university of groningen stress, gender and psychopathology
TRANSCRIPT

University of Groningen
Stress, gender and psychopathologyKuipers, Sjoukje Daouia
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2004
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Kuipers, S. D. (2004). Stress, gender and psychopathology: a multi-level analysis. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 17-07-2022

STRESS, GENDER
AND PSYCHOPATHOLOGY:
A MULTI-LEVEL ANALYSIS
Sjoukje Kuipers

STRESS, GENDER & PSYCHOPATHOLOGY: A MULTI-LEVEL ANALYSIS
The studies described in this thesis were performed at the Department of Psychiatry, Uni-versity of Groningen, P.O.Box 30.001, 9700 RB Groningen, The Netherlands. All experi-ments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC) and with the guidelines of the Animal Bioethics Committee of the University of Groningen.
This research was supported by: University of Groningen (RUG);Graduate School of Behavioral and Cognitive Neurosciences (BCN).
Financial support for the publication of this thesis was kindly provided by: Servier R&D Benelux; Eli Lilly Nederland B.V.; Boehringer Ingelheim; Sigma-Tau Ethi-farma B.V.; Astra Zeneca B.V.; Organon Nederland B.V.; Leica Microsystems B.V.; Roche Diagnostics NL; Lundbeck B.V.; Antec Leyden B.V.; Van Leersumfonds KNAW; J.E. Jur-riaanse Stichting; Dr. Ir. Van de Laar Stichting
Design & Layout: Simone Koster/www.anderkaliber.nlPrinted by: Print Partners Ipskamp B.V.© 2004 Sjoukje D. Kuipers ISBN number: 90-367-2010-9ISBN electronic: 90-367-2011-7

RIJKSUNIVERSITEIT GRONINGEN
STRESS, GENDER AND PSYCHOPATHOLOGY: A MULTI-LEVEL ANALYSIS
Proefschrift
Ter verkrijging van het doctoraat in de Medische Wetenschappen
aan de Rijksuniversiteit Groningen op gezag van de
Rector Magnifi cus, dr. F. Zwarts,in het openbaar te verdedigen op
woensdag 16 juni 2004om 13:15 uur
door
Sjoukje Daouia Kuipersgeboren op 20 augustus 1975
te Kenitra, Marokko

Promotores: Prof. Dr. J.A. den Boer Prof. Dr. G.J. ter Horst
Beoordelingcommissie: Prof. Dr. R.B. Minderaa Prof. Dr. B.H.C. Westerink
Prof. Dr. J.H.A. de Keyser

There are two things to aim for in life:fi rst to get what you want; and after that, to enjoy it.
Only the wisest of mankind achieve the second.
-Logan Pearsall Smith
For those to whom I owe this insight and greatest joys of all:Papa, Mama, Albert, Marten, and Andrea


CONTENTS PREFACE / 9Chapter 1 Introduction / 11
Introduction and scope of the thesis
I. SYSTEMIC, NEUROENDOCRINOGICAL LEVELChapter 2 Stress, gender and HPA function / 37 Repeated Stress Impairs HPA axis regulation in rats:
Indications for differential gender-dependent mechanisms
II. CELLULAR LEVELChapter 3 Stress-induced cortical defects / 69 Molecular correlates of impaired prefrontal plasticity in
response to chronic stress
Chapter 4 Stress, gender and neuronal plasticity / 93 Reduced CREB phosphorylation and calcineurin content characterize
the response to chronic stress in male rats:
Indications for sex-dependent neuroplasticity changes
III. MOLECULAR LEVELChapter 5 Stress and gender mediated neuronal impairments / 121 Immunohistochemical changes induced by repeated footshock stress:
Revelations of gender-based differences
Chapter 6 Stress, gender and synaptic vesicle proteins / 149 Chronic stress effects on synaptic-vesicle associated protein expression:
Indications for region/gender-dependent regulation
IV. IMPLICATIONS FOR PHARMACOTHERAPYChapter 7 Stress, neurogenesis and antidepressants / 171 Impact of long-term antidepressant treatment on chronic stress
induced reduction of neurogenesis in adult rats:
Revelations of sex/drug specifi c regulation CONCLUSIONChapter 8 Discussion and concluding remarks / 201
SUMMARY / SAMENVATTING / 217 / 225
ACKNOWLEDGEMENTS / 233
PUBLICATIONS / 237


PREFACE
Gender differences in the incidence of psychiatric disorders have been well recognized and documented. Epidemiological studies across a number of cultures consistently show that beginning at puberty, depression and anxiety disorders are two to three times more common in women than in men. Until recently however, women were often excluded from clinical trials of new psychotropic drugs since the menstrual cycle was thought to complicate the results of the trials. Furthermore most of the preclinical studies on the effi -cacy of new agents as well as neurochemical correlates of behavior and the mode of action of psychotropic drugs have also been conducted with male animals. Again the reason for concern being that the estrous cycle might complicate experimental results. Some of the currently prevailing theories of antidepressants’ modes of action were based solely on the results obtained with experiments with male rats.
Little progress has been seen however in recent decades with regard to the development of novel therapeutic (antidepressant) agents. In pursuit of new targets, an interesting (and obvious) possibility hence holds that fundamental differences between males and females, refl ected by their prevalence rates, may also refl ect gender differences in circuitry underly-ing pathology and thus potential therapies. As obvious as this sounds, the more surpri-sing is the lack of attention for this issue particularly since insight into this subject would benefi t both the (female) members of today’s society as well as the pharmaceutical industries.
Although the emergence of new focus areas does not necessarily indicate a paradigm shift in which each new paradigm replaces the last, there seems to be a resurgence of women’s health issues at the forefront. Today women’s health has gained widespread attention in popular magazines, television shows and the wellness industry. Not surprisingly, research in biological psychiatry and psychopharmacology is now focusing more on the analysis of sex infl uences. Discovering why these gender differences in risk exist has become perhaps one of the most intriguing and important questions in psychiatric research today: intrigu-ing because a deeper understanding of why women are more likely than men to experience anxiety and depression will provide insight into the pathophysiology of these syndromes and important because such knowledge will improve our ability to design interventions that treat and/or prevent these illnesses.


INTRODUCTION
CONTENTS CHAPTER 1
Stress and the Stress Response
• Stress and the limbic system • Stress and the HPA axis
• HPA axis regulation • Considering gender dichotomy in the stress response • Dysregulation & implication in psychopathology
Stress and Depression
• Neural circuitry of depression • Hypotheses of depression and neural dysfunction
• Monoamine theory • Neuroplasticity theory • Dysregulation of HPA axis and hippocampus • Neurotrophic hypothesis • Neurogenesis hypothesis
Pharmacotherapy
Scope of the thesis
• Multilevel Analysis • Outline of the thesis
• Systemic level and neuroendocrine aspects • Cellular level and signal transduction • Molecular level and transcriptional activity • Implications for pharmacotherapy1

12 / Introduction
Stress and the Stress Response
Stress, a response to aversive stimuli, is a concept that is diffi cult to defi ne since its inter-
pretation tends to vary across individual disciplines. In 1976, Hans Selye, a pioneer in
addressing general principles of physiology and pathophysiology in the exploration of
stress, defi ned stress as “the nonspecifi c response of the body to any demand placed upon
it” 4. He emphasized the role of an integrated response of multiple systems rather than isolated refl exes. Exposure to threat or hostile conditions, for instance results in a series of coordinated responses referred to as the “stress response” and is composed of altered beha-vior, immunologic and autonomic function and the secretion of multiple hormones including adrenocorticotropin hormone (ACTH), cortisol/corticosterone, and adrenal catecholamines. These coordinated responses are organized to enhance the probability of survival and stressors can thus be defi ned as intrinsic or extrinsic forces, that endanger or are perceived to endanger the survival of an organism 5. Nevertheless, not all states of stress are necessarily noxious or negative. Mild, brief (“acute”) and controllable chal-lenges or eustress could be perceived as pleasant or exciting stimuli and could be a positive input for the emotional and intellectual development, while the more intense, persistent (“chronic”) and uncontrollable situations of threat, or distress may lead to maladaptive responses 4,6. In general stressors can be grouped into three broad categories: (1) psycho-logical stressors based on a learned response to the threat of an impending adverse condi-tions (fear, anxiety or exposure to a novel or uncontrollable environment) (2) stressors that consist of a physical stimulus and have a strong psychological component (pain or immo-bilization) (3) stressors which challenge cardiovascular homeostasis (hemorrhage, exercise or heat). Throughout the experiments described in this thesis, use was made of repeated foot-shock application to serve as a chronic stressor. This model allows simultaneous investiga-tion of the fi rst two stressor categories, namely an aversive physical condition with a strong psychological component. As such, the term stress in this thesis will generally refer to distress, like most clinical and scientifi c contexts, unless otherwise defi ned. The emphasis was placed on cumulative and persistent exertion of the stress response since this can lead to overreaction of the stress system, a situation described primarily as the stress syndrome 7 and widely recognized as an important trigger in the expression of various psychiatric syndromes, such as major depression and anxiety disorders. The principal components of the adaptive response to stress are the sympatho-adrener-gic-noradrenergic (SAN) and the limbic-hypothalamo-pituitary-adrenal (L-HPA) systems. The SAN system implies the biosynthesis and release of adrenaline and noradrenaline, re-gulated respectively by the sympathetic division of the autonomic nervous system (ANS), and the locus coeruleus (LC), in the CNS. The L-HPA system involves limbic structures, such as the prefrontal cortex and hippocampus, in association with the HPA axis, and their respective interconnections. Both systems also participate in their mutual positive regulation, so that activation of one also involves activation of the other 8. In addition, the

Introduction / 13
stress system also includes other brain areas involved in implementation of the appropriate adaptive response 9. Although it goes beyond the scope of this thesis to describe in detail each contributing component, a selection of most relevant issues to the understanding of the data presented here is highlighted in the following paragraphs.
Stress and the limbic system
Multiple brain structures are involved in the organization of responses to aversive or stressful stimuli. Among them are the hypothalamus, hippocampus, amygdala, cingulate and prefrontal cortices, hindbrain regions such as the brainstem catecholamine cell body groups (in the nucleus of the tractus sollitarius, ventrolateral medulla and locus coeruleus), the parabrachial nucleus, cuneiform nucleus and dorsal raphe nucleus 5. Figure 1 sum-marizes a few of the well-characterized brain structures and neural circuits involved in the adaptive response to stress. Most sensory inputs or environmental stimuli, playing as external stressors are perceived by specifi c sensory receptor systems, which convey informa-tion to their respective sensory areas of the thalamus. This structure functions as a relay sta-tion so that sensory information concerning individual stimuli is then transmitted to the amygdala and sensory cortex 10,11. The sensory cortex in turn communicates either directly or via the hippocampus with the lateral amygdala through the perirhinal cortex 12-14. The amygdala is composed of several nuclei, each of which performs a different function. The lateral and basolateral nuclei of the amygdala funnel and integrate sensory input from the thalamus and cognitive information from the cortex and hippocampus, while the central amygdaloid nucleus is involved in behavioral, autonomic, and endocrine responses 5. The amygdala also innervates and is innervated by the dorsal raphe nucleus and catecholamin-ergic nuclei located in the brainstem, which in turn innervate corticotropin releasing hor-mone (CRH) neurons in the hypothalamic paraventricular nucleus (PVN) 15,16. The PVN is a particularly essential structure for proper stress response modulation as it plays a piv-otal role in the adaptive response to stressors through regulation of HPA axis activity described below.
Figure 1.
Schematic representation of the neural circuits involved in the adaptive response to stress. Sensory receptors perceive sen-sory input and convey the information to their respective sensory thalamus, pri-mary sensory cortices and higher order cortices. Basic sensory information is simultaneously transmitted to the amyg-dala from these structures particularly the prefrontal cortex (through transitional cor-tices and the hippocampus). These limbic structures regulate activation of both neural and neuroendocrine responses. The neural component involves the NA system represented by the locus coeru-leus and the adrenergic system, through the lateral hypothalamus, which acti-vates the SNS and the adrenal medulla. The neuroendocrine component involves activation of the PVN, the pituitary and the adrenal cortex, with the consequent release of CRH, ACTH and glucocorti-coids.
CHAPTER 1

14 / Introduction
Stress and the HPA axis
Living organisms survive by maintaining a complex dynamic equilibrium or homeostasis that is constantly challenged by intrinsic or extrinsic stressors. These stressors set in motion responses aimed at preserving homeostasis, including activation of the HPA axis, which in turn initiates the hormonal cascade illustrated in fi gure 2. In short, the HPA system receives and integrates various inputs indicative of stress that converge into the PVN 9,17,18. CRH, synthesized by PVN neurons, is then released into the hypophyseal portal blood and reaches the anterior pituitary 18 where it regulates the transcription of the proopiomelanocortin (POMC) gene, a common precursor for adrenocorticotropic hormone (ACTH) and related peptides, and stimulates the release of ACTH into the bloodstream 19,20. ACTH then stimulates the biosynthesis and release of glucocorticoids (cortisol in humans, corticosterone in rodents) by cells of the adrenal cortex into the sys-temic circulation 21.
HPA axis regulation
Glucocorticoids are the fi nal effectors of the HPA axis and they play a key regulatorrole in the neuroendocrine control of the HPA-axis and on the termination of the stress response by exerting negative feedback at several sites to inhibit their own release. At the pituitary level, glucocorticoids exert direct effects on the ACTH precursor POMC, infl uencing gene transcription, POMC mRNA levels and subsequent ACTH peptide stores. This occurs through glucocorticoids-receptor complex binding, translocation to the nucleus and binding to DNA sites 22. Studies have also shown that glucocorticoids interact with the CRH receptors in the anterior pituitary, acutely inhibiting the binding of CRH to its receptors and chronically decreasing CRH receptor numbers 23,24. Such direct effects of glucocorticoids on CRH receptors may account for some of the inhibitory action of glucocorticoids on ACTH release. In addition to pituitary sites of action, glucocorticoids also act at various brain sites to modulate HPA axis activity including hypothalamic 25 and some supra-hypothalamic structures 26-28.
Glucocorticoid effects are mediated by two receptor subtypes: the mineralocorticoid receptor (MR) that has a higher affi nity for corticosterone and the glucocorticoid recep-tor (GR) that possesses a lower affi nity for corticosterone 27. Consequently, MRs are pre-dominantly occupied at lower, basal corticosterone levels and GRs become occupied when corticosterone levels are high, such as during stress 29. MRs are found in some brain areas such as the hippocampus, whereas GRs have a widespread distribution and are abundantly expressed in several brain regions involved in the stress response, inclu-ding the frontal cortex, hippocampus and hypothalamic paraventricular nucleus (PVN) 27,30. These recep-tors act as ligand-dependent-transcription factors 28,31 and upon glucocorticoid binding, these receptors undergo conformational changes to facilitate their subsequent binding to DNA 32,33. The hormone-receptor complex may thereby regulate the expression of various target genes, either through activation or deactivation.

Introduction / 15
CHAPTER 1 In a formulation that may be of relevance to stress-induced disorders as depression, Sapolsky and colleagues have proposed the glucocorticoid cascade hypothesis, a model that describes the effects of chronic stress on hippocampal neurons. According to this model, repeated stress or chronic glucocorticoid administration, downregulates hippocam-pal steroid receptors, but not hypothalamic or pituitary receptors 34,35. Animals with down-regulated hippocampal glucocorticoid receptors exhibit delays in the turnoff of the gluco-corticoid response to stress and demonstrate decreased sensitivity to glucocorticoid fast feedback 35. This decrease in glucocorticoid receptors and insensitivity to negative feed-back is thought to lead to prolonged hypercortisolism, resulting in atrophy of hippocam-pal neurons and further glucocorticoid hypersecretion. Mechanisms such as these will be discussed in the following paragraphs, but serve here to illustrate how HPA system activity may be regulated through a variety of possibilities. Interestingly however, rat and human studies provide evidence that the stress response is also sexually dimorphic and that gonadal steroids play an important role in modula-ting the HPA axis, acting particularly on sensitivity to glucocorticoid negative feedback 36
through effects on brain CRH systems, pituitary responsiveness to CRH or glucocorticoid receptors. This concept, discussed in the next paragraph may provide a better understan-ding of the machinery underlying observations that women have an increased vulnerabi-lity to stress and related disorders as depression.
Figure 2 The stress response system

16 / Introduction
Considering gender dichotomy in the stress response.
Being male or female is an important individual contribution to mental and physical health. With regard to psychiatry, this perspective calls for careful consideration of sex-based differences in stress susceptibility and sex-based differences in the manifestation and prevalence of various psychiatric disorders. Ovarian steroids are known to contribute to a wide variety of neurochemical effects, which produce gender differences in multiple processes such as cognition, emotional regulation, affective style, pain sensitivity and psy-chopathology 37-39. Evidently, reproductive as well as non-reproductive dimorphisms exist between males and females and an intriguing possibility thus holds that non-reproductive gender related affective dissimilarities might underlie the increased prevalence rates of women compared to men for stress-related disorders such as post-traumatic stress disorder, major depression and other anxiety disorders. Although multiple factors could account for this differential and contribute to the increased vulnerability in women, understanding the impact of stressors is central to understanding the development of stress disorders. Since the HPA axis represents the fi nal effector in the modulation of the stress response, a large number of clinical and preclinical studies have attempted to defi ne a direct link between sex-related differences in key elements in this system and the higher female sus-ceptibility to stress-related pathology. Differences in HPA axis regulation may account for the differential response to stress observed between males and females, since the latter demonstrate a more robust HPA axis response to stress consisting of a faster onset of glucocorticoid secretion and a faster elevation of plasma adrenal steroid levels 36. Appa-rently, a steeper rise in circulating stress hormones seems to be necessary to elicit the faster glucocorticoid-mediated feedback inhibition needed in females. The mechanisms underly-ing the latter however are unclear although there is evidence that ovarian hormones are at least partly responsible for this sexual dimorphism. The HPA axis and the female gonadal system are closely intertwined and exhibit a complex bi-directional relationship. A partial estrogen response element for instance has been found on the promoter of the CRH gene 40. This not only implicates the CRH gene and thus the HPA axis as an important target of gonadal steroids, but also represents a potential mechanism by which estrogen may enhance stress responsiveness, increase gluco-corticoid levels and yield greater HPA axis resistance to glucocorticoid-mediated feedback inhibition. Accordingly estrogen has also been shown to delay ACTH and glucocorticoid shutoff 41,42. Like estrogen, progesterone also appears to be involved in the differential modulation of stress response. It has been shown for instance, that progesterone binds faster to GR than cortisol and that they bind to different sites 43,44. Furthermore female rats have been shown to have a greater number of hippocampal GRs than males, which seems to be modulated by progesterone 45. Although most progesterone-induced effects on the HPA axis are mediated by GR binding, its affi nity for MR has also been shown in a range similar to dexamethasone 46,47. Despite the fact that estrogen and progesterone have been shown to exert protective effects against hypercortisolemia, they also seem to antagonize

Introduction / 17
CHAPTER 1glucocorticoid-mediated terminating action, perhaps delaying recovery from deleterious effects of stress. Taken together they may account for the greater overall reaction seen in females and help explain gender-related differences in response to stress and relateddisorders.
Dysregulation & implication for psychopathology
After prolonged and intensive stress exposure the HPA system becomes dysregulated, lea-ding to impaired negative feedback of the HPA axis, persistent activation of this system and consequential increase in circulating glucocorticoids 48,49. Normal HPA axis acti-vity is hence altered during chronic stress, resulting in sustained increase of plasma corticoste-rone or cortisol levels 50. Since adaptive responses are meant to be acute, limited by specifi c characteristics of the stressor, an essential component of adaptation is thus protection of the organism against overreaction of this system. To maintain glucocorticoids within phys-iological ranges, the HPA axis is controlled by multiple negative-feedback loops, mediated mainly by the steroids themselves 51,52. If however the organism is unable to terminate the stress response at the end of exposure or if chronic and unavoi-dable stressful situations persist, then the sustained adaptive responses may lead to pathophysiological changes pro-duced by dysregulation of the stress syndrome. These changes may in turn develop into various types of disorders such as anxiety disorders or major depression. In this regard a signifi cant association between stress and depression is now well-documented and the parallels between some aspects of the stress response and severe depression are striking 53,54.
Hypercortisolism, for instance represents one of its most consistent biological markers 55,56.
Furthermore clinical and preclinical studies reveal that chronic exposure to stressful events is strongly associated with the development of depressive symptoms.
Stress and Depression
As opposed to most diseases of other organ systems, diagnosis of depression is not based on objective diagnostic tests, but rather on a highly variable set of symptoms. Accor-dingly, depression may be better viewed as a heterogenous syndrome rather than a single disease, as it is comprised of numerous diseases with distinct causes and pathophysiologies. Attempts have been made to establish subtypes of depression defi ned by certain sets of symptoms 57,58, yet these subtypes are based solely on symptomatic differences. For the sake of clarity throughout this thesis it must thus be noted that there is as yet no clear consensus regarding their different underlying disease states. The following paragraphs will describe underlying circuitry and dysregulation generally attributed to this syndrome, without distinction for the possible subtypes unless otherwise specifi ed.

18 / Introduction
Neural circuitry of depression
As previously described, many brain regions have been implicated in regulating emotions. It is unfortunate however, that our understanding of neural circuitry underlying normal mood is still rudimentary, since mood abnormalities are the hallmark of depression. It is likely however that many brain regions mediate the diverse symptoms of depression and depressed patients exhibit distinct pathological changes in various selective brain regions 59. These changes are observed in limbic (hippocampus, basal ganglia and amygdala) and cortical brain regions implicated in the affective and cognitive impairments observed in depression 59-63. Accordingly, brain-imaging studies also indicate impaired cerebral blood fl ow and glucose metabolism in limbic and cortical structures 64. In fact, functional imag-ing studies reveal that prefrontal cortical, ventral striatal and hippocampal volume is decreased in patients with depression 65,66. The emerging picture from these clinical studies is that cellular loss and volume decrease is associated with depression. Although it cannot be excluded that depression also has a genetic component, it is well acknowledged that neuroendocrine changes and stressful events can also lead to a depres-sive episode 67,68 and as previously stated, several studies indicate that a subset of depressed patients have glucocorticoid hypersecretion 69,70 or HPA axis hyperactivity 71. In line with the cellular loss, patients with HPA hyperactivity also exhibit distinct reductions in hip-pocampal volume 70.
Given these observations, depression research in animal models is therefore focused on understanding the changes induced in limbic and cortical brain structures by different stressors and glucocorticoids. Likewise, the focus in this thesis lies primarily in the prefron-tal cortex and hippocampus, given their crucial roles in terminating the stress response, and the fact hat they are particularly susceptible to the structural impairments induced by stress 70,72. Although their stress-induced changes may not explain all affective symptoms of depression, they might provide a cellular basis for understanding the structural and neuronal dysfunctions in these structures as well as other regions associated with stress and stress-related disorders as depression. Distinct cellular mechanisms have already been proposed to underlie some stress-induced pathological impairments and are discussed in the following paragraphs.
Hypotheses of depression and neuronal dysfunction
Unfortunately, a major impediment in depression research is the lack of validated animal models. This is in part attributable to the above-mentioned heterogeneous aspect of depression, as it constitutes diverse syndromes while varying subtypes are likely to have distinct causes and pathophysiologies. This being the case, valid animal models are needed to ascertain distinctions of these disorders. However, such animal models can only be developed once we have gained a full understanding of this disorder; thereby creating a catch twenty-two. As a result, all available animal models of depression rely on one of

Introduction / 19
CHAPTER 1two principles: (1) actions of known antidepressants or (2) responses to stress 73-76. With regard to antidepressant research, various neurotransmitter systems have been investigated with respect to their role in CNS pathophysiology and possibly origin and development of depression. This in turn has given rise to different aminergic hypotheses, commonly referred to as the “monoaminergic theory of depression”. The pathological effects of stress on HPA axis activity and crucial regulating structures as the hippocampus however have contributed to another recent hypothesis that proposes a role for neuronal plasticity in the etiology of depression and its treatment 77,78. Specifi c mechanisms responsible for media-ting the underlying neuronal dysfunction of each theory are discussed below.
Monoamine theory
For a long time the “monoamine hypothesis of depression” provided the starting point for investigators attempting to explain antidepressant action and depression pathophysio-logy. The main assumption of this hypothesis holds that depression is due to a defi ciency in the neurotransmission mediated by the biogenic amines, e.g., serotonin (5-HT), nor-epinephrine (NA), and dopamine (DA), and that antidepressants work by increasing the availability of the amines and neurotransmission in the brain. Early research focused on changes in neurotransmitter concentrations and receptor levels, but the results of these studies were at odds with the observation that the therapeutic effects of antidepressants requires chronic administration while the inhibition of serotonin and norepinephrine reuptake occurs immediately. Sequential evolution of this hypothesis hence yielded the monoamine receptor hypothesis, which proposes that the drug-induced increase in mono-aminergic neurotransmission causes changes in sensitization state of monoamine recep-tors, which may explain both the effects and the delayed onset of action of these drugs 79,80.
Although each of the above-mentioned monoamines represents a putative target, the role of serotonin is perhaps the most studied in stress and depression, given the major the-rapeutic advance in psychopharmacology provided by selective serotonin reuptake inhibi-tors (SSRIs). The serotonergic hypothesis of depression postulates that a defi cient seroto-nergic neurotransmission in the CNS may account for a higher vulnerability to this dis-order. Hence, the role of serotonin in the pathophysiology of depression has long been investigated, giving rise and supporting this hypothesis 81,82. At the molecular level, seroto-nergic neurotransmission is regulated by its rapid removal from the synaptic cleft, primar-ily through its re-uptake into the presynaptic terminals by the serotonin transporter 83.
This process exerts control on the effective concentration of the neurotransmitter at the synaptic cleft, and its availability for the interaction with both pre- and postsynaptic recep-tors 84. The therapeutic effect of antidepressants that enhance serotonergic neurotransmis-sion such as tricyclics and SSRIs are in line with this theory. SSRIs, often the treatment of choice for depression, marked a milestone in neuropsychopharmacology and rational drug design. Nevertheless, given various drawbacks inherent to this theory (see “Pharma-

20 / Introduction
cotherapy”), new approaches to understanding depression focus on the regulation of key signaling pathways involved in cellular survival and plasticity. Evidence linking stress, depression and antidepressant action suggest that depression may result from an impairment of neurons to make appropriate adaptations and/or syn-aptic connections. The following paragraph describes the cellular mechanisms that may underlie the structural impairments observed in the brains of animals used in models of depression or stress exposure as well as in patients with depression.
Neuroplasticity theory
Dysregulation of the HPA axis and hippocampus
As mentioned afore, hyperactive HPA axis activity and consequent hypercortisolemia is one of the most consistent fi ndings seen in depressed subjects 53. Glucocorticoids have been shown to affect various areas in the CNS, particularly the hippocampus, where MRs and a particularly high density of GRs have been found 85,86. When normal glucocorticoid secretion is altered, leading to increased glucocorticoid levels, this may result in down-regulation of hippocampal GRs 87. This potentially adaptive response observed in neural tissue, is apparently directed to counteract an excessive concentration of glucocorticoids, but may lead to altered negative feed-back mechanisms, resulting in increased circulating cortisol levels. This elevation may persist, even after termination of the original stimulus that gave rise to it, and could then result in degenerative changes in the hippocampus 87,88. Hippocampal alteration produced by prolonged and excessive cortisol levels, with a consequently impaired negative feed back loop, could at this stage account for the inabi-lity of glucocorticoids to regulate their own secretion during chronic stress 89,90. These observations have given rise to the hypothesis that links depression with an alteration of the L-HPA system, particularly focusing on the down-regulation of GRs at hippocampal and hypothalamic levels 54,91, with the resulting hypercortisolism. In accordance, antide-pressants could thus act through improvement of GR function, thereby leading to norma-lization of the HPA axis. Another mechanism however by which antidepressants may mediate their effects is by triggering cellular mechanisms to counteract the structural impairments that are induced by stress as refl ected by the volumetric changes associated with the prefrontal cortex and hippocampus of depressed patients. In adult rodents, both chronic stress and glucocorti-coid administration have been shown to result in distinct remodeling of the apical den-drites of hippocampal CA3 pyramidal neurons 92, which is manifested as a decrease in both number and length of apical dendrites 93. Given the complexity of stress-induced dis-orders however it is unlikely that disturbed dendritic morphology alone will fully explain the loss of volume seen in depression. Besides the changes in dendritic remodeling, stress and stress hormones also decrease the ongoing rate of cell birth and cell proliferation in the dentate gyrus granule cell layer of adult hippocampus by an undetermined mechanism

Introduction / 21
CHAPTER 194,95. Although the latter will be described in the following paragraphs, the above does sug-gest that dendritic restructuring as well as decreased cell-survival and neurogenesis may provide the cellular basis for stress impairments. In turn antidepressants might correct such impairments by targeting these features. In line with this theory, results of a chronic tianeptine study have shown reversal of stress-induced impairments and reduced hippo-campal volumes 96.
Neurotrophic hypothesis
The neurotrophic hypothesis associates the origin and development of depression with a stress-induced decrease of neurotrophic factors and proposes a role for these agents in its treatment. Neurotrophic factors were fi rst characterized for regulating neural growth and differentiation during development, but are now known to be potent regulators of plastic-ity and survival of adult neurons and glia. Complementary to the pathological effects on the hippocampus described above, the neurotrophic hypothesis of depression states that a defi ciency in neurotrophic support may contribute to hippocampal pathology during the development of depression, and that reversal of this defi ciency by antidepressant treat-ments may contribute to the resolution of depressive symptoms. Work on this hypothesis has focused primarily on brain-derived neurotrophic factor (BDNF), one of the most prevalent neurotrophic factors in the adult brain. Both acute and chronic stress decrease BDNF expression levels in the dentate gyrus and pyramidal cell layer of the hippocampus in rodents 97. This reduction appears to be mediated in part through stress-induced glucocorticoids and partly through other mechanisms, such as increased serotonergic transmission following stress 97,98. Conversely chronic (and not acute) administration of virtually all classes of antidepressants increase BDNF expression in these regions 99 while preventing stress-induced decreases in BDNF levels. Antidepres-sants’ ability to increase hippocampal BDNF levels has also been shown in human studies 100. Altered expression of BDNF in the hippocampus as well as the prefrontal cortex has been proposed as an important factor mediating atrophy and neuronal cell death associ-ated with depression 101. As BDNF is reported to enhance long-term potentiation and other forms of synaptic plasticity in the hippocampus 102-104, increased BDNF levels induced by antidepressants may promote hippocampal function. Furthermore, the time required for BDNF levels to gradually rise and exert their neurotrophic effects, might explain why antidepressant response is delayed. The neurotrophic hypothesis predicts that agents that promote BDNF function might be clinically effective antidepressants, but since no such compounds are currently available, another approach may lie in earlier intervention in this process. Antidepressant induction of BDNF is mediated largely via intracellular signaling cascades. Neurotrophins bind to tyrosine kinase (Trk) receptors and activate intracellular pathways such as the mitogen acti-vated protein kinase (MAPK) 105, or extracellular signal-regulated protein kinase (ERK) cascade. Neurotrophin-dependent MAPK plays a major role in mediating the extracellu-

22 / Introduction
lar signals to the nucleus and once activated, MAPK phosphorylates cAMP response ele-ment binding protein (CREB), which then binds to a specifi c response element called cAMP response element (CRE) of the BDNF gene and enhances transcription. A post-mortem study found decreased levels of ERK activity and expression both in hippocam-pus and cerebral cortex of depressed suicide subjects, providing additional evidence for neurotrophin dysregulation in different brain regions in stress-related disorders 106. Stress and stress-induced glucocorticoids can directly interfere with this process through bin-ding of the glucocorticoid-GR complex to CREB, thereby preventing its phosphoryla-tion and blocking the expression of target genes such as BDNF. Conversely, regulation of the MAPK/CREB signaling cascade may represent a putative target to promote a pal-liative effect. In fact, virtually all major classes of antidepressants increase levels of CREB expression and function in several brain regions including the hippocampus 107,108. Further-more, increased CREB activity in the hippocampal dentate gyrus achieved through CREB encoding viral vector injections directly into this brain region has also been shown to exert antidepressant-like effects in animal models of depression 100,109.
Taken together, observations regarding this neurotrophic hypothesis combined with the monoaminergic theory as well as structural and morphological effects of the brain associ-ated with chronic stress or depression have provided a prolifi c starting point for the devel-opment of a novel theory on the cellular basis of depression. The theory, as proposed by Jacobs, focused on the important role of serotonergic activity in both depression and the regulation of hippocampal neurogenesis 110,111. The theory, further developed by Duman and coworkers however, sees the CREB/BDNF signaling cascade as the bridging link between dendritic restructuring, decreased neuronal survival, and decreased adult hippo-campal neurogenesis in depression 112. This hypothesis, which places the potential role of neurogenesis in the context of many other cellular and biochemical alterations in depres-sion, is discussed below.
Neurogenesis hypothesis
So far, the theories suggested to explain stress-induced changes observed in prefrontocorti-cal and hippocampal areas, are based primarily on glucocorticoid-mediated cell loss, atro-phy, remodeling or compromised neuroplasticity. A failing regulation of adult hippocam-pal neurogenesis however might also contribute to this volume loss, at least in part. Adult hippocampal neurogenesis was fi rst described by Altman and Das in 1965 and has under-gone several rediscoveries since then 113-115. Recently a broader interest in this phenomenon has been sparked by reports of its possible implication in chronic stress associated impair-ments as well as antidepressants actions. Adult hippocampal neurogenesis is subject to complex regulation, and numerous factors affecting several different levels of regulation have been described. Stress however may represent one of the key factors that initiate cel-lular dysfunctions by down-regulating neurogenesis 95,116. This effect could be mediated through elevated glucocorticoid as well as NMDA-

Introduction / 23
CHAPTER 1receptor activation 117 since both are also involved in other hippocampal damage following repeated or chronic stress 72. Conversely, recent evidence demonstrates that chronic admin-istration of different classes of antidepressants increases the proliferation as well as number of newly formed neurons in the hippocampus 118. As this effect is not seen in response to other non-antidepressant psychotropic drugs, this suggests a high degree of specifi city to the effects of antidepressants 118.
Given the role of the cAMP-CREB cascade in antidepressant action, preliminary evidence also complements its involvement in hippocampal neurogenesis 119. Transgenic mice expressing a dominant negative mutant of CREB show a signifi cant decrease in the survival of newly formed hippocampal neurons 119, while in vitro studies have shown that activation of the cAMP cascade or the presence of BDNF in the surrounding environ-ment increases the differentiation and survival of neurons 120. These results suggest that up-regulation of CREB and BDNF by antidepressants could increase the differentiation and survival of hippocampal neurons in vivo. Although the precise signifi cance of hip-pocampal neurogenesis is yet to be fully elucidated, it provides a putative cellular basis for the decreased hippocampal volume observed in depressed subjects. Interestingly, cel-lular proliferation has also been shown to occur in the prefrontal and temporal cortex of adult rodents and primates 121,122, so in line with the observations in depression, these fi n-dings suggest that cortical cell loss could also result from decreased cell proliferation in this region.
Pharmacotherapy
Antidepressants can be effective agents for the treatment of depression and have been used clinically for more than 50 years. A list of therapeutic manipulations currently used to treat depressed patients is provided in table 1. Prior to SSRIs, almost all psychotropic medications were the result of chance observations. TCAs for instance were the result of an unsuccessful attempt to improve the antipsychotic effectiveness of phenothiazines 123,
while MAOIs came from a failed attempt to develop effective antitubercular medications 124. SSRIs however were a rationally designed class of psychotropic medications which hence launched a new era in psychotropic drug development 125. After imipramine, the development of subsequent SSRIs occurred over a relatively short period. Eventually fi ve SSRIs (citalopram by Lundbeck, fl uvoxamine by Solvay, fl uoxetine by Lilly, paroxetine by SmithKline-Beecham and sertaline by Pfi zer) were launched by several companies in many countries world wide, indicative of the shift from a chance dependent discovery process to one of rational drug development.

24 / Introduction
Unfortunately, after this milestone discovery, development of newer drugs has seen little progress. The following years consisted mostly of optimization of older drugs (table 2). The reason for this, being the fact that to date no clear consensus has been reached regar-ding the precise molecular and cellular mechanisms of action of these drugs.

Introduction / 25
CHAPTER 1Although the therapeutic action of antidepressants most likely involves the regulation of serotonergic and noradrenergic signal transduction pathways, given the clinical effi cacy of these drugs, intensive investigation has failed to fi nd conclusive affi rmation of a primary dysfunction in specifi c monoaminergic systems in depressed subjects. Moreover, the fol-lowing major issues have not been addressed by the monoamine hypothesis:• Effi cacy. Clinical trials have shown antidepressants to be effi cient in approximately 60% of depressed subjects 126. Besides higher tolerability and reduced side effects however, development of newer antidepressants since introduction of the fi rst tricyclic agents, has failed to yield enhanced effi cacy compared to these older drugs 127,128.
• Selectivity. Whereas SSRIs, NRIs and SNRIs clearly act through stimulation of seroto-nergic and noradrenergic systems, there is still some uncertainty regarding the specifi city of these compounds. In fact, various lines of evidence have indicated that following long-term administration, selectivity of these drugs dissipates such that numerous neurotrans-mitter systems and brain structures are affected, some of which are not even directly linked to the drugs’ pharmacological profi le. This yields the interesting possibility that this delayed non-specifi c action rather than its high specifi city might underlie antidepressants’ therapeutic effects.• Mode of action. Another indication that reduced monoamine levels are not the sole mechanism underlying depression and antidepressants is provided by the effi cacy of antidepressants that do not potentiate monoaminergic activity. Alternative mechanisms include enhancing serotonin reuptake (tianeptine) 129,130 or modulating activity of selective enzymes and/or transcription factors that are not directly linked to monoamine metabo-lism or signaling transduction pathways (lithium and valproate) 131,132.
• Delayed therapeutic action. Although many antidepressants acutely regulate mono-aminergic signal transduction, resulting in a signifi cant increase in synaptic concentra-tions of the monoamines, noradrenaline and serotonin, there is a several-week latency before onset of clinical effects of theses drugs. • Monoamine depletion studies. Experimental monoamine depletion exacerbates depressive symptoms in depressed subjects that successfully respond to SSRIs or NRIs, but fails to induce similar negative effects in medication-free symptomatic patients or healthy controls. This suggests that monoaminergic dysfunctions may play a crucial role in depres-sion and antidepressive action, but is unlikely to represent the primary cause 133,134.
Undoubtedly the monoamine theory has some merit, and while it may play a crucial role, given these inconsistencies it clearly does not act alone to fully explain the mecha-nisms underlying depression or antidepressant action. Despite another catch twenty-two which holds that a full understanding of stress-related pathology is required for the devel-opment of novel effi cacious pharmacotherapy, it cannot be denied that currently used medications are providing substantial evidence for interesting additional targets. As out-lined in the previous paragraphs, the neuroplasticity theory of depression provides plau-sible targets of drug-induced adaptive neuronal changes. Furthermore, common effects of traditional antidepressant treatment and alternative therapy such as electroconvulsive

26 / Introduction
therapy (ECT) and transcranial magnetic stimulation (TMS) in combination with stress studies, are a testimony to the effects on neural plasticity that underlie stress-induced impairments and are specifi c to all forms of antidepressant therapy. In summary of the alternative candidates, Hyman and Nestler proposed an “initia-tion and adaptation” model to describe the drug-induced neural plasticity associated with the long-term actions of antidepressants in the brain. Although the underlying detailed mechanisms are of yet unknown, the common effects of alternative therapy such as ECT and TMS and antidepressants on connectivity and synaptic plasticity in the dentate gyrus are likely to relate to affective functions of depression 135. Furthermore, data also demon-strates that chronic ECS induces sprouting of the granule cell mossy fi ber pathways in the hippocampus 136. The therapeutic effect of antidepressants and alternative therapies could result from either indirect regulation of other non-aminergic neuronal signal transduction systems or regulation of gene transcription following chronic treatment, (possibly explain-ing the often seen delayed therapeutic response). Indeed, antidepressants selectively affect certain immediate early genes and transcription factors such as c-fos 137,138 and Arc (acti-vity regulated cytoskeleton associated protein) 139. Alterations in the cAMP second messen-ger system or functional proteins related to neural plasticity such as CREB and BDNF, fur-ther suggest that changes in gene expression may also have a role in the mechanisms under-lying pathology and antidepressant action. Identifi cation and quantifi cation of altered gene expression associated with the latter can thus pave the way for the discovery of novel molecular markers useful in the diagnosis and treatment of depression.
SCOPE OF THE THESIS
Multilevel analysis
If it is generally accepted that stress may induce, exacerbate or contribute to the develop-
ment of psychiatric disorders as depression and that the stress response differs between
males and females, then mechanisms of neuronal function, dysfunction, pathology and
pharmacotherapy may not apply to both sexes equally. In this regard the chapters pre-
sented in this thesis describe the results of several experiments designed to elucidate the
concept of stress, investigate the effect of gender and highlight the implications of such
stress-gender interactions with respect to pharmacotherapy. As shown in fi gure 3 there are
three dimensions necessary to understand neuronal plasticity implicated in psychopatho-
logy and long-term actions of antidepressants, ranging from gene transcription to higher
brain functions.

Introduction / 27
Outline of the thesis
Inherent to an organism’s (neuro)physiology, information transfer of sensory input and subsequent adaptive responses are communicated through neuronal structures and require proper regulation of signal transduction as well as transcription. Naturally, their complex interactions yield considerable overlap, as one cannot function without the other. Never-theless, the chapters in this thesis have been grouped according to how the results pertain to these three respective categories:
Systemic level and neuroendocrine aspects
The studies described in this thesis provide evidence to illustrate the physiological and neu-ronal effects of stress in male and female rats. The results presented in chapter 2 entitled
“Repeated stress impairs HPA axis regulation in rats: Indications for differential gender-
dependent mechanisms” describe the effects of chronic stress exposure on various para-meters of the HPA axis and the autonomic nervous system. The results, ranging from body weight gain to adrenal morphology and steroid plasma concentrations and glucocor-ticoid receptor densities, serve largely to validate the impact of this stress paradigm by sug-gesting similar injurious effects of stress leading to prolonged HPA axis activity in both genders. Interestingly however chapter 2 also demonstrates distinct gender differences in physiology and regulation of this basic system. Several brain structures implicated in modulation of HPA axis activity are also investigated using c-fos expression as a marker of neuronal activity.
I. BRAIN CIRCUITS
II. TRANSDUCTION
III. TRANSCRIPTION
Figure 3. Three dimensions necessary to understand neural plasticity (modifi ed from Yamada and Higuchi 2002)
CHAPTER 1

Cellular level and signal transduction
The second part of this thesis addresses the impact of chronic stress exposure on various correlates of second messenger signal transduction cascades implicated in the neuro-trophic pathway. Chapter 3 entitled “Molecular correlates of impaired prefrontal plasti-city in response to chronic stress” presents the immunohistochemical effects of this stress paradigm on selected MAPK/CREB cascade members. These results, originally seen in males provide indirect indications of impaired BDNF transcription. To ascertain whether these changes were stress and/or gender specifi c, chapter 4 entitled “Reduced CREB phos-phorylation and calcineurin content characterize the response to chronic stress in male rats: Indications for sex-dependent neuroplasticity changes” describes the results as they compare to the female counterpart.
Molecular level and transcriptional activity
Chapters 5 and 6 respectively entitled “ Immunohistochemical changes induced by repeated footshock stress: Revelations of gender-based differences” and “Chronic stress effects on synaptic-vesicle associated protein expression: Indications for region/gender-dependent regulation” describe the results of chronic stress exposure on a molecular level. Chapter 5 describes gender-dependent altered regulation of transcriptional activity using cDNA gene expression analysis. Candidate genes identifi ed as the most strongly stress-affected and putatively responsible for yielding a gender-distinctive result are provided in addition to neuronal limbic activity analysis. Chapter 6 in turn illustrates an extended investigation of synaptic vesicle associated proteins using RT-PCR to corroborate identifi -cation with microarray analysis derived from chapter 5.
Implications for pharmacotherapy
Given the stress and gender dependent regulatory effects observed in chapters 2-6, on neuronal function and dysfunction, a subsequently logical and interesting question arose regarding implications of the latter for pharmacotherapeutic application. Whereas the previous chapters provide indications to affi rm association of the HPA axis dysfunction hypothesis (chapter 2) as well as impaired neuronal plasticity (chapters 3, 4, 5), synaptic
plasticity (chapter 6) and indirectly the neurotrophic theory, most antidepressant develop-ment is based on the monoaminergic theory of depression. With regard to this possibility, antidepressants of different mechanisms of action were investigated in chapter 7 entitled
“Impact of long-term antidepressant treatment on chronic stress induced reduction of neu-rogenesis in adult rats: Revelations of sex/drug specifi c regulation.” This chapter confi rms once again the importance of considering gender distinctions on all levels of stress and antidepressant research as it corroborates signifi cant stress/gender interactions regarding stress and antidepressant effects on neurogenesis, thereby providing insight into the fi nal hypothesis of depression.
28 / Introduction

CHAPTER 1
Introduction / 29
With the data presented in this thesis, an attempt was made to perform a broad coverage analysis with multi-level examination of stress-induced impairments based on current knowledge and leading hypotheses. Although not a novelty, the gender aspect was addi-tionally incorporated to examine how the latter fi ts in with current ideas, available data and more importantly, the effi cacy of pharmacotherapy. It must be noted that each chap-ter gave rise to interesting questions, calling in turn for additional studies. Nevertheless, a choice was made to start with the creation of a broad overview, upon which further investigations could be based. Although the studies presented in this thesis only scratch the surface of an ocean of underlying mechanisms, the concluding chapter provides a retrospective discussion of all the presented data and offers an, at times perhaps bold, attempt to integrate the given hypotheses with the inclusion of both genders and the con-sequences thereof.

REFERENCES1. Kessler, R.C., McGonagle,K.A., Swartz,M., Blazer, D.G. & Nelson,C.B. Sex and depression in the National Comorbidity Survey. I: Lifetime prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85-96 (1993).
2. Hrdina, P. Sex-related differences: do they matter? J. Psychiatry Neurosci. 25, 319-320 (2000).
3. Palanza, P. Animal models of anxiety and depression: how are females dif- ferent? Neurosci. Biobehav. Rev. 25, 219-233 (2001).
4. Selye, H. Forty years of stress research: principal remaining problems and misconceptions. Can. Med. Assoc. J. 115, 53-56 (1976).
5. Van de Kar, L.D. & Blair, M.L. Forebrain pathways mediating stress-induced hormone secre- tion. Front Neuroendocrinol. 20,1-48 (1999).
6. Selye, H. [Stress without distress]. Brux. Med. 56, 205-210 (1976).
7. Selye, H. A syndrome produced by diverse nocuous agents. 1936. J. Neuropsychiatry Clin. Neurosci. 10, 230-231 (1998).
8. Chrousos, G.P. et al. Glucocorticoids and glucocorticoid antagonists: lessons from RU 486. Kidney Int. Suppl 26, S18-S23 (1988).
9. Chrousos, G.P. & Gold, P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 267, 1244-1252 (1992). 10. Amiragova, M.G. Neurophysiological analysis of the development of endocrine and hyperensive reactions in prolonged emotional stress. Brain Res. 344, 303-315 (1985).
11. Pezzone, M.A., Lee, W.S., Hoffman, G.E. & Rabin, B.S. Induction of c-Fos immunoreactivity in the rat forebrain by conditioned and unconditioned aversive stimuli. Brain Res. 597, 41-50 (1992).
12. Davis, M. et al. Stress-induced activation of prefrontal cortex dopamine turn over: blockade by lesions of the amygdala. Brain Res. 664, 207-210 (1994).
13. Davis, G.W. & Murphey, R.K. Long-term regulation of short-term transmitter release proper- ties: retrograde signaling and synaptic development. Trends Neurosci. 17, 9-13 (1994).
14. LeDoux, J.E. Emotion: clues from the brain. Annu. Rev. Psychol. 46, 209-235 (1995).
15. Petrov, T., Jhamandas, J.H. & Krukoff, T.L. Electrical stimulation of the central nucleus of the amygdala induces fos-like immunoreactivity in the hypothalamus of the rat: a quantitative study. Brain Res. Mol. Brain Res. 22, 333-340 (1994).
16. Petrov, T., Krukoff, T.L. & Jhamandas, J.H. Chemically defi ned collateral projections from the pons to the central nucleus of the amygdala and hypothalamic paraventricu lar nucleus in the rat. CellTissue Res. 277, 289-295 (1994).
17. Swanson, L.W. & Sawchenko, P.E. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci. 6, 269-324 (1983).
18. Dunn, A.J. & Berridge, C.W. Physiological and behavioral responses to corticotropin-relea- sing factor administration: is CRF es. Brain Res. Rev. 15, 71-100 (1990).
19. Antoni, F.A. Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin- releasing factor. Endocr. Rev. 7, 351-378 (1986).
20. Autelitano, D.J., Blum, M., Lopingco, M., Allen, R.G. & Roberts, J.L. Corticotropin-releasing factor differentially regulates anterior and intermediate pituitary lobe proopiomelanocortin gene tran- scription, nuclear precursor RNA and mature mRNA in vivo. Neuroendocrinology 51, 123-130 (1990).
21. Axelrod, J. & Reisine, T.D. Stress hormones: their interaction and regulation. Science 224, 452-459 (1984).
22. Schachter, B.S., Johnson, L.K., Baxter, J.D. & Roberts, J.L. Differential regulation by glucocorticoids of proopiomelanoc- tin RNA levels in the anterior and intermediate lobes of the ratpituitary. Endocrinology 110, 1442-1444 (1982).
23. Childs, G.V., Morell, J.L., Niendorf, A. & Aguilera, G. Cytochemical studies of corticotropin-releasing factor (CRF) receptors in anterior lobe corticotropes: binding, glucocor ticoid regulation, and endocytosis of [biotinyl-Ser1] CRF. Endocrinology 119, 2129-2142 (1986).
24. Schwartz, J., Billestrup, N., Perrin, M., Rivier, J. & Vale, W. Identifi cation of corticotropin-releasing factor (CRF) target cells and effects of dexamethasone on binding in anterior pituitary using a fl uorescent analog of CRF. Endocrinology 119, 2376-2382 (1986).
25. de Kloet, E.R. Steroids, stability and stress. Front Neuroendocrinol. 16, 416-425 (1995).
26. Meaney, M.J. et al. Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev. Neurosci. 18, 49-72 (1996).
27. Meijer, O.C. & de Kloet, E.R. Corticosterone and serotonergic neurotransmission in the hippocampus: functional implications of central corti- costeroid receptor diversity. Crit Rev. Neurobiol. 12, 1-20 (1998).
28. Gesing, A. et al. Psychological stress increases hippocampal mineralocorticoid receptor levels: involvement of corticotropin-releasing hor- mone. J. Neurosci. 21, 4822-4829 (2001).
29. Reul, J.M., van den Bosch, F.R. & de Kloet, E.R. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treat ment: funtional implications. J. Endocrinol. 115, 459-467 (1987).
30. Jacobson, L. & Sapolsky, R. The role of the hippocampus in feedback regulation of the hy- pothalamic-pituitary-adrenocortical axis. Endocr. Rev. 12, 118-134 (1991).
31. Evans, R.M. & Arriza, J.L. A molecular framework for the actions of glucocorticoid hor- mones in the nervous system. Neuron 2, 1105-1112 (1989).
32. Beato, M. Gene regulation by steroid hormones. Cell 56, 335-344 (1989).
33. Beato, M., Chalepakis, G., Schauer, M. & Slater, E.P. DNA regulatory elements for steroid hormones. J. Steroid Biochem. 32, 737-747 (1989).
30 / Introduction

34. Sapolsky, R.M., Krey, L.C. & McEwen, B.S. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr. Rev. 7, 284-301 (1986).
35. Young, E.A. & Vazquez, D. Hypercortisolemia, hippocampal glucocorticoid receptors, and fast feedback. Mol. Psychiatry 1, 149-159 (1996).
36. Young, E.A. Sex differences and the HPA axis: implications for psychiatric disease. J. Gend. Specif. Med. 1, 21-27 (1998).
37. McEwen, B.S. Multiple ovarian hormone effects on brain struc- ture and func tion. J. Gend. Specif. Med. 1, 33-41 (1998).
38. Rubinow, D.R., Schmidt, P.J. & Roca,C.A. Estrogen-serotonin interactions: implications for affective regulation. Biol. Psychiatry 44, 839-850 (1998).
39. Woolley, C.S. Estrogen-mediated structural and functional synaptic plasticity in the female rat hippocampus. Horm. Behav. 34, 140-148 (1998).
40. Vamvakopoulos, N.C. & Chrousos, G.P. Evidence of direct estrogenic regulation of human cor- tico tropin-releasing hormone gene expression. Potential implications for the sexual dimophism of the stress response and immune/infl ammatory reaction. J. Clin. Invest 92, 1896-1902 (1993).
41. Viau, V. & Meaney, M.J. Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 129, 2503-2511 (1991).
42. Burgess, L.H. & Handa, R.J. Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor- mediated functions in female rats. Endocrinology 131, 1261-1269 (1992).
43.Rousseau, G.G., Baxter, J.D. & Tomkins, G.M. Glu cocorticoid receptors: relations between steroid binding and biological effects. J. Mol. Biol. 67, 99-115 (1972).
44. Svec, F. Comparison of glucocorticoid receptors liganded with dexa- methasone or progesterone. Proc. Soc. Exp. Biol. Med. 198, 811-817 (1991).
45. Ahima, R.S., Lawson, A.N., Osei, S.Y. & Harlan, R.E. Sexual dimorphism in regulation of type II corti costeroid receptor immunoreactivity in the rat hippocampus. Endocrinology 131, 1409-1416 (1992).
46. Arriza, J.L. et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237, 268-275 (1987).
47. Carey, M.P., Deterd, C.H., de Koning, J., Helmerhorst, F. & de Kloet, E.R. The infl uence of ovarian steroids on hypothalamic-pituitary- adrenal regulation in the female rat. J. Endocrinol. 144, 311-321 (1995).
48. Henry, J.P. Biological basis of the stress response. Integr. Physiol Behav. Sci. 27, 66-83 (1992).
49. Croes, S., Merz, P. & Netter, P. Cortisol reaction in success and failure condition in endogenous depressed patients and controls. Psychoneuroendocrinology 18, 23-35 (1993).
50. Ottenweller, J.E., Natelson, B.H., Pitman, D.L. & Drastal, S.D. Adrenocortical and behavioral responses to repeated stressors: toward an animal model of chronic stress and stress-
related mental illness. Biol. Psychiatry 26, 829-841 (1989).51. Keller-Wood M.E. & Dallman, M.F. Corticosteroid inhibition of ACTH secretion. Endocr. Rev. 5, 1-24 (1984).
52. Dallman, M.F., Makara, G.B., Roberts, J.L., Levin, N. & Blum, M. Corticotrope response to removal of releasing factors and corticosteroids in vivo. Endocrinology 117, 2190-2197 (1985).
53. Gold, P.W., Goodwin, F.K. & Chrousos, G.P. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (1). N. Engl. J. Med. 319, 348-353 (1988).
54. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23, 477-501 (2000).
55. Murphy, B.E. Steroids and depression. J. Steroid Biochem. Mol. Biol. 38, 537-559 (1991).
56. Murphy, B.E. Antiglucocorticoid therapies in major depression: a review. Psychoneuroendocrinology 22 Suppl 1, S125-S132 (1997).
57. Akiskal, H.S. et al. Re-evaluating the prevalence of and diagnostic composition within the broad clinical spectrum of bipolar disorders. J. Affect. Disord. 59 Suppl 1, S5-S30 (2000).
58. Blazer D.G. Psychiatry and the oldest old. Am. J. Psychiatry 157, 1915-1924 (2000).
59. Manji, H.K., Drevets, W.C. & Charney, D.S. The cellular neurobiology of depression. Nat. Med. 7, 541-547 (2001).
60. Ongur, D., Drevets, W.C. & Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U. S. A 95, 13290-13295 (1998).
61. Rajkowska, G. et al. Morphometric evidence for neuronal and glial prefrontal cell- pathology in major depression. Biol. Psychiatry 45, 1085-1098 (1999).
62. Rajkowska, G. Postmortem studies in mood disorders indicate altered num- bers of neurons and glial cells. Biol. Psychiatry 48, 766-777 (2000).
63. Cotter, D., Mackay, D., Landau, S., Kerwin, R. & Everall, I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry 58, 545-553 (2001).
64. Drevets, W.C. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog. Brain Res. 126, 413-431 (2000).
65.Sheline, Y.I., Wang, P.W., Gado, M.H., Csernansky, J.G. & Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. U. S. A 93, 3908-3913 (1996).
66. Bremner, J.D. et al. Hippocampal volume reduction in major depression. Am. J. Psychiatry 157, 115-118 (2000).
67. Kessler, R.C. The effects of stressful life events on depression. Annu. Rev. sychol. 48, 191-214 (1997).
68. Heim, C. & Nemeroff, C.B. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol.Psychiatry 49, 1023-1039 (2001).
Introduction / 31
CHAPTER 1

69. Gold, P.W., Licinio, J., Wong, M.L. & Chrousos, G.P. Corticotropin releasing hormone in the pathophysiology of melancholic and atypical depression and in the mechanism of action of antidepressant drugs. Ann. N. Y. Acad. Sci. 771, 716-729 (1995).
70. Sapolsky, R.M. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry 57, 925-935 (2000).
71. Arborelius, L., Owens, M.J., Plotsky, P.M. & Nemeroff,C.B. The role of corticotropin-releasing factor in depression and anxiety disorders. J. Endocrinol. 160, 1-12 (1999).
72. McEwen, B.S. Effects of adverse experiences for brain structure and function. Biol. Psychiatry 48, 721-731 (2000).
73. Willner, P. Animal models of depression: validity and applications. Adv. Biochem. Psychopharmacol. 49, 19-41 (1995).
74. Hitzemann, R. Animal models of psychiatric disorders and their relevance to alcoholism. Alcohol Res. Health 24, 149-158 (2000).
75. Porsolt, R.D. Animal models of depression: utility for transgenic research. Rev. Neurosci. 11, 53-58 (2000).
76. Lucki, I. A prescription to resist proscriptions for murine models of depression. Psychopharmacology (Berl) 153, 395-398 (2001).
77. Duman, R.S., Heninger, G.R. & Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597-606 (1997).
78. Altar, C.A. Neurotrophins and depression. Trends Pharmacol. Sci. 20, 59-61 (1999).
79. Blier, P. & de Montigny, C. Current advances and trends in the treatment of depression. Trends Pharmacol. Sci. 15, 220-226 (1994).
80. Stahl, S.M. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J. Affect. Disord. 51, 215-235 (1998).
81. Meltzer, H.Y. Role of serotonin in depression. Ann. N. Y. Acad. Sci. 600,486-499 (1990).
82. Owens, M.J. & Nemeroff, C.B. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin. Chem. 40, 288-295 (1994).
83. Amara, S.G. & Kuhar, M.J. Neurotransmitter transporters: recent progress. Annu. Rev. Neurosci. 16, 73-93 (1993).
84. Barker, E.L. & Blakely, R.D. Structural determinants of neurotransmitter transport using cross-species chimeras: studies on serotonin transporter. Methods Enzymol. 296, 475-498 (1998).
85. McEwen, B.S. Infl uences of adrenocortical hormones on pituitary and brain function. Monogr Endocrinol. 12, 467-492 (1979).
86. McEwen, B.S., de Kloet, E.R. & Rostene, W. Adrenal steroid receptors and actions in the nervous system. Physiol Rev. 66, 1121-1188 (1986).
87. Sapolsky, R.M. & McEwen, B.S. Down-regulation of neural corticosterone receptors by corti- costerone and dexamethasone. Brain Res. 339, 161-165 (1985).
88. McEwen, B.S. & Brinton, R.E. Neuroendocrine aspects of adaptation. Prog. Brain Res. 72, 11-26 (1987).
89. Chrousos, G.P. & Gold, P.W. A healthy body in a healthy mind--and vice versa--the damaging power of “uncontrollable” stress. J. Clin. Endocrinol. Metab 83, 1842-1845 (1998).
90. McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 840, 33-44 (1998).
91. Barden, N., Reul, J.M. & Holsboer, F. Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system? Trends Neurosci. 18, 6-11 (1995).
92. Magarinos, A.M., McEwen, B.S., Flugge, G. & Fuchs, E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J. Neurosci. 16, 3534-3540 (1996.
93. McEwen, B.S. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22,105-122 (1999).
94. Gould, E., Cameron, H.A., Daniels, D.C., Woolley,C.S. & McEwen,B.S. Adrenal hormones suppress cell division in the adult rat dentategyrus. J. Neurosci. 12, 3642-3650 (1992).
95. Gould, E., McEwen, B.S., Tanapat, P., Galea, L.A. & Fuchs, E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 17, 2492-2498 (1997).
96. Czeh, B. et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. U. S. A 98, 12796-12801 (2001).
97. Smith, M.A., Makino, S., Kvetnansky, R. & Post, R.M. Stress and glucocorticoids affect the expression of brain- derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15, 1768-1777 (1995).
98. Vaidya, V.A., Marek, G.J., Aghajanian, G.K. & Duman, R.S. 5-HT2A receptor-mediated regulation of brain-derived neuro- trophic factor mRNA in the hippocampus and the neocortex. J. Neurosci. 17, 2785-2795 (1997).
99. Nibuya, M., Morinobu, S. & Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539-7547 (1995).
100. Chen, B., Dowlatshahi, D., MacQueen, G.M., Wang, J.F. & Young, L.T. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 50, 260-265 (2001).
101. Duman, R.S., Malberg, J. & Thome, J. Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 46, 1181-1191 (1999).
102. Korte M. et al. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc. Natl. Acad. Sci. U. S. A 93, 12547-12552 (1996).
103. Patterson, S.L. et al. Recombinant BDNF rescues defi cits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137-1145 (1996).
104. Kang, H., Welcher, A.A., Shelton, D. & Schuman, E.M. Neurotrophins and time: different roles for TrkB signaling in
32 / Introduction

hippocampal long-term potentiation. Neuron 19, 653-664 (1997).
105. Thoenen, H. Neurotrophins and neuronal plasticity. Science 270, 593-598 (1995).
106. Dwivedi, Y. et al. Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J. Neurochem. 77, 916-928 (2001).
107. Nibuya, M., Nestler, E.J. & Duman, R.S. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 16, 2365-2372 (1996).
108. Thome, J. et al. cAMP response element-mediated gene transcription is upre- gulated by chronic antidepressant treatment. J. Neurosci. 20, 4030-4036 (2000).
109. Shirayama, Y., Chen, A.C., Nakagawa, S., Russell, D.S. & Duman, R.S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 22, 3251-3261 (2002).
110. Jacobs, B.L., Praag, H. & Gage, F.H. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol. Psychiatry 5, 262-269 (2000).
111. Jacobs, B.L. Adult brain neurogenesis and depression. Brain Behav. Immun.16, 602-609 (2002).
112. D’Sa C. & Duman, R.S. Antidepressants and neuroplasticity. Bipolar. Disord. 4, 183-194 (2002).
113. Kaplan, M.S. & Hinds, J.W. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197, 1092-1094 (1977).
114. Bayer, S.A. Neuron production in the hippocampus and olfactory bulb of the adult rat brain: addition or replacement? Ann. N. Y. Acad. Sci. 457, 163-172 (1985).
115. Cameron, H.A., Woolley, C.S., McEwen, B.S. & Gould, E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337-344 (1993).
116. Gould, E., Tanapat, P., McEwen, B.S., Flugge, G. & Fuchs, E. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc. Natl. Acad. Sci. U. S. A 95, 3168-3171 (1998).
117. Gould, E. & Cameron, H.A. Regulation of neuronal birth, migration and death in the rat dentate gyrus. Dev. Neurosci. 18, 22-35 (1996).
118. Malberg, J.E., Eisch, A.J., Nestler, E.J. & Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104-9110 (2000).
119. Duman, R.S., Nakagawa, S. & Malberg, J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 25, 836-844 (2001).
120. Palmer, T.D., Takahashi, J. & Gage, F.H. The adult rat hippocampus contains primordial neural stem cells. Mol. Cell Neurosci. 8, 389-404 (1997).
121. Gould, E., Reeves, A.J., Graziano, M.S. & Gross, C.G. Neurogenesis in the neocortex of adult primates. Science 286, 548-552 (1999).
122. Gould, E., Vail, N., Wagers, M. & Gross, C.G. Adult-generated hippocampal and neocortical neurons in macaques have a transient existence.
Proc. Natl. Acad. Sci. U. S. A 98, 10910-10917 (2001).
123. Kuhn, R. The treatment of depressive states with G22355 (imipramine hydrochloride). Am. J. Psychiatry 115, 459-464 (1958).
124. Crane, G.E. Iproniazid (marsilid) phosphate, a therapeutic agent for mental disorders and debilitating diseases. Psychiatr. Res. Rep. Am. Psychiatr. Assoc. 135, 142-152 (1957).
125. Vaswani, M., Linda, F.K. & Ramesh, S. Role of selective serotonin reuptake inhibitors in psy- chiatric disorders: a comprehensive review. Prog.Neuropsychopharmacol. Biol. Psychiatry 27, 85-102 (2003).
126. Nelson, J.C. A review of the effi cacy of serotonergic and noradrenergic reuptake inhibitors for treatment of major depression. Biol Psychiatry 46, 1301-1308 (1999).
127. Anderson, I.M. Selective serotonin reuptake inhibitors versus tricyclic anti- depressants: a meta-analysis of effi cacy and tolerability. J. Affect. Disord. 58, 19-36 (2000).
128. Geddes, J.R., Freemantle, N., Mason, J., Eccles, M.P. & Boynton, J. SSRIs versus other antidepressants for depressive disorder. Cochrane. Database. Syst. Rev CD001851 (2000).
129. Ginestet D. Effi cacy of tianeptine in major depressive disorders with or without melancholia. Eur. Neuropsychopharmacol. 7 Suppl 3, S341-S345 (1997).
130. McEwen B.S. et al. Prevention of stress-induced morphological and cognitive con- sequences. Eur. Neuropsychopharmacol. 7 Suppl 3, S323-S328 (1997).
131. Johannessen, C.U. Mechanisms of action of valproate: a commentatory. Neurochem. Int. 37, 103-110 (2000).
132. Lenox, R.H. & Hahn, C.G. Overview of the mech anism of action of lithium in the brain: fi fty-year update. J. Clin. Psychiatry 61 Suppl 9, 5-15 (2000).
133.Salomon, R.M., Miller, H.L., Krystal, J.H., Heninger, G.R. & Charney, D.S. Lack of behavioral effects of monoamine depletion in healthy subjects. Biol Psychiatry 41, 58-64 (1997).
134. Delgado, P. & Moreno, F. Antidepressants and the brain. Int. Clin Psychopharmacol. 14 Suppl 1, S9-16 (1999).
135. Stewart, C.A. & Reid, I.C. Repeated ECS and fl uoxetine administration have equivalent effects on hippocampal synaptic plasticity. Psychopharmacology (Berl) 148, 217-223 (2000).
136. Vaidya, V.A., Siuciak, J.A., Du, F. & Duman, R.S. Hippocampal mossy fi ber sprouting induced by chronic electro- convulsive seizures. Neuroscience 89, 157-166 (1999).
137. Dahmen, N., Fehr, C., Reuss, S. & Hiemke C. Stimulation of immediate early gene expression by desipramine in rat brain. Biol. Psychiatry 42, 317-323 (1997).
138.Torres, G., Horowitz, J.M., Lafl amme, N. & Rivest, S. Fluoxetine induces the transcription of genes encoding c-fos, corticotropin-releasing factor and its type 1 receptor in rat brain. Neuroscience 87, 463-477 (1998).
139. Pei,Q. et al. Serotonergic regulation of mRNA expression of Arc, an immedi- ate early gene selectively localized at neuronal dendrites. Neuropharmacology 39, 463-470 (2000).
Introduction / 33
CHAPTER 1


PART I
SYSTEMIC, NEUROENDOCRINOLOGICAL
LEVEL


REPEATED STRESS IMPAIRS HPA REGULATION IN RATS: INDICATIONS FOR DIFFERENTIAL GENDER- DEPENDENT MECHANISMS
2Stress has been associated with complex neuroendocrine abnor-
malities, including HPA axis hyperactivity and impaired neuronal
plasticity. Furthermore, adverse experiences are believed to exert
a predisposing effect in the development of a wide array of affec-
tive disorders, psychiatric conditions often characterized by cor-
tical-limbic dysfunctions and marked gender-related prevalence.
Despite advances in the understanding of stress-induced neuronal
adaptations, the neurobiological mechanisms underlying psycho-
pathology or gender-related sensitivity to stress are still poorly
understood. In this study, we thus explored the neurochemical
and neuroendocrine changes in response to repeated footshock
exposure in male and female rats, using immunohistochemical
(FOS and phospho-CREB expression) and molecular techniques
(RT-PCR). Our fi ndings collectively demonstrate the ability of
repeated stress to disrupt HPA axis regulation in a gender- and/or
region-specifi c manner and the observed differences reveal the
complex modulation of this stress response system. Sexually
dimorphic mechanisms underlying the regulation/dysregulation
of HPA axis response to stress appear to encompass glucocor-
ticoid, catecholamine, and GR/MR interplay in distinct cortical
and subcortical structures. Whereas impaired prefrontocortical
and/or hippocampal control over the HPA axis, seems to represent
a possible mechanism by which repeated stress leads to abnor-
mal regulation of the stress response in males, imbalanced GR/MR
expression, disturbed autonomic function, and abnormal amy-
gdala activity seems to refl ect events involved in the development
of HPA axis abnormalities in females. These fi ndings may in turn
provide valuable insight into the understanding of the relation-
ships between disturbed HPA function and psychopathology.
Kuipers S.D., Trentani A., Bakker P.L., Postema F., Reuvekamp P.T.W.,
Ter Horst G.J., Den Boer J.A. Endocrinology. Submitted


CHAPTER 2
Stress, gender and HPA function / 39
INTRODUCTION
Chronic stress has been associated with multiple abnormalities in the periphery and the brain. These include persistently elevated glucocorticoid concentrations, impaired immune response, and complex cortical-limbic dysfunctions 1-4. Stress, especially when persistent and severe, also plays a major role in the development of affective disorders 5, psychiatric conditions characterized by a marked gender-related prevalence 6. Women seem particularly more vulnerable to stress and psychopathology, as refl ected by their higher susceptibility to depression 7-11. Although advances have been made in the understanding of brain changes in response to stress, the neurobiological mechanisms underlying the development of psychopathology or gender-related sensitivity to stress are still poorly understood. Glucocorticoids represent the major hormonal mediators in the stress response and their release is carefully regulated by neural circuits impinging on hypothalamic neurons 12. Due to the marked sexual dimorphism observed in this region, the hypothalamus has thus been a prime focus in the search for mechanisms underlying differential male and female sensitivity to stress 13-15. The activity of the hypothalamic-pituitary-adrenal (HPA) axis however, is dynamically regulated by cortical and subcortical structures, such as the prefrontal cortex, the hippocampus and the amygdala, through positive and negative feedback mechanisms, operating over multiple time domains and at different levels 16-18. Adrenal steroids themselves exert inhibitory effects on HPA axis activity, actions that are believed to be transduced primarily by intracellular steroid receptor proteins that func-tion as hormone-activated transcription factors, namely the glucocorticoid (GR) and the mineralocorticoid receptors (MR) 19. A differential impact of adverse events on the coor-dinated activity underlying this critical stress response axis may play a central role in deter-mining gender-related susceptibility to the negative effects of stress. Despite the sex-related discrepancies and well-acknowledged role of corticosteroids in the development of stress-related pathology, only a few studies have investigated how gender actually affects the response to stress from a cellular and molecular perspective. In this study we thus examined in male and female rats, both neurochemical and neuroen-docrine adaptations to repeated footshock stress, using immunohistochemical (FOS and phospho-CREB expression) and molecular techniques (RT-PCR). Peripheral HPA axis activity as well as adrenocortical function was evaluated. Subsequent identifi cation of gen-der-related differences in the modulation of HPA axis following repeated footshock expo-sure might be of interest in an attempt to characterize differential mechanisms involved in the development of abnormal stress response regulation. This in turn might provide valu-able insight into the relationship between disturbed HPA function and psychopathology.

