
Ailevi Kanser Sendromları
Prof.Dr. Özgür ÇOĞULU
EÜTF Pediatrik Genetik Bilim Dalı
Tıbbi Genetik Anabilim Dalı

Dinleyici Dağılımı
Sıkılanlar 1
Sıkılanlar 2
İlgili olanlar

Kanser Nedir?
Hücre büyümesinin kontrol dışı olması ile karakterize bir
hastalık, 2 şekilde gerçekleşir;
Nesilden nesile geçen germline, herediter
Germ hücrelerinde rastgele oluşan mutasyonlar sonucu
Sperm
o Hücre bölünmesinin daha fazla olması nedeniyle mutasyon geliştirme riski daha
fazla
%95’i kodlama yapmayan bölgede
Ovum
Hayatın herhangi bir döneminde oluşan genetik değişiklikler,
sporadik olgular, somatik mutasyonlar
Yaklaşık 340 kadar gen
Frank TS.Hereditary cancer syndromes. Arch Pathol Lab Med. 2001 Jan;125(1):85-90.


Kanser Geliştirme Olasılığı
Normal bir hücrede mutasyon oluşması durumunda
Apoptoz
Tamir
İnsanda yaklaşık hücre sayısı 1014
Herhangi bir hücrenin kanser olabilmesi için 6 spesifik
mutasyona uğraması gerekir
Mutasyon oranı 10-6
Bir hücrenin 6 mutasyonu olma oranı 10-36!!!
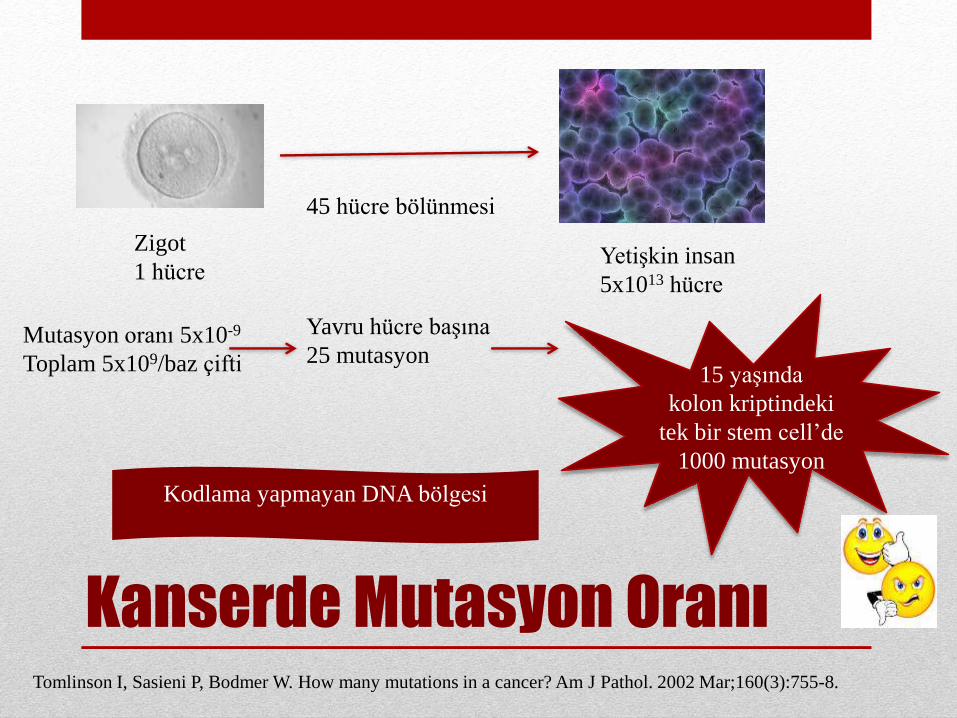
Kanserde Mutasyon Oranı
Zigot
1 hücreYetişkin insan
5x1013 hücre
45 hücre bölünmesi
Mutasyon oranı 5x10-9
Toplam 5x109/baz çifti
Yavru hücre başına
25 mutasyon15 yaşında
kolon kriptindeki
tek bir stem cell’de
1000 mutasyon
Kodlama yapmayan DNA bölgesi
Tomlinson I, Sasieni P, Bodmer W. How many mutations in a cancer? Am J Pathol. 2002 Mar;160(3):755-8.

Kanser oluşumuHer 3-4 kişiden biri Ölüm
• Erkeklerin 1/4
• Kadınların 1/5
İdealErken tanı,
Tarama,
Hedefe yönelik tedavi,
Toksitesi az ilaç kullanma
birçok kanserde yeterli ilerleme yok
KanserKelly KM, Sweet K. In search of a familial cancer risk assessment tool. Clin Genet. 2007 Jan;71(1):76-83.
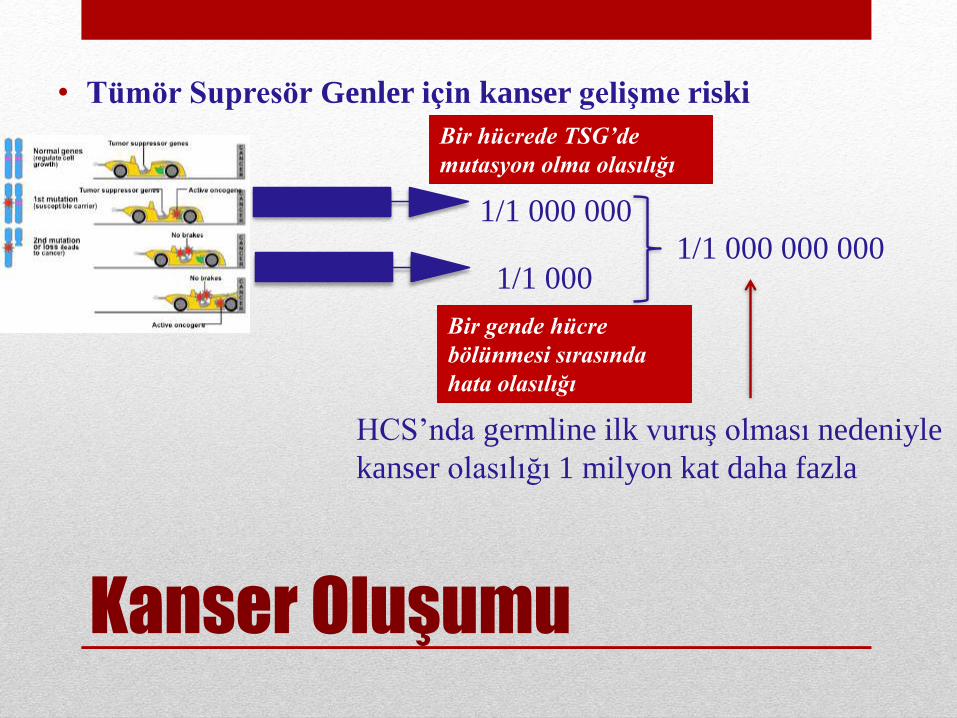
Kanser Oluşumu
• Tümör Supresör Genler için kanser gelişme riski
1/1 000 000
1/1 0001/1 000 000 000
HCS’nda germline ilk vuruş olması nedeniyle
kanser olasılığı 1 milyon kat daha fazla
Bir hücrede TSG’de
mutasyon olma olasılığı
Bir gende hücre
bölünmesi sırasında
hata olasılığı

