y-chromosome haplotype origins via biogeographical multilateration
TRANSCRIPT

Y-Chromosome Haplotype Origins via Biogeographical Multilateration Michael R. Maglio
Abstract
Current Y-chromosome migration maps only cover the broadest-brush strokes of the
highest-level haplogroups. Existing methods generalize geographic patterns based on large
population genetic frequency and diversity. New tools are required to illuminate our
nomadic, stationary and genealogical histories. Biogeographical Multilateration (BGM)
illustrates directional flow as well as chronological and physical origins at the individual
haplotype level.
Introduction
Traditional genealogy and its reliance on
paper records can only take us as far back as
records exist. This is perhaps 300 or 400
years. It could be as much as 1000 years if
we can connect to wealthy or royal families.
Y-chromosome testing can illuminate our
haplogroup origins. Genetic migration maps
(Fig. 1) show our history from over 100,000
years to about 10,000 years ago. That leaves
a large gap of time and geographic location
for our nomadic ancestors and only covers the
Fig. 1 Migration routes based on male Y-
chromosome data. Source: The Genographic Project
broad-brush strokes of the highest-level
haplogroups. Heat maps get us down to the
distribution of high level SNPs within the
haplogroup. These distributions are based on
the current locations of test populations
(Underhill et al 2001). We must be careful
not to misinterpret the genetic gradient of an
organic process (Chikhi et al 2002). How do
we get to the migration patterns at the
individual haplotype level?
We need the ability to map the four phases
of our history. The phases: historical
(current to 400 years +) – the portion of our
family history that is well documented,
stationary (~500 to ~1500 years) – our
ancestors made the rural to urban transition
(Malanima 2007), staying roughly in the same
location for centuries, nomadic (~1500 to
10,000 years) – multiple millennia of
migration and origin (10,000 years +) – the
approximate birthplace of our haplogroups.
Genealogy can cover the historical and Y-
chromosome test results reveal the origin.
New tools are required to resolve the
stationary and nomadic phases. There is a
fifth phase – Out of Africa (OoA). This phase
is common across the majority of
haplogroups.
Anthropological and genetic evidence
shows our nomadic ancestors migrating
across the Neolithic at about 1 km per year.
The agricultural revolution required our
ancestors to settle and farm. It also allowed
them to build larger families and
communities. With each generation, the
population dispersed to exploit available
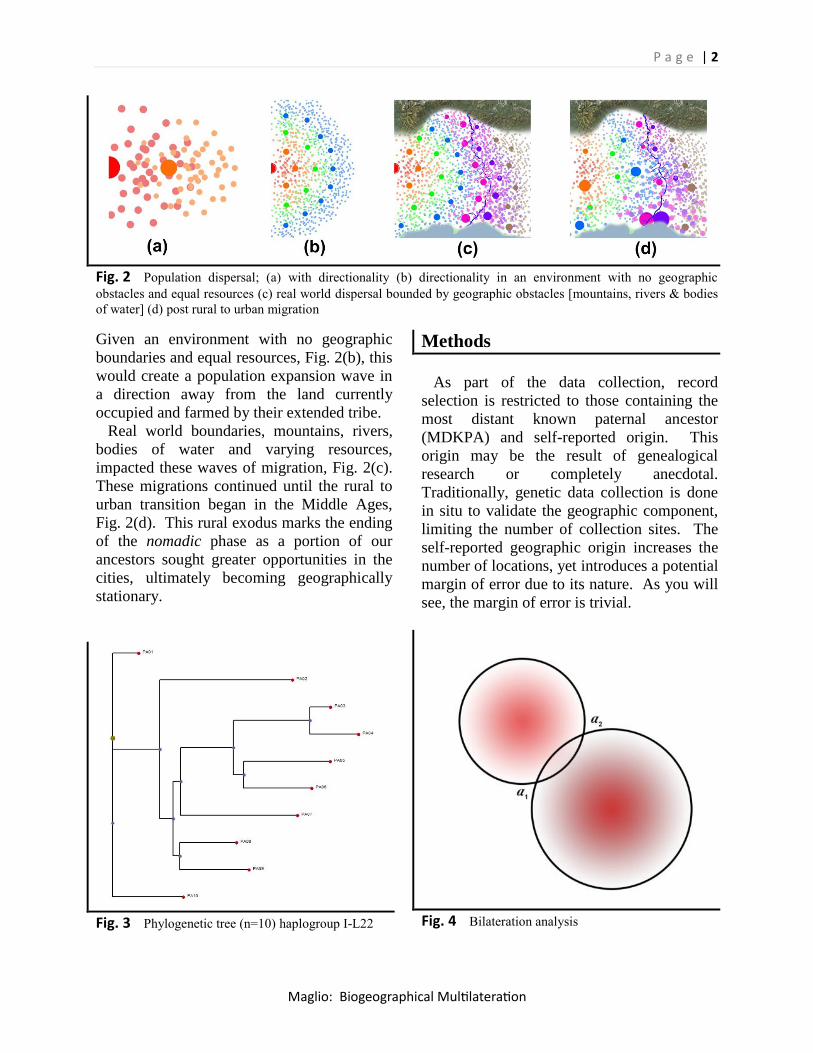
resources (Hazelwood et al 2004) Fig. 2(a).
P a g e | 2
Maglio: Biogeographical Multilateration
Fig. 2 Population dispersal; (a) with directionality (b) directionality in an environment with no geographic
obstacles and equal resources (c) real world dispersal bounded by geographic obstacles [mountains, rivers & bodies
