thèse présentée par - usto
TRANSCRIPT

MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE DES SCIENCES ET DE LA TECHNOLOGIE D’ORAN MOHAMED BOUDIAF
Faculté des sciences
Département de chimie
Spécialité : Chimie Option : Chimie des matériaux
Thèse Présentée Par :
K H E N I F I A i c h a
POUR L’OBTENTION DU DIPLOME DE DOCTORAT EN SCIENCE
THEME
ELABORATION DE MATERIAUX A BASE D’ARGILES,
CARACTERISATION, ET APPLICATION A L’ELIMINATION
DES POLLUANTS ORGANIQUES
Soutiendra le Jeudi 02 Décembre 2010 à 10h au département de chimie (4ème étage)
Devant le jury composé de :
Présidente Mme BENHARRATS N . Professeur Université USTO
Rapporteur Mr DERRICHE Z . Professeur Université USTO
Examinateur Mr KACHA S . Professeur Université Sidi Bel Abbes
Membre Invité Mr BETTAHAR N. Professeur Université USTO
Examinateur Mr GHOMARI A . Professeur Université Mostaganem
Année universitaire : 2009-2010

Résumé
L’adsorption d’un polluant organique chloré : le 2,4 DCP et d’un colorant industriel textile :
le 4GL par un mélange Bentonite/CTAB est effectuée par deux méthodes : la première en
utilisant la bentonite organophile comme adsorbant, ce matériau est préparé par échange
cationique entre le sodium résidant dans l’espace interfeuillet de l’argile de base et le cation
Cétyltriméthylamonium bromide (CTAB), cette méthode est notée two steps. La deuxième
méthode s’effectue en mettant en contact la bentonite avec un mélange des polluants organiques :
le 2,4 DCP et le 4GL et le tensioactif cationique : le CTAB, c’est la méthode one step. Ce
procédé de traitement des eaux est très intéressant, surtout pour les rejets contenant un mélange
de tensioactif cationique et de polluant organique. L’élimination du polluant organique et le
surfactant cationique se fait en une seule étape d’où le nom one step. Des études d’adsorption
sont effectuées dans ce travail afin d’évaluer l’effet de certain paramètres sur l’efficacité de la
méthode : concentration de CTAB utilisée, le temps de contact et la concentration de polluant.
Plusieurs modèles sont testés pour la cinétique d’adsorption : le modèle pseudo premier ordre,
pseudo second ordre, Elovich et le modèle de diffusion intra particulaire. Les données des
isothermes d’adsorption sont analysées en utilisant les modèles non linéaires de Freundlich, de
Langmuir et de Redlich Peterson. Les résultats de cette étude ont montré que la méthode one
step (CTAB en solution) améliore la fixation des deux polluants sur l’argile. En effet, le polluant
organique et le CTAB en solution sont éliminés rapidement par l’argile en solution. Les études
cinétiques ont montré que l’adsorption de 2,4DCP et de 4GL par les deux méthodes suit une
cinétique du pseudo second ordre. La diffusion intra particulaire n’est pas la seule étape limitant
l’adsorption. Les isothermes d’adsorption des deux polluants par la méthode one step et two steps
sont tout à fait différentes. Des interactions de type adsorbat- adsorbant et de type adsorbat-
adsorbat sont responsables de ce comportement. La caractérisation des matériaux après
adsorption des deux polluants, confirment le fait que l’adsorption de 2,4 DCP et du 4GL par la
méthode one step et two steps suit des mécanismes tout à fait différents. Les résultats de cette
étude sont très utiles pour déterminer les conditions optimales d’élimination des polluants
organiques par l’argile organophile ou par l’argile sodée quand le CTAB est en solution.
La deuxième partie de cette thèse est consacrée à un autre type d’argile, moins présents à la
surface de la terre que les argiles cationiques. Mais qui possèdent des caractéristiques fortes

intéressantes. En effet, l’élimination de deux herbicides : le Glyphosate et le Glufosinate
largement utilisés en France est effectuée par le matériau hydroxyde double lamellaire type
NiAlNO3.Une étude cinétique et isotherme est effectuée afin d’évaluer l’influence du temps de
contact et de la concentration des herbicides sur l’adsorption. Les données expérimentales sont
analysées par les modèles linéaires de Freundlich, Langmuir et Tempkin. Les analyses texturales
et structurales (DRX , IR, MEB….) du matériau NiAlNO3 après adsorption des deux polluants
ont mis en évidence un mécanisme d’adsorption à travers un échange anionique. En effet, les
deux herbicides s’adsorbent en premier lieu sur la surface des cristallites puis s’intercalent entre
les feuillets argileux.
Un capteur ampérométrique basé sur le matériau double lamellaire NiAlNO3 est réalisé
pour la détection de deux herbicides : le Glyphosate et le Glufosinate. Le matériau HDL est
préparé par deux méthodes : la méthode de coprécipitation à pH constant et la méthode de
déposition électrochimique sur une électrode de platine. Les films du matériau électrodéposé sont
caractérisés par la diffraction des rayons X : DRX, la spectroscopie IR, La spectroscopie
Raman et par microscope électronique à Balayage MEB. L’oxydation électrocatalytique des
deux herbicides : Glyphosate et Glufosinate est possible sur les centres Ni3+de la structure HDL.
Les réponses électrochimiques de l’électrode modifiée par le matériau NiAlNO3 sont enregistrées
par deux techniques : la voltamétrie cyclique et la Chrono-ampérométrie en utilisant un potentiel
de 0.49V/SCE en fonction de la concentration de l’herbicide dans un milieu basique NaOH 0.1M.
La réponse électrocatalytique montre une progression linéaire pour le Glyphosate ayant une
concentration entre 0.01 et 0.9 mM avec une limite de détection de 1µM et une sensibilité de
287 mA/m Cm². Comparé au Glyphosate, le Glufosinate montre une sensibilité moins élevée soit
178 mA/m Cm².

REMERCIEMENTS
Ce travail est le fruit d'une collaboration entre le laboratoire physico chimie des matériaux
catalyse et environnement du département de chimie, de la faculté des sciences de l’université des
sciences et technologie Mohamed Boudiaf USTOran et le laboratoire des matériaux inorganiques
LMI du l’université Blaise pascal 2 de Clermont ferrand.
Une partie de ce travail a été réalisée dans le Laboratoire des matériaux inorganiques à
Clermont Ferrand (LMI), dirigé par les professeurs Monsieur Claude Forano, madame Vanessa
Prevot et madame Christine Mousty dans le cadre d’une bourse mixte Franco-Algérienne.
Je tiens à exprimer ma reconnaissance à Monsieur le Professeur Claude Forano professeur
au laboratoire LMI pour la confiance qu’il m’a témoignée en m’accueillant au sein de son
laboratoire et m’avoir facilité mon intégration au sein de son équipe. Je voudrais également lui
exprimer mes remerciements pour avoir dirigé ce travail et les moyens mis à ma disposition tout
au long de mon séjour.
Je remercie profondément Mme Christine Mousty et Vanessa prevot, Professeurs au
laboratoire LMI pour l’intérêt constant qu’elles ont porté à ce travail en acceptant de codiriger
cette étude, pour leur disponibilité, leurs orientations et leurs remarques fructueuses. Qu’elles
trouvent ici ma profonde gratitude.
Les mots ne suffiraient pas pour exprimer toute ma reconnaissance envers Monsieur
Derriche Zoubir, Professeur à l’université d’USTO Oran, qui est à l’origine de cette étude, pour
la qualité de son encadrement, sa compétence, ses conseils, sa disponibilité et sa qualité humaine.
La liberté qu'il m'a accordée et les responsabilités qu'il m'a confiées m'ont permis d'atteindre une
maturité professionnelle que je n'aurais pas imaginée auparavant.

Je suis très sensible à l’honneur que m’ont fait Messieurs Ghomari A., Professeur à
l’université de Mostaganem et Kacha S., Professeur à l’université de Sidi bel abbés Benttahar N.
Professeur à l’université USTO pour m’avoir honoré de leur présence en acceptant d’être
rapporteurs de ce travail et de le juger.
Mes remerciements s’adressent également à Madame Benharrats N., Professeur à
l’université des sciences et de la technologie USTOran pour l’intérêt qu’elle a accordé à ce
travail en acceptant de le juger et de présider le jury.
Je n’oublie pas dans mes remerciements tout le personnel du Laboratoire des matériaux
inorganiques LMI (Enseignants, Chercheurs, Techniciens, Secrétaires) que j’ai côtoyé et qui
m’ont facilité mon intégration au sein du groupe. Mes remerciements vont également à monsieur
Mhammed Benbakkar ingénieur au laboratoire ICPAES de l’université géologie et science de la
terre de Clermont ferrand pour son aide durant toute la durée de ma formation.
Un grand merci à tous les doctorants et les docteurs que j’ai côtoyés au LMI durant cette
thèse pour l’apport de la bonne humeur et pour leur encouragement .Je pense en particulier à mes
collègues de bureau, Cheradi Khaled, Rachid Akbour, Azam Faour, Mansouri Hela, Mohamed
Hennous et Touati Souad.
Je ne pourrai terminer ces remerciements sans y associer ma famille en Algérie, mes amis
du laboratoire Physico chimie des matériaux catalyse et environnement (LPCME) de l’université
des sciences et de la technologie USTOran : Bouberka Z ; F. Yakhou, H. Hamani, K.
Benabou …. et tant d'autres sans le soutien desquels je n’aurai pu entreprendre ces études. A
toutes et à tous je leur dis merci.
Mes remerciements vont aussi à une personne très chère pour moi et qui m’a toujours
poussé vers l’avant Slimani Z. Je te souhaite une vie pleine de bonheur et de réussite.

Que ceux ou celles que j'ai pu momentanément oublier aujourd'hui, veuillent bien me
pardonner. Ils me reviendront nécessairement à ma mémoire et me feront regretter mon
ingratitudemomentanée.

DEDICACES
A, la mémoire de mon père,
A, mes deux mères,
A, ma très chère sœur, son mari et ses fils,
A, mes frères et leurs petites familles,
A, Slimani Z.
A , tous mes amis et qui me sont chers.
Aicha (Naima)

SOMMAIRE INTRODUCTION GENERALE 01
PREMIERE PARTIE : ETUDE BIBLIOGRAPHIQUE
CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUE
1. Introduction 09
2. Définition 10
3. Microstructure des argiles 10
4. Classification des argiles 13
4.1 Minéraux à 7 Å 13
4.2 Minéraux à 10 Å 13
4.3 Minéraux à 14 Å 13
4.4 Minéraux interstratifiés 13
5. Origine de la Bentonite 15
6. Caractéristiques physiques des argiles lamellaires 16
6.1 Charge des surfaces argileuses 16
6.3.1 Charge permanente 16
6.1.2 Charge variable 17
6.2 Capacité d’échange cationique 17
6.3 Propriété de gonflement 19
6.3.1 Phyllosilicates non-expansibles 20
6.3.2 Phyllosilicates expansibles 20
7. Surface spécifique 20
8. Les argiles modifiées 22
8.1 Généralités 22
8.2 Les argiles organophiles 22
8.2.1 Tensioactif ou agent d’interface 23
8.2.2 Caractéristiques structurelles des argiles organophiles 24
8.3 Famille des complexes inorgano-argileux 25
8.3 Famille des Complexes Inorgano-Organo-Argileux 26
Références bibliographiques 28

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES
(ARGILES ANIONIQUES)
1. Historique 33
2. Propriétés structurales des HDL 34
2.1 Structure 34
2.2 Nature des cations M(II) et M(III) 36
2.3 Nature de l'anion de compensation 39
2.4 Proportion d'eau dans le domaine interlamellaire 40
3. Synthèse des hydroxydes doubles lamellaires 42
3.1 Méthode de coprécipitation 43
3.1.1 Méthode de coprécipitation à faible supersaturation 46
3.1.2 Méthode de coprécipitation à supersaturation élevée 46
3.2 Méthode d’urée 47
3.3 Traitements post-synthèse 49
3.3.1 La calcination–reconstruction 49
3.3.2 Échange anionique 50
3.3.3 Délamination - réempilement 50
4. Champs d’applications des HDL 51
4.1 Catalyseurs, précurseurs de catalyseur, supports de catalyseurs 51
4.2 Charge minérale dans les polymères nanocomposites 52
4.3 Électrodes / Conducteurs ioniques 52
4.4 Précurseurs de matériaux magnétiques 53
4.5 Piégeage-Restauration environnementale 53
4.6 Usage médical 53
4.7 Hôtes pour biomolécules 54
Références bibliographiques
55

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
1. Historique et définition des biocapteurs 63
2. Les transducteurs 65
a. Les capteurs thermiques 65
2.2 Les capteurs optoélectroniques 65
2.3 Les capteurs à effet piézoélectrique 66
2.4 Les capteurs électrochimiques 67
2.4.1 Les capteurs ampérométriques 68
2.4.2 La détection potentiométrique 70
2.4.3 Les capteurs conductimétriques 71
3. Les biorécepteurs enzymatiques 72
3.1 Enzymes : les biocatalyseurs 72
3.2 Classification des enzymes 73
3.3 Les techniques d’immobilisation des enzymes 74
3.3.1 L’adsorption 75
3.3.2 L’inclusion ou le piégeage 75
3.3.3 Le couplage covalent 75
3.3.4 La réticulation et la co-réticulation 76
Références bibliographiques
77
DEUXIEME PARTIE : PARTIE EXPERIMENTALE
CHAPITRE 1 : MATERIELS ET METHODES
1. Introduction 82
2. Méthodes de caractérisation 83
2.1 La diffraction des rayons X 83
2.2 Spectroscopie infrarouge à transformée de Fourier 84
2.3 Spectrométrie d’émission plasma (ICP-AES) 84
2.4 Analyse thermogravimétrique et analyse thermique différentielle 85
2.5 Microscopie électronique à balayage 86

2.6 Zétamétrie 86
2.7 Mesures électrochimiques 86
3. Préparation des matériaux adsorbants 88
3.1 Protocole de purification de l’argile 88
3.2 Caractérisation de l’argile cationique 89
3.2.1 Tests de comportement des argiles cationiques 89
3.2.2 Mesure de la CEC 90
3.3 Préparation de l’argile organophile 92
3.4 Synthèse du matériau HDL Ni2AlNO3 93
3.4.1 Par coprécipitation 93
3.4.2 Par voie électrochimique 93
3.5 Synthèse des matériaux Ni2AlNO3 intercalés par le Glyphosate et le
Glufosinate
94
4. Application à l’adsorption 94
4.1 Les adsorbats 94
4.1.1 Le 2,4DCP 94
4.1.2 Le colorant 4GL 95
4.1.3 Les herbicides : Glyphosate et Glufosinate 96
4.4 Cinétique d’adsorption 97
4.2.1 Pour le 2,4DCP 97
4.2.2 Pour le colorant 4GL 98
4.2.3 Pour les herbicides Glyphosate et Glufosinate 98
4.3 Modèles cinétiques d’adsorption 99
4.4 Isotherme d’adsorption 100
4.4.1 Pour le 2,4DCP 100
4.4.2 Pour le colorant 4GL 100
4.4.3 Pour les herbicides Glyphosate et Glufosinate 101
4.5 Modélisation des isothermes d’adsorption 101
4.5.1 Modèle de Freundlich 101
4.5.2 Modèle de Langmuir 102
4.5.3 Modèle de Redlich Peterson 102

4.5.4 Modèle de Tempkin 103
Références bibliographique
104
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA
METHODE ONE STEP ET TWO STEPS
1. Introduction 107
2. Caractérisation des matériaux adsorbants 109
2.1 Purification de la bentonite 109
2.2 Caractérisation Du matériau purifié 109
2.2.1 Mesure de la capacité d’échange cationique 109
2.2.2 Diffraction des rayons X (DRX) et test de comportement des argiles 111
2.3 Caractérisation de l’argile organophile 113
2.3.1 Diffraction des rayons X 113
2.3.2 La spectroscopie Infrarouge 115
3. Adsorption des polluants organiques (2,4DCP et 4GL) sur l’argile
(méthode one step et two steps)
117
3.1 Effet de la concentration du CTAB sur la fixation de 2,4DCP (méthode
one step)
118
3.2 Effet de la concentration du CTAB sur la fixation du colorant 4GL
(méthode one step)
119
3.3 Effet de la concentration du CTAB intercalée sur la fixation du polluant
2,4DCP sur l’argile organophile (méthode two steps)
120
3.4 Effet de la concentration du CTAB intercalée sur la fixation du colorant
4GL sur l’argile organophile (méthode two steps)
122
3.5 Cinétique d’adsorption 123
3.5.1 Pour le polluant 2,4DCP 123
3.5.2 Pour le colorant 4GL 124
3.5.3 Modélisation cinétique 125
3.6 Etude de l’isotherme d’adsorption 132
3.6.1 Modélisation des isothermes d’adsorption 133
4. Caractérisation des matériaux après adsorption 139

4.1 Diffraction des rayons X :DRX 139
4.2 Spectroscopie IR 141
5. Conclusion 147
Références bibliographiques
148
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA
MATRICE NiAlNO3
1. Introduction 155
2. Caractérisation du matériau Ni2AlNO3 158
2.1 Diffraction des rayons X 158
2.2 La spectroscopie infrarouge 159
2.3 Le comportement thermique 160
3. Adsorption de Glyphosate et de Glufosinate sur NiAlNO3 HDL 163
3.1 Cinétique d’adsorption de Gly et de Glu sur NiAlNO3 163
3.1.1 Modélisation de la cinétique d’adsorption de Gly et Glu sur le matériau
NiAlNO3
165
3.2 Isothermes et modèles d’adsorption de Glyphosate et de Glufosinate sur
le matériau NiAlNO3
168
4. Caractérisation des matériaux après adsorption 171
4.1 Diffraction des rayons X (DRX) 171
4.2 Microscopie électronique à Balayage (MEB) 175
4.3 Spectroscopie IR 176
5. Conclusion 177
Références bibliographiques
178
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE
GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION
DE NiAl HDL
1. Introduction 183
2. Résultats et discussions 184
2.1 Caractérisation physique des films de NiAlNO3 préparé par voie 185

électrochimique
2.2 Adsorption de Glyphosate et de Glufosinate sur la matrice NiAlNO3 189
2.3 Détection électrochimique des herbicides 191
3. Conclusion 198
Références bibliographiques
199
CONCLUSION GENERALE 201
LISTE DES FIGURES 206
LISTE DES TABLEAUX 213

INTRODUCTION GENERALE

INTRODUCTION GENERALE
INTRODUCTION GENERALE
La pollution accidentelle ou volontaire des eaux et des sols, par certains produits chimiques
d’origines industrielles (hydrocarbures, phénols, colorants,...) ou agricoles (pesticides,
engrais,…) constitue une source de dégradation de l’environnement et suscite à l’heure actuelle
un intérêt particulier à l’échelle internationale.
Parmi les différents polluants des écosystèmes aquatiques, les phénols chlorés, sont
considérés comme des polluants prioritaires, car ils sont nocifs pour les organismes, même à des
concentrations très basses. L'Union européenne (UE) a classé plusieurs phénols comme
contaminants prioritaires et la directive 80/778/CEE a fixé une concentration maximale de 0,1µg
/L pour les phénols totaux dans l'eau potable. Ces contaminants se trouvent dans l’environnement
à partir des effluents industriels, eaux de ruissellement agricoles, volatilisation des herbicides
types chlorophénoxy acétique acides et par formation spontanée (par chloration de l’eau) par des
produits de désinfections. Le devenir de ces composés phénoliques dans l’environnement et leur
élimination des milieux aqueux est compliqué en raison de leur solubilité élevée et de leur faible
coefficient de partition air/eau.
De leur côté, les eaux résiduaires des industries textiles sont, elles aussi, chargées de
nombreux micropolluants organiques, notamment certains détergents et colorants. Ces derniers
sont souvent utilisés en excès pour améliorer la teinture et de ce fait les eaux de rejet se trouvent
fortement concentrées en colorants dont la faible biodégradabilité rend les traitements
biologiques difficilement applicables.
Les procédés classiques de purification ont montré leurs limites; la coagulation
floculationn-décantation ne permet pas un abattement suffisant de ces composés. Dans le cadre
de l’ozonation, l’ozone réagit parfois avec le colorant pour donner des formes oxydées également
colorées.
2

INTRODUCTION GENERALE
Actuellement, la technique d'adsorption est considérée comme un moyen efficace et un
processus attractif pour le traitement de ces eaux usées. L'utilisation de charbon actif comme
adsorbant est très connue en raison de sa grande surface spécifique, sa microstructure poreuse, sa
grande capacité d'adsorption et sa réactivité très élevée. Cependant, la régénération ou la
réutilisation du charbon actif entraîne une réduction remarquable de son efficacité, et sa
performance devient imprévisible et dépendante de la masse d’adsorbant utilisée. Ceci a conduit
de nombreux laboratoires de différents horizons à s’intéresser à la recherche de nouveaux
adsorbants à base de matériaux naturels. C’est ainsi qu’ont été utilisés des matériaux comme les
zéolithes naturelles ou synthétiques, les tufs, les cendres volcaniques et surtout les argiles dans
certains domaines comme la catalyse et le traitement des effluents.
L'utilisation de la bentonite dans le traitement des eaux usées a connu une attention accrue
et offre actuellement un marché très attractif pour le procédé de dépollution à cause de son
abondance, son faible coût et sa disponibilité dans de nombreux pays. Bien que ce matériau
(Bentonite) a prouvé son efficacité pour l’élimination des colorants basiques, il est inefficace vis
à vis des colorants acides. De même, sa capacité d'adsorption vis-à-vis des molécules organiques
qui sont très solubles, ou polaires, est très faible. Ceci est surement dû à la nature hydrophile de
ces surfaces minérales.
Toutefois, par insertion de tensio-actifs à longue chaîne cationiques, tels que les ions
d'ammonium quaternaire [R4N+], dans l'espace interfoliaire des bentonites, les propriétés
surfaciques de ces matériaux deviennent hydropobes, ce qui engendre des capacités élevées
d'adsorption vis-à-vis des polluants organiques. L’argile organophile a été utilisée pour une
grande variété d’applications environnementales.
L’application traditionnelle de la bentonite organophile dans l'élimination des polluants
organiques des eaux usées comprend deux étapes essentielles : La première est la synthèse de la
bentonite organophile, la seconde est l’utilisation de la bentonite organophile synthétisée
précédemment pour l’élimination des polluants des eaux usées. Le processus de synthèse de
bentonite organophile implique la pulvérisation de la bentonite, l’ajout d’un agent tensioactif,
3

INTRODUCTION GENERALE
une agitation pendant une durée déterminée, ensuite la récupération du matériau par
centrifugation et lavage et enfin séchage et broyage. Un adsorbant optimal pour l'élimination des
composés organiques des eaux usées doit avoir les propriétés suivantes: un faible coût, une
facilité de synthèse, une neutralité vis-à-vis de l'environnement, une grande affinité et une haute
capacité.
Afin de réduire les coûts et simplifier le processus de traitement des eaux usées par la
bentonite organophile, une seule étape incluant à la fois la synthèse de la bentonite organophile et
élimination des polluants organiques a été proposée dans ce travail. En effet, la méthode « one
step » propose le mélange entre l'adsorbat et le tensio-actif cationique en solution, puis l’ajout de
la bentonite sodée. Ainsi, les polluants organiques sont supprimés de l’eau en même temps que la
formation de l’argile organophile. Surtout que la présence d'agents de surface cationiques dans
les eaux usées d’industries de: pigments, colorants, de produits chimiques agricoles et autres peut
participer à la méthode « one step » par simple addition de bentonite ce qui éliminera à la fois les
agents de surface et les polluants organiques. En outre, la bentonite organophile résultante
pourrait être recyclée ou utilisée pour préparer des matériaux hétéro-argileux à structure poreuse.
Une autre variété d’argiles dites basiques a également retenu notre attention. Les
hydroxydes doubles lamellaires (LDH), également connu sous le nom d’hydrotalcite ou encore
argiles anioniques, sont rares dans la nature, par contre la formation dans les sols des phases
hydroxydes mixtes a été démontrée par XAFS sur la surface des phyllosilicates et de gibbsite.
Cependant, ils peuvent être facilement formés dans une large gamme de conditions de pH et
facilement synthétisés à l'échelle du laboratoire. Leur structure se compose d’un empilement de
couches brucite, où une fraction des cations divalents est remplacée par des cations trivalents. Les
couches chargées positivement sont séparés par des anions de compensation de charge et de
molécules d'eau. Ces matériaux possèdent des capacités d'échange anioniques (2-5 mmol/g) plus
importantes que les capacités d’échanges cationiques des minéraux argileux. A cet effet, ils sont
considérés comme des adsorbants potentiels pour l'élimination des substances toxiques ou des
espèces anioniques écologiquement indésirables, tels que CrO42 -, NO3
-, PO43- ainsi que les
phénols, les substances humiques, les agents de surface anioniques et les pesticides. Bien que ces
matériaux jouent un rôle important dans l'échange anionique et l'adsorption de molécules
organiques, il est possible de les utiliser comme matrices potentielles pour l’étude de la
4

INTRODUCTION GENERALE
modélisation de libération contrôlée des pesticides par l'intercalation de molécules de pesticides
entre les feuillets de l’argile.
Les produits organophosphate et organophosphonate (OP) est une famille de pesticides très
utilisée en agriculture. Par exemple le Glyphosate (N-(phosphonométhyl) glycine,Gly) et le
Glufosinate ((DL-homoalanine-4-yl)-methylphosphinic acide, Glu) sont employées comme
herbicides non sélectifs. L'utilisation intensive de ces herbicides suscite des craintes quant à leur
impact sur l'environnement et leurs risques éventuels pour la santé.
Les méthodes d’analyse utilisées pour ce type de molécules chimiques consistent
essentiellement en chromatographie phase liquide, chromatographie phase gazeuse,
électrophorèse capillaire etc…. La plupart de ces méthodes développées pour l'analyse de Glyp et
de Gluf ont besoin d’un traitement préliminaire afin de fonctionnaliser les substrats pour
permettre leur détection. En effet, les techniques de chromatographie en phase liquide utilisant
des détecteurs UV ou fluorimétrique, sont limitées à l'analyse des pesticides portant des groupes
chromophores ce qui n'est pas le cas avec le Glyp et le Gluf. Les capteurs électrochimiques
représentent donc une bonne alternative innovante en termes de détection sur place, de sensibilité,
de réponse rapide, et de taille réduite pour la détermination de ces pesticides.
Par rapport à ce contexte, le manuscrit se présente en deux grandes parties. La première étant
une synthèse bibliographique renfermant trois chapitres essentiels : Le premier chapitre et le
deuxième sont consacrés à une description générale de la structure et des propriétés de surface
des argiles cationiques et anioniques respectivement. Le dernier chapitre de la partie
bibliographie présente un aperçu général sur le mode d’obtention et de fonctionnement des
biocapteurs.
La deuxième partie de ce mémoire est essentiellement pratique, renferme quatre chapitres :
Dans le premier chapitre sont présentés les matériels et méthodes. De plus, les caractéristiques
des appareillages et produits utilisés, les méthodes de caractérisation des matrices solides, de
quantification du soluté et les protocoles expérimentaux utilisés.
5

INTRODUCTION GENERALE
Le deuxième chapitre est consacré à la présentation et discussion des différents résultats
obtenus concernant l'adsorption de deux polluants organiques : le 2,4-DCP et un colorant
industriel 4GL par des complexes de surfactants cationiques et d’argile en utilisant un processus
en une ou deux étapes. Les objectifs de cette étude sont les suivants:
• Déterminer la faisabilité de l'élimination des polluants organiques par la méthode one
step;
• Faire une comparaison entre les deux méthodes : one step et two steps vis-à-vis de deux
polluants organiques : un colorant industriel : 4GL et un polluant chloré : 2,4DCP;
• Etudier l'influence de quelques paramètres physico-chimiques tels que le temps de
contact, la concentration de l'adsorbat et du tensioactif sur l’élimination des polluants.
Le troisième chapitre est concentré à l'adsorption de deux herbicides: le Glyphosate et le
Glufosinate par un matériau LDH type Ni2AlNO3. L’adsorption de la molécule de Glyphosate
sur le matériau MgAlNO3 est déjà connue. En comparaison avec le matériau MgAlNO3, la
structure de Ni2AlNO3 est très intéressante car elle combine de bonnes propriétés électro actives
basé sur le processus redox Ni2+/ Ni3+ pour les procédés électrochimiques avec des propriétés
structurales et texturales pour l'adsorption des molécules chargées négativement. Les résultats
présentés ici sont utiles pour évaluer l'application des matériaux LDH pour le nettoyage
environnemental et à l'assainissement des sols et des eaux contaminés.
Le dernier chapitre de la partie pratique est une continuation du chapitre précédent. En effet,
des électrodes modifiées par électrodéposition d’une matrice HDL type Ni1-xAlx(OH)2NO3x
·nH2O sont appliquées comme biocapteurs pour l’analyse électrochimique de deux herbicides
largement utilisés dans l’agriculture : le Glyphosate et le Glufosinate.
6

PREMIERE PARTIE : ETUDE BIBLIOGRAPHIQUE
7

CHAPITRE 1 : GENERALITES SUR LES ARGILES
CATIONIQUES
8

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
1. Introduction Les argiles sont parmi les minéraux les plus connus à la surface de la terre et sont par
ailleurs indispensables à notre existence. En effet, depuis des millénaires l’homme a utilisé
l’argile comme matière première essentielle à ses besoins quotidiens. Grace à sa malléabilité,
l’argile peut être modelée et façonnée dans toutes les formes possibles et est notamment
utilisée dans la fabrication de la poterie. L’argile a été utilisée par nos ancêtres, et l’est
toujours, pour traiter les cheveux et les peaux de toutes natures. Chargée de toutes les vertus,
l’argile a été utilisée pour soigner. Ainsi par exemple, on a conçu des médicaments à base
d’argile afin de traiter les digestions difficiles.
Les sols contiennent toujours une grande proportion de matériaux argileux. Les
céramiques et la plupart des matériaux de construction sont fais à base d’argile. Les argiles
sont utilisées comme : adsorbants industriels, catalyseurs, échangeurs d’ions et agents de
décoloration. Les utilisations précitées dépendent des propriétés spécifiques que les argiles
possèdent. Leur pouvoir d’adsorption et leur grande affinité pour l’eau sont dû à la petite
taille des particules (2-3μm) ce qui leur confère des surfaces élevées et aussi polaires.
Les argiles sont divisées en deux grandes classes : les cationiques que l’on trouve
abondamment dans la nature et qui feront l’objet de la première partie de cette thèse. Les
argiles anioniques objets de la seconde partie de notre travail, plutôt rares dans la nature sont
simples et relativement peu couteuses à synthétiser au laboratoire.
Le domaine d’application de ces matériaux s’avère très vaste et varié. Ils ont connu
jusqu’à présent un intérêt important dans plusieurs domaines comme catalyseurs et ou
précurseurs catalytiques, échangeurs d’ions, adsorbants et dans diverses applications
médicales et cosmétiques.
9

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
2. Définition Les minéraux argileux sont des silicates hydratés (il s’agit généralement de silicates
d’aluminium, mais parfois de silicates de magnésium), dont la structure feuilletée les a ranger
dans la famille des phyllosilicates. Selon la famille du minéral argileux considérée, les
particules les plus fines peuvent être constituées d’un feuillet ou d’un assemblage de
quelques feuillets, et leur taille est très faible, de l’ordre de 2 à 5 μm. Ces dimensions sont
caractéristiques des particules argileuses et ne se retrouvent pas dans d’autres minéraux.
3. Microstructure des argiles Les argiles proviennent de l’altération et de la dégradation des roches : altération
physique sous l’effet des variations de température, et surtout altération chimique au contact
de l’eau qui permet la dégradation en particules très fines. Les conditions dans lesquelles
cette dégradation a eu lieu, ainsi que l’état d’avancement de cette dégradation peuvent
expliquer la grande diversité des argiles (Jackson & Sherman 1953, cités par Grunberger
1995). De par leur origine détritique et leur nature granulaire, la structure des sédiments
argileux est complexe ; la compréhension des mécanismes de déformation de ces matériaux,
dans lesquels la chimie de l’eau tient une place prépondérante, passe nécessairement par la
connaissance précise de la microstructure.
Avant d’entrer dans les détails de la minéralogie des argiles, il nous semble utile de
rappeler la terminologie associée pour éviter toute confusion : un terme donné est parfois
utilisé pour désigner plusieurs niveaux structuraux différents, et deux termes différents sont
parfois employés pour désigner le même niveau.
3.1 Structure de base
La cellule de base des minéraux argileux est appelée cristallite. Elle est constituée d’un
feuillet et d’un interfeuillet appelé aussi espace interfoliaire.
Chaque feuillet est lui-même formé de la superposition de deux ou trois couches
cristallisées. L’interfeuillet est constitué d’eau assurant une liaison électrochimique entre les
feuillets. Il existe différents types de liaisons interfeuillets, liées notamment à des
phénomènes de substitutions isomorphiques à la surface des cristallites.
10
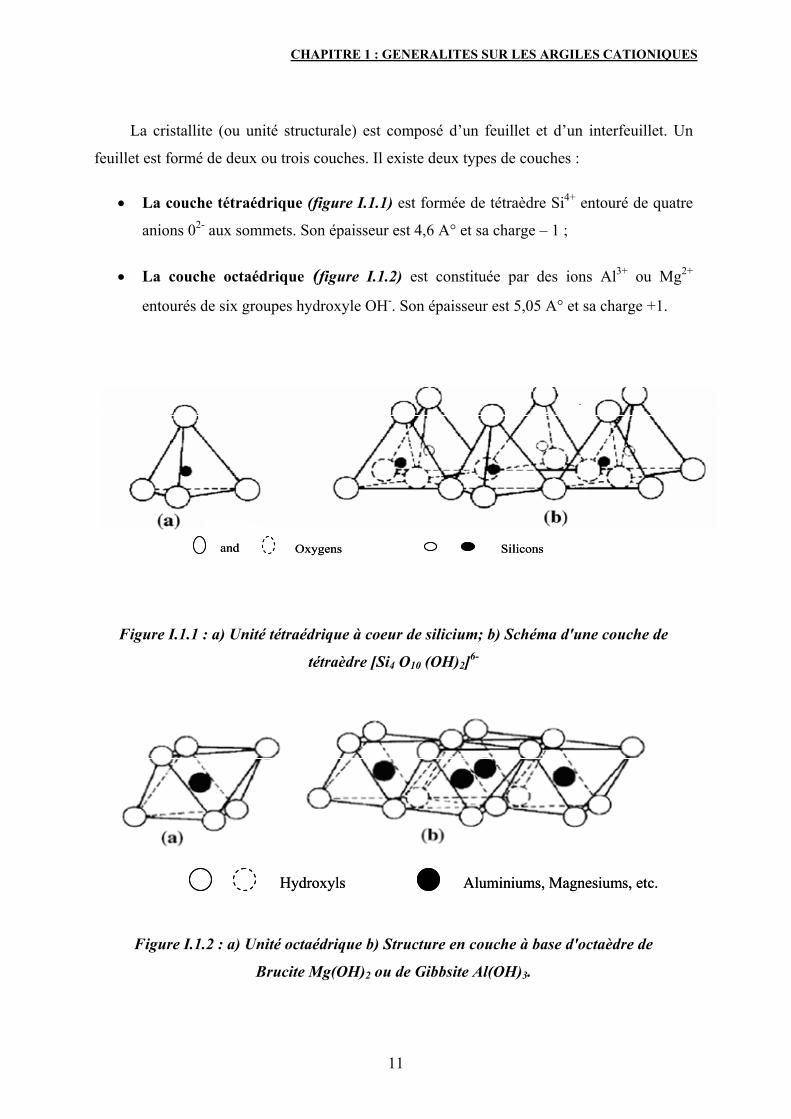
CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
La cristallite (ou unité structurale) est composé d’un feuillet et d’un interfeuillet. Un
feuillet est formé de deux ou trois couches. Il existe deux types de couches :
• La couche tétraédrique (figure I.1.1) est formée de tétraèdre Si4+ entouré de quatre
anions 02- aux sommets. Son épaisseur est 4,6 A° et sa charge – 1 ;
• La couche octaédrique (figure I.1.2) est constituée par des ions Al3+ ou Mg2+
entourés de six groupes hydroxyle OH-. Son épaisseur est 5,05 A° et sa charge +1.
and Oxygens Siliconsand Oxygens Silicons
Figure I.1.1 : a) Unité tétraédrique à coeur de silicium; b) Schéma d'une couche de
tétraèdre [Si4 O10 (OH)2]6-
Hydroxyls Aluminiums, Magnesiums, etc.Hydroxyls Aluminiums, Magnesiums, etc.
Figure I.1.2 : a) Unité octaédrique b) Structure en couche à base d'octaèdre de
Brucite Mg(OH)2 ou de Gibbsite Al(OH)3.
11

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Suivant l’ordre d’empilement des couches octaédriques (O) et tétraédriques (T), les
minéraux argileux sont classés en deux types :
• Le type de feuillets TO ou 1:1 formé d’une couche tétraédrique T et d’une couche
octaédrique O (figure I.1.3). Ces argiles, dont l’unité structurale de base est
dissymétrique, sont représentées par le groupe des kaolinites et des serpentites, ces
dernières étant beaucoup plus rares ;
• Le type de feuillets TOT ou 2 :1 formé d’une couche octaédrique O entourée de
deux couches tétraédriques T. Ces argiles, qui présentent une unité structurale de base
symétrique, comportent de nombreux groupes (illites, smectites, interstratifiés,
chlorites, vermiculites) dont la structure et les propriétés sont très variables.
Dans chacun des deux feuillets décrits précédemment, le cation peut être remplacé par
un cation de taille voisine (pour « tenir » dans le site octaédrique ou tétraédrique), mais pas
nécessairement de même valence. On parle de substitution isomorphe car les dimensions du
feuillet restent quasi inchangées. Ces substitutions entraînent alors un excès de charges
négatives à la surface des feuillets. Cette électronégativité des feuillets est une des
caractéristiques fondamentales des argiles. L’électroneutralité est obtenue par adsorption de
cations compensateurs à la surfaces des feuillets : cations (K+, Na+, Mg2+, Ca2+, Fe2+...)
provenant du milieu environnant.
Par ailleurs, les bords des cristallites possèdent aussi des charges localisées car ils
correspondent à des ruptures de liaisons. La charge des tranches de cristallites dépend alors
du pH (Grunberger 1995) : elle est négative en milieu basique, et positive en milieu acide
(en raison de la fixation de protons H+ sur des ions 02- présents sur ces bords). Nous verrons
plus loin que cela a une incidence sur l’assemblage des cristallites.
12

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Figure I.1.3 : Assemblage d’une couche octaédrique et d’une couche tétraédrique
pour une argile TO (1:1).
4. Classification des argiles Les différents groupes de minéraux argileux se différencient par l'arrangement des
couches tétraédriques et octaédriques.
On distingue 4 groupes (Caillère et al., 1982 ) : 4.1 Minéraux à 7 Å: (kaolinite, Halloysite, Dombasite, ….)
Le feuillet est constitué d'une couche tétraédrique et d’une couche octaédrique. Il est
qualifié de T:O ou de type 1:1. Son épaisseur est d’environ 7 Å.
4.2 Minéraux à 10 Å: (Pyrophyllite, illite, Montmorillonite, Saponite,…)
Le feuillet est constitué de deux couches tétraédriques et d’une couche octaédrique. Il
est qualifié de T:O:T ou de type 2:1. Son épaisseur est d’environ 10 Å.
4.3 Minéraux à 14 Å: (Chlorites)
Le feuillet est constitué de l'alternance de feuillets T:O:T et de couches octaédriques
interfoliaires.
4.4 Minéraux interstratifiés:
L’épaisseur du feuillet est variable. Ces minéraux résultent du mélange régulier ou
irrégulier d’argiles appartenant aux groupes ci-dessus (figure I.1.4).
13

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Figure I.1-4 : Représentation schématique de quelques groupes de minéraux argileux
TO (1:1) et TOT (2:1).
La structure de la couche octaédrique des smectites, ainsi que la localisation des
substitutions, ont conduit à une classification de ces minéraux (Schoonheydt, 1995). Ainsi, il
existe deux grandes catégories de smectites. Les premières sont dites dioctaédriques du fait
de l’occupation de seulement deux sites octaédriques sur trois. Parmi elles, certaines
présentent majoritairement des substitutions dans la couche octaédrique (montmorillonite),
alors que d’autres sont principalement substituées dans les couches tétraédriques (beidellite).
Typiquement l'ion en site octaédrique est alors l'aluminium, qui est remplacé par du
magnésium ou du fer, alors que le silicium tétraédrique est remplacé par de l’aluminium.
Il existe un troisième type de smectite dioctaédrique, possédant essentiellement du fer
au degré d’oxydation III, dans sa couche octaédrique, remplacé par de l’aluminium ou du
magnésium (nontronite). Les autres smectites sont trioctaédriques, car tous les sites
octaédriques sont occupés. L’ion en site octaédrique est en général le magnésium. Parmi
elles, certaines sont caractérisées par des substitutions du magnésium par le lithium dans la
couche octaédrique (hectorite), alors que pour d’autres, les substitutions ont principalement
lieu dans la couche tétraédrique, où le silicium est remplacé par de l’aluminium (saponite).
Dans les illites et la pyrophyllite, la charge provient principalement de substitutions
dans les couches silicatées, donc plus proches de la surface, ce qui confère alors à ces argiles
des propriétés d’adsorption d’ions relativement différentes de celles des smectites,
notamment au niveau de la spécificité des sites, une représentation comparative des smectites
14

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
et de l’illite/pyrophyllite est donnée sur la figure I.1.5, montrant l’agencement des feuillets
ainsi que la localisation des substitution.
Figure I.1-5. Représentation schématique de la structure feuilletée des smectites et des
illite/pyrophyllite.
5. Origine de la Bentonite L’altération et la transformation hydrothermale de cendres des tufs volcaniques riches
en verre entraînent la néoformation des minéraux argileux, qui font partie principalement du
groupe des smectites. Les roches argileuses ainsi formées portent le nom de bentonite,
d’après le gisement situé près de Fort Benton (Wyoming, Etats-Unis). Elle contient plus de
75 % de montmorillonite ; cette dernière fut découverte pour la première fois en 1847 près de
Montmorillon, dans le département de la Vienne (France). Les bentonites se caractérisent par
une capacité élevée d’adsorption, d’échange ionique et de gonflement, ainsi que par des
propriétés rhéologiques particulières (thixotropie). Elles ont de ce fait de larges applications,
toujours plus nombreuses et dans différents domaines (forage, fonderie, céramique, peinture,
pharmacie, terres décolorantes,…, etc). La majeure partie de la bentonite exploitée dans le
monde est utilisée comme liant du sable de moulage, dans l’industrie de la fonderie et aussi
pour épaissir les fluides de forage. Pour de nombreuses applications techniques, les
bentonites brutes doivent être soumises à une préparation adaptée aux exigences de leur
utilisation (activation). Ainsi, lors de l’activation alcaline, les bentonites calciques (les plus
fréquentes) sont transformées par traitement avec de la soude en bentonites de sodium, qui se
caractérisent notamment par une capacité de gonflement plus élevée.
15

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
L’activation avec des acides comme l’acide chlorhydrique augmente la porosité par
dissolution périphérique des smectites. Il en résulte un produit de haute capacité
d’adsorption.
En Algérie, les gisements de bentonite les plus importants économiquement se trouvent dans
l’oranie (ouest algérien). On relève en particulier la carrière de Maghnia (Hammam
Boughrara) dont les réserves sont estimées à un million de tonnes et de celle de Mostaganem
(M’zila) avec des réserves de deux millions de tonnes (Abdelouhab et al., 1988).
6. Caractéristiques physiques des argiles lamellaires Les propriétés des minéraux sont plus ou moins reliées à leur structure. Dans le cas des
minéraux argileux, la plupart d’entre eux présentent des similitudes de structure cristalline
(T-O, T-O-T ou T-O-T-O). Toutefois, pour une structure cristallographique identique, la
substitution isomorphe au sein du feuillet peut conduire à une variation considérable des
compositions chimiques et des propriétés physiques. Cette substitution donne lieu à
l’existence de charges (souvent négatives) à leurs surfaces, compensées par la présence de
cations compensateurs. La localisation des cations, le type et le nombre de cations sont les
paramètres principaux déterminant la différence des propriétés physiques et chimique,
notamment la capacité de gonfler en présence d’eau.
6.1 Charge des surfaces argileuses
La plupart des argiles, notamment celles du groupe smectites, se caractérisent
principalement par une surface électrique non neutre, qui est due à la fois aux substitutions
isomorphiques et à l’environnement, conduisant à deux contributions différentes (Eslinger et
Peaver, 1988):
6.1.1 Charge permanente
La charge permanente est principalement négative et située à la surface. Elle provient
des substitutions isomorphiques au sein du feuillet, résultant du remplacement des cations
métalliques par ceux d’un autre métal, de valence plus faible. Il conduit donc à un déficit de
charge en surface des feuillets, compensé par la présence des cations compensateurs tels que
Li+, Na+, Ca2+, K+ ou Mg2+.
16

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
6.1.2 Charge variable
Elle peut être positive ou négative et est située aux bords des feuillets. Elle n’apparaît
qu’en suspension. Il s’agit donc ici d’une charge dépendant du pH de la solution. En milieu
acide, l’espèce positivement chargée est prédominante, alors qu’en milieu basique, c’est
l’espèce négativement chargée qui est majoritaire.
6.2 Capacité d’échange cationique
La capacité d’échange cationique (CEC) est une mesure de la capacité d’une argile à
échanger des cations compensateurs (Bergaya et Vayer, 1997). Elle dépend à la fois de la
charge permanente et de la charge variable. Elle mesure, en effet, le nombre de cations
monovalents qu’il est possible de substituer aux cations compensateurs (Li+, Na+, Ca2+, K+
ou Mg2+) afin de compenser la charge électrique de 100 g d’argile calcinée, à pH 7. Elle
s’exprime en milliéquivalents pour 100 grammes d’argile (meq/100g). En général, pour les
argiles présentant des cations échangeables comme par exemple la Na+-montmorillonite, la
CEC peut être considérée comme équivalente à la charge totale présente sur la surface. Le
Tableau 1.1 donne les valeurs de CEC pour les principales familles argileuses (Caillère et
al.,1982).
La capacité d’échange associée peut être calculée directement si la composition des
feuillets est parfaitement connue. Les espaces qui se trouvent entre les feuillets peuvent être
vides ou remplis :
• Ils sont vides lorsque les différents feuillets sont neutres et liés entre eux par des
liaisons hydrogène dans le cas des espèces 1:1, ou par des liaisons de Van der Wals
dans le cas des minéraux 2:1 (Pédro ,1994).
• Ils sont occupés par des cations dès que les feuillets de l’édifice présentent un déficit
de charge à la suite de substitutions isomorphiques. Ces cations rétablissent l’électro-
neutralité du système et en même temps assurent la liaison entre les feuillets
adjacents, qui est ici de nature ionique. Ces cations peuvent être soit «secs» soit
hydratés. Les cations les plus fréquents sont Ca2+
, Mg2+
, K+, Na
+, Li
+.
17

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Tableau I.1.1 : Capacité d’échange cationique des principales familles argileuses (Caillère
et al.,1982)
Type d’argile Capacité d’échange cationique
(meq/100g)
Smectites 80-150
Vermiculite 120-200
illite 10-40
kaolinite 1-10
Chlorite < 10
Plusieurs méthodes pour déterminer la CEC ont été élaborées. Au début, la
détermination du CEC des argiles a été effectuée en saturant l'argile par un cation puis en
éliminons l'excès du sel utilisé pour le traitement. Ce cation est échangé par plusieurs cycles
d'échange/lavage par un autre cation (Mehlich, 1948). Les solutions recueillies sont
employées pour la détermination de la quantité du cation remplacé. Une revue rapide de la
littérature montre que ce choix est très vaste et qu’un grand nombre de cations ont déjà été
proposé pour servir d’indicateur. Ces cations ont été utilisés sous forme non complexée tels
que le calcium, le sodium, le potassium, le magnésium, le baryum, l’ammonium (Ravina et
Gurovich, 1977) mais également sous forme de complexes organiques (Pleysier et Cremers,
1975), tels que l’acétate d’ammonium, (Hendershot et Duquette, 1986), l’argent thiourée, la
cobaltihexamine (Morel, 1957; Mantin et Glaeser, 1960, Orsini et Remy,1976 ), ou encore
l’éthylènediamine de cuivre (Bergaya et Vayer, 1997), ou des ions alkylamonium (Senkayi et
al., 1985) ou cétylpyridinium (Kloppenburg, 1997).
Une autre méthode a été engendrée en saturant l'argile avec les ions NH4
+, la quantité
des ions d'ammonium adsorbée est déterminée par la méthode de Kjeldahl (Hofmann, 1939).
D'autres méthodes ont été proposées par l'utilisation de surfactants cationiques (Kloppenburg
,1997 ; Janek et Lagaly,2003). Le but était de déterminer si la quantité adsorbée pouvait être
corrélée à la valeur du CEC ou de la surface spécifique. La problématique générale de
l'utilisation des surfactants est que l'excès sera adsorbé sur l'argile, se qui nécessite alors la
détermination du point d'équilibre.
18

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
6.3 Propriété de gonflement
La propriété de gonfler en présence d’eau varie d’une famille argileuse à une autre. Les
minéraux argileux T-O n’ont pas normalement de charges présentes sur la surface. La couche
tétraédrique est totalement occupée par Si4+ et la couche octaédrique, quant à elle, est
totalement occupée par Al3+ ou Mg2+. S’il y a une substitution au sein d’une couche, il y aura
toujours une compensation par la substitution dans d’autres couches. Ainsi, la neutralité est
toujours maintenue. Cette propriété particulière rend les argiles T-O stable et leur structure,
notamment la distance entre les feuillets, n’est pas affectée par la présence d’eau. Elles n’ont
aucune capacité à gonfler et c’est précisément la raison pour laquelle on ne peut pas obtenir
de nano-composites à partir de cette famille d’argile, car les feuillets ne sont pas séparables.
Dans le cas des minéraux argileux T-O-T, il existe deux groupes présentant les
propriétés de gonflement, le groupe des vermiculites et celui des smectites. Leurs propriétés
de gonflement sont principalement gouvernées par les paramètres suivants (Moore et
Reynolds, 1997):
• la nature des cations compensateurs. Plus les cations compensateurs sont petits et peu
chargés, plus le gonflement est important. Le gonflement croît dans l’ordre des
cations suivant : K+ < Fe2+ < Ca2+ < Na+ < Li+.
• la localisation des substitutions isomorphiques au sein des feuillets. Les feuillets à
substitution octaédrique, comme les smectites par exemple, ont une charge nettement
inférieure par rapport à la substitution tétraédrique, grâce à l’effet d’écran de la
couche tétraédrique. Ceci réduit les interactions entre les feuillets, donc l’eau peut
facilement s’immiscer dans l’espace de ces derniers. Les charges d’origine
tétraédrique comme les vermiculites sont plus fortes en surface car la substitution se
localise près de la surface. Les interactions entre les feuillets sont donc plus fortes et
gênent la pénétration des molécules d’eau.
La propriété de gonflement a un lien direct avec la CEC. Le gonflement passe tout
d’abord par la présence des cations compensateurs, donc une valeur de CEC non nulle. Une
valeur de CEC trop importante se traduit par une forte force d’attraction électrostatique qui
limite le gonflement. C’est pour cette raison que les smectites, ayant une CEC modérée, sont
les argiles possédant les meilleures propriétés de gonflement.
6.3.1 Phyllosilicates non-expansibles
19

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Les feuillets d’illite et de la pyrophyllite, où la charge est compensée par du potassium,
constituent un exemple de ce type d’arrangement. Nous avons affaire à des minéraux à
espace interfoliaire anhydre et présentant des espacements constants, voisins de l’épaisseur
du feuillet (~10). Les cations interfoliaires ne sont pas en général échangeables par des
cations organiques et minéraux, se trouvant dans des solutions mises au contact du
phyllosilicate.
La présence de potassium dans l’espace interfeuillet, liant très étroitement les feuillets
entre eux et empêchant ainsi le minéral de se gonfler en présence d’eau. Le potassium
constitue par conséquent un cation difficilement échangeable.
6.3.2 Phyllosilicates expansibles
Dans ce cas les cations compensateurs sont hydratés et la présence d’un film d’eau
entre les feuillets concourt à leur écartement. On parle alors de minéraux expansibles. La
propriété essentielle de ces minéraux est de se disperser au contact de l’eau pour former des
suspensions plus ou moins stables. Les cations interfoliaires sont en général échangeables par
des cations organiques et minéraux, se trouvant dans des solutions mises au contact du
phyllosilicate (figure I.1.6).
Figure I.1.6 : Complexe de sphère externe. Le cation reste totalement hydraté. C'est le cas
des cations possédant une énergie d'hydratation élevée, comme le sodium. (Sposito, 1989)
7. Surface spécifique
Par définition, la surface spécifique d’un adsorbant est une surface par unité de masse.
Elle est généralement exprimée en m2.g
-1. Son estimation est conventionnellement fondée sur
des mesures de la quantité adsorbée qads
de l’adsorbant en question, correspondant à un
adsorbat donné ; la molécule adsorbée doit avoir une surface connue et acceptable. Il suffit à
20

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
cet effet, de déterminer la valeur de la capacité de la monocouche à partir de l’isotherme
d’adsorption.
Il est nécessaire de distinguer la surface interne et la surface externe d’une argile. La
première est la surface microporeuse Sμ
représentée par les parois des micropores ; elle peut
atteindre plusieurs mètres carrés par gramme. La deuxième est la surface non-microporeuse
ou la surface externe Sext
qui comprend les parois des mesopores et des macropores .
La Figure I.1.7 donne les valeurs caractéristiques des surfaces des grandes familles
d’argiles. La surface totale comprend la surface externe, comprise entre les particules
argileuses, et la surface interne, correspondant à l’espace interfoliaire (Eslinger et Peaver,
1988).
On remarque que la surface spécifique des argiles dépend essentiellement de la surface
interne. Pour les argiles gonflantes, la surface spécifique est largement supérieure à celle des
argiles non gonflantes.
Surface spécifique approximative de différentes argiles Surface spécifique (m²/g)
Type d’argile Interne Externe Totale Smectite 750 50 800
Vermiculite 750 < 1 750 Chlorite 0 15 15 Kaolinite 0 15 15
Illite 0 25 30
Figure I.1.7 : Valeurs de surface spécifique des particules argileuses et schéma
représentant la surface interne et la surface externe des particules d’argile (Eslinger et
Peaver, 1988).
21

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
8. Les argiles modifiées 8.1 Généralités
La modification des argiles réside dans l’intercalation entre leurs feuillets de gros
polycations métalliques simples ou mixtes dans le but d'obtenir des matériaux microporeux, à
structure rigide, avec un grand espacement interfoliaire.
De nombreux travaux sur les argiles modifiées rapportent des informations sur les
différentes méthodes de synthèse et de caractérisation texturales (Stackmeyer, 1991; Hsu et
al., 2000; Boyd et al., 1988 ; Hermosin et Cornejo, 1992;1993 ; Bouberka et al., 2008,
Khenifi et al.,2007 ;2009).
Dans le domaine de l’adsorption et malgré leurs instabilités thermiques, les complexes
organoargileux, hydrophobes et organophiles, ont été largement utilisés dans la dépollution
des eaux contaminées par certains micropolluants organiques tels que des phénols, des
pesticides, des colorants, …
Nous nous proposons de présenter en revue et pour chaque famille d’argiles modifiées,
une synthèse bibliographique des différents travaux effectués dans ces domaines et publiés
par ordre chronologique.
On peut classer les argiles modifiées en trois grandes catégories : les complexes
organoargileux, les inorgano-argileux et les organo-inorgano-argileux.
8.2 Les argiles organophiles
La majorité des polymères étant organophiles, des études visant à améliorer la
compatibilité entre la matrice et la charge ont donc été menées. Le procédé, appelé
"organomodification", consiste à modifier les montmorillonites de façon à abaisser leur
énergie de surface et donc à améliorer la mouillabilité. Le matériau composite Argile-
tensioactif est un hybride inorganique-Organique synthétisé par échanges de sodium ou de
calcium résidants dans l’espace interfoliaire de l’argile de base par des tensioactifs
cationiques. Durant la synthèse du matériau hybride argile-surfactant, les cations
échangeables sont remplacés par des agents tensioactifs cationiques. En raison de ses
propriétés variables, le matériau composite argile-tensioactif est largement utilisé dans
diverses industries comme catalyseurs ou supports de catalyseurs (Meier et al., 2001; Jiang
22

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
et al., 2001), adsorbants pour le traitement de l'eau contaminée (Stackmeyer, 1991; Hsu et
al., 2000; Boyd et al., 1988 ; Hermosin et Cornejo, 1992;1993 ; Bouberka et al., 2008,
Khenifi et al.,2007 ;2009 ), nanocomposites (Wu et Lerner, 1993), pour matériaux poreux
(Dai et al.,1999) et pour le renforcement des composites de polymères (Wang et Pinnavaia,
1998, Lan et Pinnavaia, 1994 ;Vaia et al., 1993).
8.2.1 Tensioactif ou agent d’interface
Les tensioactifs sont des molécules amphiphiles. Ils comportent deux parties de
polarités différentes. En effet, ils possèdent une partie hydrophile, la tête polaire, qui est
soluble dans l’eau et les milieux polaires, et une partie hydrophobe, constituée par une ou
plusieurs chaînes hydrocarbonées, qui est insoluble dans l’eau. Pour les tensioactifs
commerciaux, la partie apolaire est souvent constituée par des mélanges de chaînes
hydrocarbonées de différentes longueurs. Ils sont issus de trois sources principales : la
pétrochimie, les huiles végétales et les graisses animales. Ces deux dernières donnent accès à
des mélanges de chaînes hydrocarbonées aliphatiques linéaires, de 8 à 18 atomes de carbone.
Les groupements hydrocarbonés d’origine pétrochimique peuvent comporter des variations
de longueur de chaîne ou de ramifications en fonction du procédé de synthèse.
Pour la plupart des argiles organophiles commercialisées, on utilise fréquemment des
sels d’ammoniums quaternaires. Leurs structures chimiques se distinguent par le nombre de
chaînes hydrocarbonées (de 1 à 4 chaînes), et la longueur de ces dernières (variant entre 8 et
18 atomes de carbone).
La Figure I.1.8 présente un exemple de tensioactifs souvent utilisés pour modifier les
argiles.
23

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Figure I.1.8: Représentation schématique de quelques exemples de molécules de
tensioactifs.
(a) Alkylammonium (b) Alkyltriméthylammonium (c) Dialkyldiméthylammonium
(d) Alkylbis(2-hydroxyéthyl)méthylammonium (e) Alkylbenzyldiméthylammonium
( ) représente la chaîne hydrocarbonée aliphatique dont la longueur est variable :
[(CH2)n-CH3] pour (a) et [CH2-(CH2)n-CH3] pour ((b)-(e))
8.2.2 Caractéristiques structurelles des argiles organophiles
Les propriétés du matériau composite argile-tensioactif est étroitement lié à
l’environnement moléculaire des cations organiques présent dans l’espace interfoliaire des
argiles. La technique la plus utilisée pour caractériser ces matériaux composites est la
Diffraction des rayons X(DRX), qui donne l'espacement « d » basale indirectement liées au
nombre et à l'orientation moyenne des couches intercalées par rapport au plan de l'argile
(Lagaly, 1986). En se basant sur ce paramètre, divers modèles schématisant la disposition de
l'agent tensioactif ont été proposés; une monocouche latérale, une bicouche latérale, une
monocouche paraffine, une bicouche paraffine, et une pseudo triple couche (Lagaly, 1981;
Tamura, et Nakazawa, 1996).
Nous avons vu dans le paragraphe précédent que la capacité d’échange cationique
(CEC) de l’argile organophile pouvait être supérieure à la CEC de la montmorillonite-Na.
Une partie des ions ammonium est alors intercalée entre les feuillets inorganiques tandis
qu’une autre sera présente autour de ces feuillets (Kadar, 2006). Plusieurs études ont
déterminé avec précision la localisation des surfactants dans les montmorillonites
organomodifiées (Xie, 2001; Xi, 2004; Xi, 2005). Les ions s’intercalent préférentiellement
24

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
dans la région interfeuillets par échange cationique ; ils sont liés aux sites présents à la
surface du feuillet inorganique par les interactions électrostatiques. Lorsque la CEC excède la
CEC de l’argile non modifiée, les molécules de surfactants en excès se placent dans l’espace
interfeuillets et interagissent avec les cations adsorbés à la surface des feuillets inorganiques
par des forces de Van der Waals (Xi, 2004).
Par ailleurs, plusieurs publications (Lagaly, 1986; Vaia, 1994; Bonczek, 2002; Xi,
2004; Kadar, 2006) font état de l’organisation des organomodifiants intercalés. Selon la
densité, la longueur des chaînes et la température, les chaînes alkyles s’orientent
parallèlement ou s’écartent de la surface du feuillet pour former des monocouches ou des
bicouches. Il existe également une configuration "hybride" i.e. combinant les deux types de
configuration.
Selon la quantité d’organomodifiants, la longueur et la configuration de(s) chaîne(s)
alkyle(s) substituée(s), la distance interfeuillets de la montmorillonite peut augmenter (Vaia,
1994; Burgentzlé, 2004). Globalement, cet espace, initialement d’environ 10 Å dans le cas
de la montmorillonite naturelle MMT-Na, peut augmenter jusqu’à atteindre 30 Å dans le cas
de la montmorillonite modifiée par des ammoniums quaternaires.
8.3 Famille des complexes inorgano-argileux
Pour s’affranchir de l’inconvénient que représente la faible stabilité thermique des
organo-argileux, l’idée fut de synthétiser des structures pseudo chlorites qui sont des argiles
modifiées par des composés inorganiques. Ceci est réalisé à partir d’hydroxydes de cations
facilement hydroxylables tels que l’aluminium (Sapag et Mendioroz, 2001, Kloprogge et al.,
2002) et étendu ensuite au Zr (Gil et Vicente., 2000), Ti (Sychev et al., 2000), Fe (Balci et
Gokcay, 2002), et Si (Fetter et al., 1995), Ti, Ga, Nb, V et à n’importe quel oxyde métallique
en solution, qui forme une espèce polynucléaire par hydrolyse (Lahodny et Khalaf , 1994).
Ce type d’argile modifiée a été introduit par Brindley et Sempels (Brindley et Semples,
1977) en 1977 en utilisant une solution d’hydroxyde d’aluminium. Lahav et al. (Lahav et al.,
1978) et Shabatai (Shabtai, 1980) se sont intéressés à la modification des montmorillonites
par l’insertion, entre les feuillets, des polycations [Al13
O4(OH)
24(H
2O)
12]7+
.
Actuellement, il est connu que la nature du sel précurseur est primordiale dans la
modification des argiles, et le traitement est obtenu généralement par hydrolyse d’un sel
25

