the abstract book of 2nd sickle cell & thalassaemia virtual
TRANSCRIPT

www.hemaspherejournal.com
2nd Sickle Cell & Thalassaemia2nd Sickle Cell & ThalassaemiaVirtual ConferenceVirtual Conference
ABSTRACT BOOKABSTRACT BOOK

2nd Sickle Cell & Thalassaemia Virtual Conference
January 26-28, 2022
ABSTRACT BOOK

Author Information
The Abstract Book of 2nd Sickle Cell & Thalassaemia Virtual Conference is published as a supplement of HemaSphere and available online at www.hemaspherejournal.com.
HemaSphere is an open access journal powered by the European Hematology Association and dedicated to support hematology patient care, research, and education worldwide.
Please read our instructions and guidelines for preparing and submitting articles. Manuscripts must be submitted online at http://www.editorialmanager.com/hemasphere.
Article Processing Charges (APC)
Article types EHA members Non-members
Tier 1: Original articles, review articles, guidelines, & controversies 50%** €1,059* equivalent ($1,250)
Tier 2: Letters, case reports, editorials, & perspectives 50%** €530* equivalent ($625)
Tier 3: Comments, HemaTopics Free Free
All invited manuscripts*** (Invited review articles, invited guidelines, invited editorials, and invited perspectives)
Free Free
*USD/EUR equivalent rate of 1.180, please note that all payments will be in USD.**2022 Promotional Discounts on APCs: 50% discount for EHA members (first and last author of a manuscript) at the time of submission***APCs for all contributions on invitation by the Editors will be waived.
Copyright Information
(Online) ISSN: 2572-9241© 2022 the Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the European Hematology Association. This is an open access Abstract Book distributed under the Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) which allows third parties to download the articles and share them with others as long as they credit the author and the Abstract Book, but they cannot change the content in any way or use them commercially.
Rights and Permissions
Please refer to https://journals.lww.com/hemasphere/Pages/reprints.aspx for reprints and order of copies of the Abstract Book. Questions regarding permissions should be addressed to [email protected].
Abstract Book Citations: Authors, Title, HemaSphere, 2022;6:(S1), Abstract Book, DOI: http://dx.doi.org/10.1097/HS9.0000000000000566
Disclaimer
Articles published in the journal HemaSphere exclusively reflect the opinions of the authors. The authors are responsible for all content in their abstracts including accuracy of the facts, statements, citing resources, etc.

HemaSphere Editorial Board
Co-Editor-in-ChiefAndreas Engert, MDUniversity of Cologne, Germany
Co-Editor-in-ChiefJan Cools, PhDVIB-KU Leuven Center for Cancer Biology, Belgium
Associate Editors
Stephen Ansell, Mayo Clinic, Rochester, Minnesota, United StatesPaolo Corradini, Fondazione IRCCS - Istituto Nazionale dei Tumori, Milan, ItalyMartin Dreyling, Klinikum der Universität München, GermanyJeroen Eikenboom, Leiden University Medical Centre, The NetherlandsPaolo Ghia, Università Vita-Salute San Raffaele, Milan, ItalySimon Hallam, St. Bartholomew’s Hospital, Barts Health NHS Trust, London, United KingdomClaire Harrison, Guy’s and St Thomas’ NHS Foundation Trust, London, United KingdomRobert Hills, University of Oxford, United KingdomMartina Muckenthaler, University of Heidelberg, GermanyJürg Schwaller, University Hospital Basel, SwitzerlandEvangelos Terpos, University of Athens, Greece
Scientific Editors
Stephen Hibbs, St. Bartholomew’s Hospital, Barts Health NHS Trust, London, United KingdomDavid Kent, University of York, United KingdomRoger Schutgens, University Medical Center Utrecht, The NetherlandsMelania Tesio, Institute Necker des Enfantes Malades, Paris, FranceFrancesca Vinchi, Iron Research Laboratory - New York Blood Center, & Weill Cornell Medicine - Pathology and Laboratory Medicine, New York, United States
Editorial Manager, European Hematology Association
Jessica [email protected]
Editorial Coordinator, Wolters Kluwer
Samantha [email protected]

Organization
The European Hematology Association (EHA), British Society of Haematology (BSH) & Annual Academy of Sickle Cell and Thalassaemia Conference (ASCAT) joined forces again to organize the 2nd Sickle Cell & Thalassaemia Virtual Conference.
