therapeutic silencing of mir-652 restores heart function and attenuates adverse remodeling in a...
TRANSCRIPT

The FASEB Journal • Research Communication
Therapeutic silencing of miR-652 restores heartfunction and attenuates adverse remodeling in asetting of established pathological hypertrophy
Bianca C. Bernardo,*,1,2 Sally S. Nguyen,*,†,1 Catherine E. Winbanks,* Xiao-Ming Gao,*Esther J. H. Boey,* Yow Keat Tham,* Helen Kiriazis,* Jenny Y. Y. Ooi,*Enzo R. Porrello,‡ Sindhu Igoor,‡ Colleen J. Thomas,† Paul Gregorevic,*Ruby C. Y. Lin,§ Xiao-Jun Du,* and Julie R. McMullen*,2
*Baker IDI Heart and Diabetes Institute, Melbourne, Victoria, Australia; †Department of HumanBiosciences, La Trobe University, Bundoora, Victoria, Australia; ‡School of Biomedical Sciences,University of Queensland, St. Lucia, Queensland, Australia; and §Ramaciotti Centre for Genomicsand School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney,New South Wales, Australia
ABSTRACT Expression of microRNA-652 (miR-652)increases in the diseased heart, decreases in a setting ofcardioprotection, and is inversely correlated with heartfunction. The aim of this study was to assess thetherapeutic potential of inhibiting miR-652 in a mousemodel with established pathological hypertrophy andcardiac dysfunction due to pressure overload. Micewere subjected to a sham operation or transverse aorticconstriction (TAC) for 4 wk to induce hypertrophy andcardiac dysfunction, followed by administration of alocked nucleic acid (LNA)-antimiR-652 (miR-652 inhib-itor) or LNA control. Cardiac function was assessedbefore and 8 wk post-treatment. Expression of miR-652increased in hearts subjected to TAC compared tosham surgery (2.9-fold), and this was suppressed by�95% in LNA-antimiR-652-treated TAC mice. Inhibi-tion of miR-652 improved cardiac function in TACmice (fractional shortening:29�1% at 4 wk post-TACcompared to 35�1% post-treatment) and attenuatedcardiac hypertrophy. Improvement in heart functionwas associated with reduced cardiac fibrosis, less apo-
ptosis and B-type natriuretic peptide gene expression,and preserved angiogenesis. Mechanistically, we identi-fied Jagged1 (a Notch1 ligand) as a novel direct targetof miR-652. In summary, these studies provide thefirst evidence that silencing of miR-652 protects theheart against pathological remodeling and improvesheart function.—Bernardo, B. C., Nguyen, S. S.,Winbanks, C. E., Gao, X.-M., Boey, E. J. H., Tham,Y. K., Kiriazis, H., Ooi, J. Y. Y., Porrello, E. R., Igoor,S., Thomas, C. J., Gregorevic, P., Lin, R. C. Y., Du,X.-J., McMullen, J. R. Therapeutic silencing of miR-652restores heart function and attenuates adverse remod-eling in a setting of established pathological hypertro-phy. FASEB J. 28, 5097–5110 (2014). www.fasebj.org
Key Words: microRNAs � pressure overload � heart failure �LNA-therapeutics
Heart failure occurs when the heart is unable toadequately adapt to a pathological stress, such aschronic pressure overload or myocardial infarction(MI). Factors contributing toward cardiac dysfunctionand the transition to heart failure include hypertrophicgrowth of heart muscle cells (cardiomyocytes), deposi-tion of interstitial fibrosis, significant loss of cardiomy-ocytes due to apoptosis, and inadequate angiogenesis(1, 2). The prevalence of heart failure is increasing dueto an aging population and growing rates of obesity anddiabetes (3–5). Despite advances in therapeutic op-tions, the mortality rate in patients with heart failure
1 These authors contributed equally to this work.2 Correspondence: Baker IDI Heart and Diabetes Institute,
PO Box 6492, Melbourne 3004, VIC, Australia. E-mail: J.R.M.,[email protected]; B.C.B., [email protected]
doi: 10.1096/fj.14-253856This article includes supplemental data. Please visit http://
www.fasebj.org to obtain this information.
Abbreviations: Acta1, �-skeletal actin; ANOVA, analysis ofvariance; BAX, Bcl-2-associated X protein; BCL2, B-cell lym-phoma 2; BNP, B-type natriuretic peptide; ERK1/2, extracel-lular signal-regulated kinase 1/2; FS, fractional shortening;Gapdh, glyceraldehyde-3-phosphate dehydrogenase; HAEC,human aortic endothelial cell; Hprt1, hypoxanthine phos-phoribosyltransferase 1; HR, heart rate; HUVEC, humanumbilical vein endothelial cell; HW, heart weight; IVS, inter-ventricular septum; LNA, locked nucleic acid; LV, left ven-tricular; LVEDD, left ventricular end-diastolic dimension;LVESD, left ventricular end-systolic dimension; LVPW, leftventricular posterior wall; LW, lung weight; MI, myocardialinfarction; miRNA, microRNA; mRNA, messenger RNA;NRVF, neonatal rat ventricular fibroblast; NRVM, neonatalrat ventricular myocyte; PI3K, phosphoinositide 3-kinase;pH3, phosphohistone H3; qPCR, quantitative RT-PCR;Serca2a, sarcoplasmic/endoplasmic reticulum Ca2�-ATPase2a; TAC, transverse aortic constriction; TL, tibial length;WGA, wheat germ agglutinin
50970892-6638/14/0028-5097 © FASEB

remains high (6). Pharmacologic agents currently pre-scribed for heart failure largely focus on targetingmediators of maladaptive hypertrophy, but are oftenaccompanied by side effects, and are employed largelyin a palliative capacity (6, 7). Since the emergence ofmicroRNAs (miRNAs) as important regulators in car-diac pathology, their potential as a new class of thera-peutic targets for heart disease has begun to be ex-plored in preclinical models (8, 9). The translationalimplications of miRNA-based therapies was recentlyhighlighted by the first miRNA-based therapy for thetreatment of hepatitis C virus passing phase 2a clinicaltrials with encouraging outcomes (10).
Endogenous miRNAs are short noncoding RNA mol-ecules, whose primary role has been defined as post-transcriptional regulators of gene expression. MiRNAsbind to target messenger RNAs (mRNAs) usually bypartial or complete base pairing on specific miRNArecognition elements, leading to mRNA degradation,gene silencing, or translational repression (11, 12).Individual miRNAs can regulate the expression ofseveral hundred target mRNAs, which can affect vari-ous biological processes and cellular functions, ormultiple genes in a common network (13). Expressionof miRNAs is altered in settings of cardiac stress (re-viewed in ref. 12), and this has been associated with theregulation of pathophysiological processes of the heart,including apoptosis (14), fibrosis (15, 16), and cardiachypertrophy (17–19). To date, most investigators haveexamined the potential of targeting miRNAs that meettwo criteria: have relatively high expression in the heartunder basal conditions; and have expression that in-creases in a disease setting. A distinguishing feature ofthe current study is that we identified miRNAs usingthree criteria characteristic of a maladaptive response:1) expression decreases in a setting of cardiac protec-tion; 2) expression increases in a disease setting; and3) expression is inversely correlated with heart func-tion. These criteria were applied irrespective of miRNAabundance in the heart under basal settings. We previ-ously identified a number of miRNAs that were up-regulated in the heart in a mouse model of cardiacstress (MI) and down-regulated in a mouse model ofcardiac protection [i.e., physiological cardiac growthdue to increased activity of phosphoinositide 3-kinase(PI3K[p110�]) which protects the heart against ad-verse pathology in settings of stress; ref. 20]. miR-652 isnot one of the top 100 miRNAs expressed in the adultheart (21) but was of interest because its expression wassignificantly decreased in the protected PI3K heart,elevated in the diseased MI heart, and inversely corre-lated with heart function; collectively suggesting thatsilencing of miR-652 could provide benefit. Further-more, its role in the heart was unknown, and verylimited information was available regarding this miRNAin any other tissue or cell type. The aim of the currentstudy was to investigate the significance and therapeuticpotential of silencing miR-652 in a cardiac diseasesetting by chronically inhibiting miR-652 in a mouse
model with established pathological hypertrophy andcardiac dysfunction due to pressure overload.
MATERIALS AND METHODS
Experimental animals
All experiments using animals were conducted in accordancewith the Australian code for the care and use of animals forscientific purposes (National Health and Medical ResearchCouncil of Australia, 8th Ed., 2013; https://www.nhmrc.gov.au/guidelines/publications/ea28). Animal care and ex-perimental procedures were approved by the Alfred MedicalResearch and Education Precinct’s Animal Ethics Committee.All surgery was performed under anesthesia (ketamine:xyla-zine:atropine) with postoperative analgesia (carprofen).
Pressure overload
Adult (12 wk old) male C57BL/6 mice were subjected totransverse aortic constriction (TAC) or a sham operation asdescribed previously (22). Surgery was performed in weight-matched mice. A suture/band was placed around thetransverse aorta against a 26-gauge needle to ensure con-sistent banding. The TAC model induces a chronic pres-sure load on the heart and is associated with progressivepathological hypertrophy and cardiac dysfunction within 4wk of surgery (22).
Left ventricular (LV) structure and function
Transthoracic echocardiography (M-mode 2-dimensionalechocardiography) was performed on anesthetized mice(1.8% isofluorane, inhalation) using a Philips iE33 ultra-sound machine with a 15 mHz liner array transducer (Philips,Amsterdam, The Netherlands), prior to surgery (baseline), 4wk post-TAC, and 8 wk post-treatment (i.e., 12 wk post-TAC,end point). LV chamber dimensions [LV end-diastolic dimen-sion (LVEDD), LV end-systolic dimension, (LVESD)], LV wallthicknesses [LV posterior wall (LVPW), interventricular sep-tum (IVS)], heart rate (HR), and fractional shortening (FS;calculated as [(LVEDD � LVESD)/LVEDD] � 100%) wereanalyzed offline using dedicated software (ProSolv Cardiovas-cular Analyzer 3.5; ProSolv, Indianapolis, IN, USA) obtainedfrom M-mode traces.
Locked nucleic acid (LNA) oligonucleotides
The miRcury LNA microRNA inhibitor (mmu-miR-652-3pand a scrambled control; Exiqon, Skelstedet, Denmark) isLNA enhanced (LNA/DNA mixmer with �50% LNA con-tent) and contains a phosphorothioate backbone. The se-quence for mmu-miR-652-3p is 5=-ACAACCCTAGTGGCG-3=(batch number 508004) and for the scrambled control 5=-ACGTCTATACGCCCA-3= (batch number 508005).
