zinc oxide–zinc phthalocyanine interface for hybrid solar cells
TRANSCRIPT

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/235780005
ZincOxide-ZincPhthalocyanineInterfaceforHybridSolarCells
ARTICLEinTHEJOURNALOFPHYSICALCHEMISTRYC·JULY2012
ImpactFactor:4.77·DOI:10.1021/jp303781v
CITATIONS
16
READS
59
8AUTHORS,INCLUDING:
ClaudioMelis
UniversitàdeglistudidiCagliari
37PUBLICATIONS355CITATIONS
SEEPROFILE
GiulianoMalloci
ItalianNationalResearchCouncil
62PUBLICATIONS756CITATIONS
SEEPROFILE
P.Alippi
ItalianNationalResearchCouncil
63PUBLICATIONS768CITATIONS
SEEPROFILE
AlessandroMattoni
ItalianNationalResearchCouncil
88PUBLICATIONS905CITATIONS
SEEPROFILE
Availablefrom:GiulianoMalloci
Retrievedon:03February2016

Zinc Oxide−Zinc Phthalocyanine Interface for Hybrid Solar CellsGiuseppe Mattioli,*,† Claudio Melis,‡,§ Giuliano Malloci,§ Francesco Filippone,† Paola Alippi,†
Paolo Giannozzi,∥,⊥ Alessandro Mattoni,§ and Aldo Amore Bonapasta†
†Istituto di Struttura della Materia del CNR, v. Salaria Km 29,300 - C.P. 10 I-00015, Monterotondo Stazione (RM), Italy‡Department of Physics, University of Cagliari, Cittadella Universitaria, 09042 Monserrato (Ca), Italy§Istituto per l'Officina dei Materiali (CNR-IOM), UOS Cagliari SLACS, Cittadella Universitaria, I-09042 Monserrato (Ca), Italy∥Department of Chemistry, Physics, and Environment, University of Udine, via delle Scienze 208, 33100 Udine, Italy⊥DEMOCRITOS IOM-CNR National Simulation Center, 34014 Trieste, Italy
*S Supporting Information
ABSTRACT: The structural, electronic, and optical proper-ties of a hybrid interface formed by zinc phthalocyanine(ZnPc) molecules adsorbed on the (101 0) zinc oxide (ZnO)surface have been investigated by using ab initio and modelpotential theoretical methods. In particular, the attention hasbeen focused on the effects of molecular assembling on theinterface properties by considering cofacial and planarmolecular aggregates on the surface. Present results showthat planar aggregations provide a remarkable molecule-to-surface electronic coupling which can favor electron injectiontoward the substrate. Furthermore, we predict a blue shift of absorption bands in the case of cofacial aggregation and a red shift inthe case of nanostructured planar J-stripes, which are in agreement with previous phenomenological models and give a firmtheoretical support to observed relationships between red shift and molecular assembling. All together, present results indicatethat structural and electronic properties can be achieved in ZnPc-sensitized ZnO surfaces of high potential interest for improvingthe efficiency of different kinds of hybrid photovoltaic cells.
■ INTRODUCTION
Hybrid photovoltaic (HPV) cells employing both organic andinorganic components for light harvesting and energyproduction have received enormous research attention. Thenatural abundance of the organic materials and their low costmake them, indeed, potentially highly competitive with solarcells based on thin-film technology. However, drawbacks forlarge-scale applications of HPV cells are still represented bytheir low efficiency and short durability.1−3 HPV cells areformed by an electron donor (D), often an organic molecule ora π-conjugated polymer, also acting as light absorber, orsensitizer, and an inorganic substrate acting as electron acceptor(A).The most important acceptors for HPV are metal oxides, in
particular TiO2, that have been used in several HPVarchitectures including polymer-based hybrid solar cells andhybrid dye-sensitized solar cells (DSSC).1,2 Zinc oxide hasemerged more recently in the framework of photovoltaicdevices4−6 as an alternative to TiO2 because it can besynthesized with great flexibility and provides a very goodelectron mobility.7 For example, among polymer−oxidemixtures, blends of ZnO and P3HT are, up to date, the mostefficient ones.8 Furthermore, DSSC based on ZnO havereached efficiencies comparable to titania.9 The zinc oxidenanoparticles commonly used as acceptor partners in HPV
devices are often characterized by an elongated shape along the(0001) direction and a hexagonal section.10,11 Such nanorodsexpose the most stable12,13 (101 0) surface on their sides, withsmaller contribution of another low-index nonpolar surface,namely the (112 0) one, whose topmost atoms are arranged inZn−O dimers, thus showing a local morphology similar to the(101 0) surface.12 For these reasons the (101 0) surface is oftenchosen, like in the present investigation, as a suitable model torepresent the whole of ZnO surfaces in nanostructures.14,15 Asfor the organic component of HPV systems, phthalocyanine’srepresent a very important class of donor molecules with goodlight absorption extending down to the red and near-IR regionsand efficient electron transfer to the TiO2 or ZnO conductionband. Pcs (usually modified by the addition of anchoringgroups) have been used indeed as efficient dyes for DSSCdevices.16 Pcs molecules have been also used in polymer basedhybrids to complement the optical absorption of the polymer inred region of the spectrum.17 The structure of these moleculesis characterized by an aromatic macrocyclic ligand carryingclouds of π-conjugated, delocalized electrons, and a centralmetallic atom, in a typical 2+ oxidation state, playing the role of
Received: April 19, 2012Revised: June 20, 2012Published: June 27, 2012
Article
pubs.acs.org/JPCC
© 2012 American Chemical Society 15439 dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−15448

electron donor to the ligands. A prototypical example isrepresented by ZnPc16 whose main electronic features areillustrated in Figure1A.In HPV cells, the electronic properties of the D−A interface
play a key role by affecting both the optical absorption inducedby solar light and the electron injection to the substrate. Arecent, preliminary study investigating the electronic couplingbetween isolated ZnPc molecules on the ZnO surface18 hassuggested interesting potentialities of Pcs/ZnO interfaces forphotovoltaics. As a first step toward the simulation of morecomplex, realistic systems, the promises of such interfaces haveto be checked against the characteristics of the Pcs assemblingon the substrate surface as well as against its effects on theinterface properties. In this regard, it has to be taken intoaccount that Pcs tend to aggregate, as observed in gas phaseand in solution,19,20 as well as on metal and semiconductorsurfaces.21,22 Both ordered monolayers23,24 and island-growthfeatures25 have been also reported in the case of Pc absorptionon metal oxide surfaces. Hints of the tendencies of ZnPcmolecules to form planar elongated nanoclusters on the ZnOsurface (usually named and hereafter referred to as J-stripes)have been given in a recent model potential moleculardynamics (MPMD) contribution.26 These stripes are expected
to further assemble into planar islands at higher concentrations.It should be also taken into account that the aggregationphenomenon has a strong impact on the optical properties ofthe molecules, the light absorption spectrum of aggregatesbeing generally quite different from that of isolated molecules.19
In particular, by studying ZnPc/ZnO systems by UV−visabsorption spectroscopy,27 it was observed a continuous redshift of the optical absorption spectrum at increasing ZnPc’scoverage. These findings have been attributed to a molecularaggregation on the surface in the form of J-stripes basing on aphenomenological model (also known as the Davydoveffect).19,28 Such an attribution is supported by the aboveMPMD study. On the other hand, a sound theoreticaldemonstration that ZnPc stripes induce the observed redshift is still missing (at the best of the author’s knowledge).In this scenario, in the present study, we approach the
description of a ZnPc/ZnO interface by investigating the effectsof molecular aggregation and intermolecular interactions on thecoupling between ZnPc molecules and the (101 0) ZnO surface.As major results, the present findings show that: (i) Planaraggregations of ZnPc molecules in the form of dimers and J-stripes are expected to characterize the ZnPc/ZnO interface atsubmonolayer and monolayer coverages. (ii) Such planar
Figure 1. (A) Sticks and balls representation of a zinc phthalocyanine (ZnPc) molecule and |ψ|2 plots of the π highest occupied molecular orbital(HOMO) and of the π* doubly degenerate lowest unoccupied molecular orbital (LUMO) (electronic densities sampled at 0.0005 e/au3). (B) Sideand top view of the ZnO (101 0) surface. Two reference axes of the surface are labeled X, Y. The alternation of trench grooves and rows of dimersalong the X direction is shown in the lower panel. (C) Top and side views of the minimum-energy DFT configurations of an isolated ZnPc moleculeadsorbed on the ZnO (101 0) surface.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815440

aggregates are characterized by a significant ZnPc−ZnOelectronic coupling which induces the appearance of emptyelectronic levels deriving from an intimate mixing of ZnPc andZnO electronic states and strategically located within the ZnOconduction band and below the ZnPc LUMO. These levels are,therefore, specially suitable for an efficient molecule-to-surfacetransfer of photoexcited electrons. (iii) In the case of planar J-stripes, intermolecular interactions induce a red shift ofabsorption bands which agrees with the phenomenologicalmodel mentioned above and, most important, gives a firmtheoretical support to the attribution of such a red shift to theformation of J-stripes proposed in ref 27.