Abstract Reviewers
The organizers would like to thank the following experts for their time and efforts reviewing abstracts for this meeting.
Abboud M, LebanonAndemariam B, United StatesAnie K, United KingdomCampbell A, United StatesCappellini MD, ItalyChakravorty S, United KingdomColombatti R, Italyde Montalembert M, FranceInusa B, United KingdomJerlym Porter J, United StatesKesse-Adu R, United KingdomMiller R, United KingdomTaher A, LebanonTreadwell M, United States

Word of welcome
On behalf of the Annual Scientific Conference on Sickle Cell and Thalassaemia (ASCAT), the European Hematology Association (EHA), and the British Society for Haematology (BSH), we are pleased to present our 2022 Abstract Book. This year’s theme is ‘Improving the lives of people living with Sickle Cell Disease and Thalassaemia’.
The Annual Scientific Conference on Sickle Cell and Thalassaemia is one of the must attend events of the year for consultants and specialist psychologists, nurses, scientists and all relevant experts. The event is an ideal opportunity to see the latest advances in diagnosis, treatment and emerging fields in haemoglobinopathies. It is an opportunity to interact on the latest advances in clinical care, transition services and emerging new therapies including updates for curative treatment options. Abstract and poster presentations will take place during the three days of this year’s scientific meeting covering key areas in Sickle Cell and Thalassaemia. The accepted abstracts are published in this official HemaSphere supplement.
On behalf of the joint leadership of ASCAT, EHA, BSH, and the esteemed Steering Committee and abstract reviewers, we would like to welcome you to this year’s momentous conference, bringing to you a comprehensive programme on Sickle Cell Disease and Thalassemia; have a pleasant experience.
Professor Baba InusaASCAT President
Professor Marianne de MontalembertEHA Chair
Katy Amberley BSH CEO

Table of Contents
Oral presentationsBasic and translational p. 01Novel therapies, gene therapies, bone marrow transplant and emerging diagnostics p. 02Clinical and epidemiological studies p. 04Infection, autoimmunity, nutritional deficiencies p. 13Health services and outcomes research including psychology p. 14
Poster presentationsBasic and translational p. 18Novel therapies, gene therapies, bone marrow transplant and emerging diagnostics p. 19Clinical and epidemiological studies p. 20Infection, autoimmunity, nutritional deficiencies p. 28Ageing and end organ damage p. 28Health services and outcomes research including psychology p. 29

| 2022; 6:S1 1
ORAL PRESENTATIONS
Basic and translational
S101 COMBINATION OF A LUSPATERCEPT-LIKE DRUG (RAP-GRL) AND TMPRSS6-ASO IS SUPERIOR TO EITHER DRUG ALONE FOR CORRECTING β-THALASSEMIA
Guerra, A.; Demsko, P.; Sinha, S.; McVeigh, P.; Castruccio Castracani, C.; Breda, L.; Casu, C.; Rivella, S.