40 / Stress, gender and HPA function
MATERIALS AND METHODS
Animals
The experiments were performed with male (n°=24: 225-249 g; 5-6 weeks of age) and female (n°=24: 200-224 g; 5-6 weeks of age) Wistar rats. The animals were individually housed with food and water available ad libitum and maintained on a 12:12 h light/dark cycle at 21°C. All rats were handled daily for 5-8 min to minimize the non-specifi c stress response. All handling was performed in accordance with the European Communi-ties Council Directive of November 24, 1986 (86/609/EEC), and the guidelines of the Animal Bio-ethics Committee of the University of Groningen (FDC: 2509).
Chronic Stress Procedure
The rodent footshock-chamber consists of a box containing an animal space positioned on a metallic grid fl oor connected to a shock generator and scrambler. For 3 weeks, stressed rats were subjected to a daily aversive session of varying duration (between 30 and 120 min/day) in the “footshock chamber” during which 5 uncontrollable and inescapable footshocks were applied (0.8 mA in intensity and 8 sec in duration). In order to make the procedure as unpredictable as possible, starting time, inter-shock interval, and total time spent in the box were varied on a daily basis. Despite the nature of the stressor, this footshock procedure did not result in any visible injuries.
Experimental Setup
To investigate the cellular and molecular changes induced by repeated stress, rats were randomly assigned to two groups:• STRESS (n° = 24; 12 male and 12 female rats): stressed animals were stressed, according to the footshock procedure described above, for 21 consecutive days;• CTR (n°= 24; 12 male and 12 female rats): control animals followed an identical schedule in a similar setup. These rats were exposed to the footshock chamber on a daily basis but never received actual footshocks throughout the experiment.On the fi nal day of the experiment, all rats were placed in the footshock chamber for a fi nal 15-minute session without receiving any footshocks. Exposure to the box on the fi nal day was essential as it provided a way to create a stressful condition without expo-sing stressed animals to physical and painful stimuli. The response of control and stressed rats to an identical stimulus (footshock box) could then be investigated by immunohis-tochemistry and RT-PCR. To limit circadian variability in circulating corticosterone, ca-techolamine and gene expression levels, animals were sacrifi ced on subsequent days within a narrow window of 4 hours.

CHAPTER 2
Stress, gender and HPA function / 41
Physiological and endocrine measurements
To explore the changes underlying the response to repeated footshock stress, various parameters were measured. Weight gain was monitored on a daily basis and graphs were constructed to illustrate the body weight gain throughout the procedure. Upon termina-tion, adrenal and thymus glands were removed, weighed and recalculated to correct for the body weights of the animals. Blood was also collected and the serum obtained was stored at –20ºC to determine plasma corticosterone and catecholamine levels with HPLC.
Extraction and Chromatography
Catecholamines.
Noradrenaline and adrenaline were extracted from plasma using liquid/liquid extraction with 3,4-dihydroxybenzylamine as internal standard 20,21. Briefl y, plasma catecholamines were bound to diphenylborate-ethanolamine at pH 8.6 and extraction was performed with n-heptane (containing 1% octanol and 25% tetraoctylammoniumbromide). After extraction from the organic phase with diluted acetic acid, catecholamines (20 µl acetic acid extract) were analyzed using an HPLC/auto-injector (CMA, Sweden) and a Shi-madzu LC-10AD pump (Kyoto, Japan) connected to a reversed phase column (Hypersil, C18, 3µm, 150x2.0 mm), followed by an electrochemical detector (Antec Leyden, The Netherlands) working at a potential setting of 500mV vs. Ag/AgCl reference. The mobile phase consisted of 50mM acetate buffer, 150mg/l octane sulphonic acid, 150mg/l te-tramethylammonium, 15mg/ml Na
2EDTA and 3% methanol, adjusted to pH 4.1. The
fl ow-rate was 0.35ml/min. Temperature was 30°C. The detection limit was 0.1nM.
Corticosterone.
For this assay, dexamethasone was used as the internal standard. After addition of the internal standard, plasma was extracted with 3ml of diethylether, vortexed for 5 min and then centrifuged for 5 min at 3000 x g. The extraction procedure was repeated twice. The organic phase was evaporated to dryness in a 50°C waterbath. The residue was reconsti-tuted with 200µL of mobile phase and 50µL was injected into the HPLC system. The mobile phase (fl ow rate 1.0mL/min) for the determination consisted of acetonitrile in ultrapure water (27:73 v/v). The concentrations of both corticosterone and the internal standard were determined with UV detection at a wavelength of 254nm. The detection limit was 10nM.

42 / Stress, gender and HPA function
Immunohistochemistry
Twenty-four rats were terminated two hours after the beginning of the fi nal session, with an overdose of halothane preceding a transcardial perfusion with 4% paraformaldehyde solution in 0.1M sodium phosphate buffer (pH 7.4). The brains were carefully removed and post-fi xed in the same solution overnight at 4°C, before being transferred to a potas-sium phosphate buffer (KPBS 0.02M, pH 7.4) and stored at 4°C. Following cryoprotec-tion of the brains by overnight immersion in a 30% glucose solution, coronal serial sec-tions of 40µm were prepared on a cryostat microtome. Sections were collected in KPBS with sodiumazide and stored at 4°C.
FOS and phospho-CREB immunoreactivity
All stainings were performed on free-fl oating sections under continuous agitation. The sections were pre-incubated in 0.3% H
2O
2 for 15 min to reduce endogenous peroxidase
activity, before being incubated in the respective primary antibody solutions:• polyclonal rabbit anti-c-fos antibody (commercialized by Oncogene Research Products, brands of CN Biosciences, Inc, an affi liate of Merck KGaA, Darmstadt, Germany) 1:10000 dilution in KPBS 0.02M, pH 7.4, for 60-72 hr at 4°C;• polyclonal rabbit anti-phospho-CREB (commercialized by Upstate Biotechnology, Charlottesville, VA, USA: www.upstatebiotech.com) 1:1000 dilution in KPBS 0.02M, pH 7.4, for 60-72 hr at 4°C.Subsequently, sections were washed with KPBS and incubated at room temperature with secondary biotinylated goat anti-rabbit IgG antibodies (Vector Laboratories, Inc., Burlin-game, CA, USA; 1:1000 dilution in KPBS 0.02M, pH 7.4) followed by ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA, USA). After another wash, the reaction product was visualized by adding diaminobenzidine as chromogen and 1% H
2O
2
for 15 min. Finally, the sections were washed, mounted on slides, dehydrated and cover-slipped with DePex.
Antibody specifi city testing.
To control for cross-reactivity due to aspecifi c binding, immunostainings were performed by incubating the sections without the presence of one of the antibodies needed for the reaction (primary, secondary or tertiary antibody). All these reactions were negative thereby confi rming the specifi city of the antibodies.
Quantifi cation and data analysis.
FOS and phospho-CREB-labeled cells were quantifi ed using a computerized image analy-sis system by an observer who was blind to group assignment. Selected areas from regions of interest (ROIs) were digitized using a Sony (SONY Corporation, Tokyo, Japan) charge-

CHAPTER 2
Stress, gender and HPA function / 43
coupled device digital camera mounted on a LEICA Leitz DMRB microscope (LEICA, Wetzlar, Germany). Quantifi cation was carried out at x100 using at least 5 coronal serial sections (the rostro-caudal distance between consecutive sections was 0.25mm) for each area or nucleus of interest. ROIs were outlined with a digital pen and each digitized image was individually set at a threshold to subtract the background optical density. ROIs included the medial orbitofrontal (mORB: Bregma +4.85 to +3.60) and the medial pre-frontal cortex (prelimbic and infralimbic areas; mPFC: Bregma +3.60 to +1.70); the ante-rior (AC: Bregma +3.20 to +0.95) and the posterior cingulate cortex (postCING: Bregma +1.70 to -1.08); the hippocampal dentate gyrus (DG: Bregma -2.00 to –3.90); the central (CeA: Bregma –1.53 to –2.85), the lateral (LaA: Bregma –2.00 to -3.70) and the basola-teral nucleus of the amygdala (BslA: Bregma –1.78 to –3.25); the paraventricular thalamic nucleus (PVT: Bregma –1.33 to –3.90); the paraventricular hypothalamic nucleus (PVN: Bregma –1.08 to –2.00); the dorsal raphe (DR: Bregma –7.10 to –9.25) 22. The numbers of cell nuclei above background were quantifi ed using the computer-based image analysis system LEICA (LEICA Imaging System Ltd., Cambridge, England). Only cell nuclei that exceeded a defi ned threshold were detected by the system and subsequent counts were reported as number of positive cells/0.1mm2. FOS and phospho-CREB-labeled cells with gray levels below the defi ned thresholds were thus classifi ed as “negative”. This is of relevance for proper understanding of our results, since this method does not allow discrimination between negative nuclei with no immunoreactivity and nuclei with (too) low immunoreactivity. This method is thus not suitable for determining absolute protein levels. All areas were measured bilaterally and no left-right asymmetry was found 23.
Silver Staining.
To enhance distinction between adrenal cortex and medulla a silver staining was per-formed according to a previously described protocol 24. Following perfusion the adrenals were removed and immersed overnight in a phosphate buffered 30% sucrose solution (0.1M, pH7.4) at 4ºC. Forty-micrometer sections were cut on a cryostat microtome and collected into buffered 4% paraformaldehyde solution (pH 9.0). After a rinse with distil-led water, the sections were rinsed 3 x 5 min with pre-treatment solution (9% sodium hydroxide diluted 1:1 with 1.2% ammonium nitrate stock solution, pH 11.9) at room temperature. After removal of pretreatment solution, impregnation medium (9% sodium
hydroxide diluted 1.5:1 with 16% ammonium nitrate stock) was added followed by 240 µl 50% silver nitrate solution per 40ml medium. After 10 min incubation on a shaker at room temperature, the sections were rinsed 3 x 5 min with a sodium carbonate solution and incubated in developer solution (0.57g citric acid in 100 ml distilled water + 15ml 37% formaldehyde solution + 100ml 96% ethanol) for 4 minutes without agitation. After development, sections were incubated in 37.5% sodium thiosulfate silver fi xation solution for 4 min and then rinsed 3 x 5 min with distilled water. Finally sections were mounted on slides, dehydrated and coverslipped with DePex.

44 / Stress, gender and HPA function
Image analysis and volume determination.
Per rat, every section of both adrenals was used for the analysis. Using a Sony (SONY Corporation, Tokyo, Japan) charge-coupled device digital camera mounted on a LEICA Leitz DMRB microscope (LEICA, Wetzlar, Germany), the surface area of the total adre-nal, the medulla and the cortex (total – medulla area) was determined. Multiplication of area measures by 40 µm thickness of the sections, allowed calculation of the corresponding adrenal (medulla and cortex) volumes.
RNA isolation and Quantitative RT-PCR
Total RNA- and poly(A)+RNA-isolation.
To investigate the MR, GR and CRF mRNA levels, 24 rats were anesthetized with halo-thane and decapitated thirty minutes after the start of the fi nal session. The brains were dissected to separate the prefrontal cortex (Bregma +3.60 to +1.70) and the hippocampus (Bregma -2.30 to -4.16), which were subsequently quick-frozen in liquid nitrogen and stored at -80°C. Total RNA was isolated from these brain areas using Trizol (Life Tech-nology, Gaithersburg, MD, USA) according to the manufacturer’s instructions. Integrity of total RNA was confi rmed on a 2% agarose gel and fi nal concentrations were assessed spectrophotometrically. Poly(A)+RNA was in turn isolated from total RNA using Micro-FastTrack™ 2.0 Kit (Invitrogen) according to the manufacturer’s instructions.
First-strand cDNA synthesis.
First-strand cDNA was synthesized from 200ng poly(A)+RNA using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s instruc-tions. Before fi rst-strand synthesis was performed, contaminating genomic DNA was removed using Deoxyribonuclease I, Amplifi cation Grade (Invitrogen). The fi rst-strand synthesis was primed with oligo(dT), and the RNA template was not removed from the DNA:RNA hybrid.
Primers and PCR Conditions.
All primers were designed with Primer Express® Software v2.0 (Applied Biosystems) using default parameters (sequences are listed in table 1). PCR reactions were performed on an ABI Prism 7900HT Sequence Detection System. Amplifi cation mixtures (25µL divided over 3 wells, 7µl per well) for all genes contained 2ng cDNA from poly(A)+RNA template, 1 x SYBR Green PCR buffer, 3mM MgCl2, 0.2mM dATP, 0.2mM dCTP, 0.2mM dGTP 0.4mM dUTP, 0.025U/µl Amplitaq Gold, 0.01U/µl AmpErase UNG and 300nM of each primer. The cycling conditions comprised of 2 minutes AmpErase UNG activity, 10 minutes Amplitaq Gold activation, 40 cycles at 95°C for 15 seconds and 60°C

CHAPTER 2
Stress, gender and HPA function / 45
for 1 minute, and a dissociation stage: 15 seconds 95°C, 15 seconds 60°C, 15 seconds 95°C ramp rate 2%. The No Template Control contained all components except the cDNA template.
Real-time polymerase chain reaction (RT-PCR).
Real time detection was performed using an ABI PRISM 7900HT Sequence Detection System and SYBR Green PCR Core Reagents. Data analysis was performed using software provided with the instrument. The mRNAs of the different genes were relatively quanti-fi ed across groups with the Comparative CT Method. The CT value, or fractional cycle number at which the defi ned threshold is crossed above baseline, is predictive of the input amount of target and thus useful in quantifi cation of RNA 25. Here the threshold was set between 0.4 and 0.6 with a baseline range from 1 to 7. All RNA samples were measured in trio and the amount of target was normalized to an endogenous reference, the house-keeping gene, RPS29 (CT). An additional reference (ß-actin) was included as an extra control.
Statistical Analysis
Data are expressed as means ± standard error (SEM). Statistical signifi cance was determined by performing one-way analysis of variance (ANOVA) and F test of variance on the cor-responding parameter measures from experimental and control conditions. To compare
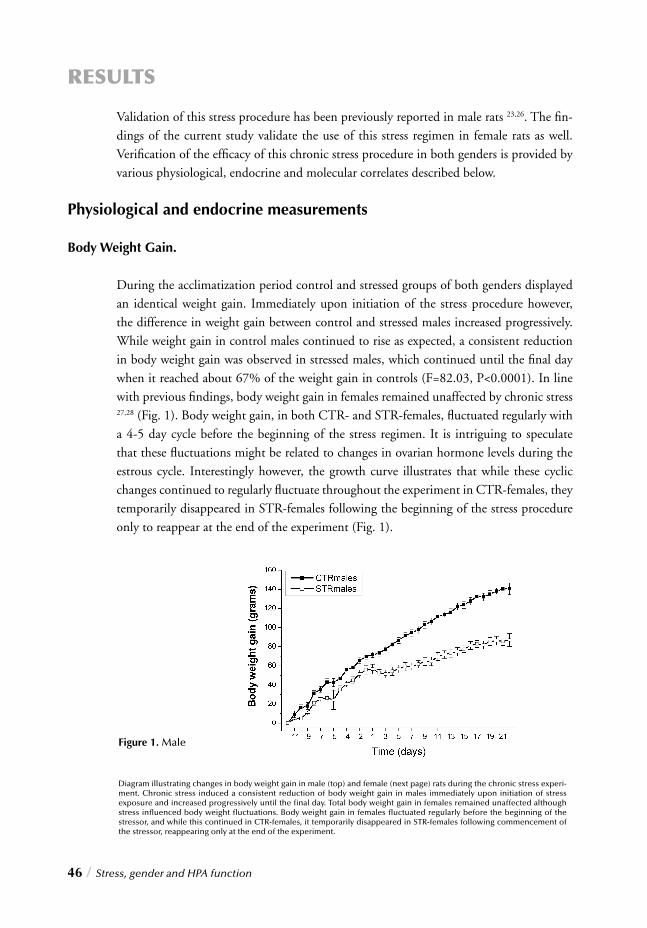
46 / Stress, gender and HPA function
RESULTS
Validation of this stress procedure has been previously reported in male rats 23,26. The fi n-dings of the current study validate the use of this stress regimen in female rats as well. Verifi cation of the effi cacy of this chronic stress procedure in both genders is provided by various physiological, endocrine and molecular correlates described below.
Physiological and endocrine measurements
Body Weight Gain.
During the acclimatization period control and stressed groups of both genders displayed an identical weight gain. Immediately upon initiation of the stress procedure however, the difference in weight gain between control and stressed males increased progressively. While weight gain in control males continued to rise as expected, a consistent reduction in body weight gain was observed in stressed males, which continued until the fi nal day when it reached about 67% of the weight gain in controls (F=82.03, P<0.0001). In line with previous fi ndings, body weight gain in females remained unaffected by chronic stress 27,28 (Fig. 1). Body weight gain, in both CTR- and STR-females, fl uctuated regularly with a 4-5 day cycle before the beginning of the stress regimen. It is intriguing to speculate that these fl uctuations might be related to changes in ovarian hormone levels during the estrous cycle. Interestingly however, the growth curve illustrates that while these cyclic changes continued to regularly fl uctuate throughout the experiment in CTR-females, they temporarily disappeared in STR-females following the beginning of the stress procedure only to reappear at the end of the experiment (Fig. 1).
Figure 1. Male
Diagram illustrating changes in body weight gain in male (top) and female (next page) rats during the chronic stress experi-ment. Chronic stress induced a consistent reduction of body weight gain in males immediately upon initiation of stress exposure and increased progressively until the fi nal day. Total body weight gain in females remained unaffected although stress infl uenced body weight fl uctuations. Body weight gain in females fl uctuated regularly before the beginning of the stressor, and while this continued in CTR-females, it temporarily disappeared in STR-females following commencement of the stressor, reappearing only at the end of the experiment.
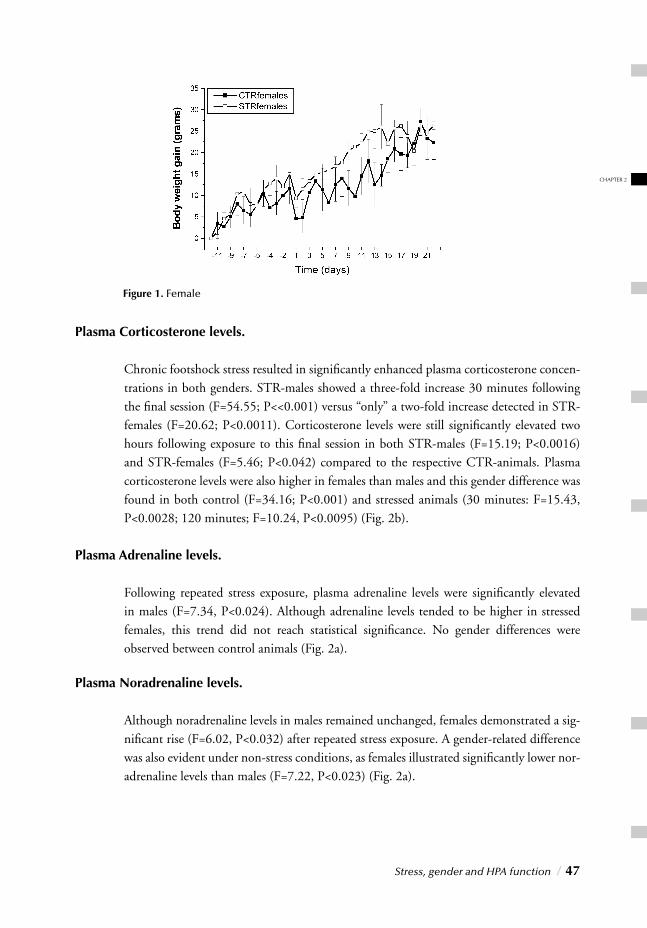
CHAPTER 2
Stress, gender and HPA function / 47
Plasma Corticosterone levels.
Chronic footshock stress resulted in signifi cantly enhanced plasma corticosterone concen-trations in both genders. STR-males showed a three-fold increase 30 minutes following the fi nal session (F=54.55; P<<0.001) versus “only” a two-fold increase detected in STR-females (F=20.62; P<0.0011). Corticosterone levels were still signifi cantly elevated two hours following exposure to this fi nal session in both STR-males (F=15.19; P<0.0016) and STR-females (F=5.46; P<0.042) compared to the respective CTR-animals. Plasma corticosterone levels were also higher in females than males and this gender difference was found in both control (F=34.16; P<0.001) and stressed animals (30 minutes: F=15.43, P<0.0028; 120 minutes; F=10.24, P<0.0095) (Fig. 2b).
Plasma Adrenaline levels.
Following repeated stress exposure, plasma adrenaline levels were signifi cantly elevated in males (F=7.34, P<0.024). Although adrenaline levels tended to be higher in stressed females, this trend did not reach statistical signifi cance. No gender differences were observed between control animals (Fig. 2a).
Plasma Noradrenaline levels.
Although noradrenaline levels in males remained unchanged, females demonstrated a sig-nifi cant rise (F=6.02, P<0.032) after repeated stress exposure. A gender-related difference was also evident under non-stress conditions, as females illustrated signifi cantly lower nor-adrenaline levels than males (F=7.22, P<0.023) (Fig. 2a).
Figure 1. Female
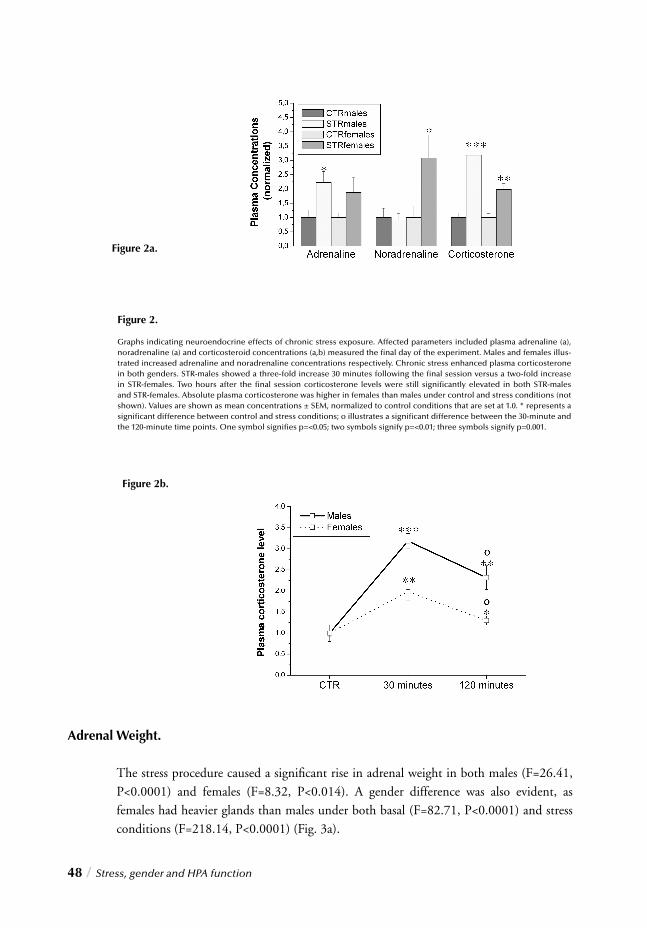
48 / Stress, gender and HPA function
Adrenal Weight.
The stress procedure caused a signifi cant rise in adrenal weight in both males (F=26.41, P<0.0001) and females (F=8.32, P<0.014). A gender difference was also evident, as females had heavier glands than males under both basal (F=82.71, P<0.0001) and stress conditions (F=218.14, P<0.0001) (Fig. 3a).
Figure 2.
Graphs indicating neuroendocrine effects of chronic stress exposure. Affected parameters included plasma adrenaline (a), noradrenaline (a) and corticosteroid concentrations (a,b) measured the fi nal day of the experiment. Males and females illus-trated increased adrenaline and noradrenaline concentrations respectively. Chronic stress enhanced plasma corticosterone in both genders. STR-males showed a three-fold increase 30 minutes following the fi nal session versus a two-fold increase in STR-females. Two hours after the fi nal session corticosterone levels were still signifi cantly elevated in both STR-males and STR-females. Absolute plasma corticosterone was higher in females than males under control and stress conditions (not shown). Values are shown as mean concentrations ± SEM, normalized to control conditions that are set at 1.0. * represents a signifi cant difference between control and stress conditions; o illustrates a signifi cant difference between the 30-minute and the 120-minute time points. One symbol signifi es p=<0.05; two symbols signify p=<0.01; three symbols signify p=0.001.
Figure 2b.
Figure 2a.
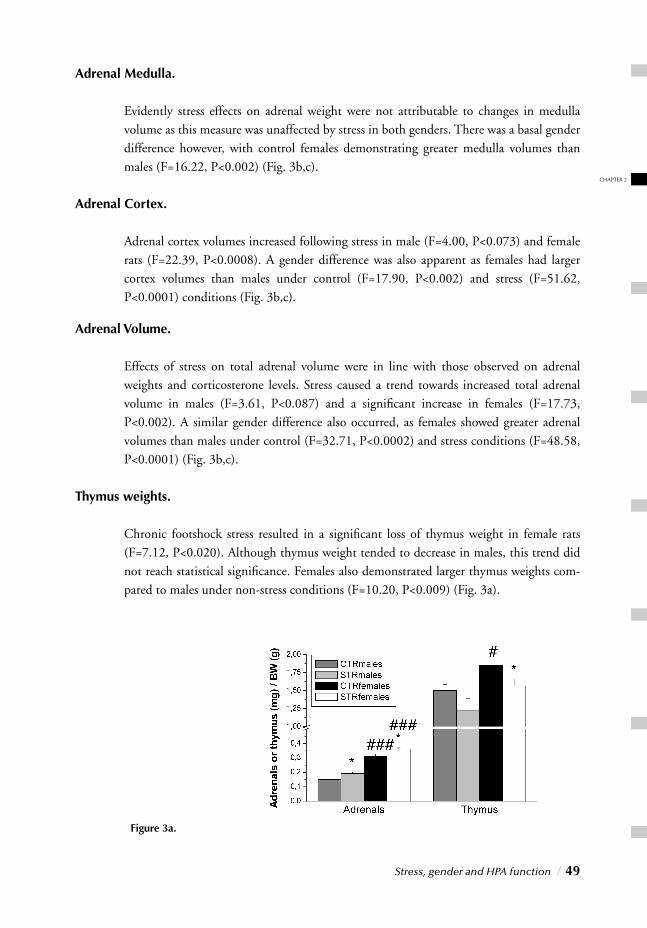
CHAPTER 2
Stress, gender and HPA function / 49
Adrenal Medulla.
Evidently stress effects on adrenal weight were not attributable to changes in medulla volume as this measure was unaffected by stress in both genders. There was a basal gender difference however, with control females demonstrating greater medulla volumes than males (F=16.22, P<0.002) (Fig. 3b,c).
Adrenal Cortex.
Adrenal cortex volumes increased following stress in male (F=4.00, P<0.073) and female rats (F=22.39, P<0.0008). A gender difference was also apparent as females had larger cortex volumes than males under control (F=17.90, P<0.002) and stress (F=51.62, P<0.0001) conditions (Fig. 3b,c).
Adrenal Volume.
Effects of stress on total adrenal volume were in line with those observed on adrenal weights and corticosterone levels. Stress caused a trend towards increased total adrenal volume in males (F=3.61, P<0.087) and a signifi cant increase in females (F=17.73, P<0.002). A similar gender difference also occurred, as females showed greater adrenal volumes than males under control (F=32.71, P<0.0002) and stress conditions (F=48.58, P<0.0001) (Fig. 3b,c).
Thymus weights.
Chronic footshock stress resulted in a signifi cant loss of thymus weight in female rats (F=7.12, P<0.020). Although thymus weight tended to decrease in males, this trend did not reach statistical signifi cance. Females also demonstrated larger thymus weights com-pared to males under non-stress conditions (F=10.20, P<0.009) (Fig. 3a).
Figure 3a.
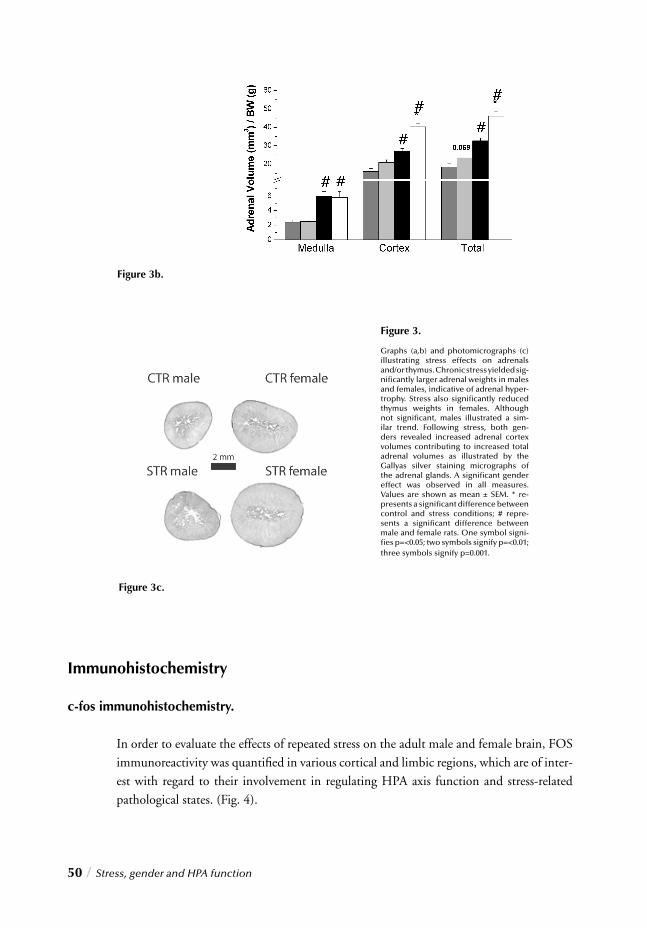
50 / Stress, gender and HPA function
Immunohistochemistry
c-fos immunohistochemistry.
In order to evaluate the effects of repeated stress on the adult male and female brain, FOS immunoreactivity was quantifi ed in various cortical and limbic regions, which are of inter-est with regard to their involvement in regulating HPA axis function and stress-related pathological states. (Fig. 4).
Figure 3.
Graphs (a,b) and photomicrographs (c) illustrating stress effects on adrenals and/or thymus. Chronic stress yielded sig-nifi cantly larger adrenal weights in males and females, indicative of adrenal hyper-trophy. Stress also signifi cantly reduced thymus weights in females. Although not signifi cant, males illustrated a sim-ilar trend. Following stress, both gen-ders revealed increased adrenal cortex volumes contributing to increased total adrenal volumes as illustrated by the Gallyas silver staining micrographs of the adrenal glands. A signifi cant gender effect was observed in all measures. Values are shown as mean ± SEM. * re-presents a signifi cant difference between control and stress conditions; # repre-sents a signifi cant difference between male and female rats. One symbol signi-fi es p=<0.05; two symbols signify p=<0.01; three symbols signify p=0.001.
Figure 3c.
Figure 3b.

CHAPTER 2
Stress, gender and HPA function / 51
In male rats, chronic stress resulted in signifi cantly decreased FOS-ir in the mPFC (F=5.17, p<0.046), the mORB (F=5.36, p<0.043), the AC (F=12.92, p<0.0049) (fi g. 5a), and the DG (F=5.17, p<0.046) (fi g. 5c). Only the PVN showed an opposite effect with signifi cantly increased FOS-ir (F=6.58, p<0.028) (fi g. 5c). Chronically stressed females in contrast, illustrated a selective increase of FOS-ir in the CeA (F=7.1, p<0.024), the LaA (F=8.25, p<0.017), the BslA (F=6.2, p<0.032) and the PVT (F=5.36, p<0.0431(fi g. 5b), while no changes were observed in any of the prefrontocortical regions examined (fi g. 5a). Like males however, chronic footshocks in females, also induced signifi cantly increased FOS-ir in PVN (F=15.78, p<0.0026) (fi g. 5c).
Figure 5b
Figure 5a.
Figure 4.
Figure 4. Photomicrographs illus-trating stress-induced c-fos immu-noreactivity in the PVN. Both gen-ders illustrated increased PVN activity, confi rming continual stress discernment without habituation in males and females.
52 / Stress, gender and HPA function
Phospho-CREB immunoreactivity.
In male rats, chronic stress exposure resulted in a general yet signifi cant reduction of phos-pho-CREB immunoreactivity in both cortical and subcortical regions (fi g. 6). Specifi cally, decreased CREB phosphorylation was observed in the mORB (F=36.06, p<<0.001), the PrL (F=16.59, p<0.002), the InfraL (F=38.18, p<<0.001) (fi g. 6a), the AC (F=10.59, p<0.009), the postCING (F=6.84, p<0.026) (fi g. 6b), the hippocampal DG (F=11.99, p<0.006), the LaA (F=24.68, p<<0.001), and the BslA (F=42.32, p<<0.001) (fi g. 6c). Sur-prisingly, no changes in CREB phosphorylation were evident in cyclic female rats with the exception of the hippocampal dentate gyrus where a marked, yet non-signifi cant, reduc-tion was observed (F=4.38, p<0.081) (fi g. 6c).
Figure 6a.
Figure 5.
Graphs illustrating gender-distinctive effects of stress on FOS-ir in structures of the frontal cortex (a), the amygdala (b) the hippocampus, thalamus, hypothalamus and the raphe (c). Values are shown as mean positive cells ± SEM, normalized to control animals that are set at 100. * represents a signifi cant difference between control and stress conditions. One symbol signifi es p=<0.05; two symbols signify p=<0.01; three symbols signify p=0.001.
Figure 5c.
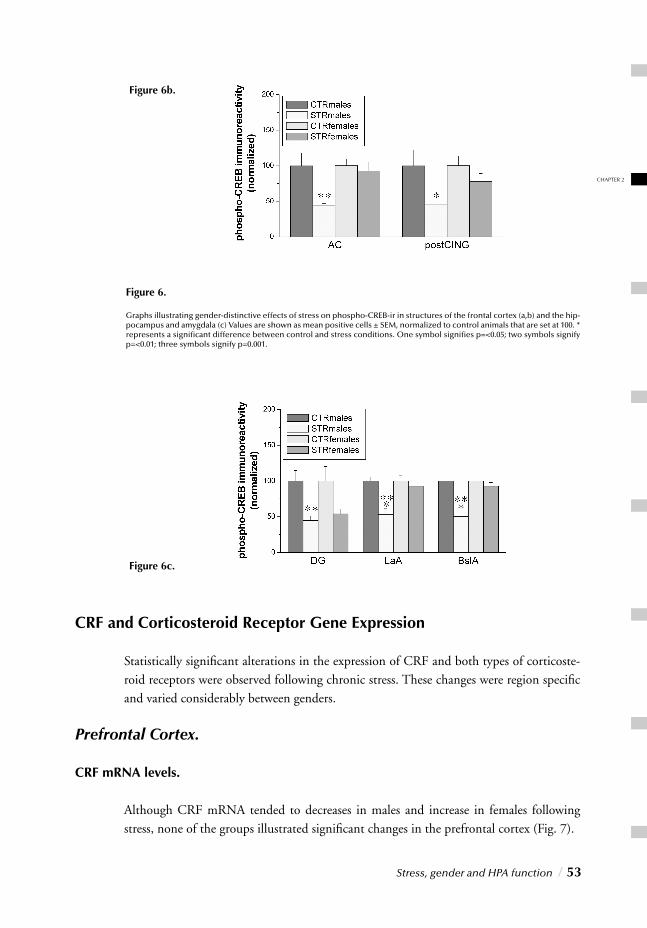
Stress, gender and HPA function / 53
CHAPTER 2
CRF and Corticosteroid Receptor Gene Expression
Statistically signifi cant alterations in the expression of CRF and both types of corticoste-
roid receptors were observed following chronic stress. These changes were region specifi c and varied considerably between genders.
Prefrontal Cortex.
CRF mRNA levels.
Although CRF mRNA tended to decreases in males and increase in females following stress, none of the groups illustrated signifi cant changes in the prefrontal cortex (Fig. 7).
Figure 6b.
Figure 6c.
Figure 6.
Graphs illustrating gender-distinctive effects of stress on phospho-CREB-ir in structures of the frontal cortex (a,b) and the hip-pocampus and amygdala (c) Values are shown as mean positive cells ± SEM, normalized to control animals that are set at 100. * represents a signifi cant difference between control and stress conditions. One symbol signifi es p=<0.05; two symbols signify p=<0.01; three symbols signify p=0.001.
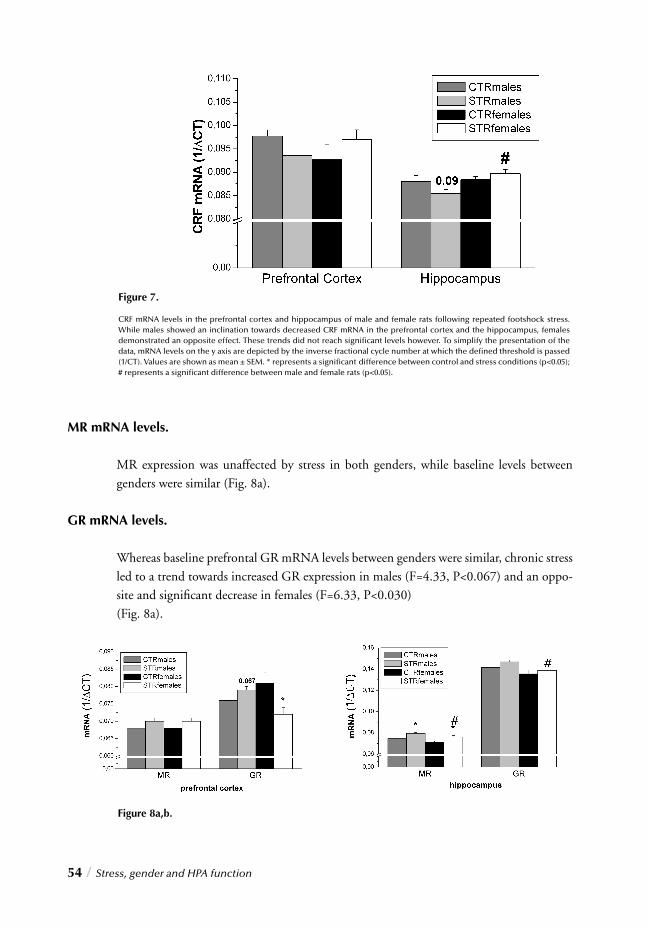
54 / Stress, gender and HPA function
MR mRNA levels.
MR expression was unaffected by stress in both genders, while baseline levels between genders were similar (Fig. 8a).
GR mRNA levels.
Whereas baseline prefrontal GR mRNA levels between genders were similar, chronic stress led to a trend towards increased GR expression in males (F=4.33, P<0.067) and an oppo-site and signifi cant decrease in females (F=6.33, P<0.030) (Fig. 8a).
Figure 7.
CRF mRNA levels in the prefrontal cortex and hippocampus of male and female rats following repeated footshock stress. While males showed an inclination towards decreased CRF mRNA in the prefrontal cortex and the hippocampus, females demonstrated an opposite effect. These trends did not reach signifi cant levels however. To simplify the presentation of the data, mRNA levels on the y axis are depicted by the inverse fractional cycle number at which the defi ned threshold is passed (1/CT). Values are shown as mean ± SEM. * represents a signifi cant difference between control and stress conditions (p<0.05); # represents a signifi cant difference between male and female rats (p<0.05).
Figure 8a,b.
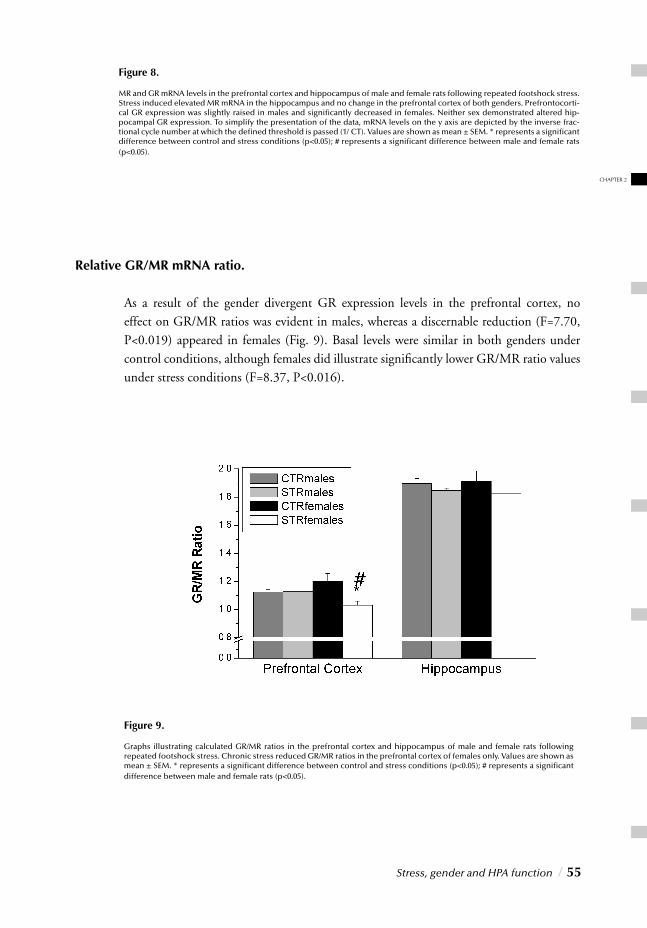
Stress, gender and HPA function / 55
CHAPTER 2
Relative GR/MR mRNA ratio.
As a result of the gender divergent GR expression levels in the prefrontal cortex, no effect on GR/MR ratios was evident in males, whereas a discernable reduction (F=7.70, P<0.019) appeared in females (Fig. 9). Basal levels were similar in both genders under control conditions, although females did illustrate signifi cantly lower GR/MR ratio values under stress conditions (F=8.37, P<0.016).
Figure 8.
MR and GR mRNA levels in the prefrontal cortex and hippocampus of male and female rats following repeated footshock stress. Stress induced elevated MR mRNA in the hippocampus and no change in the prefrontal cortex of both genders. Prefrontocorti-cal GR expression was slightly raised in males and signifi cantly decreased in females. Neither sex demonstrated altered hip-pocampal GR expression. To simplify the presentation of the data, mRNA levels on the y axis are depicted by the inverse frac-tional cycle number at which the defi ned threshold is passed (1/ CT). Values are shown as mean ± SEM. * represents a signifi cant difference between control and stress conditions (p<0.05); # represents a signifi cant difference between male and female rats (p<0.05).
Figure 9.
Graphs illustrating calculated GR/MR ratios in the prefrontal cortex and hippocampus of male and female rats following repeated footshock stress. Chronic stress reduced GR/MR ratios in the prefrontal cortex of females only. Values are shown as mean ± SEM. * represents a signifi cant difference between control and stress conditions (p<0.05); # represents a signifi cant difference between male and female rats (p<0.05).

Hippocampus
CRF mRNA levels.
Similar to effects seen in the prefrontal cortex, hippocampal CRF mRNA levels tended to be lower in males exposed to chronic stress, although this trend did not reach statis-tical signifi cance. CRF mRNA levels in females exposed to stress remained unchanged. Although basal levels were similar in both genders under control conditions, stressed females had signifi cantly higher CRF mRNA levels compared to stressed males (F=12.79, P<0.005) (Fig. 7).
MR mRNA levels.
Chronic stress induced signifi cantly increased MR mRNA levels in the hippocampus of males (F=12.36, P<0.005) as well as females (F=8.56, P<0.015). Under control condi-tions females expressed a trend towards lower levels than males (F=4.04, P<0.072) which reached signifi cance under stress conditions (F=5.02, P<=0.049) (Fig. 8b).
GR mRNA levels.
While stressed males illustrated a trend towards increased hippocampal GR mRNA levels (F=3.39, P<0.095), female levels remained unaffected by chronic stress. A basal gender difference was observed under stress conditions however as females illustrated signifi cantly lower GR mRNA levels than males (F=12.02, P<0.006) (Fig. 8b).
Relative GR/MR mRNA ratio.
The net effect of hippocampal MR and GR expression revealed unaltered GR/MR ratios in both genders. Basal levels were also similar (Fig. 9).
A summarized view of the stress effects and primary gender differences discussed here is presented in table 2.
56 / Stress, gender and HPA function

DISCUSSION
Effi cient adaptation to stressful events occurs on the psychological (emotional and cogni-tive), behavioral (fi ght and fl ight), and biological level (autonomic and neuroendocrine) and is essential for an organism’s well being. It requires complex cascades of events includ-ing neurotransmitter release 29,30, gene expression 31 and regulated phosphorylation states of specifi c proteins 32,33. This integrated process, dependent on genomic responses, is trig-gered by immediately early gene induction and transcriptional activity of nuclear hor-
Stress, gender and HPA function / 57
CHAPTER 2
Table 2.
Summarized view of sex and/or region specifi c stress effects. Symbols: Illustrate the effects of stress on male and female rats and the comparison of female versus male baseline levels. Trends toward increase (↑) decrease (↓); signifi cant increase (⇑) decrease (⇓) and no change (=) are indicated.

mone receptor ligands such as corticosterone. Following stress exposure, genomic effects initiate within minutes and culminate over hours into an integrated process that can produce long-term neuroendocrine, neurochemical, and behavioral alterations 34. Since activation of the stress system is meant to be brief, its time-limited nature renders its acute effects benefi cial rather than adverse 12. It has been speculated however, that some viduals may become more sensitive to subsequent stressors if initial stressors are too long or intense. Given that stress response systems coordinate a wide array of physiological responses, their prolonged activation could lead to the development of immune, neu-roendocrine, cardiovascular, metabolic, and psychiatric abnormalities 12,35. In this study, immunohistochemical and neuroendocrine adaptations to repeated stress were explored in order to unravel the neurobiological substrates underlying regulatory mechanisms and eventually, dysregulation of the stress response. Moreover, as gender differences have been reported in chronic stress effects in humans and animals, footshock-induced changes were investigated in male as well as female rats. The results could provide valuable insight into the neurobiological mechanisms underlying HPA axis regulation and the role of gender in the modulation of chronic stress adaptations. While repeated stress induced signs of pro-longed HPA axis hyperactivity in both males and females, interesting gender-differential cellular and molecular alterations suggest discrepant molecular and cellular events associ-ated with this common development.
Chronic stress-induced cellular and molecular alterations in male rats.
Stressed males showed marked signs of sustained HPA axis activation such as adrenal hypertrophy and elevated plasma adrenaline and corticosterone concentrations. Another indication of prolonged exposure to elevated peripheral and central glucocorticoid levels is provided by analysis of stress-induced immunohistochemical alterations. Expression and phosphorylation of CREB, a transcription factor involved in modulating neuronal plasti-city, is particularly sensitive to glucocorticoids and thus HPA axis activity. While brief stress has been associated with enhanced CREB phosphorylation 36, persistently elevated glucocorticoid levels have been related to reduced CREB expression and phosphoryla-tion and impaired cognitive functions 37,38. Reduced phospho-CREB immunoreactivity, observed here in stressed males, thus seems to substantiate the hypothesis of persistently elevated glucocorticoid concentrations, associated possibly with abnormal HPA function.
Stress-induced changes in cortical-limbic FOS-ir patterns further support this notion. Expression of c-fos, an immediate early gene, is infl uenced by altered neuronal activity and its gene product therefore provides a useful tool to mark neuronal activity 39,40. Although no changes were found in the amygdala, thalamus, and dorsal raphe, chronic stress-induced FOS-ir was signifi cantly increased in the PVN and decreased in the medial orbi-tofrontal cortex, the medial prefrontal cortex, the anterior cingulate, and the hippocampal dentate gyrus. The HPA axis is generally considered to function as a closed-loop auto-regulatory system modulated by glucocorticoid-mediated negative feedback and opera-
58 / Stress, gender and HPA function

ting over multiple time domains, at different levels and by several sources 3,12,41. Since glu-cocorticoids represent the fundamental hormonal mediators in this stress response their regulation and release is carefully regulated by coordinated interactions of this cortical and limbic circuitry. The PVN for instance plays a pivotal role in regulating HPA axis acti-vity and its increased FOS-ir substantiates a lack of habituation to this footshock proce-dure. The prefrontal cortex and the hippocampus however, modulate HPA axis activity by maintaining this system under tonic inhibition and lesions in these regions result, at least in male rats, in pronounced activation of the HPA axis 12,18. Remarkably, literature also reports chronic stress-induced prefrontocortical and hippocampal defi cits, which could ultimately compromise the ability of these regions to properly modulate HPA axis acti-vity 42,43. Although the amygdala and raphe have also been reportedly involved in stress response modulation, specifi c FOS-ir changes seen here suggest that footshock-induced HPA axis hyperactivity in males may have resulted from prefrontocortical and hippocam-pal abnormalities and subsequently impaired inhibitory feedback exerted by these struc-tures. A hypothesized mechanism by which stress could lead to the attenuation of prefron-tocortical and hippocampal negative feedback involves abnormal expression of mineralo-corticoid and glucocorticoid receptors, localized in discrete cortical-limbic regions, such as prefrontal cortex, hippocampus and hypothalamus 4. Although reports have illustrated the ability of chronic footshocks to disrupt GR-mediated functions in the prefrontal cortex and hippocampus 43, our results do not support the latter, at least not in males. Not only did we fail to detect reduced mRNA of these receptors we actually found a marked stress-induced rise of GR or MR mRNA in both mPFC and hippocampus, suggesting that HPA axis abnormalities did not result from any deleterious stress effects on the gene expression of these receptors. Although this discrepancy in results may be attributable to differences in experimental design, the mechanism involved in the attenuation of inhibitory feedback exerted by the prefrontal cortex and hippocampus remains unknown. An intriguing pos-sibility to explain this abnormal interaction between higher cortical-limbic structures and the hypothalamus might be offered by the transcription factor c-fos and its role in regu-lating glucocorticoid-induced genomic responses. Both MR and GR, are corticosteroid-activated transcription factors that modulate the expression of a variety of neuronal genes by binding to specifi c DNA sequences 44. This binding however, can be hindered by some nuclear factors such as c-fos and c-jun 45,46. Interestingly, acute stress has been shown to enhance c-fos expression in selective cortical-limbic regions 47 and it has been speculated
that following transient stressful events, such increased c-fos expression can dampen the negative feedback response and enhances HPA axis activity by interfering with glucocor-ticoid receptor activity. In contrast, reduced c-fos expression may promote glucocorticoid receptor binding, thereby promoting the negative feedback response and preventing the deleterious consequences of persistently elevated corticosteroid levels 48. It is thus intrigu-ing to speculate that reduced FOS-ir observed here in prefrontocortical and hippocampal areas may represent a neuronal adaptive mechanism specifi c for repeated adverse expe-riences. This view is also supported by signifi cantly increased MR mRNA in the hip-
Stress, gender and HPA function / 59
CHAPTER 2

the signifi cantly elevated corticosterone concentrations detected here more than 2 hours after the fi nal session. Plasma glucocorticoids have been shown to increase upon exposure to acute stress, reach a peak after 30 minutes and then progressively decrease to basal value in the next 60 minutes. Mizoguchi and colleagues however, showed that prolonged foot-shocks can change the dynamic of this physiological response by lowering the maximal peak of plasma glucocorticoid concentrations and reducing the time needed by cortico-steroids to return to basal physiological levels 42. The latter indication of reduced neuro-nal plasticity in the hippocampus and prefrontal cortex suggests that the decreased c-fos expression may also represent a condition of functional hypoactivity of these regions, rather than a mere simple adaptation to repeated footshocks. Although increased MR and GR mRNA expression following chronic stress may facilitate the rapid reduction of plasma glucocorticoids after exposure to adverse conditions (hypersuppression), it is also possible that MR/GR feedback inhibition is insuffi cient to return corticosterone to basal levels in the absence of other specifi c feedback mechanisms. Chronic stress-induced pre-frontocortical and hippocampal defects may impair these additional inhibitory actions and compromise negative feedback on HPA function, resulting in enhanced basal activity of this system and permanently elevated basal glucocorticoid levels.
Chronic stress-induced cellular and molecular changes in cyclic female rats.
Similarly to males, chronically stressed females also showed multiple neuroendocrine indi-cations of sustained HPA axis hyperactivity, such as elevated plasma norepinephrine and corticosterone concentrations, increased PVN FOS-ir, adrenal hypertrophy, and thymus hypotrophy. Particularly elevated plasma corticosterone (30 and more importantly, 120 minutes after the fi nal session) enlarged adrenal glands and reduced thymuses seem to sug-gest prolonged HPA axis activation leading to persistent glucocorticoid elevations. Despite similar neuroendocrine alterations, stress induced signifi cant gender-related differences in phospho-CREB and FOS-ir patterns. Unlike males for instance, female rats did not show marked stress-reduced phospho-CREB immunoreactivity. Dendritic atrophy in the hip-pocampus and prefrontal cortex reported after chronic stress is believed to be mediated by glucocorticoid effects on neuronal plasticity, by inhibiting CREB phosphorylation and BDNF expression 26,49,50. This gender discrepancy however might be attributed to ovarian steroids in cyclic female rats, especially since stress has been shown to permanently elevate estrogen concentrations in female rats 51. Unfortunately, here peripheral estrogen levels were not measured, although females did illustrate interesting cyclic variations in body weight gain refl ecting probable periodic hormonal fl uctuations. Whereas cyclic variations occurred in both stressed and non-stressed females during acclimatization, they continued in the non-stressed after initiation of the stressor and disappeared in the stressed, reappear-ing only at the end of the experiment. If the latter refl ects the infl uence of stress on ovarian function, it is possible that stress-induced elevated estrogen levels reduced the extent of their fl uctuation. More importantly however, estrogen exerts trophic actions in the brain.
60 / Stress, gender and HPA function

Since this requires ovarian hormone activation of specifi c intracellular cascades leading to CREB phosphorylation, stress-induced elevated estrogen may thus promote CREB phos-phorylation and partially counteract glucocorticoid-mediated inhibitory effects. In addition to phospho-CREB, gender-differential immunoreactivity patterns were also seen in c-fos. Despite marked neuroendocrine changes indicating prolonged HPA axis hyperactivity, females did not reveal FOS-ir patterns illustrating reduced prefrontocorti-cal and hippocampal functions. Impaired prefrontocortical and hippocampal inhibitory feedback to the hypothalamus may thus explain HPA axis hyperactivity in stressed male but not female rats. Remarkably however, stressed females revealed a selective induction of FOS-ir in the amygdala, a dimorphic limbic structure involved in modulating stress and emotional responses 52,53. Increased amygdala metabolism has been found in various psychiatric conditions, like depression and anxiety and its implication in psychopathol-ogy may be related to its role in regulating HPA axis activity 54,55. The amygdala, provides excitatory input to the PVN and facilitates activation of the HPA axis, so that stimuli, interpreted as threatening can elicit an immediate response characterized by enhanced vigilance, autonomic arousal, and a global catabolic state. Mobilization of these responses however requires activation of corticotropin-releasing hormone (CRH) neurons in se-veral structures, including the hypothalamus and amygdala, and stimulation of the brain stem locus coeruleus/norepinephrine system. These systems perpetuate one another, inter-act with several other neurotransmitter systems and directly activate the HPA axis and autonomic nervous system 3. Interestingly in contrast to males, stressed females showed increased CRH mRNA expression in the prefrontal cortex and hippocampus (signifi cantly higher in the latter compared to males). Although CRH mRNA was not measured in other regions involved in modulating this system (i.e. amygdala and locus coeruleus), its increased expression in the prefrontal cortex and hippocampus may represent an indi-rect indication of enhanced CRH activity in chronically stressed females. Furthermore repeated stress signifi cantly elevated plasma noradrenaline in females and its circulating levels provide a useful estimate of neurosympathetic activity. Together these fi ndings may thus suggest enhanced HPA function in response to increased CRH mRNA and auto-nomic nervous system activation. Another factor that may contribute to abnormal HPA axis activity is a disrupted MR/GR mRNA ratio. Shifts in the balance of MR/GR-mediated action in limbic struc-tures alters the set point of stress response system activity. Once this imbalance occurs, an individual loses its ability to reestablish proper internal homeostasis when challenged by adverse events. This conditions then leads to the disruption of neuroendocrine regulation and impairs behavioral adaptation, which may enhance vulnerability to subsequent insults and trigger the onset of stress-related disorders 56. Interestingly, abnormal MR/GR mRNA was observed in the hippocampus of stressed females. Combined with the previous fi n-dings, this observation suggests that abnormal MR/GR mRNA expression, together with factors such as abnormal CRH mRNA and sympathetic activity, may contribute to the development of abnormal HPA axis regulation in female rats.
Stress, gender and HPA function / 61
CHAPTER 2

Clinical implications and conclusions.
The results presented here may provide insight into the cellular and molecular mech-nisms governing males’ and females’ stress susceptibility. Impaired HPA axis regulation due to abnormal limbic function has been frequently reported in depressed subjects. More importantly, reduced prefrontocortical function, hippocampal atrophy, increased amyg-dala metabolism, and HPA axis hyperactivity represent common occurrences in depres-sion and other stress-related psychiatric disorders, while imbalanced GR/MR expression has been reported in specifi c subgroups of depressed subjects 57-59. Given the association between disrupted limbic system GR/MR ratios and HPA abnormalities often seen in stress-related disorders as depression 60-62 the importance of balanced MR and GR expres-sion for functional brain and body homeostasis has recently been re-evaluated 43,59. Inter-estingly, depression is characterized by marked gender-related prevalence with women having a greater risk of developing psychopathology. Despite ample evidence, reservations regarding gender discrepancies and stress-related pathology remain however and may be attributed to the fact that most of our current knowledge on HPA function regulation is derived from research in males 63. This issue underscores the need for future research with particular attention on the gender aspect, to better understand its role in HPA axis modulation. In doing so however some points of consideration regarding the limitations of this study should be addressed. A practical limitation discerns the fact that use was made of commercially obtained rats. As we lack information on early life conditions such as housing or sibling bonds, we cannot address the well reported infl uence of prenatal conditions on sexually dimorphic effects of stress responses in adult life 64. Another con-cern is posed by the cyclic fl uctuations of ovarian hormones in females and the role of androgens in males. As stress responses have been shown to be infl uenced by sex steroids 65 we are currently investigating this aspect in a staged design. In conclusion, these fi ndings collectively demonstrate the ability of repeated stress to disrupt HPA axis regulation in a gender- and/or region-specifi c manner. The observed dif-ferences reveal the complex modulation of this stress response system. In this regard our fi ndings suggest sexually dimorphic mechanisms underlying the regulation/dysregulation of HPA axis response to stress, which encompasses glucocorticoid, catecholamine, and GR/MR interplay in distinct cortical and subcortical structures. Impaired prefrontocor-tical and/or hippocampal control over the HPA axis, possibly caused by stress-induced reduction of neuronal plasticity seems to represent a possible mechanism by which repeated stress leads to abnormal regulation of the stress response in male rats, whereas imbalance GR/MR mRNA expression, disturbed autonomic function, and abnormal amygdala FOS activity could refl ect events involved in the development HPA axis abnor-malities in female rats. Together, these fi ndings may provide valuable neuroendocrine and molecular insight to explain discrepant gender-related susceptibility often encountered in stress-induced psychiatric disorders.
62 / Stress, gender and HPA function

