the molecular genetics of colon cancer
TRANSCRIPT

Molecular genetics of colon cancer
Principal Investigator:
Margareta Nordling
Picture from: http://hubpages.com/hub/DNA-Testing-
for-Disease-Prevention
_____________________________________________________________
Aims of the project
1. To make significant international contributions in elucidating the genetic
background of attenuated colon polyposis (AFAP), based on regional and national
patient cohorts.
2. To make personalized medicine possible for our patients, giving them a reliable
diagnosis, possibility of prognosis, adequate follow up, assessing the risk for
future generations and targeted therapy.
3. Contribute to increased understanding of variations in the genome and their effect
on the patient emphasizing the hereditary as well as the sporadic aspects of the
diseases, including investigations to find new disease-causing genes.
4. To implement advanced molecular genetics methodology in the clinic enabling
high-quality care for the patients.
____________________________________________________________

Background About 3 to 5% of people who develop colorectal cancer have an inherited genetic
susceptibility to the disease. Two syndromes are associated with the major part of
inherited colorectal cancer syndrome, hereditary non-polyposis colorectal cancer
(HNPCC) and familial adenomatous polyposis (FAP). In only 15-20% of the families
with HNPCC a deleterious mutation in one of the 3 predominant HNPCC genes can be
found. In families with FAP it is today possible to find almost all the mutations
responsible for the classical polyposis syndrome. However, in the attenuated form of the
disease (AFAP), with e.g. less polyps and higher age at onset, only a fraction of the
disease-causing mutations can be identified. The low detection rate implicates that
probably several responsible genes are still to be identified which also means that it is
important to study families from defined geographical regions as it might be that the
genetic cause of this disease is dependent on the origin of the families.
Elucidating the genetic background of hereditary cancer also has implications for the
understanding of the development of sporadic cancer. Besides the fact that germline
inactivation of APC is attributable to FAP, the gene is involved in the development of
sporadic colorectal cancers as it is inactivated in almost all sporadic colon tumours.
The gene has attracted vast interest since it encodes a multifunctional protein that
constantly is assigned new functions crucial for the function of the cell. Due to the
variable nature of the FAP syndrome extensive studies have been performed in trying to
elucidate the correlation between genotype and phenotype in this disease. The
manifestation of the disease can vary from the milder, attenuated form of disease (AFAP)
to a full classical phenotype with thousands of polyps with various degrees of
extraintestinal manifestations.
We and others have performed extensive studies of detecting mutations and performing
associations with specific genotypes and phenotypes in the FAP syndrome [1-4]. The
phenotypic expression of FAP is clearly dependant on the activity of the APC gene and
probably also on modifier genes. It has recently been shown that APC could be regulated
by alternative splicing perhaps thorough a mechanism involving cis-acting regulatory
RNA elements, trans-acting regulatory proteins and the NMD (non-sense mediated
decay) machinery. Although the functions of most spliced APC isofoms are not yet well
defined they could exert an antagonistic function versus the tumor-supressor activity of
the APC protein, thereby controlling tumorgenesis, differentiation and the development
of FAP [5].
FAP was originally assigned 100% penetrance in mutation carriers. However, the fact
that a very limited number of full mutation carriers, never develop any sign or are only
very mildly affected by the disease, has recently attracted attention. The possibility that
the FAP phenotype can be modulated by modifier genes has been proposed. To
understand how, and to what extend, the various mutations effect the function of the APC
mediated cell mechanisms, is important not only for studies on FAP, but also for the
general understanding of colon tumor development. For the same reason it is also crucial
to try to begin to understand how modifier genes can affect the disease phenotype. Today
we have the technical prerequisites to be able to perform experiments that could clarify

the differences in the genome, transcriptome and other factors affecting the expression
and function of the deleterious gene between mutation carriers that are affected by the
disease and those who are not.
The field of Clinical Genetics stands in front of a new era enabled by the introduction of
new sequencing technology. The recent introduction of Next-generation sequencing
(NGS) capable of producing millions of parallel DNA sequences is rapidly changing the
landscape of genetics, bringing new knowledge from basic biology to the genetic aspects
of personalised medicine. Already today and during the next few years there will be an
enormous focus on revealing the genetics behind hereditary disease, both rare and
common, and our unique patient material gives us a special opportunity to make
significant contributions to this research field.