In vivo delivery of LNA-antimiR oligonucleotides
At 4 wk post-TAC (after echocardiography), sham surgery andTAC mice were randomized into control or treated groups.Mice were subcutaneously administered either a LNA-controlor LNA-antimiR-652 (25 mg/kg/d) over 3 d and left for aperiod of 8 wk (Supplemental Fig. S1).
5098 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org

RNA and protein isolation
Total RNA was isolated from frozen mouse tissues and cellsusing TRI Reagent (Sigma-Aldrich, St Louis, MO, USA) andquantitated on a Nanodrop spectrometer (Thermo Scientific,Waltham, MA, USA). For protein lysates, frozen mouse ven-tricles were homogenized in a lysis buffer (10% glycerol; 137mM NaCl; 20 mM Tris-HCl, pH 7.4; 20 mM NaF; 10 mMEDTA; 1 mM EGTA; 1 mM sodium pyrophosphate; 1 mMvanadate; 1 mM PMSF; 4 �g/ml pepstatin; 4 �g/ml aprotinin;and 4 �g/ml leupeptin), and total protein concentration wasdetermined using the Bradford assay (Bio-Rad, Hercules, CA,USA).
Quantitative RT-PCR (qPCR)
For mRNA expression analysis, 2 �g of total RNA was reversetranscribed using the High Capacity RNA-to-cDNA kit (LifeTechnologies, Carlsbad, CA, USA) according to manufactur-er’s recommendations. qPCR was performed using TaqManprobes (Life Technologies) and amplified on an AppliedBiosystems 7500 real-time PCR instrument (Applied Biosystems,Foster City, CA, USA) according to manufacturer’s instructions.Hypoxanthine phosphoribosyltransferase 1 (Hprt1) was used tostandardize for cDNA concentration and data was analyzedusing the 2���Ct method of quantification. For miRNAexpression analysis, 50 ng of total RNA was reversed tran-scribed for each miRNA of interest using TaqMan MicroRNAReverse Transcription Kit (Life Technologies) according tomanufacturer’s recommendations. qPCR was performed us-ing TaqMan MicroRNA Assays according to manufacturer’sinstructions. Expression was normalized against snoU6 anddata analyzed using the 2���Ct algorithm.
Northern blotting
For mRNA analysis, 10 �g of total RNA was separated on aformaldehyde denaturing agarose gel and transferred to aHybond-N membrane (GE Healthcare, Pittsburgh, PA,USA) in 20� SSC by upward capillary transfer, and probedfor B-type natriuretic peptide (Bnp), �-skeletal actin(Acta1), sarcoplasmic/endoplasmic reticulum Ca2�-AT-Pase 2a (Serca2a), and glyceraldehyde-3-phosphate dehydro-genase (Gapdh) as described previously (23). For miRNAanalysis, 15 �g of total RNA was electrophoresed in a 20%TBE acrylamide gel (Life Technologies), transferred to aHybond-N membrane by electrophoresis, and probed withLNA oligonucleotides miR-652 and sno-U6 (Exiqon) as de-scribed previously (24).
Western blotting
Protein lysates (100 �g) were separated by SDS-PAGE, blottedonto PVDF membrane (Merck, Frankfurt, Germany), incu-bated with antibody overnight [Jagged1, 1:500, C-20, sc-6011,Santa Cruz Biotechnology, Dallas, TX, USA; �-tubulin,1:1000, 2144S, Cell Signaling Technology, Danvers, MA, USA;Bcl-2-associated X protein (BAX), 1:500, 2772, Cell SignalingTechnology; B-cell lymphoma 2 (BCL2), 1:500, 2876, CellSignaling Technology; pAkt(S473), 1:1000, 9271L, Cell Sig-naling Technology; tAkt, 1:1000, 9272S, Cell Signaling Tech-nology; pERK1/2, 1:200, sc-7383, Santa Cruz Biotechnology;tERK1/2, 1:1000, 9102, Cell Signaling Technology; pp38,1:500, 9215, Cell Signaling Technology; and p38, 1:2500,sc-535, Santa Cruz Biotechnology) and detected by chemilu-minescence. Signals were quantitated using ImageJ 1.44ppixel analysis (U.S. National Institutes of Health, Bethesda,MD, USA), and data were normalized to a control value of 1.
Luciferase assay
H9c2 cells (CellBank Australia, Westmead, NSW, Australia)were seeded at 20,000 cells/well (passages 11–16). H9c2 cells(25 mM DMEM, 10% FBS, and 1% PS) were transfected withmiRNA precursors miR-652 or an unrelated miR control(miR-27a) at a total concentration of 100 nM/well (LifeTechnologies) in 24-well plates using Lipofectamine 2000(Life Technologies). Prevalidated random sequence miRNAprecursor molecules were used as negative controls at 100nM/well (Life Technologies). The seed/target region ofJagged1 3=-UTR (120 nt covering the predicted miRNA:mRNA binding region) was cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Mad-ison, WI, USA) by GenScript (Piscataway, NJ, USA). Jagged13=UTR reporter construct (300 ng/well) and -galactosidase(100 ng/well) were also cotransfected using Lipofectamine2000. After 24 h, cells were analyzed for luciferase activityusing the Dual-Glo Luciferase Assay System (Promega). Re-nilla luciferase activity was then measured using a MicrolumatPlus luminometer (Berthold Technologies, Bad Wildbad,Germany). Luciferase activity was normalized by quantifyingexpression of -galactosidase using a -galactosidase detec-tion assay (Promega). Data are presented as the ratio of3=-UTR luciferase activity to -galactosidase expression andare representative of 4 independent experiments.
Cell culture
Primary cultures of neonatal rat ventricular myocytes(NRVMs) and neonatal rat ventricular fibroblasts (NRVFs)were prepared from 1- to 3-d-old Sprague-Dawley strain rats asdescribed previously (25). Noncardiac myocytes (fibroblastfraction) were separated from NRVMs by differential preplat-ing. MiR-652 expression was assessed by qPCR in NRVMs,NRVFs (passage 2), human aortic endothelial cells (HAECs;passage 8; Lonza, Walkersville, MD, USA), human umbilicalvein endothelial cells (HUVECs; passage 5; C-003-5C; LifeTechnologies) and H9c2 cells.
Histological analyses
Tissue samples were fixed in 4% paraformaldehyde andparaffin embedded for histological analysis at 6 �m cross-sections. To assess tissue morphology, sections were stainedwith hematoxylin and eosin (H&E). Cardiac collagen depo-sition/interstitial fibrosis (Masson’s trichrome stain), cellarea [wheat germ agglutinin (WGA) stain], angiogenesis(costained with isolectin B4 and WGA), apoptosis (TUNELstaining using a CardioTACs In Situ Apoptosis Detection Kit;4827-30-K; Trevigen, Gaithersburg, MD, USA), and cardiomy-ocyte proliferation [colocalization of phosphohistone H3(pH3) with cardiac troponin T] were assessed as describedpreviously (22, 24, 26).
Target prediction software tools
Predicted targets of miR-652 were identified using Target-Scan Mouse 6.2 (Whitehead Institute for Biomedical Re-search, Cambridge, MA, USA; http://www.targetscan.org/).Gene ontology analysis was performed using the Database forAnnotation, Visualization, and Integrated Discovery (DAVID)6.7 Panther pathway (U.S. National Institute of Allegy andInfectious Diseases, Bethesda, MD, USA; http://david.abcc.ncifcrf.gov/).
5099THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART
Statistical analyses
Statistical analyses were performed using StatView 5.0.1 (SASInstitute Inc., Cary, NC, USA). Results are presented asmeans sem. Differences between groups were identifiedusing 1-way analysis of variance (ANOVA) followed by Fisher’spost hoc tests, unless otherwise indicated. Unpaired t tests wereused when comparing 2 groups for a single measure. A valueof P � 0.05 was considered significant. All relative units areexpressed as a fold change with the relevant control groupnormalized to 1.
RESULTS
Inhibition of miR-652 attenuates pathological cardiachypertrophy and restores cardiac function
Additional analysis of miRNA expression data from ourearlier microarray screen (20) in models of cardiacstress and protection identified miR-652 as a promisingcandidate to target. miR-652 expression was increasedin hearts of mice subjected to MI (noninfarct, remotezone) which developed pathological hypertrophy; de-creased in a mouse model of cardiac protection; andinversely correlated with cardiac function (Supplemen-tal Fig. S2A, B and ref. 20). In addition, we measuredmiR-652 in hearts from mice subjected to TAC for 12wk from a previous study (27), and found miR-652expression to be elevated in a setting of pathologicalhypertrophy, which increased further with heart failure(Supplemental Fig. S2C). Collectively, these findingsled us to propose that inhibition of miR-652 may beprotective in settings of cardiac stress. In the currentstudy, we chose to assess the potential of inhibitingmiR-652 in the TAC model, which is associated with lesscardiac pathology than the MI model, and is associatedwith more homogenous changes to the heart.
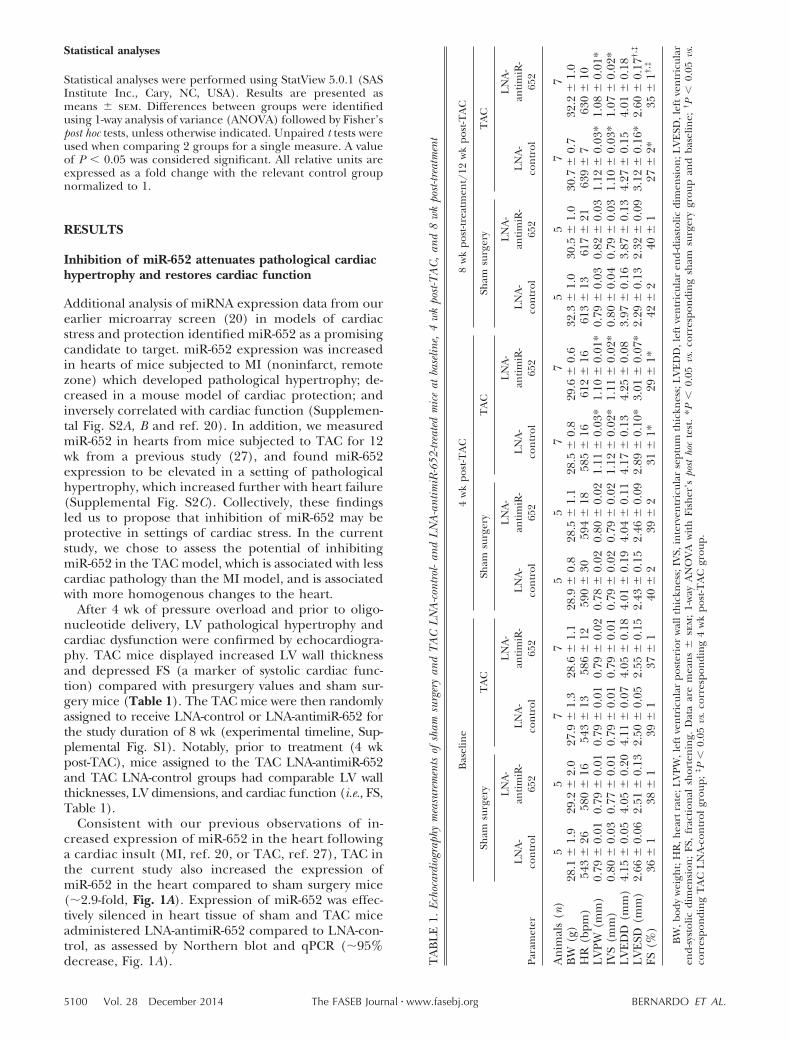
After 4 wk of pressure overload and prior to oligo-nucleotide delivery, LV pathological hypertrophy andcardiac dysfunction were confirmed by echocardiogra-phy. TAC mice displayed increased LV wall thicknessand depressed FS (a marker of systolic cardiac func-tion) compared with presurgery values and sham sur-gery mice (Table 1). The TAC mice were then randomlyassigned to receive LNA-control or LNA-antimiR-652 forthe study duration of 8 wk (experimental timeline, Sup-plemental Fig. S1). Notably, prior to treatment (4 wkpost-TAC), mice assigned to the TAC LNA-antimiR-652and TAC LNA-control groups had comparable LV wallthicknesses, LV dimensions, and cardiac function (i.e., FS,Table 1).
