造設備や機器に組み込まれているコンピュータシステムの場合は,どちらが主体で対応するかを取り決めておくことも必要です。
厚労省ガイドラインでは,「受入試験」は開発責任者(ユーザ側)が供給者に受入試験を実施させると書かれていますが,「受入試験」のテスト計画書の作成やテストの実施はどちらが主体で行うのでしょうか?
「受入試験」なので受け入れるユーザ企業がテスト計画書を作成しテストを主体的に実施するという意見もありますが,当社ではいつもサプライヤにお願いしてFATを行っています。これでは「受入試験」にならないのでしょうか?
Q2.15
A2.15 システム開発において,サプライヤではシステムテストが最終テストであり,これが完了するといつでも出荷できる状態になっています。この段階で「受入試験」が行われ,ユーザ企業としてそのシステムを自社の工場に受け入れても問題がないかどうかを確認するテストになります。このテストは通常はサプライヤの工場で実施することから,工場出荷試験(FAT)と称しています。 サプライヤはユーザからの要求仕様書をベースに,その後の機能仕様書あるいは設計仕様書に従ってシステム開発を行ってきています。しかし,ユーザ企業の業務プロセスが理解できているわけではありません。つまり,システムが要求仕様どおりに動くことはシステムテストで確認していますが,それが本来のユーザ企業の業務プロセスを満足しているかまでは確認できません。

173
第2部 CSV Q&A
2
厚労省「コンピュータ化システム適正管理ガイドライン」
一方,ユーザ側はシステムを受け入れるにあたって,自社の工場でシステムを稼働させることで目的とした業務が正しく行われるかの確認が重要です。すなわち,受入試験は“ユーザがシステムに求めた機能,つまり業務プロセスが正しく稼働するか,受け入れてもよい品質や完成度になっているかを確認するテスト”になります。そう考えると,ユーザがテスト計画書やテストスクリプトを作成し,主体となってテストを行うことが基本と考えるべきです。 厚労省ガイドラインでは「5.6 適格性評価の一部省略と引用」において,「(2) 工場出荷試験又は現地受入試験を行った場合等,その確認の方法及び記録が検証責任者によって適切と認められる場合には,適格性評価にあたって,その結果を引用しても差し支えないものとする」となっています。サプライヤの工場でのテストであり場所は異なりますが,FATをユーザ企業が適切に行ったからこそ,ユーザ企業で実施する適格性評価の一部を省略することを当局が認めたのです。 この取り組みはGAMP5のポリシーである“ダブリ作業をなくす”という考えを,厚労省ガイドラインにも盛り込んでいるところです。 ただし,近年ではVモデルのような「ウォーターフォールモデル」のシステム開発手法ではなく,「プロトタイピングモデル」で初めにシステムの試作品をサプライヤが作成し,これを基に打合せしながらシステム開発を行うようなケースでは,受入試験をユーザ主体で実施することは難しい場合もありますので,サプライヤと協議しながら実施することになると思います。(詳細は,p58参照)
製造設備や機器のバリデーションは導入時に実施していますが,CSVは実施していません。CSV対応で指摘されることになるでしょうか?Q2.16
A2.16 設備や機器がメーカ標準品であり,すでに多くの実績がある場合は設備や機器のバリデーションが正しく行われていれば問題ありません。 厚労省ガイドラインの「別紙2」では,メーカ標準品はカテゴリ3に相当します。「構成設定していないソフトウェア」と称しており,「商業ベースで販売されている既製のパッケージソフトウェアで,それ自体は業務プロセスに合わせて構成設定していないもの」となっています。また,ここは2階建てになっており「製造設備,分析機器,製造支援設備等に搭載されるシステム」と「単独のコンピュータシステム」に分かれています。 質問の場合は,「製造設備,分析機器,製造支援設備等に搭載されるシステム」になりますので,「備考3」のコメントを見ますと「3.設備に合わせて仕様の設定及び機能の検証を行うことで差し支えない。単純なシステムに関しては校正で代用することも可」とあります。 つまり,製造設備や機器であり,それが標準品であれば製造設備や機器のバリデーション時に内蔵されているシステムを稼働させてテストを行うので,そのときにシステムも含めてテストされることになるという考えに基づいています。したがって,要求仕様書やバリデーション計画書等は設備や機器として作成すればよく,IQやPQも設備や機器として実施することでよいことになっています。