Since instruments for NGS was introduced about three years ago, an increasing amount
of research publications have brought evidence for the usefulness of the technique also in
the direct practice of clinical genetics. The main sector of application for NGS in clinical
genetics (presently) is to find new disease-causing genes and to perform simultaneous
high-throughput analyses of an array of genes causing the same disease. Many more
applications such as detection of low-frequency alleles by ultra-deep NGS (meaning NGS
producing especially many sequence reads per sample) and detection of copy-number
variations are soon to come, improving the possibilities of personalized medicine both in
the case of hereditary cancer but also making analyses of somatic genetic variations in
sporadic cancer possible. These new possibilities will gradually replace the methodology
used in clinical genetics today and will also provide completely new knowledge about the
extent and significance of genetic variations in human diseases (both hereditary and
sporadic).
Material and methods Material
Patients and ethical approval
The majority of the patients included in the projects resides from the Cancer Genetic
Counselling Clinic at department of Oncology, SU/Sahlgrenska, department of Surgery,
Skaraborg Hospital and from the national Swedish Polyposis Registry, department. of
Medicine, Karolinska University Hospital.
The colon cancer project has very recently (august 2010) gained extended ethical
approval for the research connected to new sequencing and array methodology as this
methods holds the capacity to search the whole genome.
The research group and collaborations
The research group includes; one PhD student (AR) and one “biolog” both currently
working part time. We would like to extend the possibilities of having more lab personnel
and post-docs working in the project. The bioinformatics/biostatistic data generated in the
project is managed by Staffan Nilsson. Characterization of proteins will be performed by
Göran Karlsson. Targeted enrichment (e.g. exome enrichments) and NGS will initially be

outsourced to commercial labs e.g. GATC, Konstanz, Germany and others. The group of
From 2011 we will have the possibilities to perform these experiments in house (see
below).
Regional collaborations:
1. Dept of Surgery, Skaraborg Hospital (Stefan Skullman).
2. Doctors at several different hospitals in the Västra Götaland region based on
where the families live.
National collaborations;
1. The Swedish Polyposis Registry at the Dept. of Gastroenterology and Hepatology,
Karolinska University Hospital (Jan Björk).
International collaborations:
1. Barbara Graves, Dept of Oncological Sciences, University of Utah and Huntsman
Cancer Institute.
Equipment and research environment
The department of Clinical Genetics where the main part of the research is performed is
equipped with all basic and expensive instruments needed to conduct the basic research
of the project such as a variety of different PCR instruments, two ABI 3130xl sequencers
and an Affymetrix microarray scanner. Our sequencing capacity is already high but with
the availability of the facilities at Genomics Core Facility at SA, located in the same
building, we have extensive possibilities. The environment at the department of Clinical
Genetics provides extended knowledge of a variety of basic and sophisticated methods
e.g. mutation detection where a number of techniques are in daily use and various
microarray analyses etc. The Genomics Core Facility (located in the same building) also
provides access to a number of other instruments such as TaqMan for Q-PCR on the ABI
7900HT platform. In the beginning of 2011 (or earlier) an NGS platform including
instrumentation, laborative and bioinformatics support will be launched at the Genomics
Core Facility. The platform is a joint effort between the Sahlgrenska Academy and SU.
The main applicant (MN) of this research application is a member of the advisory group
for the establishment of the platform. Our department will be one of the main users of
this platform.
Methods
Mutation screening
For the next years to come the focus on development will be on NGS and its applications
but already today we have a number of different techniques for genome analyses running
in the lab that also will be used in the project. We perform mutation screening with a
variety of screening methods including e.g. MLPA (multiplex ligation-dependent probe
amplification) to detect deletions/duplications in genes and a variety of specific PCR-
based methods used to achieve as high mutation-detection frequency as possible (both on
DNA and cDNA level). Screening for mosaic mutations (using ultra-deep NGS) and

determination of lowered mRNA expression (microarray and Q-PCR) levels indicative of
mutations or epimutations are also performed. DNA sequencing (DNA and mRNA-
based) is used as a screening method but also for confirmation. We are also using MLPA
and bisulphate-modified sequencing to analyze for epigenetic modifications such as
aberrations in methylation pattern and we will also be analyzing for aberrations caused by
impaired histone acetylation. Using microarray (Affymetrix) analyses we presently detect
pathogenic CNVs and SNP in the genome. The SNP data is used for linkage analyses by
conventional methods and haplotype based homozygosity mapping. We are also using
this platform to analyze for specific transcript expression as described in Preliminary
results.