Kanser
Genetik bir hastalık
Komplike çok basamaklı olaylar zinciri
Çok sayıda mutasyonun (anahtar rol oynayan genlerde) fatal
birikimi
– Nokta mutasyonlar
– Kromozomal ya da gen düzeyinde delesyonlar
– Heterozigositenin kaybı
– Metilasyon bozuklukları
Sonucunda
– Onkogen aktivasyonu,
– Tümör supresyon inhibisyonu
– Anormal gen ekspresyonu,
– Genetik ve epigenetik anormalliklerin görülmesine kadar giden bir dizi olaylar,
– Malign fenotipin oluşumu
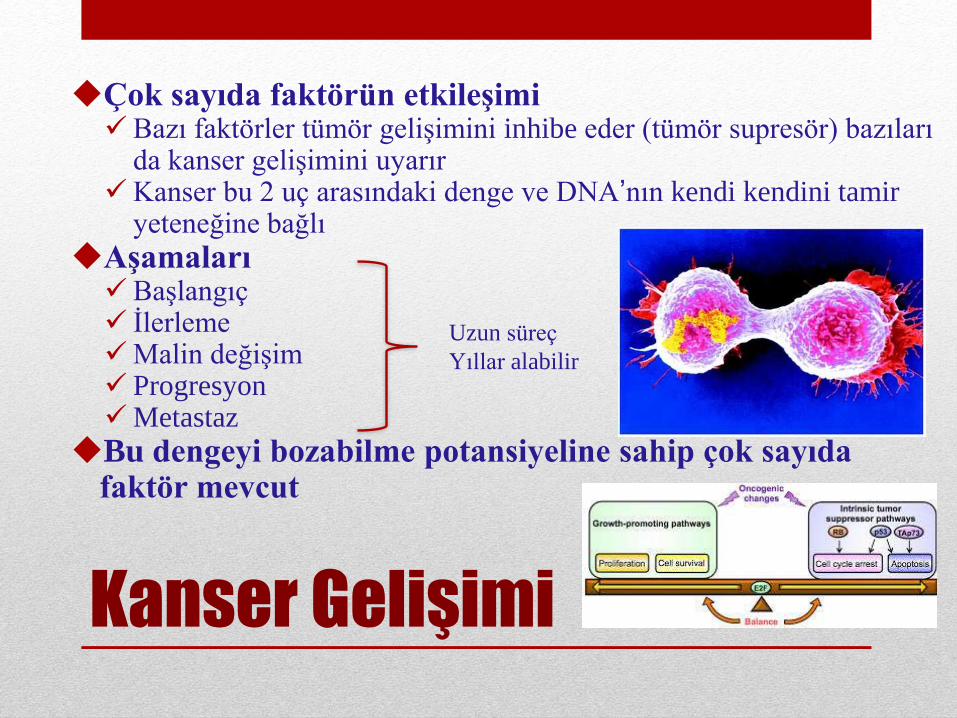
Kanser Gelişimi
Çok sayıda faktörün etkileşimiBazı faktörler tümör gelişimini inhibe eder (tümör supresör) bazıları
da kanser gelişimini uyarırKanser bu 2 uç arasındaki denge ve DNA’nın kendi kendini tamir
yeteneğine bağlı
AşamalarıBaşlangıç İlerlemeMalin değişim ProgresyonMetastaz
Bu dengeyi bozabilme potansiyeline sahip çok sayıda faktör mevcut
Uzun süreç
Yıllar alabilir

Kanser için Gerekli 6 Özellik
Eksternal büyüme sinyalleriyle bağımsız olarak
bölünebilme
Eksternal büyümeye karşı olan sinyallere karşı duyarsızlık
Apoptozdan kaçma
Ölümsüzlük
Anjiogenezin uyarılması
İnvazyon

Kansere Karşı Savunma
Mekanizması
Herhangi 3 kişiden 1’i hayatının bir döneminde kansergeliştirecek
Dolayısıyla bir şekilde savunmanın alt olma durumu söz konusu
Mevcut mutasyonun diğer mutasyonları tetiklemesi
2 mekanizma
o Mutant hücreye ait hızlı büyüme avantajı
o Genomun destabilize edilmesi
– Kromozomal instabilite
– Mikrosatellit instabilitesi
Onkogenler ve tümör supresör genler

Kanserler
Sporadik-Somatik mutasyonlar sonucu
Tek bir hücrede gelişir
Sonraki nesile geçmez
Sporadik
Herediter
Herediter kanser sendromu
Kansere yatkınlık sendromu
Herediter kanser
o 50’den fazla tipi yatkınlık nedeniyle oluşur
o Ailevi özellik söz konusu
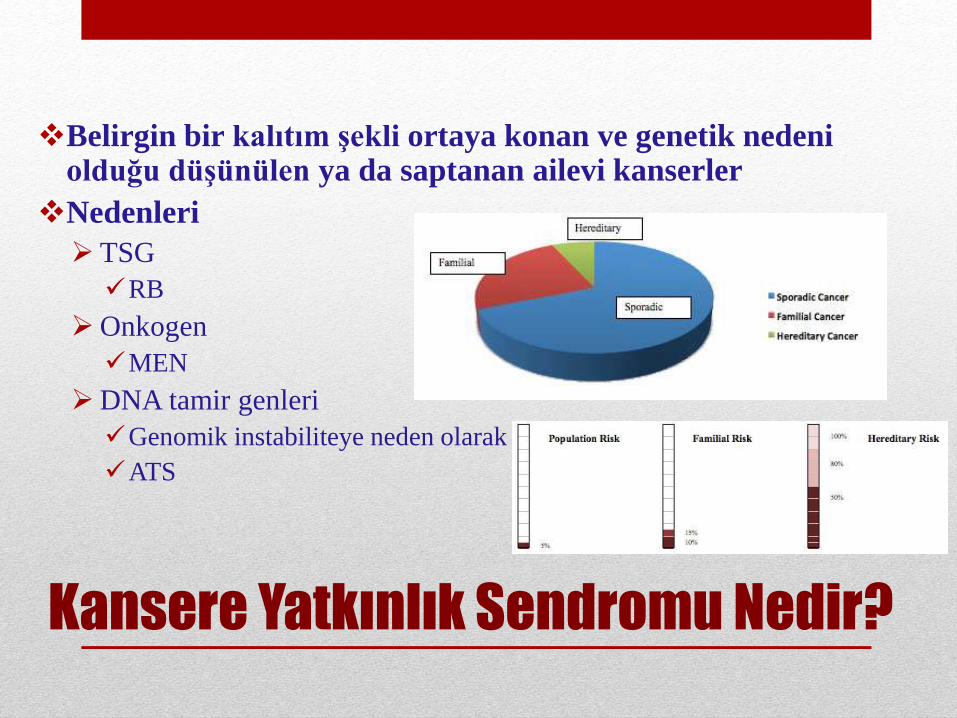
Kansere Yatkınlık Sendromu Nedir?
Belirgin bir kalıtım şekli ortaya konan ve genetik nedeniolduğu düşünülen ya da saptanan ailevi kanserler
Nedenleri
TSG
RB
Onkogen
MEN
DNA tamir genleri
Genomik instabiliteye neden olarak
ATS

Tümör supresör gen (TP53, BRCA1, BRCA2, APC, RB1)
Vücudun gardiyanları, trafiğin kontrolü!!!
Fonksiyon kaybı durumunda kanser gelişimi söz konusu
Hücre siklusunun devamını engeller
Apoptozu uyarır
Tek alleli fonksiyon için yeterli
Kanser Genetiği
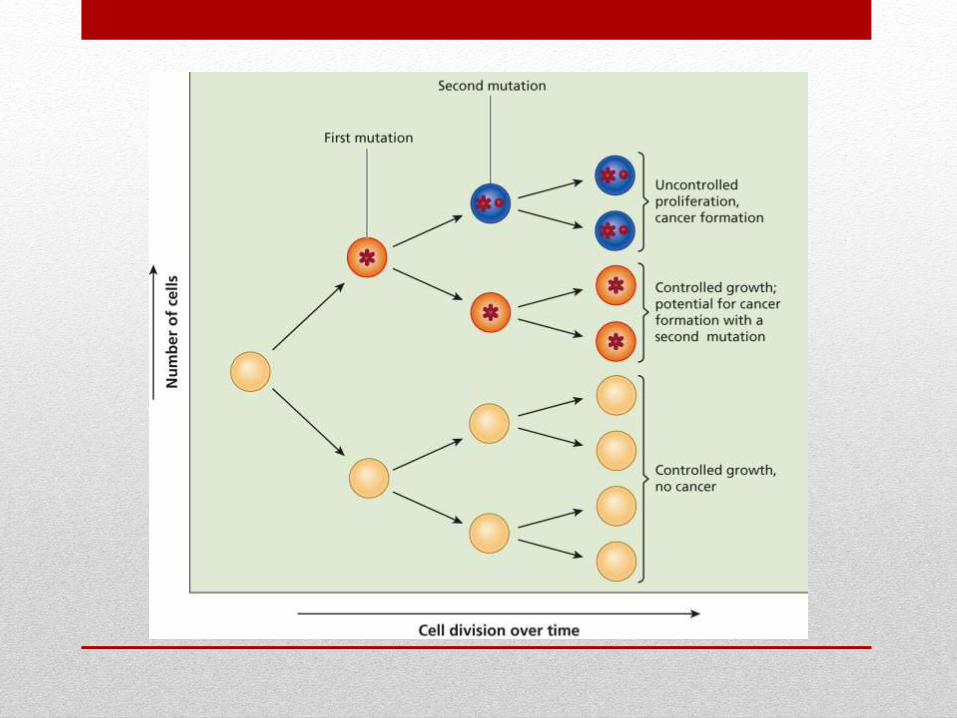