196
予測的バリデーションと回顧的バリデーションの違いは何でしょうか?Q5.1
A5.1 「予測的バリデーション」は,コンピュータ化システムを新しく導入する際に実施するバリデーションです。通常のCSVはこの予測的バリデーションの取り組みを指しています。「予測的」という表現はまだ稼働していないシステムであるということから来ています。 一方,「回顧的バリデーション」はすでに稼働しているコンピュータ化システムに対して,稼働後に実施するバリデーションです。何らかの理由で「予測的バリデーション」が実施されていない,あるいは不十分な場合に,すでに運用中であるシステムを新規に導入したように見立てて実施することになります。 PIC/Sのガイドライン等では「Retrospective validation」と定義されていますが「Legacy system(既存システム)のバリデーション」という表現を使う場合もあります。
既存の古いシステムを最新の規制に適合させるためにCSVを行う場合の適切な取り組み方とは?Q5.2
A5.2 PIC/S GMP ANNEX11や厚労省ガイドラインでも,すでに稼働中のコンピュータ化システムでCSVが未実施の場合はバリデーションが求められています。この場合の取り組みについて,PIC/Sガイドラインの16節「Retrospective validation」(回顧的バリデーション)では以下のように記載されています。
既存システムのCSV (回顧的CSV)
5

197
第2部 CSV Q&A
5
CSV CSV
予測的バリデーションと回顧的バリデーションの違いは何でしょうか?Q5.1
A5.1 「予測的バリデーション」は,コンピュータ化システムを新しく導入する際に実施するバリデーションです。通常のCSVはこの予測的バリデーションの取り組みを指しています。「予測的」という表現はまだ稼働していないシステムであるということから来ています。 一方,「回顧的バリデーション」はすでに稼働しているコンピュータ化システムに対して,稼働後に実施するバリデーションです。何らかの理由で「予測的バリデーション」が実施されていない,あるいは不十分な場合に,すでに運用中であるシステムを新規に導入したように見立てて実施することになります。 PIC/Sのガイドライン等では「Retrospective validation」と定義されていますが「Legacy system(既存システム)のバリデーション」という表現を使う場合もあります。
既存の古いシステムを最新の規制に適合させるためにCSVを行う場合の適切な取り組み方とは?Q5.2
A5.2 PIC/S GMP ANNEX11や厚労省ガイドラインでも,すでに稼働中のコンピュータ化システムでCSVが未実施の場合はバリデーションが求められています。この場合の取り組みについて,PIC/Sガイドラインの16節「Retrospective validation」(回顧的バリデーション)では以下のように記載されています。
既存システムのCSV (回顧的CSV)
5 16.1 多くの既存システム (legacy systems)は,満足で信頼性高く動作しているかもしれないが,しかしながら,それらをコンピュータ化システムバリデーション要件から除外するものではない。取るべきアプローチは,バリデーションと再適格性確認試験(requalification)の確証を提供することであり,そのシステムの回顧的バリデーションの文書化とそれを証明するためのデータと情報を提供することである。 なぜならGxPsはもう何年もの間,コンピュータ化システムのバリデーションを要求してきた。それゆえ,予測的バリデーションの確証が不足していることは,多くの規制当局によってGxPsからの深刻な逸脱と見られることが多くなっていることに注意すべきである。
回顧的バリデーションの取り組みについては,当局からガイドライン等はいっさい示されていません(当局は予測的バリデーションを要求していますので当然です)。 回顧的バリデーションは,本来そのシステムを新規に導入するときに行う「予測的バリデーション」で取り組むべき作業を,稼働後に実施するということになります。 この場合,そのシステムの文書等が何もないということであればすべてのCSV活動を回顧的に実施するということになりますが,一部の文書やエビデンスがある場合には,現在あるCSV文書と予測的バリデーションで実施すべき作業や文書とのGAP分析を行います。 つまり,何があって何が不足しているかを比較します。その結果,不足している文書や活動を後から実施するというのが基本的な取り組みになります。 回顧的バリデーションでは,ソースコードから機能仕様書等を起こすようなリバースエンジニアリングを行うこともあります。某大手製薬企業ではFDAに上市する品目に関わる生産管理システムを,1年間で約1億円かけて回顧的バリデーションを実施したケースもあります。1億円が高いかは議論のあるところですが,査察時に万が一指摘されて上市が遅れる機会損失とのバランスを考えての,トップ判断で実施したと聞いています。 回顧的バリデーションでは,一時的にシステムを停止してテストを実施するケースもある等困難な作業になることが予想されますので,予測的バリデーションを正しく実施することが基本です。
既存システムについてやるべきことの注意点として,IQ,OQが終了している場合には,運用段階以降の活動だけでよいのでしょうか? また,終了しているIQ,OQの文書を活用する注意点についても教えてください。
Q5.3
A5.3 CSVの活動は IQ,OQ等の適格性評価の前に開発段階の取り組みがあります。システムのカテゴリによっても異なりますが,要求仕様書は必要ですし,リスクアセスメントも求められていま