Consistent with our previous observations of in-creased expression of miR-652 in the heart followinga cardiac insult (MI, ref. 20, or TAC, ref. 27), TAC inthe current study also increased the expression ofmiR-652 in the heart compared to sham surgery mice(�2.9-fold, Fig. 1A). Expression of miR-652 was effec-tively silenced in heart tissue of sham and TAC miceadministered LNA-antimiR-652 compared to LNA-con-trol, as assessed by Northern blot and qPCR (�95%decrease, Fig. 1A). T
AB
LE
1.Ec
hoca
rdio
grap
hym
easu
rem
ents
ofsh
amsu
rger
yan
dT
AC
LN
A-c
ontr
ol-a
ndL
NA
-ant
imiR
-652
-trea
ted
mic
eat
base
line,
4w
kpo
st-T
AC
,an
d8
wk
post
-trea
tmen
t
Para
met
er
Bas
elin
e4
wk
post
-TA
C8
wk
post
-trea
tmen
t/12
wk
post
-TA
C
Sham
surg
ery
TA
CSh
amsu
rger
yT
AC
Sham
surg
ery
TA
C
LN
A-
con
trol
LN
A-
anti
miR
-65
2L
NA
-co
ntr
ol
LN
A-
anti
miR
-65
2L
NA
-co
ntr
ol
LN
A-
anti
miR
-65
2L
NA
-co
ntr
ol
LN
A-
anti
miR
-65
2L
NA
-co
ntr
ol
LN
A-
anti
miR
-65
2L
NA
-co
ntr
ol
LN
A-
anti
miR
-65
2
An
imal
s(n
)5
57
75
57
75
57
7B
W(g
)28
.1
1.9
29.2
2.
027
.9
1.3
28.6
1.
128
.9
0.8
28.5
1.
128
.5
0.8
29.6
0.
632
.3
1.0
30.5
1.
030
.7
0.7
32.2
1.
0H
R(b
pm)
543
26
580
16
543
13
586
12
590
30
594
18
585
16
612
16
613
13
617
21
639
7
630
10
LV
PW(m
m)
0.79
0.
010.
79
0.01
0.79
0.
010.
79
0.02
0.78
0.
020.
80
0.02
1.11
0.
03*
1.10
0.
01*
0.79
0.
030.
82
0.03
1.12
0.
03*
1.08
0.
01*
IVS
(mm
)0.
80
0.03
0.77
0.
010.
79
0.01
0.79
0.
010.
79
0.02
0.79
0.
021.
12
0.02
*1.
11
0.02
*0.
80
0.04
0.79
0.
031.
10
0.03
*1.
07
0.02
*L
VE
DD
(mm
)4.
15
0.05
4.05
0.
204.
11
0.07
4.05
0.
184.
01
0.19
4.04
0.
114.
17
0.13
4.25
0.
083.
97
0.16
3.87
0.
134.
27
0.15
4.01
0.
18L
VE
SD(m
m)
2.66
0.
062.
51
0.13
2.50
0.
052.
55
0.15
2.43
0.
152.
46
0.09
2.89
0.
10*
3.01
0.
07*
2.29
0.
132.
32
0.09
3.12
0.
16*
2.60
0.
17†,‡
FS(%
)36
1
38
139
1
37
140
2
39
231
1*
29
1*42
2
40
127
2*
35
1†,‡
BW
,bod
yw
eigh
t;H
R,h
eart
rate
;LV
PW,l
eft
ven
tric
ular
post
erio
rw
allt
hic
knes
s;IV
S,in
terv
entr
icul
arse
ptum
thic
knes
s;L
VE
DD
,lef
tve
ntr
icul
aren
d-di
asto
licdi
men
sion
;LV
ESD
,lef
tve
ntr
icul
aren
d-sy
stol
icdi
men
sion
;FS
,fr
acti
onal
shor
ten
ing.
Dat
aar
em
ean
s
sem
;1-
way
AN
OV
Aw
ith
Fish
er’s
post
hoc
test
.*P
�0.
05vs
.co
rres
pon
din
gsh
amsu
rger
ygr
oup
and
base
line;
†P
�0.
05vs
.co
rres
pon
din
gT
AC
LN
A-c
ontr
olgr
oup;
‡P
�0.
05vs
.co
rres
pon
din
g4
wk
post
-TA
Cgr
oup.
5100 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org
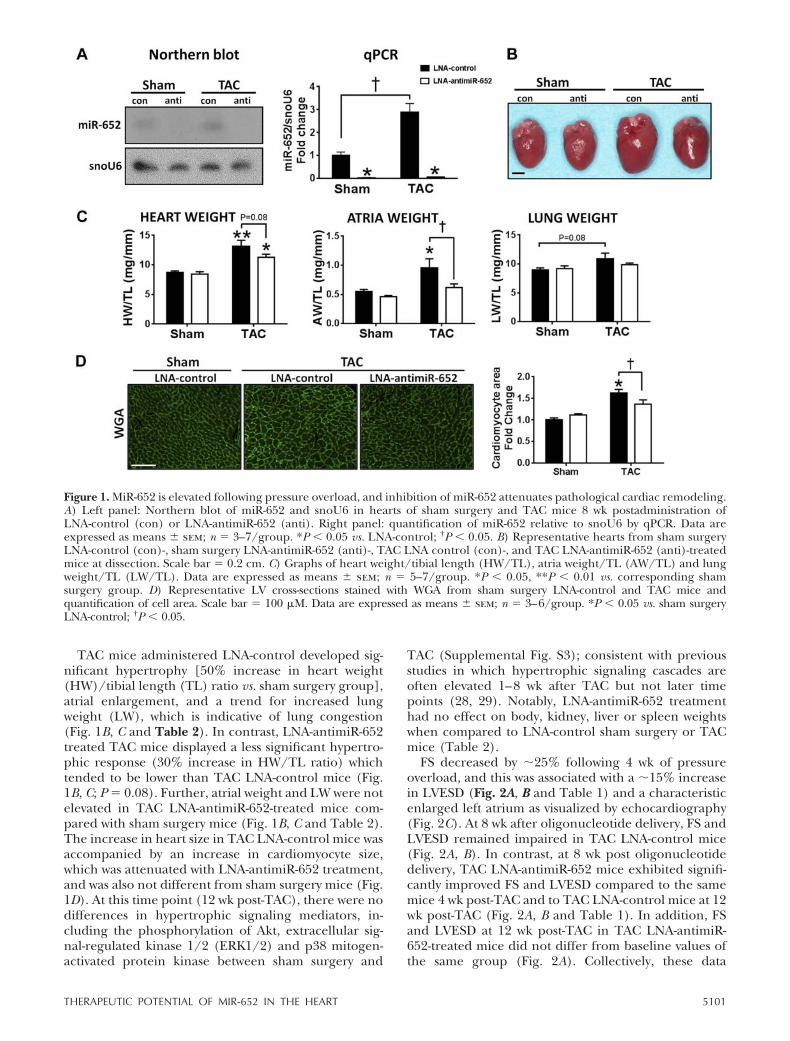
TAC mice administered LNA-control developed sig-nificant hypertrophy [50% increase in heart weight(HW)/tibial length (TL) ratio vs. sham surgery group],atrial enlargement, and a trend for increased lungweight (LW), which is indicative of lung congestion(Fig. 1B, C and Table 2). In contrast, LNA-antimiR-652treated TAC mice displayed a less significant hypertro-phic response (30% increase in HW/TL ratio) whichtended to be lower than TAC LNA-control mice (Fig.1B, C; P � 0.08). Further, atrial weight and LW were notelevated in TAC LNA-antimiR-652-treated mice com-pared with sham surgery mice (Fig. 1B, C and Table 2).The increase in heart size in TAC LNA-control mice wasaccompanied by an increase in cardiomyocyte size,which was attenuated with LNA-antimiR-652 treatment,and was also not different from sham surgery mice (Fig.1D). At this time point (12 wk post-TAC), there were nodifferences in hypertrophic signaling mediators, in-cluding the phosphorylation of Akt, extracellular sig-nal-regulated kinase 1/2 (ERK1/2) and p38 mitogen-activated protein kinase between sham surgery and
TAC (Supplemental Fig. S3); consistent with previousstudies in which hypertrophic signaling cascades areoften elevated 1–8 wk after TAC but not later timepoints (28, 29). Notably, LNA-antimiR-652 treatmenthad no effect on body, kidney, liver or spleen weightswhen compared to LNA-control sham surgery or TACmice (Table 2).
