
Proteomic Characterization
of Alternative Splicing and
Coding Polymorphism
Proteomic Characterization
of Alternative Splicing and
Coding PolymorphismNathan EdwardsCenter for Bioinformatics and Computational BiologyUniversity of Maryland, College Park

2
Mass Spectrometry for Proteomics
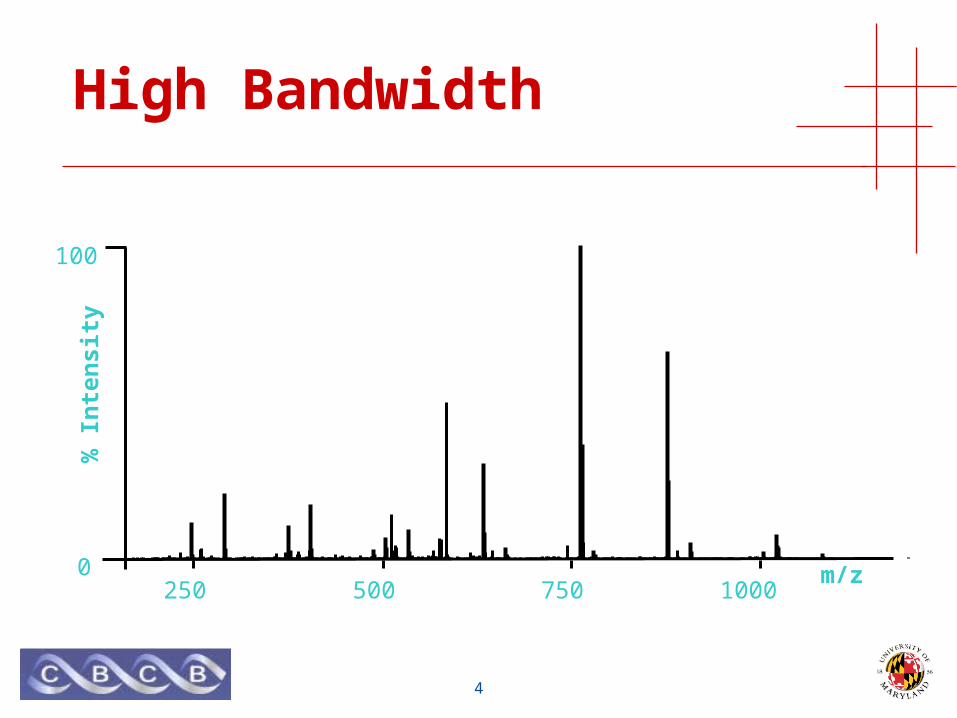
• Measure mass of many (bio)molecules simultaneously• High bandwidth
• Mass is an intrinsic property of all (bio)molecules• No prior knowledge required

3
Mass Spectrometry for Proteomics
• Measure mass of many molecules simultaneously• ...but not too many, abundance bias
• Mass is an intrinsic property of all (bio)molecules• ...but need a reference to compare to
4
High Bandwidth
100
0250 500 750 1000
m/z
% I
nte
nsit
y

