water-borne, in situ crosslinked biomaterials from phase-segregated precursors
TRANSCRIPT

Water-borne, in situ crosslinked biomaterials fromphase-segregated precursors
Brent Vernon,1* Nicola Tirelli,1 Thomas Bachi,2 David Haldimann,3 Jeffrey A. Hubbell1
1Department of Materials, Institute of Biomedical Engineering, Swiss Federal Institute of Technology (ETH),Moussonstr. 18, CH-8044 Zurich, Switzerland2Central Electron Microscopy Lab, University of Zurich, Zurich, Switzerland3Endospine AG, Cham, Switzerland
Received 15 November 2001; revised 21 March 2002; accepted 12 April 2002
Abstract: A novel process for the preparation of water-borne biomaterials for hard tissue repair from injectable pre-cursors is described, where the precursors form crosslinkedmaterials in situ under physiological conditions. The precur-sors react by means of a Michael-type addition reaction thatmakes use of addition donors such as pentaerythritol tet-rakis 3�-mercaptopropionate (QT) and addition acceptorssuch as poly(ethylene glycol) diacrylate 570 MW (PEGDA),pentaerythritol triacrylate (TA), and poly(propylene glycol)diacrylate 900 MW (PPODA). These crosslinked materials(at 75 wt% solid), prepared from water dispersions or re-verse emulsions, showed ultimate strengths in compressionof 1.8 ± 0.2 and 6.7 ± 0.5 MPa and ultimate deformations of
35 ± 2± and 37 ± 2%, respectively. Scanning electron micros-copy (SEM) shows that the morphology of the precursorstemplated the morphology of the final materials. The currentstudy indicates that it is possible to obtain injectable high-modulus materials that have appropriate mechanical prop-erties and gelation kinetics for tissue augmentation and sta-bilization applications such as mechanical stabilization ofthe intervertebral disc annulus. © 2003 Wiley Periodicals,Inc. J Biomed Mater Res 64A: 447–456, 2003
Key words: emulsions; in situ crosslinking; injectable bioma-terials; intervertebral disc; Michael-type reaction; SEM
INTRODUCTION
Injectable, in situ gelling biomaterials are attractivebecause of increased ease of use and reduced invasive-ness associated with their application as implantedmaterials for tissue augmentation and reconstruction.Examples of injectable biomaterials are plentiful. Theuse of in situ gelling materials has been investigatedfor the controlled delivery of drugs or cells.1–7In theseapplications the materials are often soft, hydrophilic,and biodegradable. On the other end of the spectrum,injectable biomaterials that harden in situ to form veryhard, stiff materials are investigated for application as
injectable bone cements or substitutes.8–10 Beyondthese examples, there is also need for injectable bio-materials that crosslink in situ possessing medium tohigh modulus (i.e., 0.5 to 50 MPa). Of particular inter-est in this study are materials applicable to the repairof defects in the intervertebral disc annulus. The an-nulus has been reported to have a Young’s modulus of5 MPa,11 and its successful mechanical stabilizationwould require a material with ultimate strength incompression of at least 8–10 MPa,12 stronger andharder than the drug-delivery materials but softer andmore resilient than the bone-replacement materials.Toward this goal, new water-borne materials havebeen designed, using water-insoluble precursors indispersion and reverse emulsion, thus combining ahigh crosslinking density in the hydrophobic phasewith a water environment.
The gelation process used in the current work isbased on the Michael-type addition of thiols onto ac-rylates 13 (See Fig. 1), a system suitable for biomateri-als applications because of its selectivity: acrylates re-act orders of magnitude faster with thiols than withamines and other nucleophiles present in biologicalsamples, where free thiols are present in negligible
*Current address: Department of Bioengineering, ArizonaState University, Tempe, Arizona
Correspondence to: N. Tirelli: e-mail: [email protected]
Contract grant sponsor: Endospine AG, Cham, Switzer-land
Contract grant sponsor: Medtech program of the SwissCommission for Technology and Innovation; contract grantnumber: 4266.1
© 2003 Wiley Periodicals, Inc.

extracellular quantities.14,15 Moreover, the reactiongenerates no toxic byproducts and, when multithioland multiacrylate precursors are used, crosslinkedmaterials are produced with negligible leachable con-tent. The Michael-type addition, being pH-activated,16
allows premixing of the reagents without reaction,and it can be started at a desired time by addition of abase. These monomeric multifunctional materials aredispersed in water at high solid content, typically 75wt%. Depending on the hydrophilic/lipophilic bal-ance, they can be dispersed with or without the help ofan emulsifier. The morphology of the fine dispersionsor emulsions templates the morphology of the finalcrosslinked material. This in situ curing process is par-ticularly suitable for conformal implants where theinjection of a simple low-viscosity and highly wetting(because of the water dispersion) liquid leads to atough solid shaped by the local in vivo environment(e.g., surrounding tissues).
The feasibility of using this Michael-type reaction togenerate materials potentially useful in the applicationof intervertebral disc annular repair has been investi-gated with the reagents shown in Figure 2. For thisreason, the current report focuses on their crosslinkingkinetics (gel points) and compression mechanicalproperties.