FS decreased by �25% following 4 wk of pressureoverload, and this was associated with a �15% increasein LVESD (Fig. 2A, B and Table 1) and a characteristicenlarged left atrium as visualized by echocardiography(Fig. 2C). At 8 wk after oligonucleotide delivery, FS andLVESD remained impaired in TAC LNA-control mice(Fig. 2A, B). In contrast, at 8 wk post oligonucleotidedelivery, TAC LNA-antimiR-652 mice exhibited signifi-cantly improved FS and LVESD compared to the samemice 4 wk post-TAC and to TAC LNA-control mice at 12wk post-TAC (Fig. 2A, B and Table 1). In addition, FSand LVESD at 12 wk post-TAC in TAC LNA-antimiR-652-treated mice did not differ from baseline values ofthe same group (Fig. 2A). Collectively, these data
Figure 1. MiR-652 is elevated following pressure overload, and inhibition of miR-652 attenuates pathological cardiac remodeling.A) Left panel: Northern blot of miR-652 and snoU6 in hearts of sham surgery and TAC mice 8 wk postadministration ofLNA-control (con) or LNA-antimiR-652 (anti). Right panel: quantification of miR-652 relative to snoU6 by qPCR. Data areexpressed as means sem; n � 3–7/group. *P � 0.05 vs. LNA-control; †P � 0.05. B) Representative hearts from sham surgeryLNA-control (con)-, sham surgery LNA-antimiR-652 (anti)-, TAC LNA control (con)-, and TAC LNA-antimiR-652 (anti)-treatedmice at dissection. Scale bar � 0.2 cm. C) Graphs of heart weight/tibial length (HW/TL), atria weight/TL (AW/TL) and lungweight/TL (LW/TL). Data are expressed as means sem; n � 5–7/group. *P � 0.05, **P � 0.01 vs. corresponding shamsurgery group. D) Representative LV cross-sections stained with WGA from sham surgery LNA-control and TAC mice andquantification of cell area. Scale bar � 100 �M. Data are expressed as means sem; n � 3–6/group. *P � 0.05 vs. sham surgeryLNA-control; †P � 0.05.
5101THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART

suggest that treatment with LNA-antimiR-652 was ableto restore cardiac function following pressure overload.
Inhibition of miR-652 attenuates cardiac stress geneexpression, fibrosis, and apoptosis
Pressure overload-induced pathological hypertrophy isassociated with the reexpression of the cardiac fetalgene program, accumulation of collagen in the extra-cellular matrix (interstitial fibrosis), apoptosis of cardi-omyocytes, and down-regulation of calcium-handlingproteins (1, 2), all of which contribute to decliningcardiac function. Hearts of TAC LNA-control mice hadincreased Bnp/Nppb (Fig. 3A) and Acta1 expression(Fig. 3A), and decreased Serca2a/Atp2a2 (Fig. 3A).Improved cardiac function in TAC LNA-antimiR-652-treated mice was associated with attenuated Bnp andActa1 expression, and more favorable Serca2a expres-sion (Fig. 3A). Cardiac dysfunction in TAC LNA-con-trol mice was also associated with increased fibrosis,which was attenuated with LNA-antimiR-652 treatment(Fig. 3B). To evaluate whether increased fibrosis andcardiac dysfunction was, in part, caused by cardiomyo-cyte loss, we determined the prevalence of apoptosis.TAC LNA-control mice exhibited higher numbers ofapoptotic cardiomyocytes compared to TAC LNA-anti-miR-652 mice (Fig. 3C, TUNEL staining). In addition,the BAX/BCL2 ratio was significantly increased in thehearts of TAC LNA-control mice compared with sham
surgery mice, and this was attenuated with LNA-anti-miR-652 treatment (Fig. 3D).
LNA-antimiR-652 treatment is associated withpreserved angiogenesis and up-regulation of amiR-652 target, Jagged1
To investigate potential mechanisms by which inhibitionof miR-652 may mediate protection in a setting of pres-sure overload, we used a target prediction software tool(TargetScan Mouse 6.2) to identify predicted targets ofmiR-652 and performed gene ontology analysis (DA-VID). We found that the Notch signaling pathway wassignificantly enriched (P � 0.016) and identified theNotch pathway ligand, Jagged1, as a predicted target ofmiR-652 (Fig. 4A). The Notch pathway has been shownto play a critical role for the maintenance of cardiacstructure and function during mammalian cardiac de-velopment and in settings of cardiac stress duringadulthood (30–34), thereby providing additional ratio-nale for investigating the regulation of Jagged1 bymiR-652 as a potential mechanism of action. BothJagged1 and Notch1 mRNA expression levels wereelevated in hearts from TAC mice treated with LNA-antimiR-652 compared with TAC LNA-control (Fig.4B). Furthermore, Jagged1 protein expression was de-creased in TAC LNA-control hearts compared to shamsurgery, and increased in TAC LNA-antimiR-652 heartscompared to TAC LNA-control; inversely correlatedwith miR-652 expression; and positively correlated with
TABLE 2. Morphological data of sham surgery and TAC LNA-control- and LNA-antimiR-652-treated mice following 8 wk of treatment
Parameter
Sham surgery TAC
LNA-control LNA-antimiR-652 LNA-control LNA-antimiR-652
Animals (n) 4–5 5 7 6–7Age (wk) 22.3 22.3 22.3 22.3BW (g) 31.8 1.1 30.3 1.2 30.8 0.7 31.8 0.9TL (mm) 16.7 0.1 16.5 0.1 16.3 0.1 16.4 0.1HW (mg) 147.4 4.1 139.6 6.9 214.7 17.1* 190.3 9.6*AW (mg) 9.0 0.5 7.6 0.4 15.6 2.6* 10.6 1.0†
LW (mg) 152.4 5.2 151.8 8.1 178.7 16.8 165.2 6.4HW/BW (mg/g) 4.6 0.2 4.6 0.2 7.0 0.6*** 6.0 0.2*AW/BW (mg/g) 0.28 0.01 0.25 0.01 0.50 0.08* 0.33 0.03†
LW/BW (mg/g) 4.8 0.2 5.0 0.1 5.8 0.4 5.2 0.1HW/TL (mg/mm) 8.7 0.3 8.5 0.4 13.1 1.0** 11.3 0.6*AW/TL (mg/mm) 0.55 0.38 0.46 0.25 0.95 0.16* 0.62 0.64†
LW/TL (mg/mm) 9.0 0.3 9.2 0.5 10.9 1.0 9.8 0.3KW (mg) 422.2 18.4 394.2 26.7 388.5 7.8 385.0 22.6LivW (g) 1.3 0.1 1.2 0.0 1.2 0.1 1.2 0.1SW (mg) 90.7 14.7 85.8 5.5 90.9 7.5 87.1 6.1KW/BW (mg/g) 13.3 0.4 13.0 0.5 12.6 0.3 12.1 0.5LivW/BW (mg/g) 41.2 3.3 41.2 1.2 39.8 3.0 39.3 2.2SW/BW (mg/g) 2.9 0.5 2.9 0.3 3.0 0.2 2.7 0.2KW/TL (mg/mm) 25.5 1.4 23.9 1.5 23.8 0.5 22.9 1.4LivW/TL (mg/mm) 83.5 5.8 75.2 1.0 75.2 5.6 77.8 3.9SW/TL (mg/mm) 4.6 0.4 5.2 0.4 5.6 0.4 5.1 0.4
BW, body weight; TL, tibia length; HW, heart weight; AW, atrium weight; LW, lung weight; KW,kidney weight; LivW, liver weight; SW, spleen weight. Data are means sem; 1-way ANOVA with Fisher’spost hoc test. *P � 0.05, **P � 0.01, ***P � 0.001 vs. corresponding sham surgery group; †P � 0.05 vs.TAC LNA-control group.
5102 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org

cardiac function (FS) in TAC mice (Fig. 4C). Todetermine whether miR-652 can directly bind to the3=-UTR of Jagged1, we performed luciferase reporterassays in H9c2 cells (a cardiomyoblast cell line). Over-expression of miR-652 inhibited luciferase activity ofthe Jagged1 3=-UTR reporter construct, but was notaffected by a negative control or another miRNA notpredicted to target the 3=-UTR of Jagged1 (Fig. 4D).These data confirmed that miR-652 can bind specifi-cally to the 3=-UTR of Jagged1 and confirms Jagged1 asa bona fide target of miR-652.
As Jagged1 and activation of the Notch pathway havebeen linked to cardiomyocyte proliferation and angio-genesis (30–32, 35–37), we investigated whether LNA-antimiR-652 treatment could induce cardiomyocyteproliferation and preserve angiogenesis following TAC.Induction of cardiomyocyte mitosis was assessed bycolocalization of pH3 with cardiac troponin T in hearts8 wk after treatment. At this time point, cardiomyocytemitoses were extremely rare and there were no obviousdifferences in pH3 staining patterns between LNA-control- and LNA-antimiR-652-treated TAC hearts(Supplemental Fig. S4A). In addition, expression of thetranscription factors CCAAT/enhancer binding pro-tein (C/EBP) and Cbp/p300-interacting transacti-vator with Glu/Asp-rich carboxy-terminal domain 4(CITED4), genes associated with cardiomyocyte prolif-eration during exercise-induced cardiac hypertrophy(38), were unchanged with LNA-antimiR-652 treat-ment (Supplemental Fig. S4B). Capillary density,measured as an indicator of angiogenesis, was signif-icantly decreased in TAC LNA-control mice com-pared to sham surgery mice (Fig. 4E). By contrast,capillary density was preserved in LNA-antimiR-652-treated TAC mice (Fig. 4E).
Inhibition of miR-652 may provide protection byacting on multiple cell types
It was not possible to assess the expression of miR-652in specific cardiac cell types in the current cohort ofsham and TAC mice. However, to explore which celltypes LNA-antimiR-652 could potentially provide bene-fit, a number of in vitro experiments were undertaken.Under basal conditions, miR-652 was expressed inisolated NRVMs and NRVFs from neonatal rat hearts,HAECs, HUVECs, and H9c2 cells; highest expressionidentified in NRVMs (Supplemental Fig. S5A). Further-more, miR-652 expression increased in NRVMs treatedwith phenylephrine (PE; hypertrophic stimulus) orhydrogen peroxide (H2O2; potent reactive oxygen spe-cies known to induce apoptosis in NRVMs; Supplemen-tal Fig. S5B).
Chronic administration of LNA-antimiR-652 had noadverse effects on tissue morphology
Systemic delivery of antimiRs with LNA modificationsare extremely stable, rapidly cleared from the plasma,and taken up by cells of multiple tissues, leading to
long-lasting silencing of miRNAs (13, 39). However,systemic inhibition may have effects in different tissues.In the present study, expression of miR-652 was signif-icantly lower in heart and lung, and tended to be lowerin kidney and liver from LNA-antimiR-652-treated micecompared to LNA-control-treated mice (SupplementalFig. S6A). However, despite chronic knockdown ofmiR-652 in these tissues, LNA-antimiR-652 administra-tion had no notable effect on these organs (other thanattenuation of HW) based on weight or morphology atdissection (Supplemental Fig. S6B and Table 2). Toinvestigate this further, histological assessment was per-formed on tissues from sham surgery mice treated withLNA-control or LNA-antimiR-652, and analysis revealedno evidence of morphological disarray (SupplementalFig. S6C).