5
Mass is fundamental!

6
Mass Spectrometry for Proteomics
• Mass spectrometry has been around since the turn of the century...• ...why is MS based Proteomics so new?
• Ionization methods• MALDI, Electrospray
• Protein chemistry & automation• Chromatography, Gels, Computers
• Protein sequence databases• A reference for comparison
7
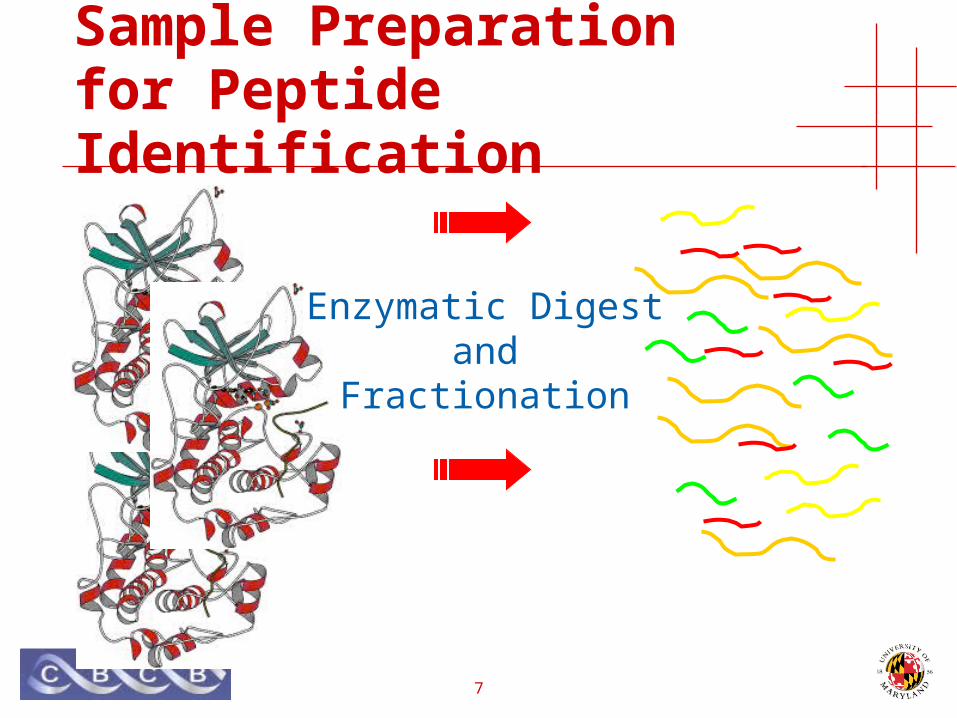
Sample Preparation for Peptide Identification
Enzymatic Digestand
Fractionation
8
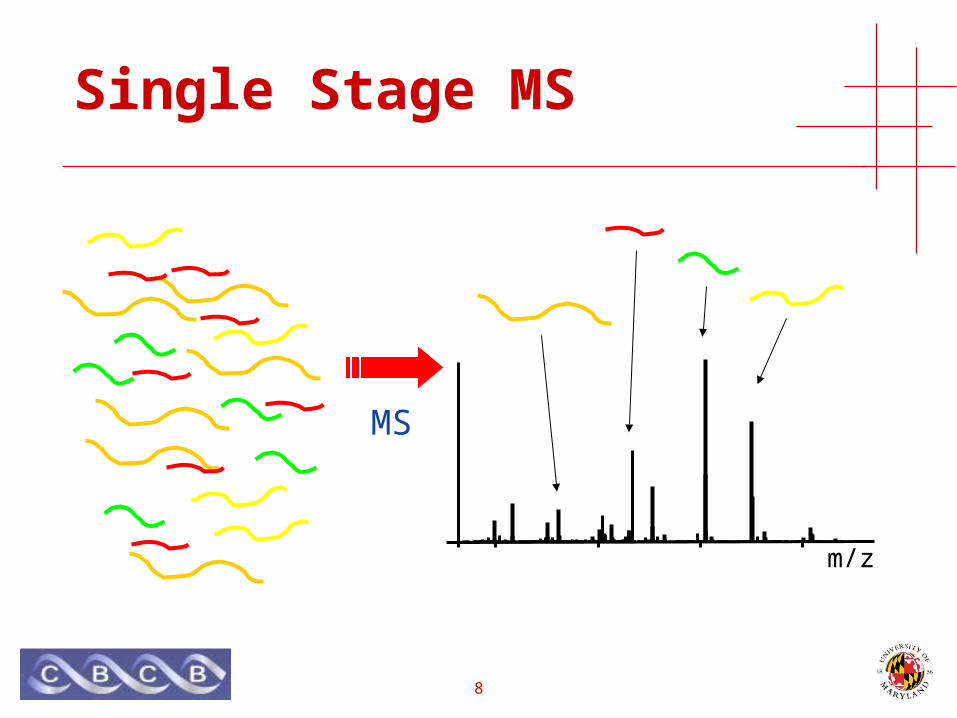
Single Stage MS
MS
m/z
9
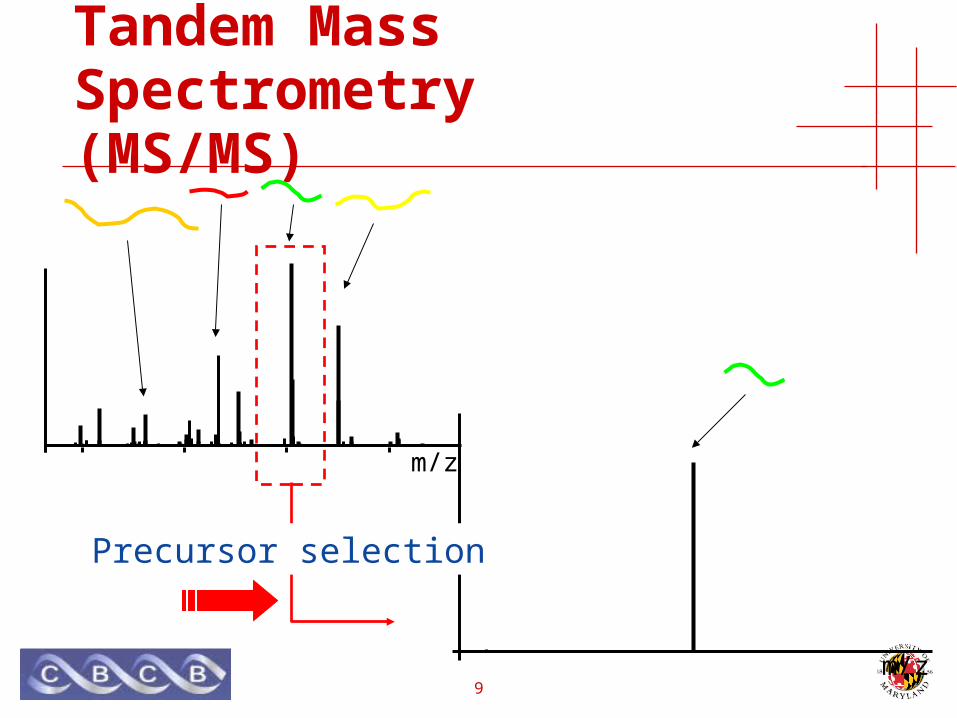
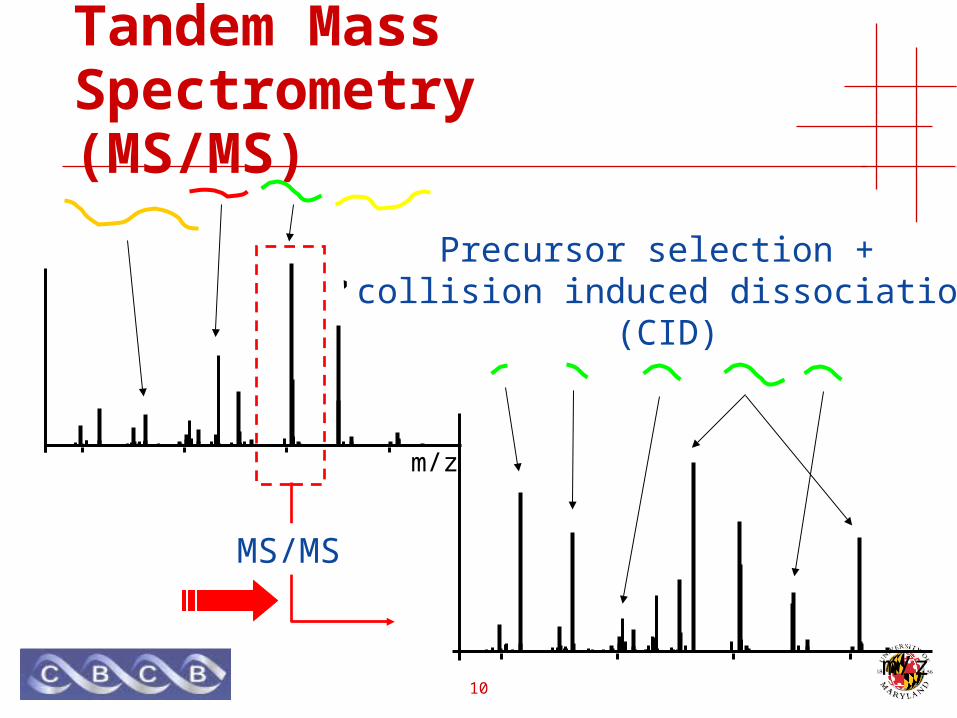
Tandem Mass Spectrometry(MS/MS)
Precursor selection
m/z
m/z
10
Tandem Mass Spectrometry(MS/MS)
Precursor selection + collision induced dissociation
(CID)
MS/MS
m/z
m/z

11
Peptide Identification
• For each (likely) peptide sequence1. Compute fragment masses2. Compare with spectrum3. Retain those that match well
• Peptide sequences from protein sequence databases• Swiss-Prot, IPI, NCBI’s nr, ...
• Automated, high-throughput peptide identification in complex mixtures

12
Why don’t we see more novel peptides?
• Tandem mass spectrometry doesn’t discriminate against novel peptides...
...but protein sequence databases do!
• Searching traditional protein sequence databases biases the results towards well-understood protein isoforms!

13
What goes missing?
• Known coding SNPs
• Novel coding mutations
• Alternative splicing isoforms
• Alternative translation start-sites
• Microexons
• Alternative translation frames

14
Why should we care?
• Alternative splicing is the norm!• Only 20-25K human genes• Each gene makes many proteins
• Proteins have clinical implications• Biomarker discovery
• Evidence for SNPs and alternative splicing stops with transcription• Genomic assays, ESTs, mRNA sequence.• Little hard evidence for translation start site