Next-generation sequencing
The NGS technique will bring us a number of new possibilities to perform genome
analyses. The NGS technique allows for whole-genome analyses and this was also the
purpose for developing the methodology. However, whole genome analysis is today not
feasible for most purposes due to cost, massive data handling and ethical considerations.
The technique is preferably used for analyzing targeted regions of the genome. Targeted
whole-genome exome sequencing where all coding exons in the genome are amplified
will be introduced as soon as possible. This method now begins to be widely used for
finding disease causing genes both in common and rare diseases. The methodology to
find new genes in this way includes massive bioinformatic handling, a service planned to
be provided by Genomics Core Facility but we also hope for possibilities to manage this
within the project. Staffan Nilsson from the dept of Statistics at Chalmers who is a
member of the research group will be important for this part of the project. Apart from
NGS a number of methods already running in the lab will be used when it comes to
characterize and select detected genetic variants.
We are already introducing ultra-deep NGS for detection of mutations that are only
present in a fraction of the cells of the patients, i.e. mosaic detection or low-frequency
allele detection. Before NGS there was no reliable method for this purpose. Our previous
study by Rohlin et al. [8] revealed detectable mutation levels down to 1 % using this
technique.
A number NGS applications will be used in the project ahead e.g. whole transcriptome
analyses and whole genome sequencing. A number of new databases built up from NGS
data, e.g. The 1000 Genomes Project
(http://en.wikipedia.org/wiki/1000_Genomes_Project, an international effort to establish
an overview of all genetic variations in humans compiled from 1000 human genomes)
and the Bejing Genome Institute (http://www.genomics.cn/en/index.php) make their data
freely available and we will also use these in the project.

Outline of the project. As suggested we will concentrate our future studies on a limited part of the project
as outlined below:
1. Identification of new genes causing AFAP
2. Studies on the genetic background of the variable expressivity of the diseases and
making an attempt to understand why some individuals are not affected by disease even
though they are full mutation carriers.
3. Contribution of inactivated tumor suppressor genes (e.g. the APC gene) to the
development of sporadic colon cancer. Does presence of low-frequency (mosaicism)
mutations in individuals contribute to the development of sporadic colon cancer?
Workplan 1. Identification of new genes
As described earlier we are interested in finding the genetic cause of disease for AFAP.
To accomplish this we have initiated analyses of patients using microarray-analyses both
for gene dosage (SNP 6.0 Array) and for transcript mapping (Exon 1.0 Array). Targeted
whole-genome exome sequencing with the NGS will be used to indentify causative genes
as outlined above. Apart from NGS a number of methods already running in the lab will
be used when it comes to characterize and select detected genetic variants. If linkage data
has to be improved we will use the high density arrays (Affymetrix 6.0 arrays) that are
now available. From in 2006 being able to genotype 10 000 SNPs on these arrays we
have today the possibility to genotype more than 900 0000 SNPs on one single array
which gives us much better possibilities to find the chromosomal location for a new
causative gene. We also have access to tumors which can be used to analyze for copy
number variants (CNV). Detection of CNVs compares intensities of CNV probes in
tumor DNA to probe intensities in normal DNA in a way to identify deleted or amplified
chromosomal regions in the genome. Deleted regions may indicate a tumor suppressor or
DNA repair gene involved in cancer development and amplified regions could be the
indication of an activated oncogene. We also use exon specific probes (as described in
section 3 below) to find varied expression patterns that could be due to an inactivated
disease-causing gene. Our intention is to investigate if patients without detectable
mutations possibly can carry new pathogenic isoforms from transcription of known
cancer-predisposing genes and also from other genes that could be implicated in tumor
development. In this way we will try to find clues to new mutations and predisposing
cancer genes. These studies are still on-going and will be continued during 2011.
