exploration du rÔle physiologique du … · 3.8 e et du choc oxydatif sur l’expression de yadb ....
TRANSCRIPT

OLIVIER FISETTE
EXPLORATION DU ROLE PHYSIOLOGIQUE
DU GENE yadB CHEZ Escherichia coli
Memoire presentea la Faculte des etudes superieures de l’Universite Lavaldans le cadre du programme de maıtrise en Biochimie
pour l’obtention du grade de Maıtre es sciences (M. Sc.)
FACULTE DES SCIENCES ET DE GENIEUNIVERSITE LAVAL
QUEBEC
AVRIL 2005
c© Olivier Fisette, 2005

ii
Resume
L’enzyme glutamyl-queuosine-ARNt synthetase, presente chez plusieurs bacteries
et codee par le gene yadB chez Escherichia coli, catalyse la formation de glutamyl-
queuosine-ARNtAsp. Le role de cet aminoacyl-ARNt est inconnu, mais la cotranscription
de yadB et de dksA suggere une implication dans la reponse generale au stress.
yadB a ete remplace dans une souche de Escherichia coli ∆lac par lacZ, sans mo-
dification des eventuels elements transcriptionnels de yadB. L’activite β-galactosidase
a ete utilisee comme rapporteur pour determiner le niveau d’expression de yadB dans
diverses conditions de croissance et de stress. Les differences phenotypiques entre les
souches sauvage et ∆yadB ont ete investiguees.
Nous avons decouvert que yadB est un gene non-essentiel chez Escherichia coli.
Sa faible expression suggere un role dans le metabolisme basal de la cellule. Cette
expression est sensiblement plus elevee pendant la phase stationnaire de croissance.
Aucune difference phenotypique n’a ete decouverte entre les souches sauvage et ∆yadB.

iii
Avant-propos
Beaucoup de gens m’ont aide pendant les deux annees qu’ont dure mes etudes
de Maıtrise en biochimie. J’aimerais utiliser ces quelques lignes pour leur offrir mes
remerciements.
Je suis avant tout grandement reconnaissant a mes parents pour leur support
indefectible tout au long de mes etudes, pour leur interet et leurs encouragements.
Je souhaite remercier mon directeur de recherche, le Professeur Jacques Lapointe,
de m’avoir accueilli dans son laboratoire, de m’avoir soutenu et encourage tout au
long de mes travaux et d’avoir su constamment stimuler mon interet (et celui des
autres etudiantes et etudiants du laboratoire) pour le domaine passionnant qu’est la
biosynthese des proteines.
Les membres de mon comite aviseur, les Professeurs Barbara Papadopoulou et
Stephane Gagne, meritent aussi mes remerciements pour leurs conseils et suggestions
qui ont contribue a definir les orientations de mon projet de recherche.
J’en profite egalement pour faire un clin d’œil aux etudiantes, etudiants et
professionnels avec qui j’ai eu la chance de travailler au Laboratoire de recherche sur la
biosynthese des proteines : Pierre-Marie Akochy, Pierre Barrau, Vanessa Baudin, David
Beaulieu, Andreane Daviau, Habib Derbali, Daniel Dubois, Louis-Patrick Gagnon,
Charlotte Habegger-Polomat et Philippe Levesque. Vos conseils et votre amitie m’ont
aide plus que je ne saurais le dire.
Enfin, je tiens a souligner le support financier qui m’a ete accorde par le Conseil
de recherche en sciences naturelles et en genie du Canada a travers l’octroi d’une bourse
d’etudes superieures.

iv
A mes parents

v
Table des matieres
Resume ii
Avant-propos iii
Table des matieres v
Liste des tableaux viii
Table des figures ix
Liste des abreviations x
1 Introduction 1
1.1 La traduction de l’information genetique . . . . . . . . . . . . . . . . . 1
1.2 Les aminoacyl-ARNt synthetases . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.2 Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2.3 Modularite . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.2.4 Specificite . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.3 Evolution des aminoacyl-ARNt synthetases de la famille GlxRS . . . . 10
1.4 La glutamyl-queuosine-ARNtAsp synthetase . . . . . . . . . . . . . . . . 12
1.4.1 Le gene yadB . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
1.4.2 Activite de YadB in vitro . . . . . . . . . . . . . . . . . . . . . 14
1.4.3 Similarites structurales entre la GluQRS et la GluRS . . . . . . 15
1.4.4 Relation avec le gene dksA . . . . . . . . . . . . . . . . . . . . . 16
1.4.5 Questions et objectifs . . . . . . . . . . . . . . . . . . . . . . . . 18
2 Materiel et methodes 19
2.1 Tampons, solutions et produits chimiques . . . . . . . . . . . . . . . . . 19
2.2 Culture cellulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.2.1 Milieux de culture et conditions de croissance . . . . . . . . . . 19
2.2.2 Agents selectifs . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.2.3 Souches . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21

Table des matieres vi
2.3 Manipulations de l’ADN . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.3.1 Plasmides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.3.2 Oligonucleotides . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.3.3 Purification d’ADN par extraction et precipitation . . . . . . . . 26
2.3.4 Preparation d’ADN plasmidique . . . . . . . . . . . . . . . . . . 27
2.3.5 Preparation d’ADN genomique . . . . . . . . . . . . . . . . . . 28
2.3.6 Reaction en chaıne par la polymerase . . . . . . . . . . . . . . . 29
2.3.7 Digestion d’ADN par des endonucleases de restriction . . . . . . 31
2.3.8 Ligature de fragments d’ADN . . . . . . . . . . . . . . . . . . . 31
2.3.9 Dephosphorylation de fragments d’ADN . . . . . . . . . . . . . 33
2.3.10 Electrophorese sur gel d’agarose . . . . . . . . . . . . . . . . . . 33
2.3.11 Purification d’ADN a partir de gels d’agarose . . . . . . . . . . 34
2.3.12 Transformation de cellules par de l’ADN plasmidique . . . . . . 35
2.4 Construction du vecteur pTOF-YB . . . . . . . . . . . . . . . . . . . . 36
2.4.1 Amplification des regions flanquant yadB . . . . . . . . . . . . . 38
2.4.2 Clonage des regions flanquantes dans pTOF24 . . . . . . . . . . 38
2.4.3 Insertion de la cassette FLK2 . . . . . . . . . . . . . . . . . . . 39
2.5 Remplacement de yadB par la cassette FLK2 . . . . . . . . . . . . . . 41
2.5.1 Transformation de E. coli TP8503 . . . . . . . . . . . . . . . . . 41
2.5.2 Integration du plasmide au chromosome . . . . . . . . . . . . . 43
2.5.3 Excision et perte du plasmide . . . . . . . . . . . . . . . . . . . 43
2.5.4 Confirmation du remplacement par PCR . . . . . . . . . . . . . 43
2.6 Deletion du gene yadB . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
2.6.1 Transformation de POK1 par pCP20 . . . . . . . . . . . . . . . 45
2.6.2 Excision de la cassette FLK2 et perte de pCP20 . . . . . . . . . 46
2.6.3 Confirmation de la deletion par PCR . . . . . . . . . . . . . . . 46
2.7 Dosage enzymatique de l’activite β-galactosidase . . . . . . . . . . . . . 47
2.8 Conditions d’expression et effets de la deletion du gene yadB . . . . . . 48
2.8.1 Croissance en conditions optimales . . . . . . . . . . . . . . . . 49
2.8.2 Croissance en milieu minimal . . . . . . . . . . . . . . . . . . . 49
2.8.3 Croissance a des temperatures non optimales . . . . . . . . . . . 49
2.8.4 Choc thermique . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
2.8.5 Choc osmotique . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
2.8.6 Choc de pH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
2.8.7 Choc oxydatif . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
3 Resultats 52
3.1 Construction de la souche POK1 . . . . . . . . . . . . . . . . . . . . . 52
3.2 Construction de la souche POK2 . . . . . . . . . . . . . . . . . . . . . 52
3.3 Thermosensibilite de la souche POK2 . . . . . . . . . . . . . . . . . . . 54
3.4 Expression et effets de la deletion de yadB . . . . . . . . . . . . . . . . 54

Table des matieres vii
3.4.1 Croissance en conditions optimales . . . . . . . . . . . . . . . . 54
3.4.2 Croissance en milieu minimal . . . . . . . . . . . . . . . . . . . 55
3.4.3 Croissance a des temperatures non optimales . . . . . . . . . . . 55
3.4.4 Choc thermique . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
3.4.5 Choc osmotique . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
3.4.6 Choc de pH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
3.4.7 Choc oxydatif . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4 Discussion 62
4.1 Expression de yadB pendant le cycle cellulaire . . . . . . . . . . . . . . 62
4.2 Cotranscription de dksA et de yadB . . . . . . . . . . . . . . . . . . . . 63
4.3 Role du Glu-Q-ARNtAsp . . . . . . . . . . . . . . . . . . . . . . . . . . 64
4.4 Lien entre les concentrations de glutamate et le role de la GluQRS . . . 65
4.5 Phenotypes des cellules ∆yadB . . . . . . . . . . . . . . . . . . . . . . 65
4.6 Analyse de la technique de remplacement employee . . . . . . . . . . . 66
4.6.1 Avantages . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
4.6.2 Inconvenients . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
4.6.3 Techniques alternatives . . . . . . . . . . . . . . . . . . . . . . . 67
5 Conclusion 69
5.1 Resume . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.2 Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
Bibliographie 71

viii
Liste des tableaux
1.1 Classification des aminoacyl-ARNt synthetases . . . . . . . . . . . . . . 5
2.1 Tampons et solutions . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.2 Milieux de culture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.3 Agents selectifs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.4 Souches de E. coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.5 Plasmides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.6 Oligonucleotides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.7 Composition des melanges reactionnels pour PCR . . . . . . . . . . . . 30
2.8 Conditions pour les reactions PCR . . . . . . . . . . . . . . . . . . . . 31
2.9 Composition des melanges reactionnels pour la restriction de l’ADN . . 31
2.10 Composition des melanges reactionnels pour la ligature de l’ADN . . . 32
2.11 Conditions pour les reactions de ligature de l’ADN . . . . . . . . . . . 32
2.12 Composition des melanges reactionnels pour la dephosphorylation de
l’ADN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33

ix
Table des figures
1.1 Le « dogme » de la biologie moleculaire . . . . . . . . . . . . . . . . . 1
1.2 Reaction d’aminoacylation des ARNt par les aaRS . . . . . . . . . . . . 3
1.3 Synthese indirecte de Gln-ARNtGln par une GluRS ND et l’AdT . . . . 4
1.4 Exemples des structures des aminoacyl-ARNt synthetases de classes I et II 7
1.5 Reactions de correction de mesacylations . . . . . . . . . . . . . . . . . 9
1.6 Evolution des aaRS de la famille GlxRS . . . . . . . . . . . . . . . . . 11
1.7 Le gene yadB de E. coli et la GluQRS ; comparaison avec la GluRS . . 13
1.8 Structure de la base hypermodifiee queuosine . . . . . . . . . . . . . . . 15
2.1 Plasmides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.2 Construction du vecteur pTOF-YB . . . . . . . . . . . . . . . . . . . . 37
2.3 Selection de pTOF-YB par cartographie de restriction . . . . . . . . . . 40
2.4 Remplacement de yadB par la cassette FLK2 . . . . . . . . . . . . . . 42
2.5 Confirmation du remplacement de yadB par PCR . . . . . . . . . . . . 44
2.6 Deletion de yadB par excision de la cassette FLK2 . . . . . . . . . . . 45
2.7 Confirmation de la deletion de yadB par PCR . . . . . . . . . . . . . . 46
3.1 Confirmation de la contruction des souches POK1 et POK2 par PCR . 53
3.2 Expression de yadB dans des conditions de croissance optimales . . . . 55
3.3 Expression de yadB en milieu minimal . . . . . . . . . . . . . . . . . . 56
3.4 Expression de yadB a des temperatures non optimales . . . . . . . . . . 57
3.5 Effet du choc thermique sur l’expression de yadB . . . . . . . . . . . . 58
3.6 Effet du choc osmotique sur l’expression de yadB . . . . . . . . . . . . 59
3.7 Effet du choc de pH sur l’expression de yadB . . . . . . . . . . . . . . . 60
3.8 Effet du choc oxydatif sur l’expression de yadB . . . . . . . . . . . . . 61
x
Liste des abreviations
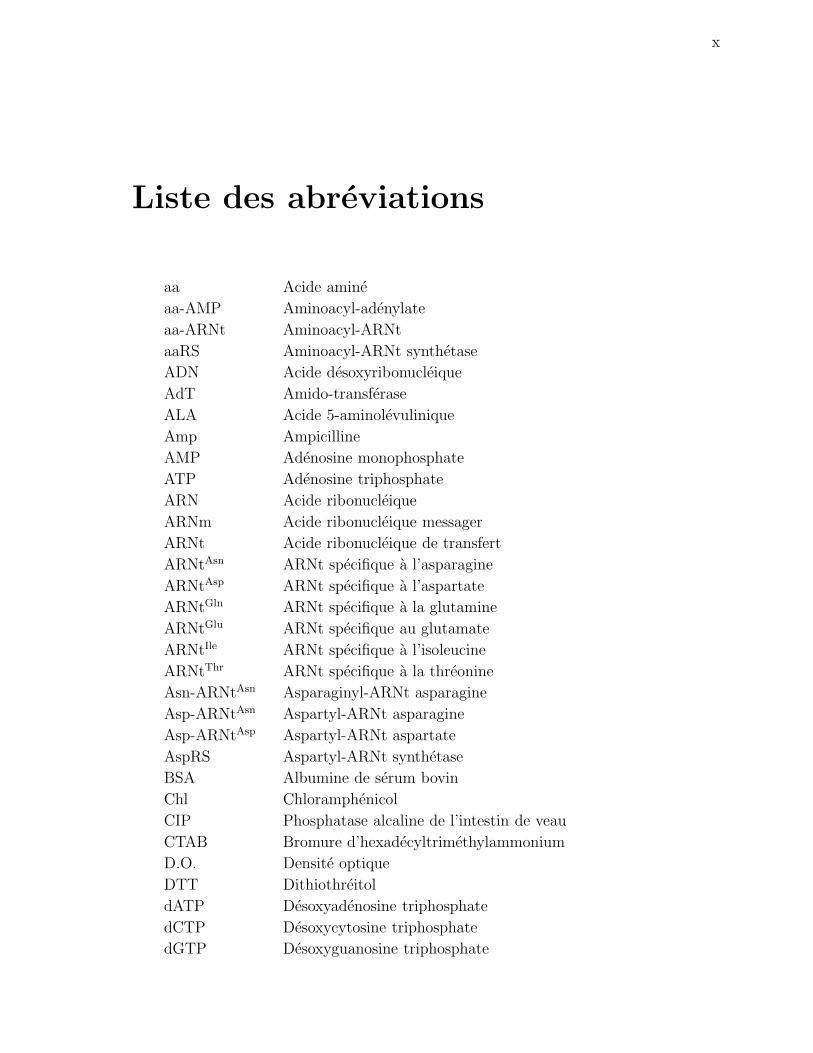
aa Acide amine
aa-AMP Aminoacyl-adenylate
aa-ARNt Aminoacyl-ARNt
aaRS Aminoacyl-ARNt synthetase
ADN Acide desoxyribonucleique
AdT Amido-transferase
ALA Acide 5-aminolevulinique
Amp Ampicilline
AMP Adenosine monophosphate
ATP Adenosine triphosphate
ARN Acide ribonucleique
ARNm Acide ribonucleique messager
ARNt Acide ribonucleique de transfert
ARNtAsn ARNt specifique a l’asparagine
ARNtAsp ARNt specifique a l’aspartate
ARNtGln ARNt specifique a la glutamine
ARNtGlu ARNt specifique au glutamate
ARNtIle ARNt specifique a l’isoleucine
ARNtThr ARNt specifique a la threonine
Asn-ARNtAsn Asparaginyl-ARNt asparagine
Asp-ARNtAsn Aspartyl-ARNt asparagine
Asp-ARNtAsp Aspartyl-ARNt aspartate
AspRS Aspartyl-ARNt synthetase
BSA Albumine de serum bovin
Chl Chloramphenicol
CIP Phosphatase alcaline de l’intestin de veau
CTAB Bromure d’hexadecyltrimethylammonium
D.O. Densite optique
DTT Dithiothreitol
dATP Desoxyadenosine triphosphate
dCTP Desoxycytosine triphosphate
dGTP Desoxyguanosine triphosphate

Liste des abreviations xi
DMSO Dimethylsulfoxide
dTTP Desoxythymidine triphosphate
E. coli Escherichia coli
E.C. Classification enzymatique
EDTA Acide ethylenediaminetetraacetique
FRT Cible de reconnaissance Flp
GDP Guanosine diphosphate
Gln-ARNtGln Glutaminyl-ARNt glutamine
GlnRS Glutaminyl-ARNt synthetase
Glu-ARNtGln Glutamyl-ARNt glutamine
Glu-ARNtGlu Glutamyl-ARNt glutamate
GluRS Glutamyl-ARNt synthetase
GluRS D Glutamyl-ARNt synthetase discriminatrice
GluRS ND Glutamyl-ARNt synthetase non discriminatrice
GTP Guanosine triphosphate
GluQ-ARNtAsp Glutamyl-queuosine-ARNtAsp
GluQRS Glutamyl-queuosine-ARNtAsp synthetase
GluRS Glutamyl-ARNt synthetase
GluTR Glutamyl-ARNt reductase
GSA Glutamate-1-semialdehyde
IleRS Isoleucyl-ARNt synthetase
Kan Kanamycine
LB Luria-Bertani
NEB New-England BioLabs
ONPG Ortho-nitro-phenyl-β-D-galactopyranoside
PEG Polyethylene glycol
PCR Reaction en chaıne par la polymerase
Pi Phosphate inorganique
Pipes Piperazine-N,N’-bis(acide 2-ethanesulfonique)
ppGpp Guanosine-3’5’-bis(diphosphate)
PPi Pyrophosphate inorganique
Q Queuosine
SDS Sulfate dodecylique de sodium
Suc Sucrose
TBE Tris borate EDTA
TE Tris EDTA
ThrRS Threonyl-ARNt synthetase
Tris 2-Amino-2-(hydroxymethyl)-1,3-propanediol
U.V. Ultraviolet

Liste des abreviations xii
Val-AMP Valyl-adenylate
Val-ARNtIle Valyl-ARNt isoleucine

Chapitre 1
Introduction
1.1 La traduction de l’information genetique
En 1958, Francis Crick enonca le « dogme » central de la biologie moleculaire [1],
qui resume les relations, recemment decouvertes a l’epoque, entre les proteines et les
acides desoxyribonucleiques (ADN) et ribonucleiques (ARN) :
L’ADN dirige sa propre replication et sa transcription en ARN qui, a son tour,
dirige sa traduction en proteines.
Cette hypothese, illustree a la figure 1.1, decrit bien les rapports entre les
catalyseurs biologiques (proteines), les vecteurs de l’information genetique (ADN) et
les effecteurs de cette information (ARN). Toutefois, il n’existe pas de correspondance
Fig. 1.1 – Le « dogme » de la biologie moleculaire

Chapitre 1. Introduction 2
structurale simple entre les bases azotees des acides nucleiques et les acides amines
(aa) des polypeptides [2]. C’est pourquoi Francis Crick suggera l’existence de molecules
adaptatrices faites d’ARN, auxquelles seraient lies enzymatiquement les acides amines
avant leur assemblage par les ribosomes. La lecture de l’information contenue dans
les codons des ARN messagers (ARNm) pourrait alors se faire par appariement de
bases. Cette « hypothese de l’adaptateur » fut confirmee par la decouverte des ARN
de transfert (ARNt) et du role de ces derniers dans la biosynthese des proteines [3].
La presence de ces adaptateurs implique l’existence d’une autre classe d’enzymes
dont la vocation est de determiner la correspondance entre les anti-codons des ARNt et
les acides amines (definissant ainsi le code genetique) par la liaison chimique specifique
de chaque adaptateur d’ARN a son acide amine correspondant. Ce role echoit aux
aminoacyl-ARNt synthetases.
1.2 Les aminoacyl-ARNt synthetases
1.2.1 Fonctions
Les aminoacyl-ARNt synthetases (aaRS), une famille d’enzymes ubiquitaires
(E.C. 6.1.1), catalysent l’esterification des acides amines aux ARNt. Chaque synthetase
est specifique a un acide amine et aux ARNt isoaccepteurs qui lui correspondent
(a deux exceptions pres qui seront presentees plus loin). Les aminoacyl-ARNt (aa-
ARNt) formes par cette reaction enzymatique servent de substrats aux ribosomes pour
l’assemblage des chaınes polypeptidiques [4].
Tel qu’indique a la figure 1.2, la reaction d’aminoacylation se deroule en deux
etapes. L’acide amine est d’abord active (figure 1.2 a) par reaction avec l’ATP, produi-
sant un aminoacyl-adenylate auquel le lien chimique reactif de l’ATP a ete transfere. Cet
aminoacyl-adenylate reste complexe a la synthetase. Notons que certaines synthetases
utilisent leur ARNt specifique comme cofacteur lors de cette etape. Par exemple, la
liaison de l’ARNt specifique au glutamate (ARNtGlu) a la glutamyl-ARNt synthetase
(GluRS), pendant l’etape d’activation de l’acide amine, modifie la conformation du site
de liaison de l’ATP, permettant la liaison de ce substrat dans une position qui favorise
la reaction avec le glutamate [5].
Lors de la seconde etape (figure 1.2 b), le deuxieme substrat de la synthetase,
soit son ARNt specifique, est complexe a l’enzyme, puis l’acide amine est lie a une

Chapitre 1. Introduction 3
Fig. 1.2 – Reaction d’aminoacylation des ARNt par les aaRS. a) Activation de l’acide
amine (formation d’un aminoacyl-adenylate) par reaction avec l’ATP. Chez certaines
synthetases, la presence de l’ARNt est necessaire pour completer cette etape de la
reaction. b) Liaison de l’acide amine a l’ARNt et production d’une molecule d’AMP.
Un ion magnesium est requis pour completer cette etape de la reaction. c) L’aminoacyl-
ARNt peut etre complexe au facteur d’initiation IF-2 ou au facteur d’elongation EF-Tu
pour servir de substrat au ribosome.
des fonctions hydroxyle de l’extremite 3’-terminale de cet ARNt. Cette seconde etape
requiert la presence d’un ion magnesium agissant comme cofacteur [6].
L’aminoacyl-ARNt ainsi forme peut alors etre complexe a un facteur d’initiation
(IF-2) ou d’elongation (EF-Tu) de la synthese proteique et, finalement, servir de substrat
au ribosome pour l’assemblage des polypeptides (figure 1.2 c) [7].
Si de nombreux organismes disposent d’une aminoacyl-ARNt synthetase pour
chacun des acides amines standards, ce n’est toutefois pas le cas de tous. Le genome de
la majorite des bacteries, par exemple, est depourvu de glutaminyl-ARNt synthetase
(GlnRS). Ces organismes possedent toutefois une GluRS dite non discriminatrice (ND)
(parce qu’elle peut charger le glutamate indifferemment sur un ARNtGlu ou un ARNtGln)
ainsi qu’une amidotransferase ARNt dependante (AdT). L’action combinee de ces deux
enzymes permet la synthese du glutaminyl-ARNtGln (Gln-ARNtGln) par une voie in-
directe, tel qu’illustre a la figure 1.3. La GluRS ND procede d’abord au chargement

Chapitre 1. Introduction 4
Fig. 1.3 – Synthese indirecte de Gln-ARNtGln par la GluRS ND et l’AdT. a) La GluRS
mesacyle l’ARNtGln avec le glutamate. b) L’AdT convertit le glutamate en glutamine,
produisant le Gln-ARNtGln.
de l’acide amine glutamate sur l’ARNtGln pour former le Glu-ARNtGln (figure 1.3 a).
Cette mesacylation est ensuite « corrigee » par l’action de l’AdT, qui transfere un ion
ammonium au glutamate pour le convertir en glutamine (figure 1.3 b). Cette voie de
synthese alternative resulte en une depense energetique superieure a celle de la voie de
synthese classique, puisque deux molecules d’ATP sont necessaires a la synthese d’un
aa-ARNt [3].
Cette voie de synthese indirecte est egalement employee par certains organismes
pour la synthese de l’Asn-ARNtAsn ; une AspRS non discriminatrice mesacyle l’ARNtAsn
pour former l’Asp-ARNtAsn, qui est ensuite converti en Asn-ARNtAsn par l’AdT [8].
En plus de leur participation a la synthese des substrats du ribosome, les
aminoacyl-ARNt synthetases jouent une variete de roles dans la biosynthese des
proteines et dans d’autres voies metaboliques, grace a leur activite d’aminoacylation
et aussi par d’autres mecanismes. La biosynthese de la porphyrine est un exemple de
l’utilisation d’un aa-ARNt dans un processus cellulaire distinct de la synthese proteique :
le Glu-ARNtGlu produit par la GluRS est reduit en glutamate-1-semialdehyde (GSA)
par la glutamyl-ARNt reductase (GluTR). Ce produit est ensuite converti en acide
5-aminolevulinique (ALA), lequel sert de precurseur a la porphyrine, elle-meme utilisee
dans le metabolisme de la chlorophylle [9]. Chez les eucaryotes, les aaRS participent a
la verification de l’integrite des ARNt en les aminoacylant apres leur maturation dans le
noyau. Seuls les ARNt aminoacyles (et donc completement matures) sont translocalises
dans le cytosol ou a lieu la synthese proteique [10]. Certaines aaRS participent aussi a
la regulation de leurs propres genes [11] ; la threonyl-ARNt synthetase (ThrRS), par
exemple, lie l’extremite 5’-terminale de son ARNm, dont la structure imite celle de
l’ARNtThr [12]. D’autres synthetases, encore, participent au controle du metabolisme
des acides amines [4].