15
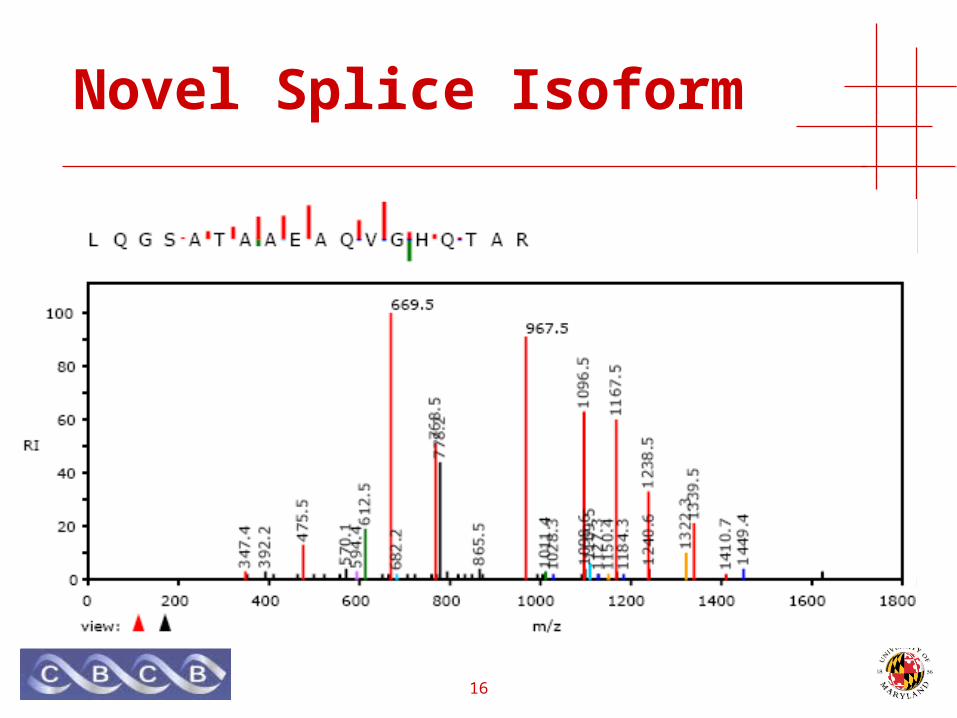
Novel Splice Isoform
• Human Jurkat leukemia cell-line• Lipid-raft extraction protocol, targeting T cells• von Haller, et al. MCP 2003.
• LIME1 gene:• LCK interacting transmembrane adaptor 1
• LCK gene:• Leukocyte-specific protein tyrosine kinase• Proto-oncogene• Chromosomal aberration involving LCK in leukemias.
• Multiple significant peptide identifications
16
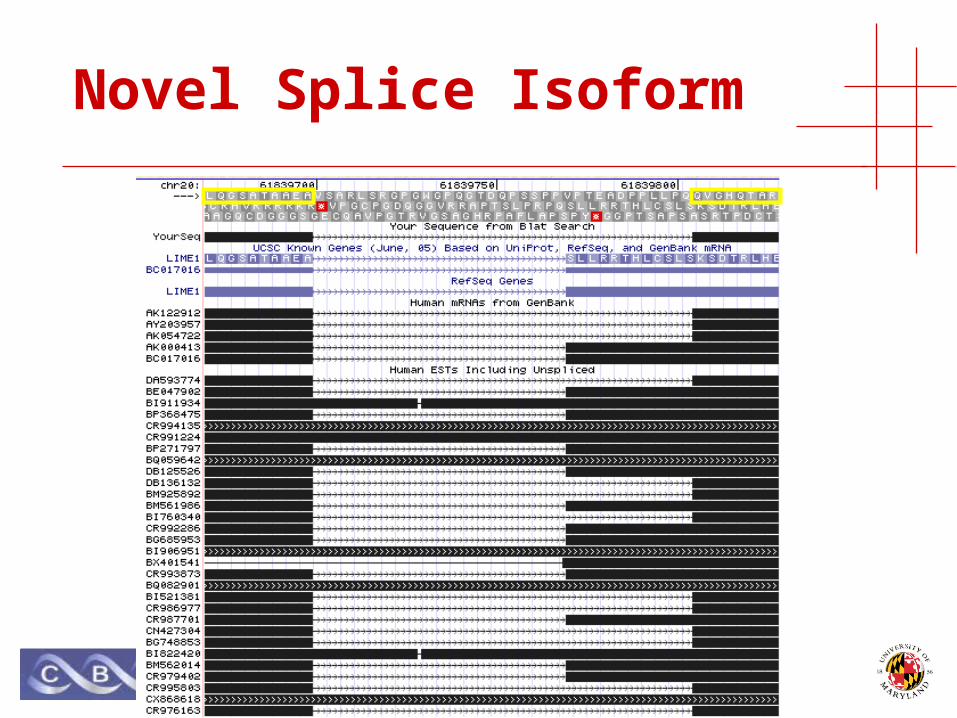
Novel Splice Isoform
17
Novel Splice Isoform
18
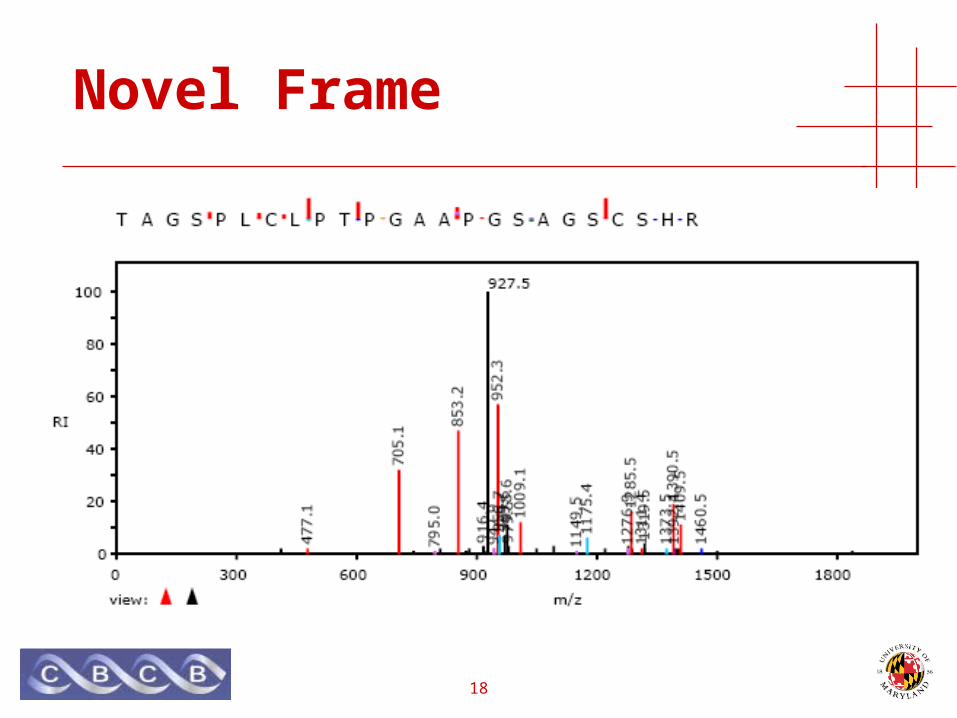
Novel Frame

19
Novel Frame

20
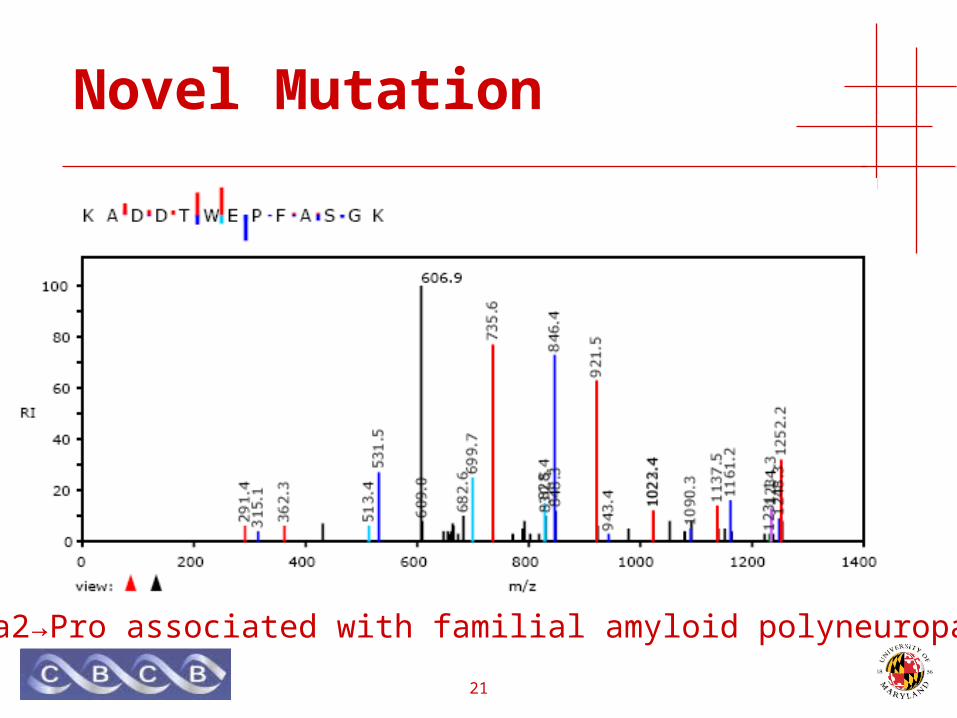
Novel Mutation
• HUPO Plasma Proteome Project• Pooled samples from 10 male & 10 female
healthy Chinese subjects• Plasma/EDTA sample protocol• Li, et al. Proteomics 2005. (Lab 29)
• TTR gene• Transthyretin (pre-albumin) • Defects in TTR are a cause of amyloidosis.• Familial amyloidotic polyneuropathy
• late-onset, dominant inheritance
21
Novel Mutation
Ala2→Pro associated with familial amyloid polyneuropathy

22
Novel Mutation

23
Searching ESTs
• Proposed long ago:• Yates, Eng, and McCormack; Anal Chem, ’95.
• Now:• Protein sequences are sufficient for protein identification• Computationally expensive/infeasible• Difficult to interpret
• Make EST searching feasible for routine searching to discover novel peptides.