of water] (d) post rural to urban migration
Given an environment with no geographic
boundaries and equal resources, Fig. 2(b), this
would create a population expansion wave in
a direction away from the land currently
occupied and farmed by their extended tribe.
Real world boundaries, mountains, rivers,
bodies of water and varying resources,
impacted these waves of migration, Fig. 2(c).
These migrations continued until the rural to
urban transition began in the Middle Ages,
Fig. 2(d). This rural exodus marks the ending
of the nomadic phase as a portion of our
ancestors sought greater opportunities in the
cities, ultimately becoming geographically
stationary.
Fig. 3 Phylogenetic tree (n=10) haplogroup I-L22
Methods
As part of the data collection, record
selection is restricted to those containing the
most distant known paternal ancestor
(MDKPA) and self-reported origin. This
origin may be the result of genealogical
research or completely anecdotal.
Traditionally, genetic data collection is done
in situ to validate the geographic component,
limiting the number of collection sites. The
self-reported geographic origin increases the
number of locations, yet introduces a potential
margin of error due to its nature. As you will
see, the margin of error is trivial.
Fig. 4 Bilateration analysis

P a g e | 3
Maglio: Biogeographical Multilateration
Data sets consist of multiple records from a
37 STR marker haplotype and corresponding
SNP (YCC 2002). In this exercise, I-L22 is
used. Time to most recent common ancestor
(TMRCA) is generated to a 95% confidence
(Walsh 2001) using FTDNA derived mutation
rates. This output is then used by the
Neighbor-joining method, which is part of the
PHYLIP package for inferring phylogenetic
relationships. A phylogenetic tree, Fig. 3, and
chronological distances are produced for each
data set. Data points are mapped using
genealogical origin and a radius drawn on a
Mercator projection calculated using the
upper value of the Neolithic migration rate of
30 km per 25 years or 1.2km/yr (Cavalli-
Sforza 2002, Hazelwood et al 2004). The
resulting intersection between pairs, Fig. 4,
represents the approximate location of the
common ancestor. A Time Difference of
Arrival (TDoA) approach is used for
detecting the origin (Peter et al 2013).
Traditional TDoA uses two or more
“beacons” with known locations and a
measurement of the time it takes to receive a
signal from each. The time is converted to a
distance. A current location can then be
deduced. In this analysis, the beacons are the
Fig. 5 Serial founder effect
paternal ancestor geographic origins. The
signal (electromagnetic wave) is the
population expansion wave measured in time,
TMRCA, converted to a distance using the
migration rate. A location for the common
ancestor can then be deduced. Bilateration
analysis of pair data proceeds through the
network of phylogenetic data illustrating
haplotype directional flow as well as
chronologic and geographic origins.