Tümör Süpresör Genler
Gatekeepers
Hücre büyümesini sınırlandırır ve hücre siklusunu, proliferasyonu,
diferansiasyonu kontrol eder
Rb, BRCA1, APC
Caretakers
DNA’yı kontrol eder ve tamir eder
p53
Landscapers
Mikroçevreyi düzenler
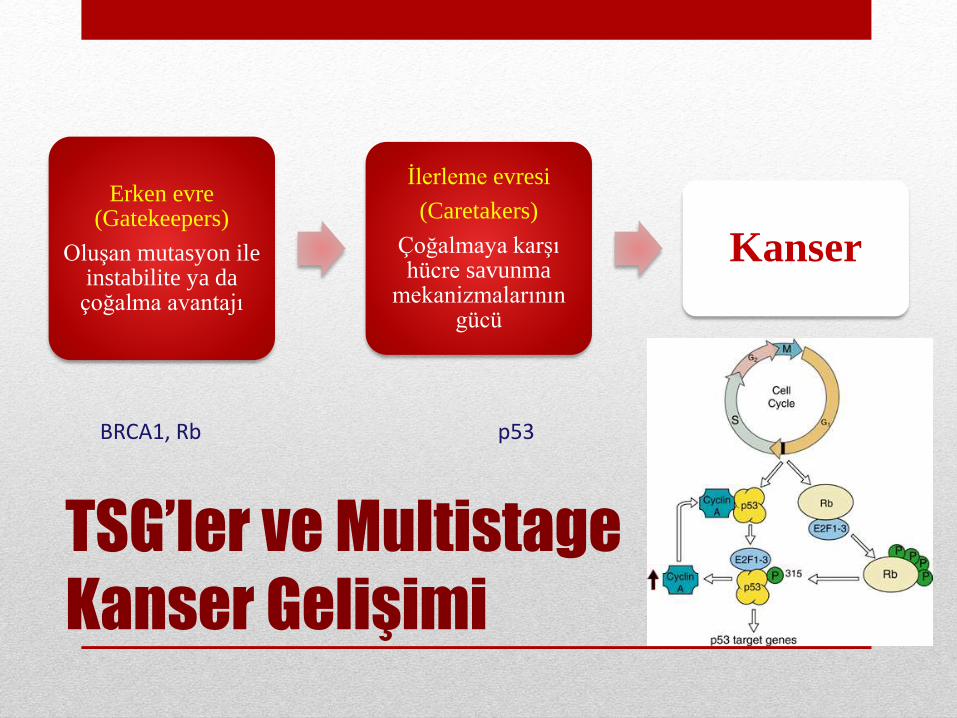
TSG’ler ve Multistage
Kanser Gelişimi
Erken evre(Gatekeepers)
Oluşan mutasyon ileinstabilite ya da çoğalma avantajı
İlerleme evresi
(Caretakers)
Çoğalmaya karşıhücre savunma
mekanizmalarınıngücü
Kanser
BRCA1, Rb p53

Kanser Genetiği
Onkogenler (Myc, Ras, HER2/neu, RET, KIT, BCR-ABL)
Kanser oluşturma kapasitesine sahip gen
Normal durumda proto-onkogen ve fizyolojik fonksiyonu var
Tek allelin farklı nedenlerle kaybı kontrolsüz hücre çoğalımı için yeterli
Kaybı mutasyon oranını arttırması bakımından önemli
80 civarında tarif edilmiş
DNA tamir mekanizmasında görevli genler

En Sık Karşılaşılan Proto-onkogenler
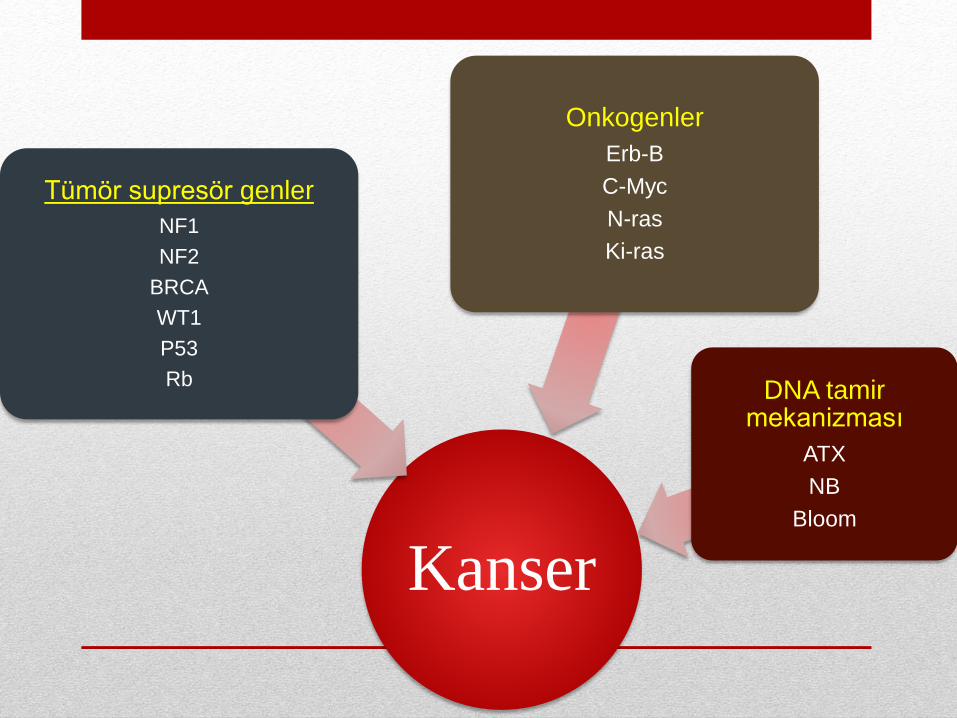
Kanser
Tümör supresör genler
NF1
NF2
BRCA
WT1
P53
Rb
Onkogenler
Erb-B
C-Myc
N-ras
Ki-ras
DNA tamirmekanizması
ATX
NB
Bloom

Kanser Genetiği
Onkogen etkilenmesi nedeniyle gelişen kalıtsal kanserler TSG’lere
oranla daha seyrek
Mutasyon nedeniyle embriyogenezde kontrolsüz hücre büyümesine
bağlı spontan terminasyon gelişir
Germline kalıtılan genler sadece kalıtsal kanser oluşumunda
sorumlu olmayıp sporadik aynı kanser tipinin gelişiminde de
sorumlu
Spermlerde aktif bölünme daha fazla olması nedeniyle ovuma göre
daha yüksek olasılıkla sonraki nesillere mutasyon aktarımı
%95’i kodlama yapmayan bölgede

Kansere Yatkınlık Sendromu
Kanserlerin yaklaşık %10’u kalıtsal gen değişikliği
Kansere karşı koruyan bir yapıda olan genlerde bir değişiklik
olması sonucu üst nesilden kalıtılır
%50 olasılıkla bu değişiklik sonraki nesile geçer
Dominant
Mutlaka kanser geliştireceği ya da her tip kanser gelişeceği
anlamına gelmez
"genetic susceptibility” genetik yatkınlık tanımlaması

Kansere Yatkınlık
Sendromlarında Sorun Nerede?
Üreme hücrelerinde mutasyon
Sonraki nesile geçer
Hücre siklusu
DNA tamiri ile ilgili genlerde etkilenme söz konusu
Bütün hücrelerde germline mutasyon mevcut
Mitotik siklus sonucu somatik hücrelerde sonraki mutasyonlardahastalık gelişir
Genellikle genetik olarak ailesel anlamda otozomal dominant; hücresel düzeyde resesif yani “two hit” hipotezi geçerli
Tespit edilmeyen hastalığa yol açan birçok mutasyon daha vardır
Turnbull C, Hodgson S. Genetic predisposition to cancer. Clin Med. 2005 Sep-Oct;5(5):491-8.
Rahner N, Steinke V. Hereditary cancer syndromes. Dtsch Arztebl Int. 2008 Oct;105(41):706-14.

Kansere Yatkınlık Sendromu
Bazılarında sadece kanser gelişimi ön plandadır
Daha nadir diğerlerinde multisistem etkilenme
• Tipik fenotip
• Nörolojik patoloji vb…
Kanser
KalıtsalYatkınlık
%10
Kanser Bulgusu
Ön Planda
HNPCC, HBOC, MEN, Rb..
SendromikBulgular Ön
Planda
Cowden, Nörofibromatozis, Costello, Bloom,
WAGR..Sporadik
%90
En İyi Bilinen Sendromlar
HNPCC
FAP
HBOC
MEN
Frank TS.Hereditary cancer syndromes. Arch Pathol Lab Med. 2001 Jan;125(1):85-90.