Children’s Hospital Of Philadelphia, Philadelphia, UNITED STATES
The hallmarks of β-thalassemia (BT) include ineffective erythropoiesis (IE), splenomegaly and iron overload (IO). Recent studies have pointed to iron restriction (IR) to improve both anemia and IO in BT (Rivella, 2019). The decrease of iron-uptake by early erythroid cells results in reduced hemichrome toxicity and prevents premature red blood cell (RBC) hemolysis. One promising IR therapy strategy, which is currently in Phase II clinical trials (NCT04059406), targets the matriptase-2 (TMPRSS6) gene using antisense oligonucleotide technology (T-ASO). As previously shown, treatment of Hbbth3/+ (th3/+) mice (a mouse model for BT-intermedia) with T-ASO improves anemia, lengthens RBC lifespan, reduces levels of erythroferrone (ERFE), decrease hemichromes and lowers reactive oxygen species (ROS), and ameliorates splenomeg-aly (Casu et al., 2016, 2020; Guo et al., 2013).Another novel therapeutic approach to improve anemia in BT targets the TGF-β pathway. Luspatercept, a TGF-β ligand trap, gained FDA approval in 2019 to treat transfusion dependent BT patients (Cappellini and Taher, 2021). In mouse models of BT, its murine analog (RAP-536) was found to promote erythropoietin (EPO)-independent maturation of late-stage erythroid cells, resulting in increased RBC parameters in a dose-dependent manner (Suragani et al., 2014). In this work we generated a Luspatercept-like protein (RAP-GRL) and used it to treat th3/+ mice in combination with T-ASO (RAP-GRL+T-ASO). Our goal was to investigate if this strat-egy would successfully target distinct morbidities associated with BT.We generated RAP-GRL against the mouse analog of Luspatercept (RAP-536). Wild-type (WT) and th3/+ mice were subcutaneously injected s.c. with 10mg/kg of purified RAP-GRL. Our results showed that treatment with RAP-GRL increased RBC levels in both WT and th3/+ mice. Next, we treated th3/+ mice with RAP-GRL (10mg/kg), T-ASO (5mg/kg), or RAP-GRL+T-ASO. The RAP-GRL+T-ASO group displayed the most pronounced increase in RBC parameters and improvements in cell morphology. Flow cytometry analysis using CD71, TER119, and CD44 antibodies of the RAP-GRL+T-ASO groups showed the greatest improvements in IE in both the bone marrow (BM) and spleen (SPL). Additionally, splenomegaly was also greatly reduced in all T-ASO and RAP-GRL+T-ASO groups compared to RAP-GRL and control groups.In conclusion our results provide pre-clinical support for combining IR and TFG-β ligand-trap strategies for the treatment of BT. Our data pro-vides evidence that IR, in conjunction with the erythroid maturation action of Luspatercept may offer additive and more effective therapeutic strategy for BT patients.
References1. Cappellini et al, Blood Adv 2021; 1:326–332. Casu et al, Blood 2021; 136:1968–793. Casu et al, Haematologica; 2016 101:e8-114. Guo et al, J Clin Invest 2013; 123:1531–415. Suragani et al, Nat Med 2014; 20:408–14
S102 OBLIGATE N-TERMINAL BUT NOT C-TERMINAL MONOFERRIC TRANSFERRIN AMELIORATES ANEMIA IN β-THALASSEMIC MICE
Guerra, A.1; Parrow, N.L.2; McVeigh, P.1; Fleming, R.E.2; Ginzburg, Y.Z.3; Rivella, S.4
1Children’s Hospital of Philadelphia, Philadelphia, UNITED STATES; 2Saint Louis University School of Medicine, St. Louis, UNITED STATES; 3Icahn School of Medicine at Mount Sinai, New York, UNITED STATES; 4Children’s Hospital Of Philadelphia, Philadelphia, UNITED STATES