Discussion
Suppose that each Neolithic generation
spawned a new set of villages, Fig. 5. This
doesn’t mean that the entire previous village
up and left. There were those that stayed,
they had an investment in land and resources.
There were those that migrated, looking for
opportunities. In a perfect scenario, there
would be a descendant from the original tribe
in each village. Across time and geography,
Fig. 6 Tracing genetic markers
genetic differentiation increased. Each
village would carry only a subset of the
genetic diversity (Deshpande et al 2009) of
the previous village and have a unique genetic
signature. Collecting and comparing y-DNA

P a g e | 4
Maglio: Biogeographical Multilateration
Fig. 7 Bilateration scenarios, (a) intersecting ranges (b) non-intersecting ranges (c) range within a range
would show us the exact path that each tribal
branch took as they migrated. Unfortunately,
there isn’t a perfect scenario. Not every
genetic branch or even every village survived.
Only a small fraction of y-DNA has been
tested and made available for comparison.
We are left with fragmented data.
The self-reported origin that accompanies
the y-DNA in this study identifies the
ancestral location in the stationary phase.
Taking into consideration that not all villages
still exist and that the rural to urban transition
consolidated the population, we should not
expect to be able to trace a genetic line
exactly back geographically. An approximate
path can be determined by walking backwards
first through STR mutations and eventually
SNP mutations, following “genetic
breadcrumbs”.
Take any two haplotypes with self-reported
origins and generate a TMRCA. Multiply the
years by 1.2 to get distance to most recent
common ancestor (DMRCA). Initial analysis
confirms the upper bounds of the Neolithic
migration rate, 1.2km/yr. Using this rate
allows the solution to converge in fewer steps.
Using a Mercator projection, plot the two
circles at the ancestral origins with the
DMRCA as the radius. The perimeter
specifies the range for the common ancestor
and the intersection(s) indicates the possible
location. Bilateration is used to visually mark
the geographic location of the common
ancestor. If necessary, the exact coordinates
could be calculated.
There are three bilateration scenarios to
consider (Cota-Ruiz et al 2012). In Fig. 7(a),
two ranges meet tangentially, creating a single
intersection or they overlap, creating two
intersections. In the case of two intersections,