Yüksek Düzeyde Penetrans
Gösteren Kanserler
• Ataksi Telenjiektazi
• Bloom
• Cowden
• Familyal adenomatozis polipozis
• Peutz Jeggers
• Rothmund Thomson
• TS
• Von Hippel Lindau
• Kseroderma Pigmentozum
• NF
• MEN
• Nijmegan Breakage
• Li Fraumeni
• Fanconi
• HNCC
• Rb
• Herediter papiller renal kanser
Nagy R, Sweet K, Eng C. Highly Penetrant Hereditary Cancer Syndromes. Oncogene. 2004 Aug 23;23(38):6445-70.
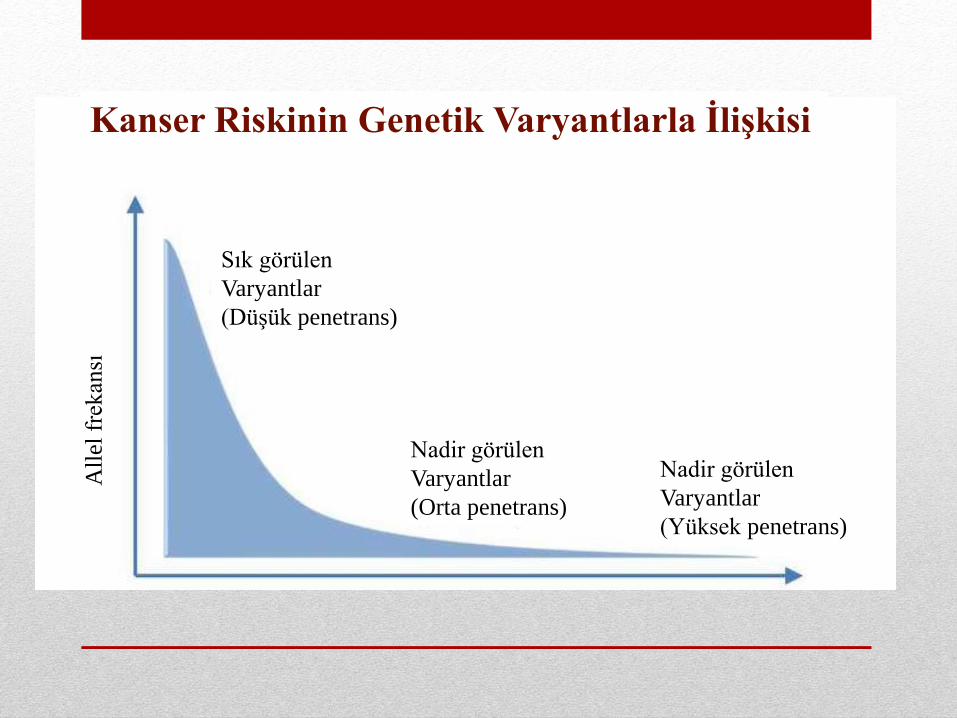
All
elfr
ekan
sı
Sık görülen
Varyantlar
(Düşük penetrans)
Nadir görülen
Varyantlar
(Orta penetrans)
Nadir görülen
Varyantlar
(Yüksek penetrans)
Kanser Riskinin Genetik Varyantlarla İlişkisi

Kansere Yatkınlık Sendromu
Tarif edilen
200
Çoğunluğu otozomal dominant !!
Artmış malignite riski ile karakterize olmalarına rağmen sıklıkla
benign tümörlere ve jeneralize hastalık tablolarına yatkınlık
yaratırlar
Cowden hastalığı
MEN
Birlikte düşünüldüğünde 70 yaşına kadar tam penetrans
Nagy R, Sweet K, Eng C. Highly Penetrant Hereditary Cancer Syndromes. Oncogene. 2004 Aug 23;23(38):6445-70.

Kansere Yatkınlık Sıklığı
Yetişkin kanserlerinin %5-10’u
Çocuklukta Kansere Yatkınlık Sendromu
370 kanser hastası-kanser enstitüsü
o Toplam %29 kanser araştırılmasına aday, riskli grup (n=109)
Ailede kanser hikayesi (%61)
Kalıtsal kanser düşündürtecek tümör (%18)
Genetik tanı gerektirten tıbbi bir nedene bağlı (%16)
Ailede başka bir tıbbi durum nedeniyle (%6)
Schiffman JD, Geller JI, Mundt E, Means A, Means L, Means V. Update on pediatric cancer predisposition syndromes.
Pediatr Blood Cancer. 2013 Aug;60(8):1247-52.
Knapke et al. Hereditary cancer risk assessment in a pediatric oncology follow-up clinic. Pediatr Blood Cancer. 2012
Jan;58(1):85-9.

Neden Herediter Kanser
Sendromunu Tespit Önemli?
Ömür boyu takip gerekir
Disiplinler arası kooperasyon gereken
Özel takip
Akrabalar taranır
Aile üyelerine bilgi vermek gerekir
Rahner N, Steinke V. Hereditary cancer syndromes. Dtsch Arztebl Int. 2008 Oct;105(41):706-14.
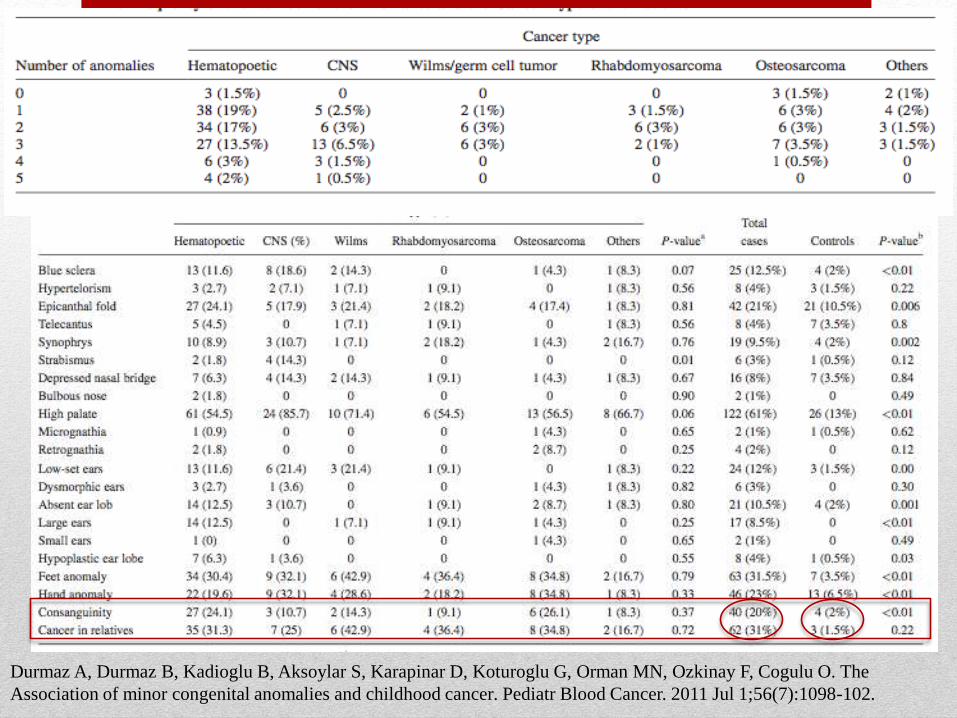
Durmaz A, Durmaz B, Kadioglu B, Aksoylar S, Karapinar D, Koturoglu G, Orman MN, Ozkinay F, Cogulu O. The
Association of minor congenital anomalies and childhood cancer. Pediatr Blood Cancer. 2011 Jul 1;56(7):1098-102.

Ne Zaman Kansere Yatkınlıktan
Şüphelenilmeli?
Aynı aile tarafında 2 ve üzerinde aynı tip kanser gelişimi
3 nesil aile ağacı analizi en önemlisi
Birden fazla jenerasyonda hastalığın görülmesi
Erken yaşlarda hastalığın ortaya çıkması 50 yaş
Bir kişide birden fazla farklı kanser gelişimi
Atipik lokalizasyon ya da bilateral olma, sağ yerleşimli kolorektal kanser
Meme kanseri gibi genetik ile ilişkisi bilinen kanser tipinin bir ailede görülmesi
Kanser yanısıra başka fenotipik bulguların eşlik etmesi ya da tipik anomalileri taşıma Overgrowth
Hemihipertrofi
Tipik tümör gelişimi Hemanjioblastoma
Feokromasitoma
Renal hücreli karsinom
Schiffman JD, Geller JI, Mundt E, Means A, Means L, Means V.Update on pediatric cancer predisposition syndromes. Pediatr Blood Cancer. 2013 Aug;60(8):1247-52.
Nagy R, Sweet K, Eng C. Highly Penetrant Hereditary Cancer Syndromes. Oncogene. 2004 Aug 23;23(38):6445-70.
Kelly KM, Sweet K. In search of a familial cancer risk assessment tool. Clin Genet. 2007 Jan;71(1):76-83.