MATERIALS AND METHODS
Materials
Pentaerythritol-tetrakis (3-mercaptopropionate) (QT) andpentaerythritol triacrylate (TA) were both obtained fromFluka (Buchs, Switzerland). Poly(ethylene glycol) diacrylate570 MW (PEGDA), poly(propylene glycol) diacrylate 900MW (PPODA 900), and poly(propylene glycol-co-polyethylene glycol-co-polypropylene glycol) 3300 MW(PEP) were all obtained from Aldrich (Buchs, Switzerland).Sorbitan monooleate (SM) was purchased from Sigma(Buchs, Switzerland). All other reagents were reagent gradeand all materials were used as received.
Synthesis of poly(ethylene glycol) hexathiol (PEGHT)
The QT (8.58 g, 17.5 mmol) was combined with 8 mL H2Oand 2 mL 1N NaOH in 100 mL of tetrahydrofuran (THF).
The PEGDA 570 (1 g, 1.75 mmol) was combined with 1 mgof 2,6-di-tertbutyl-p-chresol (radical inhibitor) in 10 mL di-chloromethane (DCM). The PEGDA solution was then di-luted with 15 mL of THF. The diluted PEG solution was thendropwise added to the stirred QT solution. After 55 min thepH was adjusted to 7 using glacial acetic acid. To removewater, the solvent was evaporated and the product was re-dissolved in 100 mL of toluene. The toluene was evaporatedand an additional 100 mL of toluene was added. About 13 gof sodium sulfate was added and then the sodium sulfatewas removed by filtering. Before precipitation in 10-fold ex-cess diethyl ether, the solution was concentrated by evapo-rating some of the toluene. The diethyl ether was decantedand the liquid product was recovered. The product wasdried under vacuum.
1H-NMR (CDCl3): � = 4.25 (t, 2H, −OCH2CH2OOC−),3.6–3.7 (40+16H, CH2CH2O and C(=O)OCH2C(CH2−)3), 2.8(m, 8H, SCH2CH2COO), 2.65 (m, 8H, SCH2CH2COO), 1.65(t, 3H, −CH2SH) ppm
Fourier transform infrared (FTIR) (thin film): 2990-2790(� C-H), 2600 (� S-H), 1735 (�C=O of saturated esters),1460 (�s CH2), 1344, 1281, 1242, (�as C−O−C), 952, 842(�s C−O−C) cm−1.
Gel permeation chromatography (GPC): Mn = 1300; Mw =1500 (THF, PEG standards)
Synthesis of poly(ethylene glycol)tetraacrylate (PEGQA)
The PEGDA 570 (20 g, 35 mmol) was combined with 20mg of 2,6-di-tertbutyl-p-chresol in 10 mL DCM. This wasthen diluted with 90 mL THF. The NaOH (3 mL, 0.2N) and1 mL of H2O were added. The QT (1g, 2.0 mmol) was dis-solved in 40 mL of THF. The QT solution was added drop-wise to the stirred PEGDA solution. After 30 min, 7 �L ofglacial acetic acid was added to neutralize the reaction. Thesolvent was evaporated and 100 mL of toluene were added.After drying over sodium sulfate, the solution was filtered,concentrated and then precipitated using 10-fold excess di-ethyl ether. After precipitation the product was dried undervacuum.
1H-NMR (CDCl3): � = 6.4 (dd, 4H, CH2=CHCOO), 6.15(dd, 4H, CH2=CHCOO trans), 5.85 (dd, 4H, CH2=CHCOOcis), 4.25 (t, 2H, −OCH2CH2OOC−), 3.6–3.7 (40+16H,CH2CH2O and C(=O)OCH2C(CH2−)3), 2.8 (m, 8H,SCH2CH2COO), 2.65 (m, 8H, SCH2CH2COO) ppm
FTIR (thin film): 2990-2790 (� C-H), 1729 (� C=O ofsaturated + unsaturated esters), 1460 (�s CH2), 1344, 1281,
Figure 1. Michael-type reaction between thiols and acrylates is used as the basis for the in situ–gelling, injectable bioma-terials studied in this work.
448 VERNON ET AL.
1242, (�as C−O−C), 952, 842 (�s C−O−C), 800 (�o.o.p. ofvinyl CH) cm-1.
GPC: Mn = 2600; Mw = 2900 (THF, PEG standards)
Material preparation
Crosslinked materials were prepared as dispersions or re-verse emulsions of precursors in modified phosphate buff-ered saline (PBS). The PBS, 10 mM solution, was obtained bymixing equal volumes of 10 mM PBS adjusted to pH 9 withthe addition of triethanolamine or 1N NaOH, respectively.
As a typical procedure for 75-wt%dispersion materials, 424mg QT and 997 mg of PEGDA 570 were combined andmixed well by vortexing. Air bubbles were removed by soni-cation. The PBS solution (473 mg) was added to the mixedprecursors. The mixture was again vortexed for about 2 minto mix well and disperse the precursors in the aqueous so-lution. Following vortexing, the mixture was again soni-cated to remove air bubbles. The materials were then al-lowed to gel at either 25°C or 37°C.