Chapitre 1. Introduction 5
Tab. 1.1 – Classification des aminoacyl-ARNt synthetasesSous-classe : Ia Ib Ic IIa IIb IIc
ArgRSa GlnRSa TrpRS GlyRSc AsnRS AlaRS
CysRS GluRSa TyrRS HisRS AspRS GlyRSc
IleRS ProRS LysRS IIb PheRS
LeuRS ThrRS
MetRS SerRS
ValRS
LysRS Iab
a Requierent la presence de leur ARNt pour l’activation de leur acide amine.
b Il existe deux types de LysRS, une appartenant a la classe I, l’autre a la classe II.
c Il existe deux types de GlyRS non apparentes.
1.2.2 Structure
Il existe deux classes d’aminoacyl-ARNt synthetases (classes I et II). Il n’y a pas
d’homologie entre les aaRS appartenant a ces deux classes, qui auraient evolue a partir
de deux proteines ancestrales differentes. Cette observation surprenante suggere que
le mecanisme de traduction aurait peut-etre evolue par la fusion de deux processus
primitifs associes a des ensembles d’acides amines differents [13]. Notons par ailleurs
que plusieurs organismes (incluant la bacterie Escherichia coli) possedent deux genes
codant pour des lysyl-ARNt synthetases, l’une appartenant a la classe I, l’autre a la
classe II [14]. Il s’agit du seul exemple connu d’une synthetase qui soit representee dans
les deux classes. Le tableau 1.1 resume la repartition des synthetases en deux classes
et en six sous-classes [4].
Si toutes les synthetases catalysent de facon similaire la reaction d’aminoacylation
[6], les synthetases des deux classes varient toutefois enormement en ce qui concerne
leurs structures et leurs facons de lier les ARNt [15]. Les differences les plus evidentes
se situent au niveau du motif structural responsable de l’activite catalytique : alors
que ce motif est un pli de Rossman (un feuillet β parallele dont chaque cote fait
contact avec deux helices α) chez les synthetases de classe I, il s’agit plutot d’un
feuillet β antiparallele chez les synthetases de classe II. Chez les enzymes de classe
I, les interactions avec la tige acceptrice de l’ARNt se font par le sillon mineur, alors
que les enzymes de classe II approchent la tige acceptrice par le sillon majeur. Le site
d’aminoacylation de l’ARNt varie egalement selon le type d’enzyme : les synthetases
de classe I esterifient l’acide amine sur la fonction hydroxyle 2’ du ribose de l’extremite
3’-terminale de l’ARNt, alors que les synthetases de classes II lient l’acide amine a
l’hydroxyle 3’ de ce meme ribose.

Chapitre 1. Introduction 6
La figure 1.4 illustre ces differences entre les deux classes d’aminoacyl-ARNt
synthetases. La GluRS (figure 1.4 a) est en conformation alongee lorsqu’elle lie son
ARNt, et etablit des contacts avec le sillon mineur de la tige acceptrice. Le domaine
N-terminal (domaine catalytique) presente le pli de Rossman caracteristique des syn-
thetases de classe I. A l’oppose, l’aspartyl-ARNt synthetase (AspRS) (figure 1.4 b)
adopte une conformation courbee dans le complexe synthetase ARNt, et approche cet
ARNt par l’autre cote, interagissant avec le sillon majeur de la tige acceptrice. Enfin,
on remarque que le domaine catalytique est forme d’un feuillet β.
1.2.3 Modularite
Une importante caracteristique structurale des aminoacyl-ARNt synthetases est
leur modularite. Les aaRS ancestrales n’etaient probablement constituees que d’un seul
domaine, soit le cœur catalytique responsable de l’esterification des acides amines aux
ARNt. Au cours de l’evolution, des domaines supplementaires se sont greffes a ce cœur,
augmentant l’efficacite de ces enzymes. Ces modules sont hautement independants, tel
que demontre par de nombreuses etudes structurales [19].
Ces domaines additionnels ont des fonctions variees. La plus evidente est l’etablis-
sement d’interactions avec les determinants et antideterminants des ARNt, ameliorant
la specificite de la reconnaissance de ces derniers par la synthetase [20]. Une autre
fonction repandue est l’hydrolyse de produits de mesacylation ; les aminoacyl-ARNt
synthetises par le cœur catalytique peuvent etre deplaces vers ce domaine avant de
quitter la synthetase, permettant ainsi de verifier leur nature et, si necessaire, de
les hydrolyser avant qu’ils ne puissent etre reconnus par les facteurs d’initiation et
d’elongation de la traduction [21]. Enfin, certains domaines supplementaires peuvent
transmettre un signal vers le domaine catalytique, permettant d’induire des change-
ments conformationnels dans le site actif en reponse a la liaison de l’ARNt [22].
L’existence d’enzymes de correction des mesacylations homologues aux domaines
d’edition des aaRS [21] et l’independance importante des domaines des aaRS montrent
que les synthetases modernes ont evolue de facon divergente par duplication et fusion de
genes. Ce sont ces evenements de duplication qui auraient permis l’incorporation au code
genetique de l’ensemble des acides amines standards et, particulierement, celle des acides
amines glutamine et asparagine, souvent consideres comme les derniers acides amines a
avoir ete acquis par les organismes. La pression selective resultant de la duplication
d’un gene d’aaRS aurait force la specialisation concomitante des deux paralogues,
ou la transformation rapide d’un des genes en un gene cryptique [23]. La presence
de nombreux paralogues des aaRS dans les genomes des organismes contemporains

Chapitre 1. Introduction 7
(a) La glutamyl-ARNt synthetase (une aaRS de classe I) de Thermus
thermophilus (en vert) complexee a son ARNt (en mauve) [16]. Le
motif structural (en bleu) contenant le site catalytique est un pli de
Rossman. m : Cote du sillon mineur (situe derriere l’ARN par rapport
a l’observateur). L’helice d’ARN est depliee apres la liaison a l’enzyme.
(b) L’aspartyl-ARNt synthetase (une aaRS de classe II) de E. coli
(en vert) complexee a son ARNt (en mauve) [17]. Le motif structural
(en bleu) contenant le site catalytique est un feuillet β anti-parallele.
M : Cote du sillon majeur (situe devant l’ARN par rapport a
l’observateur).
Fig. 1.4 – Exemples des structures des aminoacyl-ARNt synthetases de classes I et II.
Images stereoscopiques realisees avec le logiciel Pymol [18].

Chapitre 1. Introduction 8
atteste cette hypothese. Le gene yadB, presente plus loin (section 1.4.1), est un de ces
paralogues, et a d’abord ete considere comme un gene cryptique resultant de l’evolution
des aminoacyl-ARNt synthetases de la classe Ib (section 1.3).
1.2.4 Specificite
La fidelite de la biosynthese des proteines depend grandement de l’aminoacylation
correcte des ARNt. Un ARNt mesacyle peut etre utilise lors de la synthese de nouvelles
proteines par le ribosome, lequel ne discrimine pas les ARNt correctement et incor-
rectement charges [24]. La specificite des aminoacyl-ARNt synthetases est le facteur
determinant dans l’aminoacylation correcte des ARNt.
La reconnaissance specifique des ARNt par les aaRS se fait principalement par le
biais de contacts avec la tige acceptrice et la region de l’anticodon. Dans le premier cas,
le domaine implique est celui responsable de l’activite catalytique. Dans le second cas, il
s’agit plutot d’un des domaines supplementaires presentes plus tot (section 1.2.3). La
preponderance des contacts avec la tige acceptrice suggere que les ancetres des ARNt
modernes etaient probablement constitues d’une mini-helice, laquelle aurait evolue par
fusion avec d’autres ARN [25]. Des contacts entre les aaRS et d’autres regions des
ARNt, telles que la tige D, certaines paires de bases et certains nucleotides modifies ont
egalement ete observes.
La specificite de certaines synthetases est augmentee par un mecanisme d’ajuste-
ment induit impliquant l’ARNt. Dans le cas de la GluRS, la liaison de l’ARNt modifie
la conformation du site actif, ce qui favorise la liaison de l’ATP dans un site qui permet
la reaction d’activation du glutamate (section 1.2.1). A l’inverse, lorsque l’ARNt est
absent, l’ATP est lie dans un site ou la distance qui separe son phosphate α de la
fonction carboxyle α du glutamate est trop importante pour que la reaction puisse avoir
lieu. Ce mecanisme d’ajustement induit permet a la GluRS de reagir a la concentration
d’ARNtGlu libre dans la cellule et de limiter la mesacylation d’ARNt non specifiques [5].
Le nombre eleve de contacts specifiques pouvant etre etablis avec les ARNt facilite
leur reconnaissance. Dans le cas des acides amines, la reconnaissance est plus difficile :
s’il est facile de distinguer certaines chaınes laterales (par exemple la cysteine, dont la
structure est tres particuliere [26]), d’autres sont difficiles a discriminer puisque qu’elles
different tres peu entre elles ; par exemple, l’isoleucine ne differe de la valine que par la
presence d’un groupe methyle supplementaire. Afin d’assurer la fidelite de l’aminoacy-
lation, les aaRS ont developpe deux mecanismes de correction, illustres a la figure 1.5
[27]. Le premier de ces mecanismes (figure 1.5 c) est la correction avant transfert :

Chapitre 1. Introduction 9
Fig. 1.5 – Reactions de correction de mesacylations. Exemple pour l’isoleucyl-ARNt
synthetase. a) Mesactivation de la valine par l’IleRS. b) Mesacylation de la valine sur
l’ARNt de l’isoleucine. c) Correction pre-transfert : l’aminoacyl-adenylate incorrect est
hydrolyse avant son esterification a l’ARNt. d) Correction post-transfert : l’aminoacyl-
ARNt incorrect est hydrolyse.
l’acide amine active sous forme d’un aminoacyl-adenylate complexe a la synthetase
est hydrolyse avant d’etre charge sur l’ARNt. L’isoleucyl-ARNt synthetase (IleRS), par
exemple, dispose de deux sites catalytiques distincts qui agissent successivement comme
des filtres moleculaires. Le premier site, responsable de l’activation de l’isoleucine, exclut
les acides amines plus volumineux que l’isoleucine (et ne peut donc exclure la valine),
alors que le second site, qui possede une activite hydrolytique, ne peut lier que les petits
acides amines (esterifies a l’ARNt) tels que la valine [28].
Le second mecanisme de correction (figure 1.5 d), la correction post-transfert,
consiste en l’hydrolyse des aminoacyl-ARNt indesirables. Tel que mentionne plus tot
(section 1.2.3), cette fonction est remplie par des domaines structuraux autres que
celui qui assure l’aminoacylation. Certaines synthetases utilisent les deux mecanismes de
correction. C’est le cas de l’IleRS qui, meme si elle active une valine pour 150 isoleucines
in vivo, parvient a corriger ses erreurs pour limiter le remplacement de l’isoleucine par
la valine dans les proteines a une frequence de moins de une erreur pour 3 000 [29].
Ces determinants et antideterminants pour la reconnaissance des ARNt et des
acides amines, combines aux mecanismes d’edition, procurent aux synthetases une
fiabilite elevee : leur taux d’erreur moyen est de une pour 10 000 [4].
Enfin, les concentrations relatives des acides amines, des ARNt et des aminoacyl-
ARNt synthetases in vivo contribuent egalement a la specificite des reactions d’ami-
noacylation grace a l’etablissement de mecanismes de competition. Ces competitions
s’etablissent entre differentes synthetases pour l’utilisation d’un ARNt dont les

Chapitre 1. Introduction 10
elements d’identite sont ambigus, et entre differents ARNt pouvant servir de substrats
a une meme synthetase. Globalement, ces mecanismes de competition augmentent la
specificite de l’aminoacylation par rapport aux valeurs mesurees dans les experiences
in vitro ou seule la specificite des aaRS est prise en compte [30].
1.3 Evolution des aminoacyl-ARNt synthetases de
la famille GlxRS
Les aminoacyl-ARNt synthetases glutamyl-ARNt synthetase et glutaminyl-ARNt
synthetase (GlnRS) sont les aaRS les plus rapprochees d’un point de vue phylogenetique
et forment ensemble la famille GlxRS (sous-classe Ib), elle-meme divisee en deux sous-
familles de genes. Etonnamment, cette division en sous-familles ne separe pas les GluRS
des GlnRS, mais rassemble plutot d’un cote les GluRS bacteriennes et organellaires,
et de l’autre les GluRS archæbacteriennes et eucaryotes ainsi que les GlnRS. Ces
relations evolutives non-canoniques impliquent egalement un transfert lateral du gene
de la GlnRS a partir des eucaryotes vers quelques bacteries [23].
L’evolution de la famille des GlxRS, illustree a la figure figure 1.6, debuta
par la formation de deux lignees distinctes par evolution d’une GluRS ancestrale
(figure 1.6 a). Cette derniere etait formee d’un seul domaine structural servant a
l’aminoacylation des ARNt glutamate et glutamine. La voie indirecte de formation
du Gln-ARNtGln par transamidation existait donc avant l’apparition de la GlnRS.
Cette separation en deux lignees concorde avec l’evolution de l’arbre de la vie, soit la
formation d’une lignee menant a l’apparition des bacteries et d’une autre rassemblant
les ancetre des eucaryotes et des archæ.
Les deux groupes de GluRS ainsi formes acquirent par la suite un domaine supple-
mentaire permettant la reconnaissance de l’anticodon des ARNt. Dans le cas des GluRS
bacteriennes, ce domaine est forme d’une cage d’helices α (figure 1.6 b), alors que,
dans le cas des eucaryotes et des archæ, il est forme d’un barril β (figure 1.6 c).
La separation entre les archæ et les eucaryotes (figure 1.6 d) eut lieu avant l’ap-
parition de la GlnRS. L’emergence de la GlnRS se produisit sans doute immediatement
apres cette scission, puisque tous les eucaryotes disposent de l’enzyme GlnRS. Cette
apparition se fit par duplication du gene de la GluRS eucaryotique (figure 1.6 e) et
par specialisation concomitante des produits des deux genes : l’un d’eux a perdu la
capacite de reconnaıtre l’ARNtGln pour devenir une GluRS discriminatrice, alors que

Chapitre 1. Introduction 11
Fig. 1.6 – Evolution des aaRS de la famille GlxRS. a) Scission entre les bacteries et les
eucaryotes et archæ. b) Acquisition d’un domaine α de reconnaissance de l’ARNt par
les GluRS bacteriennes. c) Acquisition d’un domaine β de reconnaissance de l’ARNt
par les GluRS eucaryotes et archæbacteriennes. d) Scission entre les eucaryotes et les
archæ. e) Duplication et evolution de la GluRS chez les eucaryotes pour donner la
GlnRS. f) Acquisition de la GlnRS par quelques bacteries par transfert horizontal. g)
Specialisation de la GluRS bacterienne qui devient discriminatrice chez les bacteries qui
ont acquis une GlnRS par transfert horizontal.

Chapitre 1. Introduction 12
l’autre a change de specificite pour reconnaıtre la glutamine plutot que le glutamate,
tout en perdant la capacite d’aminoacyler l’ARNtGlu.
La presence d’une GlnRS dans un nombre restreint de bacteries (incluant E.
coli) s’explique par le transfert horizontal du gene de la GlnRS eucaryote vers ces
organismes (figure 1.6 f). La presence simultanee des mecanismes direct et indirect
d’aminoacylation de l’ARNtGln n’etait probablement pas viable pour des organismes
deja tres specialises ; c’est pourquoi le transfert a probablement ete accompagne de la
specialisation immediate de la GluRS, qui a perdu la capacite de reconnaıtre l’ARNtGln
(figure 1.6 g).
Enfin, notons que les plupart des cellules eucaryotes disposent egalement d’une
ou de plusieurs GluRS bacteriennes, ces dernieres se trouvant dans les organelles qui
ont ete acquises par endosymbiose d’organismes qui etaient les ancetres des bacteries
modernes.
1.4 La glutamyl-queuosine-ARNtAsp synthetase
1.4.1 Le gene yadB
L’evolution complexe de la famille GlxRS presentee ci-haut (section 1.3) im-
plique plusieurs phenomenes de duplication et de specialisation de genes. Il ne serait
donc pas surprenant de retrouver dans les genomes des organismes contemporains des
reliques de cette evolution sous la forme de genes cryptiques ou de pseudogenes. Chez
E. coli, le gene yadB, codant pour la glutamyl-queuosine-ARNtAsp synthetase (dont on
ne connaissait pas la fonction jusqu’a tout recemment), ressemble a un tel gene : il est
homologue au gene gltX codant pour la GluRS, mais sa sequence est plus courte et ne
correspond qu’aux domaines N-terminaux de la GluRS ; de surcroıt, yadB n’aminoacyle
pas l’ARNtGlu in vivo a un niveau substantiel puisque l’inactivation de la GluRS de E.
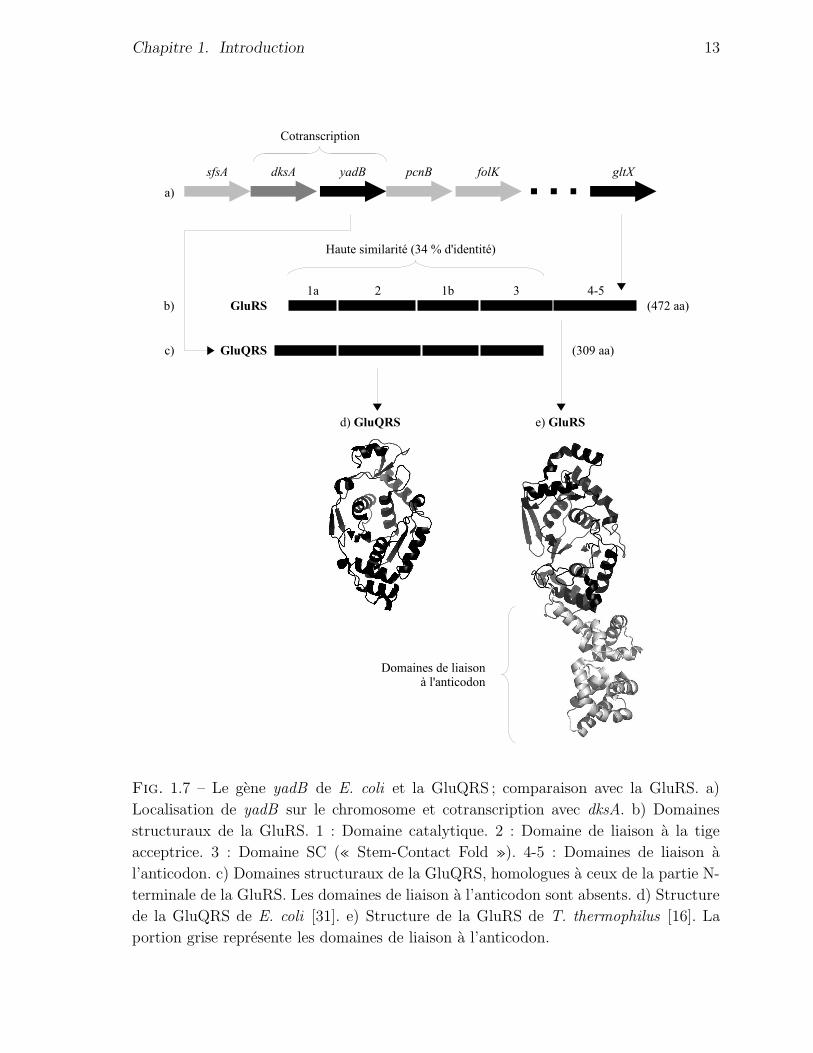
coli est letale. La figure 1.7 detaille la structure du gene yadB et compare son produit
d’expression a la GluRS.
Il a toutefois ete demontre que yadB est exprime chez E. coli. Il ne s’agit donc
pas d’un gene cryptique : l’ARN messager de yadB a pu etre amplifie a partir de
preparations d’ARN total de E. coli en phase exponentielle de croissance, et le produit
d’expression YadB a ete detecte par transfert Western. YadB pourrait donc avoir un
role metabolique, tout au moins chez E. coli.
Chapitre 1. Introduction 13
Fig. 1.7 – Le gene yadB de E. coli et la GluQRS ; comparaison avec la GluRS. a)
Localisation de yadB sur le chromosome et cotranscription avec dksA. b) Domaines
structuraux de la GluRS. 1 : Domaine catalytique. 2 : Domaine de liaison a la tige
acceptrice. 3 : Domaine SC (« Stem-Contact Fold »). 4-5 : Domaines de liaison a
l’anticodon. c) Domaines structuraux de la GluQRS, homologues a ceux de la partie N-
terminale de la GluRS. Les domaines de liaison a l’anticodon sont absents. d) Structure
de la GluQRS de E. coli [31]. e) Structure de la GluRS de T. thermophilus [16]. La
portion grise represente les domaines de liaison a l’anticodon.