■ THEORETICAL FRAMEWORKPrevious model potential molecular dynamics MPMD inves-tigations of ZnPc molecules adsorbed on the ZnO surface26,29
have provided equilibrium geometries for single molecules,dimers, and molecular stripes, used as a first guess for thepresent ab initio DFT calculations. The model potential for theZnO wurtzite crystal was described by the sum of a Coulomband a Buckingham-type two-body potential.30,31 The AMBERforce field,32 including both bonding (stretching, bending,torsional) and nonbonding (van der Waals plus Coulomb)contributions, has been used to describe ZnPc molecules.29
Interatomic forces between atoms of the ZnPc and the ZnOsubstrate were calculated by including electrostatic anddispersive interactions. Coulomb terms involved interactionsbetween atomic partial charges of the molecule and the ZnOions. Dispersive interactions of the Lennard-Jones type weretaken from the AMBER database.32 All the simulations wereperformed by using DL_POLY33 (version 3). The assemblingof ZnPc molecules at room temperature has been studied byusing the metadynamics technique, which allows to acceleraterare events and to sample the free energy as a function ofsuitable collective variables,34 e.g., the distance between thecenter of mass of two ZnPc molecules in Figure 5.Ab initio DFT calculations have been performed by using the
Quantum-ESPRESSO package.35 Total energies have beencalculated by using ultrasoft pseudopotentials36 for all atomsbut Zn, whose electronic core was represented by a norm-conserving pseudopotential.37 The Zn 3d shell has beenembedded into the pseudopotential. A similar approach hasbeen used and discussed in detail in the case of the Ga atom inthe wurtzite GaN crystal,38 probably the ZnO closest relative.Extensive calculations have been performed to carefully checkthis approach; the achieved results are presented and discussedin detail within the Supporting Information. Kohn−Shameigenfunctions have been expanded on a plane-wave basis set;satisfactorily converged results have been achieved by usingcutoffs of 35 Ry on the plane waves and of 280 Ry on theelectronic density as well as by sampling the first Brillouin zonewith the Γ point only. Negligible differences on the structuraland electronic properties of the investigated molecule−surfacesystems have been obtained by using a 2 × 2 × 1 Γ-centered k-point mesh. Several ZnO (101 0) surface cells have beenmodeled by adding ≈15 Å of empty space to different crystalslabs all formed by six atomic layers of bulk ZnO parallel to the(101 0) crystal plane. In the case of a single ZnPc moleculeadsorbed on the ZnO surface, almost identical results have beenachieved in the case of 4 × 6 (288 atoms), 4 × 8 (384 atoms,used also in the case of periodic ZnPc stripes), 4 × 9 (432atoms, used also in the case of isolated ZnPc dimers), and 5 × 9(540 atoms, 26 Å × 29 Å wide) surface cells. A 6 × 8 (288
atoms) rhombic surface cell has been used to simulate a close-packed ZnPc monolayer. Similar surface cells, only four atomiclayers thick, have been used in the case of time-dependentdensity functional perturbation theory (TDDFPT) calculations(see below).Geometry optimization procedures have been performed by
fully relaxing the positions of all of the atoms in a supercell,except for the atoms of the bottom layer of the semiconductor.The electronic properties of the ZnPc/ZnO system have beeninvestigated by analyzing the electronic eigenvalues calculatedat the Γ point. Particular care has been given to the simulationof dispersive interactions. In this regard, the exchange-correlation functional has been constructed by adding an abinitio nonlocal van der Waals correlation contribution39,40 tothe semilocal gradient-corrected PBE functional.41 Thisimproved functional addresses the problem of the lack ofdescription of long-range electronic correlation interactions instandard generalized gradient approximation (GGA) func-tionals. Such a drawback leads to a drastic underestimate ofdispersion forces, efficiently corrected here by using the abovefunctional.42
A further drawback of the GGA-PBE functional regards theKohn−Sham eigenvalues related to isolated (e.g., molecules)and extended (e.g., surface slabs) systems, which can be affectedat different extents by short-range electronic interaction errorsleading, in particular, to underestimate the band gap or theHOMO−LUMO gap.43,44 In turn, such a drawback can affectthe relative alignment of molecule and surface electroniceigenvalues which is crucial to provide a reliable estimate of theelectronic and optical properties of a D−A interface, like theZnPc/ZnO one. In this regard, we have performed furthercalculations in order to carefully compare GGA-PBE resultswith results obtained by using a GGA+U correction,45,46 as wellas the screened hybrid HSE06 functional,47,48 particularlyaccurate to simulate the properties of hybrid organic−inorganicsystems.49 We can anticipate that the picture given by PBEresults closely agrees with the GGA+U and HSE ones, thusjustifying the choice of a PBE approach in the case of the largemolecule−surface systems investigated here. The quantitativeextent of this agreement, together with the GGA+U and HSEresults, is discussed in detail in the Supporting Information.Optical absorption spectra ranging from the near-IR to the
near-UV regions have been calculated in the case of a gas-phaseZnPc molecule, a gas-phase ZnPc dimer, the most stable singleZnPc/ZnO interacting system, and a periodic ZnPc stripeinteracting with the ZnO surface by using a recent approach tothe solution of the Bethe−Salpeter equation within theframework of time-dependent density matrix perturbationtheory (TDDFPT).50,51 In detail, absorption frequencies, andthe related oscillator strenght, are calculated as linear responseof the whole charge density of the system to the oscillatingelectric perturbation; A Lorentzian broadening of 0.14 eV hasbeen applied to all the oscillators. Such an approach is expectedto provide reliable results when applied to large systems,including sensitized metal oxides.18,52 A careful analysis isrequired for one or more ZnPc molecules interacting with theZnO surface. More specifically, the contribution of the ZnOslab has to be separately calculated and subtracted by theZnPc/ZnO spectra because of the generally acknowledgedinaccurate contribution of periodic systems to TDDFTcalculations, when performed at the adiabatic generalizedgradient approximation (A-GGA) level.53,54 In the case ofZnPc/ZnO systems, the simulated spectra show therefore the
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815441
changes occurring in the ZnPc spectrum due to the adsorptionof one or more molecules on the ZnO surface. A reduced 192atoms four-layer 4 × 6 (3 × 8) ZnO slab has been used in thecase of TDDFPT calculations to simulate the molecule/surface(periodic stripe/surface) system. The DFT results indicate thatthe structural and electronic properties of the ZnPc/ZnOsystem calculated with the reduced slab are in a goodagreement with the ones obtained by using the above six-layer slabs.