Dispersion-type materials with barium sulfate inorganicparticles (as a radiocontrast agent) were prepared accordingto the protocol described above. Before addition of the acti-vating buffer (pH 9.0 PBS), 10 wt% of 0.8 �m BaSO4 particleswere added to the mixed precursors. The activating buffer
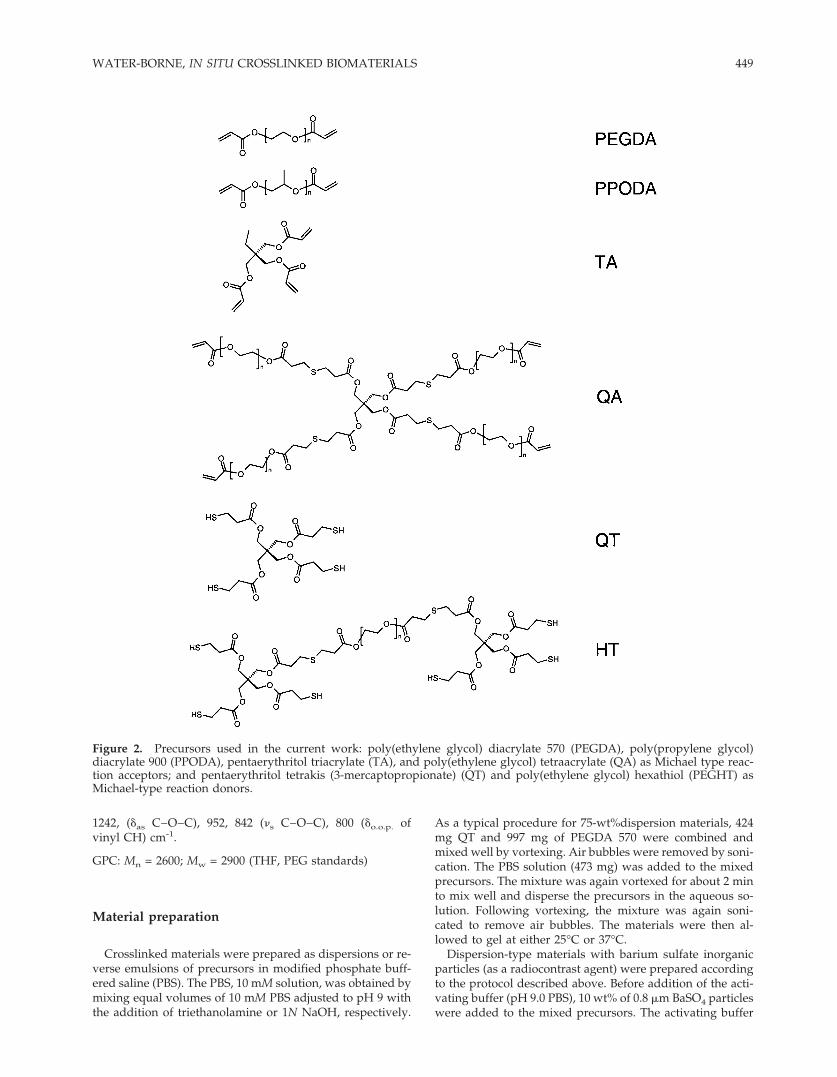
Figure 2. Precursors used in the current work: poly(ethylene glycol) diacrylate 570 (PEGDA), poly(propylene glycol)diacrylate 900 (PPODA), pentaerythritol triacrylate (TA), and poly(ethylene glycol) tetraacrylate (QA) as Michael type reac-tion acceptors; and pentaerythritol tetrakis (3-mercaptopropionate) (QT) and poly(ethylene glycol) hexathiol (PEGHT) asMichael-type reaction donors.
449WATER-BORNE, IN SITU CROSSLINKED BIOMATERIALS

was then added and the mixture was vortexed and allowedto crosslink.
Other dispersion-type materials were also prepared byadding SM to the PBS 9.0 buffer at 4 wt% before adding themixed precursors, QT, and PEGDA 570. Dispersion-typematerials at 75 wt% solid were similarly prepared using theprereacted precursors, PEGHT and PEGQA at 1:1 thiol toacrylate ratio.
Reverse-emulsion materials were also prepared. As a typi-cal procedure in preparing the reverse emulsion–type mate-rials, 1223 mg QT (2.5 mmol) was mixed with 993 mg TA (3.3mmol) and 90 mg PEP. The PBS buffer, with 0.1N NaOH,(738 mg) was added and the mix vortexed thoroughly. Themixture was then allowed to crosslink.
Crosslinking kinetics—shear rheology
The dispersions or reverse emulsions at 75 wt% contentwere placed between two parallel plates (separated by 100�m) of a CVO120 Rheometer (Bohlin Instruments) at 25°C.A constant oscillatory strain of 3 × 10−1 at 1 Hz was applied.The gel point was defined as the time when the phase anglewas equal to 45° (i.e., loss modulus equal to the storagemodulus). The mean gel time and mean rate of change in thephase angle at gelation for five samples were reported foreach of these three materials.
Cross-linking kinetics—attenuated totalreflection–infrared spectroscopy (ATR-IR)
The QT and PEGDA (75 wt%) dispersion gels were pre-pared with 1:1 thiol to acrylate ratios and 2:1 ratios to dem-onstrate the 1:1 consumption of thiols and acrylates by quan-tifying the S-H stretching vibration at 2600 cm−1 and theolefin out-of-plane C-H bending vibration at 800 cm−1. Bothpeaks were normalized in intensity, referring to the peak ofC-H stretching vibrations, the intensity of which should notchange dramatically during the reaction.
Morphology
The physical morphology of 75 wt% QT/PEGDA disper-sions and QT/PPODA reverse emulsions was investigatedusing scanning electron microscopy (SEM) on a JEOL 255electron microscope. Dispersion materials (75 wt% solid)were also imaged using light microscopy at 100× in brightfield.