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
métallique par une base forte (ou un acide fort) selon la nature du métal choisi. Après
calcination, à différentes températures, les polycations insérés se transforment en grappes
d’oxydes métalliques rigides et résistants, confèrent à ces solides une stabilité thermique
élevée, et une surface microporeuse développée (Schoonheydt et al.,1999).
De leur coté, Lahav et al. (Lahav et al., 1978) ont pu, grâce à l’analyse par DRX,
confirmer l’intercalation des polycations d’aluminium avec des espacements basaux de
l’ordre de 18 Å à température ambiante et qui se transforment en oxyde Al2O
3 après
calcination à 500 °C.
Plusieurs études ont rapporté l’utilisation des argiles inorganiques préparées par
intercalation de polycations métalliques tel que Al13 pour l’adsorption de polluants
organiques (Bouras, 1999; Bouberka et al., 2006). Bouras et al. ont étudié l’adsorption d’un
colorant anionique sur une argile intercalée par le Ti et co- adsorbée par un tensioactif. Les
résultats de cette étude ont aboutit à la conclusion suivante : une modification appropriée de
l’argile conduit à des résultats d’adsorption prometteurs (Bouras et al., 2002). L’affinité de
l’argile vis-à-vis des polluants organiques semble augmenter avec : augmentation de la
charge des cations interfoliaires, augmentation de la surface spécifique et diminution de la
densité de charge surfacique. Les argiles intercalées et pontées possèdent la majorité de ces
caractéristiques. Par conséquence ce type d’argile montre une grande affinité vis-à-vis des
polluants organiques comparé au matériau de base. Des argiles intercalées et pontées à Al,
Ti, et Zr ont été utilisées récemment pour la fixation des polluants organiques anioniques.
Matthes et Kahr (Matthes et Kahr, 2000) ont trouvé que la capacité d’adsorption maximale
de l’atrazine sur une argile intercalée au Zr est presque égale à celle du charbon actif GAC.
L’argile intercalée au chrome montre une surface spécifique importante comparée aux autres
matériaux. A cet effet Bouberka et al. (Bouberka et al., 2006) ont testé l’efficacité de ce
matériau vis-à-vis d’un colorant industriel 4GL et ont trouvé des résultats intéressants.
8.4 Famille des Complexes Inorgano-Organo-Argileux
Les premiers travaux publiés sur l’application des complexes organo-inorgano-argileux
ou argiles pontées mixtes ont montré le caractère très hydrophobe de cette nouvelle
génération de matériaux adsorbants. C’est ainsi que Zielke et al. (1988) ; Michot et al.
(1991, 1992, 1993) ;Srinivasan et Fogler (1990 a, b), Montarges et al. (1998) ont consacré
26

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
leurs premières recherches essentiellement sur les tests d'adsorption de certains composés
phénolés sur des smectites pontées à l’aluminium et coadsorbées par des molécules
tensioactives cationiques. A travers les différents résultats obtenus, ils ont montré que la co-
adsorption de toutes ces argiles pontées avec des molécules à chaînes longues augmentent
l'hydrophobie de ces matériaux au même titre que les charbons actifs utilisés et que plus la
chaîne hydrocarbonée est longue et plus l’hydrophobie et l’organophilie sont élevées.
Shu et al. (1997) ont utilisé trois matériaux adsorbants différents dans l’adsorption de
certains chlorophénols et nitrophénols et ont constaté que la montmorillonite pontée au
zirconium et co-adsorbée par un tensioactif non ionique (Tergitol 15S-5) adsorbe
d’importantes quantités de ces micropolluants organiques en comparaison avec une silicalite
et une zéolite béta.
Pour leur part, Jiang et al. (2002) ont montré qu’une montmorillonite (Aldrich)
intercalée par des polycations d’aluminium et modifiée par co-adsorption avec de
l’hexadécyltriméthyl ammonium HDTMA adsorbe beaucoup plus de phénol que les autres
matrices organo- et inorganomontmorillonites préparée
Bouras et al. (Bouras et al., 1998 et 1999a, 2001) ont préparé plusieurs matrices à
base de piliers d’Al(III), Fe(III), Ti(IV) et du tensioactif (CTAB ou CTAC) qu’ils ont testé
pour l’adsorption de composés phénoliques . Bouberka et al., ont étudié l’adsorption d’un
colorant industriel 4GL sur une argile intercalée par un polycation d’aluminium ou de
chrome et co adsorbé par un tensioactif cationique (Bouberka et al., 2009).
27

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
Références bibliographiques
• Abdelouahab C., Ait Amar H., Obretenov T. Z. & Gaid A. (1988), analyses, 16,
292-299.
• Balci S. and Gokcay E. (2002), Mater. Chem. And Phys., 76, 46-51.
• Bergaya F. and Vayer M. (1997), Appl. Clay Sci, 12, 275-280.
• Bonczek J. L., Harris W. G., Nkedi-Kizza P. (2002), Clays Clay Miner., 50(1), 11-
17.
• Bouberka Z., Khenifi A., Benderdouche N. and Derriche Z. (2006), J. Hazard.
Mater., B133, 154–161.
• Bouberka Z., Khenifi A., Sekrane F., Bettahar N. and Derriche Z. (2008), J.
Chem. Eng., 136, 295–305.
• Bouberka Z., Khenifi A., Ait Mahamed H., Haddou B., Belkaid N., Bettahar N.
and Derriche Z. (2009) J. Hazard. Mater., 162, 378–385.
• Bouras O., Houari, M. & Khalaf H. (2001), Environ. Technol., 22, 69-74.
• Bouras O., Khalaf H., Houari M. & Moustiri F. (1999 B), Jordan International
Chemical Engineering Conference Iii, Amman (Jordan), 27-29 Sept. 1999, 225-241.
• Bouras, O., Houari, M. & Khalaf H. (1999 A), Toxicol. Environ. Chem., 70, 221-
227.
• Bouras O., Chami T., Houari M., Khalaf H., Bollinger J.C., Baudu M. (2002),
Environ. Technol. 23, 405-411.
• Boyd S.A., Mortland M.M. and Chiou C.T. (1988), Soil Sci. Soc. Am. J. 52, 652–
657.
• Brindley G. W. and Semples R. E. (1977), Clay. Miner., 12, 229-237.
• Burgentzlé D., Duchet J., Gerard J. F., Jupin A. and B. Fillon (2004), j. Colloid.
Interface. Sci., 278(1), 26-39
• Caillère S., Hénin S. and M. Rautureau (1982), 2ème édition, Masson,
• Dai J.C., Xiao Z.J and L. Ye (1999), J. Inorg. Mater. 14(1), 90-94.
• Eslinger E. and D. Peaver (1988), Clay minerals for petroleum geologists and
engineers, SEPM Short course n°22, Soc. Economic paleontologists and mineralogists,
Tulsa, USA.
• Fetter G., Tichit D., Menorval L.C. and Figueras F. (1995), Appl. Catal. A:
General, 126, 165-176.
28

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
• Gil A. and Vicente M.A. (2000), Micropor. Mesopor. Mat., 34, 115-125.
• Grim R.E. (1953), Clay mineralogy, Édité par R.R. Shrock (McGraw-Hill series in
Geology, Londres), et références incluses.
• Grunberger D., (1995), Thèse Université de Montpellier II, Sciences et Techniques
du Languedoc, 20 Décembre 1995.
• He H., Galy J and Gérard J. F. (2005), J. Phys. Chem., 109(27), 13301-13306.
• Heinz H., Koerner H., Anderson K. L., Vaia R. A. and Farmer B. L. (2005),
Chem. Mat, 17(23), 5658-5669.
• Heinz H., Vaia R. A. And Farmer, B. L. (2006), J. Chem. Phys., 124, 224713
• Helmy A.K., Ferreiro E.A., De Bussetti S.G. and Peinemann N. (1998), Colloid
Polym. Sci., 276, 539-543.
• Hendershot W.H. and Duquette M. (1986). Soil Sci. Soc. Am. J., 50, 605-608.
• Hermosin M.C. and J. Cornejo (1992), Chemosphere 24, 1493–1503.
• Hermosin M.C. and Cornejo J. (1993), J. Environ. Qual. 22, 325–331.
• Hofmann U. and Giese K. (1939), J. Colloid., 21-36.
• Hsu Y.H., Wang M.K., Pai C.W. and Wang Y.S. (2000), Appl. Clay Sci., 16, 147–
159.
• Janek M. and Lagaly G. (2003), Colloid. Polym. Sci., 281, 293-301.
• Jiang Y.S., Zhang J. and Fang S.S. (2001), Acta Petrol. Mineral., 20, 445-454.
• Jiang J. Q., Cooper C. & Ouki S. (2002), Chemosphere, 47, 711- 716.
• Kadar F., Szazdi L., Fekete E. and Pukanszky B. (2006), Langmuir, 22(18), 7848-
7854.
• Khalaf H., Bouras O., & Perrichon V. (1997), Microporous Mater., 8, 141-150.
• Khenifi A., Bouberka Z., Bentaleb K., Hamani H. and Derriche Z. (2009), Chem.
Eng. J.,146, 345–354.
• Khenifi A., Bouberka Z., Sekrane F., Kameche M. and Derriche Z. (2007),
Adsorption, 13, 149–158.
• Kloppenburg S. (1997) Kolloidchemische Steuerung der Porosität aggregierter
Tonminerale, Dissertation, Universität Kiel.
• Kloprogge J.T., Evans R., Hickey L. and Frost R.L. (2002). Appl. Clay. Sci., 20,
157–163.
• Lagaly G. (1986), Solid State Ionics, 22(1), 43-51.
29

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
• Lagaly G. (1981), Clay Miner., 16, 1-21
• Lahav N., Shani U. and Shabtai J. (1978), Clays Clay Miner., 26, 107-115.
• Lahodny S. and Khalaf H. (1994), Appl. Clay Sci., 8, 405-415.
• Lan T. and Pinnavaia T.J. (1994), Chem. Mater., 6, 2216-2219.
• Larpent C., (1995), Tensioactifs, Techniques de l’ingénieur, Ref. K342.
• Matthes W. and Kahr G. (2000), Clays Clay Miner., 48, 593–602.
• Mantin I. and Glaeser R. (1960), Bull Gr. Fr. Argiles, 12, 83-88.
• Mehlich A., (1948), Soil Sci., 66, 429-445.
• Meier L.P. and Kahr G., (1999), Clays Clay Miner, 47, 386-388.
• Meier P., Nueesch R. and Madsen F.T., (2001), J. Colloid Interface Sci., 238, 24-
32.
• Michot L. J. & Pinnavaia, J. J., (1991), Clays Clay Miner., 39, 634-641.
• Michot L. J. & Pinnavaia, J. J., (1992), Chem. Mater., 4, 1433-1437.
• Michot L. J., Barres O., Hegg E. L. & Pinnavaia, J. J., (1993), Langmuir, 9, 1794-
1800.
• Montarges E., Moreau A. & Michot L. J. (1998), Appl. Clay Sci., 13, 165-185.
• Moore D.M. and R.C. Reynolds (1997), 2nd edition, Oxford University Press.
• Morel (1957), Bull Gr. Fr. Argiles, 12, 3-8.
• Orsini L. and Remy J.C., (1976), Science du sol, 4, 269-275.
• Osman M.A., Ploetze M. and P. Skrabal (2004), J. Phys. Chem., B 108, 2580-
2588.
• Pédro G. (1994),Eds. Duchaufour Ph. et Southier B. Masson, Paris 665p.
• Pleysier J. and Cremers A. (1975), Farad. Trans, 71, 256-264.
• Ravina I. and Gurovich E. (1977), Soil Sci. Soc. Am. J., 41, 319-322.
• Sapag K. and Mendioroz S. (2001), Colloids Surf., 187-188,141-149.
• Schoonheydt R.A. (1995), Edité par D.J. Vaughan et R.A.D. Pattrick (Chapman &
Hall, Londres), chapitre 9 (Clay mineral surfaces), 303-332.
• Schoonheydt R.A., Pinnavaia T., Lagaly G. and Gangas N. (1999), Pure Appl.
Chem., 71, 2367-2371.
• Senkayi A.L., Dixon J.B., Hossner L.R. and Kippenberger L.A. (1985), Soil Sci.
Soc. Am. J, 49 1054-1060.
• Shabtai J. (1980), US Patent, 4, 238,364.
30

CHAPITRE 1 : GENERALITES SUR LES ARGILES CATIONIQUES
• Shu H. T., Li D., Scala A. A. & Yi Y.M. (1997), Separ. Purif. Technol., 11, 27-36.
• Sposito G. (1989), Chimia, 43, 169-176.
• Srinivasan K. R. & Fogler S. H. (1990 A), Clays Clay Miner., 38, 277- 286.
• Srinivasan K. R. & Fogler S. H. (1990 B), Clays Clay Miner., 38, 287-293.
• Stackmeyer M.R. (1991), Appl. Clay Sci., 6, 39-57.
• Sychev M., Shubina T., Rozwadowski M., Sommen A.P.B., De Beer V.H.J. and
Van Santen R.A. (2000), Microp. Mesop. Mater, 37, 187-200.
• Tamura K. and Nakazawa H. (1996), Clays Clay Miner., 44, 501-505.
• Vaia R. A., Ishii H. and Giannelis E.P. (1993), Chem. Mater.,5, 1694-1696.
• Vaia R.A., Teukolsky R. K. and Giannelis, E. P. (1994), Chem. Mat., 6(7), 1017-
1022.
• Wang Z. and Pinnavaia T.J. (1998), Chem. Mater., 10, 3769-3771.
• Wu J.H. and Lerner M.M. (1993), Chem. Mater., 5, 835-838.
• Xi Y., Ding Z., He H. and Frost, R. L. (2004), J. Colloid Interface Sci., 277(1),
116–120.
• Xi Y., Frost R. L., He H., Kloprogge T. and Bostrom, T. (2005), Langmuir,
21(19), 8675-8680.
• Xie W., Gao Z., Pan W. P., Hunter D., Singh A. and Vaia, R. (2001), Chem.
Mater.,13(9), 2979-2990.
• Zielke R. C. & Pinnavaia J. J. (1988), Clays Clay Miner., 36, 403-408.
31

CHAPITRE 2 : LES HYDROXYDES DOUBLES
LAMELLAIRES (ARGILES ANIONIQUES)
32

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES
(ARGILES ANIONIQUES)
1. Historique
Les matériaux type-hydrotalcites (ou hydroxydes doubles lamellaires, HDL)
appartiennent à une classe de composés lamellaires inorganiques à caractère basique avec une
forte capacité à l'insertion d'anions. Découverts en Suède au milieu du XIXième siècle, ils
doivent leur nom au fait qu'une fois broyés ils ressemblent à du talc.
En 1915, E. Manasse (professeur de minéralogie à l'Université de Florence) proposa la
première formule pour le composé hydrotalcite naturel. Cette formule Mg6Al2(OH)16CO3,
4H2O est la première à mettre clairement en évidence l'importance de la présence d'ions
carbonates dans la structure (Manasse,1915).
La composition des hydrotalcites résulte donc d'un mélange d'hydroxy carbonates
d'aluminium et de magnésium et se trouvent dans le milieu naturel, sous forme de feuillets
déformés ou de masse fibreuse. Une structure identique à celle des hydrotalcites, appelée
pyroaurite, car elle a l'aspect de l'or une fois chauffée, mais composée d'hydrocarbonates de
magnésium et de fer, a été découverte à la même période.
S'appuyant sur une étude réalisée en 1930 sur la base d'expérimentations de diffraction
des rayons X, Aminoff et Broomè, montrèrent l'existence de deux polytypes d'hydrotalcite
(Aminoff, 1930), le premier de symétrie rhomboédrique (3R) appelé "Hydrotalcite", le second
de symétrie hexagonale (2H) appelé "manasseite" en l'honneur de E. Manasse.
La synthèse de composés de type hydrotalcite fut réalisée, pour la première fois, en
1942 par Feitknecht (Feitknecht, 1942) . Il appela ces composés "doppelschichtstrukturen"
(structures doubles couches) encore appelés hydroxydes doubles lamellaires (HDL),
considérant l'empilement simple de feuillets de brucite Mg(OH)2 et de feuillets Al(OH)3.
33

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Cette structure simple fut démentie dans les années 60 par Allmann (Allmann, 1968) et
Taylor (Taylor, 1969) qui se sont appuyés sur une analyse cristallographique d'un
monocristal. Ils conclurent que les deux cations (Mg2+ et Al3+) étaient situés sur la même
couche et montrèrent la présence d’ions carbonates et de l'eau dans le domaine interlamellaire.
Un temps assez long est écoulé entre la découverte des composés Hydrotalcites et la
publication de leur structure. Ceci est dû à leur caractère non stœchiométrique et à la difficulté
de former un monocristal adapté à des analyses de diffractions des rayons X. En effet, les
derniers travaux publiés par Allmann et Taylor étaient essentiellement consacrés à la structure
pyroaurite (pour laquelle des monocristaux étaient disponibles). Les Hydrotalcites ont été,
quant à elles, étudiées bien plus tard.
Ce n'est qu'au début des années 70 que les premières applications dans des domaines tels
que la catalyse ou l'échange d'ions ont été proposées pour les composés Hydrotalcites. En
1971, Miyata et coll. (Miyata et al., 1971) publièrent les premiers travaux sur les
Hydrotalcites utilisées comme catalyseurs basiques. En 1975, Bröcker et Kaempfer (Bröcker
et Kaempfer, 1975), puis Miyata (Miyata, 1977) en 1977, utilisèrent ces composés comme
catalyseurs d'hydrogénation.
Les hydrotalcites appartiennent à la famille des argiles anioniques qui sont moins
représentées dans la nature que les argiles cationiques. Les composés de type hydrotalcite ont
trouvé de nombreuses applications comme catalyseurs (hydrogénation) (Bröcker et Kainer,
1971) ;polymérisation, (Nakatsuka et al., 1979 ; Kohjiya et al., 1981), échangeurs d'ions
(Reichle, 1986), absorbants (Chanakya, 1987), retardateurs de flamme (Sutker et al., 1987),
antiacides (Miyata, 1977, 1985), antipeptines (Miyata, 1977; Pawlacz et al.,1985), tamis
moléculaires ou stabilisateurs en pharmacologie (Miyata et Kumura, 1973).
2. Propriétés structurales des HDL 2.1 Structure
Afin de mieux appréhender l'architecture des Hydrotalcites (ce nom sera pris comme
générique pour tous les composés isomorphes du composé référence de composition
Mg6Al2(OH)16CO3, 4H2O), rappelons les caractéristiques de la structure de type brucite
(hydroxyde de magnésium, Mg(OH)2) dont elle dérive. Celle-ci est constituée par un
34

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
enchaînement d'octaèdres dont les centres sont occupés par des ions Mg2+ et les sommets par
des groupements hydroxyles. Les octaèdres partagent leurs arêtes et forment ainsi une chaîne
infinie de feuillets. Les feuillets sont empilés les uns au-dessus des autres et leur cohésion est
assurée par des liaisons hydrogène.
Pour les structures dérivées de l’Hydrotalcite, une partie des ions Mg2+ de la brucite est
remplacée par des cations trivalents (ex: Al3+ dans le cas des Hydrotalcites et Fe3+ pour les
pyroaurite) générant ainsi une charge positive. Cette charge positive est compensée par des
anions (ions carbonates dans le cas de l'Hydrotalcite naturelle par exemple) qui se répartissent
de manière aléatoire dans le domaine interlamellaire, assurant ainsi la neutralité électrique de
l'ensemble (Cavani et al., 1991; Li et Duan, 2006; Evans et Duan, 2006). Dans ce domaine
interlamellaire se trouve également des molécules d'eau (Figure I.2.1).
Figure I.2.1 : Représentation schématique de la structure de matériaux de type hydrotalcite
d'après A. de Roy et coll. (de Roy et al., 1992).
Les anions de compensation (organiques et inorganiques) et l'eau peuvent "circuler"
assez librement dans le domaine interlamellaire après rupture des liaisons hydrogène. Les
groupements hydroxyles des feuillets de brucite sont, directement ou par l'intermédiaire de
l'eau, liés aux anions de compensation par des liaisons hydrogène (Allmann, 1970). En ce qui
concerne la structure cristalline des Hydrotalcites, les paramètres de la cellule unité a et c
dépendent de la nature des cations divalents et trivalents (donc de leur rayon ionique).
35

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
L'espacement de base (c’) représente l'épaisseur totale du feuillet brucitique plus la
région intercalaire. Les HDL ayant une structure type Hydrotalcite peuvent être alors définis
par la formule suivante:
[M(II)1-xM(III)
x(OH)2]x+(A-nx/n).mH2O.
• M(II) un métal divalent tel Mg2+, Fe2+, Co2+, Ni2+....
• M(III) un métal trivalent comme Al3+, Cr3+, Fe3+…
• An- étant l'anion de compensation (CO32-, Cl-, NO3
-,….).
La nature de MII+ et MIII+, x, et An- peut varier dans une large gamme, donnant ainsi
naissance à une grande classe de matériaux iso structurels avec des propriétés physico-
chimiques variables (Evans et Duan, 2006).
Le matériau de base de ces argiles anioniques est l'hydrotalcite naturelle minérale de
formule Mg6Al2 (OH)16CO3.4H2O. Différents types d’empilement des feuillets brucitiques
ont été observés. elles peuvent être empilées, soit avec deux couches par unité de cellule avec
une symétrie hexagonale (manasseite) ou avec trois couches par unité de cellule dans une
symétrie rhomboédrique (hydrotalcite), ou en un arrangement moins symétrique (Carrado et
al., 1988).
Pour obtenir une structure de type Hydrotalcite, il est nécessaire que la valeur x soit
comprise entre 0,1 et 0,5, mais l’obtention d’une structure pure n’est possible que pour des
valeurs x restreintes entre 0,20 et 0,33 (Thevenot et al., 1989). En effet, pour des valeurs x
hors de cet intervalle, on obtient soit des hydroxydes, soit des composés de structures
différentes (mélange de phases) (Gastuche et al., 1967; Mascalo et Marino, 1980). Lorsque
l'ion trivalent est Al3+ et l'ion divalent est Mg2+, des valeurs élevées de x entraînent la
formation de phases Al(OH)3 et, inversement, des valeurs faibles de x entraîne la formation de
Mg(OH)2.
2.2 Nature des cations M(II) et M(III)
Les cations ayant un rayon ionique voisin de celui du magnésium, peuvent conduire à la
formation d'HDL. Ainsi, ils sont capables de se substituer au magnésium et de s'insérer dans
les espaces placés au centre des octaèdres formés par les groupements hydroxyles dans les
couches de type brucite. Le tableau I.2.1 regroupe tous les cations divalents et trivalents
susceptibles d'intervenir dans une structure de type HDL, comme recensés dans la
36
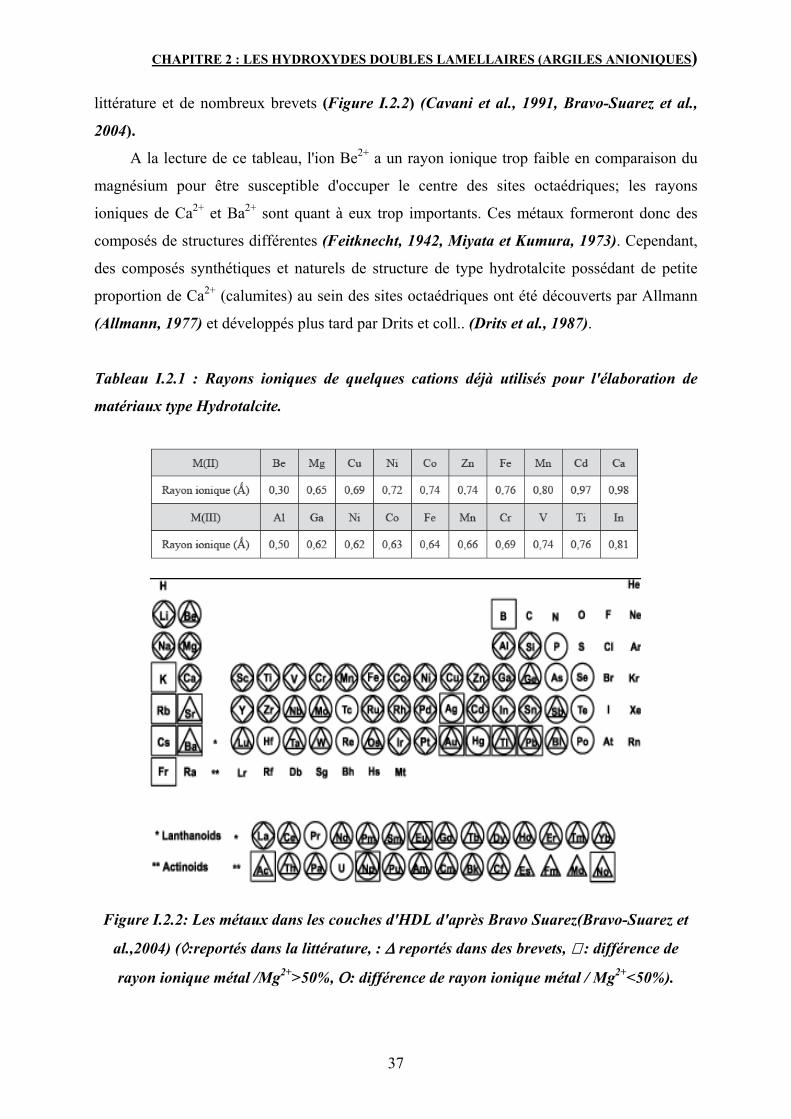
CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
littérature et de nombreux brevets (Figure I.2.2) (Cavani et al., 1991, Bravo-Suarez et al.,
2004).
A la lecture de ce tableau, l'ion Be2+ a un rayon ionique trop faible en comparaison du
magnésium pour être susceptible d'occuper le centre des sites octaédriques; les rayons
ioniques de Ca2+ et Ba2+ sont quant à eux trop importants. Ces métaux formeront donc des
composés de structures différentes (Feitknecht, 1942, Miyata et Kumura, 1973). Cependant,
des composés synthétiques et naturels de structure de type hydrotalcite possédant de petite
proportion de Ca2+ (calumites) au sein des sites octaédriques ont été découverts par Allmann
(Allmann, 1977) et développés plus tard par Drits et coll.. (Drits et al., 1987).
Tableau I.2.1 : Rayons ioniques de quelques cations déjà utilisés pour l'élaboration de
matériaux type Hydrotalcite.
Figure I.2.2: Les métaux dans les couches d'HDL d'après Bravo Suarez(Bravo-Suarez et
al.,2004) (◊:reportés dans la littérature, : Δ reportés dans des brevets, : différence de
rayon ionique métal /Mg2+>50%, Ο: différence de rayon ionique métal / Mg2+<50%).
37

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Les HDL peuvent contenir tous les types de cations métalliques divalents (de Mg2+ à
Mn2+), à l'exception des ions Cu2+ qui malgré un rayon ionique convenable ne forment des
HDL purs qu’en compagnie d’un autre cation divalent M(II) (ex: Mg2+, Co2+ ou Zn2+). Pour
cela le rapport Cu2+ /M(II) doit être inférieur ou égal à 1 (Busetto et al., 1984). Ce
comportement du cuivre (comme des ions d'autres métaux: Cr2+, Mn3+, Ni3+) provient de
l'effet Jahn Teller: les ions de configuration électronique « d9 » conduisent à la formation
d'octaèdres déformés. Tant que le rapport Cu2+/M(II) est inférieur ou égal à 1, les ions Cu2+ se
placent dans les sites octaédriques non déformés de la couche de type brucite. Lorsque ce
rapport est supérieur à 1, une partie des ions Cu2+ peut se placer dans des octaèdres déformés
et donner ainsi des hydroxydes de cuivre dont l'énergie est préférentielle (d'un point de vue
stabilisation électronique) à celle des structures HDL (West, 1990).
Concernant les cations trivalents, à l'exception de V3+ et Ti3+ qui ne sont pas stables à
l’air, tous les cations possédant un rayon ionique compris entre 0,5 et 0,8 Ǻ conduisent à la
formation de d’hydroxydes doubles lamellaires. Quelques valeurs possibles de x (MII/MIII)
recensées dans la littérature sont répertoriées dans le tableau I.2.2.
Tableau I.2.2 : Valeurs de x permettant l'obtention de phases HDL pures.
Intervalle de valeurs de x Composé Auteurs
0,25-0,44 MgAlOH-tHT* Pausch et coll.(Pausch et al.,1986)
0,23-0,33 MgAlOH-tHT* Mascolo et coll. (Mascalo et Marino, 1980)
0,20-0,33 MgAlClO4-tHT* Brindley et coll.(Brindley et Kikkawa,1979)
0,17-0,33 MgAlCO3-HT* Gastuche et coll. (Gastuche et al., 1967)
0,20-0,337 MgAlCO3-HT* Miyata et coll.(Miyata, 1980)
0,10-0,34 MgAlCO3-HT* Miyata et coll.
0,15-0,33 MgAlCO3-HT* Sato et coll.( Sato et al.,1988)
0,17-0,33 NiAlCO3-tHT* Brindley et coll. (Brindley et Kikkawa,1979)
0,25-0,34 NiAlCO3-tHT* Kruissink et coll. (Kruissink et al.,1981)
0,20-0,41 NiAlCO3-tHT* Sato et coll. ( Sato et al.,1988)
* HT signifiant structure hydrotalcite et tHT structure type hydrotalcite
38

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
2.3 Nature de l'anion de compensation
Il n'y a pratiquement aucune contrainte quant à la nature de l'anion qui va compenser
l'excès de charge positive du feuillet type brucite tant qu'il ne forme pas de complexes forts
avec les cations présents. Le problème essentiel réside dans la préparation de composés purs
et bien cristallisés. A ce titre, la préparation d’hydroxydes doubles lamellaires contenant des
anions de compensation différents de CO32- est difficile car il faut s'affranchir de toute
contamination de CO2 provenant de l'atmosphère ambiante. Un autre type de difficulté
intervient avec l'instabilité de certains anions dans la gamme de pH imposée durant la
synthèse des matériaux hydrotalcites. Ci-dessous est reportée une liste non exhaustive des
anions (organiques et inorganiques) pouvant enrichir la composition des HDL :
• Des anions inorganiques: F-, Cl-, Br-, I-, (ClO4)-, (NO3)-, (ClO3)-, (IO3)-, OH-, (CO3)2-,
(SO4)2-, (S2O3)2-, (WO4)2-, (CrO4)2-, [Fe(CN)6]3- …..
• Des hétéropolyacides : (PMo12O40)3-, (PW12O40)3- .....
• Des acides organiques : adipique, oxalique, succinique, malonique .......
• Des composés lamellaires : le minéral chlorite ((Mg2Al(OH)6)+.[Mg3(OH)2/Si3AlO10]-.
Le nombre, la taille, l'orientation, et la force des liaisons entre les anions et les
groupements hydroxyles de la couche type brucite déterminent l'épaisseur du domaine
interlamellaire.
Le tableau I.2.3 (Miyata, 1983) regroupe quelques valeurs de c' en fonction des anions
de compensation. Rappelons que la valeur du paramètre de maille a est indépendante de la
nature de l'anion puisqu'elle est déduite de l'arrangement périodique des atomes des feuillets
de type brucite. D'après les valeurs de c', l'épaisseur du domaine interlamellaire semble
augmenter avec la taille de l'anion de compensation. Cependant, ce seul paramètre ne peut
expliquer les faibles valeurs obtenues pour c' dans le cas des ions CO32- et OH-, les fortes
valeurs pour NO3- et par exemple la différence notable entre ClO4
- et SO42- qui ont un rayon
ionique équivalent.
39

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Tableau I.2.3 : Valeur de c' en fonction de l'anion de compensation
Par ailleurs, Brindley et coll (Brindley et Kikkawa, 1980) ont montré que pour certains
HDL, c' dépendait également de l'état d'hydratation du matériau. Dans le cas de l'anion NO3-,
sa monovalence implique une présence en plus grande quantité pour compenser la charge
positive. Cependant, sa taille relativement importante entraîne un arrangement très dense dans
le domaine interlamellaire et donc de fortes répulsions électroniques lorsque sa concentration
augmente. Une expansion interlamellaire intervient alors naturellement.
2.4 Proportion d'eau dans le domaine interlamellaire
Des molécules d'eau sont également situées dans le domaine interlamellaire, dans les
sites inoccupés par les anions de compensation. Généralement, la quantité d'eau est
déterminée par la perte de masse mesurée grâce à des analyses thermogravimétriques (Bish et
Livingstone, 1981; Sato et al.,1988; Rives, 2002; Yang et al., 2002; Tichit et al.,1998;
Miyata,1975). Cependant, il est également possible de retrouver une estimation quantitative
"maximale" d'eau en se basant sur le nombre de sites présents dans le domaine interlamellaire
auxquels sont soustraits les sites occupés par les anions. Plusieurs modes de calcul de cette
estimation quantitative sont envisageables et différentes formules ont été rapportées dans la
littérature et peuvent être utilisées en l'état.
40

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Sur la base des travaux de Miyata (Miyata, 1975 ;1977), une formule générale assez
simple peut être avancée:
nxNm ×−=1 (I.2.1)
Avec :
N = nombre de sites occupés par les anions, n = charge de l'anion, x = M(III)/(M(II)+M(III))
(rapport atomique). En considérant l'exemple d'une Hydrotalcite avec des ions carbonates en
tant qu'anions de compensation, la formule se simplifie à :
2
31 xm ×−= (I.2.2)
D'après Taylor (Taylor, 1969), cette formule doit être extrapolée à l'aide d'une constante
pour mieux rendre compte de la réalité.
dxm +×−=2
31 (I.2.3)
Avec :
d= 0.125
Mascolo et coll.(Mascalo et Marino, 1980) préconisent dans le cas de MgAlOH
d'utiliser la relation suivante :
xm −= 81.0 (I.2.4)
Dans tous les cas et quelle que soit la formule adoptée, une augmentation de la valeur x
entraîne une diminution de la quantité d'eau dans le domaine interlamellaire. Par exemple, en
se basant sur les travaux de Miyata (Miyata, 1975 ; 1977), pour le composé
Mg0,75Al0,25(OH)2CO3, m est égal à 0,625. Ramené à des valeurs entières, cela donne la
formule suivante: Mg6Al2(OH)16CO3.5H2O. La valeur relative m calculée à partir des travaux
de Miyata (m relative = 5) est donc supérieure à celle mesurée pour l'Hydrotalcite naturelle m
relative = 4. Comme l'atteste ces résultats, ces méthodes de calcul ne sont donc pas totalement
fiables et il est donc très difficile d'estimer, à priori, la quantité d'eau réelle localisée dans le
domaine interlamellaire.
41

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
3. Synthèse des hydroxydes doubles lamellaires À la différence des argiles cationiques, les hydroxydes doubles lamellaires sont aisément
préparés en laboratoire par chimie douce (Rives, 2001; DeRoy et al., 2001; He et al., 2006;
Williams et O’Hare, 2006). La synthèse des matrices hydroxydes doubles lamellaires a
commencé dans les années 1930 avec Feitknecht (Feitknecht, 1942; Feitknecht et al., 1935),
qui précipite une solution aqueuse diluée de sels métalliques avec une base. Gastuche (1967)
(Brown et Van Oosterwyck-Gastuche, 1967; Van Oosterwyck-Gastuche et al., 1967)
poursuivit en utilisant des solutions plus concentrées et en éliminant les électrolytes résiduels
par dialyse. Puis d’autres chercheurs ont examiné les méthodes de synthèse de ces matériaux,
Brindley et coll. (Brindley et Kikkawa, 1979), Taylor et coll. (Taylor, 1984) et en particulier
Miyata (1975) (Miyata, 1975) qui a amélioré l’homogénéité des produits en effectuant la
coprécipitation à pH constant.
Plusieurs méthodes de synthèse sont disponibles pour la préparation d'argiles anioniques
(Braterman et al., 2004). En effet, la précipitation à pH constant (Bröcker et al.,1972;
Kruissink et al.,1979), à pH variable (Feitknecht, 1942; Ross et Kodama, 1967; Courty et
al., 1985), l'échange d'anions (Bish et Brindley,1977; Woltermann, 1984; Bish, 1980), la
synthèse par voie sol-gel (Lopez et al.,1996), la méthode par chimie douce (Delmas et
Borthomieu, 1993), la croissance hydrothermale (Mascalo et Marino,1980;Martin et
Pearson, 1996), la méthode urée (Adachi-Pagano et al., 2003; Ogawa, et Kaiho, 2002; Shaw
et Bordeaux, 1955) ou encore la synthèse électrochimique (Indira et al.,1994) permettent
d'élaborer des HDL. Cependant, il apparaît que la coprécipitation est la technique la mieux
adaptée et la plus simple pour préparer de grandes quantités d'argiles anioniques avec très peu
de contraintes expérimentales. Pour nos travaux, ce mode de synthèse a été retenu.
Afin d'obtenir des structures pures de type hydrotalcite, les proportions en anions et
cations doivent globalement répondre aux conditions suivantes :
0,2 ≤ M(III)/[M(III)+M(II)] ≤ 0,4 ;
1/n ≤An-/M(III) ≤ 1.
L'anion de compensation introduit dans l'HDL doit être l'espèce majoritaire dans les
solutions de réactifs (afin que son insertion dans le domaine interlamellaire soit optimale) et
doit avoir une grande affinité avec le composé lamellaire. De plus, les plus grandes
précautions doivent être prises afin d'éviter que les anions des solutions métalliques ne
42

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
contaminent les HDL élaborés. La synthèse d'HDL devient particulièrement critique lorsque
l'anion de compensation n'est pas (CO3)2-. En effet, le dioxyde de carbone de l'air ambiant
peut très facilement s'insérer dans la structure du fait de son fort pouvoir oxydant ; il faut donc
généralement avoir recours à des techniques d'échange d'ions sous atmosphère contrôlée (flux
de diazote).
3.1 Méthode de coprécipitation
Cette méthode de synthèse, développée par Miyata (Miyata, 1975), est la plus utilisée
actuellement et consiste à précipiter de manière contrôlée une solution de sels des métaux par
une solution basique (Bases de Brønsted : NaOH, LiOH, NH4OH, KOH). L’ajout simultané
des deux réactifs permet de maintenir le pH constant. Lorsque l’anion à intercaler est le
chlorure, la réaction globale de cette synthèse est donnée comme suit :
Dans cette méthode, il est possible d’agir sur les réactifs et sur les conditions
expérimentales. La composition du produit obtenu va ainsi dépendre de la nature, du rapport
des cations métalliques, et des concentrations des solutions de départ. Elle va également être
conditionnée par le solvant (Malherbe, 1997), la température (Thévenot, 1994), et
l’atmosphère du milieu de synthèse (air ou azote). Une addition lente des réactifs est
généralement favorable à une bonne cristallinité des phases. Le pH doit être optimisé pour
chaque système MII/MIII (tableau I.2.4), afin de permettre la précipitation conjointe des
cations trivalent et divalent (Figure I.2.3) (Boclair et Braterman, 1999; Boclair et al.,
1999).
La réaction s’effectue sous agitation magnétique ou mécanique pour homogénéiser le
milieu, et sous flux d’azote pour éviter la pollution par les ions carbonate. Afin d’éliminer les
résidus de synthèse, le produit final séparé par centrifugation et mis en suspension dans l’eau
décarbonatée, cette étape est répétée plusieurs fois.
43
CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Tableau I.2.4 :pH de synthèse pour quelques compositions
Figure I.2.3 : Courbes de titration de solutions 2:1 MgCl2:FeCl3 et MgCl2:AlCl3, d’après
(Boclair et Braterman, 1999)
Le solide est finalement séché à l’air à température ambiante où à l’étuve, ou sous vide
dans un dessiccateur. Cette étape de lavage et séchage est commune à tous les types de
synthèse décrits plus loin. En suivant ce protocole, le milieu est rapidement sursaturé en base,
provoquant une nucléation continue des hydroxydes métalliques simultanément à la
croissance des particules déjà formées et à l’agrégation par mûrissement d’Oswald. La
coprécipitation conduit donc généralement à la formation de plaquettes hexagonales de tailles
variables, dont les dimensions varient suivant les compositions. Ainsi, des particules d’un
diamètre de ∼200 nm et de∼ 20 nm d’épaisseur sont observées pour une matrice Zn–Al, et de
∼ 80 nm de diamètre pour Mg–Al. Une amélioration de la cristallinité est possible pour la
phase Mg–Al en maintenant le milieu réactionnel à 65 °C, ou en réalisant un traitement
44

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
hydrothermal post-synthèse. Ces plaquettes ont tendance à s’agencer de manière à former des
roses des sables ou encore des châteaux de cartes (Figure I.2.4).
Une modification de la méthode de coprécipitation a été récemment développée par
Zhao et coll. (Zhao et al., 2002) afin de séparer les étapes de nucléation et de croissance
cristalline. Les solutions de sels et de base sont ajoutées dans un « colloid mill » tournant à
3000 rpm durant 2 min. Les précurseurs sont intimement mélangés entre un stator et un rotor
conique distants de 10 μm. La nucléation s’effectue donc dans un mince film liquide entre les
deux. Le mélange est ensuite laissé 13h à 100 °C avant d’être filtré, lavé, et séché. À l’issue
de cette étape de mûrissement, des particules hexagonales uniformes de 60-80 nm de diamètre
sont obtenues. La faible polydispersité en taille peut être expliquée par deux causes :
• le caractère turbulent du fluide imposé lors de la nucléation qui empêche
l’agglomération.
• la faible durée de cette étape (2 min) ne permet pas la croissance des particules, les
germes étant tous formés au même moment en grand nombre.
Figure I.2.4 : Clichés MEB de phases Mg–Al (a) et Cu–Cr (b) synthétisées par
coprécipitation
Concernant la méthode de synthèse dite de coprécipitation, deux procédures distinctes
peuvent être utilisées: l'une dite "à faible supersaturation" et l'autre "à supersaturation élevée"
45

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
3.1.1 Méthode de coprécipitation à faible supersaturation:
Cette méthode est la plus utilisée pour la synthèse d'HDL. Les agents de précipitation
basiques (NaOH et Na2CO3) sont lentement mélangés aux sels métalliques, le pH étant
maintenu constant. Les conditions généralement retenues sont les suivantes: la gamme de pH
entre 7 et 10 avec une température pouvant varier entre 25°C et 70°C et des débits
d'écoulement des solutions qui restent très faibles. Ces conditions de synthèse favorisent des
structures mieux cristallisées, les cristallites finales étant relativement de petite taille (Marchi
et al., 1986;1988; Courty et al., 1982). A titre d'exemple, Bröcker et coll. (Bröcker et al.,
1972) réalisèrent par cette méthode une des toutes premières synthèses de l'HDL NiAlCO3 en
1972 ou encore Woltermann (Woltermann, 1984) qui synthétisa ZnAlNO3 dans les mêmes
conditions.
3.1.2 Méthode de coprécipitation à supersaturation élevée:
Les nitrates métalliques sont ajoutés aux agents de précipitation très rapidement à pH
constant. Les synthèses réalisées dans ces conditions donnent généralement des matériaux peu
cristallisés. Il y a une vingtaine d'années, Marchi et coll. (Marchi et al., 1986 ; 1988), lors de
la préparation d'un composé trimétallique (Cu-Co-Al), ont obtenu un précipité amorphe par
addition de sels de nitrates à une solution contenant du NaHCO3 alors que dans des conditions
de synthèse à faible supersaturation, ils obtenaient une structure type HDL. Malgré ces
résultats très différents, les premiers matériaux HDL ont été synthétisés en utilisant des
conditions de super saturation élevée (French, 1971).
Le tableau I.2.5 résume les facteurs prédominants lors des synthèses d' HDL et les
caractéristiques intrinsèques importantes de ces matériaux. Le mauvais contrôle d'un de ces
paramètres a des conséquences sensibles sur l'état de cristallinité, la nature et la pureté du
matériau élaboré.
46

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Tableau I.2.5 : Facteurs influençant l’architecture de type hydrotalcite (Vaccari,1998;
Cavani et al., 1991)
3.2 Méthode d’urée
L’intérêt de la méthode urée (Cai et al., 1994; Costantino et al., 1998; Oh et al., 2002;
Adachi-Pagano et al., 2003; Ogawa et Kaiho, 2002; Rao et al., 2005) réside dans le contrôle
de la cinétique de précipitation de la matrice HDL par l’utilisation de l’urée comme base
retardante. Les processus de nucléation et croissance sont généralement confondus lors d’une
coprécipitation classique car la solution est rapidement sursaturée en soude. Le contrôle de
l’hydrolyse thermique de l’urée, en évitant la sursaturation, permet de séparer ces deux étapes
et de favoriser une meilleure homogénéité en taille des cristallites et une plus grande
cristallinité. Une faible sursaturation induit une faible vitesse de nucléation, donc des
cristallites en nombre inférieur, mais de plus grande taille, >40 μm (Kayano et Ogawa, 2006).
L’urée, base de Brønsted très faible (pKb=13,8), se décompose au-dessus de 70 °C,
selon Shaw et Bordeaux (Shaw et al., 1955), d’abord en formant du cyanate d’ammonium
(NH4CNO) puis en s’hydrolysant en carbonate d’ammonium. La précipitation est contrôlée
par la vitesse de décomposition de l’urée, qui augmente avec l’élévation de la température
(multipliée par 200 entre 60 °C et 100 °C). Cette hydrolyse régule le pH entre 7 et 9, selon la
température.
Cette méthode est particulièrement adaptée à la synthèse des phases hydroxydes doubles
lamellaires car les produits de décomposition de l’urée (hydroxydes et carbonate) sont les
constituants des HDL. Costantino et coll. (Costantino et al., 1998) ont étudié l’influence de la
température et des concentrations sur la structure et les compositions des produits obtenus. La
méthode urée est une méthode qui permet de synthétiser des HDL Mg−Al−CO3 à forte densité
47

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
de charge, mais la production d’ions carbonate constitue un inconvénient majeur lorsque
d’autres anions doivent être intercalés.
Les produits synthétisés par la méthode urée sont souvent constitués de plaquettes
isolées, de taille relativement mono-disperse, généralement entre 1 μm et 5 μm suivant les
conditions. Les dimensions de ces cristallites peuvent atteindre 20 μm en diminuant la
concentration des sels, cette grande taille étant permise par la faible vitesse d’hydrolyse de
l’urée qui mène à un faible taux de sursaturation. L’épaisseur de ces cristallites est variable
suivant les compositions et le temps de réaction. Les particules présentent une forme
hexagonale dans le cas d’une phase MgAl, et plus arrondies pour les phases ZnAl. Elles
peuvent s’assembler par les faces principales sous forme de roses des sables (NiAl) ou de
colonnes (ZnAl), ou bien rester relativement isolées (MgAl et CoAl) (Figure I.2.5).
Figure I.2.5: Clichés MEB de phases Mg-Al (a) et Zn-Al (b) synthétisées par la méthode
urée
(Thèse de S. Vial 2005)
Une modification de cette méthode a été mise au point au laboratoire (Vial, 2005;Vial et
al., 2006 a et b). Elle consiste en un procédé de type « biominéralisation », en utilisant un
catalyseur enzymatique, l’uréase, pour effectuer l’hydrolyse de l’urée à température ambiante.
Les particules produites sont de petite taille, et se présentent sous forme d’agrégats assez mal
cristallisés (Figure I.2.6). La charge négative portée par l’uréase dans les conditions de
synthèse favorise la formation de ces agrégats HDL/uréase qui vont réduire l’activité de
l’enzyme et limiter la croissance cristalline. Cette méthode de synthèse permet de préparer des
phases hybrides HDL/uréase, intéressantes pour élaborer des biocapteurs pour la détection de
l’urée (Vial et al., 2006 c).
48

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Figure I.2.6 :Cliché MEB d’une phase Mg-Al obtenue par décomposition enzymatique de
l’urée (Thèse de S. Vial 2005)
À ces principales méthodes de synthèse s’en ajoutent d’autres rencontrées
ponctuellement : hydrolyse induite (Taylor, 1984), hydrolyse en milieu polyol (Prevot et al.,
2005), méthode sel+oxyde (Boehm et al., 1977), procédé sol/gel (Aramendía, et al., 2002). . .
3.3 Traitements post-synthèse
3.3.1 La calcination–reconstruction
Les phases HDL ont la propriété de pouvoir se reconstruire après une calcination
modérée par simple remise en suspension dans une solution aqueuse (Reichle, 1986; Hibino
et al., 1995). Cette aptitude est entre autre utilisée pour intercaler des molécules volumineuses
difficiles à intercaler par d’autres moyens (Bigey, 1998; Dimotakis et Pinnavaia,1990;
Chibwe et Jones, 1989). La méthode concerne plus particulièrement les phases HDL à base
d’aluminium et de magnésium. Elle consiste à chauffer le composé à une température
inférieure à celle de formation irréversible des oxydes stœchiométriques (MgO et MgAl2O4),
mais suffisamment élevée pour volatiliser l’anion intercalé et former des oxydes mixtes pré-
spinelle, soit 450 °C pour l’Hydrotalcite. Ces oxydes métastables sont très réactifs et
retrouvent ensuite la structure de départ par simple remise en suspension dans une solution
aqueuse de l’anion à intercaler. La réaction de reconstruction peut se faire à température
ambiante, à reflux ou encore en conditions hydrothermales.
L’équilibre du processus de calcination/reconstruction pour une phase [Mg3Al] peut être
décrit par :
49

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Un des avantages de cette méthode, par rapport à l’échange anionique, est qu’elle
permet d’utiliser des précurseurs carbonatés. Ces matrices sont d’ailleurs préférables dans ce
processus car l’anion carbonate se décompose à faible température.
3.3.2 Échange anionique
C’est une transformation topotactique qui conserve la structure en feuillet, elle est
généralement effectuée par simple mise en suspension du précurseur dans la solution de
l’anion à échanger, sous agitation et atmosphère contrôlée. L’échange anionique est un moyen
simple de remplacer l’espèce anionique intercalée lors de la synthèse par une autre. Il est
cependant nécessaire que l’affinité de l’anion à intercaler avec la matrice soit plus grande que
celle de l’anion de départ. Miyata a proposé une échelle d’affinité pour des anions mono- et
divalents :
Les anions à densité de charge élevée sont plus stables dans le domaine interlamellaire.
Il est possible de combiner l’échange anionique avec la coprécipitation pour pouvoir
intercaler des anions autres que ceux des sels, cela consiste à effectuer la coprécipitation dans
une solution concentrée de l’anion à intercaler, on parle dans ce cas d’échange direct. La loi
d’action de masse doit bien évidemment être en faveur de l’anion à intercaler dans ce cas.
3.3.3 Délamination - réempilement
Ce n’est que très récemment (2000), qu’à pu mettre admis en évidence la possibilité de
délaminer des phases HDL. À cette fin, les forces d’attraction entre les feuillets sont réduites
par l’intercalation d’un surfactant (dodécylsulfate par exemple) (Jose dos Reis et al., 2004;
Pavan et al., 1999), puis le matériau est mis en suspension dans un solvant polaire tel que le
butanol (Adachi-Pagano et al.,2000) ou le Formamide (Venugopal et al., 2006). La mise à
reflux du système conduit alors à une suspension colloïdale (Figure I.2.7). La délamination
est plus facile dans les solvants polaires tels que les alcools, et pour des rapports MII/MIII
petits, mais elle ne semble pas sensible à la nature des cations. La stabilité de la suspension
colloïdale obtenue augmente avec la taille du surfactant.
50

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Figure I.2.7 : Morphologie d’une phase [ZnAl–DDS] délaminée dans l’éthanol (à gauche)
et le butanol (à droite) (MET) (Adachi-Pagano et al.,2000)
Une délamination rapide, à température ambiante, a été également décrite par Hibino
(Hibino et Jones, 2001). Son approche est basée sur l’emploi d’un couple acide
aminé/solvant polaire, en effet, les fortes liaisons hydrogènes entre l’acide aminé et le solvant
polaire permettent la pénétration de plus grands volumes de solvant et finalement la
délamination. Le couple présentant les meilleurs résultats est formé par la glycine et le
formamide. La matrice HDL– glycine est simplement mise en suspension dans le formamide
quelques minutes pour obtenir une suspension colloïdale. Des travaux plus récents montrent
qu’il est possible d’étendre cette méthode à d’autres amino-acides que la glycine (Hibino,
2004), et également à des phases nitrates (Liu et al., 2006).
4. Champs d’applications des HDL Les phases hydroxydes doubles lamellaires suscitent beaucoup d’intérêt de part leurs
propriétés originales et font l’objet d’études dans des domaines très variés. Certaines de leurs
applications sont citées ci-après, et vont de la catalyse au milieu médical (Rives,2001; Li et
Duan, 2006), elles utilisent chacune des propriétés différentes de ces phases.
4.1 Catalyseurs, précurseurs de catalyseur, supports de catalyseurs
Les oxydes simples et mixtes produits lors de la calcination ou la réduction des phases
hydroxydes doubles lamellaires sont utilisés dans le domaine de la catalyse, en tant que
catalyseurs et précurseurs ou support de catalyseurs (Vaccari, 1999). Ces oxydes possèdent en
effet des propriétés basiques, une dispersion homogène et stable thermiquement des ions
métalliques, ainsi que des surfaces spécifiques relativement élevées, atout majeur en catalyse
où les réactions ont lieu aux interfaces. Par exemple, des phases Mg–Al calcinées sont
utilisées comme support de métaux de transition pour la réduction catalytique sélective de NO
51

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
par NH3 (Pasel et al., 1998) et la déshydrogénation du n-butane (Lòpez et al., 1995). Les
phases non calcinées peuvent être utilisées comme catalyseurs par exemple de l’époxydation
du styrène (Mg–Al) (Kirm et al., 2004), de la condensation de Knoevenagel (Ni–Al)
(Costantino et al., 2003), ou bien de l’hydroxylation du phénol (Co–Ni–Al) (Rives et al.,
2003).
4.2 Charge minérale dans les polymères nanocomposites
Les travaux sur les nanocomposites polyamide/argiles ont été initiés par les chercheurs
du groupe Toyota dans les années 90. Ces recherches ont été relayées par l’importance
croissante, technologique et marchande, des matériaux polymères qui nécessite d’améliorer
leurs performances, et de nos jours de nombreuses études portent sur les matériaux
nanocomposites polymère/charge minérale, en particulier pour améliorer les propriétés
mécaniques, mais aussi en tant que retardateur de feu (ignifugation).
Les matériaux lamellaires, en particulier les hydroxydes doubles lamellaires (Leroux et
Besse, 2001) apparaissent comme étant des additifs de choix pour la stabilisation thermique,
la photoprotection, l’élimination des résidus de catalyseurs de polymérisation, l’inhibition de
la corrosion, la prévention de la décoloration, ou encore l’amélioration de l’adhérence
(Leroux et Besse, 2004). Par exemple, utilisés comme additifs dans le polyéthylène, les HDL
permettent d’augmenter l’absorption des rayons IR des films protecteurs des serres.
Parallèlement, la résistance au feu du PVC et d’autres polymères est améliorée lors de
l’incorporation de particules d’HDL (Camino et al., 2001).
4.3 Électrodes / Conducteurs ioniques
Plusieurs études menées sur l’utilisation des hydroxydes doubles lamellaires en tant
qu’électrolytes et conducteurs protonique ont mis en évidence que ces matériaux possèdent
une conduction protonique élevée (DeRoy et al., 1985; DeRoy et Besse, 1989; DeRoy et
Besse, 1991; Lal et Howe, 1981). Ces bonnes performances de conduction ionique peuvent
être attribuées aux échanges protoniques entre les feuillets hydroxylés et les molécules d’eau
interfoliaire, ainsi qu’à la mobilité des anions insérés. Ces capacités ont permis la réalisation
d’un capteur d’humidité à partir de ces phases (Morandi et al., 2006). De fait, les phases HDL
52

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
sont également intéressants comme support pour électrodes modifiées (Itaya, et al., 1987;
Therias et Mousty,1995).
4.4 Précurseurs de matériaux magnétiques
La calcination de phases HDL de type MII–FeII–FeIII–SO4 permet de synthétiser des
ferrites spinelles MFe2O4 (Li et al., 2004). Ces matériaux présentent une magnétisation à
saturation supérieure à celle des spinelles produits par les méthodes de synthèse céramique
traditionnelles bien que leurs compositions soient identiques. En effet, l’utilisation
d’hydroxydes doubles lamellaires comme précurseurs garantit une répartition homogène des
cations métalliques à l’échelle atomique. De plus, des températures de traitement thermique
moins élevées sur des temps plus courts sont nécessaires pour la décomposition des HDL,
comparativement à la synthèse classique des spinelles.
4.5 Piégeage-Restauration environnementale
En raison de leur propriété d’échange anionique élevée et l’importante charge de surface
des feuillets, les argiles anioniques sont aussi des matrices intéressantes pour la restauration
environnementale. Elles sont en effet utilisées pour le piégeage d’espèces polluantes
inorganiques telles que des oxoanions, des phosphates, des anions oxométalates (sélénates,
chromates. . .). Les phases HDL se sont également avérées être de bons supports pour
immobiliser des polluants organiques des milieux aquatiques comme des phénols, des
pesticides (MCPA, Dicamba), des colorants, des substances humiques. . . (Forano, 2004).
4.6 Usage médical
De part leur caractère basique, les phases HDL sont aussi présentes dans le domaine
médical, comme agents antiacides et antipepsiniques. Ces phases sont utilisées pour maîtriser
l’action de l’acide chlorhydrique et de la pepsine dans l’estomac. C’est un traitement efficace
de l’ulcère gastrique (Miyata, 1977; Playle et al., 1974). Ils se révèlent également efficaces
pour la prévention et le traitement des diverses maladies associées aux carences en fer
(Reichle, 1986), ou encore comme inhibiteur de caries en tant qu’additif aux pâtes dentaires.
Des molécules actives (vitamines, médicaments) sensibles à l’environnement (lumière,
oxygène) peuvent être intercalées et donc protégées dans une matrice HDL avant d’être
insérées dans le corps. Ce confinement a été utilisé dans le cas de l’anti-inflammatoire
53

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Ibuprofen, intercalé dans une phase [Mg2AlCl] pour le stabiliser et permettre une diffusion de
manière contrôlée, afin de limiter les effets secondaires (Ambrogi et al., 2001).
El-Toni et coll. ont constaté que des nanocomposites HDL Zn–Al intercalées par 4,4’-
diaminostilbène-2,2’-acide disulfonique recouverts de silice présentaient de bonnes propriétés
comme filtres UV et étaient appropriés à des formulations de crèmes solaires (El-Toni et al.,
2004). Cette capacité à former une barrière aux UV est rapportée également par Duan et coll.
au sujet de phases Zn–Al intercalées par le salicylate (Jiao et al., 2002).
4.7 Hôtes pour biomolécules
Les HDL présentent également des propriétés adéquates pour être hôtes de
biomolécules. En plus d’être biocompatibles (Kwak et al., 2004), ils possèdent en effet une
structure ouverte qui peut intercaler beaucoup de biomolécules anioniques (ADN, ATP,
acides aminés. . .). De plus, leur préparation étant effectuée par chimie douce, cela permet le
maintien de l’intégrité chimique et biologique de la biomolécule. L’immobilisation de
fragments d’ADN a été réalisée par Choy (Desigaux et al.,2006; Choy et al., 2000) dont
l’objectif d’élaborer des vecteurs de gène pour la thérapie génique par simple échange
anionique. L’étude montre que la molécule ne se détériore pas, et que le matériau hybride
pénètre bien à l’intérieur de la cellule avant que la matrice HDL ne se dissolve, en raison du
pH acide, et libère les fragments d’ADN. Récemment, des fragments plus longs, 6000 à 8000
paires de bases, ont pu être introduits dans une structure HDL par coprécipitation (Desigaux
et al.,2006).
L’immobilisation d’enzymes sur des solides est également étudiée pour des applications
en biotechnologie. L’objectif est de protéger efficacement leur activité contre les processus de
dénaturation. Des travaux récents ont montré que l’uréase a une forte affinité avec les phases
HDL et qu’il est possible de produire des biocapteurs en immobilisant l’enzyme sur la matrice
HDL (Vial, 2005 ;Vial et al., 2006).
54

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
Références bibliographiques
• Adachi-Pagano M., Forano C. and Besse J.-P. (2003), J. Mater. Chem., 13(8),
1988–1993.
• Adachi-Pagano M., Forano C., and Besse J.-P. (2000),Chem. Commun., 1, 91–92.
• Allmann R. (1968), Acta Cryst., B24, 972-977.
• Allmann R. (1970), Chimia, 24, 99-108.
• Allmann R. and Jhb N. (1977), Miner. Mh., 3, 136-144.
• Ambrogi G., Fardella V., Grandolini G., and Perioli L. (2001), Int. J. Pharm., 220,
23–32.
• Aminoff G. and Broomè B (1930), Kungl. Sven. Vetensk. Handl. 9, 3, n°.5, 23.
• Aramendía M.A., Borau V., Jiménez C., Marinas J.M., Ruiz J.R. and Urbano
F.J. (2002), J. Solid State Chem., 168(1), 156–161.
• Bigey L. (1998)., Thèse de Doctorat, Université Blaise Pascal, Clermont-Ferrand,
• Bish D. L. (1980), Bull. Miner., 103, 170-175.
• Bish D.L. and Livingstone A. (1981), Miner. Mag., 44, 339-343.
• Bish D.L. and Brindley G.W. (1977), Amer. Min.,62, 458-464.
• Boclair J.W. and Braterman P.S. (1999), Chem. Mater., 11(2), 298–302.
• Boclair J.W., Braterman P.S., Jiang J., Lou S., and Yarberry F. (1999), Chem.
Mater., 11(2), 303–307.
• Boehm H.P., Steinle J. and Vieweger C. (1977), Angewandte Chemie, , 89(4), 259–
260.
• Braterman P. S., Xu Z. P. and F. Yarberry (2004), In Handbook of Layered
Materials; S. M. Auerbach, K.A. Carrado, P. K. Dutta, Eds.; Marcel Dekker: New York, 373.
• Bravo-Suarez J. J., Paez-Mozo E. A. and Oyama S. T. (2004), Quim. Nova, 27 (4),
601-614.
• Brindley G. W. and Kikkawa S. (1979), Am. Mineral., 64(7-8), 836 -843.
• Brindley G.W. and Kikkawa S. (1980), Clays Clay Miner, 28, 87-91.
• Bröcker F.J., Dethlefsen W., Kaempfer K. K., Marosi L., Scwarzmann M.,
Triebskorn B. and Zirker G. (1972), German Patent (BASF AG) 2, 255 909.
• Bröcker F.J. and Kaempfer K. (1975), Chem Ing. Techn., 47 513.
• Bröcker F. J. and Kainer L. (1970, 1971), German Patent (BASF AG) 2,024,282,
and UK Patent(BASF AG) 1, 342, 020.
55

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• Brown G. and Van Oosterwyck-Gastuche M. C. (1967), Clay Miner, 7(2), 193–
201.
• Busetto C., Del Piero G., Manara G., Trifiro F. and Vaccari A. (1984), J. Catal.
85, 260-266.
• Cai H., Hillier A. C., Franklin K. R., Nunn C. C., and Ward M. D. (1994),
Science, 266, 1551–1555.
• Camino G., Maffezzoli A., Braglia M., De Lazzaro M., and Zammarano M.
(2001), Polym. Degrad. Stab., 74(3), 457–464.
• Carrado K.A., Kostapapas A. and Suib S.L. (1988), Solid State Ion. 26 (2), 77–86.
• Cavani F., Trifiro F. and Vaccari A. (1991), Catal. Today, 11, 173–301.
• Chanakya M. (1987), US Patent 4, 656, 156.
• Chibwe K. and Jones W. (1989), Chem. Mater., 1, 489–490.
• Choudary B.M., Prassad A. D., Bhuma V. and Swapna S. (1992), J. Org. Chem.,
57, 5841-5844.
• Choy J. H, Kwak S. Y., Jeong Y. J., and Park J. S.(2000), Angew. Chem. Int. Ed.,
39(22), 4041– 4045.
• Costantino U., Marmottini F., Nocchetti M., and Vivani R. (1998), Eur. J. Inorg.
Chem., 1439–1446.
• Costantino U., Curini M., Montanari F., Nocchetti M., and Rosati. O. (2003), J.
Mol. Catal. A: Chem., 195(1), 245–252.
• Courty Ph., Durand D., Freund E. and Sugier A. (1982), J. Mol. Catal., 17, 241-
254.
• Courty Ph. and Marcilly Ch. (1985), in G. Poncelet, P. Grange, P. A. Jacobs (Eds),
Prepartaion of catalysts III, Elsevier, Vol. 7, 1.
• Delmas C. and Borthomieu Y. (1993), J. Solid State Chem., 104, 345-352.
• Del Piero G., Di Conca M., Trifiro F. and Vaccari A. (1989), in C. Morterra, A.
Zecchina and G. Costa (Editors), Structure and Reactivity of Surfaces, Elsevier, Amsterdam, ,
239.
• Dimotakis E. D. and Pinnavaia T. J. (1990), Inorg. Chem., 29(13), 2393–2394.
• DeRoy A., , Besse J.-P. and Bondot P. (1985), Mater. Res. Bull., 20(9), 1091–1098.
• DeRoy A. and Besse J.-P. (1989), Solid State Ionics, , 35(1-2), 35–43.
• DeRoy A. and Besse J.-P. (1991), Solid State Ionics, 46, 95–101.
56