Transferrin (TF) is a bilobed 80kD glycoprotein with N- and C-lobe iron binding sites. TF circulates as four forms: unbound to iron (apo-TF), iron bound to the N-lobe (monoferric N-TF), the C-lobe (monoferric C-TF), or to both lobes (diferric-TF). Most circulating TF under physiological conditions is monoferric. The iron-bound TF forms interact with TF receptor-1 (TFR1), which is ubiquitously expressed and serves as the main mechanism for cel-lular iron delivery. Iron-bound TF also interacts with TF receptor-2 (TFR2) which is expressed on hepatocytes, erythroblasts, and bone cells. Whereas TFR1 serves primarily as a cargo receptor, TFR2 serves primarily to influence cellular signaling events regulating hepcidin expression, erythropoiesis, and bone formation. We pro-posed that different transferrin forms provide differential signaling properties in this regulation. We thus generated TF mutant mice in which all iron-containing TF was either monoferric N (TfmonoN) or monoferric C (TfmonoC). Compared with TfmonoC mice, the TfmonoN mice demonstrated increased RBC production and increased hepci-din expression relative to iron status (Parrow et al. Blood). Based on observations in β-thalassemic mice treated with exogenous TF (Li et al. Nat Med), we hypothesized that β-thalassemic mice obli-gate for monoN TF would demonstrate improved erythropoietic and iron parameters compared with β-thalassemic mice obligate for monoC TF.To address this hypothesis, we crossed Hbbth3/+ mice, a mouse model of β-thalassemia intermedia (BT), with TfmonoN and TfmonoC mice. Compared with Hbbth3/+Tf+/+mice, in Hbbth3/+TfmonoN mice demon-strated significantly increased RBC counts, elevated hemoglobin, improved erythrocyte morphology (Figure 1A-B), decreased sple-nomegaly, fewer bone marrow erythroblasts, and improvement of ineffective erythropoiesis (as measured by the ratio of progenitors to RBC in the bone marrow). Additionally, serum erythroferrone (ERFE) was significantly reduced and hepcidin levels were increased in Hbbth3/+TfmonoN relative to Hbbth3/+Tf+/+ controls. Conversely, hema-tological parameters from Hbbth3/+TfmonoC mice were comparable to Hbbth3/+Tf+/+ mice. Similarly, Hbbth3/+TfmonoC mice had no improvements in markers of ineffective erythropoiesis in the bone marrow com-pared with Hbbth3/+Tf+/+ mice.In summary, we demonstrate that the differential regulatory effects of monoN and monoC TF on erythropoiesis are relevant not only in steady-state, but also in the ineffective erythropoiesis that is char-acteristic of β-thalassemia. Because both monoN and monoC TF forms can deliver only one iron atom per TF-TFR1 binding event, our findings suggest that the improvements observed only in the Hbbth3/+TfmonoN mice were not due to iron restriction alone. We are now elucidating the mechanisms by which the two TF lobes exert their differential effects on ineffective erythropoiesis and exploring the translational potential of obligate monoN TF in the treatment of β-thalassemia.
References1. Li et al, Nat Med 2010; 16:1772. Parrow et al, Blood 2019; 134:1373
Abstract Book for the 2nd Sickle Cell & Thalassaemia Virtual Conference
LWW
Abstract

ASCAT2022
2 | 2022; 6:S1
S103 TRIAL IN PROGRESS: A PHASE 2, OPEN-LABEL STUDY EVALUATING THE SAFETY AND EFFICACY OF THE PKR ACTIVATOR ETAVOPIVAT (FT-4202) IN PATIENTS WITH THALASSEMIA OR SICKLE CELL DISEASE
Lal, A.1; Brown, C.2; Coates, T.3; Kalfa, T.4; Kwiatkowski, J.L.5; Brevard, J.6; Trenor, C.6; Wood, K.6; Sheth, S.7
1University of California San Francisco, San Francisco, UNITED STATES; 2Children’s Healthcare of Atlanta, Atlanta, UNITED STATES; 3University of Southern California Keck School of Medicine, Los Angeles, UNITED STATES; 4Cincinnati Children’s Hospital Medical Center, Cincinnati, UNITED STATES; 5Children’s Hospital of Philadelphia and Perelman School of Medicine, University of Pennsylvania, Philadelphia, UNITED STATES; 6Forma Therapeutics, Inc, Watertown, UNITED STATES; 7Weill Cornell Medicine, New York City, UNITED STATES