DISCUSSION
It is well recognized that hundreds of miRNAs aredysregulated in settings of cardiac pathology (40–42).Numerous studies have focused on targeting miRNAsthat have relatively high expression in the heart underbasal conditions, which increases further in a cardiacdisease setting (8). In contrast, we used selection crite-ria that ranked candidate miRNAs based on differentialexpression in settings of cardiac stress and protection(20) and correlation with cardiac function. We previ-ously demonstrated the therapeutic potential of target-ing one of these miRNAs, miR-34 (24, 27). In thecurrent study, we assessed the effect of inhibitinganother candidate miRNA, miR-652. Very little isknown regarding the function of miR-652 in any tissue,and the role in the heart has not been explored.Furthermore, given the relatively low expression ofmiR-652 in the heart under basal conditions comparedwith other miRNAs (21), the role of miR-652 wasunlikely to be examined without a selection criteriasuch as that described here. We assessed the role ofsilencing miR-652 in a mouse model with establishedpathological cardiac hypertrophy and dysfunction dueto pressure overload, which over time is associated withincreased apoptosis and fibrosis, and progresses toheart failure (43). Here, we provide the first evidencethat inhibition of miR-652 in a mouse model withpreexisting cardiac hypertrophy and dysfunction is ableto attenuate pathological heart growth, attenuate apo-ptosis and fibrosis, preserve angiogenesis, and improveheart function. Furthermore, we identified and vali-dated Jagged1 as a target of miR-652. Notably, knock-down of miR-652 for 8 wk was not associated with anynotable toxicity in other tissues, including the kidney,liver, or lungs.
To investigate molecular mechanisms by which si-lencing miR-652 might be providing protection in asetting of pressure overload, we used target predictionsoftware tools. These analyses identified a gene that isinvolved in the Notch signaling pathway, Jagged1 (aNotch1 ligand). We defined Jagged1 as a candidate
5103THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART

target of particular interest because of the known roleof the Notch pathway in mammalian cardiac develop-ment, and maintenance of structural and functionalintegrity during the response to increased workload inthe heart (30–34). Furthermore, cardiac-specific over-expression of Jagged1 in mice attenuated pathologicalremodeling (i.e., attenuated HW, fibrosis and cardiacstress gene expression) following pressure overload(33). This finding provided further rationale for closerscrutiny of Jagged1, as the reported phenotype in miceoverexpressing Jagged1 is reminiscent of what we ob-served in LNA-antimiR-652-treated TAC mice. In hu-mans, mutations in the JAG1 gene cause Alagille syn-drome, where patients present with congenital heartdisease and vascular disease (44, 45). In the presentstudy, Jagged1 and Notch1 expression increased inhearts of LNA-antimiR-652-treated mice, and we exper-imentally validated Jagged1 as a bona fide target ofmiR-652.
Next, we examined potential mechanisms via whichincreased Jagged1 might mediate protection in a set-ting of pressure overload, including cardiomyocyteproliferation/regeneration, angiogenesis, and inhibi-tion of apoptosis. Jagged1 and/or the Notch pathwayhave been reported to regulate each of these processesin the cardiovascular system (30–32, 35). In the currentstudy, there was no apparent induction of cardiomyo-cyte proliferation with LNA-antimiR-652 treatment.Thus, myocyte proliferation is unlikely to play a signif-
icant (if any) role in contributing to the improvedoutcome observed in TAC LNA-antimiR-652-treatedmice. This finding is consistent with a previous reportsuggesting that Jagged1 is important for proliferationof immature cardiomyocytes but is insufficient to drivecell cycle reentry in fully mature cardiomyocytes (37).Furthermore, previous studies, which have identifiedproliferating cardiomyocytes in the adult heart follow-ing endurance exercise training, have typically re-ported activation within the first 2 wk following injury(38). In the present study, LNA treatment was notcommenced until 4 wk after injury, and samples wereanalyzed 8 wk post-TAC. This may further explain whyno induction of proliferation was observed. A potentialmechanism by which LNA-antimiR-652 treatment mayhave restored cardiac function in the absence of achange in cardiomyocyte proliferation, is by improvingcardiomyocyte contractility. This was beyond the scopeof the current study but would be of interest to assess infuture work.
Reduced angiogenesis in cardiac stress models isknown to contribute to the transition to heart failure(46, 47), and gene therapy interventions that increaseor preserve angiogenesis following a cardiac insult areassociated with improved cardiac outcome (22, 24, 48).In the current report, capillary density was decreased inTAC LNA-control mice compared to sham surgerymice, but preserved with LNA-antimiR-652 treatment.Consistent with increased Jagged1 and Notch1 contrib-
Figure 2. Administration of LNA-antimiR-652 improves car-diac function in mice with established cardiac dysfunctiondue to TAC. A) Quantification of FS and LVESD at baseline(presurgery), 4 wk post-sham surgery/TAC (prior to LNA-oligonucleotide delivery), and 8 wk post-treatment (12 wk
post-sham surgery/TAC). Data are expressed as means sem. Sham surgery, n � 5/group; TAC, n � 7/group. AntimiRdelivery commenced at 4 wk post-sham surgery/TAC (indicated by arrow). *P � 0.05 vs. corresponding baseline and shamsurgery group; †P � 0.05. B) Representative M-mode echocardiograms from TAC mice before and after treatment. Arrowsindicate differences in LV chamber dimensions. C) Representative image of enlarged left atrium (LA; echocardiography:long-axis 2-dimensional image) in TAC LNA-control mice and attenuation in TAC LNA-antimiR-652-treated mice.
5104 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org
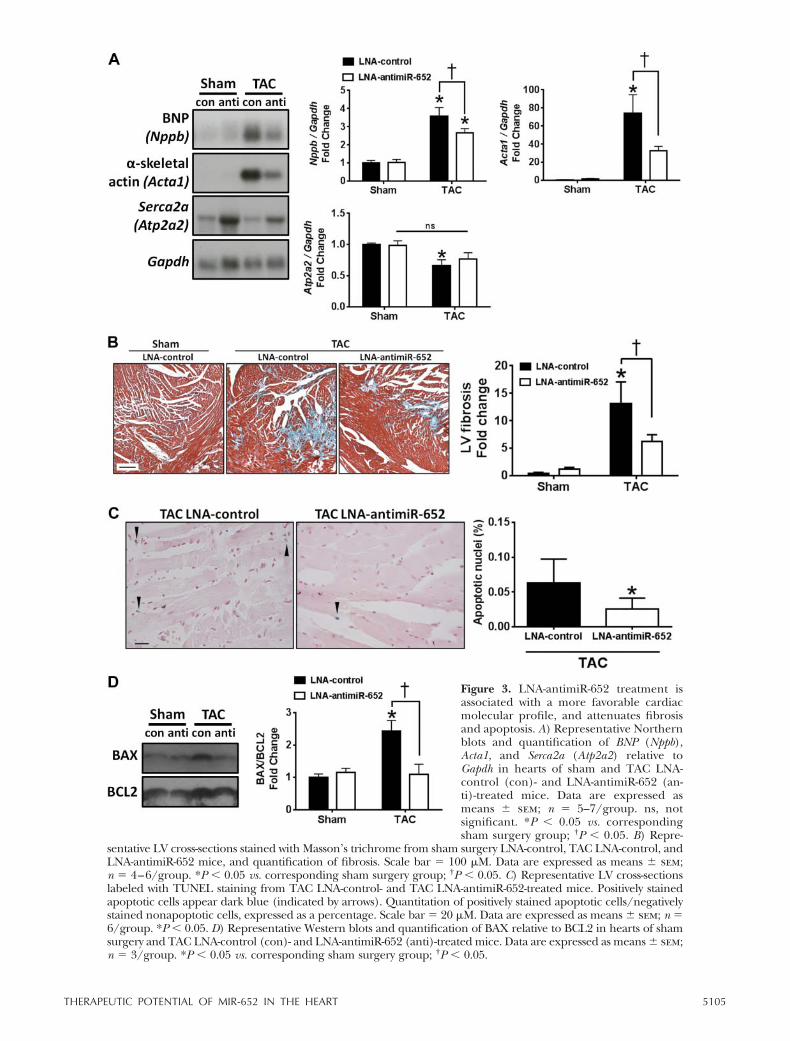
Figure 3. LNA-antimiR-652 treatment isassociated with a more favorable cardiacmolecular profile, and attenuates fibrosisand apoptosis. A) Representative Northernblots and quantification of BNP (Nppb),Acta1, and Serca2a (Atp2a2) relative toGapdh in hearts of sham and TAC LNA-control (con)- and LNA-antimiR-652 (an-ti)-treated mice. Data are expressed asmeans sem; n � 5–7/group. ns, notsignificant. *P � 0.05 vs. correspondingsham surgery group; †P � 0.05. B) Repre-
sentative LV cross-sections stained with Masson’s trichrome from sham surgery LNA-control, TAC LNA-control, andLNA-antimiR-652 mice, and quantification of fibrosis. Scale bar � 100 �M. Data are expressed as means sem;n � 4–6/group. *P � 0.05 vs. corresponding sham surgery group; †P � 0.05. C) Representative LV cross-sectionslabeled with TUNEL staining from TAC LNA-control- and TAC LNA-antimiR-652-treated mice. Positively stainedapoptotic cells appear dark blue (indicated by arrows). Quantitation of positively stained apoptotic cells/negativelystained nonapoptotic cells, expressed as a percentage. Scale bar � 20 �M. Data are expressed as means sem; n �6/group. *P � 0.05. D) Representative Western blots and quantification of BAX relative to BCL2 in hearts of shamsurgery and TAC LNA-control (con)- and LNA-antimiR-652 (anti)-treated mice. Data are expressed as means sem;n � 3/group. *P � 0.05 vs. corresponding sham surgery group; †P � 0.05.