additional haplotype samples are used to
disambiguate. Multiple bilaterations turn into
a multilateration analysis. In Fig. 7(b), two
ranges do not intersect. This may indicate
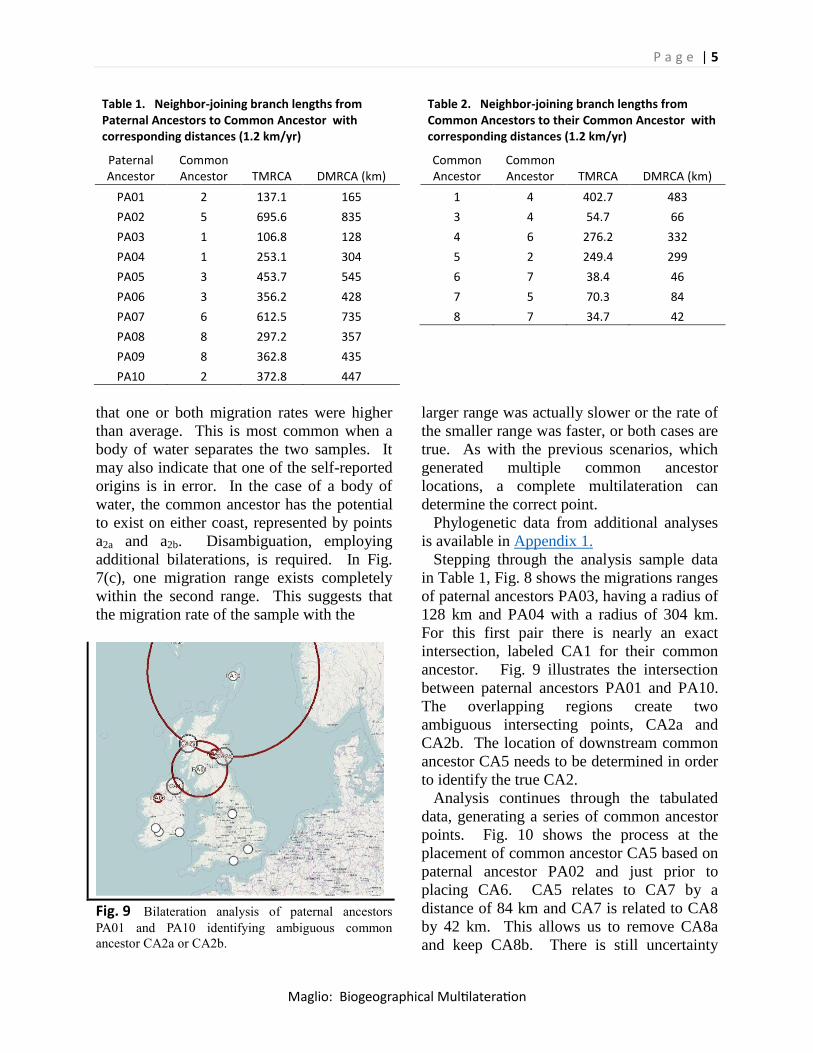
Fig. 8 Bilateration analysis of paternal ancestors
PA03 and PA04 identifying common ancestor CA1.
P a g e | 5
Maglio: Biogeographical Multilateration
Table 1. Neighbor-joining branch lengths from Paternal Ancestors to Common Ancestor with corresponding distances (1.2 km/yr)
Paternal Ancestor
Common Ancestor TMRCA DMRCA (km)
PA01 2 137.1 165
PA02 5 695.6 835
PA03 1 106.8 128
PA04 1 253.1 304
PA05 3 453.7 545
PA06 3 356.2 428
PA07 6 612.5 735
PA08 8 297.2 357
PA09 8 362.8 435
PA10 2 372.8 447
Table 2. Neighbor-joining branch lengths from Common Ancestors to their Common Ancestor with corresponding distances (1.2 km/yr)
Common Ancestor
Common Ancestor TMRCA DMRCA (km)
1 4 402.7 483
3 4 54.7 66
4 6 276.2 332
5 2 249.4 299
6 7 38.4 46
7 5 70.3 84
8 7 34.7 42
that one or both migration rates were higher
than average. This is most common when a
body of water separates the two samples. It
may also indicate that one of the self-reported
origins is in error. In the case of a body of
water, the common ancestor has the potential
to exist on either coast, represented by points
a2a and a2b. Disambiguation, employing
additional bilaterations, is required. In Fig.
7(c), one migration range exists completely
within the second range. This suggests that
the migration rate of the sample with the
Fig. 9 Bilateration analysis of paternal ancestors
PA01 and PA10 identifying ambiguous common
ancestor CA2a or CA2b.
larger range was actually slower or the rate of
the smaller range was faster, or both cases are
true. As with the previous scenarios, which
generated multiple common ancestor
locations, a complete multilateration can
determine the correct point.
Phylogenetic data from additional analyses
is available in Appendix 1.
Stepping through the analysis sample data
in Table 1, Fig. 8 shows the migrations ranges
of paternal ancestors PA03, having a radius of
128 km and PA04 with a radius of 304 km.
For this first pair there is nearly an exact
intersection, labeled CA1 for their common
ancestor. Fig. 9 illustrates the intersection
between paternal ancestors PA01 and PA10.
The overlapping regions create two
ambiguous intersecting points, CA2a and
CA2b. The location of downstream common
ancestor CA5 needs to be determined in order
to identify the true CA2.