■ RESULTS AND DISCUSSION
Isolated ZnPc on the ZnO Surface. The most stablenonpolar (101 0) ZnO surface exhibits trench groovesalternated with rows of dimers both oriented along thedirection labeled X in Figure 1B. The minimum-energyconfiguration of an adsorbed molecule, hereafter referred toas A configuration,18,29 is shown in Figure 1C. The moleculebinds to the surface by a combination of two joinedinteractions: the formation of a chemical bond involving themolecular Zn and one of the 3-fold coordinated O atomsbelonging to the surface dimers rows and a “face-to-face”interaction involving the broad molecular macrocyclic systemand the rather flat ZnO surface primarily due to strong van derWaals forces. These two bonding contributions result in a ZnPcadsorption energy, Eads, equal to 3.8 eV.On the side of the electronic properties of the ZnPc/ZnO
system in the A configuration, the relevant “face-to-face”interaction induces a high polarization of electronic charge atthe molecule−surface interface. More specifically, a fraction ofthe ZnPc charge density is shared with the ZnO surface inagreement with the donor tendency of the molecule and theacceptor tendency of the surface;18 see, e.g., the values of the
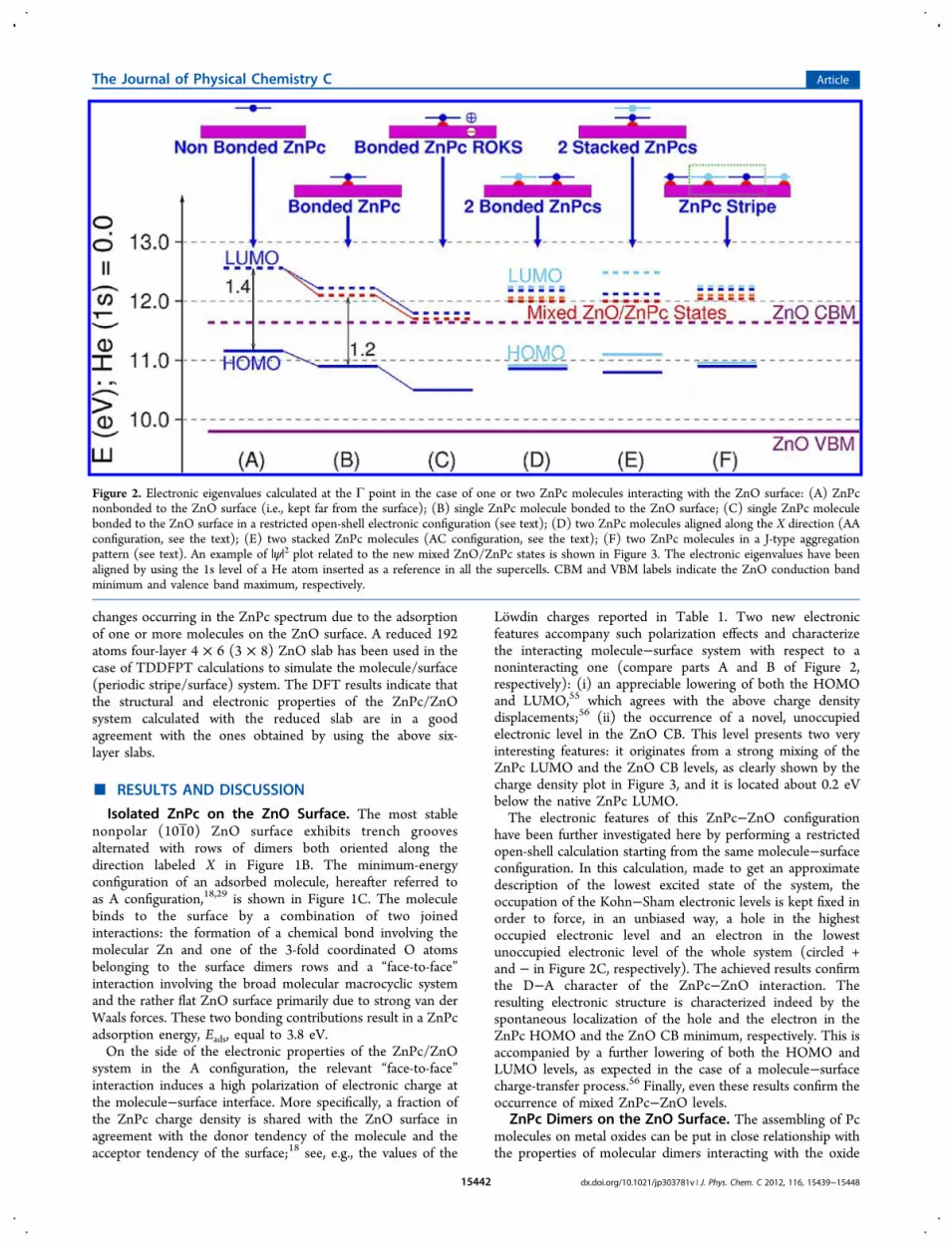
Lowdin charges reported in Table 1. Two new electronicfeatures accompany such polarization effects and characterizethe interacting molecule−surface system with respect to anoninteracting one (compare parts A and B of Figure 2,respectively): (i) an appreciable lowering of both the HOMOand LUMO,55 which agrees with the above charge densitydisplacements;56 (ii) the occurrence of a novel, unoccupiedelectronic level in the ZnO CB. This level presents two veryinteresting features: it originates from a strong mixing of theZnPc LUMO and the ZnO CB levels, as clearly shown by thecharge density plot in Figure 3, and it is located about 0.2 eVbelow the native ZnPc LUMO.The electronic features of this ZnPc−ZnO configuration
have been further investigated here by performing a restrictedopen-shell calculation starting from the same molecule−surfaceconfiguration. In this calculation, made to get an approximatedescription of the lowest excited state of the system, theoccupation of the Kohn−Sham electronic levels is kept fixed inorder to force, in an unbiased way, a hole in the highestoccupied electronic level and an electron in the lowestunoccupied electronic level of the whole system (circled +and − in Figure 2C, respectively). The achieved results confirmthe D−A character of the ZnPc−ZnO interaction. Theresulting electronic structure is characterized indeed by thespontaneous localization of the hole and the electron in theZnPc HOMO and the ZnO CB minimum, respectively. This isaccompanied by a further lowering of both the HOMO andLUMO levels, as expected in the case of a molecule−surfacecharge-transfer process.56 Finally, even these results confirm theoccurrence of mixed ZnPc−ZnO levels.