Mechanical properties
Mechanical properties in compression were investigatedfor dispersion-type materials with and without added sur-factant or radiopaque inorganic particles, for dispersion-type materials using prereacted precursors, and for reverseemulsion–type materials. The effect of the thiol-to-acrylate
ratio on material ultimate strength and ultimate deformationwas also investigated in the dispersion materials by adjust-ing the relative quantities of QT and PEGDA 570, keepingthe solid content at 75 wt%.
Mechanical properties were evaluated for the reverseemulsion–type materials, varying the solid content and thecrosslinking density (by substituting TA and PPODA 900).
Each gelling mixture was poured into polyethylene tubeswith an internal diameter of 8 mm and allowed to cure for24 h. Following gelation, the material was cut into 6-mm-long sections, using a metal frame to standardize the samplesize and ensure sectioning perpendicular to the axis of thecylindrical sample. After 24 h in 10 mM PBS, the gels werecompressed with a Zwick Z005/TN2S mechanical tester(Zwick GmbH, Ulm, Germany) with a 5 kN load cell at 10mm/min to get �max (ultimate strength) and �max (ultimatedeformation) data. At least three samples for each materialtype were tested.
Statistical analysis
Results are reported as mean ± standard deviation. Statis-tical significance was determined using the Student t test.Statistical significance is assumed for p < 0.05.
RESULTS
Crosslinking kinetics
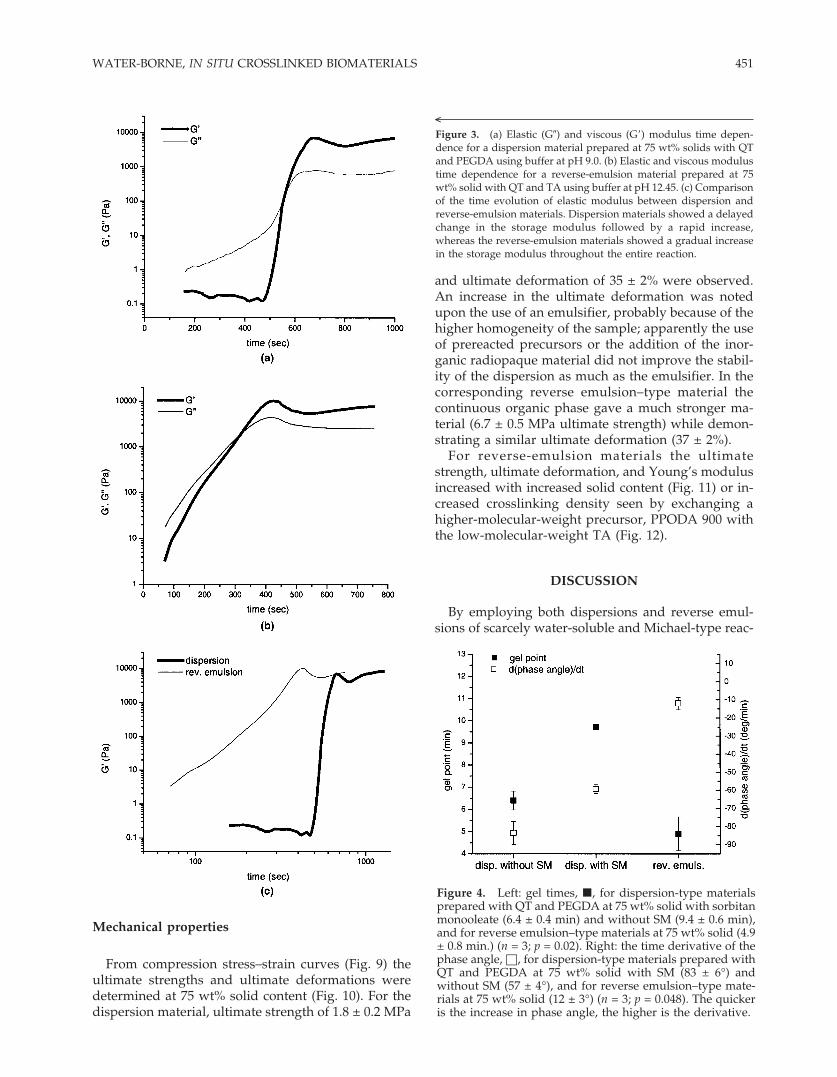
Typical rheological measurements are presented inFigure 3(a,b) for a 75-wt% dispersion or reverse-emulsion material, respectively. Differences in gelpoint and rate of change of the phase angle at the gelpoint are shown in Figure 4. The addition of SM de-creased the gel point from 9.4 ± 0.6 min to 6.4 ± 0.4 min(n = 3, p = 0.001). The reverse-emulsion materialshowed an even shorter gel point, 4.9 ± 0.8 min, butwith a more gradual transition: d(phase angle)/dt ofonly 12 ± 3°/min compared with 57 ± 4°/min (fordispersion-type materials without SM) or 83 ± 6°/min(dispersion-type materials with SM).
Monitoring the depletion of thiols and acrylates as afunction of time (Fig. 5) showed that both with 1:1 and2:1 thiol-to-acrylate formulations the two groups fol-lowed similar depletion kinetics. Additionally, at 1:1ratio, no unreacted groups were visible at the end ofthe reaction, whereas in the 2:1 case the thiols werereduced to half their original concentration, whereasthe acrylates were reduced to nearly zero at the end ofthe reaction.