REFERENCES1. Mayberg, H.S. Limbic-cortical dysregulation: a proposed model of depression. J. Neuropsychiatry Clin. Neurosci. 9, 471-481 (1997).
2. Leonard, B. Stress, depression and the activation of the immune system. World J. Biol. Psychiatry 1, 17-25 (2000).
3. Tsigos, C. & Chrousos, G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 53, 865-871 (2002).
4. de Kloet, R.E. Hormones, brain and stress. Endocr. Regul. 37, 51-68 (2003).
5. Kendler, K.S., Karkowski, L.M. & Prescott, C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 156, 837-841 (1999).
6. Kessler, R.C., McGonagle, K.A., Swartz, M., Blazer, D.G. & Nelson, C.B. Sex and depression in the National Comorbidity Survey. I: Life time prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85-96 (1993).
7. Jorm, A.F. Sex and age differences in depression: a quantitative synthesis of published research. Aust. N. Z. J. Psychiatry 21, 46-53 (1987).
8. Breslau, N., Davis, G.C., Peterson, E.L. & Schultz, L. Psychiatric sequelae of posttraumatic stress disorder in women. Arch. Gen. Psychiatry 54, 81-87 (1997).
9. Desai, H.D. & Jann, M.W. Major depression in women: a review of the literature. J. Am. Pharm. Assoc. (Wash.) 40, 525-537 (2000).
10. Bachmann, C.G., Linthorst, A.C., Holsboer, F. & Reul, J.M. Effect of chronic administration of selective glucocorticoid receptor antagonists on the rat hypothalamic-pituitary-adreno cortical axis. Neuropsychopharmacology 28, 1056-1067 (2003).
11. Veenema, A.H., Meijer, O.C., de Kloet, E.R. & Koolhaas, J.M. Genetic selection for coping style predicts stressor susceptibility. J. Neuroendocrinol. 15, 256-267 (2003).
12. McEwen, B.S. Effects of adverse experiences for brain structure and function. Biol. Psychiatry 48, 721-731 (2000).
13. Torpy, D.J., Papanicolaou, D.A. & Chrousos, G.P. Sexual dimorphism of the human stress response may be due to estradiol-mediated stimulation of hypothalamic corticotro pin-releasing hormone synthesis. J. Clin. Endocrinol. Metab 82, 982 (1997).
14. Young, E.A. Sex differences and the HPA axis: implications for psychiatric disease. J. Gend. Specif. Med. 1, 21-27 (1998).
15. Rhodes, M.E. & Rubin, R.T. Functional sex differences (‘sexual diergism’) of central nervous system cholinergic systems, vaso pressin, and hypothalamic-pituitary-adrenal axis activity in mammals: a selective review. Brain Res. Brain Res. Rev. 30, 135-152 (1999).
16. Herman, J.P. & Cullinan, W.E. Neurocircuitry of stress: central control of the hypothalamo- pituitary-adrenocortical axis. Trends Neurosci. 20, 78-84 (1997).
17. Lopez, J.F., Akil, H. & Watson, S.J. Neural circuits mediating stress. Biol. Psychiatry 46, 1461-1471 (1999).
18. Sullivan, R.M. & Gratton, A. Prefrontal cortical regulation of hypothalamic-pituitary-adrenal function in the rat and implications for psychopathology: side matters. Psychoneuroendocrinology 27, 99-114 (2002).
19. de Kloet, E.R., Vreugdenhil, E., Oitzl, M.S. & Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 19, 269-301 (1998).
20. Smedes, F., Kraak, J.C. & Poppe, H. Simple and fast solvent extraction system for selective and quantitative isolation of adrenaline, noradrenaline and dopa mine from plasma and urine. J. Chromatogr. 231, 25-39 (1982).
21. Remie, R., Knot, H.J., Bos, E.A. & Zaagsma, J. Characterization of presynaptic beta-adrenoceptors facilitating endogenous noradrenaline release in the portal vein of perma nently cannulated, freely moving rats. Eur. J. Pharmacol. 157, 37-43 (1988).
22. Swanson LW. Brain maps: structure of the rat brain. 1998. Amsterdam, Elsevier.
23. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphos phorylation in medial prefrontocortical dendrites: implications for stress- related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
24. Ter Horst, G.J. et al. Silver staining of traumatized neurons: application of a Gallyas procedure in experimental cerebral hypoxia / ischaemia research. Neuroscience Protocols 95-050-02, 1-13. 1995. 25. Heid, C.A., Stevens, J., Livak, K.J. & Williams, P.M. Real time quantitative PCR. Genome Res. 6, 986-994 (1996).
26. Kuipers, S.D., Trentani, A., Den Boer, J.A. & Ter Horst, G.J. Molecular correlates of impaired prefrontal plasticity in response to chronic stress. J. Neurochem. 85, 1312-1323 (2003).
27. Duncko, R., Kiss, A., Skultetyova, I., Rusnak, M. & Jezova, D. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 26, 77-89 (2001).
28. Westenbroek, C., Den Boer, J.A. & Ter Horst, G.J. Gender-specifi c effects of social housing on chronic stress- induced limbic Fos expression. Neuroscience 121, 189-199 (2003).
29. Kalen, P., Lindvall, O. & Bjorklund, A. Electrical stimulation of the lateral habenula increases hippo campal noradrenaline release as monitored by in vivo microdialysis. Exp. Brain Res. 76, 239-245 (1989).
30. Kalen, P., Strecker, R.E., Rosengren, E. & Bjorklund, A. Regulation of striatal serotonin release by the lateral habenula- dorsal raphe pathway in the rat as demonstrated by in vivo microdialysis: role of excitatory amino acids and GABA. Brain Res. 492, 187-202 (1989).
31. Stamp, J. & Herbert, J. Corticosterone modulates autonomic responses and adaptation of central immediate-early gene expression to repeated restraint stress. Neuroscience 107, 465-479 (2001).
32. Mayr, B. & Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599-609 (2001).
33. Sweatt, J.D. The neuronal MAP kinase cascade: a biochemical signal integra tion system subserving synaptic plasticity and memory. J. Neurochem. 76, 1-10 (2001).
34. Hajos-Korcsok, E. et al. Rapid habituation of hippocampal serotonin and norepineph rine release and anxiety-related behaviors, but not plasma corti- costerone levels, to repeated footshock stress in rats.
Stress, gender and HPA function / 63
CHAPTER 2

Pharmacol. Biochem. Behav. 74, 609-616 (2003).
35. Eriksen, H.R., Olff, M., Murison, R. & Ursin, H. The time dimension in stress responses: relevance for survival and health. Psychiatry Res. 85, 39-50 (1999).
36. Bilang-Bleuel, A., Rech, J., De Carli, S., Holsboer, F. & Reul, J.M. Forced swimming evokes a biphasic response in CREB phos phorylation in extrahypothalamic limbic and neocortical brain structures in the rat. Eur. J. Neurosci. 15, 1048-1060 (2002).
37. McEwen, B.S. & Sapolsky, R.M. Stress and cognitive function. Curr. Opin. Neurobiol. 5, 205-216 (1995).
38. Focking, M., Holker, I. & Trapp, T. Chronic glucocorticoid receptor activation impairs CREB tran- scriptional activity in clonal neurons. Biochem. Biophys. Res. Commun. 304, 720-723 (2003).
39. Kovacs, K.J. c-Fos as a transcription factor: a stressful (re)view from a func- tional map. Neurochem. Int. 33, 287-297 (1998).
40. Dube, T., Brunson, T., Nehlig, A. & Baram, T.Z. Activation of specifi c neuronal circuits by corticotropin releasing hormone as indicated by c-fos expression and glucose metabolism. J. Cereb. Blood Flow Metab 20, 1414-1424 (2000).
41. Plotsky, P.M., Owens, M.J. & Nemeroff, C.B. Psychoneuroendocrinology of depression. Hypothalamic- pituitary-adrenal axis. Psychiatr. Clin. North Am. 21, 293-307 (1998).
42. Mizoguchi, K. et al. Chronic stress differentially regulates glucocorticoid negative feedback response in rats. Psychoneuroendocrinology 26, 443-459 (2001).
43. Mizoguchi, K., Ishige, A., Aburada, M. & Tabira, T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience 119, 887-897 (2003).
44. Datson, N.A., van der, P.J., de Kloet, E.R. & Vreugdenhil, E. Identifi cation of corticosteroid-responsive genes in rat hippo- campus using serial analysis of gene expression. Eur. J. Neurosci. 14, 675-689 (2001).
45. Schule, R. et al. Functional antagonism between oncoprotein c-Jun and the glu- cocorticoid receptor. Cell 62, 1217-1226 (1990).
46. Yang-Yen, H.F. et al. Transcriptional interference between c-Jun and the glucocorti coid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 62, 1205-1215 (1990).
47. Trentani, A. et al. Immunohistochemical changes induced by repeated footshock stress: revelations of gender-based differences. Neurobiol. Dis. 14, 602-618 (2003).
48. Senba, E. & Ueyama, T. Stress-induced expression of immediate early genes in the brain and peripheral organs of the rat. Neurosci. Res. 29, 183-207 (1997).
49. Duman, R.S., Heninger, G.R. & Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597-606 (1997).
50. Nestler, E.J. et al. Neurobiology of depression. Neuron 34, 13-25 (2002).
51. Shors, T.J., Pickett, J., Wood, G. & Paczynski, M. Acute stress persistently enhances estrogen levels in the female rat. Stress. 3, 163-171 (1999).
52. Cahill, L. et al. Sex-related difference in amygdala activity during emotionally infl uenced memory storage. Neurobiol. Learn. Mem. 75, 1-9 (2001).
53. Zald, D.H. The human amygdala and the emotional evaluation of sensory stimuli. Brain Res. Brain Res. Rev. 41, 88-123 (2003).
54. Tebartz, V.E., Woermann, F., Lemieux, L. & Trimble, M.R. Increased amygdala volumes in female and depressed humans. A quantitative magnetic resonance imaging study. Neurosci. Lett. 281, 103-106 (2000).
55. Frodl, T. et al. Enlargement of the amygdala in patients with a fi rst episode of major depression. Biol. Psychiatry 51, 708-714 (2002).
56. de Kloet, E.R. Stress in the brain. Eur. J. Pharmacol. 405, 187-198 (2000).
57. Checkley, S. The neuroendocrinology of depression and chronic stress. Br. Med. Bull. 52, 597-617 (1996).
58. Parker, K.J., Schatzberg, A.F. & Lyons, D.M. Neuroendocrine aspects of hypercortisolism in major depres- sion. Horm. Behav. 43, 60-66 (2003).
59. Young, E.A., Lopez, J.F., Murphy-Weinberg, V., Watson, S.J. & Akil, H. Mineralocorticoid receptor function in major depression. Arch. Gen. Psychiatry 60, 24-28 (2003).
60. Yau, J.L., Olsson, T., Morris, R.G., Meaney, M.J. & Seckl, J.R. Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: relationship with spatial learning in young and aged rats. Neuroscience 66, 571-581 (1995).
61. de Kloet, E.R., Oitzl, M.S. & Joels, M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 22, 422-426 (1999).
62. Liberzon, I., Lopez, J.F., Flagel, S.B., Vazquez, D.M. & Young, E.A. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: relevance to post-traumatic stress dis order. J. Neuroendocrinol. 11, 11-17 (1999).
63. Palanza, P. Animal models of anxiety and depression: how are females dif- ferent? Neurosci. Biobehav. Rev. 25, 219-233 (2001).
64. Weinberg, J. Prenatal ethanol effects: sex differences in offspring stress responsiveness. Alcohol 9, 219-223 (1992).
65. Atkinson, H.C. & Waddell, B.J. Circadian variation in basal plasma corticosterone and adreno- corticotropin in the rat: sexual dimorphism and changes across the estrous cycle. Endocrinology 138, 3842-3848 (1997).
64 / Stress, gender and HPA function

Stress, gender and HPA function / 65
CHAPTER 2


PART II
CELLULAR LEVEL

3

3
MOLECULAR CORRELATES OF IMPAIRED PREFRONTAL PLASTICITY IN RESPONSE TO CHRONIC STRESS
Disturbed adaptations at the molecular and cellular levels fol-
lowing stress could represent compromised neural plasticity that
contributes to the pathophysiology of stress-induced disorders.
Evidence illustrates atrophy and cell death of stress-vulnerable
neurons in the prefrontal cortex. Reduced plasticity may be
realized through the destabilized function of selective proteins
involved in organizing the neuronal skeleton and translating
neurotrophic signals. To elucidate the mechanisms underlying
these effects, rats were exposed to chronic footshock stress. Pat-
terns of c-fos, phospho-extracellular-regulated protein kinases 1/2
(ERK1/2), calcineurin and phospho-cyclic-AMP response-element
binding protein (CREB) expression were subsequently investi-
gated. The results indicate chronic stress-induced impairments in
prefrontal and cingulate signal transduction cascades underlying
neuronal plasticity. The medial prefrontal cortex, demonstrated
functional hyperactivity and dendritic phospho-ERK1/2 hyper-
phosphorylation, while reduced c-fos and calcineurin immuno-
reactivity occurred in the cingulate cortex. Signifi cantly reduced
phospho-CREB expression in both cortical regions, considering
its implication in brain-derived neurotrophic factor (BDNF) tran-
scription, suggests reduced synaptic plasticity. This data confi rms
the damaging effect of stress on cortical activity, on a molecular
level. Due to the association of these markers in the regulation of
BDNF signaling, these fi ndings suggest a central role for intracel-
lular neurotrophin transduction members in the pathways under-
lying cellular actions of stress in the brain.
Kuipers S.D., Trentani A., Den Boer J.A., Ter Horst G.J.
Journal of Neurochemistry, 2003, 85 : 1312-1323


Stress-induced cortial defects / 71
INTRODUCTION
An emerging hypothesis suggests that the pathogenesis and treatment of stress-related dis-orders is likely to involve disturbed neuronal pathways. Depression is one of the most common and perhaps most studied stress-related psychiatric disorders, and many believe that it could result from an inability to make the appropriate adaptive response to stress or other aversive stimuli. Numerous clinical fi ndings substantiate such a link between stress and loss of neuronal plasticity in the development of psychopathology 1-3. It has also been reported that depressed subjects exhibit dysfunctions in the cortical-limbic system, the neural network of highly interconnected forebrain structures responsible for the modula-tion of mood, emotions and the stress response 4. Cortical neuronal and glial pathology 5,6, hippocampal atrophy 7,8, and hypothalamic-pituitary-adrenal axis hyperactivity 9 are exemplary features often associated with this disorder. Despite much investigation how-ever, the neurobiological mechanisms underlying these cortical-limbic defects, and conse-quently the ontogeny of depression, remain greatly allusive. Several of the mechanisms which could mediate stress-induced deleterious effects involve hyperactivity of the hypothalamic-pituitary-adrenocortical (HPA) axis, as long-term exposure to elevated glucocorticoid levels has been associated with altered neu-rotransmitter function 3,10 and reduced synaptic plasticity 1,11,12. Prolonged stress exposure may affect brain plasticity by destabilizing the function of selective proteins involved in the organization and maintenance of the neuronal cytoskeleton and the translation of neurotrophic signals 13. Accordingly, several theories associating neurotrophin abnormali-ties to depression have recently been proposed 14,15. The transduction of neurotrophic signals requires the coordinated interaction of several protein kinases, phosphatases and transcription factors in the cascade, including extracellular signal-regulated kinase (ERKs), calcineurin and cyclic AMP (cAMP) response element binding protein (CREB) 16,17. Stress however has been shown to affect this coordination by altering the expression and func-tion of these enzymes 13. Consistently, clinical results seem to confi rm preliminary obser-vations from experimental models of depression regarding the relevance of neurotrophins in depression 15. Decreased plasma brain derived neurotrophic factor (BDNF) levels for instance have also been reported in depressed individuals 18. This data thus seems to sup-port the association between defects in the neurotrophin signaling transduction pathway
and stress-related neuropsychiatric disorders. In an attempt to elucidate the neurobiological substrates mediating the deleterious effects of prolonged stress, adult male rats were exposed to chronic footshock stress. This 3-week design offers a closer resemblance to the chronicity of stressful life events often reported by patients to precede the onset of a depressive episode. Subsequent patterns of frontocortical FOS-ir (molecular marker of neuronal activation) 19-21, phospho-ERK1/2, calcineurin and phospho-CREB (markers of neuronal plasticity and survival) 16,22 were then immunohistochemically characterized. The results indicate chronic stress-induced
CHAPTER 3

72 / Stress-induced cortial defects
defects in the prefrontal and cingulate cortices at the molecular and cellular levels. The present article provides preclinical data to support the deleterious effects of chronic stress on cortical activity. The results confi rm morphological and functional abnormalities associated with long-term stress exposure and suggest a central role for neurotrophin intracellular signaling members in the pathway underlying the deleterious effects of stress on brain function.
MATERIALS AND METHODS
Animals
The experiments were performed on male Wistar rats (n=18 weighing 212-240 g at the beginning of the experiment). Eighteen rats were used in this experiment. Twelve rats were stressed (6 chronic and 6 acute), while 6 served as controls. The animals were individually housed (cages 45 x 28 x 20 cm) with food and water available ad libitum while maintained on a 12 h light/dark cycle at 20°C. All rats were weighed (09:00) and handled daily for 5-8 min to minimize the effects of non-specifi c stress during the actual experiment. The experiments were performed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and the guidelines of the Animal Bio-ethics Committee of the University of Groningen (FDC: 2509).
Chronic stress procedure
The rodent test-chamber consists of a box containing an animal space positioned on a metallic grid fl oor connected to a shock generator and scrambler. A light, on the inner chamber wall, was used for conditioning. The set-up allows for a treatment of 12 rats per session. Test rats were subjected to a daily footshock protocol for 21 days (chronic experi-ment) or 3 days (acute experiment), receiving one session per day during which 5 inesca-pable footshocks were applied (0.8 mA in intensity and 8 sec in duration: unconditioned stimulus; US). During the daily sessions, starting time, shock order, inter-shock interval and total time spent in the box were randomized in order to make the procedure as unpre-dictable as possible. Each footshock was preceded by a 10 sec light pulse and a 5 sec rest in order to condition the rats to this signal (conditioned stimulus; CS). Control rats fol-
lowed a similar schedule in an identical setup but were exposed to the CS only, without receiving any shocks. On the fi nal day of the experiment (21st for the chronic, 3rd for the acute) all rats received a session of 5 CSs without USs. The coupling of CSs to USs was fundamental on the fi nal day of the experiment as it allowed the investigation of protein expression patterns induced by the conditioned stress experienced by the rats rather than footshock-related physical pain, as the latter can activate closely related neurocircuits 23.

Stress-induced cortial defects / 73
Physiological and neuroendocrine correlates of the chronic stress response
To defi ne the dynamics of the chronic stress response, various physiological and neuroen-docrine parameters were measured. Weight gain was monitored on a daily basis through-out the experiment, and upon termination, adrenals were removed and weighed. Graphs were constructed in which body and adrenal weights were recorded and calculated to serve as a reference to verify the severity of stress perceived by the animals. In addition, upon termination blood samples were taken and stored at -20°C after the last sessions to deter-mine plasma corticosterone and adrenaline levels with HPLC.
Extraction and Chromatography
Catecholamines.
Noradrenaline and adrenaline were extracted from plasma using liquid/liquid extraction with 3,4-dihydroxybenzylamine as internal standard 24,25. Briefl y, plasma catecholamines were bound to diphenylborate-ethanolamine at pH 8.6. Extraction was performed with n-heptane (containing 1% octanol and 25 % tetraoctylammoniumbromide). Next, cat-echolamines were extracted from the organic phase with diluted acetic acid.Catecholamines (20 µl acetic acid extract) were analyzed using an HPLC/auto-injector (CMA, Sweden) and a Shimadzu LC-10AD pump (Kyoto, Japan) connected to a reversed phase column (Hypersil, C18, 3 µm, 150x2.0 mm), followed by an electrochemical detector (Antec Leyden, The Netherlands) working at a potential setting of 500 mV vs. Ag/AgCl reference. The mobile phase consisted of 50 mM acetate buffer, 150 mg/l octane sulphonic acid, 150 mg/l tetramethylammonium, 15 mg/ml Na
2EDTA and 3% metha-
nol, adjusted to pH 4.1. The fl ow-rate was 0.35 ml/min. Temperature was 30°C. The detection limit was 0.1 nM.
Corticosterone.
For this assay, dexamethasone was used as internal standard. After addition of the internal standard, plasma was extracted with 3 ml of diethylether, vortexed for 5 min and then cen-trifuged for 5 min at 3000 x g. The extraction procedure was repeated twice. The organic phase was evaporated to dryness in a 50°C waterbath. The residue was reconstituted with 200 µL of mobile phase and 50 µL was injected into the HPLC system. The mobile phase (fl ow rate 1.0 mL/min) for the determination consisted of acetonitrile in ultrapure water (27:73 v/v). The concentration of both corticosterone and the internal standard was determined with UV detection at a wavelength of 254 nm. The detection limit was 10 nM.
CHAPTER 3

74 / Stress-induced cortial defects
Immunohistochemistry
Two hours after the start of the fi nal session, the rats were deeply anaesthetized with halo-thane and transcardially perfused with 4% paraformaldehyde solution in 0.1M sodium phosphate buffer (pH 7.4). The brains were carefully removed and post-fi xed in the same solution overnight at 4°C, before being transferred to a potassium phosphate buffer (KPBS 0.02M, pH 7.4) and stored at 4°C. Following cryoprotection of the brains by over-night immersion in a 30% glucose solution, coronal serial sections of 40µm were prepared on a cryostat microtome. Sections were collected in KPBS with sodiumazide and stored at 4°C. All stainings were performed on free-fl oating sections under continuous agitation.
c-fos immunohistochemistry (FOS-ir).
The sections were preincubated in 0.3% H2O
2 in H
2O for 15 min to reduce endogenous
peroxidase activity, before being incubated in a primary polyclonal rabbit anti-c-fos anti-body (Oncogene Research Products; Darmstadt, Germany; 1:10000 dilution) for 48 hr at 4°C. Sections were subsequently rinsed with KPBS and incubated at room tempera-ture with biotinylated goat anti-rabbit IgG (Vector Laboratories, Inc., Burlingame, CA; 1:1000 dilution) followed by the ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA). After another rinse, the reaction product was visualized by adding diaminobenzidine as chromogen and 1% H
2O
2 in H
2O for 15 min. Finally, sections were
washed, mounted on slides, dehydrated and coverslipped with DePex.
c-fos analysis - relative regional expression.
In view of the fact that many clinical studies perform functional investigations using ima-ging techniques, an attempt was made to perform a calculation, which could serve as a more reliable marker for extrapolating c-fos measurements to clinical fi ndings. Imaging analysis scale down global activity to physiologically realistic values to identify regional differences that are not explicable by changes due to total activation patterns. With this in mind we attempt to simulate this global normalization technique in order to reduce potential bias. By considering the customary absolute c-fos values alongside their calcu-lated contributions relative to the entire limbic system, (taking into account the size or analyzed areas per region) we hope to obtain a better indication of the response of indi-vidual brain regions relative to the network as a whole rather than limiting the analysis to their absolute levels of activation. The neural network of forebrain regions often termed the cortical-limbic system is com-prised of various structures besides the prefrontal cortex and cingulate (table 1). Specifi c relay of information between these components determines its effi cacy in modulating mood and emotions and regulating the stress response. In order to perform this calcula-tion, we determined the average regional surface (ARS) of all regions of interest (ROIs), which was calculated by determining the mean surface area of each region across all the

animals. The c-fos positive cell densities of each region were then multiplied by the aver-age regional surface for all animals (regional cell density
rat n * ARS), in order to correct
for possible differences in quantifi ed areas between different rats. This provides c-fos posi-tive cell numbers across a similar cortical-limbic quantifi ed surface area in all rats, sui-table for comparison. By adding the number of c-fos positive cells of every region (regional cell density
rat n * ARS) together for each animal we obtained the total number of cortical-
limbic c-fos positive cells (TOT rat n
). To acquire the relative regional activation values of each animal (% regional activation
rat n), the “regional cell density
rat n * ARS” was divided
by the TOT rat n
. Table 1 provides a sample calculation to illustrate the formulas used.
Phospho-CREB immunohistochemistry.
The sections were pre-incubated in 0.3% H2O
2 in H
2O for 15 min to reduce endog-
enous peroxidase activity, before being incubated in a primary polyclonal rabbit anti-phospho-CREB antibody (commercialized by New England BioLabs, Inc., Beverly, MA, USA; 1:300 dilution in KPBS 0.02 M, pH 7.4) overnight at room temperature. Subse-quently, sections were rinsed with KPBS and incubated at room temperature with bio-tinylated goat anti-rabbit IgG (Vector Laboratories, Inc., Burlingame, CA; 1:1000 dilu-tion in KPBS 0.02 M, pH 7.4) followed by the ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA). After another rinse, the reaction product was visualized by adding diaminobenzidine as chromogen and 1% H
2O
2 in H
2O for 15 min. Finally, the
sections were washed, mounted on slides, dehydrated and coverslipped with DePex.
Phospho-ERK1/2 immunohistochemistry.
The staining was performed on free-fl oating sections under continuous agitation. The sec-tions were pre-incubated in 0.3% H
2O
2 in H
2O for 15 min and thereafter incubated in
a primary monoclonal mouse anti-phospho-ERK1/2 antibody (commercialized by New England BioLabs, Inc., Beverly, MA, USA; 1:5000 dilution in KPBS 0.02 M, pH 7.4) overnight at room temperature. Subsequently, sections were washed with KPBS and incu-bated at room temperature with biotinylated goat anti-mouse IgG (Vector Laboratories, Inc., Burlingame, CA, USA; 1:1000 dilution in KPBS 0.02 M, pH 7.4) followed by the ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA, USA). The reac-tion product was visualized by adding diaminobenzidine as chromogen and 1% H
2O
2 in
H2O for 15 min. Finally, the sections were washed, mounted on slides, dehydrated and
coverslipped with DePex.
Calcineurin immunohistochemistry.
The sections were preincubated in 0.3% H2O
2 in PBS (0.1M, pH 7.4) for 30 min to
reduce endogenous peroxidase activity followed by a blocking step with 3% normal rabbit
Stress-induced cortial defects / 75
CHAPTER 3

serum for 2 hrs. Sections were subsequently incubated in a primary polyclonal goat anti-calcineurin antibody (Santa Cruz Biotechnology, Inc.; 1:250 dilution in PBS 0.1M pH 7.4, 0.25% triton, 3% normal rabbit serum) overnight at room temperature. Afterwards sections were washed with PBS, and incubated at room temperature with biotinylated rabbit anti-goat IgG (Vector Laboratories, Inc., Burlingame, CA; 1:1000 dilution in PBS 0.1M pH 7.4, 0.25% triton, 3% normal rabbit serum) followed by the ABC complex (Vector Laboratories, Inc., Burlingame, CA). After another wash, visualization of the reac-tion product was accomplished by adding diaminobenzidine as chromogen and 1% H
2O
2
in H2O for 30 min. Finally sections were washed, mounted on slides and coverslipped
with DePex.
Antibody specifi city testing.
To control for cross-reactivity due to aspecifi c binding, negative controls were included by incubating several sections and performing the immunostaining without one of the anti-bodies needed for the reaction (primary, secondary, or tertiary). All reactions were negative thereby confi rming the specifi city of the antibodies.
Quantifi cation and data analysis.
Immunohistochemical positive nuclei and dendrites were counted using a computerized imaging analysis system. The selected areas from regions of interest (Fig. 3) were digitized with a Sony charge-coupled device digital camera (SONY Corporation, Tokyo, Japan) mounted on a LEICA Leitz DMRB microscope (LEICA, Wetzlar, Germany). FOS, phos-pho-CREB, phospho-ERK1/2 and calcineurin immunoreactivity was blindly quantifi ed 26 using a computerized image analysis system (LEICA Imaging System Ltd, Cambridge, UK). All quantifi cations of immunopositive cells were carried out at x100 magnifi cation using at least fi ve coronal serial sections (the rostro-caudal distance between consecutive sections was 0.04 mm) for each area analyzed. In each rat, the average regional surface (ARS) quantifi ed in the medial prefrontal cortex (mPFC; Bregma +3.60 to +1.70) was 1.5 mm2 for phospho-CREB and phospho ERK1/2, 3.5 mm2 for calcineurin, and 6.5 mm2 for c-fos. The average area quantifi ed in the cingulate cortex (CING; Bregma +3.20 to +0.95) was 1.0 mm2 for phospho-CREB and phospho-ERK1/2, 2.1 mm2 for calcineurin, and 2.9 mm2 for c-fos 27. Additional regions quantifi ed to calculate relative FOS immu-noreactivity included: 1.25 mm2 for the hippocampal dentate gyrus (DG; Bregma –2.00 to –3.90), 1.18 mm2 for the hippocampal CA1 (CA1; Bregma –2.45 to –4.60), 1.50 mm2 for the paraventricular hypothalamic nucleus (PVN; Bregma –1.08 to –2.00), 2.00 mm2 for the dorsomedial hypothalamic nucleus (DMH; Bregma –2.45 to –3.70), 1.00 mm2 for the central nucleus of the amygdala (CeA; Bregma –1.53 to-2.85), 1.40 mm2 for the lat-eral nucleus of the amygdala (LaA; Bregma –2.00 to –3.70), 1.60 mm2 for the basolateral nucleus of the amygdala (BslA; Bregma –1.78 to –3.25), 2.00 mm2 for the medial nucleus
76 / Stress-induced cortial defects

of the amygdala (MeA; Bregma –1.78 to –3.25), 0.80 mm2 for the centromedial thalamic nucleus (CMT; Bregma –1.53 to –3.90), 1.60 mm2 for the dorsomedial thalamic nucleus (DMT; Bregma –2.00 to –3.90) and 1.80 mm2 for the paraventricular thalamic nucleus (PVT; Bregma –1.33 to –3.90). The areas of interest were acquired and digitized using LEICA Imaging software (LEICA Imaging System Ltd) to reduce quantifi cation errors. After image acquisition, all measure-ments were performed bilaterally and no left-right asymmetry of FOS, phospho-ERKs, phospho-CREB and calcineurin immunoreactivity were found. FOS positive nuclei were quantifi ed and the densities were reported as absolute numbers of positive cells/0.1mm2 for each region and as relative FOS-ir of each region compared to the entire limbic system. Phospho-CREB-densities were reported as the number of positive nuclei/0.1 mm2. Phos-pho-ERK1/2-stained dendrites were quantifi ed, as previously described 13, as the number of horizontal (H) and vertical (V) intersections (H + V contacts) between positive den-drites and an imaginary detection grid (composed of 514 horizontal x 698 vertical lines) present in the quantifi cation fi eld. Phospho-ERK1/2-positive dendrites were reported as the number of positive cell H + V intersections/0.1 mm2. Calcineurin immunoreactivity was expressed as grey value intensity (GVI) 28 of the selected region after correction with corpus callosum as background value and internal control for each section.
Statistics
The data are expressed as means ± standard error (SEM). Statistical signifi cance was deter-mined by performing one-way analysis of variance (ANOVA) and F test of variance on the number of immunoreactive cells of individual brain regions from experimental and control conditions. To compare the cell counts from individual brain regions, values between males and females, and/or control and experimental conditions, t tests for equal or unequal variance were performed. P<0.05 was defi ned as the level of signifi cance between groups. Calculations were made using Jandel SigmaStat statistical software.
RESULTS
Physiological and neuroendocrine correlates of the chronic stress response
To defi ne the dynamics of the chronic stress response, several physiological and neuroen-docrine parameters were measured including body weight gain during the experiment, adrenal gland weight, and plasma corticosterone and adrenaline levels upon termination.
Stress-induced cortial defects / 77
CHAPTER 3
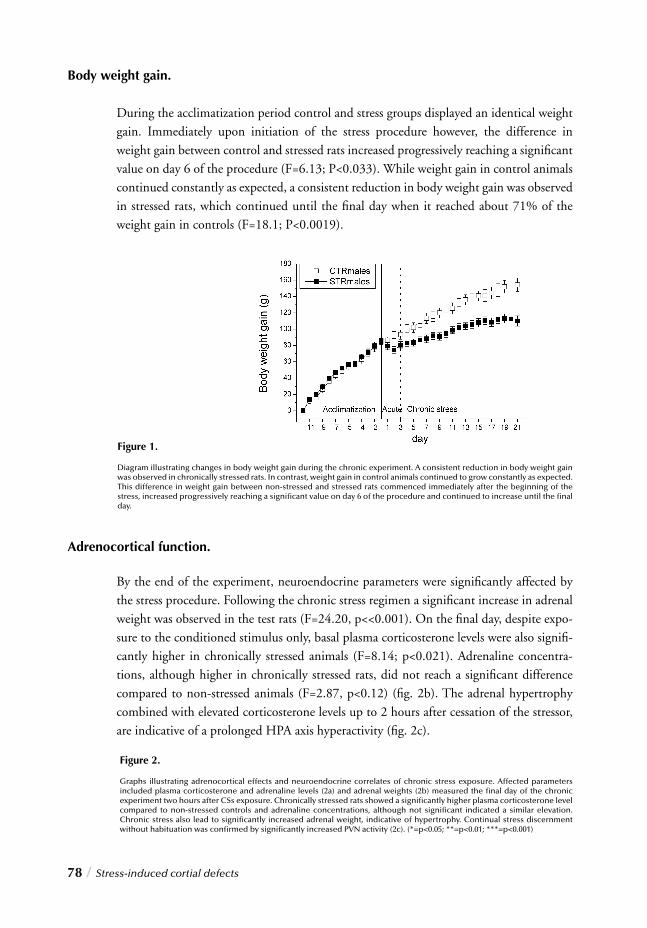
78 / Stress-induced cortial defects
Body weight gain.
During the acclimatization period control and stress groups displayed an identical weight gain. Immediately upon initiation of the stress procedure however, the difference in weight gain between control and stressed rats increased progressively reaching a signifi cant value on day 6 of the procedure (F=6.13; P<0.033). While weight gain in control animals continued constantly as expected, a consistent reduction in body weight gain was observed in stressed rats, which continued until the fi nal day when it reached about 71% of the weight gain in controls (F=18.1; P<0.0019).
Adrenocortical function.
By the end of the experiment, neuroendocrine parameters were signifi cantly affected by the stress procedure. Following the chronic stress regimen a signifi cant increase in adrenal weight was observed in the test rats (F=24.20, p<<0.001). On the fi nal day, despite expo-sure to the conditioned stimulus only, basal plasma corticosterone levels were also signifi -cantly higher in chronically stressed animals (F=8.14; p<0.021). Adrenaline concentra-tions, although higher in chronically stressed rats, did not reach a signifi cant difference
compared to non-stressed animals (F=2.87, p<0.12) (fi g. 2b). The adrenal hypertrophy combined with elevated corticosterone levels up to 2 hours after cessation of the stressor, are indicative of a prolonged HPA axis hyperactivity (fi g. 2c).
Figure 1.
Diagram illustrating changes in body weight gain during the chronic experiment. A consistent reduction in body weight gain was observed in chronically stressed rats. In contrast, weight gain in control animals continued to grow constantly as expected. This difference in weight gain between non-stressed and stressed rats commenced immediately after the beginning of the stress, increased progressively reaching a signifi cant value on day 6 of the procedure and continued to increase until the fi nal day.
Figure 2.
Graphs illustrating adrenocortical effects and neuroendocrine correlates of chronic stress exposure. Affected parameters included plasma corticosterone and adrenaline levels (2a) and adrenal weights (2b) measured the fi nal day of the chronic experiment two hours after CSs exposure. Chronically stressed rats showed a signifi cantly higher plasma corticosterone level compared to non-stressed controls and adrenaline concentrations, although not signifi cant indicated a similar elevation. Chronic stress also lead to signifi cantly increased adrenal weight, indicative of hypertrophy. Continual stress discernment without habituation was confi rmed by signifi cantly increased PVN activity (2c). (*=p<0.05; **=p<0.01; ***=p<0.001)
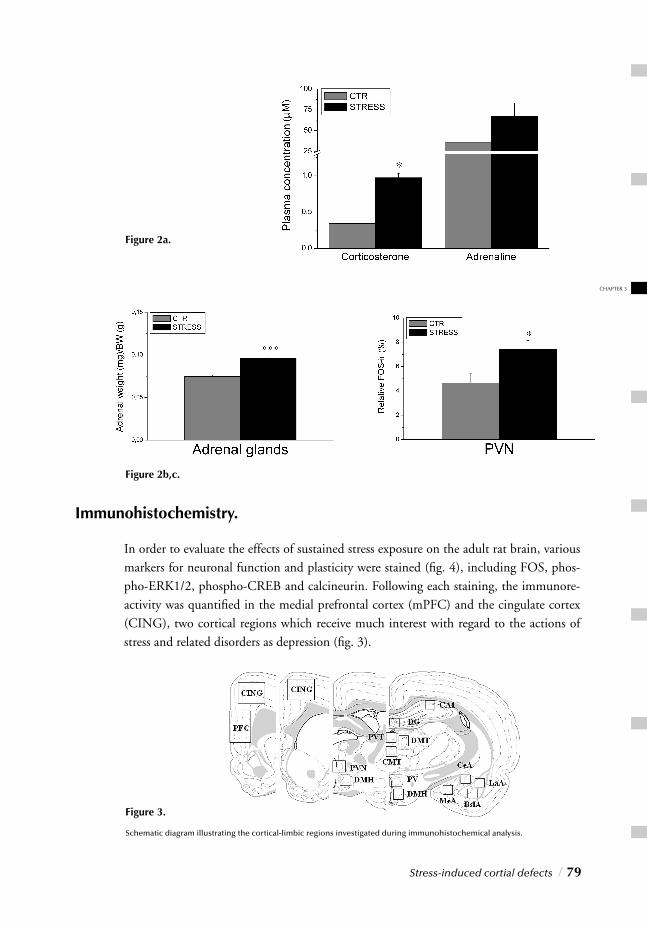
Stress-induced cortial defects / 79
CHAPTER 3
Immunohistochemistry.
In order to evaluate the effects of sustained stress exposure on the adult rat brain, various markers for neuronal function and plasticity were stained (fi g. 4), including FOS, phos-pho-ERK1/2, phospho-CREB and calcineurin. Following each staining, the immunore-activity was quantifi ed in the medial prefrontal cortex (mPFC) and the cingulate cortex (CING), two cortical regions which receive much interest with regard to the actions of stress and related disorders as depression (fi g. 3).
Figure 3.
Schematic diagram illustrating the cortical-limbic regions investigated during immunohistochemical analysis.
Figure 2a.
Figure 2b,c.
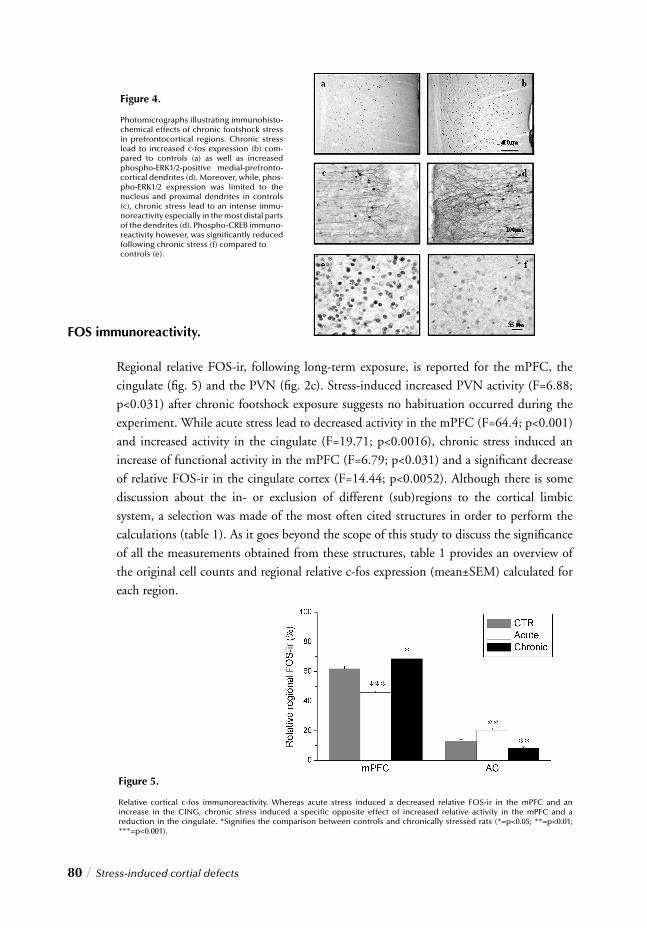
80 / Stress-induced cortial defects
Figure 4.
Photomicrographs illustrating immunohisto-chemical effects of chronic footshock stress in prefrontocortical regions. Chronic stress lead to increased c-fos expression (b) com-pared to controls (a) as well as increased phospho-ERK1/2-positive medial-prefronto-cortical dendrites (d). Moreover, while, phos-pho-ERK1/2 expression was limited to the nucleus and proximal dendrites in controls (c), chronic stress lead to an intense immu-noreactivity especially in the most distal parts of the dendrites (d). Phospho-CREB immuno-reactivity however, was signifi cantly reduced following chronic stress (f) compared to controls (e).
FOS immunoreactivity.
Regional relative FOS-ir, following long-term exposure, is reported for the mPFC, the cingulate (fi g. 5) and the PVN (fi g. 2c). Stress-induced increased PVN activity (F=6.88; p<0.031) after chronic footshock exposure suggests no habituation occurred during the experiment. While acute stress lead to decreased activity in the mPFC (F=64.4; p<0.001) and increased activity in the cingulate (F=19.71; p<0.0016), chronic stress induced an increase of functional activity in the mPFC (F=6.79; p<0.031) and a signifi cant decrease of relative FOS-ir in the cingulate cortex (F=14.44; p<0.0052). Although there is some discussion about the in- or exclusion of different (sub)regions to the cortical limbic system, a selection was made of the most often cited structures in order to perform the calculations (table 1). As it goes beyond the scope of this study to discuss the signifi cance of all the measurements obtained from these structures, table 1 provides an overview of the original cell counts and regional relative c-fos expression (mean±SEM) calculated for each region.
Figure 5.
Relative cortical c-fos immunoreactivity. Whereas acute stress induced a decreased relative FOS-ir in the mPFC and an increase in the CING, chronic stress induced a specifi c opposite effect of increased relative activity in the mPFC and a reduction in the cingulate. *Signifi es the comparison between controls and chronically stressed rats (*=p<0.05; **=p<0.01; ***=p<0.001).
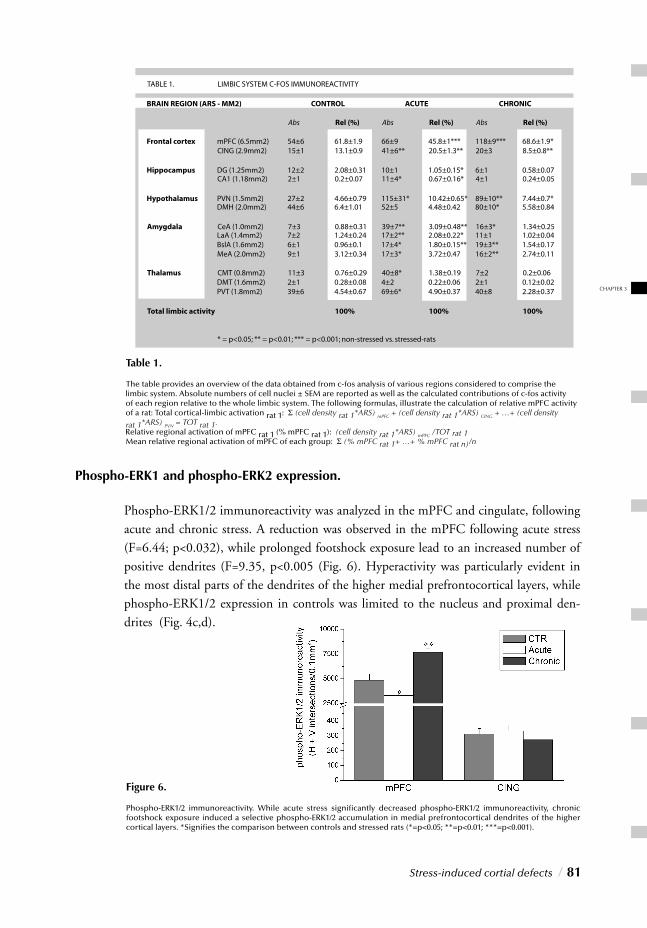
Phospho-ERK1 and phospho-ERK2 expression.
Phospho-ERK1/2 immunoreactivity was analyzed in the mPFC and cingulate, following acute and chronic stress. A reduction was observed in the mPFC following acute stress (F=6.44; p<0.032), while prolonged footshock exposure lead to an increased number of positive dendrites (F=9.35, p<0.005 (Fig. 6). Hyperactivity was particularly evident in the most distal parts of the dendrites of the higher medial prefrontocortical layers, while phospho-ERK1/2 expression in controls was limited to the nucleus and proximal den-drites (Fig. 4c,d).
Stress-induced cortial defects / 81
CHAPTER 3
Figure 6.
Phospho-ERK1/2 immunoreactivity. While acute stress signifi cantly decreased phospho-ERK1/2 immunoreactivity, chronic footshock exposure induced a selective phospho-ERK1/2 accumulation in medial prefrontocortical dendrites of the higher cortical layers. *Signifi es the comparison between controls and stressed rats (*=p<0.05; **=p<0.01; ***=p<0.001).
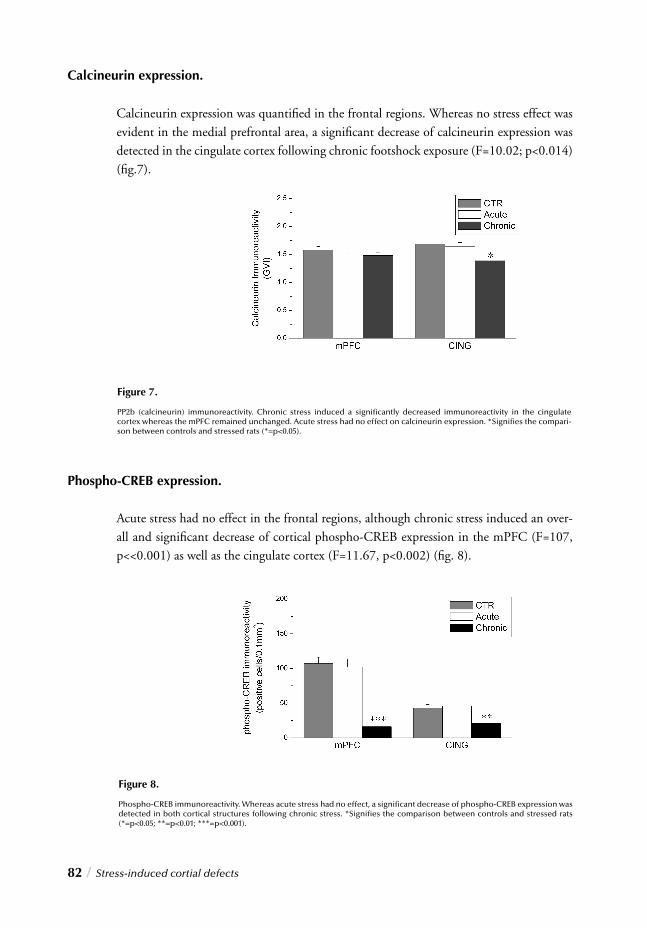
82 / Stress-induced cortial defects
Figure 7.
PP2b (calcineurin) immunoreactivity. Chronic stress induced a signifi cantly decreased immunoreactivity in the cingulate cortex whereas the mPFC remained unchanged. Acute stress had no effect on calcineurin expression. *Signifi es the compari-son between controls and stressed rats (*=p<0.05).
Calcineurin expression.
Calcineurin expression was quantifi ed in the frontal regions. Whereas no stress effect was evident in the medial prefrontal area, a signifi cant decrease of calcineurin expression was detected in the cingulate cortex following chronic footshock exposure (F=10.02; p<0.014) (fi g.7).
Phospho-CREB expression.
Acute stress had no effect in the frontal regions, although chronic stress induced an over-all and signifi cant decrease of cortical phospho-CREB expression in the mPFC (F=107, p<<0.001) as well as the cingulate cortex (F=11.67, p<0.002) (fi g. 8).
Figure 8.
Phospho-CREB immunoreactivity. Whereas acute stress had no effect, a signifi cant decrease of phospho-CREB expression was detected in both cortical structures following chronic stress. *Signifi es the comparison between controls and stressed rats (*=p<0.05; **=p<0.01; ***=p<0.001).

Stress-induced cortial defects / 83
CHAPTER 3
DISCUSSION
It is well established that stress-induced neuronal activity greatly infl uences the organiza-tion and function of circuits in the brain yet the molecular signals that translate activity into structural and functional changes in connections remain largely obscure. This study illustrates the effects of long-term stress exposure on frontocortical functions in adult male rats using multiple markers of neuronal activity and synaptic plasticity. The differential regulation of phospho-ERK1/2, calcineurin and phospho-CREB in this region provides evidence that neural plasticity in the adult central nervous system is infl uenced by stress, at least in part through these candidate proteins. Moreover, the decreased relative FOS immunoreactivity in the cingulate and its increased expression in the medial prefrontal cortex, confi rm that the chronic stress response involves these regions, although it also suggests that stress may exert its infl uence through different regional mechanisms. These structures are of particular interest with respect to the pathogenesis of mood disorders due to their connections with the amygdala, hypothalamus, and periaqueductal gray, which have been implicated in emotional behavior and responses to stress 29,30,31. The link between stress and induced pathology has been well established 1,32,33,34 and previous stud-ies have shown that it may result from disturbed adaptations at the cellular and molecu-lar levels. At the cellular level, preclinical studies have reported atrophy, cell death and decreased neurogenesis in stress-vulnerable neurons of the hippocampus 35,36, while clini-cal studies provide evidence for pathophysiology in the prefrontal cortex, as depressed patients demonstrate altered blood fl ow, metabolism and volume of this region 33,37. The predictive validity of this model for studying stress-related disorders in humans is sup-ported by the similar action of prolonged stress exposure on the modeled adrenocortical symptoms found in depressed patients 38. These entail a persistent HPA axis activation throughout the stress challenge (suggesting no habituation occurred), PVN hyperactivity (fi g. 2c), reduced weight gain (fi g. 1), elevated corticosterone levels and adrenal hypertro-phy (fi g. 2ab). Moreover, previous behavioral fi ndings illustrate that rats exposed to this stress paradigm also demonstrate signifi cantly altered activity in an open fi eld, a widely used test to study anxiety 39. It has been suggested that damage in stress-vulnerable neurons may be attributed to increased glucocorticoid levels and decreased neurotrophin levels 36. This in turn could potentiate stress-induced cellular alterations in neurotrophin-related intracellular mechanisms in individuals genetically predisposed to depression 14. Chronic antidepressant administration has been shown to counteract stress-induced reductions of BDNF, and seem to exert their effects by stimulating appropriate adaptive changes to oppose these adverse effects and loss of neural plasticity 36,40. Since stress and antidepressants both regulate specifi c neurotrophin-related target genes, thereby affecting structural neuronal plasticity, this has lead to the hypothesis that stress-induced pathology and in turn its treatment involve the regulation of the BDNF signaling cascade 15. Although not fully understood the molecular mechanisms underlying this process include adaptations in

84 /Stress-induced cortial defects
the cAMP transduction cascade, including ERK, CREB and calcineurin 13. Here we demonstrate altered expression of these BDNF signaling cascade members by stress, indicating that the regulation of these proteins is a molecular correlate of prefrontal metaplastic changes as well as of functional and morphological alterations after stress exposure in vivo.
Medial prefrontocortical hyperactivation and dendritic ERK1/2 hyper- phosphorylation
The mPFC revealed signifi cant chronic stress-induced molecular and cellular abnormalities. Signifi cantly increased relative FOS-ir in this region indicates prefrontocortical functional hyperactivity (fi g. 5). Since the mPFC plays a key role in modulating higher brain functions and integrating cognitive and emotional processes with complex stress and behavioral responses 41, this region represents one of the highest hierarchical structures involved in receiving and processing sensory input from various cortical and subcortical regions. This position may therefore expose the mPFC to functional hyperactivity, particularly when sensory input is strongly increased as during long-term stress. These fi ndings are in agreement with previously obtained molecular data, as gene expression patterns demonstrating a general upregulation of prefrontocortical gene expression following chronic stress are in line with mPFC hyperfunction illustrated by relative FOS-ir 13,42. The increased relative FOS-ir in this region following chronic footshock stress coincides with signifi cant ERK1/2 hyperphosphorylation in the distal dendrites (fi g. 6) whereas acute challenge induced a signifi cant reduction of both markers. ERK, also known as mitogen-activated protein kinase (MAPK), is a family of serine/threonine protein kinases implicated in signal transduction from cell surface to nuclei. ERKs serve to coordinate responses to extracellular signals in adult neurons and function in proper maintenance of synaptic plasticity, learning and memory processes 43,44. Intricate regulation of their dual phosphorylation states determines the effi ciency of ERK mediated activities as perturbed ERK signaling has been associated with cytoskeletal destabilization 45,46, neuronal dysfunc-tion and even death 47,48. Since stress is believed to infl uence brain functions through its effects on neuronal plasticity and neurogenesis, hyperphosphorylated ERK1/2 observed here might represent neurochemical evidence for its deleterious action on the regulation of intracellular signaling cascades involved in modulating synaptic plasticity. Disturbed ERK phosphorylation offers one potential explanation for impaired neuronal plasticity associated with mPFC dysfunction. Recent data supporting this possibility report that prolonged ERK cascade activation in cortical neurons through glutamate exposure lead to excitotoxic degeneration or neuronal death 48. Since studies show that prefrontocortical atrophy also results from long-term stress and sustained exposure to elevated glucocorti-coid levels 34, our data seem in accordance as prefrontocortical ERK1/2 hyperphosphory-lation only characterized animals exposed to chronic stress as opposed to acute (3 days) or

CHAPTER 3
Stress-induced cortial defects / 85
even subchronic (10 days) exposure 13. A direct indication in support of the deleterious effects of chronic stress on prefronto-cortical function is provided by the marked reduction of cortical phospho-CREB expres-sion (fi g. 8). Activation of this transcription factor is heavily implicated in synaptic plasti-city, learning and survival 49. As a target of calcium/calmodulin- and ERK1/2- signaling cascades, phosphorylation of CREB regulates the transcription of specifi c target genes including the neurotrophin BDNF 50. Through downregulating phospho-CREB expres-sion or infl uencing its phosphorylation state, chronic stress may thus compromise PFC plasticity required for proper response and/or adaptation to (stressful) stimuli.
Cingulate hypofunction and reduced calcineurin expression
Contrary to the functional hyperactivation observed in the mPFC, long-term stress expo-sure induced a signifi cantly reduced relative FOS-ir in the cingulate. Like mPFC hyperac-tivity, cingulate hypoactivation was also selective for chronic stress exposure, as acute stress signifi cantly increased its activity (fi g. 5). In line with observations characterizing several stress-related neuropsychiatric diseases such as post-traumatic stress disorder and depres-sion, reduced cingulate activity appears to be the result of neuronal or glial abnormalities 33,51. Reversal of this condition by antidepressants leads to improved symptomatology and clinical recovery 52. The stress-induced hypofunction observed here coincided with signifi -cantly reduced calcineurin (PP2B) expression. This calcium-dependent protein phospha-tase is involved in modulating intracellular calcium transduction pathways and is impli-cated in various aspects of synaptic plasticity 53-56. It is widely believed that calcium plays a primary role in the development of neuronal cell injury in different pathological states of the brain, as abnormal calcium homeostasis has been associated with reduced synaptic plasticity, neuronal defects and excitotoxic degeneration 57. Calcium-mediated cascades are also fundamental for the regulation of gene expression underlying short- and long-term neuronal plasticity 49,58-60. Initial altered calcineurin activity could lead to detained CREB dephosphorylation and thus increased phospho-CREB immunoreactivity. How-ever, due to the involvement of several other phosphatases the regulation of CREB and its phosphorylation can ultimately return to basal levels. Upon continuation the situa-tion differs as long-term defects in calcineurin regulation may lead to abnormal calcium homeostasis and consequently disrupted neuronal functions, potentially contributing to
reduced CREB phosphorylation (fi g. 8) and possibly disturbed BDNF expression.
Activity-dependent regulation of prefrontocortical plasticity
In short, these fi ndings demonstrate that long-term footshock stress induces functional hyperactivity and ERK1/2 hyperphosphorylation in the mPFC, and reduced function and calcineurin immunoreactivity in the cingulate. Although stress appears to target distinct intracellular pathways in these regions, the fi nal downstream target seems to

be common in both areas, since reduced phospho-CREB immunoreactivity and subse-quently decreased BDNF expression are detrimental for the modulation of neuronal plas-ticity. The maintenance of cortical plasticity however requires activity-dependent changes in synaptic strength, which stipulate long-lasting biochemical alterations in the postsynaptic neuron 61-64. Neurotrophins like BDNF play a central role in coupling these changes to lasting effects on synaptic function 59,65. The induction of BDNF transcription is regu-lated in part by intracellular calcium although this process is dependent on its route of entry into the cell, the integrity of intracellular pathways responsible for transmitting calcium-mediated signals and CREB phosphorylation 22,58,66. In the cingulate therefore the combined disturbances of calcium-dependent calcineurin and phospho-CREB expression may represent profound intracellular disturbances ultimately impairing multiple neuronal functions in addition to synaptic plasticity. This view is in line with neuronal and/or glial defects repeatedly observed in the cingulate of depressed patients 6. Similarly, impaired plasticity in the mPFC may also be attributable to the activity dependent nature of CREB-mediated transcription and thus BDNF release from cortical dendritic terminals. Since this process is subject to the availability of phospho-CREB 59,67,68, it infers that neuro-trophin is released only when needed. Yet, while stress-induced hyperfunction of the mPFC can initially cause massive BDNF release, prolonged discharge following chronic exposure can deplete reserves and exhaust the system leading to BDNF withdrawal 13. Preclinical data confi rm the ability of stress to reduce BDNF levels through decreased phospho-CREB expression in these regions 13,69-71. This neurotrophin depletion may in turn disrupt the intracellular transduction pathway causing dendritic ERK1/2 hyperphos-phorylation as seen in the mPFC. Since ERKs are essential for regulating cytoskeletal integrity 72, dendritic phospho-ERK1/2 accumulation may cause hyperphosphorylation of cytoskeletal proteins consequently weakening dendritic structure, namely in the synap-tic terminals where these proteins are particularly abundant 45,73,74. Abnormal dendritic traffi cking, signal transmission, and neurotransmitter release can follow, contributing to reduced prefrontocortical plasticity and in turn reduced CREB phosphorylation, thereby completing a vicious circle. Evidently, neuronal plastic function is an intricate process requiring careful regulation of multiple factors. Crucial to proper functioning is a close modulation of the phosphory-lation state of synaptic proteins by protein kinases and phosphatases 75-77. Maintenance of
their ratios as well as Ca2+ dependent mechanisms may thus prove essential for sustained homeostasis. Such regulatory processes are part of the stress response in the brain and their effects could be modifi ed by the decreased expression of calcineurin and increased expression of ERK1/2 after chronic footshock exposure as observed in this study. Altered expression patterns of such proteins may also be associated with frontocortical impair-ment that occurs during prolonged stress, as well as with stress-induced plastic changes such as adaptation and sensitization. In line with the reduced plasticity markers observed here, previous gene expression analysis illustrates decreased expression of genes associated
86 /Stress-induced cortial defects

primarily with synaptic plasticity including SNAP, synaptophysin, and synapsin1a/1b 13. Current studies are being performed to further highlight the roles of these correlates in the chronic stress response. In conclusion, our fi ndings demonstrate that phospho-ERK1/2, calcineurin and phospho-CREB are stress-responsive compounds, which raise the possi-bility that alterations in the expression of these or other synaptic proteins might be impor-tant in producing some of the physiological and pathophysiological effects of stress in the frontal regions. Although it remains to be shown whether or not these effects are part of a normal adaptive response to chronic footshocks, the nature of these compounds and their changes suggest that plasticity is adversely effected. The lack of or opposed effects seen under acute conditions suggest that these changes are specifi c for chronic exposure. Ne-vertheless, although seemingly negative, these adaptations could also represent a normal response to long-term footshock stress. These fi ndings prove particularly relevant to such clinical disorders as depression and posttraumatic stress disorder that are sensitive to stress and involve changes in neural and synaptic plasticity.
ACKNOWLEDGEMENTS
The technical assistance of Folkert Postema and Kor Venema is gratefully acknowledged.
CHAPTER 3
Stress-induced cortial defects / 87

REFERENCES1. Bremner, J.D. Does stress damage the brain? Biol. Psychiatry 45, 797-805 (1999).
2. Honer, W.G. et al. Synaptic and plasticity-associated proteins in anterior frontal cortex in severe mental illness. Neuroscience 91, 1247-1255 (1999).
3. Sheline, Y.I. 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol. Psychiatry 48, 791-800 (2000).
4. Drevets, W.C. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog. Brain Res. 126, 413-431 (2000).
5. Ongur, D., Drevets, W.C. & Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U. S. A 95, 13290-13295 (1998).
6. Rajkowska, G. et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085-1098 (1999).
7. Sheline, Y.I., Wang, P.W., Gado, M.H., Csernansky, J.G. & Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. U. S. A 93, 3908-3913 (1996).
8. Sapolsky, R.M. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry 57, 925-935 (2000).
9. Sapolsky, R.M. Stress, Glucocorticoids, and Damage to the Nervous System: The Current State of Confusion. Stress. 1, 1-19 (1996).
10. McEwen, B.S. Allostasis, allostatic load, and the aging nervous system: role of excitatory amino acids and excitotoxicity. Neurochem. Res. 25, 1219-1231 (2000).
11. Magarinos, A.M., McEwen, B.S., Flugge, G. & Fuchs, E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J. Neurosci. 16, 3534-3540 (1996).
12. McEwen, B.S. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105-122 (1999).
13. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphos- phorylation in medial prefrontocortical dendrites: implications for stress- related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
14. Duman, R.S., Heninger, G.R. & Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597-606 (1997).
15. Altar, C.A. Neurotrophins and depression. Trends Pharmacol. Sci. 20, 59-61 (1999).
16. Grewal, S.S., York, R.D. & Stork, P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 9, 544-553 (1999).
17. Miyata, K. et al. Involvement of the brain-derived neurotrophic factor/TrkB pathway in neuroprotecive effect of cyclosporin A in forebrain ischemia. Neuroscience 105, 571-578 (2001).
18. Karege, F. et al. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 109, 143-148 (2002).
19. Campeau, S. et al. Elicitation and reduction of fear: behavioural and neuroendo- crine indices and brain induction of the immediate-early gene c-fos. Neuroscience 78, 1087-1104 (1997).
20. Chaudhuri, A. Neural activity mapping with inducible transcription factors. Neuroreport 8, v-ix (1997).
21. Dube, T., Brunson, T., Nehlig, A. & Baram, T.Z. Activation of specifi c neuronal circuits by corticotropin relea- sing hormone as indicated by c-fos expression and glucose metabolism. J. Cereb. Blood Flow Metab 20, 1414-1424 (2000).
22. Finkbeiner, S. et al. CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031-1047 (1997).
23. Ter Horst, G.J., Meijler, W.J., Korf, J. & Kemper, R.H. Trigeminal nociception-induced cerebral Fos expression in the conscious rat. Cephalalgia 21, 963-975 (2001).
24. Remie, R., Knot, H.J., Bos, E.A. & Zaagsma, J. Characterization of presynaptic beta-adrenoceptors facilitating endogenous noradrenaline release in the portal vein of perma- nently cannulated, freely moving rats. Eur. J. Pharmacol. 157, 37-43 (1988).
25. Smedes, F., Kraak, J.C. & Poppe, H. Simple and fast solvent extraction system for selective and quantitative isolation of adrenaline, noradrenaline and dopa- mine from plasma and urine. J. Chromatogr. 231, 25-39 (1982).
26. West, M.J. New stereological methods for counting neurons. Neurobiol. Aging 14, 275-285 (1993).
27. Swanson LW. Brain maps: structure of the rat brain. 2002. Amsterdam,Elsevier.
28. Lukyanetz, E.A. Evidence for colocalization of calcineurin and calcium channels in dorsal root ganglion neurons. Neuroscience 78, 625-628 (1997).
29. Fuster, J.M. Prefrontal neurons in networks of executive memory. Brain Res. Bull. 52, 331-336 (2000).
30. Miller, E.K. The prefrontal cortex and cognitive control. Nat. Rev. Neurosci. 1, 59-65 (2000).
31. Austin, M.P., Mitchell, P. & Goodwin, G.M. Cognitive defi cits in depression: possible implications for functional neuropathology. Br. J. Psychiatry 178, 200-206 (2001).
32. McEwen, B.S. Effects of adverse experiences for brain structure and function. Biol. Psychiatry 48, 721-731 (2000).
33. Rajkowska, G. Postmortem studies in mood disorders indicate altered num- bers of neurons and glial cells. Biol. Psychiatry 48, 766-777 (2000).
34. Wellman, C.L. Dendritic reorganization in pyramidal neurons in medial pre- frontal cortex after chronic corticosterone administration. J. Neurobiol. 49, 245-253 (2001).
35. Magarinos, A.M., Verdugo, J.M. & McEwen, B.S. Chronic stress alters synaptic terminal structure in hippocam- pus. Proc. Natl. Acad. Sci. U. S. A 94, 14002-14008 (1997).
88 /Stress-induced cortial defects

36. Duman, R.S., Malberg, J. & Thome, J. Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 46, 1181-1191 (1999).
37. Drevets, W.C. et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824-827 (1997).
38. Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuro-psychopharmacology 23, 477-501 (2000).
39. Westenbroek ,C. et al. Gender-specifi c effects of social housing in rats after chronic mild stress exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 21-30 (2003).
40. Malberg, J.E., Eisch, A.J., Nestler, E.J. & Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104-9110 (2000).
41. Drevets, W.C. Neuroimaging studies of mood disorders. Biol. Psychiatry 48, 813-829 (2000).
42. Trentani A, Den Boer, J.A., Korf J & Ter Horst, G.J. Gender-related sensitivity to the effects of chronic stress in rats. Society for Neuroscience Abstracts . 2000.
43. Magarinos, A.M. & McEwen, B.S. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience 69, 83-88 (1995).
44. Magarinos, A.M. & McEwen, B.S. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69, 89-98 (1995).
45. Runden, E. et al. Regional selective neuronal degeneration after protein phos- phatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J. Neurosci. 18, 7296-7305 (1998).
46. Vaillant, A.R. et al. Signaling mechanisms underlying reversible, activity- dependent dendrite formation. Neuron 34, 985-998 (2002).
47. Satoh, T. et al. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci. Lett. 288, 163-166 (2000).
48. Stanciu, M. et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200-12206 (2000).
49. Bading, H. Transcription-dependent neuronal plasticity the nuclear calcium hypothesis. Eur. J. Biochem. 267, 5280-5283 (2000).
50. Tao, X., Finkbeiner, S., Arnold, D.B., Shaywitz, A.J. & Greenberg, M.E. Ca2+ infl ux regulates BDNF transcription by a CREB family tran- scription factor-dependent mechanism. Neuron 20, 709-726 (1998).
51. Drevets, W.C., Ongur, D. & Price, J.L. Neuroimaging abnormalities in the subgenual prefrontal cortex: implications for the pathophysiology of familial mood disorders. Mol. Psychiatry 3, 220-221 (1998).
52. Mayberg, H.S. Limbic-cortical dysregulation: a proposed model of depression. J. Neuropsychiatry Clin. Neurosci. 9, 471-481 (1997).
53. Victor, R.G., Thomas, G.D., Marban, E. & O’Rourke, B. Presynaptic modulation of cortical synaptic activity by cal- cineurin. Proc. Natl. Acad. Sci. U. S. A 92, 6269-6273 (1995).
54. Morioka, M., Hamada, J., Ushio, Y. & Miyamoto, E. Potential role of calcineurin for brain ischemia and traumatic injury. Prog. Neurobiol. 58, 1-30 (1999).
55. Winder, D.G. & Sweatt, J.D. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2, 461-474 (2001).
56. Zeng, H. et al. Forebrain-specifi c calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107, 617-629 (2001).
57. Mattson, M.P. & Chan, S.L. Dysregulation of cellular calcium homeostasis in Alzheimer’s disease: bad genes and bad habits. J. Mol. Neurosci. 17, 205-224 (2001).
58. Shieh, P.B., Hu, S.C., Bobb, K., Timmusk, T. & Ghosh, A. Identifi cation of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20, 727-740 (1998).
59. Shieh, P.B. & Ghosh, A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J. Neurobiol. 41, 127-134 (1999).
60. West, A.E. et al. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U. S. A 98, 11024-11031 (2001).
61. Baudry, M. Synaptic plasticity and learning and memory: 15 years of progress. Neurobiol. Learn. Mem. 70, 113-118 (1998).
62. Barth, A.L. et al. Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J. Neurosci. 20, 4206-4216 (2000).
63. Segal, M. Rapid plasticity of dendritic spine: hints to possible functions? Prog. Neurobiol. 63, 61-70 (2001).
64. Dudai, Y. Molecular bases of long-term memories: a question of persistence. Curr. Opin. Neurobiol. 12, 211-216 (2002).
65. Black, I.B. Trophic regulation of synaptic plasticity. J. Neurobiol. 41, 108-118 (1999).
66. Tao, X., West, A.E., Chen, W.G., Corfas, G. & Greenberg, M.E. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 33, 383-395 (2002).
67. Finkbeiner, S. CREB couples neurotrophin signals to survival messages. Neuron 25, 11-14 (2000).
68. Conti, .C., Cryan, J.F., Dalvi, A., Lucki, I. & Blendy, J.A. cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepres- sant drugs. J. Neurosci. 22, 3262-3268 (2002).
69. Smith, M.A. Hippocampal vulnerability to stress and aging: possible role of neurotrophic factors. Behav. Brain Res. 78, 25-36 (1996).
70. Ueyama, T. et al. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci. Res. 28, 103-110 (1997).
71. Rasmusson, A.M., Shi, L. & Duman, R. Downregulation of BDNF mRNA in the Hippocampal Dentate Gyrus after Re-exposure to Cues Previously Associated with Footshock. Neuropsychopharmacology 27, 133-142 (2002).
CHAPTER 3
Stress-induced cortial defects / 89