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• DeRoy A, Forano C., El Malki K., Besse J. P. and Occelli M. L. (1992)., H. E.
Robson Editions, Expanded Clays and Other Microporous Solids, Vol.2, Reinhold, New
York,
• DeRoy A, Forano C. and. Besse J.-P. (2001), Layered Double Hydroxides. Rives, V.
Ed, Nova Science Publishers, Inc, , pages 1–37.
• Desigaux L., Ben Belkacem M., Richard P., Cellier J., Leone P., Cario L., Leroux
F., Taviot-Gueho C. and Pitard B. (2006), Nano Lett., 6(2), 199–204.
• Drits V.A., Sokolova T.N. and Cherkashin V.I. (1987), Clays Clay Min, 35, 401-
417.
• El-Toni M., Yin S. and Sato T. (2004), J. Solid State Chem., 177(9), 3197–3201.
• Evans, D.G. and Duan, X. (2006), chem. Commun: 486-495.
• Feitknecht W., Fischer G., and Helu G. (1935), Helv. Chim. Acta, 18, 555–569.
• Feitknecht W. (1942), Helv. Chim. Acta, 25, 555–569.
• Feitknecht W. (1942), Helv. Chim. Acta, 25, 131-137.
• Forano C. (2004), Environmental remediation involving layered double hydroxides".
Clay Surfaces – Fundamentals and Applications, F. Wypych, K.G. Satyanarayana, Elsevier
Academic Press ,425–458.
• French Patent (1971), 2, 091, 785, to BASF AG.
• Gastuche M.C., Brown G. and Mortland M. (1967), Clay Minerals, 7, 177-192.
• He J., Wei M., Li B., Kang Y., Evans D. G. and Duan X. (2006), Structure and
Bonding. Duan, X. and Evans, D. G. Eds, Springer, 119, 89–119.
• Hibino T., Yamashita Y., Kosuge K. and Tsunashima A. (1995), Clays Clay
Miner., 43(4), 427–432.
• Hibino T. and Jones W. (2001), J. Mater. Chem., 11(5), 1321–1323.
• Hibino T. (2004), Chem. Mater., 16(25), 5482–5488.
• Indira L., Dixit M. and Kamath P. V. (1994), J. Power Sources, 52, 93-97.
• Itaya K., Chang H. C. and Uchida I. (1987), Inorg. Chem., 26(4), 624–626.
• Jiao Q. Z., Zhao Y., Xie H., Evans D. G., and Duan X.(2002), Chin. J. Appl.
Chem., 19, 1011-1013.
• Jose dos Reis M., Silverio F., Tronto J. and Valim J. B. (2004), J. Phys. Chem.
Solids, 65(2-3), 487–492.
• Kayano M. and Ogawa M. (2006), Clays Clay Miner., 54(3), 382–389.
57

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• Kirm Medina F., Rodriguez X., Cesteros Y., Salagre P., and Sueiras J.E ( 2004),
Applied Catalysis A: General, , 272(1), 175–185.
• Kohjiya S., Sato T., Nakayama T. and Yamashita S. (1981), Makromol. Chem.
Rapid Commun. 2, 231-233.
• Kruissink E. C., Alzamora L. E., Orr S., Doesburg E. B. M., Van Reijen L. L.,
Ross J. R. H. and Van Veen G. (1979), in B. Delmon, P. Grange, P. Jacobs, G. Poncelet
(Eds), Preparation of Catalysts II, Elsevier, Amsterdam, 143.
• Kruissink E.C.,. Van Reijen L.L and Ross J.R.H. (1981), J. Chem. Soc., Faraday
Trans. 1, 77, 649-663.
• Kwak S. Y., Kriven W. M.,. Wallig M. A and Choy J. H. (2004), Biomaterials,
25(28), 5995–6001.
• Lal M. and Howe A. T. (1981), J. Solid State Chem., 39(3), 368–376.
• Leroux F. and. Besse J.-P (2001), Chem. Mater., 13(10), 3507– 3515.
• Leroux F. and Besse. J.-P. (2004), Layered double hydroxide/ polymer
nanocomposites". Clay Surfaces -Fundamentals and Applications, F. Wypych, K.G.
Satyanarayana, Elsevier Academic Press, 459–495.
• Li F. and Duan X. (2006), Applications of layered double hydroxides". Structure and
Bonding. Duan, X. and Evans, D. G. Eds, Springer, 119, 193–223.
• Li F. and Duan, X. (2005), Struct. Bond. 119 (1), 193–223.
• Li F., Liu J., Evans D. G. and Duan X. (2004), Chem. Mater., 16(8), 1597–1602.
• Liu Z., Ma R., Osada M., Iyi N., Ebina Y., Takada K. and Sasaki T. (2006), J.
Am. Chem. Soc., 128(14), 4872–4880.
• Lopez T., Bosch P., Ramos E., Gomez R., Navara O., Acosta D. And Figueras F.
(1996), Langmuir, 12, 189-192.
• Lòpez Nieto J. M., Dejoz A. and Vazquez M. I. (1995), Applied Catalysis A, 132,
41–59.
• Malherbe F. (1997), Thèse de Doctorat, Université Blaise Pascal, Clermont-Ferrand.
• Manasse E. (1915), Atti. Soc. Toscana Sc. Nat., Proc.Verb.,24, 92.
• Marchi A.J., Sedran A.G. and Apesteguia C.R. (1986), in Prepr. IVth Int. Symp.
On Scientific Bases for the Preparation of Heterogeneous Catalysts, Louvain-la-Neuve(B), ,
H-7.
58

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• Marchi A.J, Di Cosimo J.I. and Apesteguia C.R. (1988), in F. Cossio, O.
Bermudez, G. Del Angel and R. Gomez (Editors), Proc.XI IberoAmer. Symp. on Catalysis,
IMP, Mexico D.F., Vol. 1, 25.
• Martin E. and Pearson A. (1996), U. S. Patent 5, 514, 361.
• Mascalo G. and Marino O. (1980), Miner. Mag., 43, 619-621.
• Miyata S., Kumura T., Hattori H. and Tanabe K. (1971), Nippon Kagaku Zasshi,
92, 514-519.
• Miyata S. (1975), Clays Clay Miner., 31(23), 369-375.
• Miyata S. (1977), Kagaku Gijutsushi Mol, 15(10), 32–37.
• Miyata S. (1980), Clays Clay Miner., 28, 50-56.
• Miyata S. (1983), Clays Clay Miner., 31, 305-311.
• Miyata S. (1985), US Patent 4, 514, 389.
• Miyata S. and Kumura T. (1973), Chem. Letters, 843-848.
• Morandi S., Prinetto F., Di Martino M., Ghiotti G., Lorret O., Tichit D., Malagu
C., Vendemiati B., and Carotta M. C. (2006), Sens. Actuators, B: Chemical, 118(1-2), 215–
220.
• Nakatsuka T., Kawasaki H., Yamashita S., Kohjiya S. (1979), Bull. Chem. Soc.
Japan, 52, 2449-2450.
• Ogawa M. and Kaiho H. (2002), Langmuir, 18(11), 4240–4242.
• Oh J.-M, Hwang S. H. and Choy J. H. (2002), Solid State Ionics, 151, 285– 291.
• Pasel J., Kazner P., Montanari B., Gazzano M., Vaccari A., Makowski W.,
Lojewski T., Dziembaj H. and Papp R. (1998), Appl Catal B, 18, 199–213.
• Pausch I., Lohse H.H., Scürmann K. and Allmann R. (1986), Clays Clay Miner.,
34, 507-510.
• Pavan P. C., Crepaldi E. L., de A. Gomes G., and Valim J. B. (1999), Colloids
Surf. A: Physicochemical and Engineering Aspects, 154(3), 399– 410.
• Pawlaczyk J., Kokot Z. And Rafinska A. (1985), Acta Pol. Pharm., 42, 153-158.
• Playle C., Gunning S. R, and Llewellyn A. F. (1974), Pharm Acta Helv, 49(9-10),
298– 302.
• Prevot V., Forano C., and Besse J.-P. (2005), Chem. Mater., 17(26), 6695–6701.
• Rao M. M., Reddy B. R., Jayalakshmi M., Swarna V. and Sridhar B. (2005),
Mater. Res. Bull., 40, 347–359.
59

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• Reichle W.T. (1986), Chemtech, , 16, 58–63.
• Rives V. (2001), Layered double hydroxides: present and future. Nova Science
Publishers, New York,
• Rives V. (2002), Mater. Chem. And Phys., 75, 19-25.
• Rives V., Prieto O., Dubey A. and Kannan S. (2003), J. Catal., 220(1), 161–171.
• Ross G. J. and Kodama H. (1967), Amer. Min., 52, 1037-1040.
• Sato T., Fujita H., Endo T. and Shimada M. (1988), React. Solids, 5, 219-228.
• Shaw W. M. H. R and Bordeaux. John J. (1955), J. Am. Chem. Soc., 77, 4729–
4733.
• Sutker B. J. (1987), in W. Gerhartz, Y. S. Yamamoto, B. Elvers, J. F. Rounsaville,
G. Schulz (Editors), Ullmann's Encyclopedia of Industrial Chemistry, VCH, Veinheim (D),
A11, 123.
• Taylor H.F.W. (1969), Miner. Mag., 37, 338-342.
• Taylor R. M. (1984), Clay Miner, 19(4), 591–603.
• Tichit D., Bennani M. N., Figueras F. and Ruiz J. R. (1998), Langmuir, 14, 2086-
2091.
• Therias S. and Mousty C. (1995), Appl. Clay Sci., 10, 147–162.
• Thevenot F., Szymanski R. and Chaumette P. (1989), Clays Clay Miner., 37, 396-
402.
• Thévenot F. (1994), Thèse de Doctorat, Université Blaise Pascal, Clermont-Ferrand,
• Vaccari A. (1999), Appl. Clay Sci., 14(4), 161–198.
• Van Oosterwyck-Gastuche M. C., Brown G. and Mortland M. M., Clay Miner,
1967, 7(2), 177–92.
• Vial S. (2005), Thèse de Doctorat, Université Blaise Pascal, Clermont-Ferrand.
• Vial S., Prevot V., and Forano C. (2006), J. Phys. Chem. Solids, , 67, 1048–1053.
• Vial S., Ghanbaja J. and Forano C. (2006), Chem. Commun., 3, 290–292.
• Vial S., Forano C., Shan D., Mousty C., Barhoumi H., Martelet C. and Jaffrezic
N. (2006), Mater. Sci. Eng., C, 26, 387–393.
• Venugopal B.R., Shivakumara C. and Rajamathi M. (2006), J. Colloid Interface
Sci., 294, 234–239.
• West R. (1990), Solid State Chemistry and its application, Wiley, New York, USA.
• Williams G. R. and O’Hare D. (2006), J. Mater. Chem., 16(30), 3065–3074
60

CHAPITRE 2 : LES HYDROXYDES DOUBLES LAMELLAIRES (ARGILES ANIONIQUES)
• Woltermann G.M. (1984), US Patent 4,454, 244, to Ashland Oil.
• Yang W., Kim Y., Liu P. K. T., Sahimi M. and Tsotsis T. T. (2002), Chem. Eng.
Sci., 57, 2945-2953.
• Zhao Y., Li F., Zhang R., Evans D. G. and Duan X. (2002), Chem. Mater., 14,
4286–4291.
61

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE
BASE ET GENERALITES
62

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET
GENERALITES
1. Historique et définition des biocapteurs L’histoire des capteurs chimiques, parmi lesquels les biocapteurs, représente un sous
groupe très connu, débute en 1960 suite aux premiers travaux réalisés par Clark et Lyon pour
la détection du glucose (Lyons, 1962). Ces études ont été approfondies vers 1967 suite à la
naissance du premier biocapteur réalisé par Updicke et Hicks (Updicke et Hicks, 1967). Ce
biocapteur est une électrode à enzyme pour le dosage du glucose basée sur l’immobilisation
de la glucose- oxydase dans un gel de polyamide. A la naissance du premier capteur
biologique, la réalisation de ces dispositifs était simple et basée sur l’immobilisation physique
de l’enzyme soluble à la surface d’un transducteur électrochimique. Quelques années plus
tard, ces systèmes sont devenus de plus en plus complexes suite au couplage direct de
l’enzyme avec le transducteur ce qui peut réduire partiellement ou totalement son activité
biologique. Pour cette raison, il était important de mettre au point des méthodes
d'immobilisation de l’enzyme qui perturbent le moins possible ses propriétés catalytiques.
Plusieurs définitions et terminologies ont été utilisées pour définir un biocapteur
(Higson et al., 1994 ; Keese et Giaever, 1994) ; de façon générale, un capteur biochimique ou
capteur biologique est un dispositif analytique qui se situe à l’interface entre la physique, la
chimie et la biologie. Les composantes essentielles du biocapteur incluent un élément de
reconnaissance moléculaire qui constitue le biorécepteur et une composante physique qui
constitue ce qu’on appelle le transducteur. L’élément sensible est constitué par un matériau
biologique chargé de reconnaître sélectivement l’espèce spécifique qu’on désire analyser
(analyte). En outre, plusieurs entités biologiques sont susceptibles d’être employées telles que
les enzymes, les anticorps, les antigènes, les cellules, les tissus, l’ADN.….. . En général, le
transducteur joue le rôle d'un traducteur en convertissant les interactions ou les
reconnaissances physico-chimiques entre le biorécepteur et la substance cible en un signal
électrique, optique ou chimique (Figure I.3.1).
63

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
Depuis le premier biocapteur développé par Clark et Lyon au début des années 1960
(Clark, 1962), la recherche sur leur développement a connu ces dernières années un effort
considérable en raison de leurs nombreuses applications potentielles, que ce soit dans le
domaine médical, agro-alimentaire, ou du contrôle environnemental.
Dans ce dernier cas, les biocapteurs sont considérés comme une solution alternative
particulièrement intéressante, aux techniques analytiques traditionnelles telles que la
chromatographie ou la spectrométrie (la chromatographie en phase gazeuse, couplée à la
spectrométrie de masse (GC-MS), la chromatographie liquide haute performance (HPLC),
couplée à la spectrométrie de masse (HPLC-MS), la spectrométrie d’absorption atomique
(SAA) ou encore la technique de spectrométrie d’émission Plasma ICP-AES (ICP-MS). De
plus, leur petite taille, leur facilité d’utilisation ou encore la possibilité qu’ils offrent de
réaliser des mesures sur site en font des outils particulièrement intéressants.
Figure I.3.1:Principe de fonctionnement des biocapteurs
L’élément sensible ou biorécepteur constitue l’élément clé dans la conception d’un
biocapteur. Plusieurs entités biologiques ont été testées pour l’élaboration d’une grande
variété de capteurs biologiques. Parmi ces capteurs, on rencontre les capteurs enzymatiques,
les capteurs immunologiques, les capteurs cellulaires et les capteurs ADN ou bio puces qui
sont respectivement, à base d’enzymes, d’anticorps ou d’antigènes, de cellules et d’ADN
(acide
64

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
désoxyribonucléique). Il est aussi important de présenter les principales classes de
biomolécules utilisées comme récepteurs pour la réalisation de biocapteurs.
2. Les transducteurs Le transducteur est l’élément physique qui sert à exploiter la modification biochimique
issue d’une interaction entre un analyte et le biorécepteur pour la transformer en signal
électrique. Le type de transducteur sera choisi en fonction des modifications biochimiques se
produisant au niveau du biorécepteur. Cette adéquation entre le transducteur et l’élément
biologique permettra d’obtenir un signal sensible, facilement exploitable et avec un minimum
de bruit de fond. Plus le bruit de fond sera faible, plus il assurera un seuil de détection bas et
améliorera les performances du biocapteur (Tran-Minh, 1992 ; Comtat et Bergel, 1997).
2.1 Les capteurs thermiques
Les capteurs thermométriques appelés aussi capteurs enthalpimétriques sont destinés à
déterminer la concentration d’un substrat par la variation d’enthalpie associée à la réaction
enzymatique (Ramanathan et Danielsson, 2001). Cette méthode fait essentiellement appel
aux réactions exo ou endothermiques. Le changement de température, ΔT, est déterminé par
un microcalorimètre et est relié aux variations d’enthalpie, ΔH, et la capacité de chaleur du
réacteur, Cp par la relation suivante:
Cp
HnT Δ×=Δ (I.3.1)
Avec n étant le nombre de moles de substrat ayant réagit.
Pour ce type de biocapteur, il n’est pas nécessaire de mesurer le produit de la réaction,
seule la chaleur dégagée au cours de la réaction est utilisée dans la mesure.
2.2 Les capteurs optoélectroniques
La reconnaissance biologique à la surface du transducteur se traduit par des
changements des propriétés optiques de celui-ci. Ces appareils sont constitués d’une ou
plusieurs fibres optiques pour le rayon incident et pour le faisceau lumineux qui sera mesuré.
Les biocapteurs à
65

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES fibre optique offrent de nombreux avantages; ils ne sont pas influencés par le champ
électrique, ils sont utilisables pour les mesures en continu, et ils maintiennent les propriétés
chimiques des échantillons pendant la mesure, ils ne demandent ni signal de référence ni
armature électrique. De plus, leur sensibilité est facilement adaptable à la gamme de mesure
désirée. Mais ils sont perturbés par la lumière naturelle, ils sont alors normalement mis en
oeuvre dans l’obscurité. Ce type de biocapteur permet de détecter de nombreux pesticides
(Jeong-Woo et al., 2003 ; Hsiao-Chung, 2003 ; Oroszlan et al., 1993 ; Brecht et al., 1995 ;
Schipper et al.,1997 ; Meusel et al., 1998 ; Skládal et al.,1999 ; Barzen et al.,2002). Toutefois, la gamme de détection de ces pesticides n’est pas encore très intéressante de l’ordre
de dizaine de ppm à la centaine de ppm. Il est nécessaire donc de les développer davantage
pour une bonne application dans l’environnement.
2.3 Les capteurs à effet piézoélectrique
Les transducteurs piézoélectriques mesurent des variations de masse à leur surface. Le
principe de ces transducteurs repose sur les propriétés piézoélectriques des matériaux utilisés.
L’effet piézo direct correspond au phénomène qui a lieu lorsqu’un solide cristallin est soumis
à une contrainte mécanique appliquée sur ses faces: la déformation du cristal s’accompagne
d’une polarisation électrique dont l’amplitude est proportionnelle à la contrainte appliquée. La
piézoélectricité traduit donc l’interdépendance des propriétés électriques et mécaniques de
certains matériaux. A l’inverse, si une différence de potentiel est appliquée entre les faces
d’un matériau piézo, cela fait apparaître des contraintes au sein du matériau qui induit sa
déformation: c’est l’effet piézo inverse, à la base du fonctionnement des transducteurs piézo.
L’application d’un champ électrique alternatif au sein d’un cristal de quartz provoque sa
déformation, le cristal se met à vibrer et une onde acoustique est générée au sein du matériau.
Quand le quartz est inséré dans un circuit électronique approprié, la vibration peut être
entretenue. Des variations de masse à la surface du cristal vont modifier la fréquence de
vibration selon l’équation de Sauerbrey (Sauerbrey, 1959 ; Ader et Callum, 1983) et ces
vibrations de fréquence (Δf) seront enregistrées.
66

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
AmFf Δ
×××−=Δ − ²103.2 6 (I.3.2)
Où f est la fréquence de résonance (en Hz), F la fréquence fondamentale du cristal (en Hz),
Δm la variation de masse (en g), A : la surface de l’électrode (en cm2).
P. Skádal a utilisé un capteur de ce type pour la détection du virus de l’hépatite type C.
Son capteur utilise une électrode résonante à 10 MHz. Le biorécepteur utilisé ici est un brin
d’ADN (Skládal et al., 2004). En utilisant des électrodes similaires, d’autres auteurs se sont
intéressés à détecter d’autres types de virus (Uttenthaler et al., 1998 ; Zhou et al., 2002).
Dans les études de contrôle environnemental, les biocapteurs enzymatiques à effet
piézoélectrique servent à détecter indirectement des pesticides ou des métaux lourds dans les
eaux par inhibition de l’activité enzymatique en présence de ces polluants (Tombelli et al.,
2000 ; Horácĕk et Skládal, 2000). Par exemple le système utilisé dans le travail de Alexander
Makower et ses collègues permet de détecter 2 ppm de DiisopropulFluoroPhosphate (DFP)
(Makower et al., 2003).
2.4 Les capteurs électrochimiques
Nous allons classer les capteurs électrochimiques selon leur mode de transduction:
potentiométrique, conductométrique ou ampérométrique : figure I.3.2.
Figure I.3.2 : Détection électrochimique
Les capteurs pontentiométriques et ampérométriques sont les plus répandus mais il y a
peu de travaux portant sur les capteurs conductimétrique. Toutefois, depuis les années 1980s,
67

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
les capteurs conductimétrique commencent à être à leur tour de plus en plus utilisés compte
tenu de leur facilité d’élaboration.
2.4.1 Les capteurs ampérométriques
L’ampérométrie est une technique qui repose sur la détermination de l’intensité de
courant qui traverse une cellule électrochimique à un potentiel imposé. Elle est fonction de la
concentration des corps électroactifs qui seront oxydés ou réduits à une électrode indicatrice,
la seconde étant en général une électrode de référence. Il est donc possible, après étalonnage,
de déterminer la concentration de certains corps présents, par la mesure de l’intensité. Il existe
trois générations de biocapteurs ampérométriques.
Dans la première génération de biocapteurs ampérométriques développé par L. Clark,
l’analyte enzymatiquement généré est directement oxydé ou réduit au niveau de l’électrode.
Par exemple dans le cas où le biorécepteur est la glucose oxydase (GOD), la réaction rédox du
glucose est donnée par l’équation suivante :
D’après cette réaction on voit que, le capteur peut mesurer soit la consommation
d’oxygène, figure I.3.3a. soit la production d’eau oxygénée, figure I.3.3b. (Clark, 1962 ;
Guilbault et Lubrano, 1973). De tels systèmes sont encore exploités et développés bien que
certains problèmes persistent.
a b
S: Substrat, P: Produit
a b
S: Substrat, P: Produit Figure I.3.3 : Capteurs ampérométriques de première génération
a) détection de la consommation d'oxygène b) détection de la production en eau
oxygénée
68

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
La deuxième génération de biocapteurs ampérométriques, figure I.3.4, a été développée
pour réduire les phénomènes d’interférences en abaissant le potentiel de détection des espèces
électroactives.
Figure I.3.4 : Biocapteur ampérométrique de deuxième génération.
M est le médiateur sous forme oxydée (ox)ou réduite(réd)
Pour ce faire on utilise des médiateurs rédox artificiels (Cass, 1984; Turner et al., 1987)
comme le ferrocène, les quinones, le ferricyanure, le phénazine-méthosulfate, le
tétrathiafulvalène (TTF), et le tétracyanoquinodiméthane (TCNQ). Ces médiateurs peuvent
être simplement absorbés à la surface de l’électrode, emprisonnés dans les mailles de
polymère (Heller, 1992 ; Koide et Yokoyama, 1999), du sol-gel (Pankratove et Lev, 1995 ;
Walcarius, 1998) ou greffés sur celui-ci.
Enfin, une troisième génération de biocapteur est à l’étude. Dans cette génération,
l’enzyme est directement connectée à l’électrode afin de simplifier le transfert d’électrons. Ce
système permet alors de s’affranchir des problèmes d’interférences rencontrés lors de dosage
dans des milieux complexe (Schuhmann, 1993 ; Lötzbeyer et al., 1996). Très récemment
L.A. Dang a présenté un nouveau copolymère conducteur multifonctonnel (juglone-co-
juglone-3-acide thioacétique ou poly (JUG-co-JUGA)) permettant à la fois le greffage
covalent des enzymes (Pyruvate Oxydase (PyOD)) et le transfert d’électrons entre les sites
actifs de l’enzyme et l’électrode. Le biocapteur à pyruvate à base de ce copolymère JUG-co-
JUGA peut fonctionner à 0,1 V/ECS, un potentiel faible pour que la réponse du biocapteur
soit indépendante des interférences en solution conduisant à une bonne sélectivité du
biocapteur (Dang, 2004).
69

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
Dans le même groupe que L.A. Dang, J. Haccoun a utilisé le même copolymère pour
développer le biocapteur ampérométrique à base de la Lactase Oxydase (LOD) pour la
détermination du substrat L-Lactate. Le biocapteur à base de ce copolymère conducteur
travaillant à très faible potentiel d’oxydation -0,1 V/ECS (le plus bas que tous les médiateurs
immobilisés utilisés jusqu’à présent) permet une gamme de détection relativement large de
concentration en substrat (0,05 – 0,8mM) avec une sensibilité très remarquable (S=110 ± 20
μA/mM/cm2). De plus, il est peu sensible à la présence d’oxygène en solution (Haccoun,
2004).
L’ampérométrie est le mode le plus utilisé pour les biocapteurs enzymatiques. La
majorité des dispositifs commercialisés (Matsushita Ltd, 1984 a et b; Mitsubishi Ltd, 1985e ;
Kessler, et Hoeper, 1985) sont des électrodes ampérométriques. De nombreux brevets ont été
déposés sur ce type de biocapteurs et les applications sont très variables (Klitgaard et
Pedersen, 1984; Corenman et al., 1985 ; Kriebisch et Netz, 1985 ; Chang et Ramsey,1985).
2.4.2 La détection potentiométrique
La potentiométrie est une méthode électrochimique basée sur la mesure de la différence
de potentiel entre une électrode de mesure et une électrode de référence. La détermination des
potentiels des électrodes permet de mesurer directement la concentration de l’analyte à doser.
Dans ce type de système, un équilibre local est établi à la surface du capteur et conduit à la
génération d’un potentiel proportionnel au logarithme de la concentration (activité) de
l’échantillon selon la loi de Nernst (équation 3):
red
oxredox
aa
fnTREEp ln/
0 ×××
+= (I.3.3)
où EP représente le potentiel du couple rédox; E°Ox/Red le potentiel normal standard du couple
rédox; R la constante des gaz parfaits (8.314 J . K-1.mol-1); aOx/aRed le rapport de l’activité de
l’espèce déterminant le potentiel à l’état oxydé et à l’état réduit; T la température absolue en
Kelvin.
70

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES 2.4.3 Les capteurs conductimétriques
La conductimétrie est une technique électrochimique alternative à l’ampérométrie et à la
potentiométrie (Bard et Faulkner, 1980). La conductimétrie permet de mesurer les variations
(consommation ou production) d’espèces chargées générées au cours des réactions
enzymatiques. La conductance d’un corps est donnée par l’équation 4;
λ
γ AG ×= (I.3.4)
La constante γ (en S.cm-1) qui est la constante caractéristique d’un produit connu, est la
conductance ou conductivité spécifique; A/λ (en cm) la constante géométrique de la cellule.
La mesure de la conductance d’un électrolyte s’effectue en immergeant dans la solution une
cellule de mesure comprenant deux électrodes dont la surface A et la longueur λ sont données.
L’étalonnage ou le contrôle de la cellule sont effectués en mesurant sa conductance Ge pour
un électrolyte de conductivité γe connue : k = Ge/γe. Lorsque l’on connaît la constante de
conductivité de la cellule k, on peut déterminer la conductivité γ d’un électrolyte quelconque,
en mesurant la conductance G de la cellule immergée dans cet électrolyte : γ= G/k.
La mesure de conductance est considérée comme relativement non-sélective.
Cependant, des nombreux brevets récents montrent de nouvelles approches dans le
développement des biocapteurs conductimétriques notamment dans le domaine biomédical
pour la détermination du glucose dans le sang et l’urine. Par exemple un biocapteur proposé
par Toshiba Corp. dans lequel on a mis une membrane semi conductrice de polyacétylène
dopé de sels d’iode entre deux électrodes. Dans ce biocapteur, un mélange de deux enzymes
de glucose oxydase et de peroxydase ont été immobilisée sur cette membrane polymérique.
En présence du glucose, la réaction catalysée par les enzymes conduit à un changement de
conductance de la membrane (Hu et Vogelhut, 1985). Par ailleurs, la structure proposée dans
le travail de Hu, K. W. J. et Vogelhut utilise une matrice de polypyrrole tetrachlororuthenate
étalée sur une électrode en or dans laquelle est immobilisée du glucose oxydase (Toshiba
Corp, 1985). Le biocapteur de Kuroda et Osawa (Kuroda et Osawa, 1985) utilisant l’enzyme
creanine deaminase permet, la détermination de la créatinine, un des paramètres qui permet
d’estimer le fonctionnement des reins.
71

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
L’application des biocapteurs conductimétriques n’est pas limitée au domaine
biomédical. P.C.J. Roach et al., ont réalisé un système comprenant une électrode en diméthyl
silicone poreux (10x1 mm de diamètre-épaisseur chez ImmobaSilTM, Ashy Scientific,
Coalville, Leics., UK) comme électrode de travail et deux électrodes de platine. On a
immobilisée sur l’électrode de travail une membrane Rhodococcus ruber NCMB 40757,
préparée par un mélange d’acétonitrile, d’hydrochlorure de thiamine, de pantothénate de
calcium, d’hydrochlorure de pyridoxine, de d-biotine et d’un tampon KH2PO4, dans laquelle
se trouve l’enzyme nitrilase. Le système permet une large gamme de détection très linéaire de
l’acrylonitrile: 106 - 2650 ppm (Roach et al., 2003). Le système développé par Zhylyak G.A
et S. Dzyadevych permet quant à lui de déterminer les métaux lourds avec une gamme de
détection très intéressante (Hg2+: 1μM; Cu2+ : 2μM; Cd2+: 5μM; Co2+: 10 μM, Pb2+: 20μM;
Sr2+:100 μM) en utilisant l’enzyme uréase (Zhylyak et al., 1995). Les chercheurs s’intéressent
de plus en plus à développer ce type de capteur (Limbut et al.,2004 ; Frances et al., 2000 ;
Norman et al.,1996 ; Piletsky et al.,1995 ; Pak et al., 2001) pour la détection de polluants en
milieu aqueux car leur fabrication est beaucoup plus simple par rapport aux autres types de
capteurs (Evangelyn et al.,2003).
3. Les biorécepteurs enzymatiques Parmi les biorécepteurs, les enzymes sont très largement utilisées dans le domaine des
biocapteurs. Elles sont des biocatalyseurs très spécifiques (Tableau I.3.1).
Les réactions qu’elles catalysent sont relativement simples: l’hydrolyse, l’oxydation, la
décarboxylation de liaisons peptides par exemple. Les enzymes, jusqu’à maintenant, ont été
employées dans les biocapteurs pour la détermination d’environ 80 substrats
3.1 Enzymes : les biocatalyseurs
L’enzyme est une molécule protéinique de structure très complexe constituée d’acides
aminés formant des chaînes reliées par des liaisons peptidiques. Les enzymes jouent le rôle de
biocatalyseur et assure le déroulement de toutes les réactions métaboliques. Les enzymes
accélèrent 103 à 106 fois la réaction correspondante qui se déroulerait spontanément.
72

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
En abaissant l’énergie d’activation de la réaction qu’elle catalyse, une enzyme abaisse le
niveau énergétique de l’état de transition et accélère ainsi la réaction. Les enzymes ont une
stéréospécificité tellement forte qu’elles effectuent des réactions leur permettant de choisir
parmi des différents énantiomères ou de discriminer d’autres groupes pratiquement identiques
entre eux.
Tableau I.3.1 : Exemples d’enzymes inhibées par différents inhibiteurs (Bilitewski et
Turner, 2000)
3.2 Classification des enzymes
Les enzymes sont répertoriées en 6 catégories (Stolkowski, 1983) :
• les hydrolases ;
• les oxydoréductases ;
• les transférases ;
• les lyases;
• les isomérases
• les liages.
73

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES 3.3 Les techniques d’immobilisation des enzymes
L’immobilisation de l’élément biologique, en particulier celle des enzymes, au contact
du transducteur ou placé dans une position optimale vis-à-vis du transducteur, est nécessaire
pour une bonne détection du signal biochimique issu du biorécepteur. Les enzymes peuvent
être immobilisées par des méthodes chimiques et/ou physiques. Les qualités demandées à la
membrane enzymatique sont nombreuses:
• stabilité: la capacité à conserver l’activité de l’enzyme pendant le temps nécessaire des
mesures;
• sélectivité: l’aptitude de l’enzyme à reconnaître un seul analyte défini;
• reproductibilité: l’aptitude de l’enzyme à retrouver l’activité initiale après des
mesures;
• sensibilité: la capacité à détecter une concentration faible de l’analyte;
• accessibilité de l’espèce de reconnaissance: la capacité de l’analyte de passer
facilement à travers la couche sensible vers l’enzyme.
Figure I.3.5 : Exemples de méthodes d'immobilisation d'enzyme
Plusieurs méthodes d’immobilisation permettent de réaliser une association
biorécepteur-transducteur n’empêchant pas l’accès du polluant au biorécepteur (Hartmeier,
1988 ; Mulchandani et Rogers, 1988). Ces méthodes d’immobilisation (figure I.3.5) sont
résumées dans les paragraphes suivants.
74

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
3.3.1 L’adsorption
L’adsorption est due à des interactions de type ionique, hydrophobe ou encore des
liaisons hydrogène entre l’enzyme et la surface du transducteur par l’intermédiaire d’un
matériau actif. Les matériaux actifs peuvent être des résines échangeuses d’ions anionique ou
cationique, du charbon actif, des gels de silice, de l’argile, de l’oxyde d’aluminium, du verre
poreux ou des céramiques (Scheller et Schubert,1992). Cette technique permet une
immobilisation simple et non dénaturante de l’enzyme. Malgré sa simplicité de mise en
œuvre, elle demeure peu utilisée pour la conception des biocapteurs parce que les enzymes
immobilisées peuvent facilement se désorber sous l’action des variations de pH, de la
température, de la concentration en substrat ou de la force ionique (Car et Bowers, 1980). La
stabilité et la durée de vie des biocapteurs sont donc diminuées.
3.3.2 L’inclusion ou le piégeage
La méthode d’inclusion consiste à incorporer l’enzyme dans un gel insoluble. Ce gel
peut être constitué d’une matrice organique (polymère) (Bratov et al., 1995 ; Marty et
al.,1992 ; Wan et al.,1999) ou inorganique (argiles) (Besombes, 1994). Dans les deux cas,
l’enzyme est mélangée au matériau, puis déposé à la surface d’un biocapteur sous certaines
conditions. L’enzyme se trouve piégée mécaniquement à l’intérieur des matrices
polymériques ou inorganiques. La maille de la matrice assure de manière purement physique
la rétention de l’enzyme tout en permettant la diffusion du substrat jusqu’au site actif de
l’enzyme grâce à une porosité du gel suffisante. Toutefois cette technique est parfois limitée
par la taille des pores du gel qui favorise le relargage des enzymes de faible poids
moléculaire. L’activité de l’enzyme est dépendante du microenvironnement local de l’enzyme
immobilisé (pH, force ionique, diffusion moléculaire, etc.)
3.3.3 Le couplage covalent
Le couplage covalent d’enzyme sur un transducteur nécessite la présence d’un
groupement fonctionnel sur la surface de celui-ci. Ces groupements sont en général –COOH, -
NH2, -OH, ou –SH ; ils sont peu réactifs chimiquement et il convient de les activer pour qu’il
réagit dans des
75

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
conditions douces avec des groupements fonctionnels de l’enzyme, n’intervenant pas dans le
processus de reconnaissance moléculaire.
3.3.4 La réticulation et la co-réticulation
La réticulation consiste, à l’aide d’un agent réticulant, à créer des liaisons chimiques qui
renforcent la cohésion de la membrane. Quant à la coréticulation, elle est basée sur le même
principe que la réticulation en utilisant à la fois un agent réticulant et une protéine inerte pour
faciliter ou améliorer la réticulation. L’utilisation de cette protéine permet, par une meilleure
répartition des masses des différentes protéines, une meilleure maîtrise de l’activité
enzymatique sans altérer les propriétés mécaniques des membranes obtenues. Cependant, à
cause de la réaction chimique, une perte d’activité enzymatique de la membrane co-réticulée
peut être observée pendant les mesures.
Le choix de la technique d’immobilisation dépend du type d’élément biologique utilisé
(enzyme, cellule entière…), du transducteur et de l’environnement dans lequel le biocapteur
fonctionne. Il dépend aussi des stabilités mécaniques et chimiques de la matrice et de son coût
(Bowers, 1986).
76

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES
Références bibliographique
• Ader J.F. and Mc Callum J.J. (1983), Analyst 108, 1169-1189.
• Bard A. J. and Faulkner L.R. (1980), Electrochemical Methods, J. Willey & Sons.
• Barzen C., Brecht A. and Gauglitz G. (2002), Biosens. Bioelectron. 17, 289-295.
• Besombes J.L. (1994), Thèse de Doctorat de l’Université de Savoie, spécialité chimie.
• Bilitewski U. and Turner A.P.F. (2000), Biosensor for environmental monitoring.
Eds., Amsterdam, Harwood Academic Publishers.
• Bowers L.D. (1986), Anal. Chem. 58, 523-530.
• Brecht A., Piehler J. and Lang G. (1995), Anal. Chim. Acta. 311, 289-299.
• Bratov A., Abramova N., Munor J., Carlos Domínguez, Alegret S. and Bartrolí J.
(1995), Anal. Chem. 67, 3589-3595.
• Car P.V., and Bowers L.D (1980), Immobilized Enzymes in Analytical and Clinical
Chemistry: Willey, NewYork
• Cass A.E. G., Graham D., Graeme D. F., and Allen H. H (1984), Anal. Chem. 56, 667-671. • Chang J. J. and Ramsey M. (1985),US Patent Application US 4499981.
• Clark L.C. and Lyon, C. (1962), Ann. NY Acad. Sci. 102, 29-45.
• Comtat M. and Bergel, A. (1997), Biofur. 171, 33-36.
• Corenman J. E., New W., Goodman D. E. & Yelderman M. (1985). Perinatal
Oximeter (Nellcor Inc.). European Patent Applicatjon EP 135840.
• Dang L. A. (2004), Thèse de doctorat, Université Paris 7-Denis Diderot.
• Evangelyn A. C. (2003), United States Patent Application US O7449910.
• Frances H. A., Zheng W. and Michaels A. S. (2000), J. Membr. Sci, 167, 227–239.
• Guilbault, G.G. and Lubrano G.J. (1973), Anal. Chim. Acta, 64, 439-455.
• Haccoun J. (2004), Thèse de l'Université Paris 7-Denis Diderot.
• Hartmeier W.(1988), Immobilized biocatalysts: An introduction., Berlin, Springer-
Verlag,
• Heller A. (1992), J. Phys. Chem., 96, 3579-3587.
• Higson S. P. J.,Reddy S. M. R and Vadgama P. M. (1994)., Engineering Science
and Education Journal, 41-48.
• Horácĕk J. and Skládal P. (2000), Anal. Chim. Acta, 412, 37-45.
77

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES • Tsai H.-C., Doong R.-A., Chiang H.-C. and Chen K.-T. (2003), Anal. Chim. Acta.,
481, 75–84.
• Hu K. W. J. & Vogelhut P. O. (1985), European Patent Application EP 140322.
• Choi J.-W., Kim Y.-K.,. Oh B.-K, Song S.-Y. and Lee W. H. (2003), Biosens.
Bioelectron., 18, 591-597.
• Keese C. R. and Giaever I. (1994), IEEE Engineering in Medicine and Biology,
June/July 1994, 435-445.
• Kessler M. and Hoeper J. (1985), FRG Patent Application DE 3332745.
• Klitgaard P. C. and Pedersen K. G., (1984), European Patent Application EP
127148.
• Koide A. and Yokoyama K. (1999), J. Electronal.Chem., 468, 193-201.
• Kriebisch R. and Netz V. (1985), GDR Patent Application DD 218188.
• Kuroda M. & Osawa H. (1985), Japanese Patent Application JP 60151560.
• Limbut W., Thavarungkul P., Kanatharana P., Asawatreratanakul P., Limsakul
C. and Wongkittisuks B. (2004), Biosens. Bioelectron. 19, 813-820.
• Lötzbeyer T., Schuhmann W. and Schmidt H.-L. (1996), Sens. Actuat. B 33, 50-54.
• Lyons L. C. (1962), Ann. NY Acad., Sci, 102, 29-45.
• Makower A., Hálámek J., Skládal P., Kernchen F. and Scheller F. W. (2003),
Biosens. Bioelectron. 18, 1329-1337.
• Marty J. L., Mionetto N. and Rouillon R. (1992), Anal. Letters, 25, 81,389-398.
• Matsushita Ltd (1984), Japanese Patent Application JP 59217152.
• Matsushita Ltd (1984b), Biosensor.Japanese Patent Application JP 59151052.
• Matsushita Ltd (1985a), Japanese Patent Application JP 6025448.
• Meusel M., Trau D., Katerkamp A., Meier F., Polzius R. and Camman K.
(1998),Actuators B. 51, 249-255.
• Mitsubishi Ltd (1985e), Japanese Patent Application JP 6093344.
• Mulchandani A. and Rogers K.R. (1998), Principles of enzyme biosensors. Dans
Mulchandani A., and Rogers K.R., Eds. Totowa, Humana Press.
• Norman F., Sheppard J., Mears D. J. and Guiseppi-Elie A. (1996), Biosens.
Bioelectron, 11 (10), 967-979.
• Oroszlan P., Duveneck G.L., Ehrat M. and Widmerr H.M. (1993), Fiber Sens.
Actuators B. 11, 301-305
• Pak S. C., Penrose W. and Hesketh P. J. (2001), Biosens. Bioelectron.16, 371-379.
78

CHAPITRE 3 : LES BIOCAPTEURS: PRINCIPE DE BASE ET GENERALITES • Pankratove I., and Lev O. (1995), J. Electronal.Chem., 393, 35-41.
• Piletsky S.A., Piletskaya E.V., Elgersma A.V., Yano K., and Karube I. (1995),
Biosens. Bioelectron.10, 959-964.
• Ramanathan K. and Danielsson B. (2001), Biosens. Bioelectron.,16, 417-423.
• Roach P.C.J., Ramsden D.K., Hughes J. and Williams P. (2003), Biosens.
Bioelectron. 19, 73-78.
• Sauerbrey G. (1959), J. Phys., 155, 206-222.
• Scheller F. and Schubert F. (1992), Biosensors.,11, 24–25.
• Schipper E.F., Bergervoet A.J.H., Kooyman R.P.H. and Greve J. (1997), Anal.
Chim. Acta. 341, 171-176.
• Schuhmann W., Kranz C., Huber J. and Wohlschlager H. (1993), Synth. Met. 61,
31-35.
• Skládal P., Deng A. and Kolar V. (1999), Anal. Chim. Acta. 399, 29-36.
• Skládal P, dos Santos Riccardi C, Yamanaka H, da Costa PI. (2004), J Virol
Methods, 117, 145-151.
• Stolkowski J. (1983), Les enzymes, Que sais-je ?, 6ème édition.
• Tombelli S., Mascini M., Sacco C. and Turner A. P.F. (2000), Anal. Chim. Acta,
418, 1-9.
• Toshiba Corp. (1985), Japanese Patent Application JP 6095343.
• Tran-Minh, C. (1991), Les biocapteurs. Principes, constructions et applications,
Masson, Paris, 158.
• Turner A.P.F., Hendry S.P. and Cardosi M.F. (1987), World Biotechnol. 1, 125-
127.
• Updicke S. J. and Hicks G. (1967), Nature, 214, 986-988.
• Uttenthaler E., Köblinger C. and Drost S. (1998), Anal. Chim. Acta, 362, 91-100.
• Walcarius A. (1998), Electroanal.,10 (18), 1217-1235.
• Wan K., Chovelon J.M, Jaffrezic-Renault N. and Soldatkin A.P. (1999), Sensors
and Actuators B 58, 399-408.
• Zhou X., Liu L., Hu M., Wang L. and Hu J. (2002), J. Pharm. Biomed. Anal., 27,
341–345.
• Zhylyak G.A., Dzyadevich S.V., Korpan Y.I., Soldakin A.P. and Elskaya A.V.
(1995), Actuators B24-25, 145-148.
79

DEUXIEME PARTIE : PARTIE EXPERIMENTALE
80

CHAPITRE 1 : MATERIELS ET METHODES
81

CHAPITRE 1 : MATERIELS ET METHODES
CHAPITRE 1 : MATERIELS ET METHODES
1. Introduction Ce chapitre a pour objectif de présenter d’une part les matériaux utilisés dans le cadre de
cette étude et d’autre part les outils employés, ainsi que les diverses techniques utilisées pour
caractériser ces matériaux.
Nous présentons en premier lieu les différentes techniques de caractérisation des
matériaux adsorbants : la diffraction des rayons X (DRX), la spectroscopie infrarouge (IR), la
spectrométrie d’émission plasma (ICP AES), l’analyse thermogravimétrique couplée avec
l’analyse thermique différentielle (ATG-DTG) et la microscopie électronique à balayage
(MEB). Nous donnerons également un aperçu sur la mesure du potentiel zêta de quelques
matériaux.
Ensuite, on passera à la synthèse des matériaux adsorbants. Pour l’argile cationique, on
va présenter le mode de purification et d’obtention d’argile sodée. La préparation de l’argile
organophile se fait par échange cationique avec un tensioactif à longue chaine alkyle : le
CTAB. Tandis que pour l’argile anionique (HDL), on va présenter ici deux modes de
préparations de la matrice HDL : NiAlNO3 : Par coprécipitation et par voie électrochimique.
Notons que la caractérisation électrochimique est effectuée en utilisant la méthode de la
voltamétrie cyclique ainsi que la méthode de voltampéromètrie. Les matériaux hybrides HDL-
molécules organiques sont préparés par coprécipitation et par échange anionique.
La dernière partie de ce chapitre sera consacrée à l’étude de la cinétique et l’isotherme
d’adsorption d’une variété de polluants organiques sur les matériaux adsorbants. Une
modélisation cinétique et isotherme suit chaque étude en utilisant différents modèles
proposés dans la littérature.
82

CHAPITRE 1 : MATERIELS ET METHODES
2. Méthodes de caractérisation 2.1 La diffraction des rayons X
Toutes les mesures par diffraction des rayons X ont été effectuées sur un diffractomètre
de poudre SIEMENS D501 équipé d’un tube à anticathode de cuivre et d’un monochromateur
arrière à lame de graphite. Ce monochromateur isole le doublet Kα1+ Kα2 du cuivre; le
détecteur ne reçoit donc les photons issus de ces transitions que s’ils résultent d’interactions
élastiques du rayonnement avec la matière.
L’échantillon pulvérulent est étalé sur une lame de verre dépoli ou mieux, saupoudré par
tamisage pour limiter les phénomènes d’orientations préférentielles des cristallites. De même,
l’emploi d’un porte échantillon tournant permet d’améliorer la nature isotrope de la statique
d’orientation de ces cristallites.
Les diffractogrammes de routine ont été réalisés dans les conditions opératoires
suivantes :
• Domaine angulaire en 2θ : 2°-70°
• Incrément angulaire en 2θ : 0.08°
• Temps d’intégration par comptage : 4s
Les conditions d’acquisition des données semblent être un compromis acceptable entre
la durée des mesures (environ 1heure), une résolution suffisante par rapport à la largeur
moyenne des raies et une bonne statistique de comptage rendue nécessaire par l’existence
d’un fond continu relativement important.
Les radiations émises par les plans atomiques qui sont en phase vont engendrer un
faisceau cohérent qui pourra être détecté.
La condition pour que les radiations soient en phase s’exprime par la loi de Bragg :
θλ sin2 ××=× hkldn (II.1.1)
où
n : Nombre entier correspondant à l’ordre de la diffraction, λ : Longueur d’onde du
rayonnement utilisé (A°), d : distance entre les plans réticulaires d’une même famille
désignée conventionnellement par les indices de Miller h,k,l (Å) et θ : Angle de diffraction
(°).
83

CHAPITRE 1 : MATERIELS ET METHODES
La diffraction des rayons X (DRX) des poudres et des couches minces d'échantillons
d’HDL préparés par voie électrochimique sont enregistrés en utilisant un diffractogramme
type Philips X’Pert Pro utilisant un rayonnement Kα du cuivre (λ=0.15414 nm), travaillant
dans les conditions suivantes :
• Domaine angulaire en 2θ : 2°-70°
• Incrément angulaire en 2θ : 0.02°
• Temps d’intégration par comptage : 75 s
2.2 Spectroscopie infrarouge à transformée de Fourier
Les mesures en spectroscopie Infrarouge ont été obtenues à l’aide d’un spectromètre à
transformée de Fourier type Nicolet 5700. Les échantillons sont conditionnés sous forme de
pastilles de 13 mm de diamètre, constitué de 2 mg de produit dilué dans 200 mg de KBr. Les
résultats sont présentés en absorbance pour des nombres d’onde compris entre 4000 et 400
Cm-1.
2.3 Spectrométrie d’émission plasma (ICP-AES)
La composition chimique des échantillons a été déterminée par spectrométrie d’émission
Plasma ICP-AES (Voïnovitch, 1988) avec un spectromètre d’émission atomique type Perkin-
Elmer Optima 3000XL.
La spectrométrie atomique d’émission, utilisant la technique du plasma d’argon induit
par des hautes fréquences (Inductively Coupled Plasma-Atomic Emission Spectrometry
ICPAES) comme source d’ionisation et d’excitation, a pour principale application l’analyse
d’échantillons sous forme liquide. Le terme plasma désigne des gaz partiellement ionisés. La
source ICP apparaît comme une flamme blanche, brillante et très intense. Sa température (>
7000 K), plus élevée que celle de la spectrométrie de flamme (3000 K), est à l’origine de
l’intérêt qu’elle suscite en analyse. En effet, à ces températures, les espèces moléculaires sont
parfaitement dissociées en atomes excités et/ou ions libres. Ceci permet l’analyse d’un grand
nombre d’éléments avec une très bonne sensibilité (limite de détection dans l’eau de l’ordre
de μg/l pour beaucoup d’éléments). Comme la plupart des techniques physico-chimiques,
c’est une méthode comparative : les concentrations élémentaires de l’échantillon à analyser
sont déterminées à partir de courbes d’étalonnage réalisées quotidiennement avec des
solutions contenant des quantités connues des éléments à doser.
84

CHAPITRE 1 : MATERIELS ET METHODES
L’échantillon à doser, préalablement mis en solution, est pompé et converti en aérosol
(ensemble de très fines particules liquides dans un flux gazeux) par nébulisation. Cet aérosol
est ensuite transporté jusqu’au plasma où il est vaporisé, atomisé, excité et/ou ionisé. Chaque
atome ou ion excité émet une radiation caractéristique lors de son retour à l’état fondamental.
L’intensité de chacune des longueurs d’onde émise est directement proportionnelle à la
quantité d’atomes de l’espèce impliquée. Un photomultiplicateur, placé dans le champ,
convertit l’intensité lumineuse observée pour chaque longueur d’onde en un courant
électrique d’intensité proportionnelle à la concentration en élément dosé. Un appareil ICPAES
classique comprend donc les éléments suivants :
• un système d’introduction d’échantillon qui permet d’amener l’échantillon au plasma,
• une torche,
• un générateur haute fréquence,
• un système optique qui analyse le spectre émis,
• des systèmes de détection et de traitement du signal qui permettent les déterminations
qualitatives et quantitatives à partir du rayonnement émis.
Notons que cette analyse est utilisée pour le dosage des éléments métalliques constituant
la phase NiAlNO3, ainsi que pour les herbicides Glyphosate et Glufosinate par dosage de
phosphore total.
2.4 Analyse thermogravimétrique et analyse thermique différentielle
Le principe de la thermogravimétrie est basé sur la mesure de la perte de masse en
fonction de la température et du temps. Plusieurs facteurs interviennent pour contrôler la
perte de masse, notons par exemple, les propriétés physico-chimiques des matériaux étudiés
et les conditions opératoires. Au cours de la manipulation, il y a plusieurs réactions qui se
produisent à différentes températures lors de la décomposition des matériaux. Pour mieux
comprendre, en régime dynamique, l’évolution de la structure en fonction du temps et de la
température certaines grandeurs doivent être définies :
• Le pourcentage de perte de masse est exprimé par la relation suivante :
100%0
×=mmt (II.1.2)
85

CHAPITRE 1 : MATERIELS ET METHODES mt : la masse de l’échantillon à l’instant t, m0 : la masse initiale de l’échantillon
• La fonction dérivée est déterminée par la relation suivante :
1001
1 ×⎟⎟⎠
⎞⎜⎜⎝
⎛−−
=−
−
tt
tt
TTmmdérivé (II.1.3)
Les thermogrammes ont été enregistrés à l’aide d’une microbalance SETARAM TG-
DTA92 couplé avec un analyseur à spectroscopie de masse type Thermostar 300 Balzers
Instruments permettant d’opérer sous atmosphère contrôlée, et d’effectuer simultanément
l’analyse thermo-gravimétrique et l’analyse thermique différentielle. Les essais ont été
effectués sur des quantités de poudre de masse initiale 15 mg environ contenus dans des
creusets d’alumine pour des températures allant de 20 à 1100°C et une vitesse de chauffe de
5°/mn.
2.5 Microscopie électronique à balayage
Les clichés de microscopie électronique ont été réalisés sur un microscope type JEOL
5190. Les échantillons pulvérulents sont saupoudrés sur les porte-échantillons puis métallisé à
l’or pendant quelques minutes. Les observations sont effectuées sous une accélération de
voltage de 15keV Au sein du laboratoire CASIMIR (Clermont-Ferrand).
2.6 Zétamétrie
Le potentiel zêta représente la charge que la particule acquiert grâce aux ions qui
l’entourent quand elle est en solution. Le potentiel zêta (ξ) des particules argileuses permet
une estimation de la charge de surface portée par ceux-ci et par conséquent, peut conduire à
une interprétation des résultats obtenus lors de l’adsorption de composés organiques. Le
potentiel Zêta est mesuré par un zétamètre type Malvern.
2.7 Mesures électrochimiques
La technique de voltampérométrie cyclique
La connaissance des caractéristiques fondamentales d’une réaction électrochimique se
fait au moyen de la mesure des variations du courant en fonction du potentiel appliqué aux
bornes d’une cellule d’électrolyse. La détermination expérimentale de la relation entre le
86

CHAPITRE 1 : MATERIELS ET METHODES courant et le potentiel d’électrode se traduit par l’obtention de figures appelées
voltampérogrammes.
Le principe général de la voltampérométrie est donc l’obtention d’une réponse (le
courant) du système étudié à l’excitation (le potentiel) responsable de la réaction
électrochimique désirée. Cette opération est réalisée en effectuant une exploration par
imposition et variation progressive du potentiel d’électrode (balayage de potentiel).
Pour pouvoir imposer le potentiel d’électrode, le faire varier et produire ainsi des
réactions électrochimiques, il faut opérer dans une cellule d’électrolyse comportant trois
électrodes auxquelles un circuit extérieur se trouve connecté. Le potentiel de l’électrode
principale où doivent avoir lieu les réactions que l’on désire produire, électrode dite
indicatrice (ou encore, électrode de travail) peut être contrôlé avec l’aide d’une électrode de
référence. L’électrolyse se manifeste alors par la circulation d’un courant électrique, dont
l’intensité peut être mesurée sur le circuit extérieur, à l’aide de la troisième électrode dite
contre-électrode (le potentiel de la contre-électrode n’ayant en général pas besoin d’être lui-
même contrôlé). Celle des deux électrodes qui est traversée par le courant dans le sens
correspondant à un processus de réduction est désignée par le terme de cathode. L’autre
électrode, traversée par le courant dans le sens qui correspond à un processus d’oxydation, est
désignée par le terme d’anode. Une inversion du sens du courant dans le circuit (au cours de
la variation du potentiel) intervertit les rôles des deux électrodes.
Pour imposer à l’électrode indicatrice un potentiel bien contrôlé, on utilise un appareil
électronique d’asservissement appelé potentiostat, aux bornes duquel les trois électrodes sont
connectées. Cet appareil fournit automatiquement la tension électrique entre l’électrode
indicatrice et la contre-électrode, nécessaire pour que la tension entre l’électrode indicatrice et
l’électrode de référence soit maintenue égale à une valeur de consigne affichée à l’appareil.
Pour décrire un voltampérogramme, on effectue alors un balayage de potentiel en modifiant
progressivement la tension de consigne contrôlée par le potentiostat, au moyen d’un système
de pilotage automatique. La mesure automatique du courant peut aussi être effectuée et portée
en fonction du potentiel, point par point. L’automatisation complète du tracé permet
l’enregistrement du voltampérogramme. Outre cet aspect essentiel lié à la procédure
87

CHAPITRE 1 : MATERIELS ET METHODES expérimentale de détermination des voltampérogrammes, il est important de considérer les
différents dispositifs d’électrode indicatrice.
La voltamétrie cyclique et la chronoampérométrie sont réalisés à l’aide d’un
potentiostat/galvanostat type Autolab PGSTAT100 opérant à l’air, en utilisant une cellule à un
seul compartiment contenant trois électrodes. Tous les potentiels sont mesurés par rapport à
une électrode saturée en calomel (SCE) (électrode de référence), un fil de platine est utilisé
comme contre électrode, et une plaque de platine comme électrode de travail (A = 0.196 cm2).
Avant toute utilisation, les plaques de platine (électrodes) sont polies avec des particules
d’alumine (0.05µm), lavées à l’eau et à l’éthanol aux ultrasons et finalement rincées à l’eau et
séchées.
3. Préparation des matériaux adsorbants 3.1 Protocole de purification de l’argile
De point de vue pratique, ce procédé de purification comporte deux étapes essentielles :
1. Sédimentation
La sédimentation consiste à disperser une masse donnée de l'échantillon de bentonite
brute naturelle dans un certain volume d'eau distillée. Cette méthode est basée sur la loi de
Stockes qui relie la taille des particules à leur vitesse de sédimentation (Haouzi, 1997).
ησρ 9/)²(2 −××= rgv (II.1.4)
Où :
v : vitesse de sédimentation, r : rayon de la particule sphérique, ρ : masse volumique de la
particule, σ : masse volumique de l’eau, η : la viscosité de l’eau .
La vitesse de sédimentation s’écrit alors en fonction du diamètre d des particules :
²dkv ×= (II.1.5)
Expérimentalement, on effectue un délitage pour désagréger l’argile sous l’action de
l’eau. On agite pendant 2 heures pour disperser les feuillets, puis on laisse sédimenter pendant
8 heures pour récupérer les fractions argileuses à 2µm.
88

CHAPITRE 1 : MATERIELS ET METHODES
2. Traitement chimique
Le but de cette opération est l’élimination des impuretés existantes dans l’argile comme
la matière organique, les sulfures de fer, les hydroxydes et oxydes d’aluminium. En premier
lieu, la suspension contenant la fraction argileuse à 2µm est attaquée par une solution d’acide
chlorhydrique 0.05M pendant 2 heures sous forte agitation pour détruire les carbonates. Des
lavages à l’eau distillée sont effectués afin d’éliminer les chlorures. Ensuite, La matière
Organique contenue dans l’argile est éliminée par un traitement à l’eau oxygénée. La
suspension attaquée par HCl est traitée par H2O2 à 30 volumes pendant 2 heures. Après
sédimentation, la suspension récupérée est lavée avec de l’eau distillée à chaud pour éliminer
et décomposer l’eau oxygénée résiduelle. Une saturation par le sodium est effectuée pour
assurer une homogénéisation de l’argile. Ceci se fait par échange avec une solution NaCl 1M,
pendant 4 heures. Ce traitement est répété 3 fois, puis des lavages à l’eau distillée sont
effectués pour éliminer les chlorures excédentaires jusqu’à test négatif par AgNO3, l’argile
obtenue est notée argile sodée.
3.2 Caractérisation de l’argile cationique
3.2.1 Tests de comportement des argiles cationiques
Ces tests ont essentiellement pour but de faire varier la distance d001 des minéraux
argileux de type (2 :1) en jouant sur les espèces interfoliaires. La réalisation pratique de ces
différents tests conditionnera la détermination du type d’argile.
a. Gonflement avec les polyalcools
Ce test est réalisé pour connaître l’aptitude au gonflement des divers types de smectites.
Un liquide polaire, l’éthylène glycol remplace l’eau interfoliaire et forme des complexes à une
ou plusieurs couches d’épaisseur connue d’où l’augmentation de la distance interfoliaire qui
passe de 10 A° à 17 A° après traitement.
b. Traitement au KCl
Ce traitement est généralement utilisé pour différencier les smectites des vermiculites.
Le minéral est traité au minimum trois fois par une solution KCl (1N) pendant 1heure, séché à
25°C ensuite chauffé à 110°C. A cet effet, les smectites et les vermiculites donnent des
distances basales de 12 et 10 A° respectivement, on observe ensuite, pour différencier les
divers types de smectites l’aptitude au gonflement avec l’éthylène glycol.
89

CHAPITRE 1 : MATERIELS ET METHODES c.Traitement à l’éthylène- glycol
L’éthylène- glycol est le plus souvent utilisé à l’état de vapeur. Il suffit pour cela de
placer les lames contenant l’échantillon dans un dessiccateur où viennent se loger les vapeurs
de L’éthylène- glycol. Il est cependant possible de l’utiliser à l’état liquide.
d. Test de Hoffmann Klemen »
Ce test permet de distinguer, parmi les smectites dioctaédriques, une montmorillonite
d’une beidellite en utilisant la méthode suivante : saturation de l’argile par le lithium grâce à
des traitements répétés par une solution LiCl (1N) pendant 2 heures à 60°C, rincer à l’eau et
disposer l’échantillon sur une lame de verre puis laisser sécher à température ambiante,
chauffer à 300°C pendant une nuit environ, la lame ainsi préparée est traitée par L’éthylène-
glycol vapeur pendant 12 heures.
Un diffractogramme de rayon X est réalisé. Si on observe des espacements normaux à
17A°, l’argile utilisée est une beidellite, car la montmorillonite perd son aptitude au
gonflement du fait de la suppression de la charge d’origine octaédrique par la migration du
lithium.
3.2.2 Mesure de la CEC :
Plusieurs méthodes ont été proposées pour estimer la capacité d’échange cationique
d’une argile. Les différents procédés consistent à remplacer les cations compensateurs
contenus dans l’espace interfoliaire par d’autres éléments, puis de doser les concentrations
résiduaires
Dans notre travail nous avons déterminé la CEC de notre argile purifiée par deux
méthodes essentielles : la méthode de bleu de méthylène et par titrage conductimétrique.
• Méthode du bleu de méthylène (Brindley et Thompson, 1970)
Elle consiste à échanger le sodium de l’argile par le cation de bleu de méthylène, puis à
doser le bleu de méthylène restant dans le surnageant par colorimétrie.
Cette méthode dépend du pH de la suspension et du cation compensateur ; en effet, de
très faibles valeurs de CEC sont obtenues lorsqu’elle est déterminée dans un milieu acide,
alors que ces valeurs deviennent très importantes lorsqu’on se place dans un milieu basique.
Ces variations sont dues aux charges (positives dans le 1er cas et négatives dans le deuxième)
90