2. Transcriptome studies using the Affymetrix GeneChip® Human Exon 1.0 ST
array in patients with variable expressivity of the diseases
The Human Exon 1.0 ST arrays contain approximately four probes per exon and roughly
40 probes per each known gene in the genome (over 5 million probes grouped into over
300,000 transcript clusters) and enables two complementary levels of analysis, total gene

expression from all known genes and alternative splicing from each specific gene. With
this platform it is possible to distinguish between different isoforms.
We will investigate if there are differences in expression from the whole genome in
families where a defined variable expression of disease is observed between family
members. In this way we will try to find out which genes that are able of modulating the
disease to be more severe or to be “silenced”. We will try to pin-point different genes or
isoforms that has an affect on the development of the disease in an attempt to try to
understand which mechanisms that triggers disease on-set and which that suppress
disease development. We have made the first analyses and are continuously collecting
samples for the study
3. APC mosaicism
We are using ultra-deep NGS to detect mosaic mutations in tumour, blood and normal
cell mucosa from patients with sporadic colon cancer. Our previous study by Rohlin et al.
[8] revealed detectable mutation levels down to 1 % using this technique. An additional
subject to study with this in background is the presence of epimutations that are
frequently mosaic within the individual, and inheritance in these cases is often weak.
Previous work and preliminary results Our previous work in the project is described in references [1, 3, 4, 6, 7, 8, 9].
1a. Identification of a novel mutation in Family 1 of the Swedish Polypos isRegistry.
In collaboration with The Swedish Polyposis Registry we have for several years been
working with the largest FAP family in Sweden in trying to find the disease-causing
mutation. This mutation has confounded us for many years and exhausted our
competence regarding mutation detection. Thanks to the high-density CNV arrays
(Affymetrix 6.0) that now are available we finally found the elusive disease-causing
mutation in spring this year. Initially we found one deletion far up-stream of the gene in a
region of no known regulatory significance for the APC gene. After a whole lot of
pondering and work we realized that the deletion was connected to a second deletion
separated by only 1500 base pairs impossible to detect on the array data. This second
deletion included approximately half of promoter 1B of the APC gene (Fig. 1). Apart
from being the first disease-causing mutation ever to be found in this region of the gene
the finding led to several new insights about the regulation of the APC gene that is
presented in the attached manuscript by Rohlin et al. [9]. Briefly, we had earlier observed
lowered expression of the disease causing allele in affected patients which led us to the
conclusion that promoter 1B is more important for the APC gene then generally
considered (which also was verified with expression studies). We could also suggest that
this lowered expression could protect carriers from development of desmoid tumours
which is a major cause of morbidity and mortality in FAP patients. Studies of adenomas
from the family also revealed interesting observations regarding the function of
inactivation of APC in tumorigenesis. As our project focuses on direct clinically relevant
issues the future studies regarding the function of APC in this family will be carried out
in collaboration with Prof Barbara Foulkes at Huntsman Cancer Institute, Utah. The APC

gene was identified at this institution many years ago and as the gene today is the most
studied gene in colon tumors both hereditary and sporadic, the Utah group has shown
considerable interest in our work.
Figure 1. Schematic illustration of the deletion in Family 1. The second deletion (Del 2)
includes 320 bp of promoter 1B (586 bp as a whole) of the APC gene.
1b. Identification of new genes
As described earlier we are interested in finding the genetic cause of disease for AFAP.
To accomplish this we have initiated analyses of patients using microarray-analyses both
for gene dosage (SNP 6.0 Array) and for transcript mapping (Exon 1.0 Array). Using
these methods we identified two genes (Fig. 2) with potential to be involved in the
disease. We will continue our studies of a large family from the Västra Götaland region
where we have localised a possible deleterious gene on chromosome 5 with a LOD score
of 2.7 and we will use the NGS approach including targeted amplification of exons with
subsequent bioinformatic filtering against known sequence variants to try to find the
causative gene. We are also continuously collecting families with this disease to use for
new-gene. These studies are still on-going and will be continued during 2011.
2. Patients with variable expressivity of the FAP diseases
We have made initial analyses using Affymetrix GeneChip® Human Exon 1.0 ST Array
on family members with reduced expression of the disease compared to members with a
classical polyposis. The analyses of these experiments are on going.