Morphology
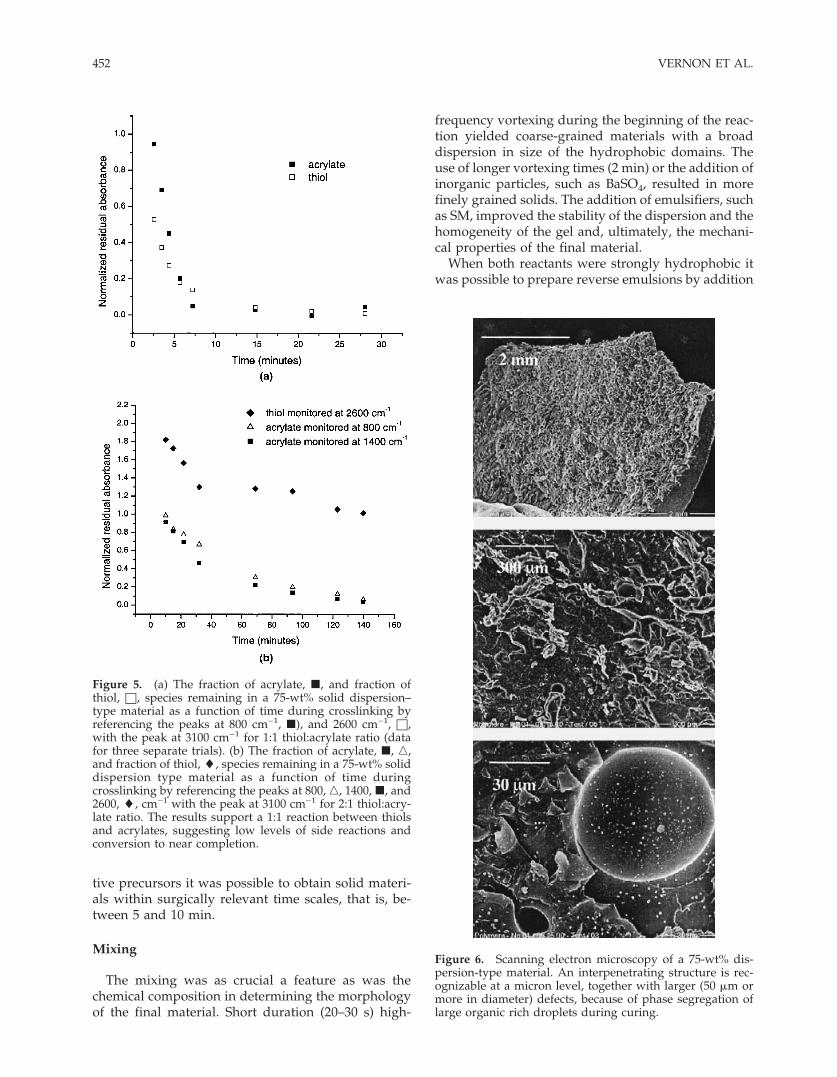
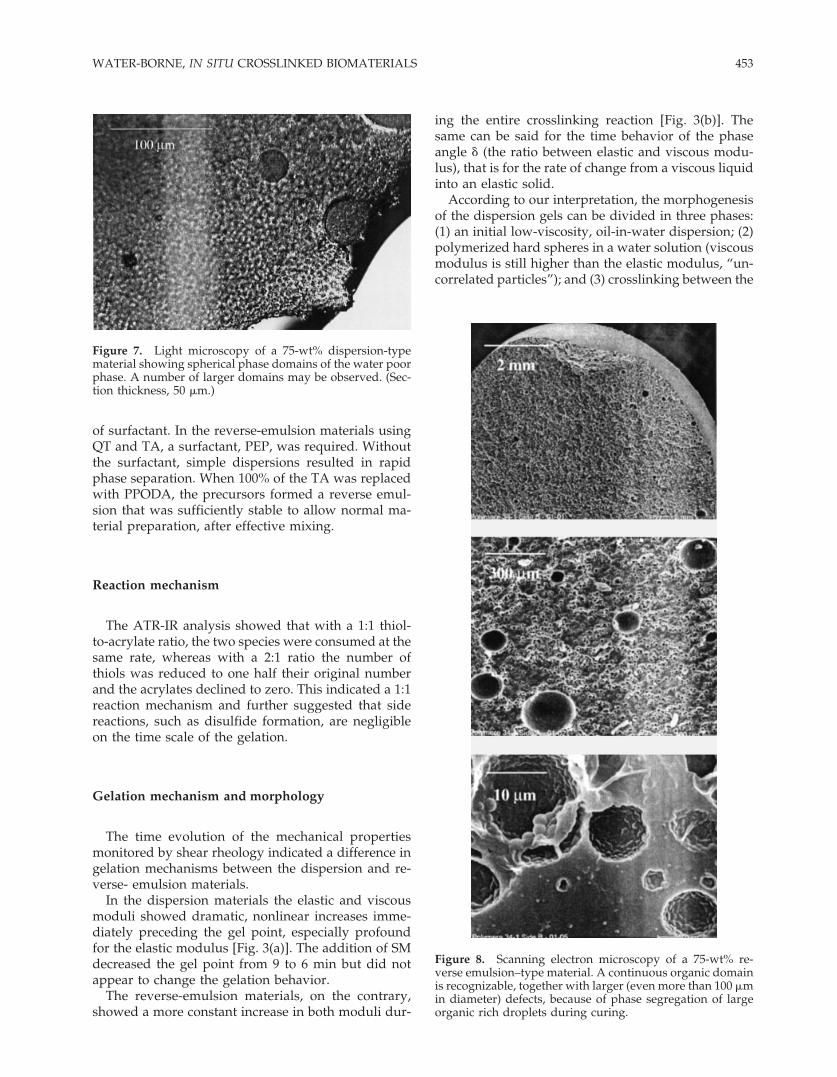
The SEM images showed the course and likely in-terpenetrated structure of the dispersion materials(Fig. 6; the corresponding optical microscopy pictureis in Fig. 7), and the continuous organic phase of thereverse-emulsion ones (Fig. 8).