Kansere Yatkınlık Yapan Sendromlarda
Şüphelenilecek Fizik Bulgular
Fizik Bulgular Sendromlar
Café au Lait NF, Silver Russell, Bloom
Avuç içi ve ayak altında çukurluklar, milia
Gorlin
Anjiofibrom, shagreen lekesi TS
Telenjiektazi Ataksi Telenjiektazi
Makrosefali Banayan-Riley-Ruvalcaba, Cowden, Sotos, Gorlin, Simpson-Golabi-Behmel
Mikrosefali Bloom, Fanconi, Dubowitz
Multipl lipom, hemanjiom Banayan-Riley-Ruvalcaba
Makroglossi, kulak memesi yarıklanmsı BWS
Hiperpigmentasyon NF, Fanconi, Bloom
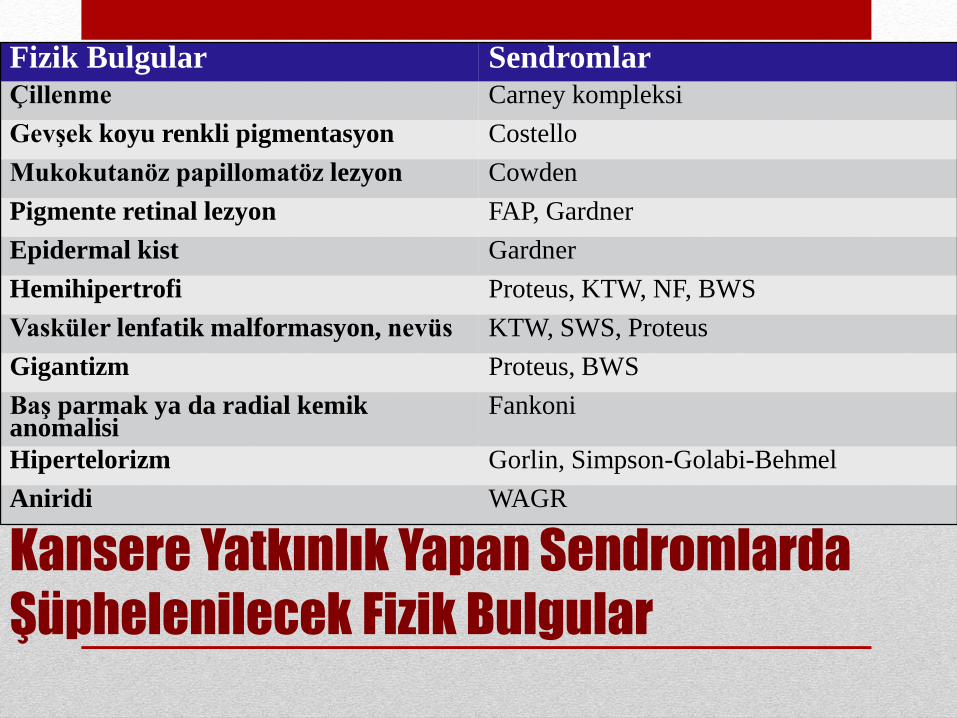
Fizik Bulgular SendromlarÇillenme Carney kompleksi
Gevşek koyu renkli pigmentasyon Costello
Mukokutanöz papillomatöz lezyon Cowden
Pigmente retinal lezyon FAP, Gardner
Epidermal kist Gardner
Hemihipertrofi Proteus, KTW, NF, BWS
Vasküler lenfatik malformasyon, nevüs KTW, SWS, Proteus
Gigantizm Proteus, BWS
Baş parmak ya da radial kemikanomalisi
Fankoni
Hipertelorizm Gorlin, Simpson-Golabi-Behmel
Aniridi WAGR
Kansere Yatkınlık Yapan Sendromlarda
Şüphelenilecek Fizik Bulgular

Tümör İlgili Sendrom
Adrenokortikal karsinom, meme kanseri LFS
Wilms WAGR, BWS, Denys-Drash, SGB
Hemanjioblastoma VHL
Berrak hücreli böbrek kanseri VHL, TS
Feokromasitoma VHL, MEN 2B, NF1
Hepatoblastoma BWS, FAP
Menenjiom, vasküler swannom NF2
Kutanöz bazal hücreli karsinom Gorlin, XP
Testiküler tm Proteus
Nb SGBS, Costello,
Melanom XP, Werner, Proteus
Troid kanseri BRRS
Kansere Yatkınlık Yapan Sendrom
Düşündürecek Tümörler

Tümör İlgili Sendrom
Medüller troid karsinomu MEN 2
Rabdomyosarkom Herediter RB, LFS, NF1, BWS
Optik gliom, nörofibrom NF1
Böbrek ya da SSS’ne ait rabdoid tümör Rabdoid tümör yatkınlık sendromu
Nöroblastom NF1, BWS
Embriyonel rabdomyosarkom NF1, Costello
Retinoblastom RB
Multipl bazal hücreli karsinom Gorlin
Lösemi Trizomi 21, NF, Fanconi
Beyin tm LiFraumeni, XP, Bloom
Mesane Costello
Meme Cowden, LiFraumeni,Bloom,HBOC,BRRS
Erkeklerde meme ca Klinefelter,HBOC
Kardiyak rabdomyom TS
Kansere Yatkınlık Yapan Sendrom
Düşündürecek Tümörler

Sık Görülen KYS’ları ve
İzlem
Tümör İzlem
NF Yıllık izlem ve göz takibi; düzenli TA takibi; ayrıntılı sistem muayenesi
BWS 3 ayda bir abdominal USG, AFP; 6 ayda bir karın muayenesi
MEN 1 Takip 5-10 yaş arası başlar, AKŞ, Ca, PTH, insülin, prolaktin, IGF1; yıllık USG; 3-5
yılda bir pankreatik ve hipofiz MRI; yıllık abdominal ve kafa MRI
MEN 2 2A için 5 yaşına kadar, 2B için ilk yıl proflaktik troidektomi; yıllık katekolamin düzey
tayini ve 3 yılda bir MRI
LFS Adrenokortikal karsinom için her 3-4 ayda bir karın USG, tam kan ve idrar analizi;
meme ca için kontrol 18 yaşında başlar, yılda 2 defa muayene, 20-25 yaşlarında yılda
bir mamografi, osteosarkom için yılda bir tüm vücud MRI
VHL Retinal hemanjiom için yıllık göz muayenesi; SSS hemanjioblastoması için ergenlik
dönemi ile birlikte 2 yılda bir beyin omurilik MRI; Renal kontrol için yılda bir 5
yaşından sonra USG; Pankreas karsinomu için 20 yaşından itibaren MR;
feokromasitoma için TA ve katekolamin takibi
FAP Hepatoblastoma için 3-5 yaşına kadar 3 ayda bir AFP ve karın USG; kolon kanseri
için 10 yaşından itibaren yılda bir kolonoskopi