CHAPITRE 1 : MATERIELS ET METHODES qui apparaissent sur la surface du minéral sous l’influence du pH. Quant à l’influence du
cation compensateur, elle est expliquée par Brindley et Thompson (Brindley et Thompson,
1970), comme étant un problème stérique observé pour les cations échangeables de valence
élevée. La détermination de la CEC par cette méthode n’est donc appropriée que pour un
échantillon sodé à pH neutre. La valeur de la CEC est déterminée à partir du plateau de
l’isotherme correspondant à une adsorption maximale du bleu de méthylène. Alors que la
surface totale de l’argile est calculée au point de floculation optimum correspondant à une
déviation de 45° par rapport à la droite (point d’intersection entre les deux droites). Le mode
opératoire équivalent à cette méthode se présente comme suit :
Dans une série d’erlenmeyer, nous introduisons un volume constant d’une suspension
argileuse de concentration égale à 1g/L et des quantités variables de bleu de méthylène, les
différents mélanges sont maintenus sous agitation normale pendant 1heure à température
ambiante. Après équilibre les suspensions sont centrifugées et les solutions de bleu de
méthylène résiduaire sont dosées par spectrophotométrie UV-Visible à une longueur d’onde
de 665nm. L’isotherme est obtenue en traçant la quantité de bleu de méthylène fixée en
fonction de la concentration résiduelle.
• Méthode par titrage conductimétrique (Kahr et Madsen, 1995; Chiu et al., 1990)
La méthode conductimétrique est basée sur le principe d’échange de cations.
Initialement le sodium présent dans l’argile est échangé par le barium. Puis un autre échange
est effectué entre le barium et le magnésium en titrant la suspension argileuse saturée en
barium par une solution de sulfate de magnésium. L’enregistrement de la courbe donnant la
conductivité électrique en fonction du volume du réactif permet la détermination du point
équivalent correspondant à la CEC. Cette courbe montre deux phases bien distinctes : la
première phase montrant un faible changement dans la conductivité à cause de la formation
du sulfate de barium insoluble. La deuxième phase montre une augmentation de la
conductivité de la solution avec augmentation du volume du sulfate de magnésium. Le point
d’équivalence est déterminé à l’intersection des deux courbes et correspond à la capacité
d’échange cationique (Kahr et Madsen, 1995; Chiu et al., 1990). Le mode opératoire est
présenté ci-dessous :
1g d’argile sèche est mis en suspension dans 100 ml d’eau distillée suivi d’une agitation
pendant 20 minutes. Une solution de 150 ml de BaCl2 tamponnée à pH= 8,2 par la
91

CHAPITRE 1 : MATERIELS ET METHODES triéthanolamine est ajoutée à la suspension argileuse, le mélange est maintenu sous agitation
à 70 °C pendant 4 heures. L’opération est répétée afin d’assurer une saturation complète de
l’échantillon, ce dernier est lavé plusieurs fois jusqu’au test négatif au nitrate d’argent
(absence de chlorure), et finalement séché à 60 °C. 0,5 g d’argile échangée par le barium est
transféré dans un bêcher de 150 ml auquel est ajouté 50 ml d’eau distillée. Un titrage
conductimétrique classique est effectué au moyen d’une solution de sulfate de magnésium.
3.3 Préparation de l’argile organophile
L’argile organophile est préparée par échange cationique entre le sodium initialement
présent dans l’espace interfoliaire de la bentonite et le cation ammonium quaternaire par
rapport à un pourcentage bien défini de la capacité d’échange cationique CEC, suivant la
méthode décrite par Bartlet-Hunt et al.( Bartlet-Hunt et al., 2003): la quantité de cations
organiques ajoutée à la bentonite est déterminée par la relation suivante :
XGMWMCEC
Mfcationclay
cation
×××= (II.1.6)
Où
f : Fraction de la capacité d’échange cationique correspondante au cation organique (sans
dimension) ; Mcation : masse du cation organique (g) nécessaire pour atteindre la fraction f ;
CEC: capacité d’échange cationique de l’argile (Méq/g); Mclay: masse de l’argile (g);
GMWcation: masse molaire du cation organique (g/mol); X: nombre de charge molaire par
équivalent (mol/equivalent).
Théoriquement, la capacité d'échange cationique représente la quantité maximale de
cations organiques pouvant être échangés à la surface de l'argile. La teneur totale de cations
organiques échangés coïncide bien avec la valeur théorique calculée (Redding et al., 2002).
La synthèse de l’argile organophile est réalisée par la procédure suivante: 1 g de
bentonite sodée est dispersée dans 100 ml d'eau déminéralisée, à laquelle une quantité bien
déterminée de bromure de cétyltriméthylammonium (CTAB) est lentement ajoutée. La
concentration de CTAB ajoutée varie entre 17 à 120 par rapport à la capacité d’échange
cationique de l’argile initiale (CEC). Les suspensions sont agitées pendant 24 h, puis lavées
jusqu’à test négatif d’anions bromure (AgNO3), séché à 60°C et broyé dans un mortier en
agate. La méthode de préparation de l’argile organophile est appelée méthode two steps
(deux étapes).
92

CHAPITRE 1 : MATERIELS ET METHODES
Le Cetyltrimethylammonium bromide: CTAB (99%), de formule chimique C19H42NBr
fournis par ACROS ORGANICS se compose de 16 chaines de carbone attachées à une tête de
triméthyl ammonium quaternaire contenant une charge permanente (+1).
3.4 Synthèse du matériau HDL Ni2AlNO3
3.4.1 Par coprécipitation
Le matériau HDL Ni2AlNO3 est préparé par la méthode de coprécipitation à pH constant
sous azote et forte agitation magnétique, afin de minimiser la contamination par le CO2
atmosphérique. A cet effet, une solution de sel métallique d’aluminium et de nickel avec un
rapport molaire Ni/Al = 2 ayant une concentration totale de 1M est ajoutée goute à goute à un
récipient contenant 100 ml d’eau décarbonatée. Une base NaOH de concentration 2M est
ajoutée de la même façon afin de maintenir le pH de coprécipitation totale à 10±0.1. Le temps
d’addition est estimé à 5 heures. Le solide est séparé de la solution par trois cycles de lavages
et de centrifugations, séché et broyé.
3.4.2 Par voie électrochimique
Des films très fins de matériau NiAlNO3 sont déposés sur la surface des plaques de
platine (A = 0.196 cm2) par réduction cathodique d’une solution contenant 0.0225M de
Ni(NO3)2 et 0.0075M de Al(NO3)3, le tout est préparé dans une solution KNO3 de
concentration 0.3M. L’électrodéposition est effectuée sous un potentiel de -0.9V/SCE pendant
des temps variables allant de 10 jusqu’à 120 secondes suivant la réaction illustrée sur la figure
II.1.1. L’électro activité des électrodes modifiées par les films de NiAlNO3 est caractérisée
par la voltamétrie cyclique à 10 Mv/s dans une solution NaOH 0.1M en absence et en
présence de Glyphosate et de Glufosinate. La détection ampérométrique des deux herbicides
est effectuée à 0.49 V/SCE avec une électrode à disque rotatif tournant à 500 tr/mn.
NO3
- + H2O + 2é NO2-+2OH-
(1-x) Ni2+ + x Al3+ + 2OH- + x NO3- + m H2O [Ni 1-x Al x (OH)2]x (NO3)x,m H2O
Figure II.1.1 : Schéma réactionnel de la formation du matériau NiAlNO3 par voie
électrochimique
93

CHAPITRE 1 : MATERIELS ET METHODES 3.5 Synthèse des matériaux Ni2AlNO3 intercalés par le Glyphosate et le Glufosinate
Le matériau Ni2AlNO3 intercalé par le Glyphosate noté : Ni2AlGly ou le Glufosinate
noté Ni2AlGlu sont préparés de la même manière que le matériau précurseur, à l’exception
qu’on ajoute dans le récipient un excès d’herbicide Glyphosate ou Glufosinate de façon à
obtenir un rapport en équivalence de Gly/Al ou Glu/Al = 2.5. Des matériaux Ni2AlGly et
Ni2AlGlu sont préparés aussi par échange anionique à pH=7 à partir d’une solution
d’herbicide de concentration initiale égale à 300 mg/L et en utilisant un rapport
adsorbant/adsorbant égale à 1 mg/ml. Les matériaux sont lavés trois fois par de l’eau
décarbonatée et séchés à l’air.
4. Application à l’adsorption 4.1 Les adsorbats
Nous avons examiné l’adsorption, sur l’argile organophile, l’argile en présence de
CTAB et sur une HDL de type NiAlNO3, d’un certain nombre de micropolluants organiques
susceptibles de polluer les eaux. Les adsorbats utilisés sont des polluants définis comme
prioritaires par l’EPA. Le choix de ces molécules a été motivé, d'une part, par leur caractère
nocif pour l’environnement et, d'autre part, par la facilité de leur dosage dans l'eau. Ils
possèdent, de plus, des groupements chimiques variés. Toutes les solutions et les dispersions
sont de grade chimique et sont préparées avec de l’eau MilliQ.
4.1.1 Le 2,4DCP
Le 2,4-Dichlorophénol (2,4-DCP) utilisée dans cette étude est un réactif de grade
analytique de pureté 95% fournie par Sigma Chemical Co. Les solutions de 2,4DCP sont
préparées dans 0.1 mol/L de KCl afin d’éviter la floculation et d’avoir une solution
électrolytique de concentration constante. Les caractéristiques principales de 2,4
dichlorophénol sont résumées dans le tableau II.1.1. A cause de la variation de l’adsorption
en fonction du pH dans les régions proche du pKa des chloropénols. Ceux-ci étant des acides
organiques faibles avec un Kow élévé et un pKa de 7.83, l’utilisation d’un milieu tampon
pour éviter les changements de pH s’avère plus important que la variation de la capacité
d’adsorption due à l’utilisation de cet tampon à un pH donné. Il a été démontré au cours des
études précédentes, que l'adsorption des composés phénoliques est réduite de façon
remarquable lorsque le pH est supérieur au pKa (Kim et al., 1996). Dans cette étude la valeur
94

CHAPITRE 1 : MATERIELS ET METHODES du pKa de 2,4DCP est toujours supérieure aux valeurs du pH. Par conséquent, la dissociation
des composés phénoliques vers les phénolates, n’est pas significative.
Tableau II.1.1 : Caractéristiques physiques et chimiques de 2,4DCP
Composé Formule chimique Masse molaire
(g/mol)
Solubilité dans l’eau
(mg/L) Log KOW pKa
2,4DCP C6H3Cl2OH 163.01 4500 3.23 7.85
4.1.2 Le colorant 4GL
Beaucoup de colorants utilisés dans l’industrie textile peuvent être toxiques vis-à-vis de
l’homme et entraîne ainsi des effluents nuisibles à l’environnement. Les groupements
électrophiles ou radicalaires attaquent les bases puriques et pyrimidiques de l’ADN et causent
par conséquent une altération du code génétique avec mutation et risque de cancer.
L’augmentation de la couleur rend l’eau des effluents industriels correspondant
impropre aux usages domestiques et industriels et réduit la transmittance de la lumière ce qui
limite la croissance des plantes aquatiques et provoque indirectement un préjudice pour la
pisciculture.
Le colorant choisi pour cette étude est un colorant industriel utilisé pour teindre les
fibres textiles. Il est synthétisé et commercialisé sous un nom commercial codé : jaune
supranol 4GL (Kacha et al. 1997) et utilisé sans purification. Sa longueur d’onde maximale
d’absorption 410 nm est déterminée par un spectrophotomètre de type SAFAS.
Le dosage des solutions de 2,4DCP et du colorant 4GL est effectué par
spectrophotométrie UV-visible. Le spectrophotomètre que nous avons utilisé est un appareil
qui permet de mesurer directement les densités optiques. Les analyses sont effectuées sur un
spectrophotomètre à double faisceaux type SAFAS piloté par un ordinateur. Les longueurs
d’onde maximales sont obtenues directement par balayage automatique entre 200 et 800 nm.
4.1.3 Les herbicides : Glyphosate et Glufosinate
Le Glyphosate (N-phosphonométhylglycine, Gly) et le Glufosinate (DL-homoalanin-4-
(méthyl) phosphonique acide, Glu) (Figure II.1.2) sont utilisés dans le monde entier comme
herbicides non sélectifs pour lutter contre les mauvaises herbes à feuilles larges (Zimdahl,
1993). Ils interférent avec la capacité des plantes à former des acides aminés, en plus ils
95

CHAPITRE 1 : MATERIELS ET METHODES réagissent négativement sur la photosynthèse et la respiration. Plus précisément, Le
Glyphosate inhibe le synthase 5-énolpyruvylshikimate-3-phosphate, une enzyme impliquée
dans la voie de biosynthèse des acides aminés aromatiques, ce qui fait obstacle à la
production des protéines en raison d'un manque de tyrosine, de phénylalanine et de
tryptophane (Rubin et al., 1982). Le Glufosinate inhibe la glutamine synthétase, une enzyme
responsable de catalyser la réaction entre l'ammoniac et le glutamate pour former la glutamine
(Bayer et al., 1972).
Figure II.1.2: Structures chimiques du Glyphosate et de Glufosinate.
Les deux herbicides Glyphosate et Glufosinate de pureté 99% sont fournis par Aldrich
Chemicals. Les caractéristiques principales de ces deux herbicides sont présentées dans le
tableau II.1.2.
Le dosage de ces deux herbicides est effectué par analyse du phosphore total par ICP
AES , en utilisant un appareil type Perkin-Elmer Optima 3000XL.
Tableau II.1.2 : Caractéristiques générales des deux herbicides Glyphosate et Glufosinate.
Herbicide Formule
chimique
Masse molaire
(g/mol)
Solubilité dans l’eau
à 25°C (g/L) pKa
Glyphosate C3H8NO5P 169.07 12 pK1<2, pK2=2.6,
pK3=5.6, pK4=10.6
Glufosinate C5H12NO4P 198.27 500 pK1=2, pK2=2.9,
pK3=9.8
96

CHAPITRE 1 : MATERIELS ET METHODES
Pour tous les composés étudiés, nous avons utilisé la même méthode qui consiste à
préparer d'abord une solution-mère de concentration donnée, à partir de laquelle nous
préparons, par dilutions successives, une série de solutions de concentrations bien
déterminées. Celles-ci sont, par la suite, analysées par spectrophotométrie UV-visible, ou
ICP-AES. Nous établissons ainsi la droite d’étalonnage représentant la densité optique DO, au
maximum de la bande d'absorption, en fonction de la concentration C et qui obéit à la relation
de Beer Lambert.
4.2 Cinétique d’adsorption
L’équilibre thermodynamique entre l’adsorbat en phase liquide et l’adsorbat fixé sur le
solide est atteint avec une vitesse qui dépend non seulement de la vitesse avec laquelle les
constituants diffusent dans l’adsorbant et dans le fluide mais aussi de l’interaction adsorbant-
adsorbat. L’étude de l’adsorption d’un composé sur un adsorbant nous permet d’examiner
l’influence du temps de contact sur sa rétention.
4.2.1 Pour le 2,4DCP
Les expériences d'adsorption sont réalisées en utilisant la méthode des Erlens séparés, et
sont effectuées au laboratoire en mettant en contact un volume déterminé d’une solution de
2,4DCP avec une masse équivalente de bentonite sodée ou organophile.
Afin de réaliser l’étude cinétique, 5g d’adsorbant (bentonite sodée dans le cas de one
step ou bentonite organophile dans le cas de two steps) est mise en contact avec un 1L de
solution de 2,4DCP de concentration égale à 5mmol/L, les mélanges sont agités à une vitesse
de 3000 tr/mn. Durant un test d’adsorption préliminaire, un effet négligeable de la vitesse
d’agitation est relevé. Des prélèvements de 2.5ml de solution sont effectués à l’aide d’une
seringue de 10ml pendant des intervalles de temps bien définis, ces solutions sont centrifugées
et la concentration de 2,4DCP résiduelle est mesurée par Spectroscopie UV-Visible à une
longueur d’onde de 285 nm (Sathishkumar et al., 2007).
4.2.2 Pour le colorant 4GL
L’étude de l’influence du temps de contact sur la quantité fixée du colorant 4GL est
effectuée en mettant en contact 0.2 g d’argile organophile avec 100 ml de solution de
colorant de concentration initiale égale à 100 mg/L. Les mélanges sont agités pendant des
97

CHAPITRE 1 : MATERIELS ET METHODES durées variables allant de 2 à 60 mn, le matériau est ensuite séparé par centrifugation et les
surnagents dosés à 410 nm.
4.2.3 Pour les herbicides Glyphosate et Glufosinate
Des suspensions de matériau HDL type Ni2AlNO3 sont mises en agitation continue dans
des tubes en verre. Afin d’obtenir des dispersions homogènes, les échantillons sont dispersés
dans 25 ml d’eau décarbonatée et dé ionisée et laissés sous agitation pendant 24 heures avant
que les molécules d’herbicides soient ajoutées. Le pH est ajusté à 7 par addition d’HCl 0.1 M
ou NaOH 0.1 M. L’effet du temps de contact sur la fixation de ces deux herbicides est
effectué en maintenant constant la concentration de l’herbicide soit 200 mg/L, un rapport
adsorbat/adsorbant = 1ml/mg et une température de 25°C. Après un temps de contact bien
déterminé, le matériau est séparé par centrifugation et le surnageant est analysé par mesure de
phosphore totale par ICPAES.
Des solutions de polluants organiques à différentes concentrations sont préparées à
chaque expérience afin de réaliser la courbe d’étalonnage.
Durant les expériences d’adsorption, la quantité de polluant perdue de la solution est supposée
être adsorbée sur le matériau argileux. La quantité fixée est déterminée par la relation
suivante:
m
VCeCiqe ×−=
)( (II.1.7)
Où
qe : la quantité fixée en mmol/g, Ci : la concentration initiale de l’adsorbat en mmol/L ;
Ce : la concentration résiduelle (concentration à l’équilibre) de l’adsorbat en mmol/L ;
V : le volume de la solution en ml, m : la masse de l’adsorbant en g .
4.3 Modèles cinétiques d’adsorption
Afin d’étudier l’étape limitante de l’adsorption des polluants organiques présentés
précédemment par les matériaux adsorbants, quatre modèles cinétiques sont proposés : la
cinétique du pseudo premier Ordre, du pseudo second ordre, d’Elovich et la diffusion intra
particulaire. La conformité entre les résultats expérimentaux et les modèles cinétiques est
exprimée par le facteur de corrélation empirique R². Une valeur élevée de R² indique que le
modèle appliqué décrit convenablement la cinétique d’adsorption
98

CHAPITRE 1 : MATERIELS ET METHODES
La cinétique du pseudo premier ordre est décrite par la relation suivante (Lagregen,
1898):
303.2
log)( 1 tkqeqtqeLog ×−=− (II.1.8)
Où
qe et qt sont les quantités fixés en mmol/g à l’équilibre et au temps t respectivement, k1:
constante de vitesse du pseudo premier ordre (mn1-).
La cinétique du pseudo second ordre (Ho et McKay, 1999) est régie par l’équation
suivante :
tqeqekqt
t×+
×= )1(
²1
2
(II.1.9)
qe et qt sont les quantités fixés en mmol/g à l’équilibre et au temps t respectivement, k2:
constante de vitesse du pseudo second ordre (g/(mmol mn)).
La cinétique d’adsorption est aussi vérifiée par le modèle d’Elovich (Chien et Clayton,
1980).
tqt ln)ln( ×+××= ββαβ (II.1.10)
Où
α est la vitesse d’adsorption initiale en mmol/g mn, β: la constante de désorption en g/mmol.
Les constantes d’Elovich sont calculées en traçant (qt) en fonction de ln (t).
Le modèle de diffusion intra particulaire est testé dans ce travail. La diffusion intra
particulaire est originaire de la deuxième loi de Fick. Webber et Morris ont confirmés que si
la quantité fixée varie en fonction de la racine carré du temps, ceci veut dire que la diffusion
intra particulaire est l’étape limitante dans l’adsorption (Weber et al., 1963). Ainsi, les
vitesses d’adsorption sont souvent mesurées en déterminant la variation de la capacité
d’adsorption en fonction de la racine carrée du temps (Juang et al., 2000). Ho et McKay ont
démontré que la courbe qt en fonction de la racine carrée du temps doit passer par l’origine,
si la diffusion intra particulaire est la seule étape limitant l’adsorption (Ho et McKay, 1998).
Quand la courbe ne passe pas par l’origine, ceci peut indiquer le contrôle de la couche limite
de la diffusion sur l’adsorption et peut indiquer aussi que la diffusion intra particulaire n’est
99

CHAPITRE 1 : MATERIELS ET METHODES pas l’étape limitant et que l’adsorption est contrôlée par d’autres phénomènes opérant
simultanément.
En se basant sur les travaux de Weber et al, (Weber et al., 1963), le coefficient de
diffusion intra particulaire kp est défini par la relation suivante :
tkpqt ×= (II.1.11)
Où
kp est la constante vitesse de la diffusion intra particulaire (mmol/g mn0.5) et peut être
calculée à partir du tracé de la courbe qt(mmol/g) en fonction de t (mn0.5).
4.4 Isotherme d’adsorption
4.4.1 Pour le 2,4DCP
Les isothermes d’adsorption sont effectuées à 25°C en utilisant un bain thermostat. A
cet effet, 50 ml de solution de 2,4DCP à différentes concentrations allant de 1 à 20 mmol/L
sont mises en contact avec 0.25 g de bentonite (sodée ou organophile) pendant une durée de
40 mn. Le solide est séparé par centrifugation et le surnageant est dosé par UV-Visible.
4.4.2 Pour le colorant 4GL
Les isothermes d’adsorption sont effectuée dans un bain thermostat à 25°C en
mélangeant 100 ml de solution de colorant de concentration variable allant de 200 à 1400
mg/L avec une masse d’argile organophile égale à 0.2 g pendant une durée d’une heure. Pour
toutes les expériences d’adsorption, après agitation le matériau est séparé par centrifugation et
le surnageant est dosé par UV-Visible à la longueur d’onde correspondante.
4.4.3 Pour les herbicides Glyphosate et Glufosinate
L’étude de l’influence de la concentration initiale des herbicides sur la capacité
d’adsorption du matériau Ni2AlNO3 est effectuée en faisant varier la concentration initiale des
herbicides entre 0 et 300 mg/L et en maintenant constant le rapport solide /liquide = 1mg/ml,
le volume totale de la solution= 50 ml et un pH =7. Après un temps de contact de 24 heures,
le matériau est centrifugé et le surnageant dosé par analyse élémentaire de phosphore par
ICPAES.
100

CHAPITRE 1 : MATERIELS ET METHODES 4.5 Modélisation des isothermes d’adsorption
4.5.1 Modèle de Freundlich
Les données d’adsorption obtenues à partir des isothermes sont analysées par le modèle
de Freundlich. Ce dernier a développé une équation empirique pour décrire l’isotherme
d’adsorption (Freundlich, 1906). Sa théorie est basée sur le fait que la surface hétérogène de
l’adsorbant est constituée de différents sites d’adsorption, l’adsorption dans chaque site suit le
modèle de Langmuir. Freundlich a démontré que le rapport entre la quantité de soluté
adsorbée sur une masse de l’adsorbant et la concentration de soluté en solution n’est pas
constant pour différentes concentrations. Ce modèle ne prédit aucune saturation de la surface
de l’adsorbant par le soluté, donc un recouvrement infinis de la surface de l’adsorbant,
indiquant une adsorption en multicouches (Donat et al., 2005).Le modèle de Freundlich
suppose que si la concentration du soluté en solution (Ce) est élevée à la puissance n, la
quantité adsorbée étant qe, donc Cen est constant pour une température donnée.
L’isotherme d’adsorption de Freundlich est donnée par l’équation suivante :
(II.1.12) nf CeKqe ×=
Où
Kf : est la constante de Freundlich traduisant la capacité d’adsorption en mmol/g ou en mg/g,
n : représente l’intensité d’adsorption adsorbat-adsorbant, et peut indiquer la réversibilité de
l’isotherme d’adsorption, pour n =0 système irréversible, pour 1>n>0 le système est
favorable et pour n>1 le système est non favorable (Alley, 2000).
L’équation précédente peut être arrangée sous sa forme linéaire pour donner :
(II.1.13) )log()log()log( CenKqe f ×+=
4.5.2 Modèle de Langmuir
L’équation du modèle de Langmuir (Langmuir, 1916) initialement obtenus des études
cinétiques est basée sur l’hypothèse que la surface de l’adsorbant est constituée d’un nombre
définie de sites d’adsorption homogènes ayant la même énergie d’adsorption. Chaque site ne
peut fixer qu’une seule molécule de substrat, de la même manière que les autres sites et qu’il
101

CHAPITRE 1 : MATERIELS ET METHODES n’a ya pas d’interactions entre les molécules adsorbées. Le modèle d’adsorption de Langmuir
sert à déterminer la capacité maximale d’adsorption, lorsqu’elle n’est pas atteinte durant
l’expérience. Elle est basée sur l’hypothèse physique suivante : la capacité d’adsorption
maximale atteinte est traduite par une monocouche d’adsorption et que l’énergie d’adsorption
est distribuée de façon homogène sur toute la surface de l’adsorbant. L’équation de Langmuir
est donnée par la formule suivante :
Ceb
Cebqqe×+××
=1max (II.1.14)
Où :
b : le coefficient d’adsorption à l’équilibre (L/g), qmax : la quantité d’adsorption maximale
déterminée par ce modèle (mmol/g ou mg/g), Ce : la concentration à l’équilibre (mmol/L ou
mg/L) et qe : la quantité fixée à l’équilibre en (mmol/g ou mg/g).
La forme linéaire de l’équation de Langmuir est donnée par la formule suivante :
maxmax
1qCe
qbqeCe
+×
= (II.1.15)
4.5.3 Modèle de Redlich Peterson
Le modèle de Redlich Peterson combine les éléments des deux modèles précédents
(Freundlich et Langmuir). Il est appliqué surtout quand le mécanisme d’adsorption est hybride
( adsorption homogène et hétérogène en même temps) et qu’elle ne suit pas une isotherme de
type monocouche idéale (Redlich et Peterson, 1959). L’équation de Redlich Peterson est
souvent utilisée comme modèle compris entre celui de Freundlich et de Langmuir. Elle est
donnée par l’équation suivante :
βCeaCekqe
R
R
×+×
=1
(II.1.16)
Où:
kR et aR sont les constantes de l’isotherme de Redlich Peterson, β est un exposant variant entre
0 et 1. Le modèle de Redlich Peterson comprend trois paramètres et peut être appliqué en
système homogène ou hétérogène.
102

CHAPITRE 1 : MATERIELS ET METHODES
La forme linéaire de cette équation est donnée par la relation suivante :
( ) (CeaqeCek RR lnln1ln ×+=⎟⎟
⎠
⎞⎜⎜⎝
⎛−× β ) (II.1.17)
4.5.4 Modèle de Tempkin
L’équation de l’isotherme de Tempkin (Tempkin et Pyzhev, 1940) contient un facteur
qui prend en compte les interactions adsorbat- adsorbant. Il suppose que la chaleur
d'adsorption de toutes les molécules dans la couche adsorbée décroît linéairement avec le taux
de recouvrement de l’adsorbant à cause des répulsions adsorbat-adsorbat et que l'adsorption
consiste en une distribution uniforme de l'énergie de liaison maximale (Kavitha et
Namasivayam ,2007). En outre, il suppose que la diminution de la chaleur d'adsorption est
linéaire plutôt que logarithmique, comme le laisse entendre l'équation de Freundlich. Le
modèle de Tempkin est souvent représenté par la relation suivante (Aharoni et Sparks,
1991) :
)ln()ln( CeBABqe TTT ×+×= (II.1.18)
Où :
T
T bTRB ×
= (II.1.19)
La constante BT est reliée à l’énergie de l’adsorption, R : constante des gaz parfaits, T :
température en °K ; AT : représente la constante de liaison à l’équilibre (L/mn) correspondant
à l’énergie de liaison maximale (Pearce et al., 2003). Le tracé de qe en fonction de ln (Ce)
permet la détermination des constantes de Tempkin.
103

CHAPITRE 1 : MATERIELS ET METHODES
Références bibliographiques
• Aharoni C. and Sparks D.L. (1991). In: Sparks, D.L., Suarez, D.L. (Eds.), Rate of
Soil Chemical Processes. Soil Science Society of America, Madison, WI, pp. 1–18.
• Alley E.R. (2000), Water Quality Control Handbook, 8, McGraw Hill, p125.
• Bartelt-Hunt S.L., Burns S.E. and Smith J.A. (2003), J. Colloid Interface Sci., 266,
251–258.
• Bayer E., Gugel K.H., Haegele K. and Zachner Z. (1972), Helv. Chem. Acta 55,
224–239.
• Bouberka Z., Kacha S., Kameche M., Elmaleh S. and Derriche Z. (2005), Hazard.
Mater. B 119, 117–124.
• Brindley G.W. and Thompson T.D. (1970), J. Chem., 8, 405–415.
• Chien S.H. and Clayton W.R. (1980), J. Am. Soil Sci. Soc., 44, 265–268.
• Chiu Y.C., Huang L.N., Uang C.M. and Huang J.F. (1990), J. Colloids Surf. 46,
327–337
• Donat R., Akdogan A., Erdem E. and Cetisli H.J. (2005), Colloid Interface Sci.,
286, 43–52.
• Freundlich H.M.F. (1906), Z. Phys. Chem.,57, 385–470.
• Haouzi A . (1997)., Thèse De Doctorat, Université Montpellier II.
• Ho Y.S. and Mc Kay G. (1998), J. Chem. Eng. 70, 115–124.
• Juang R.S., Wu F.C. and Tseng R.L. (2000), J. Colloid Interface Sci., 227, 437–444.
• Kacha S., Ouali S., Elmaleh M.S. (1997), Rev. Sci. Eau 10, 233–247.
• Kahr G. and Madsen F.T. (1995), Appl. Clay Sci. 9, 327–336.
• Kavitha D. and Namasivayam C. (2007), Bioresour. Technol. 98, 14–21.
• Kim Y.S., Song D.I., Jeon Y.W. and Choi S.j. (1996), Sep. Sci. Technol. 31, 2815–
2830.
• Lagergren S. (1898), Kungliga svenska vetenskapsakademiens, Handlingar band. 24
1–39.
• Langmuir (1916), J. Am. Chem. Soc. 38, 2221–2295.
• Pearce C.I., Lioyd J.R., Guthrie J.T. (2003), Dyes Pigments 58, 179–196.
• Redding A.Z., Burns S.E., Upson R.T. and Anderson E.F. (2002), J. Colloid.
Interface Sci. 250, 261–264.
• Redlich O. and. Peterson D.L (1959), J. Phys. Chem. 63, 1024–1029.
104

CHAPITRE 1 : MATERIELS ET METHODES
• Rubin J.L., Gains C.G. and Jensen R.A. (1982). Plant Physiol. 7, 833–839.
• Sathishkumar M., Binupriya A.R., Kavitha D. and Yun S.E. (2007), Bioresour.
Technol. 98, 866–873.
• Tempkin M.J. and Pyzhev V. (1940), Acta Physiochim. URSS 12, 217–222.
• Voïnovitch I. A. (1988), "Analyse des sols, roches et ciments: méthodes choisies",
Edition Masson, p. 348, Paris.
• Weber W.J., Morris J.C. (1963), J. Sanit. Eng. Div. Am. Soc. Civ. Eng., 89 (17), 31-60. • Zimdahl R.L.(1993). Fundamentals of Weed Science. Academic Press, London.
105

CHAPITRE 2 : ADSORPTION DES POLLUANTS
ORGANIQUES PAR LA METHODE ONE STEP ET
TWO STEPS
106

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
CHAPITRE 2 : ADSORPTION DE 2.4DCP ET DE 4GL PAR LA
METHODE ONE STEP ET TWO STEPS
1. Introduction L’application traditionnelle de la bentonite organophile dans l'élimination des polluants
organiques des eaux usées comprend deux étapes essentielles : La première est la synthèse de
la bentonite organophile, la seconde est l’utilisation de la bentonite organophile synthétisée
précédemment pour l’élimination des polluants des eaux usées. Le processus de synthèse de
bentonite organophile implique la pulvérisation de la bentonite, l’ajout d’un agent tensioactif
,une agitation pendant une durée déterminée, ensuite la récupération du matériau par
centrifugation et lavage et enfin séchage et broyage (Smith et Jaffe, 1991; Smith et Galan,
1995).
Des quantités énormes d’énergie et d’eau sont requises dans le procédé de synthèse de
l’argile organophile, ce qui pourrait être un facteur limitant son application dans le traitement
des eaux usées. En outre, Il a été observé que la bentonite organophile présente une difficulté
lors de sa séparation de l’eau, ce qui pose un autre obstacle à son application industrielle (Zhu
et al., 2000; Zhu et al.,2005). Ainsi, la bentonite organophile n'a pas été appliquée dans le
processus de traitement des eaux en raison des coûts élevés (Zhu et al., 2005).Un adsorbant
optimal pour l'élimination des composés organiques des eaux usées doit avoir les propriétés
suivantes: un faible coût, une facilité de synthèse, une neutralité vis-à-vis de l'environnement,
une grande affinité et une haute capacité.
Afin de réduire les coûts et simplifier le processus de traitement des eaux usées par la
bentonite organophile, une seule étape incluant à la fois la synthèse de la bentonite
organophile et élimination des polluants organiques a été proposée dans ce travail. En effet, la
méthode « one step » propose le mélange entre l'adsorbat et le tensio-actif cationique en
solution, puis l’ajout de la bentonite sodée. Ainsi, les polluants organiques sont supprimés de
l’eau en même temps que la formation de l’argile organophile (Jianfeng et Lizhong, 2007).
107

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Surtout que la présence d'agents de surface cationiques dans les eaux usées d’industries de:
pigments, colorants, de produits chimiques agricoles et autres peut réaliser la méthode « one
step » par simple addition de bentonite ce qui éliminera à la fois les agents de surface et les
polluants organiques (Saleh, 2006).
L’utilisation des surfactants cationiques dans l’industrie est courante pour l'élimination
des polluants organiques. Ainsi, le processus pourrait efficacement réduire le coût de l'eau et
d'électricité pour la synthèse des bentonites organophiles. Dans tous les cas, la méthode one
step pourrait devenir un moyen simple, efficace et économiquement acceptable comme
alternative aux méthodes classiques de traitement des eaux de rejet. En outre, la bentonite
organophile résultante pourrait être recyclée ou utilisée pour préparer des matériaux hétéro-
argileux à structure poreuse (Zhu et al., 2005).
Dans ce travail, les propriétés d'adsorption de la bentonite organophile et de bentonite
seule en présence de mélange de tensioactifs et de polluants organiques ont été étudiées. A cet
effet, nous nous sommes intéressés à l’étude de l'adsorption de deux polluants organiques : le
2,4-DCP et un colorant industriel, le 4GL par des complexes de surfactants cationiques et
d’argile en utilisant deux méthodes : la première, quand le tensioactif cationique est ajouté
dans la solution contenant le polluant et le mélange est adsorbé par la bentonite ce qu’on a
appelé : la méthode one step. L’adsorption du polluant par la bentonite organophile est
appelée la méthode : two steps. Les objectifs de cette étude sont les suivants:
• Déterminer la faisabilité de l'élimination des polluants organiques par la méthode one
step;
• Faire une comparaison entre les deux méthodes : one step et two steps vis-à-vis de
deux polluants organiques : un colorant industriel : le 4GL et un polluant chloré : le
2,4DCP;
• Etudier l'influence de quelques paramètres pysico-chimiques tels que le temps de
contact, la concentration de l'adsorbat et du tensioactif sur l’élimination des polluants.
108

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
2. Caractérisation des matériaux adsorbants
2.1 Purification de la bentonite
La bentonite utilisée dans ce travail provient d’une carrière située à Maghnia (ouest de
l’Algérie) et nous a été fourni par l’entreprise ENOF (une société spécialisée dans la
fabrication des produits non ferriques et des substances utiles). Elle contient des impuretés
tels que : le quartz, la cristobalite et la calcite. Les principales caractéristiques de cette
bentonite sont regroupées dans le Tableau II.2.1.
Tableau II.2.1: Analyse chimique de la bentonite naturelle utilisée (% en poids)
SiO2
Al203
Fe2O3
MgO
CaO
Na2O
K2O PAF
% 54.90 27.71 2.82 1.85 0.08 3.14 0.08 9.4
PAF: perte au feu à 900 °C.
Le traitement préliminaire de la bentonite naturelle par homo-ionisation sodique
consiste, non seulement, à la débarrasser de toutes les phases cristallines (quartz, feldspath,
calcite, ...), mais aussi à remplacer tous les cations échangeables de natures diverses par des
cations de sodium tous identiques. Il permet aussi d’avoir des fractions granulométriques bien
définies, de taille inférieure à 2 micromètres (< 2 μm), qui correspondent à la montmorillonite
homo-ionique sodique.
Le protocole de purification de la bentonite est détaillé dans la partie : matériels et
méthodes.
2.2 Caractérisation Du matériau purifié
2.2.1 Mesure de la capacité d’échange cationique
• La méthode au Bleu de méthylène
La courbe donnant l’isotherme d’adsorption du bleu de méthylène sur l’argile sodée est
présentée sur la figure II.2.1.
À partir du plateau de l’isotherme (Figure II.2.1), nous déduisons la valeur de la CEC
qui est égale à 103 meq /100gr d’argile. Au point de floculation optimum correspondant à
l’intersection des deux droites, nous avons la quantité de bleu de méthylène nécessaire à la
formation d’une monocouche. Connaissant le nombre de molécules de bleu de méthylène et
109
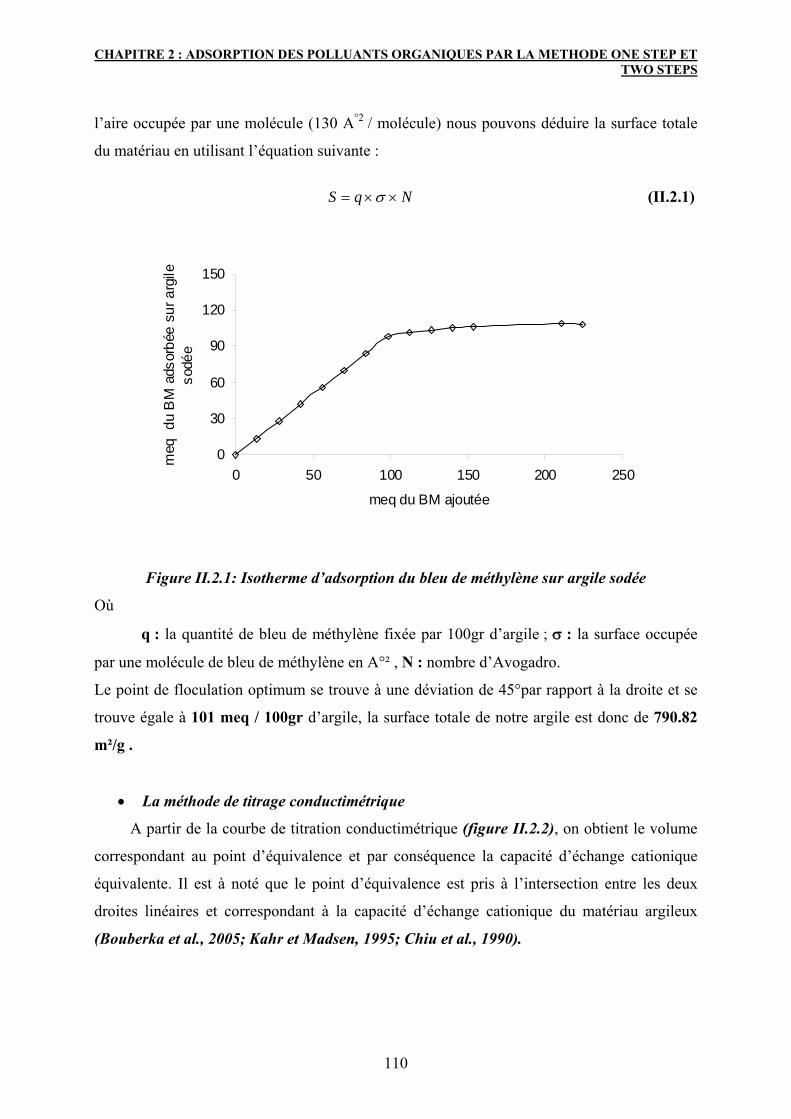
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
l’aire occupée par une molécule (130 A°2 / molécule) nous pouvons déduire la surface totale
du matériau en utilisant l’équation suivante :
NqS ××= σ (II.2.1)
0
30
60
90
120
150
0 50 100 150 200 250
meq du BM ajoutée
meq
du
BM
ads
orbé
e su
r arg
ile
sodé
e
Figure II.2.1: Isotherme d’adsorption du bleu de méthylène sur argile sodée
Où
q : la quantité de bleu de méthylène fixée par 100gr d’argile ; σ : la surface occupée
par une molécule de bleu de méthylène en A°² , N : nombre d’Avogadro.
Le point de floculation optimum se trouve à une déviation de 45°par rapport à la droite et se
trouve égale à 101 meq / 100gr d’argile, la surface totale de notre argile est donc de 790.82
m²/g .
• La méthode de titrage conductimétrique
A partir de la courbe de titration conductimétrique (figure II.2.2), on obtient le volume
correspondant au point d’équivalence et par conséquence la capacité d’échange cationique
équivalente. Il est à noté que le point d’équivalence est pris à l’intersection entre les deux
droites linéaires et correspondant à la capacité d’échange cationique du matériau argileux
(Bouberka et al., 2005; Kahr et Madsen, 1995; Chiu et al., 1990).
110
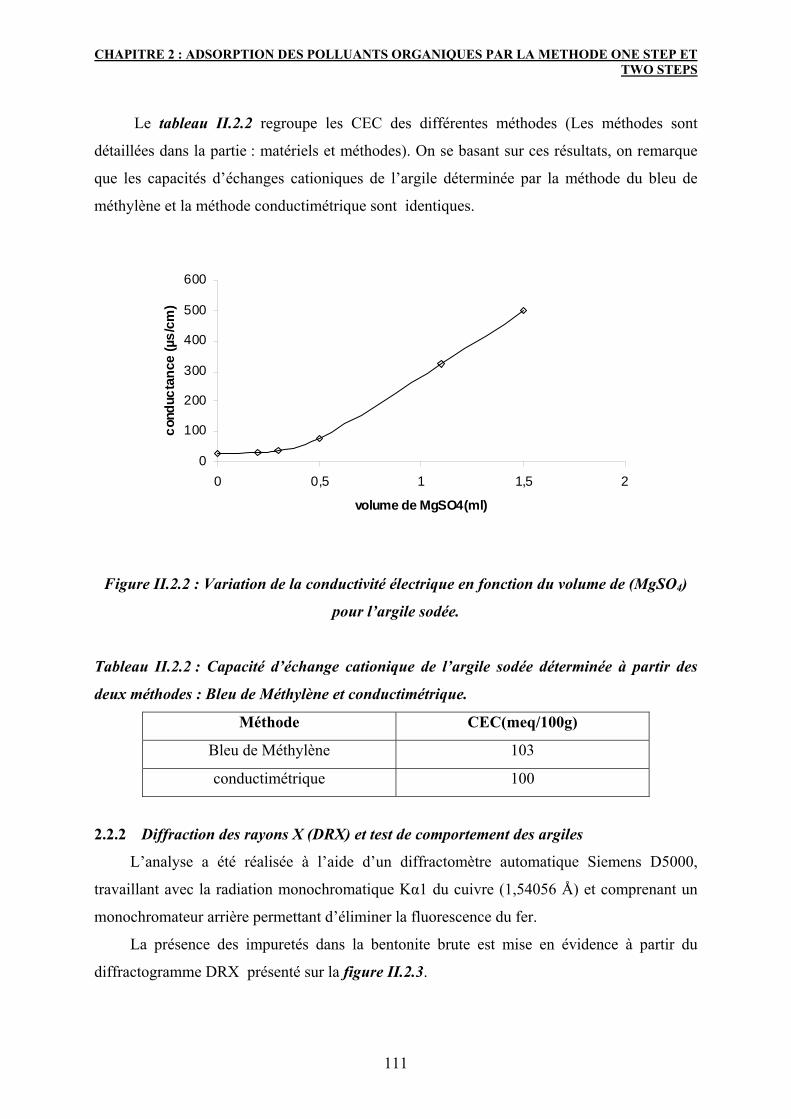
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Le tableau II.2.2 regroupe les CEC des différentes méthodes (Les méthodes sont
détaillées dans la partie : matériels et méthodes). On se basant sur ces résultats, on remarque
que les capacités d’échanges cationiques de l’argile déterminée par la méthode du bleu de
méthylène et la méthode conductimétrique sont identiques.
0
100
200
300
400
500
600
0 0,5 1 1,5 2
volume de MgSO4(ml)
cond
ucta
nce
(µs/
cm)
Figure II.2.2 : Variation de la conductivité électrique en fonction du volume de (MgSO4)
pour l’argile sodée.
Tableau II.2.2 : Capacité d’échange cationique de l’argile sodée déterminée à partir des
deux méthodes : Bleu de Méthylène et conductimétrique.
Méthode CEC(meq/100g)
Bleu de Méthylène 103
conductimétrique 100
2.2.2 Diffraction des rayons X (DRX) et test de comportement des argiles
L’analyse a été réalisée à l’aide d’un diffractomètre automatique Siemens D5000,
travaillant avec la radiation monochromatique Kα1 du cuivre (1,54056 Å) et comprenant un
monochromateur arrière permettant d’éliminer la fluorescence du fer.
La présence des impuretés dans la bentonite brute est mise en évidence à partir du
diffractogramme DRX présenté sur la figure II.2.3.
111
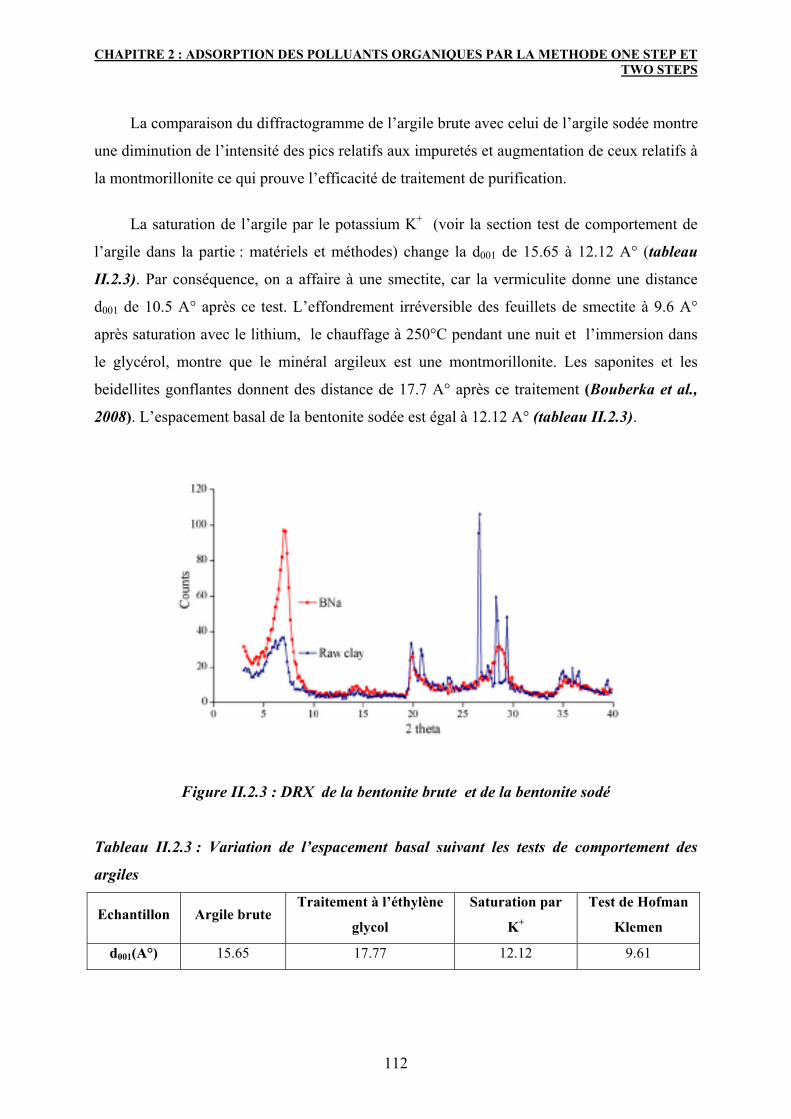
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
La comparaison du diffractogramme de l’argile brute avec celui de l’argile sodée montre
une diminution de l’intensité des pics relatifs aux impuretés et augmentation de ceux relatifs à
la montmorillonite ce qui prouve l’efficacité de traitement de purification.
La saturation de l’argile par le potassium K+ (voir la section test de comportement de
l’argile dans la partie : matériels et méthodes) change la d001 de 15.65 à 12.12 A° (tableau
II.2.3). Par conséquence, on a affaire à une smectite, car la vermiculite donne une distance
d001 de 10.5 A° après ce test. L’effondrement irréversible des feuillets de smectite à 9.6 A°
après saturation avec le lithium, le chauffage à 250°C pendant une nuit et l’immersion dans
le glycérol, montre que le minéral argileux est une montmorillonite. Les saponites et les
beidellites gonflantes donnent des distance de 17.7 A° après ce traitement (Bouberka et al.,
2008). L’espacement basal de la bentonite sodée est égal à 12.12 A° (tableau II.2.3).
Figure II.2.3 : DRX de la bentonite brute et de la bentonite sodé
Tableau II.2.3 : Variation de l’espacement basal suivant les tests de comportement des
argiles
Echantillon Argile brute Traitement à l’éthylène
glycol
Saturation par
K+
Test de Hofman
Klemen
d001(A°) 15.65 17.77 12.12 9.61
112

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
2.3 Caractérisation de l’argile organophile
2.3.1 Diffraction des rayons X
La bentonite modifiée par un cation ammonium quaternaire à longue chaine alkyle
comme le CTAB donne naissance à une argile organophile. Comme il est indiqué sur le
tableau II.2.4, les cations du CTAB sont intercalés dans l’espace interfoliaire de la bentonite
et la distance basale augmente progressivement avec la concentration du CTAB ajoutée.
Quand la concentration du CTAB ajoutée dépasse la capacité d’échange cationique de l’argile
de base, la distance basale n’augmente plus, ce résultat est en accord avec ceux de Volzone
et al., 2006. La molécule du CTAB est formée de 16 chaines de carbone attachées à une tête
d’ammonium quaternaire contenant une charge permanente (+1). Le positionnement de la
molécule organique entre les feuillets de l’argile change en fonction de la quantité de CTAB
ajoutée (Lagaly, 1984; Lee et Kim, 2002). En effet, pour une faible concentration de CTAB
équivalente à 0.35 de la capacité d’échange cationique de l’argile de base, l’espacement basal
est égal à 14 A° (tableau II.2.4), correspondant à la formation d’une monocouche de CTAB
dans l’espace interfeuillet (Bonczek et al., 2002). Une addition supplémentaire de CTAB
conduit à une augmentation de l’espacement basal jusqu’à atteindre une valeur de 20 A°.
Tableau II.2.4 : Distances inter-réticulaires de l’argile sodée et organophile à différentes
concentrations de CTAB
Concentration de CTAB utilisée pour modifier l’argile exprimée par rapport à la CEC
(%CEC)
%CEC BNa 17 34 51 68 85 100 117
d001(A°) 12.12 13.76 14.34 15.44 16.44 18.34 19.87 19.02
L’orientation des ions d’alkylammoniums entre les feuillets argileux est variable. Les
ions d’alkylammoniums peuvent former une monocouche (13.7 A°), une double couche
(17.7A°) ou une pseudo triple couche (21.7 A°) ou encore un complexe paraffine (> 22 A°)
(Jaynes et Boyd, 1991). La bentonite organophile obtenue avec une concentration de CTAB
égale à 100% de la capacité d’échange cationique de l’argile de base, montre un espacement
basal de 19.87 A° (figure II.2.4). Cet espacement basal correspond à une double couche de
CTAB entre les feuillets de l’argile. En effet, en supposant que l’épaisseur de la couche TOT
de la montmorillonite soit égale à 9.7A° (Harris et al., 1999), l’espacement basal de 19.87 A°
correspond à une hauteur de galerie de 10.17 A°. Quand un ion ammonium quaternaire est
113
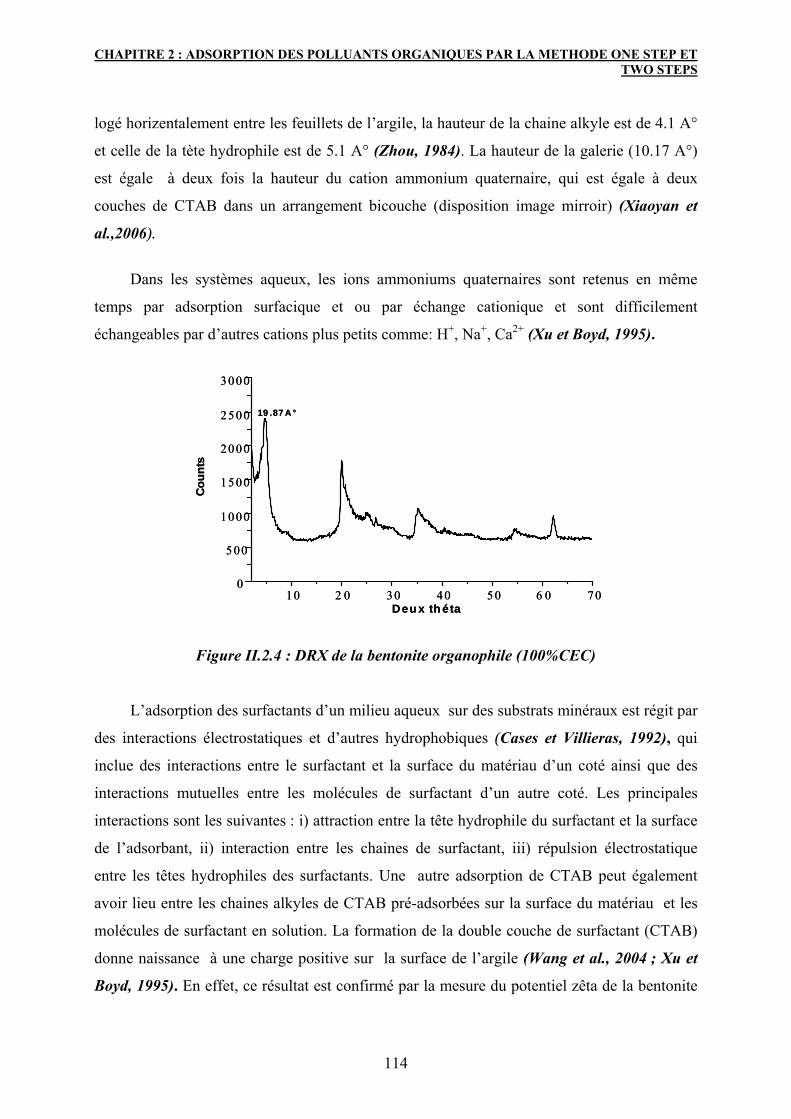
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
logé horizentalement entre les feuillets de l’argile, la hauteur de la chaine alkyle est de 4.1 A°
et celle de la tète hydrophile est de 5.1 A° (Zhou, 1984). La hauteur de la galerie (10.17 A°)
est égale à deux fois la hauteur du cation ammonium quaternaire, qui est égale à deux
couches de CTAB dans un arrangement bicouche (disposition image mirroir) (Xiaoyan et
al.,2006).
Dans les systèmes aqueux, les ions ammoniums quaternaires sont retenus en même
temps par adsorption surfacique et ou par échange cationique et sont difficilement
échangeables par d’autres cations plus petits comme: H+, Na+, Ca2+ (Xu et Boyd, 1995).
10 2 0 30 40 50 6 0 700
500
1000
1500
2000
2500
3000
19 .87 A °
Cou
nts
Deu x th éta10 2 0 30 40 50 6 0 70
0
500
1000
1500
2000
2500
3000
19 .87 A °
Cou
nts
Deu x th éta
Figure II.2.4 : DRX de la bentonite organophile (100%CEC)
L’adsorption des surfactants d’un milieu aqueux sur des substrats minéraux est régit par
des interactions électrostatiques et d’autres hydrophobiques (Cases et Villieras, 1992), qui
inclue des interactions entre le surfactant et la surface du matériau d’un coté ainsi que des
interactions mutuelles entre les molécules de surfactant d’un autre coté. Les principales
interactions sont les suivantes : i) attraction entre la tête hydrophile du surfactant et la surface
de l’adsorbant, ii) interaction entre les chaines de surfactant, iii) répulsion électrostatique
entre les têtes hydrophiles des surfactants. Une autre adsorption de CTAB peut également
avoir lieu entre les chaines alkyles de CTAB pré-adsorbées sur la surface du matériau et les
molécules de surfactant en solution. La formation de la double couche de surfactant (CTAB)
donne naissance à une charge positive sur la surface de l’argile (Wang et al., 2004 ; Xu et
Boyd, 1995). En effet, ce résultat est confirmé par la mesure du potentiel zêta de la bentonite
114

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
sodée et organophile où on remarque une inversion de la charge surfacique qui passe d’une
valeur de –32 mV vers une valeur totalement positive égale à +33mV.
La valeur expérimentale de la teneur en carbone organique (17.67%) est équivalente à
100% CEC (Bouberka et al., 2008). Les caractéristiques générales de la bentonite sodée et
organophile sont présentés dans le tableau II.2.5.
Tableau II.2.5 : Caractéristiques générales de la bentonite sodée et organophile Proprièté Echantillon
Bentonite sodée Bentonite organophile
Formule chimique du cation - C19H42N+
Masse molaire du cation (g/mol) - 364.46
Capacité d’échange cationique(meq/100g) 101.25 -
Teneur en carbon organique (%) 0.05 17.67
2.3.2 spectroscopie Infrarouge
Les bandes caractéristiques de la montmorillonite (figure II.2.5) sont situées à 3625,
3415 et 3230cm−1(Van der Marel et Beutelspacher, 1976). Elles sont attribuées à des unités
OH internes dans la structure de la montmorillonite et à l’eau d’hydratation du cation
interfoliaire (sodium). Une bande moins intense est remarquée vers 3230 Cm-1 est attribué à
l'hydrogène de l'eau lié à d'autres molécules d'eau dans l’espace interfoliaire de la
montmorillonite (Liu et al.,2009).
Les bandes entre 500 et 2000 cm-1 caractéristiques de la montmorillonite sodée sont à
1645, 1050, 1000, 925, 910, 885 et 795cm-1. Ces bandes sont attribuées aux vibrations
d’élongation et de flexion des liaisons Si-O et Si-O-Si. La bande à 1445cm-1est attribuée au
mode de déformation de l'eau intercalaire de l’argile (Boufatit et al.,2007). Les bandes situées
à 525, 468 et 425 cm-1 sont attribuées respectivement aux vibrations de déformation des
liaisons SiOAlVI, SiOMgVI et Si-O-Fe. Le partage du groupement OH entre les atomes Fe et
Al en position octaédrique peut déplacer les vibrations Al-OH vers les basses fréquences aux
environs de 815 et 915 cm-1. Ainsi, les vibrations Mg-O et Mg-OH (confondues avec celle de
SiO) sont localisées respectivement à 530 cm-1 et 560 cm-1 (Farmer, 1974 ; Salerno et al.,
2001).
115

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Les bandes observées entre 2800 et 3000 cm-1 sur le spectre IR de l’argile organophile
sont affectés aux vibrations d’élongation symétriques et antisymétriques de CH2 (Zhou et al.,
2007). L’intensité de la bande de déformation de l’eau diminue après échange avec le CTAB,
ceci est probablement dû à un changement dans la composition de l’argile de base. Après un
échange cationique avec le CTAB, le matériau de base hydrophile adopte un caractère
hydrophobe, l’eau interfoliaire est perdue et le surfactant cationique occupe l’espace
interfoliaire (Williams et Fleming, 1966 ; Liu et al., 2009). La bande située vers 2800 cm-1
est attribuée aux vibrations de valence des liaisons CH2-CH3. Les vibrations de valence de la
liaison C-H sont localisées à 3100 cm-1.
La bande située vers 2926 cm-1 est due aux vibrations de valence des groupes CH3-N.
les bandes caractéristiques de la liaison C-N qui se situent entre 910 et 1000 cm-1 sont
masquées par des vibrations de déformation Al-OH (926 cm-1) de la montmorillonite.
Les bandes situées entre (1400 et 1500 cm-1) sont attribuées aux vibrations de
déformation des groupes CH3 situés à 1480 cm-1. Les bandes caractéristiques des ammoniums
quaternaires avec effet de masquage par les vibrations de déformation Al-OH (926 cm-1) sont
localisées normalement vers 920 et 726 cm-1.
116
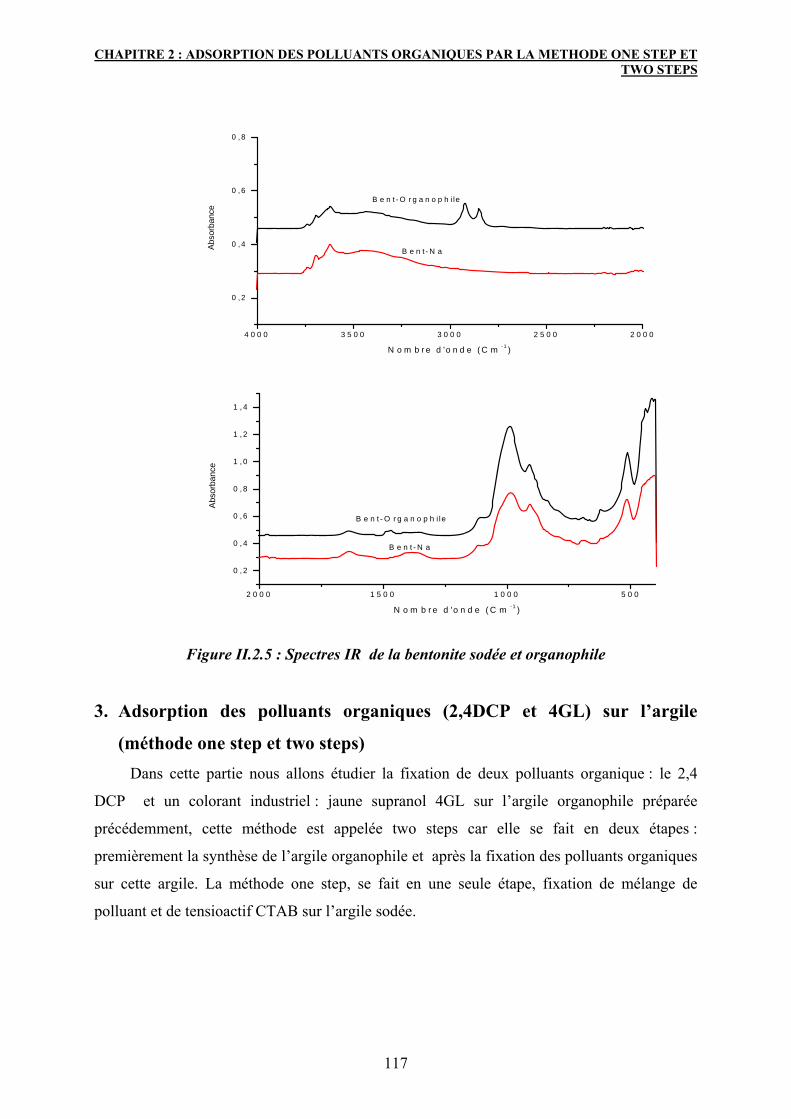
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0
0 , 2
0 , 4
0 , 6
0 , 8
B e n t - O r g a n o p h i le
B e n t - N aAbso
rban
ce
N o m b r e d 'o n d e ( C m - 1 )
2 0 0 0 1 5 0 0 1 0 0 0 5 0 0
0 , 2
0 , 4
0 , 6
0 , 8
1 , 0
1 , 2
1 , 4
B e n t - O r g a n o p h i le
B e n t - N a
Abso
rban
ce
N o m b r e d 'o n d e ( C m - 1 )
Figure II.2.5 : Spectres IR de la bentonite sodée et organophile
3. Adsorption des polluants organiques (2,4DCP et 4GL) sur l’argile
(méthode one step et two steps) Dans cette partie nous allons étudier la fixation de deux polluants organique : le 2,4
DCP et un colorant industriel : jaune supranol 4GL sur l’argile organophile préparée
précédemment, cette méthode est appelée two steps car elle se fait en deux étapes :
premièrement la synthèse de l’argile organophile et après la fixation des polluants organiques
sur cette argile. La méthode one step, se fait en une seule étape, fixation de mélange de
polluant et de tensioactif CTAB sur l’argile sodée.
117
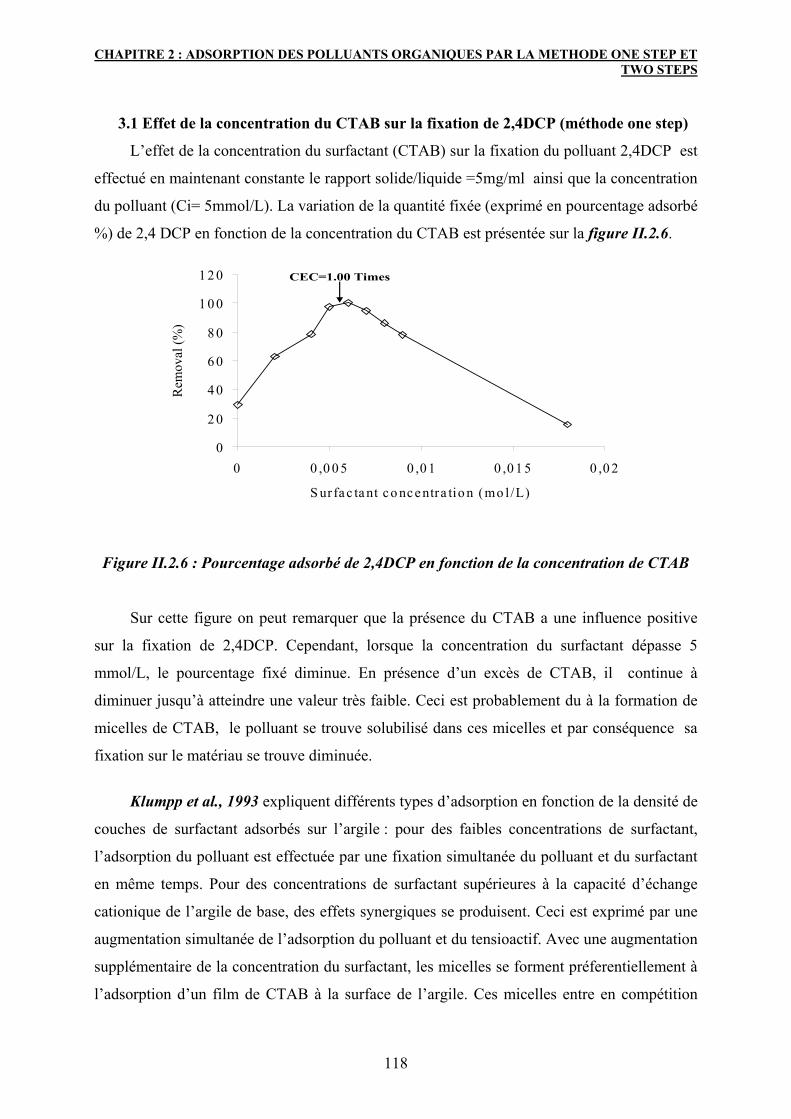
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
3.1 Effet de la concentration du CTAB sur la fixation de 2,4DCP (méthode one step)
L’effet de la concentration du surfactant (CTAB) sur la fixation du polluant 2,4DCP est
effectué en maintenant constante le rapport solide/liquide =5mg/ml ainsi que la concentration
du polluant (Ci= 5mmol/L). La variation de la quantité fixée (exprimé en pourcentage adsorbé
%) de 2,4 DCP en fonction de la concentration du CTAB est présentée sur la figure II.2.6.
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0
0 0 ,0 0 5 0 ,0 1 0 ,0 1 5 0 ,0 2
S ur fa c ta nt c o nc e ntra tio n (mo l /L)
Rem
oval
(%)
CEC=1.00 Times
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0
0 0 ,0 0 5 0 ,0 1 0 ,0 1 5 0 ,0 2
S ur fa c ta nt c o nc e ntra tio n (mo l /L)
Rem
oval
(%)
CEC=1.00 Times
Figure II.2.6 : Pourcentage adsorbé de 2,4DCP en fonction de la concentration de CTAB
Sur cette figure on peut remarquer que la présence du CTAB a une influence positive
sur la fixation de 2,4DCP. Cependant, lorsque la concentration du surfactant dépasse 5
mmol/L, le pourcentage fixé diminue. En présence d’un excès de CTAB, il continue à
diminuer jusqu’à atteindre une valeur très faible. Ceci est probablement du à la formation de
micelles de CTAB, le polluant se trouve solubilisé dans ces micelles et par conséquence sa
fixation sur le matériau se trouve diminuée.
Klumpp et al., 1993 expliquent différents types d’adsorption en fonction de la densité de
couches de surfactant adsorbés sur l’argile : pour des faibles concentrations de surfactant,
l’adsorption du polluant est effectuée par une fixation simultanée du polluant et du surfactant
en même temps. Pour des concentrations de surfactant supérieures à la capacité d’échange
cationique de l’argile de base, des effets synergiques se produisent. Ceci est exprimé par une
augmentation simultanée de l’adsorption du polluant et du tensioactif. Avec une augmentation
supplémentaire de la concentration du surfactant, les micelles se forment préferentiellement à
l’adsorption d’un film de CTAB à la surface de l’argile. Ces micelles entre en compétition
118