450 VERNON ET AL.
Mechanical properties
From compression stress–strain curves (Fig. 9) theultimate strengths and ultimate deformations weredetermined at 75 wt% solid content (Fig. 10). For thedispersion material, ultimate strength of 1.8 ± 0.2 MPa
and ultimate deformation of 35 ± 2% were observed.An increase in the ultimate deformation was notedupon the use of an emulsifier, probably because of thehigher homogeneity of the sample; apparently the useof prereacted precursors or the addition of the inor-ganic radiopaque material did not improve the stabil-ity of the dispersion as much as the emulsifier. In thecorresponding reverse emulsion–type material thecontinuous organic phase gave a much stronger ma-terial (6.7 ± 0.5 MPa ultimate strength) while demon-strating a similar ultimate deformation (37 ± 2%).
For reverse-emulsion materials the ultimatestrength, ultimate deformation, and Young’s modulusincreased with increased solid content (Fig. 11) or in-creased crosslinking density seen by exchanging ahigher-molecular-weight precursor, PPODA 900 withthe low-molecular-weight TA (Fig. 12).
DISCUSSION
By employing both dispersions and reverse emul-sions of scarcely water-soluble and Michael-type reac-
Figure 4. Left: gel times, �, for dispersion-type materialsprepared with QT and PEGDA at 75 wt% solid with sorbitanmonooleate (6.4 ± 0.4 min) and without SM (9.4 ± 0.6 min),and for reverse emulsion–type materials at 75 wt% solid (4.9± 0.8 min.) (n = 3; p = 0.02). Right: the time derivative of thephase angle, □, for dispersion-type materials prepared withQT and PEGDA at 75 wt% solid with SM (83 ± 6°) andwithout SM (57 ± 4°), and for reverse emulsion–type mate-rials at 75 wt% solid (12 ± 3°) (n = 3; p = 0.048). The quickeris the increase in phase angle, the higher is the derivative.
<Figure 3. (a) Elastic (G�) and viscous (G�) modulus time depen-dence for a dispersion material prepared at 75 wt% solids with QTand PEGDA using buffer at pH 9.0. (b) Elastic and viscous modulustime dependence for a reverse-emulsion material prepared at 75wt% solid with QT and TA using buffer at pH 12.45. (c) Comparisonof the time evolution of elastic modulus between dispersion andreverse-emulsion materials. Dispersion materials showed a delayedchange in the storage modulus followed by a rapid increase,whereas the reverse-emulsion materials showed a gradual increasein the storage modulus throughout the entire reaction.
451WATER-BORNE, IN SITU CROSSLINKED BIOMATERIALS
tive precursors it was possible to obtain solid materi-als within surgically relevant time scales, that is, be-tween 5 and 10 min.
Mixing
The mixing was as crucial a feature as was thechemical composition in determining the morphologyof the final material. Short duration (20–30 s) high-
frequency vortexing during the beginning of the reac-tion yielded coarse-grained materials with a broaddispersion in size of the hydrophobic domains. Theuse of longer vortexing times (2 min) or the addition ofinorganic particles, such as BaSO4, resulted in morefinely grained solids. The addition of emulsifiers, suchas SM, improved the stability of the dispersion and thehomogeneity of the gel and, ultimately, the mechani-cal properties of the final material.
When both reactants were strongly hydrophobic itwas possible to prepare reverse emulsions by addition
Figure 6. Scanning electron microscopy of a 75-wt% dis-persion-type material. An interpenetrating structure is rec-ognizable at a micron level, together with larger (50 �m ormore in diameter) defects, because of phase segregation oflarge organic rich droplets during curing.
Figure 5. (a) The fraction of acrylate, �, and fraction ofthiol, □, species remaining in a 75-wt% solid dispersion–type material as a function of time during crosslinking byreferencing the peaks at 800 cm−1, �), and 2600 cm−1, □,with the peak at 3100 cm−1 for 1:1 thiol:acrylate ratio (datafor three separate trials). (b) The fraction of acrylate, �, �,and fraction of thiol, �, species remaining in a 75-wt% soliddispersion type material as a function of time duringcrosslinking by referencing the peaks at 800, �, 1400, �, and2600, �, cm−1 with the peak at 3100 cm−1 for 2:1 thiol:acry-late ratio. The results support a 1:1 reaction between thiolsand acrylates, suggesting low levels of side reactions andconversion to near completion.
452 VERNON ET AL.
of surfactant. In the reverse-emulsion materials usingQT and TA, a surfactant, PEP, was required. Withoutthe surfactant, simple dispersions resulted in rapidphase separation. When 100% of the TA was replacedwith PPODA, the precursors formed a reverse emul-sion that was sufficiently stable to allow normal ma-terial preparation, after effective mixing.