Chapitre 1. Introduction 14
Cette idee est renforcee par des etudes phylogenetiques. Celles-ci montrent que
des homologues de yadB sont presents dans de nombreuses bacteries et que les mo-
tifs caracteristiques des synthetases de classe I y sont conserves. Notons par ailleurs
qu’aucun homologue de yadB n’a ete identifie chez les eucaryotes et les archæ.
YadB est manifestement plus proche evolutivement des GluRS bacteriennes et
organellaires (type α) que des GluRS eucaryotes et archæbacteriennes (type β). La
presence d’un homologue de yadB dans toutes les bacteries disposant d’une GlnRS
permet de croire que yadB aurait pu jouer un role lors du transfert horizontal de
cette enzyme vers ces bacteries, en fournissant une voie metabolique temporaire pour
l’incorporation du glutamate dans les proteines pendant la transition de la GluRS non
discriminatrice vers la GluRS discriminatrice. Voir [32] pour ce paragraphe et les trois
precedents.
1.4.2 Activite de YadB in vitro
La similarite elevee de YadB avec la GluRS (figure 1.7 b et c) suggerait que
cette enzyme encore non caracterisee aurait pu proceder au chargement du gluta-
mate sur l’ARNtGlu. Des mesures d’echange ATP-PPi montrent que, contrairement
a la GluRS, YadB n’a pas besoin de son ARNt pour l’activation du glutamate. Des
experiences d’aminoacylation realisees avec de l’ARNtGlu pur montrent par ailleurs que
YadB n’aminoacyle pas cet ARN [31].
Toutefois, d’autres experiences montrent que YadB catalyse une activite d’amino-
acylation lorsqu’il est en presence d’un extrait d’ARNt total de E. coli. Des experiences
d’aminoacylation realisees avec d’autres ARNt isoaccepteurs purifies ont finalement
demontre que le chargement du glutamate se fait sur l’ARNtAsp. De plus, il a ete claire-
ment demontre que les deux acides amines (glutamate et aspartate) peuvent etre charges
simultanement sur cet ARNt, ce qui suggere l’existence d’un site d’aminoacylation du
glutamate distinct de celui de l’aspartate.
La facilite avec laquelle le Glu-ARNtAsp peut etre hydrolyse indique que le site
de chargement du glutamate est sans doute similaire en termes de proprietes chimiques
a l’extremite 3’ de l’ARN (site du chargement de l’aspartate). L’incapacite de YadB
d’utiliser un transcrit de l’ARNtAsp produit in vitro (et donc depourvu de bases mo-
difiees) comme substrat etait un indice qu’une base modifiee pourrait servir de site de
chargement. Des experiences de spectroscopie de masse ont permis de decouvrir que la
base hypermodifiee queuosine, presente a la premiere position de l’anticodon, est le site
d’aminoacylation. La structure de cette base modifiee est presentee a la figure 1.8. La

Chapitre 1. Introduction 15
N
HN
NARNt
H2N
O N
OHHO
H2CH
Fig. 1.8 – Structure de la base hypermodifiee queuosine
purification d’ARNtAsp a partir d’une souche incapable d’echanger la guanosine 34 de
l’ARNt par la queuosine confirme ces donnees, l’enzyme etant incapable de charger cet
ARNt. Voir [33] pour ce paragraphe et le precedent.
Ces proprietes etonnantes suggeraient de renommer YadB pour mieux refleter
sa fonction biochimique : on designe maintenant cette enzyme sous le nom glutamyl-
queuosine-ARNtAsp synthetase (GluQRS) [34].
1.4.3 Similarites structurales entre la GluQRS et la GluRS
Il existe des similarites marquees entre la tige acceptrice de l’ARNtGlu et la tige
de l’anticodon de l’ARNtAsp chez les organismes possedant une GluQRS [34]. Ces
ressemblances soulevent des questions sur l’evolution des ARNt et des synthetases, et
renforcent l’hypothese selon laquelle une paire d’aaRS ancestrales (une enzyme de classe
I et une autre de classe II) auraient lie un ARNt primitif ; dans un tel complexe ternaire,
chaque synthetase aurait etabli des interactions avec un cote different de l’ARN [35].
Dans le cas present, on peut supposer que la GluRS et l’AspRS (des aaRS de classe
I et II respectivement) auraient lie un meme ARNt, peut-etre un ancetre des ARNt
specifiques au glutamate et a l’aspartate.
Les ressemblances entre ARNtGlu et ARNtAsp ne suffisent toutefois pas a expliquer
comment la GluQRS peut lier un ARNt dans une position si differente de celle employee
par la GluRS, enzyme avec laquelle la GluQRS partage 34 % d’identite. Une partie de
l’explication reside sans doute dans l’absence, dans la GluQRS, de regions homologues
aux domaines C-terminaux de reconnaissance de l’anticodon que l’on observe dans
les GluRS (figure 1.7 d et e) : sans ces deux domaines, la seule portion de l’ARNt
avec laquelle l’enzyme peut etablir des contacts est la tige acceptrice. Or, un nombre
important de determinants et antideterminants de l’identitie des ARNt se retrouvent
dans la region de l’anticodon.

Chapitre 1. Introduction 16
Notons egalement le remplacement, dans tous les genes homologues a yadB, d’une
threonine hautement conservee (potentiellement impliquee dans la selection de l’acide
amine par la GluRS et la GlnRS) par un residu hydrophobe, soit la valine, la leucine ou
l’isoleucine. La specificite de la GluQRS pour le L-glutamate est pourtant aussi elevee
que celle de la GluRS. Il s’agit d’un indice structural supplementaire suggerant que le
mode d’action de la GluQRS est significativement different de celui de la GluRS [32].
La structure de la GluQRS libre a ete determinee a une resolution de 1,5 A [31].
Elle ressemble fortement a celle des domaines N-terminaux de la GluRS. Bien qu’un
modele ait ete propose pour la liaison de l’ARNt a l’enzyme [34], ce modele repose
essentiellement sur des contraintes theoriques et intuitives et ne permet pas de formuler
d’hypothese solide decrivant l’orientation de l’ARNt lors de l’approche par la GluQRS.
1.4.4 Relation avec le gene dksA
L’amplification de l’ARNm de yadB a demontre que, chez E. coli, les genes dksA et
yadB sont coexprimes (figure 1.7 a). La presence d’un operon dksA yadB est egalement
suggeree par la sequence du chromosome, ou l’on trouve une sequence promotrice en
amont de dksA, mais aucune immediatement en amont de yadB ; aucune sequence
terminatrice ne semble separer les deux genes [32].
Bien que leur coexpression n’ait pas ete verifiee pour d’autres organismes que E.
coli, ces genes montrent une relation syntenique dans le genome de plusieurs bacteries,
par exemple Pseudomonas æruginosa. Cette relation suggere qu’ils pourraient etre
impliques dans la meme voie metabolique.
dksA a d’abord ete identifie en tant que suppresseur dosable d’une mutation
inactivant dnaK. La surexpression de dksA dans la souche E. coli ∆dnaK palie la
thermosensibilite causee par l’inactivation [36].
Chez P. æruginosa, dksA est un regulateur important de plusieurs facteurs ex-
tracellulaires de virulence. La synthese de rhamnolipides et l’expression de l’elastase
LasB (une protease impliquee dans l’infection de l’hote) sont pratiquement nulles dans
les cellules ou dksA a ete delete. Des experiences ayant demontre que les niveaux de
transcription des genes lasB et rhlAB (une enzyme impliquee dans la synthese des
rhamnolipides) ne varient pas entre la souche sauvage et la souche ∆dksA, il semble
que dksA soit un regulateur post-transcriptionnel [37].

Chapitre 1. Introduction 17
Le gene dksA est aussi implique dans la reponse a des stress environnementaux
varies chez certains pathogenes. Des etudes realisees avec la bacterie Salmonella typhi-
murium associent dksA a la resistance de cette bacterie a de faibles pH et a sa survie
pendant la phase stationnaire. Le mecanisme d’action de dksA chez S. typhimurium
n’est pas connu, mais serait lie a l’accumulation du facteur sigma alternatif σS de
l’ARN polymerase. DksA serait requis pour la traduction du gene rpoS ; son inactivation
empeche la synthese du facteur σS, ce qui diminue la virulence de la bacterie chez la
souris [38].
Un mecanisme semblable de regulation du gene rpoS par dksA a ete observe
chez Shigella flexneri. Une sensibilite accrue au choc oxydatif et a un milieu acide a
ete observee chez un mutant dksA. Ces effets ne seraient toutefois que partiellement
attribuables a une production insuffisante de RpoS ; DksA agirait aussi par le biais
d’un mecanisme independant de RpoS. Globalement, l’inactivation de dksA conduit a
une pathogenicite reduite, les cellules mutantes etant incapables de se propager dans le
milieu intercellulaire dans des cultures tissulaires [39].
Des informations supplementaires sur le mecanisme de regulation de RpoS par
dksA ont ete obtenues par l’etude des stress nutritifs chez E. coli. On sait qu’un manque
de nutriments induit, chez les bacteries, la synthese de guanosine-3’5’-bis(diphosphate)
(ppGpp). Ce metabolite agit comme un signal de l’appauvrissement du milieu en acides
amines libres. Son absence empeche l’expression de RpoS, maintenant la cellule en phase
exponentielle de croissance. L’action regulatrice de dksA impliquerait la mediation du
signal ppGpp conduisant a l’induction de rpoS [40].
La determination de la structure de DksA [41], couplee a des etudes recentes sur
les interactions entre l’ARN polymerase et l’effecteur ppGpp, permet de comprendre
le role de DksA au niveau moleculaire. Le modele propose suggere que DksA forme
un complexe avec l’ARN polymerase et stabilise la liaison du ppGpp a ce complexe
enzymatique. Cette interaction serait necessaire a la fixation du nucleotide tri-phosphate
(NTP) initial permettant l’initiation de la transcription des genes des ARN ribosomiaux
(ARNr). Ce modele est confirme par l’invariabilite du taux de transcription des genes
des ARNr dans une souche de E. coli ∆dksA ; a l’inverse, dans la souche sauvage, le taux
de transcription des genes des ARNr est influence, entre autres, par la concentration
d’acides amines libres et par la phase de croissance cellulaire. La meme etude rapporte
que l’effet de dksA est maximal pendant la phase stationnaire de croissance [42].

Chapitre 1. Introduction 18
1.4.5 Questions et objectifs
Le role du Glu-Q-ARNtAsp synthetise par la GluQRS est encore inconnu. Les
etudes realisees jusqu’a maintenant excluent toutefois la possiblite d’une utilisation
classique de cet aminoacyl-ARNt dans la synthese proteique ; le chargement du glu-
tamate sur l’ARNtAsp ressemble davantage a une modification de l’ARN qu’a une
aminoacylation conventionnelle.
L’eventuelle relation entre dksA et yadB est egalement inconnue, mais l’existence
d’un operon liant l’expression de ces deux genes laisse penser que yadB pourrait, lui
aussi, etre implique dans un mecanisme de reponse generale au stress et dans le passage
a la phase stationnaire de croissance.
La presente etude visait, dans un premier temps, a determiner si yadB est un gene
essentiel chez E. coli. Pour ce faire, une souche mutante a ete construite par deletion
du gene yadB. La viabilite des cellules a ete verifiee dans des conditions de culture
optimales et dans des conditions minimales. Les differences phenotypiques entre les
souches sauvage et mutante ont ete investiguees.
Dans un second temps, le gene yadB a ete remplace sur le chromosome de E. coli
par un gene rapporteur (lacZ ). Ce dernier a ete utilise pour determiner les conditions
d’expression de yadB par dosage enzymatique du nombre de copies du rapporteur
(la β-galactosidase) dans des cultures liquides. Il a ainsi ete possible de verifier que
l’expression de yadB est liee au passage a la phase stationnaire de croissance. Nous
avons aussi essaye d’induire l’expression du gene en reproduisant in vitro certains stress
cellulaires.

Chapitre 2
Materiel et methodes
2.1 Tampons, solutions et produits chimiques
La composition des tampons et autres solutions utilisees dans les experiences
decrites plus loin est donnee au tableau 2.1. Toutes les solutions ont ete preparees avec
de l’eau purifiee par un systeme NANOpure (qui combine les methodes d’ultra-filtration
et de photo-oxydation aux rayons U.V.) de la compagnie Barnstead. Les solutions ont
ete sterilisees par filtration, a l’aide de membranes de pores de 0,22 µm de diametre. Des
produits chimiques de grade extra-pur ont ete employes dans la confection de toutes
les solutions. Les fournisseurs de ces produits sont les compagnies LabMat, Anachemia,
Fischer Scientific et BDH.
2.2 Culture cellulaire
2.2.1 Milieux de culture et conditions de croissance
Quatre milieux de culture ont ete utilises : Luria-Bertani (LB), M9 enrichi, SOB et
SOC. La composition de ces milieux est donnee dans le tableau 2.2. Les milieux ont ete
prepares en dissolvant les composants (provenant des fournisseurs Difco, Fischer Scien-
tific et LabMat) dans de l’eau demineralisee (milieux LB, SOB et SOC) ou NANOpure
(milieu M9), avant d’autoclaver le milieu pendant 20 minutes a 121 ◦C a une pression de
15 psi. Pour les milieu M9, SOB et SOC, les supplements nutritifs (chlorure et sulfate
de magnesium, glucose, thiamine, leucine et proline) ont ete prepares independamment,

Chapitre 2. Materiel et methodes 20
Tab. 2.1 – Tampons et solutionsSolution Composition (a 1 X) Reference
Reactif ONPGaONPG 4,0 mg/ml
[43]Solution preparee dans le tampon Z
Tampon de charge
pour gels d’agarose
Ficoll 400 2,0 %
[44]
EDTA 10 mM
SDS 0,1 %
Bleu de bromophenol 0,25 %
pHb (ajuste avec NaOH) 8,0
Tampon TB
Pipes 10 mM
[45]
CaCl2 15 mM
KCl 250 mM
MnCl2c 55 mM
pHb (ajuste avec KOH) 6,7
Tampon TBE
Tris 90 mM
[44]Acide borique 90 mM
EDTA 3,2 mM
pHb (non ajuste) ∼ 8, 3
Tampon TE
Tris 10 mM
[46]EDTA 1 mM
pHb (ajuste avec HCl) 8,5
Tampon Z
NaH2PO4 60 mM
[43]
NaH2PO4 40 mM
KCl 10 mM
MgSO4 1 mM
2-mercaptoethanold 50 mM
pHb (ajuste avec HCl ou NaOH) 7,0a Prepare immediatement avant son utilisation.
b Mesure a 22 ◦C.
c Ajoute en dernier apres ajustement du pH.
d Ajoute immediatement avant usage.

Chapitre 2. Materiel et methodes 21
filtres pour les steriliser, puis rajoutes au milieu une fois ce dernier autoclave et refroidi
a une temperature de moins de 50 ◦C. L’ajout des acides amines leucine et proline
est requis par l’auxotrophie des souches TP8503, POK1 et POK2. (Les genotypes et
phenotypes des souches utilisees sont presentes a la section 2.2.3.) Les antibiotiques
appropries (section 2.2.2) ont ete ajoutes sterilement aux milieux apres que ceux-ci
aient ete autoclaves et que leur temperature ait redescendu en dessous de 50 ◦C.
Les milieux SOB et SOC n’ont ete employes que pour la transformation de cellules
de E. coli par de l’ADN plasmidique (section 2.3.12). Le milieu minimal M9 a ete
employe pour l’etude des conditions d’expression et des effets de la deletion du gene
yadB (section 2.8.2). Le milieu LB a aussi ete utilise pour les etudes sur l’expression et
les effets de la deletion de yadB, ainsi que pour toutes les autres experiences de biologie
moleculaire (section 2.3).
A moins d’indication contraire, les cultures ont ete realisees a une temperature de
37 ◦C, dans des tubes de verre de 18 mm (cultures de 5,0 ml) ou dans des erlenmeyers
de 250 ml (cultures de 50 ml) soumis a une agitation lineaire de 250 rpm.
2.2.2 Agents selectifs
Lorsque necessaire, les antibiotiques ampicilline, chloramphenicol et kanamycine,
ainsi que le sucrose, ont ete utilises comme agents de selection, selon les concentrations
indiquees dans le tableau 2.3. Tous les antibiotiques ont ete fournis par la compagnie
Boehringer Mannheim.
2.2.3 Souches
Les souches de E. coli employees dans le cadre de cette etude sont repertoriees
dans le tableau 2.4. Toutes les souches ont ete conservees a -80 ◦C dans un congelateur
sans cycle de degivrage, apres ajout de 10 % de glycerol a une culture liquide fraıchement
saturee.
Les souches E. coli XL1-Blue et TOP10 ont ete utilisees pour tous les clonages qui
ont ete necessaires a la construction du vecteur permettant le remplacement du gene
yadB. Ces procedures sont decrites en details plus loin (section 2.4). Les souches
EDCM, la souche DH5α/pCP20 et la souche TP8503 ont ete obtenues aupres des
chercheurs ayant mis au point la methode de remplacement et de deletion utilisee [48].

Chapitre 2. Materiel et methodes 22
Tab. 2.2 – Milieux de cultureMilieu Composant Concentration Reference
LB
Hydrolysat de caseine (tryptone) 10,0 g/l
[47]Extraits de levure 5,0 g/l
NaCl 2,5 g/l
Agara 15,0 g/l
M9
Na2HPO4 6,0 g/l
[47]
KH2PO4 3,0 g/l
NH4Cl 1,0 g/l
NaCl 0,5 g/l
CaCl2 3,0 mg/l
Agara 15,0 g/l
MgSO4 1,0 mM
Glucose 0,2 %
Thiamine 50 ppm
L-Leucine 0,25 g/l
L-Proline 0,25 g/l
SOB
Hydrolysat de caseine (tryptone) 20,0 g/l
[45]
Extraits de levure 5,0 g/l
NaCl 10 mM
KCl 2,5 mM
MgCl2 10 mM
MgSO4 10 mM
SOC
Hydrolysat de caseine (tryptone) 20,0 g/l
[45]
Extraits de levure 5,0 g/l
NaCl 10 mM
KCl 2,5 mM
MgCl2 10 mM
MgSO4 10 mM
Glucose 20 mMa Seulement pour les cultures en milieu solide.
Tab. 2.3 – Agents selectifs
Produit Abreviation ConcentrationGene de Gene de Phenotypes
resistance sensibilite Resistant Sensible
Ampicilline Amp 100 µg/ml bla ApR ApS
Chloramphenicol Chl 35 µg/ml cat CmR CmS
Kanamycine Kan 50 µg/ml aph KmR KmS
Sucrose Suc 10 % sacB SucR SucS

Chapitre 2. Materiel et methodes 23
Les plasmides presents dans les souches EDCM et dans la souche DH5α/pCP20 sont
decrits plus loin (section 2.3.1). La souche TP8503 a ete utilisee dans la construction
de la souche POK1, ou le gene yadB est remplace (section 2.5) par la cassette FLK2
portant le gene rapporteur lacZ, et dans la construction de la souche POK1, ou ce gene
est delete (section 2.6). Les souches POK1 et POK2 ont ete utilisees pour determiner
les conditions d’expression et les effets de la deletion (section 2.8) du gene yadB.
Apres la reception des cellules EDCM, DH5α/pCP20 et TP8503 par courrier,
sous forme de cultures en gelose profonde, chaque souche a ete etalee sur les geloses
appropriees (LB-Chl-Kan pour EDCM70, LB-Amp-Kan pour EDCM81, LB-Amp-Chl
pour DH5α/pCP20 et LB pour TP8503). Apres incubation a 37 ◦C pour les souches
EDCM81 et TP8503, et a 30 ◦C pour les souches EDCM70 et DH5α/pCP20 (afin de
permettre la replication des plasmides dont l’origine de replication est thermosensible),
un clone de chaque souche a ete repique en milieu liquide. Apres 18 heures de crois-
sance, un echantillon de 1,0 ml de chaque souche a ete preleve et conserve a -80 ◦C
dans les conditions indiquees ci-dessus. La presence des plasmides d’interet dans les
cellules a ensuite ete verifiee par preparation de l’ADN plasmidique (section 2.3.4) et
electrophorese sur gel d’agarose (section 2.3.10).
2.3 Manipulations de l’ADN
2.3.1 Plasmides
Les plasmides employes dans le cadre de ce travail sont listes dans le tableau 2.5.
Les plasmides pTOF24, pTOF30 et pET-dksA-yadB ont ete utilises pour la construction
du vecteur pTOF-YB (section 2.4), ce dernier ayant servi au remplacement du gene
yadB par la cassette FLK2 porteuse du rapporteur lacZ, resultant en la creation de
la souche POK1 (section 2.5). Le plasmide pTOF-YA est un intermediaire dans la
construction de pTOF-YB ; il contient les regions flanquant le gene yadB, mais la cas-
sette FLK2 n’y a pas encore ete inseree. Le plasmide pCP20 exprime la flippase de levure
(recombinase Flp), qui a permis d’obtenir une souche ∆yadB (POK2) (section 2.6)
par excision de la cassette FLK2 presente dans le genome de la souche POK1. Le detail
des plasmides pTOF24, pTOF30, pTOF-YA et pTOF-YB est presente a la figure 2.1.