ZnPc Dimers on the ZnO Surface. The assembling of Pcmolecules on metal oxides can be put in close relationship withthe properties of molecular dimers interacting with the oxide
Figure 2. Electronic eigenvalues calculated at the Γ point in the case of one or two ZnPc molecules interacting with the ZnO surface: (A) ZnPcnonbonded to the ZnO surface (i.e., kept far from the surface); (B) single ZnPc molecule bonded to the ZnO surface; (C) single ZnPc moleculebonded to the ZnO surface in a restricted open-shell electronic configuration (see text); (D) two ZnPc molecules aligned along the X direction (AAconfiguration, see the text); (E) two stacked ZnPc molecules (AC configuration, see the text); (F) two ZnPc molecules in a J-type aggregationpattern (see text). An example of |ψ|2 plot related to the new mixed ZnO/ZnPc states is shown in Figure 3. The electronic eigenvalues have beenaligned by using the 1s level of a He atom inserted as a reference in all the supercells. CBM and VBM labels indicate the ZnO conduction bandminimum and valence band maximum, respectively.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815442
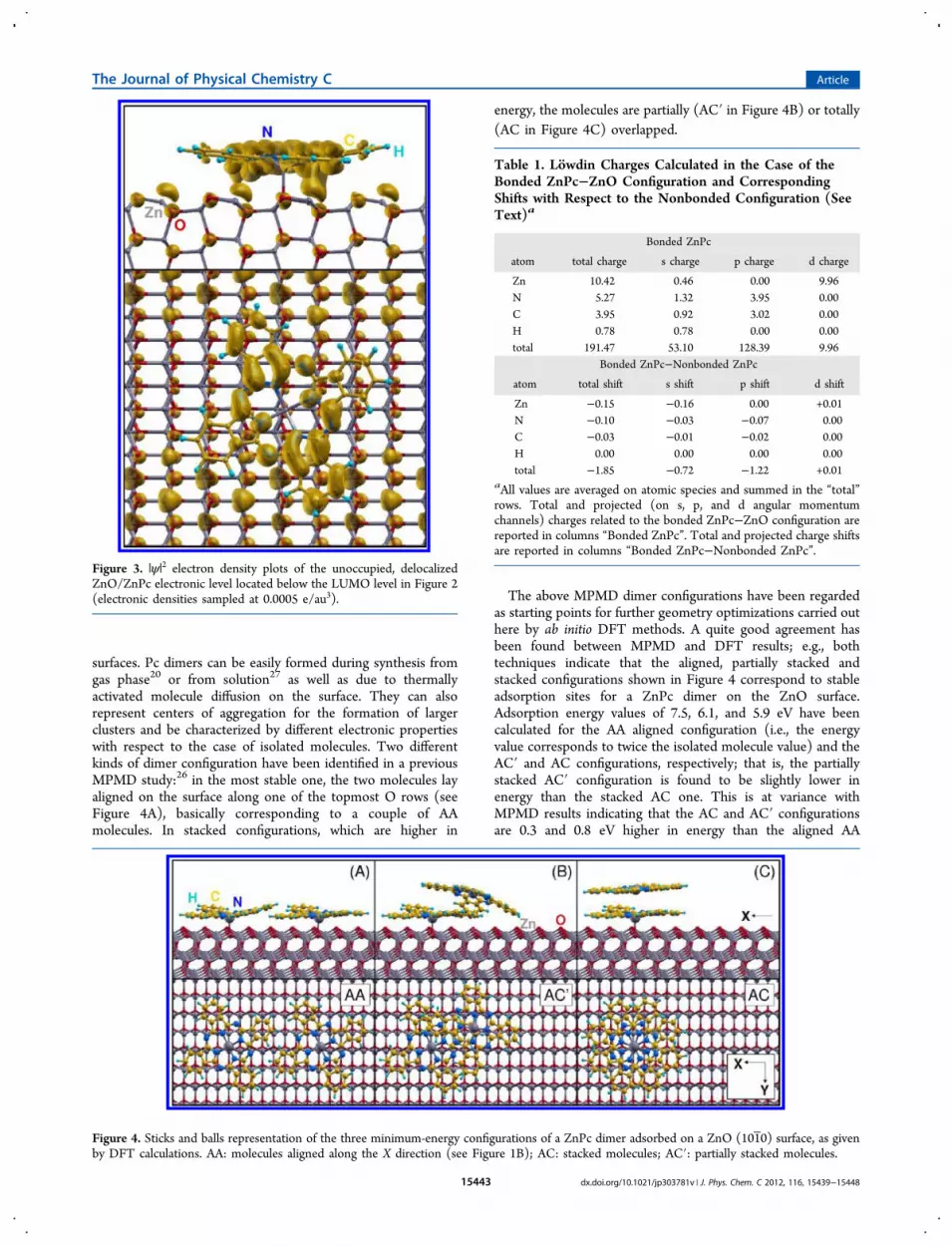
surfaces. Pc dimers can be easily formed during synthesis fromgas phase20 or from solution27 as well as due to thermallyactivated molecule diffusion on the surface. They can alsorepresent centers of aggregation for the formation of largerclusters and be characterized by different electronic propertieswith respect to the case of isolated molecules. Two differentkinds of dimer configuration have been identified in a previousMPMD study:26 in the most stable one, the two molecules layaligned on the surface along one of the topmost O rows (seeFigure 4A), basically corresponding to a couple of AAmolecules. In stacked configurations, which are higher in
energy, the molecules are partially (AC′ in Figure 4B) or totally(AC in Figure 4C) overlapped.
The above MPMD dimer configurations have been regardedas starting points for further geometry optimizations carried outhere by ab initio DFT methods. A quite good agreement hasbeen found between MPMD and DFT results; e.g., bothtechniques indicate that the aligned, partially stacked andstacked configurations shown in Figure 4 correspond to stableadsorption sites for a ZnPc dimer on the ZnO surface.Adsorption energy values of 7.5, 6.1, and 5.9 eV have beencalculated for the AA aligned configuration (i.e., the energyvalue corresponds to twice the isolated molecule value) and theAC′ and AC configurations, respectively; that is, the partiallystacked AC′ configuration is found to be slightly lower inenergy than the stacked AC one. This is at variance withMPMD results indicating that the AC and AC′ configurationsare 0.3 and 0.8 eV higher in energy than the aligned AA
Figure 3. |ψ|2 electron density plots of the unoccupied, delocalizedZnO/ZnPc electronic level located below the LUMO level in Figure 2(electronic densities sampled at 0.0005 e/au3).
Figure 4. Sticks and balls representation of the three minimum-energy configurations of a ZnPc dimer adsorbed on a ZnO (101 0) surface, as givenby DFT calculations. AA: molecules aligned along the X direction (see Figure 1B); AC: stacked molecules; AC′: partially stacked molecules.