Reaction mechanism
The ATR-IR analysis showed that with a 1:1 thiol-to-acrylate ratio, the two species were consumed at thesame rate, whereas with a 2:1 ratio the number ofthiols was reduced to one half their original numberand the acrylates declined to zero. This indicated a 1:1reaction mechanism and further suggested that sidereactions, such as disulfide formation, are negligibleon the time scale of the gelation.
Gelation mechanism and morphology
The time evolution of the mechanical propertiesmonitored by shear rheology indicated a difference ingelation mechanisms between the dispersion and re-verse- emulsion materials.
In the dispersion materials the elastic and viscousmoduli showed dramatic, nonlinear increases imme-diately preceding the gel point, especially profoundfor the elastic modulus [Fig. 3(a)]. The addition of SMdecreased the gel point from 9 to 6 min but did notappear to change the gelation behavior.
The reverse-emulsion materials, on the contrary,showed a more constant increase in both moduli dur-
ing the entire crosslinking reaction [Fig. 3(b)]. Thesame can be said for the time behavior of the phaseangle � (the ratio between elastic and viscous modu-lus), that is for the rate of change from a viscous liquidinto an elastic solid.
According to our interpretation, the morphogenesisof the dispersion gels can be divided in three phases:(1) an initial low-viscosity, oil-in-water dispersion; (2)polymerized hard spheres in a water solution (viscousmodulus is still higher than the elastic modulus, “un-correlated particles”); and (3) crosslinking between the
Figure 7. Light microscopy of a 75-wt% dispersion-typematerial showing spherical phase domains of the water poorphase. A number of larger domains may be observed. (Sec-tion thickness, 50 �m.)
Figure 8. Scanning electron microscopy of a 75-wt% re-verse emulsion–type material. A continuous organic domainis recognizable, together with larger (even more than 100 �min diameter) defects, because of phase segregation of largeorganic rich droplets during curing.
453WATER-BORNE, IN SITU CROSSLINKED BIOMATERIALS
hard spheres to form continuous gel (“correlated par-ticles”). The slow initial increase of the viscous modu-lus should correspond to phases (1) and (2), and thesuccessive quick development of the elastic modulusshould correspond to the interconnection of the hardparticles (phase3).
As seen in the literature, crosslinked micelles17 orminiemulsions18 tend to form noninteracting micro- ornano-particles. These dispersion-type materials, re-ported here, were able to form crosslinked biphasicsolids because of two factors. First, the surface prop-erties of the PEGDA 570 stabilized the particles (effectincreased by adding an emulsifier) and hindered ag-gregation and bulk phase separation. Second, the
small but existing water solubility of the PEGDA 570and of the deprotonated QT allowed the reaction toproceed (with a slower kinetics) also in the waterphase.
More homogeneous-sized grains with a different in-terfacial composition were obtained with an emulsi-fier. This interpretation was also supported by SEMand optical microscopy, which showed an irregularmorphology for the dispersion materials (Figs. 6 and7). These materials exist as a system of phase domains(likely a water-poor phase within a water-rich phase)whose size and heterogeneity also depend on thelength and intensity of mixing during the gelation pe-riod.
A continuous, homogeneous organic phase withsmall aqueous phase domains interspersed was re-corded for the reverse-emulsion gels (Fig. 8).
Figure 9. Typical stress–strain behavior of the reverseemulsion–type material (75 wt%solid with QT and TA).
Figure 10. Ultimate strength and ultimate deformation ofvarious materials at 75 wt% solid. A, QT and PEGDA 570dispersion; B, QT and PEGDA 570 dispersion with SM (1wt%); C, QT and PEGDA 570 dispersion with BaSO4 (10-wt% barium sulfate 0.8-�m particles); D, PEGHT and QAdispersion; E, QT and PPODA reverse emulsion; and F, QTand TA reverse emulsion (n = 3).
Figure 11. Mechanical behavior as a function of solid con-tent in reverse emulsion–type materials, with QT and TA. (a)Ultimate stress and ultimate deformation values. (b)Young’s moduli at 1 MPa (n = 3, except at 83 wt% solid intop frame, where n = 1).
454 VERNON ET AL.
Mechanical properties
All the materials evaluated in this study exhibitedstrain hardening (increasing Young’s modulus withstrain). The dramatic difference in the ultimate stressbetween the reverse-emulsion and suspension gels(up to 4 times higher ultimate stress was observed forthe reverse-emulsion gels) are ascribed to the differentconnectivity of the hard, crosslinked organic domainsand to variations in the crosslinking density. Thevariation in the crosslinking density in reverse emul-sions, obtained by substituting PPODA with TA [seeFig. 11(b)], caused significant increases in their ulti-mate stress, especially at higher TA content. The lowermodulus for the dispersion gels was likely caused bythe viscous behavior of the water-based matrix, andthe failure of the dispersion gels at lower loadings is
ascribed to the continuity of the water (softer) domain,which allowed easier crack propagation.
By comparing the mechanical properties of materi-als obtained from dispersions with and without sur-factant and reverse emulsions, it would appear thatthe average grain diameter and the size dispersion(reduced by the presence of an emulsifier) stronglyinfluenced the failure behavior, whereas the elasticmodulus was mainly dependent on the chemical com-position (crosslinking density).