5105THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART
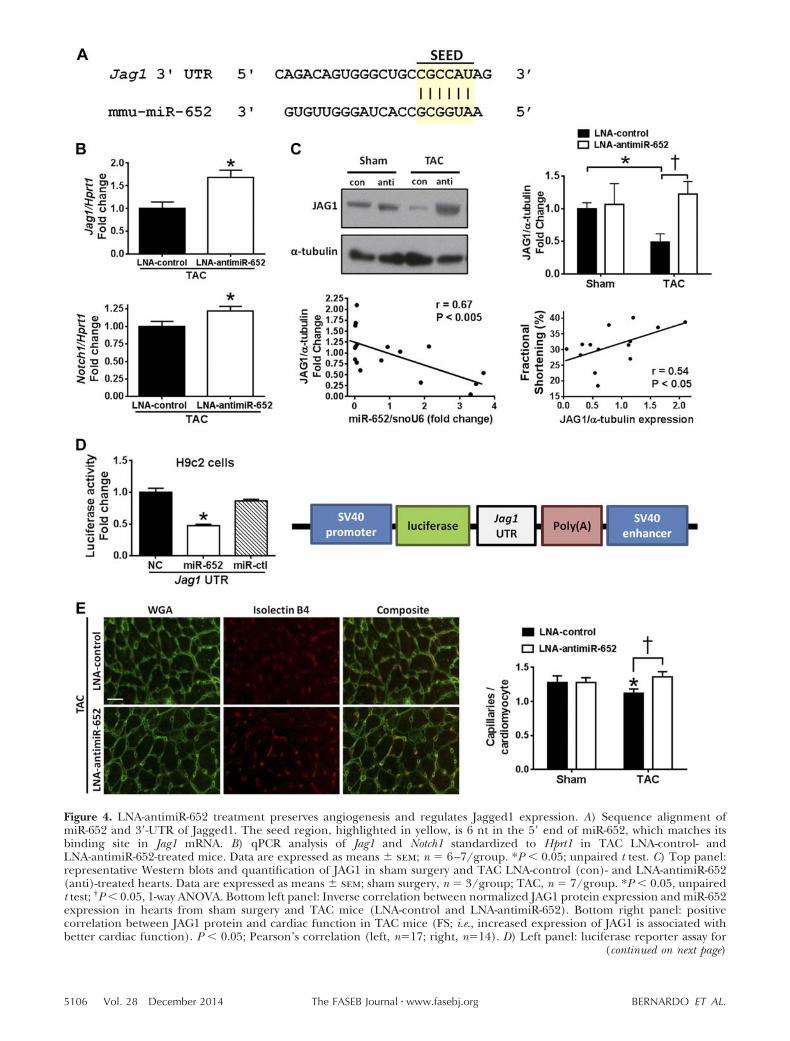
Figure 4. LNA-antimiR-652 treatment preserves angiogenesis and regulates Jagged1 expression. A) Sequence alignment ofmiR-652 and 3=-UTR of Jagged1. The seed region, highlighted in yellow, is 6 nt in the 5= end of miR-652, which matches itsbinding site in Jag1 mRNA. B) qPCR analysis of Jag1 and Notch1 standardized to Hprt1 in TAC LNA-control- andLNA-antimiR-652-treated mice. Data are expressed as means sem; n � 6–7/group. *P � 0.05; unpaired t test. C) Top panel:representative Western blots and quantification of JAG1 in sham surgery and TAC LNA-control (con)- and LNA-antimiR-652(anti)-treated hearts. Data are expressed as means sem; sham surgery, n � 3/group; TAC, n � 7/group. *P � 0.05, unpairedt test; †P � 0.05, 1-way ANOVA. Bottom left panel: Inverse correlation between normalized JAG1 protein expression and miR-652expression in hearts from sham surgery and TAC mice (LNA-control and LNA-antimiR-652). Bottom right panel: positivecorrelation between JAG1 protein and cardiac function in TAC mice (FS; i.e., increased expression of JAG1 is associated withbetter cardiac function). P � 0.05; Pearson’s correlation (left, n�17; right, n�14). D) Left panel: luciferase reporter assay for
(continued on next page)
5106 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org
uting to the preservation of angiogenesis in TACLNA-antimiR-652-treated mice, Notch signaling playsa critical role in the regulation of angiogenesis (49),Jagged1-knockout mice are embryonically lethal dueto defects in the vasculature (50), and overexpressionof Jagged1 promoted angiogenesis in the mouseretina (36).
Finally, another feature of pathological hypertrophyis cardiomyocyte apoptosis (2). The gradual loss ofcardiomyocytes over time contributes to the eventualfailure of the heart. Notch signaling plays a critical rolein maintaining the balance between cell proliferation,differentiation, and apoptosis (31). Mice lackingNotch1 in the heart demonstrated increased apoptoticcell death in response to hemodynamic overload (30).LNA-antimiR-652 treatment in TAC mice was associ-
ated with a decreased rate of cardiomyocyte apoptosiscompared to LNA-control mice. Thus, Jagged1 activa-tion of the Notch signaling pathway may represent aprotective mechanism enabling cell survival. This, inpart, would contribute to the improved cardiac out-come observed in TAC LNA-antimiR-652 mice.
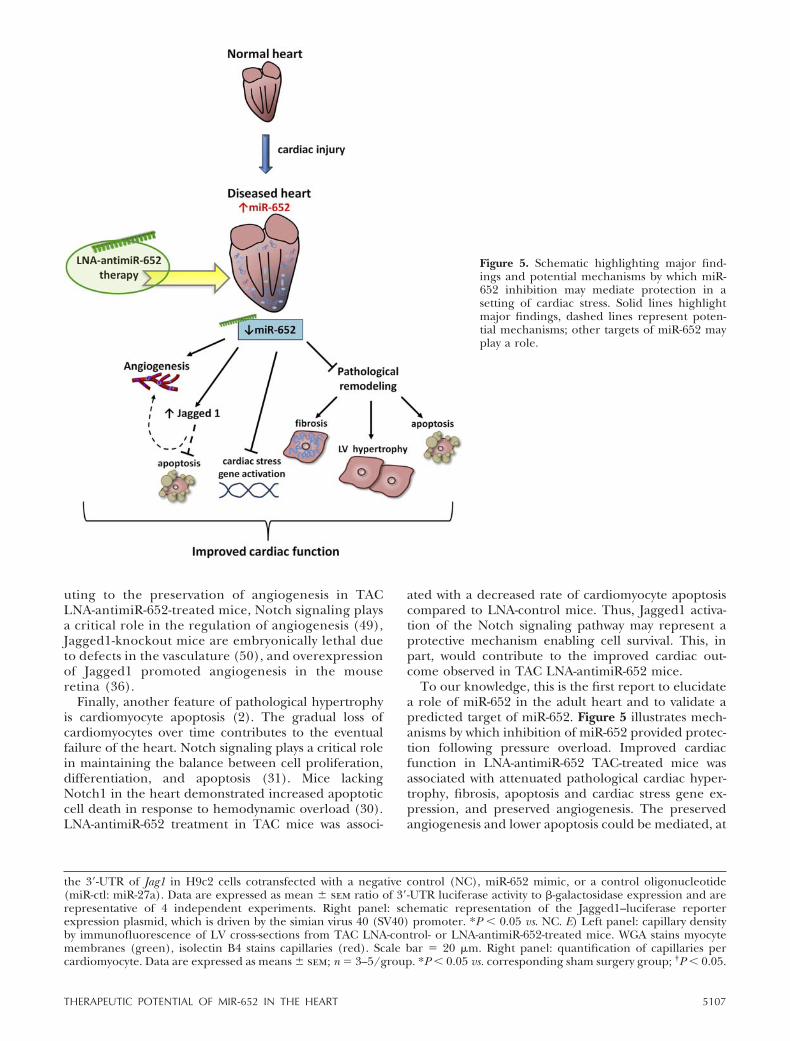
To our knowledge, this is the first report to elucidatea role of miR-652 in the adult heart and to validate apredicted target of miR-652. Figure 5 illustrates mech-anisms by which inhibition of miR-652 provided protec-tion following pressure overload. Improved cardiacfunction in LNA-antimiR-652 TAC-treated mice wasassociated with attenuated pathological cardiac hyper-trophy, fibrosis, apoptosis and cardiac stress gene ex-pression, and preserved angiogenesis. The preservedangiogenesis and lower apoptosis could be mediated, at
the 3=-UTR of Jag1 in H9c2 cells cotransfected with a negative control (NC), miR-652 mimic, or a control oligonucleotide(miR-ctl: miR-27a). Data are expressed as mean sem ratio of 3=-UTR luciferase activity to -galactosidase expression and arerepresentative of 4 independent experiments. Right panel: schematic representation of the Jagged1–luciferase reporterexpression plasmid, which is driven by the simian virus 40 (SV40) promoter. *P � 0.05 vs. NC. E) Left panel: capillary densityby immunofluorescence of LV cross-sections from TAC LNA-control- or LNA-antimiR-652-treated mice. WGA stains myocytemembranes (green), isolectin B4 stains capillaries (red). Scale bar � 20 �m. Right panel: quantification of capillaries percardiomyocyte. Data are expressed as means sem; n � 3–5/group. *P � 0.05 vs. corresponding sham surgery group; †P � 0.05.
Figure 5. Schematic highlighting major find-ings and potential mechanisms by which miR-652 inhibition may mediate protection in asetting of cardiac stress. Solid lines highlightmajor findings, dashed lines represent poten-tial mechanisms; other targets of miR-652 mayplay a role.
5107THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART

least in part, via an up-regulation of the validatedmiR-652 target, Jagged1. Consistent with Jagged1 me-diating some degree of protection, Jagged1 proteinexpression was depressed in hearts from LNA-controlTAC mice compared with sham surgery but not de-creased in LNA-antimiR-652-treated TAC mice, andJagged1 protein expression was positively correlatedwith heart function in TAC mice. However, givenmiRNAs have numerous target genes, we cannot ex-clude the possibility that other target genes are contrib-uting to the favorable phenotype.
Cardiovascular disease is complex, and the multifac-eted pathophysiology of heart failure limits the effec-tiveness of current therapeutics. A potential advantageof targeting miRNAs is that individual miRNAs cantarget hundreds of genes that may regulate multiplebiological networks. Here, we show that inhibitingmiR-652 had a favorable effect by regulating hypertro-phy, fibrosis, apoptosis and angiogenesis. However, asrecognized in the cancer field, a combination of drugsthat target multiple pathways may be the best therapeu-tic approach. Previous studies have shown that target-ing miRNA families or multiple miRNAs simultaneouslycan be more efficacious than targeting individual miR-NAs alone (24, 26, 27, 51). Recent studies have high-lighted the successful preclinical use of miRNA thera-peutics in various cardiac disease settings, including MIand ischemia-reperfusion injury (19, 24, 26, 51). Giventhe successful completion of a phase 2a clinical trial formiravirsen, a miRNA inhibitor for the treatment ofhepatitis C virus (10), there is potential for the success-ful development of miRNA therapeutics for cardiovas-cular disease. Furthermore, miRNAs are attractivedruggable targets because of their stability and con-served sequence (52). In the current study, we observedno adverse consequences of systemic delivery of anti-sense oligonucleotides in other tissues, assessed 8 wkafter administration. However, we cannot exclude thepossibility that knockdown of miR-652 over a longertime frame may cause pathology. Thus, a targeteddelivery approach may be preferable. Tissue-specificlong-lasting repression of miRNAs in vivo in othertissues has already been achieved with the use of viralvectors (53, 54). The potential relevance of miR-652 inhuman heart disease is highlighted by analysis fromexisting profiling datasets demonstrating increased ex-pression of miR-652 in left atrial appendage tissue frompatients with valvular heart disease compared tohealthy left atrial appendage tissue, and a trend(P�0.068) for increased miR-652 expression in hearts(LV) from heart failure patients with hypertrophiccardiomyopathy (compared to a nonfailing cohort;Supplemental Fig. S7 and refs. 55, 56). Finally, giventhe antifibrotic properties of LNA-antimiR-652 treat-ment, this therapy could be applied in other diseasesettings where fibrosis is a common feature (e.g., renalfibrosis and hepatic cirrhosis; ref. 57).