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
avec les couches de CTAB adsorbées vis-à-vis du polluant organiques. La diminution de la
quantité fixée du polluant au-delà de la concentration micellaire critique du tensioactif est
probablement due à la solubilisation des molécules de polluant dans les micelles en phase
acqueuse (Esumi et al., 1996). Ces phénomènes sont également observés dans d’autres
études de co-adsorption (Aloulou et al., 2004). Il a été observé que la quantité co-adsorbé du
polluant diminue quand la concentration du surfactant atteint la CMC(concentration
micellaire critique). Cependant, la concentration du CTAB à laquelle le pourcentage du
polluant commence à diminuer est faible par rapport à la CMC.
En comparaison avec d’autres cas de co-adsorption signalés dans la littérature (Okamoto
et al., 2004; Adak et al., 2006). Quand la concentration du CTAB augmente, le volume
vacant dans l’espace interfoliaire de l’argile se trouve diminué pour adsorber d’autres
molécules. Par conséquent, la quantité adsorbée du polluant diminue à cause de
l’encombrement stérique. Dans la suite de ce travail, nous avons choisi une concentration de
CTAB de 5mmol/L correspondante à 100% CEC,et pour laquelle la fixation est maximale.
3.2 Effet de la concentration du CTAB sur la fixation du colorant 4GL (méthode one
step)
L’étude de l’influence de la concentration du CTAB en solution sur la fixation du
colorant 4GL est présentée sur la figure II.2.7. On remarque pour une concentration initiale
de 100 mg/L, une adsorption presque totale du colorant, ceci est réalisée pour une faible
concentration de CTAB égale à 0.2 mmol/L. En augmentant la concentration du CTAB
jusqu’à 5 mmol/L, la quantité fixée reste constante soit 100% du colorant adsorbé. Au-delà de
cette concentration, il y’a une diminution importante de la quantité fixée. Le même
comportement est observé pour le polluant 2,4DCP.
119
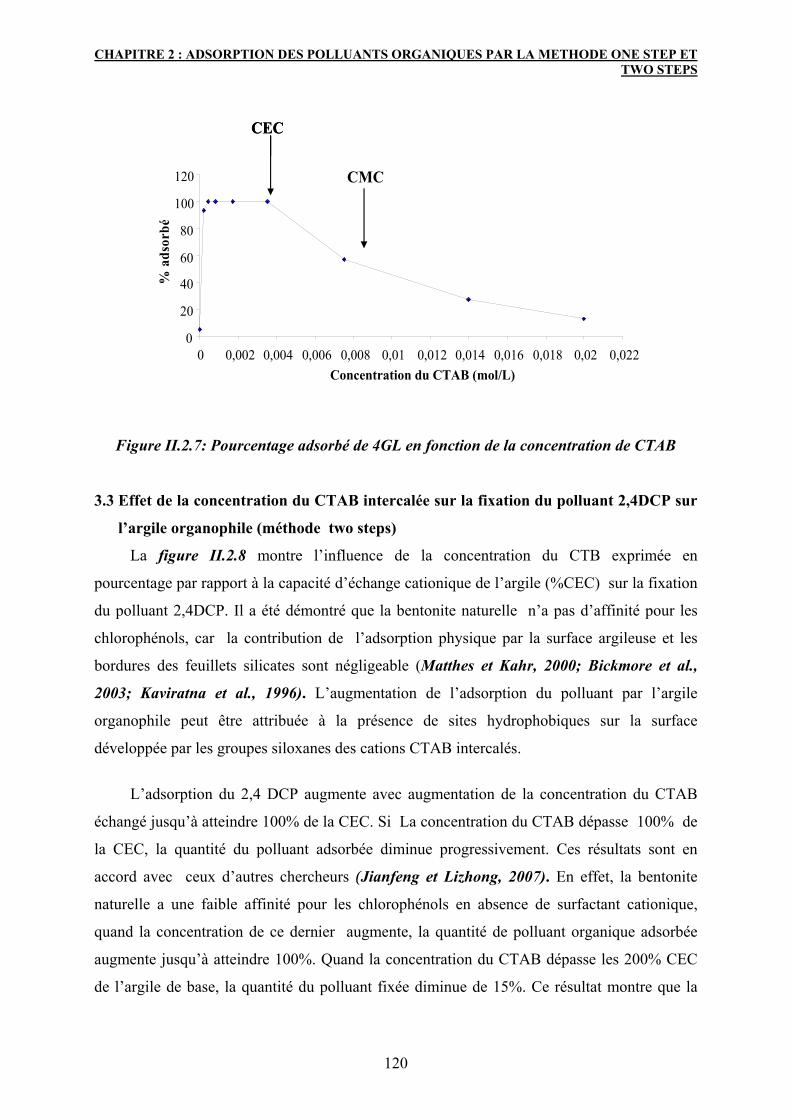
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
0
20
40
60
80
100
120
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014 0,016 0,018 0,02 0,022Concentration du CTAB (mol/L)
% a
dsor
bé
CEC
CMC
0
20
40
60
80
100
120
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014 0,016 0,018 0,02 0,022Concentration du CTAB (mol/L)
% a
dsor
bé
CEC
CMC
Figure II.2.7: Pourcentage adsorbé de 4GL en fonction de la concentration de CTAB
3.3 Effet de la concentration du CTAB intercalée sur la fixation du polluant 2,4DCP sur
l’argile organophile (méthode two steps)
La figure II.2.8 montre l’influence de la concentration du CTB exprimée en
pourcentage par rapport à la capacité d’échange cationique de l’argile (%CEC) sur la fixation
du polluant 2,4DCP. Il a été démontré que la bentonite naturelle n’a pas d’affinité pour les
chlorophénols, car la contribution de l’adsorption physique par la surface argileuse et les
bordures des feuillets silicates sont négligeable (Matthes et Kahr, 2000; Bickmore et al.,
2003; Kaviratna et al., 1996). L’augmentation de l’adsorption du polluant par l’argile
organophile peut être attribuée à la présence de sites hydrophobiques sur la surface
développée par les groupes siloxanes des cations CTAB intercalés.
L’adsorption du 2,4 DCP augmente avec augmentation de la concentration du CTAB
échangé jusqu’à atteindre 100% de la CEC. Si La concentration du CTAB dépasse 100% de
la CEC, la quantité du polluant adsorbée diminue progressivement. Ces résultats sont en
accord avec ceux d’autres chercheurs (Jianfeng et Lizhong, 2007). En effet, la bentonite
naturelle a une faible affinité pour les chlorophénols en absence de surfactant cationique,
quand la concentration de ce dernier augmente, la quantité de polluant organique adsorbée
augmente jusqu’à atteindre 100%. Quand la concentration du CTAB dépasse les 200% CEC
de l’argile de base, la quantité du polluant fixée diminue de 15%. Ce résultat montre que la
120

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
quantité du CTAB présente dans l’argile a une grande influence sur l’adsorption des
chlorophénols.
Dans les systèmes aqueux, les cations ammoniums quaternaires sont retenus par échange
cationique sur la surface externe de l’argile ainsi que par l’espace interfoliaire et se trouvent
difficilement déplacés par d’autres cations comme: H+, Na+, or Ca2+ (Zhang et al., 1993; Xu
et Boyd, 1995a,b; Sulivan et al., 1998). L’adsorption de surfactant sur les argiles est contrôlée
par des interactions électrostatiques et hydrophobiques (Cases et Villieras, 1992), qui inclue
en même temps des interactions entre la surface du matériau et le surfactant et aussi des
interactions entre les molécules de surfactant lui-même. Ces dernières peuvent être résumées
comme suit : i) interaction entre la tète hydrophile du surfactant et la surface argileuse, ii)
interaction entre les chaines hydrophobes de surfactant, iii) répulsion électrostatique entre les
têtes hydrophiles de surfactant. Généralement une isotherme à deux étapes est rencontrée lors
de l’adsorption des surfactant cationiques sur les argiles (Trompette et al., 1994;
Bijsterbosch, 1974; Cases et al.,1986; Chen et al., 1992; Pashley et Israelachvili, 1981;
Patzko et Dekany, 1993; Xu et Boyd, 1995; Mizutani et al., 1995; Brahim et al., 1992 ).
Xu et Boyd, 1995a ont trouvé que le surfactant cationique s’adsorbe initialement par
échange cationique entre les feuillets argileux, ceci cause une agrégation extensive de l’argile.
Quand la concentration du CTAB augmente, ce dernier tend à s’adsorber sur les surfaces
externes des agrégats par échange cationique et liaisons hydrophobiques. Cette dernière
développe une charge positive à la surface argileuse.
A partir de ces résultats, nous allons essayer de comparer la cinétique et l’isotherme
d’adsorption de 2,4DCP sur la bentonite sodée en présence de CTAB (méthode one step) et
sur la bentonite organophile (argile échangée à 100%CEC par le CTAB): méthode two steps.
121
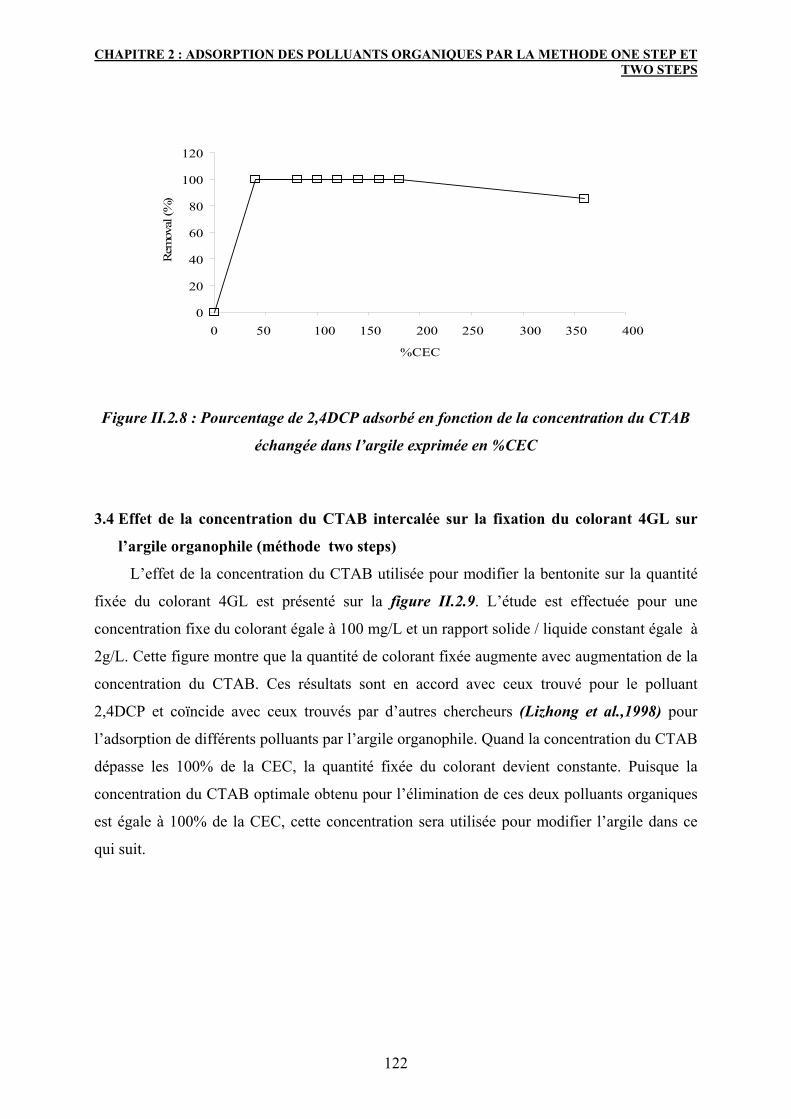
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
0
20
40
60
80
100
120
0 50 100 150 200 250 300 350 400
%CEC
Rem
oval
(%)
0
20
40
60
80
100
120
0 50 100 150 200 250 300 350 400
%CEC
Rem
oval
(%)
Figure II.2.8 : Pourcentage de 2,4DCP adsorbé en fonction de la concentration du CTAB
échangée dans l’argile exprimée en %CEC
3.4 Effet de la concentration du CTAB intercalée sur la fixation du colorant 4GL sur
l’argile organophile (méthode two steps)
L’effet de la concentration du CTAB utilisée pour modifier la bentonite sur la quantité
fixée du colorant 4GL est présenté sur la figure II.2.9. L’étude est effectuée pour une
concentration fixe du colorant égale à 100 mg/L et un rapport solide / liquide constant égale à
2g/L. Cette figure montre que la quantité de colorant fixée augmente avec augmentation de la
concentration du CTAB. Ces résultats sont en accord avec ceux trouvé pour le polluant
2,4DCP et coïncide avec ceux trouvés par d’autres chercheurs (Lizhong et al.,1998) pour
l’adsorption de différents polluants par l’argile organophile. Quand la concentration du CTAB
dépasse les 100% de la CEC, la quantité fixée du colorant devient constante. Puisque la
concentration du CTAB optimale obtenu pour l’élimination de ces deux polluants organiques
est égale à 100% de la CEC, cette concentration sera utilisée pour modifier l’argile dans ce
qui suit.
122
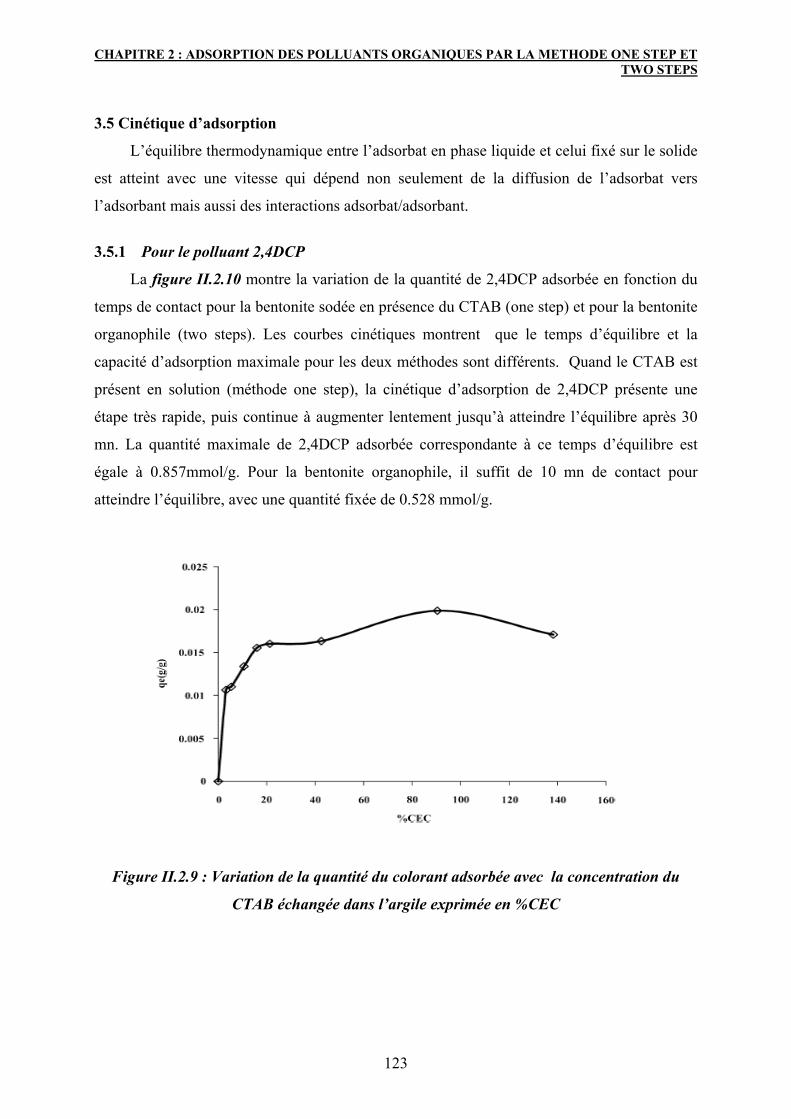
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
3.5 Cinétique d’adsorption
L’équilibre thermodynamique entre l’adsorbat en phase liquide et celui fixé sur le solide
est atteint avec une vitesse qui dépend non seulement de la diffusion de l’adsorbat vers
l’adsorbant mais aussi des interactions adsorbat/adsorbant.
3.5.1 Pour le polluant 2,4DCP
La figure II.2.10 montre la variation de la quantité de 2,4DCP adsorbée en fonction du
temps de contact pour la bentonite sodée en présence du CTAB (one step) et pour la bentonite
organophile (two steps). Les courbes cinétiques montrent que le temps d’équilibre et la
capacité d’adsorption maximale pour les deux méthodes sont différents. Quand le CTAB est
présent en solution (méthode one step), la cinétique d’adsorption de 2,4DCP présente une
étape très rapide, puis continue à augmenter lentement jusqu’à atteindre l’équilibre après 30
mn. La quantité maximale de 2,4DCP adsorbée correspondante à ce temps d’équilibre est
égale à 0.857mmol/g. Pour la bentonite organophile, il suffit de 10 mn de contact pour
atteindre l’équilibre, avec une quantité fixée de 0.528 mmol/g.
Figure II.2.9 : Variation de la quantité du colorant adsorbée avec la concentration du
CTAB échangée dans l’argile exprimée en %CEC
123
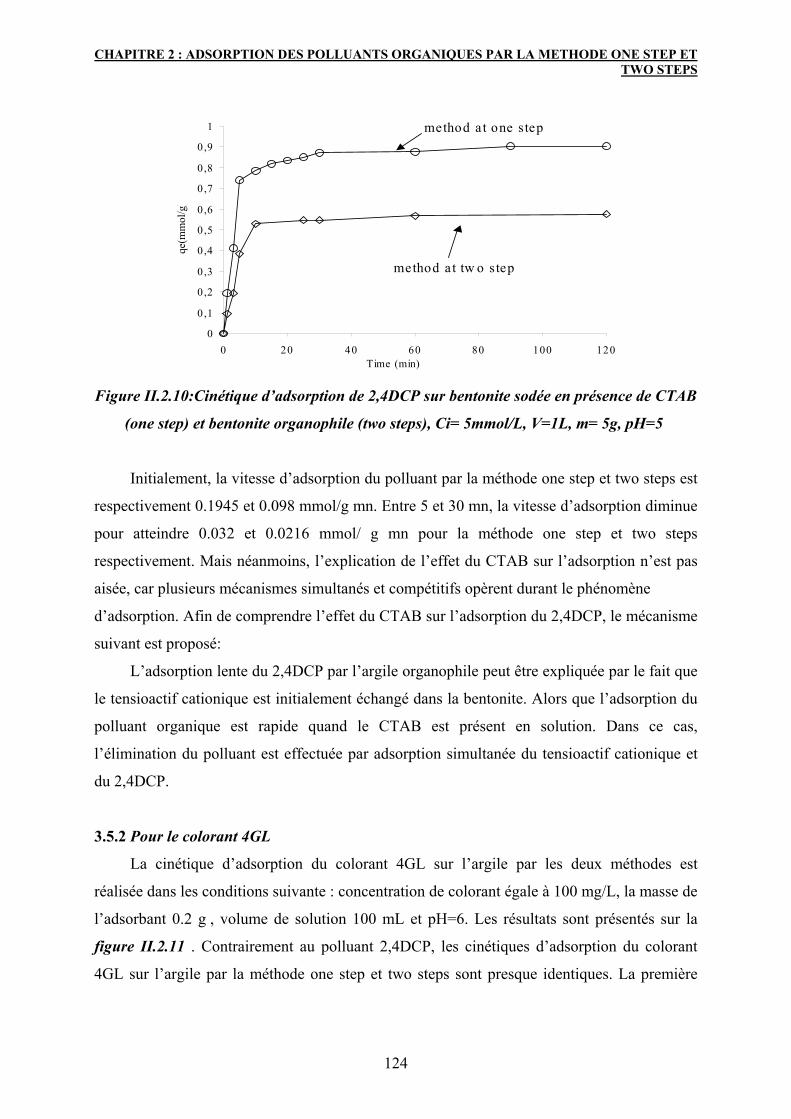
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
0
0 ,1
0 ,2
0 ,3
0 ,4
0 ,5
0 ,6
0 ,7
0 ,8
0 ,9
1
0 20 40 60 80 100 12Time (min)
qe(m
mol
/g
0
)
method at tw o step
method at one step
Figure II.2.10:Cinétique d’adsorption de 2,4DCP sur bentonite sodée en présence de CTAB
(one step) et bentonite organophile (two steps), Ci= 5mmol/L, V=1L, m= 5g, pH=5
Initialement, la vitesse d’adsorption du polluant par la méthode one step et two steps est
respectivement 0.1945 et 0.098 mmol/g mn. Entre 5 et 30 mn, la vitesse d’adsorption diminue
pour atteindre 0.032 et 0.0216 mmol/ g mn pour la méthode one step et two steps
respectivement. Mais néanmoins, l’explication de l’effet du CTAB sur l’adsorption n’est pas
aisée, car plusieurs mécanismes simultanés et compétitifs opèrent durant le phénomène
d’adsorption. Afin de comprendre l’effet du CTAB sur l’adsorption du 2,4DCP, le mécanisme
suivant est proposé:
L’adsorption lente du 2,4DCP par l’argile organophile peut être expliquée par le fait que
le tensioactif cationique est initialement échangé dans la bentonite. Alors que l’adsorption du
polluant organique est rapide quand le CTAB est présent en solution. Dans ce cas,
l’élimination du polluant est effectuée par adsorption simultanée du tensioactif cationique et
du 2,4DCP.
3.5.2 Pour le colorant 4GL
La cinétique d’adsorption du colorant 4GL sur l’argile par les deux méthodes est
réalisée dans les conditions suivante : concentration de colorant égale à 100 mg/L, la masse de
l’adsorbant 0.2 g , volume de solution 100 mL et pH=6. Les résultats sont présentés sur la
figure II.2.11 . Contrairement au polluant 2,4DCP, les cinétiques d’adsorption du colorant
4GL sur l’argile par la méthode one step et two steps sont presque identiques. La première
124
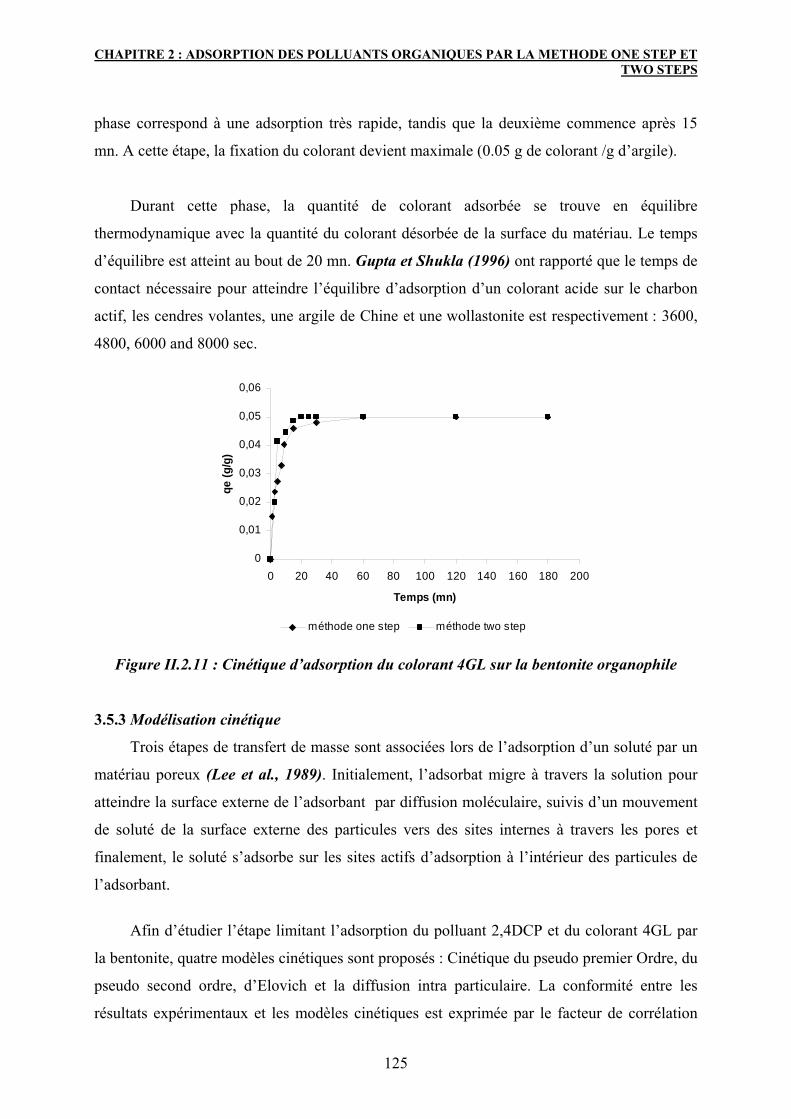
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
phase correspond à une adsorption très rapide, tandis que la deuxième commence après 15
mn. A cette étape, la fixation du colorant devient maximale (0.05 g de colorant /g d’argile).
Durant cette phase, la quantité de colorant adsorbée se trouve en équilibre
thermodynamique avec la quantité du colorant désorbée de la surface du matériau. Le temps
d’équilibre est atteint au bout de 20 mn. Gupta et Shukla (1996) ont rapporté que le temps de
contact nécessaire pour atteindre l’équilibre d’adsorption d’un colorant acide sur le charbon
actif, les cendres volantes, une argile de Chine et une wollastonite est respectivement : 3600,
4800, 6000 and 8000 sec.
0
0,01
0,02
0,03
0,04
0,05
0,06
0 20 40 60 80 100 120 140 160 180 200
Temps (mn)
qe (g
/g)
méthode one step méthode two step
Figure II.2.11 : Cinétique d’adsorption du colorant 4GL sur la bentonite organophile
3.5.3 Modélisation cinétique
Trois étapes de transfert de masse sont associées lors de l’adsorption d’un soluté par un
matériau poreux (Lee et al., 1989). Initialement, l’adsorbat migre à travers la solution pour
atteindre la surface externe de l’adsorbant par diffusion moléculaire, suivis d’un mouvement
de soluté de la surface externe des particules vers des sites internes à travers les pores et
finalement, le soluté s’adsorbe sur les sites actifs d’adsorption à l’intérieur des particules de
l’adsorbant.
Afin d’étudier l’étape limitant l’adsorption du polluant 2,4DCP et du colorant 4GL par
la bentonite, quatre modèles cinétiques sont proposés : Cinétique du pseudo premier Ordre, du
pseudo second ordre, d’Elovich et la diffusion intra particulaire. La conformité entre les
résultats expérimentaux et les modèles cinétiques est exprimée par le facteur de corrélation
125

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
empirique R². Une valeur élevée de R² indique que le modèle appliqué décrit convenablement
la cinétique d’adsorption.
La cinétique du pseudo premier ordre est décrite par la relation suivante (Lagregen,
1898):
303.2
log)( 1 tkqeqtqeLog ×−=− (II.2.2)
Où
qe et qt sont les quantités fixés en mmol/g à l’équilibre et au temps t respectivement, k1:
constante de vitesse du pseudo premier ordre (mn1-).
La cinétique du pseudo second ordre (Ho et McKay, 1999) est régis par l’équation
suivante :
tqeqekqt
t×+
×= )1(
²1
2
(II.2.3)
qe et qt sont les quantités fixés en mmol/g à l’équilibre et au temps t respectivement, k2:
constante de vitesse du pseudo second ordre (g/(mmol mn)).
La cinétique d’adsorption de 2,4 DCP est aussi testée par le modèle d’Elovich (Chien et
Clayton, 1980).
tqt ln)ln( ×+××= ββαβ (II.2.4)
Où
α est la vitesse d’adsorption initiale en mmol/g mn, β: la constante de désorption en g/mmol.
Les constantes d’Elovich sont calculées en traçant (qt) en fonction de ln (t).
La figure II.2.12 montre les formes linéaires de la cinétique du pseudo premier ordre
et du pseudo second ordre de l’adsorption de 2,4DCP sur la bentonite en utilisant les deux
méthodes (one step et two steps). Les constantes de vitesses k1 et k2 obtenues à partir des
équations 2 et 3 sont rassemblées dans le tableau II.2.6. Le modèle du pseudo-premier ordre
est applicable uniquement au début de la cinétique. Tandis que le modèle du pseudo-second
ordre donne de bons résultats durant toute l’expérience et pour les deux méthodes. Ceci
indique que l’étape limitante d’adsorption du 2,4DCP sur la bentonite organophile et sodée
en présence du CTAB est chimique. Des résultats similaires ont été publiés par l’équipe de Ho
126
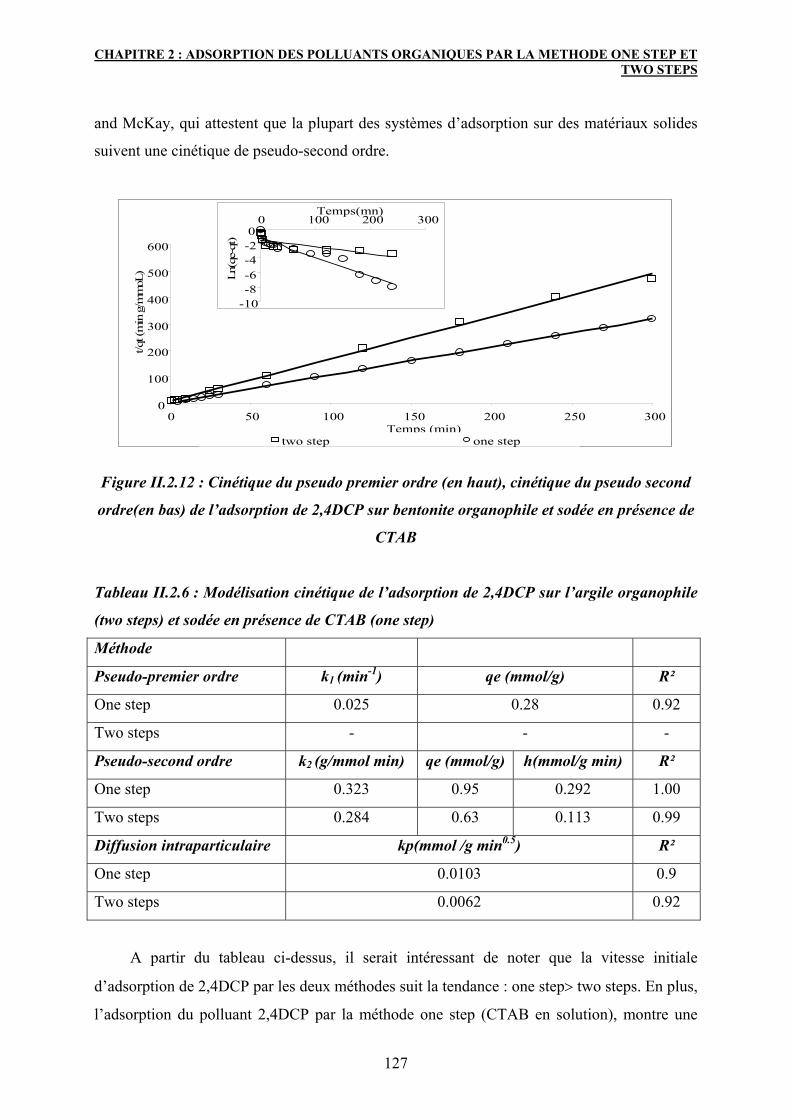
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
and McKay, qui attestent que la plupart des systèmes d’adsorption sur des matériaux solides
suivent une cinétique de pseudo-second ordre.
0
100
200
300
400
500
600
0 50 100 150 200 250 300Temps (min)
t/qt(
min
g/m
moL
)
two step one step
-10-8-6-4-20
0 100 200 300Temps(mn)
Ln(q
e-qt
)
0
100
200
300
400
500
600
0 50 100 150 200 250 300Temps (min)
t/qt(
min
g/m
moL
)
two step one step
-10-8-6-4-20
0 100 200 300Temps(mn)
Ln(q
e-qt
)
Figure II.2.12 : Cinétique du pseudo premier ordre (en haut), cinétique du pseudo second
ordre(en bas) de l’adsorption de 2,4DCP sur bentonite organophile et sodée en présence de
CTAB
Tableau II.2.6 : Modélisation cinétique de l’adsorption de 2,4DCP sur l’argile organophile
(two steps) et sodée en présence de CTAB (one step)
Méthode
Pseudo-premier ordre k1 (min-1) qe (mmol/g) R²
One step 0.025 0.28 0.92
Two steps - - -
Pseudo-second ordre k2 (g/mmol min) qe (mmol/g) h(mmol/g min) R²
One step 0.323 0.95 0.292 1.00
Two steps 0.284 0.63 0.113 0.99
Diffusion intraparticulaire kp(mmol /g min0.5) R²
One step 0.0103 0.9
Two steps 0.0062 0.92
A partir du tableau ci-dessus, il serait intéressant de noter que la vitesse initiale
d’adsorption de 2,4DCP par les deux méthodes suit la tendance : one step> two steps. En plus,
l’adsorption du polluant 2,4DCP par la méthode one step (CTAB en solution), montre une
127

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
capacité d’adsorption nettement plus élevée que celle obtenue pour l’argile organophile. Ceci
implique des interactions très fortes entre le polluant organique et la surface de l’adsorbant
quand le CTAB est présent en solution. Ceci est probablement dû à l’effet synergique produit
par la présence du surfactant en même temps que le polluant en solution. Pour des adsorptions
chimiques sur des adsorbants hétérogènes, l’équation d’Elovich donne parfois de meilleures
informations par rapport à la cinétique du pseudo second ordre. Le tracé de la forme linéaire
de l’équation d’Elovich donne des coefficients de corrélation empiriques variant entre 0.78 et
0.83 (résultats non présentés). Ces valeurs sont inférieures à ceux obtenus par le modèle du
pseudo second ordre. Le modèle d’Elovich ne prédit aucun mécanisme d’adsorption, mais il
se trouve utile pour décrire l’adsorption sur des matériaux hétérogènes (Bhattacharyya et
Sharma, 2004 ; Sparks, 1986 ; Annadurai et al., 2002). Des résultats similaires ont été
donnés par O¨zacar et al.( O¨zacar et al., 2008) lors de l’adsorption du plomb sur une résine
de Valonia.
La modélisation de la cinétique de l’adsorption de 4GL sur l’argile en utilisant la
méthode one step et two steps montre que les deux modèles sont applicables à nos résultats
expérimentaux. En effet, avec des coefficients de corrélation empiriques proche de l’unité et
des capacités d’adsorption presque égales aux capacités d’adsorption maximales, on peut
déduire que l’adsorption du 4GL sur l’argile suit une cinétique du pseudo premier ordre et du
pseudo second ordre en même temps, ce résultat coïncide avec celui trouvé récemment par
Zhu et Ma (Zhu et Ma, 2008) concernant l’adsorption d’un colorant acide Orange II, par la
bentonite en présence du CTAB en solution. Contrairement à ce qui est remarqué pour le
polluant 2,4DCP, la vitesse d’adsorption du colorant 4GL suit la tendance Two steps >one
step. L’application du modèle d’Elovich à nos résultats expérimentaux donne des coefficients
de corrélation empiriques très faibles, ce qui induit que l’adsorption du colorant 4GL sur
l’argile par les deux méthodes ne suit pas le modèle d’Elovich.
Le modèle de diffusion intra particulaire a été testé dans ce travail. En se basant sur les
travaux de Weber et al, (Weber et al., 1963), le coefficient de diffusion intra particulaire kp
est défini par la relation suivante :
tkpqt ×= (II.2.5)
Où
128

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
kp est la constante vitesse de la diffusion intra particulaire (mmol/g mn0.5) et peut être
calculée à partir du tracé de la courbe qt(mmol/g) en fonction de t (mn0.5).
A partir de la figure II.2.14 et la figure II.2.15 on peut remarquer que l’adsorption des
deux polluants : 2,4DCP et 4GL se fait en deux étapes. L’étape la plus lente contrôle le
phénomène d’adsorption. Généralement, la diffusion intra particulaire est l’étape limitante
dans un système d’adsorption en Batch. Les coefficients de diffusion intra particulaire relatifs
à nos expériences sont rassemblés dans le tableau II.2.6 et II.2.7. Les coefficients de
corrélation empirique étant supérieur à 0.85, ils restent toujours inférieurs à ceux trouvés pour
le modèle du pseudo second ordre. Les deux phases présentes sur la courbe de la diffusion
intra particulaire indiquent que l’adsorption se fait en deux étapes : adsorption surfacique et
diffusion intra particulaire. La première étape de la courbe indique un contrôle de la double
couche et la seconde étape correspond à la diffusion intra particulaire ou diffusion dans les
pores de l’adsorbant. Des résultats semblables ont été reportés par Sankar et al (Sankar et al.,
1999) lors de l’adsorption d’un colorant acide et un colorant direct sur charbon actif et par
Sivaraj et al. lors de l’adsorption d’un colorant acide violet sur des pépites d’oranges (Sivaraj
et al., 2001). La pente de la seconde partie de la courbe est définie comme étant la constante
de diffusion intra particulaire kp et l’intersection de cette droite avec l’axe des y donne l’effet
de la double couche diffuse sur l’adsorption. Une valeur importante de cette intersection
montre une grande contribution de l’adsorption surfacique sur le phénomène de diffusion intra
particulaire.
129
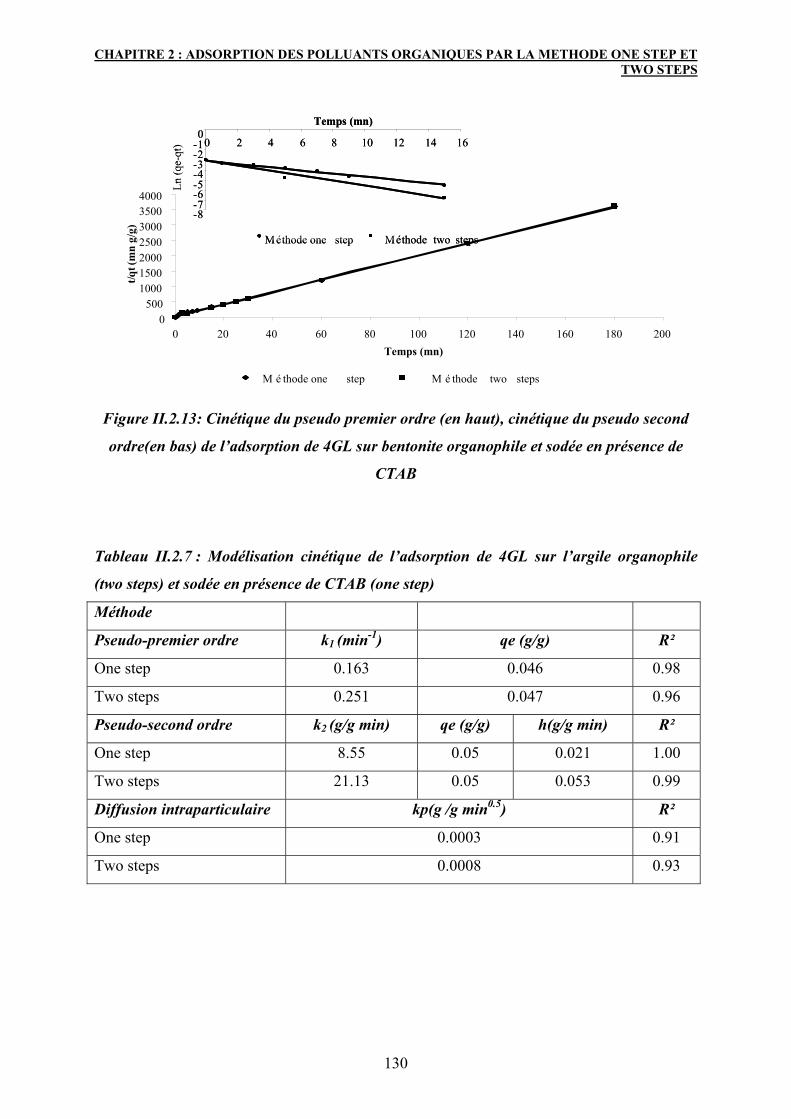
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
-
Méthode one step Méthode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(
mn
g/g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
Ln (q
e-qt
)
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
-
Méthode one step Méthode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(
mn
g/g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
-
Méthode one step Méthode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
-
Méthode one step Méthode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(m
n g/
g)
M é thode one step M é thode two steps
0500
1000150020002500300035004000
0 20 40 60 80 100 120 140 160 180 200Temps (mn)
t/qt(
mn
g/g)
M é thode one step M é thode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
-8-7-6-5-4-3-2-10
0 2 4 6 8 10 12 14 16
Temps (mn)
Méthode one step Méthode two steps
Ln (q
e-qt
)
Figure II.2.13: Cinétique du pseudo premier ordre (en haut), cinétique du pseudo second
ordre(en bas) de l’adsorption de 4GL sur bentonite organophile et sodée en présence de
CTAB
Tableau II.2.7 : Modélisation cinétique de l’adsorption de 4GL sur l’argile organophile
(two steps) et sodée en présence de CTAB (one step)
Méthode
Pseudo-premier ordre k1 (min-1) qe (g/g) R²
One step 0.163 0.046 0.98
Two steps 0.251 0.047 0.96
Pseudo-second ordre k2 (g/g min) qe (g/g) h(g/g min) R²
One step 8.55 0.05 0.021 1.00
Two steps 21.13 0.05 0.053 0.99
Diffusion intraparticulaire kp(g /g min0.5) R²
One step 0.0003 0.91
Two steps 0.0008 0.93
130
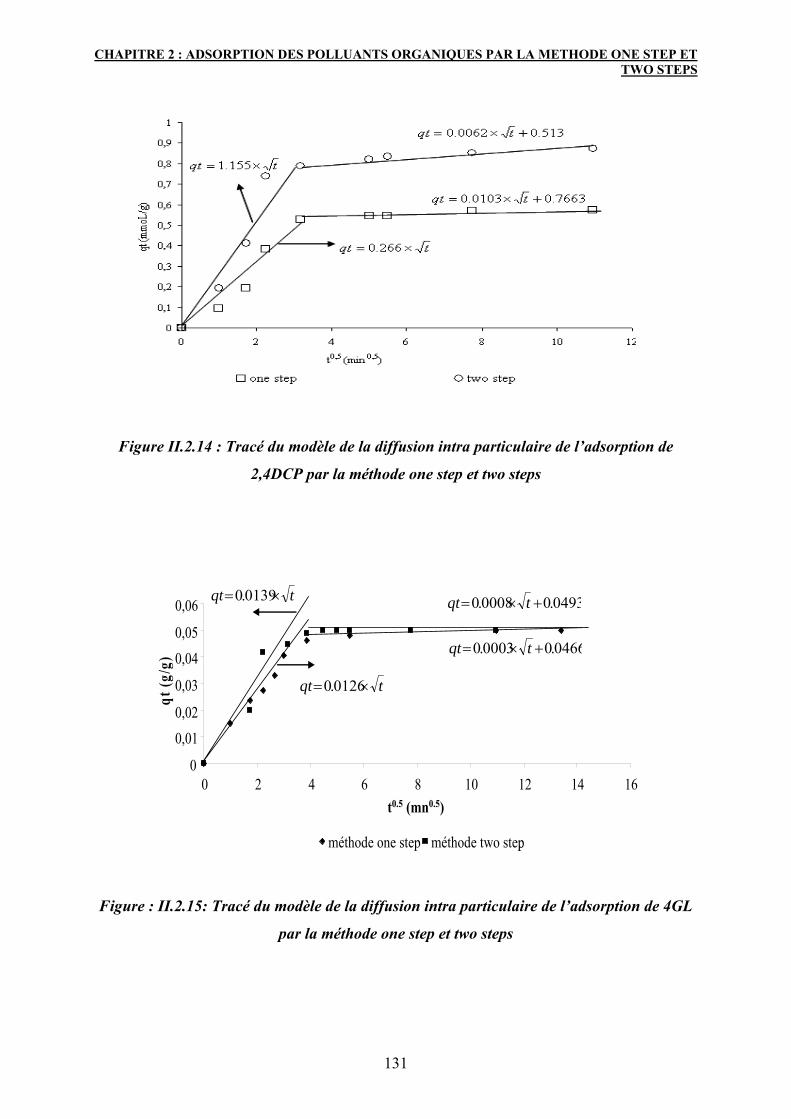
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Figure II.2.14 : Tracé du modèle de la diffusion intra particulaire de l’adsorption de
2,4DCP par la méthode one step et two steps
0
0,010,02
0,03
0,040,05
0,06
0 2 4 6 8 10 12 14 1t0.5 (mn0.5)
qt(g
/g)
6
méthode one step méthode two step
tqt ×= 0126.0
0466.00003.0 +×= tqt
tqt ×= 0139.0 0493.00008.0 +×= tqt
0
0,010,02
0,03
0,040,05
0,06
0 2 4 6 8 10 12 14 1t0.5 (mn0.5)
qt(g
/g)
6
méthode one step méthode two step
tqt ×= 0126.0
0466.00003.0 +×= tqt
tqt ×= 0139.0 0493.00008.0 +×= tqt
Figure : II.2.15: Tracé du modèle de la diffusion intra particulaire de l’adsorption de 4GL
par la méthode one step et two steps
131

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
3. Etude de l’isotherme d’adsorption
L’adsorption d’une substance d’une phase vers la surface d’une autre phase dans un
système donné, conduit à une distribution thermodynamique de cette substance entre les deux
phases jusqu’à atteindre l’équilibre. Cette distribution peut être exprimée en termes
d’isotherme d’adsorption (Kinniburgh, 1986).
Les isothermes d’adsorption du polluant 2,4DCP par l’argile organophile et l’argile
sodée en présence du CTAB sont présentées sur la figure II.2.16 (a et b). Les résultats
expérimentales d’adsorption de 2,4DCP par les deux méthodes (one step et two steps)
donnent des résultats tout à fait différents. La comparaison des deux courbes d’adsorption,
met en évidence le fait que la présence du CTAB en solution change complètement l’allure de
l’isotherme d’adsorption et augmente de façon importante la capacité d’adsorption de l’argile
de base.
En se basant sur la classification des isothermes d’adsorption donné par Giles et al
(Giles et al., 1960) concernant la pente initiale et la forme finale de la courbe, l’isotherme
d’adsorption de 2,4DCP sur l’argile sodée en présence de CTAB (méthode one step) est de
type Langmuir sous groupe 4.C’est une isotherme de type multicouches, typique de celle
obtenue par des interactions modérées entre adsorbat et adsorbant, suivie par un effet
coopératif des molécules adsorbées (Gregg et Sing, 1982). Cette courbe est tout à fait
différente de l’isotherme d’adsorption du même polluant sur l’argile organophile. En effet,
dans ce cas on trouve une isotherme de type Langmuir sous groupe 2 (Langmuir
monocouche), mais la formation de la monocouche s’effectue à la même concentration à
l’équilibre (Ce) pour les deux cas. Quand la concentration du polluant est faible
(0.05mmol/L), une faible interaction entre le polluant et la surface argileuse est remarquée
quand le CTAB est présent en solution.
En augmentant la concentration du polluant, un effet coopératif de l’adsorption fait
augmenter la capacité d’adsorption jusqu’à atteindre la valeur de 2.5mmol/g. Dans le dernier
cas, des interactions s’établissent entre les alkyls ammonium et la surface de la bentonite,
mais quand la molécule de CTAB est adsorbée sur la surface argileuse, les interactions
bentonites -CTAB font augmenter la capacité d’adsorption. Cependant, la capacité
d’adsorption de l’argile organophile est plus faible et ne peut atteindre que 1.5 mmol/g.
132

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Le polluant 2,4DCP est faiblement adsorbé par la bentonite seule (Mc Bride, 1987). Ceci est
dû à l’adsorption préférentielle de l’eau sur la surface argileuse. Il existe deux types
d’interactions entre les produits organiques polaires et les organo bentonites (argiles
organophiles) : l’adsorption et le partage, et sont étroitement liés à la teneur en matière
organique et aux caractéristiques minérales de la surface argileuse.
L’adsorption surfacique peut inclure l’échange ionique, la protonation, les liaisons
hydrogènes et d’autres interactions argileuses (Sposito, 1984; Bleam, 1990). Le partage
comprend l’interaction entre la matière organique et le polluant organique. Lorsqu’une grosse
molécule organique comme le CTAB réagit avec la bentonite, le phénomène de partage joue
un rôle important. L’adsorption des chlorophénols par les sols a été décrite en supposant un
partage hydrophobique des espèces neutres par la matière organique du sol (Divincenzo et
Sparks, 2001) ou décrite par He et al, (He et al., 2006) comme étant un mécanisme
d’adsorption lié au remplissage de vide de la matière organique du sol facilité par des liaisons
d’hydrogènes. Comme résultat de l’échange cationique, le cation hydrophile dans l’espace
intercalaire est remplacé et l’argile devient hydrophobe. Les smectites organophiles sont
largement utilisées pour l’élimination des polluants organiques. Pour mieux comprendre, il
suffit de regarder l’espace intercalaire comme étant un solvant organique susceptible de
retenir les molécules organiques (Boyd et al., 1988; Smith et Jaffe, 1991; Smith et al., 1990;
Boyd, 1992 ).
3.6.1 Modélisation des isothermes d’adsorption
Les données d’adsorption obtenues à partir de ces isothermes sont analysées par les
modèles de Freundlich (Freundlich, 1906), de Langmuir (Langmuir, 1916) et de Redlich
Peterson (Redlich et Peterson, 1959). Ces modèles sont discutés en détails dans la section
matériels et méthodes et leurs équations respectives sont données ci-dessous.
Modèle de Freundlich (II.2.6) nf CeKqe ×=
Modèle de Langmuir Ceb
Cebqqe×+××
=1max (II.2.7)
Modèle de Redlich Peterson βCeaCekqe
R
R
×+×
=1
(II.2.8)
133