24
Searching Expressed Sequence Tags (ESTs)
Pros• No introns!• Primary splicing
evidence for annotation pipelines
• Evidence for dbSNP• Often derived from
clinical cancer samples
Cons• No frame• Large (8Gb)• “Untrusted” by
annotation pipelines• Highly redundant• Nucleotide error
rate ~ 1%

25
Compressed EST Peptide Sequence Database
• For all ESTs mapped to a UniGene gene:• Six-frame translation• Eliminate ORFs < 30 amino-acids• Eliminate amino-acid 30-mers observed once• Compress to C2 FASTA database
• Complete, Correct for amino-acid 30-mers
• Gene-centric peptide sequence database:• Size: < 3% of naïve enumeration, 20774 FASTA entries• Running time: ~ 1% of naïve enumeration search• E-values: ~ 2% of naïve enumeration search results

26
Compressed EST Peptide Sequence Database
• For all ESTs mapped to a UniGene gene:• Six-frame translation• Eliminate ORFs < 30 amino-acids• Eliminate amino-acid 30-mers observed once• Compress to C2 FASTA database
• Complete, Correct for amino-acid 30-mers
• Gene-centric peptide sequence database:• Size: < 3% of naïve enumeration, 20774 FASTA entries• Running time: ~ 1% of naïve enumeration search• E-values: ~ 2% of naïve enumeration search results
27
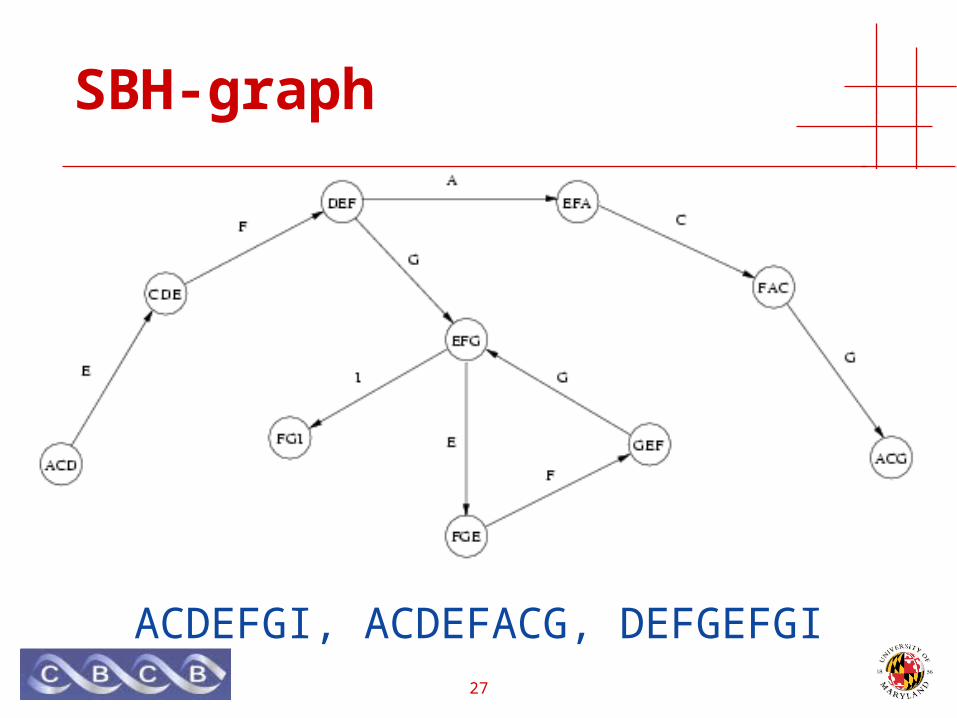
SBH-graph
ACDEFGI, ACDEFACG, DEFGEFGI
28
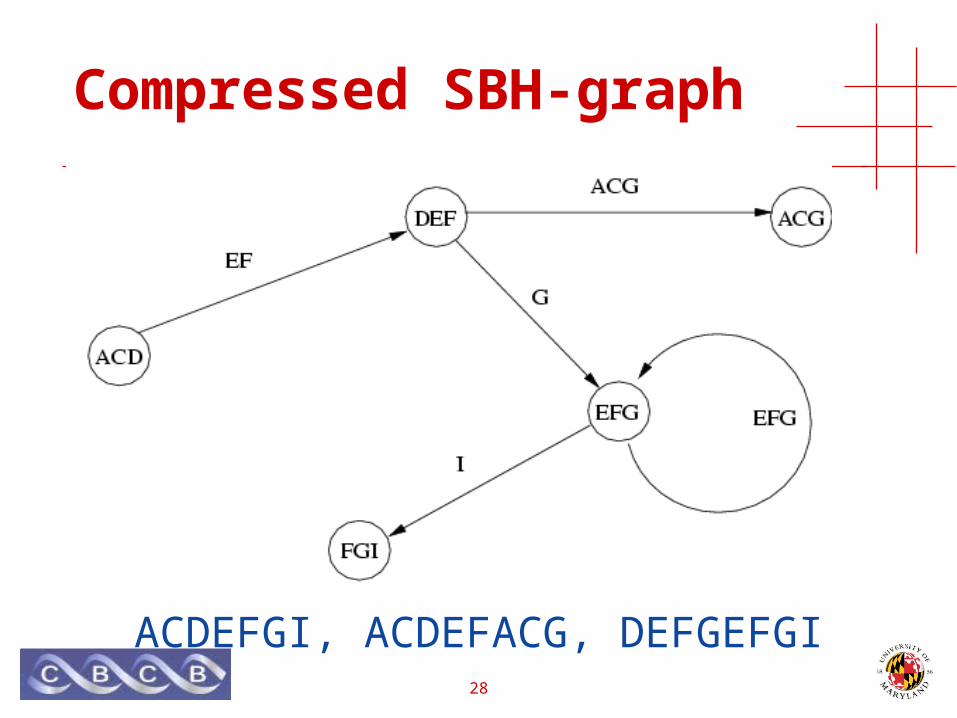
Compressed SBH-graph
ACDEFGI, ACDEFACG, DEFGEFGI
29
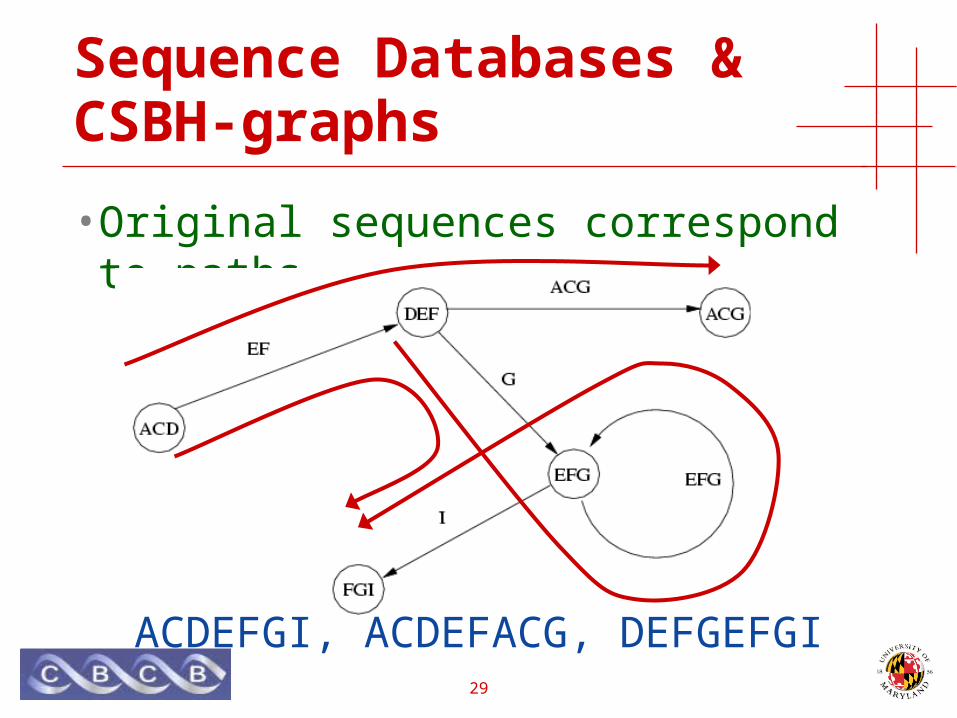
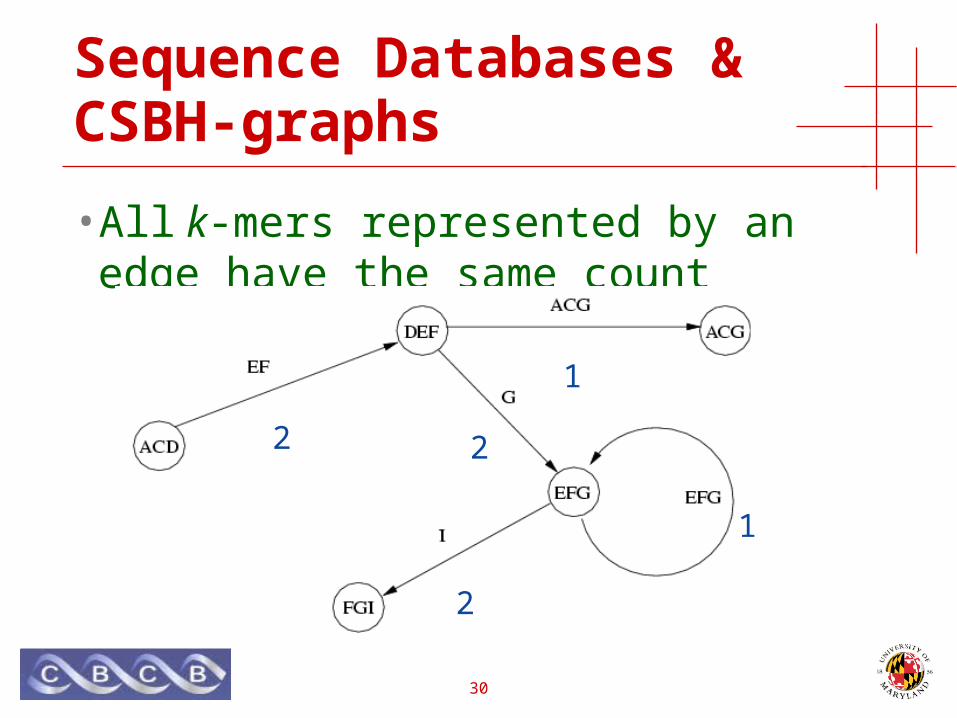
Sequence Databases & CSBH-graphs
• Original sequences correspond to paths
ACDEFGI, ACDEFACG, DEFGEFGI
30
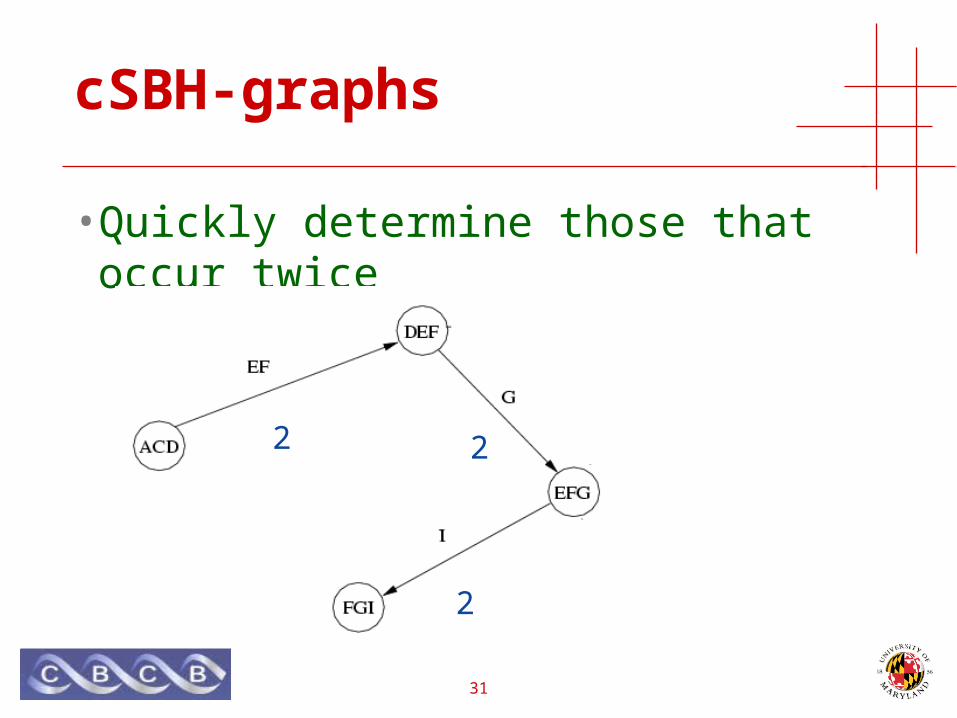
Sequence Databases & CSBH-graphs
• All k-mers represented by an edge have the same count
2 2
1
2
1
31
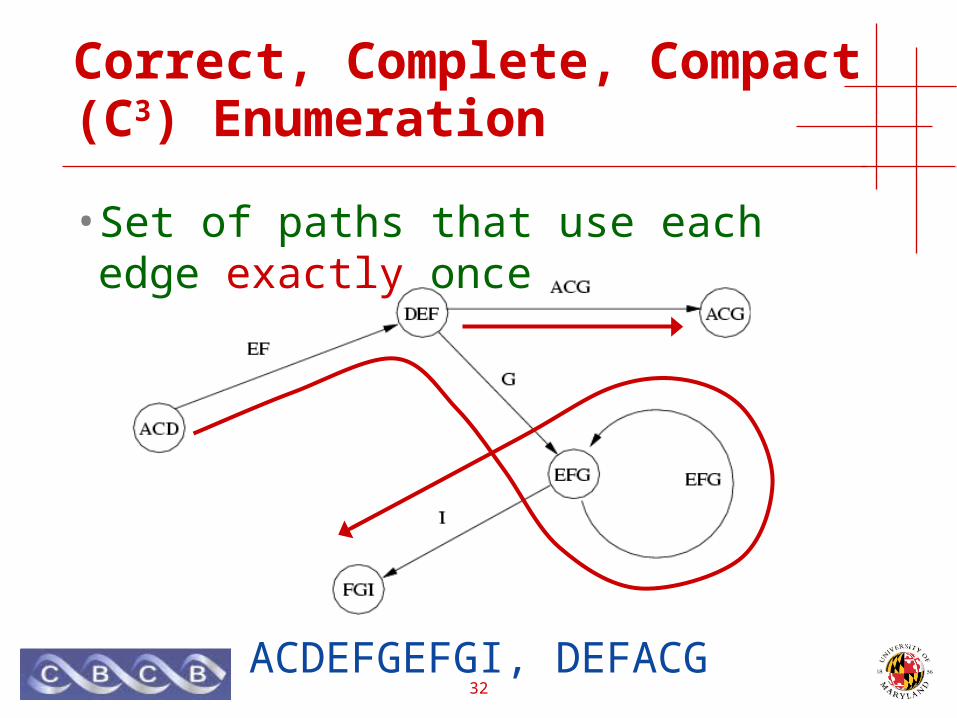
cSBH-graphs
• Quickly determine those that occur twice
2 2
1
2
32
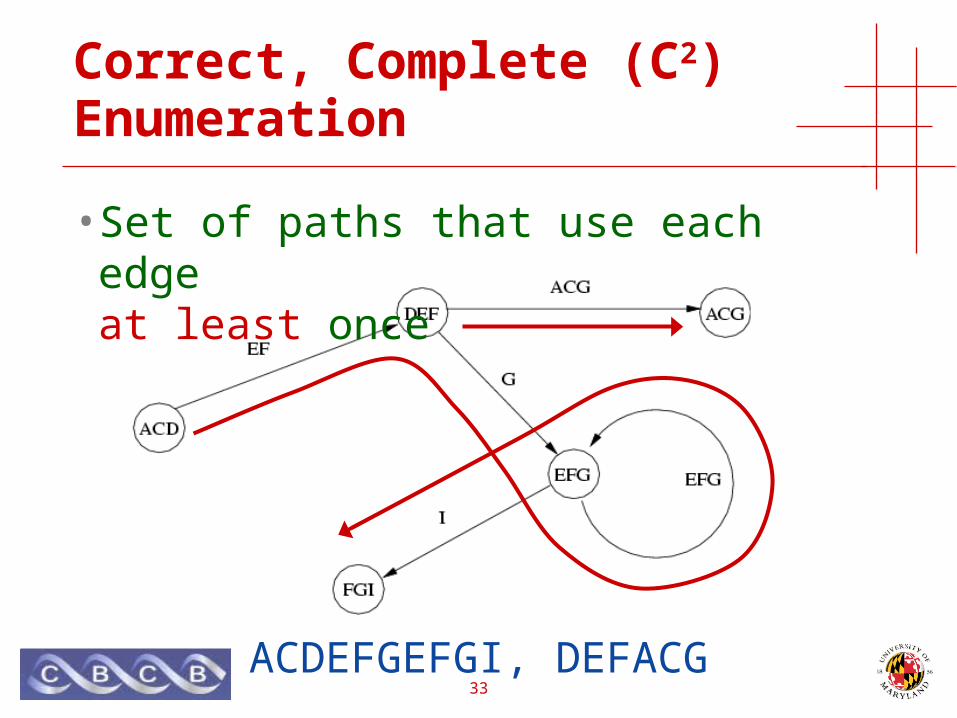
Correct, Complete, Compact (C3) Enumeration
• Set of paths that use each edge exactly once
ACDEFGEFGI, DEFACG
33
Correct, Complete (C2) Enumeration
• Set of paths that use each edge at least once
ACDEFGEFGI, DEFACG