There are several limitations to this report. Thecurrent study has not defined which cardiac cell typesin the adult diseased heart express miR-652 and the cell
type in which miR-652 inhibition is providing benefit.However, given that miR-652 is expressed in neonatalrat cardiomyocytes and fibroblasts and human endothe-lial cell lines, inhibition of miR-652 has the potential toprovide protection in multiple cardiac cell types, andshould be examined in future studies. Next, the currentdata suggest that LNA-antimiR-652 is providing somedegree of protection by increasing Jagged1, althoughadditional work will be required to assess the degree ofbenefit and the cell types in which Jagged1 is providingprotection. Finally, we cannot exclude the possibilitythat at least some of the cardioprotective effects ofLNA-antimiR-652 were due to off-target or indirecteffects.
In summary, these studies provide the first demon-stration that inhibition of miR-652 can attenuate path-ological remodeling and improve cardiac function in amouse model of pressure overload with preexistingpathological hypertrophy and dysfunction, and revealthe first validated target of miR-652, Jagged1. Further-more, the approach of identifying and targeting candi-date miRNAs that are differentially regulated in settingsof protection and disease (regardless of basal expres-sion) may also prove valuable in identifying candidatemiRNAs for other diseases, including metabolic disor-ders such as obesity and diabetes.
This study was funded by National Health and MedicalResearch Council of Australia project grant 586603 (to J.R.Mand R.C.Y.L), and also supported in part by the VictorianGovernment’s Operational Infrastructure Support Program.X.J.D. and J.R.M. are National Health and Medical ResearchCouncil Senior Research Fellows (APP1043026 and 586604).J.R.M., R.C.Y.L., and P.G. are supported by an AustralianResearch Council Future Fellowship (FT0001657), a Univer-sity of New South Wales Vice Chancellor Research Fellowship,and a R.D. Wright Biomedical Research Fellowship from theNational Health and Medical Research Council (1046782),respectively. E.R.P. is supported by grants and fellowshipsfrom the National Health and Medical Research Council andthe National Heart Foundation (APP1033815 and 635530).The authors declare no conflicts of interest.
REFERENCES
1. Bernardo, B. C., Ooi, J. Y. Y., and McMullen, J. R. (2012) The yinand yang of adaptive and maladaptive processes in heart failure.Drug Disc. Today Ther. Strateg. 9, e163–e172
2. Bernardo, B. C., Weeks, K. L., Pretorius, L., and McMullen, J. R.(2010) Molecular distinction between physiological and patho-logical cardiac hypertrophy: experimental findings and thera-peutic strategies. Pharmacol. Ther. 128, 191–227
3. Huynh, K., Bernardo, B. C., McMullen, J. R., and Ritchie, R. H.(2014) Diabetic cardiomyopathy: mechanisms and new treat-ment strategies targeting antioxidant signaling pathways. Phar-macol. Ther. 142, 375–415
4. Lavie, C. J., Milani, R. V., and Ventura, H. O. (2009) Obesity andcardiovascular disease: risk factor, paradox, and impact ofweight loss. J. Am. Coll. Cardiol. 53, 1925–1932
5. Heidenreich, P. A., Trogdon, J. G., Khavjou, O. A., Butler, J.,Dracup, K., Ezekowitz, M. D., Finkelstein, E. A., Hong, Y.,Johnston, S. C., Khera, A., Lloyd-Jones, D. M., Nelson, S. A.,Nichol, G., Orenstein, D., Wilson, P. W. F., and Woo, Y. J. (2011)Forecasting the future of cardiovascular disease in the UnitedStates: a policy statement from the American Heart Association.Circulation 123, 933–944
5108 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org

6. Dickstein, K., Cohen-Solal, A., Filippatos, G., McMurray, J. J.,Ponikowski, P., Poole-Wilson, P. A., Stromberg, A., van Veldhu-isen, D. J., Atar, D., Hoes, A. W., Keren, A., Mebazaa, A.,Nieminen, M., Priori, S. G., and Swedberg, K.; European Societyof Cardiology Committee for Practice Guidelines (2008) ESCguidelines for the diagnosis and treatment of acute and chronicheart failure 2008: the task force for the diagnosis and treatmentof acute and chronic heart failure 2008 of the European Societyof Cardiology. Developed in collaboration with the Heart Fail-ure Association of the ESC (HFA) and endorsed by the Euro-pean Society of Intensive Care Medicine (ESICM). Eur. J. HeartFail. 10, 933–989
7. Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey, D. E.,Drazner, M. H., Fonarow, G. C., Geraci, S. A., Horwich, T.,Januzzi, J. L., Johnson, M. R., Kasper, E. K., Levy, W. C.,Masoudi, F. A., McBride, P. E., McMurray, J. J. V., Mitchell, J. E.,Peterson, P. N., Riegel, B., Sam, F., Stevenson, L. W., Tang,W. H. W., Tsai, E. J., and Wilkoff, B. L. (2013) 2013 ACCF/AHAguideline for the management of heart failure: a report of theAmerican College of Cardiology Foundation/American HeartAssociation Task Force on Practice Guidelines. Circulation 128,1810–1852
8. Dangwal, S., and Thum, T. (2014) microRNA therapeutics incardiovascular disease models. Annu. Rev. Pharmacol. Toxicol. 54,185–203
9. van Rooij, E., and Olson, E. N. (2012) MicroRNA therapeuticsfor cardiovascular disease: opportunities and obstacles. Nat. Rev.Drug Discov. 11, 860–872
10. Janssen, H. L., Reesink, H. W., Lawitz, E. J., Zeuzem, S.,Rodriguez-Torres, M., Patel, K., van der Meer, A. J., Patick, A. K.,Chen, A., Zhou, Y., Persson, R., King, B. D., Kauppinen, S.,Levin, A. A., and Hodges, M. R. (2013) Treatment of HCVinfection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694
11. Bernardo, B. C., Charchar, F. J., Lin, R. C. Y., and McMullen,J. R. (2012) A MicroRNA guide for clinicians and basic scien-tists: background and experimental techniques. Heart Lung Circ.21, 131–142
12. Hata, A. (2013) Functions of MicroRNAs in cardiovascularbiology and disease. Annu. Rev. Physiol. 75, 69–93
13. Ooi, J. Y. Y., Bernardo, B. C., and McMullen, J. R. (2014) Thetherapeutic potential of microRNAs regulated in settings ofphysiological cardiac hypertrophy. Future Med. Chem. 6, 205–222
14. Wang, X., Zhang, X., Ren, X. P., Chen, J., Liu, H., Yang, J.,Medvedovic, M., Hu, Z., and Fan, G. C. (2010) MicroRNA-494targeting both proapoptotic and antiapoptotic proteins protectsagainst ischemia/reperfusion-induced cardiac injury. Circulation122, 1308–1318
15. Thum, T., Gross, C., Fiedler, J., Fischer, T., Kissler, S., Bussen,M., Galuppo, P., Just, S., Rottbauer, W., Frantz, S., Castoldi, M.,Soutschek, J., Koteliansky, V., Rosenwald, A., Basson, M. A.,Licht, J. D., Pena, J. T., Rouhanifard, S. H., Muckenthaler,M. U., Tuschl, T., Martin, G. R., Bauersachs, J., and Engelhardt,S. (2008) MicroRNA-21 contributes to myocardial disease bystimulating MAP kinase signalling in fibroblasts. Nature 456,980–984
16. Van Rooij, E., Sutherland, L. B., Thatcher, J. E., DiMaio, J. M.,Naseem, R. H., Marshall, W. S., Hill, J. A., and Olson, E. N.(2008) Dysregulation of microRNAs after myocardial infarctionreveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci.U. S. A. 105, 13027–13032
17. van Rooij, E., Sutherland, L. B., Qi, X. X., Richardson, J. A., Hill,J., and Olson, E. N. (2007) Control of stress-dependent cardiacgrowth and gene expression by a microRNA. Science 316, 575–579
18. Callis, T. E., Pandya, K., Seok, H. Y., Tang, R.-H., Tatsuguchi, M.,Huang, Z.-P., Chen, J.-F., Deng, Z., Gunn, B., Shumate, J., Willis,M. S., Selzman, C. H., and Wang, D.-Z. (2009) MicroRNA-208ais a regulator of cardiac hypertrophy and conduction in mice. J.Clin. Invest. 119, 2772–2786
19. Care, A., Catalucci, D., Felicetti, F., Bonci, D., Addario, A., Gallo,P., Bang, M. L., Segnalini, P., Gu, Y. S., Dalton, N. D., Elia, L.,Latronico, M. V. G., Hoydal, M., Autore, C., Russo, M. A., Dorn,G. W., Ellingsen, O., Ruiz-Lozano, P., Peterson, K. L., Croce,C. M., Peschle, C., and Condorelli, G. (2007) MicroRNA-133controls cardiac hypertrophy. Nat. Med. 13, 613–618
20. Lin, R. C. Y., Weeks, K. L., Gao, X.-M., Williams, R. B. H.,Bernardo, B. C., Kiriazis, H., Matthews, V. B., Woodcock, E. A.,Bouwman, R., Mollica, J. P., Speirs, H. J., Dawes, I. W., Daly, R. J.,Shioi, T., Izumo, S., Febbraio, M. A., Du, X.-J., and McMullen,J. R. (2010) PI3K(p110�) protects against myocardial infarction-induced heart failure/Identification of PI3K-regulated miRNAsand mRNAs. Arterioscler. Thromb. Vasc. Biol. 30, 724–732
21. Matkovich, S. J., Hu, Y., and Dorn, G. W. (2013) Regulation ofcardiac microRNAs by cardiac microRNAs. Circ. Res. 113, 62–71
22. Weeks, K. L., Gao, X., Du, X. J., Boey, E. J., Matsumoto, A.,Bernardo, B. C., Kiriazis, H., Cemerlang, N., Tan, J. W., Tham,Y. K., Franke, T. F., Qian, H., Bogoyevitch, M. A., Woodcock,E. A., Febbraio, M. A., Gregorevic, P., and McMullen, J. R.(2012) Phosphoinositide 3-kinase p110alpha is a master regula-tor of exercise-induced cardioprotection and PI3K gene therapyrescues cardiac dysfunction. Circ. Heart. Fail. 5, 523–534