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Les tracés des formes non linéaires de ces trois modèles sont présentés sur la figure
II.2.16. Les paramètres ajustables obtenues à partir de ces modèles (qmax, b, Kf, n, kR, aR et β)
sont listés dans le tableau II.2.8.
En se basant sur les valeurs du coefficient de corrélation empiriques R², on remarque
que le modèle de Freundlich donne de bons résultats par rapport à celui de Langmuir et de
Redlich Peterson. Le coefficient de Freundlich « n » donne des informations concernant
l’hétérogénéité de la surface et l’affinité entre l’adsorbat et l’adsorbant. Une valeur de « n »
de 0.32 et 0.51 indique une adsorption favorable pour les deux méthodes (one step et two
steps). Par contre, les valeurs de la capacité d’adsorption maximale sont calculées à partir du
modèle de Langmuir, qui donne des résultats plus proches des valeurs expérimentales. Les
capacités d’adsorption maximales de 2,4DCP sur l’argile organophile (two steps) et l’argile
sodée quand le CTAB est présent en solution (one step) sont respectivement : 2.293 et 3.546
mmol/g.
Les isothermes d’adsorption du colorant 4GL sur la bentonite par la méthode one step
et two steps sont présentées sur la figure II.2.17. L’isotherme d’adsorption du colorant sur la
bentonite organophile (two steps) est de type L, correspondant à la formation d’une
monocouche de colorant sur la surface du matériau avec une capacité d’adsorption de 0.5 g de
colorant /g d’argile organophile. Contrairement à ce qui est prévu, l’isotherme d’adsorption
du colorant sur la bentonite quand le CTAB est présent en solution (one step) est totalement
différente. En effet, c’est une isotherme de type H traduisant une grande affinité entre
l’adsorbat et l’adsorbant et donnant une capacité d’adsorption de l’ordre de 0.6 g/g d’argile.
Par conséquence, on peut en déduire que le mécanisme de fixation du colorant 4GL sur la
bentonite en présence de CTAB en solution est tout à fait différent à celui quand le CTAB est
échangée dans l’argile. Afin de mieux comprendre le mécanisme d’adsorption du colorant
4GL par les deux méthodes, une modélisation non linéaire des isothermes d’adsorption de
ce colorant est effectuée en utilisant les modèles précédents. Les résultats de cette
modélisation sont tracés sur la figure II.2.17 et résumés dans le tableau II.2.9.
134
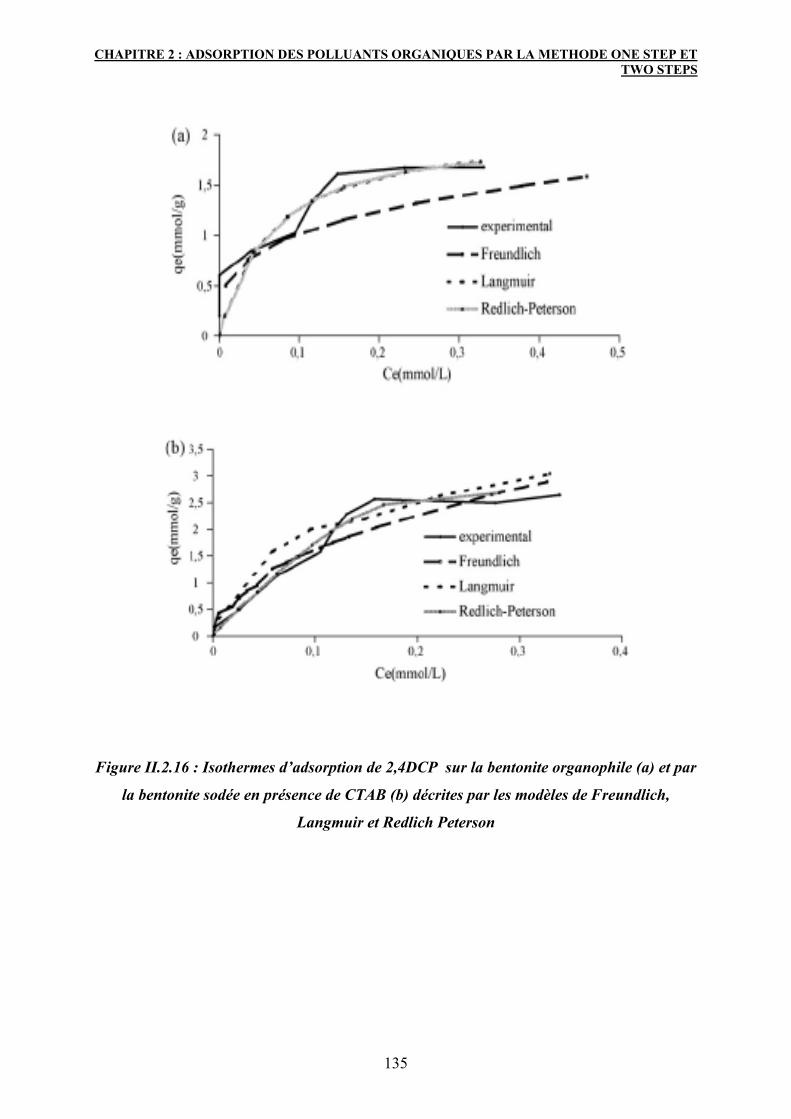
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Figure II.2.16 : Isothermes d’adsorption de 2,4DCP sur la bentonite organophile (a) et par
la bentonite sodée en présence de CTAB (b) décrites par les modèles de Freundlich,
Langmuir et Redlich Peterson
135
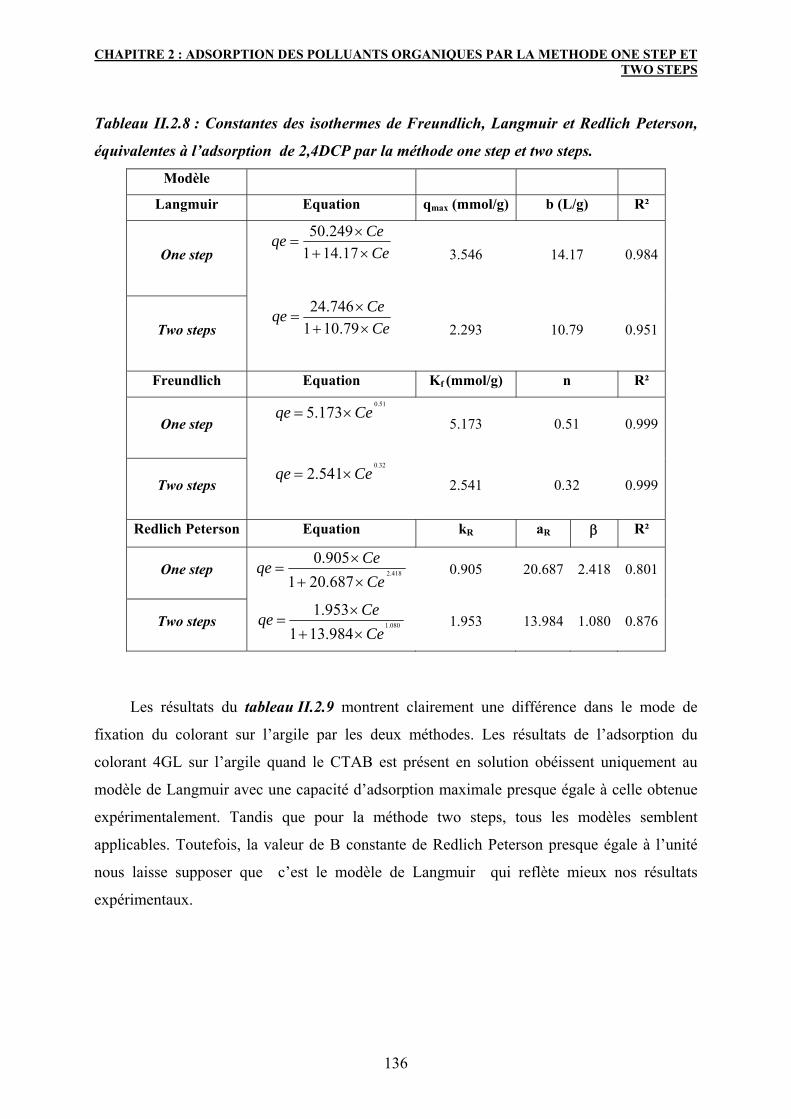
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Tableau II.2.8 : Constantes des isothermes de Freundlich, Langmuir et Redlich Peterson,
équivalentes à l’adsorption de 2,4DCP par la méthode one step et two steps.
Modèle
Langmuir Equation qmax (mmol/g) b (L/g) R²
One step CeCeqe×+×
=17.141
249.50
3.546 14.17 0.984
Two steps CeCeqe×+×
=79.101
746.24
2.293 10.79 0.951
Freundlich Equation Kf (mmol/g) n R²
One step 51.0
173.5 Ceqe ×=
5.173 0.51 0.999
Two steps 32.0
541.2 Ceqe ×=
2.541 0.32 0.999
Redlich Peterson Equation kR aR β R²
One step 418.2
687.201905.0
CeCeqe×+×
= 0.905 20.687 2.418 0.801
Two steps 080.1
984.131953.1
CeCeqe×+×
= 1.953 13.984 1.080 0.876
Les résultats du tableau II.2.9 montrent clairement une différence dans le mode de
fixation du colorant sur l’argile par les deux méthodes. Les résultats de l’adsorption du
colorant 4GL sur l’argile quand le CTAB est présent en solution obéissent uniquement au
modèle de Langmuir avec une capacité d’adsorption maximale presque égale à celle obtenue
expérimentalement. Tandis que pour la méthode two steps, tous les modèles semblent
applicables. Toutefois, la valeur de B constante de Redlich Peterson presque égale à l’unité
nous laisse supposer que c’est le modèle de Langmuir qui reflète mieux nos résultats
expérimentaux.
136
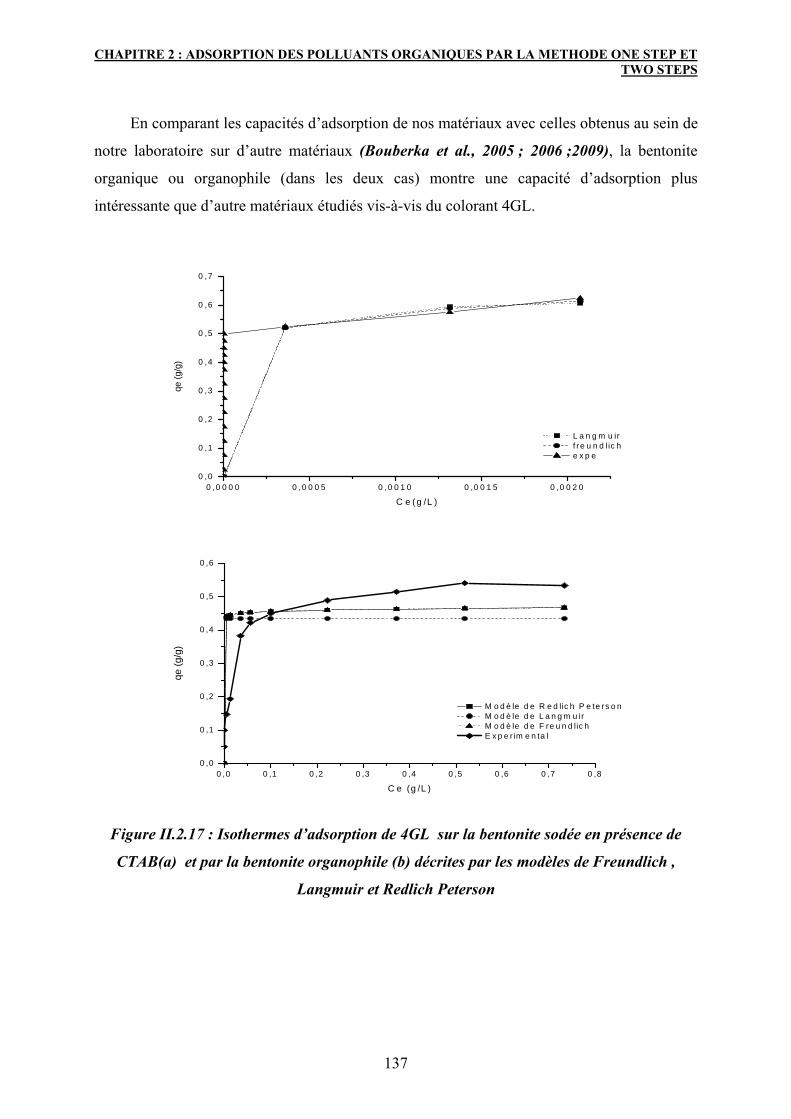
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
En comparant les capacités d’adsorption de nos matériaux avec celles obtenus au sein de
notre laboratoire sur d’autre matériaux (Bouberka et al., 2005 ; 2006 ;2009), la bentonite
organique ou organophile (dans les deux cas) montre une capacité d’adsorption plus
intéressante que d’autre matériaux étudiés vis-à-vis du colorant 4GL.
0 ,0 0 0 0 0 ,0 0 0 5 0 ,0 0 1 0 0 ,0 0 1 5 0 ,0 0 2 00 ,0
0 ,1
0 ,2
0 ,3
0 ,4
0 ,5
0 ,6
0 ,7
qe (g
/g)
C e (g /L )
L a n g m u ir f r e u n d l ic h e x p e
0 ,0 0 ,1 0 ,2 0 ,3 0 ,4 0 ,5 0 ,6 0 ,7 0 ,80 ,0
0 ,1
0 ,2
0 ,3
0 ,4
0 ,5
0 ,6
qe (g
/g)
C e (g /L )
M o d è le d e R e d lic h P e te rs o n M o d è le d e L a n g m u ir M o d è le d e F re u n d lic h E x p e r im e n ta l
Figure II.2.17 : Isothermes d’adsorption de 4GL sur la bentonite sodée en présence de
CTAB(a) et par la bentonite organophile (b) décrites par les modèles de Freundlich ,
Langmuir et Redlich Peterson
137
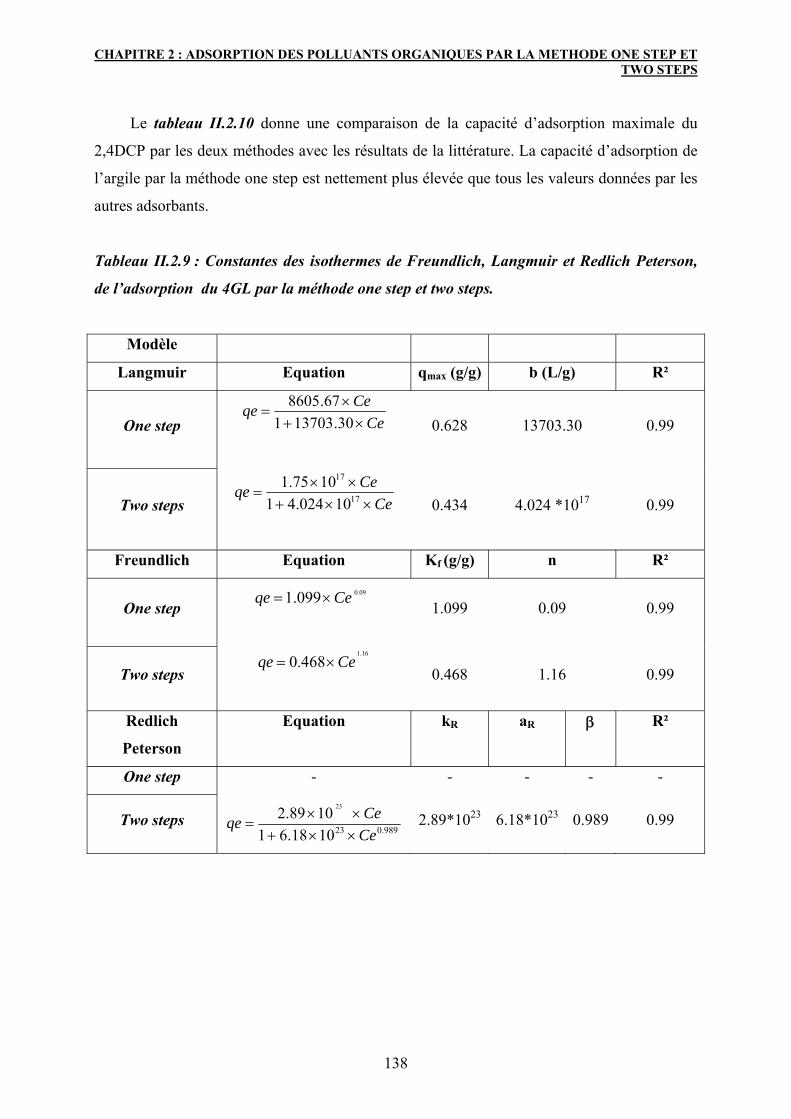
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Le tableau II.2.10 donne une comparaison de la capacité d’adsorption maximale du
2,4DCP par les deux méthodes avec les résultats de la littérature. La capacité d’adsorption de
l’argile par la méthode one step est nettement plus élevée que tous les valeurs données par les
autres adsorbants.
Tableau II.2.9 : Constantes des isothermes de Freundlich, Langmuir et Redlich Peterson,
de l’adsorption du 4GL par la méthode one step et two steps.
Modèle
Langmuir Equation qmax (g/g) b (L/g) R²
One step CeCeqe×+
×=
30.13703167.8605
0.628 13703.30 0.99
Two steps CeCeqe××+
××= 17
17
10024.411075.1
0.434 4.024 *1017 0.99
Freundlich Equation Kf (g/g) n R²
One step 09.0099.1 Ceqe ×=
1.099 0.09 0.99
Two steps 16.1
468.0 Ceqe ×=
0.468 1.16 0.99
Redlich
Peterson
Equation kR aR β R²
One step - - - - -
Two steps 989.0231018.61
1089.2 23
CeCeqe
××+××
= 2.89*1023 6.18*1023 0.989 0.99
138
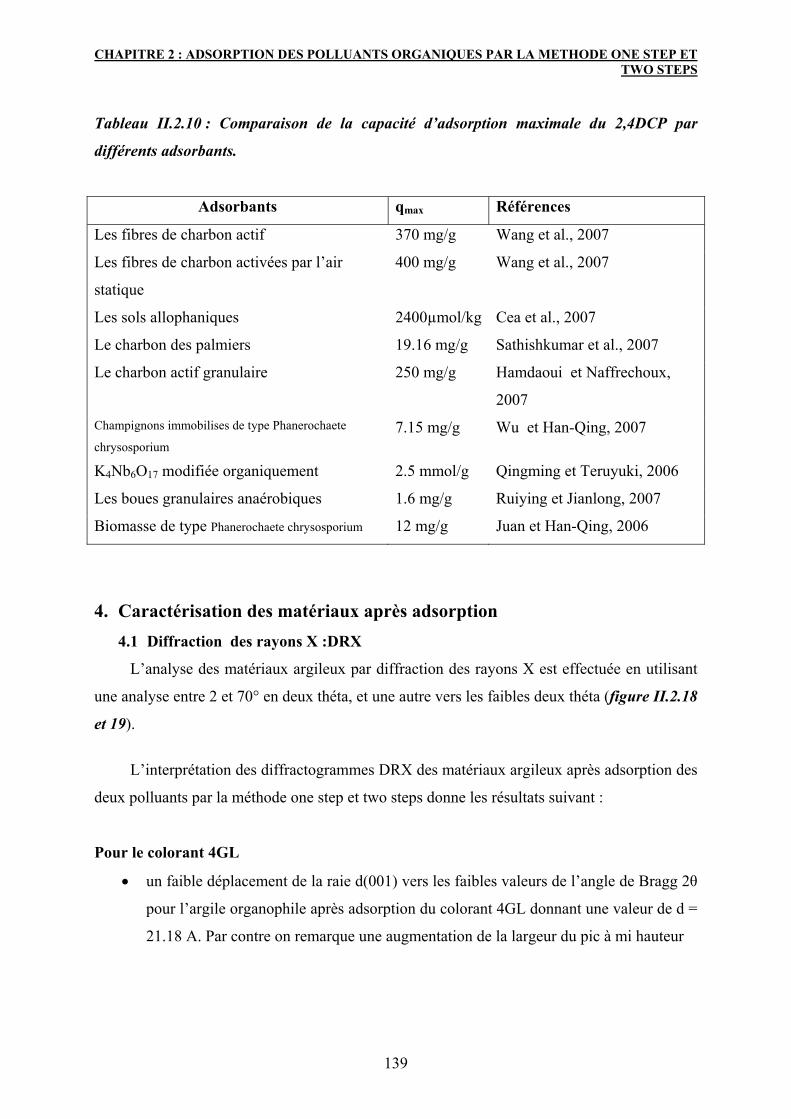
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Tableau II.2.10 : Comparaison de la capacité d’adsorption maximale du 2,4DCP par
différents adsorbants.
Adsorbants qmax Références
Les fibres de charbon actif 370 mg/g Wang et al., 2007
Les fibres de charbon activées par l’air
statique
400 mg/g Wang et al., 2007
Les sols allophaniques 2400µmol/kg Cea et al., 2007
Le charbon des palmiers 19.16 mg/g Sathishkumar et al., 2007
Le charbon actif granulaire 250 mg/g Hamdaoui et Naffrechoux,
2007 Champignons immobilises de type Phanerochaete chrysosporium
7.15 mg/g Wu et Han-Qing, 2007
K4Nb6O17 modifiée organiquement 2.5 mmol/g Qingming et Teruyuki, 2006
Les boues granulaires anaérobiques 1.6 mg/g Ruiying et Jianlong, 2007
Biomasse de type Phanerochaete chrysosporium 12 mg/g Juan et Han-Qing, 2006
4. Caractérisation des matériaux après adsorption 4.1 Diffraction des rayons X :DRX
L’analyse des matériaux argileux par diffraction des rayons X est effectuée en utilisant
une analyse entre 2 et 70° en deux théta, et une autre vers les faibles deux théta (figure II.2.18
et 19).
L’interprétation des diffractogrammes DRX des matériaux argileux après adsorption des
deux polluants par la méthode one step et two steps donne les résultats suivant :
Pour le colorant 4GL
• un faible déplacement de la raie d(001) vers les faibles valeurs de l’angle de Bragg 2θ
pour l’argile organophile après adsorption du colorant 4GL donnant une valeur de d =
21.18 A. Par contre on remarque une augmentation de la largeur du pic à mi hauteur
139

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• qui passe de 3.06 ° (2θ) pour l’argile organophile à 5.03° (2θ) pour le même
matériau après adsorption du colorant 4GL ;
• Pour le matériau argile sodée après adsorption du colorant 4GL en présence du CTAB
en solution, on remarque deux catégories de pics : le premier donnant une distance
égale à celle obtenue par l’argile organophile tandis que le deuxième situé à 2.5° en 2θ
(d= 35 A) dû à l’intercalation du CTAB et du colorant en même temps.
Pour le polluant 2,4DCP
• Contrairement à ce qui est remarqué pour le colorant 4GL, l’argile après adsorption
du polluant 2,4DCP montre deux séries de pics dans les deux cas (one step et two
steps) ceci est probablement dû à une intercalation d’espèces de dimensions
différentes entres les feuillets argileux (Liu et al.,2009) ;
• Pour le premier matériau après adsorption du polluant, le premier pic correspondant à
l’argile organophile tandis que l’autre situé vers les faibles deux théta et donnant une
distance de d= 27.69 A correspond à l’intercalation de la molécule de 2,4DCP entre
les feuillets ;
• En comparaison avec le premier matériau, le deuxième montre une grande
intercalation du polluant entre les feuillets argileux. en effet, on remarque un décalage
supplémentaire vers les faibles deux thêta jusqu’à atteindre une valeur de 40 A°, ainsi
qu’une diminution de l’intensité du pic correspondant à l’argile organophile.
En comparant les résultats de l’adsorption one step (CTAB en solution) avec ceux de
Zhu et Ma, 2008 concernant l’adsorption du colorant acide, orange II en présence de CTAB
par l’argile, ces auteurs ont trouvé que l’espacement basal passe de 15.5 A° pour la bentonite
seule à 48.5 A° pour le même matériau après adsorption du colorant et une concentration de
CTAB équivalente à 200% CEC de l’argile de base. Toutefois, l’espacement basal de la
bentonite intercalée à 200% CEC par le CTAB est de 41.2 A, ce qui est plus faible que la
valeur trouvée quand le CTAB et le colorant sont adsorbés. Ces résultats sont en accord avec
nos travaux et confirment le fait que le colorant s’est adsorbé dans l’espace intercalaire de
l’argile (Chen et al., 2005). L’augmentation de l’espace intercalaire prouve que le surfactant
a formé une structure étendu avec un angle d’inclinaison avec le feuillet argileux.
140

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
L’adsorption du CTAB se fait sous sa forme cationique soit CTA+ par des interactions
électrostatiques entre le feuillet chargé négativement et la molécule chargée positivement.
Lorsque le polluant organique est adsorbé, les ions bromures de CTAB s’échangent avec
les molécules anioniques du polluant et par conséquence l’espacement basal augmente pour
atteindre 35 A° pour le colorant 4GL et 40A° pour le polluant 2,4DCP (Zhu et Ma, 2008).
Nous proposons par la suite un schéma donnant la fixation des polluants organiques et CTAB
sur l’argile pour la méthode one step et two steps (figure II.2.20)
En conclusion de cette partie on peut dire que l’adsorption simultanée du polluant et du
CTAB conduit à la formation de l’argile organophile avec une forte adsorption du polluant.
Le mécanisme d’adsorption des deux polluants par la méthode one step et two steps est tout à
fait différent. Ceci est probablement dû à la disposition et l’orientation des chaines de
tensioactifs entre les feuillets argileux.
4. Spectroscopie IR
Les spectres IR des matériaux argileux après adsorption des polluants organiques sont
présentés sur les figure II.2.21 et 22.Pour mieux voir les changements dû à l’adsorption des
polluants organiques sur les phases argileuses, les spectres sont séparés en deux parties une
couvrant la gamme 4000-2000 Cm-1 et l’autre entre 2000 et 400 Cm-1.
La bande correspondante à la vibration d’élongation OH du phénol est située à 3607cm-
1(OH libre) ou à 3250 cm-1(liaison H intermoléculaires) dans le spectre infrarouge de 2,4DCP.
Dans cette région, l’eau interfoliaire de l’argile absorbe les radiations IR, il ne sera pas
possible de détecter la bande OH de DCP dans l’argile organophile (Ovadyahu et al.,1996).
De même la liaison C-Cl présente deux pics sur le spectre IR de 2,4DCP vers 1101 et 1091
Cm-1. Malheureusement, il est impossible de détecter ces bandes après adsorption de 2,4DCP
car elles sont masquées par les vibrations Si-O de l’argile (Robinson, 1995). Après adsorption
du polluant 2,4DCP, les bandes à 3625, 3230 et 3415 cm-1 existent encore, mais leurs
intensités varient en fonction de l'existence de CTAB et de 2,4DCP comparés à la
montmorillonite pure. La bande à 3575 cm-1 augmente en intensité, mais l'intensité de celle à
3230 cm-1 diminue. Cela peut être dû à une interaction entre le CTAB et le OH de la
structure interne de la montmorillonite. L’intensité de la bande à 2930 Cm-1 augmente de
façon importante quand le CTAB est présent en solution. Ceci est probablement du à une
141

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
augmentation de la quantité de 2,4DCP adsorbée (méthode one step) par rapport à l’argile
organophile.
La bande à 1480cm-1 est observée après adsorption du polluant. Cette bande est la bande
infrarouge caractéristique du phénol (Daimay et al.,1991). Elle est attribuée à la vibration de
flexion de la liaison C-H. Les vibrations d’élongations des liaisons Si-O et Si-O-Si sont
localisées à 885 cm-1et 795 cm-1 respectivement. La présence d’une bande à 920 cm-1 est due
à une vibration de déformation OH liée à Al3 + (Tabak et al.,2007). Cette même bande se
déplace vers les grandes nombre d’onde ceci est causé par l'existence d’une quantité
importante de molécules organiques. Il est important de noter que l'intensité de la bande à
920cm-1 augmente quand la montmorillonite réagi avec le 2,4DCP adsorbé.
La structure non connue du colorant 4GL empêche l’interprétation du spectre IR du
matériau argileux après adsorption de ce colorant. Mais néanmoins les remarques concernant
l’adsorption du 2,4 DCP et la modification de la structure de l’argile organophile restent
toujours valables.
142
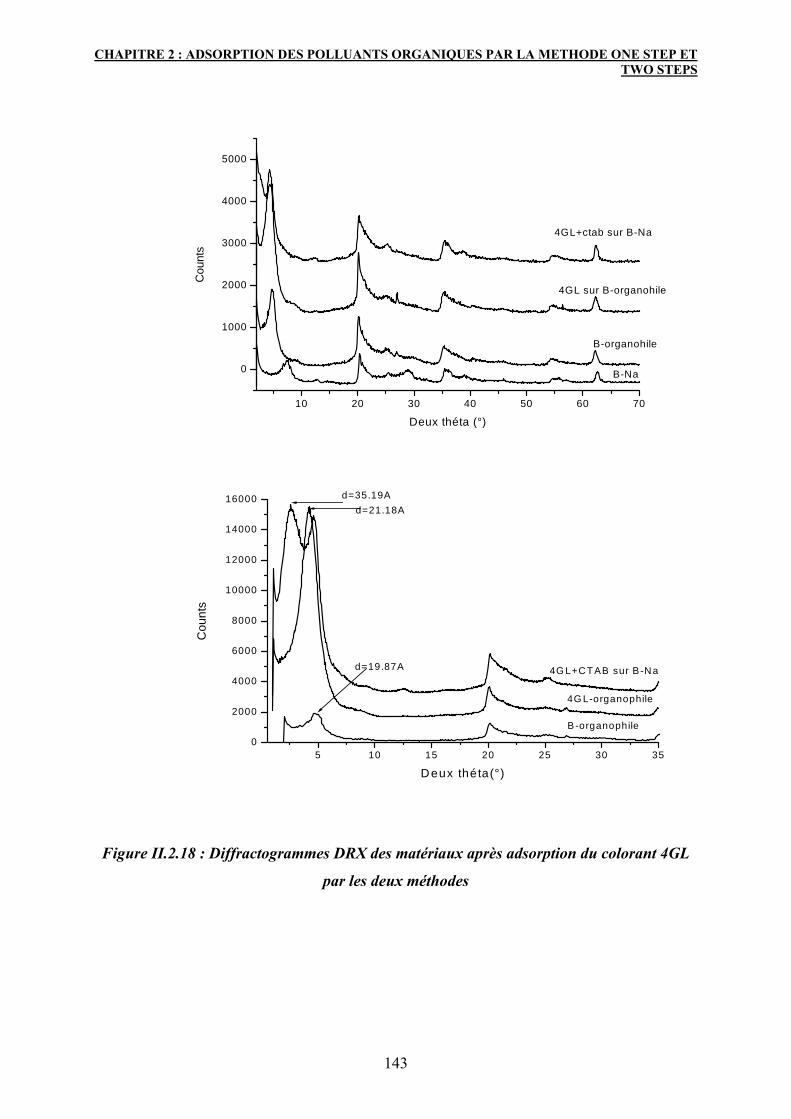
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
10 20 30 40 50 60 70
0
1000
2000
3000
4000
5000
4GL+ctab sur B-Na
4GL sur B-organohile
B-organohile
B-Na
Cou
nts
Deux théta (°)
5 10 15 20 25 30 350
2000
4000
6000
8000
10000
12000
14000
16000 d=35.19Ad=21.18A
d=19.87A 4G L+CTAB sur B -Na
4G L-organophile
B-organophile
Cou
nts
Deux théta(°)
Figure II.2.18 : Diffractogrammes DRX des matériaux après adsorption du colorant 4GL
par les deux méthodes
143
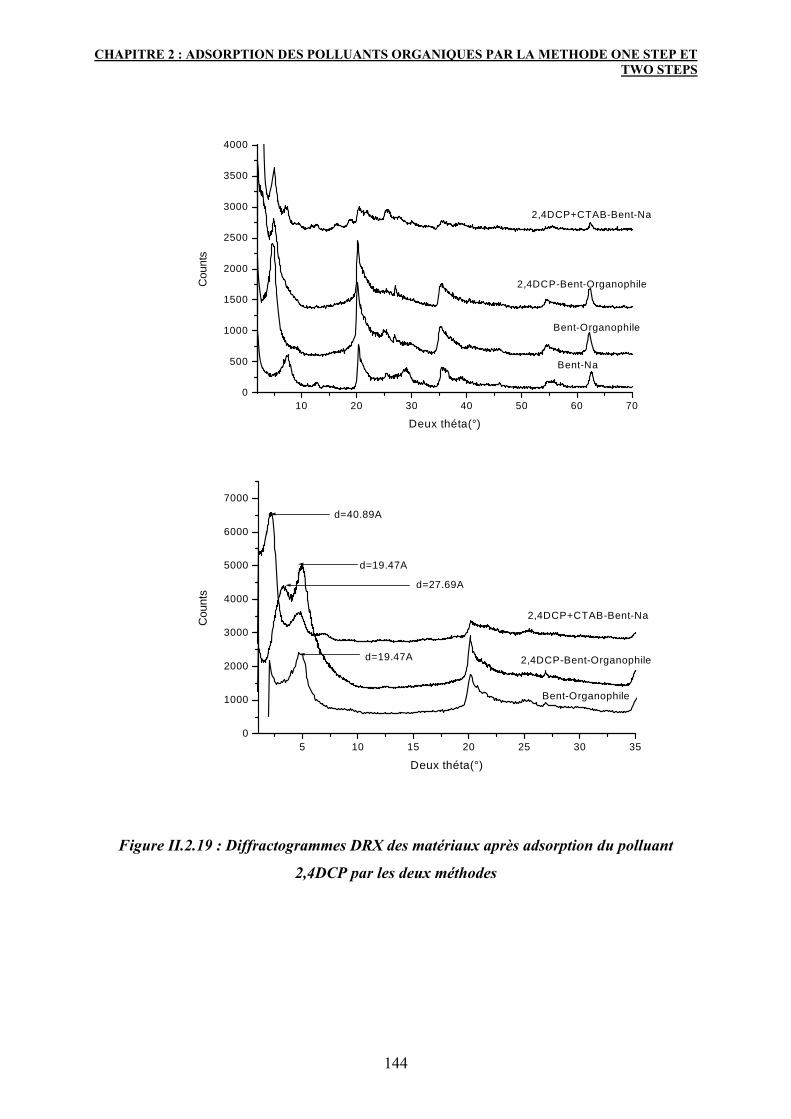
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
10 20 30 40 50 60 700
500
1000
1500
2000
2500
3000
3500
4000
2,4DCP+CTAB-Bent-Na
2,4DCP-Bent-Organophile
Bent-Organophile
Bent-Na
Cou
nts
Deux théta(°)
5 10 15 20 25 30 350
1000
2000
3000
4000
5000
6000
7000d=40.89A
d=27.69A
d=19.47A
d=19.47A
2,4DCP+CTAB-Bent-Na
2,4DCP-Bent-Organophile
Bent-Organophile
Cou
nts
Deux théta(°)
Figure II.2.19 : Diffractogrammes DRX des matériaux après adsorption du polluant
2,4DCP par les deux méthodes
144

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
19.47A°19.47A°
A A21 A°
A AA A21 A°
(1) (2)
A
A
A
A
A
A35-40 A°
A
A
A
A
A
A
A
A
A
A
A
A35-40 A°
(3) (4)
A A= 2,4DCP ou 4GLA A= 2,4DCP ou 4GL
Figure II.2.20 : Représentation schématique de (de la gauche vers la droite) : 1)
argile organophile, 2) Adsorption de polluant organique sur argile organophile, 3)
adsorption de polluant organique et de CTAB sur argile sodée, 4) molécule de CTAB.
145
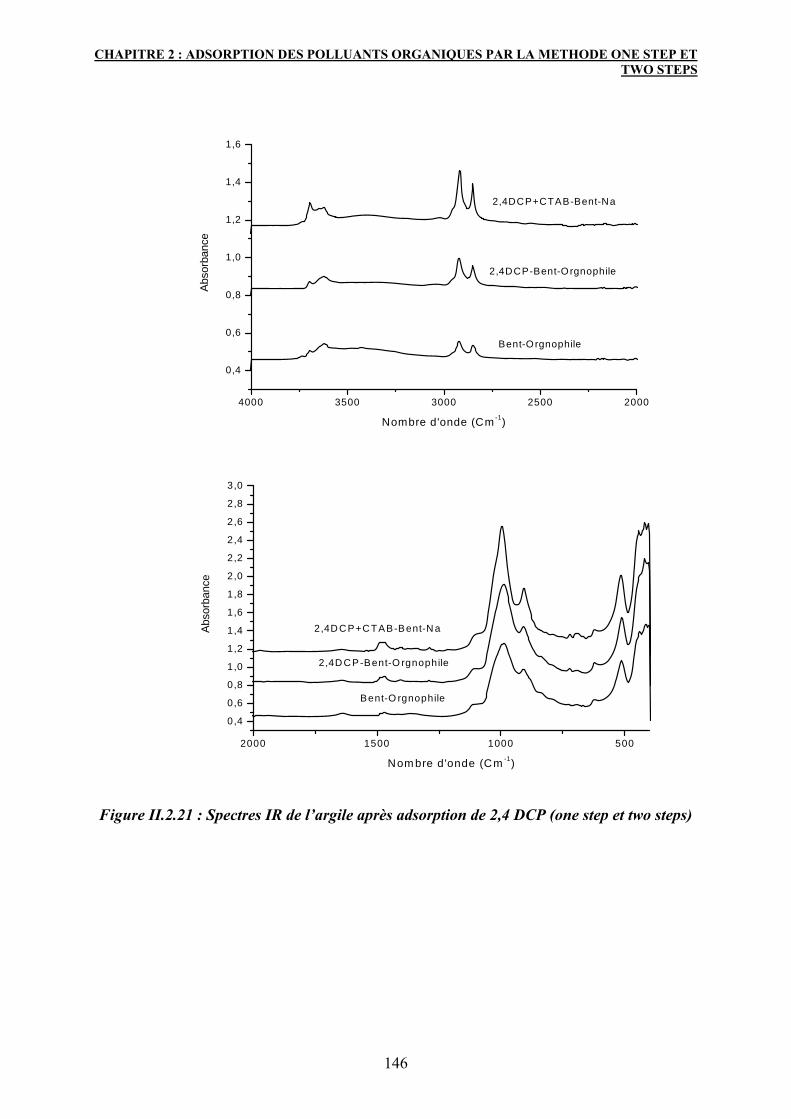
CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
4000 3500 3000 2500 2000
0,4
0,6
0,8
1,0
1,2
1,4
1,6
2,4DCP+CTAB-Bent-Na
2,4DCP-Bent-Orgnophile
Bent-Orgnophile
Abso
rban
ce
Nombre d'onde (Cm -1)
2000 1500 1000 500
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
2,4D CP+CTAB-Bent-N a
2,4D CP-Bent-O rgnophile
Bent-O rgnophile
Abso
rban
ce
Nom bre d'onde (Cm -1)
Figure II.2.21 : Spectres IR de l’argile après adsorption de 2,4 DCP (one step et two steps)
146

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
5. Conclusion Dans ce travail nous nous sommes intéressés à l’étude de l’adsorption de deux polluants
organiques: le 2,4DCP et un colorant industriel textile le 4GL par une bentonite Algérienne
purifiée au sein du laboratoire, en utilisant deux méthodes : la première méthode sur l’argile
organophile (bentonite échangée à 100% CEC par le CTAB), et la seconde méthode par la
bentonite sodée quand le CTAB se trouve en solution.
Les résultats de cette étude ont montré que la méthode one step (CTAB en solution)
améliore la fixation des deux polluants sur l’argile. En effet, le polluant organique et le CTAB
en solution sont éliminés rapidement par l’argile en solution. Les études cinétiques ont
montré que l’adsorption de 2,4DCP et de 4GL par les deux méthodes suit une cinétique du
pseudo second ordre. La diffusion intra particulaire n’est pas la seule étape limitant
l’adsorption, Les isothermes d’adsorption des deux polluants par la méthode one step et two
steps sont tout à fait différentes. Des interactions de type adsorbat- adsorbant et de type
adsorbat- adsorbat sont responsables de ce comportement.
La caractérisation des matériaux après adsorption des deux polluants, confirment le fait
que l’adsorption de 2,4 DCP et du 4GL par la méthode one step et two steps suit des
mécanismes tout à fait différents. Les résultats de cette étude sont très utiles pour déterminer
les conditions optimales d’élimination des polluants organiques par l’argile organophile ou
par l’argile sodée quand le CTAB est en solution.
En conclusion, on peut dire que la méthode one step est une méthode efficace (cinétique
très rapide, capacité d’adsorption élevée, facilité de réalisation) pour l’élimination des
polluants organiques des eaux usées, ce qui ouvre la perspective de son utilisation dans le
traitement des rejets industriels.
147

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
Références bibliographiques
• Adak A. P. and Bandyopadhyay M. (2006), J. Colloid Surface A 277, 63–68.
• Allen S.J. and Brown P.A. (1995), J. Chem. Technol. Biotechnol., 62, 17–24.
• Al-Asheh S. and Duvniajak Z. (1997)., J. Hazard. Mater., 56, 35–51.
• Aloulou F., Boufi S. and Chakchouk M. (2004), Colloid Polym. Sci., 282, 699–707.
• Annadurai G., Juang R.S. and Lee D.J. (2002), J. Hazard. Mater. 92, 263–274.
• Antizar-Ladislao B. and Galil N.I. (2004), Water Res. 38, 267–276.
• Arami M., Yousefi Limaee N., Mohammad Mahmoodi N. and Salman Tabrizi N.
(2005): J. Colloid Interface Sci., 288, 371–376.
• Barrer R.M. and Macleod D.M (1955), Trans. Faraday Soc. 51, 1290–1300.
• Bhattacharyya K.G. and Sharma A. (2004), J. Hazard. Mater. B 113, 97–109.
• Bickmore B.R., Rosso K.M., Nagy K.L., Cygan R.T. and Tadanier C.J. (2003),
Clays Clay Miner. 51,359–371.
• Bijsterbosch B. H (1974), J. Colloid Interface Sci. 47, 186-198.
• Bleam W.F. (1990), Clays Clay Miner., 38, 527–536.
• Bonczek J.L., Harris W.G. and Nkedi-Kizza P. (2002), Clay Clay Miner., 50, 11–
17.
• Bouberka Z., Kacha S., Kameche M., Elmaleh S. and Derriche Z. (2005)., Hazard.
Mater. J. B 119, 117–124.
• Bouberka Z., Khenifi A., Sekrane F., Bettahar N. and Derriche Z. (2008), J.
Chem. Eng., 136, 295–305.
• Boufatit M., Ait-Amar H. and McWhinnie W.R. (2007), Desalination, 206, 394–
406.
• Boyd S.A., Mortland M.M. and Chiou C.T. (1988), J. Soil Sci. Soc. Am. 52, 652–
657.
• Boyd S. (1992), Method of Removing Hydrocarbon Contaminants from Air and Water
with Organophilic, Quaternary Ammonium Ion-Exchanged Smectite Clay, EPO 474386B1.
• Brahim B., Labbe P. and Reverdy G. (1992)., Langmuir 8, 1908-1918.
• Brower G.R. and Reed G.D. (1987), Economical pre-treatment for colour removal
from textile dye wastes. In: Proceedings of 41st Purdue University Industrial Waste
Conference, vol. 41, pp. 612–616.
148

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• Cases J. M., Cunin P., Grillet Y., Poinsignon C. and Yvon J. (1986), Clay Miner.
21, 55-68.
• Cases J.M. and Villieras F. (1992), Langmuir 8, 1251–1264.
• Chen Y. L., Chen S., Frank C. and Israelachvili J. (1992), J. Colloid Interface Sci.
153, 244-265.
• Chen B., Zhu L., Zhu J. and Xing B. (2005), Environ. Sci. Technol. 39 (16) 6093–
6100.
• Chien S.H. and Clayton W.R. (1980), J. Am. Soil Sci. Soc. 44, 265–268.
• Chiu Y.C., Huang L.N., Uang C.M. and Huang J.F. (1990), J. Colloid Surf. 46,
327-337.
• Chung Y.C., Li Y.H. and Chen C.C. (2005), J. Environ. Sci. Health 40, 1775–1790.
• Crini G. (2006), Bioresour. Technol. 97 (9), 1061–1085.
• Daimay L.V., Norman B.C., William G.F. and Jeanette G.G. (1991), Academic
Press, California, pp. 45–58.
• Degs Y.A., Khraisheh M.A.M., Allen S.J. and Ahmad M.N. (2000), Water Res. 34,
927–931.
• Divincenzo J. and Sparks D.L. (2001), Arch. Environ. Contam. Toxicol. 40, 445–
450.
• El Guendi M.S (1995), Adsorpt. Sci. Technol. 13, 295–303.
• Esumi K., Goino M. and Koide Y. (1996), J. Colloid Interface Sci. 183, 539–545.
• Farmer V. C. (1974), The Infrared Spectra of Minerals, Mineralogical Society,
Monograph 4, London, 539 p.
• Freundlich H.M.F. (1906), Z. Phys. Chem. 57, 385–470.
• Gardiner D.K. and Borne B.J. (1978), Soc. Dyes Colourists J. 94, 339–348.
• Giles C.H., MacEwan T.H., Nakhwa S.N. and Smith D. (1960), J. Chem. Soc. 3,
3973–3993.
• Gregg S.J. and Sing K.S.W. (1982), Adsorption, Surface Area and Porosity,
Academic Press, New York.
• Gupta G.S. and Shukla S.P. (1996), Adsorpt. Sci. Technol. 13, 15–26.
• Hamdaoui O. and Naffrechoux E. (2007), J. Hazard. Mater. 147, 401–411.
• Harper M. and Purnell C. (1990), Environ. Sci. Technol. 24, 55– 62.
149

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• Harris D. J., Bonagamba T. .J. and Schmidt-Rohr K, (1999), Macromolecules. 32,
6718-6724.
• He Y., Xu J., Wang H., Ma Zh. andChen J. (2006), Environ. Res. 101, 362–372.
• Hermosin M.C. and Cornejo J. (1992), Chemosphere 24, 1493–1503.
• Ho Y.S. and McKay G. (1999), Process. Biochem. 34, 451–465.
• Ho Y.S. and Mc Kay G. (1998), J. Chem. Eng. 70, 115–124.
• Horning R.H. (1977), Tex. Chem. Colourists 9, 24–27.
• Hsu Y.H., Wang M.K., Pai C.W. and Wang Y.S. (2000), Appl. Clay Sci. 16, 147–
159.
• Hu Q.H., Qiao S.Z., Haghseresht F., Wilson M.A. and Lu G.Q. (2006), Ind. Eng.
Chem. Res. 45 (2), 733–738.
• Jaynes W.F. and Boyd S.A. (1991), J. Soil. Sci. Soc. Am. 55, 43–48.
• Jianfeng Ma. And Lizhong Z. (2007), Chemosphere 68, 1883–1888.
• Jordan J.W. (1949)., Phys. Chem. J. 53, 294–306.
• Juan W. and Han-Qing Y. (2006), J. Hazard. Mater. 137, 498–508.
• Juang R.S., Wu F.C. and Tseng R.L. (2000), J. Colloid Interface Sci. 227, 437–444.
• Kahr G. and Madsen F.T. (1995), Appl. Clay Sci. 9, 327-336.
• Kaviratna P.D., Pinnavaia J.T. and Schroeder P.A. (1996), J. Phys. Chem. Solids
57, 1897–1906.
• Khattri S.D. and Singh M.K. (1999), Adsorpt. Sci. Technol. 17, 269–278.
• Khenifi A., Bouberka Z., Sekrane F., Kameche M. and Derriche Z. (2007),
Adsorption 13, 149–158.
• Khenifi A., Bouberka Z., Bentaleb K., Hamani H. and Derriche Z. (2009), Chem
Eng J, 146, 345–354.
• Kinniburgh D.G. (1986), Environ. Sci. Technol. 20, 895–904.
• Klumpp E., Heitmann H. and Schwuger M.J. (1993), Colloids Surf. A
Physicochem. Eng. Asp. 78, 93–98.
• Konduru R., Ramakrishna K.R. and Viraraghavan T. (1997), Water Sci. Technol.
36, 189–196.
• Lagaly G. (1984), Philos. Trans. R. Soc. London A311, 315–332.
• Lagergren S. (1898), Zur theorie der sogenannten adsorption geloster Stoffe,
Kungliga svenska vetenskapsakademiens, Handlingar band. 24, 1–39.
150

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• Langmuir S. (1916), J. Am. Chem. Soc. 38, 2221–2295.
• Lee J.F., Mortland M.M., Boyd S.A. and Chiou C.T. (1989), J. Chem. Soc. Faraday
Trans. 85, 2953–2962.
• Lee S.Y. and Kim S.J. (2002), Clay Clay Miner. 50, 435–445.
• Liu R., Frost R. L. and Martens W. N. (2009), Mater Chem Phys, 113, 707–713.
• Lizhong Z., Xiaogang R. and Shaobin Y. (1998)., Environ. Sci. Technol. 32, 3374–
3378.
• Liversidge M., Lloyd G.J., Wase D.A.J. and Forster C.F.(1997), Process. Biochem.
32, 473–477.
• Mahlok J.L., Shindala A.M., Griff E.C. and Barnett W.A. (1975), Am. Dye Stuff
Report 64, 24–46.
• Cea M., Seaman J.C., Jara A. A., Fuentes B., Mora M.L. and Diez M.C. (2007),
Chemosphere, 67 1354–1360.
• Matthes W. and Kahr G. (2000), Clays Clay Miner. 48, 593–602.
• Mc Bride M.B. (1987), Soil Sci. Soc. Am. 51, 1466–1472.
• Mc Kay G. and Allen S.J. (1980), Can. Chem. Eng. J. 58, 521–525.
• Mizutani T., Takano T. and Ogoshi H. (1995), Langmuir 11, 880-884.
• Morais L.C., Freitas O.M., Goncalves E.P., Vasconcelos L.T. and Gonzalez Beca
C.G. (1999), Water Res. 33, 979–988.
• Namasivayam C., Prabha D., Kumutha M. (1998), Biores. Technol. 64, 77–79.
• Okamoto N., Yoshimura T. and Esumi K. (2004), J. Colloid Interface Sci. 275,
612–617.
• Ovadyahu D., Shoval S., Lapides I. and Yariv S. (1996), Thermochim. Acta,
282/283, 369-383.
• Özacar M., ¸ Engil I. A. S andTürkmenler H. (2008), Chem. Eng. J. 143, 32–42.
• Pashley R. M. and Israelachvili J. N (1981), Colloids Surf. 2, 169-187.
• Patzko A. and Dekany I (1993)., Colloids Surf. 71, 299-307.
• Poots V.J.P., Mc Kay G. and Healy J.J. (1976), Water Res. 10, 1061–1066.
• Qingming W. and Teruyuki N. (2006), Micropor. Mesopor. Mater. 96, 84–92.
• Raghavacharya C. (1997)., Chem. Eng. World 32, 53–58.
• Redlich O. and Peterson D.L. (1959), J. Phys. Chem. 63, 1024–1029.
151

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• Robinson J.W. (1995), Undergranduate Instrumental Analysis, 5th ed., Marcel
Dekker, New York, p. 242.
• Rodriguez I., Llompart M.P. and Cela R. (2000), J. Chromatogr., 885, 291–304.
• Ruiying G. and Jianlong W. (2007), J. Hazard. Mater. 145, 398–403.
• Sabbah I. and Rebhum M. (1997),Water. Environ. Res. 69, 1032–1038.
• Saleh M.M. (2006), Water Res. 40, 1052–1060.
• Salerno P., Asenjo M. B. & Mendioroz S. (2001), Thermochim. Acta, 379, 101-109.
• Sankar M., Sekaran G., Sadulla S. and Ramasami T. (1999), J. Chem. Technol.
Biotechnol. 74, 334–337.
• Sathishkumar M., Binupriya A.R., Kavitha D. and Yun S.E. (2007), Bioresour.
Technol. 98, 866–873.
• Sinkkonen S. and Paasivirta J. (2000), Chemosphere: (Oxf.) 40, 619–626.
• Sivaraj R., Namasivayam C. and Kadirvelu K. (2001), Waste Manage. 21, 105–
110.
• Smith J.A. and Galan A. (1995), Environ. Sci. Technol. 29, 685–692.
• Smith J., Jaffe P. and Chiou C. (1990), Environ. Sci. Technol. 24, 1167–1172.
• Smith J.A. and Jaffe P.R. (1991), Environ. Sci. Technol. 25, 2054–2058.
• Smith J.A., Tuck D.M., Jaffe P.R. and Mueller R.T. (1991): Effect of surfactants
on the mobility of nonpolar organic contaminants in porous media in organic substances and
sediments in water. In: Baker, R.A. (ed.) pp. 201–230. Lewis
• Sposito G. (1984) (Ed.), The Surface Chemistry of Soils, 234, Oxford Univ. Press,
Oxford,
• Sparks D.L. (1986), Kinetics of Reaction in Pure and Mixed Systems, Soil Physical
Chemistry, CRC Press, Boca Raton, pp. 12–18.
• Sullivan E. J., Carey J.W. and Bownam R.S (1998), J. Colloid Interface Sci. 206,
369-380;
• Sun G. and Xu X. (1997), Ind. Eng. Chem. Res. 36, 808–812.
• Tabak B. A., Aygun S.F. and Koksal E. (2007), Journal of Thermal Analysis and
Calorimetry 87 (2), 375–381.
• Trompette J. L., Zajac J., Keh E., and Partyka S. (1994), Langmuir. 10, 812-818.
• Van der Marel H.W., Beutelspacher H., Altas of Infrared Spectroscopy of Clay
Minerals and Their Admixtures, 1976, pp. 113–115.
152

CHAPITRE 2 : ADSORPTION DES POLLUANTS ORGANIQUES PAR LA METHODE ONE STEP ET TWO STEPS
• Volzone C., Rinaldi J.O., Ortiga J. (2006), J. Environ. Manag., 79, 247–252.
• Wang C.C., Juang L.C., Lee C.K., Hsu T.C., Lee J.F. and Chao H.P. (2004), J.
Colloid Interface Sci., 280, 27–35.
• Wang J.P., Feng H.M. and Yu H.Q. (2007a), J. Hazard. Mater. 144, 200–207.
• Wang J.P., Chen Y.Z., Feng H.M., Zhang S.J. and H.Q. Yu (2007b), Colloid
Interface Sci. 313, 80–85.
• Weber W.J., Morris J.C. and J. Sanit (1963), Eng. Div. Am. Soc. Civ. Eng. 89, 31–
60.
• Williams H. D. & Fleming I. (1966), Spectroscopic Methods In Organic Chemistry,
Mcgraw-Hill, 222 P.
• Wu J. and Han-Qing Y. (2007), Bioresour. Technol. 98, 253–259.
• Xiaoyan W. , Hongping H., Jianxi Z., Yang J., Chaohui Y. and Feng D (2006), J.
Colloid Interface Sci. 299 , 754–760.
• Xu S. and Boyd S. A. (1995a), Environ. Sci. Technol. 29, 312-320.
• Xu, S. and Boyd S. A. (1995 b), Environ. Sci. Technol. 29, 3022-3028;
• Xu S., and Boyd S. A. (1995)., Langmuir 11, 2508-2514.
• Zhang Z.Z., Sparks D.L. and Scrivner N.C (1993), Environ. Sci. Technol. 27, 1625-
1631.
• Zhou G. D. (1984), Inorganic Structural Chemistry, Science Press, Beijing, p. 118 (in
Chinese)
• Zhou Q., Frost R.L, He H., Xi Y. and Liu H. (2007), J. Colloid Interface Sci, 307
(2) 357–363.
• Zhu L., Chen B., Tao S., Chiou C. and Chen B. (2000), Environ. Sci. Technol. 34
2997–3002.
• Zhu L.Z., Tian S.L. and Shi Y. (2005), Clay Clay Miner. 53, 123–136.
• Zhu L. and Ma J. (2008), Chem Eng J, 139, 503–509.
153

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET
DE GLUFOSINATE PAR LA MATRICE NiAlNO3
154

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE
GLUFOSINATE PAR LA MATRICE NiAlNO3
1. Introduction L’interaction entre l’eau et les particules des sols joue un rôle crucial pour la définition
des propriétés physiques et chimiques des sols. Pratiquement toutes les réactions chimiques
et biologiques qui se produisent dans les sols se produisent à l’interface solide-eau. Comme
exemples de substances couramment trouvées dans les sols, on peut citer : les produits de
dissolution des sols minéraux, les ions métalliques, les polluants organiques, les nutriments
pour les végétaux et les produits agricoles chimiques comme les pesticides. Le Glyphosate
(N-phosphonométhylglycine, Gly) et le Glufosinate (DL-homoalanin-4-(méthyl)
phosphonique acide, Glu) (Figure II.3.1) sont utilisés dans le monde entier comme herbicides
non sélectifs pour lutter contre les mauvaises herbes à feuilles larges (Zimdahl, 1993). Ils
interférent avec la capacité des plantes à former des acides aminés, en plus ils réagissent
négativement sur la photosynthèse et la respiration. Plus précisément, Le Glyphosate inhibe
le synthase 5-énolpyruvylshikimate-3-phosphate, une enzyme impliquée dans la biosynthèse
des acides aminés aromatiques, ce qui fait obstacle à la production des protéines en raison
d'un manque de tyrosine, de phénylalanine et de tryptophane (Rubin et al., 1982). Le
Glufosinate inhibe la glutamine synthétase, une enzyme responsable de catalyser la réaction
entre l'ammoniac et le glutamate pour former la glutamine (Bayer et al., 1972).
Figure II.3.1 : Structures chimiques du Glyphosate et de Glufosinate.
155

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Ces composés possèdent non seulement une capacité très élevée pour former des
complexes solides avec les métaux de transition en solution aqueuse, mais aussi montrent une
grande affinité pour la surface d'aluminium et de l’oxyde de fer. Tous ces propriétés jouent un
rôle très important dans le devenir et le transport de ces composés dans l'environnement
(Barja et al., 2001 ; Franz et al., 1997). Après la mort des plantes, les herbicides peuvent être
absorbée par le sol et peut pénétrer dans l'environnement aquatique en raison de leur solubilité
dans l'eau. En effet, les données bibliographiques suggèrent que les produits chimiques ayants
une grande affinité pour la matrice du sol sont sensibles au lessivage (Flury, 1996;
Vereecken, 2005). Par conséquent, de faibles concentrations de Glyphosate et de Glufosinate
ont été trouvés dans les aquifères peu profonds, ce qui remet en question l'idée commune que
ces produits ont une mobilité réduite dans les sols. En plus, le phosphate, introduit par les
engrais, est appliqué en continu sur le sol parce que le phosphore est l'un des éléments
essentiels pour la croissance des plantes. En raison de la diminution de la production agricole
résultant de l’augmentation de la population, le phosphate et d’autres engrais sont appliqués
de façon abusive en agriculture. De cette façon, le phosphate peut s'accumuler et risque
d’occuper les sites d'adsorption des sols (Gimsing et Borggaard, 2002; Gimsing et al.,
2004b). Ceci peut conduire à une réduction des capacités des sols à retenir le Glyphosate et le
Glufosinate, puisque le phosphate et ces herbicides semblent entrer en compétition envers les
mêmes cites d’adsorption (De Jonge et al., 2001; Dion et al., 2001; Gimsing et al., 2004a;
Gimsing et al., 2007). Même si ces herbicides sont de toxicité relativement faible pour les
hommes et les animaux, leur devenir dans l'environnement n’a pas été évalué avec précision
malgré leur grande utilisation (Hudson, 2000).
Bien que les stocks naturels d’hydroxydes doubles lamellaires (LDH), également
connu sous le nom d’hydrotalcite ou encore argiles anioniques, sont rares dans la nature, la
formation dans les sols des phases hydroxydes mixtes a été démontrée par XAFS sur la
surface des phyllosilicates et de gibbsite (Scheidegger et al., 1997). En effet, ils peuvent être
facilement formés dans une large gamme de conditions de pH et facilement synthétisés à
l'échelle du laboratoire (Miyata, 1975). Leur structure se compose d’un empilement de
couches brucite, où une fraction des cations divalents est remplacée par des cations trivalents.
Les couches chargées positivement sont séparés par des anions de compensation de
charge et de molécules d'eau (De Roy et al., 1992; Cavani et al., 1991; Atwood et al., 1997).
156

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Comme nous l’avons déjà expliquer dans la partie bibliographique, les LDH ont la formule
générale suivante : M(II)1-xM(III)
x(OH)2]x+(A-nx/n).mH2O, où MII :est le métal divalent, MIII :le
métal trivalent, x : le rapport molaire MII/MII+MIII variant entre 0.2 et 0.33, et A constitue
l’anion interfoliaire. Les LDH ont généralement une surface relativement grande (30-150
m2/g), une densité de charge élevée, qui change à partir de 50 A2 par charge (surface
équivalente) pour un rapport molaire de 4, à 25 A2 par charge pour un rapport de 2.
Ces matériaux possèdent des capacités d'échange anioniques (2-5 mmol/g) plus
importantes que les capacités d’échanges cationiques des minéraux argileux (Miyata, 1975;
Moujahid et al., 2003). A cet effet, ils sont considérés comme des adsorbants potentiels pour
l'élimination des substances toxiques ou des espèces anioniques écologiquement indésirables,
tels que CrO42 -, NO3
-, PO43- ainsi que les phénols, les substances humiques, les agents de
surface anioniques et les pesticides (Rhee et al., 1997; Parker et al., 1995, Forano, 2004).
En vertu d’une substitution anionique topotactique, la molécule organique négative est
introduite dans la matrice inorganique, tout en conservant la structure globale. Bien que ces
matériaux jouent un rôle important dans l'échange anionique et l'adsorption de molécules
organiques, il est possible de les utiliser comme matrices potentielles pour l’étude de la
modélisation de libération contrôlée des pesticides par l'intercalation de molécules de
pesticides entre les feuillets de l’argile.
A cet effet, notre travail s’est concentré sur l'adsorption de deux herbicides: le
Glyphosate et le Glufosinate par un matériau LDH type Ni2AlNO3. L’adsorption de la
molécule de Glyphosate sur le matériau MgAlNO3 a été effectuée par Li et al., 2005. En
comparaison avec le matériau MgAlNO3, la structure de Ni2AlNO3 est très intéressante car
elle combine de bonnes propriétés électro actives basé sur le processus redox Ni2+/ Ni3+ pour
les procédés électrochimiques avec des propriétés structurales et texturales pour l'adsorption
des molécules chargées négativement. A notre connaissance, l'adsorption de ces herbicides
sur le matériau NiAlNO3 n'a pas été effectuée et publiée jusqu'à présent. Les résultats
présentés ici sont utiles pour évaluer l'application des matériaux LDH pour le nettoyage
environnemental et à l'assainissement des sites, des sols et des eaux contaminés.
157

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
2. Caractérisation du matériau Ni2AlNO3 Le matériau Ni2AlNO3 est préparé par la méthode de coprécipitation à pH constant sous
azote, afin de minimiser la contamination par le CO2 atmosphérique. La méthode de synthèse
est détaillée dans la partie matériaux et méthodes. Afin de comprendre le comportement de la
matrice NiAlNO3 vis-à-vis des deux herbicides : Glyphosate et Glufosinate, une série de
caractérisation est effectuée.
2.1 Diffraction des rayons X
Le diffractogramme DRX du matériau Ni2AlNO3 (Figure II.3.2.) montre les
caractéristiques typiques des HDL sans impuretés (Cavani et al., 1991). Le diffractogramme
DRX présente une série de pic (00l) symétriques vers les faibles deux téta. La distance inter
réticulaire de la raie 003, est relié à l’écartement des feuillets (distance inter feuillet), et
permet un calcul approché du paramètre de maille c par la relation suivante : c/3=d003. La raie
(110) située au voisinage de 60° en 2θ, détermine la distance interatomique métal-métal du
feuillet et le paramètre de maille a : a/2=d110 (Vaysse, 2001 ; Solin et al., 1996). Les deux
réflexions à bas 2θ (2θ=10,36 et 20,60 °) sont attribués aux raies basales (003) et (006),
correspondant à un empilement de feuillet type brucite, le reste correspondant à des pics non
basales (01l et 11l )
L'espacement basal (d003) est égale à 0,86 nm, ce qui est en accord avec les valeurs
rapportées par d'autres auteurs (Lv et al., 2009) concernant des HDL nitrates. Un important
élargissement des raies de diffraction RX se produit pour les matériaux solides ayants une
cristallinité très faible, associés à un trouble structurel élevé dû à des effets turbo statiques et
stériques. Les largeurs à mi hauteur sont particulièrement importantes : 3.28 et 4.48° en 2θ
pour les raies 003 et 006 respectivement. A cause de ces données cristallographiques très
pauvres, la détermination des paramètres de maille n’a pas été effectuée et l'indexation est
proposée en supposant un groupe d'espace R-3 m pour une maille hexagonale.
158
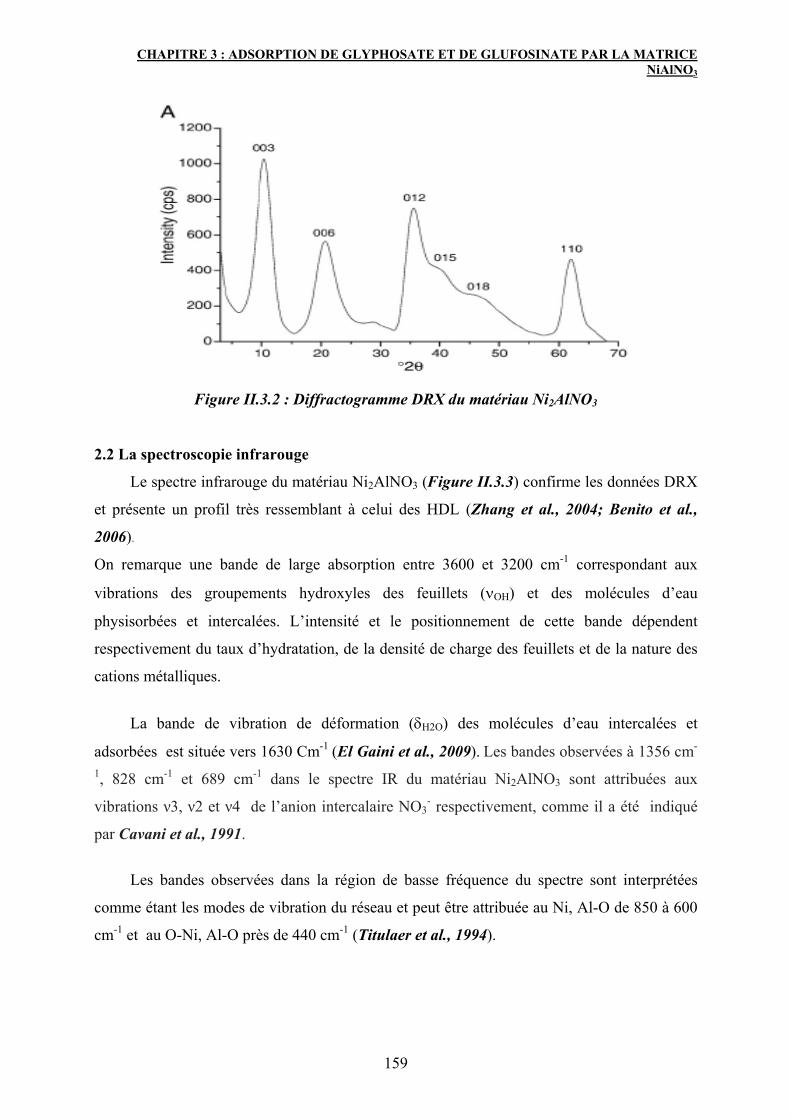
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.2 : Diffractogramme DRX du matériau Ni2AlNO3
2.2 La spectroscopie infrarouge
Le spectre infrarouge du matériau Ni2AlNO3 (Figure II.3.3) confirme les données DRX
et présente un profil très ressemblant à celui des HDL (Zhang et al., 2004; Benito et al.,
2006).
On remarque une bande de large absorption entre 3600 et 3200 cm-1 correspondant aux
vibrations des groupements hydroxyles des feuillets (νOH) et des molécules d’eau
physisorbées et intercalées. L’intensité et le positionnement de cette bande dépendent
respectivement du taux d’hydratation, de la densité de charge des feuillets et de la nature des
cations métalliques.
La bande de vibration de déformation (δH2O) des molécules d’eau intercalées et
adsorbées est située vers 1630 Cm-1 (El Gaini et al., 2009). Les bandes observées à 1356 cm-
1, 828 cm-1 et 689 cm-1 dans le spectre IR du matériau Ni2AlNO3 sont attribuées aux
vibrations ν3, ν2 et ν4 de l’anion intercalaire NO3- respectivement, comme il a été indiqué
par Cavani et al., 1991.
Les bandes observées dans la région de basse fréquence du spectre sont interprétées
comme étant les modes de vibration du réseau et peut être attribuée au Ni, Al-O de 850 à 600
cm-1 et au O-Ni, Al-O près de 440 cm-1 (Titulaer et al., 1994).
159
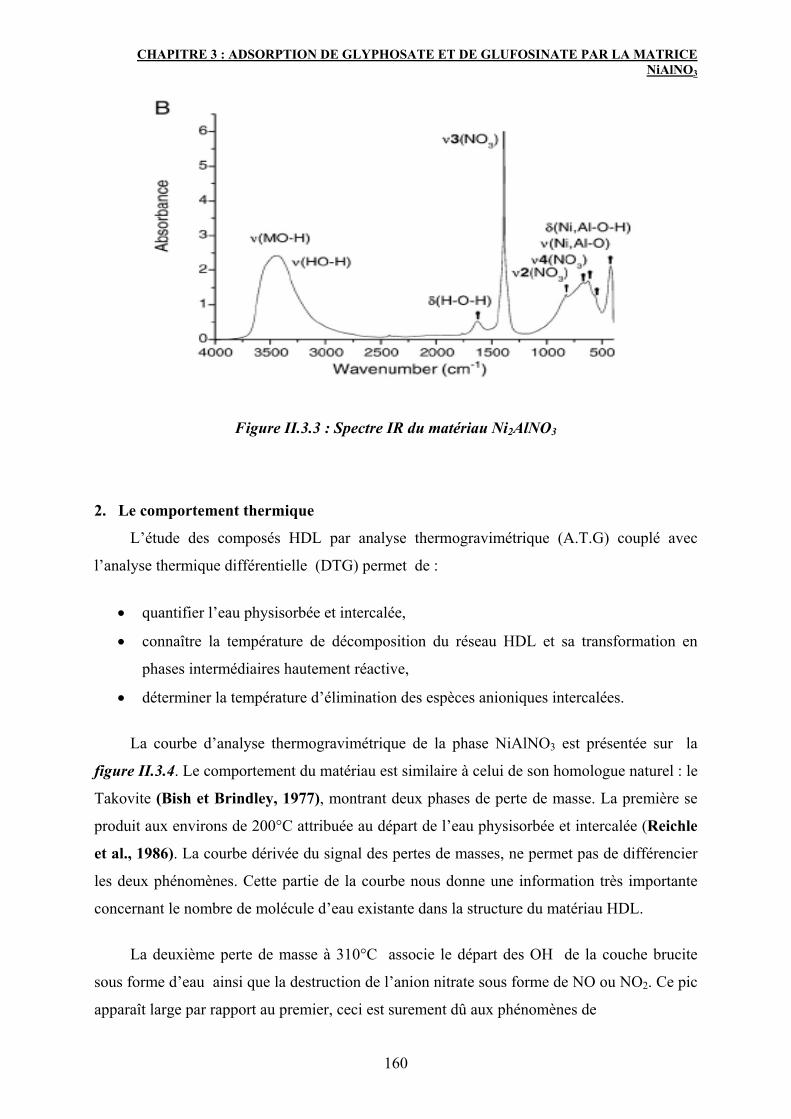
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.3 : Spectre IR du matériau Ni2AlNO3
2. Le comportement thermique
L’étude des composés HDL par analyse thermogravimétrique (A.T.G) couplé avec
l’analyse thermique différentielle (DTG) permet de :
• quantifier l’eau physisorbée et intercalée,
• connaître la température de décomposition du réseau HDL et sa transformation en
phases intermédiaires hautement réactive,
• déterminer la température d’élimination des espèces anioniques intercalées.
La courbe d’analyse thermogravimétrique de la phase NiAlNO3 est présentée sur la
figure II.3.4. Le comportement du matériau est similaire à celui de son homologue naturel : le
Takovite (Bish et Brindley, 1977), montrant deux phases de perte de masse. La première se
produit aux environs de 200°C attribuée au départ de l’eau physisorbée et intercalée (Reichle
et al., 1986). La courbe dérivée du signal des pertes de masses, ne permet pas de différencier
les deux phénomènes. Cette partie de la courbe nous donne une information très importante
concernant le nombre de molécule d’eau existante dans la structure du matériau HDL.
La deuxième perte de masse à 310°C associe le départ des OH de la couche brucite
sous forme d’eau ainsi que la destruction de l’anion nitrate sous forme de NO ou NO2. Ce pic
apparaît large par rapport au premier, ceci est surement dû aux phénomènes de
160