Sendrom İlişkili Tümör İzlem
AT Hematolojik, Meme AFP
Bloom WT, Osteosarkom, hematolojik BWS taraması
Cowden Meme, troid, endometriyum Yıllık, 18 yaş sonrası, 30’dan sonra mamografi, 18
sonrası USG
Rb Rb, Osteosarkom Her ay (0-3/12), 2 ayda bir anestezi ile (3/12-7/12),
3 ayda bir (7/12-18/12), 6 ayda bir (3 yaşa kadar),
yıllık (7 yaşa kadar)
Gorlin Medulloblastom, Bazal hücreli karsinom Sık cilt muayenesi, Nörolojik değerlendirme 6 ayda
bir 3 yaşa kadar, sonra yılda 1, 7 yaşına kadar
Meme ca. Meme, Over, Pankreas, Prostat, melanom 18 yaşından itibaren her ay elle muayene, klinik
muayene 25 y. dan sonra
HNPCC Kolon, Endometriyum, Üriner sistem 25 y. dan sonra yılda 2 defa kolon muayenesi
Tbs Astrositom, Böbrek, Rabdomyom Göz muayenesi, Renal USG, CT, MRI, EKO, EEG
Nf Swannom, Menengiom 10 y.dan sonra yılda bir MR, sık işitme ve görme
testi
Werner Troid, Menengiom, Melanom, Sarkom Yıllık fizik muayene, idrar analizi
XP Deri, Korneal, Melanom, Lösemi Her 3-6 ayda bir cilt ve göz muayenesi
Sık Görülen KYS’ları ve İzlem
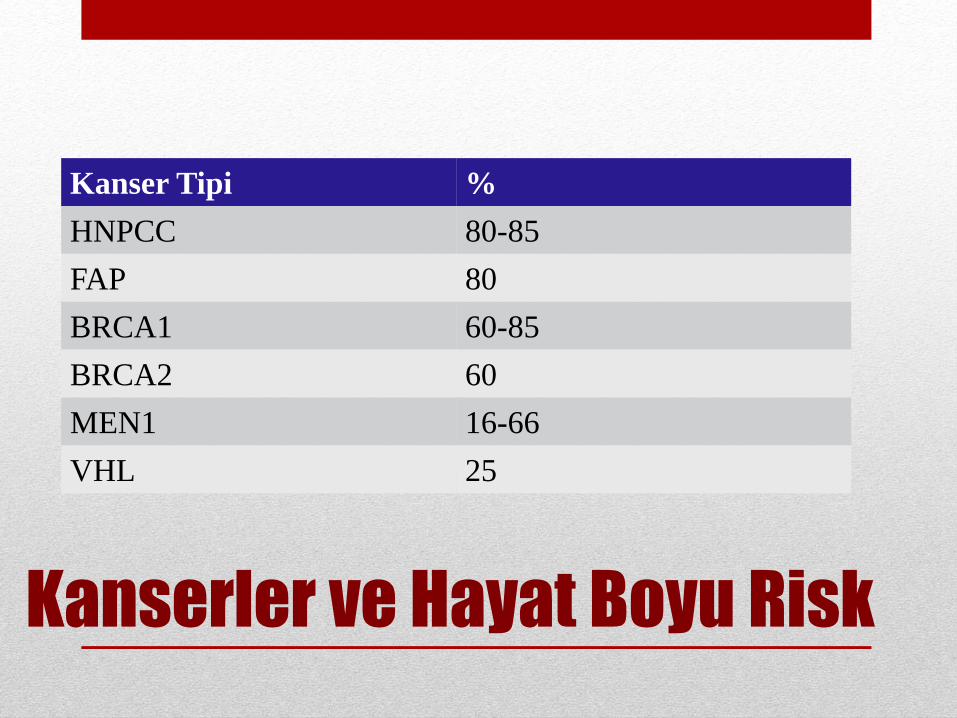
Kanserler ve Hayat Boyu Risk
Kanser Tipi %
HNPCC 80-85
FAP 80
BRCA1 60-85
BRCA2 60
MEN1 16-66
VHL 25

Proflaktik Cerrahi
Hastalık temelinde spesifik düşünülmekle
birlikte koruyucu tedavi bakımından
öneriliyor
Bilimoria MM.Prophylactic surgery in hereditary cancer syndromes: an ounce of prevention may be the only cure. J Surg Oncol. 2002
Mar;79(3):131-3.

Kansere Yatkınlık Sendrom Taraması
Genel kural
Çocuklukta başlayan bir hastalık durumu
Etkili ve güvenli tarama ve tedavi imkanlarının bulunması durumundaçocuklara test yapılır
Test öncesi olayın psikolojik boyutu mutlaka ele alınmalı
Çocuk
Aile
Bazı sendromlara özgü protokoller mevcut
VHL
MEN
Peutz jeggers
Pten hjamartom sendromu
BWS…
Schiffman JD, Geller JI, Mundt E, Means A, Means L, Means V.Update on pediatric cancer predisposition syndromes. Pediatr Blood Cancer. 2013
Aug;60(8):1247-52.

Kanser Genetik Testi Kararında
Önemli Noktalar
Test spesifik, güvenilir ve sonuçları değerlendirilebilir olmalı
Sonuçlarına göre ilgili bireyler bu sonuçtan yarar
sağlayabilmeli
Sonuçlara göre test sonrası genetik danışmanlıkta riskler
tartışılabilir, önlemler alınabilir olmalı
Çocukluk yaş grubunda kanser testinin kendisi tarafından karar
verilebilir yaşa kadar bekletilebilirliğinin değerlendirilmesi,
gerekirse ebeveynlerin karar vermesi

Kansere Yatkınlık Yaratan Sendromlar
Kromozomal anomaliler
• Down sendromu
Geçici myeloproliferatif bozukluk-%10 DS
Akut lösemi-10-20 kat artmış risk
5 yaşına kadar %2, 30 yaşına kadar %3
ALL, %2; AML (özellikle M7), %10
M7 riski 400 kat fazla
Hangi genler sorumlu???
• 329 gen içinde RUNX1, CRF2-4, IFNAR
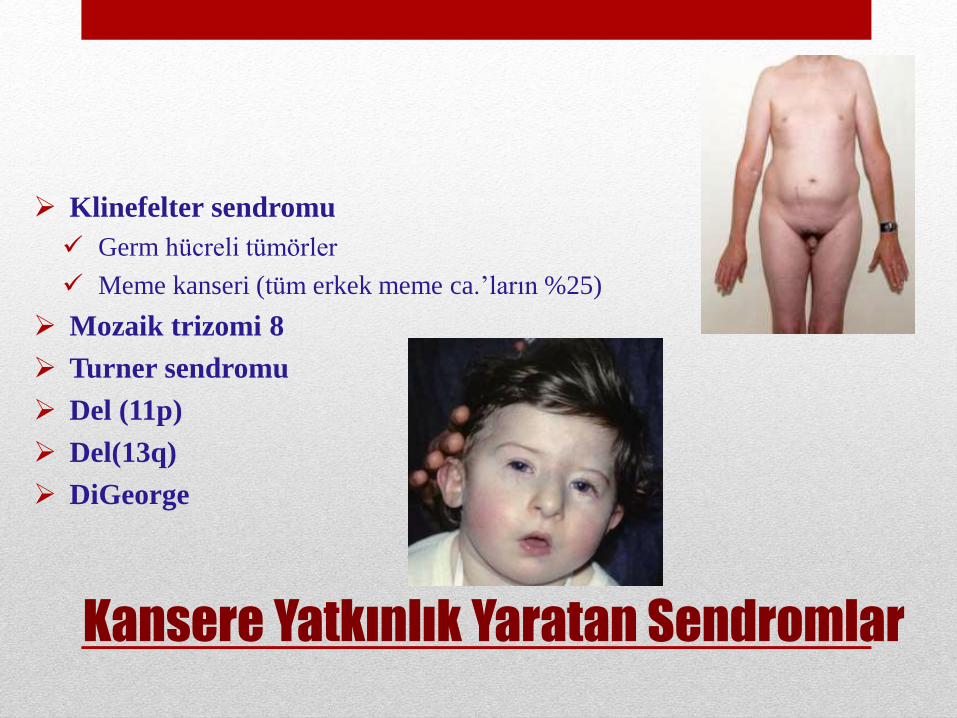
Klinefelter sendromu
Germ hücreli tümörler
Meme kanseri (tüm erkek meme ca.’ların %25)
Mozaik trizomi 8
Turner sendromu
Del (11p)
Del(13q)
DiGeorge
Kansere Yatkınlık Yaratan Sendromlar

Konstitusyonel Anöploidinin
Kanser Yapıcı Etkisi
Direkt onkojenik etki
Trizomi 21-ALL
Trizomi 8—AML
Trizomi 18--Wilms
Mikroçevreye bağlı
Trizomi 8’de hematopoetik progenitör proliferasyonu gibi
Makroçevre değişiklikleri
Hormonal değişiklikler-cinsiyet kromozomal anomalilerde

Otozomal Dominant Bozukluklar
1 allelin bozukluğu ile karakterize
TSG için “2 hit” olması gerekirken klinik düzeyde dominant geçiş
söz konusu
Retinoblastom
Li Fraumeni
MEN
Famiyal kolon kanser sendromları
Herediter Breast/Ovarian Kanser
Fakomatozlar
• TS, NF, VHL, AT, IP
Kansere Yatkınlık Yaratan Sendromlar

Fakomatozlar
Nörokutanöz sendromlar
SSS patolojisi
Cilt bulguları
Retinal bulgular
Nörofibromatozis
Sturge weber
Von Hippel Lindau
Ataksi Telenjiektazi
İnkontinentia Pigmenti…

Nörofibromatozis 1
Friedrich von Recklinghausen tarafından 1882 yılında tanımlanmış
1/2500, NF1, 17q11.2
OD kalıtım ancak sorumlu gen nörofibromin, tümör supresör protein
RAS proteinin aktivasyonunu sınırlar
Çok geniş ekspresyon
Olguların yarısı yeni mutasyon
Yıllar içinde nörofibromlar oluşur ve artabilir
Komplikasyon durumunda ya da
Malignite durumunda cerrahi
Optik gliom
%50’si NF
Çok yavaş ilerler
6 yaşına kadar oluşmalı, yoksa risk çok düşer
Takip
Yıllık fizik muayene, göz muayenesi, IQ
Ergenlikte skolyoz takibi