Chapitre 2. Materiel et methodes 24
Tab. 2.4 – Souches de E. coli
Souchea PlasmideGenotypes et Elements conferes Source ou
phenotypes par un plasmide reference
TOP10
F− mcrA ∆(mrr-hsdRMS-mcrBC )
Invitrogenφ80lacZ∆M15 ∆lacX74 deoR
recA1 araD139 ∆(ara-leu)7697
galU galK rpsL endA1 nupG
XL1-Blue
recA1 endA1 gyrA96 thi-1 hsdR17
StratagenesupE44 relA1 lac [F’ proAB
lac1 qZ∆M15 Tn10 ]
TP8503 ∆(lac-proB) leu thi-1 supE42 fhuA [48]
EDCM70
(DH5α)pTOF24
φ80lacZ∆M15 recA1 endA1 gyrA96aph cat
sacB repAts [48]thi-1 hsdR17 (r−κ
m+κ
) supE44
relA1 deoR ∆(lacZYA-argF)U169
EDCM81
(DH5α)pTOF30
φ80lacZ∆M15 recA1 endA1 gyrA96bla FRT-lacZ -
aph-FRT[48]thi-1 hsdR17 (r−κ m+
κ ) supE44
relA1 deoR ∆(lacZYA-argF)U169
DH5α pCP20
φ80lacZ∆M15 recA1 endA1 gyrA96bla cat flp
repAts [48]thi-1 hsdR17 (r−κ
m+κ
) supE44
relA1 deoR ∆(lacZYA-argF)U169
POK1 ∆(lac-proB) leu thi-1 supE42 fhuAsection 2.5
(TP8503) yadB〈〉(FRT-lacZ -aph-FRT)
POK2 ∆(lac-proB) leu thi-1 supE42 fhuAsection 2.6
(TP8503) ∆yadBa Lorsqu’une souche a ete construite a partir d’une autre souche, cette derniere est indiquee entre parentheses.
Tab. 2.5 – PlasmidesPlasmide Elements genetiques Description Reference
pTOF24 aph cat sacB repAts
Vecteur pour le clonage de yadB
[48]et pour l’insertion de la
cassette FLK2
pTOF30 bla FRT-lacZ-aph-FRT Source de la cassette FLK2 [48]
pCP20 bla cat flp repAts
Exprime la recombinase Flp
[48]permettant l’excision de la
cassette FLK2
pET-dksA-yadBdksA-yadB-pncB’ Source du gene yadB et de ses D. Dubois
lacI aph regions flanquantes (non publie)
pTOF-YAsacB repAts cat dksA’ - Intermediaire dans la construction
section 2.4yadB’ -’yadB-pncB’ du vecteur pTOF-YB
pTOF-YB
sacB repAts cat dksA’ - Vecteur pour le remplacement du
section 2.4yadB’ -FRT-lacZ -aph- gene yadB par la cassette
FRT-’yadB -pncB’ FLK2

Chapitre 2. Materiel et methodes 25
(a) pTOF24 - Vecteur servant a la
construction de pTOF-YB
(b) pTOF30 - Source de la cassette
FLK2
(c) pTOF-YA - Intermediaire dans la
construction de pTOF-YB
(d) pTOF-YB - Vecteur pour le rem-
placement de yadB
Fig. 2.1 – Plasmides

Chapitre 2. Materiel et methodes 26
Tab. 2.6 – OligonucleotidesNom Site de restriction Sequencea Tm
b
Ci NotI ccgttccaagcggccgcaagagcgAGCGTCGAGCAAATCCTTCAGTCA 64
Co SalI aaaaagtcgacGCTTCGTATCCCGCTTTATTGAGC 69
Ni NotI cgctcttgcggccgcttggaacggCAGAGAGCCAAAATGAAGCTCGCC 67
No PstI aaaaactgcagATCGCACCGTTACACATATGCAGG 69
Cext TCCGTGAAGTAGCGGGTAAT 60
Next CCGGGCGAAGAGTATATGAA 60
F1 GGCCTCTTCGCTATTACGC 60
F2 CTGCCTCGGTGAGTTTTCTC 60a Les bases complementaires a la sequence genomique sont en majuscules. Les sites de restriction sont soulignes.
b Temperature de fusion
2.3.2 Oligonucleotides
Les oligonucleotides employes dans les experiences decrites plus loin sont detailles
au tableau 2.6. Les oligonucleotides No et Ni ont ete utilises pour amplifier par PCR
la region 5’ du gene yadB et une partie du gene en amont (dksA). De maniere similaire,
les oligonucleotides Co et Ci ont ete utilises pour l’amplification de la portion 3’ de yadB
et d’une partie du gene en aval (pncB). La paire No et Co a servi a amplifier le fragment
resultant de la ligature des regions 5’ et 3’. Ce fragment a servi a la construction du
vecteur pTOF-YB presente plus tot (section 2.3.1).
Les oligonucleotides Next et Cext ont ete utilises, soit ensemble, soit en combi-
naison avec les oligonucleotides F1 et F2, afin de confirmer le remplacement de yadB
par la cassette FLK2 (section 2.5.4) dans la souche POK1, et la deletion de yadB
(section 2.6.3) dans la souche POK2. Tous les oligonucleotides ont ete synthetises
par la compagnie Invitrogen et purifies par desalage. Le programme eprimer3, qui
fait partie de la suite logicielle EMBOSS [49], a ete utilise pour faciliter l’ingenierie des
oligonucleotides. Le programme remap, du meme ensemble logiciel, a ete utilise pour
choisir des sites de restriction compatibles avec les sequences a cloner.
2.3.3 Purification d’ADN par extraction et precipitation
L’ADN obtenu par digestion (section 2.3.7), dephosphorylation (section 2.3.9)
et ligature (section 2.3.8) a ete purifie par extraction au phenol et au chloroforme et
par precipitation a l’acetate de sodium et a l’ethanol, selon un protocole etabli [50] :

Chapitre 2. Materiel et methodes 27
1. Ajouter a l’ADN en solution dans un microtube 1,0 volume d’une solution de
phenol / chloroforme / alcool isoamylique (25 : 24 : 1).
2. Vortexer vigoureusement pendant 10 secondes, puis centrifuger 15 secondes a
10 000 × g dans une microcentrifugeuse. Les proteines denaturees par le phenol
se retrouvent dans la phase organique, alors que les acides nucleiques restent dans
la phase aqueuse.
3. Prelever la phase superieure (aqueuse) et la transferer dans un autre microtube.
4. Ajouter 1,0 volume de tampon TE 1 X a la phase organique.
5. Vortexer vigoureusement pendant 10 secondes, puis centrifuger 15 secondes a
10 000 × g dans une microcentrifugeuse.
6. Prelever la phase superieure (aqueuse) et l’ajouter a la premiere phase aqueuse
qui a ete prelevee. Cette deuxieme extraction permet d’augmenter le rendement
de la purification.
7. Ajouter a la phase aqueuse 1,0 volume d’une solution de phenol / chloroforme /
alcool isoamylique (25 : 24 : 1).
8. Vortexer vigoureusement pendant 10 secondes, puis centrifuger 15 secondes a
10 000 × g dans une microcentrifugeuse.
9. Prelever la phase superieure (aqueuse) et la transferer dans un autre microtube.
Cette extraction supplementaire reduit la possibilite de contamination par des
proteines.
10. Ajouter 0,1 volume d’acetate de sodium 3 M pH 5,2. Melanger.
11. Ajouter 2,0 volumes d’ethanol 100 % froid. Melanger en vortexant. L’ADN en
solution commence a precipiter.
12. Placer a -80 ◦C pendant 20 minutes afin de favoriser la precipitation complete de
l’ADN.
13. Centrifuger 10 minutes a 10 000 × g.
14. Enlever le surnageant.
15. Ajouter 500 µl d’ethanol 80%.
16. Centrifuger 5 minutes a 10 000 × g. Ce lavage permet d’eliminer les sels.
17. Enlever le surnageant. Secher sous vide.
18. Resuspendre l’ADN dans le tampon TE 1 X.
2.3.4 Preparation d’ADN plasmidique
L’extraction d’ADN plasmidique a partir de cellules de E. coli a ete faite avec
la trousse commerciale QIAprep Spin Miniprep Kit de la compagnie QIAGEN. Les

Chapitre 2. Materiel et methodes 28
preparations ont ete realisees a partir de 5,0 ml de cultures bacteriennes inoculees
environ 18 heures plus tot. Les croissances ont ete realisees a 30 ◦C afin de permettre la
replication des plasmides contenant une origine de replication thermosensible (repAts).
Le protocole employe est celui recommande par QIAGEN [51]. Les cellules sont
d’abord culotees par centrifugation, puis resuspendues dans un faible volume de tampon
contenant de la RNAse, afin de degrader les ARN. Les cellules sont lysees a l’aide
d’une solution alcaline contenant du NaOH et du SDS. A l’interieur d’une etroite plage
de pH (12,0 a 12,5), l’ADN circulaire de faible poids moleculaire (ADN plasmidique)
n’est pas denature, alors que l’ADN de plus haut poids moleculaire (ADN genomique)
l’est [52]. Le lysat est neutralise a l’aide d’un tampon contenant de l’acide acetique
et du chlorure de guanidinium (agent denaturant). La renaturation rapide de l’ADN
genomique conduit a sa precipitation, alors que l’ADN plasmidique demeure solubilise
dans le surnageant. Les proteines et l’ADN genomique precipites et les debris cellulaires
sont enleves par centrifugation, et le surnageant contenant l’ADN plasmidique est
applique sur une colonne contenant une resine de silice sur laquelle l’ADN s’adsorbe a
bas pH et a concentration saline elevee. Apres le passage sur la colonne de tampons de
lavage, l’ADN plasmidique purifie est elue a l’aide d’un tampon de pH eleve (8,5) dont
la concentration en sels est faible.
Pour chaque preparation, un controle est realise avec une souche de E. coli qui ne
porte aucun plasmide. Les plasmides obtenus et le controle sont analyses par electro-
phorese sur gel d’agarose (section 2.3.10).
2.3.5 Preparation d’ADN genomique
L’ADN genomique de E. coli a ete prepare selon une methode publiee [53] :
1. Centrifuger pendant 2 minutes a 10 000 × g, 1,5 ml d’une culture saturee afin de
culoter les cellules.
2. Resuspendre les cellules dans 570 µl de tampon TE 1 X.
3. Ajouter 3 µl de proteinase K 20 mg/ml et 30 µl de SDS 10 %. Melanger et incuber
une heure a 37 ◦C. Cette etape vise a solubiliser les parois des cellules et a degrader
les proteines.
4. Ajouter 100 µl de NaCl 5,0 M et melanger. Cet ajout empeche que les acides
nucleiques ne precipitent lors de l’ajout de CTAB a l’etape suivante ; a faible
concentration en sels, il y aurait formation de complexes CTAB • acides
nucleiques.

Chapitre 2. Materiel et methodes 29
5. Ajouter 80 µl d’une solution de CTAB 10 % / NaCl 0,7 M. Melanger et incuber
10 minutes a 65 ◦C. Le CTAB forme un precipite avec les debris cellulaires, les
proteines denaturees et les polysaccharides.
6. Ajouter 1,0 volume de chloroforme / alcool isoamylique (49 : 1). Extraire la phase
superieure (phase aqueuse) contenant l’ADN.
7. Ajouter 1,0 volume de phenol / chloroforme / alcool isoamylique (25 : 24 : 1).
Extraire la phase superieure (aqueuse) contenant l’ADN.
8. Precipiter l’ADN en rajoutant 0,6 volume d’isopropanol. Il n’est pas necessaire de
rajouter un sel d’acetate pour precipiter l’ADN puisque la concentration saline
est deja elevee.
9. Centrifuger 5 minutes a 10 000 × g, puis laver le culot avec de l’ethanol 70 %
pour enlever les sels.
10. Resuspendre dans 100 µl de tampon TE 1 X.
2.3.6 Reaction en chaıne par la polymerase
La technique de PCR a ete utilisee lors de la construction du vecteur pTOF-
YB pour amplifier les regions chromosomiques flanquant le gene yadB (section 2.4.1).
Cette technique a aussi ete employee afin de demontrer l’integration de la cassette FLK2
dans le chromosome (section 2.5.4) et la deletion du gene yadB (section 2.6.3).
Les melanges reactionnels ont ete assembles tel qu’indique au tableau 2.7. Les
concentrations en MgSO4 et en DMSO ainsi que la temperature d’hybridation et le
temps d’elongation optimums ont ete determines experimentalement pour chaque paire
d’amorces utilisee. Ces conditions sont indiquees au tableau 2.8. Le protocole suivant
a ete utilise pour toutes les experiences de PCR [54] :
1. Les melanges reactionnels sont assembles dans des microtubes de 0,7 ml a parois
fines. L’enzyme est ajoutee apres tous les autres composants. Le contenu des tubes
est rapidement melange.
2. Environ 25 µl d’huile minerale est deposee a la surface du melange. Les tubes sont
centrifuges quelques secondes.
3. Les melanges reactionnels sont introduits dans un thermocycleur.
4. L’ADN est denature a 95 ◦C pendant 1 minute.
5. Le cycle denaturation / hybridation / elongation suivant est repete 30 fois :
(a) Denaturation de l’ADN a 95 ◦C pendant 30 secondes.

Chapitre 2. Materiel et methodes 30
Tab. 2.7 – Composition des melanges reactionnels pour PCRTampon Venta 1 X
MgSO4 2-8 mM
DMSO 0-2,5 %
dATP 50 µM
dCTP 50 µM
dGTP 50 µM
dTTP 50 µM
Oligonucleotide 1 500 nM
Oligonucleotide 2 500 nM
ADN gabarit 1 ng
Polymerase Venta 1 U
pH 8,8
Volumeb 100 µla Provenant de New England BioLabs.
b Ajuste avec de l’eau NANOpure sterile.
(b) Hybridation des oligonucleotides au gabarit d’ADN pendant 30 secondes. La
temperature d’hybridation varie selon la paire d’oligonucleotides utilisee.
(c) Elongation des oligonucleotides a 76 ◦C. Le temps d’elongation varie selon la
longueur du gabarit d’ADN utilise.
6. Une etape d’elongation supplementaire de 7 minutes a 76 ◦C est realisee afin
d’obtenir seulement des produits PCR pleine longueur.
7. Le melange reactionnel est refroidi a 4 ◦C.
Un thermocycleur DNA Thermal Cycler de la compagnie Perkin Elmer a ete
employe pour toutes les experiences de PCR.
Les produits PCR obtenus ont ete purifies grace a la trousse commerciale QIAquick
PCR Purification Kit de la compagnie QIAGEN. Le protocole employe est celui propose
par QIAGEN [46]. Le melange reactionnel PCR est dilue 6 X dans une solution aqueuse
de propanol et de chlorure de guanidinium. Ce melange est depose sur une colonne
contenant une resine de silice sur laquelle l’ADN s’adsorbe a bas pH et a concentration
saline elevee. Apres le passage sur la colonne d’un tampon de lavage, le produit PCR
purifie est elue a l’aide d’un tampon de pH eleve (8,5) dont la concentration en sels
est faible.
Pour chaque experience PCR, une reaction controle a ete realisee ou l’enzyme n’a
pas ete ajoutee. Les produits des reactions PCR, incluant le controle, sont analyses par
electrophorese sur gel d’agarose (section 2.3.10).

Chapitre 2. Materiel et methodes 31
Tab. 2.8 – Conditions pour les reactions PCRAmorces [MgSO4] [DMSO] Th.
a tel.b
mM % ◦C min
Ci - Co 4 0 50 1
Ni - No 4 0 50 1
No - Co 2 2,5 45 2
Next - F1 2 0 55 2
Cext - F2 4 1,0 60 2
Next - Cext 6 0 55 3a Temperature d’hybridation
b Temps d’elongation
Tab. 2.9 – Composition des melanges reactionnels pour la restriction de l’ADNTampon de restrictiona 1 X
BSA 100 µg/ml
ADN ∼ 1 µg
Enzyme de restriction 1 U/µg ADN
pH 7,9
Volumeb 25 µla Fourni par NEB. Le tampon varie selon l’enzyme employee.
b Ajuste avec de l’eau NANOpure sterile.
2.3.7 Digestion d’ADN par des endonucleases de restriction
Toutes les enzymes de restriction utilisees dans le cadre de ce projet (PstI, SalI,
NotI) proviennent de la compagnie New-England BioLabs. La composition des melanges
reactionnels est donnee au tableau 2.9. Tous les melanges reactionnels ont ete incubes
une heure a 37 ◦C, tel que recommande par le fournisseur.
Apres la digestion, l’ADN est isole par extraction au phenol et au chloroforme,
precipite a l’acetate de sodium et a l’ethanol (section 2.3.3), et resuspendu dans 20 µl
de tampon TE 1 X. Pour chaque experience, un controle sans enzyme a ete realise.
Les produits des reactions de digestion et de la reaction controle sont analyses par
electrophorese sur gel d’agarose (section 2.3.10).
2.3.8 Ligature de fragments d’ADN
Les fragments d’ADN obtenus par restriction ont ete ligatures a l’aide de la
ligase du phage T4. L’enzyme a ete obtenue aupres de la compagnie Invitrogen. La
composition des melanges reactionnels est donnee au tableau 2.10. La proportion des

Chapitre 2. Materiel et methodes 32
Tab. 2.10 – Composition des melanges reactionnels pour la ligature de l’ADNTris • Cl 50 mM
MgCl2 10 mM
ATP 1 mM
DTT 1 mM
PEG-8000 5 %
ADN total 1,0 µg
ADN Ligase T4 1 U
pH 7,5
Volumea 20 µla Ajuste avec de l’eau NANOpure sterile.
Tab. 2.11 – Conditions pour les reactions de ligature de l’ADNF1a F2 `F1
b `F2 Site(s) Ratio −PO3−4
c
pb pb F1 : F2
’yadB-pncB’ ’dksA-yadB’ 448 368 NotI 1 : 1 Non
pTOF24∆aph ’dksA-yadB’-’yadB-pncB’ 5619 800 PstI SalI 1 : 3 Non
pTOF-YA FRT-lacZ -aph-FRT 6413 4578 NotI 1 : 2 Ouia F1 : fragment 1 ; F2 : fragment 2
b` : longueur du fragment
c−PO3−
4: dephosphorylation du fragment 1
deux fragments d’ADN ajoutes au milieu est variable et depend de la taille de chacun
des fragments. Ces proportions sont indiquees au tableau 2.11. Ce tableau indique
egalement quels fragments ont ete prealablement dephosphoryles (section 2.3.9). Tous
les melanges reactionnels de ligature ont ete incubes une heure a 25 ◦C. Ces conditions
correspondent a celles suggerees dans la litterature [55].
L’ADN resultant de la ligature des regions flanquant le gene yadB (section 2.4.1)
a ete isole par extraction au phenol et au chloroforme, precipite a l’acetate de sodium
et a l’ethanol, et resuspendu dans 20 µl de tampon TE 1 X. L’ADN circulaire produit
par les autres reactions de ligature a ete utilise directement pour transformer E. coli
XL1-Blue ou TOP10 (section 2.3.12). Pour chaque reaction de ligature, une reaction
controle sans ligase T4 a ete realisee. Les produits de toutes les reactions, incluant
les controles, ont ete analyses par electrophorese sur gel d’agarose (section 2.3.10).
Lorsque l’ADN a ete utilise directement pour transformer E. coli (section 2.3.12), le
produit de la reaction controle a ete utilise comme controle negatif lors de la reaction
de transformation.

Chapitre 2. Materiel et methodes 33
Tab. 2.12 – Composition des melanges reactionnels pour la dephosphorylation de l’ADNNaCl 100 mM
Tris • Cl 50 mM
MgCl2 10 mM
DTT 1 mM
ADN ∼ 1 µg
CIP 0,5 U/µg ADN
pH 7,9
Volumea 20 µla Ajuste avec de l’eau NANOpure sterile.
2.3.9 Dephosphorylation de fragments d’ADN
Le vecteur recombinant dans lequel la cassette FLK2 a ete inseree a ete
dephosphoryle avant la ligature a l’aide de phosphatase alcaline de l’intestin de
veau (CIP) selon une procedure publiee [56]. L’enzyme provient de la compagnie
New-England BioLabs. La composition du melange reactionnel est indiquee au
tableau 2.12. Le melange reactionnel a ete incube une heure a 37 ◦C. L’ADN
dephosphoryle a ete isole par extraction au phenol et au chloroforme, precipite a
l’acetate de sodium et a l’ethanol (section 2.3.3), et resuspendu dans 20 µl de tampon
TE 1 X.
Un controle negatif sans CIP a ete realise. Les produits de la reaction et de
son controle ont tous les deux ete utilises dans une reaction de ligature de l’ADN
(section 2.3.8). Les deux melanges de ligature ont ensuite ete utilises pour transformer
E. coli (section 2.3.12).
La dephosphorylation n’a pas ete utilisee pour les deux autres reactions de ligature.
Dans le cas de la fusion des regions flanquant yadB, ce n’etait pas necessaire car l’ADN
lineaire obtenu a ensuite ete amplifie par PCR ; une tres faible quantite d’ADN ligature
etait donc suffisante, et le fragment d’interet amplifie a ete purifie sur gel d’agarose. Dans
le cas de l’insertion des regions flanquant yadB dans le vecteur pTOF24, la technique
du clonage positionnel a ete employee ; le vecteur ne pouvait donc pas se refermer sur
lui-meme.
2.3.10 Electrophorese sur gel d’agarose
La technique d’electrophorese sur gel d’agarose [44] a ete utilisee pour verifier
la presence de plasmides dans des souches de E. coli (section 2.3.4), pour analyser

Chapitre 2. Materiel et methodes 34
les resultats des amplifications par PCR (section 2.3.6), pour confirmer la nature de
fragments de restriction (section 2.3.7) et pour purifier ces fragments, ainsi que pour
verifier le succes des reactions de ligature (section 2.3.8) et de dephosphorylation
(section 2.3.9).
Toutes les electrophoreses ont ete realisees avec des gels d’agarose 1,0 % prepares
dans le tampon TBE 1 X. Les echantillons (∼100 ng d’ADN) sont prepares dans un
volume total de 15 µl dans le tampon de charge pour gel d’agarose 1 X (section 2.1).
La migration se fait a 7 V/cm dans le tampon TBE 1 X, jusqu’a ce que le bleu de
bromophenol present dans le tampon de charge ait parcouru les deux tiers du gel de
8,5 cm (environ 30 minutes). Le gel est ensuite trempe dans une solution de bromure
d’ethidium 2,0 µg/ml et agite faiblement (50 rpm) pendant 10 minutes, puis rince a
l’eau froide pour eliminer l’exces de bromure d’ethidium.
L’ADN est ensuite visualise par exposition au rayonnement U.V. d’un transillu-
minateur, et le gel est photographie. Ces etapes ont ete realisees avec un appareil de
photodocumentation Chemi Genius2 Bio Imaging System, de la compagnie Syngene.
2.3.11 Purification d’ADN a partir de gels d’agarose
Certains fragment d’ADN resultant de reactions de digestion (section 2.3.7)
ou d’amplification PCR (section 2.3.6) ont ete purifies sur gel d’agarose apres leur
resolution par electrophorese et leur visualisation au bromure d’ethidium tel que decrit
ci-haut (section 2.3.10). Les bandes d’interet ont ete excisees a l’aide d’un scalpel,
puis la trousse commerciale QIAquick Gel Extraction Kit de QIAGEN a ete utilisee
pour recuperer l’ADN.
Le protocole suggere par QIAGEN a ete utilise [46]. On ajoute au morceau
d’agarose excise du gel un tampon contenant du thiocyanate de guanidinium (agent
denaturant). La solution contenant l’agarose est chauffee a 50 ◦C pour dissoudre l’aga-
rose et ainsi liberer l’ADN. L’ADN en solution est depose sur une colonne contenant
une resine de silice sur laquelle l’ADN s’adsorbe a bas pH et a concentration saline
elevee. Apres le passage sur la colonne d’un tampon de lavage, l’ADN purifie est elue a
l’aide d’un tampon de pH eleve (8,5) dont la concentration en sels est faible.