Background: Sickle cell disease (SCD) and thalassemia are hemoglob-inopathies characterized by lifelong anemia. In SCD, a single β-globin gene mutation results in sickle hemoglobin (HbS) that polymerizes upon deoxygenation, causes RBCs to sickle and leads to various complications. In thalassemia, α- and/or β-globin gene mutation(s) result in reduced or absent adult Hb, ineffective erythropoiesis, and downstream complica-tions. The resultant anemias, exacerbated by impaired RBC health, are associated with lower ATP levels than in healthy RBCs. Supportive care and agents like hydroxyurea are used most in SCD, with some patients (pts) on regular transfusions. Regular or episodic transfusions, with their own set of complications, are the mainstay of treatment for thalassemia. Etavopivat, an investigational, once-daily, selective, erythrocyte pyruvate kinase (PKR) activator increases ATP and decreases 2,3 diphosphoglyc-erate (2,3-DPG). In a Phase 1 study, etavopivat 300–600 mg once daily in pts with SCD not regularly transfused was well-tolerated, improved hematologic markers, decreased hemolysis, and improved markers of RBC health [1,2]. Etavopivat 200 and 400 mg once daily (dose levels predicted to provide the desired PD response profiles) are being evalu-ated in a Phase 2/3 study of pts with SCD not on chronic transfusions (The Hibiscus Study, NCT04624659).Aims: Describe the design of a Phase 2, open-label, multicenter study (NCT04987489) evaluating the efficacy and safety of etavopivat in pts with: SCD on chronic transfusions (Cohort A), transfusion-dependent thalassemia (Cohort B), and non–transfusion-dependent thalassemia (Cohort C).Methods: Up to 20 pts (12–65 y) will be enrolled in each of the 3 cohorts described above. Key eligibility criteria are outlined in the Table. Pts will receive etavopivat 400 mg once daily for 48-wks (Figure). Pts will
provide written informed consent. Baseline assessments will include medical, disease, transfusion, and medication histories. Transfusions received during the study (every ~3–5 wks) will be recorded and include Hb values before and ≥15 min after transfusion, transfusion dates, num-ber of RBC units, volume of packed RBCs, and hematocrit of the trans-fused unit (if available). If a pt has an increase in pre-transfusion Hb of ≥1.0 g/dL versus their baseline pre-transfusion Hb, the investigator may delay transfusion 1 wk or reduce the number of RBC units transfused. In pts with SCD, RBC exchange transfusions may also be performed. The primary endpoints are outlined in the Figure. Secondary and exploratory endpoints include the proportion of pts with a reduction in transfusions over 12 wks of ≥33% and ≥50%, respectively, and a reduction in trans-fusions over 12, 24, and 48 wks (Cohorts A/B); and Hb response at Wks 24 and 48, and changes from baseline in Hb over 12, 24, and 48 wks (Cohort C). The following additional endpoints will be assessed (all cohorts): changes from baseline in quality of life (using the SF-36 and PROMIS); changes from baseline in serum ferritin levels at 12, 24, and 48 wks; liver iron at 48 wks; 2,3-DPG and ATP; PK; and safety. All pri-mary endpoints will be analyzed using a 1-sided test at α=0.025.Summary: Etavopivat is a novel, investigational, once-daily, selective PKR activator with potential to improve RBC health and lifespan. This Phase 2 study will assess the safety of etavopivat and its impact on Hb levels and transfusion burden in pts (12–65 y) with SCD or thalassemia.
References1. Brown et al, ASH Annual Meeting 2021; Abstr #147091.2. Kalfa et al, ASH Annual Meeting 2021; Abstr #147078.