Analysis continues through the tabulated
data, generating a series of common ancestor
points. Fig. 10 shows the process at the
placement of common ancestor CA5 based on
paternal ancestor PA02 and just prior to
placing CA6. CA5 relates to CA7 by a
distance of 84 km and CA7 is related to CA8
by 42 km. This allows us to remove CA8a
and keep CA8b. There is still uncertainty
P a g e | 6
Maglio: Biogeographical Multilateration
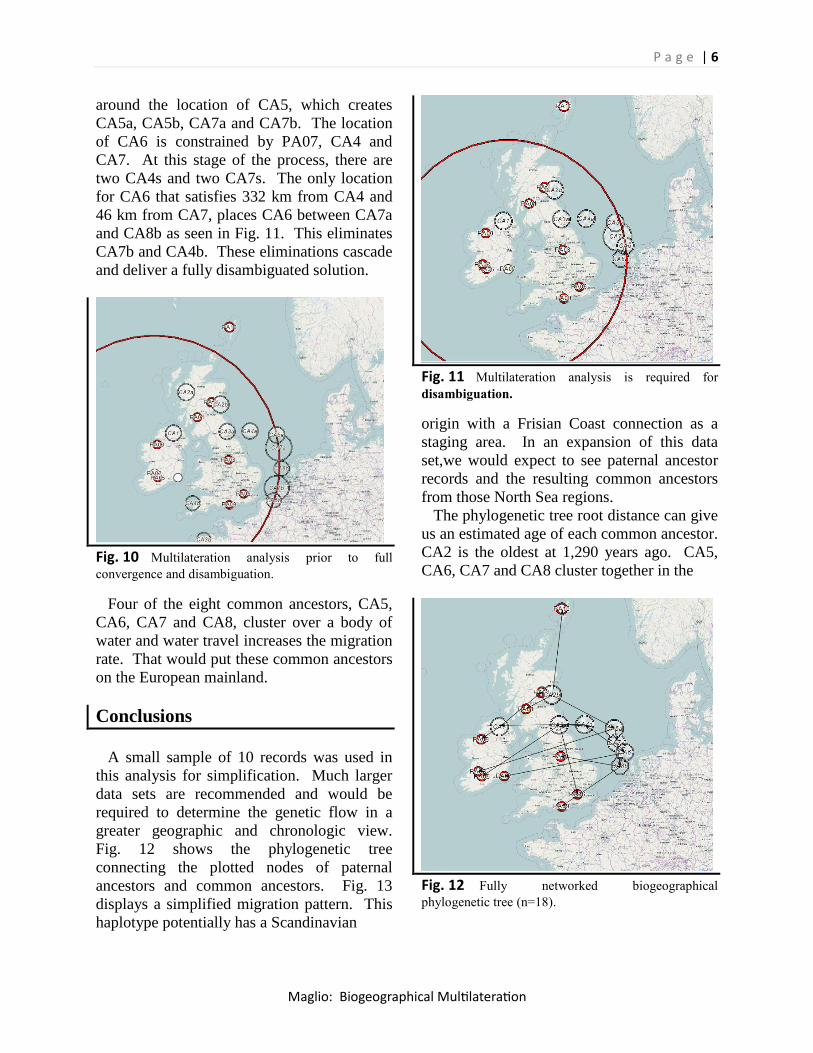
around the location of CA5, which creates
CA5a, CA5b, CA7a and CA7b. The location
of CA6 is constrained by PA07, CA4 and
CA7. At this stage of the process, there are
two CA4s and two CA7s. The only location
for CA6 that satisfies 332 km from CA4 and
46 km from CA7, places CA6 between CA7a
and CA8b as seen in Fig. 11. This eliminates
CA7b and CA4b. These eliminations cascade
and deliver a fully disambiguated solution.
Fig. 10 Multilateration analysis prior to full
convergence and disambiguation.
Four of the eight common ancestors, CA5,
CA6, CA7 and CA8, cluster over a body of
water and water travel increases the migration
rate. That would put these common ancestors
on the European mainland.