Nörofibromatozis 2
OD
Yarısı de novo
1/35000 NF2
NF2, 22q12, Merlin proteini, tümör supresör gen
Çok az oranda malign tümör gelişir
En önemli bulgu işitme kusuru
Schwannomas, skin tag benzeri
Özellikle vestibüler
Diğer bölgelerde de görülebilir
Café au lait daha nadir
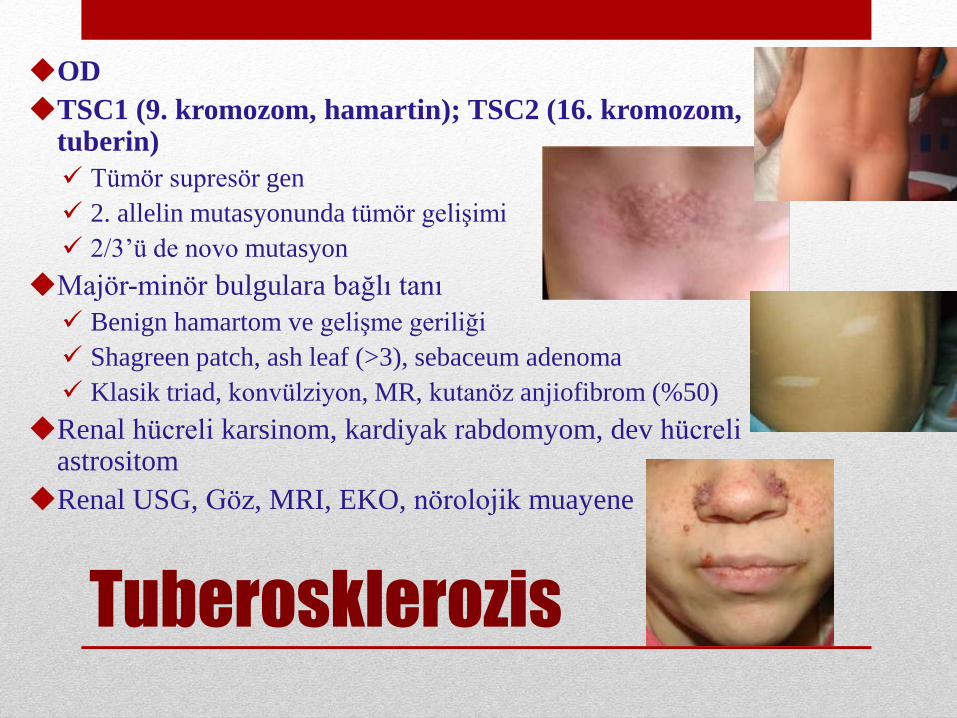
Tuberosklerozis
OD
TSC1 (9. kromozom, hamartin); TSC2 (16. kromozom, tuberin)
Tümör supresör gen
2. allelin mutasyonunda tümör gelişimi
2/3’ü de novo mutasyon
Majör-minör bulgulara bağlı tanı
Benign hamartom ve gelişme geriliği
Shagreen patch, ash leaf (>3), sebaceum adenoma
Klasik triad, konvülziyon, MR, kutanöz anjiofibrom (%50)
Renal hücreli karsinom, kardiyak rabdomyom, dev hücreliastrositom
Renal USG, Göz, MRI, EKO, nörolojik muayene

Li Fraumeni
En çok bilinen, 1969 yılında tanımlanmış
Otozomal dominant
Yumuşak doku sarkomları, meme kanseri, lösemi, osteosarkom,
beyin tümörü, GİS karsinomu
P53 mutasyonu
İleri erken yaş tümörleri ile karakterize ancak
Çocuklukta gelişebilir
16 yaşına kadar %42 risk
Risk altındaki yetişkinler 20’li yaşlardan itibaren yıllık mamogram,
MRI
Çocukluk çağında kolon kanseri görülebileceğinden kolonoskopi
BT önerilmez

P53
Özellikle (kalıtsal kanser düşündürecek diğer
bulguların varlığında) p53 mutasyonu taranması
gereken tümörler;
Çocukluk çağı adrenokortikal karsinom-%50-100
Beyin tümörü-%2-10
Osteosarkom-%3
Rabdomyosarkom-%9
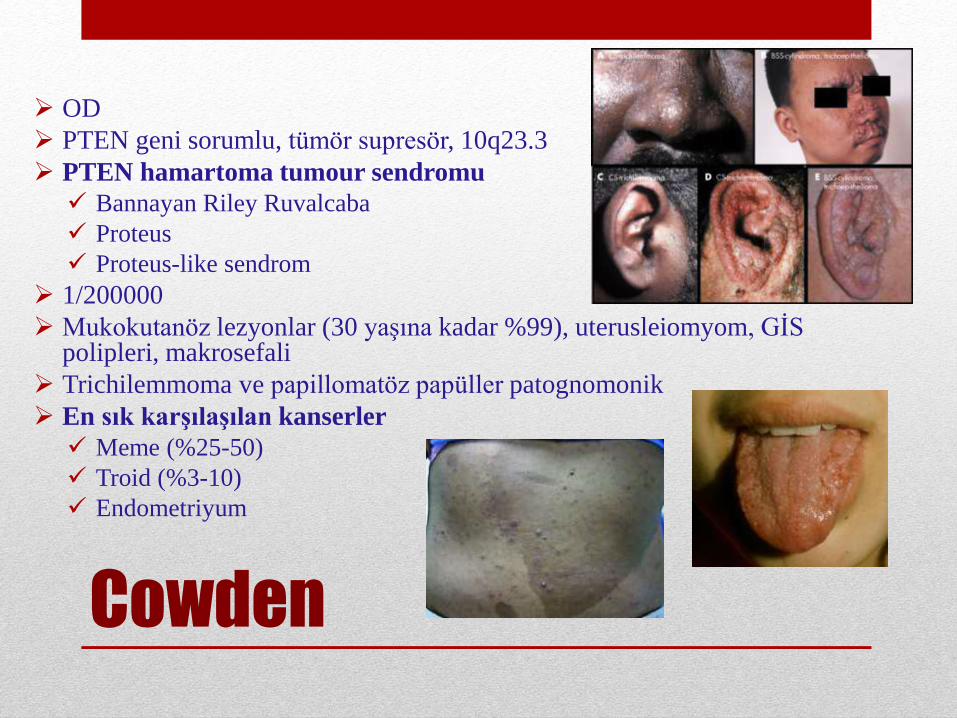
Cowden
OD
PTEN geni sorumlu, tümör supresör, 10q23.3
PTEN hamartoma tumour sendromu
Bannayan Riley Ruvalcaba
Proteus
Proteus-like sendrom
1/200000
Mukokutanöz lezyonlar (30 yaşına kadar %99), uterusleiomyom, GİS polipleri, makrosefali
Trichilemmoma ve papillomatöz papüller patognomonik
En sık karşılaşılan kanserler
Meme (%25-50)
Troid (%3-10)
Endometriyum
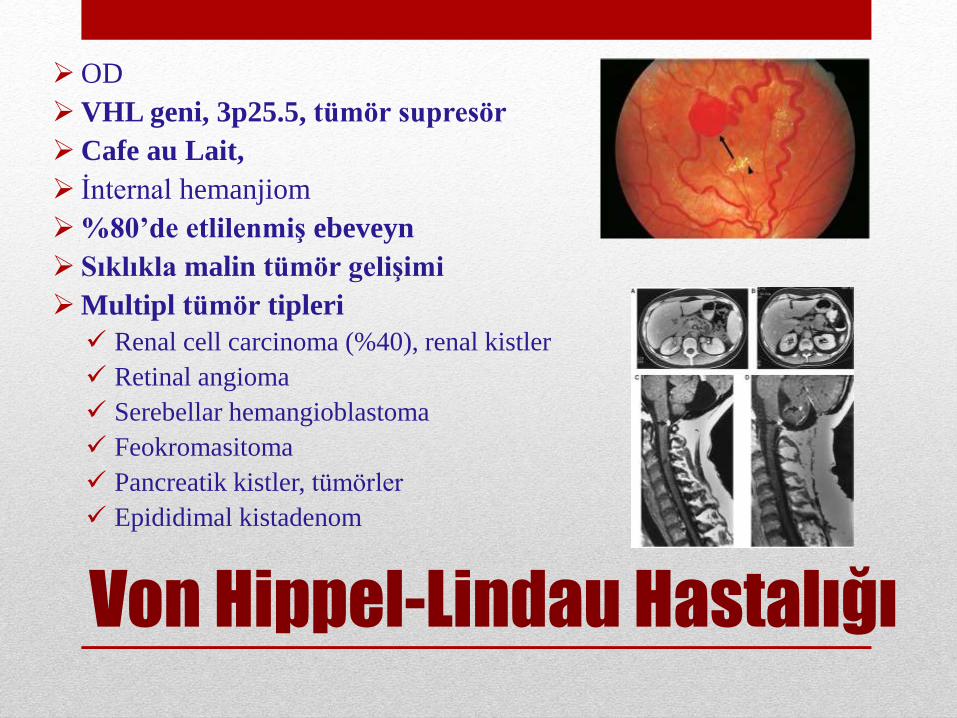
Von Hippel-Lindau Hastalığı
OD
VHL geni, 3p25.5, tümör supresör
Cafe au Lait,
İnternal hemanjiom
%80’de etlilenmiş ebeveyn
Sıklıkla malin tümör gelişimi
Multipl tümör tipleri
Renal cell carcinoma (%40), renal kistler
Retinal angioma
Serebellar hemangioblastoma
Feokromasitoma
Pancreatik kistler, tümörler
Epididimal kistadenom