Chapitre 2. Materiel et methodes 35
2.3.12 Transformation de cellules par de l’ADN plasmidique
Afin de pouvoir conserver et produire les plasmides recombinants pTOF-YA et
pTOF-YB crees dans le cadre de cette recherche (section 2.4), des cellules competentes
de E. coli (XL1-Blue ou TOP10) ont ete transformees avec ces plasmides (obtenus par
ligature de fragments d’ADN). pTOF-YB a par la suite ete utilise pour transformer
des cellules competentes de la souche TP8503, afin de remplacer, sur le chromosome
bacterien, le gene yadB par la cassette FLK2 (section 2.5). Des cellules competentes
de la souche POK1 ont ete transformees par pCP/20 afin d’obtenir une souche ∆yadB
(section 2.6).
Les cellules de E. coli XL1-Blue, TP8503 et POK1 ont ete traitees au DMSO pour
les rendre competentes, d’apres le protocole suivant [45] :
1. Ensemencer 50 ml de milieu SOB avec un inoculum provenant d’une culture
saturee (au moins 18 heures de croissance).
2. Incuber a 18 ◦C jusqu’a ce que la densite optique atteigne 0,6 (a une longueur
d’onde de 600 nm). Placer dans la glace pendant 10 minutes.
3. Prelever 20 ml de culture et centrifuger a 2 500 × g a 4 ◦C pendant 5 minutes.
4. Resuspendre dans 6,4 ml de tampon TB froid.
5. Placer dans la glace pendant 10 minutes.
6. Centrifuger a 2 500 × g a 4 ◦C pendant 5 minutes.
7. Resuspendre dans 1,6 ml de tampon TB froid contenant 7 % de DMSO.
8. Placer dans la glace pendant 10 minutes.
9. Aliquoter des portions de 100 µl dans des microtubes froids, et congeler aussitot
dans l’azote liquide.
10. Conserver a -80 ◦C. Utiliser a l’interieur d’un delais de 60 jours.
A cause du faible rendement de la reaction de ligature produisant pTOF-YB, il
n’a pas ete possible de transformer des cellules XL1-Blue traitees au DMSO par ce
plasmide. En lieu et place, des cellules hypercompetentes (1 • 109 cfu/µg ADN) E. coli
TOP10 fournies par Invitrogen ont ete utilisees. Toutes les transformations (XL1-Blue
par pTOF-YA, TOP10 par pTOF-YB, TP8503 par pTOF-YB, POK1 par pCP20) ont
ete realisees selon un protocole publie [45] modifie :
1. Degeler un microtube de cellules competentes a la temperature de la piece.

Chapitre 2. Materiel et methodes 36
2. Ajouter au moins 1,0 ng d’ADN plasmidique dans un volume d’au moins 20 µl. S’il
s’agit du produit d’une reaction de ligature, diluer le melange reactionnel 5 X avec
de l’eau NANOpure sterile avant de l’ajouter aux cellules. Melanger delicatement
le tube.
3. Conserver sur la glace pendant 30 minutes.
4. Plonger dans un bain a 42 ◦C pendant 30 secondes. Ne pas agiter.
5. Plonger dans la glace pendant 5 minutes. Ne pas agiter.
6. Ajouter 0,8 ml de milieu SOC prechauffe a 30 ◦C (et non pas a 37 ◦C, afin de
permettre la replication des plasmides portant une origine de replication thermo-
sensible).
7. Incuber a 30 ◦C pendant une heure avec une agitation de 250 rpm.
8. Etaler des volumes de 10 µl et 100 µl sur les geloses selectives appropriees.
Au moins trois clones sont repiques en milieu liquide pour chaque transformation.
La presence du plasmide d’interet dans ces transformants est verifiee par preparation
de l’ADN plasmidique. Apres cette verification, des echantillons des transformants
selectionnes sont prepares et conserves a -80 ◦C tel que decrit precedemment
(section 2.2.3). Pour chaque experience de transformation, deux controles sont
realises : un controle negatif, auquel aucun ADN plasmidique n’est ajoute, et un
controle positif, realise en ajoutant aux cellules competentes 1,0 ng du plasmide
controle pUC19 (procure chez Invitrogen). 50 µl du controle negatif est etale sur une
gelose selective identique a celle utilisee pour la reaction de transformation. 50 µl du
controle positif est etale sur une gelose LB-Amp.
2.4 Construction du vecteur pTOF-YB
Cette section explique en details la construction du vecteur pTOF-YB ayant
servi au remplacement du gene yadB par la cassette FLK2 sur le chromosome de
E. coli TP8503. Cette construction a ete faite selon une methode publiee [48] se
deroulant en trois etapes illustrees a la figure 2.2 et detaillees dans les sous-sections
suivantes. Les regions flanquant le gene yadB sont d’abord amplifiees par PCR et
ligaturees (section 2.4.1). Ces regions sont ensuite inserees dans le vecteur pTOF24
(section 2.4.2). Enfin, la cassette FLK2 est excisee du vecteur pTOF30 et inseree
entre les portions aval et amont des regions flanquantes (section 2.4.3). Les plasmides,
amorces PCR et techniques utilises pour realiser cette construction ont ete decrits plus
tot (section 2.3).

Chapitre 2. Materiel et methodes 37
Fig. 2.2 – Construction du vecteur pTOF-YB

Chapitre 2. Materiel et methodes 38
2.4.1 Amplification des regions flanquant yadB
Le plasmide pET-dksA-yadB contient une courte region du chromosome de E.
coli K12, incluant les genes dksA (situe en amont de yadB) et yadB. Cette region
chromosomique comprend egalement la portion 5’ du gene pncB (situe en aval de
yadB). Dans la premiere etape de la construction de pTOF-YB (figure 2.2 a1), deux
sequences d’environ 400 paires de bases contenues dans cette region ont ete amplifees
independamment a partir de pET-dksA-yadB. La premiere de ces sequences (designee
N puisqu’elle contient la portion du gene yadB codant pour la partie N-terminale de
la GluQRS) comprend environ 300 paires de bases de l’extremite 3’ de dksA, la region
intergenique dksA-yadB et environ 100 paires de bases de l’extremite 5’ de yadB. Elle a
ete amplifiee a l’aide des amorces Ni et No. La deuxieme sequence (designee C ) contient
environ 180 paires de bases de l’extremite 3’ de yadB, la region intergenique yadB -pncB
et environ 160 paires de bases de l’extremite 5’ de pncB. Elle a ete amplifiee grace aux
amorces Ci et Co. Les extremites du gene yadB ont ete conservees afin de ne pas modifier
les eventuels signaux transcriptionnels qui peuvent etre presents dans ces regions.
Les amorces utilisees pour l’amplification de ces deux regions ont ete concues
pour inserer le site de restriction PstI a l’extremite 5’ de la sequence N (portion 5’ et
region en amont de yadB) et le site SalI a l’extremite 3’ de la sequence C (portion
3’ et region en aval de yadB). Les amorces ont egalement ete concues pour inserer des
sites de restriction NotI a l’extremite 3’-terminale de la sequence N et a l’extremite
5’-terminale de la sequence C.
Les sequences N et C ont ensuite ete digerees avec l’endonuclease de restriction
NotI puis ligaturees (figure 2.2 a2). Trois produits de ligature sont possibles : N-C,
N-N et C-C. Le melange de ces trois sequences a ete utilise comme gabarit dans une
seconde reaction PCR (figure 2.2 a3), ou les amorces No et Co ont ete utilisees. Les
trois fragments du melange ont ete amplifies, puis N-C a ete purifie par electrophorese
sur gel d’agarose.
2.4.2 Clonage des regions flanquantes dans pTOF24
Le vecteur pTOF24 (figure 2.1 a) a ete digere avec PstI et SalI pour en retirer
le fragment aph. pTOF24∆aph a ete purifie sur gel d’agarose, puis ligature avec les
regions flanquantes N-C (aussi digerees avec PstI et SalI) pour construire pTOF-YA
(figure 2.2 b).

Chapitre 2. Materiel et methodes 39
pTOF-YA a ete utilise pour transformer E. coli XL1-Blue. Les transformants ont
ete selectionnes sur gelose LB-Chl. Le remplacement du fragment aph par le locus delete
et les regions flanquantes de yadB a ete confirme par la sensibilite des transformants a
la kanamycine.
2.4.3 Insertion de la cassette FLK2
La cassette FLK2 a ete excisee de pTOF30 (figure 2.1 b) par digestion avec
l’endonuclease NotI, puis purifiee sur gel d’agarose. pTOF-YA a aussi ete digere par
NotI, et dephosphoryle. La cassette et le vecteur ont ensuite ete ligatures (figure 2.2 c).
La cassette peut s’inserer soit dans la meme direction que yadB, soit dans la direction
opposee. L’objectif de la construction etant d’obtenir une region homologue au chro-
mosome ou le rapporteur lacZ est sous le controle des elements transcriptionnels de
yadB, il est necessaire que la cassette FLK2 soit inseree de telle facon que ses elements
genetiques soient dans le meme sens que ceux des regions flanquantes du gene yadB. La
construction ou la cassette est inseree dans le sens voulu est designee pTOF-YB.
La reaction de ligature contenant pTOF-YB et l’autre construction (ou la cassette
FLK2 est dans le sens non approprie) a ete utilisee pour transformer des cellules de E.
coli TOP10 hypercompetentes (le faible rendement de la reaction de ligature n’ayant
pas permis de transformer avec succes des cellules de E. coli XL1-Blue traitees au
DMSO pour les rendre competentes). Les transformants ont ete selectionnes sur gelose
LB-Chl-Kan. La resistance des transformants a la kanamycine confirme la presence de
la cassette FLK2 dans le vecteur.
Afin de selectionner un transformant ou la cassette FLK2 a ete inseree dans le
vecteur dans le sens desire, l’ADN plasmidique extrait de plusieurs transformants a
ete analyse par cartographie de restriction avec l’enzyme PstI : l’ADN plasmidique
est digere et les fragments resultant sont resolus par electrophorese sur gel d’agarose.
Tel qu’indique a la figure 2.3, la position des sites de restriction PstI sur le vecteur
pTOF-YB est telle que 3 fragments de 1235, 3717 et 6070 paires de bases resulteront
de la digestion. Par ailleurs, le patron de digestion du plasmide ou la cassette FLK2
est integree dans le sens non desire contiendra 3 bandes de 500, 1235 et 9287 paires de
bases.
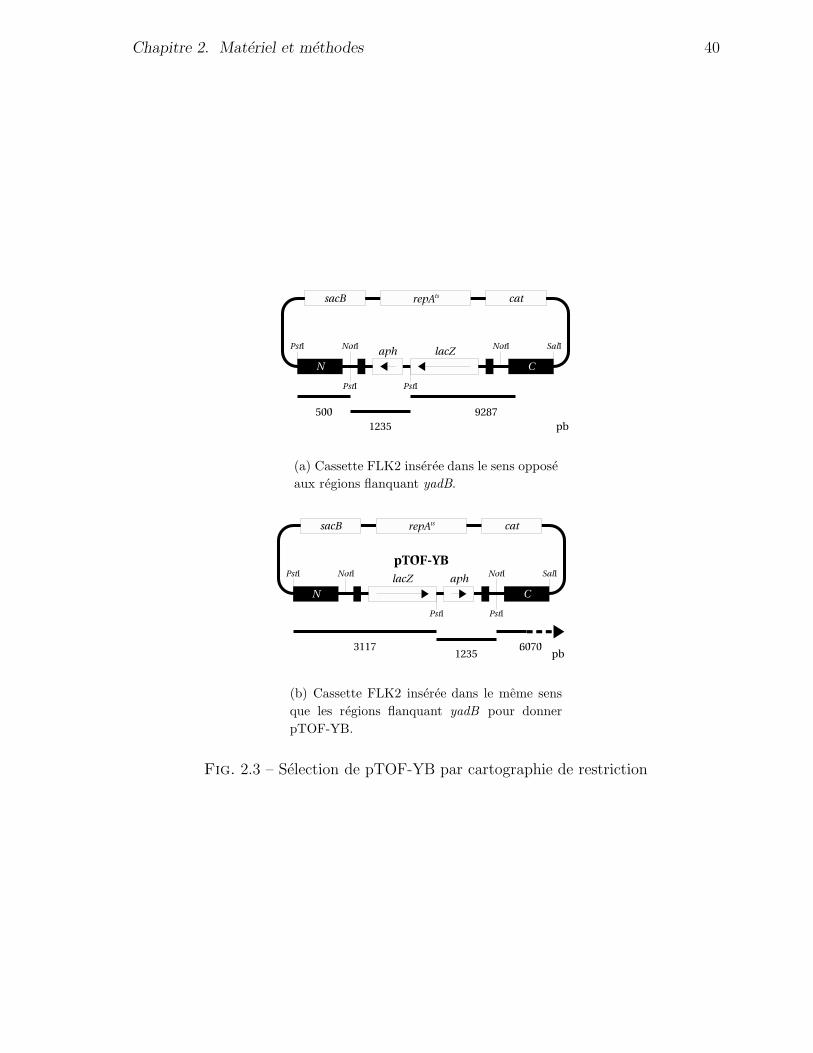
Chapitre 2. Materiel et methodes 40
(a) Cassette FLK2 inseree dans le sens oppose
aux regions flanquant yadB.
(b) Cassette FLK2 inseree dans le meme sens
que les regions flanquant yadB pour donner
pTOF-YB.
Fig. 2.3 – Selection de pTOF-YB par cartographie de restriction

Chapitre 2. Materiel et methodes 41
2.5 Remplacement de yadB par la cassette FLK2
Le vecteur pTOF-YB, dont la construction a ete detaillee a la section precedente,
a ete utilise pour remplacer le gene yadB par la cassette FLK2 sur le chromosome de
E. coli. Ce remplacement a ete fait dans la souche TP8503, ou l’operon lac est delete.
L’absence de lacZ dans cette souche permet d’utiliser la copie de ce gene presente sur
la cassette FLK2 comme un indicateur de l’expression du gene remplace par la cassette,
en l’occurrence yadB. La souche resultante est designee POK1.
Le remplacement est fait de maniere a laisser les deux extremites du gene yadB
intactes (c’est pourquoi ces extremites ont ete amplifiees lors de la creaction de pTOF-
YB (section 2.4.1)). Ainsi, les eventuels elements transcriptionnels de yadB ne sont pas
modifies, et lacZ est sous le controle de ces elements. Le gene lacZ de la cassette FLK2
possede son propre site de liaison au ribosome, et n’est pas suivi d’un terminateur, ce qui
minimise l’influence de la cassette FLK2 sur la transcription des elements genetiques
en aval. La fin du gene yadB etant encore presente sur le chromosome, un eventuel
terminateur n’est pas delete lors de l’insertion de la cassette.
Le remplacement a ete fait en exploitant le phenomene de recombinaison homo-
logue medie par la recombinase RecA. La presence de regions homologues au chromo-
some sur le vecteur pTOF-YB a permis d’utiliser RecA pour integrer la casette FLK2
au chromosome par recombinaison.
Les trois etapes necessaires au remplacement sont detaillees dans les sous-sections
suivantes et illustrees a la figure figure 2.4. Le plasmide pTOF-YB est d’abord uti-
lise pour transformer E. coli TP8503 (section 2.5.1). Un premier phenomene de
recombinaison homologue conduit a l’integration du plasmide dans le chromosome
(section 2.5.2). Une seconde recombinaison excise le plasmide et le gene yadB, ne
laissant que la cassette FLK2 dans le chromosome ; le plasmide est immediatement
perdu (section 2.5.3). A chacune de ces etapes, les elements genetiques provenant de
pTOF-YB permettent la selection des cellules. L’integration de la cassette est finalement
confirmee par PCR (section 2.5.4).
2.5.1 Transformation de E. coli TP8503
Le vecteur pTOF-YB a ete utilise pour transformer la souche E. coli TP8503
(figure 2.4 a). Les transformants ont ete selectionnes sur gelose LB-Chl-Kan a 30 ◦C
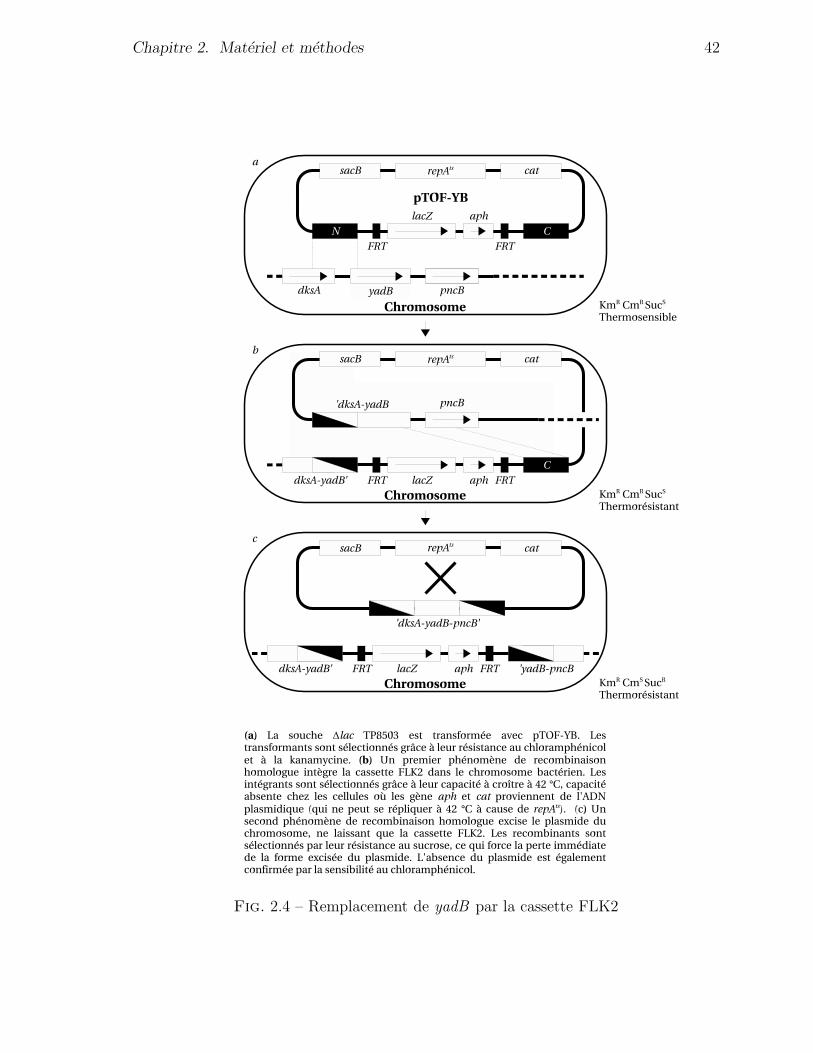
Chapitre 2. Materiel et methodes 42
Fig. 2.4 – Remplacement de yadB par la cassette FLK2

Chapitre 2. Materiel et methodes 43
(afin de permettre la replication du plasmide, qui ne peut se faire a 37 ◦C a cause de
l’origine de replication thermosensible repAts).
2.5.2 Integration du plasmide au chromosome
Une culture de TP8503/pTOF-YB (incubee a 30 ◦C) a ete utilisee pour strier des
geloses LB-Chl-Kan. Ces dernieres ont ete incubees a 42 ◦C. La replication de pTOF-
YB etant impossible a 42 ◦C, les seules cellules a conserver les phenotypes CmR et KmR
sont celles ou le plasmide s’est integre au chromosome par recombinaison homologue
(figure 2.4 b). Les integrants produisent des colonies de taille normale, alors que
les autres cellules ne produisent que des micro-colonies. Un integrant a ete repique
en milieu liquide, puis strie a nouveau sur gelose, toujours en maintenant la pression
selective (temperature de 42 ◦C, antibiotiques chloramphenicol et kanamycine).
2.5.3 Excision et perte du plasmide
Un integrant a ete repique en milieu liquide LB et incube a 30 ◦C jusqu’a satu-
ration, ce qui retire toute pression selective et permet a un deuxieme phenomene de
recombinaison homologue d’avoir lieu (figure 2.4 c).
Des dilutions sequentielles de la culture ont ete etalees sur geloses LB-Suc-Kan,
lesquelles ont ete incubees a 42 ◦C. Le sucrose est toxique pour les cellules ou le plasmide
est encore integre au chromosome, a cause de la presence du gene de sensibilite sacB.
Seules les cellules ayant excise le plasmide de leur chromosome par recombinaison
peuvent survivre. La temperature elevee ne permet pas la replication de la forme excisee
du plasmide, et puisque sacB est present dans ce plasmide, celui-ci est toxique pour
les cellules. Par consequent, les seules colonies observables sont celles ou le plasmide a
ete excise du chromosome puis elimine de la cellule. L’antibiotique kanamycine assure
la selection de cellules ou la cassette FLK2 est encore presente. La sensibilite au
chloramphenicol a ete utilisee pour confirmer l’absence du plasmide.
2.5.4 Confirmation du remplacement par PCR
Un clone KmR CmS a ete selectionne. Afin de demontrer le remplacement du locus
yadB par la cassette FLK2, l’ADN genomique du clone a ete utilise comme gabarit dans

Chapitre 2. Materiel et methodes 44
Fig. 2.5 – Confirmation du remplacement de yadB par PCR
deux reactions d’amplification par PCR tel qu’indique a la figure 2.5. La premiere
reaction utilise les amorces Next et F1. Next s’hybride a l’interieur du gene dksA, alors
que F1 s’hybride dans le gene lacZ present sur la cassette FLK2 ; la region amplifiee
comprend une partie du gene dksA et la portion N-terminale de la cassette FLK2. La
region ou s’hybride Next est situee en amont de la region ou s’hybride l’amorce No (qui
a ete utilisee pour amplifier la region en amont de yadB lors de la construction de
pTOF-YB). Next a ete concue ainsi pour qu’elle ne puisse pas s’hybrider a pTOF-YB
et l’amplifier. Ainsi, l’amplification par PCR ne peut avoir lieu que si la cassette est
presente dans le chromosome. La deuxieme reaction utilise les amorces Cext et F2, qui
s’hybrident respectivement a l’interieur de pncB (sur le chromosome) et de aph (sur
la cassette) pour amplifier la jonction C-terminale entre la cassette et le chromosome.
L’amorce Cext a ete concue pour s’hybrider en aval de la region ou s’hybride Co afin
qu’elle ne puisse pas amplifier l’ADN de pTOF-YB. Les fragments anticipes ont des
longueurs de 748 (portion N-terminale de FLK2) et de 1546 (portion C-terminale)
paires de bases. Des controles negatifs ont ete realises avec les meme amorces et l’ADN
genomique de TP8503. La souche obtenue par remplacement de yadB par la cassette
FLK2 dans TP8503 est designee E. coli POK1.
2.6 Deletion du gene yadB
La cassette FLK2 integree au chromosome dans la souche POK1 a ete excisee par
recombinaison homologue specifique pour ne laisser qu’une courte sequence de 93 paires
de bases qui ne change pas le cadre de lecture dans le chromosome. La souche ∆yadB
ainsi obtenue est nommee POK2.