Table 1. Lowdin Charges Calculated in the Case of theBonded ZnPc−ZnO Configuration and CorrespondingShifts with Respect to the Nonbonded Configuration (SeeText)a
Bonded ZnPc
atom total charge s charge p charge d charge
Zn 10.42 0.46 0.00 9.96N 5.27 1.32 3.95 0.00C 3.95 0.92 3.02 0.00H 0.78 0.78 0.00 0.00total 191.47 53.10 128.39 9.96
Bonded ZnPc−Nonbonded ZnPc
atom total shift s shift p shift d shift
Zn −0.15 −0.16 0.00 +0.01N −0.10 −0.03 −0.07 0.00C −0.03 −0.01 −0.02 0.00H 0.00 0.00 0.00 0.00total −1.85 −0.72 −1.22 +0.01
aAll values are averaged on atomic species and summed in the “total”rows. Total and projected (on s, p, and d angular momentumchannels) charges related to the bonded ZnPc−ZnO configuration arereported in columns “Bonded ZnPc”. Total and projected charge shiftsare reported in columns “Bonded ZnPc−Nonbonded ZnPc”.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815443

configuration, respectively. Such a difference between MPMDand DFT adsorption energies can be ascribed to the delicateinterplay between covalent bonding and dispersion forces in themolecule−surface interaction. On the other hand, it should benoted that the two techniques find an identical value in the caseof a gas-phase ZnPc dimer, 1.6 eV, in line also with previousresults.20 Finally, in the case of the topmost molecule, DFTcalculations give adsorption energy values of 2.1 and 2.3 eV forthe stacked and partially stacked configurations, respectively. Acomparison of these values with that estimated for the gas-phase dimers indicates that in both configurations the topmostZnPc molecules are still weakly interacting with the underlyingZnO surface.On the side of the electronic properties, the stable AA
configuration is characterized by charge density distributionsand electronic levels hardly distinguishable from those relatedto the A configuration of a single molecule. Both moleculesundergo indeed a strong coupling with the ZnO surface whichleads to similar changes of the electronic structure, that is, alowering of the HOMO and LUMO levels (see Figure 2D),and the occurrence of mixed ZnPc/ZnO levels like thoseshown in Figure 3. It is worth noticing that, in this dimerconfiguration, the closeness of the two molecules does notinduce any mixing between their electronic states; that is, thereis no evidence of any intermolecular electronic interaction.In the case of the stacked AC configuration, the upper
molecule is only weakly coupled with the ZnO surface; itselectronic levels (see Figure 2E) are mainly aligned with theones corresponding to a ZnPc molecule kept far from thesurface and noninteracting with the electronic states of the ZnOsubstrate. The lower molecule shows instead an evolution ofthe electronic states consistent with a strong coupling with thesurface. This dimer configuration is also characterized by somemixing between the electronic states of the two moleculeswhich, however, is significantly smaller than that occurring ingas-phase dimers with the same “face-to-face” arrangement. Inthe AC′ case, an intermediate behavior between the AA and ACconfigurations has been found (not reported in Figure 2).The above results show the existence of marked differences
between the electronic properties of AC and AA dimers.Accordingly, the electronic and optical properties of a ZnPc/ZnO interface may depend on their relative abundance. AlignedAA dimers are energetically more stable and are expected to bemore abundant at equilibrium. However, stacked dimers caneasily form in solution or in gas phase19,20,27 and can beeventually adsorbed on the ZnO surface as AC dimers. At finitetemperature this AC configuration can also evolve into thelower energy AA configuration by overcoming a free energybarrier. Thus, the height of such barrier, which controls the AClifetime and relative population, becomes a delicate parameteraffecting the molecular assembling on the ZnO surface. In orderto take into account possible effects of dimer formation on theproperties of the ZnPc/ZnO system, we have investigated somefeatures of the dimer dynamics in a combined MPMD-DFTapproach. In this regard, as detailed in the SupportingInformation, we have preliminarily checked the accuracy ofthe MPMD approach by comparing the energy barriersestimated along a same path connecting AC and AAconfigurations by MPMD and by DFT-based nudged elasticband (NEB) simulations. The achieved results show a goodagreement between the two methods, thus strengthening thedescription of the dynamical behavior of ZnPc dimers given bymetadynamics. Then, we have considered a process where the
top molecule of the AC dimer slips down to the AAconfiguration and performed a new MPMD-based metady-namics calculation choosing as a collective variable the distancebetween the centers of two ZnPc molecules adsorbed on theZnO surface (details on this kind of simulations are givenelsewhere26). The corresponding one-dimensional free energyprofile (see Figure 5) is characterized by three minima
corresponding to the previously discussed AC, AC′, and AAdimers. The AC configuration is separated from the AAconfiguration by a free energy barrier of ∼0.4 eV, which is thesum of two contributions: a free energy barrier of 0.2 eV, whichbrings the AC dimer to the AC′ configuration; a further freeenergy barrier of 0.2 eV which allows the top molecule tofurther move, reaching the AA configuration. It is important tonote that the reverse reaction from AA to AC is quite unlikelyto occur because of the large free energy barrier (1.0 eV). Alltogether, the above results indicate that adhesive molecule−surface interactions dominate over cohesive molecule−molecule ones and suggest a dissolution of ZnPc dimers, thusfavoring the formation of a two-dimensional ZnPc monolayerinstead of three-dimensional structures.
J-Type Aggregates on the Surface. According to thementioned MPMD-based analysis,26 the energetically favoredaggregate of a large number of ZnPc molecules interacting withthe ZnO surface is represented by close-packed linear stripes ofZnPc along the X direction (see Figure 6). Such a J-stripeconfiguration, also observed in the case of a similar CuPc/TiO2interface,24 has been proposed as responsible of red-shiftedabsorption features observed in the ZnO/ZnPc system.27
Regarding DFT calculations, we simulate the structural andelectronic properties of J-stripes by enforcing periodicboundary conditions around an AA dimer along the X-axis;this corresponds to a supercell (enclosed into the red rectanglein Figure 6A) containing two molecules. A Zn−Zn distance of13 Å along the X-axis, corresponding to a ×4 periodicity alongthe surface Zn−O dimers row, indicates that nearest-neighborsmolecules are close packed at the minimum distance allowed bythe intrinsic dimensions of the system. At the same time, 21 Åseparate instead a molecule from its periodically repeated imagealong the Y-axis, ensuring negligible interaction effects. It isworth noticing that, due to the use of periodic boundaryconditions, in the following discussion of the J-stripes
Figure 5. Curve in the figure indicates the free energy sampled by aMPMD-based metadynamics calculation along the minimum freeenergy path related to the Zn−Zn intermolecular distance. The AA,AC, and AC′ sketched configurations are shown in Figure 4 anddescribed in the text.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815444