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
déshydroxylation et de dénitration qui ont eu lieu simultanément durant cette étape. Il faut
noter que la stabilité thermique des HDL est liée à celle des feuillets brucitiques. En plus, une
petite perte de masse se produit au environ de 600°C qui est attribuée à l’élimination complète
de l’anion interfoliaire.
L’analyse thermique différentielle ATD confirme les pertes de masse par l’apparition de
bandes endothermiques. Par contre, on n’observe aucun pic exothermique qui indique une
reconstruction d’une nouvelle phase et confirme que le matériau HDL subis simplement une
décomposition thermique (Kannan et al., 1996). A la suite de cette étude, nous proposons un
schéma de décomposition thermique pour le matériau NiAlNO3 (figure II.3.5).
La figure II.3.6 Indique la variation des diffractogrammes DRX du matériau HDL avec
la température de calcination. La calcination est effectuée à chaque température pendant une
heure sous air. Le matériau NiAlNO3 est stable jusqu’à une température de 200°C donnant un
DRX semblable à celui des HDL. Cependant une diminution des intensités des raies ainsi
qu’un décalage de la raie 003 vers des thêtas plus élevées est remarqué. Ceci est probablement
dû au départ de l’eau physisorbée et intercalée d’où la diminution de la distance intercalaire.
En général, deux étapes essentielles peuvent être observées lors de la transformation de
phase LDH sous chauffage: (i) diminution de l’espace intercalaire, conséquence du départ de
l’eau et (ii) la disparition des pics de diffraction LDH et la formation des phases oxydes
(Kovanda et al., 2009). Une calcination au-delà de cette température (> 300°C) conduit à la
formation d’une phase NiO faiblement cristallisée.
Bien que les oxydes métalliques mixtes obtenus dans la gamme de température 400-800
soient mal cristallisés, ils sont caractérisés par des propriétés inhabituelles, telles qu’une
stabilité thermique élevée, une bonne dispersion et une grande surface spécifique. La
cristallinité des oxydes augmente avec la température de calcination (jusqu'à 900°C), ceci se
traduit par l’augmentation des intensités de pics et leurs finesses sur le diffractogramme DRX.
Un nouvel accroissement de la température de calcination (> 1000) conduit à une séparation
entre les phases NiO et NiAI204 (Kannan et al., 1996).
161
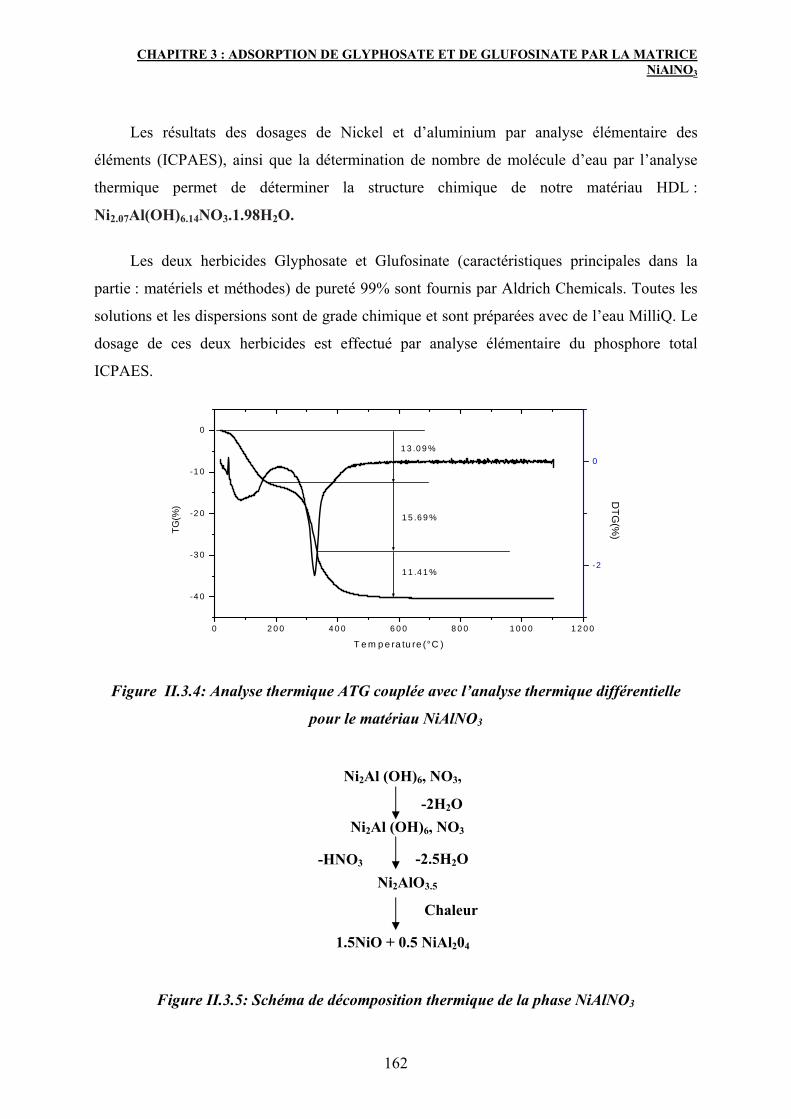
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Les résultats des dosages de Nickel et d’aluminium par analyse élémentaire des
éléments (ICPAES), ainsi que la détermination de nombre de molécule d’eau par l’analyse
thermique permet de déterminer la structure chimique de notre matériau HDL :
Ni2.07Al(OH)6.14NO3.1.98H2O.
Les deux herbicides Glyphosate et Glufosinate (caractéristiques principales dans la
partie : matériels et méthodes) de pureté 99% sont fournis par Aldrich Chemicals. Toutes les
solutions et les dispersions sont de grade chimique et sont préparées avec de l’eau MilliQ. Le
dosage de ces deux herbicides est effectué par analyse élémentaire du phosphore total
ICPAES.
0 2 0 0 4 0 0 6 0 0 8 0 0 1 0 0 0 1 2 0 0
-4 0
-3 0
-2 0
-1 0
0
1 1 .4 1 %
1 5 .6 9 %
1 3 .0 9 %
T e m p e ra tu re (°C )
TG(%
)
-2
0
DTG
(%)
Figure II.3.4: Analyse thermique ATG couplée avec l’analyse thermique différentielle
pour le matériau NiAlNO3
Ni2Al (OH)6, NO3,
Ni2Al (OH)6, NO3
-2H2O
Ni2AlO3.5
-2.5H2O -HNO3
Chaleur
1.5NiO + 0.5 NiAl204
Figure II.3.5: Schéma de décomposition thermique de la phase NiAlNO3
162
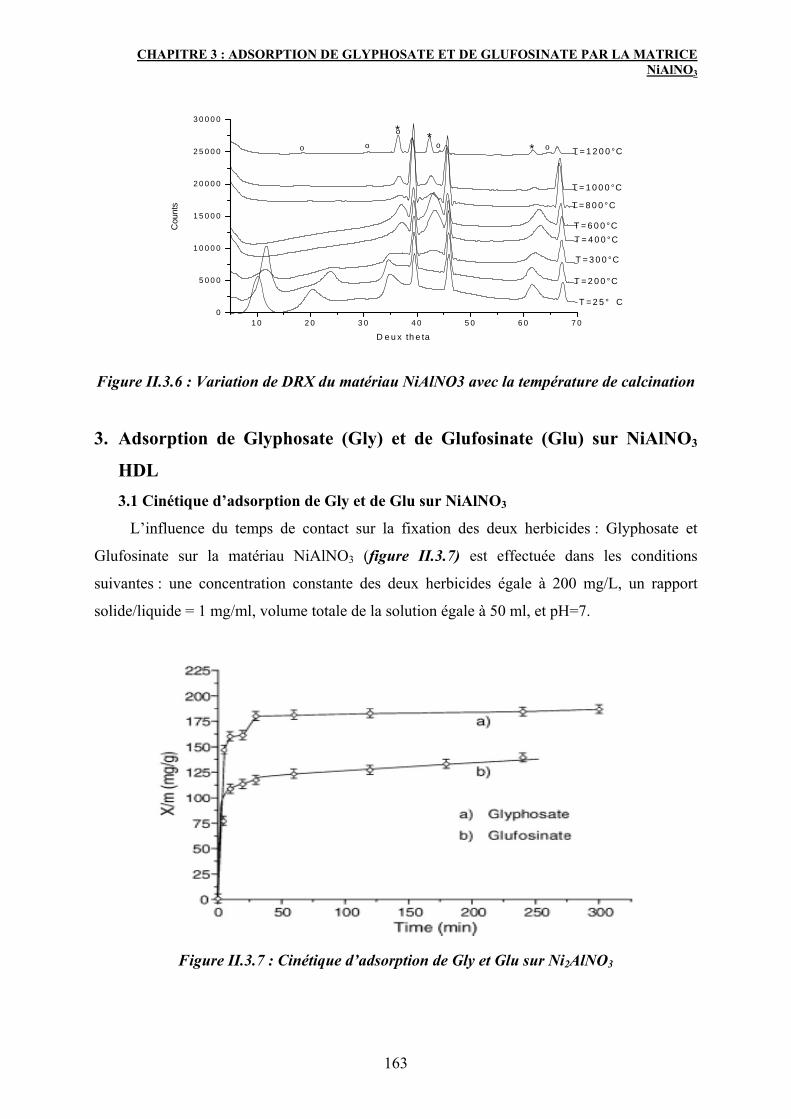
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
1 0 2 0 3 0 4 0 5 0 6 0 7 00
5 0 0 0
1 0 0 0 0
1 5 0 0 0
2 0 0 0 0
2 5 0 0 0
3 0 0 0 0
***
o
o
ooo T = 1 2 0 0 °C
T = 1 0 0 0 °C
T = 8 0 0 °C
T = 6 0 0 °CT = 4 0 0 °C
T = 3 0 0 °C
T = 2 0 0 °C
T = 2 5 ° C
Cou
nts
D e u x th e ta
Figure II.3.6 : Variation de DRX du matériau NiAlNO3 avec la température de calcination
3. Adsorption de Glyphosate (Gly) et de Glufosinate (Glu) sur NiAlNO3
HDL 3.1 Cinétique d’adsorption de Gly et de Glu sur NiAlNO3
L’influence du temps de contact sur la fixation des deux herbicides : Glyphosate et
Glufosinate sur la matériau NiAlNO3 (figure II.3.7) est effectuée dans les conditions
suivantes : une concentration constante des deux herbicides égale à 200 mg/L, un rapport
solide/liquide = 1 mg/ml, volume totale de la solution égale à 50 ml, et pH=7.
Figure II.3.7 : Cinétique d’adsorption de Gly et Glu sur Ni2AlNO3
163

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
La figure II.3.7 montre clairement des taux d’adsorptions tout à fait différents pour les
deux herbicides, une première étape très rapide suivie par une autre moins rapide après 30 mn
de contact. A ce stade là, l’adsorption des deux polluants atteint le maximum : 180 mg/g pour
le Glyphosate et 140 mg/g pour le Glufosinate. Après 30 mn de contact, la quantité maximale
reste constante indiquant un état d’équilibre entre l’adsorbat fixé sur le matériau et celui en
solution. Ainsi, une adsorption initiale très rapide des phénols sur le matériaux HDL a été
rapportée dans la littérature (Hermosin et al., 1993; Hermosin et al., 1996). Li et al., 2005
ont trouvé que la première étape d’adsorption de Glyphosate sur le matériau Mg2AlNO3 est
complète au bout d’un temps relativement long :150 mn (30 mn dans notre cas) pour une
concentration de polluant égale à 25 mg/L et une masse d’adsorbant égale à 100 mg. Ceci est
probablement dû aux propriétés physico-chimiques différentes des deux matériaux HDL qui
influent de façon considérable sur la cinétique d'adsorption. En effet, les deux matériaux
affichent des propriétés de surface différentes en termes de basicité et de morphologie.
Les feuillets du matériau Mg2AlNO3 montrent une basicité plus élevée que celles du
matériau Ni2AlNO3. Le pH dune suspension de 1g/L des deux matériaux est égal à 8.9 pour le
Mg2AlNO3 et uniquement 7.5 pour la matrice Ni2AlNO3. La présence d’une concentration
élevée des ions OH- à l’interface solide liquide du matériau MgAlNO3 empêche la diffusion
des deux herbicides. En plus, les particules du matériau NiAlNO3 montrent une faible
tendance à l’agrégation par rapport au deuxième matériau. La taille moyenne des particules de
Mg2AlNO3 et de Ni2AlNO3 est égale à 250 et 100 nm respectivement.
3.1.1 Modélisation de la cinétique d’adsorption de Gly et Glu sur le matériau NiAlNO3
La modélisation de la cinétique d’adsorption des deux herbicides sur le matériau HDL
est effectuée en utilisant les modèles décrits précédemment (chapitre matériels et méthodes) :
pseudo premier ordre, pseudo second ordre, modèle d’Elovich et le modèle de diffusion intra
particulaire. La figure II.3.8 montre les formes linéaires du modèle pseudo premier ordre et
pseudo second ordre de l’adsorption des deux herbicides sur le matériau NiAlNO3. Les
valeurs des constantes de vitesses k1 et k2 correspondantes à ces deux modèles sont résumées
dans le tableau II.3.1
164
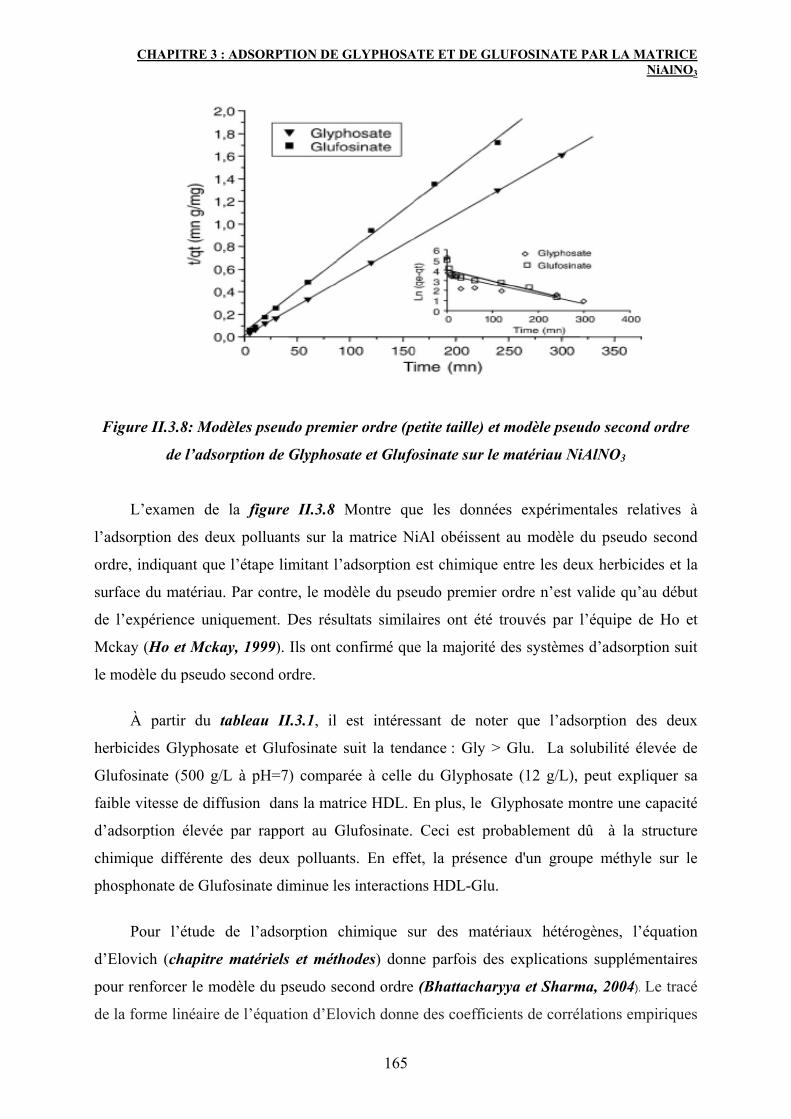
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.8: Modèles pseudo premier ordre (petite taille) et modèle pseudo second ordre
de l’adsorption de Glyphosate et Glufosinate sur le matériau NiAlNO3
L’examen de la figure II.3.8 Montre que les données expérimentales relatives à
l’adsorption des deux polluants sur la matrice NiAl obéissent au modèle du pseudo second
ordre, indiquant que l’étape limitant l’adsorption est chimique entre les deux herbicides et la
surface du matériau. Par contre, le modèle du pseudo premier ordre n’est valide qu’au début
de l’expérience uniquement. Des résultats similaires ont été trouvés par l’équipe de Ho et
Mckay (Ho et Mckay, 1999). Ils ont confirmé que la majorité des systèmes d’adsorption suit
le modèle du pseudo second ordre.
À partir du tableau II.3.1, il est intéressant de noter que l’adsorption des deux
herbicides Glyphosate et Glufosinate suit la tendance : Gly > Glu. La solubilité élevée de
Glufosinate (500 g/L à pH=7) comparée à celle du Glyphosate (12 g/L), peut expliquer sa
faible vitesse de diffusion dans la matrice HDL. En plus, le Glyphosate montre une capacité
d’adsorption élevée par rapport au Glufosinate. Ceci est probablement dû à la structure
chimique différente des deux polluants. En effet, la présence d'un groupe méthyle sur le
phosphonate de Glufosinate diminue les interactions HDL-Glu.
Pour l’étude de l’adsorption chimique sur des matériaux hétérogènes, l’équation
d’Elovich (chapitre matériels et méthodes) donne parfois des explications supplémentaires
pour renforcer le modèle du pseudo second ordre (Bhattacharyya et Sharma, 2004). Le tracé
de la forme linéaire de l’équation d’Elovich donne des coefficients de corrélations empiriques
165

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
qui varient entre 0.94 et 0.97. Ces valeurs sont plus faibles que ceux trouvés pour le modèle
du pseudo second ordre. Des résultats similaires ont été rapportés par Ozacar et al., 2008
concernant l’adsorption du plomb sur une résine de Valonia.
Le modèle de diffusion intra particulaire a également été testé. Comme on l’a déjà vu
dans le chapitre précédent, la diffusion intra particulaire est originaire de la deuxième loi de
Fick. Weber et Morris affirment que si la diffusion intra particulaire est l’étape limitant
l’adsorption, alors la quantité fixée varie avec la racine carré du temps (Weber et al., 1963 ;
Juang et al.,2000). Ho et Mc Kay (Ho et Mc Kay, 1998) démontrent que si le tracé de la
quantité fixée en fonction de la racine carré du temps passe par l’origine alors on peut dire que
la diffusion intra particulaire est la seule étape limitant l’adsorption. Dans le cas ou la droite
ne passe pas par l’origine, cela veut dire que la diffusion intra particulaire n’est pas la seule
étape limitant l’adsorption mais il existe d’autres phénomènes qui agissent simultanément sur
la vitesse d’adsorption. Le tracé du modèle de la diffusion intra particulaire pour l’adsorption
de Glyphosate et de Glufosinate sur le matériau NiAlNO3 est présenté sur la figure II.3.9
A partir de la figure II.3.9 on peut remarquer que la diffusion intra particulaire
comprend deux phases, la phase la plus lente contrôle la vitesse d’adsorption. Généralement,
la diffusion intra particulaire est l’étape limitant l’adsorption dans un système en Batch, les
coefficients de corrélation empiriques relatifs à l’adsorption de Glyphosate et de Glufosinate
sont rassemblés dans le tableau II.3.1 ils sont légèrement plus faibles que ceux du pseudo
second ordre.
La présence de deux étapes dans la figure II.3.9, représentant la diffusion intra
particulaire montre que l’adsorption des deux herbicides par le matériau NiAlNO3 est régit par
une diffusion à la surface du matériau plus une diffusion intra particulaire (Cheung et al.,
2007; Wu et al., 2000, 2005). La première phase de la courbe indique un effet de la couche
limite tandis que la deuxième est due à la diffusion dans les pores ou à la diffusion intra
particulaire.Des résultats similaires sont rapportés par (Sankar et al., 1999 ; Sivaraj et al.
2001).
La pente de la deuxième partie de la courbe donne le coefficient de diffusion intra
particulaire kp (mg/g mn0.5) et l’intersection de la droite avec l’axe des Y donne l’effet de la
couche limite. Les valeurs de kp pour l’adsorption de Glyphosate et de Glufosinate sont
respectivement 0.54 et 2.18 mg/g mn0.5.
166
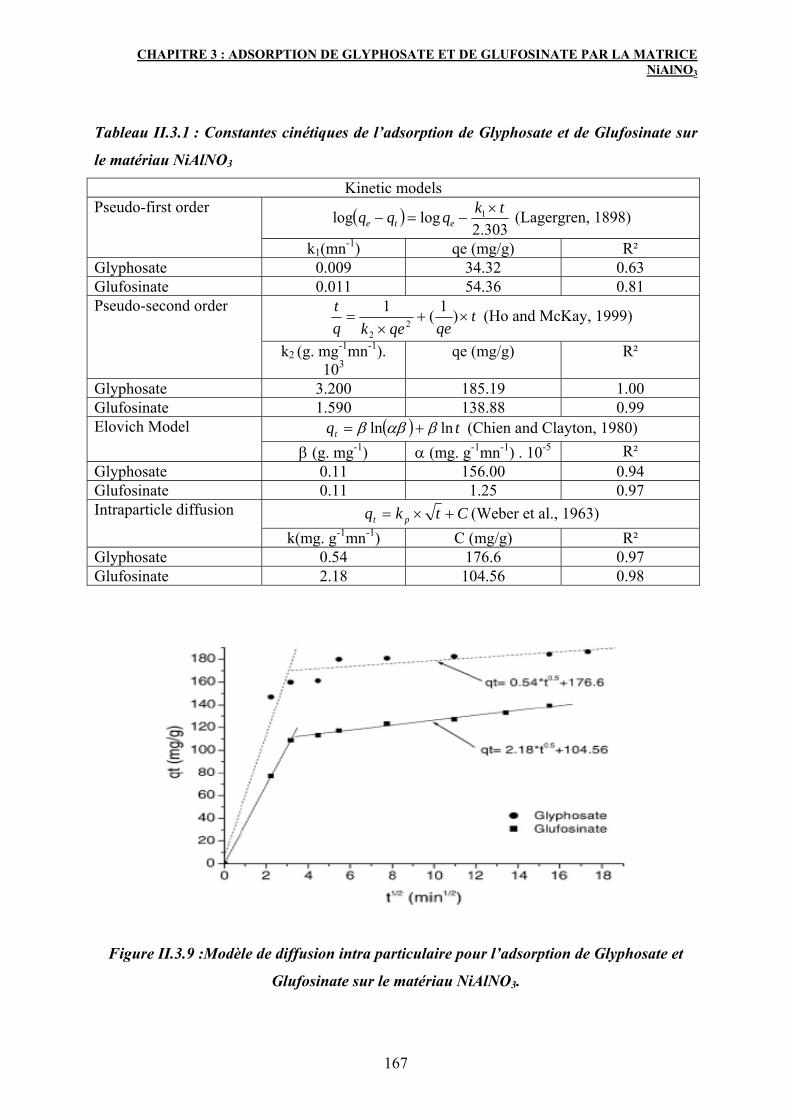
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Tableau II.3.1 : Constantes cinétiques de l’adsorption de Glyphosate et de Glufosinate sur
le matériau NiAlNO3
Kinetic models
( )303.2
loglog 1 tkqqq ete×
−=− (Lagergren, 1898) Pseudo-first order
k (mn-1) qe (mg/g) R² 1Glyphosate 0.009 34.32 0.63 Glufosinate 0.011 54.36 0.81
tqeqekq
t×+
×= )1(1
22
(Ho and McKay, 1999) Pseudo-second order
-1 -1k2 (g. mg mn ). 10
qe (mg/g) R² 3
Glyphosate 3.200 185.19 1.00 Glufosinate 1.590 138.88 0.99
( ) tqt lnln βαββ += (Chien and Clayton, 1980) Elovich Model -1 -1 -1β (g. mg ) α (mg. g mn ) . 10-5 R²
Glyphosate 0.11 156.00 0.94 Glufosinate 0.11 1.25 0.97
Ctkq pt +×= (Weber et al., 1963) Intraparticle diffusion -1 -1k(mg. g mn ) C (mg/g) R²
Glyphosate 0.54 176.6 0.97 Glufosinate 2.18 104.56 0.98
Figure II.3.9 :Modèle de diffusion intra particulaire pour l’adsorption de Glyphosate et
Glufosinate sur le matériau NiAlNO . 3
167

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
3. Isothermes et modèles d’adsorption de Glyphosate et de Glufosinate sur le matériau
NiAlNO 3
Les isothermes d’adsorption de Glyphosate et de Glufosinate sur le matériau NiAlNO3
sont présentées sur la figure II.3.10. En se basant sur la classification de Giles (Giles et al.,
1960), les deux isothermes dont tout à fait différentes. En fonction de la pente initiale de la
courbe d’isotherme ainsi que sa partie finale, l’isotherme d’adsorption de Glyphosate sur le
matériau NiAlNO3 est de type H, indiquant une grande affinité entre l’adsorbat et l’adsorbant.
L’isotherme est différente de celle de Glufosinate sur le même matériau. En effet, l’isotherme
apparaît de type L sous groupe 2 (monocouche de Langmuir), qui tende vers la forme S pour
les grandes concentrations à l’équilibre. Des résultats similaires sont obtenus par Li et al.,
2005 concernant l’adsorption de Glyphosate sur le matériau MgAlNO . 3
Ces auteurs ont supposé que l’adsorption de ce polluant sur le matériau HDL est
composée de plusieurs mécanismes. Dans la première étape la molécule est adsorbée sur la
surface du matériau par des interactions électrostatiques et des liaisons d’hydrogène. La
première couche adsorbée change le caractère hydrophile du matériau argileux qui devient
hydrophobe. Par conséquence, des interactions hydrophobiques s’ajoutent aux liaisons
hydrogène et l’ensemble fait augmenter la capacité d’adsorption du matériau HDL, en plus
d’un échange anionique se produisant pour les grandes concentrations à l’équilibre.
Les résultats expérimentaux de ces isothermes sont analysés par trois modèles :
Freundlich (Freundlich, 1906), Langmuir (Langmuir, 1916) et Tempkin (Tempkin et
Pyzhev, 1940).Notons que ces modèles sont détaillés dans la partie matériels et méthodes.
Les tracés des modèles de Langmuir et Freundlich sont présentés sur la figure II.3.11.
Les constantes équivalentes à chaque modèle sont résumées dans le tableau II.3.2
En se basant sur la valeur des constantes de corrélation empiriques R² (tableau II.3.2),
on remarque que le modèle de Langmuir donne de meilleurs résultats par rapport aux autres
modèles appliqués. Ces résultats sont en accord avec ceux trouvés par l’équipe de Li et al.,
2005 concernant l’adsorption de Glyphosate sur le matériau MgAlNO . 3
Le fait que le modèle de Langmuir traduit bien nos résultats expérimentaux peut être due
à une distribution homogènes des sites actifs d’adsorption sur la surface du matériau solide,
puisque le modèle de Langmuir suppose que la surface de l’adsorbant est constitué de sites
168
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
homogènes. Les capacités d’adsorption de Glyphosate et de Glufosinate correspondantes à
une saturation maximale de la monocouche sont respectivement 172.4 et 175.4 mg/g. La
valeur de la constante de Langmuir « b » est plus importante pour le Glyphosate indiquant
une forte interaction entre l’herbicide et le matériau Ni AlNO2 3 par rapport au Glufosinate. En
effet, à pH = 7,0 les deux herbicides affichent des charges négatives différentes, Gly2- et Glu-.
Selon Miyata (1983), la sélectivité du matériau HDL est plus importante pour l’anion
divalent que pour l’anion monovalent. En outre, la structure moléculaire zwitéréonique de la
molécule de Gly2- + 2- (-OOC-CH -NH -CH -P = OO2 2 2 ) semble beaucoup plus favorable à des
interactions attractives que la structure de Glu- +(-OOC-CH (NH2) - (CH2)2-P = O(CH )O-3 )
(regarder les structures moléculaires des deux herbicides dans la partie matériels et méthodes).
La présence simultanée dans la structure de Glufosinate d’un groupe méthyle sur le
phosphonate ainsi qu’un ammonium peut limiter son attraction vers les feuillets chargés
positivement du matériau HDL.
Tableau II.3.2: Constantes de Freundlich, Langmuir et Tempkin pour l’adsorption de
Glyphosate et de Glufosinate sur le matériau NiAlNO 3
Models Constants
Freundlich K (mg/g) n R²
Glyphosate 39.7 0.36 0.91
Glufosinate 27.3 0.44 0.94
Langmuir Qmax (mg/g) b (L/g) R²
Glyphosate 172.4 14.5 1.00
Glufosinate 175.4 0.11 0.99
Tempkin BT (L/g) AT (L/min) R²
Glyphosate 23.8 24.77 0.97
Glufosinate 21.1 17.29 0.97
169
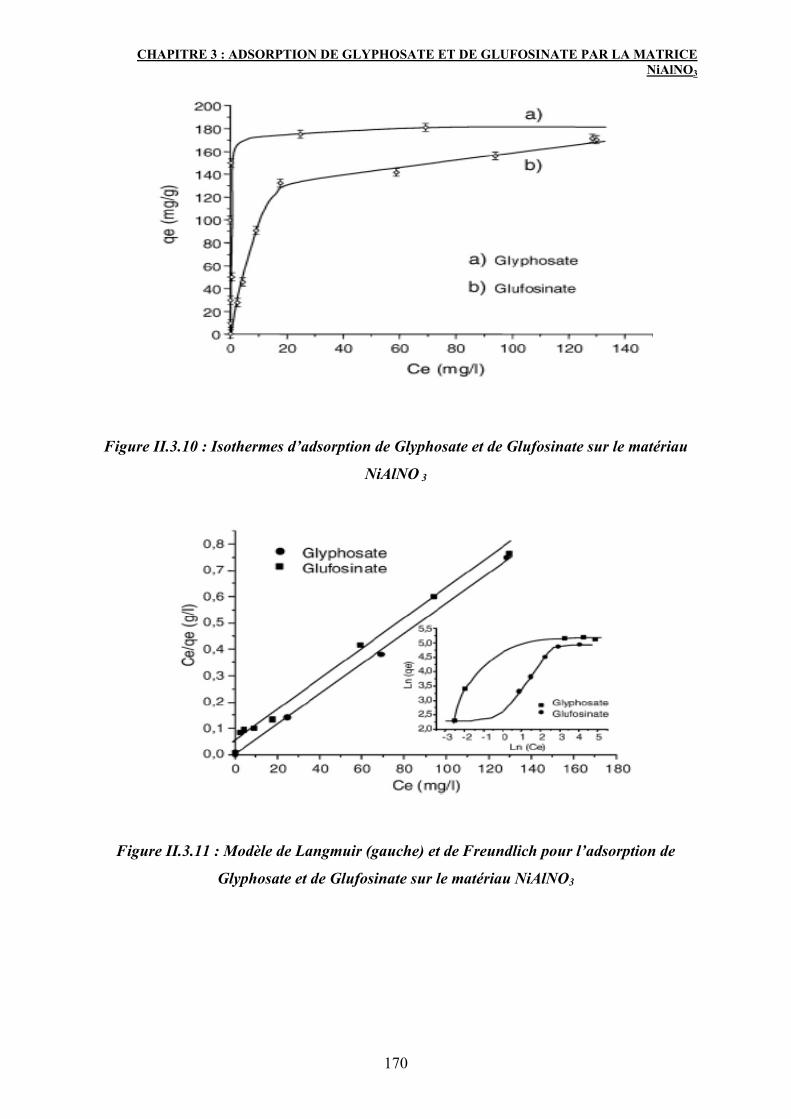
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.10 : Isothermes d’adsorption de Glyphosate et de Glufosinate sur le matériau
NiAlNO 3
Figure II.3.11 : Modèle de Langmuir (gauche) et de Freundlich pour l’adsorption de
Glyphosate et de Glufosinate sur le matériau NiAlNO 3
170

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
4. Caractérisation des matériaux après adsorption
4.1 Diffraction des rayons X (DRX)
Les diffractogrammes DRX du matériau NiAlNO3 après adsorption du Glyphosate et du
Glufosinate à différentes concentrations initiales sont présentés sur la figure II.3.12.Pour de
faibles concentrations des deux herbicides, on remarque que les diffractogrammes restent
inchangeables. Ceci met en évidence le fait que le Glyphosate et le Glufosinate sont adsorbés
sur la surface externe du matériau sans aucun changement structural de la matrice HDL. Par
contre, pour des concentrations de Glyphosate supérieures à 50 mg/L et celles de Glufosinate
dépassants 150 mg/L, on remarque l’apparition de nouvelles raies 00l correspondants à une
augmentation de l’espacement basal du matériau argileux. Ces nouveaux pics traduisent la
formation du matériau NiAlNO intercalé par le Glyphosate et le Glufosinate. 3
Notons que l’intercalation par le Glyphosate se produise à des concentrations plus
faibles que celles du Glufosinate.
Afin de mieux comprendre la nature des changements structuraux et les mécanismes
d’adsorption de Glyphosate et de Glufosinate, les DRX du matériau NiAlNO3 intercalé par le
Glyphosate et le Glufosinate en utilisant la méthode de coprécipitation et la méthode
d’échange anionique sont présentés sur la figure II.3.13.
De toute évidence les DRX des matériaux NiAlGly et NiAlGlu sont typiques de ceux
des HDL surtout pour des valeurs 2θ supérieure à 35°.Cependant, les matériaux préparés par
co précipitation et échange anionique montrent des DRX différents.
Les matériaux hybrides NiAlGly et NiAlGlu préparés par coprécipitation montrent des
DRX identiques avec des séries de raies harmoniques 00l mal définies. Les trois premières
raies 00l observées entre 3 et 30° sont identifiées afin de déterminer la distance basale du
matériau NiAlGly et NiAlGlu soit 12.4 et 12.5 A° respectivement. D’un point de vue
structural, les deux phases coprécipitées donnent des espacements basales identiques à cause
des dimensions presque égales des deux molécules Gly3- (7.4 A°) et Glu2- (7.2 A°). Ces
valeurs sont en accord avec l’orientation perpendiculaire des anions dans le domaine
intercalaire comme le montre la figure II.3.14.
171

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
En effet, l'espacement basal calculé à partir de l’épaisseur de la couche brucite, Mg
(OH)2 (4,8 Å), est compatible avec les valeurs expérimentales. Notons que la modélisation
structurale des deux molécules : Glyphosate et Glufosinate est effectuée en utilisant un
programme de stimulation semi empirique ChemBioDraw Ultra version 11.0 MM2.
Pour les matériaux hybrides préparés par échange anionique, les raies 00l du matériau de
base sont présents sur les diffractogrammes DRX , traduisant un échange anionique partiel
des molécules de Gly et de Glu. Des raies 00l supplémentaires sont observées sur le
diffractogrammes des matériaux NiAlGly et NiAlGlu préparés par échange anionique. Deux
phénomènes sont inclus dans l’adsorption des molécules de Gly et de Glu : adsorption et
intercalation. Dans le cas du matériau NiAlGly préparé par échange anionique, les trois raies
situées à 8.30, 12.7 et 22.6 (°2θ) sont attribuées aux diffractions 00l correspondant à un
espacement basal de 20.4 A° .Donc la molécule de Glyphosate adopte un arrangement en
double couche à l’intérieur des feuillets HDL. Pour le matériau NiAlGlu, la pauvreté des
données structurales empêche la détermination de la distance basale.
La différence entre les phases hybrides NiAlGly et NiAlGlu préparées par
coprécipitation et par échange anionique, peut être expliquée par une différence dans les
conditions de pH : pH = 10,0 et 7.0, pour la co-précipitation et l'échange anionique
respectivement. Les deux herbicides : le Glyphosate (pK <2, pK =2.6, pK =5.6 et pK1 2 3 4=10.6)
(Li et al., 2004) et le Glufosinate (pK1=2, pK =2.9, pK2 3=9.8) sont deux molécules
amphotères. Suivant leurs constantes de dissociations, la molécule de Glyphosate se balance
entre la forme Gly+ et Gly3- + tandis que la molécule de Glufosinate entre Glu et Glu2- . A pH
7 (échange anionique), les deux molécules se trouvent sous la forme : Gly2- et Glu-
respectivement et par conséquence l’intercalation sera plus difficile que dans le cas de pH
10 (coprécipitation).
172
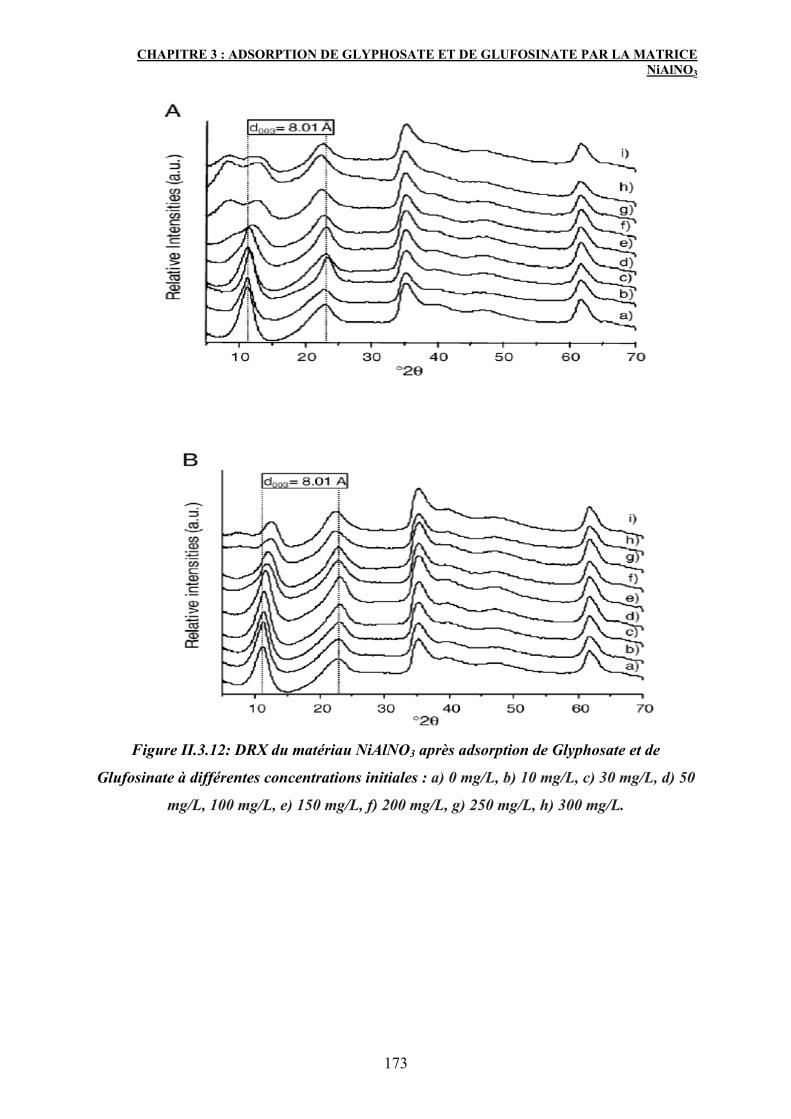
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.12: DRX du matériau NiAlNO3 après adsorption de Glyphosate et de
Glufosinate à différentes concentrations initiales : a) 0 mg/L, b) 10 mg/L, c) 30 mg/L, d) 50
mg/L, 100 mg/L, e) 150 mg/L, f) 200 mg/L, g) 250 mg/L, h) 300 mg/L.
173
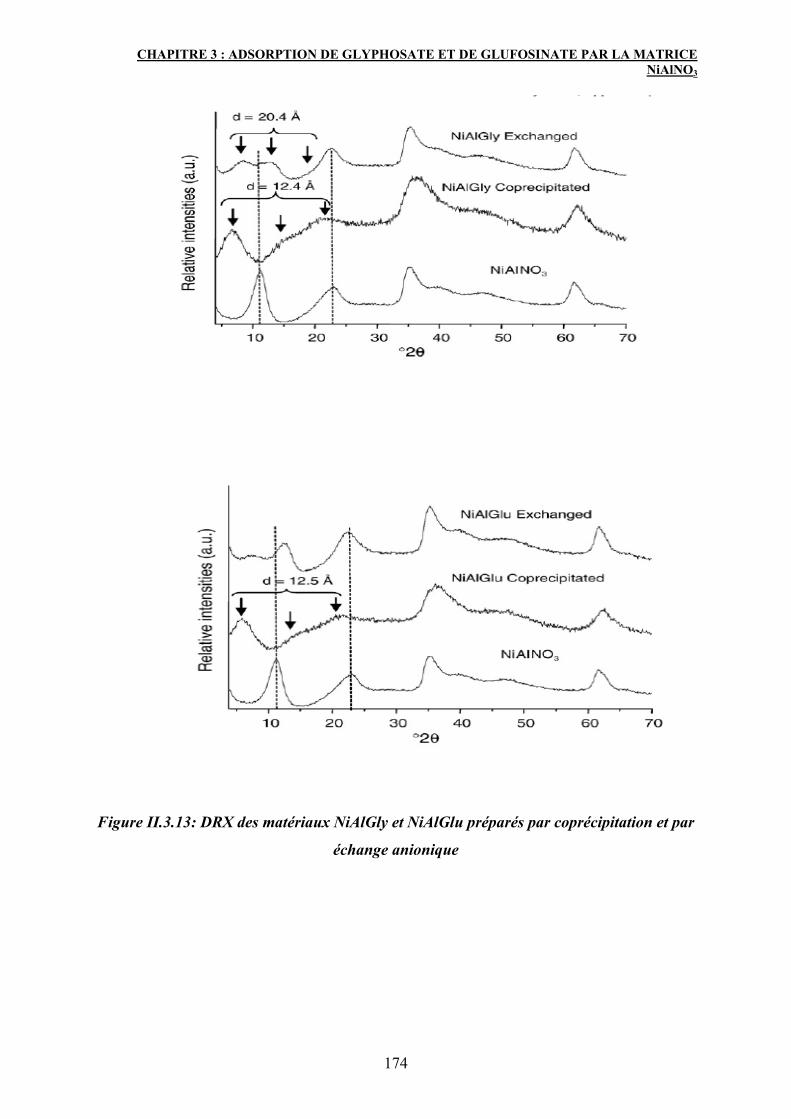
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.13: DRX des matériaux NiAlGly et NiAlGlu préparés par coprécipitation et par
échange anionique
174

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.14: Représentation schématique des phase NiAlGly et NiAlGlu coprécipitées
4.2 Microscopie électronique à Balayage (MEB)
Comme le montre les photos de microscope électronique à balayage (MEB), la
morphologie du matériau HDL est aussi influencée par l’adsorption de Glyphosate et de
Glufosinate (figure II.3.15). Les photos MEB du matériau de base montre un faible niveau
de cristallinité de cette matrice où les plaquettes se précipitent pour former des petits agrégats
pseudo sphériques tout en conservant la morphologie rose de sable.
La taille des particules primaires est inférieure à 20 et 100 nm pour la petite et la grande
dimension respectivement. L’adsorption de Glyphosate et de Glufosinate sur le matériau
NiAlNO3 modifie clairement la surface de la matrice HDL, la morphologie rose de sable est
fortement affectée. Malheureusement, la distinction des particules isolées n’est plus possible,
la forme balance vers une agrégation plus compacte. Cette transformation est plus prononcée
dans le cas du Glufosinate.
175

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Figure II.3.15 : Photos MEB du matériau NiAlNO3 avant et
après adsorption de Glyphosate et de Glufosinate (Ci = 300mg/L)
4. Spectroscopie IR
La caractérisation de la phase NiAlNO3 après adsorption de Glyphosate et de
Glufosinate est aussi effectuée par spectroscopie IR, les résultats sont présentés sur la figure
II.3.16. D’après les résultats de la figure précédente, on remarque que l’intensité des pics
situés à 1056 cm−1 and 1103 cm−1 caractéristiques des vibrations d’élongations de la liaison P-
O du Glyphosate et Glufosinate respectivement, augmente avec augmentation de la
concentration des herbicides. Il en est de même pour les pics situés à 1573 cm−1 and 1561
cm−1 caractéristiques des vibrations d’élongation de la liaison C-O des molécules respectives .
176
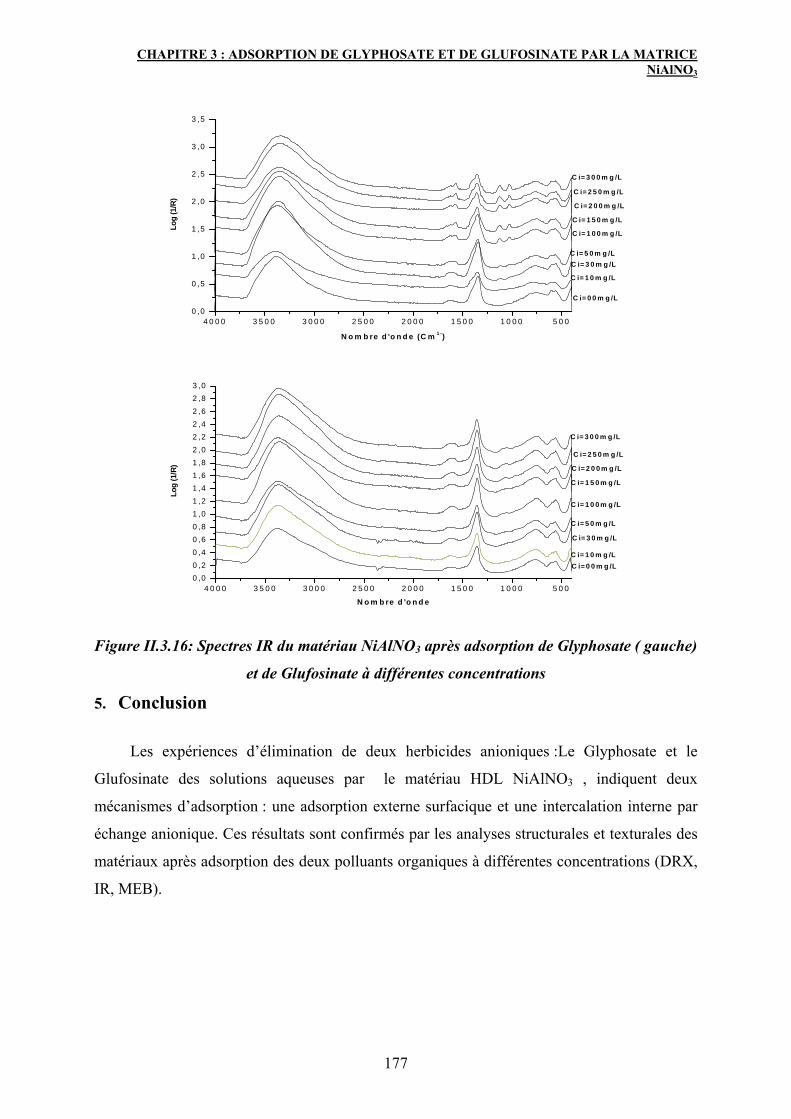
CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0 1 5 0 0 1 0 0 0 5 0 00 ,0
0 ,5
1 ,0
1 ,5
2 ,0
2 ,5
3 ,0
3 ,5
C i= 3 0 0 m g /L
C i= 2 5 0 m g /L
C i= 2 0 0 m g /L
C i= 1 5 0 m g /L
C i= 1 0 0 m g /L
C i= 5 0 m g /LC i= 3 0 m g /L
C i= 1 0 m g /L
C i= 0 0 m g /L
Log
(1/R
)
N o m b re d 'o n d e (C m 1 -)
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0 1 5 0 0 1 0 0 0 5 0 00 ,00 ,20 ,40 ,60 ,81 ,01 ,21 ,41 ,61 ,82 ,02 ,22 ,42 ,62 ,83 ,0
C i= 1 0 m g /L
C i= 3 0 0 m g /L
C i= 2 5 0 m g /L
C i= 2 0 0 m g /L
C i= 1 5 0 m g /L
C i= 1 0 0 m g /L
C i= 5 0 m g /L
C i= 3 0 m g /L
C i= 0 0 m g /L
Log
(1/R
)
N o m b re d 'o n d e
Figure II.3.16: Spectres IR du matériau NiAlNO3 après adsorption de Glyphosate ( gauche)
et de Glufosinate à différentes concentrations
5. Conclusion
Les expériences d’élimination de deux herbicides anioniques :Le Glyphosate et le
Glufosinate des solutions aqueuses par le matériau HDL NiAlNO3 , indiquent deux
mécanismes d’adsorption : une adsorption externe surfacique et une intercalation interne par
échange anionique. Ces résultats sont confirmés par les analyses structurales et texturales des
matériaux après adsorption des deux polluants organiques à différentes concentrations (DRX,
IR, MEB).
177

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
La cinétique d’adsorption des deux herbicides sur le matériau HDL suit un modèle du
pseudo second ordre. La diffusion intra particulaire n’est pas la seule étape limitant
l’adsorption des deux herbicides, en effet l’adsorption surfacique et la diffusion opèrent de
façon simultanée lors des interactions de Glyphosate et de Glufosinate avec le matériau HDL.
Les isothermes d’adsorption sont de type H et L pour le Glyphosate et le Glufosinate
respectivement et suivent le modèle de Langmuir. Les résultats de ce travail indiquent le rôle
important du matériau HDL NiAlNO3 comme adsorbant potentiel pour l’élimination des
polluants organo-phosphates et organo-phosphonates.
178

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
Références bibilographiques
• Atwood J.L., Davies J.E. and MacNicol D.D. (1997). In: Vogtle, F. (Ed.),
Comprehensive Supramolecular Chemistry. Pergamon, Elmsford, New York. Chapter 7.
• Barja B.C., Herszaje J., Dos Santos Afonso M. (2001), Polyhedron 20, 1821–1830 .
• Bhattacharyya K.G. and Sharma A. (2004), J. Hazard. Mater. B 113, 97–109.
• Bayer E., Gugel K.H., Haegele K., Zachner Z. (1972), Helv. Chem. Acta 55, 224–
239.
• Benito P., Labajos F.M., Rocha J. And Rives V. (2006), Micropor. Mesopor. Mater.
94, 148–158.
• Bish D.L. and Brindley G.W. (1977), Am. Mineral., 62, 458-464.
• Cavani F., Trifiro F. and Vaccari A. (1991), Catal. Today 11, 173–307.
• Cheung W.H., Szeto Y.S. and McKay G. (2007), Bioresour. Technol. 98, 2897–
2904.
• Chien S.H. and Clayton W.R. (1980), J. Am. Soil Sci. Soc. 44 (809), 265–268.
• De Jonge H., De Jonge L.W., Jacobsen O.H., Yamaguchi T. and Moldrup P.,
(2001), Soil Sci. 166, 230–238.
• De Roy A., Forano C., El Malki K. and Besse J.-P.(1992),Expanded clays and other
microporous solids. In: Occelli, M.L., Robson, H. (Eds.), Synthesis of Microporous Materials.
Van Nostrand Reinhold, New York. Chapter 7.
• Dion H.M., Harsh J.B., Hill Jr. H.H. (2001), J. Radioanal. Nucl. Chem. 49, 385–
390.
• El Gaini L., Lakraimi M., Sebbar E., Meghea A. and Bakasse M.(2009). Journal
Hazard Mater 161, 627–632.
• Flury M. (1996), J. Environ. Qual. 25, 25–45.
• Forano C (2004). Environmental remediation involving layered double hydroxides.
In: Wypych, F., Satyanarayana, K.G. (Eds.), Clay Surfaces: Fundamentals and applications.
Elsevier, p. 553. Chapter 15.
• Franz J., Mao M. and Siroski J. (1997). Glyphosate: a unique global herbicide; ACS
Monograph 189. American Chemical Society, Washington, DC.
• Freundlich H.M.F. (1906), Z. Phys. Chem. 57, 385–470.
• Giles C.H., MacEwan T.H., Nakhwa S.N. and Smith D. (1960), J. Chem. Soc. 3,
3973–3993.
179

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
• Gimsing A.L. and Borggaard O.K.(2002), Clay Miner. 37, 509–515.
• Gimsing A.L., Borggaard O.K. and Bang M. (2004a), Eur. J. Soil Sci. 55, 183–191.
• Gimsing A.L., Borggaard O.K., Sestoft P. (2004b), Environ. Sci. Technol. 38,
1718–1722.
• Gimsing A.L., Szilas C. and Borggaard O.K. (2007), Geoderma 138, 127–132.
• Hermosin M.C., Pavlovic I., Ulibarri M.A. and Cornejo J. (1993), J. Environ. Sci.
Health, Part A 28, 1875–1888.
• Hermosin M.C., Pavlovic I., Ulibarri M.A. and Cornejo J. (1996), Water Res. 30,
171–177.
• Ho Y.S. and Mc Kay G. (1998), J. Chem. Eng. 70, 115–124.
• Ho Y.S. and McKay G. (1999), Process. Biochem. 34, 451–465.
• Hudson H.R. (2000). In: Hudson, H.R., Kukhar, V. (Eds.), Aminoalkanephosphonic
and Aminoalkanephosphinic Acids: Chemistry and Biological Activity. Wiley, Chichester,
pp. 443–465. Chapter 13.
• Juang R.S., Wu F.C. and Tseng R.L. (2000), J. Colloid Interface Sci. 227, 437–444.
• Kannan S., Narayanan A., Swamy C. S. (1996). J Mat Sci 31, 2353-2360.
• Khenifi A., Derriche Z., Mousty C., Prévot V. and Forano C. (2010), Appl Clay
Sci, 47, 362–371.
• Kovanda F., Rojka T., Bezdicka P., Jiratova K., Obalova L., Pacultova K., Bastl
Z. and Grygar T. (2009), Journal of Solid State Chemistry, 182, 27–36.
• Lagergren S. (1898). Zur theorie der sogenannten adsorption geloster Stoffe,
Kungliga svenska vetenskapsakademiens. Handlingar band. 24, 1–39.
• Langmuir I. (1916), J. Am. Chem. Soc. 38, 2221–2295.
• Li F., Zhang L., Evans D.G., Forano C. and Duan X. (2004), Thermochimica Acta
424, 15–23.
• Li F., Wang Y., Yang Q., Evans D.G., Forano C. and Duan X. (2005), J. hazard
mater, B125, 89–95.
• Lv L., Sun P., Gu Z., Du H., Pang X., Tao X., Xu R., Xu L. (2009, J Hazard Mater,
161, 1444–1449.
• Miyata S. (1975), Clays Clay Miner., 23, 369–375.
• Miyata S. (1983), Clays Clay Miner. 31, 305–311.
180

CHAPITRE 3 : ADSORPTION DE GLYPHOSATE ET DE GLUFOSINATE PAR LA MATRICE NiAlNO3
• Moujahid E.M., Inacio J., Besse J.-P. and Leroux F. (2003), Microporous
Mesoporous Mater. 57, 37–46.
• Ozacar M., Ayhan I.S. and Türkmenler E.H. (2008), J. Chem. Ing 143, 32–42.
• Parker L.M., Milestone N.B. and Newman R.H. (1995), Ind. Eng. Chem. Res. 34,
1196–1202.
• Reichle W.T. , Kang S.Y. & Everehardt D.S. (1986), J. Catal., 101, 352-359.
• Rhee S.W., Kang M.J., Kim H., Moo C.H. (1997), Environ. Technol. 18, 231–236.
• Rubin J.L., Gains C.G. and Jensen R.A. (1982), Plant Physiol. 7, 833–839.
• Sankar M., Sekaran G., Sadulla S. and Ramasami T. (1999), J. Chem. Technol.
Biotechnol. 74, 334–337.
• Scheidegger A.M., Lamble G.M., Sparks D.L. (1997), J. Colloid and Interf. Sci.
186, 118–128.
• Sivaraj R., Namasivayam C. and Kadirvelu K. (2001), Waste Manage. 21, 105–
110.
• Solin S. A., Hines D. R., Seidler G. T. & Treacy M. M. J. (1996), J. Phys. Chem.
Solids, 57, 1043-1048.
• Tempkin M.J. and Pyzhev V. (1940), Acta Physiochim. URSS 12, 217–222.
• Titulaer M.K., Jansen J.B.H. and Geus J.W. (1994), Clays Clay Miner. 42, 249–
258.
• Vaysse C (2001), thèse doctorat, Université Bordeaux 1.
• Vereecken H. (2005), Pest Manag. Sci. 61, 1139–1151.
• Weber W.J., Morris J.C. and Sanit J. (1963), Eng. Div. Am. Soc. Civ. Eng. 89, 31–
60.
• Wu Feng-Chin, Tseng, Ru-Ling, Juang, Ruey-Shin, 2000. Comparative adsorption
of metal and dye on flake and bead-types of chitosans prepared from fishery wastes. J.
Hazardous Materials B73, 63–75.
• Wu F.-C., Tseng R.-L. and Juang R.-S. (2005), J. Colloid Interface Sci. 283, 49–56.
• Zhang L., Feng L., Evans D.G., Duan X. (2004), Mater. Chem. Phys. 87, 402–410.
• Zimdahl R.L. (1993). Fundamentals of Weed Science. Academic Press, London.
181

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE
GLYPHOSATE ET DE GLUFOSINATE PAR UNE
ELECTRODE MODIFIEE PAR
ELECTRODEPOSITION DE NiAl HDL.
182

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE
GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR
ELECTRODEPOSITION DE NiAl HDL.
1. Introduction Les produits organophosphate et organophosphonate (OP) sont une famille de pesticides
très utilisée en agriculture. Par exemple le Glyphosate (N-(phosphonométhyl) glycine,Gly) et
le Glufosinate ((DL-homoalanine-4-yl)-methylphosphinic acide, Glu) sont employées comme
herbicides non sélectifs. L'utilisation intensive de ces herbicides suscite des craintes quant à
leur impact sur l'environnement et leurs risques éventuels pour la santé.
Les méthodes d’analyse utilisées pour ce type de molécules chimiques sont présentées
par Stalikas et Konidari, 2001. Elles consistent essentiellement à l’analyse par
chromatographie phase liquide, chromatographie phase gazeuse, électrophorèse capillaire
etc…. La plupart de ces méthodes développées pour l'analyse de Glyp et de Gluf ont besoin
d’un traitement préliminaire afin de fonctionnaliser les substrats pour permettre leur
détection. En effet, les techniques de chromatographie en phase liquide utilisant des
détecteurs UV ou fluorimétrique, sont limitées à l'analyse des pesticides portant des groupes
chromophores ce qui n'est pas le cas avec le Glyp et le Gluf. Les capteurs électrochimiques
représentent donc une bonne alternative innovante en termes de détection sur place, de
sensibilité, de réponse rapide, et de taille réduite pour la détermination de ces pesticides (Sato
et al.,2001 ; Ruiz Simoes, 2006; Songa et al., 2009 a et b).
Les capteurs et les biocapteurs à bases d’électrodes modifiées par des argiles ont été
utilisés pour la détermination des polluants, tels que les pesticides, les produits dérivés du
phénol, les cyanures, les métaux, etc. (Mousty, 2004). En particulier les hydroxydes doubles
lamellaires appelées aussi des argiles anioniques ou hydrotalcites, montrant un arrangement
structural à deux dimensions (2D) ont été largement utilisés pour la fabrication des capteurs
électrochimiques ou biochimiques. Par exemple, les HDL contenants un métal de transition
comme le CoII II, le Ni , ou le MnII, qui subit une réaction d'oxydo-réduction réversible dans
183

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
une large gamme de potentiel, sont décrits comme des matériaux de revêtement pour
électrodes en raison de leurs propriétés de transport de charge et de catalyse redox en
particulier dans un milieu basique. Les électrodes modifiées par la matrice HDL NiAl-X sont
appliquées à la détection électro catalytique des alcools mono et polyvalents (tels que le
méthanol, l'éthanol, le propanol, le glucose, le fructose, le saccharose, etc.) ainsi que pour les
amines aromatiques ou aliphatiques (Ballarin et al., 2000 et 2005 Scavetta et al., 2006).
Contrairement aux argiles cationiques, les matériaux HDL peuvent être facilement
préparés en laboratoire. Différentes méthodes de synthèse des HDL sont connues jusqu’à
présent (Rives, 2001 ; Duan et Evans, 2006), on peut citer par exemple: la méthode de
coprécipitation à pH constant, la coprécipitation en utilisant une base retardateur tel que l'urée
(Adachi-Pagano et al., 2003) ou la méthode de préparation par les polyols (Prevot et al.,
2005). L’électro synthèse des HDL par génération électrochimique d'hydroxyle en utilisant la
réduction cathodique des ions nitrates a également été décrite par différents auteurs comme
étant une méthode efficace pour la préparation de couches minces HDL appropriées pour des
applications de détection (Indira et Kamath, 1994 ; Scavetta et al., 2007 et 2009 ; Yarger et
al.,2008).
Dans ce travail, des électrodes modifiées par électrodéposition d’une matrice HDL type
Ni1-xAlx(OH)2NO3x ·nH2O sont appliquées à l’analyse électrochimique de Glyp et de Gluf.
Les matrices HDL type NiAl sont des matériaux intéressants car ils combinent des propriétés
électro actives excellentes sur la base du processus redox Ni2+ 3+ /Ni avec des propriétés
structurales et texturales adaptées à l'adsorption des herbicides chargés négativement. Pour
mieux comprendre les interactions entre les herbicides et le matériau HDL, une isotherme
d’adsorption est réalisée.
2. Résultats et discussions Le Glyphosate, le Glufosinate ainsi que les sels métalliques utilisés dans ce travail sont
fournis par Sigma Aldrich et sont utilisés sans purification.
Afin de réaliser les isothermes d’adsorption, le matériau NiAlNO3 est préparé par la
méthode de coprécipitation à pH constant décrite précédemment (Chapitre matériels et
184

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
méthodes). La cinétique et l’isotherme d’adsorption des deux herbicides sur le matériau
NiAlNO sont effectuées de la même manière que dans le chapitre précédent. 3
2.1 Caractérisation physique des films de NiAlNO3 préparé par voie
électrochimique
Des films de NiAlNO3 sont déposés sur des plaques de platines (expérience détaillée
dans la partie matériels et méthodes) suivant le schéma réactionnel ci dessous (Scavetta et al.,
2007).
- - - NO + H O + 2é NO +2OH3 2 2
(1-x) Ni2+ + x Al3+ + 2OH- - + x NO + m H O [Ni 3 2 1-x Al x (OH)2] (NO ) ,m H O x 3 x 2
Figure II.4.1: Schéma réactionnel de la formation du matériau NiAlNO3 par voie
électrochimique
Trois temps d’éléctrodéposion sont utilisés: 60, 120 et 200 secondes. Une analyse DRX
est effectuée sur ces films et comparée au matériau NiAlNO3 préparé par coprécipitation
(Figure II.4.2).
Les raies de diffraction DRX caractéristiques de la structure HDL (réseau hexagonal
avec une symétrie rhomboédrique R-3m) sont clairement visibles sur le diffractogramme X du
matériau de référence (Figure II.4.2 a) donnant les paramètres de maille suivants : a = 0,1499
nm, c = 2.5689 nm et un espacement basal d = 0,8563 nm. Ces raies ne sont pas visibles sur
les diffractogrammes des films électrodéposés à des durées différentes, il en est de même pour
le film déposé à 200s (figure II.4.2 c-d). Les films obtenus avec des temps
d’électrodéposition très faibles donnent des matériaux NiAlNO3 amorphes contenant de
petites particules qui génèrent des hautes diffusions de rayons X pour les faibles deux thêta.
Pour résoudre ce problème et pouvoir mener à bien la caractérisation du matériau HDL
obtenu par éléctrodéposition, dix répétitions sont effectuées à 200s, la poudre obtenue par
grattage de la plaque de platine est analysée par DRX (figure II.4.2 b). Ce dernier est typique
du matériau à faible cristallinité NiAl HDL présenté par la littérature (Scavetta et al., 2007).
Les spectres IR et Raman du film obtenu par électrodéposition est comparé au matériau
préparé par coprécipitation (figures II. 4.3 A et B).
185