72. Veeranna, Shetty, K.T., Takahashi, M., Grant, P. & Pant, H.C. Cdk5 and MAPK are associated with complexes of cytoskeletal proteins in rat brain. Brain Res. Mol. Brain Res. 76, 229-236 (2000).
73. Olsen, M.K., Reszka, A.A. & Abraham, I. KT5720 and U-98017 inhibit MAPK and alter the cytoskeleton and cell morphology. J. Cell Physiol 176, 525-536 (1998).
74. Perry, G. et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 10, 2411-2415 (1999).
75. Schulman, H. Protein phosphorylation in neuronal plasticity and gene expression. Curr. Opin. Neurobiol. 5, 375-381 (1995).
76. Tokuda, M. & Hatase, O. Regulation of neuronal plasticity in the central nervous system by phosphorylation and dephosphorylation. Mol. Neurobiol. 17, 137-156 (1998).
77. Turner, K.M., Burgoyne, R.D. & Morgan, A. Protein phosphorylation and the regulation of synaptic mem- brane traffi c. Trends Neurosci. 22, 459-464 (1999).
90 /Stress-induced cortial defects

CHAPTER 3
Stress-induced cortial defects / 91


4
REDUCED CREB PHOSPHORYLATION AND CALCINEURIN CONTENT CHAR-ACTERIZE THE RESPONSE TO CHRONIC STRESS IN MALE RATS: INDICATIONS FOR SEX-DEPENDENT NEUROPLASTICITY CHANGES
Disturbed molecular and cellular adaptations play a central role in
the development of stress-induced neuronal abnormalities. Stress
has been proposed to impair neuroplasticity by selectively target-
ing expression and/or activity levels of key intracellular signaling
cascade members involved in transduction of neurotrophin sig-
nals. To elucidate stress effects on expression and phosphoryla-
tion levels of specifi c neurotrophin cascade members, rats were
exposed to prolonged footshock stress after which phospho-
CREB and calcineurin immunoreactivity as well as calcineurin and
CREB gene expression were investigated. The hippocampus and
prefrontal cortex revealed unaltered CREB and calcineurin mRNA
levels yet reduced phospho-CREB and calcineurin immunoreac-
tivity in males, while stressed females illustrated comparatively
attenuated alterations. Considering the critical role of CREB and
calcineurin in regulating BDNF signaling these fi ndings seem to
confi rm the deleterious action of stress on neuronal plasticity. Fur-
thermore, this data provides new insights into the mechanisms
governing distinct sex-dependent discrepancies often encoun-
tered in response to stress.
Kuipers S.D., Trentani A., Bakker P.L., Veenstra R., Ter Horst G.J,
Den Boer J.A. European Journal of Neuroscience. Submitted


CHAPTER 4
Stress, gender and neuronal plasticity / 95
INTRODUCTION
Disturbed molecular and cellular adaptations represent common predisposing factors for the occurrence of structural and functional neuronal abnormalities. Stress, particularly when persistent and severe, can play a primary role in this pathophysiological process. Common features often observed following prolonged stress exposure for instance include hypothalamic-pituitary-adrenal (HPA) axis hyperactivity and atrophy of apical dendrites in the medial prefrontal cortex and hippocampus 1-9. In turn these implicated prefrontal cortex and hippocampus regions, primary targets of subsequent glucocorticoid-mediated damaging effects, also modulate this stress response by actively inhibiting HPA axis activity 10,11. Although to date very little is known about the cellular and molecular mechanisms responsible for stress-induced abnormalities, it has recently been suggested that impaired neuronal function might result from stress’ selective infl uence on expression and/or acti-vity levels of key members of intracellular signaling cascades underlying neuronal plasti-city, particularly those involved in neurotrophin signal transduction 12-17. Recent efforts to understand the neurobiological substrates underlying neuronal plasticity have demon-strated the importance of cAMP response element binding protein (CREB) 18-21. Phos-phorylated (activated) CREB promotes the transcription of target genes such as brain derived neurotrophic factor (BDNF) 22 leading to structural changes that increase synaptic effi cacy and ultimately produce long-lasting alterations of cellular function 23-25 . Proper maintenance of synaptic plasticity however, requires a carefully regulated balance of phos-phorylation and dephosphorylation. While kinase activity represents a crucial component of a gate that regulates neuronal plasticity by phosphorylating critical elements such as CREB 26, activation of specifi c phosphatases as calcineurin (PP2B) oppose this action by dephosphorylating target proteins 23. The coordinated activity of protein kinases and phos-phatases in response to specifi c stimuli thus represents a crucial feature for proper neuronal functioning, especially since abnormal regulation of these intracellular proteins has been associated with neuronal dysfunctions and in severe cases cell death 27,28.
We have recently suggested that abnormal CREB phosphorylation and calcineurin expression may account for the development of stress-induced neuronal dysfunctions in prefrontal regions of male rats 17,28. In the present study, we extend our investigation of the cellular and molecular adaptations in response to prolonged stress to include both the prefrontal cortex and the hippocampus, in order to determine whether similar mecha-nisms underlie stress-induced disturbances in both regions. Using this same stress para-digm, our prior fi ndings have also corroborated marked sex differences in the activity of the hippocampo-prefrontocortical pathway as well as the HPA axis response. Since gender is known to represent a critical aspect in both stress sensitivity and psychopathology 29,30,

96 / Stress, gender and neuronal plasticity
female rats were also included in this investigation. The results presented here may thus provide new insights into the neurobiological substrates underlying gender-related dis-crepancies and the occurrence of neuronal abnormalities in response to sustained stress.
MATERIALS AND METHODSAnimals
The experiments were performed on male (n°=24: 225-249 g) and female (n°=24: 200-224 g) Wistar rats. The animals were individually housed with food and water avai-lable ad libitum and maintained on a 12:12 hr light/dark cycle at 21°C. All rats were handled daily for 5-8 min to minimize the non-specifi c stress response. All handling was performed in accordance with the European Communities Council Directive of Novem-ber 24, 1986 (86/609/EEC) and the guidelines of the Animal Bio-ethics Committee of the University of Groningen (FDC: 2509).
Stress Procedure
The rodent test-chamber consists of a box containing an animal space positioned on a metallic grid fl oor connected to a shock generator and scrambler. For 3 weeks, stressed rats were subjected to one aversive session of varying duration (between 30 and 120 min/day) in the “footshock box” during which 5 uncontrollable and inescapable footshocks were applied (0.8 mA in intensity and 8 sec in duration). During these daily sessions, starting time, inter-shock interval, and total time spent in the box were randomized in order to make the procedure as unpredictable as possible.
Experimental Setup
To investigate the cellular and molecular changes induced by sustained footshock expo-sure, rats were randomly assigned to two groups:• STRESS (n° = 24; 12 male and 12 female rats): stressed rats were exposed to the foot-shock and stressed according to the footshock procedure described above for 21 consecu-tive days;• CTR (n° = 24; 12 male and 12 female rats): control animals followed an identical sche-dule in a similar setup, but did not receive any actual footshocks throughout the experiment.On the fi nal (21st) day of the experiment all rats were placed in the box for 15 minutes, although no footshocks were delivered. It is important to note that exposure to the box on the fi nal day of the experiment was critical as it established a link between a harm-less stimulus (environment in which footshocks were applied or “footshock box”) and aversive events (“footshocks”). This provided a way to create a stress condition without

CHAPTER 4
Stress, gender and neuronal plasticity / 97
the unwanted side effects of direct changes induced by physical or painful stimuli. The response of control and stress rats to an identical, painless stimulus could then be inves-tigated by evaluating patterns of gene expression (CREB and calcineurin mRNA), pro-tein synthesis (FOS and calcineurin immunoreactivity) and phosphorylation (phospho-CREB immunoreactivity). As stressed animals could learn to associate this environment with aversive footshocks, this design also allowed the opportunity to study the neuroen-docrine in addition to immunohistochemical changes underlying the “psychological stress response” without exposure to actual footshock stress.
Physiological and endocrine measurements
On the fi nal day of the experiment, rats used for immunohistochemistry were terminated two hours after the beginning of the fi nal session, while those used for molecular biology were sacrifi ced 15 minutes following the end of the last stimulus. To explore their response to repeated stress, various physiological and neuroendocrine parameters were measured. Weight gain was monitored on a daily basis and graphs were constructed to illustrate body weight gain changes throughout the procedure. Thymus and adrenal glands were also weighed and recalculated to correct for the body weight of the animals. Blood samples drawn upon termination, were stored at -20°C and used to determine plasma corticoste-rone and adrenaline concentrations with HPLC. Constructed graphs serve to verify the severity of stress perceived by the animals.
Extraction and Chromatography
Catecholamines.
Noradrenaline and adrenaline were extracted from plasma using liquid/liquid extraction with 3,4-dihydroxybenzylamine as internal standard 31,32. Briefl y, plasma catecholamines were bound to diphenylborate-ethanolamine at pH 8.6. Extraction was performed with n-heptane (containing 1% octanol and 25 % tetraoctylammoniumbromide). Next, cate-cholamines were extracted from the organic phase with diluted acetic acid. Catecho-lamines (20 µl acetic acid extract) were analyzed using an HPLC/auto-injector (CMA, Sweden) and a Shimadzu LC-10AD pump (Kyoto, Japan) connected to a reversed phase column (Hypersil, C18, 3 µm, 150x2.0 mm), followed by an electrochemical detector (Antec Leyden, The Netherlands) working at a potential setting of 500 mV vs. Ag/AgCl reference. The mobile phase consisted of 50 mM acetate buffer, 150 mg/l octane sul-phonic acid, 150 mg/l tetramethylammonium, 15 mg/ml Na
2EDTA and 3% methanol,
adjusted to pH 4.1. The fl ow-rate was 0.35 ml/min. Temperature was 30°C. The detec-tion limit was 0.1 nM.

98 / Stress, gender and neuronal plasticity
Corticosterone.
For this assay, dexamethasone was used as internal standard. After addition of the internal standard, plasma was extracted with 3 ml of diethylether, vortexed for 5 min and then centrifuged for 5 min at 3000 x g. The extraction procedure was repeated twice. The organic phase was evaporated to dryness in a 50°C waterbath. The residue was reconstituted with 200 µL of mobile phase and 50 µL was injected into the HPLC system. The mobile phase (fl ow rate 1.0 mL/min) for the determination consisted of acetonitrile in ultrapure water (27:73 v/v). The concentration of both corticosterone and the internal standard was determined with UV detection at a wavelength of 254 nm. The detection limit was 10 nM.
Immunohistochemistry
Two hours after the start of the fi nal session, rats were deeply anaesthetized with halothane and transcardially perfused with 4% paraformaldehyde solution in 0.1M sodium phos-phate buffer (pH 7.4). The brains were carefully removed and post-fi xed in the same solu-tion overnight at 4°C, before being transferred to a potassium phosphate buffer (KPBS 0.02M, pH 7.4) and stored at 4°C. Following cryoprotection of the brains by overnight immersion in a 30% glucose solution, coronal serial sections of 40µm were prepared on a cryostat microtome. Sections were collected in KPBS with sodiumazide and stored at 4°C. All stainings were performed on free-fl oating sections under continuous agitation.
FOS and phospho-CREB immunohistochemistry.
The sections were preincubated in 0.3% H2O
2 for 15 min to reduce endogenous peroxi-
dase activity, before being incubated in primary polyclonal rabbit anti-FOS (Oncogene Research Products, brands of CN Biosciences, Inc, an affi liate of Merck KGaA, Darm-stadt, Germany; 1:10000 dilution in KPBS 0.02 M, pH 7.4) or anti-phospho-CREB antibodies (Upstate Biotechnology, Charlottesville, VA, USA: www.upstatebiotech.com; 1:1000 dilution in KPBS 0.02 M, pH 7.4) for 60-72 hr at 4°C. Subsequently, sections were washed with KPBS and incubated at room temperature with biotinylated goat anti-rabbit IgG (Vector Laboratories, Inc., Burlingame, CA, USA; 1:1000 dilution) followed by ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA, USA). After another wash, the reaction product was visualized by adding diaminobenzidine as chro-mogen and 1% H
2O
2 for 15 min. Then, the sections were washed, mounted on slides,
dehydrated and coverslipped with DePex.
Calcineurin immunohistochemistry.
The sections were preincubated in 0.3% H2O
2 in PBS (0.1M, pH 7.4) for 30 min to
reduce endogenous peroxidase activity followed by a blocking step with 3% normal rabbit

CHAPTER 4
Stress, gender and neuronal plasticity / 99
serum for 2 hrs. Sections were subsequently incubated in a primary polyclonal goat anti-calcineurin antibody (Santa Cruz Biotechnology, Inc.; 1:250 dilution in PBS 0.1M pH 7.4, 0.25% triton, 3% normal rabbit serum) overnight at room temperature. Afterwards sections were washed with PBS, and incubated at room temperature with biotinylated rabbit anti-goat IgG (Vector Laboratories, Inc., Burlingame, CA; 1:1000 dilution in PBS 0.1M pH 7.4, 0.25% triton, 3% normal rabbit serum) followed by the ABC complex (Vector Laboratories, Inc., Burlingame, CA). After another wash, visualization of the reac-tion product was accomplished by adding diaminobenzidine as chromogen and 1% H
2O
2
in H2O for 30 min. Finally sections were washed, mounted on slides and coverslipped
with DePex.
Antibody specifi city testing.
To control for cross-reactivity due to aspecifi c binding, negative controls were included by incubating several sections and performing the immunostaining without one of the anti-bodies needed for the reaction (primary, secondary, or tertiary). All reactions were negative thereby confi rming the specifi city of the antibodies.
Image analysis and counting procedure (semi-quantitative analysis).
FOS, phospho-CREB, and calcineurin immunoreactivity was quantifi ed using a com-puterized imaging analysis system by an observer who was blind to group assignment. Selected areas from regions of interest (ROIs) included the prefrontal cortex (prelimbic (PrL) and infralimbic (InfraL); Bregma +3.60 to +1.70), the anterior cingulate (AC: Bregma +3.20 to +0.95), and the posterior cingulate cortex (postCING; Bregma +3.20 to +0.95); the hippocampal dentate gyrus (DG: Bregma –2.00 to –3.90), the hippocampal CA1 (CA1; Bregma –2.45 to –4.60) and the hippocampal CA3 (CA3; Bregma –2.45 to –4.60); the paraventricular hypothalamic nucleus (PVN Bregma –1.08 to –2.00). Coor-dinates are with reference to the rat Swanson’s brain atlas 33. The ROIs were acquired and digitized using a Sony (SONY Corporation, Tokyo, Japan) charge-coupled device digital camera mounted on a LEICA Leitz DMRB microscope (LEICA, Wetzlar, Germany) to reduce quantifi cation errors. After image acquisition, all measurements were performed bilaterally and no left-right asymmetry of FOS, phospho-CREB, or calcineurin immu-noreactivity was found. When counting the immunoreactive cell nuclei, at least 4-5 sec-tions per region were analyzed. Using the computer-based LEICA image analysis system (LEICA Imaging System Ltd., Cambridge, England), quantifi cation of FOS, phospho-CREB, and calcineurin immunoreactivity was performed according to prior descriptions 17,28,34. For FOS and phospho-CREB, each digitized image was individually set at a thresh-old to subtract the background optical density. The numbers of positive nuclei above background were quantifi ed and the densities were reported as the number of positive nuclei/0.1 mm2. Calcineurin immunoreactivity was expressed as gray value intensity (GVI) of the selected region after correction with corpus callosum as background value and internal control for each section 17.

100 / Stress, gender and neuronal plasticity
RNA isolation and Quantitative RT-PCR.
Total RNA- and poly (A)+RNA-isolation.
Thirty minutes after the start of the fi nal session, rats used for the molecular biology were anesthetized with halothane and decapitated. The brains were dissected to separate the prefrontal cortex and the hippocampus, which were subsequently quick-frozen in liquid nitrogen and stored at -80°C. Total RNA was isolated from these brain areas by using Trizol (Life Technology, Gaithersburg, MD, USA) according to the manufacturer’s instructions. Integrity of total RNA was confi rmed on a 2% agarose gel and fi nal con-centrations were assessed spectrophotometrically. Poly(A)+RNA was in turn isolated from total RNA using Micro-FastTrack™ 2.0 Kit (Invitrogen) according to the manufacturer’s instructions.
First-strand cDNA synthesis.
First-strand cDNA was synthesized from 200 ng poly(A)+RNA using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s instructions. Before fi rst-strand synthesis was performed, contaminating genomic DNA was removed using Deoxyribonuclease I, Amplifi cation Grade (Invitrogen). The fi rst-strand synthesis was primed with oligo(dT), and the RNA template was not removed from the DNA:RNA hybrid.
Primers and PCR Conditions.
All primers used were designed with Primer Express® Software v2.0 (Applied Biosystems) using default parameters (sequences are listed in table 1). PCR reactions were performed on an ABI Prism 7900HT Sequence Detection System. Amplifi cation mixtures (25 µL divided over 3 wells, 7 µl per well) for all genes contained 2 ng cDNA from poly(A)+RNA template, 1 x SYBR Green PCR buffer, 3 mM MgCl2, 0.2 mM dATP, 0.2 mM dCTP, 0.2 mM dGTP 0.4 mM dUTP, 0.025U/µl Amplitaq Gold, 0.01 U/µl AmpErase UNG and 300 nM of each primer. The cycling conditions comprised of 2 minutes AmpErase UNG activity, 10 minutes Amplitaq Gold activation, 40 cycles at 95°C for 15 seconds and 60°C for 1 minute, and a dissociation stage: 15 seconds 95°C, 15 seconds 60°C, 15 seconds 95°C ramp rate 2%. The No Template Control contained all components except the cDNA template.

CHAPTER 4
Stress, gender and neuronal plasticity / 101
Real-time polymerase chain reaction (RT-PCR).
Real time detection was performed using an ABI PRISM 7900HT Sequence Detection System and SYBR Green PCR Core Reagents. Data analysis was performed using software provided with the instrument. The mRNAs of the different genes were relatively quanti-fi ed across groups with the Comparative CT Method. The CT value, or fractional cycle number at which the defi ned threshold is crossed above baseline, is predictive of the input amount of target and thus useful in quantifi cation of RNA. Here the threshold was set between 0.4 and 0.6 with a baseline range from 1 to 7. All RNA samples were measured in trio and the amount of target was normalized to an endogenous reference, the housekee-ping gene, RPS29 (∆CT). An additional reference (ß-actin) was included as an extra con-trol.
Statistical Analysis
Data are expressed as means ± standard error (SEM). Statistical signifi cance was deter-mined by performing one-way analysis of variance (ANOVA) and F test of variance on the corresponding parameter measures from experimental and control conditions. To compare values between male and female, and/or control and experimental conditions t tests for equal or unequal variance were performed. P<0.05 was defi ned as the level of signifi cance between groups. Calculations were made using Jandel SigmaStat statistical software.
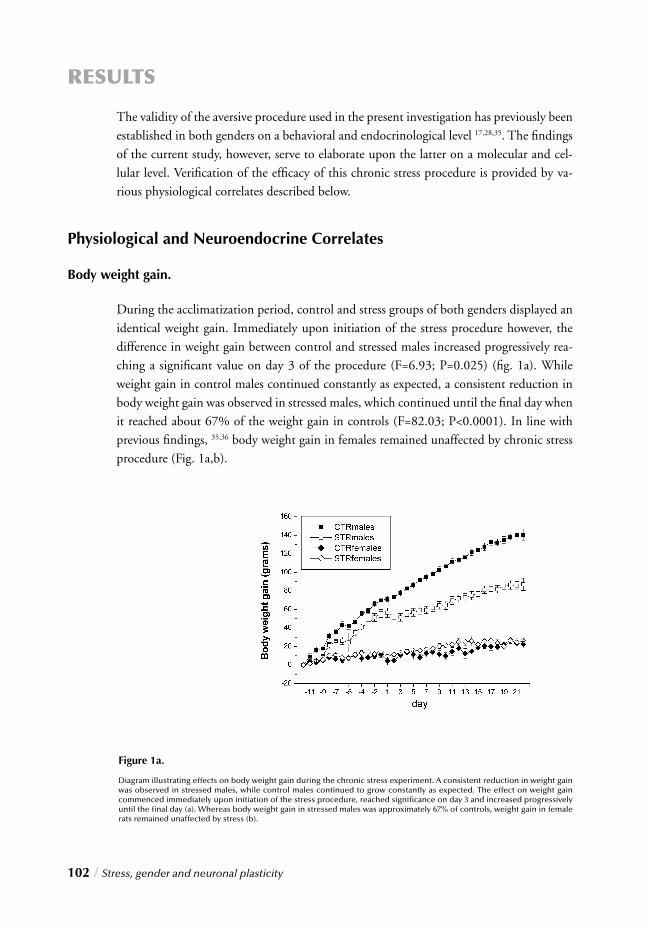
102 / Stress, gender and neuronal plasticity
RESULTS
The validity of the aversive procedure used in the present investigation has previously been established in both genders on a behavioral and endocrinological level 17,28,35. The fi ndings of the current study, however, serve to elaborate upon the latter on a molecular and cel-lular level. Verifi cation of the effi cacy of this chronic stress procedure is provided by va-rious physiological correlates described below.
Physiological and Neuroendocrine Correlates
Body weight gain.
During the acclimatization period, control and stress groups of both genders displayed an identical weight gain. Immediately upon initiation of the stress procedure however, the difference in weight gain between control and stressed males increased progressively rea-ching a signifi cant value on day 3 of the procedure (F=6.93; P=0.025) (fi g. 1a). While weight gain in control males continued constantly as expected, a consistent reduction in body weight gain was observed in stressed males, which continued until the fi nal day when it reached about 67% of the weight gain in controls (F=82.03; P<0.0001). In line with previous fi ndings, 35,36 body weight gain in females remained unaffected by chronic stress procedure (Fig. 1a,b).
Figure 1a.
Diagram illustrating effects on body weight gain during the chronic stress experiment. A consistent reduction in weight gain was observed in stressed males, while control males continued to grow constantly as expected. The effect on weight gain commenced immediately upon initiation of the stress procedure, reached signifi cance on day 3 and increased progressively until the fi nal day (a). Whereas body weight gain in stressed males was approximately 67% of controls, weight gain in female rats remained unaffected by stress (b).
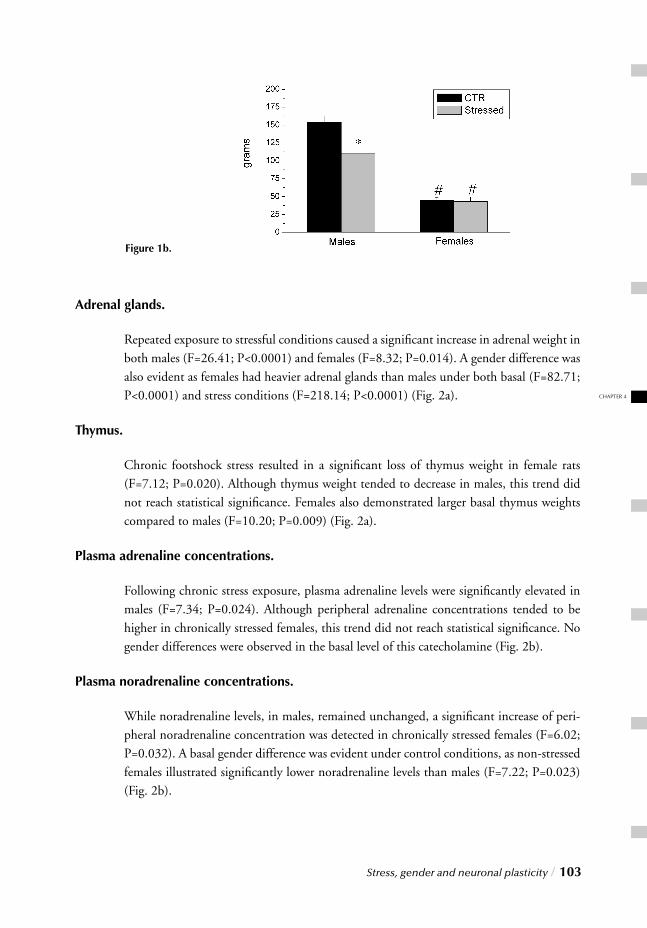
CHAPTER 4
Stress, gender and neuronal plasticity / 103
Adrenal glands.
Repeated exposure to stressful conditions caused a signifi cant increase in adrenal weight in both males (F=26.41; P<0.0001) and females (F=8.32; P=0.014). A gender difference was also evident as females had heavier adrenal glands than males under both basal (F=82.71; P<0.0001) and stress conditions (F=218.14; P<0.0001) (Fig. 2a).
Thymus.
Chronic footshock stress resulted in a signifi cant loss of thymus weight in female rats (F=7.12; P=0.020). Although thymus weight tended to decrease in males, this trend did not reach statistical signifi cance. Females also demonstrated larger basal thymus weights compared to males (F=10.20; P=0.009) (Fig. 2a).
Plasma adrenaline concentrations.
Following chronic stress exposure, plasma adrenaline levels were signifi cantly elevated in males (F=7.34; P=0.024). Although peripheral adrenaline concentrations tended to be higher in chronically stressed females, this trend did not reach statistical signifi cance. No gender differences were observed in the basal level of this catecholamine (Fig. 2b).
Plasma noradrenaline concentrations.
While noradrenaline levels, in males, remained unchanged, a signifi cant increase of peri-pheral noradrenaline concentration was detected in chronically stressed females (F=6.02; P=0.032). A basal gender difference was evident under control conditions, as non-stressed females illustrated signifi cantly lower noradrenaline levels than males (F=7.22; P=0.023) (Fig. 2b).
Figure 1b.
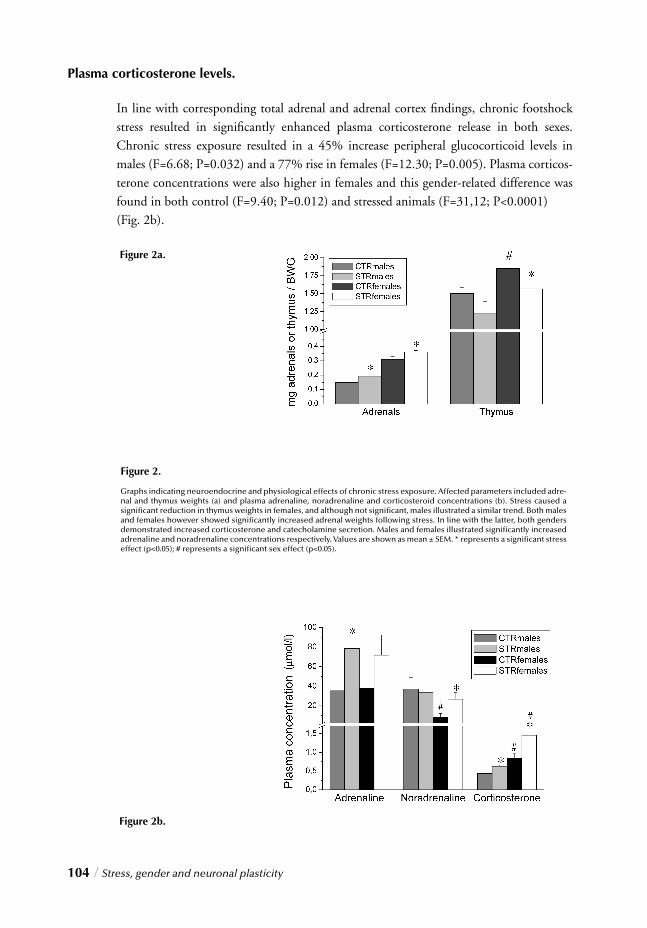
104 / Stress, gender and neuronal plasticity
Figure 2b.
Plasma corticosterone levels.
In line with corresponding total adrenal and adrenal cortex fi ndings, chronic footshock stress resulted in signifi cantly enhanced plasma corticosterone release in both sexes. Chronic stress exposure resulted in a 45% increase peripheral glucocorticoid levels in males (F=6.68; P=0.032) and a 77% rise in females (F=12.30; P=0.005). Plasma corticos-terone concentrations were also higher in females and this gender-related difference was found in both control (F=9.40; P=0.012) and stressed animals (F=31,12; P<0.0001) (Fig. 2b).
Figure 2.
Graphs indicating neuroendocrine and physiological effects of chronic stress exposure. Affected parameters included adre-nal and thymus weights (a) and plasma adrenaline, noradrenaline and corticosteroid concentrations (b). Stress caused a signifi cant reduction in thymus weights in females, and although not signifi cant, males illustrated a similar trend. Both males and females however showed signifi cantly increased adrenal weights following stress. In line with the latter, both genders demonstrated increased corticosterone and catecholamine secretion. Males and females illustrated signifi cantly increased adrenaline and noradrenaline concentrations respectively. Values are shown as mean ± SEM. * represents a signifi cant stress effect (p<0.05); # represents a signifi cant sex effect (p<0.05).
Figure 2a.
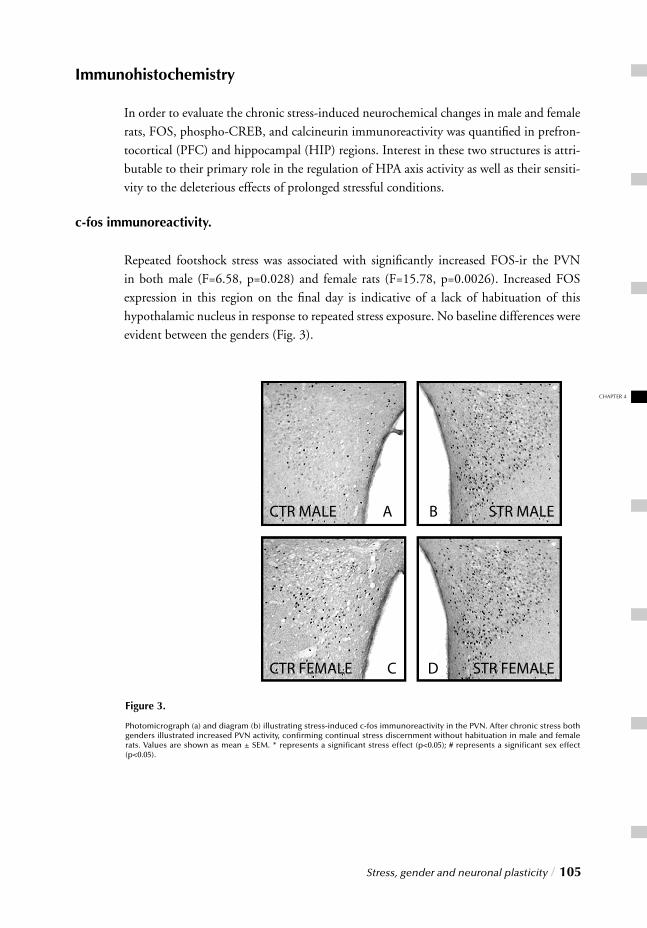
CHAPTER 4
Stress, gender and neuronal plasticity / 105
Immunohistochemistry
In order to evaluate the chronic stress-induced neurochemical changes in male and female rats, FOS, phospho-CREB, and calcineurin immunoreactivity was quantifi ed in prefron-tocortical (PFC) and hippocampal (HIP) regions. Interest in these two structures is attri-butable to their primary role in the regulation of HPA axis activity as well as their sensiti-vity to the deleterious effects of prolonged stressful conditions.
c-fos immunoreactivity.
Repeated footshock stress was associated with signifi cantly increased FOS-ir the PVN in both male (F=6.58, p=0.028) and female rats (F=15.78, p=0.0026). Increased FOS expression in this region on the fi nal day is indicative of a lack of habituation of this hypothalamic nucleus in response to repeated stress exposure. No baseline differences were evident between the genders (Fig. 3).
Figure 3.
Photomicrograph (a) and diagram (b) illustrating stress-induced c-fos immunoreactivity in the PVN. After chronic stress both genders illustrated increased PVN activity, confi rming continual stress discernment without habituation in male and female rats. Values are shown as mean ± SEM. * represents a signifi cant stress effect (p<0.05); # represents a signifi cant sex effect (p<0.05).
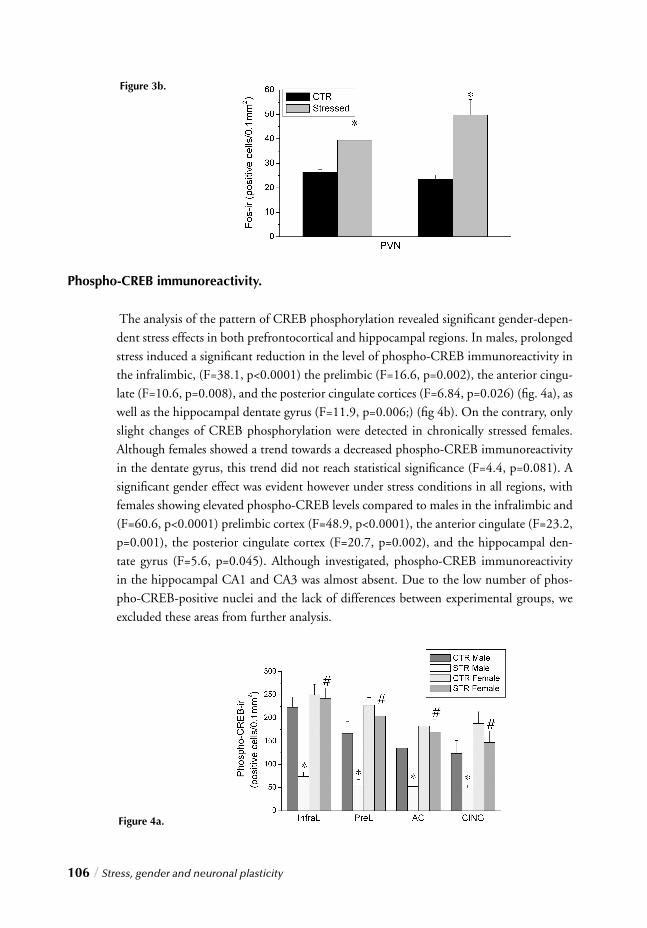
106 / Stress, gender and neuronal plasticity
Figure 4a.
Phospho-CREB immunoreactivity.
The analysis of the pattern of CREB phosphorylation revealed signifi cant gender-depen-dent stress effects in both prefrontocortical and hippocampal regions. In males, prolonged stress induced a signifi cant reduction in the level of phospho-CREB immunoreactivity in the infralimbic, (F=38.1, p<0.0001) the prelimbic (F=16.6, p=0.002), the anterior cingu-late (F=10.6, p=0.008), and the posterior cingulate cortices (F=6.84, p=0.026) (fi g. 4a), as well as the hippocampal dentate gyrus (F=11.9, p=0.006;) (fi g 4b). On the contrary, only slight changes of CREB phosphorylation were detected in chronically stressed females. Although females showed a trend towards a decreased phospho-CREB immunoreactivity in the dentate gyrus, this trend did not reach statistical signifi cance (F=4.4, p=0.081). A signifi cant gender effect was evident however under stress conditions in all regions, with females showing elevated phospho-CREB levels compared to males in the infralimbic and (F=60.6, p<0.0001) prelimbic cortex (F=48.9, p<0.0001), the anterior cingulate (F=23.2, p=0.001), the posterior cingulate cortex (F=20.7, p=0.002), and the hippocampal den-tate gyrus (F=5.6, p=0.045). Although investigated, phospho-CREB immunoreactivity in the hippocampal CA1 and CA3 was almost absent. Due to the low number of phos-pho-CREB-positive nuclei and the lack of differences between experimental groups, we excluded these areas from further analysis.
Figure 3b.
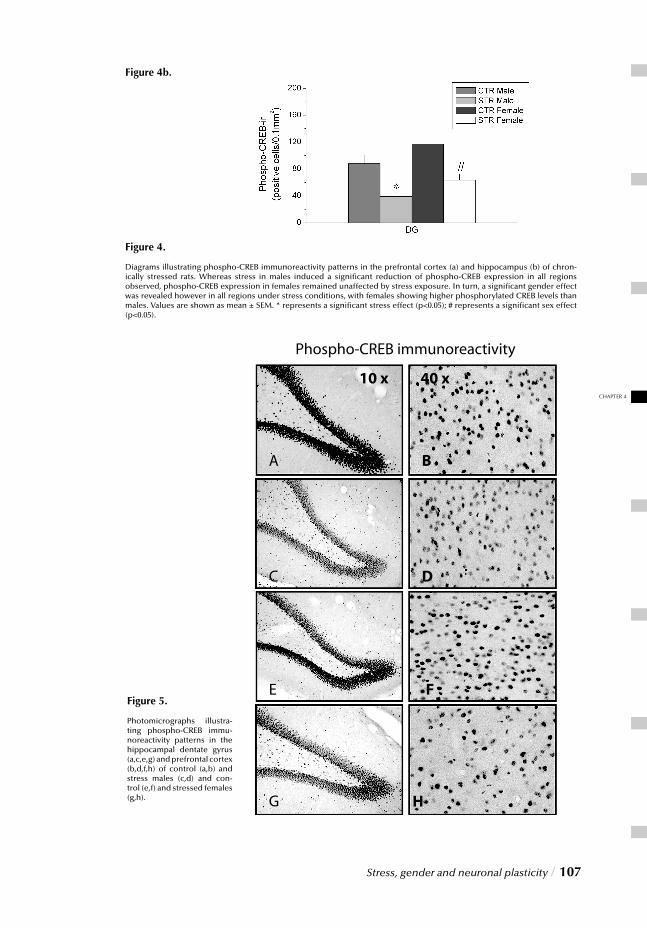
CHAPTER 4
Stress, gender and neuronal plasticity / 107
Figure 4b.
Figure 4.
Diagrams illustrating phospho-CREB immunoreactivity patterns in the prefrontal cortex (a) and hippocampus (b) of chron-ically stressed rats. Whereas stress in males induced a signifi cant reduction of phospho-CREB expression in all regions observed, phospho-CREB expression in females remained unaffected by stress exposure. In turn, a signifi cant gender effect was revealed however in all regions under stress conditions, with females showing higher phosphorylated CREB levels than males. Values are shown as mean ± SEM. * represents a signifi cant stress effect (p<0.05); # represents a signifi cant sex effect (p<0.05).
Figure 5.
Photomicrographs illustra-ting phospho-CREB immu-noreactivity patterns in the hippocampal dentate gyrus (a,c,e,g) and prefrontal cortex (b,d,f,h) of control (a,b) and stress males (c,d) and con-trol (e,f) and stressed females (g,h).
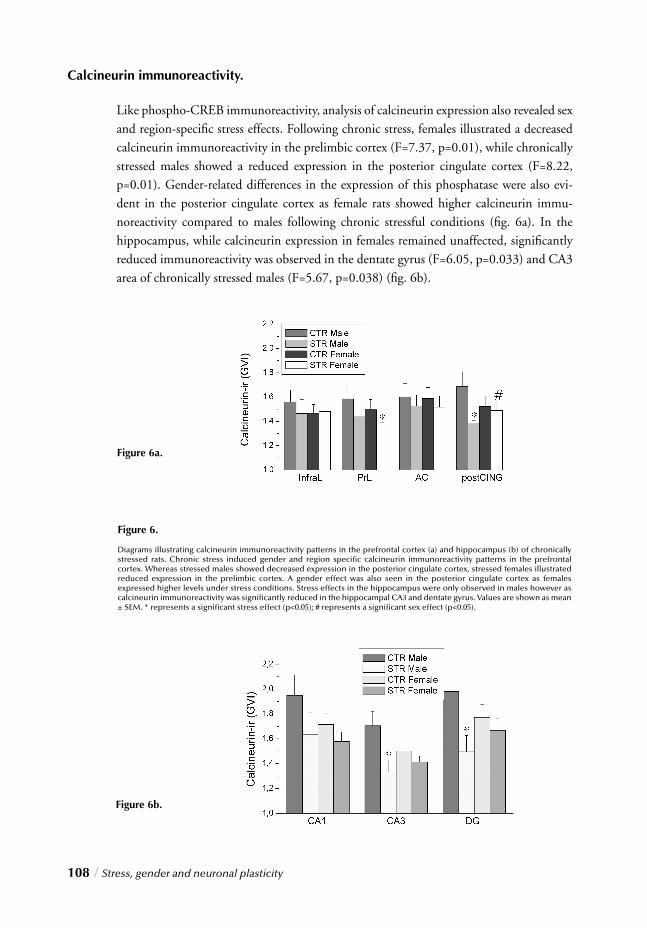
108 / Stress, gender and neuronal plasticity
Calcineurin immunoreactivity.
Like phospho-CREB immunoreactivity, analysis of calcineurin expression also revealed sex and region-specifi c stress effects. Following chronic stress, females illustrated a decreased calcineurin immunoreactivity in the prelimbic cortex (F=7.37, p=0.01), while chronically stressed males showed a reduced expression in the posterior cingulate cortex (F=8.22, p=0.01). Gender-related differences in the expression of this phosphatase were also evi-dent in the posterior cingulate cortex as female rats showed higher calcineurin immu-noreactivity compared to males following chronic stressful conditions (fi g. 6a). In the hippocampus, while calcineurin expression in females remained unaffected, signifi cantly reduced immunoreactivity was observed in the dentate gyrus (F=6.05, p=0.033) and CA3 area of chronically stressed males (F=5.67, p=0.038) (fi g. 6b).
Figure 6a.
Figure 6b.
Figure 6.
Diagrams illustrating calcineurin immunoreactivity patterns in the prefrontal cortex (a) and hippocampus (b) of chronically stressed rats. Chronic stress induced gender and region specifi c calcineurin immunoreactivity patterns in the prefrontal cortex. Whereas stressed males showed decreased expression in the posterior cingulate cortex, stressed females illustrated reduced expression in the prelimbic cortex. A gender effect was also seen in the posterior cingulate cortex as females expressed higher levels under stress conditions. Stress effects in the hippocampus were only observed in males however as calcineurin immunoreactivity was signifi cantly reduced in the hippocampal CA3 and dentate gyrus. Values are shown as mean ± SEM. * represents a signifi cant stress effect (p<0.05); # represents a signifi cant sex effect (p<0.05).
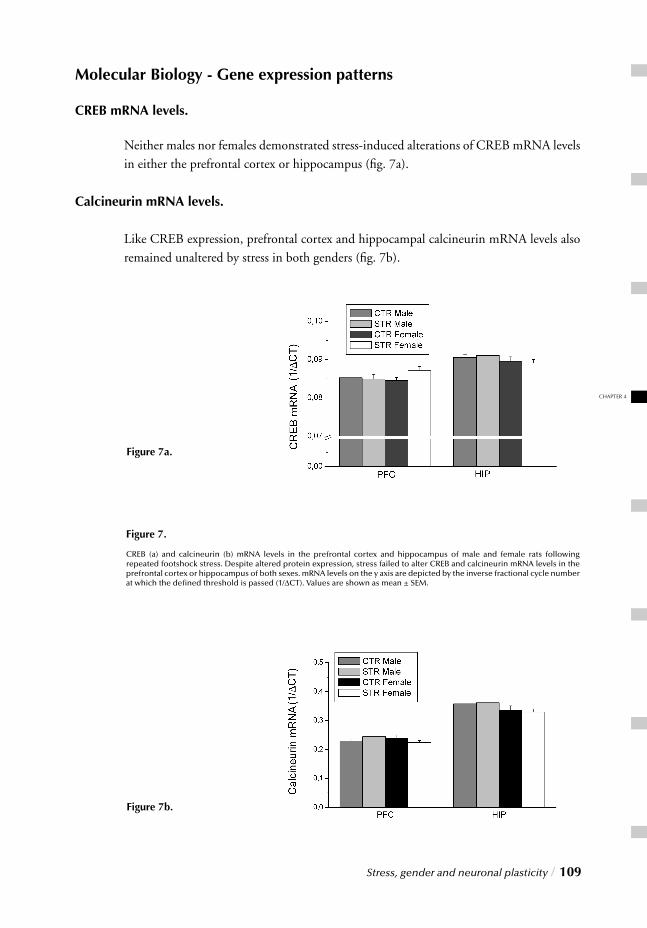
CHAPTER 4
Stress, gender and neuronal plasticity / 109
Molecular Biology - Gene expression patterns
CREB mRNA levels.
Neither males nor females demonstrated stress-induced alterations of CREB mRNA levels in either the prefrontal cortex or hippocampus (fi g. 7a).
Calcineurin mRNA levels.
Like CREB expression, prefrontal cortex and hippocampal calcineurin mRNA levels also remained unaltered by stress in both genders (fi g. 7b).
Figure 7.
CREB (a) and calcineurin (b) mRNA levels in the prefrontal cortex and hippocampus of male and female rats following repeated footshock stress. Despite altered protein expression, stress failed to alter CREB and calcineurin mRNA levels in the prefrontal cortex or hippocampus of both sexes. mRNA levels on the y axis are depicted by the inverse fractional cycle number at which the defi ned threshold is passed (1/∆CT). Values are shown as mean ± SEM.
Figure 7a.
Figure 7b.

110 / Stress, gender and neuronal plasticity
DISCUSSION
The phosphatase calcineurin and the transcription factor CREB have been established as essential components of the intracellular transduction apparatus involved in the regulation of neuronal plasticity 37,38. Stress however, particularly when persistent and severe, has been associated with impairment of neuroplasticity, as illustrated by reduced neuroge-nesis in the dentate gyrus 39,40 and atrophy of apical dendrites in selective prefrontocortical and hippocampal areas 4,7,41. Although our understanding of the exact mechanisms underlying the deleterious effects of stress on neuronal integrity is limited, it is intriguing to speculate that altered expression and/or phosphorylation of key proteins could account for stress-induced disruption of the coordinated regulation of synaptic plasticity thereby promoting structural and functional abnormalities. Interestingly, despite numerous reports of hip-pocampal atrophy following prolonged stress exposure in males, ample evidence also sup-ports a gender discrepancy with females being more sensitive to stress than males 42-45. In the present study, neurochemical changes in response to repeated stress were thus investi-gated in male and female rats, on a cellular and molecular level with immunohistochemis-try and quantitative RT-PCR, respectively. This data in turn may ultimately help to eluci-date two fundamental questions: Is gender a critical determinant for the impact of chronic stress on neuronal plasticity? If so, how do male and female rat reactivity to chronic stress compare at the cellular and molecular level? Various physiological and neuroendocrine parameters served to verify the severity of the stress procedure in rats of both genders. These included reduced body weight gain in males (fi g. 1a,b) and prolonged HPA axis hyperactivity in both sexes following chronic stress (fi g. 2,3). The latter was in turn substantiated by several adrenocortical parameters inclu-ding signifi cantly elevated plasma corticosterone and catecholamine levels (adrenaline in males and noradrenaline in females) (fi g. 2a), reduced thymus weights (in females) (fi g. 2b), enlarged adrenal glands (fi g. 2b), and increased FOS-ir in the paraventricular hypo-thalamic nucleus (fi g. 3). On a molecular level, sustained stress and subsequently elevated glucocorticoid levels have been associated with reduced expression of BDNF 12,13,46. Since CREB is known to play a crucial role in modulating BDNF transcription 47, a possible mechanism by which stress could limit availability of this fundamental neurotrophin is through decreased CREB transcription. Alternatively, chronic stress can also impair neuronal integrity by
affecting transcription of other specifi c genes involved in the modulation of synaptic plas-ticity such as calcineurin. Despite the marked neuroendocrine effects observed here fol-lowing stress however, quantitative RT-PCR analysis revealed unaltered CREB and calci-neurin mRNA levels in the frontal cortex and hippocampus of both genders (fi g. 7). This data therefore appears to refute a direct effect of chronic stress on transcript levels of these two genes as a possible mechanism by which stress impairs neuronal plasticity. Analysis of phospho-CREB and calcineurin immunoreactivity however proved more revealing. Whereas males illustrated stress-induced reduction of calcineurin immunoreac-

CHAPTER 4
Stress, gender and neuronal plasticity / 111
tivity and CREB phosphorylation in the prefrontal cortex and hippocampus (particularly the dentate gyrus and the cingulate cortex) female rats exposed to the same stressor, dis-played comparatively attenuated changes (fi g. 4,6). The CREB protein family is critical for induction of gene expression in all eukaryotic cells 24,25 and the crucial step underlying its ability to initiate the transcription of several target genes lies in its phosphorylation at serine 133 25. CREB phosphorylation is considered a molecular switch to transform short-lasting into long-lasting changes of synaptic plasticity 48, especially in the hippocampus, enabling long-term memory storage in mammals 23. Previous data from a similar stress experiment support a facilitatory role for CREB in the modulation of neuronal plasticity following acute stress in prefrontocortical regions 17. However, while acute stress has been reported to promote learning acquisition and memory formation 49,50, sustained stress has been associated with cognitive impairment and emotional dysregulation 51-53. Accordingly chronic stress caused signifi cantly reduced phospho-CREB immunoreactivity in the same cortical regions 17,28. In line with these prior fi ndings, the current results in males confi rm a stress-induced reduction of CREB phosphorylation in several subregions of the prefrontal cortex including the infralimbic, prelimbic, anterior and posterior cingulate cortex (fi g, 4a). Moreover this decrease was not only limited to cortical areas as this study revealed similar fi ndings in the hippocampal dentate gyrus (fi g. 4b), suggesting that the negative impact of chronic stress on CREB phosphorylation extends beyond the prefrontal cortex. Neurotrophins regulate various neuronal functions, including survival, differentia-tion, and plasticity. Although phosphorylated CREB mediates the transduction of neu-rotrophin signals and BDNF transcription 47, exposure of neurons to BDNF in turn also stimulates CREB phosphorylation 22. This can occur via at least two signaling path-ways: a Ras-MAPK-dependent pathway 26 and a calcium/calmodulin-dependent kinase-regulated pathway, activated by the release of intracellular calcium 22. Calcium infl ux also regulates protein phosphatase 2B (calcineurin) activity, another key component of the calcium/calmodulin-dependent kinase-regulated pathway 54.
Interestingly, functional interactions between calcium/calmodulin kinase and the MAPK cascade have been reported and both calcineurin and CREB appear to represent common links between these two intracellular pathways 55-57. More specifi cally, calcineu-rin plays a pivotal role in modulating neuronal plasticity by actively regulating the phos-phorylation state of several kinases and transcription factors such as Elk1 and CREB 58,59. In line with the decreased CREB phosphorylation, a signifi cant reduction of calcineurin
immunoreactivity was also observed in the frontal cortex (posterior cingulate) (fi g. 6a) and hippocampus (DG and CA3) (fi g. 6b) of stressed males. Given the fundamental role of this phosphatase in the maintenance of neuronal plasticity, persistent stress-induced reduction of calcineurin levels, particularly in the hippocampus (fi g 6b), may lead to abnormal activation of different intracellular transduction members, ultimately disrupting CREB phosphorylation. As noted however, an interesting fi nding emerged with regard to the gender-dependent nature of these results, as signifi cant gender divergent regulation of calcineurin immuno-

112 / Stress, gender and neuronal plasticity
Impaired regulation of HPA axis activity for instance, common to both genders may con-stitute the mechanism by which chronic stress leads to persistently elevated glucocorticoid levels and PVN activation, although the data shows gender-dependent means of attain-ment. Impaired prefrontocortical and/or hippocampal control over the HPA axis seem to represent the mechanism by which repeated footshocks lead to abnormal stress response regulation in male rats, whereas imbalanced GR/MR expression, disturbed autonomic function, and abnormal amygdala activity could instead illustrate the events involved in the development of abnormal HPA axis responses in female rats (fi gure 1,2). On a more cellular/molecular level, stress-induced effects of neuronal plasticity and synaptic integrity seem to represent a possible mechanism by which this effect is realized in males given the changes observed in their general gene expression, specifi c second mes-senger signalling cascade members and synaptic vesicle associated proteins. Considering the potential role of these proteins and functional neurotransmission in BDNF activity, the data may represent indirect indications for impaired neurotrophic plasticity in males. In view of the lesser effects observed on these parameters in females, it raises the interest-ing possibility that the differential impact of stress on neuronal plasticity may depend on gender-related mechanisms that either potentiate or suppress the deleterious infl uences of adverse events. Dependent upon the interpretation, in the case of the latter, ovarian hormones and their established neurotrophic effects, may protect the female brain against the insults of chronic stress by stimulating specifi c intracellular cascades underlying neu-ronal plasticity and preventing the reduced neuronal plasticity (CREB phosphorylation) observed in chronically stressed males. On the other hand, these neurotrophic effects might have a paradoxically damaging effect if these “blocked” neuronal/synaptic changes are required in order to attain an adaptive response. After all, neurogenesis/proliferation was predominantly affected in these females. Whatever the pathway however, both dys-regulations seen in the genders are known to contribute to fi nal volumetric changes often reported in stress and psychopathology. The fi nal question regarding the sensitivity of the sexes remains and unfortunately it is not as unambiguous as one might desire. In view of the data presented here and illustrated in fi gures 1 and 2, the answer to this seems to depend on the parameter of interest, since both sexes appear to be affected albeit through different mechanisms. In view of the future of stress research, it would be of interest from a clinical perspective to further pursue the neurobiological substrates and neurochemical profi les underlying effects of stress in both genders. As illustrated by the antidepressant data, each gender should be evaluated independently to determine the stress and/or treatment response and thus new avenues of therapeutic approach. The data presented here offer ample indications to argue for further investigations of the role of steroid hormones in multi-level research. With this in mind the paramount message is that fi ndings associated with one gender cannot be automati-cally extrapolated to the other; and that which may appear “negative” in one could repre-sent a physiological condition in the other and vice versa.

CHAPTER 4
Stress, gender and neuronal plasticity / 113
that the deleterious action of prolonged footshock exposure involve primarily post-trans-scriptional changes, at least in the male prefrontal cortex and hippocampus. Specifi cally chronic stress appeared to alter CREB phosphorylation and calcineurin levels without affecting coincident transcription of the respective genes. In turn this lack of altered gene expression may ultimately deprive cortical and hippocampal neurons of what is most essential, namely a continuous supply of newly synthesized CREB and calcineurin to antagonize the negative impact of prolonged stress exposure. Although one can only spe-culate the latter, current endeavors are being pursued to further explore this notion.

114 / Stress, gender and neuronal plasticity
REFERENCES1. Checkley, S. The neuroendocrinology of depression and chronic stress. Br. Med. Bull. 52, 597-617 (1996).
2. Fuchs, E. & Flugge, G. Stress, glucocorticoids and structural plasticity of the hippo- campus. Neurosci Biobehav Rev 23, 295-300 (1998).
3. Bremner, J.D. Does stress damage the brain? Biol. Psychiatry 45, 797-805 (1999).
4. McEwen, B.S. Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105-122 (1999).
5. Mizoguchi, K. et al. Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J. Neurosci. 20, 1568-1574 (2000).
6. Sheline, Y.I. 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol. Psychiatry 48, 791-800 (2000).
7. Wellman, C.L. Dendritic reorganization in pyramidal neurons in medial pre frontal cortex after chronic corticosterone administration. J. Neurobiol. 49, 245-253 (2001).
8. Lee, A.L., Ogle, W.O. & Sapolsky, R.M. Stress and depression: possible links to neuron death in the hippocampus. Bipolar. Disord. 4, 117-128 (2002).
9. Mizoguchi, K., Ishige, A., Aburada, M. & Tabira, T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience 119, 887-897 (2003).
10. Young, E.A. & Vazquez, D. Hypercortisolemia, hippocampal glucocorticoid receptors, and fast feedback. Mol. Psychiatry 1, 149-159 (1996).
11. Sullivan, R.M. & Gratton, A. Prefrontal cortical regulation of hypothalamic-pituitary-adrenal function in the rat and implications for psychopathology: side matters. Psychoneuroendocrinology 27, 99-114 (2002).
12. Smith, M.A., Makino, S., Kvetnansky, R. & Post, R.M. Effects of stress on neurotrophic factor expression in the rat brain. Ann. N. Y. Acad. Sci. 771, 234-239 (1995).
13. Ueyama, T. et al. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci. Res. 28, 103-110 (1997).
14. Nibuya, M., Takahashi, M., Russell, D.S. & Duman, R.S. Repeated stress increases catalytic TrkB mRNA in rat hippocam- pus. Neurosci. Lett. 267, 81-84 (1999).
15. D’Sa, C. & Duman, R.S. Antidepressants and neuroplasticity. Bipolar. Disord. 4, 183-194 (2002).
16. Gerges, N.Z., Aleisa, A.M., Schwarz, L.A. & Alkadhi, K.A. Chronic psychosocial stress decreases calcineurin in the den- tate gyrus: a possible mechanism for preservation of early ltp. Neuroscience 117, 869-874 (2003).
17. Kuipers, S.D., Trentani, A., Den Boer, J.A. & Ter Horst, G.J. Molecular correlates of impaired prefrontal plasticity in res- ponse to chronic stress. J. Neurochem. 85, 1312-1323 (2003).
18. Murphy, D.D. & Segal, M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element
binding protein. Proc. Natl. Acad. Sci. U. S. A 94, 1482-1487 (1997).
19. Alonso, M., Vianna, M.R., Izquierdo, I. & Medina, J.H. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol. Neurobiol. 22, 663-674 (2002).
20. Ying, S.W. et al. Brain-derived neurotrophic factor induces long-term potentia tion in intact adult hippocampus: requirement for ERK activa tion coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 22, 1532-1540 (2002).
21. Waltereit, R. & Weller, M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 27, 99-106 (2003).
22. Finkbeiner, S. et al. CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031-1047 (1997).
23. Abel, T. & Kandel, E. Positive and negative regulatory mechanisms that mediate long- term memory storage. Brain Res. Brain Res. Rev. 26, 360-378 (1998).
24. Silva, A.J., Kogan, J.H., Frankland, P.W. & Kida, S. CREB and memory. Annu. Rev. Neurosci. 21, 127-148 (1998).
25. Shaywitz, A.J. & Greenberg, M.E. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821-861 (1999).
26. Grewal, S.S., York, R.D. & Stork, P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol 9, 544-553 (1999).
27. Stanciu, M. et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200-12206 (2000).
28. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphosp horylation in medial prefrontocortical dendrites: implications for stress-related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
29. Kessler, R.C., McGonagle, K.A., Swartz, M., Blazer, D.G. & Nelson, C.B. Sex and depression in the National Comorbidity Survey. I: Life time prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85-96 (1993).
30. Ustun, T.B. Cross-national epidemiology of depression and gender. J. Gend. Specif. Med. 3, 54-58 (2000).
31. Remie, R., Knot, H.J., Bos, E.A. & Zaagsma, J. Characterization of presynaptic beta-adrenoceptors facilitating endogenous noradrenaline release in the portal vein of perma- nently cannulated, freely moving rats. Eur. J. Pharmacol. 157, 37-43 (1988).
32. Smedes, F., Kraak, J.C. & Poppe, H. Simple and fast solvent extraction system for selective and quantitative isolation of adrenaline, noradrenaline and dopa- mine from plasma and urine. J. Chromatogr. 231, 25-39 (1982).
33. Swanson LW. Brain maps: structure of the rat brain. Elsevier, Amsterdam (1998).
34. Lukyanetz, E.A. Evidence for colocalization of calcineurin and calcium channels in dorsal root ganglion neurons. Neuroscience 78, 625-628

CHAPTER 4
Stress, gender and neuronal plasticity / 115
(1997).
35. Westenbroek, C. et al. Gender-specifi c effects of social housing in rats after chronic mild stress exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 21-30 (2003).
36. Duncko, R., Kiss, A., Skultetyova, I., Rusnak, M. & Jezova, D. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 26, 77-89 (2001).
37. Mulkey, R.M., Endo, S., Shenolikar, S. & Malenka, R.C. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486-488 (1994).
38. Winder, D.G., Mansuy, I.M., Osman, M., Moallem, T.M. & Kandel, E.R. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell 92, 25-37 (1998).
39. Czeh, B. et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. U. S. A 98, 12796-12801 (2001).
40. Duman, R.S., Nakagawa, S. & Malberg, J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 25, 836-844 (2001).
41. Fuchs, E. et al. Psychosocial stress, glucocorticoids, and structural alterations in the tree shrew hippocampus. Physiol Behav. 73, 285-291 (2001).
42. Wood, G.E. & Shors, T.J. Stress facilitates classical conditioning in males, but impairs classical conditioning in females through activational effects of ovarian hormones. Proc. Natl. Acad. Sci. U. S. A 95, 4066-4071 (1998).
43. Shors, T.J., Chua, C. & Falduto, J. Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J Neurosci 21, 6292-6297 (2001).
44. Luine, V. Sex differences in chronic stress effects on memory in rats. Stress. 5, 205-216 (2002).
45. Bowman, R.E., Beck, K.D. & Luine, V.N. Chronic stress effects on memory: sex differences in perfor- mance and monoaminergic activity. Horm. Behav. 43, 48-59 (2003).
46. Rasmusson, A.M., Shi, L. & Duman, R. Downregulation of BDNF mRNA in the hippocampal dentate gyrus after re-exposure to cues previously associated with foo tshock. Neuropsychopharmacology 27, 133-142 (2002).
47. Tao, X., Finkbeiner, S., Arnold, D.B., Shaywitz, A.J. & Greenberg, M.E. Ca2+ infl ux regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709-726 (1998).
48. West, A.E. et al. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U. S. A 98, 11024-11031 (2001).
49. Buchanan, T.W. & Lovallo, W.R. Enhanced memory for emotional material following stress-level cortisol treatment in humans. Psychoneuroendocrinology 26, 307-317 (2001).
50. Shors, T.J. Acute stress rapidly and persistently enhances memory forma- tion in the male rat. Neurobiol Learn Mem 75, 10-29 (2001).
51. Krugers, H.J. et al. Exposure to chronic psychosocial stress and corticosterone in the rat: effects on spatial discrimination learning and hippo- campal protein kinase Cgamma immunoreactivity. Hippocampus 7, 427-436 (1997).
52. McEwen, B.S. Effects of adverse experiences for brain structure and function. Biol Psychiatry 48, 721-731 (2000).
53. Pavlides, C., Nivon, L.G. & McEwen, B.S. Effects of chronic stress on hippocampal long-term potentia- tion. Hippocampus 12, 245-257 (2002).
54. Shibasaki, F., Hallin, U. & Uchino, H. Calcineurin as a multifunctional regulator. J. Biochem. (Tokyo) 131, 1-15 (2002).
55. Curtis, J. & Finkbeiner, S. Sending signals from the synapse to the nucleus: possible roles for CaMK, Ras/ERK, and SAPK pathways in the regulation of synaptic plasticity and neuronal growth. J. Neurosci. Res. 58, 88-95 (1999).
56. Egea, J., Espinet, C. & Comella, J.X. Calcium infl ux activates extracellular-regulated kinase/mitogen- activated protein kinase pathway through a calmodulin-sensi tive mechanism in PC12 cells. J. Biol. Chem. 274, 75-85 (1999).
57. Wu, G.Y., Deisseroth, K. & Tsien, R.W. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensi- tive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. U. S. A 98, 2808-2813 (2001).
58. Bito, H., Deisseroth, K. & Tsien, R.W. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87, 1203-1214 (1996).
59. Tian, J. & Karin, M. Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin). J. Biol. Chem. 274, 15173-15180 (1999).
60. Naftolin, F., Leranth, C., Horvath, T.L. & Garcia-Segura, L.M. Potential neuronal mechanisms of estrogen actions in synapto- genesis and synaptic plasticity. Cell Mol. Neurobiol. 16, 213-223 (1996).
61. Cordoba Montoya, D.A. & Carrer, H.F. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats. Brain Res. 778, 430-438 (1997).
62. Murphy, D.D., Cole, N.B. & Segal, M. Brain-derived neurotrophic factor mediates estradiol-induced dendritic spine formation in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A 95, 11412-11417 (1998).
63. Woolley, C.S Estrogen-mediated structural and functional synaptic plasticity in the female rat hippocampus. Horm. Behav. 34, 140-148 (1998).
64. Bi, R., Foy, M.R., Vouimba, R.M., Thompson, R.F. & Baudry, M. Cyclic changes in estradiol regulate synaptic plasticity through the MAP kinase pathway. Proc. Natl. Acad. Sci. U. S. A 98, 13391-13395 (2001).
65. Wise, P.M., Dubal, D.B., Wilson, M.E., Rau, S.W. & Liu, Y. Estrogens: trophic and protective factors in the adult brain. Front Neuroendocrinol. 22, 33-66 (2001).
66. Toran-Allerand, C.D., Singh, M. & Setalo, G., Jr. Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol. 20, 97-121 (1999).