3. APC mosaicism As we established that very low levels of mosaic mutations could be detected [8] we
undertook a study on a material constituting of blood, tumour and normal tissue from
patients with sporadic colon cancer. The reason was that we previously had identified a
mosaic mutation in a patient with a very mild FAP disease [6], resembling sporadic colon
cancer, so our interest was to examine if sporadic colon cancer could be caused by
constitutional mosaic mutations. In fact, ultra-deep NGS did find very low levels of
mutation in blood and normal tissue in approximately 10% of the patients. The analyses
where performed by the Metagenomic facility at KTH, Stockholm. As these results are
very interesting and certainly will be questioned we decided to reanalyze some of the
samples at a different facility. These studies are now ongoing.
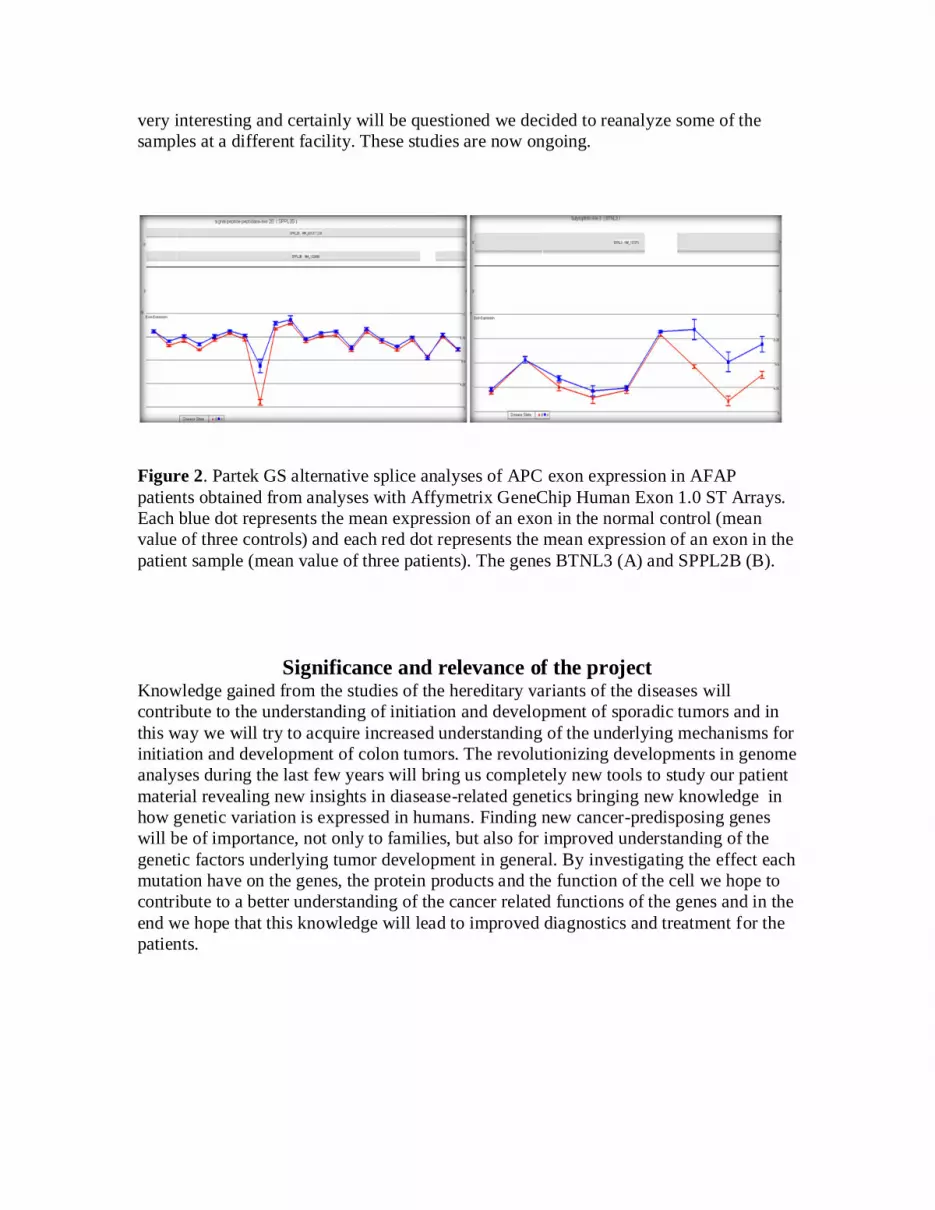
Figure 2. Partek GS alternative splice analyses of APC exon expression in AFAP
patients obtained from analyses with Affymetrix GeneChip Human Exon 1.0 ST Arrays.