properties it will be assumed that present results can be appliedto stripes long enough to minimize boundary effects.On the side of the structural properties of this J-type system,
both the Zn−O molecule-surface bond and ZnPc orientationresult to be quite similar to those found for the AA dimer(compare Figures 6 and 4). On the side of the electronicproperties, the position of electronic eigenvalues with respect tothe ZnO bands, sketched in Figure 2F, is also almost identicalto the AA dimer one in Figure 2D. Thus, this J-type assemblingdoes not affect the main features of the molecule−surfaceelectronic coupling which characterize isolated molecules andAA dimers. Relevant differences have been found insteadconcerning the nature and spatial distribution of the J-stripeelectronic states. In addition to the above mixing of moleculeand surface electronic states occurring in the case of thenonperiodic interacting systems (i.e., the AA dimer config-uration), a periodically repeated molecular structure inducesindeed a strong mixing of molecular orbitals between the twoZnPc molecules forming the recurring unit in the stripe. As an
example, electron density plots of HOMO orbitals, correspond-ing to the eigenvalues represented by blue and light bluecontinuous lines in Figure 2F, clearly show an electronic chargedistribution on both molecules (see Figure 6B). Similar featurescharacterize LUMO orbitals in addition to the above-discussedmixing with the ZnO conduction band. This mixing ofmolecular electronic states affect the optical properties of J-type stripes, which result to be different from those of isolatedmolecules and dimers interacting with the ZnO surface, as itwill described in the next section. The orbital mixing betweenmolecules arranged in the J-type configuration indicates theoccurrence of intermolecular interactions. Moreover, suchinteractions represent an intrinsic property of such a moleculararrangement. The same orbital mixing has been found indeedfor ZnPc molecules arranged in the same J-type geometry andnot interacting with the ZnO surface.In order to gain further insight on the effects of
intermolecule interactions, a closer packing of ZnPc moleculeshas been also considered (see Figure 6C). In such a molecular
Figure 6. (A) Sticks and balls representation of a J-type aggregate of ZnPc molecules along the X direction (see Figure 1B) of the ZnO surface. Thesimulated periodic supercell, containing two ZnPc molecules, has been enclosed into a red rectangle. (B) Electron density plots of the mixed HOMOorbitals of two ZnPc molecules arranged in a J-type aggregate. (C) Sticks and balls representation of a close-packed ZnPc monolayer adsorbed on theZnO surface. The simulated periodic supercell, containing two ZnPc molecules, has been enclosed into a red rhombus. (D) Electron density plots ofthe mixed HOMO orbitals of ZnPc molecules in a close-packed monolayer. The distances (in Å) between nearest-neighbors molecules along the Xand Y directions have been highlighted.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815445

arrangement, basically, the J-type stripes are arranged in a close-packed configuration both along the X and Y axes in order tosample intermolecular interactions in both directions. This hasbeen achieved by setting up a rhombic surface periodic unitcontaining two molecules which are nearest neighbors bothalong the X axis and Y axis, as shown in Figure 6C. The abovearrangement can be also considered as a model of an orderedclose-packed ZnPc monolayer adsorbed on the ZnO surface,with intramolecular distances of 13.0 and 15.5 Å along the Xand Y axes, respectively. Present DFT results on thatarrangement clearly show the occurrence, in addition to theexpected mixing of molecule and surface electronic states, of astrong mixing of molecular orbitals between the two ZnPcmolecules forming the recurring unit; see, e.g., the electroniccharge distributions in Figure 6D. Thus, such an orbital mixing,whose extent is larger than that of the isolated J-stripe, dependsonly on the configuration of the neighboring molecules on theZnO surface.Optical Properties of ZnPc Molecules and ZnPc/ZnO
Systems. In the case of gas-phase, isolated ZnPc molecules,the absorption spectrum is characterized by two strong featuresknown as Q-band and Soret band, common to almost allphthalocyanine and porphyrin molecules, which fall in the redpart of the visible region (1.88 eV) and in the near-UV region(3.58 eV), respectively.57 Such absorption peaks are very wellreproduced by our TDDFPT calculations, as shown in Figure7B. When a single isolated ZnPc molecule is chemisorbed onthe surface, both the spectral features are red-shifted withrespect to the gas-phase spectrum (compare Figure 7B andFigure 7C). The Q-band, in particular, undergoes a 0.10 eV (37nm) red shift, in a full agreement with the above DFTindications of a smaller HOMO−LUMO energy differencearising from the ZnPc/ZnO interaction.In the cases of ZnPc dimers and stripes, the close proximity
of two or more macrocyclic rings can lead to a strong couplingof their electronic states (the Davydov effect mentioned above),which induces a splitting of the related excited states.19,28 This,in turn, is responsible for relevant shifts of the absorptionbands. In particular, a blue shift of the Q-band is predicted bythe Davydov model in the case of a cofacial alignment of two ormore molecules; a red shift of the Q-band, on the contrary, isexpected in the case of coplanar dimers or stripes, as long as astrong coupling of electronic states still occurs. The resultsachieved here for ZnPc molecules, both in gas phase and wheninteracting with the ZnO surface, agree with the above Davydovmodel. In the former case, a gas phase ZnPc dimer (cofacialalignment) undergoes indeed a 0.06 eV (21 nm) blue shift ofthe Q-band, as shown in Figure 7A. In the case of ZnPcmolecules interacting with the ZnO surface, besides the isolatedZnPc molecule, we have considered a J-type aggregation wherea coplanar alignment is accompanied by a strong mixingbetween ZnPc molecular orbitals (shown in Figure 6) whichadds to the mixing between molecular and surface states. Thecorresponding TDDFPT absorption spectra, shown in Figure7C,D, confirm the existence of significant relationships betweenaggregation patterns, the electronic properties of molecularaggregates on the surface, as described by “single particle” DFTcalculations, and their optical properties, as estimated by a“many body” TDDFPT approach. In the case of the J-typeconfiguration, a 0.18 eV (71 nm) red shift of the Q-band isindeed calculated, larger than that found for an isolatedmolecule adsorbed on the ZnO surface, thus indicating that theintermolecular mixing characterizing the J-stripes enhances the