CONCLUSION
This work has demonstrated the possibility of usingthese precursors as dispersions or reverse emulsionsto produce injectable materials that are capable of cur-ing into solids at the implant site. This system useschemical reactions that are self-selective, reducing po-tential side reactions with other species found in thebody. Although we have previously demonstratedthat such reactions could be used to make highly swol-len hydrogels,14 this work focused on materials thatare much stronger, potentially suitable for load-bearing applications, such as augmentation of collag-enous or cartilaginous tissues, including the annulusof the spinal disc. It was possible to tailor materialproperties and the crosslinking kinetics of these ma-terials. It was also possible to modulate domain size,and thus failure characteristics, by the addition of sur-factants and by varying mixing time. Although themechanical properties and gelation kinetics indicatepotential for use in the targeted applications, biocom-patibility and stability studies must be carried out tofully realize this potential. These studies are currentlyunder way.
The authors acknowledge Mr. Klaus Marquardt for theSEM investigation. This work was funded in part by Endo-spine AG (Cham, Switzerland) and the Medtech program ofthe Swiss Commission for Technology and Innovation (proj-ect no. 4266.1).
References
1. Jeong B, Bae YH, Kim SW. Drug release from biodegradableinjectable thermosensitive hydrogel of PEG-PLGA-PEG tri-block copolymers. J Controlled Release 2000;63:155–163.
2. Jain RA, Rhodes CT, Railkar AM, Malick AW, Shah NH. Con-trolled release of drugs from injectable in situ formed biode-gradable PLGA microspheres: effect of various formulationvariables. Eur J Pharm Biopharm 2000; 50:257–262.
3. Ismail FA, Napaporn J, Hughes JA, Brazeau GA. In situ gelformulations for gene delivery: release and myotoxicity stud-ies. Pharm Dev Technol 2000;5:391–397.
4. Eliaz RE, Kost J. Characterization of a polymeric PLGA-
Figure 12. Mechanical behavior as a function of theamount of TA in reverse emulsion–type materials with QT,PPODA, and TA at 75 wt% solid. (a) Ultimate stress andultimate deformation values. (b) Young’s moduli at 1 MPa (n= 3).
455WATER-BORNE, IN SITU CROSSLINKED BIOMATERIALS

injectable implant delivery system for the controlled release ofproteins. J Biomed Mater Res 2000;50:388–396.
5. Chenite A, Chaput C, Wang D, Combes C, Buschmann MD,Hoemann CD, Leroux JC, Atkinson BL, Binette F, Selmani A.Novel injectable neutral solutions of chitosan form biodegrad-able gels in situ. Biomaterials 2000;21:2155–2261.
6. Cappello J, Crissman JW, Crissman M, Ferrari FA, Textor G,Wallis O, Whitledge JR, Zhou X, Burman D, Aukerman L, Ste-dronsky ER. In-situ self-assembling protein polymer gel sys-tems for administration, delivery, and release of drugs. J Con-trolled Release 1998;53:105–117.
7. Vernon B, Kim SW, Bae YH. Thermoreversible copolymer gelsfor extracellular matrix of a biohybrid pancreas. J Biomater Res2000;51:69–79.
8. Turczyn R, Weiss P, Lapkowski M, Daculsi G. In situ self hard-ening bioactive composite for bone and dental surgery. J Bio-mater Sci Polym Ed 2000;11:217–223.
9. McKoy BE, An YH. An injectable cementing screw for fixationin osteoporotic bone. J Biomed Mater Res 2000;53:216–220.
10. Weiss P, Gauthier O, Bouler JM, Grimandi G, Daculsi G. In-jectable bone substitute using a hydrophilic polymer. Bone1999;25(Suppl 2):67S–70S.
11. Meakin JR, Hukins WL. Replacing the nucleus pulposus of theintervertebral disk: prediction of suitable properties of a re-
placement material using finite element analysis. J Mater SciMater Med 2001;12:207–213.
12. Humzah MD, Soames RW. Human intervertebral disc: Struc-ture and function. Anat Rec 1988;220:337–356.
13. Hurd CD, Gershbein LL. Reactions of mercaptans with acrylicand methacrylic derivatives. J Am Chem Soc 1947;69:2328–2335.
14. Elbert DL, Pratt AB, Lutolf MP, Halstenberg S, Hubbell JA.Protein delivery from materials formed by self-selective con-jugate addition reactions. J Controlled Release 2001;76:11–25.
15. Friedman M, Cavins JF, Wall JS. Relative nucleophilic reactivi-ties of amino groups and mercaptide ions in addition reactionswith �,�-unsaturated compounds. J Am Chem Soc 1965;87:3672–3682.
16. Lutolf M, Tirelli N, Cerritelli S, Colussi L, Hubbell JA. System-atic modulation of (Michael-type) reactivity of thiols throughthe use of charged amino acids. Bioconjugate Chem 2001;12:1051–1056.
17. Butun V, Wang XS, Banez MVD, Robinson KL, Billingham NC,Armes SP, Tuzar Z. Synthesis of shell cross-linked micelles athigh solids in aqueous media. Macromolecules 2000;33:1–3.
18. Landfester K, Tiarks F, Hentze HP, Antonietti M. Polyadditionin miniemulsions: A new route to polymer dispersions. Mac-romol Chem Phys 2000;201:1–5.
456 VERNON ET AL.