Chapitre 2. Materiel et methodes 45
Fig. 2.6 – Deletion de yadB par excision de la cassette FLK2
L’excision de la cassette est rendue possible grace a la presence de regions FRT aux
deux extremites de la cassette FLK2. Ces regions contiennent une sequence reconnue
specifiquement par la recombinase Flp de la levure. Cette enzyme catalyse la recom-
binaison homologue des regions FRT, ce qui conduit a l’excision de la cassette sous
forme d’ADN circulaire incapable de replication, tel que detaille dans les sous-sections
suivantes. La figure 2.6 illustre le processus.
2.6.1 Transformation de POK1 par pCP20
Le vecteur pCP20, qui exprime constitutivement la recombinase Flp de la levure
(gene flp), a ete utilise pour transformer E. coli POK1 (figure 2.6 a). Les transfor-

Chapitre 2. Materiel et methodes 46
(a) Amplification du locus yadB dans E.
coli TP8503.
(b) Amplification de la region du locus
yadB delete dans E. coli POK2.
Fig. 2.7 – Confirmation de la deletion de yadB par PCR
mants ont ete selectionnes sur geloses LB-Amp-Chl-Kan a 30 ◦C (pCP20 contient les
genes bla et cat de resistance a l’ampicilline et au chloramphenicol, ainsi qu’une origine
de replication thermosensible).
2.6.2 Excision de la cassette FLK2 et perte de pCP20
Un transformant a ete strie sur une gelose LB incubee a 42 ◦C, ce qui assure la perte
du plasmide. Une dizaine de colonies ont ete repiquees sur des geloses LB, LB-Amp,
LB-Chl et LB-Kan incubees a 30 ◦C. Les clones provenant d’une cellule ou la cassette
FLK2 a ete excisee et ou pCP20 a ete perdu sont sensibles aux trois antibiotiques
(figure 2.6 b).
2.6.3 Confirmation de la deletion par PCR
La deletion du locus yadB dans la souche POK2 a ete confirmee par PCR. De
l’ADN genomique provenant de TP8503 et de POK2 a ete employe comme gabarit
dans deux reactions d’amplification utilisant les amorces No et Co. Tel qu’illustre a la
figure 2.7, le fragment PCR obtenu avec l’ADN de POK2 sera plus court (1012 paires
de bases) que celui obtenu avec l’ADN de TP8503 (1547 paires de bases), ce qui est du
a la deletion de yadB, dont il ne reste que les extremites. (Le fragment amplifie chez
POK2 contient aussi la courte cicatrice de 93 paires de bases laissee par la recombinaison
catalysee par la recombinase Flp).

Chapitre 2. Materiel et methodes 47
2.7 Dosage enzymatique de l’activite β-galactosidase
La souche E. coli POK1, dont la construction a ete detaillee a la section 2.5, a
servi a l’etude de l’expression du gene yadB dans differentes conditions de croissance
et de stress. Ces conditions seront presentees en detail a la section 2.8. Les mesures
de l’expression ont ete rendues possibles grace au gene rapporteur lacZ.
L’expression de lacZ et, donc, de yadB, a ete determinee par dosage de l’activite
β-galactosidase en culture liquide, tel que decrit dans la litterature [43]. Des echantillons
de cultures ont ete preleves a des intervalles determines tel que decrit a la section 2.8.
Le protocole suivant a ete utilise pour determiner l’activite β-galactosidase des cultures
au moment de l’echantillonnage :
1. Prelever un echantillon de 1,6 ml de culture et le placer sur la glace afin de stopper
la croissance cellulaire.
2. Prelever 1,0 ml d’echantillon pour en determiner la densite cellulaire (densite
optique a 600 nm).
3. Prelever 0,5 ml d’echantillon, et ajouter a 0,5 ml de tampon Z 2 X (section 2.1)
froid dans un microtube de 1,5 ml. Le tampon Z ajuste le pH a 7,0 (l’activite
β-galactosidase etant mesuree a ce pH) et limite l’oxydation de l’enzyme grace au
2-mercaptoethanol.
4. Ajouter 2 gouttes de chloroforme et 1 goutte de SDS 0,1 % et vortexer vigoureu-
sement pendant 10 secondes. Le SDS et le chloroforme dissolvent partiellement les
membranes cellulaires, ce qui libere les enzymes des cellules. Les faibles concen-
trations de chloroforme et de SDS utilisees ne sont pas suffisantes pour denaturer
la β-galactosidase.
5. Centrifuger pendant 2 minutes a 10 000 × g. Les debris cellulaires et l’ADN sont
culotes, laissant les enzymes en solution.
6. Transferer 800 µl de la phase superieure (aqueuse) dans une eprouvette de verre
sterile.
7. Incuber 5 minutes a 28 ◦C.
8. Ajouter 200 µl de reactif ONPG (section 2.1). Agiter. Noter le moment de
l’ajout. L’ONPG est un analogue chromogenique du substrat naturel de la β-
galactosidase, le lactose. L’hydrolyse de l’ONGP libere une molecule d’o-nitro-
phenol, qui absorbe la lumiere de facon importante a 420 nm.
9. Lorsqu’une coloration jaune s’est developpee, mesurer l’absorbance a 420 nm et
noter le moment de cette mesure.

Chapitre 2. Materiel et methodes 48
Le niveau d’activite β-galactosidase est calcule d’apres la formule suivante :
1000 × D.O.λ=420
t × v × D.O.λ=600
= Activite β-galactosidase
Ou t est le temps en minutes et v le volume de culture utilise pour le dosage. L’activite
ainsi calculee est exprimee en unites Miller.
Ce niveau d’expression est directement comparable a celui d’un autre echantillon
et d’une autre culture, puisqu’il est exprime independamment du temps de reaction
et de la densite cellulaire de la culture. Une activite d’environ 1 Miller correspond a
l’expression constitutive de lacZ dans l’operon lac dans une culture non induite. Une
activite d’environ 1 000 Miller correspond a l’expression de lacZ dans une culture ou
l’operon lac est induit au maximum.
Les mesures de densites cellulaires ont ete realisees avec un spectrophotometre
GeneQuant Pro (Biochrom). Les mesures d’absorbance de l’o-nitrophenol ont ete
realisees avec un spectrophotometre Ultrospec III (Pharmacia). Dans les deux cas, des
cuvettes de plastique jetables BioRad ont ete utilisees.
2.8 Conditions d’expression et effets de la deletion
du gene yadB
La souche POK1, construite par remplacement de yadB par la cassette FLK2
(section 2.5), a ete utilisee pour etudier l’expression du gene yadB dans diverses
conditions de croissance et de stress. Des differences phenotypiques dues a la deletion
de yadB ont egalement ete recherchees, par comparaison des cellules POK1 et TP8503.
Si des variations de phenotypes avaient ete observees, la souche POK2, ou yadB a
ete delete (section 2.6), aurait servi a confirmer ces differences. (L’absence de cas-
sette FLK2 dans POK2 permet d’exclure la possibilite qu’une difference phenotypique
observee entre POK1 et TP8503 soit due a la presence de la cassette.)
La technique utilisee pour la mesure du niveau d’expression de yadB a deja ete
decrite (section 2.7). Les conditions etudiees sont les suivantes : croissance en condi-
tions optimales (milieu riche, 37 ◦C) ou en milieu minimal (a 37 ◦C), croissance a des
temperatures non optimales (32 ◦C et 42 ◦C, en milieu riche) et croissance interrompue
par un choc thermique, un choc osmotique, un choc de pH ou un choc oxydatif. Les
procedures detaillees pour chacune de ces etudes sont presentees dans les sous-sections
suivantes.

Chapitre 2. Materiel et methodes 49
Dans tous les cas, 50 ml de milieu a ete inocule avec 10 µl d’une preculture
saturee realisee dans des conditions identiques a celles qui seront employees pour la
culture. Les experiences sont toujours realisees avec les souches POK1 et TP8503 (cette
derniere servant de controle negatif pour le dosage de lacZ et de repere pour detecter
les variations de phenotypes dues a la deletion de yadB). Toutes les experiences ont
ete realisees en triplicata, a l’exception de celles impliquant le choc de pH et le choc
oxydatif, qui n’ont ete realisees qu’une seule fois.
En plus de ces experiences, la thermosensibilite de la souche TP8503 et celle de
la souche POK2 ont ete comparees en incubant des geloses LB striees par ces souches
a des temperatures de 4 ◦C, 22 ◦C, 30 ◦C, 37 ◦C, 42 ◦C, 48 ◦C et 56 ◦C.
2.8.1 Croissance en conditions optimales
Les cultures sont realisees en milieu LB, soumises a une agitation de 250 rpm et
incubees a 37 ◦C tel que detaille precedemment (section 2.2.1). A chaque heure, la
densite optique des cultures est mesuree, et l’activite β-galactosidase est dosee pour
determiner le niveau d’expression de yadB. Les mesures sont interrompues lorsque les
cultures atteignent la saturation. Une mesure supplementaire est realisee apres 24 heures
de croissance.
2.8.2 Croissance en milieu minimal
Les cultures sont realisees en milieu M9 et incubees a 37 ◦C. A chaque heure, la
densite optique des cultures est mesuree, et l’activite β-galactosidase est dosee pour
determiner le niveau d’expression de yadB. Les mesures sont interrompues lorsque les
cultures atteignent la saturation. Une mesure supplementaire est realisee apres 24 heures
de croissance.
2.8.3 Croissance a des temperatures non optimales
Les cultures sont realisees en milieu LB et incubees a 42 ◦C ou a 32 ◦C. A chaque
heure, la densite optique des cultures est mesuree, et l’activite β-galactosidase est
dosee pour determiner le niveau d’expression de yadB. Les mesures sont interrompues

Chapitre 2. Materiel et methodes 50
lorsque les cultures atteignent la saturation. Une mesure supplementaire est realisee
apres 24 heures de croissance.
2.8.4 Choc thermique
Les cultures sont realisees en milieu LB et incubees a 37 ◦C. A chaque heure, la
densite optique des cultures est mesuree, et l’activite β-galactosidase est dosee pour
determiner le niveau d’expression de yadB. Lorsque la densite optique atteint 1,0, la
culture est refroidie dans un bain d’eau glacee pendant une heure, puis sa temperature
est amenee a 42 ◦C par immersion dans un bain d’eau (a 42 ◦C) pendant deux minutes.
La culture est ensuite a nouveau incubee a 37 ◦C. La densite optique et l’activite β-
galactosidase sont mesurees immediatement apres le choc thermique, puis a intervalles
de 15 minutes pendant une heure. Par la suite, la densite cellulaire et l’activite β-
galactosidase sont mesurees a toutes les heures. Les mesures sont interrompues lorsque
les cultures atteignent la saturation. Une mesure supplementaire est realisee apres
24 heures de croissance.
2.8.5 Choc osmotique
Les cultures sont realisees en milieu LB et incubees a 37 ◦C. Lorsque la densite
optique atteint 1,0, les cellules sont culotees par centrifugation a 2 500 × g pendant 15
minutes, puis resuspendues dans 50 ml de sucrose 20 %. Apres 10 minutes d’incubation,
les cellules sont a nouveau culotees, puis resuspendues dans 50 ml d’eau distillee. Apres
10 minutes d’incubation, les cellules sont finalement culotees et resuspendues dans
50 ml de milieu LB. La densite optique et l’activite β-galactosidase sont mesurees
immediatement apres le choc osmotique, puis a intervalles de 15 minutes pendant une
heure. L’experience est alors terminee.
2.8.6 Choc de pH
Contrairement aux autres experiences ou deux cultures sont realisees (une pour
la souche POK1, une autre pour TP8503), l’etude de l’effet du choc de pH requiert 5
paires de cultures de E. coli POK1 et TP8503. (Une de ces paires sert de controles.)
Les cultures sont realisees en milieu LB et incubees a 37 ◦C. Lorsque la densite optique
atteint 1,0, le pH des cultures est ajuste a 5, 6, 8 ou 9. Une culture de POK1 et une autre

Chapitre 2. Materiel et methodes 51
de TP8503 sont ajustees a chacune de ces valeurs de pH. (Le pH des cultures controles
est d’environ 7 et n’est pas ajuste.) La densite optique et l’activite β-galactosidase sont
mesurees immediatement apres le choc de pH, puis a intervalles de 15 minutes pendant
une heure. L’experience est alors terminee. Pour toutes les valeurs de pH utilisees, le pH
du milieu reactionnel (tampon Z) utilise pour mesurer l’expression de yadB a ete verifie
pour s’assurer qu’il n’ait pas ete modifie substantiellement par le pH de la culture.
2.8.7 Choc oxydatif
Contrairement aux autres experiences ou deux cultures sont realisees (une pour
la souche POK1, une autre pour TP8503), l’etude de l’effet du choc oxydatif requiert
6 paires de cultures de E. coli POK1 et TP8503. (Une de ces paires sert de controles.)
Les cultures sont realisees en milieu LB et incubees a 37 ◦C. Lorsque la densite optique
atteint 1,0, la concentration en H2O2 est ajustee a 2,5 mM, 5 mM, 10 mM, 20 mM, ou
40 mM. Une culture de POK1 et une autre de TP8503 sont ajustees a chacune de ces
concentrations. (Les cultures controles ne contiennent pas de H2O2.) La densite optique
et l’activite β-galactosidase sont mesurees immediatement apres le choc oxydatif, puis
a intervalles de 15 minutes pendant une heure. L’experience est alors terminee.

Chapitre 3
Resultats
3.1 Construction de la souche POK1
La construction de la souche POK1 a ete confirmee par PCR tel que decrit plus
tot (section 2.5.4). La figure 3.1 a illustre cette confirmation ; les puits 2 et 3
montrent l’amplification des jonctions 5’ et 3’ (respectivement) de la cassette FLK2
et du chromosome, alors que les puits 4 et 5 montrent une absence d’amplification dans
la souche TP8503. Les tailles attendues (respectivement 748 et 1546 paires de bases)
correspondent a ce qui est observe.
L’obtention d’une souche POK1 viable aussi bien en milieu riche qu’en milieu
minimal demontre que yadB est un gene non-essentiel chez E. coli.
3.2 Construction de la souche POK2
La souche POK2 a ete construite, et la deletion du gene yadB dans cette souche
a ete confirmee tel que decrit precedemment (section 2.6.3). Comme l’illustre la
figure 3.1 b, la region amplifiee sur le chromosome de POK2 (puit 2) est sensiblement
plus courte que la region correspondante dans la souche TP8503 (puit 3), indiquant
la deletion du locus d’interet. Les tailles observees correspondent a celles prevues, soit
1012 et 1547 paires de bases pour POK2 et TP8503 respectivement.

Chapitre 3. Resultats 53
(a) POK1 : Amplification des
jonctions 5’ et 3’ entre la cassette
FLK2 et le chromosome. Puit 1 :
Echelle moleculaire d’ADN 1 Kb.
Puit 2 : Jonction 5’ amplifiee
chez POK1. Puit 3 : Jonction
3’ amplifiee chez POK1. Puit
4 : Absence d’amplification pour
la jonction 5’ dans la souche
controle TP8503. Puit 5 : Absence
d’amplification pour la jonction 3’
dans la souche TP8503.
(b) POK2 : Amplification du
locus du gene yadB et de ses
regions flanquantes. Puit 1 :
Echelle moleculaire d’ADN 1 Kb.
Puit 2 : Amplification dans la
souche TP8503, ou le gene yadB
est intact. Puit 3 : Amplification
dans POK2 ; le fragment obtenu
est sensiblement plus court, indi-
quant la deletion du gene.
Fig. 3.1 – Confirmation de la contruction des souches POK1 et POK2 par PCR

Chapitre 3. Resultats 54
Le fait que la souche POK2 soit viable, aussi bien en milieu riche qu’en milieu
minimal, confirme que yadB n’est pas un gene essentiel a la croissance de E. coli.
3.3 Thermosensibilite de la souche POK2
La thermosensibilite de la souche ∆yadB POK2 a ete comparee a celle de la souche
TP8503 selon le protocole decrit precedemment (section 2.8). Aucune difference n’a
pu etre reperee entre les deux souches. La croissance est observee a 4 ◦C, 22 ◦C, 30 ◦C,
37 ◦C, 42 ◦C et 48 ◦C pour les deux souches, autant en milieu LB qu’en milieu minimal.
L’absence de yadB n’a donc pas d’effet sur la croissance de E. coli a des temperatures
non permissives.
3.4 Expression et effets de la deletion de yadB
L’expression de yadB et la vitesse de la croissance des cellules ou yadB a ete
inactive par remplacement ont ete etudiees tel que decrit au chapitre precedent
(section 2.8).
3.4.1 Croissance en conditions optimales
Comme l’indique la figure 3.2, aucune difference phenotypique n’est apparente
au niveau de la croissance entre les souches TP8503 et POK1.
On remarque egalement que l’expression de yadB augmente significativement
lorsque la progression de la densite cellulaire cesse d’etre exponentielle (ce qui cor-
respond a la fin de la linearite de la courbe sur la figure, l’echelle etant logarithmique).
Cette augmentation a lieu lors de la transition de la phase exponentielle a la phase
stationnaire, et est maximale environ 4 heures apres le debut de la phase stationnaire.
Des experiences de PCR [32] ont demontre que l’ARNm de yadB est present dans la
cellule au debut de la phase exponentielle de croissance ; les resultats de la presente
experience indiquent toutefois que cette transcription du gene en ARN ne se fait qu’a
un niveau tres faible (indetectable avec la methode utilisee ici). L’expression observee
pendant la phase stationnaire, bien que plus importante, est tout de meme relativement
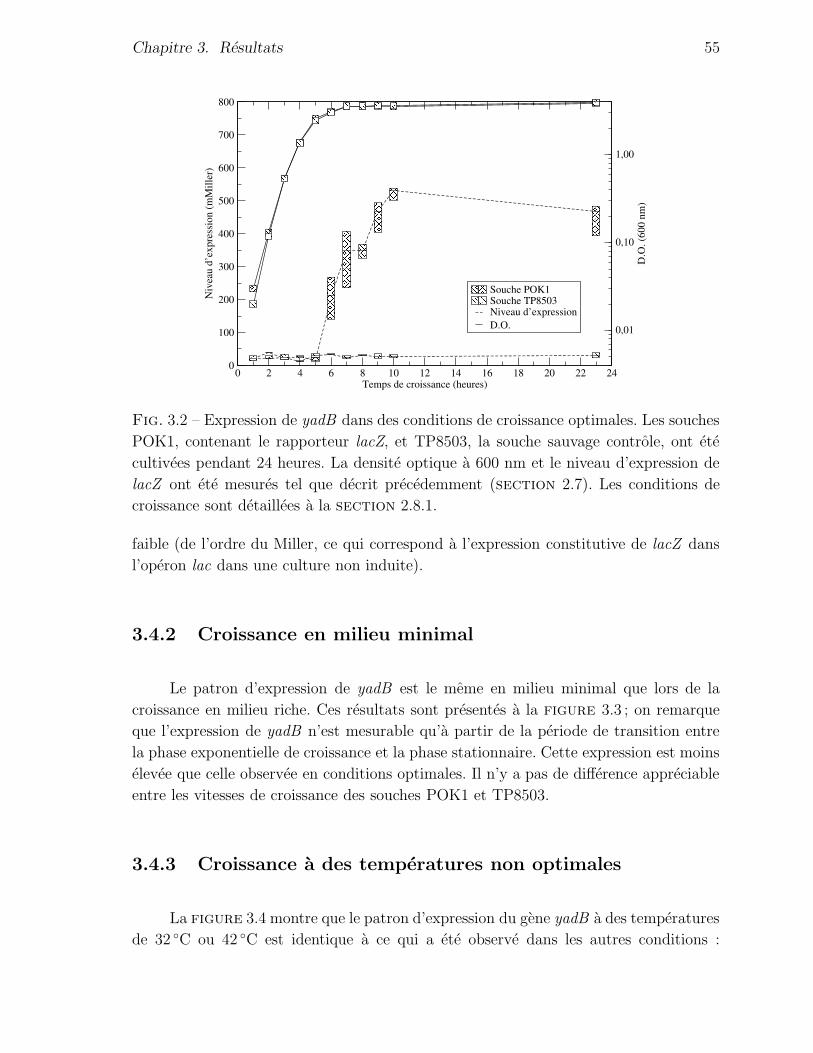
Chapitre 3. Resultats 55
Fig. 3.2 – Expression de yadB dans des conditions de croissance optimales. Les souches
POK1, contenant le rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete
cultivees pendant 24 heures. La densite optique a 600 nm et le niveau d’expression de
lacZ ont ete mesures tel que decrit precedemment (section 2.7). Les conditions de
croissance sont detaillees a la section 2.8.1.
faible (de l’ordre du Miller, ce qui correspond a l’expression constitutive de lacZ dans
l’operon lac dans une culture non induite).
3.4.2 Croissance en milieu minimal
Le patron d’expression de yadB est le meme en milieu minimal que lors de la
croissance en milieu riche. Ces resultats sont presentes a la figure 3.3 ; on remarque
que l’expression de yadB n’est mesurable qu’a partir de la periode de transition entre
la phase exponentielle de croissance et la phase stationnaire. Cette expression est moins
elevee que celle observee en conditions optimales. Il n’y a pas de difference appreciable
entre les vitesses de croissance des souches POK1 et TP8503.
3.4.3 Croissance a des temperatures non optimales
La figure 3.4 montre que le patron d’expression du gene yadB a des temperatures
de 32 ◦C ou 42 ◦C est identique a ce qui a ete observe dans les autres conditions :
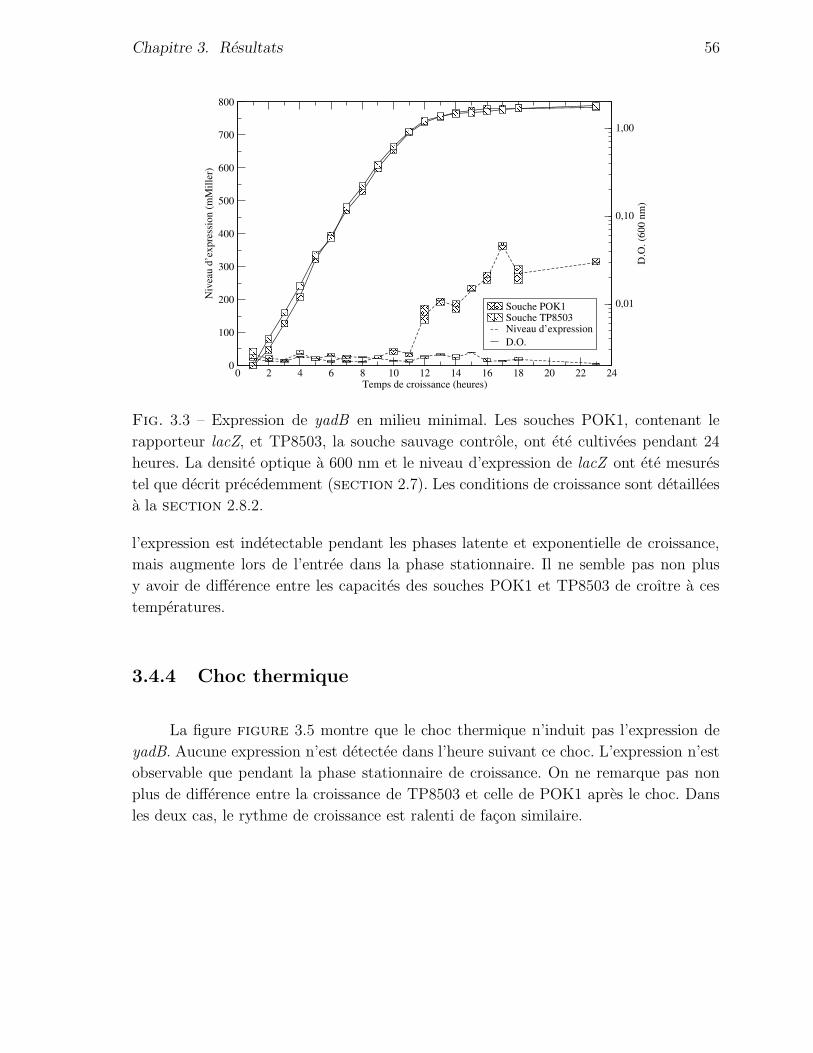
Chapitre 3. Resultats 56
Fig. 3.3 – Expression de yadB en milieu minimal. Les souches POK1, contenant le
rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete cultivees pendant 24
heures. La densite optique a 600 nm et le niveau d’expression de lacZ ont ete mesures
tel que decrit precedemment (section 2.7). Les conditions de croissance sont detaillees
a la section 2.8.2.
l’expression est indetectable pendant les phases latente et exponentielle de croissance,
mais augmente lors de l’entree dans la phase stationnaire. Il ne semble pas non plus
y avoir de difference entre les capacites des souches POK1 et TP8503 de croıtre a ces
temperatures.
3.4.4 Choc thermique
La figure figure 3.5 montre que le choc thermique n’induit pas l’expression de
yadB. Aucune expression n’est detectee dans l’heure suivant ce choc. L’expression n’est
observable que pendant la phase stationnaire de croissance. On ne remarque pas non
plus de difference entre la croissance de TP8503 et celle de POK1 apres le choc. Dans
les deux cas, le rythme de croissance est ralenti de facon similaire.