Conclusions
A small sample of 10 records was used in
this analysis for simplification. Much larger
data sets are recommended and would be
required to determine the genetic flow in a
greater geographic and chronologic view.
Fig. 12 shows the phylogenetic tree
connecting the plotted nodes of paternal
ancestors and common ancestors. Fig. 13
displays a simplified migration pattern. This
haplotype potentially has a Scandinavian
Fig. 11 Multilateration analysis is required for
disambiguation.
origin with a Frisian Coast connection as a
staging area. In an expansion of this data
set,we would expect to see paternal ancestor
records and the resulting common ancestors
from those North Sea regions.
The phylogenetic tree root distance can give
us an estimated age of each common ancestor.
CA2 is the oldest at 1,290 years ago. CA5,
CA6, CA7 and CA8 cluster together in the
Fig. 12 Fully networked biogeographical
phylogenetic tree (n=18).

P a g e | 7
Maglio: Biogeographical Multilateration
next age range of 830 to 975 years ago. The
haplogroup, dates and locations are all
consistent with the Norse and Viking raids on
the British Isles.
Fig. 13 Simplified genetic flow.
In the event that haplotype data does not
have self-reported origins, biogeographical
multilateration (BGM) has the potential to
narrow the range as the analysis can be used
to solve for an unknown location. Finding the
previously unidentified historical homeland
can aide in genealogical research. Clustering
of common ancestor data may indicate the
stationary phase sites. As the common
ancestor sites span the continent, we can see
the intermediate nomadic locations that
connect to the origins of our haplogroups.
BGM can be a major tool in developing
genetic migration patterns at the individual
haplotype level to bridge the gap between the
modern era and the maps of our Neolithic
origins.
Acknowledgements
I thank all of the DNA donors who have
made their results publically accessible for
review. Special thanks to Dean McGee for
making his DNA analysis website available.
Web Resources
Y-Utility: Y-DNA Comparison Utility,
http://www.mymcgee.com/tools/yutility.ht
ml?mode=ftdna_mode
References
Cavalli-Sforza LL (2002). Demic diffusion as
the basic process of human expansions.
Examining the farming/language dispersal
hypothesis. Cambridge: McDonald
Institute for Archaeological Research, 79-
88.
Chikhi L, Nichols RA, Barbujani G, &
Beaumont MA (2002). Y genetic data
support the Neolithic demic diffusion
model. Proceedings of the National
Academy of Sciences, 99(17), 11008-
11013.
Cota-Ruiz J, Rosiles JG, Sifuentes E, &
Rivas-Perea P (2012). A low-complexity
geometric bilateration method for
localization in wireless sensor networks
and its comparison with least-squares
methods. Sensors, 12(1), 839-862.
Deshpande O, Batzoglou S, Feldman MW, &
Cavalli-Sforza LL (2009). A serial
founder effect model for human
settlement out of Africa. Proceedings of
the Royal Society B: Biological Sciences,
276(1655), 291-300.
Hazelwood L, Steele J (2004) Spatial
dynamics of human dispersals -
Constraints on modelling and
archaeological validation. J ARCHAEOL
SCI , 31 (6) 669 - 679
Malanima P (2007) Decline or Growth?
European Cities and Rural Economies
1300-1600. University of Vienna
Peter BM, & Slatkin M (2013). Detecting
range expansions from genetic data. arXiv
preprint arXiv:1303.7475.
Underhill PA, Passarino G, Lin AA, Shen P,
Mirazon Lahr M, Foley RA, Oefner PJ &
Cavalli-Sforza LL (2001). The

P a g e | 8
Maglio: Biogeographical Multilateration
phylogeography of Y chromosome binary
haplotypes and the origins of modern
human populations. Annals of human
genetics, 65(1), 43-62.
Walsh B (2001). Estimating the time to the
most recent common ancestor for the Y
chromosome or mitochondrial DNA for a
pair of individuals. Genetics, 158(2), 897-
912.
Y-Chromosome-Consortium (2002) A
nomenclature system for the tree of
human Y-chromosomal binary
haplogroups. Genome Res 12:339–348