34
Patching the CSBH-graph
• Use artificial edges to fix unbalanced nodes

35
Compressed EST Database
• Gene centric compressed EST peptide sequence database• 20,774 sequence entries• ~8Gb vs 223 Mb• ~35 fold compression
• 22 hours becomes 15 minutes• E-values improve by similar factor!
• Makes routine EST searching feasible• Search ESTs instead of IPI?

36
“Novel Peptide” Computational Infrastructure
• Binaries (C++)• cSBH-graph construction
• Condor grid-enabled• Eulerian path k-mer enumeration
• Suitable for large graphs
• Data-model for peptide identification• Spectra (>5 million)• Peptide identifications
• Mascot, SEQUEST, X!Tandem, NIST • Genomic context of peptides

37
“Novel Peptide” Computational Infrastructure
• Condor grid-enabled MS/MS search• Mascot, X!Tandem, (Inspect, OMSSA)
• TurboGears python web-stack• SQLObject Object-Relational-Manager• MVC web-application framework• Suitable for AJAX & web-services too
• Integration with UCSC genome browser• caBIG compatible web-services
• Java applet for viewing spectra
38
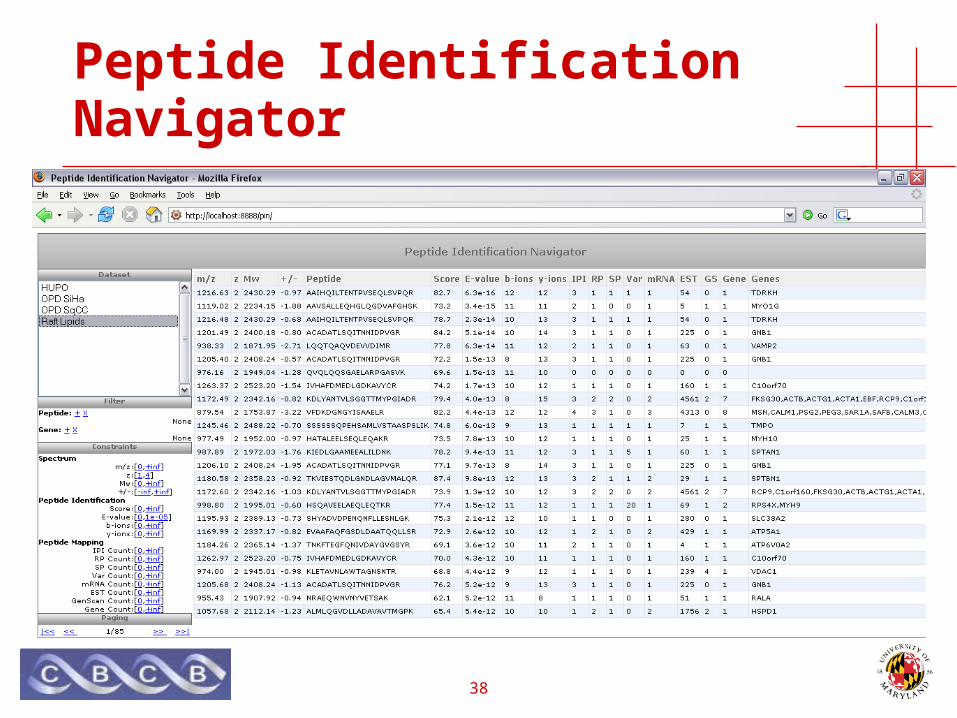
Peptide Identification Navigator
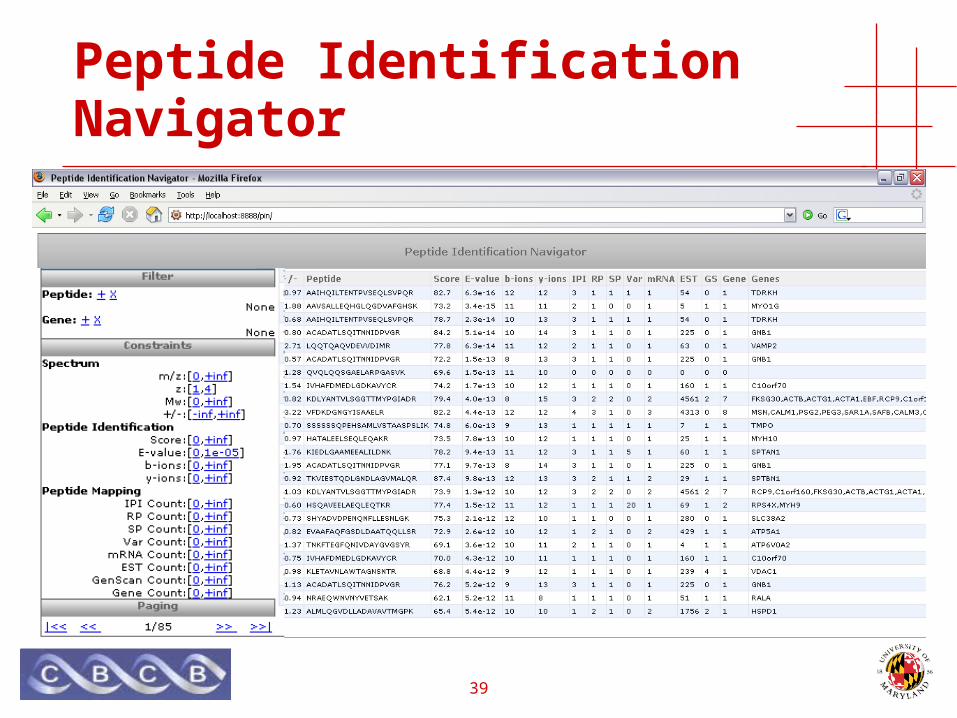
39
Peptide Identification Navigator
40
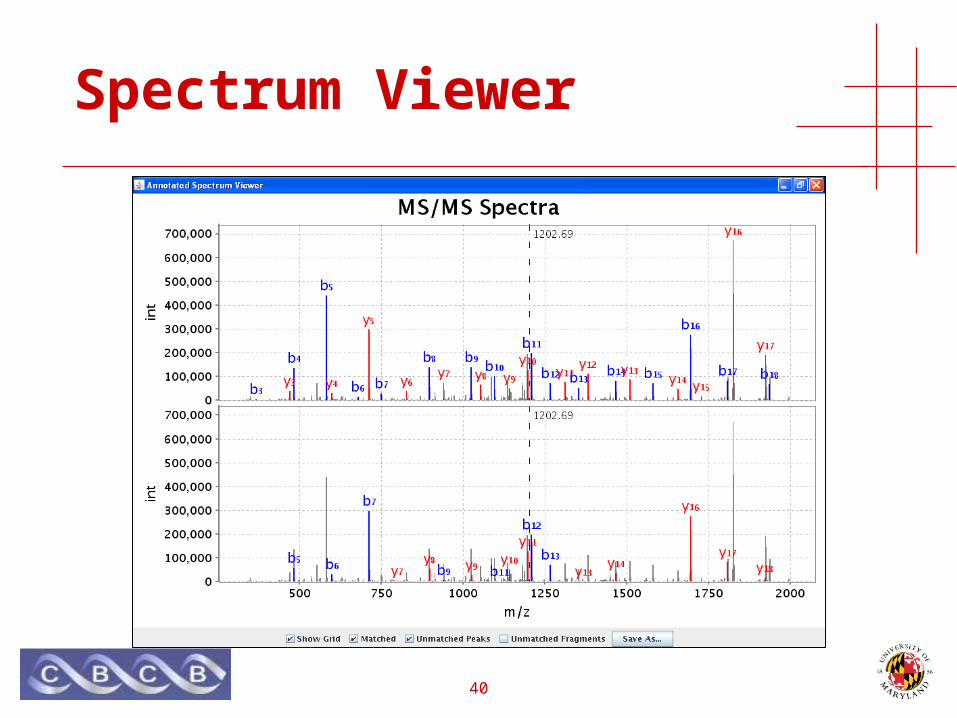
Spectrum Viewer
41
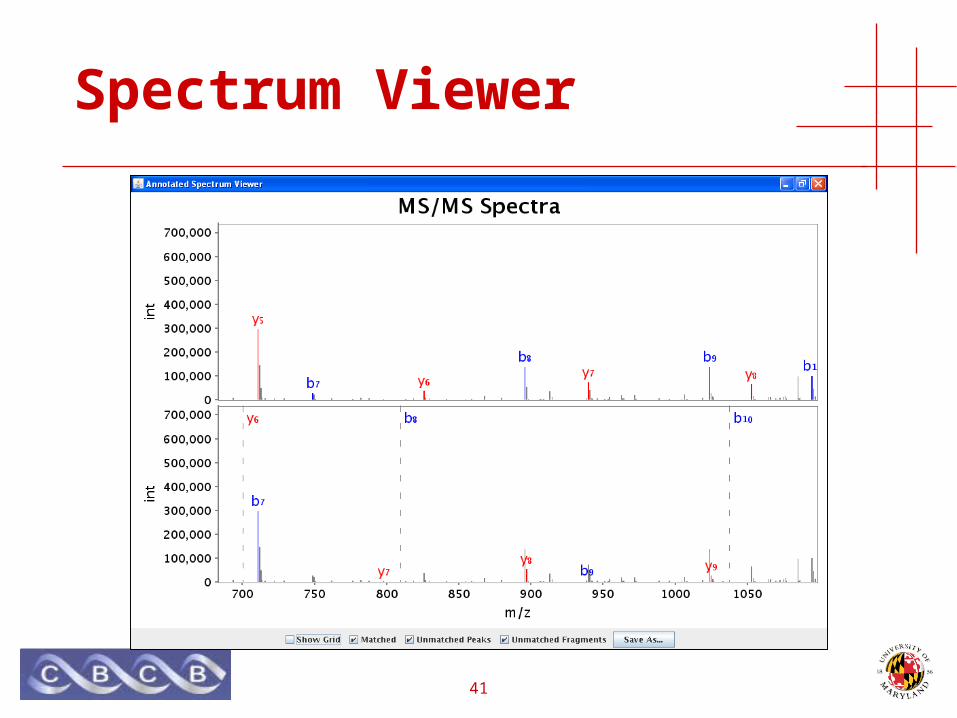
Spectrum Viewer

42
Back to the lab...
• Current LC/MS/MS workflows identify a few peptides per protein• ...not sufficient for protein isoforms
• Need to raise the sequence coverage to (say) 80%• ...protein separation prior to LC/MS/MS
analysis• Potential for database of splice sites of
(functional) proteins!
43
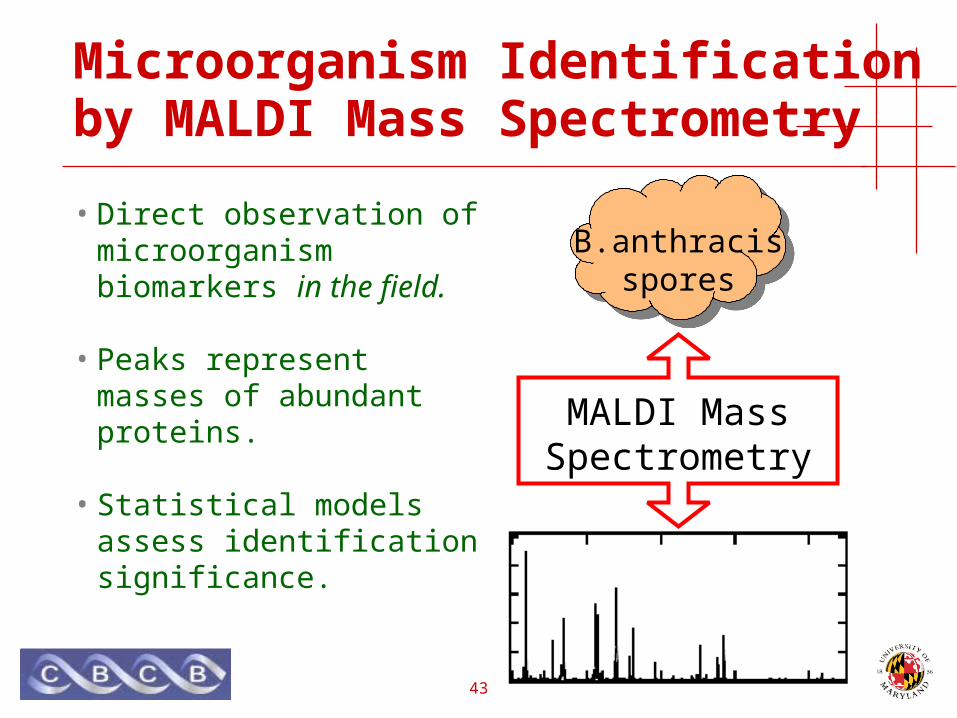
Microorganism Identification by MALDI Mass Spectrometry
• Direct observation of microorganism biomarkers in the field.
• Peaks represent masses of abundant proteins.
• Statistical models assess identification significance.
B.anthracisspores
MALDI Mass Spectrometry