Chapitre 3. Resultats 57
(a) Croissance a 42 ◦C
(b) Croissance a 32 ◦C
Fig. 3.4 – Expression de yadB a des temperatures non optimales. Les souches POK1,
contenant le rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete cultivees
pendant 24 heures. La densite optique a 600 nm et le niveau d’expression de lacZ ont
ete mesures tel que decrit precedemment (section 2.7). Les conditions de croissance
sont detaillees a la section 2.8.3.
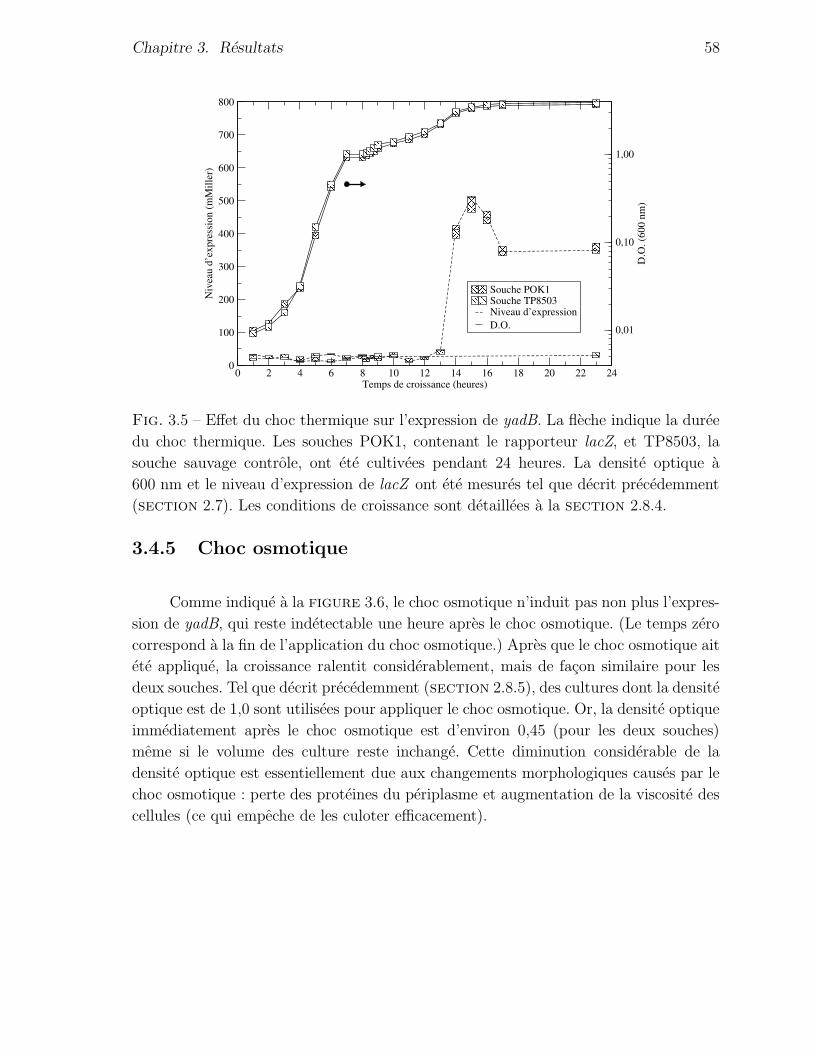
Chapitre 3. Resultats 58
Fig. 3.5 – Effet du choc thermique sur l’expression de yadB. La fleche indique la duree
du choc thermique. Les souches POK1, contenant le rapporteur lacZ, et TP8503, la
souche sauvage controle, ont ete cultivees pendant 24 heures. La densite optique a
600 nm et le niveau d’expression de lacZ ont ete mesures tel que decrit precedemment
(section 2.7). Les conditions de croissance sont detaillees a la section 2.8.4.
3.4.5 Choc osmotique
Comme indique a la figure 3.6, le choc osmotique n’induit pas non plus l’expres-
sion de yadB, qui reste indetectable une heure apres le choc osmotique. (Le temps zero
correspond a la fin de l’application du choc osmotique.) Apres que le choc osmotique ait
ete applique, la croissance ralentit considerablement, mais de facon similaire pour les
deux souches. Tel que decrit precedemment (section 2.8.5), des cultures dont la densite
optique est de 1,0 sont utilisees pour appliquer le choc osmotique. Or, la densite optique
immediatement apres le choc osmotique est d’environ 0,45 (pour les deux souches)
meme si le volume des culture reste inchange. Cette diminution considerable de la
densite optique est essentiellement due aux changements morphologiques causes par le
choc osmotique : perte des proteines du periplasme et augmentation de la viscosite des
cellules (ce qui empeche de les culoter efficacement).
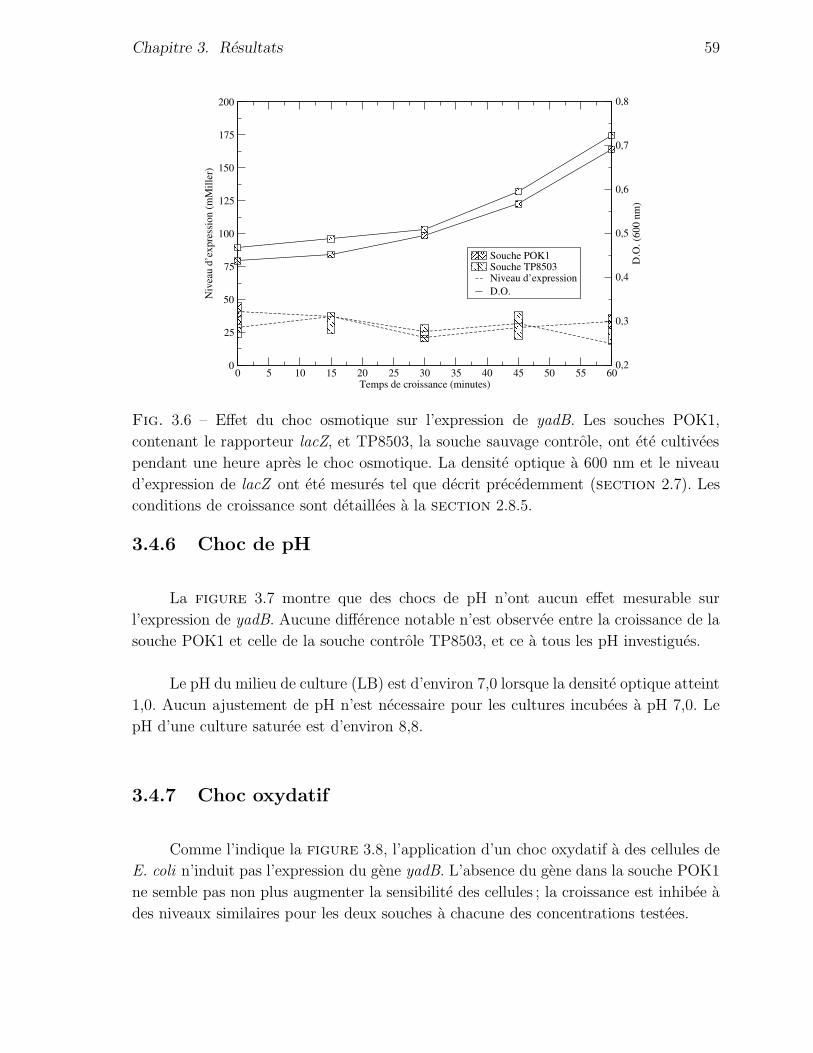
Chapitre 3. Resultats 59
Fig. 3.6 – Effet du choc osmotique sur l’expression de yadB. Les souches POK1,
contenant le rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete cultivees
pendant une heure apres le choc osmotique. La densite optique a 600 nm et le niveau
d’expression de lacZ ont ete mesures tel que decrit precedemment (section 2.7). Les
conditions de croissance sont detaillees a la section 2.8.5.
3.4.6 Choc de pH
La figure 3.7 montre que des chocs de pH n’ont aucun effet mesurable sur
l’expression de yadB. Aucune difference notable n’est observee entre la croissance de la
souche POK1 et celle de la souche controle TP8503, et ce a tous les pH investigues.
Le pH du milieu de culture (LB) est d’environ 7,0 lorsque la densite optique atteint
1,0. Aucun ajustement de pH n’est necessaire pour les cultures incubees a pH 7,0. Le
pH d’une culture saturee est d’environ 8,8.
3.4.7 Choc oxydatif
Comme l’indique la figure 3.8, l’application d’un choc oxydatif a des cellules de
E. coli n’induit pas l’expression du gene yadB. L’absence du gene dans la souche POK1
ne semble pas non plus augmenter la sensibilite des cellules ; la croissance est inhibee a
des niveaux similaires pour les deux souches a chacune des concentrations testees.

Chapitre 3. Resultats 60
(a) Souche POK1
(b) Controle - Souche TP8503
Fig. 3.7 – Effet du choc de pH sur l’expression de yadB. Les souches POK1, contenant
le rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete cultivees pendant
une heure apres le choc de pH. La densite optique a 600 nm et le niveau d’expression
de lacZ ont ete mesures tel que decrit precedemment (section 2.7). Les conditions de
croissance sont detaillees a la section 2.8.6.

Chapitre 3. Resultats 61
(a) Souche POK1
(b) Controle - Souche TP8503
Fig. 3.8 – Effet du choc oxydatif sur l’expression de yadB. Les souches POK1, contenant
le rapporteur lacZ, et TP8503, la souche sauvage controle, ont ete cultivees pendant
une heure apres le choc oxydatif. La densite optique a 600 nm et le niveau d’expression
de lacZ ont ete mesures tel que decrit precedemment (section 2.7). Les conditions de
croissance sont detaillees a la section 2.8.7.

Chapitre 4
Discussion
4.1 Expression de yadB pendant le cycle cellulaire
La presente etude demontre que yadB est exprime au moins pendant la phase
stationnaire de croissance. Des experiences de PCR [32] ont permis d’amplifier l’ARNm
de yadB a partir de cellules recoltees en phase exponentielle, ce qui indique que yadB
est egalement faiblement exprime pendant cette phase de croissance. Toutefois, les
valeurs mesurees dans les presentes experiences pendant la phase exponentielle sont
indistinguables du bruit de fond observe avec la souche controle ∆lac TP8503. L’ex-
pression detectee par PCR pourrait donc correspondre a l’expression basale du gene
yadB reprime.
L’expression plus elevee de yadB pendant la phase stationnaire de croissance
procure un indice sur le role physiologique de la GluQRS. De nombreux changements
environnementaux sont associes a cette phase de croissance, par exemple : depletion des
nutriments du milieu, changements de pH, choc oxydatif. Le phenomene de passage a
l’etat stationnaire est complexe, et yadB pourrait etre impliquee soit dans la reponse a
un stress environnemental particulier, soit dans le mecanisme global de regulation des
genes pendant la phase stationnaire. Notre incapacite a induire l’expression de yadB
en soumettant les cellules a des stress similaires a ceux qui accompagnent la phase
stationnaire suggere un role global. Notons par ailleurs que nos resultats montrent
que yadB n’est pas implique dans la reponse au choc thermique ou dans la vitesse de
croissance des cellules a des temperatures non optimales.
L’expression de yadB demeure faible, meme dans les conditions ou le gene est
exprime le plus fortement. Ce niveau d’expression (entre 500 et 1000 mMiller lorsque

Chapitre 4. Discussion 63
mesure avec le rapporteur β-galactosidase) est environ 500 fois plus faible que ce qui
a ete mesure (par la meme technique) pour le gene yacK (aussi designe cueO) dont
le produit d’expression est implique dans le metabolisme du cuivre [48]. L’expression
du gene yacK est induite par l’accumulation de cuivre dans le milieu. Son niveau
d’expression faible suggere que yadB pourrait etre implique dans le metabolisme basal
de la cellule plutot que dans la reponse a un stress particulier.
Les changements morphologiques (par exemple la diminution de la taille des
cellules) qui ont lieu lors de l’entree dans la phase stationnaire peuvent influencer le
rapport entre le nombre de copies du rapporteur et la densite cellulaire mesures, ce qui
limite la fiabilite de la technique employee ici. Toutefois, le rapport entre les valeurs
mesurees en phase stationnaire et le bruit de fond permet d’affirmer qu’il existe une
augmentation d’un facteur d’au moins 10 lors de l’entree dans la phase stationnaire, ce
qui ne peut etre entierement explique par des changements morphologiques.
L’expression sensiblement plus faible de yadB en milieu minimal (environ
300 mMiller) qu’en milieu riche (environ 500 mMiller) souligne le caractere non
essentiel du gene pour E. coli. Un grand nombre de genes qui ne sont pas necessaires au
metabolisme de base de la cellule sont sensiblement moins exprimes dans des conditions
ou la disponibilite des nutriments est moindre.
4.2 Cotranscription de dksA et de yadB
Bien que l’objectif de cette etude ait ete de doser le niveau d’expression du gene
yadB, l’absence de promoteur canonique immediatement en amont de ce gene suggere
que c’est plutot l’expression de l’operon dksA-yadB qui a ete mesuree. Toutefois, les
resultats obtenus different de ceux d’une autre equipe qui a determine le nombre de
copies de DksA pendant l’ensemble du cycle cellulaire et qui rapporte que ce niveau est
stable [41]. Nos resultats montrent que l’expression de yadB n’est pas constante, mais
augmente significativement pendant la phase stationnaire de croissance.
Cette disparite entre les resultats pourrait etre expliquee par la presence d’un
element de regulation situe entre les genes dksA et yadB. La transcription de dksA
pourrait alors se faire a un niveau constant, mais la propagation de l’onde transcrip-
tionnelle dans le gene yadB pourrait etre regulee en fonction de la phase de croissance.
Des experiences supplementaires seront necessaires pour verifier cette hypothese.

Chapitre 4. Discussion 64
4.3 Role du Glu-Q-ARNtAsp
Bien que le role de l’aminoacyl-ARNt synthetise par la GluQRS soit encore in-
connu, la position de la base hypermodifiee queuosine chargee par la GluQRS sur
l’ARNtAsp laisse croire que cette modification de l’ARN pourrait avoir une influence sur
la traduction de l’information genetique. La queuosine etant situee a la premiere position
de l’anticodon, une modification de sa structure est susceptible d’affecter l’appariement
entre codon et anticodon sur le ribosome. L’importance des bases hypermodifiees a la
premiere position de l’anticodon pour l’appariement codon / anticodon sur le ribosome
a deja ete demontree pour d’autres ARNt [57].
Il n’existe qu’un seul type de gene d’ARNtAsp chez la plupart des bacteries (bien
que plusieurs copies de ce gene soient frequemment presentes dans les genomes). Le
decodage des deux codons aspartate (GAU et GAC) se fait donc par correspondance
avec un seul ARNt, ce qui implique un flottement (« wobble ») au niveau du troisieme
nucleotide des codons aspartate lors de l’appariement a l’ARNt sur le ribosome. La
presence de la queuosine a cette position rend ce flottement possible.
On sait que la queuosine est glutamylee in vivo dans l’ARNtAsp [33] et, donc,
que cette glutamylation n’empeche pas le decodage des codons aspartate. L’obtention
d’une souche ∆yadB viable demontre par ailleurs que la glutamylation n’est pas requise
pour la lecture des codons aspartate. Toutefois, il n’est pas exclu que la lecture d’un
ou des deux codons puisse etre influencee par la glutamylation de la queuosine, par
stabilisation ou destabilisation des interactions codon / anti-codon. Un tel phenomene
aurait un impact certain sur la traduction de l’ARNm par le ribosome. Considerant la
courte demi-vie du Glu-Q-ARNtAsp, la glutamylation de la queuosine pourrait participer
a la regulation post-transcriptionnelle de l’expression des genes.
Il a deja ete demontre que l’occurrence des codons correspondant a des ARNt
isoaccepteurs dont les concentrations physiologiques sont faibles est inferieure a la
moyenne dans les genes fortement exprimes [58]. Un mecanisme similaire pourrait
exister ou une categorie de genes presenterait une densite elevee en un certain codon
aspartate dont la lecture serait soit facilitee, soit rendue plus difficile par la gluta-
mylation de la queuosine de l’ARNtAsp. Des observations preliminaires indiquent a
ce sujet que plusieurs genes bacteriens dont l’expression est liee a la virulence et au
phenomene d’infection contiennent un nombre eleve de codons GAU ainsi qu’un ratio
eleve de codons GAU par rapport aux codons GAC. Ces genes incluent rns, qui, chez
E. coli, regule la production de CS1 et CS2 (respectivement une toxine et une structure

Chapitre 4. Discussion 65
favorisant l’adherence aux cellules hotes) [59] et virF qui, chez S. flexneri, joue un role
central dans la regulation de l’expression des genes de virulence lors de l’infection [60].
L’implication de yadB dans la regulation post-transcriptionnelle pourrait ega-
lement expliquer certaines observations qui suggeraient que dksA jouait un role de
regulateur post-transcriptionnel, alors que l’elucidation de sa structure et de son role
physiologique rendent cette hypothese peu plausible [41]. Les informations suggerant
un role de regulateur post-transcriptionnel pour dksA ont ete obtenues a l’aide d’une
souche bacterienne ou dksA a ete delete [37]. Or, il est possible que cette deletion ait
egalement affecte yadB, situe immediatement en aval et faisant partie du meme operon.
4.4 Lien entre les concentrations de glutamate et le
role de la GluQRS
Tel que mentionne plus tot (section 1.4.4), le ppGpp est utilise par les bacteries
comme un senseur indirect du manque de nutriments (la synthese du ppGpp etant
induite par de faibles niveaux d’aminoacylation de l’ARNt). DksA reagit a l’accumula-
tion de ce metabolite en regulant la transcription des genes des ARN stables (ARNt et
ARNr).
De facon analogue, la GluQRS pourrait hypothetiquement etre influencee par
le niveau de glutamate libre. Des concentrations extracellulaires de glutamate elevees
favorisent la reponse au choc osmotique et aux bas pH chez E. coli et d’autres bacteries
enteriques, ces phenomenes etant directement relies aux conditions environnementales
auxquelles sont confrontees ces bacteries lors de l’infection de l’intestin [61]. La
GluQRS pourrait servir d’effecteur des fluctuations de la concentration de glutamate
en reagissant aux concentrations elevees par la glutamylation d’une plus grande
proportion d’ARNtAsp.
4.5 Phenotypes des cellules ∆yadB
L’hypothese presentee ci-haut (sections 4.3 et 4.4) concorde avec nos resultats
selon lesquels l’expression de yadB augmente lors du passage a la phase stationnaire
de croissance. En effet, le glutamate est une composante essentielle des mecanismes de
resistance aux conditions acides, et ces mecanismes sont particulierement actifs pen-

Chapitre 4. Discussion 66
dant la phase stationnaire [61]. Toutefois, notre incapacite a identifier un ou plusieurs
phenotypes associes a la deletion de yadB limite la confiance que l’on peut accorder a
cette hypothese. Si yadB procede bien a la regulation post-transcriptionnelle de genes
associes a la reponse a certaines conditions environnementales associees au stress ou a
l’infection d’un hote, il devrait etre possible de mesurer une decroissance de la viabilite
des cellules mutees dans ces conditions.
Cette absence de resultats s’explique peut-etre par la methodologie employee pour
mesurer les differences phenotypiques. Des cultures ont ete realisees sur une echelle d’une
journee ; les cellules restent donc en phase stationnaire pendant moins de 24 heures. De
plus, seule la vitesse de croissance des cellules a ete mesuree, puisque l’objectif vise etait
la caracterisation du patron d’expression du gene yadB.
Afin de determiner les effets physiologiques de l’absence de yadB, des experiences
supplementaires seront necessaires. Celles-ci devront mesurer la viabilite des cellules
en phase stationnaire dans des conditions variees. Ces mesures devraient se faire sur
une periode de plusieurs jours, et etre realisees par comptage des cellules viables par
dilutions sequentielles et etalement sur geloses. Aussi, des observations microscopiques
devraient etre effectuees sur des cellules mutantes a divers stades du cycle cellulaire.
4.6 Analyse de la technique de remplacement
employee
4.6.1 Avantages
La methode de remplacement utilisee dans cette etude presente de nombreux
avantages par rapport aux autres techniques qui auraient pu etre employees pour obtenir
des informations similaires. D’abord, sa simplicite permet de la mettre en œuvre sans
avoir recours a des equipements sophistiques ou couteux. Elle est aussi tres fiable,
puisque son deroulement peut etre suivi a chaque etape grace a des phenotypes varies
(resistance a differents antibiotiques ou au sucrose, thermosensibilite). De plus, le test
enzymatique permettant de doser l’expression genique est facilement echelonnable : il
peut aisement etre automatise, et une variete d’analogues du lactose sont disponibles,
incluant des composes dont les produits d’hydrolyse sont fluorescents, ce qui permet la
mise au point de tests extremement sensibles.