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Les mesures par spectroscopie Raman ont été effectuées en utilisant un appareil type
JobinT64000 Yvon Horiba équipé d’un détecteur CCD refroidit par N2 liquide. Une ligne
laser Ar à 515.5 nm est utilisée comme source d'excitation avec une puissance de 20 mM.
Les deux échantillons (coprécipité et electrodéposé) présentent des bandes
caractéristiques de la matrice HDL NiAl, avec des modes de déformation et d’élongation
typiques au plan NiAl-O pour des nombres d’ondes inférieures à 610 Cm-1 superposés aux
vibrations D3h de l’anion nitrate intercalaire. Les vibrations majoritaires de l’anion nitrate se
résument comme suit : υ1(NO3) à 1055 et 1044cm−1(Figure II.4.3 A), υ (NO2 3) à 823 et
825cm−1 (Figure II.4.3 B), pour le matériau coprécipité et électrodéposé respectivement.
Notons la disparition de la bande υ (NO4 3) pour le matériau électrodéposé, cette extinction est
probablement dû à une orientation préférentielle des plaquettes NiAl-LDH à la surface de la
plaque de platine. La bande à 981 Cm-1 n’est pas encore identifiée.
Les propriétés texturales des différents dépôts sont étudiées par spectroscopie
électronique à balayage (MEB). La figure II.4.4 donne les images MEB des films obtenus
avec des temps d’électrodéposition différents (t= 60, 120, 200 s). De toute évidence, le temps
d’électrodéposition a un grand effet sur la morphologie des films. A 60 s, une membrane
dense et homogène ressemblante à un gel faite de nano particules de taille inférieure à 50 nm
est obtenue. Un agrandissement de la figure II.4.4 a montre la présence de mésopores
dispersés dans le film continue. L'épaisseur de ce film dense (<150 nm) peut être estimée à
partir de la Figure II.4.4.c. l’électrogénération des OH- sur la surface totale de l'électrode
favorise la formation de ce film dense. Lorsque le temps d’électrodéposition atteint 120 s, le
film devient moins compacte, la croissance des plaquettes se fait d’une façon homogène dans
une direction perpendiculaire à l'électrode conduisant à un réseau de feuillets reliées entre eux
et couvrant la surface totale de l’électrode (Figure II.4.4 d et e). Les plaquettes ont tendance
à s'agréger ensemble pour donner des formes pseudo-sphériques des particules secondaires.
Enfin, pour un temps d’électrodéposition de 200 s, des particules sphériques, issues
d’agrégation de plaquettes donnant une morphologie rose de sable , sont formés tous le long
de la surface de la membrane dense (figure II.4.4 i). Une forte ressemblance avec la
morphologie du matériau HDL coprécipité NiAl-NO (Figure II.4.4 J et k) est mise en 3
186

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
évidence par la suite. En effet, la coprécipitation électrogénéré du matériau NiAlNO3 n’est
plus localisée à la surface de l’électrode et la croissance des plaquettes est plus ressemblante
au matériau coprécipité du départ.
Figure II.4.2: DRX de : a)matériau préparé par coprécipitation, b) NiAlNO3 obtenu après
10 électrodéposition à 200 secondes, c)film de NiAlNO3 éléctrodéposé sur Pt à 60s, d) à
120s, e) à 200s (*Pt,∇ : impuretés du KNO ). 3
2.2 Adsorption de Glyphosate et de Glufosinate sur la matrice NiAlNO3
Les isothermes d’adsorption des deux herbicides Glyphosate et Glufosinate sont
effectuées (figure II.4.5) sur le matériau coprécipité NiAlNO3 afin de mesurer la quantité de
l’anion adsorbée et de savoir si l’adsorption et/ou l’intercalation peuvent affecter la réponse
électrochimique du biocapteur. En effet, le Glyphosate (pK = 0,78 (1er phosphonique), pKa1 a2
=2,29 (carboxylate), pK =5.96 (2èmephosphonique),et pKa3 a4 =10.98 (amine)) et le
Glufosinate (pK =2,0 (phosphonique), pK =2,9 (carboxylate) et pKa1 a2 a3 =9,8 (amine)) sont
chargées négativement sous les conditions de pH d’analyse électrochimique (pH 13,0),
donnant les formes suivantes : Glyp3- et Gluf2- . Par conséquent, le Glyp3- et le Gluf2-peuvent
être absorbées dans les films électrogénérés au cours de leur détection électrochimique à
cause des propriétés d’échange anionique de la matrice HDL NiAlNO3. L'adsorption du
Glyphosate et de Glufosinate par les sols (Laitinen et al., 2008 ; Pessagno et al., 2008 ;
187
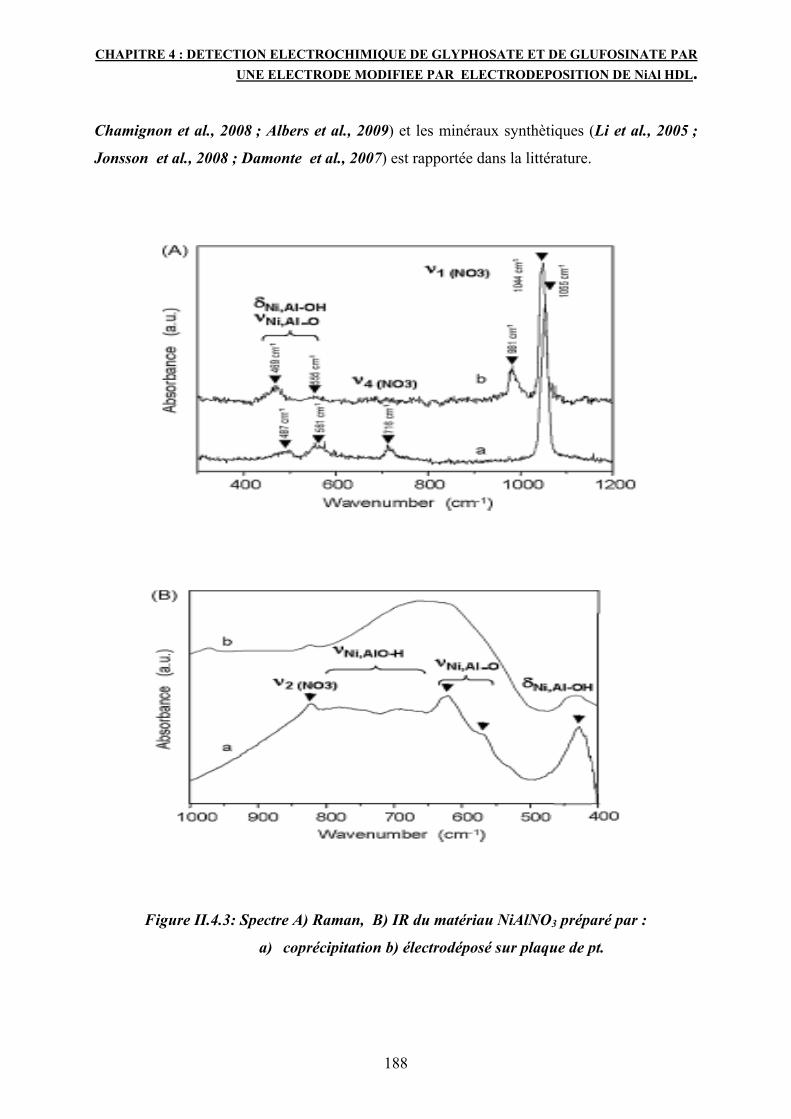
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Chamignon et al., 2008 ; Albers et al., 2009) et les minéraux synthètiques (Li et al., 2005 ;
Jonsson et al., 2008 ; Damonte et al., 2007) est rapportée dans la littérature.
Figure II.4.3: Spectre A) Raman, B) IR du matériau NiAlNO préparé par : 3
a) coprécipitation b) électrodéposé sur plaque de pt.
188

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Figure II.4.4 :Images MEB des films NiAlNO3 électrodéposé à 60 secondes (a,b,c), à 120
secondes(d,e,f), à 200 secondes (g,h,i), du matériau coprécipité (j), du matériau coprécipité
et adsorbé par le Glyphosate (k) et adsorbé par le Glufosinate (l).
Afin de déterminer le temps nécessaire pour atteindre l'équilibre d’adsorption de
Glyphosate et de Glufosinate sur le matériau NiAlNO3, les Cinétiques d'adsorption des deux
herbicides sont effectuées et présentées sur la figure II.4.5 .Cette dernière montre clairement
qu’il n’a pas d'adsorption de Gluf à pH 13.0, alors qu’une capacité d’adsorption de 170 mg/g
est atteinte à pH 7 (Khenifi et al., 2010) (regarder les résultats présentés dans le chapitre
précédent). Contrairement au Glufosinate, le Glyphosate est adsorbé avec un temps
d’équilibre de 175 min (figure II.4.5 A).
L’isotherme d’adsorption de Glyp (figure II.4.5 B) suit le modèle de Langmuir avec un
coefficient de corrélation empirique très satisfaisant (R²= 0.97). La valeur de la capacité
d’adsorption maximale tirée à partir de ce modèle ainsi que le coefficient « b » de Langmuir
189
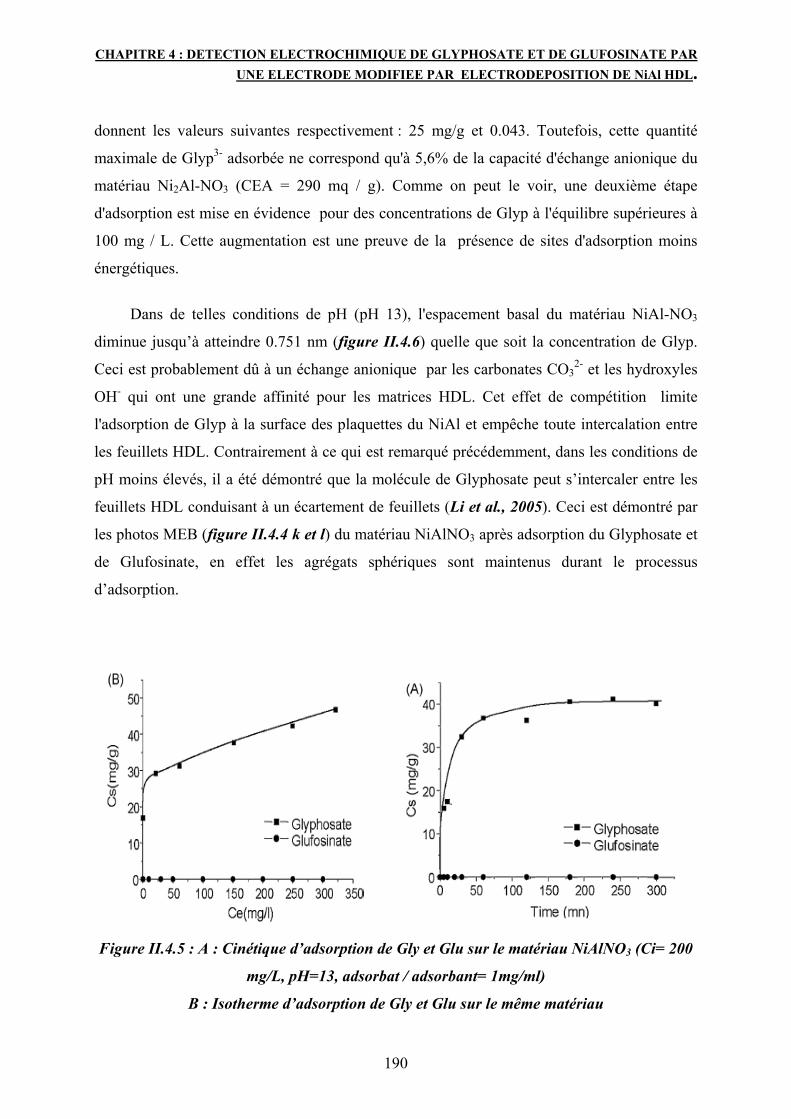
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
donnent les valeurs suivantes respectivement : 25 mg/g et 0.043. Toutefois, cette quantité
maximale de Glyp3- adsorbée ne correspond qu'à 5,6% de la capacité d'échange anionique du
matériau Ni Al-NO2 3 (CEA = 290 mq / g). Comme on peut le voir, une deuxième étape
d'adsorption est mise en évidence pour des concentrations de Glyp à l'équilibre supérieures à
100 mg / L. Cette augmentation est une preuve de la présence de sites d'adsorption moins
énergétiques.
Dans de telles conditions de pH (pH 13), l'espacement basal du matériau NiAl-NO3
diminue jusqu’à atteindre 0.751 nm (figure II.4.6) quelle que soit la concentration de Glyp.
Ceci est probablement dû à un échange anionique par les carbonates CO 2-3 et les hydroxyles
OH- qui ont une grande affinité pour les matrices HDL. Cet effet de compétition limite
l'adsorption de Glyp à la surface des plaquettes du NiAl et empêche toute intercalation entre
les feuillets HDL. Contrairement à ce qui est remarqué précédemment, dans les conditions de
pH moins élevés, il a été démontré que la molécule de Glyphosate peut s’intercaler entre les
feuillets HDL conduisant à un écartement de feuillets (Li et al., 2005). Ceci est démontré par
les photos MEB (figure II.4.4 k et l) du matériau NiAlNO3 après adsorption du Glyphosate et
de Glufosinate, en effet les agrégats sphériques sont maintenus durant le processus
d’adsorption.
Figure II.4.5 : A : Cinétique d’adsorption de Gly et Glu sur le matériau NiAlNO3 (Ci= 200
mg/L, pH=13, adsorbat / adsorbant= 1mg/ml)
B : Isotherme d’adsorption de Gly et Glu sur le même matériau
190
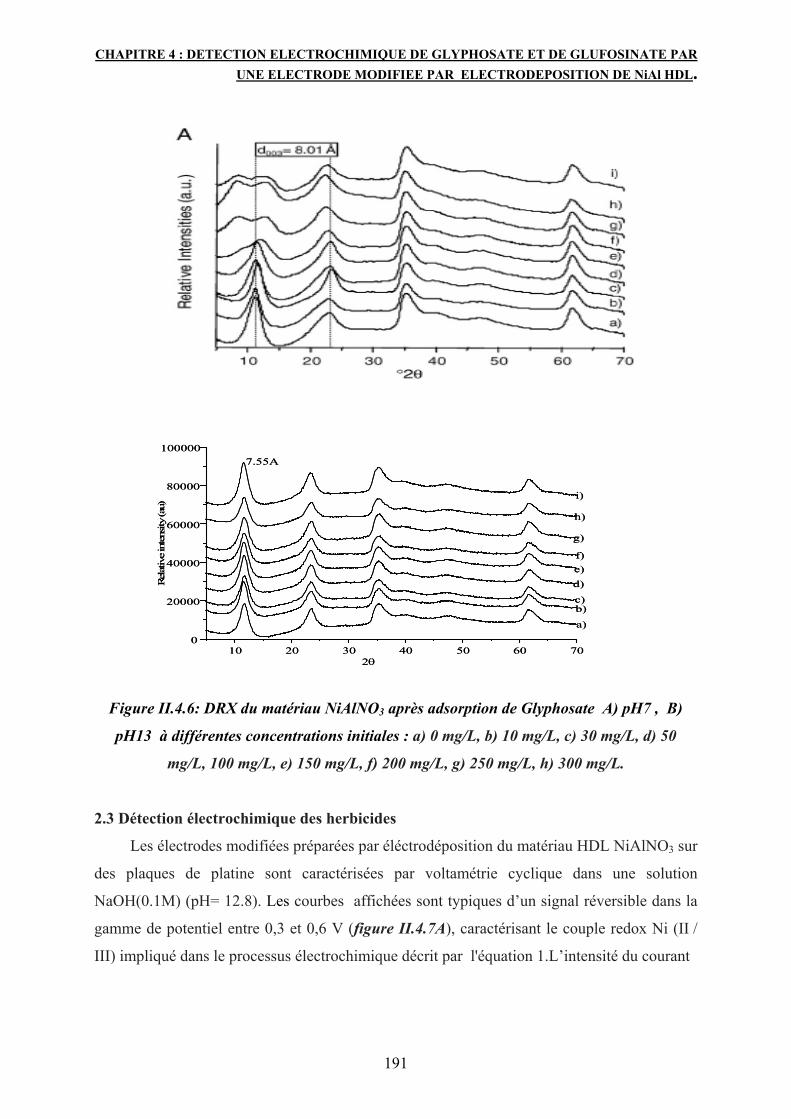
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
10 20 30 40 50 60 700
20000
40000
60000
80000
100000
i)
h)
g)
f)e)d)
c)b)
a)
7.55A
Rel
ativ
e in
tens
ity(a
u)
2θ10 20 30 40 50 60 70
0
20000
40000
60000
80000
100000
i)
h)
g)
f)e)d)
c)b)
a)
7.55A
Rel
ativ
e in
tens
ity(a
u)
2θ
Figure II.4.6: DRX du matériau NiAlNO3 après adsorption de Glyphosate A) pH7 , B)
pH13 à différentes concentrations initiales : a) 0 mg/L, b) 10 mg/L, c) 30 mg/L, d) 50
mg/L, 100 mg/L, e) 150 mg/L, f) 200 mg/L, g) 250 mg/L, h) 300 mg/L.
2.3 Détection électrochimique des herbicides
Les électrodes modifiées préparées par éléctrodéposition du matériau HDL NiAlNO3 sur
des plaques de platine sont caractérisées par voltamétrie cyclique dans une solution
NaOH(0.1M) (pH= 12.8). Les courbes affichées sont typiques d’un signal réversible dans la
gamme de potentiel entre 0,3 et 0,6 V (figure II.4.7A), caractérisant le couple redox Ni (II /
III) impliqué dans le processus électrochimique décrit par l'équation 1.L’intensité du courant
191

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
augmente avec le temps d’électrodéposition. Le nombre de sites électroactifs de Nickel est
estimé en intégrant le pic anodique à une vitesse de scan faible égale à 10 mV/S. La charge
anodique (Qa) augmente rapidement jusqu’à atteindre 60 secondes, puis tend à se stabiliser
pour des temps d’électrodéposition élevés (Figure II.4.7 B, courbe a).L’augmentation de
l’intensité du courant enregistrée entre 10 et 60 S d’électrodéposition est due à une
augmentation des sites de Ni durant le processus électrochimique. Quand le temps
d’électrodéposition dépasse 60 S, le film HDL de NiAl devient plus épais et moins homogène
Comme le montre les photos MEB (figure II.4.4). Le transport de charge et des ions
hydroxyles assurant l’électroneutralité est gêné par l’épaisseur du film formé, limitant ainsi le
nombre de sites électroactifs de nickel. Cette observation peut être reliée aux changements
morphologiques observés sur les photos MEB, et montre l’intérêt de préparation de couches
minces homogènes de matériau HDL NiAl par électrodéposition à la surface de l'électrode
par rapport à des couches plus épaisses obtenues à des temps plus long ou aux dépôts par
sédimentation de matériau NiAlNO préparé par la méthode de coprécipitation. 3
Quand le Glyphosate est ajouté à la solution électrolytique, une augmentation de
l’intensité du courant anodique est observée sur les courbes de la voltamétrie cyclique de
l’électrode NiAl, et l’intensité du pic dépend de la concentration initiale de Glyphosate
(figure II.4.8).Nous avons vérifié que l’oxydation directe de Glyp n’as pas lieu dans cette
gamme de potentiel sur une électrode de platine nue (figure II.4.8 B).
En se basant sur des résultats antérieurs relatifs à l’électroxidation des amines
aliphatiques et aromatiques sur une électrode de platine modifiée par électrodéposition du
matériau HDL NiAl (Carpani et Tonelli, 2006; Scavetta et al., 2006), l’augmentation de
l’intensité du courant anodique relatif aux sites de Ni (II) est due à une oxydation électro
catalytique de Glyphosate, suivant les réactions ci-dessous :
Ni(II)Al-HDL + OH- Ni(III)Al(OH)-HDL + é (1)
Hebred + Ni(III)Al(OH)-HDL Hebox + Ni(II)Al-HDL (2)
Ni(II)Al-HDL + OH- Ni(III)Al(OH)-HDL + é (1)
Hebred + Ni(III)Al(OH)-HDL Hebox + Ni(II)Al-HDL (2)
La réaction 2 est considérée comme étant l’étape limitante permettant la régénération du
Ni(II) durant l’oxydation du composé aminé.
192
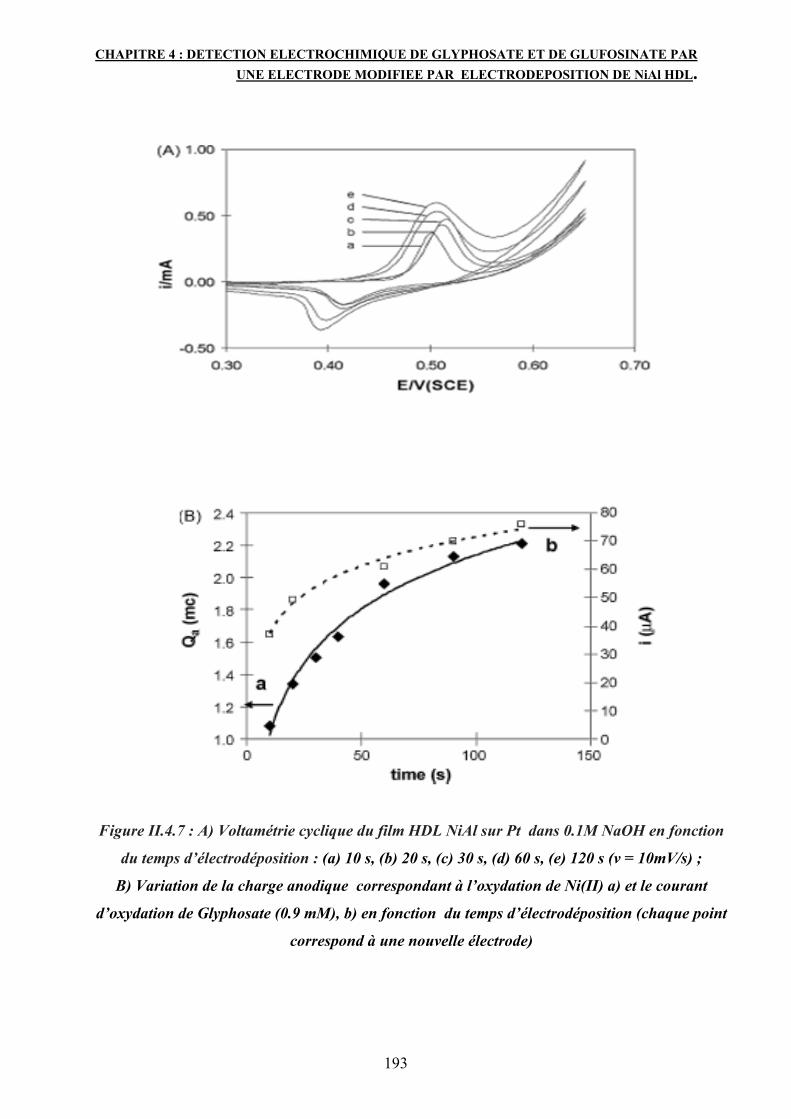
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Figure II.4.7 : A) Voltamétrie cyclique du film HDL NiAl sur Pt dans 0.1M NaOH en fonction
du temps d’électrodéposition : (a) 10 s, (b) 20 s, (c) 30 s, (d) 60 s, (e) 120 s (v = 10mV/s) ;
B) Variation de la charge anodique correspondant à l’oxydation de Ni(II) a) et le courant
d’oxydation de Glyphosate (0.9 mM), b) en fonction du temps d’électrodéposition (chaque point
correspond à une nouvelle électrode)
193
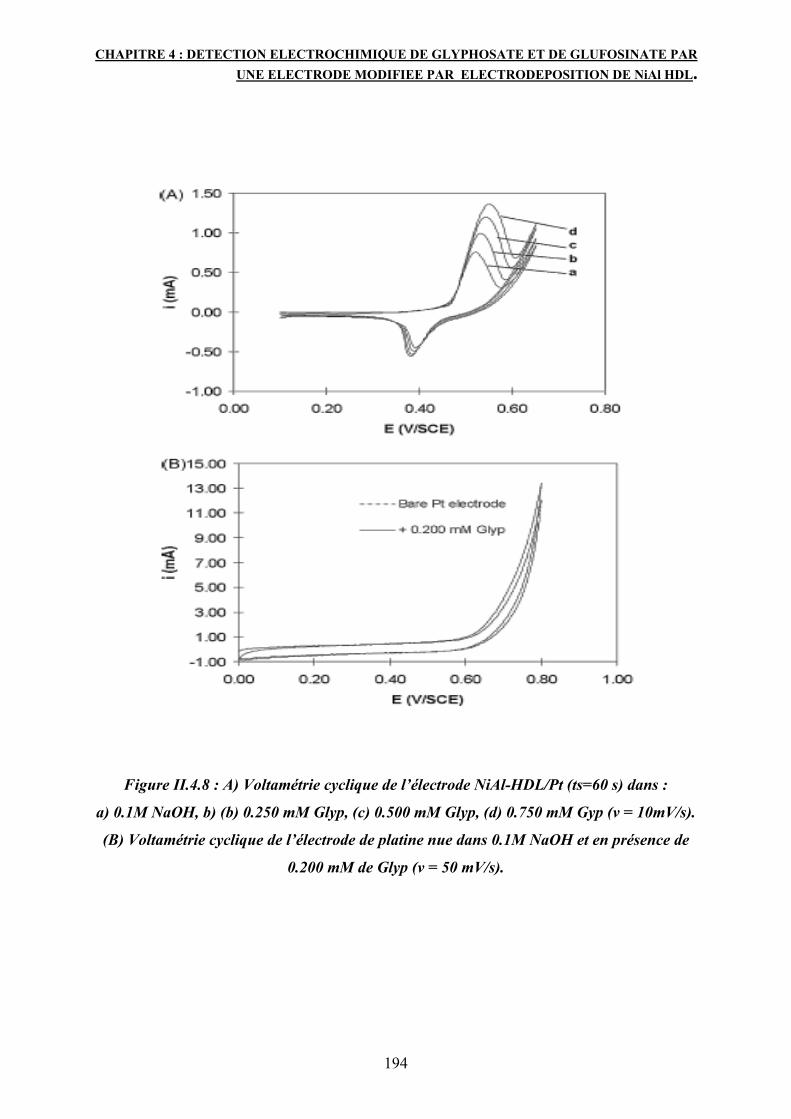
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Figure II.4.8 : A) Voltamétrie cyclique de l’électrode NiAl-HDL/Pt (ts=60 s) dans :
a) 0.1M NaOH, b) (b) 0.250 mM Glyp, (c) 0.500 mM Glyp, (d) 0.750 mM Gyp (v = 10mV/s).
(B) Voltamétrie cyclique de l’électrode de platine nue dans 0.1M NaOH et en présence de
0.200 mM de Glyp (v = 50 mV/s).
194

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
La détermination de Glyphosate et de Glufosinate est réalisée sous des conditions
hydrodynamiques grâce à une électrode à disque rotatif (500 tr/mn) opérant à un potentiel de
E = 0.49 V avec des additions successives des herbicides à la solution électrolytique. app
Le capteur présente une réponse stable et rapide avec un temps moyen de réponse égale
à 20 s (figure II.4.9). Les courbes de calibrations sont obtenues en traçant le courant
d’équilibre mesurée après soustraction de sa ligne de base relative en fonction de la
concentration du substrat. Les électrodes de platine préparées à cinq temps différents
d’électrodéposition (ts = 10, 20, 60, 90, 120 s) sont testées dans ce travail. Comme il a été
montré sur la figure II.4.7 B (courbe b), le courant correspondant à 0.9 mM de Glyphosate
suit la même tendance que Qa en fonction du temps de l’électrodéposition. Ceci confirme le
rôle des sites électro actifs de nickel dans l’électroxidation de Glyphosate. Les électrodes de
platine modifiées par le matériau HDL NiAl électrodéposé pendant 60 s sont choisis pour les
expériences de reproductibilité, puisque la configuration de ces films est la plus reproductible
et stable avec le temps (Scavetta et al., 2006).
Trois électrodes sont préparées dans les conditions optimisées précédentes et la
reproductibilité des capteurs est vérifiée. La déviation standard relative déterminée pour une
électrode donnée (RSD) est égale à 7.1% pour neuf additions de 0.0125 mM de Glyphosate.
Ces biocapteurs présentent une progression linéaire entre 0.01 et 0.9 Mm et une sensibilité de
287± 12 mA/ M cm² (tableau II.4.1). La limite de détection (LoD) déterminée avec un
rapport signal/ bruit égale à 3 est de 1µM.
La même procédure est appliquée pour la détection de Glufosinate. Dans ce cas, la
sensibilité diminue pour atteindre 178 mA/ M cm² à travers une évolution linéaire entre 0.01
et 0.3 Mm et une limite de détection égale à 5µM. D’un point de vue structural, le Glyphosate
et le Glufosinate sont deux herbicides similaires contenant un groupement amine primaire ou
secondaire (Voir le schéma structural des deux herbicides dans le chapitre précédent). Leur
détection électrochimique est basée sur l’oxydation de l’amine présent dans leur structure
respective (Carpani et Tonelli, 2006 ;Sato et al., 2001).
Cependant, l’adsorption des deux herbicides sur la surface du matériau HDL NiAl est
assez différente, ceci peut modifier l’efficacité électrocatalytique des sites actifs de Ni.
Contrairement à nos résultats, un effet opposé avec une haute sensibilité pour le Glufosinate
195
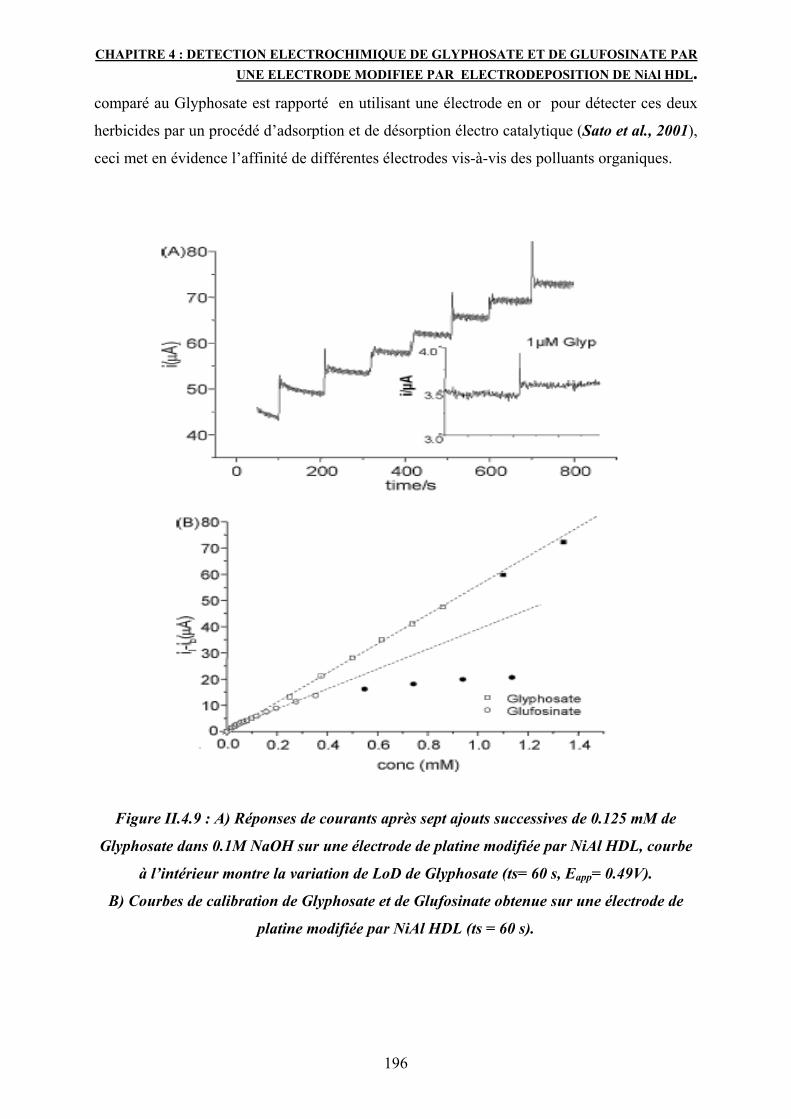
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
comparé au Glyphosate est rapporté en utilisant une électrode en or pour détecter ces deux
herbicides par un procédé d’adsorption et de désorption électro catalytique (Sato et al., 2001),
ceci met en évidence l’affinité de différentes électrodes vis-à-vis des polluants organiques.
Figure II.4.9 : A) Réponses de courants après sept ajouts successives de 0.125 mM de
Glyphosate dans 0.1M NaOH sur une électrode de platine modifiée par NiAl HDL, courbe
à l’intérieur montre la variation de LoD de Glyphosate (ts= 60 s, Eapp= 0.49V).
B) Courbes de calibration de Glyphosate et de Glufosinate obtenue sur une électrode de
platine modifiée par NiAl HDL (ts = 60 s).
196
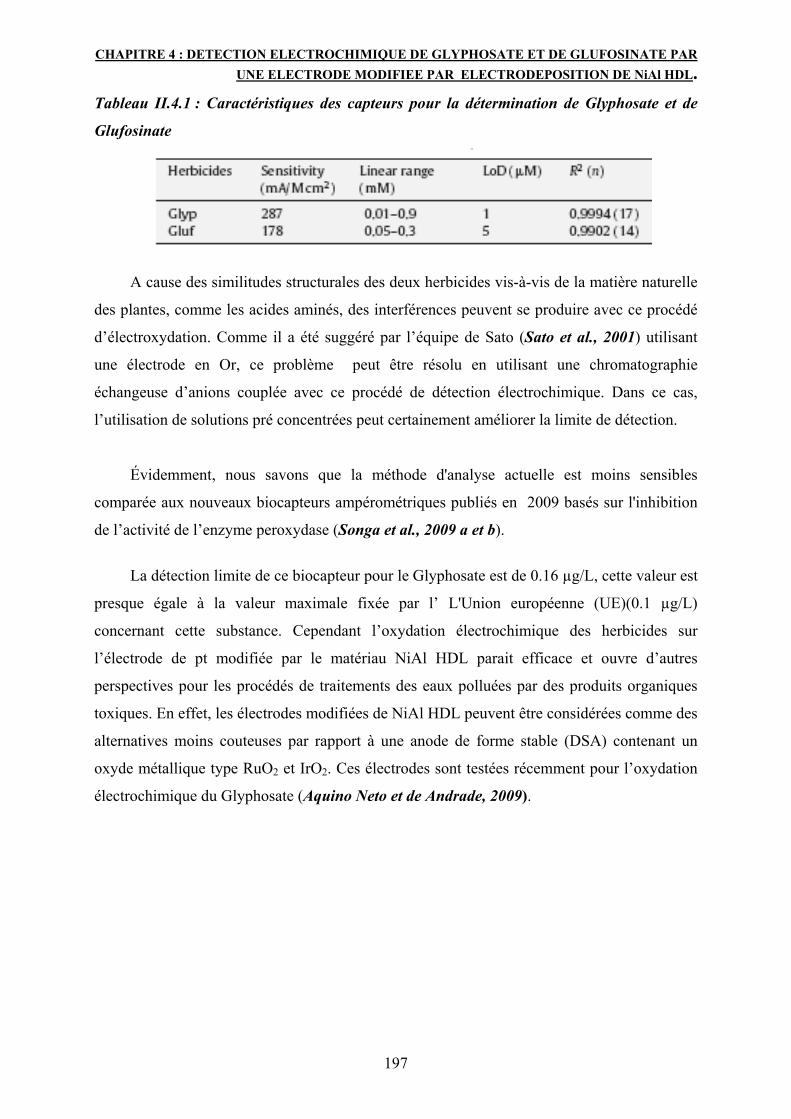
CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Tableau II.4.1 : Caractéristiques des capteurs pour la détermination de Glyphosate et de
Glufosinate
A cause des similitudes structurales des deux herbicides vis-à-vis de la matière naturelle
des plantes, comme les acides aminés, des interférences peuvent se produire avec ce procédé
d’électroxydation. Comme il a été suggéré par l’équipe de Sato (Sato et al., 2001) utilisant
une électrode en Or, ce problème peut être résolu en utilisant une chromatographie
échangeuse d’anions couplée avec ce procédé de détection électrochimique. Dans ce cas,
l’utilisation de solutions pré concentrées peut certainement améliorer la limite de détection.
Évidemment, nous savons que la méthode d'analyse actuelle est moins sensibles
comparée aux nouveaux biocapteurs ampérométriques publiés en 2009 basés sur l'inhibition
de l’activité de l’enzyme peroxydase (Songa et al., 2009 a et b).
La détection limite de ce biocapteur pour le Glyphosate est de 0.16 µg/L, cette valeur est
presque égale à la valeur maximale fixée par l’ L'Union européenne (UE)(0.1 µg/L)
concernant cette substance. Cependant l’oxydation électrochimique des herbicides sur
l’électrode de pt modifiée par le matériau NiAl HDL parait efficace et ouvre d’autres
perspectives pour les procédés de traitements des eaux polluées par des produits organiques
toxiques. En effet, les électrodes modifiées de NiAl HDL peuvent être considérées comme des
alternatives moins couteuses par rapport à une anode de forme stable (DSA) contenant un
oxyde métallique type RuO et IrO2 2. Ces électrodes sont testées récemment pour l’oxydation
électrochimique du Glyphosate (Aquino Neto et de Andrade, 2009).
197

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
3. Conclusion Dans cette étude, des électrodes modifiées par le matériau NiAlHDL sont préparées par
électrodéposition et leur efficacité pour la détection électro catalytique de deux herbicides le
Glyphosate et le Glufosinate est testée. Leur détection électrochimique est basée sur
l’oxydation de groupe amine présent dans leurs structures respectives par les sites actifs de
Ni(III).La morphologie ainsi que l’efficacité électrocatalytique des films d’HDL obtenus sont
fortement dépendantes du temps d’électrodéposition. Dans les conditions de pH de l’analyse
électrochimique (pH =13), l’adsorption des herbicides est très faible : l’intercalation de Gly3-
est empêchée grâce à la compétition avec les carbonates tandis que la molécule Glu2- n’est
pas adsorbée. Une étude plus détaillée sur l’adsorption des deux herbicides à différents pH sur
le matériau NiAlNO3 est en cours dans notre laboratoire. Puisque la modification de la surface
des électrodes avec ces films électroactifs du matériau NiAl HDL est très simple et rapide,
leur utilisation pour la détermination électrochimique des deux herbicides Glyphosate et
Glufosinate offre une alternative aux autres méthodes d’analyses et procédés de traitements
des eaux polluées par des produits organiques toxiques.
198

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
Références bibliographiques
• Adachi-Pagano M., Forano C. and Besse J.-P. (2003), J. Mater. Chem., 13 1988–
1993.
• Aquino Neto S. and De Andrade A.R. (2009), Electrochim. Acta 54, 2039–2045.
• Albers C.N., Banta G.T., Hansen P.E., J.O.S. (2009), Environ. Pollut. 157, 2865–
2870.
• Ballarin B., Seeber R., Tonelli D. and Zanardi C. (2000), Electroanalysis 12, 434–
441.
• Ballarin B., Berrettoni M., Carpani I., Scavetta E. and Tonelli D. (2005), Anal.
Chim. Acta 538, 219–224.
• Carpani I and Tonelli D. (2006), Electroanalysis 18, 2421–2425.
• Chamignon C., Haroune N., Forano C., Delort A.-M., Besse-Hoggan P. and
Combourieu B. (2008), Eur. J. Soil Sci. 59, 572–583.
• Damonte M., Torres Sanchez R.M., Dos Santos Afonso M. (2007), Appl. Clay Sci.
36, 86–94.
• Duan X. and Evans D.G., Structure and Bonding: Layered Double Hydroxides,
Springer, Berlin/Heidelberg, 2006.
• Indira L. and Kamath P.V. (1994), J. Mater. Chem. 4, 1487–1490.
• Jonsson C.M., Persson P., Sjoeberg S. and Loring J.S. (2008), Environ. Sci.
Technol. 42, 2464–2469.
• Khenifi, A., Derriche Z., Mousty C., Prévot V. and Forano C. (2010), Applied
Clay Science 47, 362–371.
• Laitinen P., Siimes K., Ramo S., Jauhiainen L., Eronen L., Oinonen S.,
Hartikainen H. (2008), J. Environ. Qual. 37, 830–838.
• Li F.,. Wang Y, Yang Q., Evans D.G., Forano C. and Duan X. (2005), J. Hazard.
Mater. B125, 89–95.
• Mousty C. (2004), Appl. Clay Sci. 27, 159–177.
• Pessagno R.C., Torres Sanchez R.M. and Dos Santos Afonso M. (2008), Environ.
Pollut. 153, 53–59.
• Prevot V., Forano C. and Besse J.-P. (2005), Chem. Mater. 17, 6695–6701.
• Rives V. (2001), Layered Doubles Hydroxides: Present and Future, Nova Science
Publisher,
199

CHAPITRE 4 : DETECTION ELECTROCHIMIQUE DE GLYPHOSATE ET DE GLUFOSINATE PAR UNE ELECTRODE MODIFIEE PAR ELECTRODEPOSITION DE NiAl HDL.
New York, USA,
• Ruiz Simoes F., Capparelli Mattoso L.H. and Vaz C.M.P. (2006), Sensor Lett., 4,
319–324.
• Sato K., Jin J.-Y., Takeuchi T., Miwa T., Suenami K., Takekoshi Y. and Kanno
S. (2001), J. Chromatogr. A 919, 313–320.
• Scavetta E., Ballarin B., Berrettoni M., Carpani I., Giorgetti M. And Tonelli D.
(2006), Electrochim. Acta 51, 2129–2134.
• Scavetta E., Mignani A., Prandstraller D. and Tonelli D. (2007), Chem. Mater. 19,
4523–4529.
• Scavetta E., Ballarin B., Gazzano M. And Tonelli D. (2009), Electrochim. Acta 54,
1027–1033.
• Songa E.A., Somerset V., Waryo T., Baker P.G.L. and Iwuoha E.I. (2009a), Pure
Appl. Chem. 81, 123–139.
• Songa E.A., Waryo T., Jahed N., Baker P.G.L., Kgarebe B.V. and Iwuoha E.I.
(2009b), Electroanalysis 21, 671–674.
• Stalikas C.D. and Konidari C.N. (2001), J. Chromatogr. A 907, 1–19.
• Yarger M.S., Steinmiller E.M.P. and Choi K.-S. (2008), Inorg. Chem. 47, 5859–
5865.
200

CONCLUSION GENERALE
201

CONCLUSION GENERALE
CONCLUSION GENERALE
Cette présente étude a pour objectif principal le mode de préparation et d’application des
argiles cationiques et anioniques dans l'adsorption de certains micropolluants organiques
susceptibles d’être présents dans les eaux.
La première partie de ce travail porte sur l’adsorption de deux polluants organiques: le
2,4DCP et un colorant industriel textile le 4GL par une bentonite Algérienne purifiée en
utilisant deux méthodes : la première méthode par l’argile organophile (bentonite échangée à
100% CEC par le CTAB), et la seconde méthode le polluant organique est éliminé en même
temps que le CTAB par l’argile sodée.
En effet, afin de réduire les coûts et simplifier le processus de traitement des eaux usées
par la bentonite organophile, une seule étape incluant à la fois la synthèse de la bentonite
organophile et l’élimination des polluants organiques a été proposée dans ce travail. La
méthode « one step » propose le mélange entre l'adsorbat et le tensio-actif cationique en
solution, puis l’ajout de la bentonite sodée. Ainsi, les polluants organiques sont supprimés de
l’eau en même temps que la formation de l’argile organophile. L’étude de l’influence de la
concentration du CTAB en solution sur la fixation des polluants organiques montre que la
meilleure fixation est obtenue pour une concentration de tensioactif correspondante à 100%
de la CEC de l’argile de base. En effet, au-delà de cette concentration, la quantité fixée
diminue rapidement. Ceci est probablement du à la formation de micelles de CTAB, le
polluant se trouve solubilisé dans ces micelles et par conséquence sa fixation sur le matériau
se trouve diminuée.
Les résultats de cette étude ont montré que la méthode one step (CTAB en solution)
améliore la fixation des deux polluants sur l’argile. En effet, le polluant organique et le CTAB
en solution sont éliminés rapidement par l’argile en solution. Les temps d’équilibre
correspondant à l’adsorption du polluant 2,4 DCP par la méthode one step et two steps sont
respectivement 30 mn et 10 mn pour des quantités fixées de 0.857 et 0.528 mmol/g.
L’adsorption lente du 2,4DCP par l’argile organophile peut être expliquée par le fait que le
tensioactif cationique est initialement échangé dans la bentonite. Alors que l’adsorption du
polluant organique est rapide quand le CTAB est présent en solution. Dans ce cas,
l’élimination du polluant est effectuée par adsorption simultanée du tensioactif cationique et
202

CONCLUSION GENERALE
du 2,4DCP. Contrairement au polluant 2,4DCP, les cinétiques d’adsorption du colorant 4GL
sur l’argile par la méthode one step et two steps sont presque identiques. En effet, l’équilibre
est atteint au bout de 20mn avec une quantité fixée de 0.05 g de colorant /g d’argile. Les
modélisations cinétiques ont montré que l’adsorption de 2,4DCP et de 4GL par les deux
méthodes suit une cinétique du pseudo second ordre et que la diffusion intra particulaire n’est
pas la seule étape limitant l’adsorption.
Les isothermes d’adsorption des deux polluants par la méthode one step et two steps
sont tout à fait différentes. l’isotherme d’adsorption de 2,4DCP sur l’argile sodée en présence
de CTAB (méthode one step) est de type multicouches, typique de celle obtenue par des
interactions modérées entre adsorbat et adsorbant, suivie par un effet coopératif des molécules
adsorbées. Cette courbe est tout à fait différente de l’isotherme d’adsorption du même
polluant sur l’argile organophile. En effet, dans ce cas on trouve une isotherme de type
Langmuir. Les résultats expérimentaux suivent le modèle de Langmuir avec des capacités
d’adsorption égale à 3.546 et 2.293 mmol/g pour la méthode one step et two steps
respectivement. Il en est de même pour le colorant 4GL, l’isotherme d’adsorption par la
méthode one step et de type H. Tandis que pour la méthode two steps, l’isotherme est de type
L. Les capacités d’adsorption du colorant 4GL déterminées par le modèle de Langmuir
donnent des valeurs de 0.6 et 0.5 g/g pour la méthode one step et two steps respctivement.
Des interactions de type adsorbat- adsorbant et de type adsorbat- adsorbat sont responsables
de ce comportement.
En conclusion, on peut dire que la méthode one step est une méthode très efficace
(cinétique très rapide, capacité d’adsorption élevée, facilité de réalisation) pour l’élimination
des polluants organiques des eaux usées, ce qui ouvre des perspectives intéréssantes pour
l’environnement.
Une autre variété d’argiles dites basiques a également retenu notre attention. Ces
matériaux sont rares dans la nature mais ils peuvent être facilement synthétisés à l'échelle du
laboratoire. Ces matériaux possèdent des capacités d'échange anioniques (2-5 mmol/g) plus
importantes que les capacités d’échanges cationiques des minéraux argileux. A Cet effet,
l’élimination de deux herbicides anioniques :Le Glyphosate et le Glufosinate des solutions
aqueuses par le matériau HDL NiAlNO3 est étudié. Le choix de ce matériau n’est pas
aléatoire, la structure de cette matrice est très intéressante car elle combine de bonnes
203

CONCLUSION GENERALE
2+ 3+propriétés électro actives basé sur le processus redox Ni / Ni pour les procédés
électrochimiques avec des propriétés structurales et texturales pour l'adsorption des molécules
chargées négativement.
L’adsorption des deux herbicides : Le Glyphosate et le Glufosinate indiquent deux
mécanismes essentiels : une adsorption externe surfacique et une intercalation interne par
échange anionique. Ces résultats sont confirmés par les analyses structurales et texturales des
matériaux après adsorption des deux polluants organiques à différentes concentrations (DRX,
IR, MEB).
La cinétique d’adsorption des deux herbicides sur le matériau HDL suit un modèle de
pseudo second ordre. La diffusion intra particulaire n’est pas la seule étape limitant
l’adsorption des deux herbicides, en effet l’adsorption surfacique et la diffusion opèrent de
façon simultanée lors des interactions de Glyphosate et de Glufosinate avec le matériau HDL.
Les isothermes d’adsorption sont de type H et L pour le Glyphosate et le Glufosinate
respectivement et suivent le modèle de Langmuir. De même, il est bon de signaler que la
solubilité dans l’eau de ces molécules organiques ainsi que leurs structures chimiques sont
apparemment les principaux facteurs qui gouvernent la rétention de ces micropolluants. Les
résultats de ce travail indiquent le rôle important du matériau HDL NiAlNO3 comme
adsorbant potentiel pour l’élimination des polluants organo-phosphates et organo-
phosphonates..
Les produits organophosphate et organophosphonate (OP) est une famille de pesticides
très utilisée en agriculture. Les méthodes d’analyse utilisées pour la détection de ces
molécules organiques sont en chromatographie en phase liquide, la chromatographie en
phase gazeuse, l’électrophorèse capillaire etc… Les techniques de chromatographie en phase
liquide utilisant des détecteurs UV ou fluorimétrique, sont limitées à l'analyse des pesticides
portant des groupes chromophores ce qui n'est pas le cas avec le Glyphosate et le
Glufosinate. Les capteurs électrochimiques représentent donc une bonne alternative innovante
en termes de détection in situe, de sensibilité, de réponse rapide, et de taille réduite pour la
détermination de ces pesticides.
204

CONCLUSION GENERALE
Dans la dernière partie de cette étude, des électrodes modifiées par le matériau HDL
NiAl sont préparées par électrodéposition et leur efficacité pour la détection électro
catalytique de deux herbicides le Glyphosate et le Glufosinate est testée. Leur détection
électrochimique est basée sur l’oxydation de groupe amine présent dans leurs structures
respectives par les sites actifs de Ni(III).
La morphologie ainsi que l’efficacité électrocatalytique des films d’HDL obtenus sont
fortement dépendantes du temps d’électrodéposition. Sous les conditions de pH de l’analyse
électrochimique (pH =13), l’adsorption des herbicides est très faible : l’intercalation de Gly3-
est empêchée grâce à la compétition avec les carbonates tandis que la molécule Glu2- n’est
pas adsorbée.
205

LISTE DES FIGURES
LISTE DES FIGURES
206

LISTE DES FIGURES
LISTE DES FIGURES
Figure I.1.1 : a) Unité tétraédrique à coeur de silicium; b) Schéma d'une couche de tétraèdre
[Si4 O10 (OH) ]6-. 2
Figure I.1.2 : a) Unité octaédrique b) Structure en couche à base d'octaèdre de Brucite
Mg(OH) . ou de Gibbsite Al(OH)2 3
Figure I.1.3 : Assemblage d’une couche octaédrique et d’une couche tétraédrique pour une
argile TO (1:1).
Figure I.1-4 : Représentation schématique de quelques groupes de minéraux argileux TO
(1:1) et TOT (2:1).
Figure I.1-5. Représentation schématique de la structure feuilletée des smectites et des
illite/pyrophyllite.
Figure I.1.6 : Complexe de sphère externe. Le cation reste totalement hydraté. C'est le cas
des cations possédant une énergie d'hydratation élevée, comme le sodium. (Sposito, 1989).
Figure I.1.7 : Valeurs de surface spécifique des particules argileuses et schéma représentant
la surface interne et la surface externe des particules d’argile (Eslinger et Peaver, 1988).
Figure I.1.8: Représentation schématique de quelques exemples de molécules de
tensioactifs.
(a) Alkylammonium (b) Alkyltriméthylammonium (c) Dialkyldiméthylammonium , (d)
Alkylbis(2-hydroxyéthyl)méthylammonium, (e) Alkylbenzyldiméthylammonium, ( )
représente la chaîne hydrocarbonée aliphatique dont la longueur est variable : [(CH2)n-CH3]
pour (a) et [CH2-(CH2)n-CH3] pour ((b)-(e))
Figure I.2.1 : Représentation schématique de la structure de matériaux de type hydrotalcite
d'après A. de Roy et coll. (de Roy et al., 1992).
Figure I.2.2: Les métaux dans les couches d'HDL d'après Bravo Suarez(Bravo-Suarez et
al.,2004) (◊:reportés dans la littérature, : Δ reportés dans des brevets, : différence de rayon
ionique métal /Mg2+>50%, Ο: différence de rayon ionique métal / Mg2+<50%).
Figure I.2.3 : Courbes de titration de solutions 2:1 MgCl :FeCl et MgCl2 3 2:AlCl3, d’après
(Boclair et Braterman, 1999).
Figure I.2.4 : Clichés MEB de phases Mg–Al (a) et Cu–Cr (b) synthétisées par
coprécipitation.
Figure I.2.5: Clichés MEB de phases Mg-Al (a) et Zn-Al (b) synthétisées par la méthode
urée (Thèse de S. Vial 2005).
207

LISTE DES FIGURES
Figure I.2.6 :Cliché MEB d’une phase Mg-Al obtenue par décomposition enzymatique de
l’urée (Thèse de S. Vial 2005).
Figure I.2.7 : Morphologie d’une phase [ZnAl–DDS] délaminée dans l’éthanol (à gauche) et
le butanol (à droite) (MET) (Adachi-Pagano et al., 2000).
Figure I.3.1: Principe de fonctionnement des biocapteurs.
Figure I. 3.2 : Détection électrochimique.
Figure I.3.3 : Capteurs ampérométriques de première génération , a) détection de la
consommation d'oxygène, b) détection de la production en eau oxygénée.
Figure I.3.4 : Biocapteur ampérométrique de deuxième génération. M est le médiateur sous
forme oxydée (ox)ou réduite(réd).
Figure I.3.5 : Exemples de méthodes d'immobilisation d'enzyme.
Figure II.1.1 : Schéma réactionnel de la formation du matériau NiAlNO3 par voie
électrochimique.
Figure II.2.1: Isotherme d’adsorption du bleu de méthylène sur argile sodée.
Figure II.2.2 : Variation de la conductivité électrique en fonction du volume de (MgSO4)
pour l’argile sodée.
Figure II.2.3 : DRX de la bentonite brute et de la bentonite sodé.
Figure II.2.4 : DRX de la bentonite organophile (100%CEC).
Figure II.2.5 : Spectres IR de la bentonite sodée et organophile.
Figure II.2.6 : Pourcentage adsorbé de 2,4DCP en fonction de la concentration de CTAB.
Figure II.2.7: Pourcentage adsorbé de 4GL en fonction de la concentration de CTAB.
Figure II.2.8 : Pourcentage de 2,4DCP adsorbé en fonction de la concentration du CTAB
échangée dans l’argile exprimée en %CEC.
Figure II.2.9 : Variation de la quantité du colorant adsorbée avec la concentration du CTAB
échangée dans l’argile exprimée en %CEC.
Figure II.2.10: Cinétique d’adsorption de 2,4DCP sur bentonite sodée en présence de CTAB
(one step) et bentonite organophile (two steps), Ci= 5mmol/L, V=1L, m= 5g, pH=5
Figure II.2.11 : Cinétique d’adsorption du colorant 4GL sur la bentonite organophile.
Figure II.2.12 : Cinétique du pseudo premier ordre (en haut), cinétique du pseudo second
ordre(en bas) de l’adsorption de 2,4DCP sur bentonite organophile et sodée en présence de
CTAB.
Figure II.2.13: Cinétique du pseudo premier ordre (en haut), cinétique du pseudo second
ordre(en bas) de l’adsorption de 4GL sur bentonite organophile et sodée en présence de
CTAB.
208

LISTE DES FIGURES
Figure II.2.14 : Tracé du modèle de la diffusion intra particulaire de l’adsorption de 2,4DCP
par la méthode one step et two steps.
Figure : II.2.15: Tracé du modèle de la diffusion intra particulaire de l’adsorption de 4GL par
la méthode one step et two steps.
Figure II.2.16 : Isothermes d’adsorption de 2,4DCP sur la bentonite organophile (a) et par la
bentonite sodée en présence de CTAB (b) décrites par les modèles de Freundlich, Langmuir et
Redlich Peterson.
Figure II.2.17 : Isothermes d’adsorption de 4GL sur la bentonite sodée en présence de
CTAB(a) et par la bentonite organophile (b) décrites par les modèles de Freundlich ,
Langmuir et Redlich Peterson.
Figure II.2.18 : Diffractogrammes DRX des matériaux après adsorption du colorant 4GL par
les deux méthodes.
Figure II.2.19 : Diffractogrammes DRX des matériaux après adsorption du polluant 2,4DCP
par les deux méthodes.
Figure II.2.20 : Représentation schématique de (de la gauche vers la droite) : 1) argile
organophile, 2) Adsorption de polluant organique sur argile organophile, 3) adsorption de
polluant organique et de CTAB sur argile sodée, 4) molécule de CTAB.
Figure II.2.21 : Spectres IR de l’argile après adsorption de 2,4 DCP (one step et two steps).
Figure II.3.1 : Structures chimiques du Glyphosate et de Glufosinate.
Figure II.3.2 : Diffractogramme DRX du matériau Ni2AlNO . 3
Figure II.3.3 : Spectre IR du matériau Ni2AlNO . 3
Figure II.3.4: Analyse thermique ATG couplée avec l’analyse thermique différentielle pour
le matériau NiAlNO . 3
Figure II.3.5: Schéma de décomposition thermique de la phase NiAlNO . 3
Figure II.3.6 : Variation de DRX du matériau NiAlNO3 avec la température de calcination.
Figure II.3.7 : Cinétique d’adsorption de Gly et Glu sur Ni AlNO . 2 3
Figure II.3.8: Modèles pseudo premier ordre (petite taille) et modèle pseudo second ordre de
l’adsorption de Glyphosate et Glufosinate sur le matériau NiAlNO . 3
Figure II.3.9 : Modèle de diffusion intra particulaire pour l’adsorption de Glyphosate et
Glufosinate sur le matériau NiAlNO . 3
Figure II.3.10 : Isothermes d’adsorption de Glyphosate et de Glufosinate sur le matériau
NiAlNO . 3
Figure II.3.11 : Modèle de Langmuir (gauche) et de Freundlich pour l’adsorption de
Glyphosate et de Glufosinate sur le matériau NiAlNO . 3
209

LISTE DES FIGURES
Figure II.3.12: DRX du matériau NiAlNO3 après adsorption de Glyphosate et de Glufosinate
à différentes concentrations initiales : a) 0 mg/L, b) 10 mg/L, c) 30 mg/L, d) 50 mg/L, 100
mg/L, e) 150 mg/L, f) 200 mg/L, g) 250 mg/L, h) 300 mg/L.
Figure II.3.13: DRX des matériaux NiAlGly et NiAlGlu préparés par coprécipitation et par
échange anionique.
Figure II.3.14: Représentation schématique des phase NiAlGly et NiAlGlu coprécipitées.
Figure II.3.15 : Photos MEB du matériau NiAlNO3 avant et après adsorption de Glyphosate
et de Glufosinate (Ci = 300mg/L).
Figure II.3.16: Spectres IR du matériau NiAlNO3 après adsorption de Glyphosate ( gauche)
et de Glufosinate à différentes concentrations.
Figure II.4.1: Schéma réactionnel de la formation du matériau NiAlNO3 par voie
électrochimique.
Figure II.4.2: DRX de : a)matériau préparé par coprécipitation, b) NiAlNO3 obtenu après 10
électrodéposition à 200 secondes, c)film de NiAlNO3 éléctrodéposé sur Pt à 60s, d) à 120s, e)
à 200s (*Pt,∇ : impuretés du KNO ). 3
Figure II.4.3: Spectre A) Raman, B) IR du matériau NiAlNO3 préparé par : a)
coprécipitation b) électrodéposé sur plaque de pt.
Figure II.4.4 :Images MEB des films NiAlNO3 électrodéposé à 60 secondes (a,b,c), à 120
secondes(d,e,f), à 200 secondes (g,h,i), du matériau coprécipité (j), du matériau coprécipité et
adsorbé par le Glyphosate (k) et adsorbé par le Glufosinate (l).
Figure II.4.5 : A : Cinétique d’adsorption de Gly et Glu sur le matériau NiAlNO3 (Ci= 200
mg/L, pH=13, adsorbat / adsorbant= 1mg/ml) B : Isotherme d’adsorption de Gly et Glu sur le
même matériau.
Figure II.4.6: DRX du matériau NiAlNO3 après adsorption de Glyphosate A) pH7 , B)
pH13 à différentes concentrations initiales : a) 0 mg/L, b) 10 mg/L, c) 30 mg/L, d) 50 mg/L,
100 mg/L, e) 150 mg/L, f) 200 mg/L, g) 250 mg/L, h) 300 mg/L.
Figure II.4.7 : A) Voltamétrie cyclique du film HDL NiAl sur Pt dans 0.1M NaOH en fonction
du temps d’électrodéposition : (a) 10 s, (b) 20 s, (c) 30 s, (d) 60 s, (e) 120 s (v = 10mV/s) ;
B) Variation de la charge anodique correspondant à l’oxydation de Ni(II) a) et le courant
d’oxydation de Glyphosate (0.9 mM), b) en fonction du temps d’électrodéposition (chaque
point correspond à une nouvelle électrode).
Figure II.4.8 : A) Voltamétrie cyclique de l’électrode NiAl-HDL/Pt (ts=60 s) dans : a) 0.1M
NaOH, b) (b) 0.250 mM Glyp, (c) 0.500 mM Glyp, (d) 0.750 mM Gyp (v = 10mV/s).
210

LISTE DES FIGURES
(B)Voltamétrie cyclique de l’électrode de platine nue dans 0.1M NaOH et en présence de
0.200 mM de Glyp (v = 50 mV/s).
Figure II.4.9 : A) Réponses de courants après sept ajouts successives de 0.125 mM de
Glyphosate dans 0.1M NaOH sur une électrode de platine modifiée par NiAl HDL, courbe à
l’intérieur montre la variation de LoD de Glyphosate (ts= 60 s, Eapp= 0.49V).
B) Courbes de calibration de Glyphosate et de Glufosinate obtenue sur une électrode de
platine modifiée par NiAl HDL (ts = 60 s).
211

LISTE DES TABLEAUX
LISTE DES TABLEAUX
212

LISTE DES TABLEAUX
LISTE DES TABLEAUX
Tableau I.1.1 : Capacité d’échange cationique des principales familles argileuses (Caillère et
al., 1982).
Tableau I.2.1 : Rayons ioniques de quelques cations déjà utilisés pour l'élaboration de
matériaux type Hydrotalcite.
Tableau I.2.2 : Valeurs de x permettant l'obtention de phases HDL pures.
Tableau I.2.3 : Valeur de c' en fonction de l'anion de compensation.
Tableau I.2.4 : pH de synthèse pour quelques compositions.
Tableau I.2.5 : Facteurs influençant l’architecture de type hydrotalcite (Vaccari,1998;
Cavani et al., 1991).
Tableau I.3.1 : Exemples d’enzymes inhibées par différents inhibiteurs (Bilitewski et Turner,
2000).
Tableau II.1.1 : Caractéristiques physiques et chimiques de 2,4DCP.
Tableau II.1.2 : Caractéristiques générales des deux herbicides Glyphosate et Glufosinate.
Tableau II.2.1: Analyse chimique de la bentonite naturelle utilisée (% en poids).
Tableau II.2.2 : Capacité d’échange cationique de l’argile sodée déterminée à partir des deux
méthodes: Bleu de Méthylène et conductimétrique.
Tableau II.2.3 : Variation de l’espacement basal suivant les tests de comportement des
argiles.
Tableau II.2.4 : Distances inter-réticulaires de l’argile sodée et organophile à différentes
concentrations de CTAB.
Tableau II.2.5 : Caractéristiques générales de la bentonite sodée et organophile.
Tableau II.2.6 : Modélisation cinétique de l’adsorption de 2,4DCP sur l’argile organophile
(two steps) et sodée en présence de CTAB (one step).
Tableau II.2.7 : Modélisation cinétique de l’adsorption de 4GL sur l’argile organophile (two
steps) et sodée en présence de CTAB (one step).
Tableau II.2.8 : Constantes des isothermes de Freundlich, Langmuir et Redlich Peterson,
équivalentes à l’adsorption de 2,4DCP par la méthode one step et two steps.
Tableau II.2.9 : Constantes des isothermes de Freundlich, Langmuir et Redlich Peterson, de
l’adsorption du 4GL par la méthode one step et two steps.
213

LISTE DES TABLEAUX
Tableau II.2.10 : Comparaison de la capacité d’adsorption maximale du 2,4DCP par
différents adsorbants.
Tableau II.3.1 : Constantes cinétiques de l’adsorption de Glyphosate et de Glufosinate sur le
matériau NiAlNO . 3
Tableau II.3.2: Constantes de Freundlich, Langmuir et Tempkin pour l’adsorption de
Glyphosate et de Glufosinate sur le matériau NiAlNO . 3
Tableau II.4.1 : Caractéristiques des capteurs pour la détermination de Glyphosate et de
Glufosinate.
214