Otozomal Resesif Bozukluklar
Daha ağır
Daha erken yaş
Her 2 ebeveynden de hatalı allel gelmeli
XP
Cockayne
TTD (trikotiyodistrofi)
AT
Fankoni
Kansere Yatkınlık Yaratan
Sendromlar
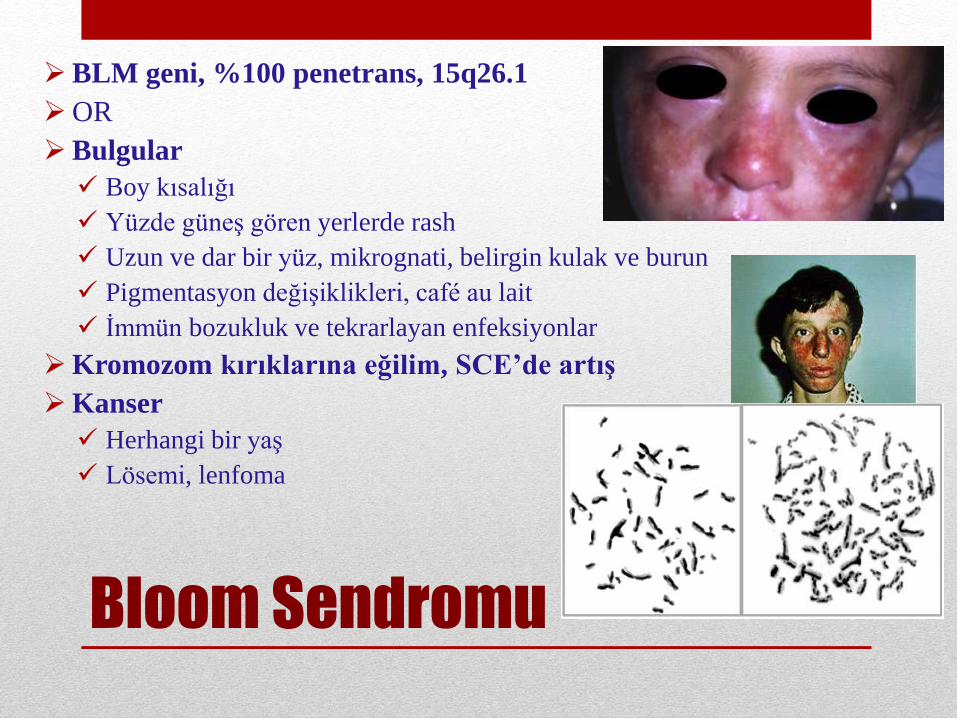
Bloom Sendromu
BLM geni, %100 penetrans, 15q26.1
OR
Bulgular
Boy kısalığı
Yüzde güneş gören yerlerde rash
Uzun ve dar bir yüz, mikrognati, belirgin kulak ve burun
Pigmentasyon değişiklikleri, café au lait
İmmün bozukluk ve tekrarlayan enfeksiyonlar
Kromozom kırıklarına eğilim, SCE’de artış
Kanser
Herhangi bir yaş
Lösemi, lenfoma

Fanconi Anemisi
1/300000, OR, FANCA
FA proteinleri A,B,C,E,F,G,L,M birleşerek kompleks oluşur
FANCD2 üzerinden BRCA1, FANCD1,ATR,BLM gibi proteinlerin tamirinde
Bulgular Boy kısalığı
Hiperpigmentasyon
Özellikle baş parmak olmak üzere parmak anomalisi
Kemik iliği yetersizliği
Kansere yatkınlık Lösemi (AML)-1/10
Cilt kanseri
GIS kanseri, oral kanserler
Wilms tümörü
Genitoüriner sistem tümörü-vulvar kanserler
DNA hasarı yapan ajanlara hassasiyet ve çok sayıda tamir edilememiş kırık
DEB
Mitomisin C

Ataksi Telenjiektazi
ATM, 11q22.3
DNA hasarına yanıt defektif ve çift zincir kırıkları tamir edilemez, apoptozgerçekleşmez ve telomer stabilize değil
Bulgular
Progresif serebellar ataksi-erken çocuklukta başlar, yürümeyle birlikte
Ciltte ve korneada telenjiektazi-5 yaşına kadar belirgin
İmmünite bozukluğu (humoral)
Kromozom instabilitesi ve ionizan radyasyona duyarlık
UV duyarlılığı yok
DNA iplikçik tamiri kusurlu ve UV iplikçikleri etkilemez
Kanser ile ilişkisi
Hematolojik malignite
Taşıyıcılarda meme kanseri
Yetişkin yaşlara kadar yaşam

Wilms tümörü ilişkili sendromlar
WAGR
Denys-Drash
Frasier
BWS
Kansere Yatkınlık Yaratan Sendromlar
Overgrowth sendromlar
Simpson Golabi Behmel
Wilms, Nb
GPC3
Proteus
Gonadal, meningiom, parotid adenom
PTEN
Sotos
Hematolojik
NSD1
Costello
Rabdomyosarkom, Nb, mesane kanseri
HRAS
Bannayan-Riley-Ruvalcaba
Cowden
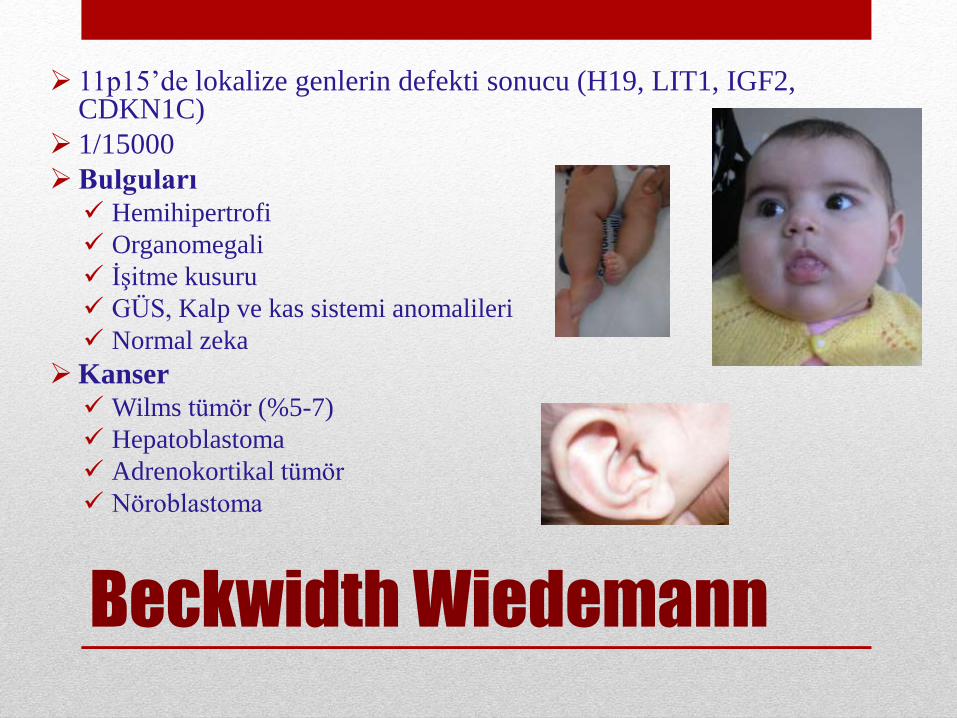
Beckwidth Wiedemann
11p15’de lokalize genlerin defekti sonucu (H19, LIT1, IGF2, CDKN1C)
1/15000
Bulguları Hemihipertrofi
Organomegali
İşitme kusuru
GÜS, Kalp ve kas sistemi anomalileri
Normal zeka
Kanser Wilms tümör (%5-7)
Hepatoblastoma
Adrenokortikal tümör
Nöroblastoma

Desai AN et al. Cilin Genet, 2012
Diagnostik amaçlı NGS
48 örneğe kadar multiplex
2761 gen, 40000 ekzon
Hedefli panel, kanser
EÜTF Tıbbi Genetik AD

Teşekkür Ederim