116 / Stress, gender and neuronal plasticity
67. Kim, J.S. et al. Enhancement of rat hippocampal long-term potentiation by 17 beta-estradiol involves mitogen-activated protein kinase- dependent and -independent components. Neurosci. Lett. 332, 65-69 (2002).
68. Sharrow, K.M., Kumar, A. & Foster, T.C. Calcineurin as a potential contributor in estradiol regulation of hippocampal synaptic function. Neuroscience 113, 89-97 (2002).
69. Carlstrom, L., Ke, Z.J., Unnerstall, J.R., Cohen, R.S. & Pandey, S.C. Estrogen modulation of the cyclic AMP response element- binding protein pathway. Effects of long-term and acute treat ments. Neuroendocrinology 74, 227-243 (2001).
70. Wade, C.B. & Dorsa, D.M. Estrogen activation of cyclic adenosine 5’-monophosphate response element mediated transcription requires the extracel- lularly regulated kinase/mitogen-activated protein kinase pathway. Endocrinology 144, 832-838 (2003).

CHAPTER 4
Stress, gender and neuronal plasticity / 117


PART III
MOLECULARLEVEL


5
IMMUNOHISTOCHEMICAL CHANGES INDUCED BY REPEATED FOOTSHOCK STRESS: REVELATIONS OF GENDER-BASED DIFFERENCES
As a growing amount of literature has proven, adverse experi-
ences, particularly when severe and persistent, play a pivotal role
in the development of neuronal dysfunctions and psychopatho-
logy. In the present study, the neurochemical changes induced
by acute and repeated footshock exposure were investigated at
the molecular and cellular level, using c-fos and phospho-ERK1/2
immunoreactivity, and gene expression arrays. Marked gender-
related differences were found following both acute and pro-
longed footshock exposure. Acute aversive conditioning resulted
in signifi cant immunohistochemical changes that might be criti-
cally involved in the modulation of fear-related responses, espe-
cially in males. Prolonged footshock exposure, in contrast, was
associated with sustained hypothalamic-pituitary-adrenal axis
hyperactivity, differential gender-related patterns of cortical-limbic
activity, and abnormal neuronal plasticity, especially in medial pre-
frontocortical regions. This data may provide additional insights
into the understanding of the neural circuits underlying the effects
of acute and repeated footshock exposure and clarify some of the
mechanisms involved in the development of stress-related neuro-
nal abnormalities.
Kuipers S.D.,* Trentani A.,* te Meerman G.J., Beekman J., Ter Horst G.J., Den Boer J.A. (* Co-fi rst authorship) Neurobiology of Disease, 2003, 14: 602-618


Stress and gender mediated neuronal impairments / 123
CHAPTER 5
INTRODUCTION
Recent advances have been made in understanding the changes of neuronal plasticity in
response to stress. Acute stressful experiences, for instance, facilitate the consolidation of
new memories and promote cognitive processes 1,2 while adverse experiences, particularly
when severe and persistent, may contribute to the development of neuronal dysfunctions
and psychopathology 3. At the cellular level, evidence has emerged to indicate dendritic
atrophy and neuronal loss in response to stress 4-7. At the molecular level, it has been
suggested that these abnormalities, detected mostly in the hippocampus and prefrontal
cortex, result from a decreased neuronal plasticity associated with persistent elevation of
glucocorticoid levels 8-10. Although in the short run, adrenal steroids play a critical role
in the acquisition of fear-conditioned responses 11,12, prolonged exposure to elevated glu-
cocorticoid levels results in functional and structural abnormalities and cognitive impair-
ment 4,7,13,14. It is of interest to note that although considerable progress has been made in
the elucidation of neurobiological substrates underlying the acute stress response, chronic
stress-induced neurochemical changes remain poorly understood. Furthermore, although
gender represents a critical aspect in both sensitivity to stress and psychopathology 15,16,
most of the clinical and preclinical research concerning stress-related neuronal abnormali-
ties have been conducted in males 17.
With this in mind, the cellular and molecular changes associated with short-term (2 days)
and prolonged footshock stress (20 days) were investigated in male and cyclic female rats,
in an attempt to gain new insights into the neuronal circuits modulating the response
to acute and chronic footshock stress as well as the mechanisms underlying the biphasic
effects of stress on cognitive and emotional processes. By using identical settings (5 foot-
shocks delivered randomly during 30-minute sessions) but extending the length of expo-
sure from 2 to 20 days, these footshock procedures might prove a potentially useful model
for investigating the dynamics of stress-induced disruption of cognitive processing in a
gender comparative setting. Footshock-induced neurochemical changes were examined
using molecular and immunohistochemical techniques, including c-fos immunoreactiv-
ity (FOS-ir), phospho-ERK1/2 expression, and gene expression microarrays as markers
of neuronal activity 18-20 and neuronal plasticity 21-23. This data may thus contribute to
the understanding of the mechanisms underlying gender-related differences in emotional
processing and their relationship with the development of stress-induced cortical-limbic
dysfunctions.

124 / Stress and gender mediated neuronal impairments
MATERIALS AND METHODS
Animals
The experiments were performed using adult male (n=28: 212-240 g at the beginning of the experiments) and cyclic female Wistar rats (n=25: 195-212 g). The animals were housed individually (cages 45 x 28 x 20 cm) with food and water available ad libitum, and maintained on a 12/12-hr light/dark cycle. All rats were weighed and handled daily for 5-8 min to minimize the non-specifi c stress response. The experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and with the guidelines of the Animal Bio-Ethics Committee of the University of Groningen (FDC: 2509).
Footshock procedure
The rodent test-chamber consists of a box containing an animal space placed on a grid fl oor connected to a shock generator and scrambler. Test-rats received one session of 30 min/day in the footshock box during which 5 inescapable footshocks were given (0.8 mA in intensity and 8 sec in duration: unconditioned stimulus; US) with different inter-shock intervals in order to make the procedure as unpredictable as possible. Each footshock was preceded by 10 sec of light (conditioned stimulus; CS). Control rats followed the same schedule in an identical setup but were exposed to CSs only without receiving any shocks. This conditioning procedure was followed for 2 (acute conditioning) or 20 days (chronic conditioning). The fi nal day of each experiment (3rd for the acute and 21st for the chronic) all rats were placed in the footshock box and exposed to 5 CSs only, without receiving any painful footshocks. This allowed investigation of the pattern of protein expression (FOS-ir) and phosphorylation (phospho-ERK1/2) induced by identical and painless sti-muli hereby avoiding exposure of animals to physical stress.
Acute experiment.
Six male and six cyclic female rats were used in the acute experiment. Test-rats were con-ditioned for two consecutive days. The third and last day all rats were subjected to 5 CSs.
Chronic experiment.
Eleven males and ten females were used in this experiment. Test-rats were conditioned for 20 consecutive days. On the fi nal (21st) day all rats received 5 CSs without consequent footshocks.

Stress and gender mediated neuronal impairments / 125
CHAPTER 5
Control rats.
Eleven males and nine females were used as control animals. These were exposed to the same stimuli as the stressed rats (as they were housed in the same room and similarly exposed to the footshock box and CSs). They did not however receive USs during the entire duration of the experiment.
Physiological and neuroendocrine changes following repeated footshock exposure
To defi ne the dynamics of the response to repeated footshock stress, various physiologi-cal and neuroendocrine parameters were measured. Weight gain was monitored on a daily basis throughout the experiment, and upon termination, plasma corticosterone concen-trations were measured and adrenal glands were removed and weighed. Blood samples were also drawn by transcardial puncture immediately upon termination and stored at -20°C. These samples were then used to determine plasma corticosterone and adrenaline concentrations with HPLC. Graphs were constructed to serve as a reference to verify the severity of stress perceived by the animals.
Extraction and Chromatography
Adrenaline.
Adrenaline was extracted from plasma using liquid/liquid extraction with 3,4-dihydroxy-benzylamine as internal standard 24,25. Briefl y, plasma adrenaline was bound to diphenyl-borate-ethanolamine at pH 8.6. The extraction was performed with n-heptane (contain-ing 1% octanol and 25% tetraoctylammoniumbromide). Finally, adrenaline was extracted from the organic phase with diluted acetic acid. Adrenaline (20 µl acetic acid extract) was analysed using an HPLC/auto-injector (CMA, Sweden) and a Shimadzu LC-10AD pump (Kyoto, Japan) connected to a reversed phase column (Hypersil, C18, 3µm, 150x2.0mm), followed by an electrochemical detector (Antec Leyden, The Netherlands) working at a potential setting of 500mV vs. Ag/AgCl reference. The mobile phase consisted of 50mM acetate buffer, 150mg/l octane sulphonic acid, 150mg/l tetramethylammonium, 15mg/ml Na
2EDTA and 3% methanol, adjusted to pH 4.1. The fl ow-rate was 0.35ml/min. Tem-
perature was 30°C. The detection limit of the method was 0.1nM.
Corticosterone.
For the assay, dexamethasone was used as internal standard. After addition of the internal standard, plasma was extracted with 3ml of diethylether, vortexed for 5 min and then cen-trifuged for 5 min at 3000 x g. The extraction procedure was repeated twice. The organic phase was evaporated to dryness in a 50°C waterbath. The residue was reconstituted with

126 / Stress and gender mediated neuronal impairments
200µL of mobile phase and 50µL was injected into the HPLC system. The mobile phase (fl ow rate 1.0mL/min) for the determination consisted of acetonitrile in ultrapure water
(27:73 v/v). The concentration of both corticosterone and internal standard was deter-
mined with UV detection at a wavelength of 254nm. The detection limit of the method
was 10nM.
Histological procedure - Immunohistochemistry
Two hours after the beginning of the fi nal session, the rats were terminated with an over-
dose of halothane, which preceded a transcardial perfusion with 4% paraformaldehyde
solution in 0.1M sodium phosphate buffer (pH 7.4). The brains were carefully removed
and post-fi xed in the same solution overnight at 4°C, before being transferred to a potas-
sium phosphate buffer (KPBS 0.02 M, pH 7.4) and stored at 4°C. Following cryoprotec-
tion of the brains by overnight immersion in a 30% glucose solution, coronal serial sec-
tions of 40 µm were prepared on a cryostat microtome. Sections were collected in KPBS
with sodiumazide and stored at 4°C.
c-fos and phospho-ERK1/2 immunoreactivity.
The stainings were performed on free-fl oating sections under continuous agitation. The
sections were preincubated in 0.3% H2O
2 for 15 min to reduce endogenous peroxidase
activity, before being incubated in primary monoclonal mouse anti-phospho-ERK1/2
(New England Biolabs, Inc., Beverly, MA, USA; www.neb.com; 1:5000 dilution in KPBS
0.02 M, pH 7.4, overnight at room temperature) or polyclonal rabbit anti-FOS anti-
body (Oncogene Research Products, brands of CN Biosciences, Inc, an affi liate of Merck
KGaA, Darmstadt, Germany; 1:10000 dilution in KPBS 0.02 M, pH 7.4, 60-72 hours at
4˚C) depending on the primary antibody host. Subsequently, sections were washed with
KPBS and incubated at room temperature with biotinylated goat anti-mouse or goat anti-
rabbit IgG (Vector Laboratories, Inc., Burlingame, CA, USA; 1:1000 dilution) followed
by ABC complex (Vector ABC kit, Vector Laboratories, Burlingame, CA, USA). After
another wash, the reaction product was visualized by adding diaminobenzidine as chro-
mogen and 1% H2O
2 for 15 min. Thereafter, sections were washed, mounted on slides,
dehydrated and coverslipped with DePex.
Antibody specifi city testing.
To control for cross-reactivity due to aspecifi c binding, immunostainings were performed
by incubating several sections without the presence of one of the antibodies needed for
the reaction (primary, secondary or tertiary antibody). All these reactions were negative
thereby confi rming the specifi city of all antibodies used.

Stress and gender mediated neuronal impairments / 127
CHAPTER 5
Image analysis and counting procedure (semi-quantitative analysis).
FOS immunoreactive cells were quantifi ed in 14 different brain regions or subregions with
reference to the rat Swanson’s brain atlas 26 while the quantifi cation of phospho-ERK1/2
immunoreactivity was limited to the medial prefrontal cortex (prelimbic and infralimbic
areas) where an abnormal phosphorylation of these enzymes was found in chronically
stressed male rats 10. This quantifi cation was performed by an observer who was blind to
group assignment. For counting of the immunoreactive cell nuclei, at least 4-5 sections
per brain area were analyzed. ROIs included prefrontal (prelimbic and infralimbic area;
mPFC: Bregma +3.60 to +1.70) and cingulate cortices (AC: Bregma +3.20 to +0.95); hip-
pocampal CA1 (CA1: Bregma –2.45 to –4.60) and dentate gyrus (DG: Bregma -2.00 to
–3.90); central (CeA: Bregma –1.53 to –2.85), lateral (LaA: Bregma –2.00 to -3.70), baso-
lateral (BslA: Bregma –1.78 to –3.25) and medial (MeA: Bregma –1.78 to –3.25) nuclei
of the amygdala; paraventricular (PVT: Bregma –1.33 to –3.90), dorsomedial (DMT:
Bregma –2.00 to –3.90) and centromedial (CMT: Bregma –1.53 to –3.90) thalamic
nuclei; paraventricular (PVN: Bregma –1.08 to –2.00) and dorsomedial (DMH: Bregma
–2.45 to –3.70) hypothalamic nuclei; dorsal (DR: Bregma –7.10 to –9.25) and medial
(MR: Bregma –9.25 to –10.35) raphe nuclei 26. The areas from structures of interest
(ROI) were digitized using a Sony (SONY Corporation, Tokyo, Japan) charge-coupled
device digital camera mounted on a LEICA Leitz DMRB microscope (LEICA, Wetzlar,
Germany) at x100 magnifi cation. Each digitized image was individually set at a threshold
to subtract the background optical density, and the numbers of cell nuclei above back-
ground were counted using the computer-based image analysis system LEICA (LEICA
Imaging System Ltd., Cambridge, England). After image acquisition, FOS positive nuclei
and phospho-ERK1/2-stained dendrites were quantifi ed. All areas were measured bilater-
ally (no left-right asymmetry for FOS and phospho-ERKs immunoreactivity was found)
and therefore the resulting data was reported as number of positive cells/0.1mm2 (FOS-ir)
or number of horizontal (H) and vertical (V) intersections (H+V contacts) between posi-
tive dendrites and an imaginary detection grid (composed by 514 horizontal x 698 ver-
tical lines) present in the quantifi cation fi eld (H+V intersections/0.1mm2) 10. The abso-
lute regional FOS and phospho-ERK1/2 densities (mean±standard error (SEM)) for each
region were reported (see Table 1). F tests of variance were run on numbers of immunore-
active cell nuclei from individual brain regions from experimental and control conditions.
That value determined whether t tests for equal or unequal variance were performed to
compare the cell counts from individual brain regions of control and experimental condi-
tions. P < 0.05 was defi ned as the level of signifi cance between groups.

128 / Stress and gender mediated neuronal impairments
Histological procedure - Molecular biology
Tissue and RNA Preparation.
Thirty minutes after the start of the fi nal session, rats used for molecular biology were anesthetized with halothane and decapitated. The prefrontal cortex was dissected, quick-frozen in liquid nitrogen and stored at -80°C. Total RNA was isolated from the prefrontal cortex of each animal by using Trizol (Life Technology, Gaithersburg, MD, USA) accord-ing to the manufacturer’s instructions. Integrity of total RNA was confi rmed on an agarose gel and fi nal concentrations were assessed spectrophotometrically.
cDNA microarray.
RNA was extracted from the prefrontal cortex of 4 or 5 rats within the groups participat-ing in the chronic experiment and their controls. RNA (2-5mg/rat), subsequently con-verted into a 32P-labeled fi rst-strand cDNA, was used to hybridize cDNA microarrays (rat
s

Stress and gender mediated neuronal impairments / 129
CHAPTER 5
atlas cDNA array 1.2; Clontech, Palo Alto, CA, USA). Use of a broad coverage array instead of a stress array was intentionally chosen because of our interest in the role of tran-scription factors and second messengers in stress-induced neuronal dysfunction, which could involve the expression of numerous candidate genes. In this microarray, plasmid and bacteriophage DNAs are included as negative controls, along with several housekee-ping cDNAs as positive controls. A complete list of the genes and controls spotted on the array, as well as array coordinates and GenBank accession numbers, is available at Clon-tech’s web site, (http://www.clontech.com). In order to suppress non-specifi c background each membrane was prehybridized for 30 min at 68ºC in 5ml of hybridization solution (ExpressHyb, Clontech) with continuous agitation. Hybridization was subsequently car-ried out by the addition of the denatured, labeled cDNA to the prehybridization solution at 68ºC for overnight incubation to reach a fi nal probe concentration of 2-5 x 106cpm/ml. Membranes were stringently washed with continuous agitation at 68ºC in 2 x SSC, 1% SDS (4x30 min) and then in 0.1 SSC, 0.5% SDS (30 min). After a fi nal rinse in 0.1 x SSC (5 min), membranes were mounted on Whatman paper, plastic-wrapped, exposed to x-ray fi lm overnight at -80ºC followed by exposure to a phosphoimager screen for 3 days.
PhosphoImaging analysis.
Membranes were scanned using a Molecular Dynamics STORM PhosphoImager (Mole-cular Dynamics, Inc., Sunnyvale, CA, USA), and images were analyzed by ImageQuant (Molecular Dynamics, Inc., Sunnyvale, CA, USA). According to the Array manufacturer, Clontech, the radioactive cDNA signal is linear for RNAs present at levels of 0.01-3% of the total RNA population. An admonition of this quantitative analysis however, is that the accuracy for the extremely low abundant genes may not be reliable due to the detection limitation of this technique. The expression level for each gene was measured by the phos-phoimager in arbitrary signal intensity units. This original raw output was subsequently used to perform the statistical analysis.
Statistics.
To perform the following analysis, use was made of specifi c Delphi 5.0 data transforma-tion software to convert data from Clontech format to SPSS readable datasets (available on demand) and SPSS 11.0 software. Data from the 19 hybridizations was analyzed by log transforming the datapoints. Subsequently, factor analysis was performed to identify the common component in the (log) data. This common component serves as the ref-erence value, comparable to a control labeling in two-dye array analysis. Subtraction of this common component yields a log ratio value, used for further analysis. The data was checked for systemic positional effects, using analysis of variance on blocks of spots. A signifi cant effect was found in the y coordinate of the genes, but since this only explained 1% of the variance, a correction step for this effect would have minimal infl uence on data quality, and was thus omitted. To further analyze the data, a reference set of genes was

130 / Stress and gender mediated neuronal impairments
chosen against which to test hypotheses with regard to the experimental variation. The genes selected for inclusion in the analysis set were those which displayed the greatest variation compared to other genes, while the reference set contained the genes with the least amount of variation between arrays. To verify the sensitivity of the results for this selection procedure, the criterion for inclusion of genes in the test set versus the reference set was variation in log expression ratio over a range of standard deviations (>1.5, >1.8, >1.9, >2.5). In order to identify the strongest alterations of individual genes a plot was made of the common variation or expression of the genes (fi rst principal component of the subset of genes selected for analysis) against the interarray differences or variation (second principal component). A test of signifi cance of individual gene expression alterations was not done, because the observed variation was quite small and the required multiple test-ing correction would have given negative test results. The outliers in table 3 are genes most likely responsible for the observed experimental effects. The statistical analysis was performed on the average log expression ratio of the genes included in the test set, because we reasoned that only an aggregate of individual genes would yield enough information about the response of the subgroups.
RESULTS
Physiological and neuroendocrine changes following repeated foot-shock exposure
To defi ne the dynamics of the response to prolonged footshock stimulation, various phy-
siological and neuroendocrine parameters were measured, including body weight gain
throughout the experiment, plasma corticosterone concentrations and adrenal weights
upon termination.
Body weight gain
Body weights were measured daily during the acclimatization period and the chronic stress
procedure in control and stressed rats (fi g. 1a). During the acclimatization period both
groups showed an identical weight gain. Immediately after the initiation of stress exposure
however, a consistent reduction in body weight gain was observed in stressed males, while
weight gain in non-stressed animals continued constantly as expected. The difference in
weight gain between non-stressed and stressed males increased progressively reaching a
signifi cant value on day 6 of the procedure (F=6.13, p<0.033) and continued to increase
until the fi nal day (F=18.09, p<0.0019). No differences were found in chronically stressed
females. This fi nding was in accordance with previous preclinical data showing that stress
exposure does not affect body weight gain in female rats as much as it does in males 27.
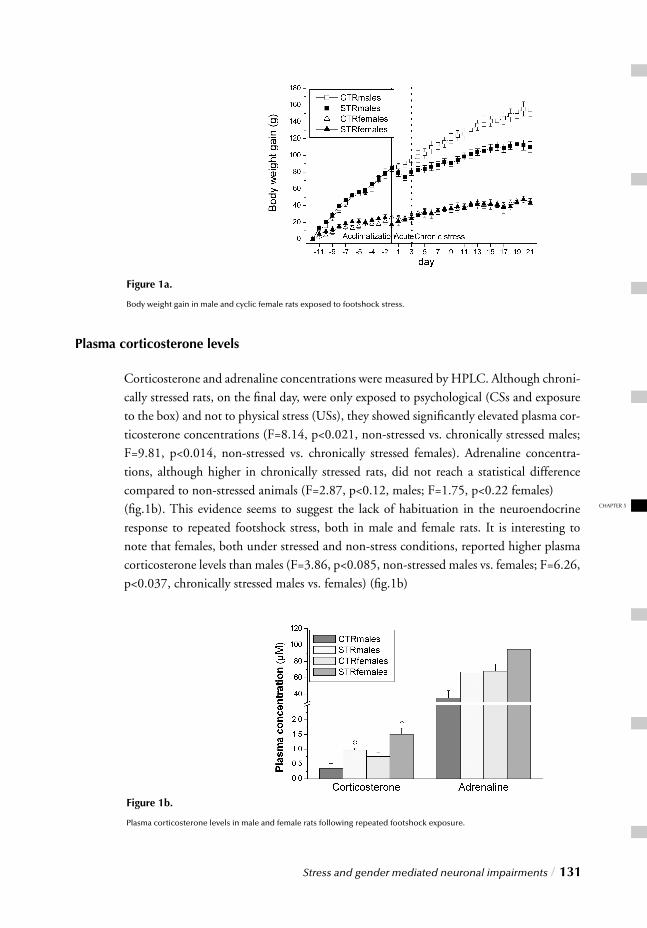
Stress and gender mediated neuronal impairments / 131
CHAPTER 5
[Fig. 1a]
Plasma corticosterone levels
Corticosterone and adrenaline concentrations were measured by HPLC. Although chroni-cally stressed rats, on the fi nal day, were only exposed to psychological (CSs and exposure to the box) and not to physical stress (USs), they showed signifi cantly elevated plasma cor-ticosterone concentrations (F=8.14, p<0.021, non-stressed vs. chronically stressed males; F=9.81, p<0.014, non-stressed vs. chronically stressed females). Adrenaline concentra-tions, although higher in chronically stressed rats, did not reach a statistical difference compared to non-stressed animals (F=2.87, p<0.12, males; F=1.75, p<0.22 females) (fi g.1b). This evidence seems to suggest the lack of habituation in the neuroendocrine response to repeated footshock stress, both in male and female rats. It is interesting to note that females, both under stressed and non-stress conditions, reported higher plasma corticosterone levels than males (F=3.86, p<0.085, non-stressed males vs. females; F=6.26, p<0.037, chronically stressed males vs. females) (fi g.1b)
[Fig. 1b]
Figure 1b.
Plasma corticosterone levels in male and female rats following repeated footshock exposure.
Figure 1a.
Body weight gain in male and cyclic female rats exposed to footshock stress.
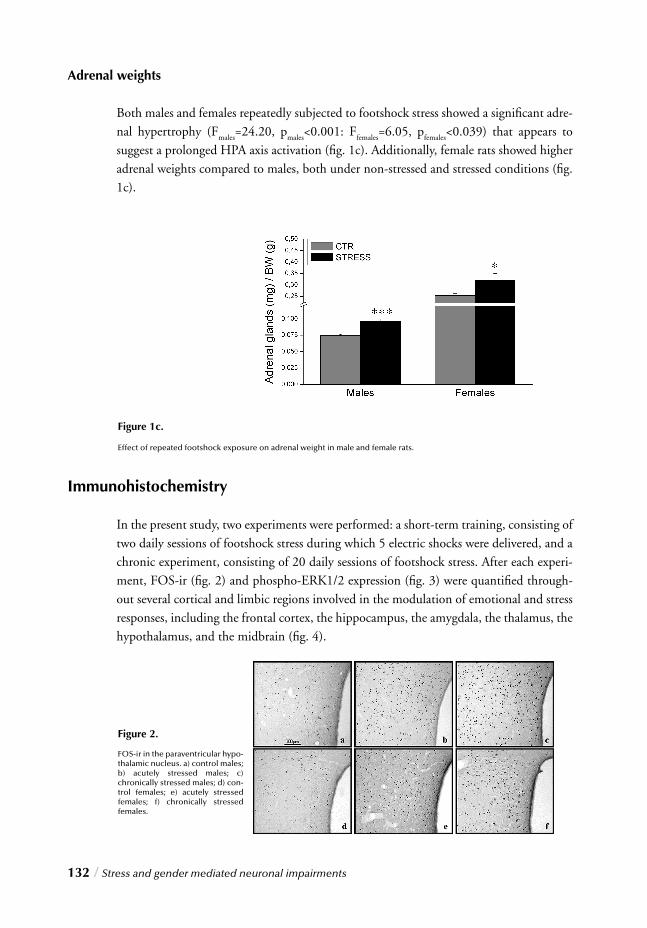
132 / Stress and gender mediated neuronal impairments
Adrenal weights
Both males and females repeatedly subjected to footshock stress showed a signifi cant adre-nal hypertrophy (F
males=24.20, p
males<0.001: F
females=6.05, p
females<0.039) that appears to
suggest a prolonged HPA axis activation (fi g. 1c). Additionally, female rats showed higher adrenal weights compared to males, both under non-stressed and stressed conditions (fi g. 1c).
Immunohistochemistry
In the present study, two experiments were performed: a short-term training, consisting of two daily sessions of footshock stress during which 5 electric shocks were delivered, and a chronic experiment, consisting of 20 daily sessions of footshock stress. After each experi-ment, FOS-ir (fi g. 2) and phospho-ERK1/2 expression (fi g. 3) were quantifi ed through-out several cortical and limbic regions involved in the modulation of emotional and stress responses, including the frontal cortex, the hippocampus, the amygdala, the thalamus, the hypothalamus, and the midbrain (fi g. 4).
Figure 1c.
Effect of repeated footshock exposure on adrenal weight in male and female rats.
Figure 2.
FOS-ir in the paraventricular hypo-thalamic nucleus. a) control males; b) acutely stressed males; c) chronically stressed males; d) con-trol females; e) acutely stressed females; f) chronically stressed females.
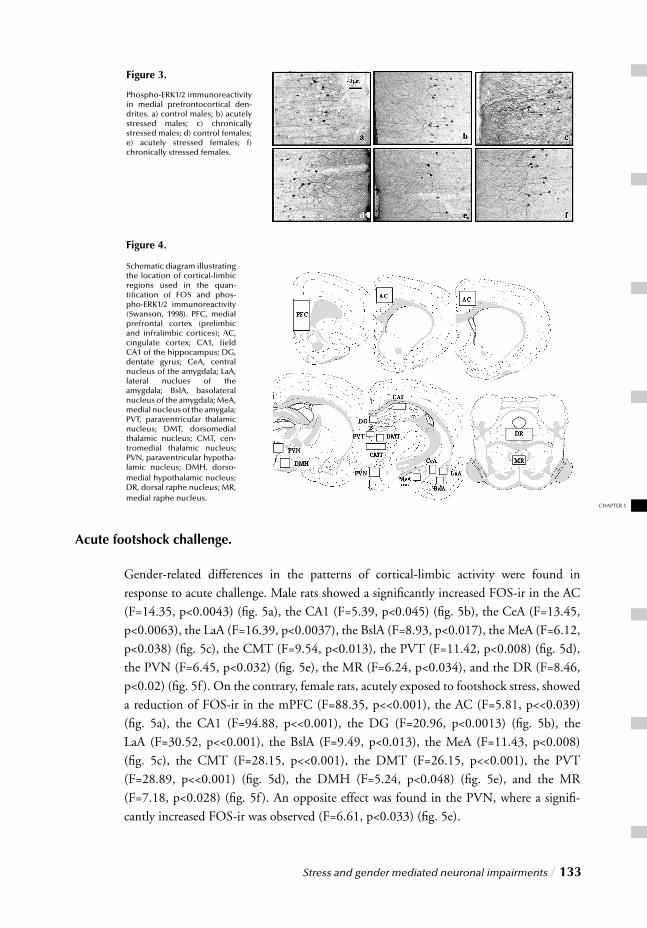
Stress and gender mediated neuronal impairments / 133
CHAPTER 5
Figure 3.
Phospho-ERK1/2 immunoreactivity in medial prefrontocortical den-drites. a) control males; b) acutely stressed males; c) chronically stressed males; d) control females; e) acutely stressed females; f) chronically stressed females.
Acute footshock challenge.
Gender-related differences in the patterns of cortical-limbic activity were found in response to acute challenge. Male rats showed a signifi cantly increased FOS-ir in the AC (F=14.35, p<0.0043) (fi g. 5a), the CA1 (F=5.39, p<0.045) (fi g. 5b), the CeA (F=13.45, p<0.0063), the LaA (F=16.39, p<0.0037), the BslA (F=8.93, p<0.017), the MeA (F=6.12, p<0.038) (fi g. 5c), the CMT (F=9.54, p<0.013), the PVT (F=11.42, p<0.008) (fi g. 5d), the PVN (F=6.45, p<0.032) (fi g. 5e), the MR (F=6.24, p<0.034), and the DR (F=8.46,
p<0.02) (fi g. 5f ). On the contrary, female rats, acutely exposed to footshock stress, showed a reduction of FOS-ir in the mPFC (F=88.35, p<<0.001), the AC (F=5.81, p<<0.039) (fi g. 5a), the CA1 (F=94.88, p<<0.001), the DG (F=20.96, p<0.0013) (fi g. 5b), the LaA (F=30.52, p<<0.001), the BslA (F=9.49, p<0.013), the MeA (F=11.43, p<0.008) (fi g. 5c), the CMT (F=28.15, p<<0.001), the DMT (F=26.15, p<<0.001), the PVT (F=28.89, p<<0.001) (fi g. 5d), the DMH (F=5.24, p<0.048) (fi g. 5e), and the MR (F=7.18, p<0.028) (fi g. 5f ). An opposite effect was found in the PVN, where a signifi -cantly increased FOS-ir was observed (F=6.61, p<0.033) (fi g. 5e).
Figure 4.
Schematic diagram illustrating the location of cortical-limbic regions used in the quan-tifi cation of FOS and phos-pho-ERK1/2 immunoreactivity (Swanson, 1998). PFC, medial prefrontal cortex (prelimbic and infralimbic cortices); AC, cingulate cortex; CA1, fi eld CA1 of the hippocampus; DG, dentate gyrus; CeA, central nucleus of the amygdala; LaA, lateral nuclues of the amygdala; BslA, basolateral nucleus of the amygdala; MeA, medial nucleus of the amygala; PVT, paraventricular thalamic nucleus; DMT, dorsomedial thalamic nucleus; CMT, cen-tromedial thalamic nucleus; PVN, paraventricular hypotha-lamic nucleus; DMH, dorso-medial hypothalamic nucleus; DR, dorsal raphe nucleus; MR, medial raphe nucleus.
134 / Stress and gender mediated neuronal impairments
Unexpectedly, acute footshock exposure caused a signifi cant decrease of phospho-ERK1/2 immunoreactivity in medial prefrontocortical dendrites in male rats while no changes were found in cyclic females (fi g. 6).
Figure 5.
Patterns of FOS-ir in controls, acutely and chronically stressed male and female rats. The symbol * expresses the comparison of FOS-ir between controls and stressed rats (*=p<0.05; **=p<0.01; ***=p<0.001).
Males Femalesa
b
c
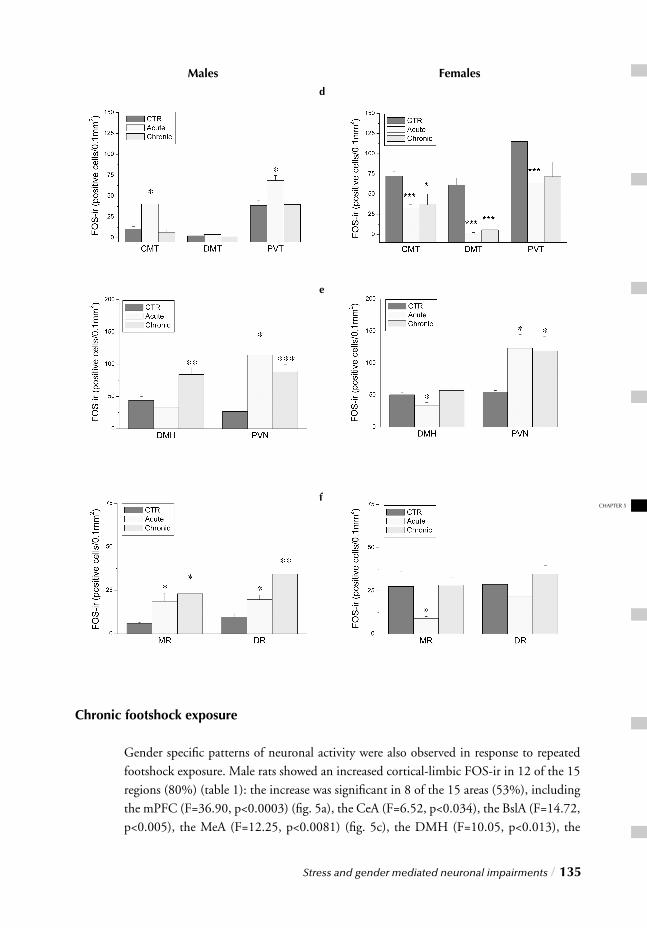
Stress and gender mediated neuronal impairments / 135
CHAPTER 5
Chronic footshock exposure
Gender specifi c patterns of neuronal activity were also observed in response to repeated footshock exposure. Male rats showed an increased cortical-limbic FOS-ir in 12 of the 15 regions (80%) (table 1): the increase was signifi cant in 8 of the 15 areas (53%), including the mPFC (F=36.90, p<0.0003) (fi g. 5a), the CeA (F=6.52, p<0.034), the BslA (F=14.72, p<0.005), the MeA (F=12.25, p<0.0081) (fi g. 5c), the DMH (F=10.05, p<0.013), the
Males Femalesd
e
f
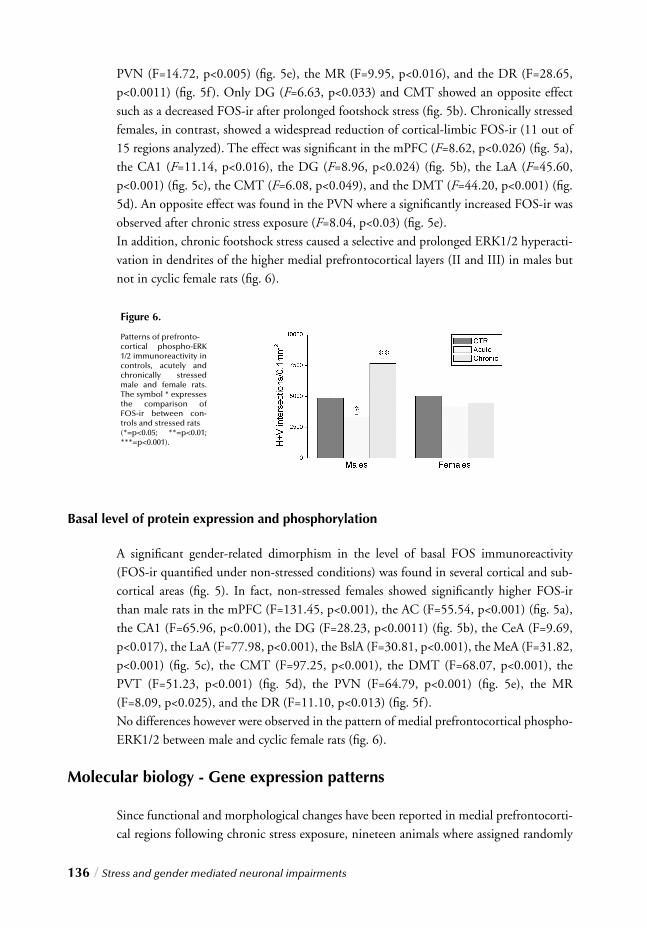
136 / Stress and gender mediated neuronal impairments
PVN (F=14.72, p<0.005) (fi g. 5e), the MR (F=9.95, p<0.016), and the DR (F=28.65, p<0.0011) (fi g. 5f ). Only DG (F=6.63, p<0.033) and CMT showed an opposite effect such as a decreased FOS-ir after prolonged footshock stress (fi g. 5b). Chronically stressed females, in contrast, showed a widespread reduction of cortical-limbic FOS-ir (11 out of 15 regions analyzed). The effect was signifi cant in the mPFC (F=8.62, p<0.026) (fi g. 5a), the CA1 (F=11.14, p<0.016), the DG (F=8.96, p<0.024) (fi g. 5b), the LaA (F=45.60, p<0.001) (fi g. 5c), the CMT (F=6.08, p<0.049), and the DMT (F=44.20, p<0.001) (fi g. 5d). An opposite effect was found in the PVN where a signifi cantly increased FOS-ir was observed after chronic stress exposure (F=8.04, p<0.03) (fi g. 5e). In addition, chronic footshock stress caused a selective and prolonged ERK1/2 hyperacti-vation in dendrites of the higher medial prefrontocortical layers (II and III) in males but not in cyclic female rats (fi g. 6).
Basal level of protein expression and phosphorylation
A signifi cant gender-related dimorphism in the level of basal FOS immunoreactivity (FOS-ir quantifi ed under non-stressed conditions) was found in several cortical and sub-cortical areas (fi g. 5). In fact, non-stressed females showed signifi cantly higher FOS-ir than male rats in the mPFC (F=131.45, p<0.001), the AC (F=55.54, p<0.001) (fi g. 5a), the CA1 (F=65.96, p<0.001), the DG (F=28.23, p<0.0011) (fi g. 5b), the CeA (F=9.69, p<0.017), the LaA (F=77.98, p<0.001), the BslA (F=30.81, p<0.001), the MeA (F=31.82, p<0.001) (fi g. 5c), the CMT (F=97.25, p<0.001), the DMT (F=68.07, p<0.001), the PVT (F=51.23, p<0.001) (fi g. 5d), the PVN (F=64.79, p<0.001) (fi g. 5e), the MR (F=8.09, p<0.025), and the DR (F=11.10, p<0.013) (fi g. 5f ). No differences however were observed in the pattern of medial prefrontocortical phospho-ERK1/2 between male and cyclic female rats (fi g. 6).
Molecular biology - Gene expression patterns
Since functional and morphological changes have been reported in medial prefrontocorti-cal regions following chronic stress exposure, nineteen animals where assigned randomly
Figure 6.
Patterns of prefronto-cortical phospho-ERK 1/2 immunoreactivity in controls, acutely and chronically stressed male and female rats. The symbol * expresses the comparison of FOS-ir between con-trols and stressed rats (*=p<0.05; **=p<0.01; ***=p<0.001).
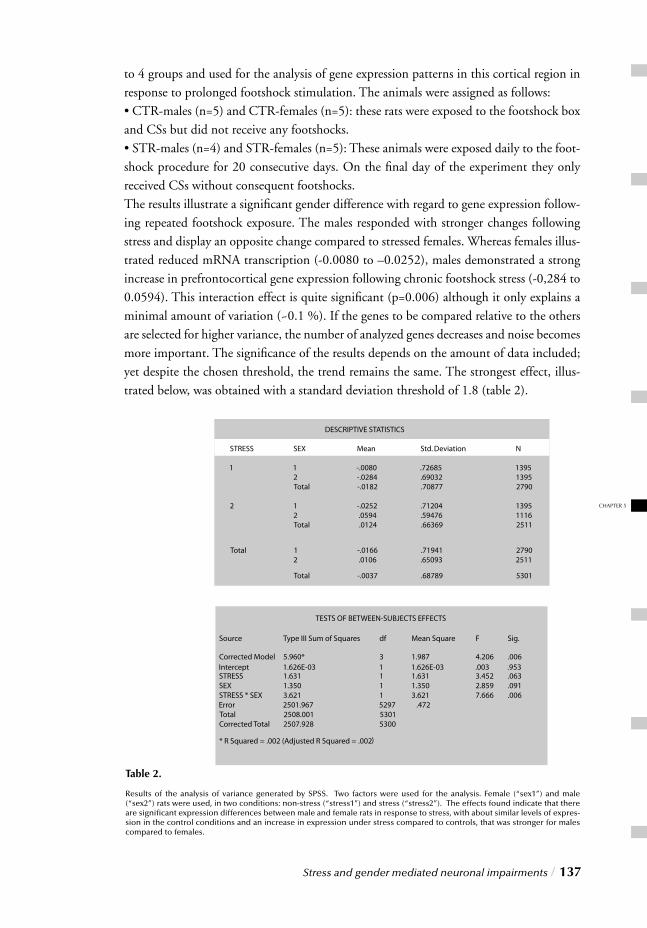
Stress and gender mediated neuronal impairments / 137
CHAPTER 5
to 4 groups and used for the analysis of gene expression patterns in this cortical region in response to prolonged footshock stimulation. The animals were assigned as follows:• CTR-males (n=5) and CTR-females (n=5): these rats were exposed to the footshock box and CSs but did not receive any footshocks. • STR-males (n=4) and STR-females (n=5): These animals were exposed daily to the foot-shock procedure for 20 consecutive days. On the fi nal day of the experiment they only received CSs without consequent footshocks.The results illustrate a signifi cant gender difference with regard to gene expression follow-ing repeated footshock exposure. The males responded with stronger changes following stress and display an opposite change compared to stressed females. Whereas females illus-trated reduced mRNA transcription (-0.0080 to –0.0252), males demonstrated a strong increase in prefrontocortical gene expression following chronic footshock stress (-0,284 to 0.0594). This interaction effect is quite signifi cant (p=0.006) although it only explains a minimal amount of variation (~0.1 %). If the genes to be compared relative to the others are selected for higher variance, the number of analyzed genes decreases and noise becomes more important. The signifi cance of the results depends on the amount of data included; yet despite the chosen threshold, the trend remains the same. The strongest effect, illus-trated below, was obtained with a standard deviation threshold of 1.8 (table 2).
)
Table 2.
Results of the analysis of variance generated by SPSS. Two factors were used for the analysis. Female (“sex1”) and male (“sex2”) rats were used, in two conditions: non-stress (“stress1”) and stress (“stress2”). The effects found indicate that there are signifi cant expression differences between male and female rats in response to stress, with about similar levels of expres-sion in the control conditions and an increase in expression under stress compared to controls, that was stronger for males compared to females.
138 / Stress and gender mediated neuronal impairments
These opposing fi ndings in gene expression seem to confi rm FOS-ir data concerning a sex-related dimorphism in the pattern of protein expression and possibly, neuronal activ-ity in response to repeated footshock exposure. In support of recent studies that docu-mented atrophy of prefrontocortical dendrites in chronically stressed male rats 7, the pres-ent investigation reports an abnormal pattern of prefrontocortical ERK1/2 phosphoryla-tion in chronically stressed males (fi g. 6). Due to the pivotal role played by ERK1 and 2 in this neuronal function, the expression arrays were further analyzed to identify changes in genes that have been reported to modulate synaptic plasticity in the medial prefrontal areas and might be affected by prolonged footshock exposure. When regression factor score 1 was plotted against factor 2 a skewed distribution was evident (table 3).
In line with the gender effects, the majority of outliers were located on the negative part of the X axis, coinciding with the greatest variation between highly expressive genes in males and low expressive genes in females. It is of interest to note that the expression of several
genes involved in the modulation of neuronal plasticity, such as synapsins II, SNAP25, calmodulin, and ERK2, was differentially affected by repeated footshock stimulation in male compared to female rats (table 4)
Table 3.
Plot of the fi rst factor against the second factor illustrates similar variation across the entire range of expression with distribution skewed to <0.

Stress and gender mediated neuronal impairments / 139
CHAPTER 5

140 / Stress and gender mediated neuronal impairments
DISCUSSION
Upon analysis of neuroendocrine and immunohistochemical changes induced by acute and prolonged footshock stress, important gender-related differences emerged in the pat-terns of cortical-limbic FOS-ir and prefrontocortical phospho-ERK1/2 expression. The choice of investigating the level of expression and phosphorylation of these specifi c pro-teins was made upon reviewing their specifi c cellular functions. Changes of FOS-ir have been widely used as molecular marker of neuronal activity 18-20. The analysis of FOS-ir has become a molecular tool to investigate complex processes, such as learning 28-34 and memory 35-38 as well as the neurocircuits activated by stress 39-48. The extracellular signal-regulated kinase (ERK) is a member of a family of serine/threonine protein kinases impli-cated in the transduction of neurotrophic signals from the cell surface to the nucleus 21. The ERK cascade plays a central role during neurodevelopment in the regulation of cell growth, proliferation, and differentiation but, interestingly, several family members, including ERK1 and ERK2, are also widely expressed by post-mitotic neurons in the mammalian nervous system 49. This evidence has suggested that ERKs might contribute to the regulation of important functions in the adult brain, including neuronal plasticity, learning, and memory 50,51. A critical step in the regulation of ERK-mediated activities is the dual phosphorylation of these kinases that leads to their transient activation and translocation from the cytoplasm to the nucleus 52. Only phosphorylated ERKs (phospho-ERKs) are able to interact with and activate cytoplasmic and nuclear targets, and modu-late such critical neuronal functions 21. Changes in the levels of ERK1/2 phosphorylation may thus provide crucial indications concerning the ability of stress to infl uence neuronal plasticity.
Immunohistochemical changes in response to acute footshock challenge
Acute footshock exposure activated, in male rats, cortical and subcortical structures, including the cingulate cortex (fi g. 5a), the hippocampal CA1 (fi g. 5b), the central, lateral, basolateral, and medial nucleus of the amygdala (fi g. 5c), the centromedial and paraven-tricular nucleus of the thalamus (fi g. 5d), the paraventricular hypothalamic nucleus (fi g. 5e), the median and dorsal raphe nucleus (fi g. 5f ). In contrast, in female rats, acute foot-shock stress was associated with a signifi cant reduction of FOS-ir in most of the above-mentioned cortical-limbic regions (fi g. 5), with the only exception of the PVN where, similarly to males, a marked increase of neuronal activation was observed (fi g. 5e). Acute emotional experiences have been reported to promote learning and memory 53-55. A growing body of evidence has pinpointed in particular the amygdala and the hip-pocampus as core components of the brain’s fear system 56-62. Thus, the increased FOS-ir reported by males in the amygdala (CeA, LaA, BslA, and MeA) (fi g. 5c) and hippocampus (CA1) (fi g. 5b) may support the participation of these limbic structures in the modulation of acute fear-related responses. Surprisingly, a different pattern of cortical-limbic FOS-ir

Stress and gender mediated neuronal impairments / 141
CHAPTER 5
was observed in female rats following short-term aversive challenge. In contrast to male rats, acutely stressed females showed a signifi cant reduction of FOS expression in both amygdala (LaA, BslA and MeA) and hippocampus (CA1) compared to non-stressed ani-mals (fi g. 5b,c). This differential pattern of neuronal activation was not limited to these two limbic structures but also involved the anterior cingulate cortex (fi g. 5a), the tha-lamus (centromedial and paraventricular nuclei) (fi g. 5d), and the midbrain (median raphe nucleus) (fi g. 5f ). Interestingly, a decreased phospho-ERK1/2 immunoreactivity was observed in medial prefrontocortical dendrites of acutely stressed males while no changes in the level of kinase phosphorylation were found in cyclic females (fi g. 6). A criti-cal step in ERK-mediated facilitation of neuronal plasticity involves their dual phosphory-lation followed by translocation from the cytoplasm to the nucleus 52. Reduced phospho-ERK1/2 immunoreactivity, observed in the medial prefrontocortical dendrites of males, might thus illustrate the translocation of these enzymes from the periphery to the nucleus (fi g. 3a,b, 6) and support the molecular changes underlying the consolidation of fear-related memories. In recent years a cumulative body of evidence concerning the existence of morphologi-cal 63-69 and functional differences 70-75 between the male and female brain has emerged. Cognitive processes, such as learning and memory, as well as behavioral responses to stress are infl uenced by sex 70,74,76-81. The gender-related patterns of neuronal activity observed in the present study following acute challenge might represent sex-specifi c coping strategies under aversive conditions. That males and females may differ in their coping strategies has been proposed by Taylor and colleagues 82. Taylor stated that the male response to stress in humans along with some animal species, is characterized by a “fi ght-or-fl ight” response, whereas the female response is more typically characterized by a pattern termed “tend-and befriend” 82. These gender-related behavioral responses may refl ect the involvement of different neural pathways and our results might offer indirect immunohistochemical evidence linking such gender-related coping styles with differential patterns of neuronal activity. A closer look at our data reveals that the divergent response to acute footshock challenge appears to be strongly related to the different level of basal FOS-ir (FOS-ir in non-stressed animals) (fi g. 5). Non-stressed females reported, in fact, an overall higher neuronal activation than males (up to 5-7 times higher), especially in frontocortical areas (fi g. 5a). In line with these fi ndings, neuroimaging investigations have also found gender-related diversities in brain activity patterns in humans, as women illustrated higher values than men 71,83,84. Furthermore, Esposito and colleagues reported substantial gender-related differences in the frontal lobe rCBF during performance of a variety of cognitive tasks with women showing signifi cantly higher activation 70. Although functional brain imag-ing studies have illustrated sex-related differences in global as well as regional brain activ-ity, reports of differential activation in the frontal lobes have been particularly prevalent 71,85. The results presented here may thus provide new insights into gender-related differ-ences in the neuronal circuitry engaged in the acute stress response and the molecular mechanisms underlying its modulation.

Gender-related dimorphism following repeated footshock exposure
Brief elevations of glucocorticoid levels play a critical role in the modulation of fear-related responses 86, promoting learning acquisition and memory consolidation 2,55,87. This benefi cial effect of adrenal steroids however, is only temporary as prolonged exposure to high glucocorticoid concentrations has been shown to impair cognitive processes, pos-sibly through the deleterious effects of stress hormones on neuronal plasticity 4,6,7,10,88-90. It is intriguing to speculate that the gender-related differences in FOS-ir and ERK1/2 phos-phorylation, detected following repeated footshock stress, may illustrate the deleterious effects of prolonged exposure to a hostile environment, on functional and structural integ-rity of the brain. The latter is supported by physiological and neuroendocrine evidence, such as the reduction of body weight gain (fi g. 1a, non-stressed vs. chronically stressed males), the signifi cant elevation of corticosterone levels (fi g. 1b), the hyperactivity of the PVN (fi g. 5e) and, more importantly, the adrenal hypertrophy, observed in both males and cyclic females (fi g. 1c). Footshocks have been reported to strongly activate the PVN, elevating plasma corticosteroid concentrations 44. Given the pivotal role of the PVN in the regulation of the HPA axis 91,92, these neuroendocrine changes seem to substantiate a lack of habituation and, possibly, an abnormal HPA axis activation in response to repeated stress. An intriguing possibility is that repeated footshock exposure promotes functional and morphological impairments by persistently elevating corticosteroid concentrations in the brain 7,10,90. Prolonged exposure to high glucocorticoid levels is also known to exert a deleterious infl uence on important neuronal functions, such as neurogenesis and synap-tic plasticity 4,93,94, and cause functional and morphological abnormalities in vulnerable regions, such as the hippocampus 13,95-97 and the prefrontal cortex 7,10. Functional cortical-limbic alterations included, in chronically stressed males, a reduced neuronal activation in the DG and an increased FOS-ir in the mPFC (fi g. 5a), amygdala (CeA, BslA and MeA) (fi g. 5c), hypothalamus (DMH and PVN) (fi g. 5e) and raphe (MR and DR) (fi g. 5f ). The abnormal ERK1/2 phosphorylation in medial prefrontocortical dendrites (fi g. 6) may, instead, document a stress-related structural impairment. In female rats, on the contrary, prolonged aversive stimulation was associated with a general reduction of FOS-ir in most cortical and subcortical regions, including the mPFC (fi g. 5a), the hippocampus (CA1 and DG) (fi g. 5b), the LaA (fi g. 5c), and the thalamus (CMT and DMT) (fi g. 5d). No changes in the level of phospho-ERK1/2 immunoreactivity were observed (fi g. 6).
These gender-related patterns of neuronal activity were also confi rmed at the molecular level, as gene expression analysis illustrates a general up-regulation of gene transcription in the frontal lobe of chronically stressed males, while a differential response was observed in females. It is interesting to note that, in addition to the gender-related dimorphism observed in the level of FOS and phospho-ERK1/2 immunoreactivity following repeated footshock exposure, differential gene expression patterns were also detected between male and female rats, including various key genes underlying neuronal plasticity (table 4).
142 / Stress and gender mediated neuronal impairments

CHAPTER 5
Conclusions and limitations
In this study, c-fos expression was used as a marker of neuronal activation. The use of this immediate early gene as activation marker may raise some doubts. Although c-fos may be considered an imprecise and unspecifi c marker, its reliability in the investigation of the neurocircuits underlying various processes, including learning and memory, has been confi rmed by numerous reports 18,30,31,37,98-105. In addition, the differential responses follow-ing chronic stressful stimulation, and consequently the reliability of FOS analysis, were confi rmed at a molecular level by gene expression analysis. An additional confounding element in the interpretation of the immunohistochemical data is represented by the hor-monal state of the female animals. In fact, since the neurocircuits regulating the stress response are prime targets for ovarian hormone action 106-108, estrogen and progesterone may play a central role in determining the gender-related dimorphisms observed here. In a parallel study specifi cally performed to assess the roles of these steroids in central stress integration however, the immunohistochemical changes associated with sustained stress exposure were analyzed in both cyclic and ovariectomized female rats. Surprisingly, no dif-ferences in FOS-ir between cyclic and ovariectomized females were found following long-term aversive stimulation in any of the cortical and subcortical regions examined 109 sug-gesting that, although important, ovarian hormones did not account for all the differences observed between males and females. In conclusion, in the present study we have reported signifi cant immunohistochemical differences, following acute and repeated footshock exposure, in the patterns of FOS and phospho-ERK1/2 immunoreactivity in both male and cyclic female rats. In addition, a marked gender-related dimorphism was also found in the number of cortical-limbic FOS positive nuclei, the level of ERK1/2 phosphorylation, and the patterns of gene expres-sion. These fi ndings might be indicative of different mechanisms underlying neuronal processing and provide additional evidence supporting the existence of functional gender-based brain dimorphisms. More importantly, the functional dimorphism observed follow-ing repeated footshock stimulation, instead, might illustrate gender-related differences in vulnerability to the deleterious effects of stress. Differences in the molecular mechanisms underlying cognitive and/or emotional processes, sex-related coping strategies, together with gender-related levels of basal neuronal activity, may represent important predisposing factors involved in the differential response of male and female rats to acute and chronic
stressful experiences.
ACKNOWLEDGEMENTS
I would like to thank G. Harms (Dept of Nuclear Medicine-Radiopharmacology), R. Veenstra (Dept of Pathology), P. Reuvekamp, M. Jongsma, F. Postema and K. Venema (Dept of Psychiatry) for their skillful technical assistance.
Stress and gender mediated neuronal impairments / 143

REFERENCES1. Shors, T.J., Weiss, C. & Thompson, R.F. Stress-induced facilitation of classical conditioning. Science 257, 537-539 (1992).
2. Beylin, A.V. & Shors, T.J. Stress enhances excitatory trace eyeblink conditioning and opposes acquisition of inhibitory conditioning. Behav. Neurosci. 112, 1327-1338 (1998).
3. Kendler, K.S., Karkowski, L.M. & Prescott,C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 156, 837-841 (1999).
4. McEwen, B.S. Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105-122 (1999).
5. Rajkowska, G. Postmortem studies in mood disorders indicate altered num- bers of neurons and glial cells. Biol. Psychiatry 48, 766-777 (2000).
6. Fuchs, E. et al. Psychosocial stress, glucocorticoids, and structural alterations in the tree shrew hippocampus. Physiol Behav. 73, 285-291 (2001).
7. Wellman, C.L. Dendritic reorganization in pyramidal neurons in medial pre- frontal cortex after chronic corticosterone administration. J Neurobiol 49, 245-253 (2001).
8. Herbert, J. Neurosteroids, brain damage, and mental illness. Exp. Gerontol. 33, 713-727 (1998).
9. Garcia, R. Stress, synaptic plasticity, and psychopathology. Rev. Neurosci. 13, 195-208 (2002).
10. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphos phorylation in medial prefrontocortical dendrites: implications for stress-related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
11. Sandi, C. The role and mechanisms of action of glucocorticoid involve- ment in memory storage. Neural Plast. 6, 41-52 (1998).
12. Korte, S.M. Corticosteroids in relation to fear, anxiety and psychopathology. Neurosci. Biobehav. Rev. 25, 117-142 (2001).
13. Magarinos, A.M., Verdugo, J.M. & McEwen, B.S. Chronic stress alters synaptic terminal structure in hippocampus. Proc. Natl. Acad. Sci. U. S. A 94, 14002-14008 (1997).
14. de Kloet, E.R., Oitzl, M.S. & Joels, M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 22, 422-426 (1999).
15. Kessler, R.C., McGonagle, K.A., Swartz, M., Blazer, D.G. & Nelson, C.B. Sex and depression in the National Comorbidity Survey. I: Life time prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85-96 (1993).
16. Ustun, T.B. Cross-national epidemiology of depression and gender. J. Gend. Specif. Med. 3, 54-58 (2000).
17. Palanza, P. Animal models of anxiety and depression: how are females dif ferent? Neurosci. Biobehav. Rev. 25, 219-233 (2001).
18. Campeau, S. et al. Elicitation and reduction of fear: behavioural and neuroendo- crine indices and brain induction of the immediate-early gene
c-fos. Neuroscience 78, 1087-1104 (1997).
19. Chaudhuri, A. Neural activity mapping with inducible transcription factors. Neuroreport 8, (1998).
20. Dube, T., Brunson, T., Nehlig, A. & Baram, T.Z. Activation of specifi c neuronal circuits by corticotropin relea- sing hormone as indicated by c-fos expression and glucose metabolism. J. Cereb. Blood Flow Metab 20, 1414-1424 (2000).
21. Grewal, S.S., York, R.D. & Stork,P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol 9, 544-553 (1999).
22. Sweatt, J.D. The neuronal MAP kinase cascade: a biochemical signal integra- tion system subserving synaptic plasticity and memory. J. Neurochem. 76, 1-10 (2001).
23. Hebert, A.E. & Dash, P.K. Extracellular signal-regulated kinase activity in the entorhinal cortex is necessary for long-term spatial memory. Learn. Mem. 9, 156-166 (2002).
24. Smedes, F., Kraak, J.C. & Poppe, H. Simple and fast solvent extraction system for selective and quantitative isolation of adrenaline, noradrenaline and dopa- mine from plasma and urine. J. Chromatogr. 231, 25-39 (1982).
25. Remie, R., Knot, H.J., Bos, E.A. & Zaagsma, J. Characterization of presynaptic beta-adrenoceptors facilitating endogenous noradrenaline release in the portal vein of perma- nently cannulated, freely moving rats. Eur. J. Pharmacol. 157, 37-43 (1988).
26. Swanson LW. Brain maps: structure of the rat brain. Elsevier, Amsterdam (1998).
27. Duncko, R., Kiss, A., Skultetyova, I., Rusnak, M. & Jezova, D. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 26, 77-89 (2001).
28. Beck, C.H. & Fibiger, H.C. Conditioned fear-induced changes in behavior and in the expression of the immediate early gene c-fos: with and without diazepam pretreatment. J. Neurosci. 15, 709-720 (1995).
29. Dragunow, M. A role for immediate-early transcription factors in learning and memory. Behav. Genet. 26, 293-299 (1996).
30. Gall, C.M., Hess, U.S. & Lynch, G. Mapping Brain Networks Engaged by, and Changed by, Learn ing. Neurobiol Learn Mem 70, 14-36 (1998).
31. Radulovic, J., Kammermeier, J. & Spiess, J. Relationship between fos production and classical fear condi tioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci 18, 7452-7461 (1998).
32. Bertaina-Anglade, V., Tramu, G. & Destrade, C. Differential learning-stage dependent patterns of c-Fos protein expression in brain regions during the acquisition and memory consolidation of an operant task in mice. Eur. J. Neurosci. 12, 3803-3812 (2000).
33. Stanciu, M., Radulovic, J. & Spiess, J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos produc- tion. Brain Res. Mol. Brain Res. 94, 15-24 (2001).
34. Ishida, Y. et al. Conditioned-fear stress increases Fos expression in monoami nergic and GABAergic neurons of the locus coeruleus and dorsal raphe nuclei. Synapse 45, 46-51 (2002).
144 / Stress and gender mediated neuronal impairments

35. Tischmeyer, W. & Grimm, R. Activation of immediate early genes and memory formation. Cell Mol. Life Sci. 55, 564-574 (1999). 36. Wan, H., Aggleton, J.P. & Brown, M.W. Different contributions of the hippocampus and perirhinal cortex to recognition memory. J. Neurosci. 19, 1142-1148 (1999).
37. Vann, S.D., Brown, M.W., Erichsen, J.T. & Aggleton, J.P. Fos imaging reveals differential patterns of hippocampal and parahippocampal subfi eld activation in rats in response to dif- ferent spatial memory tests. J Neurosci 20, 2711-2718 (2000).
38. Tronel, S. & Sara, S.J. Mapping of olfactory memory circuits: region-specifi c c-fos activation after odor-reward associative learning or after its retrieval. Learn. Mem. 9, 105-111 (2002).
39. Cullinan, W.E., Herman, J.P., Battaglia, D.F., Akil, H. & Watson, S.J. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience 64, 477-505 (1995).
40. Matsuda, S. et al. Persistent c-fos expression in the brains of mice with chronic social stress. Neurosci. Res. 26, 157-170 (1996).
41. Campeau, S. & Watson, S.J. Neuroendocrine and behavioral responses and brain pattern of c-fos induction associated with audiogenic stress. J. Neuroendocrinol. 9, 577-588 (1997).
42. Bhatnagar, S. & Dallman, M. Neuroanatomical basis for facilitation of hypothalamic-pituitary- adrenal responses to a novel stressor after chronic stress. Neuroscience 84, 1025-1039 (1998).
43. Kovacs, K.J. c-Fos as a transcription factor: a stressful (re)view from a func- tional map. Neurochem. Int. 33, 287-297 (1998).
44. Li, H.Y. & Sawchenko, P.E. Hypothalamic effector neurons and extended circuitries acti vated in “neurogenic” stress: a comparison of footshock effects exerted acutely, chronically, and in animals with controlled glu cocorticoid levels. J. Comp Neurol. 393, 244-266 (1998).
45. Bruijnzeel, A.W. et al. Long-term sensitization of Fos-responsivity in the rat central nervous system after a single stressful experience. Brain Res. 819, 15-22 (1999).
46. Sawchenko, P.E., Li, H.Y. & Ericsson, A. Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog. Brain Res. 122, 61-78 (2000).
47. Pacak, K. & Palkovits, M. Stressor specifi city of central neuroendocrine responses: impli cations for stress-related disorders. Endocr. Rev. 22, 502-548 (2001).
48. Stamp, J. & Herbert, J. Corticosterone modulates autonomic responses and adaptation of central immediate-early gene expression to repeated restraint stress. Neuroscience 107, 465-479 (2001).
49. Flood, D.G. et al. Immunolocalization of the mitogen-activated protein kinases p42MAPK and JNK1, and their regulatory kinases MEK1 and MEK4, in adult rat central nervous system. J Comp Neurol. 398, 373-392 (1998).
50. Atkins, C.M., Selcher, J.C., Petraitis, J.J., Trzaskos, J.M. & Sweatt, J.D. The MAPK cascade is required for mammalian associative learn- ing. Nat. Neurosci 1, 602-609 (1998).
51. Impey, S. et al. Stimulation of cAMP response element (CRE)-mediated tran- scription during contextual learning. Nat. Neurosci. 1, 595-601 (1998).
52. Khokhlatchev, A.V. et al. Phosphorylation of the MAP kinase ERK2 promotes its homodi- merization and nuclear translocation. Cell 93, 605-615 (1998).
53. Buchanan, T.W. & Lovallo, W.R. Enhanced memory for emotional material following stress-level cortisol treatment in humans. Psychoneuroendocrinology 26, 307-317 (2001).
54. Marti, O., Garcia, A., Velles, A., Harbuz, M.S. & Armario, A. Evidence that a single exposure to aversive stimuli triggers long- lasting effects in the hypothalamus-pituitary-adrenal axis that consolidate with time. Eur. J. Neurosci. 13, 129-136 (2001).
55. Shors, T.J. Acute stress rapidly and persistently enhances memory forma tion in the male rat. Neurobiol Learn Mem 75, 10-29 (2001).
56. Maren, S. Long-term potentiation in the amygdala: a mechanism for emo- tional learning and memory. Trends Neurosci 22, 561-567 (1999).
57. Buchel, C. & Dolan, R.J. Classical fear conditioning in functional neuroimaging. Curr. Opin. Neurobiol 10, 219-223 (2000).
58. Fanselow, M.S. Contextual fear, gestalt memories, and the hippocampus. Behav. Brain Res. 110, 73-81 (2000).
59. Gewirtz, J.C., McNish, K.A. & Davis, M. Is the hippocampus necessary for contextual fear conditioning? Behav. Brain Res. 110, 83-95 (2000).
60. LeDoux, J.E. Emotion circuits in the brain. Annu Rev Neurosci 23, 155-184 (2000).
61. Blair, H.T., Schafe, G.E., Bauer, E.P., Rodrigues, S.M. & LeDoux, J.E. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8, 229-242 (2001).
62. Schafe, G.E., Nader, K., Blair, H.T. & LeDoux, J.E. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 24, 540-546 (2001).
63. Davatzikos, C. & Resnick, S.M. Sex differences in anatomic measures of interhemispheric con- nectivity: correlations with cognition in women but not men. Cereb. Cortex 8, 635-640 (1998).
64. Rabinowicz, T., Dean, D.E., Petetot, J.M. & de Court. Gender differences in the human cerebral cortex: more neu- rons in males; more processes in females. J Child Neurol 14, 98-107 (1999).
65. Courten-Myers, G.M. The human cerebral cortex: gender differences in structure and function. J. Neuropathol. Exp. Neurol. 58, 217-226 (1999).
66. Rhodes, M.E. & Rubin, R.T. Functional sex differences (‘sexual diergism’) of central nervous system cholinergic systems, vasopressin, and hypothalamic- pituitary-adrenal axis activity in mammals: a selective review. Brain Res Brain Res Rev 30, 135-152 (1999).
67. Nopoulos, P., Flaum, M., O’Leary, D. & Andreasen, N.C. Sexual dimorphism in the human brain: evaluation of tissue volume, tissue composition and surface anatomy using mag- netic resonance imaging. Psychiatry Res 98, 1-13 (2000).
68. Yucel, M. et al. Hemispheric and gender-related differences in the gross mor phology of the anterior cingulate/paracingulate cortex in normal volunteers: an MRI morphometric study. Cereb. Cortex 11, 17-25 (2001).
69. Gur, R.C., Gunning-Dixon, F., Bilker, W.B. & Gur, R.E. Sex Differences in Temporo-limbic and Frontal Brain Volumes of Healthy Adults. Cereb. Cortex 12, 998-1003 (2002).
Stress and gender mediated neuronal impairments / 145
CHAPTER 5