Chapitre 4. Discussion 67
Ces caracteristiques font de cette methode un outil interessant pour des etudes a
grande echelle de genes dont la fonction est inconnue. Une banque de mutants de E. coli
construits par le remplacement systematique des genes dont la fonction est inconnue
est d’ailleurs deja en cours [M. Masters, communication personnelle].
4.6.2 Inconvenients
La presente etude souligne une des faiblesses de la methode de remplacement uti-
lisee. La faible expression de yadB, couplee a l’utilisation de l’analogue chromogenique
peu sensible ONPG, n’a permis d’obtenir qu’une difference d’un facteur 10 entre le
bruit de fond et les valeurs d’expression les plus elevees. L’utilite de la methode est
donc discutable pour la caracterisation du patron d’expression de genes faiblement
exprimes, a moins d’avoir recours a des analogues plus sensibles.
Soulignons egalement que le dosage enzymatique de l’activite β-galactosidase
ne fournit aucune information sur le nombre de copies du produit du gene remplace
dans la cellule a un moment donne ; seul le niveau d’expression est mesure, ce qui ne
permet pas de juger de la demi-vie de la proteine. De plus, les valeurs d’expression
mesurees peuvent etre faussees par les changements metaboliques causes par le rem-
placement. Par exemple, tout phenomene d’auto-regulation sera rendu impossible par
l’absence de la proteine originale. L’expression mesuree dans un tel cas serait differente
de celle qui aurait lieu dans la cellule sauvage. Enfin, les phenomenes de regulation
post-transcriptionnelle ne peuvent etre mesures par cette technique. Meme si, chez les
bacteries, ce type de regulation est beaucoup moins frequent que la regulation de la
transcription, il est tout de meme important ; des techniques complementaires, dont
certaines sont mentionnees ci-dessous (section 4.6.3), devraient donc etre utilisees
afin d’identifier les mecanismes controlant le niveau intracellulaire de GluQRS.
4.6.3 Techniques alternatives
Le transfert Northern est une autre technique qui peut etre utilisee pour detecter
et quantifier l’expression d’un gene. L’avantage principal de la technique utilisee ici par
rapport au transfert Northern est la possibilite de tester un nombre eleve de conditions
et de faire des echantillonnages a des temps rapproches. Avec la technique du transfert
Northern, realiser un nombre aussi grand de mesures aurait ete malaise. Toutefois,
le transfert Northern a l’avantage de ne pas modifier le metabolisme de la cellule,
contrairement au remplacement de gene.

Chapitre 4. Discussion 68
L’immunobuvardage (ou transfert Western) permet d’obtenir des informations sur
le nombre de copies du produit d’expression d’un gene dans les cellules. Il permet donc
d’integrer des informations sur la demi-vie des proteines. Toutefois, cette technique,
tout comme le transfert Northern, n’est pas utilisable avec un nombre de mesures aussi
eleve que ce qui a ete realise dans la presente etude.
Enfin, la technique de PCR en temps reel permet de mesurer de facon quantitative
et tres precise les niveaux d’ARNm d’un certain gene. La technique peut etre utilisee a
grande echelle avec un nombre eleve d’echantillons. Toutefois, l’instrumentation associee
a cette technique est complexe, onereuse et necessite une expertise considerable.

Chapitre 5
Conclusion
5.1 Resume
Le gene yadB code pour l’enzyme glutamyl-queuosine-ARNtAsp synthetase
(GluQRS), dont la fonction est la glutamylation de l’ARNtAsp. Le role physiologique
de l’aminoacyl-ARNt ainsi forme est inconnu. La GluQRS agit comme une enzyme de
modification de l’ARNt plutot que comme une aminoacyl-ARNt synthetase classique,
puisqu’elle lie le glutamate a la base hypermodifiee queuosine de l’anticodon de son
ARNt correspondant plutot que sur une fonction hydroxyle de l’extremite 3’-terminale.
Chez E. coli et, probablement, d’autres bacteries, yadB forme un operon avec le
gene dksA, qui est implique dans la regulation de la transcription des ARNr et dont
le produit d’expression a une activite maximale pendant la phase stationnaire de
croissance.
Nous avons demontre par deletion de gene que yadB est non-essentiel chez E.
coli, aussi bien en milieu riche qu’en milieu minimal. Une souche ou le gene yadB a ete
remplace par le gene rapporteur lacZ a egalement ete construite. Ce remplacement a
ete realise de facon a conserver les elements transcriptionnels de yadB. Le dosage de
l’activite β-galactosidase a permis de determiner le patron d’expression de yadB dans
des conditions de croissance variees. Nous avons decouvert que l’expression de yadB
est tres faible pendant la phase exponentielle, mais augmente significativement lors du
passage a la phase stationnaire. Nous avons tente d’induire l’expression de yadB en
reproduisant des stress associes a la phase stationnaire, mais sans succes. Il semble que
l’expression de yadB soit reliee au phenomene global de passage a l’etat stationnaire de
croissance.

Chapitre 5. Conclusion 70
La GluQRS pourrait agir comme regulateur post-transcriptionnel pour un en-
semble de genes impliques dans la phase stationnaire ou dans certains phenomenes
de virulence. Cette regulation pourrait se faire par le biais du decodage preferentiel
d’un codon aspartate par le ribosome. Ce decodage preferentiel serait lui-meme du a
la glutamylation de la queuosine de l’ARNtAsp, qui pourrait influencer le phenomene
de flottement qui a lieu a la troisieme position du codon dans l’appariement codon /
anticodon. Cette hypothese repose sur l’occurrence anormalement elevee (par rapport
aux valeurs moyennes pour le genome) dans de nombreux genes associes a la virulence
du codon GAU, alors que le codon GAC est y considerablement sous-utilise.
5.2 Perspectives
Afin de reperer les eventuels phenotypes dus a la deletion de yadB, des etudes de
viabilite en phase stationnaire seront realisees, ainsi que des examens microscopiques
des cellules, qui considereront l’ensemble du cycle cellulaire. Le rapport entre les ARNm
des genes yadB et dksA pendant les phases exponentielle et stationnaire de croissance
sera determine par transfert Northern. Ces observations permettront de verifier s’il
existe un element de regulation de l’expression de yadB immediatement en amont de ce
gene. Des anticorps ciblant DksA et la GluQRS etant disponibles, le nombre de copies
de ces deux proteines sera mesure pendant les phases exponentielle et stationnaire.
A long terme, il serait interessant de realiser des etudes proteomiques portant
sur les effets de la deletion de yadB sur l’expression des autres genes de E. coli. Par
comparaison de gels d’electrophorese en deux dimensions realises a partir des proteines
des cellules ∆yadB et des cellules sauvages, il pourrait etre possible d’identifier des
genes dont le patron d’expression est modifie en l’absence de yadB.

Bibliographie
[1] Crick, F. (1958) On Protein Synthesis, in : Symposium of the society for
experimental biology XII. p. 153, New-York : Academic Press.
[2] Crick, F. (1968) The origin of the genetic code. Journal of Molecular Biology
38(3) : 367–379.
[3] Wilcox, M. & Nirenberg, M. (1968) Transfer RNA as a cofactor coupling
amino acid synthesis with that of protein. Proceedings of the National Academy of
Sciences of the United States of America 61(1) : 229–236.
[4] Ibba, M. & Soll, D. (2000) Aminoacyl-tRNA synthesis. Annual Reviews of
Biochemistry 69 : 617–650.
[5] Sekine, S., Nureki, O., Dubois, D. Y., Bernier, S., Chevevert, R.,
Lapointe, J. & Vassylyev, D. G. (2003) ATP binding by glutamyl-tRNA
synthetase is switched to the productive mode by tRNA binding. European
Molecular Biology Organization Journal 22(3) : 676–688.
[6] Arnez, J. G. & Moras, D. (1997) Structural and functional considerations of
the aminoacylation reaction. Trends in Biochemical Sciences 22 : 211–216.
[7] Clark, B. F. C., Kjeldgaard, M., Barciszewski, J. & Sprinzl, M. (1995)
Recognition of Aminoacyl-tRNAs by Protein Elongation Factors, in : tRNA -
Structure, Biosynthesis, and Function. D. Soll & U. L. RajBhandary ed., p. 423-
442, Washington, D. C. : ASM Press.
[8] Curnow, A. W., Ibba, M. & Soll, D. (1996) tRNA-dependent asparagine
formation. Nature 382(6592) : 589–590.
[9] Kumar, A. M., Schaub, U., Soll, D. & Ujwal, M. L. (1996) Glutamyl-transfer
RNA : at the crossroad between chlorophyll and protein biosynthesis. Trends in
Plant Science 1(11) : 371–376.
[10] Lund, E. & Dahlberg, J. E. (1998) Proofreading and Aminoacylation of tRNAs
Before Export from the Nucleus. Science 282(5386) : 2082–2085.
[11] Putzer, H., Grunberg-Manago, M. & Springer, M. (1995) Bacterial
Aminoacyl-tRNA Synthetases : Genes and Regulation of Expression, in : tRNA -

Bibliographie 72
Structure, Biosynthesis, and Function. D. Soll & U. L. RajBhandary ed., p. 423-
442, Washington, D. C. : ASM Press.
[12] Sankaranarayanan, R., Dock-Bregeon, A. C., Romby, P., Caillet, J.
& Springer, M. (1999) The structure of threonyl-tRNA synthetase-tRNAThr
complex enlightens its repressor activity and reveals an essential zinc ion in the
active site. Cell 97(3) : 371–381.
[13] Woese, C. E., Olsen, G. J., Ibba, M. & Soll, D. (2000) Aminoacyl-tRNA
Synthetases, the Genetic Code, and the Evolutionary Process. Microbiology and
Molecular Biology Reviews 64(1) : 202–236.
[14] Onesti, S., Miller, A. D. & Brick, P. (1995) The crystal structure of the
lysyl-tRNA synthetase (LysU) from Escherichia coli. Structure 3(2) : 163–176.
[15] Eriani, G., Delarue, M., Poch, O., Gangloff, J. & Moras, D. (1990)
Partition of tRNA synthetases into two classes based on mutually exclusive sets of
sequence motifs. Nature 347(6289) : 203–206.
[16] Sekine, S., Nureki, O., Vassylyev, D. G. & Yokoyama, S. (2001) Structural
basis for anticodon recognition by discriminating glutamyl-tRNA synthetase.
Nature Structural Biology 8(3) : 203–206.
[17] Eiler, S., Dock-Bregeon, A., Moulinier, L., Thierry, J. C. & Moras, D.
(1999) Synthesis of aspartyl-tRNAAsp in Escherichia coli - a snapshot of the second
step. European Molecular Biology Organization Journal 18(22) : 6532–6541.
[18] DeLano, W. L. (2002) The PyMOL Molecular Graphics System,
http://www.pymol.org.
[19] Delarue, M. & Moras, D. (1993) The aminoacyl-tRNA synthetase family :
modules at work. Bioessays 15(10) : 675–687.
[20] Giege, R., Puglisi, J. D. & Florentz, C. (1993) tRNA structure and
aminoacylation efficiency. Progress in Nucleic Acid Research and Molecular Biology
45 : 129–206.
[21] Ahel, I., Korencic, D., Ibba, M. & Soll, D. (2003) Trans-editing of mischarged
tRNAs. Proceedings of the National Academy of Sciences of the United States of
America 100(26) : 15 422–15 427.
[22] Rogers, M. J., Adachi, T., Inokuchi, H. & Soll, D. (1994) Functional
communication in the recognition of tRNA by Escherichia coli glutaminyl-tRNA
synthetase. Proceedings of the National Academy of Sciences of the United States
of America 91(1) : 291–295.
[23] Siatecka, M., Rozek, M., Barciszewski, J. & Mirande, M. (1998) Modular
Evolution of the Glx-tRNA synthetase family. European Journal of Biochemistry
256(1) : 80–87.

Bibliographie 73
[24] Chapeville, F., Lipmann, F., Ehrenstein, G., Weisblum, B., Ray, W. J.
& Benzer, S. (1962) On the role of soluble ribonucleic acid in coding for amino
acids. Proceedings of the National Academy of Sciences of the United States of
America 48(1) : 1086–1092.
[25] Tamura, K. & Schimmel, P. (2001) Oligonucleotide-directed peptide synthesis
in a ribosome- and ribozyme-free system. Proceedings of the National Academy of
Sciences of the United States of America 98(4) : 1393–1397.
[26] Fersht, A. R. & Dingwall, C. (1979) Cysteinyl-tRNA synthetase from
Escherichia coli does not need an editing mechanism to reject serine and alanine.
High binding energy of small groups in specific molecular interactions. Biochemistry
18(7) : 1245–1249.
[27] Jakubowski, H. & Goldman, E. (1992) Editing of errors in selection of amino
acids for protein synthesis. Microbiological Reviews 56(3) : 412–429.
[28] Schmidt, E. & Schimmel, P. (1994) Mutational isolation of a sieve for editing
in a transfer RNA synthetase. Science 264(5156) : 265–267.
[29] Ibba, M. & Soll, D. (1999) Quality control mechanisms during translation.
Science 286(5446) : 1893–1897.
[30] Sherman, J. C., Rogers, M. J. & Soll, D. (1992) Competition of aminoacyl-
tRNA synthetases for tRNA ensures the accuracy of aminoacylation. Nucleic Acids
Research 20(11) : 2847–2852.
[31] Campanacci, V., Dubois, D. Y., Becker, H. D., Kern, D., Spinelli, S.,
Valencia, C., Pagot, F., Salomoni, A., Grisel, S., Vincentelli, R., Bignon,
C., Lapointe, J., Giege, R. & Cambillau, C. (2004) The Escherichia coli YadB
Gene Product Reveals a Novel Aminoacyl-tRNA Synthetase Like Activity. Journal
of Molecular Biology 337(1) : 273–283.
[32] Dubois, D. Y., Blaise, M., Becker, H. D., Campanacci, V., Keith,
G., Giege, R., Cambillau, C., Lapointe, J. & Kern, D. (2004) An
aminoacyl-tRNA synthetase-like protein encoded by the Escherichia coli yadB
gene glutamylates specifically tRNAAsp. Proceedings of the National Academy of
Sciences of the United States of America 101(20) : 7530–7535.
[33] Salazar, J. C., Ambrogelly, A., Crain, P. F., McCloskey, J. A. & Soll,
D. (2004) A truncated aminoacyl-tRNA synthetase modifies RNA. Proceedings
of the National Academy of Sciences of the United States of America 101(20) :
7536–7541.
[34] Blaise, M., Becker, H. D., Keith, G., Cambillau, C., Lapointe, J., Giege,
R. & Kern, D. (2004) A minimalist glutamyl-tRNA synthetase dedicated to
aminoacylation of the tRNAAsp QUC anticodon. Nucleic Acids Research 32(9) :
2768–2775.

Bibliographie 74
[35] Ribas De Pouplana, L. & Schimmel, P. (2001) Two classes of tRNA
synthetases suggested by sterically compatible dockings on tRNA acceptor stem.
Cell 104 : 191–193.
[36] Kang, P. J. & Craig, E. A. (1990) Identification and Characterization of a New
Escherichia coli Gene That Is a Dosage-Dependent Suppressor of a dnaK Deletion
Mutation. Journal of Bacteriology 172(4) : 2055–2064.
[37] Jude, F., Kohler, T., Branny, P., Perron, K., Mayer, M. P., Comte,
R. & van Delden, C. (2003) Posttranscriptional Control of Quorum-Sensing-
Dependent Virulence Genes by DksA in Pseudomonas aeruginosa. Journal of
Bacteriology 185(12) : 3558–3566.
[38] Webb, C., Moreno, M., Wilmes-Riesenberg, M., Curtiss III, R. & Foster,
J. W. (1999) Effects of DksA and ClpP protease on sigma S production and
virulence in Salmonella typhimurium. Molecular Microbiology 34(1) : 112–113.
[39] Mogull, S. A., Runyen-Janecky, L. J., Hong, M. & Payne, S. M.
(2001) dksA Is Required for Intercellular Spread of Shigella flexneri via an Rpos-
Independent Mechanism. Infection and Immunity 69(9) : 5742–5751.
[40] Brown, L., Gentry, D., Elliot, T. & Cashel, M. (2002) DksA Affects ppGpp
Induction of RpoS at a Translational Level. Journal of Bacteriology 184(16) :
4455–4465.
[41] Perederina, A., Svetlov, V., Vassylyeva, M. N., Tahirov, T. H.,
Yokosama, S., Artsimovitch, I. & Vassylyev, D. G. (2004) Regulation
through the Secondary Channel - Structural Framework for ppGpp-DksA
Synergism during Transcription. Cell 118(1) : 297–309.
[42] Paul, B. J., Barker, M. M., Ross, W., Schneider, D. A., Webb, C.,
Foster, J. W. & Gourse, R. L. (2004) DksA : A Critical Component of
the Transcription Initiation Machinery that Potentiates the Regulation of rRNA
Promoters by ppGpp and the Initiating NTP. Cell 118(1) : 311–322.
[43] Miller, J. H. (1992) β-galactosidase assay, in : A Short Course in Bacterial
Genetics, p. 72-75, Cold Spring Harbor : Cold Spring Harbor Laboratory Press.
[44] Moore, D. (1988) Resolution and recovery of large DNA fragments, in : Current
Protocols in Molecular Biology, vol. 1. Frederick M. Ausubel ed., p. 2.5.1-2.5.9,
New-York : John Wiley & Sons.
[45] Inoue, H., Nojima, H. & Okayama, H. (1990) High efficiency transformation
of Escherichia coli with plasmids. Gene 96 : 23–28.
[46] QIAGEN (2002) QIAquick Spin Handbook .
[47] Lech, K. & Brent, R. (1998) Media Preparation and Bacteriological Tools, in :
Current Protocols in Molecular Biology, vol. 1. Frederick M. Ausubel ed., p. 1.1.1-
1.1.4, New-York : John Wiley & Sons.

Bibliographie 75
[48] Merlin, C., McAteer, S. & Masters, M. (2002) Tools for Characterization of
Escherichia coli Genes of Unknown Function. Journal of Bacteriology 184(16) :
4573–4581.
[49] Rice, P., Longden, I. & Bleasby, A. (2000) EMBOSS : The European
Molecular Biology Open Software Suite. Trends in Genetics 16(6) : 276–277.
[50] Moore, D. (1998) Purification and concentration of DNA from aqueous solutions,
in : Current Protocols in Molecular Biology, vol. 1. Frederick M. Ausubel ed.,
p. 2.1.1-2.1.4, New-York : John Wiley & Sons.
[51] QIAGEN (2004) QIAprep Miniprep Handbook .
[52] Birnboim, H. C. & Doly, J. (1979) A rapid alkaline procedure for screening
recombinant plasmid DNA. Nucleic Acids Research 7(6) : 1513–1523.
[53] Moore, D. (1999) Preparation of Genomic DNA from Bacteria, in : Current
Protocols in Molecular Biology, vol. 1. Frederick M. Ausubel ed., p. 2.4.1-2.4.5,
New-York : John Wiley & Sons.
[54] Kramer, M. F. & Coen, D. M. (1998) The Polymerase Chain Reaction, in :
Current Protocols in Molecular Biology, vol. 2. Frederick M. Ausubel ed., p. 15.1.1-
15.1.15, New-York : John Wiley & Sons.
[55] Tabor, S. (1987) DNA Ligases, in : Current Protocols in Molecular Biology, vol. 1.
Frederick M. Ausubel ed., p. 3.14.2-3.14.4, New-York : John Wiley & Sons.
[56] Tabor, S. (1987) Phosphatases and Kinases, in : Current Protocols in Molecular
Biology, vol. 1. Frederick M. Ausubel ed., p. 3.10.1-3.10.5, New-York : John Wiley
& Sons.
[57] Yasukawa, T., Suzuki, T., Ishii, N., Ohta, S. & Watanabe, K. (2001)
Wobble modification defect in tRNA disturbs codon-anticodon interaction in a
mitochondrial disease. European Molecular Biology Organization Journal 20(17) :
4794–4802.
[58] Ikemura, T. (1981) Correlation between the abundance of Escherichia coli
transfer RNAs and the occurrence of the respective codons in its protein genes.
Journal of Molecular Biology 146(1) : 1–21.
[59] Caron, J., Coffield, L. M. & Scott, J. R. (1989) A plasmid-encoded regulatory
gene, rns, required for expression of the CS1 and CS2 adhesins of enterotoxigenic
Escherichia coli. Proceedings of the National Academy of Sciences of the United
States of America 86(3) : 963–967.
[60] Durand, J. M., Dagberg, B., Uhlin, B. E. & Bjork, G. R. (2000) Transfer
RNA modification, temperature and DNA superhelicity have a common target in
the regulatory network of the virulence of Shigella flexneri : the expression of the
virF gene. Molecular Microbiology 35(4) : 924–935.

Bibliographie 76
[61] Lin, J., Smith, M. P., Chapin, K., Baik, H. S., Bennett, G. N. & Foster,
J. W. (1996) Mechanisms of Acid Resistance in Enterohemorrhagic Escherichia
coli. Applied and Environmental Microbiology 62(9) : 3094–3100.