red-shifting induced by the molecule−surface coupling. Thisresult is fully consistent with the red shift predicted by theDavydov model. Most important, it gives a firm theoreticalsupport to the results of the mentioned experimental studyinvestigating the properties of ZnPc-impregnated ZnO nano-crystals.27 This study reports indeed a pronounced red shift ofthe Q-band in the case of heavily loaded ZnO nanoparticles, bysuggesting also a key role of J-type aggregates in the opticalproperties of the hybrid interface. It is worth noticing that, inthe case of the monolayer coverage formed by dense ZnPcstripes, the above DFT results suggest an absorption spectrumfor such an arrangement quite similar to that calculated for theJ-type aggregation, possibly characterized by an even morepronounced red shift of the Q-band. The role of the molecularaggregation in the optical properties can be further acknowl-edged by the fact that AA dimers, although arranged in acoplanar fashion, do not show any appreciable intermolecularmixing of their degenerate HOMO orbitals.59 All together, theabove results indicate that a direct link can be establishedbetween the structure of molecular aggregates and the
Figure 7. TDDFPT absorption spectra of (A) a gas-phase ZnPc dimer,(B) an isolated gas-phase ZnPc molecule, (C) a ZnPc moleculeadsorbed on the ZnO surface, and (D) two ZnPc molecules arrangedin a J-type aggregate on the ZnO surface. The absorption energy valueof the typical phthalocyanine Q-band is reported in both eV and nm(the latter one in parentheses) to ensure the best comparison withexperimental measurements. The vertical dotted line is a guide to theeye which indicates the extent of red and blue shifts of the Q-band.(C) and (D) spectra involve the contribution of ZnO surface slabsunderlying the ZnPc molecules. As detailed in the TheoreticalFramework section, such a contribution has been subtracted out fromthe spectra and the resulting thin black lines have been smoothed byusing spline functions.58
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815446

simulated (and measured) absorption spectra, thus permittingto achieve a sound, theoretical picture of the properties of aZnPc/ZnO interface.Sensitization of the ZnO Surface. The dynamical
behavior of ZnPc molecules on the ZnO surface indicates ageneral tendency of ZnPc molecules to form planar aggregateson the surface, up to a monolayer coverage, rather thanmultilayered islands. Moreover, isolated molecules, aligneddimers, and J-stripes are expected to characterize submonolayercoverages of the ZnO surface. In the case of isolated molecules,a ZnPc molecule exhibits a strong chemical and electroniccoupling with the zinc oxide, characterized by: (i) Theoccurrence of a strong molecule−surface interaction, due tothe combined actions of chemical bonding and dispersionforces; this condition is noticeably realized without requiring amolecular functionalization for anchoring the molecule to thesurface. (ii) The appearance of a new unoccupied and mixedZnPc/ZnO orbital close to the ZnO conduction bandminimum and slightly below the ZnPc LUMO, as shown inFigure 2. The same kind of structural and electronic couplingcharacterizes ZnPc aligned dimers and J-stripes. In addition,intermolecular interactions occurring between ZnPc moleculesarranged in J-stripes enhance the red shift of the Q-band. Alltogether, these results permit a deeper understanding of theproperties of the ZnPc/ZnO interface and, at the same time,shed a few beams of light on the complex mechanisms ofgeneration, diffusion, and splitting of excited charge carriersunderlying the sensitization of the ZnO surface by means of theadsorption of ZnPc molecules. These molecules could actindeed as dyes for harvesting solar light up to the near-infraredregion in novel, solid-state architectures of dye sensitized solarcells. In this regard, a key role can be played by the mixedZnPc−ZnO orbitals, sketched in Figure 2 and shown in Figure3, laying just below the “native” LUMO. In fact, in the case ofe−−h+ pairs photogenerated inside the ZnPc molecules by π →π* HOMO−LUMO excitation processes (the so-called Q-bands, dominating the visible absorption spectra of phthalo-cyanine molecules57), such mixed orbitals may provide an easyroute to transfer the excited electrons to the ZnO conductionband. The same orbitals could also be involved in a directinjection from the ZnPc HOMO to the ZnO. The red shifteffects related to the formation of J-stripes could furtherimprove the light harvesting of such sensitized surfaces, whichis extended to the near-IR spectral region. It can be observedthat even the electronic properties of a possible secondmolecular layer can favor molecule to surface electron-transferprocesses: e−h pairs photogenerated into this layer could beindeed efficiently split, with electrons raised to the LUMOfalling downward into the ZnO conduction band and holesraising upward and transferred to a suitable transport medium,as required in DSSCs.
■ CONCLUSIONSIn the present study, we have performed an ab initio theoreticalinvestigation of the electronic and optical properties of thehybrid interface formed by ZnPc molecules adsorbed on theZnO surface. Such interface properties have been investigatedby considering the evolution of the properties of singlemolecules induced by the molecular assembling on the ZnOsurface. Dimers and stripes have been considered as basic unitsfor molecular assembling, while the dynamical behavior ofdimers has been investigated in detail in a combined DFT andMPMD approach. As already suggested by previous MPMD
calculations, and confirmed by the present results, ZnPcmolecules show a general tendency to form planar aggregateson the surface rather than multilayered islands. PreviousMPMD results also indicated that isolated molecules, aligneddimers, and J-stripes should characterize the ZnPc/ZnOinterface at submonolayer coverages. These indications havebeen taken into account here by considering also a fullmonolayer, formed by packed J-stripes, as a possible model ofthe ZnPc/ZnO interface. As major results, we have found that:(i) The above different kinds of molecular arrangement presentsimilar electronic properties deriving by a same kind of strongmolecule−surface coupling. The main features of such acoupling are represented by the existence of unoccupiedelectronic levels which, due to their nature and location, canplay a key role in the ignition of photogenerated electrons fromthe molecule to the surface, that is, in processes at the core ofthe hybrid photovoltaic cell operation. (ii) Intermolecularinteractions induce a blue shift of absorption bands in the caseof cofacial alignment of two molecules (stacked dimer) and ared shift in the case of planar J-stripes. This agrees with theDavydov phenomenological model and, most important, gives afirm theoretical support to the relationships between the redshift of absorption lines and the formation of J-type aggregatesproposed in ref 27. All together, the present results revealstructural and electronic properties of the ZnPc/ZnO interfacewhich make ZnPc-sensitized ZnO surfaces of high potentialinterest for improving the efficiency of different kinds of hybridphotovoltaic cells.