70. Esposito, G., Van Horn, J.D., Weinberger, D.R. & Berman, K.F. Gender differences in cerebral blood fl ow as a function of cog nitive state with PET. J Nucl. Med 37, 559-564 (1996).
71. George, M.S., Ketter, T.A., Parekh, P.I., Herscovitch, P. & Post, R.M. Gender differences in regional cerebral blood fl ow during transient self-induced sadness or happiness. Biol Psychiatry 40, 859-871 (1996).
72. Nyberg, L., Habib, R. & Herlitz, A. Brain activation during episodic memory retrieval: sex differen- ces. Acta Psychol (Amst) 105, 181-194 (2000).
73. Schneider, F., Habel, U., Kessler, C., Salloum, J.B. & Posse, S. Gender differences in regional cerebral activity during sadness. Hum. Brain Mapp. 9, 226-238 (2000).
74. Cahill, L. et al. Sex-related difference in amygdala activity during emotionally infl uenced memory storage. Neurobiol Learn Mem 75, 1-9 (2001).
75. Iidaka, T. et al. Neural interaction of the amygdala with the prefrontal and tem poral cortices in the processing of facial expressions as revealed by fMRI. J Cogn Neurosci 13, 1035-1047 (2001).
76. Kimura D. Sex, sexual orientation and sex hormones infl uence human cog nitive function. Curr Opin Neurobiol 6, 259-263 (1996).
77. Wood, G.E. & Shors, T.J. Stress facilitates classical conditioning in males, but impairs classical conditioning in females through activational effects of ovarian hormones. Proc. Natl. Acad. Sci. U. S. A 95, 4066-4071 (1998).
78. Young, E.A. Sex differences and the HPA axis: implications for psychiatric disease. J Gend. Specif. Med 1, 21-27 (1998).
79. Gur, R.C. et al. Sex differences in brain gray and white matter in healthy young adults: correlations with cognitive performance. J Neurosci 19, 4065-4072 (1999).
80. Speck, O. et al. Gender differences in the functional organization of the brain for working memory. Neuroreport 11, 2581-2585 (2000).
81. Troisi, A. Gender differences in vulnerability to social stress: a Darwinian perspective. Physiol Behav 73, 443-449 (2001).
82. Taylor, S.E. et al. Biobehavioral responses to stress in females: tend-and- befriend, not fi ght-or-fl ight. Psychol. Rev. 107, 411-429 (2000).
83. Devous, M.D., Sr, Stokely, E.M., Chehabi, H.H. & Bonte, F.J. Normal distribution of regional cerebral blood fl ow measured by dynamic single-photon emission tomography. J Cereb Blood Flow Metab 6, 95-104 (1986).
84. Baxter, L.R. Jr et al. Cerebral glucose metabolic rates in normal human females versus normal males. Psychiatry Res 21, 237-245 (1987).
85. Andreason, P.J., Zametkin, A.J., Guo, A.C., Baldwin, P. & Cohen, R.M. Gender-related differences in regional cerebral glucose metab olism in normal volunteers. Psychiatry Res 51, 175-183 (1994).
86. Koob, G.F. Corticotropin-releasing factor, norepinephrine, and stress. Biol. Psychiatry 46, 1167-1180 (1999).
87. Liu, L., Tsuji, M., Takeda, H., Takada, K. & Matsumiya, T. Adrenocortical suppression blocks the enhancement of memory storage produced by exposure to psychological stress in rats. Brain Res. 821, 134-140 (1999).
88. Schaaf, M.J., de Jong, J., de Kloet, E.R. & Vreugdenhil, E. Downregulation of BDNF mRNA and protein in the rat hip pocampus by corticosterone. Brain Res. 813, 112-120 (1998).
89. Magarinos, A.M., Deslandes, A. & McEwen, B.S. Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur. J. Pharmacol. 371, 113-122 (1999).
90. Bisagno, V., Ferrini, M., Rios, H., Zieher, L.M. & Wikinski, S.I. Chronic corticosterone impairs inhibitory avoidance in rats: possible link with atrophy of hippocampal CA3 neurons. Pharmacol. Biochem. Behav. 66, 235-240 (2000).
91. Herman, J.P., Adams, D. & Prewitt, C. Regulatory changes in neuroendocrine stress-integrative cir cuitry produced by a variable stress paradigm. Neuroendocrinology 61, 180-190 (1995).
92. Herman, J.P., Tasker, J.G., Ziegler, D.R. & Cullinan, W.E. Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol. Biochem.Behav. 71, 457-468 (2002).
93. Gould, E. & Tanapat, P. Stress and hippocampal neurogenesis. Biol Psychiatry 46, 1472-1479 (1999).
94. Czeh, B. et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. U. S. A 98, 12796-12801 (2001).
95. Fuchs, E., Uno, H. & Flugge, G. Chronic psychosocial stress induces morphological alterations in hippocampal pyramidal neurons of the tree shrew. Brain Res 673, 275-282 (1995).
96. McKittrick, C.R. et al. Chronic social stress reduces dendritic arbors in CA3 of hip pocampus and decreases binding to serotonin transporter sites. Synapse 36, 85-94 (2000).
97. Pavlides, C., Nivon, L.G. & McEwen, B.S. Effects of chronic stress on hippocampal long-term potentia tion. Hippocampus 12, 245-257 (2002).
98. Rosen, J.B., Fanselow, M.S., Young, S.L., Sitcoske, M. & Maren, S. Immediate-early gene expression in the amygdala following footshock stress and contextual fear conditioning. Brain Res 796, 132-142 (1998).
99. Morrow, B.A., Elsworth, J.D., Inglis, F.M. & Roth, R.H. An antisense oligonucleotide reverses the footshock-induced expression of fos in the rat medial prefrontal cortex and the subsequent expression of conditioned fear-induced immobility. J Neurosci 19, 5666-5673 (1999).
100. Savonenko, A., Filipkowski, R.K., Werka, T., Zielinski, K. & Kaczmarek, L. Defensive conditioning-related functional heterogeneity among nuclei of the rat amygdala revealed by c-Fos mapping. Neuroscience 94, 723-733 (1999).
101. Hall, J., Thomas, K.L. & Everitt, B.J. Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur J Neurosci 13, 1453-1458 (2001). 102. Wan, H. et al. Fos imaging reveals differential neuronal activation of areas of rat temporal cortex by novel and familiar sounds. Eur J Neurosci 14, 118-124 (2001).
103. Guzowski, J.F. Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fl u orescent imaging approaches. Hippocampus 12, 86-104 (2002).
146 / Stress and gender mediated neuronal impairments

104. Halasz, J., Liposits, Z., Kruk, M.R. & Haller, J. Neural background of glucocorticoid dysfunction-induced abnormal aggression in rats: involvement of fear- and stress- related structures. Eur. J. Neurosci 15, 561-569 (2002).
105. He, J., Yamada, K. & Nabeshima, T. A role of Fos expression in the CA3 region of the hippocampus in spatial memory formation in rats. Neuropsychopharmacology 26, 259-268 (2002).
106. Chrousos, G.P., Torpy, D.J. & Gold, P.W. Interactions between the hypothalamic-pituitary-adrenal axis and the female reproductive system: clinical implications. Ann. Intern. Med. 129, 229-240 (1998).
107. Shors, T.J., Chua, C. & Falduto, J. Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J.Neurosci 21, 6292-6297 (2001).
108. Figueiredo, H.F., Dolgas, C.M. & Herman, J.P. Stress activation of cortex and hippocampus is modulated by sex and stage of estrus. Endocrinology 143, 2534-2540 (2002).
109. Trentani, A., Den Boer, J.A., Korf, J. & Ter Horst, G.J. Gender-related sensitivity to the effects of chronic stress in rats. Society for Neuroscience Abstracts . 2000.
Stress and gender mediated neuronal impairments / 147
CHAPTER 5


6
CHRONIC STRESS EFFECTS ON SYNAPTIC VESICLE-ASOCIATED PROTEINS EXPRESSION:INDICATIONS FOR REGION/GENDER- DEPENDENT REGULATION
Stress-induced neuronal dysfunctions have been associated with
altered synaptic protein expression. In this study, chronic stress-
induced changes in expression levels of several synaptic vesicle-
associated proteins were thus investigated, in the hippocampus
and prefrontal cortex of male and female rats, by microarray
and RT-PCR analysis. The results illustrate gender- and region-spe-
cifi c regulation of membrane-associated synaptic protein expres-
sion in response to stress. Specifi cally, chronically stressed males
revealed increased transcription levels of several synaptic pro-
teins, including synaptophysin, synaptotagmin and synapsin in
prefrontocortical and hippocampal areas. Although no changes
were observed in stressed females, they consistently expressed
lower mRNA levels of these proteins under stress conditions com-
pared to males. These fi ndings support the hypothesis that altered
expression of selective synaptic proteins may constitute a poten-
tial mechanism underlying stress-induced impairment of neuro-
nal plasticity. Moreover, these data suggest the possibility that
differentially affected synaptic protein expression may contribute
to gender distinctive mechanisms of dysregulation that character-
ize certain affective disorders.
Kuipers S.D., Trentani A., te Meerman G.J., Bakker P.L., Reuvekamp
P.T.W., Den Boer J.A., Ter Horst G.J. Molecular Psychiatry. Submitted


CHAPTER 6
Stress, gender and synaptic vesicle proteins / 151
INTRODUCTION
Stress, particularly when prolonged and severe, has been associated with multiple morpho-logical and functional abnormalities in cortical and subcortical structures of both humans and animals. Consistent neuroimaging fi ndings in support of the latter include hippocam-pal atrophy and reduced prefrontocortical metabolism following chronic stress and stress-related disorders 1-5. Stress-induced structural and functional impairments have in turn been implicated in cognitive defi cits and signifi cant behavioral alterations 6-11. Reduced neuronal plasticity however has also been reported in response to stress and is believed to represent a common feature in a wide variety of psychiatric disorders. Since impaired synaptic plasticity may depend upon stress-induced alterations on the molecular level, it is thus notable that investigation of these underlying mechanisms has been only ten-dentious. Stress, for instance, has been reported to affect the expression of neurotrophic factors as brain derived neurotrophic factor (BDNF) and neurotrophin 3 (NT3) 12,13
and adverse events have also been speculated to impair neuronal plasticity by affecting expression of various synaptic vesicle-associated proteins (SVaPs) 14,15. The latter raises an intriguing possibility that abnormal expression of these proteins may contribute to the molecular basis of stress-induced neuronal plasticity changes as well as behavioral and cognitive alterations associated with prolonged stress exposure. Proper regulation of SVaP expression is in fact essential for effi cient modulation of neurotransmitter release while depleted or dysfunctional neurotransmission has been implicated in the occurrence of various affective disorders. Abnormal expression and/or function of SVaP has thus been identifi ed as a possible predisposing factor for the pathophysiology of psychiatric disorders such as schizophrenia and depression 16-18. In this study, we examined chronic footshock-induced alterations in synaptotagmin, synaptophysin, synapsin I and synapsin II expres-sion in the prefrontal cortex and hippocampus, to investigate whether dysregulated tran-scription of specifi c synaptic vesicle-associated proteins could account, at least in part, for the often documented neuronal plasticity impairments in response to repeated stress. The experiments were performed in both male and female rats in view of the well-estab-lished gender-related prevalence of stress-related psychiatric illnesses, which illustrate that women are more likely to develop depressive or anxiety disorders than men 19,20. Although recent reports have attributed this differential, prevalence of psychopathology to sex-
related differences in behavioral response to stress 21, considerably less attention has been given to gender-related differences in the neurobiological mechanisms underlying stress and psychopathology, particularly at a molecular level. The present data illustrate a region- and gender-dependent regulation of SVaP expression in response to repeated stress, which may offer additional insights into the understanding of neurobiological substrates under-lying the differential gender-related susceptibility to adverse events and psychopathology.

152 / Stress, gender and synaptic vesicle proteins
MATERIAL AND METHODS
Animals
The experiments were performed with male (n°=48: 225-249 g) and female (n°=48: 200-224 g) Wistar rats. The animals were individually housed with food and water avail-able ad libitum and maintained on a 12:12 h light/dark cycle at 21°C. All rats were hand-led daily for 5-8 min to minimize the nonspecifi c stress response. All handling was per-formed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and the guidelines of the Animal Bio-ethics Committee of the University of Groningen (FDC: 2509).
Stress Procedure
The rats were subjected to a daily footshock stress protocol for 3 weeks as previously described 22,23. In short, the rats were placed in a rodent test-chamber with a metallic grid fl oor connected to a shock generator and scrambler. Rats in the stress group (n° = 24; 12 male and 12 female rats) were subjected to a single daily session of varying duration (between 30 and 120 min/day) in the “footshock box” during which fi ve uncontrollable and inescapable footshocks were applied (0.8 mA in intensity and 8 sec in duration). In order to make the procedure as unpredictable as possible, starting time, inter-shock inter-val, and total time spent in the box were varied on a daily basis. To investigate the cellular and molecular changes induced by sustained footshock stress, non-stressed control rats (n° = 24; 12 male and 12 female rats) followed an identical schedule in a similar setup, but did not receive any footshocks throughout the experiment.On the fi nal (21st) day of the experiment all rats were placed in the footshock box for 15 minutes without receiving any electric shocks. Exposure to the footshock chamber on the fi nal day was essential as it established a link between a harmless stimulus (environment in which footshocks were applied or “footshock box”) and aversive events (“footshocks”). This provided a way to create a stress condition without directly exposing the stressed ani-mals to physical or painful stimuli. The response of control and stress rats to an identical, stimulus could then be investigated by evaluating gene expression changes (microarrays and quantitative RT-PCR). To limit circadian variability in circulating hormone levels, animals were sacrifi ced on subsequent days within a narrow window of 4 hours.
Physiological parameters
Upon termination, adrenal glands were removed, weighed and recalculated to correct for the body weight of the animals. Blood was also collected upon termination and the serum obtained was stored at –20ºC to determine plasma corticosterone levels with HPLC. These parameters served to verify the effi cacy of the chronic stress procedure.

Stress, gender and synaptic vesicle proteins / 153
CHAPTER 6
Corticosterone assay.
Plasma corticosterone levels were measured by HPLC as previously described 22,23. For quantifi cation of corticosterone concentration dexamethasone was used as internal stan-dard. Plasma was extracted with 3ml of diethylether, vortexed for 5 min, and then centri-fuged for 5 min at 3000 x g. The extraction procedure was repeated twice. The organic phase was evaporated to dryness in a 50˚C waterbath. The detection limit of corticoste-rone was 10 nM.
Tissue and RNA preparation
Total RNA- and poly(A)+RNA-isolation.
Thirty minutes after the start of the fi nal session, 24 rats were anesthetized with halothane and decapitated. The brains were dissected to separate the prefrontal cortex and the hip-pocampus, which were subsequently quick-frozen in liquid nitrogen and stored at -80°C. Total RNA was isolated from these brain areas by using Trizol (Life Technology, Gaith-ersburg, MD, USA) according to the manufacturer’s instructions. Integrity of total RNA was confi rmed on a 2% agarose gel and fi nal concentrations were assessed spectrophoto-metrically. Poly(A)+RNA was in turn isolated from total RNA using Micro-FastTrack™ 2.0 Kit (Invitrogen) according to the manufacturer’s instructions.
cDNA gene expression arrays
RNA (2-5µg/rat) extracted from the prefrontal cortex (of 4-5 animals per group) was sub-sequently converted into a 32P-labeled fi rst-strand cDNA and used to hybridize cDNA microarrays (rat atlas cDNA array 1.2; Clontech, Palo Alto, CA, USA). In this microar-ray, plasmid and bacteriophage DNAs are included as negative controls, along with several housekeeping cDNAs as positive controls. A complete list of genes and controls spotted on the array, as well as array coordinates and GenBank accession numbers, is available at the Clontech web site (http://www.clontech.com). To suppress non-specifi c background each membrane was prehybridized for 30 min at 68ºC in 5ml of hybridization solution (ExpressHyb, Clontech) with continuous agitation. Hybridization was subsequently car-ried out by the addition of the denatured, labeled cDNA to the prehybridization solution at 68ºC for overnight incubation to reach a fi nal probe concentration of 2-5 x 106cpm/ml. Membranes were stringently washed with continuous agitation at 68ºC in 2 x SSC, 1% SDS (4x30 min) and then in 0.1 SSC, 0.5% SDS (30 min). After a fi nal rinse in 0.1 x SSC (5 min), membranes were mounted on Whatman paper, plastic-wrapped, exposed to x-ray fi lm overnight at -80ºC followed by exposure to a phosphoimager screen for 3 days.

154 / Stress, gender and synaptic vesicle proteins
PhosphoImaging analysis.
Membranes were scanned using a Molecular Dynamics STORM PhosphoImager (Molec-ular Dynamics, Inc., Sunnyvale, CA, USA), and images were analyzed by ImageQuant (Molecular Dynamics, Inc., Sunnyvale, CA, USA). According to the Array manufacturer, Clontech, the radioactive cDNA signal is linear for RNAs present at levels of 0.01-3% of the total RNA population. An admonition of quantitative analysis however, is that the accuracy for extremely low abundant genes may not be reliable due to the detection limita-tion of this technique. Here the expression level for each gene was measured by the phos-phoimager in arbitrary signal intensity units and the original raw output was subsequently used to perform the statistical analysis.
Statistical analysis.
A full account of the microarray analysis has been previously described 24. In short, use was made of specifi c Delphi 5.0 data transformation software and SPSS 11.0. After log transformation, factor analysis was performed on the data points to identify the common component in the (log) data. Subtraction of this common component yields a log ratio value, used for further analysis. After checking for systemic positional effects, a reference set of genes was chosen against which to test hypotheses with regard to the experimental variation. Genes selected for inclusion in the analysis set were those, which displayed the greatest variation compared to other genes; the reference set contained the genes with the least amount of variation between arrays. To verify sensitivity of the results for this selec-tion procedure, the criterion for inclusion of genes in the test set versus the reference set was variation in log expression ratio over a range of standard deviations (>1.5 - >2.5). The common variation or expression of the genes (fi rst principal component) plot against the interarray differences or variation (second principal component) allowed identifi cation of the strongest alterations of individual genes. The outliers, previously published 24 are genes most likely responsible for the observed experimental effects.
Real-time quantitative polymerase chain reaction (qRT-PCR)
First-strand cDNA synthesis.
First-strand cDNA was synthesized from 200 ng poly(A)+RNA using SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s instructions. Before fi rst-strand synthesis was performed, contaminating genomic DNA was removed using Deoxyribonuclease I, Amplifi cation Grade (Invitrogen). The fi rst-strand synthesis was primed with oligo(dT), and the RNA template was not removed from the DNA:RNA hybrid.

Stress, gender and synaptic vesicle proteins / 155
CHAPTER 6
Primers and PCR Conditions.
All primers were designed with Primer Express® Software v2.0 (Applied Biosystems) using default parameters (sequences are listed in table 1). PCR reactions were performed on an ABI Prism 7900HT Sequence Detection System. Amplifi cation mixtures (25 µL divided over 3 wells, 7 µl per well) for all genes contained 2 ng cDNA from polyA+RNA template, 1 x SYBR Green PCR buffer, 3 mM MgCl2, 0.2 mM dATP, 0.2 mM dCTP, 0.2 mM dGTP 0.4 mM dUTP, 0.025U/ml Amplitaq Gold, 0.01 U/ml AmpErase UNG and 300 nM of each primer. The cycling conditions comprised of 2 minutes AmpErase UNG activity, 10 minutes Amplitaq Gold activation, 40 cycles at 95°C for 15 seconds and 60°C for 1 minute, and a dissociation stage: 15 seconds 95°C, 15 seconds 60°C, 15 seconds 95°C ramp rate 2%. The No Template Control contained all components except the cDNA template.
Real-time polymerase chain reaction (RT-PCR).
Real time detection was performed using an ABI PRISM 7900HT Sequence Detection System and SYBR Green PCR Core Reagents. Data analysis was performed using software provided with the instrument. The mRNAs of the different genes were relatively quanti-fi ed across groups with the Comparative CT Method. The CT value, or fractional cycle number at which the defi ned threshold is crossed above baseline, is predictive of the input amount of target and thus useful in quantifi cation of RNA 25. Here the threshold was set

156 / Stress, gender and synaptic vesicle proteins
between 0.4 and 0.6 with a baseline range from 1 to 7. All RNA samples were measured in triplo and the amount of target was normalized to an endogenous reference, the house-keeping gene, RPS29 ( CT). An additional reference ( -actin) was included as an extra control.
Statistical Analysis.
Data are expressed as means ± standard error (SEM). Statistical signifi cance was deter-mined by performing one-way analysis of variance (ANOVA) and F test of variance on the corresponding parameter measures from experimental and control conditions. To com-pare values between male and female, and/or control and experimental conditions t tests for equal or unequal variance were performed. P<0.05 was defi ned as the level of signifi -cance between groups. Calculations were made using Jandel SigmaStat statistical software.
RESULTS
Adrenal Weight
Chronic footshock exposure caused signifi cant adrenal hypertrophy in both male (29%; F=26.41; P<0.0001) and female rats (17%; F=8.32; P=0.014). A gender-related difference in the weight of adrenal glands was also evident as females had heavier glands than males under both basal (107%; F=82.71; P<0.0001) and stress conditions (87%; F=218.14; P<0.0001) (Fig. 1a).
Plasma Corticosterone
In line with corresponding adrenal weights, chronic footshock stress resulted in signifi -cantly enhanced plasma corticosterone concentrations in both sexes. Males expressed a 45% (F=6.68; P=0.032) increase compared to basal levels and females exhibited a 77% rise (F=12.30; P=0.005). Plasma corticosterone was also higher in females than males and this gender-related difference was found under both control (F=9.40; P=0.012) and stress conditions (F=31,12; P<0.0001) (Fig. 1b).
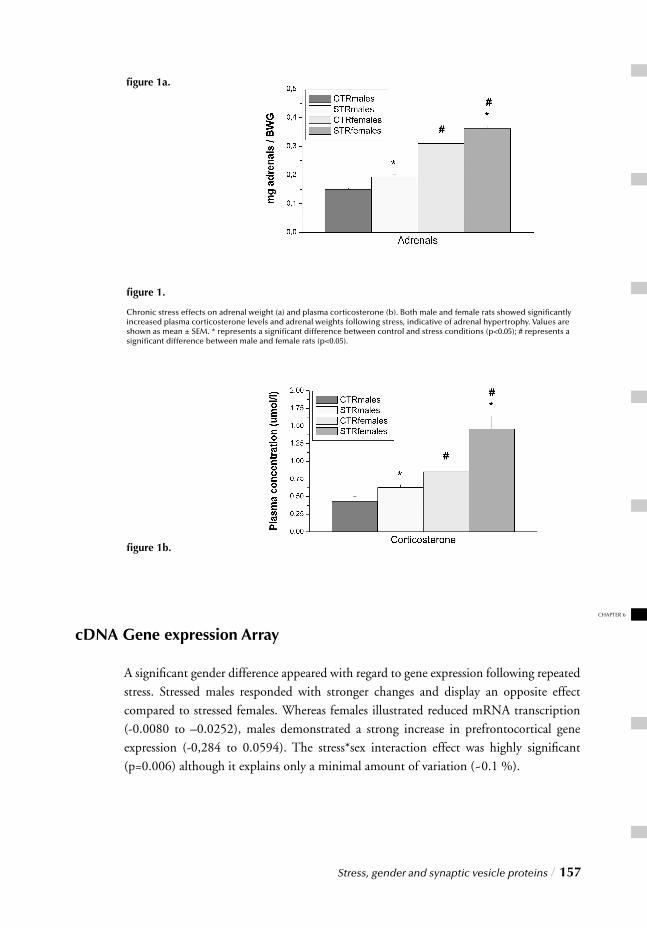
Stress, gender and synaptic vesicle proteins / 157
CHAPTER 6
cDNA Gene expression Array
A signifi cant gender difference appeared with regard to gene expression following repeated
stress. Stressed males responded with stronger changes and display an opposite effect compared to stressed females. Whereas females illustrated reduced mRNA transcription (-0.0080 to –0.0252), males demonstrated a strong increase in prefrontocortical gene expression (-0,284 to 0.0594). The stress*sex interaction effect was highly signifi cant (p=0.006) although it explains only a minimal amount of variation (~0.1 %).
fi gure 1.
Chronic stress effects on adrenal weight (a) and plasma corticosterone (b). Both male and female rats showed signifi cantly increased plasma corticosterone levels and adrenal weights following stress, indicative of adrenal hypertrophy. Values are shown as mean ± SEM. * represents a signifi cant difference between control and stress conditions (p<0.05); # represents a signifi cant difference between male and female rats (p<0.05).
fi gure 1a.
fi gure 1b.
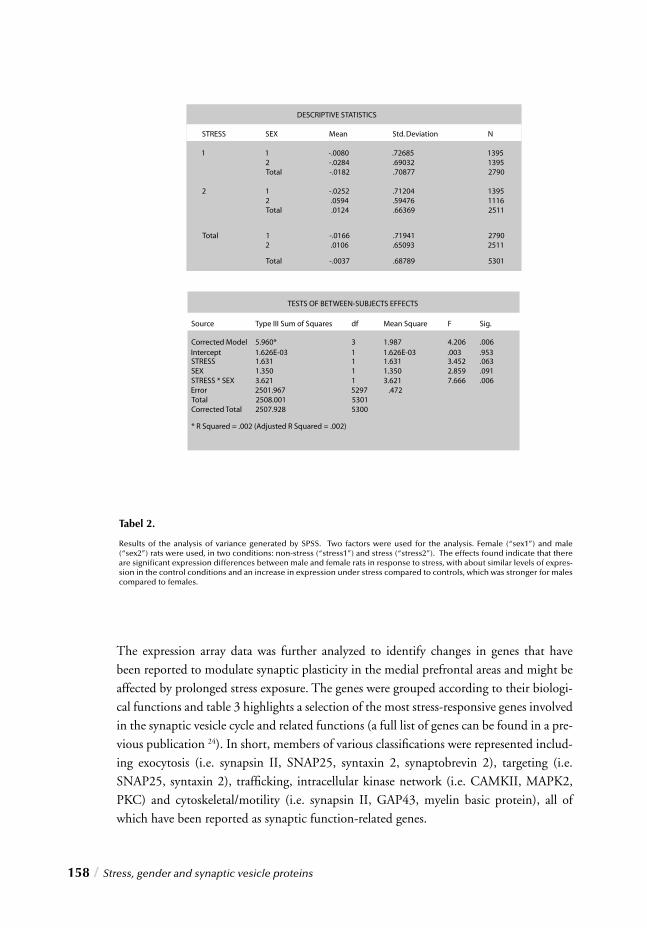
158 / Stress, gender and synaptic vesicle proteins
The expression array data was further analyzed to identify changes in genes that have been reported to modulate synaptic plasticity in the medial prefrontal areas and might be affected by prolonged stress exposure. The genes were grouped according to their biologi-cal functions and table 3 highlights a selection of the most stress-responsive genes involved in the synaptic vesicle cycle and related functions (a full list of genes can be found in a pre-vious publication 24). In short, members of various classifi cations were represented includ-ing exocytosis (i.e. synapsin II, SNAP25, syntaxin 2, synaptobrevin 2), targeting (i.e. SNAP25, syntaxin 2), traffi cking, intracellular kinase network (i.e. CAMKII, MAPK2, PKC) and cytoskeletal/motility (i.e. synapsin II, GAP43, myelin basic protein), all of which have been reported as synaptic function-related genes.
Tabel 2.
Results of the analysis of variance generated by SPSS. Two factors were used for the analysis. Female (“sex1”) and male (“sex2”) rats were used, in two conditions: non-stress (“stress1”) and stress (“stress2”). The effects found indicate that there are signifi cant expression differences between male and female rats in response to stress, with about similar levels of expres-sion in the control conditions and an increase in expression under stress compared to controls, which was stronger for males compared to females.
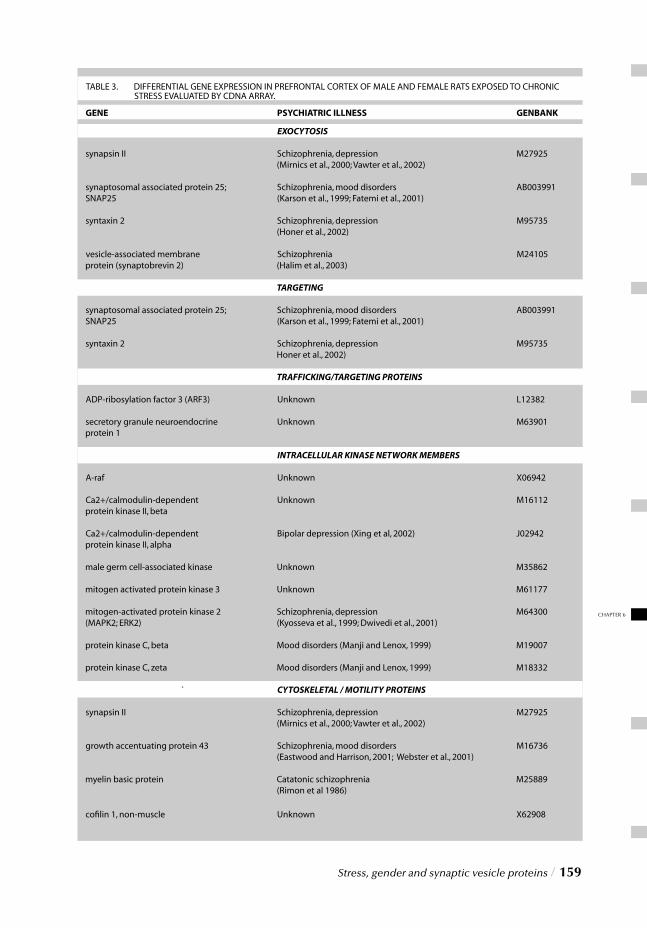
Stress, gender and synaptic vesicle proteins / 159
CHAPTER 6

160 / Stress, gender and synaptic vesicle proteins
Synaptic Protein Expression
Signifi cant alterations in the expression of synaptic vesicle associated proteins were observed following repeated stress. Although males tended to show increased expression, females generally showed tendencies towards unaltered or reduced expression levels. The specifi c changes appear region specifi c however and vary considerably between genders.
Prefrontal Cortex
Synaptotagmin mRNA levels. Prefrontal synaptotagmin mRNA levels showed gender distinctive stress effects as males illustrated signifi cantly increased expression (p<0.021; F=8.198) and females a trend towards decreased expression (p<0.088; F=3.579). This opposite stress response between genders reached signifi cance when males and females were compared under stress conditions (p<0.002; F=19.612) (fi g. 2).
Synaptophysin mRNA levels. Following chronic stress, males also indicated a strong trend towards increased synaptophysin mRNA levels (p<0.053; F=4.955). Although females did not illustrate a signifi cant stress effect, their trend towards reduced expression reached signifi cance when compared to stressed male levels (p<0.006; F=11.932) (fi g. 2).
Synapsin I mRNA levels. Although a gender effect was observed under stress conditions (p<0.005; F=12.811), males and females illustrated only a trend towards increased (p<0.079; F=3.917) and decreased (p<0.189; F=1.99) synapsin I expression respectively (fi g. 2).
Synapsin II mRNA levels. In contrast to the other synaptic proteins, synapsin II expres-sion revealed neither a stress effect nor a gender effect in the prefrontal cortex (fi g. 2).
Figure 2.
Stress effects on synaptic vesicle associated protein expression in the prefrontal cortex. Chronic stress increased synaptotagmin and syn-aptophysin mRNA in the prefrontal cortex of males whereas it did not signifi cantly affect SvaP expression in females. Stressed females illustrated signifi cantly less synaptotagmin, syn-aptophysin, and synapsin I mRNA levels compared to stressed males however. mRNA levels on the y axis are depicted by the inverse fractional cycle number at which the defi ned threshold is passed (1/ CT). Values are shown as mean ± SEM. * represents a signfi cant difference between control and stress conditions (p<0.05); # repre-sents a signifi cant difference between male and female rats (p<0.05).

Stress, gender and synaptic vesicle proteins / 161
CHAPTER 6
Hippocampus
Synaptotagmin mRNA levels. Following stress hippocampal synaptotagmin mRNA levels were increased in males (p<0.035; F=5.968) and unaltered in females such that a gender difference was only observed under stress conditions (p<0.016; F=8.503) (fi g. 3).
Synaptophysin mRNA levels. Like synaptotagmin, synaptophysin mRNA levels were also increased in males (p<0.041; F=5.456) and unaffected in females following stress. Similarly, a gender effect was evident under stress conditions with females expressing lower levels than males (p<0.019; F=7.784) (fi g. 3).
Synapsin I mRNA levels. Following stress, hippocampal synapsin I expression was ele-vated in males (p<0.014; F=8.750) as well as females although the latter did not reach signifi cant levels (p<0.059; F=4.536) (fi g. 3).
Synapsin II mRNA levels.In contrast to the other synaptic proteins, synapsin II expres-sion following stress was unaltered in males as well as females. A gender effect was evident however with females expressing lower levels than males under both control (p<0.037; F=5.834) and stress conditions (p<0.004; F=14.370) (fi g. 3).
Figure 3.
Stress effects on synaptic vesicle associated proteins expression in the hippocampus. Chronic stress increased synaptotagmin, synap-tophysin, and synapsin I mRNA in the hippocampus of males whereas it did not signifi cantly affect SvaP expression in females. Stressed females illustrated sig-nifi cantly less synaptotagmin, synaptophysin, and synapsin II mRNA levels compared to stressed males however. Values are shown as mean ± SEM. * rep-resents a signfi cant difference between control and stress con-ditions (p<0.05); # represents a signifi cant difference between male and female rats (p<0.05).

162 / Stress, gender and synaptic vesicle proteins
DISCUSSION
An important mechanism by which nerve cells modulate the amount of neurotransmit-ter release in response to discrete stimuli is through controlled coordination of the relative number of vesicles in the releasable and reserve pool 15. Action potential arrival at a synapse triggers the opening of voltage gated Ca2+ channels, transient calcium infl ux into the pre-synaptic terminal, repolarization of the invading action potential, synaptic vesicle fusion with the presynaptic plasma membrane, exocytosis and fi nally neurotransmitter release 15. Stress has been reported to disturb this delicate equilibrium, affecting neurotransmit-ter release and causing neuronal defects 18. These observations have led to the hypothesis that stress-induced disruption of neurotransmitter release at the synaptic terminals, pos-sibly mediated by attendant calcium overload, may play a key role in the development of neuronal impairments. In the present investigation, microarray analysis revealed the ability of chronic stress to infl uence transcriptional activity in a gender-related manner. Specifi cally, male rats illustrated a generally increased level of mRNA transcription follow-ing stress, whereas females revealed relatively decreased gene expression. This gender dif-ference in the ability of chronic stress to affect transcriptional activity, especially in male rats is in line with previous data illustrating sex-related dimorphism in patterns of protein expression (calcineurin), phosphorylation (phospho-CREB) and possibly neuronal activ-ity (c-fos) in response to chronic stress (Kuipers et al., unpublished observations) 24. In support of recent studies that document atrophy of prefrontocortical dendrites in response to chronic stress 26, our fi ndings also substantiate abnormal gene expression of prefronto-cortical MAPK1/2-CREB signaling cascade members in chronically stressed rats (Table 3) 22,23. Given the pivotal role of this cascade in neuronal plasticity, the expression arrays were further analyzed to identify changes in genes that have been reported to modulate synaptic plasticity in the medial prefrontal areas and might be affected by prolonged stress exposure. Interestingly, “stress-responsive genes” underlying this marked sex-dependent infl uence of stress on gene expression codify for selective proteins that play a central role in the regulation of specifi c synaptic functions including synaptophysin, synaptotagmin and synapsin I. After microarray identifi cation of these genes, RT-PCR was applied to fur-ther investigate their specifi c transcriptional changes and confi rm these potential targets of stress. The data suggest that stress represents a major factor in the regulation of the expres-sion of these specifi c genes. More importantly, their abnormal regulation could account,
at least in part, for reduced neuronal plasticity and neuronal dysfunctions observed in response to repeated stress. Synaptophysin and synaptotagmin are major integral proteins of synaptic vesicles and are essential for vesicle-cytoplasmic membrane fusion and neu-rotransmitter release 18,27-29. Similarly, synapsins belong to a family of highly conserved phosphoproteins that are specifi cally associated with the cytoplasmic surface of the synap-tic vesicle membrane. By means of changes in their phosphorylation state, they control the fraction of synaptic vesicle available for release, thereby regulating synaptic vesicle life cycle 30 and the effi ciency of neurotransmitter release 31. Abnormal expression of these

Stress, gender and synaptic vesicle proteins / 163
CHAPTER 6
three intrinsic synaptic proteins is consistent with the idea that stress-induced neuronal impairments might be associated with abnormal neurotransmitter release. This is sup-ported by the fact that several stress-related psychiatric illnesses have been associated with altered synaptic vesicular docking protein levels specifi cally in the hippocampal and pre-frontal regions 16-18. Given the importance of a well-regulated synaptic vesicle life cycle for effi cient neu-rotransmitter release, stress-induced alterations in the expression of such proteins in crit-ical brain regions thus support their potential involvement in stress-related psychopa-thology. Dysfunctions in prefrontocortical and hippocampal areas have been reported in response to repeated stress 23,26,32,33 and have also been associated with cognitive and emo-tional defi cits observed in stress-related disorders such as depression 10 or schizophrenia 34. Some of the most consistent neuroimaging fi ndings in support of this hypothesis include decreased hippocampal volume 35,36 and reduced prefrontocortical metabolism 2,37. Studies aiming to identify the molecular and cellular correlates underlying stress-induced pathol-ogy have identifi ed both a reduction in the number of cells as well as atrophy of apical dendrites in these regions 9,26,38. Although such studies have provided suggestions for vari-ous accountable mechanisms, defi nite consensus on this matter is still lacking. In view of stress-mediated effects on synaptic integrity, the nature of these presynaptic components and their potential role in neurotransmitter release, an interesting possibility hence holds that stress-induced disruption of synaptic protein expression and functions may represent a critical contributing factor in the occurrence of cortical-limbic dysfunctions. Interest-ingly and in support of the latter, microarray analysis also revealed stress-responsivity of closely related genes such as various kinases implicated in the modulation of cytoskeleton integrity and synaptic traffi c (CAMK-II), as well as various cytoskeletal proteins essential for the structural synapse integrity (GAP-43) and regulation of the synaptic vesicle cycle and exocytosis (members of SNARE complex) (table 3). Calcium/calmodulin-dependent protein kinase II (CAMKII), suggested as a potential calcium sensor, modulates the phos-phorylation of synaptic proteins 15,39-41 and serves as an essential regulator of the stress response in the brain. Synaptic proteins such as GAP-43 and the SNARE complex are crucial for proper synaptic functioning as they interact with microtubules and neurofi la-ments, which defi ne shape and are important for the guidance of organelles toward synap-tic terminals where they accumulate 29,42. Since synaptic proteins defi ne the basic machin-ery for bridging membranes, mediate the fusion between synaptic vesicle and cell mem-brane and, ultimately, regulate neurotransmitter release, dysregulation in their expression might account for some the deleterious consequences of stress on the brain, including disturbed neuronal plasticity 43. Neuronal abnormalities could thus be attributable to a stress-induced decrease in the expression of specifi c synaptic elements as this may impair synaptic integrity, cause abnormal neurotransmission and compromise neuronal plasticity. Considering the gen-eral increase in SVaPs expression observed in males (and unchanged levels in females) however, it is intriguing to speculate that these changes could refl ect a compensatory

164 / Stress, gender and synaptic vesicle proteins
increase in synaptic protein transcription to recompense reduced SVaP levels lowered by the persistent exposure to adverse conditions. In schizophrenia for instance, reduced presynaptic proteins such as SNAP25, synaptophysin, and synapsins have been reported in several brain regions including the prefrontal cortex and hippocampus 16,44-47. More-over, preclinical reports have also revealed reduced synaptophysin mRNA levels in response to stress 18. Abnormal synapsin, synaptophysin and synaptotagmin expression observed in this study may support the deleterious infl uence of stress on expression of these synaptic proteins. Notably, this effect was more evident in male rats, as illustrated by their increased synaptic protein expression while females demonstrated an overall lack of change. If reduced synaptic protein expression in such disorders refl ects decreased synaptic integrity, it thus raises an interesting possibility that the increased expression seen in males and not females might represent gender-divergent forms of adaptation and/or sensitivity to stress. If we assume that the latter refl ects gender-differential sensitivity to stress, then discrepant gene expression of synaptic proteins might represent an interesting underlying mechanism. It must also be noted however that clarity regarding the nature of these protein changes remains unclear. Despite preclinical and post-mortem studies document-ing decreased SVaP expression in response to stress and psychopathology, reports of increased expression have also been made. A post-mortem study, for instance, has illus-trated increased levels of SNAP25 in the hippocampus of depressed subjects, whereas pre-clinical investigations have shown increased synaptotagmin expression following stress 18. The present data thus illustrate the complex role of stress in regulating the transcription of these proteins. One could assume that changes in the level of expression of specifi c genes codifying for selective SVaPs might refl ect potentially impaired neurotransmission effi cacy and disrupted neuronal plasticity. Alternatively however, if increased transcription of synaptophysin, synaptotagmin and synapsin mRNA levels seen here in males refl ects a compensatory adaptation to correct for decreased protein levels, then this response may also represent an important aspect of an effi cacious adaptation to counter stress-induced plasticity impairments. Given the discrepancy observed in gene expression analysis in which stressed males respond with increased transcriptional activity relative to stressed females, these gender-related activity changes may also account for the increased synapsin I but not synapsin II expression observed in stressed males. High levels of synapsin II rela-tive to synapsin I expression has been associated with excitatory synapses, while relatively high level of synapsin I expression is thought to be associated with inhibitory synapses 41,48,49. This would suggest that defects in turnover rate of synaptic vesicles at inhibitory synapses but not at excitatory synapses can produce disintegration of certain neural cir-cuits culminating in presumable removal of inhibitory transmission as has been suggested in mutant mice lacking synapsin I 50. Considering the overall increased transcriptional activity observed in males, their enhanced synapsin I expression might thus represent a compensatory inhibitory response, to possibly limit excessive stress-induced excitation. Together these data provide evidence for distinct gender-dependent synaptic regulatory responses to stress. More specifi cally these fi ndings demonstrate that synaptic vesicle pro-

Stress, gender and synaptic vesicle proteins / 165
CHAPTER 6
teins are stress-responsive genes that are regulated in a sex- and region-dependent manner. If negatively infl uenced, disturbed regulation and altered expression of these or other syn-aptic vesicle-associated proteins might be related to some of the physiological and patho-physiological effects of stress in the prefrontal cortex and hippocampus. In turn, stress-induced impairment of synaptic protein regulation could represent one of the molecular mechanisms underlying disorders such as depression and schizophrenia, psychiatric con-ditions characterized by abnormal neuronal plasticity 32,51,52. Although many questions remain, the investigation of putative regulators of synaptic formation in parallel with broad coverage analysis of implicated gene expression provides a useful approach to fur-ther investigate impaired synaptic functions following chronic stress. Clearly there is much to be learned about gender-related mechanisms underlying the regulation of neuronal plasticity under prolonged stressful conditions and it remains an open question whether synaptic proteins are mechanistically related to differential neurotransmission in males and females. This work provides strong indications in support of gender-distinctive syn-aptic protein expression upon stress and future studies will no doubt continue to elucidate the role of these synaptic protein interactions following sustained stress.
ACKNOWLEDGEMENTS
The authors appreciate the skillful assistance of Folkert Postema, Kor Venema, Fokko Bosker (Department of Psychiatry, University of Groningen), Minke Jongsma (University Center of Pharmacy, University of Groningen), and Geert Harms (Department of Patho-logy, University of Groningen). The authors would also like to acknowledge the helpful suggestions and expertise of Karoly Mirnics (Departments of Psychiatry and Neurobio-logy, University of Pittsburg).

166 / Stress, gender and synaptic vesicle proteins
REFERENCES1. Sheline, Y.I., Wang, P.W., Gado, M.H., Csernansky, J.G. & Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. U. S. A 93, 3908-3913 (1996).
2. Drevets, W.C. et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824-827 (1997).
3. Sheline, Y.I., Sanghavi, M., Mintun, M.A. & Gado, M.H. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depres- sion. J. Neurosci. 19, 5034-5043 (1999).
4. Kumar, A., Bilker, W., Lavretsky, H. & Gottlieb, G. Volumetric asymmetries in late-onset mood disorders: an attenuation of frontal asymmetry with depression severity. Psychiatry Res. 100, 41-47 (2000). 5. Lai, T., Payne, M.E., Byrum, C.E., Steffens, D.C. & Krishnan, K.R. Reduction of orbital frontal cortex volume in geriatric depres- sion. Biol. Psychiatry 48, 971-975 (2000).
6. Foy, M.R., Stanton, M.E., Levine, S. & Thompson, R.F. Behavioral stress impairs long-term potentiation in rodent hip- pocampus. Behav. Neural Biol. 48, 138-149 (1987).
7. Kim, J.J. & Yoon, K.S. Stress: metaplastic effects in the hippocampus. Trends Neurosci. 21, 505-509 (1998).
8. Magarinos, A.M., Verdugo, J.M. & McEwen, B.S. Chronic stress alters synaptic terminal structure in hippocam- pus. Proc. Natl. Acad. Sci. U. S. A 94, 14002-14008 (1997).
9. Rajkowska, G. Postmortem studies in mood disorders indicate altered num- bers of neurons and glial cells. Biol. Psychiatry 48, 766-777 (2000).
10. Drevets, W.C. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog. Brain Res. 126, 413-431 (2000).
11. Swaab, D.F., Fliers, E., Hoogendijk, W.J., Veltman, D.J. & Zhou, J.N. Interaction of prefrontal cortical and hypothalamic systems in the pathogenesis of depression. Prog. Brain Res. 126, 369-396 (2000).
12. Smith, M.A., Makino, S., Kvetnansky, R. & Post, R.M. Stress and glucocorticoids affect the expression of brain- derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15, 1768-1777 (1995).
13. Lipska, B.K., Khaing, Z.Z., Weickert, C.S. & Weinberger, D.R. BDNF mRNA expression in rat hippocampus and prefrontal cortex: effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur. J. Neurosci. 14, 135-144 (2001).
14. Janz, R. et al. Essential roles in synaptic plasticity for synaptogyrin I and syn aptophysin I. Neuron 24, 687-700 (1999).
15. Turner, K.M., Burgoyne, R.D. & Morgan, A. Protein phosphorylation and the regulation of synaptic mem- brane traffi c. Trends Neurosci. 22, 459-464 (1999).
16. Eastwood, S.L. & Harrison, P.J. Decreased synaptophysin in the medial temporal lobe in schizophrenia demonstrated using immunoautoradiography. Neuroscience 69, 339-343 (1995).
17. Honer, W.G. et al. Synaptic and plasticity-associated proteins in anterior frontal cortex in severe mental illness. Neuroscience 91, 1247-1255 (1999).
18. Thome, J. et al. Stress differentially regulates synaptophysin and synaptotagmin expression in hippocampus. Biol. Psychiatry 50, 809-812 (2001).
19. Klein, L.C. & Corwin, E.J. Seeing the unexpected: how sex differences in stress responses may provide a new perspective on the manifestation of psychi- atric disorders. Curr. Psychiatry Rep. 4, 441-448 (2002).
20. Kessler, R.C. Epidemiology of women and depression. J. Affect. Disord. 74, 5-13 (2003).
21. Taylor, S.E. et al. Biobehavioral responses to stress in females: tend-and- befriend, not fi ght-or-fl ight. Psychol. Rev. 107, 411-429 (2000).
22. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphos- phorylation in medial prefrontocortical dendrites: implications for stress-related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
23. Kuipers, S.D., Trentani, A., Den Boer, J.A. & Ter Horst, G.J. Molecular correlates of impaired prefrontal plasticity in response to chronic stress. J. Neurochem. 85, 1312-1323 (2003).
24. Trentani, A. et al. Immunohistochemical changes induced by repeated footshock stress: revelations of gender-based differences. Neurobiol. Dis. 14, 602-618 (2003).
25. Heid, C.A., Stevens, J., Livak, K.J. & Williams, P.M. Real time quantitative PCR. Genome Res. 6, 986-994 (1996).
26. Wellman, C.L. Dendritic reorganization in pyramidal neurons in medial pre frontal cortex after chronic corticosterone administration. J. Neurobiol. 49, 245-253 (2001).
27. Matthew, W.D., Tsavaler, L. & Reichardt, L.F. Identifi cation of a synaptic vesicle-specifi c membrane protein with a wide distribution in neuronal and neurosecretory tissue. J. Cell Biol. 91, 257-269 (1981).
28. Sudhof, T.C., Lottspeich, F., Greengard, P., Mehl, E. & Jahn, R. A synaptic vesicle protein with a novel cytoplasmic domain and four transmembrane regions. Science 238, 1142-1144 (1987).
29. Greengard, P., Valtorta, F., Czernik, A.J. & Benfenati, F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780-785 (1993).
30. Murthy, V.N. Spreading synapsins. Nat. Neurosci. 4, 1155-1157 (2001).
31. Chi, P., Greengard, P. & Ryan, T.A. Synapsin dispersion and reclustering during synaptic activity. Nat. Neurosci. 4, 1187-1193 (2001).
32. McEwen, B.S. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105-122 (1999).
33. Mizoguchi, K., Ishige, A., Aburada, M. & Tabira, T. Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience 119, 887-897 (2003).
34. Kuperberg, G. & Heckers, S. Schizophrenia and cognitive function. Curr. Opin. Neurobiol. 10, 205-210 (2000).
35. Copolov, D. et al. Neurobiological fi ndings in early phase schizophrenia. Brain Res. Brain Res. Rev. 31, 157-165 (2000).

Stress, gender and synaptic vesicle proteins / 167
CHAPTER 6
36. Sheline, Y.I. 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol. Psychiatry 48, 791-800 (2000).
37. Carter, C.S. et al. Functional hypofrontality and working memory dysfunction in schizophrenia. Am. J. Psychiatry 155, 1285-1287 (1998).
38. McEwen, B.S. The neurobiology of stress: from serendipity to clinical rele- vance. Brain Res. 886, 172-189 (2000).
39. Rubenstein, J.L., Greengard, P. & Czernik, A.J. Calcium-dependent serine phosphorylation of synaptophysin. Synapse 13, 161-172 (1993).
40. Hilfi ker, S., Pieribone, V.A., Nordstedt, C., Greengard, P. & Czernik, A.J. Regulation of synaptotagmin I phosphorylation by multiple pro- tein kinases. J. Neurochem. 73, 921-932 (1999).
41. Hilfi ker, S. et al. Synapsins as regulators of neurotransmitter release. Philos. Trans. R. Soc. Lond B Biol. Sci. 354, 269-279 (1999).
42. Burgoyne, R.D. Control of exocytosis in adrenal chromaffi n cells. Biochim. Bio-phys. Acta 1071, 174-202 (1991).
43. Weickert, C.S. & Weinberger, D.R. A candidate molecule approach to defi ning developmental pathology in schizophrenia. Schizophr. Bull. 24, 303-316 (1998).
44. Jorgensen, O.S. & Riederer, P. Increased synaptic markers in hippocampus of depressed patients. J. Neural Transm. 64, 55-66 (1985).
45. Browning, M.D., Dudek, E.M., Rapier, J.L., Leonard, S. & Freedman, R. Signifi cant reductions in synapsin but not synaptophysin spe- cifi c activity in the brains of some schizophrenics. Biol. Psychiatry 34, 529-535 (1993).
46. Glantz, L.A. & Lewis, D.A. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specifi city [corrected and republished article originally appeared in Arch Gen Psychiatry 1997 Jul;54(7):660-9]. Arch. Gen. Psychiatry 54, 943-952 (1997).
47. Jorgensen, O.S. SNAP-25 is the major immunoreactive component of the brain- specifi c D3 protein. Neuroreport 7, 73-76 (1995).
48. Mandell, J.W., Czernik, A.J., De Camilli, P., Greengard, P. & Townes-Anderson, E. Differential expression of synapsins I and II among rat retinal synapses. J. Neurosci. 12, 1736-1749 (1992).
49. Iwakuma, M. et al. Antisense in vivo knockdown of synaptotagmin I and synapsin I by HVJ-liposome mediated gene transfer modulates ischemic injury of hippocampus in opposing ways. Neurosci. Res. 45, 285-296 (2003).
50. Terada, S., Tsujimoto, T., Takei, Y., Takahashi, T. & Hirokawa, N. Impairment of inhibitory synaptic transmission in mice lacking synapsin I. J. Cell Biol. 145, 1039-1048 (1999).
51. Gage, F.H. Structural plasticity: cause, result, or correlate of depression. Biol. Psychiatry 48, 713-714 (2000).
52. Manji, H.K., Drevets, W.C. & Charney, D.S. The cellular neurobiology of depression. Nat. Med. 7, 541-547 (2001).


PART IV
IMPLICATIONS FOR
PHARMACOTHERAPY


7
IMPACT OF LONG-TERM ANTIDEPRES-SANT TREATMENT ON CHRONIC STRESS INDUCED REDUCTION OF NEUROGENESIS IN ADULT RATS: REVELATIONS OF SEX/DRUG SPECIFIC REGULATION
Introduction: Growing amounts of evidence have established a
link between stressful life events, reduced hippocampal neuro-
plasticity and development or exacerbation of depression and
new theories are beginning to consider impaired adult hippo-
campal neurogenesis as a critical step in this loss of plasticity.
Objective: To investigate the role of stress and/or effects of con-
comitant antidepressant treatments on hippocampal cell prolifera-
tion. Methods: BrdU immunoreactivity changes were investigated
in response to repeated footshock stress and antidepressant
administration in male and female rats. Results: Repeated stress
revealed a suppressive effect on hippocampal neurogenesis/
survival, particularly in cyclic female rats, refl ecting possible gen-
der-differential mechanisms governing these complex processes.
Gender also appears to be a major variable in determining antide-
pressant ability to attenuate stress-induced impairment of adult
cell proliferation since tianeptine, reboxetine and citalopram
counteracted chronic stress-induced reduction of hippocampal
BrdU-labeled cells in males but not females. Conclusion: Ovarian
hormones and serotonin may play a critical role in these diffe-
rences since persistently elevated estrogen levels and disrupted
serotonin neurotransmission may inhibit both production and
survival of hippocampal cells. Together these fi ndings may offer
new insights into the mechanisms involved in the deleterious
neuronal effects of chronic stress, the molecular events underly-
ing antidepressants’ therapeutic action and the role of gender in
response to stress, psychopathology and pharmacotherapy.
Kuipers S.D., Trentani A., Koch T., Ter Horst G.J., Den Boer J.A.
Neuropsychopharmacology. Submitted


CHAPTER 7
Stress, neurogenesis and antidepressants / 173
INTRODUCTION
For most of the past century, it was believed that the adult brain was incapable of ge-nerating new neurons. Recent data however, have led to the widespread recognition that neurogenesis, is not restricted to embryonic development, but continues into adulthood in specifi c regions of the mammalian brain such as the subgranular zone of the dentate gyrus 1-3. Various factors appear to affect this process however. Stress for instance, has been reported to suppress adult cell proliferation, possibly through elevation of adrenal corticosteroids, stimulation of glutamatergic neurotransmission and/or decline in seroto-nergic activity, 4-6. In contrast, increased cell proliferation, has been observed following stimulation of serotonergic function (possibly modulated by 5-HT1A receptor) and estro-gen treatment 7,8. The impact of stress, directly or indirectly, on neuron production is of particular interest during chronic aversive conditions due to the cumulative infl uence of adverse events on neurogenesis. Although not the only structure to exhibit neurogenesis throughout adulthood, since cell proliferation also occurs in the olfactory bulb, the subventricular zone and the neocor-tex 2,9, the dentate gyrus is of particular interest since: Neurogenesis occurs robustly in this area (up to 9000 new neurons per day in mice and rats), a feature common in all mam-malian species 10; Most studies on adult neurogenesis have been conducted in the dentate gyrus; This region plays a central role in modulating the stream of information processing from extrahippocampal regions to various cell fi elds of the hippocampus 5, explaining why any disrupted dentate gyrus activity may result in abnormal hippocampal functioning. Proper hippocampal functioning is in turn essential given its central role in the pathophys-iology and treatment of depression 11,12, a psychiatric disorder characterized by cortical-limbic abnormalities (i.e. hippocampal atrophy and HPA axis hyperacti-vity) and marked gender-related prevalence (with women more prone to psychopathology than men) 11,13-15. Stress however, particularly when severe and persistent, also plays a crucial role in the development of depression. Although the biological origins of this disorder have been dif-fi cult to grasp, it has been suggested to result from disrupted neuronal plasticity 16,17 and new theories linking stress, neurogenesis and depression, are beginning to consider stress-induced impairment of adult hippocampal neurogenesis as a critical step in this loss of plasticity 5,18. A large body of evidence has in fact established a link between stressful life events, reduced hippocampal neuroplasticity and development or exacerbation of depres-sion 19,20. At the cellular level, evidence indicates neuronal atrophy and cell loss following stress and depression 21-24 and at the molecular level, it has been suggested that these cellu-lar defi ciencies, occurring primarily in the hippocampus, result from impaired neuroplas-ticity 25-27. It has thus also been speculated that up-regulation of neurogenesis could block or reverse effects of stress on hippocampal neurons and ameliorate depressive symptoms. Interestingly, treatment with antidepressant such as selective serotonin reuptake inhibi-tors (SSRIs), have indeed been reported to enhance cell proliferation in the adult brain 28,29. Together these observations have led to the hypothesis that antidepressants may exert

174 / Stress, neurogenesis and antidepressants
their effects by stimulating neurogenesis and neuroplasticity, thereby promoting adaptive changes in neuronal systems that counteract negative stress effects and facilitate recovery from depression 5. To investigate the effects of stress and/or concomitant antidepressant treatment on hip-pocampal cell proliferation/survival, BrdU immunoreactivity changes were investigated following repeated footshocks and antidepressant administration (tianeptine, citalopram and reboxetine). Peripheral tryptophan levels and adrenal activity were also examined. Furthermore, since gender represents a crucial aspect in stress sensitivity and psychopa-thology development, neuroendocrine and immunohistochemical adaptations were inves-tigated in both male and female rats 13,30. These results may provide novel insight into effects of prolonged stress on adult neurogenesis as well as the mechanisms underlying antidepressants’ mode of action in the brain.
MATERIALS AND METHODS
Animals
Male (n=48: 212-240 g) and cyclic female Wistar rats were used (n=51: 195-212 g). The animals were individually housed (cages 45 x 28 x 20 cm) with food and water ad libitum. They were maintained on a 12/12-hr light/dark cycle and weighed (9:00 am) and handled daily for 5-8 min to minimize non-specifi c stress response. The experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and the guidelines of the Animal Bio-ethics Committee of the Uni-versity of Groningen (FDC: 2509).
Footshock procedure
The rodent footshock-chamber consists of a box containing an animal space placed on a grid fl oor connected to a shock generator and scrambler. Stressed rats received one daily session of varying duration (between 30 and 120 min/day) in the footshock-chamber. During each session 5 inescapable and uncontrollable footshocks were given (0.8 mA in
intensity and 8 sec in duration). In order to make the procedure as unpredictable as pos-sible, starting time, inter-shock interval, and total time spent in the box were varied on a daily basis. This stress procedure did not result in any visible injuries to the rats.

CHAPTER 7
Stress, neurogenesis and antidepressants / 175
Experimental Setup
To investigate the immunohistochemical and neuroendocrine alterations induced by sus-tained stress exposure, long-term antidepressant treatments, and the interactions between experimental conditions, a 2 X 2 procedure was applied. Per gender rats were randomly assigned to eight groups:
1. CTR-vehicle (n˚=6): these rats received daily exposre to the footshock box and vehicle injections. They were not subjected to any actual footshocks during the experiment; 2. STRESS-vehicle (n˚=6): these rats received daily footshocks and vehicle injections; 3. CTR-tianeptine (n˚=6): these rats were used as controls to identify neurochemical adaptations induced by long-term tianeptine administration. They received daily exposure to the footshock box and tianeptine injections, but not any actual footshocks; 4. STR-tianeptine (n˚=7): these animals received daily footshocks and tianeptine injections;5. CTR-citalopram (n˚=6): these rats received daily exposure to the footshock box and citalopram injections but no footshocks; 6. STR-citalopram (n˚=7): stressed rats received daily exposure to the aversive procedure and citalopram injections;7. CTR-reboxetine (n˚=6): these animals were exposed to the footshock box and received daily injections of reboxetine, without any actual footshocks; 8. STR-reboxetine (n˚=7): these rats received daily footshocks and reboxetine injections;On the fi nal day of the experiment all rats were placed in the footshock box for a 15 minute session. During this fi nal session however, no footshocks were delivered. It is also important to note that no injections were administered prior to this fi nal session, allowing a washout period of about 24 hours.
Pharmacological profi le, mode of action, dosage, and route of administration
Citalopram. This SSRI’s mechanism of action is presumed to be linked to the potentia-tion of serotonergic activity in the CNS resulting from inhibition of neuronal reuptake of
serotonin (5 HT) 31,32. Dosage. Citalopram, kindly provided by Lundbeck B.V. (The Netherlands), was dissolved in saline (0.9% NaCl) at a concentration of 20mg*ml-1 and injected intraperitoneously (i.p.) at the dosage of 20mg*kg-1*day-1, for a 21 day-period, 30-45 minutes before expo-sure to the footshock procedure. The daily dose of 20mg*kg-1*day-1 was chosen after reviewing recent pharmacological studies using this antidepressant in a long-term setting 33,34. These studies suggest that an administered dosage of 20mg*kg-1 will provide a suf-fi cient plasma concentration of about 250-300 nmol*l-1 34,35, regardless of the mode of administration (oral vs. osmotic pumps).