Each blue dot represents the mean expression of an exon in the normal control (mean
value of three controls) and each red dot represents the mean expression of an exon in the
patient sample (mean value of three patients). The genes BTNL3 (A) and SPPL2B (B).
Significance and relevance of the project Knowledge gained from the studies of the hereditary variants of the diseases will
contribute to the understanding of initiation and development of sporadic tumors and in
this way we will try to acquire increased understanding of the underlying mechanisms for
initiation and development of colon tumors. The revolutionizing developments in genome
analyses during the last few years will bring us completely new tools to study our patient
material revealing new insights in diasease-related genetics bringing new knowledge in
how genetic variation is expressed in humans. Finding new cancer-predisposing genes
will be of importance, not only to families, but also for improved understanding of the
genetic factors underlying tumor development in general. By investigating the effect each
mutation have on the genes, the protein products and the function of the cell we hope to
contribute to a better understanding of the cancer related functions of the genes and in the
end we hope that this knowledge will lead to improved diagnostics and treatment for the
patients.

References: 1. Bjork J, Akerbrant H, Iselius L, Bergman A, Engwall Y, Wahlstrom J, Martinsson T,
Nordling M, Hultcrantz R: Periampullary adenomas and adenocarcinomas in familial
adenomatous polyposis: cumulative risks and APC gene mutations. Gastroenterology 2001, 121(5):1127-1135.
2. Jass JR: Colorectal polyposes: from phenotype to diagnosis. Pathology, research and
practice 2008, 204(7):431-447. 3. Kanter-Smoler G, Bjork J, Fritzell K, Engwall Y, Hallberg B, Karlsson G, Gronberg H,
Karlsson P, Wallgren A, Wahlstrom J, Hultcrantz R, Nordling M: Novel findings in
Swedish patients with MYH-associated polyposis: mutation detection and clinical
characterization. Clin Gastroenterol Hepatol 2006, 4(4):499-506. 4. Nordling M, Engwall Y, Wahlstrom J, Wiklund L, Eriksson MA, Gustavsson B, Fasth S,
Larsson PA, Martinsson T: Novel mutations in the APC gene and clinical features in
Swedish patients with polyposis coli. Anticancer Res 1997, 17(6D):4275-4280.
5. De Rosa M, Scarano MI, Panariello L, Carlomagno N, Rossi GB, Tempesta A, Borgheresi P, Renda A, Izzo P: Three submicroscopic deletions at the APC locus and
their rapid detection by quantitative-PCR analysis. Eur J Hum Genet 1999, 7(6):695-703.
6. Kanter-Smoler G, Fritzell K, Rohlin A, Engwall Y, Hallberg B, Bergman A, Meuller J, Gronberg H, Karlsson P, Bjork J, Nordling M: Clinical characterization and the
mutation spectrum in Swedish adenomatous polyposis families. BMC medicine 2008,
6:10. 7. Meuller J, Kanter-Smoler G, Nygren AO, Errami A, Gronberg H, Holmberg E, Bjork J,
Wahlstrom J, Nordling M: Identification of genomic deletions of the APC gene in
familial adenomatous polyposis by two independent quantitative techniques. Genet Test 2004, 8(3):248-256.
8. Rohlin A, Wernersson J, Engwall Y, Wiklund L, Bjork J, Nordling M: Parallel
sequencing used in detection of mosaic mutations: comparison with four diagnostic
DNA screening techniques. Hum Mutat 2009, 30(6):1012-1020. 9. Rohlin et al., Alteration of promoter 1B of APC in classical adenomatous polyposis-
predictor of low desmoid risk. Submitted