23. Shioi, T., Kang, P. M., Douglas, P. S., Hampe, J., Yballe, C. M.,Lawitts, J., Cantley, L. C., and Izumo, S. (2000) The conservedphosphoinositide 3-kinase pathway determines heart sizein mice. EMBO J. 19, 2537–2548
24. Bernardo, B. C., Gao, X. M., Winbanks, C. E., Boey, E. J., Tham,Y. K., Kiriazis, H., Gregorevic, P., Obad, S., Kauppinen, S., Du,X. J., Lin, R. C., and McMullen, J. R. (2012) Therapeuticinhibition of the miR-34 family attenuates pathological cardiacremodeling and improves heart function. Proc. Natl. Acad. Sci.U. S. A. 109, 17615–17620
25. Ng, D. C. H., Ng, I. H. W., Yeap, Y. Y. C., Badrian, B., Tsoutsman,T., McMullen, J. R., Semsarian, C., and Bogoyevitch, M. A.(2011) Opposing actions of extracellular signal-regulated kinase(ERK) and signal transducer and activator of transcription 3(STAT3) in regulating microtubule stabilization during car-diac hypertrophy. J. Biol. Chem. 286, 1576–1587
26. Porrello, E. R., Mahmoud, A. I., Simpson, E., Johnson, B. A.,Grinsfelder, D., Canseco, D., Mammen, P. P., Rothermel, B. A.,Olson, E. N., and Sadek, H. A. (2013) Regulation of neonataland adult mammalian heart regeneration by the miR-15 family.Proc. Natl. Acad. Sci. U. S. A. 110, 187–192
27. Bernardo, B. C., Gao, X.-M., Tham, Y. K., Kiriazis, H., Winbanks,C. E., Ooi, J. Y. Y., Boey, E. J. H., Obad, S., Kauppinen, S.,Gregorevic, P., Du, X.-J., Lin, R. C. Y., and McMullen, J. R.(2014) Silencing of miR-34a attenuates cardiac dysfunction in asetting of moderate, but not severe, hypertrophic cardiomyop-athy. PLoS. ONE. 9, e90337
28. Li, X. M., Ma, Y. T., Yang, Y. N., Liu, F., Chen, B. D., Han, W.,Zhang, J. F., and Gao, X. M. (2009) Downregulation of survivalsignalling pathways and increased apoptosis in the transition ofpressure overload-induced cardiac hypertrophy to heart failure.Clin. Exp. Pharmacol. Physiol. 36, 1054–1061
29. Lei, B., Chess, D. J., Keung, W., O’Shea, K. M., Lopaschuk, G. D.,and Stanley, W. C. (2008) Transient activation of p38 MAPkinase and up-regulation of Pim-1 kinase in cardiac hypertrophydespite no activation of AMPK. J. Mol. Cell. Cardiol. 45, 404–410
30. Croquelois, A., Domenighetti, A. A., Nemir, M., Lepore, M.,Rosenblatt-Velin, N., Radtke, F., and Pedrazzini, T. (2008)Control of the adaptive response of the heart to stress via theNotch1 receptor pathway. J. Exp. Med. 205, 3173–3185
31. Gude, N., and Sussman, M. (2012) Notch signaling and car-diac repair. J. Mol. Cell. Cardiol. 52, 1226–1232
32. Gude, N. A., Emmanuel, G., Wu, W., Cottage, C. T., Fischer, K.,Quijada, P., Muraski, J. A., Alvarez, R., Rubio, M., Schaefer, E.,and Sussman, M. A. (2008) Activation of Notch-mediated pro-tective signaling in the myocardium. Circ. Res. 102, 1025–1035
33. Nemir, M., Metrich, M., Plaisance, I., Lepore, M., Cruchet, S.,Berthonneche, C., Sarre, A., Radtke, F., and Pedrazzini, T.(2012) The Notch pathway controls fibrotic and regenerativerepair in the adult heart. [E-pub ahead of print] Eur. Heart J.doi: 10.1093/eurheartj/ehs269
34. Niessen, K., and Karsan, A. (2008) Notch signaling in car-diac development. Circ. Res. 102, 1169–1181
35. Campa, V. M., Gutierrez-Lanza, R., Cerignoli, F., Diaz-Trelles,R., Nelson, B., Tsuji, T., Barcova, M., Jiang, W., and Mercola, M.(2008) Notch activates cell cycle reentry and progression inquiescent cardiomyocytes. J. Cell Biol. 183, 129–141
36. Benedito, R., Roca, C., Sorensen, I., Adams, S., Gossler, A.,Fruttiger, M., and Adams, R. H. (2009) The notch ligands Dll4and Jagged1 have opposing effects on angiogenesis. Cell 137,1124–1135
5109THERAPEUTIC POTENTIAL OF MIR-652 IN THE HEART

37. Collesi, C., Zentilin, L., Sinagra, G., and Giacca, M. (2008)Notch1 signaling stimulates proliferation of immature cardio-myocytes. J. Cell Biol. 183, 117–128
38. Boström, P., Mann, N., Wu, J., Quintero, P. A., Plovie, E. R.,Panáková, D., Gupta, R. K., Xiao, C., MacRae, C. A., Rosenzweig,A., and Spiegelman, B. M. (2010) C/EBP beta controls exercise-induced cardiac growth and protects against pathological car-diac remodeling. Cell 143, 1072–1083
39. Obad, S., dos Santos, C. O., Petri, A., Heidenblad, M., Broom,O., Ruse, C., Fu, C., Lindow, M., Stenvang, J., Straarup, E. M.,Hansen, H. F., Koch, T., Pappin, D., Hannon, G. J., andKauppinen, S. (2011) Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 43, 371–378
40. Ikeda, S., Kong, S. W., Lu, J., Bisping, E., Zhang, H., Allen, P. D.,Golub, T. R., Pieske, B., and Pu, W. T. (2007) Altered microRNAexpression in human heart disease. Physiol. Genomics 31, 367–373
41. Ikeda, S., and T. Pu, W. (2010) Expression and function ofmicroRNAs in heart disease. Curr. Drug Targets 11, 913–925
42. Kumarswamy, R., and Thum, T. (2013) Non-coding RNAs incardiac remodeling and heart failure. Circ. Res. 113, 676–689
43. Rockman, H. A., Ross, R. S., Harris, A. N., Knowlton, K. U.,Steinhelper, M. E., Field, L. J., Ross, J., Jr., and Chien, K. R.(1991) Segregation of atrial-specific and inducible expression ofan atrial natriuretic factor transgene in an in vivo murine modelof cardiac hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 88, 8277–8281
44. Oda, T., Elkahloun, A. G., Pike, B. L., Okajima, K., Krantz, I. D.,Genin, A., Piccoli, D. A., Meltzer, P. S., Spinner, N. B., Collins,F. S., and Chandrasekharappa, S. C. (1997) Mutations in thehuman Jagged1 gene are responsible for Alagille syndrome. Nat.Genet. 16, 235–242
45. Li, L., Krantz, I. D., Deng, Y., Genin, A., Banta, A. B., Collins,C. C., Qi, M., Trask, B. J., Kuo, W. L., Cochran, J., Costa, T.,Pierpont, M. E., Rand, E. B., Piccoli, D. A., Hood, L., andSpinner, N. B. (1997) Alagille syndrome is caused by mutationsin human Jagged1, which encodes a ligand for Notch1. Nat.Genet. 16, 243–251
46. Taimeh, Z., Loughran, J., Birks, E. J., and Bolli, R. (2013)Vascular endothelial growth factor in heart failure. Nat. Rev.Cardiol. 10, 519–530
47. Shiojima, I., Sato, K., Izumiya, Y., Schiekofer, S., Ito, M., Liao, R.,Colucci, W. S., and Walsh, K. (2005) Disruption of coordinatedcardiac hypertrophy and angiogenesis contributes to the transi-tion to heart failure. J. Clin. Invest. 115, 2108–2118
48. Meloni, M., Marchetti, M., Garner, K., Littlejohns, B., Sala-Newby, G., Xenophontos, N., Floris, I., Suleiman, M. S.,Madeddu, P., Caporali, A., and Emanueli, C. (2013) Localinhibition of MicroRNA-24 improves reparative angiogenesisand left ventricle remodeling and function in mice with myo-cardial infarction. Mol. Ther. 21, 1390–1402
49. Gridley, T. (2007) Notch signaling in vascular developmentand physiology. Development 134, 2709–2718
50. Xue, Y., Gao, X., Lindsell, C. E., Norton, C. R., Chang, B., Hicks,C., Gendron-Maguire, M., Rand, E. B., Weinmaster, G., andGridley, T. (1999) Embryonic lethality and vascular defects inmice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8,723–730
51. Hullinger, T. G., Montgomery, R. L., Seto, A. G., Dickinson,B. A., Semus, H. M., Lynch, J. M., Dalby, C. M., Robinson, K.,Stack, C., Latimer, P. A., Hare, J. M., Olson, E. N., and van Rooij,E. (2012) Inhibition of miR-15 protects against cardiac ischemicinjury/novelty and significance. Circ. Res. 110, 71–81
52. Stenvang, J., Petri, A., Lindow, M., Obad, S., and Kauppinen, S.(2012) Inhibition of microRNA function by antimiR oligonucle-otides. Silence 3, 1
53. Winbanks, C. E., Beyer, C., Hagg, A., Qian, H., Sepulveda, P. V.,and Gregorevic, P. (2013) miR-206 represses hypertrophy ofmyogenic cells but not muscle fibers via inhibition of HDAC4.PLoS One 8, e73589
54. Ebert, M. S., and Sharp, P. A. (2010) MicroRNA sponges:progress and possibilities. RNA 16, 2043–2050
55. Cooley, N., Cowley, M. J., Lin, R. C. Y., Marasco, S., Wong, C.,Kaye, D. M., Dart, A. M., and Woodcock, E. A. (2012) Influenceof atrial fibrillation on microRNA expression profiles in left andright atria from patients with valvular heart disease. Physiol.Genomics 44, 211–219
56. Leptidis, S., El Azzouzi, H., Lok, S. I., de Weger, R., Olieslagers,S., Kisters, N., Silva, G. J., Heymans, S., Cuppen, E., Berezikov,E., De Windt, L. J., and da Costa Martins, P. (2013) A deepsequencing approach to uncover the miRNOME in the hu-man heart. PLoS One. 8, e57800
57. Friedman, S. L., Sheppard, D., Duffield, J. S., and Violette, S.(2013) Therapy for fibrotic diseases: nearing the starting line.Sci. Transl. Med. 5, 167sr161
Received for publication March 26, 2014.Accepted for publication August 4, 2014.
5110 Vol. 28 December 2014 BERNARDO ET AL.The FASEB Journal � www.fasebj.org