176 / Stress, neurogenesis and antidepressants
Tianeptine. Tianeptine is an atypical antidepressant, both structurally and in terms of its neurochemical profi le. It is devoid of sedative effects and induces slight stimulation of locomotor activity. In monkeys, it decreases aggressive and emotive states and improves individual behavior 36. Pharmacological studies have shown that, unlike other antidepres-sants, tianeptine stimulates the uptake of serotonin and increases 5-hydroxyindoleacetic acid levels in the brain 37, although recent hypotheses also suggest modulation of exci-tatory amino acid transmission by targeting the phosphorylation-state of glutamate recep-tors at the CA3 synapse 38. Dosage. Tianeptine, kindly provided by the Institut de Recherches Internationales Servier (Paris, France), was dissolved in saline (0.9% NaCl) at a concentration of 10mg*ml-1, and injected intraperitoneously (i.p.) at the dosage of 10mg*kg-1*day-1, for a 21 day-period, 30-45 minutes before exposure to the footshock procedure. The daily dose of 10mg*kg-
1*day-1 was chosen based on the indications provided by the drug manufacturer and li-terature review, as the effective dosage suffi cient to prevent behavioral and neurochemical abnormalities in a chronic setting 38-43. Reboxetine. Reboxetine, a potent and selective norepinephrine reuptake inhibitor with-out any affi nity for neurotransmitter receptors, displays an antidepressant profi le in both animal tests and in clinical trials. Unlike desipramine or imipramine, reboxetine has a weak affi nity for muscarinic, histaminergic H1, adrenergic alpha1, and dopaminergic D2 receptors and low toxicity in animals. Dosage. Reboxetine, a racemic mixture of R,R- and S,S-([2-[alpha [2-ethoxyphenoxy] benzyl]-morpholine sulfate]) and (+)-(S,S)-reboxetine methanesulfon, was kindly pro-vided by Pharmacia B.V. (The Netherlands). The noradrenaline reuptake inhibitor was dissolved in saline (0.9% NaCl) at a concentration of 20mg*ml-1, and injected intraperito-neously (i.p.) at the dosage of 20mg*kg-1*day-1, for a 21 day-period, 30-45 minutes before exposure to the footshock procedure. The daily doses of 20mg*kg-1*day-1 was chosen based on its reported low half-life and recent preclinical studies investigating the behavioral and neurochemical changes induced by prolonged administration of this antidepressant using similar dosages in rats 44-47.
Route of administration
In contrast to studies in which osmotic pumps were used to deliver drugs, here intraperi-
toneous injections were chosen as the route of administration. Although osmotic pumps allow a more constant administration, the antidepressant concentration loaded into each pump requires prior estimation based on each individual animal’s weight. The current experimental design however presents problems in establishing body weight since the female cycle as well as antidepressant treatment have been shown to modify female body weight growth by reducing food intake 48,49. Furthermore, use was made of a chronic aver-sive paradigm, which has also been reported to strongly affect normal weight gain in ani-mals 50. Given the diffi culties in reliably predicting body weight changes in cyclic female

CHAPTER 7
Stress, neurogenesis and antidepressants / 177
rats during chronic stress and long-term antidepressant administration, drugs were deliv-ered by i.p. injections to guarantee a fi xed and constant dosage throughout the experi-ment, regardless of effects due to experimental conditions.
Physiological and neuroendocrine correlates of the chronic stress response
To defi ne the physiological and neuroendocrine changes induced by prolonged footshock exposure and/or antidepressant treatment, various parameters were measured. Weight gain was monitored on a daily basis throughout the experiment and, upon termination, adrenal glands were removed and weighed. Furthermore, blood samples were taken after the fi nal session and stored at -20°C to determine plasma tryptophan concentrations.
Plasma tryptophan concentrations: extraction and chromatography
Tryptophan is the precursor for the neurotransmitter 5-hydroxytryptamine (5-HT). This amino acid is present in the blood in bound and free form. Elevation of the free fraction of L-tryptophan in plasma provides an increased amount of the precursor molecule to pass across the blood-brain barrier. This higher penetration and increased rate of entry into the brain activates synthesis of brain serotonin, leading to higher levels of 5-HT. Since serotonin has been found to have a potential neurotrophic role in the brain, any altera-tion in its metabolism could refl ect a permanent change in brain neuroplasticity and/or neurogenesis.
Instrumentation.
The quantifi cation of total plasma tryptophan was performed as previously described 51 with methods based on solid-phase extraction (SPE) and gradient high performance liquid chromatography (HPLC) with fl uorometric detection. In short, the on-line SPE unit used was a Prospekt (Spark Holland, Emmen, The Netherlands) consisting of a SPE control-ler unit, a solvent delivery unit, and an autosampler. Samples were injected with a Midas autosampler (Spark Holland BV). The HPLC pump used was a Gynkotek Series P580A binary high-pressure gradient pump (Softron GmbH); detection was performed with a Jasco 920 FP spectrofl uorometer (excitation wavelength, 285 nm; emission wavelength, 340 nm). Detector output was integrated using ChromPerfect (Ver. 3.51) integration soft-ware (Nelson).
Gradient.
The binary gradient system consisted of eluent A (50 mmol/L potassium dihydrogen phosphate in water adjusted to pH 3.3 with phosphoric acid) and eluent B (700 mL/L

178 / Stress, neurogenesis and antidepressants
eluent A–300 mL/L acetonitrile). Before use, all eluents were fi ltered through a 0.45 µm membrane fi lter (Schleicher & Schuell) and degassed at reduced pressure for 5 min. Gra-dient elution was performed according to the following elution program: 0–2 min, 80% A, 20% B; 2–10 min, 25% A, 75% B; 10–10.1 min, 0% A, 100% B; 10.1–15 min, 0% A, 100% B; 15–15.1 min, 80% A, 20% B; 15.1–20 min, 80 A, 20% B. The gradients applied were linear; the fl ow rate was 0.6 mL/min. Chromatography was performed at ambient temperature.
Analysis and quantifi cation.
Plasma tryptophan levels were quantifi ed by calculating the peak-area ratios relative to that of the internal standard (5-MTP) in samples. Sample peak-area ratios were compared with the peak-area ratios obtained for the calibration solutions at fi ve different concentra-tions. Calibrators were freshly prepared by diluting stock solutions to concentrations of 5–20 µmol/L with 0.01 mol/L acetic acid. Peak identity was verifi ed by the response of the retention time to different chromatographic conditions and the addition of calibrators 51.
Immunohistochemistry
All rats were terminated 120 minutes following exposure to the fi nal session, with an over-dose of halothane followed by a transcardial perfusion with 4% paraformaldehyde solu-tion in 0.1M sodium phosphate buffer (pH 7.4). The brains were carefully removed and post-fi xed in the same solution overnight at 4°C, before being transferred to a potassium phosphate buffer (KPBS 0.02 M, pH 7.4) and stored at 4°C. Following cryoprotection of the brains by overnight immersion in a 30% glucose solution, coronal serial sections of 40 mm were prepared on a cryostat microtome. Sections were collected in KPBS with sodiumazide and stored at 4°C.
BrdU injections and immunohistochemistry
From day 13 to 16, animals received daily i.p. injections of 5-bromo-2’-deoxyuridine (BrdU; 100 mg/kg; Sigma) to label dividing cells. Since cyclic females were not synchro-nized before the experiment and rates of neurogenesis have been reported to differ in vari-ous phases of the estrous cycle, BrdU was administered for four consecutive days. This way all females received BrdU during one entire cycle, independent of their specifi c estrous phases at the time of injection. All rats followed the same schedule and were perfused 6 days following the last BrdU injection. This survival time allows for the completion of several cell cycles (up to 9) and was chosen based on recent studies that report death of a large portion of new cells before reaching maturity 52. In fact, indirect evidence suggests a marked decrease in newly generated adult dentate gyrus cells between 1 and 2 weeks after

CHAPTER 7
Stress, neurogenesis and antidepressants / 179
BrdU labeling 52,53. BrdU immunohistochemistry was performed as previously described 54. In short, BrdU labeling requires the following pretreatment steps: DNA denaturation (50% formamide/2*SSC (pH 7.0), 65°C, 120 min), and acidifi cation (2 M HCl, 30 min). Primary anti-body concentration was a monoclonal mouse anti-BrdU (Roche, 1:800) and secondary antibody concentration was goat anti mouse (Jackson ImmunoResearch, 1:400). The reaction product was visualized by adding diaminobenzidine as chromogen and 1% H
2O
2
for 15 min. Finally, the sections were washed, mounted on slides, dehydrated and cover-slipped with DePex.
BrdU-labeling analysis
A modifi ed unbiased stereology protocol was used that has been reported to successfully quantify BrdU labeling. Quantifi cation of positive cell nuclei was performed using at least 8 sections of a specifi c portion of the hippocampal dentate gyrus (DG: Bregma -2.00 to -3.50). Every seventh section (an average of 7) through the rostral/caudal extent of the dentate gyrus was collected and coded before processing for immunohistochemistry to ensure objectivity. All BrdU positive cells in the granule cell layer and the subgranular zone (defi ned as a two-cell-body-wide zone along the border of the granule cell layer) were counted regardless of size or shape using a computerized imaging analysis system (LEICA Imaging System Ltd., Cambridge, England). To enable counting of cell clusters, cells were examined under ×400 magnifi cation and ×1000. The total number of BrdU positive cells was estimated by multiplying the number of cells counted in every 7th section by 7 and reported as total number of cells (mean and SE) 55.
Statistics
Results were analyzed by performing F tests of variance. That value determined whether t tests for equal or unequal variance were performed to compare the various experimental and control conditions. P < 0.05 was defi ned as the level of signifi cance between groups.
RESULTS
Body weight gain
Stress.
In male rats prolonged stress exposure resulted in a signifi cant reduction of total body weight gain (tBWG) by the end of the experiment (day 22) (F=9.79, p<0.0031) (fi g. 1). This stress-induced reduction was evident in every treatment group including vehicle (CTR-vehicle vs. STR-vehicle; F=11.18, p<0.0086), tianeptine (CTR-tianeptine vs. STR-tianeptine; F=9.75, p<0.011), citalopram (CTR-citalopram vs. STR-citalopram; F=6.00, p<0.034) and reboxetine (CTR-reboxetine vs. STR-reboxetine; F=13.48, p<0.0043) (fi g.
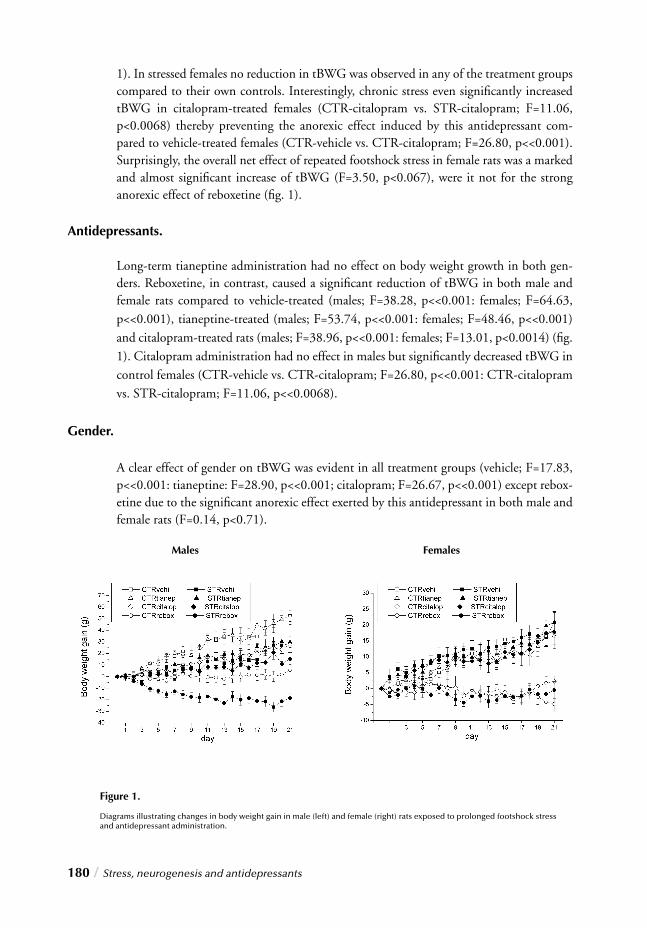
180 / Stress, neurogenesis and antidepressants
1). In stressed females no reduction in tBWG was observed in any of the treatment groups compared to their own controls. Interestingly, chronic stress even signifi cantly increased tBWG in citalopram-treated females (CTR-citalopram vs. STR-citalopram; F=11.06, p<0.0068) thereby preventing the anorexic effect induced by this antidepressant com-pared to vehicle-treated females (CTR-vehicle vs. CTR-citalopram; F=26.80, p<<0.001). Surprisingly, the overall net effect of repeated footshock stress in female rats was a marked and almost signifi cant increase of tBWG (F=3.50, p<0.067), were it not for the strong anorexic effect of reboxetine (fi g. 1).
Antidepressants.
Long-term tianeptine administration had no effect on body weight growth in both gen-ders. Reboxetine, in contrast, caused a signifi cant reduction of tBWG in both male and female rats compared to vehicle-treated (males; F=38.28, p<<0.001: females; F=64.63, p<<0.001), tianeptine-treated (males; F=53.74, p<<0.001: females; F=48.46, p<<0.001) and citalopram-treated rats (males; F=38.96, p<<0.001: females; F=13.01, p<0.0014) (fi g. 1). Citalopram administration had no effect in males but signifi cantly decreased tBWG in control females (CTR-vehicle vs. CTR-citalopram; F=26.80, p<<0.001: CTR-citalopram vs. STR-citalopram; F=11.06, p<<0.0068).
Gender.
A clear effect of gender on tBWG was evident in all treatment groups (vehicle; F=17.83, p<<0.001: tianeptine: F=28.90, p<<0.001; citalopram; F=26.67, p<<0.001) except rebox-etine due to the signifi cant anorexic effect exerted by this antidepressant in both male and female rats (F=0.14, p<0.71).
Figure 1.
Diagrams illustrating changes in body weight gain in male (left) and female (right) rats exposed to prolonged footshock stress and antidepressant administration.
Males Females

CHAPTER 7
Stress, neurogenesis and antidepressants / 181
Adrenal weight
Stress.
Repeated footshock stress caused a marked enlargement of adrenal glands in both male (CTR-vehicle vs. STR-vehicle: F=5.16, p<0.047) and female rats (CTR-vehicle vs. STR-vehicle: F=6.23, p<0.032). All antidepressants were able to attenuate chronic stress-induced adrenal hypertrophy in both male and female rats. This stress-reducing effect was particularly evident for citalopram (which even signifi cantly reduced adrenal weight compared to its own controls; CTR-citalopram vs. STR-citalopram, F=5.14, p<0.047) and reboxetine in male rats (CTR-reboxetine vs. STR-reboxetine, F=0.002, p<0.96) while tianeptine (CTR-tianeptine vs. STR-tianeptine, F=1.15, p<0.31) and citalopram (CTR-citalopram vs. STR-citalopram, F=2.03, p<0.18) were more effective in females (fi g. 2). Interestingly tianeptine was the only antidepressant able to signifi cantly attenuate chronic stress-induced adrenal enlargement compared to vehicle-treated female rats (STR-vehicle vs. STR-tianeptine, F=7.49, p<0.019).
Antidepressants.
Long-term antidepressant treatments, particularly citalopram and reboxetine, resulted in enlarged adrenals in male rats (vehicle vs. tianeptine: F=2.86, p<0.10; vehicle vs. citalo-pram: F=8.99, p<0.007; vehicle vs. reboxetine: F=8.25, p<0.009) (fi g. 2). This effect was even stronger when non-stressed, vehicle treated males were compared with the non-stressed, antidepressant treated rats (CTR-vehicle vs. CTR-tianeptine: F=5.75, p<0.038; CTR-vehicle vs. CTR-citalopram: F=38.56, p<<0.001; CTR-vehicle vs. CTR-reboxetine: F=18.11, p<0.0017). In female rats, no signifi cant effects were found with the exception of tianeptine, which led to an opposite effect, namely reduced adrenal gland size (vehicle vs. tianeptine: F=7.90, p<0.01; CTR-vehicle vs. CTR-tianeptine: F=2.80, p<0.13).
Gender.
A signifi cant effect of gender was observed amongst all treatment groups, with female rats showing signifi cantly larger adrenals than males (vehicle: F=54.79, p<<0.001; tianeptine: F=9.67, p<0.005; citalopram: F=32.25, p<<0.001; reboxetine: F=7.72, p<0.011) (fi g. 2).
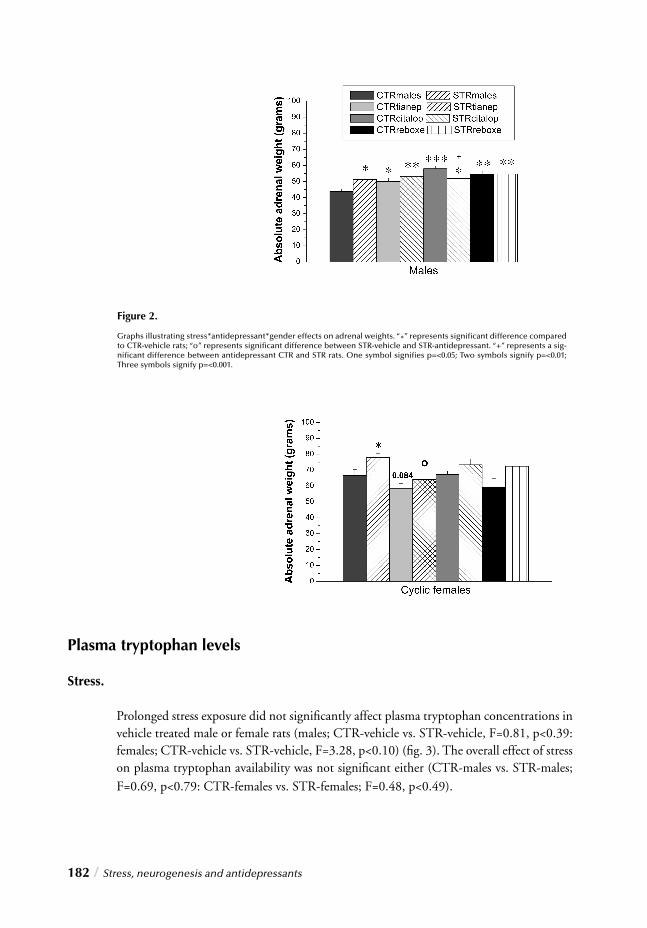
182 / Stress, neurogenesis and antidepressants
Plasma tryptophan levels
Stress.
Prolonged stress exposure did not signifi cantly affect plasma tryptophan concentrations in vehicle treated male or female rats (males; CTR-vehicle vs. STR-vehicle, F=0.81, p<0.39: females; CTR-vehicle vs. STR-vehicle, F=3.28, p<0.10) (fi g. 3). The overall effect of stress on plasma tryptophan availability was not signifi cant either (CTR-males vs. STR-males; F=0.69, p<0.79: CTR-females vs. STR-females; F=0.48, p<0.49).
Figure 2.
Graphs illustrating stress*antidepressant*gender effects on adrenal weights. “*” represents signifi cant difference compared to CTR-vehicle rats; “o” represents signifi cant difference between STR-vehicle and STR-antidepressant. “+” represents a sig-nifi cant difference between antidepressant CTR and STR rats. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001.
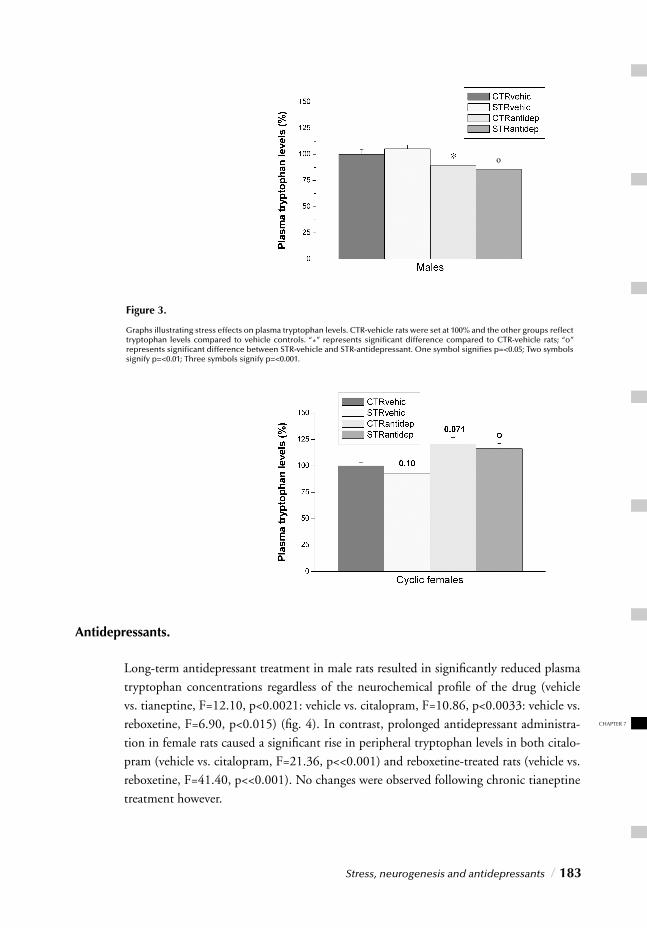
CHAPTER 7
Stress, neurogenesis and antidepressants / 183
Antidepressants.
Long-term antidepressant treatment in male rats resulted in signifi cantly reduced plasma tryptophan concentrations regardless of the neurochemical profi le of the drug (vehicle vs. tianeptine, F=12.10, p<0.0021: vehicle vs. citalopram, F=10.86, p<0.0033: vehicle vs. reboxetine, F=6.90, p<0.015) (fi g. 4). In contrast, prolonged antidepressant administra-tion in female rats caused a signifi cant rise in peripheral tryptophan levels in both citalo-pram (vehicle vs. citalopram, F=21.36, p<<0.001) and reboxetine-treated rats (vehicle vs. reboxetine, F=41.40, p<<0.001). No changes were observed following chronic tianeptine treatment however.
Figure 3.
Graphs illustrating stress effects on plasma tryptophan levels. CTR-vehicle rats were set at 100% and the other groups refl ect tryptophan levels compared to vehicle controls. “*” represents signifi cant difference compared to CTR-vehicle rats; “o” represents signifi cant difference between STR-vehicle and STR-antidepressant. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001.
o
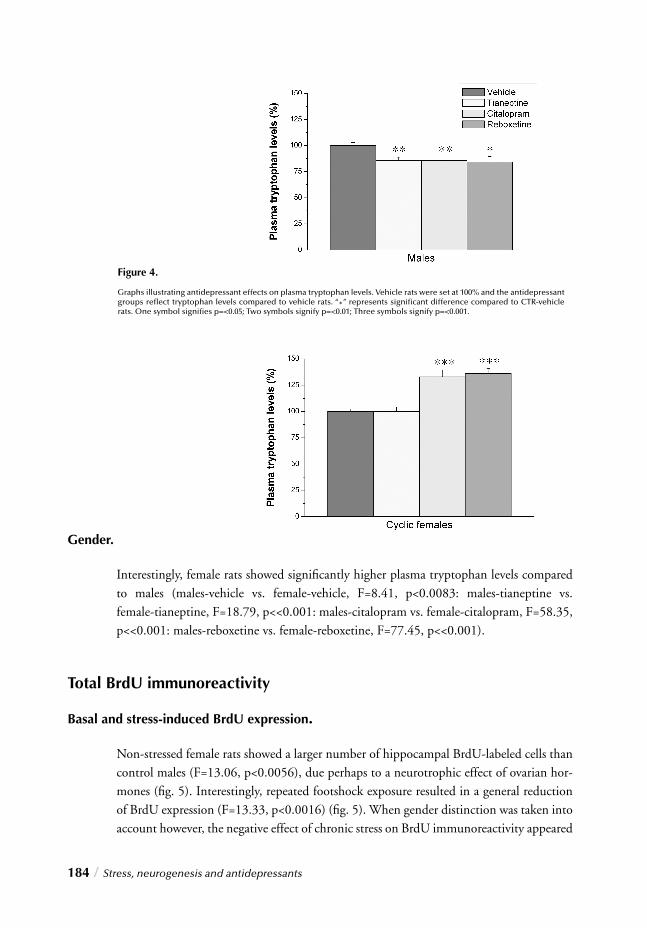
184 / Stress, neurogenesis and antidepressants
Gender.
Interestingly, female rats showed signifi cantly higher plasma tryptophan levels compared to males (males-vehicle vs. female-vehicle, F=8.41, p<0.0083: males-tianeptine vs. female-tianeptine, F=18.79, p<<0.001: males-citalopram vs. female-citalopram, F=58.35, p<<0.001: males-reboxetine vs. female-reboxetine, F=77.45, p<<0.001).
Total BrdU immunoreactivity
Basal and stress-induced BrdU expression.
Non-stressed female rats showed a larger number of hippocampal BrdU-labeled cells than control males (F=13.06, p<0.0056), due perhaps to a neurotrophic effect of ovarian hor-mones (fi g. 5). Interestingly, repeated footshock exposure resulted in a general reduction of BrdU expression (F=13.33, p<0.0016) (fi g. 5). When gender distinction was taken into account however, the negative effect of chronic stress on BrdU immunoreactivity appeared
Figure 4.
Graphs illustrating antidepressant effects on plasma tryptophan levels. Vehicle rats were set at 100% and the antidepressant groups refl ect tryptophan levels compared to vehicle rats. “*” represents signifi cant difference compared to CTR-vehicle rats. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001.
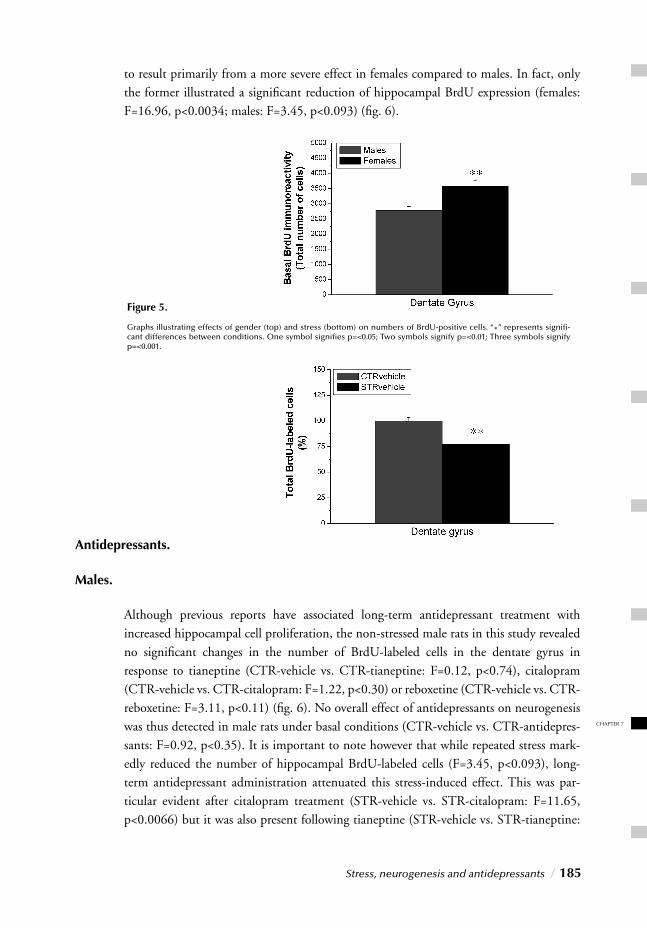
CHAPTER 7
Stress, neurogenesis and antidepressants / 185
to result primarily from a more severe effect in females compared to males. In fact, only the former illustrated a signifi cant reduction of hippocampal BrdU expression (females: F=16.96, p<0.0034; males: F=3.45, p<0.093) (fi g. 6).
Antidepressants.
Males.
Although previous reports have associated long-term antidepressant treatment with increased hippocampal cell proliferation, the non-stressed male rats in this study revealed no signifi cant changes in the number of BrdU-labeled cells in the dentate gyrus in response to tianeptine (CTR-vehicle vs. CTR-tianeptine: F=0.12, p<0.74), citalopram (CTR-vehicle vs. CTR-citalopram: F=1.22, p<0.30) or reboxetine (CTR-vehicle vs. CTR-reboxetine: F=3.11, p<0.11) (fi g. 6). No overall effect of antidepressants on neurogenesis was thus detected in male rats under basal conditions (CTR-vehicle vs. CTR-antidepres-sants: F=0.92, p<0.35). It is important to note however that while repeated stress mark-edly reduced the number of hippocampal BrdU-labeled cells (F=3.45, p<0.093), long-term antidepressant administration attenuated this stress-induced effect. This was par-ticular evident after citalopram treatment (STR-vehicle vs. STR-citalopram: F=11.65, p<0.0066) but it was also present following tianeptine (STR-vehicle vs. STR-tianeptine:
Figure 5.
Graphs illustrating effects of gender (top) and stress (bottom) on numbers of BrdU-positive cells. “*” represents signifi -cant differences between conditions. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001.
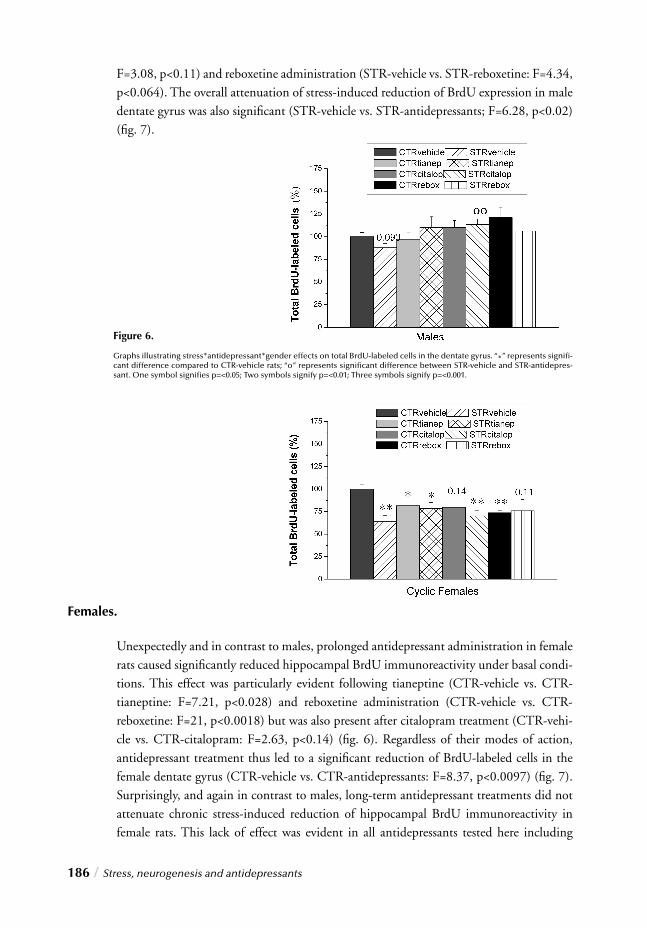
186 / Stress, neurogenesis and antidepressants
Figure 6.
Graphs illustrating stress*antidepressant*gender effects on total BrdU-labeled cells in the dentate gyrus. “*” represents signifi -cant difference compared to CTR-vehicle rats; “o” represents signifi cant difference between STR-vehicle and STR-antidepres-sant. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001.
F=3.08, p<0.11) and reboxetine administration (STR-vehicle vs. STR-reboxetine: F=4.34, p<0.064). The overall attenuation of stress-induced reduction of BrdU expression in male dentate gyrus was also signifi cant (STR-vehicle vs. STR-antidepressants; F=6.28, p<0.02) (fi g. 7).
Females.
Unexpectedly and in contrast to males, prolonged antidepressant administration in female rats caused signifi cantly reduced hippocampal BrdU immunoreactivity under basal condi-tions. This effect was particularly evident following tianeptine (CTR-vehicle vs. CTR-tianeptine: F=7.21, p<0.028) and reboxetine administration (CTR-vehicle vs. CTR-reboxetine: F=21, p<0.0018) but was also present after citalopram treatment (CTR-vehi-cle vs. CTR-citalopram: F=2.63, p<0.14) (fi g. 6). Regardless of their modes of action, antidepressant treatment thus led to a signifi cant reduction of BrdU-labeled cells in the female dentate gyrus (CTR-vehicle vs. CTR-antidepressants: F=8.37, p<0.0097) (fi g. 7). Surprisingly, and again in contrast to males, long-term antidepressant treatments did not attenuate chronic stress-induced reduction of hippocampal BrdU immunoreactivity in female rats. This lack of effect was evident in all antidepressants tested here including
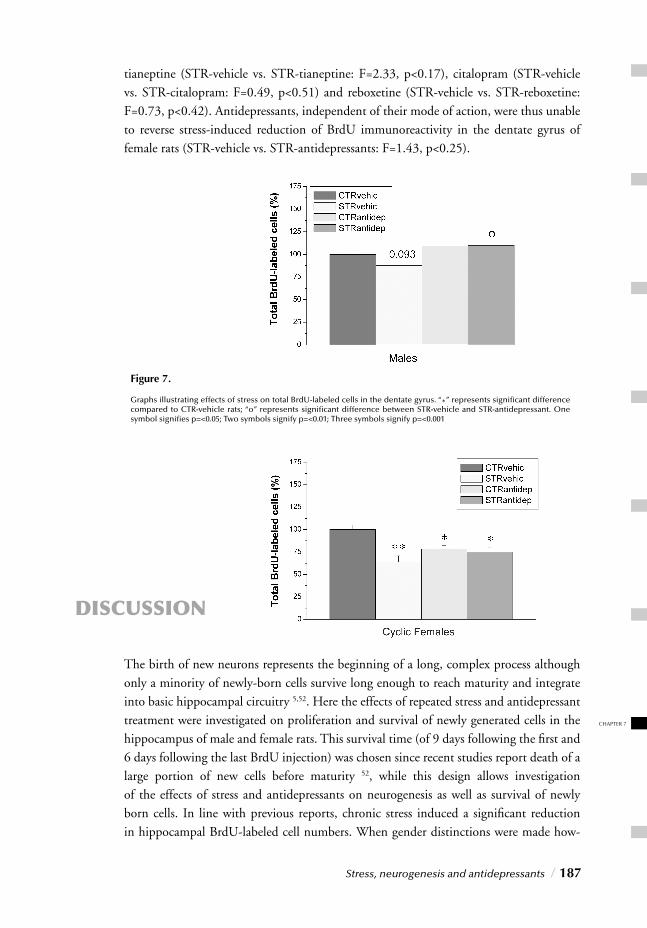
CHAPTER 7
Stress, neurogenesis and antidepressants / 187
tianeptine (STR-vehicle vs. STR-tianeptine: F=2.33, p<0.17), citalopram (STR-vehicle vs. STR-citalopram: F=0.49, p<0.51) and reboxetine (STR-vehicle vs. STR-reboxetine: F=0.73, p<0.42). Antidepressants, independent of their mode of action, were thus unable to reverse stress-induced reduction of BrdU immunoreactivity in the dentate gyrus of female rats (STR-vehicle vs. STR-antidepressants: F=1.43, p<0.25).
DISCUSSION
The birth of new neurons represents the beginning of a long, complex process although only a minority of newly-born cells survive long enough to reach maturity and integrate into basic hippocampal circuitry 5,52. Here the effects of repeated stress and antidepressant treatment were investigated on proliferation and survival of newly generated cells in the hippocampus of male and female rats. This survival time (of 9 days following the fi rst and 6 days following the last BrdU injection) was chosen since recent studies report death of a large portion of new cells before maturity 52, while this design allows investigation of the effects of stress and antidepressants on neurogenesis as well as survival of newly born cells. In line with previous reports, chronic stress induced a signifi cant reduction in hippocampal BrdU-labeled cell numbers. When gender distinctions were made how-
Figure 7.
Graphs illustrating effects of stress on total BrdU-labeled cells in the dentate gyrus. “*” represents signifi cant difference compared to CTR-vehicle rats; “o” represents signifi cant difference between STR-vehicle and STR-antidepressant. One symbol signifi es p=<0.05; Two symbols signify p=<0.01; Three symbols signify p=<0.001

188 / Stress, neurogenesis and antidepressants
ever, a strong sex-related stress effect emerged. While males illustrated a marked, yet non-signifi cant decrease in newly born cells, females illustrated signifi cantly less hippocampal BrdU-labeled cells after repeated stress. Remarkably however, long-term antidepressant treatment, regardless of their action mechanisms, attenuated footshock-mediated sup-pression of neurogenesis/survival in males, yet surprisingly, yielded signifi cantly reduced neurogenesis/survival in females, both with and without concurrent stress exposure. The dynamic process of adult neurogenesis is both positively and negatively regulated by neuronal activity and environmental factors. Stress exerts a particularly critical role 10 since hormones and other physiological agents mediating stress effects may have pro-tective, adaptive results in the short run, but can accelerate pathophysiology when over-stimulated or disturbed 15. In this study, repeated footshocks caused signifi cant adrenal enlargement in both genders, while previous studies have shown enhanced basal corticos-terone levels following this regimen in male and female rats 27,56. Given this testimony to the ability of this procedure to induce prolonged HPA axis hyperactivity, it is interes-ting to note that glucocorticoids are reported to exert a suppressive effect on dentate gyrus neurogenesis and that basal adrenal steroid levels are inversely related to the rate of hip-pocampal cell proliferation 57,58. Although the regulatory role of stress steroids on granule cell proliferation has been well established, the absence of mineralocorticoid and gluco-corticoid receptors in most granule cell precursors 53 suggests that glucocorticoid-mediated effects might occur through indirect infl uences and involve other factors. An interesting candidate for this stress-mediated inhibition of hippocampal neurogenesis is glutamate. Since activation of NMDA receptors has been associated with suppressed hippocampal cell proliferation 59 several studies suggest that new dentate gyrus cell production is directly regulated by excitatory input through these receptors 60. Together, the inhibitory effects of glucocorticoids and NMDA receptor activity on granule cell genesis suggest that chronic stress, by persistently elevating levels of basal corticosterone and hippocampal glutamate release 10,61, may represent a likely candidate to explain the reduced number of BrdU-labeled cells in chronically stressed rats. Although the molecular mechanisms underlying stress-induced suppression of hip-pocampal cell proliferation are still a matter of debate, recent reports have also implicated serotonin in the regulation of adult hippocampal neurogenesis 7. Serotonin, one of the most widely distributed neurotransmitters in the central nervous system, is involved in multiple processes, such as modulation of stress and affective responses. This has led to the suggestion that stress may predispose development of depression by disrupting serotonin-mediated neurogenesis modulation. Interestingly however the more recent “cellular neu-roplasticity hypothesis” does not exclude the older “monoamine theory of depression”. The former in fact appears to support the latter. Whereas depression might be due to dysregulated molecular mechanisms that govern neuronal cell proliferation and survival, antidepressants might mediate their effects by attenuating stress-induced impairment of monoaminergic function. Long-term administration and stimulation of monoaminergic-mediated neurotransmission, could ultimately lead to potentiation of neurogenesis and

CHAPTER 7
Stress, neurogenesis and antidepressants / 189
activation of intracellular pathways underlying neuronal plasticity and survival 62. In line with suppressive chronic stress effects on neurogenesis, our results illustrate the ability of repeated footshocks to reduce BrdU-labeled cell numbers in the male dentate gyrus. More importantly however all antidepressants, despite their classifi cation, yielded attenuation of this inhibitory effect in males. Although citalopram, a selective serotonin reuptake inhibitor 63,64 was particularly effective, serotonin is unlikely to be the only factor involved in modulating neurogenesis, since antidepressants characterized by other pharmacologi-cal profi les (tianeptine and reboxetine) also prevented/reversed stress-mediated inhibition of neurogenesis/survival. Interestingly however, in males, all antidepressants signifi cantly reduced plasma tryptophan concentrations while attenuating stress-induced reduction of hippocampal BrdU-labeled cells. Tryptophan, an essential amino acid, is an indispensable precursor for serotonin syn-thesis 65. Given our incapacity to synthesize this substrate, we rely on diet for its provi-sion, although most of the tryptophan consumed through food (about 99%) is used for protein synthesis and only a “negligible” amount reaches the brain where it is converted to 5-hydroxy-tryptophan, and subsequently serotonin 65. Central serotonin synthesis and function is thus strictly dependent on circulating tryptophan levels 66-68. Although altered central serotonin synthesis is unlikely to affect peripheral tryptophan availability, persis-tently reduced circulating tryptophan concentrations can affect central serotonin synthe-sis. Since the tryptophan transporter across the blood brain barrier is not saturated by circulating tryptophan concentrations under physiological conditions 69, elevated peri-pheral tryptophan levels may lead to enhanced penetration of this amino acid into the CNS. Alternatively, persistently reduced circulating tryptophan may lower rates of sero-tonin synthesis in the brain. Remarkably, however, despite induction of persistently low-ered plasma tryptophan in males, all antidepressants markedly enhanced hippocampus neurogenesis/survival, thereby counteracting the suppressive infl uence of repeated stress. Considering their different pharmacological profi les, these fi ndings suggest that antide-pressants’ effects on adult cell proliferation/survival may constitute a more complex me-chanism than mere potentiation of serotonergic neurotransmission. Chronic stress also inhibited cell proliferation/survival in females although the footshock-mediated effects on neurogenesis appeared to be more severe in females than males. In contrast to males however, long-term antidepressants (particularly citalopram and reboxetine), did not attenuate stress-induced reduction of BrdU-labeled cells in females, despite signifi cantly elevated circulating tryptophan levels. Surprisingly, anti-depressants alone without concurrent stress exposure even inhibited cell proliferation/survival. Although the mechanisms underlying this gender dichotomy remain obscure, it is intriguing to speculate that these differences in response to prolonged antidepressant treatment may be related to differential gender-related mechanisms governing production and survival of hippocampal cells 2. Therefore, by affecting gender-specifi c pathways or similar pathways in a gender-specifi c manner, antidepressants may be able to counteract deleterious stress effects on neurogenesis/survival in male but not female rats.

190 / Stress, neurogenesis and antidepressants
Recent investigations confi rm sex-related differences in rates of adult neurogenesis with females producing more new dentate gyrus cells than males 8. Accordingly, signifi -cantly more BrdU-labeled cells were seen in non-stressed females than non-stressed males. This gender difference has been attributed to modulatory effects of ovarian hormones and female rats do indeed exhibit naturally occurring fl uctuations in new cell numbers across the estrous cycle with maximal proliferation during proestrous, the stage with highest estrogen levels 8. This higher rate of cell proliferation during proestrous may thus account for the sex-related differences in new dentate gyrus cell production/survival. Interestingly however, while short-term estrogen treatment (up to 4 hours) has been shown to enhance neurogenesis, persistently elevated estradiol levels (up to 48 hours) have been associated with suppressed cell proliferation in female meadow voles and rats 70,71. This suggests that it might not be exposure to elevated ovarian steroids but periodic fl uctuations in circula-ting sex steroid levels, that underlie estrogen-mediated stimulation of hippocampal cell proliferation. Infl uence of prolonged stress on ovarian axis activity, may thus provide an interesting possibility to explain the stronger impact of stress in cyclic females compared to males. Shors and colleagues, have shown that stress persistently elevates estrogen levels in cyclic females and reduces the amplitude of cyclic fl uctuations during estrous 72 and as previously mentioned, recent studies also illustrate ovarian steroids’ ability to affect neu-rogenesis in a dual manner 70,71. The signifi cant reduction of BrdU-labeled cells following stress in these cyclic females might thus be related to a possible stress-induced elevation of ovarian hormones. Our results also illustrate the inability of antidepressants in cyclic females, to attenuate stress-induced reduction of hippocampal BrdU-labeled cell num-bers, due perhaps to their incapacity to rapidly normalize stress-induced changes in estro-gen release. With regard to the inhibition of cell proliferation/survival observed following long-term antidepressant treatments in the absence of concurrent stress exposure, we can only speculate that this effect may be related to transitory changes in estrogen release in response to the antidepressants themselves. In summary these results support a role for stress in psychopathology development. Stress-induced inhibition of neurogenesis/survival may impair hippocampal neuroplasti-city, compromise hippocampal ability to cope with novelty and complexity, and ultimately lead to inadequate information processing at the interface between systems involved in learning and affect regulation 9. Insuffi cient reactions to incoming stimuli could over-whelm the system, result in hippocampal “shut down” and due to interconnections between hippocampus and other cortical-limbic structures, could also affect functioning of a larger network and contribute to the array of symptoms associated with depression such as cognitive defi cits and abnormal affective regulation. In this “cellular neuroplasti-city hypothesis of depression”, impaired neurogenesis and depressive symptoms might be mutually reinforcing, leading to the vicious circle that often characterizes this disorder 18. This model however fails to address the temporal relationship between impaired neuro-genesis and psychopathology development, since altered neurogenesis/survival could re-present either a cause, consequence or correlate of depression 73. One limitation of this

CHAPTER 7
Stress, neurogenesis and antidepressants / 191
design concerns the inability to simultaneously investigate effects on neurogenesis and survival. Since only cells that reach maturity are incorporated into hippocampal circuitry, determining BrdU-labeled cell numbers between 6 and 9 days after BrdU injections might still provide valuable information on the infl uence of stress and antidepressants on hippo-campal integrity. Another limitation regards the risk of losing BrdU-labeled cells to dilu-tion during this 6-9 day period. BrdU-labeled cells that continue to divide dilute their incorporated BrdU until levels in daughter cells are too low for immunohistochemical detection and “disappear”. Continued generation of new cells can either add new BrdU-labeled cells, as one labeled precursor becomes two labeled cells, or paradoxically, decrease the number of BrdU-labeled cells, when one labeled precursor becomes two unlabeled daughter cells. This possibility was explored however and it has been demonstrated that any decrease in BrdU-labeled cells more than 4 days after BrdU injections is very unlikely to refl ect dilution of BrdU and can thus be attributed to cell death 52.
Conclusions
In conclusion we report a suppressive effect of repeated stress on hippocampal neurogenesis/survival, particularly evident in females and refl ecting possible gender-differ-ential mechanisms underlying these complex processes. The data also support gender as a major variable in the ability of long-term antidepressant treatments to attenuate stress-induced impairments of adult cell proliferation. While tianeptine, reboxetine and (parti-cularly) citalopram counteracted chronic stress-induced reduction in hippocampal BrdU-labeled cells in male rats, these same antidepressants were unable to exert a similar effect in cyclic females. Ovarian hormones and serotonin may play a critical role in these dif-ferences. Persistently elevated estrogen levels and disrupted serotonin neurotransmission may inhibit both the production of hippocampal cells as well as their survival. Together these fi ndings may offer new insights into the mechanisms involved in exertion of delete-rious effects of chronic stress on the brain, the molecular events underlying antidepres-sants’ therapeutic action and the role of gender in response to stress, psychopathology and pharmacotherapy.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the assistance of F. Postema, K. Venema, M. Jongsma,
J. Korf (Dept of Psychiatry) and I.P. Kema (Dept. of Pathology and Laboratory Medicine).

192 / Stress, neurogenesis and antidepressants
REFERENCES1. Altman, J. & Das, G.D. Autoradiographic and histological evidence of postnatal hip- pocampal neurogenesis in rats. J. Comp Neurol. 124, 319-335 (1965).
2. Gould, E. & Gross, C.G. Neurogenesis in adult mammals: some progress and problems. J. Neurosci. 22, 619-623 (2002).
3. van Praag, H. et al. Functional neurogenesis in the adult hippocampus. Nature 415, 1030-1034 (2002).
4. Gould, E., Tanapat, P., Rydel, T. & Hastings, N. Regulation of hippocampal neurogenesis in adulthood. Biol. Psychiatry 48, 715-720 (2000).
5. Jacobs, B.L. Adult brain neurogenesis and depression. Brain Behav. Immun. 16, 602-609 (2002).
6. Malberg, J.E. & Duman, R.S. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fl uoxetine treatment. Neuropsychopharmacology 28, 1562-1571 (2003).
7. Gould, E. Serotonin and hippocampal neurogenesis. Neuropsychopharmacology 21, 46S-51S (1999).
8. Tanapat, P., Hastings, N.B., Reeves, A.J. & Gould, E. Estrogen stimulates a transient increase in the number of new neu- rons in the dentate gyrus of the adult female rat. J. Neurosci. 19, 5792-5801 (1999).
9. Kempermann, G. Why new neurons? Possible functions for adult hippocampal neuro- genesis. J. Neurosci. 22, 635-638 (2002).
10. Gould, E. & Tanapat, P. Stress and hippocampal neurogenesis. Biol. Psychiatry 46, 1472-1479 (1999).
11. Sheline, Y.I., Wang, P.W., Gado, M.H., Csernansky, J.G. & Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. U. S. A 93, 3908-3913 (1996).
12. Sapolsky, R.M. Glucocorticoids and hippocampal atrophy in neuropsychiatric dis- orders. Arch. Gen. Psychiatry 57, 925-935 (2000).
13. Kessler, R.C., McGonagle, K.A., Swartz, M., Blazer, D.G. & Nelson, C.B. Sex and depression in the National Comorbidity Survey. I: Lifetime prevalence, chronicity and recurrence. J. Affect. Disord. 29, 85-96 (1993).
14. Bremner, J.D. et al. Hippocampal volume reduction in major depression. Am. J. Psychiatry 157, 115-118 (2000).
15. Gold, P.W., Drevets, W.C. & Charney, D.S. New insights into the role of cortisol and the glucocorticoid recep- tor in severe depression. Biol. Psychiatry 52, 381-385 (2002).
16. Duman, R.S., Heninger, G.R. & Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597-606 (1997).
17. Manji, H.K., Drevets, W.C. & Charney, D.S. The cellular neurobiology of depression. Nat. Med. 7, 541-547 (2001).
18. Kempermann, G. & Kronenberg, G. Depressed new neurons--adult hippocampal neurogenesis and a
cellular plasticity hypothesis of major depression. Biol. Psychiatry 54, 499-503 (2003).
19. Vaidya, V.A. & Duman, R.S. Depresssion--emerging insights from neurobiology. Br. Med. Bull. 57, 61-79 (2001).
20. Garcia, R. Stress, synaptic plasticity, and psychopathology. Rev. Neurosci. 13, 195-208 (2002).
21. Magarinos, A.M., Verdugo, J.M. & McEwen, B.S. Chronic stress alters synaptic terminal structure in hippocampus. Proc. Natl. Acad. Sci. U. S. A 94, 14002-14008 (1997).
22. Rajkowska, G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry 48, 766-777 (2000).
23. Lee, A.L., Ogle, W.O. & Sapolsky, R.M. Stress and depression: possible links to neuron death in the hip- pocampus. Bipolar. Disord. 4, 117-128 (2002).
24. McEwen, B.S. Mood disorders and allostatic load. Biol. Psychiatry 54, 200-207 (2003).
25. Nestler, E.J. et al. Neurobiology of depression. Neuron 34, 13-25 (2002).
26. Rasmusson, A.M., Shi, L. & Duman, R. Downregulation of BDNF mRNA in the hippocampal dentate gyrus after re-exposure to cues previously associated with footshock. Neuropsychopharmacology 27, 133-142 (2002).
27. Kuipers, S.D., Trentani, A., Den Boer, J.A. & Ter Horst, G.J. Molecular correlates of impaired prefrontal plasticity in response to chronic stress. J. Neurochem. 85, 1312-1323 (2003).
28. Malberg, J.E., Eisch, A.J., Nestler, E.J. & Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104-9110 (2000).
29. Duman, R.S., Nakagawa, S. & Malberg, J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 25, 836-844 (2001).
30. Ustun, T.B. Cross-national epidemiology of depression and gender. J. Gend. Specif. Med. 3, 54-58 (2000).
31. Hyttel, J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int. Clin. Psychopharmacol. 9 Suppl 1, 19-26 (1994).
32. Goodnick, P.J. & Goldstein, B.J. Selective serotonin reuptake inhibitors in affective disorders--I. Basic pharmacology. J. Psychopharmacol. 12, S5-20 (1998).
33. Wikell, C. et al. Pharmacokinetic and pharmacodynamic responses to chronic administration of the selective serotonin reuptake inhibitor citalo- pram in rats. Clin. Neuropharmacol. 22, 327-336 (1999).
34. Kugelberg, F.C., Apelqvist, G., Carlsson, B., Ahlner, J. & Bengtsson, F. In vivo steady-state pharmacokinetic outcome following clinical and toxic doses of racemic citalopram to rats. Br. J. Pharmacol. 132, 1683-1690 (2001).
35. Melzacka, M., Rurak, A., Adamus, A. & Daniel, W. Distribution of citalopram in the blood serum and in the central nervous system of rats after single and multiple dosage. Pol. J. Pharmacol. Pharm. 36, 675-682 (1984).
36. Mocaer, E., Rettori, M.C. & Kamoun, A. Pharmacological antidepressive effects and tianeptine-induced 5-HT uptake increase. Clin. Neuropharmacol. 11 Suppl 2, S32-S42 (1988).

Stress, neurogenesis and antidepressants / 193
37. Wilde, M.I. & Benfi eld, P. Tianeptine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic effi cacy in depression and coexisting anxiety and depression. Drugs 49, 411-439 (1995).
38. Kole, M.H., Swan, L. & Fuchs, E. The antidepressant tianeptine persistently modulates glutamate receptor currents of the hippocampal CA3 commissural associa- tional synapse in chronically stressed rats. Eur. J. Neurosci. 16, 807-816 (2002).
39. Whitton, P.S., Sarna, G.S., O’Connell, M.T. & Curzon, G. The effect of the novel antidepressant tianeptine on the concentra- tion of 5-hydroxytryptamine in rat hippocampal dialysates in vivo. Neuropharmacology 30, 1-4 (1991).
40. Delbende, C. et al. Effect of chronic treatment with the antidepressant tianeptine on the hypothalamo-pituitary-adrenal axis. Eur J Pharmacol 251, 245-251 (1994).
41. Frankfurt, M., McKittrick, C.R., McEwen, B.S. & Luine, V.N. Tianeptine treatment induces regionally specifi c changes in mono amines. Brain Res 696, 1-6 (1995).
42. McEwen, B.S. et al. Prevention of stress-induced morphological and cognitive conse- quences. Eur. Neuropsychopharmacol. 7 Suppl 3, S323-S328 (1997).
43. Conrad, C.D., LeDoux, J.E., Magarinos, A.M. & McEwen, B.S. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav. Neurosci. 113, 902-913 (1999).
44. Connor, T.J., Kelliher, P., Harkin, A., Kelly, J.P. & Leonard, B.E. Reboxetine attenuates forced swim test-induced behavioural and neurochemical alterations in the rat. Eur J Pharmacol 379, 125-133 (1999).
45. Sacchetti, G. et al. Studies on the acute and chronic effects of reboxetine on extracellular noradrenaline and other monoamines in the rat brain. Br. J Pharmacol 128, 1332-1338 (1999).
46. Invernizzi, R.W. et al. Chronic treatment with reboxetine by osmotic pumps facilitates its effect on extracellular noradrenaline and may desensitize alpha(2)-adrenoceptors in the prefrontal cortex. Br. J Pharmacol 132, 183-188 (2001).
47. Mori, S., Popoli, M., Brunello, N., Racagni, G. & Perez, J. Effect of reboxetine treatment on brain cAMP- and calcium/ calmodulin-dependent protein kinases. Neuropharmacology 40, 448-456 (2001).
48. Fichtner, C.G. & Braun, B.G. Hyperphagia and weight loss during fl uoxetine treatment. Ann. Pharmacother. 28, 1350-1352 (1994).
49. Fava, M. Weight gain and antidepressants. J. Clin. Psychiatry 61 Suppl 11, 37-41 (2000).
50. Trentani, A., Kuipers, S.D., Ter Horst, G.J. & Den Boer, J.A. Selective chronic stress-induced in vivo ERK1/2 hyperphosphory- lation in medial prefrontocortical dendrites: implications for stress- related cortical pathology? Eur. J. Neurosci. 15, 1681-1691 (2002).
51. Kema, I.P. et al. Profi ling of tryptophan-related plasma indoles in patients with carci- noid tumors by automated, on-line, solid-phase extraction and HPLC with fl uorescence detection. Clin. Chem. 47, 1811-1820 (2001).
52. Dayer, A.G., Ford, A.A., Cleaver, K.M., Yassaee, M. & Cameron, H.A. Short-term and long-term survival of new neurons in the rat dentate gyrus. J. Comp Neurol. 460, 563-572 (2003).
53. Cameron, H.A., Woolley, C.S., McEwen, B.S. & Gould, E. Differentiation of newly born neurons and glia in the dentate gyrus
of the adult rat. Neuroscience 56, 337-344 (1993).
54. Kuhn, H.G., Dickinson-Anson, H. & Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027-2033 (1996).
55. Czeh, B. et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treat ment with tianeptine. Proc. Natl. Acad. Sci. U. S. A 98, 12796-12801 (2001).
56. Trentani, A. et al. Immunohistochemical changes induced by repeated footshock stress: revelations of gender-based differences. Neurobiol. Dis. 14, 602-618 (2003).
57. Schlessinger, A.R., Cowan, W.M. & Gottlieb, D.I. An autoradiographic study of the time of origin and the pattern of granule cell migration in the dentate gyrus of the rat. J. Comp Neurol. 159, 149-175 (1975).
58. Sapolsky, R.M. & Meaney, M.J. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res. 396, 64-76 (1986).
59. Cameron, H.A., McEwen, B.S. & Gould, E. Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J. Neurosci. 15, 4687-4692 (1995).
60. Gould, E., Cameron, H.A. & McEwen, B.S. Blockade of NMDA receptors increases cell death and birth in the developing rat dentate gyrus. J. Comp Neurol. 340, 551-565 (1994).
61. Moghaddam, B., Bolinao, M.L., Stein-Behrens, B. & Sapolsky, R. Glucocorticoids mediate the stress-induced extracellular accumula- tion of glutamate. Brain Res. 655, 251-254 (1994).
62. D’Sa, C. & Duman, R.S. Antidepressants and neuroplasticity. Bipolar. Disord. 4, 183-194 (2002).
63. Hyttel, J. Citalopram--pharmacological profi le of a specifi c serotonin uptake inhibitor with antidepressant activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 6, 277-295 (1982).
64. Milne, R.J. & Goa, K.L. Citalopram. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs 41, 450-477 (1991).
65. Martinez, A., Knappskog, P.M. & Haavik, J. A structural approach into human tryptophan hydroxylase and its implications for the regulation of serotonin biosynthesis. Curr. Med. Chem. 8, 1077-1091 (2001).
66. Tagliamonte, A., Biggio, G., Vargiu, L. & Gessa, G.L. Free tryptophan in serum controls brain tryptophan level and sero- tonin synthesis. Life Sci. II 12, 277-287 (1973).
67. Gessa, G.L. & Tagliamonte, A. Possible role of free serum tryptophan in the control of brain trypto- phan level and serotonin synthesis. Adv. Biochem. Psychopharmacol. 11, 119-131 (1974).
68. Curzon, G. Infl uence of plasma tryptophan on brain 5HT synthesis and seroto- nergic activity. Adv. Exp. Med. Biol. 133, 207-219 (1981).
69. Pardridge, W.M. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem. Res. 23, 635-644 (1998).
70. Ormerod, B.K. & Galea, L.A. Reproductive status infl uences cell proliferation and cell survival in the dentate gyrus of adult female meadow voles: a possible regula tory role for estradiol. Neuroscience 102, 369-379 (2001).
CHAPTER 7

194 / Stress, neurogenesis and antidepressants
71. Ormerod, B.K., Lee, T.T. & Galea, L.A. Estradiol initially enhances but subsequently suppresses (via adrenal steroids) granule cell proliferation in the dentate gyrus of adult female rats. J. Neurobiol. 55, 247-260 (2003).
72. Shors, T.J., Pickett, J., Wood, G. & Paczynski, M. Acute stress persistently enhances estrogen levels in the female rat. Stress. 3, 163-171 (1999).
73. Gage, F.H. Structural plasticity: cause, result, or correlate of depression. Biol. Psychiatry 48, 713-714 (2000).

Stress, neurogenesis and antidepressants / 195
CHAPTER 7


CONCLUSION


8DISCUSSION AND CONCLUDING REMARKS


CHAPTER 8
Discussion / 201
DISCUSSION
The chapters constituting this thesis, present a wide array of data (fi gure 1), which
although broad-natured, illustrate one obviously consistent fi nding. Irrespective of the
more specifi c cellular, molecular and physiological effects, the subsections of this thesis
present invariably recurring gender-distinctive observations upon multiple levels. Be it
signal transduction, gene transcription, synaptic plasticity, neurotransmission, neuroge-
nesis or physiological effects on adrenal steroids or body weight regulation, each of the
chapters provide interesting fi ndings which in themselves warrant further investigation,
particularly with regard to their implication for pharmacotherapeutic developments since
also here (section IV) similar observations were evident. Unfortunately however, although
undoubtedly interesting and challenging, it goes beyond the scope of this thesis to elabo-
rate upon each of these studies and perform additional in-depth experimentation. In this
dissertation, preference was instead given to a broad large-scale analysis rather than a
detailed examination of individual aspects. Given this, an attempt was made to create a
higher overview of the results and to integrate the data into the overall picture. In order
to do so fi gure 1, a schematic representation of the data, serves to summarize the results
presented in this thesis. Although the following discussion and concluding remarks may
at times provide rather bold interpretations of these fi ndings, this is done in an attempt
to place them within the greater framework of our current knowledge and postulated
hypotheses, regarding the pathophysiology of stress-exposure, neuronal dysfunction and
the often-associated disorder, depression.
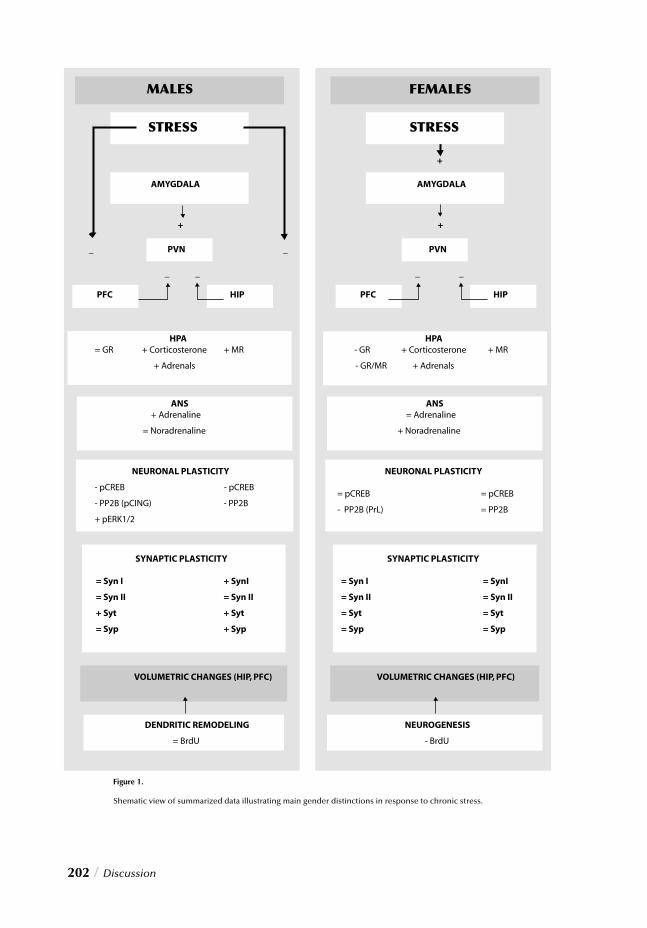
202 / Discussion

CHAPTER 8
Discussion / 203
Stress and neuronal dysfunction
A unifying hypothesis.
As stated in introductory paragraphs, exposure to adverse conditions initiates a series
of adaptive responses organized to defend the stability of the internal environment
and enhance an organism’s survival. This orchestrated process, referred to as the “stress
response”, involves various mechanisms, which allow the body to make the necessary
physiological and metabolic adjustments required to cope with the demands of a homeo-
static challenge. Allostasis, the process of adaptation to daily life events, is promoted by
stress through maintained stability or homeostasis 1,2. When mediators of allostasis, such as glucocorticoids, catecholamines, neurotransmitters (such as serotonin and glutamate) and neuropeptides, are released in response to stress, they promote adaptation and their effects are generally benefi cial. Nevertheless, conditions characterized by failed shutoff when no longer needed, or overuse in response to excessive challenge may lead to cumula-tive changes that could result in wear and tear, or “allostatic load”, on the body and brain 2,3. Recent decades have brought many attempts to identify the neurobiological substrates and molecular mechanisms that mediate stress-induced neuronal dysfunctions, yet our knowledge regarding the cascades of events that ultimately lead to the development of pathophysiology remains limited. This thesis provides an attempt to integrate data regard-ing cellular, molecular and physiological changes in response to repeated footshock stress in an effort to shed some light on the neurobiological mechanisms underlying chronic stress-mediated adaptations/maladaptations, while highlighting a role for gender in its determinism. As different hypotheses have already been proposed however, the following paragraphs will provide a brief review of their primary concepts and evaluate how the data presented here integrate with these hypotheses. For sake of clarity however, the allo-static load hypothesis described by McEwen 1 shall be maintained as the general model, common to and underlying these alternative potential theories, to which to relate their interactions and/or overlap.
The “glucocorticoid hypothesis” of neuronal dysfunctions.
One of the most-documented stress-induced biochemical abnormalities regards the gluco-corticoids, the primary physiologic mediators of allostasis. These adrenal hormones exert their cellular effects by acting on selective receptors present in various tissues and organs, but their actions, although initially adaptive, may become damaging if their release is not properly regulated. Persistent exposure to elevated glucocorticoid concentrations (allo-static state) has been previously reported in response to chronic stress and in accordance, elevated peripheral corticosterone levels and adrenal hypertrophy were also observed here following prolonged footshock stress in both male and female rats. These results, indica-tive of possibly abnormal HPA axis activity, thus establish the ability of this paradigm to

204 / Discussion
induce regulatory abnormalities in this vital stress axis. Chapter 2 illustrates that modulation of HPA axis responses to stress appeared to include the effects of glucocorticoids, catecholamines, and GR/MR interplay in distinct cortical and subcortical structures. Interestingly however, repeated footshock stress dis-rupted HPA axis regulation in a gender- and/or region-specifi c manner. These observed distinctions are a testimony to the complex modulation of this stress response system and suggest sexually dimorphic mechanisms to underlie its regulation/dysregulation. Collec-tively, impaired prefrontocortical and/or hippocampal control over the HPA axis seem to represent the mechanism by which repeated footshocks lead to abnormal stress response regulation in male rats, whereas imbalanced GR/MR expression, disturbed autonomic function, and abnormal amygdala activity could instead illustrate the events involved in the development of abnormal HPA axis responses in female rats. Impaired regulation of HPA axis activity may thus constitute the mechanism by which chronic stress leads to persistently elevated glucocorticoid levels. More importantly how-ever, the subsequent state of permanently elevated corticosterone concentrations (allo-static state) has been suggested to mediate the deleterious effects of stress on neuronal plas-ticity. Accordingly and with reference to the alternatively discussed theories of neuronal dysfunction, there are three types of plasticity that are affected by adrenal steroids:• Glucocorticoids reversibly and biphasically regulate the excitability of neuronal cells and infl uence the magnitude of long-term potentiation 4,5;• Glucocorticoids participate along with excitatory amino acids in the reversible stress-induced remodeling of hippocampal apical dendrites; an effect that has interestingly only been reported in male animals and not in the female counterpart 6,7;• Glucocorticoids participate along with excitatory amino acids in regulating neuroge-nesis of dentate gyrus granule neurons 8,9. Although effects of stress on LTP and neuronal excitability were not investigated, the data presented in this thesis provide direct evidence to substantiate the detrimental role of adverse conditions on neuronal plasticity. Specifi cally, chronic stress in male rats was asso-ciated with signifi cantly reduced phosphorylated CREB immunoreactivity in both corti-cal and subcortical regions, as illustrated in chapters 2-4. CREB phosphorylation, crucial for its ability to bind DNA and modulate gene expression, is essential for the transcription of BDNF. Reduced BDNF availability resulting from decreased phospho-CREB might thus in turn result in reduced neuronal plasticity, an indispensable feature when faced with prolonged stressful conditions. In fact, sustained stressful experiences have indeed been associated with reduced neurotrophin expression, a condition that might disturb the dynamic equilibrium between intracellular signaling cascades 10,11. In contrast to males, chronically stressed females failed to illustrate reduced CREB phosphorylation, raising an intriguing possibility that the differential impact of stress on neuronal plasticity may depend on gender-related mechanisms that either potentiate or suppress the deleterious infl uences of adverse events. In this case, ovarian hormones and their established neurotrophic effects, may protect the female brain against the injuri-

CHAPTER 8
ous insults of chronic stress by stimulating specifi c intracellular cascades underlying neu-ronal plasticity and preventing the reduced CREB phosphorylation observed in chroni-cally stressed males.
In addition to negative stress effects on CREB phosphorylation, a suppressive effect of
repeated stress on hippocampal neurogenesis/survival was also seen. This effect, although
present in both males and females, was particularly evident in the latter and may also be
accountable to ovarian hormones since stress has been reported to persistently increase
estrogen release in intact females 12. While stress-induced elevations of estrogen may pre-vent footshock-mediated phospho-CREB reduction, it also appears to contribute to the disruption of adult neurogenesis, possibility through inhibited production of new hip-pocampal cells or reduced survival. Ovarian steroids thus seem to simultaneously exert both positive and negative infl uences on critical neuronal processes. These fi ndings in turn refl ect the gender-related differential mechanisms that govern these complex processes. Whereas reduced phosphorylated CREB in males may substantiate deleterious stress effects on neuronal plasticity, decreased neurogenesis in females may substantiate its deleterious impact on neurogenesis. Both fi ndings illustrate the complex interrelation between impaired HPA axis regulation and the neuroplasticity and neurogenesis hypothe-ses. Another well-documented phenomenon observed in chronically stressed animals how-ever regards the alternative theory of reversible atrophy of apical dendrites (also known as dendritic remodeling) in selective prefrontocortical and hippocampal fi elds. Dendritic remodeling however also seems to be related to the deleterious infl uence of stress either directly or indirectly via neuronal plasticity 13,14. Although the precise mechanisms under-lying this morphological alteration remain fragmentary, it is important to note that this structural impairment has been reported to occur predominantly in male animals. Given the previous fi ndings, stress-induced suppression of CREB phosphorylation seen in males only may thus represent a marker of neuronal plasticity impairment underlying possibly ongoing dendritic remodeling. This supposition is further substantiated by the more direct immunohistochemical evidence of phospho-ERK1/2 accumulation in prefrontocortical dendrites and reduced hippocampal calcineurin immunoreactivity, as well as molecular fi ndings of abnormal synaptic vesicle-associated proteins expression in these regions in response to sustained stress. In line with previous reports of dendritic remodeling in males, most of these morphological alterations like phospho-CREB were only detected in the prefrontal cortex and hippocampus of chronically stressed males; this in contrast to females who failed to demonstrate any indications of such a process, despite the fact that they were exposed to identical adverse events. In summary, it is important to acknowledge the fact that glucocorticoids play a fun-damental role in stress-induced impairments, either directly or indirectly through impair-ment of neuronal plasticity, suppression of hippocampal neurogenesis/survival or den-dritic atrophy/remodeling. Despite numerous investigations that point to adrenal steroids as primary mediators in these processes however, it is important to recognize that the indi-rect role played by these hormones is not always alone but often in association with addi
Discussion / 205

206 / Discussion
tional players. In fact, sole exposure to persistently elevated glucocorticoids is not always enough to induce these effects since chronic stress-induced dendritic remodeling and sup-pression of hippocampal cell proliferation have also been shown to occur in the absence of prolonged HPA axis hyperactivity 15. During such processes their deleterious effects may thus also depend upon interactions with multiple other mediators of the stress response, including excitatory amino acids such as glutamate.
The “glutamate hypothesis” of neuronal dysfunctions.
Although extracellular glutamate levels are signifi cantly elevated in the hippocampus fol-lowing stress exposure 16, adrenalectomy can prevent this stress-induced rise 17, thereby suggesting a role for adrenal hormone participation in the modulation of glutamate release 15. The ability of some pharmacotherapeutic agents to interfere with systems as serotonin and glutamate are also in line with this notion and some of the mechanisms go-verning neuronal dysfunction also share additional common mediators besides glucocorticoids. Excessive glutamate release, for instance, appears to be a fundamental factor underlying inhibition of cell proliferation in the dentate gyrus as well as the remodeling of dendrites 9,18. More importantly, persistently elevated extracellular glutamate levels may also explain some of the neuronal abnormalities observed following prolonged stress exposure such as ERK1/2 hyperphosphorylation in prefrontocortical dendrites and reduced calcineurin immunoreactivity in addition to suppressed hippocampal neurogenesis. Whereas the interactions between HPA axis hyperactivity and alternative theories thus attest the fact that multiple hypotheses act concurrently to induce neuronal defects, glucocorticoids like-wise, do not act alone to promote the effects underlying these respective theories but simultaneously with other mediators such as glutamate. Throughout attempts to identify the intracellular changes that mediate stress-induced neuronal defects, glutamate emerged as a critical player in the modulation of deleterious stress effects on neuronal plasticity. This excitatory amino acid has been suggested to lead to neuronal dysfunctions through excitotoxicity and oxidative stress, both of which are associated with alterations in multiple signaling systems including the MAP kinase cas-cade and Ca2+/calmodulin-dependent protein kinase pathway. Excessive extracellular glu-tamate, for instance, inhibits cystine uptake, a precursor for glutathione synthesis. Cellular glutathione synthesis thus declines and reactive oxygen species (ROS) accumulate, gener-ating a condition of oxidative stress, which in turn activates the MAPK cascade, stimulates ERK1/2 and contributes to cellular dysfunctions as shown in neurons 19. Accordingly, persistent ERK1/2 phosphorylation has been reported in response to increased extracel-lular glutamate concentrations 20. Furthermore, while prolonged activation following oxi-dative stress has been associated with neuronal dysfunctions and cell death, inhibition of ERK1/2 phosphorylation by MAPK inhibitors has been shown to block glutamate-mediated neuronal defects 21,22. Besides prolonged ERK1/2 kinase activity however, a role for abnormal phosphatase activity has also been proposed to mediate neuronal dysfunction, as phosphatase inhibi-

CHAPTER 8
tion has been associated with persistent ERK phosphorylation, cellular cytoskeleton dis-ruption and ultimately, impaired neuronal functions 23. Interestingly, and in line with these reports, the results in this thesis demonstrate reduced hippocampal calcineurin immunoreactivity as well as prolonged phospho-ERK1/2 accumulation in prefrontocorti-cal dendrites following chronic stress. These observations yield the interesting possibility that chronic stress impairs the function of specifi c protein phosphatases but in a region-dependent manner. Although abnormal phosphatase regulation could lead to persistent activation of specifi c protein kinases such as ERK1 and ERK2, reduced calcineurin was found in the hippocampus whereas abnormal ERK1/2 phosphorylation occurred in pre-frontocortical dendrites. Nevertheless, these alterations may represent similar disturbances in the same cascade underlying this pathophysiological process. Whereas chronic stress may impair the regulation of critical intracellular pathways by inhibiting activity of specifi c region-dependent protein phosphatases leading to prolonged kinase activity and neuronal defects, alterations in this signal transduction apparatus may in turn play a fundamental role in the prefrontocortical and hippocampal dendritic atro-phy. Stress-induced phospho-ERK1/2 accumulation in males, was in fact, particularly evi-dent in the distal parts of prefrontocortical dendrites, which may represent a key event in the cascade leading to dendritic remodeling following repeated exposure to adverse condi-tions. Furthermore, perturbed ERK signaling has been related to cytoskeleton destabiliza-tion, attributable in part to the ability of these kinases to phosphorylate various synaptic proteins. In line with the gender-distinctive phospho-CREB, phospho-ERK1/2, and cal-cineurin fi ndings as well as reports of dendritic remodeling, this notion is further sup-ported by altered expression of various synaptic vesicle-associated proteins in chronically stressed male but not female rats. Like glucocorticoids, the interregulatory effects of glutamate also extend to the neu-rogenesis hypothesis 8,24,25. Repeated footshock exposure caused marked suppression of adult neurogenesis in both genders, yet particularly in stressed females. Although gluco-corticoids have been proposed as major players involved in the inhibitory action of stress on hippocampal cell proliferation, this effect is indirectly due to the fact that granule cell precursors lack glucocorticoid receptors. The predominant excitatory neurotransmit-ter glutamate, however, exerts a direct regulatory effect on neurogenesis. Elevation of extracellular glutamate concentrations as well as administration of glutamate analogs has been associated with the suppression of cell proliferation, whereas glutamate antagonists enhance hippocampal neurogenesis 24. These inhibitory infl uences of glutamate appear to depend on the stimulation of NMDA receptors. Chronic stress-mediated enhance-ment of glutamate release in the hippocampus may elevate extracellular levels of this neu-rotransmitter, leading to activation of ionotropic receptors and ultimately inhibition of neurogenesis in both male and female rats. Interestingly, the stronger suppressive effects of chronic stress on hippocampal cell proliferation in female rats may be related to interac-tions between persistently elevated glutamate and estrogen levels. To recapitulate, repeated footshock stress appears to promote specifi c gender- and
Discussion / 207

208 / Discussion
region-dependent molecular and cellular adaptations/maladaptations. Despite differences
in gender specifi c adaptations however, glucocorticoid-glutamate interactions may repre-
sent primary mediating factors for development of neuronal abnormalities in rats of both
gender since the results presented here illustrate 1) the role of glucocorticoids and the
HPA axis activity 2) the interaction of the latter with the neuronal dysfunction hypotheses
of dendritic remodeling, neuronal plasticity, and neurogenesis 3) the effects of glucocorti-
coids in relation to the mediator glutamate and 4) the direct and indirect regulatory inter-
actions between the latter and the hypotheses of dendritic remodeling, neuronal/synaptic
plasticity and neurogenesis.
Stress and psychopathology
Gender differences extend beyond sole stress sensitivity. In fact, the central role played
by stress in psychopathology has lead to marked gender dimorphisms in the prevalence
of a wide range of psychiatric disorders. Depression and anxiety, for instance, are more
than twice as common in women than in men, although the neurobiological mechanisms
underlying this gender-differential susceptibility remain obscure. As stated in chapter 1,
the latter may be partly due to the fact that the pathophysiological mechanisms involved
in the treatment of such disorders are also poorly understood. However, given the marked
gender differences observed in adaptations/maladaptations to chronic stress, an intrigu-
ing possibility holds that similar differences may also characterize the molecular and cel-
lular dysfunctions underlying the development of these disorders in men and women. If
this holds true and gender-specifi c mechanisms are involved in the dimorphic prevalence
of psychopathologies, then antidepressants used to ameliorate these conditions may also
have different effects in men and women. Several studies have in fact reported that women
respond better to SSRIs while men react better to tricyclic agents 26,27. This supports the
hypothesis of gender-differential response to antidepressant medications and indirectly
substantiates the possibility of gender-dimorphic pathophysiological mechanisms under-
lying psychopathology in men and women. However, since women still benefi t from tri-
cyclic agents while men show high response rates to SSRIs, this does not disprove the
original hypothesis and suggests that antidepressants may act at different levels in men and
women, to infl uence gender-specifi c targets and lead to the correction of those molecular
or cellular defects responsible for the occurrence and maintenance of psychiatric disorders.
In an attempt to verify the validity of the presented results against the above and current
hypotheses of depression and psychopathology, the fi nal section of this thesis discusses
data regarding the ability of different antidepressants to attenuate stress-related neuronal
changes.