44
Key Principles
• Protein mass from protein sequence• No introns, few PTMs
• Specificity of single mass is very weak• Statistical significance from many peaks
• Not all proteins are equally likely to be observed• Ribosomal proteins, SASPs
45
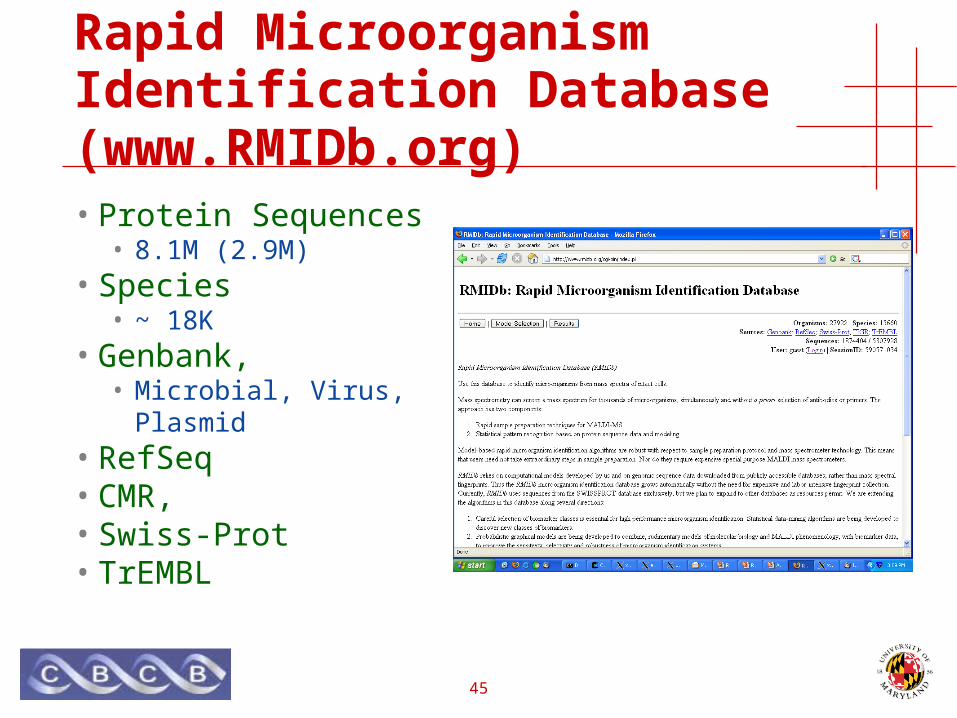
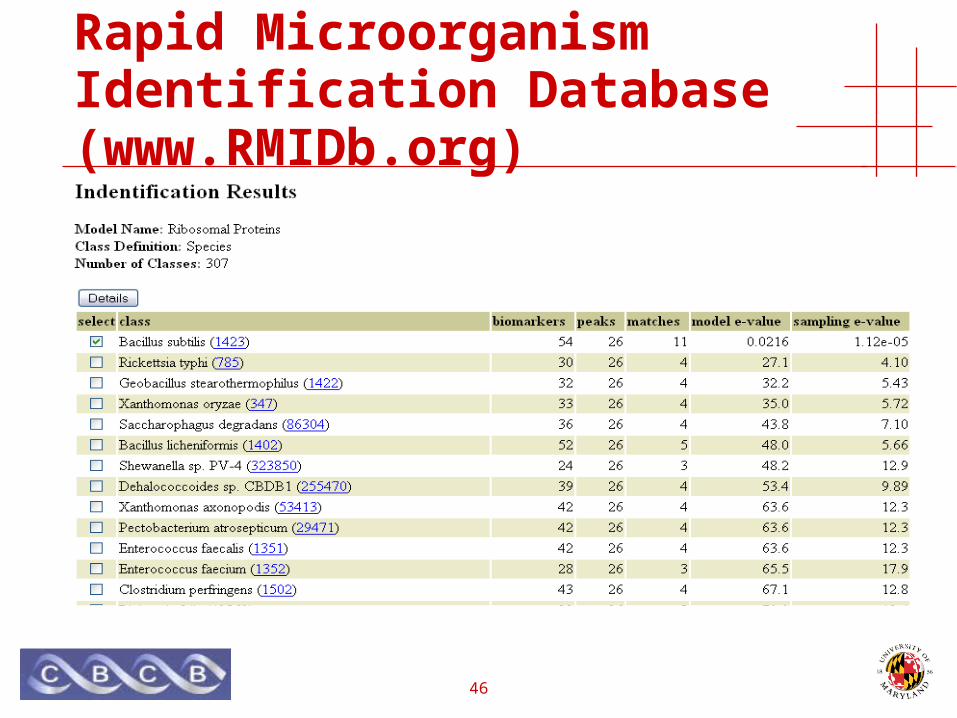
Rapid Microorganism Identification Database (www.RMIDb.org)
• Protein Sequences• 8.1M (2.9M)
• Species• ~ 18K
• Genbank,• Microbial, Virus, Plasmid
• RefSeq• CMR,• Swiss-Prot• TrEMBL
46
Rapid Microorganism Identification Database (www.RMIDb.org)

47
Informatics Issues
• Need good species / strain annotation• B.anthracis vs B.thuringiensis
• Need correct protein sequence• B.anthracis Sterne α/β SASP• RefSeq/Gb: MVMARN... (7442 Da)• CMR: MARN... (7211 Da)
• Need chemistry based protein classification

48
Conclusions
• Proteomics can inform genome annotation• Eukaryotic and prokaryotic • Functional vs silencing variants
• Peptides identify more than just proteins• Untapped source of disease biomarkers
• Compressed peptide sequence databases make routine EST searching feasible

49
Future Research Directions
• Identification of protein isoforms:• Optimize proteomics workflow for isoform
detection• Identify splice variants in cancer cell-lines
(MCF-7) and clinical brain tumor samples• Aggressive peptide sequence enumeration• dbPep for genomic annotation• Open, flexible informatics infrastructure for
peptide identification

50
Future Research Directions
• Proteomics for Microorganism Identification• Specificity of tandem mass spectra• Revamp RMIDb prototype• Incorporate spectral matching
• Primer design• k-mer sets as FASTA sequence databases• Uniqueness oracle for exact and inexact match• Integration with Primer3• Tiling, multiplexing, pooling, & tag arrays

51
Acknowledgements
• Chau-Wen Tseng, Xue Wu• UMCP Computer Science
• Catherine Fenselau, Steve Swatkoski• UMCP Biochemistry
• Calibrant Biosystems
• PeptideAtlas, HUPO PPP, X!Tandem
• Funding: National Cancer Institute


![Integrating transcriptome and proteome profiling ... · Proteomic diversity of a eukaryote is largely attributed to alternative mRNA splicing [4]. The number of possible splice variants](https://cdn.vdocuments.mx/doc/165x107/5e1066a1a9bcaa2462626ad8/integrating-transcriptome-and-proteome-profiling-proteomic-diversity-of-a-eukaryote.jpg)