■ ASSOCIATED CONTENT*S Supporting InformationAssessment of theoretical methods (ZnO and ZnO/ZnPcsystems); details on the ZnO/ZnPc interaction (isolatedmolecule and ZnPc dimers). This material is available free ofcharge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge computational support by CYBERSAR(Cagliari, Italy), CINECA (Casalecchio di Reno, Italy; grantIscrA_SIMuLATe), and CASPUR (Rome, Italy; grants std11-478 and std11-666). We acknowledge the platform computa-tion of the Italian Institute of Technology (IIT) and financialsupport under Project IIT SEED “POLYPHEMO” and ProjectCNR “EFOR”. A.M. acknowledges Regione Autonoma dellaSardegna for funding under L.R. 2007. G.M. is glad to thank A.Paoletti for the useful discussions on phthalocyanine UV−visspectra.
■ REFERENCES(1) Gratzel, M. Acc. Chem. Res. 2009, 42, 1788−1798.(2) Mayer, A. C.; Scully, S. R.; Hardin, B. E.; Rowell, M. W.;McGehee, M. D. Mater. Today 2007, 10, 28−33.(3) Bredas, J.-L.; Norton, J. E.; Cornil, J.; Coropceanu, V. Acc. Chem.Res. 2009, 42, 1691−1699.(4) Kim, J. J.; Kim, K. S.; Jung, G. Y. J. Mater. Chem. 2011, 21, 7730−7735.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815447

(5) Karst, N.; Rey, G.; Doisneau, B.; Roussel, H.; Deshayes, R.;Consonni, V.; Ternon, C.; Bellet, D. Mater. Sci. Eng., B 2011, 176,653−659.(6) Xu, C.; Wang, Z. L. Adv. Mater. 2011, 23, 873−877.(7) Ozgur, U.; Alivov, Y. I.; Liu, C.; Teke, A.; Reshchikov, M. A.;Dooan, S.; Avrutin, V.; Cho, S.-J.; Morkoc, H. J. Appl. Phys. 2005, 98,041301.(8) Oosterhout, S. D.; Wienk, M. M.; van Bavel, S. S.; Thiedmann,R.; Koster, L. J. A.; Gilot, J.; Loos, J.; Schmidt, V.; Janssen, R. A. J. Nat.Mater. 2010, 8, 818−824.(9) Guerin, V. M.; Magne, C.; Pauporte, T.; Bahers, T. L.; Rathousky,J. ACS Appl. Mater. Interfaces 2010, 2, 3677−3685.(10) Greene, L. E.; Law, M.; Tan, D. H.; Montano, M.; Goldberger,J.; Somorjai, G.; Yang, P. Nano Lett. 2005, 5, 1231−1236.(11) Greene, L. E.; Yuhas, B. D.; Law, M.; Zitoun, D.; Yang, P. Inorg.Chem. 2006, 45, 7535−7543.(12) Diebold, U.; Vogel Koplitz, L.; Dulub, O. Appl. Surf. Sci. 2004,237, 336−342.(13) Cooke, D. J.; Marmier, A.; Parker, S. C. J. Phys. Chem. B 2006,110, 7985−7991.(14) Dag, S.; Wang, L.-W. Nano Lett. 2008, 8, 4185−4190.(15) Labat, F.; Ciofini, I.; Hratchian, H. P.; Frisch, M.; Raghavachari,K.; Adamo, C. J. Am. Chem. Soc. 2009, 131, 14290−14298.(16) Garcìa-Iglesias, M.; Cid, J.-J.; Yum, J.-H.; Forneli, A.; Vazquez,P.; Nazeeruddin, M. K.; Palomares, E.; Gratzel, M.; Torres, T. EnergyEnviron. Sci. 2011, 4, 189−194.(17) Boucle, J.; Ackermann, J. Polym. Int. 2012, 61, 355−373.(18) Mattioli, G.; Filippone, F.; Alippi, P.; Giannozzi, P.; AmoreBonapasta, A. J. Mater. Chem. 2012, 22, 440−446.(19) Cook, M. J. Spectroscopy of New Materials; John Wiley & SonsLtd.: New York, 1993; Chapter 3.(20) Marom, N.; Tkatchenko, A.; Scheffler, M.; Kronik, L. J. Chem.Theory Comput. 2010, 6, 81−90.(21) Papageorgiou, N.; Salomon, E.; Angot, T.; Layet, J.-M.;Giovannelli, L.; Le Lay, G. Prog. Surf. Sci. 2004, 77, 139−170.(22) Chen, X.; Fu, Y.-S.; Ji, S.-H.; Zhang, T.; Cheng, P.; Ma, X.-C.;Zou, X.-L.; Duan, W.-H.; Jia, J.-F.; Xue, Q.-K. Phys. Rev. Lett. 2008,101, 197208.(23) Godlewski, S.; Tekiel, A.; Prauzner-Bechcicki, J. S.; Budzioch, J.;Gourdon, A.; Szymonski, M. J. Chem. Phys. 2011, 134, 224701.(24) Wang, Y.; Ye, Y.; Wu, K. J. Phys. Chem. B 2006, 110, 17960−17965.(25) Yu, S.; Ahmadi, S.; Palmgren, P.; Hennies, F.; Zuleta, M.;Gothelid, M. J. Phys. Chem. C 2009, 113, 13765−13771.(26) Melis, C.; Raiteri, P.; Colombo, L.; Mattoni, A. ACS Nano 2011,5, 9639−9647.(27) Ingrosso, C.; Petrella, A.; Cosma, P.; Curri, M.; Striccoli, M.;Agostiano, A. J. Phys. Chem. B 2006, 110, 24424−24432.(28) Hush, N. S.; Woolsey, I. S. Mol. Phys. 1971, 21, 465−474.(29) Melis, C.; Colombo, L.; Mattoni, A. J. Phys. Chem. C 2011, 115,18208−18212.(30) Matsui, M.; Akaogi, M. Mol. Simul. 1991, 6, 239−244.(31) Wolf, D.; Keblinski, P.; Phillpot, S. R.; Eggebrecht, J. J. Chem.Phys. 1999, 110, 8254−8282.(32) Lin, F.; Wang, R. J. Chem. Theory Comput. 2010, 6, 1852−1870.(33) Smith, W.; Forester, T. R. J. Mol. Graphics 1996, 14, 136−141.(34) Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi,D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.;Parrinello, M. Comput. Phys. Commun. 2009, 180, 1961−1972.(35) Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.;Cavazzoni, C.; Ceresoli, D.; Chiarotti, G. L.; Cococcioni, M.; Dabo, I.;et al. J. Phys.: Condens. Matter 2009, 21, 395502.(36) Vanderbilt, D. Phys. Rev. B 1990, 41, 7892−7895.(37) Troullier, N.; Martins, J. L. Phys. Rev. B 1991, 43, 1993−2006.(38) Van de Walle, C. G.; Neugebauer, J. J. Appl. Phys. 2004, 95,3851−3879.(39) Dion, M.; Rydberg, H.; Schroder, E.; Langreth, D. C.;Lundqvist, B. I. Phys. Rev. Lett. 2004, 92, 246401.(40) Roman-Perez, G.; Soler, J. M. Phys. Rev. Lett. 2009, 103, 096102.
(41) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77,3865−3868.(42) Langreth, D. C.; Lundqvist, B. I.; Chakarova-Kack, S. D.;Cooper, V. R.; Dion, M.; Hyldgaard, P.; Kelkkanen, A.; Kleis, J.; Kong,L.; Li, S.; Moses, P. G.; Murray, E.; Puzder, A.; Rydberg, H.; Schroder,E.; et al. J. Phys.: Condens. Matter 2009, 21, 084203.(43) Cohen, A. J.; Mori-Sanchez, P.; Yang, W. Science 2008, 321,792−794.(44) Mori-Sanchez, P.; Cohen, A. J.; Yang, W. Phys. Rev. Lett. 2008,100, 146401.(45) Anisimov, V. I.; Aryasetiawan, F.; Liechtenstein, A. I. J. Phys.:Condens. Matter 1997, 9, 767−808.(46) Cococcioni, M.; de Gironcoli, S. Phys. Rev. B 2005, 71, 035105.(47) Heyd, J.; Scuseria, G. E.; Ernzerhof, M. J. Chem. Phys. 2003, 118,8207−8215.(48) Heyd, J.; Scuseria, G. E.; Ernzerhof, M. J. Chem. Phys. 2006, 124,219906.(49) Janesko, B. G.; Henderson, T. M.; Scuseria, G. E. Phys. Chem.Chem. Phys. 2009, 11, 443−454.(50) Rocca, D.; Lu, D.; Galli, G. J. Chem. Phys. 2010, 133, 164109.(51) Malcioglu, O. B.; Gebauer, R.; Rocca, D.; Baroni, S. Comput.Phys. Commun. 2011, 182, 1744−1754.(52) Rocca, D.; Gebauer, R.; De Angelis, F.; Nazeeruddin, M. K.;Baroni, S. Chem. Phys. Lett. 2009, 475, 49−53.(53) Kim, Y.-H.; Gorling, A. Phys. Rev. Lett. 2002, 89, 096402.(54) Izmaylov, A. F.; Scuseria, G. E. J. Chem. Phys. 2008, 129,034101.(55) We use the “LUMO” label, well defined in the case of anisolated ZnPc molecule, even when dealing with molecules interactingwith the ZnO surface, because also in this case an analysis of the totaland projected density of states (DOS and PDOS, respectively) permitsto identify an electronic level showing the same main features of theLUMO in the isolated molecule (e.g., a high degree of localization onthe ZnPc C and N atoms), which is therefore still referred to as theZnPc LUMO. A similar DOS and PDOS analysis has been performedin the case of the mixed ZnPc/ZnO electronic state shown in Figures 2and 3. Both the DOS and PDOS related to the LUMO and to the newmixed electronic level are provided as Supporting Information.(56) The issue has been extensively discussed in: Mattioli, G.;Filippone, F.; Giannozzi, P.; Caminiti, R.; Amore Bonapasta, A. Chem.Mater. 2009, 21, 4555−4567.(57) Edwards, L.; Gouterman, M. J. Mol. Spectrosc. 1970, 33, 292−310.(58) Reinsch, C. H. Num. Math. 1967, 10, 177−183.(59) For these reasons we have not considered isolated AA dimers asworth of a TDDFPT investigation; we expect an absorption spectrumvery similar to the one shown in Figure 7C.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp303781v | J. Phys. Chem. C 2012, 116, 15439−1544815448