CHAPTER 8
The monoamine hypothesis of depression.
Despite extensive investigation to defi ne the pathophysiological mechanisms underlying complex, heterogeneous affective disorders, the etiological factors underlying develop-ment of depression and anxiety are not yet fully understood. The monoamine hypothesis of depression, coined over 30 years ago, suggests defi cient central nervous system mono-amine neurotransmission as the underlying biological basis for depression, particularly serotonin and norepinephrine. Since stimulation of the monoaminergic system has indeed been associated with improvement or resolution of depressive symptoms, various classes of antidepressants were developed to increase monoamine levels within the synaptic cleft, by either inhibiting their degradation or blocking their reuptake. With respect to aforementioned efforts to evaluate theories of neuronal dysfunction, citalopram-, tianeptine- and reboxetine-induced adaptations were examined in male and female rats to establish their role in correcting neuronal impairments and determine potential implications of the monoamine hypothesis in psychopathological development. Regardless of intensive investigations however, no conclusive affi rmation of a primary dys-function in specifi c monoaminergic systems in subjects with major depressive disorders has yet emerged. Likewise the data presented here fail to confi rm a primary dysfunction in a specifi c monoaminergic system, although the data do illustrate a clear potential for monoaminergic modulation (via antidepressant administration) to exert strong infl uences on neuronal function. Interestingly the therapeutic effect seems to be largely attributable to indirect effects on neuronal plasticity as detailed below.
The neuroplasticity hypothesis of depression.
Since the monoamine hypothesis left various questions unaddressed (effi cacy, selectivity, mode of action and therapeutic delay of antidepressants), scientists searched for alterna-tive theories to explain the pathophysiological processes underlying depression and other related disorders. The data illustrated in this thesis support the different theories proposed for neuronal dysfunctions and are in line with recent theories linking stress and depression via neuroplasticity 28,29. Specifi cally the results illustrate how stress affects functional and structural neuronal integrity by inducing alterations at the molecular and cellular levels; molecular changes included modifi ed gene expression, protein synthesis and phosphory-lation and cellular changes included dendritic remodeling and/or atrophy, reduced neu-rogenesis, and perhaps neuronal death. These effects sketch a theoretical mechanism by which sustained stress may reduce neuronal plasticity and lead ultimately to selective cor-tical-limbic abnormalities. This in turn supports the notion of stress-induced depression through negative neuroplasticity effects, but how then does the monoamine hypo-thesis fi t into this framework? To answer this question it must fi rst be reiterated that these forms of neuronal plasticity are crucial for proper functioning of the brain as illustrated by the fact that
Discussion / 209

210 / Discussion
numerous psychiatric disorders are characterized by reduced hippocampal neurogenesis and neuronal atrophy. The results presented here illustrate inhibitory effects of chronic stress on these parameters. Reduced BrdU-immunoreactivity could indicate decreased neurogenesis/survival while altered phospho-CREB, phospho-ERK1/2 and calcineurin levels may suggest overall disturbed neuronal plasticity. The fi ndings support a potentially pathologic role of stress in neuronal impairment, but more importantly, illustrate gender specifi city with regards to explicit cellular plasticity impairments; signifi cantly reduced BrdU-immunoreactivity occurred in females whereas reduced CREB phosphorylation, phospho-ERK1/2 accumulation, and abnormal calcineurin levels developed in males. Interestingly however, pharmacotherapeutic agents based on monoamine modulation also counteracted some stress-induced effects in a sex specifi c manner as they reversed stress-induced reduction of BrdU-immunoreactivity in males but not females. Together these fi ndings support the initial hypothesis regarding differential gender-dependent mecha-nisms underlying the injurious infl uence of repeated stress on the brain. The fact that anti-depressant treatment resulted in specifi c gender-dimorphic adaptations also supports the subsequent hypothesis that these gender-differential neurobiological mechanisms might dictate distinct pharmacotherapeutic responses in males and females. The fact that mono-aminergic-modulating drugs exert gender specifi c actions on stress-induced adaptations/maladaptations however is a tribute to the potential implication of these monoaminergic neurotransmitters in the pathophysiological mechanisms of stress as well as their potential role in mediating therapeutic response. Despite conceptual differences, the monoamine and cellular neuroplasticity hypotheses of depression do not exclude one another. In fact, like the theories proposed for neuronal dysfunction, there seems to be some overlap. For instance, even though the exact mecha-nisms underlying stress-induced suppression of neuroplasticity are not yet fully known, recent reports have implicated serotonin in the regulation of neuronal plasticity as well as adult hippocampal cell proliferation 30. While stimulation of serotonergic activity posi-tively affects neurogenesis and neuronal plasticity, it is interesting that stress negatively affects these very same processes. Although most substantial evidence to support a role for monoamine systems is provided by antidepressant modes of action, circumstantial fi n-dings such as these also support a role for monoamine defi ciencies in depression. Despite their monoaminergic properties, antidepressants act through different me-chanisms to exert their therapeutic effect and thereby illustrate the interrelation of the hypotheses of depression. While some antidepressants act directly on the cause of neuro-nal dysfunction (i.e. glutamate and glucocorticoids), others act indirectly through mecha-nisms that stimulate compensatory adaptations. The former class may include tianeptine, previously classifi ed as a serotonin reuptake enhancer but recently emerging as a glutama-tergic agent. If this latter mode of action constitutes the primary mechanism for tianep-tine’s therapeutic effects, it raises an interesting possibility that tianeptine’s ability to pre-vent stress-induced reduction of hippocampal neurogenesis in males might hold direct refe-rence to the previously discussed glutamate-mediated deleterious effects on neuronal

CHAPTER 8
Discussion / 211
plasticity. Antidepressants such as citalopram or reboxetine, respectively selective sero-tonin and norepinephrine reuptake inhibitors, may instead exert their benefi cial effects via indirect mechanisms. Since reports of specifi c serotonergic and noradrenergic neurotrans-mission defi ciencies in depression are not unequivocally explicit, enhanced monoaminer-gic activity might not prevent or correct abnormalities underlying psychopathology deve-lopment but rather promote new adaptations that recompense impaired brain functions caused by those dysfunctions. Although stress acts in part through elevated glucocorticoid and extracellular glu-tamate levels to suppress neurogenesis and induce dendritic remodeling, monoamine reuptake inhibitors might not interfere directly with such glutamate- and corticosteroid-mediated effects to compensate stress-induced dysfunctions. They may instead poten-tiate monoamine functions to indirectly promote the birth of new hippocampal cells or stimulate dendritogenesis in cortical and subcortical regions. Such compensatory rather than preventive/corrective effects might also explain why depressed subjects successfully treated with these antidepressants may experience a relapse of symptoms upon termina-tion of medication. The fact that all antidepressants examined here attenuated stress-induced reduction of hippocampal cell proliferation in male rats only and not in females is interesting, but at the same time puzzling. The prospect remains that this effect might be mediated by stress-induced elevation of ovarian hormones. To the best of our current knowledge however, none of the antidepressants included in these studies have shown any established effect on the reproductive axis under both basal and stress conditions. By the same token although not measured, it might suggest that the antidepressants were also unable to prevent stress-induced elevations of estrogen release and thus ineffective in pre-venting inhibition of hippocampal neurogenesis.
CONCLUDING REMARKS
To conclude upon these results and the discussions of the previous paragraphs, it is evident that there is no clear single hypothesis governing stress-induced effects/defects and subse-quent psychopathology. Looking back, it appears considerable overlap and integrated con-nections exist between the glucocorticoid and glutamate hypotheses of neuronal dysfunc-tion as well as the monoaminergic and neuroplasticity theories of depression. Figure 2 illustrates how each system is in some way interlinked with the other, although the precise manner of interregulation may vary. In this thesis the deleterious effects often associated with stress and depression are present but dependent upon circumstances such as gender. Markers of disturbed neuronal plasticity for instance are evident in males, whereas indica-tions of HPA axis overactivity and compromised neurogenesis are predominant in females. Nevertheless, despite these complex interrelations, it seems there are gender-specifi c paths that are preferentially affected and regardless of the divergent stress effects on multi levels, albeit systemic, molecular or cellular, the fi nal result is in both cases deleterious.

212 / Discussion
Impaired regulation of HPA axis activity for instance, common to both genders may con-stitute the mechanism by which chronic stress leads to persistently elevated glucocorticoid levels and PVN activation, although the data shows gender-dependent means of attain-ment. Impaired prefrontocortical and/or hippocampal control over the HPA axis seem to represent the mechanism by which repeated footshocks lead to abnormal stress response regulation in male rats, whereas imbalanced GR/MR expression, disturbed autonomic function, and abnormal amygdala activity could instead illustrate the events involved in the development of abnormal HPA axis responses in female rats (fi gure 1,2). On a more cellular/molecular level, stress-induced effects of neuronal plasticity and synaptic integrity seem to represent a possible mechanism by which this effect is realized in males given the changes observed in their general gene expression, specifi c second mes-senger signalling cascade members and synaptic vesicle associated proteins. Considering the potential role of these proteins and functional neurotransmission in BDNF activity, the data may represent indirect indications for impaired neurotrophic plasticity in males. In view of the lesser effects observed on these parameters in females, it raises the interest-ing possibility that the differential impact of stress on neuronal plasticity may depend on gender-related mechanisms that either potentiate or suppress the deleterious infl uences of adverse events. Dependent upon the interpretation, in the case of the latter, ovarian hormones and their established neurotrophic effects, may protect the female brain against the insults of chronic stress by stimulating specifi c intracellular cascades underlying neu-ronal plasticity and preventing the reduced neuronal plasticity (CREB phosphorylation) observed in chronically stressed males. On the other hand, these neurotrophic effects might have a paradoxically damaging effect if these “blocked” neuronal/synaptic changes are required in order to attain an adaptive response. After all, neurogenesis/proliferation was predominantly affected in these females. Whatever the pathway however, both dys-regulations seen in the genders are known to contribute to fi nal volumetric changes often reported in stress and psychopathology. The fi nal question regarding the sensitivity of the sexes remains and unfortunately it is not as unambiguous as one might desire. In view of the data presented here and illustrated in fi gures 1 and 2, the answer to this seems to depend on the parameter of interest, since both sexes appear to be affected albeit through different mechanisms. In view of the future of stress research, it would be of interest from a clinical perspective to further pursue the neurobiological substrates and neurochemical profi les underlying effects of stress in both genders. As illustrated by the antidepressant data, each gender should be evaluated independently to determine the stress and/or treatment response and thus new avenues of therapeutic approach. The data presented here offer ample indications to argue for further investigations of the role of steroid hormones in multi-level research. With this in mind the paramount message is that fi ndings associated with one gender cannot be automati-cally extrapolated to the other; and that which may appear “negative” in one could repre-sent a physiological condition in the other and vice versa.
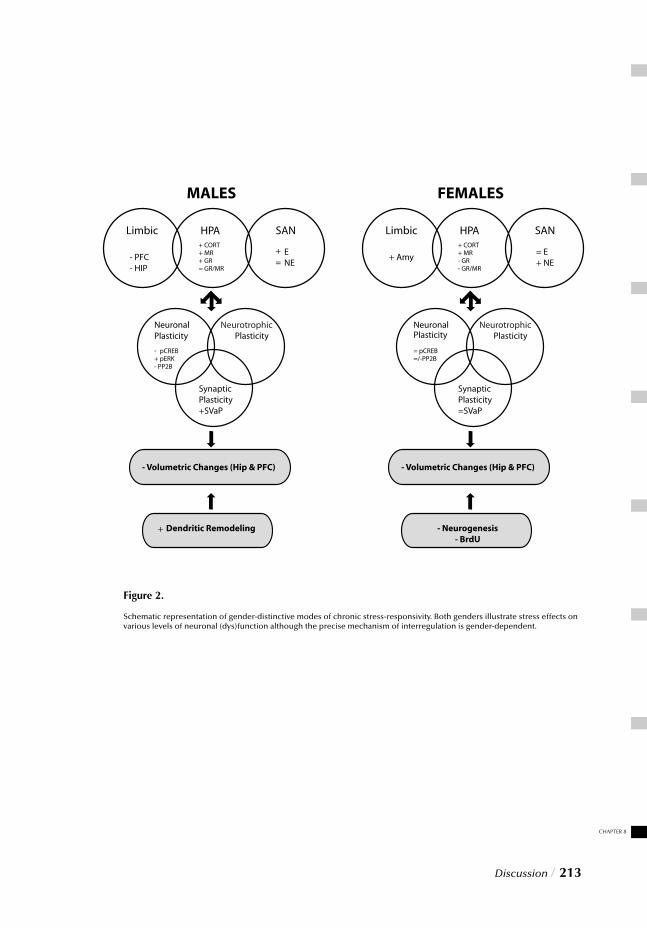
CHAPTER 8
Discussion / 213
Figure 2.
Schematic representation of gender-distinctive modes of chronic stress-responsivity. Both genders illustrate stress effects on various levels of neuronal (dys)function although the precise mechanism of interregulation is gender-dependent.

REFERENCES1. McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 840, 33-44 (1998).
2. McEwen, B.S. Mood disorders and allostatic load. Biol. Psychiatry 54, 200-207 2003).
3. Goldstein, D.S. & McEwen, B. Allostasis, homeostats, and the nature of stress. Stress. 5, 55-58 (2002).
4. Diamond, D.M., Bennett, M.C., Fleshner, M. & Rose, G.M. Inverted-U relationship between the level of peripheral corti- costerone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2, 421-430 (1992).
5. Pavlides, C. & McEwen, B.S. Effects of mineralocorticoid and glucocorticoid receptors on long-term potentiation in the CA3 hippocampal fi eld. Brain Res. 851, 204-214 (1999).
6. Magarinos, A.M. & McEwen, B.S. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 69, 89-98 (1995).
7. McEwen, B.S. & Magarinos, A.M. Stress and hippocampal plasticity: implications for the patho physiology of affective disorders. Hum. Psychopharmacol. 16, S7-S19 (2001).
8. Cameron, H.A., Tanapat, P. & Gould, E. Adrenal steroids and N-methyl-D-aspartate receptor activation regulate neurogenesis in the dentate gyrus of adult rats through a common pathway. Neuroscience 82, 349-354 (1998).
9. Gould, E. & Tanapat, P. Stress and hippocampal neurogenesis. Biol. Psychiatry 46, 1472-1479 (1999).
10. Smith, M.A., Makino, S., Kvetnansky, R. & Post, R.M. Stress and glucocorticoids affect the expression of brain- derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15, 1768-1777 (1995).
11. Duman, R.S. Novel therapeutic approaches beyond the serotonin receptor. Biol. Psychiatry 44, 324-335 (1998).
12. Shors, T.J., Pickett, J., Wood, G. & Paczynski, M. Acute stress persistently enhances estrogen levels in the female rat. Stress. 3, 163-171 (1999).
13. Conrad, C.D., LeDoux, J.E., Magarinos, A.M. & McEwen, B.S. Repeated restraint stress facilitates fear conditioning indepen dently of causing hippocampal CA3 dendritic atrophy. Behav. Neurosci. 113, 902-913 (1999).
14. Magarinos, A.M., Deslandes, A. & McEwen, B.S. Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur. J. Pharmacol. 371, 113-122 (1999).
15. McEwen, B.S. Allostasis, allostatic load, and the aging nervous system: role of excitatory amino acids and excitotoxicity. Neurochem. Res. 25, 1219-1231 (2000).
16. Moghaddam, B., Bolinao, M.L., Stein-Behrens, B. & Sapolsky, R. Glucocorticoids mediate the stress-induced extracellular accu- mulation of glutamate. Brain Res. 655, 251-254 (1994).
17. Lowy, M.T., Gault, L. & Yamamoto, B.K. Adrenalectomy attenuates stress-induced elevations in extracel lular glutamate concentrations in the hippocampus. J. Neuro-chem. 61, 1957-1960 (1993).
18. McEwen, B.S. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105-122 (1999).
19. Coyle, J.T. & Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689-695 (1993).
20. Jiang, Q., Gu, Z., Zhang, G. & Jing, G. Diphosphorylation and involvement of extracellular signal-re- gulated kinases (ERK1/2) in glutamate-induced apoptotic-like death in cultured rat cortical neurons. Brain Res. 857, 71-77 (2000).
21. Satoh, T. et al. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci. Lett. 288, 163-166 (2000).
22. Stanciu, M. et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200-12206 (2000).
23. Runden, E. et al. Regional selective neuronal degeneration after protein phos phatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J. Neurosci. 18, 7296-7305 (1998).
24. Cameron, H.A., McEwen, B.S. & Gould, E. Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J. Neurosci. 15, 4687-4692 (1995).
25. Gould, E., McEwen, B.S., Tanapat, P., Galea, L.A. & Fuchs, E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 17, 2492-2498 (1997).
26. Yonkers, K.A., Kando, J.C., Cole, J.O. & Blumenthal, S. Gender differences in pharmacokinetics and pharmacodyna- mics of psychotropic medication. Am. J. Psychiatry 149, 587-595 (1992).
27. Bies, R.R., Bigos, K.L. & Pollock, B.G. Gender differences in the pharmacokinetics and pharmacody namics of antidepressants. J. Gend. Specif. Med. 6, 12-20 (2003).
28. Duman, R.S., Malberg, J. & Thome, J. Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 46, 1181-1191 (1999).
29. Jacobs, B.L. Adult brain neurogenesis and depression. Brain Behav. Immun. 16, 602-609 (2002).
30. Gould, E. Serotonin and hippocampal neurogenesis. Neuropsychopharmacology 21, 46S-51S (1999).
214 / Discussion

CHAPTER 8
Discussion / 215


Summary / 217
SUMMARY
Feelings of restlessness, hopelessness, irritability and fatigue, diffi culties concentrating and sleep disturbances. These are but a few of the symptoms that characterize stress-related affective disorders as anxiety and depression. These psychiatric disorders can be very drain-ing on those who have the disease as well as their families. Victims of these disorders often experience diffi culties functioning in various aspects of their lives, such as work, school, family life, etc. As a result, these disorders have been described as the leading cause of disability worldwide. In this regard it is thus important to pursue improved medications, alternative treatments, causes and eventual cures for such disorders. Unfortunately how-ever, in recent decades little progress has been seen with respect to the development of novel therapeutic (antidepressant) agents. Interestingly, these psychiatric disorders have been characterized by a marked gender-related prevalence. In fact epidemiological studies across a number of cultures consistently show that beginning at puberty, depression and anxiety disorders are two to three times more common in women than in men. Although this is perhaps the reason why the female sex has often been termed “neurotic”, this is not necessarily fair. In the search for new targets, an interesting avenue of approach thus lies in elucidating the fundamental dif-ferences between male and female psychophysiology. Scientists have long known of the anatomical differences between the sexes, but only recently have they begun to uncover signifi cant biological and physiological differences between the sexes. Sex differences have been found everywhere ranging from bone matter composition and pain experience to drug metabolism and the rate of neurotransmitter synthesis in the brain. Naturally, gender differences may thus also be refl ected in the circuitry underlying pathology and thus potential therapies. In view of these observations, it is therefore remarkable that until recently women were often excluded from clinical trials of new psychotropic drugs, while most of the preclinical studies on the effi cacy of new compounds and their neurochemi-cal modes of action were conducted with male animals. The primary reason for this lack of equal representation lies in concerns associated with the menstrual cycle, which may complicate results of experimental trials. With regard to future development of new pharmacotherapy, this viewpoint is unfor-tunate however since insight into new therapeutic targets requires a better understanding of the pathology; and this holds true for both genders if we are to gain a full understand-ing of these disorders. Elucidating the pathophysiology however is realized in part through investigating current medications’ modes of action, while most of the currently prevail-ing theories of antidepressants’ modes of action are based solely on the results obtained with experiments with male rats. Research progression thus calls for a paradigm shift, which replaces male-oriented investigation with that of dual-gender comparative analyse

218 / Summary
Fortunately today, women’s health has gained widespread attention in popular magazines, television shows and the wellness industry. John Gray’s “Men are from Mars Women are from Venus” is a testimony to the media interest and acknowlegdement of the role of the sexes throughout society. Research in biological psychiatry and psychopharmacology is now starting to focus more on the analysis of sex infl uences. Discovering why these gender differences in risk exist has in fact become one of the most intriguing and important questions in psychiatric research today: intriguing because a deeper understanding of why women are more likely than men to experience anxiety and depression will provide insight into the pathophysiology of these syndromes and important because such knowledge will improve our ability to design interventions that treat and/or prevent these illnesses.
Oftentimes however psychiatric disorders like depression can develop or exacerbate as a result of stress in the form of stressful life events (loss of a job, spouse etc), particularly when severe or long lasting. As a matter of fact, preclinical studies reveal that chronic stress exposure in rats induces marked degeneration of brain structures responsible for regulat-ing mood and emotions; fi ndings that are also in line with neuroimaging results observed in depressed patients. Chronic stress, may thus contribute directly to the development of neuronal dysfunctions and psychopathology. Although considerable progress has been made in unraveling the neurobiological substrates of the acute stress response, which can facilitate consolidation of new memories and promote cognitive processes, it is thus ironic that chronic stress-induced neurochemical changes are still poorly understood. Also con-trary to expectation is the fact that most research on stress-related neuronal abnormali-ties (like psychotropic drugs) have been conducted in males since the genders differ con-siderabye in their sensitivity to stress and related disorders, making it questionable if the mechanisms of stress response regulation, neuronal function, dysfunction, pathology and pharmacotherapy apply equally to both sexes. Therefore the experiments described in this thesis were designed to elucidate the concept of stress and simulate chronic stress-induced impairments so that these questions may be addressed. In order to do so, male and female rats were given daily sessions of footshock stress for a period of 21 days after which stress and gender comparative interactions were investigated. There are different dimensions that are necessary to understand the impact of stress on an animal’s physiology, ranging from systemic endocrinological functions to its state of neuroplasticity or the ability of the brain to shape or mold itself by expansion or contrac-tion of neuronal processes due to injury or adverse circumstances. Notably, this ability of the brain to adapt is also refl ected at multiple levels; cellular and molecular, including the functional state of emotional brain structures, as well as proper regulation of signal trans-duction, gene transcription and protein expression. Fortunately the resemblance between many chronic stress induced impairments seen in laboratory animals and psychiatrically ill patients is striking:

Summary / 219
Systemic level and neuroendocrine aspects
Depression is often associated with hypercortisolemia, persistently elevated levels of cor-tisol, the stress hormone secreted by the adrenal glands. Similarly, studies with chroni-cally stressed rats also reveal increased concentrations in the blood of corticosterone (the equivalent of cortisol in humans). This fi nding has been attributed to an overactivity of the hypothalamus-pituitary adrenal or HPA axis. This is the system that initiates activa-tion of the stress response and without which the body is unable to exert a proper adaptive response in the face of threat or danger. Acute activation of this system can be important for the survival of an animal as it prompts the body for the well known “fi ght or fl ight” response. During this process, the autonomic nervous system is stimulated to prepare for escape or a confrontation; heart beat rises, blood pressure increases, catecholamines such as adrenaline are released as well as glucocorticoids. Although short-term activation can be benefi cial for an organism, long term stimulation of this system due to prolonged stress exposure can lead to HPA axis hyperactivity. Subsequently excessive release of glucocorti-coids can have detrimental effects in the long run however. Glucocorticoids are the mediators of the stress response and through specifi c receptors, located in key regulatory brain structures, they modulate feedback regulation so that the response can be terminated when the stressor is eliminated. When chronically stimulated however the feedback becomes disturbed leading to potentially injurious effects on the body’s homeostasis or balance. This in turn has been reported to be a major contributing factor to the atrophy or degeneration that occurs in the prefrontal cortex (hypofrontality) and hippocampus (dendritic atrophy). Depressed patients reveal reduced prefrontal cortex blood fl ow and metabolism while chronically stressed rats have revealed reduced dendritic branching in the hippocampus. Proper functioning of these regions is important, not only due to their regulatory role in instructing HPA axis activity, but also because of their role in regulating mood and emotional responses. Given their interconnections with other limbic structures, insults to these regions can thus contribute to a dysregulated stress response, as well as disturbed neurotransmission, often associated with affective illnesses as depression.
The studies described in part I of this thesis provide evidence to illustrate the phy-siological and neuronal effects of stress in rats of both gender. Chapter 2 describes results ranging from body weight gain and adrenal morphology to corticosteroid and catecho-lamine plasma concentrations and glucocorticoid receptor densities. These fi ndings not only validate the impact of this stress paradigm on both sexes, they also confi rm an effect of stress on HPA axis activity. Interestingly however, although the results suggest an inju-rious net effect of stress leading to prolonged HPA axis activity in both genders, the data also demonstrate distinct gender differences in physiology and regulation of this basic system. Several brain structures implicated in modulating HPA axis activity that were investigated with markers of neuronal activity (FOS) revealed that the brain circuits pre-ferentially affected by stress is largely dependent upon the sex of the animals.

220 / Summary
Cellular level and signal transduction
Part II of this thesis discusses how chronic stress exposure affected various members of second messenger cascades involved in the transduction of neurotrophin signals, particu-larly brain derived neurotrophic factor (BDNF). Neurotrophins are growth factors some-times seen as a type of “elixir” for the brain. They are essential for the development and survival of neurons and become especially important in a compromised brain, to counter damage induced for instance by chronic stress. Chronic stress as well as depression have in fact been associated with reduced levels of this protein as well as phosphorylated CREB, the transcription factor responsible for it’s expression, while local infusion of BDNF into the brain has been associated with antidepressant-like effects. Likewise, antidepressants have indeed been reported to raise levels of this protein and its signaling member pCREB. Chapter 3 describes immunohistochemical evidence for stress effects on selected MAPK/CREB cascade members (ERK1/2, pCREB, PP2B). These results, seen fi rst in males, pro-vide indirect indications of impaired BDNF transcription and thus decreased neuroplas-ticity in prefrontal regions of the brain. To determine whether these changes were stress and/or gender specifi c, chapter 4 describes the results found in the female counterpart. Interestingly, males appeared more susceptible to this type of insult since females showed little or no disturbance of this cascade.
Molecular level and gene expression
Part III of this thesis describes chronic stress effects on a more molecular level. While stress effects on limbic structure activity can be achieved through differential protein expression, the latter can in turn be dependent upon transcriptional activity and gene expression. To investigate this aspect, a technique called cDNA microarrays were used to establish gene expression profi les of circa 1200 genes following chronic stress. The most interesting fi nding described in chapter 5 was a strong gender dependent aspect underly-ing regulation of transcriptional activity and patterns of neuronal activity. Compared to females, males showed a strong upregulation of genes in response to stress. These micro-arrays were then further investigated to identify genes most strongly affected and most likely responsible for this gender-distinctive result. A group of genes encoding for synap-tic vesicle associated proteins appeared particularly affected, and were further investigated based on their role in regulating synaptic plasticity, which requires both neurotrophins and synaptic activity. Loss of either can lead to cognitive impairments since new informa-tion cannot be properly stored or recalled later. The results obtained by microarray analy-sis were corroborated by RT-PCR and chapter 6 describes the results which were in line with gene expression data; males, but not females, indicated increased expression of several of these proteins (synaptophysin, synaptotagmin, synapsin I,II). Although we can only speculate about the meaning of these fi ndings, it is evident that stress clearly has a gender-specifi c effect on synaptic function. It is important however to learn to recognize and

Summary / 221
understand synaptic dysfunction following stress since this may decrease effi cient neuro-transmission and even neurotrophin production, which eventually precedes overt neuro-degeneration seen in psychiatric disorders as depression.
Implications for pharmacotherapy
Since we saw strong effects of stress and gender on neuronal function and dysfunction, an interesting and logical question would regard the implications of these factors for drug application and development. Chapters 2-6 provide indications to support the HPA axis dysfunction hypothesis (chapter 2) as well as impaired neuronal plasticity (chapters 3, 4, 5), synaptic plasticity (chapter 6) and indirectly the neurotrophic theory. Development of most new antidepressants however is based on the monoaminergic theory of depression. To explore this fi nal possibility, antidepressants of different mechanisms of action were investigated in chapter 7 of part IV. The results reveal striking gender differences with regard to antidepressant effects on systemic tryptophan levels, the precursor for serotonin, and neurogenesis, the birth of new neurons. Although males showed decreased trypto-phan levels, neurogenesis/proliferation was unaffected (or slightly raised) by antidepres-sants; this was in contrast to females that showed increased tryptophan levels but consi-derably reduced neurogenesis. This effect was also common to all antidepressants (citalo-pram, tianeptine and reboxetine), despite their mechanisms of action. In contrast to the effects seen on signal transduction and (synaptic protein) gene expression in males, it appears women are more prone to stress induced effects on neurogenesis and survival of brain cells. The results confi rm once again the importance of considering gender distinc-tions on all levels of stress research. More importantly however, these gender differences evidently also extend onto the application of antidepressants.
To summarize the above, it seems that no matter where we look, stress exerts strong effects on the physiology and psychophysiology of an organism. Although benefi cial in the short run it is not surprising that long term stress can disturb the natural equilibrium state of an animal, given it’s effects on the extended range of pararameters (systemic, endocrinologi-cal, cellular, molecular; see fi gure below). The most interesting observation however was that females consistently showed an opposite or different effect than males. With regard to the question of which sex is more or less susceptible to stress, it appears this answer is thus
less clear-cut than we would hope. While stress seems to affect transcriptional activity and cellular signal transduction in males, females appear more vulnerable to the negative stress effects on cell birth and/or survival. The manner in which HPA axis activity is affected in males and females is a testimony to this gender-differential regulation, since both sexes seem to indicate prolonged HPA axis overactivity, albeit in a sex-specifi c manner. The question we should therefore focus on is not which sex is more vulnerable, but how do the sexes differ in their vulnerabilities. It is more insight into this matter that may ultimately lead to way to new and improved drug development that considers sex specifi c neural circuits implicated in stress response regulation.

222 / Summary
Perhaps one day we can even speak of customized drug design, targeted to meet gender specifi c disturbances.
Figure 1.
Although the neuronal effects of stress stand central to this investigation, the data illustrate that stress also exerts interregula-tory effects on multiple levels of an organism’s physiology (and vice versa).

Summary / 223


Nederlandse samenvatting / 225
NEDERLANDSE SAMENVATTING
Angst en depressie, affectieve stoornissen die gecorreleerd zijn met chronische stress en/of zo genaamde “life events” zoals uit epidemiologische studies blijkt, worden gek-enmerkt door rusteloosheid, hopeloosheid, irritatie, vermoeidheid, en concentratie- en slaapstoornissen. Deze psychiatrische stoornissen beïnvloeden de levenskwaliteit van de patiënt zelf maar in belangrijke mate ook dat van zijn of haar omgeving. Wereldwijd worden deze aandoeningen beschouwd als de belangrijkste oorzaak van invaliditeit. In 2020, zo schat de World Health Organization zijn angst en depressie de op een na grootste aandoening na de cardiovasculaire ziektes. Men verwacht zelfs dat de affectieve stoornissen voor vrouwen dan de voornaamste oorzaak van ziekte zullen zijn. Helaas, is er in de laatste decennia relatief weinig voortgang geboekt met de ontwikkeling van nieuwe geneesmid-delen. Sekse verschillen in het voorkomen van affectieve stoornissen zijn een belangrijk epi-demiologisch gegeven dat de afgelopen jaren systematisch is genegeerd in het klinische en preklinische wetenschappelijke onderzoek. Epidemiologische studies tonen dat in ver-schillende culturen vanaf de puberteit angst en depressie gemiddeld 2 tot 3 keer vaker voorkomt bij vrouwen dan bij mannen. Dit is misschien wel de reden waarom vrouwen vaak worden gekarakteriseerd als zijnde ‘neurotisch’ maar dit is waarschijnlijk niet terecht naar mag blijken uit de bevindingen die worden gerapporteerd in dit proefschrift. Onder-zoek naar fundamentele verschillen in ontstaan van psychopathologie bij mannen en vrou-wen zou een belangrijke bijdrage kunnen leveren aan de ontwikkeling van nieuwe antide-pressiva. De anatomische verschillen van mannen en vrouwen zijn al voldoende bekend maar recent zijn wetenschappers ook geïnteresseerd geraakt in neurobiologische en fysio-logische sekse verschillen. De samenstelling van botweefsel, pijnverwerking, het metabo-lisme van geneesmiddelen, en snelheid van neurotransmitter productie in de hersenen bli-jken sterk te verschillen bij mannen en vrouwen. Het ligt dan voor de hand om aan te nemen dat sekse verschillen ook zijn gerepresenteerd in de manier waarop psychopatholo-gie zich ontwikkeld. Het is dus bijzonder dat nog tot vrij recent voor klinische geneesmid-delen studies, het vrouwelijke geslacht een exclusie criterium was en in het preklinische onderzoek uitsluitend mannelijke proefdieren werden gebruikt. De belangrijkste reden voor deze benadering is dat de hormonale cyclus van de vrouw de interpretatie van de uitkomsten van de studie zou bemoeilijken. Voor de ontwikkeling van nieuwe farmacotherapeutische benaderingen voor affec-tieve stoornissen is een goed begrip van de pathofysiologie noodzakelijk zodat sekse-(a)specifi eke aangrijpingspunten voor geneesmiddelen in de cellulaire fysiologie kunnen worden geïdentifi ceerd. Hierin past ook het gebruik van de huidige generatie geneesmid-delen die soms nog deels onbegrepen werkingsmechanismen hebben. Hypothesen over

226 / Nederlandse samenvatting
het ontstaan van affectieve stoornissen zullen moeten worden herzien of aangepast want de hedendaagse theorieen zijn vooral gebaseerd op onderzoek in mannelijke proefdieren en het wordt steeds duidelijker dat resultaten verkregen met vrouwelijke proefdieren slecht kunnen worden verklaard met deze hypothesen. Dit vereist een andere manier van denken van de onderzoekers, van een ‘mannen wereld’ naar studies waarin men actief de sekse verschillen bestudeert. In de populaire pers is de gezondheid van de vrouw al langer onder-werp van discussie en ook in de biologische psychiatrie en de psychofarmacologie mag dit onderwerp zich verheugen in een groeiende belangstelling.
De rol van stress bij het ontstaan van psychiatrische stoornissen als angst en depressie is een belangrijk uitgangspunt voor preklinisch onderzoek. Bijna alle onderzoekers maken op een of andere manier gebruik van dit gegeven voor de bestudering in diermodellen van inductie en behandeling van de pathobiologie van affectieve stoornissen. Preklinische studies tonen dat chronische stress aanleiding is voor het ontstaan van neurodegeneratieve verschijnselen in het limbische systeem; het netwerk in de hersenen voor regulatie van de gemoedstoestand en emoties. Soortgelijke resultaten zijn verkregen met in vivo beeldvor-mend onderzoek bij patienten, ook daar werden afwijkingen gevonden in het limbische systeem, in het bijzonder in de prefrontale cortex en hippocampus. Chronische stress is dus zeer waarschijnlijk een belangrijke oorzaak voor het ontstaan van neuronale disfunctie en psychopathologie. Er is een aanzienlijke hoeveelheid informatie beschikbaar over het neuronale substraat van de acute stress response van mens en dier, die leidt tot verbete-ring van geheugen en cognitieve processen, maar er is nog relatief weinig kennis over de neurobiologie van chronische stress verwerking. Een bijkomend probleem is dat er een groot sekse verschil is in stressgevoeligheid en het voorkomen van stress-gerelateerde psy-chiatrische stoornissen waardoor we grote vraagtekens kunnen plaatsen bij de mate van overeenkomst van stress response mechanismen, neuronale functie en disfunctie, ontstaan van psychopathologie, en effectiviteit van farmacotherapie bij mannen en vrouwen. In dit proefschrift zijn een serie experimenten beschreven die tot doel hadden overeenkomsten en verschillen in stressverwerking bij mannelijke en vrouwelijke proefdieren op het niveau van de fysiologie, moleculaire biologie, en neurobiologisch substraat te karakteriseren. De stressor die werd gebruikt was het verblijf van 15-120 minuten in een ruimte van 20x20x40 cm waar de dieren in die periode enkele zwakke ‘footshocks’ kregen toegediend gedurende 21 dagen zodat de dieren deze box als stressvol gaan ervaren. Op de laatste dag van het onderzoek werden geen ‘shocks’ meer gegeven maar werd voor het neuro-moleculair biologische onderzoek van sekse-verschillen in stress pathofysiologie het ‘psy-chologische aspect’ van het verblijf in de stressvolle omgeving als stimulus gebruikt. Stressverwerking vindt plaats op verschillende niveau’s, in de cardiovasculaire fysiologie en de afgifte van stress hormonen zoals corticosteron en adrenaline, en op het vlak van de neuroplasticiteit die o.a. wordt gekenmerkt door veranderingen van aantallen synaptische contacten in neuronale netwerken en vorming van nieuwe neuronen. Op deze manier past het brein zich aan aan de veranderende omstandigheden. Deze aanpassing vereist

een complex samenspel van cellulaire en moleculaire processen die gezamenlijk zorgen voor veranderingen van de signaaltransductie, de gen- en eiwit-expressie, en uiteindelijk aanpassing van het functioneren van het limbische systeem. De mechanismen betrokken bij het ontstaan van stressgerelateerde pathofysiologie bij mensen en dieren tonen grote overeenkomsten en zullen hierna worden besproken.
Stresshormonen
Hypercortisolemie, verhoogde spiegels van het stresshormoon cortisol (corticosteron bij de rat) in het bloed, komt veel voor bij depressieve patiënten. Proefdieren tonen na chro-nische stress ook een verhoging van de plasma corticosteron spiegels evenals een vergrot-ing van de bijnier, het orgaan wat de afgifte van corticosteron regelt. Deze neuroendocri-ene veranderingen worden toegeschreven aan veranderingen in het functioneren van de hypothalamus-hypofyse-bijnieras (HPA-as). De HPA-as initieert een fysiologische stress respons die nodig is om op adequate wijze te kunnen reageren op dreigend gevaar. Een acute activering van dit HPA-as systeem is onderdeel van de survival reactie die ook bekend staat als de “fi ght or fl ight” respons en die daarnaast veranderingen in de cardio-vasculaire fysiologie initieert via het autonome zenuwstelsel, zoals een verhoging van hart-slag, bloeddruk, en plasma adrenaline spiegels die van belang zijn voor het aangaan of ver-mijden van een gevecht. Een kort durende HPA-as activatie en het autonome zenuwstel-sel heeft duidelijke voordelen voor de overleving van het organisme/individu maar steeds meer onderzoek toont dat de langdurige activatie van het HPA-as systeem en chronische verhoging van de plasma cortisol (corticosteron) spiegels in het bloed schadelijk is. Het stresshormoon cortisol (corticosteron) passeert de bloed hersenbarriere en in de hersenen bevinden zich receptoren voor dit stresshormoon die er voor moeten zorgen dat er een negatieve feedback loop wordt geactiveerd waardoor de activiteit van de HPA-as wordt afgeremd wanneer de stressor is verdwenen. Bij chronische stress is dit feedback mechanisme verstoord waardoor corticosteroiden schade in de hersenen kunnen veroorza-ken zoals atrofi e van neuronen in de prefrontale cortex en hippocampus. Depressieve pati-enten tonen in deze hersengebieden een vermindering van de lokale activiteit en doorbloe-ding (hypofrontaliteit) en chronisch gestresste ratten een vermindering van de aantallen vertakkingen van dendrieten in de hippocampus. Een verstoorde functie van de prefron-tale cortex en hippocampus is niet alleen belangrijk voor de regulatie van de HPA-as maar deze gebieden zijn ook onderdeel van het limbische systeem en dus betrokken bij de controle van de gemoedstoestand en emoties. Een kleine verstoring ergens in de pre-frontale cortex en/of hippocampus kan zo de aanleiding zijn voor ontregeling van de neu-rotransmitter afgifte in andere delen van de hersenen met als gevolg een verstoorde stress respons. Deel I van het proefschrift beschrijft de effecten van chronische stress op fysiologische en neurobiologische mechanismen van mannelijke en vrouwelijke ratten. De neuroen-docriene effecten worden besproken in hoofdstuk 2, waarbij aandacht is voor veranderin-
Nederlandse samenvatting / 227

gen in bijniergewichten, lichaamsgewicht, plasma corticosteron spiegels en glucocorticoid receptor expressie in de hersenen. We concluderen dat het stress paradigma een sekse onaf-hankelijke impact heeft op het functioneren van de HPA-as (hyperactiviteit) maar dat er sekse specifi eke factoren een rol spelen bij het ontstaan van deze functieverstoring. Vooral in de patronen van neuronale activatie in de hersenen werden bij mannetjes en vrouwtjes ratten verschillen waargenomen. Voor de karakterisering van patronen van hersenactiviteit werd gebruik gemaakt van een kwantitatieve analyse van de Fos expressie; een eiwit dat wordt geproduceerd in actieve zenuwcellen.
Cellulaire signaaltransductie systemen
Deel II van het proefschrift beschrijft de effecten van chronische stress in cellulaire sig-naaltransductie cascades die een rol spelen bij handhaving van neuroplasticiteit. Groeifac-toren zoals brain derived nerve growth factor (BDNF) zijn van belang voor de ontwikkel-ing en plasticiteit van het zenuwstelsel. Stressvolle omstandigheden eisen adaptatie van het zenuwstelsel en bij dit proces van aanpassing spelen groeifactoren als BDNF een cruciale rol. Chronische stress en depressie zijn recent in verband gebracht met een vermindering van de afgifte van BDNF en de geactiveerde vorm van de transcriptiefactor CREB (cAMP Response Element Binding protein). Enkele studies tonen in proefdieren een “antide-pressief effect” van intracerebrale BDNF injecties terwijl bij patiënten antidepressiva de BDNF concentratie en expressie van phospho-CREB lijken te verhogen. Hoofdstuk 3 beschrijft de effecten van chronische stress in de MAPK/CREB cascade van mannelijke ratten en toont met behulp van immunocytochemische kleuringen van hersenweefsel dat deze cascade in de prefrontale cortex is verstoord; een aanwijzing voor een disfunc-tionele BDNF cascade met als gevolg een gereduceerde neuroplasticiteit. In hoofdstuk 4 worden de effecten beschreven die zijn waargenomen in vrouwtjes ratten en onderzocht in hoeverre de gevonden defecten in de BDNF signaaltransductie cascade sekse specifi ek zijn. Mannelijke dieren blijken het meest gevoelig te zijn voor een verstoring van de MAPK/CREB cascade en in vrouwtjes werden in deze cascade slechts kleine stress-geïnduceerde veranderingen waargenomen.
Moleculaire niveau en genexpressie
Deel III van het proefschrift beschrijft de effecten van chronische stress op het moleculaire niveau, in het bijzonder veranderingen in de gen expressie in de prefrontale cortex. Hier-voor is gebruik gemaakt van zogenaamde cDNA micro-arrays die het mogelijk maken om tegelijkertijd de veranderingen in de expressie van 1200 genen te bestuderen. Ook in de genexpressie na chronische stress werden grote sekse verschillen gevonden (hoofdstuk 5). Mannetjes toonden een up-regulatie (toename) van gen-expressie in de prefrontale cortex na chronische stress maar bij vrouwtjes was dit effect vrijwel afwezig; een bevestiging van de beschreven immunocytochemische studies. Deze resultaten waren aanleiding voor een
228 / Nederlandse samenvatting

nauwkeuriger karakterisering van de bij de stress respons betrokken genen. Vooral genen die van belang zijn voor handhaving van de synaptische plasticiteit (synaptic vesicle associ-ated proteins (SVAP)) bleken in het bijzonder gevoelig te zijn voor de invloed van stress. Veranderingen in expressiepatronen van deze synaptic vesicle associated proteins zijn in verband gebracht met een verminderd cognitief functioneren door problemen met de opslag en recall van informatie. Hoofdstuk 6 beschrijft de uitkomsten van een RT-PCR analyse van enkele SVAP’s. Bij mannetjes, maar opnieuw niet bij de vrouwtjes, bleek de expressie van synaptophysin, synaptotagmin, en synapsin I/II te zijn verhoogd na chro-nische stress, een duidelijke aanwijzing dat stress sekse-specifi eke gevolgen heeft voor handhaving van de synaptische plasticiteit; een proces dat een cruciale rol speelt bij afgifte van neurotransmitters, effi ciëntie van de signaaloverdracht in neuronale netwerken, en zelfs voor de afgifte van groeifactoren.
Farmacotherapeutische effecten
In de hiervoor beschreven experimenten worden belangrijke sekse-verschillen gerap-porteerd in stressverwerking die van belang zouden kunnen zijn voor het gebruik en ontwikkeling van antidepressiva. De ontwikkeling van nieuwe antidepressiva is vooral gebaseerd op de zogenaamde mono-amine hypothese van depressie; een theorie die ervan uitgaat dat verstoring van de monoamine huishouding in het brein ten grondslag ligt aan het ontstaan van depressie en dat herstel van deze functie belangrijk is voor de behandel-ing van deze psychiatrische aandoening. In hoofdstuk 7 (Deel IV) worden opzienbarende sekse verschillen beschreven van antidepressiva met verschillende werkingsmechanismen op de serum tryptofaan spiegels, de precursor van serotonine, en de neurogenese, de vorm-ing van nieuwe neuronen in de gyrus dentatus van de hippocampus. Chronische toedien-ing van antidepressiva resulteerde bij mannetjes in een verlaagde serum tryptofaan spiegel en een niet signifi cante verandering van de neurogenese maar bij vrouwtjes ratten steeg de serum tryptofaan spiegel en was er een signifi cante daling van de neurogenese. Alle gebruikte antidepressiva toonden dit effect alhoewel hun veronderstelde werkingsmecha-nismen verschilde. Op basis van deze resultaten komen wij tot de conclusie dat in man-netjes ratten stress de synaptische plasticiteit en signaaltransductie verstoort terwijl bij vrouwtjes vooral de neurogenese en overleving van hersencellen gevoelig lijkt te zijn voor de invloed van stress. De resultaten bevestigen opnieuw het idee dat sekse een essentiele
component is voor de stress-gerelateerde pathofysiologische processen die ten grondslag liggen aan het ontstaan van affectieve stoornissen en dat dit aspect gevolgen heeft voor ontwikkeling van nieuwe antidepressiva en identifi catie van nieuwe targets in de cellulaire fysiologie voor farmacotherapie.
Samenvattend: stress beïnvloedt op een sekse-specifi eke wijze een breed scala aan fysiolo-gische, endocrinologische, cellulaire en moleculaire mechanismen (fi guur 1). Dat vrou-welijke proefdieren bijna altijd een tegengesteld of in elk geval ander effect van chro-
Nederlandse samenvatting / 229

230 / Nederlandse samenvatting
nische stress verwerking toonden dan mannelijke dieren is opvallend. De vraag welke sekse het meest stress gevoelig is, is hiermee nog niet beantwoord, en misschien nu door dit onderzoek zelfs wel minder duidelijk geworden dan het voorheen was. Stress beïnvloedt bij mannetjes voornamelijk gen-expressie en neurotransmissie processen, maar vrouwtjes lijken juist meer gevoelig te zijn voor veranderingen in neurogenese en overleving van zenuwcellen. In beide seksen is er sprake van een langdurige HPA-as activatie, maar de wijze waarop die verandering tot stand komt verschilt bij mannetjes en vrouwtjes ratten. Dus is niet de vraag interessant welke sekse het meest stress gevoelig is maar in het bij-zonder hoe ze daarin verschillen. Inzicht in deze problematiek zou ons uiteindelijk verder kunnen brengen op weg naar een nieuwe en meer effi ciënte farmacotherapie waarin reken-ing wordt gehouden met het feit dat verschillende neuronale circuits en signaaltransductie mechanismen betrokken zijn bij de regulatie van de stress response.
Figuur 1.
Het onderzoek in dit proefschrift concentreerde zich vooral op de neuronale mechanismen die worden beïnvloed door chro-nische stress, maar de fi guur toont dat dit slechts een klein onderdeel is van het totale stressverwerkingssysteem waarin ook centrale en perifere moleculaire, cellulaire en fysiologische mechanismen zijn betrokken.

Nederlandse samenvatting / 231


Acknowledgements / 233
ACKNOWLEDGEMENTS
As I sit down to write these fi nal pages, I realize this project has fi nally come to its end. As
challenging yet wonderful as it was however, it could not have been done without the help
of a lot of other people. For this I would like to express my thanks and gratitude.
First, to my promoters Prof. Dr. Hans den Boer and Prof. Dr. Gert ter Horst. I will always
be grateful to the both of you for the opportunity to have worked in your department, for
your guidance and most of all your friendship. Without doubt it has been a most fruitful
and learning experience. Not only has our “working together” provided an excellent base
for my scientifi c future, it has also opened many new doors. Hopefully one day we can
pick up where we left off and continue our collaboration. Until then, I will look back with
fond memories and nostalgia on my time as an AiO under your supervision, characterized
always by latitude, trust, stimulation, enthusiasm and continual support. I could not have
wished it any other way.
As happy as I am, to have completed this thesis, it is with equal sadness that I realize my
time with such wonderful colleagues has reached its end. To Tineke S., Tineke K. and
Folkert I owe a very big thanks. During my “pregraduate” training you provided me with
my fi rst introduction to science and evidently the impression you made was great enough
to get me to come back for more; fi rst for a course, then for an internship, and fi nally for
a PhD! At fi rst I couldn’t even manage a single immunostaining, and now I have a whole
thesis full of them! Thanks to you! More importantly however, I am grateful for your kind-
ness and constant readiness to help, on all matters (regarding science and beyond). To Kor,
Peter, and Petra your technical assistance was invaluable throughout my experiments. I
really could not have done it without you. To Jaap, although we didn’t have the chance to
work very closely, I appreciate your continuous interest, friendly notes, and help with data
interpretations. To my fellow colleagues/friends from the 7th, Andrea, Charmaine, Chris-
tel, Dirk, Gabor, Gea, and Marjolein, thank you for the good times and “social support”
throughout the years. Likewise to the colleagues/friends from the 6th fl oor, Annemiek,
Fokko, Franske, Ingrid, Marjan, Marthijn, Mbemba, Peter Paul and Sascha thanks for the
fun, the laughs and friendly tête-à-tête. Joanna, Marieke, Minke and Simone you were
fantastic “room slash dinner” mates! …a little nuts perhaps, but I will really miss you guys
and our (oftentimes hilarious) discussions! To Jacqueline and Margo, thanks for your help,
advice and always pleasant nature. To all the others, without mentioning names, thank
you very much.

234 / Acknowledgements
All of you have contributed in your own way to a wonderful, warm working atmosphere
that I will miss for a very long time.
A special word of thanks is extended to my pregraduate students, Niels (Ingen Housz),
Sonja (Janmaat) and Raoul (Ribot). You know, like no other, the extent of discipline and
perseverance that accompanies long-term experimentation. Oftentimes throughout these
projects however, we ourselves became subject to “chronic stressful conditions”, but your
patience and enthusiasm made it that much easier to get the job done. I am grateful to
your hard work, invested time and efforts, which contributed greatly to a bulk of the data
presented in this thesis. I wish you all the luck in your futures.
I would also like to thank the members of the Pathology group for their pleasant com-
pany, advice and help whenever I needed it. I am particularly indebted to Rense Veenstra,
Elvira Oosterom, Geert Harms and Lorian Menkema, who were there to witness my ini-
tial “take-off”, four years ago, which would have been much slower and a lot more frus-
trating without their generous and excellent technical support. By the same token, I am
indebted to Gerard te Meerman and Karoly Mirnics for providing invaluable assistance
during perhaps the most challenging aspect of this project, the microarray analysis. Fortu-
nately we can look back on a pleasurable and successful collaboration. Thank you.
I am grateful to the BCN for their support throughout my PhD endeavours, with a special
note of thanks to Tinie Alma, Diana Koopmans, Rob Visser and the PhD council. Not
alone in their contribution however, I am also grateful for the professional and/or fi nancial
assistance from the industries that contributed toward the completion of this thesis with
a special note of appreciation to Dr. Carmen Munoz, for her steadfast help and coopera-
tion.
Last but not least I am grateful to those who have done so much more than help me
fulfi ll this course of study. I am thankful to my friends and family, Appie, Beppe, Chris,
Eleonora, Els, Giuseppe, Ids, Ingrid, JD, Marjan, Marjolein, Markus, Marten, Monique,
Ralph, Ronnie, Sonia, Thomas and Tarzan for providing a solid source of merriment and
relaxation and at times the much needed distraction from PhD-associated struggles and
tribulations. A special mention goes to Henne and Ineke, my beautiful paranymphs and
very dear friends throughout this academic journey. I consider myself lucky with friends
like you and I’m sure the best is yet to come!
To papa, mama, Albert and Marten, I thank you for always being there for me, enabling
me to be who I am today and where I am today. It is your continual belief in me, to
which I thank all of my accomplishments. As far back as I can remember (temporally and

Acknowledgements / 235
geographically), you have been the constant source of love, support and encouragement,
making everything possible. There are not enough words to describe all the thanks you
deserve, but perhaps these three come close: I love you!
…and then to save the best for last, Andrea, the one person who was absolutely indis-
pensable throughout this project. Your continuous input, effort, clever suggestions, and
optimistic nature were invaluable and it goes without saying that without you this thesis
would not have turned out as it did. More precious than that however, were your friend-
ship, love and dedication throughout the years. From the moment we met you became
my colleague, friend and confi dant and eventually my dearest companion. In breve, tu sei
tutto per me. Ti voglio bene!
Thanks again to all of you!
Sjoukje


PUBLICATIONS
1. Kuipers S.D*., Trentani A*., te Meerman G.J., ter Horst G.J., Den Boer J.A. (2003) Immunohistochemical changes induced by repeated footshock stress: revelations of gen-der-based differences. Neurobiology of Disease, 14, 602-618. (*co-fi rst author)
2. Kuipers S.D., Trentani A., Ter Horst G.J., Den Boer J.A. Looking beyond the receptor: molecular biological approaches to antidepressant therapy. In “Current and Future Developments In Psychopharmacology”, in press.
3. Kuipers S.D., Trentani A., Den Boer J.A., Ter Horst G.J. (2003) Molecular correlates of impaired prefrontal plasticity in response to chronic stress.Journal of Neurochemistry, 85, 1312-1323.
4. Westenbroek C., Ter Horst G.J., Kuipers S.D., Trentani A., Den Boer J.A. (2003) Gender-specifi c effects of social housing in rats after chronic mild stress exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry, 27, 21-30.
5. Trentani A., Kuipers S.D., Ter Horst G.J., Den Boer J.A. (2002) Selective chronic stress-induced in vivo ERK1/2 hyperphosphorylation in medial prefron-tocortical dendrites: implications for stress-related cortical pathology? European Journal of Neuroscience, 15, 1681-91.
6. Trentani A., Kuipers S.D., Ter Horst G.J., Den Boer J.A. (2002)Intracellular signaling transduction dysregulation in depression and possible future targets for antidepressant therapy. In “Handbook of Depression and Anxiety” Marcel Dekker Inc., New York, Basel.
7. Sebens J.B., Kuipers S.D., Koch T., Ter Horst G.J., Korf J. (2000) Limited participation of 5-HT(1A) and 5-HT(2A/2C) receptors in the clozapine-induced Fos-protein expression in rat forebrain regions. European. Journal of Pharmacology, 408, 11-7.
8. Bosker F.J., Westerink B.H.C. Cremers T.I.F.H., Gerrits M., Kuipers S.D., van der Pompe G., ter Horst G.J., den Boer J.A. and Korf J.,
“Future antidepressants, what is in the pipeline and what is missing?”. CNS Drugs, in press.
Publications / 237


Publications / 239
SUBMITTED OR IN PREPARATION
1. Kuipers S. D., Trentani A., Bakker P.L., Postema F., Reuvekamp P.T.W., Ter Horst G.J., Den Boer J.A. Repeated Stress Impairs HPA axis regulation: Indications for differential gender-depen-dent mechanisms. Endocrinology.
2. Kuipers S.D., Trentani A., Bakker P.L., Veenstra R., Ter Horst G.J., Den Boer J.A. Reduced CREB phosphorylation and calcineurin content characterize the response to chronic stress in male rats: Indications for sex-dependent neuroplasticity changes. European Journal of Neuroscience.
3. Kuipers S. D., Trentani A., te Meerman G.J., Bakker P.L., Reuvekamp P.T.W., Den Boer J.A., Ter Horst G.J. Chronic stress effects on synaptic-vessicle associated protein expression: Indications for region/gender-dependent regulation. Molecular Psychiatry.
4. Kuipers S. D., Trentani A., Koch, T., Ter Horst G.J., Den Boer J.A. Impact of long-term antidepressant treatment on chronic stress induced reduction of neu-rogenesis in adult rats: Revelations of sex/drug specifi c regulation. Neuropsychopharmacology.
5. Kuipers S. D., Trentani A., Korf J., Kema, I.P., Den Boer J.A., Ter Horst G.J. Long-term antidepressant administration affects peripheral tryptophan availability and impairs body weight regulation in rats: evidence for gender-specifi c effects. Neuropharmacology.
6. Trentani A., Kuipers S.D., Ter Horst G.J., Den Boer J.A. Investigating the neurobiological substrates underlying the deleterious infl uences of chronic stress and the benefi cial effects of tianeptine in the female rat brain. European Journal of Neuroscience.
7. Trentani A., Kuipers S.D., Ter Horst G.J, Den Boer J.A. Does long-term citalopram treatment overcome chronic stress-induced reduction of neu-roplasticity in the female rat brain? Neuroscience.
8. Trentani A., Kuipers S.D., Ter Horst G.J, Den Boer J.A. Long-term reboxetine administration attenuates stress-induced reduction of neuronal plas-ticity by enhancing CREB phosphorylation in the female rat brain. Biological Psychiatry.