Novel therapies, gene therapies, bone marrow transplant and emerging diagnostics
S104 A SEVERE MOUSE MODEL OF ALPHA-THALASSEMIA SHOWS ABNORMAL IRON METABOLISM, ERYTHROPOIESIS AND COAGULATION, AND CAN BE RESCUED BY A NOVEL GENE THERAPY APPROACH
Chappell, M1; Breda, L1; Guerra, A1; Ghiaccio, V1; Fedorky, M1; Jarocha, D1; Gollomp, K1; Teawtrakul, N2; Glentis, S3; Kattamis, A3; Rivella, S1
1CHOP, Philadelphia, UNITED STATES; 2Khon Kaen University, Khon Kaen, THAILAND; 3University of Athens, Athens, GREECE
Background: Clinical presentation of deletional a-thal varies from an asymptomatic condition (one inactivated a-globin gene) to a complete knockout (Hb Bart’s Hydrops Fetalis). In patients with severe a-thal, a blood transfusion independent state is achievable through allogeneic bone marrow transplantation.Aims: The aims of this study are to develop a novel adult mouse model of a-thal and a gene therapy approach for this disease.Methods: We generated adult animals that do not produce a-globin chains (a-KO) through transplantation of homozygous B6.129S7-Hbatm1Paz/J fetal liver cells (FLC; isolated at E14.5) into WT recipient mice.To generate a gene therapy tool, we screened multiple lentiviral vectors to identify the variant capable of producing the highest human a-glo-bin protein per copy. The selection was conducted in murine erythro-leukemia cells and human umbilical cord derived erythroid progenitor (HUDEP) cells, modified by knocking out all the human a-globin genes.Results: The a-KO animals demonstrate a worsening phenotype, par-adoxically showing elevated hematocrit, high reticulocyte count and a high number of red blood cells (RBC) which expressed only b-globin chains (HbH). RBC show aberrant morphology and aggregation of b-globin tetramers on RBC membranes. Due to severe inability of these RBC to deliver oxygen, the mice eventually succumb to anemia, show-ing splenomegaly and other organ pathologies, including vaso-occlusive events, associated with neutrophil infiltration, fibrinogen staining, von Willebrand factor (vWF) released, platelet recruitment and activation. These animals also show iron deposition in the liver and kidney, in agree-ment with very low levels of hepcidin expression in the liver, and ele-vated erythropoietin (EPO) in the kidney.We identified ALS20a, a vector where a-globin is under control of the b-globin promoter and its locus control region, as the most efficient vec-tor. One copy of ALS20α produces exogenous a-globin at a level compa-rable to that produced by one endogenous a-globin gene. These results suggest that a relatively low VCN could result in dramatic therapeutic

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 3
benefits. Use of ALS20a resulted in correction of the disease phenotype in a dose-dependent manner in a-KO mice. At VCN<1 we observe a delay in death proportional to the VCN value, while at VCN>1 we observe phenotypic normalization, including Hb, hepcidin and EPO levels.We tested ALS20a in CD34 cells isolated from four patients with both deletional and non-deletional HbH disease. We measured the change of b/a-globin mRNA ratio (b/aR) and protein level by HPLC in erythro-blasts derived from these cultures. For the specimen with mutational HbH, the initial b/aR matches that of healthy controls, as the mutations do not eliminate the ability for the gene to produce aberrant mRNA transcripts, and decreased with increasing VCN. Erythroblasts with dele-tional HbH have a b/aR approximately 3x higher than normal cells, decreasing in a dose dependent manner with increasing VCN. HPLC detection of HbH (β4), a hallmark of HbH disease, is observed in hemo-lysis products from all non-transduced a-thal erythroblasts. A ~50% reduction of HbH is detected in the very same specimens upon integra-tion of ALS20α (VCN between 1 and 2).Conclusion: We generated an adult mouse model of lethal a-thal and, in preliminary experiments, we rescue it with ALS20a. Furthermore, ALS20a successfully improves a-globin levels in patient cells.
References1. Harteveld at al, Orphanet J Rare Dis. 2010; 5:13.2. Mettananda et al, Blood. 2015; 125(24):3694–7013. Higgs, Cold Spring Harb Perspect Med. 2013; Jan 1;3(1)
S105 ADDING AZATHIOPRINE/HYDROXYUREA PRECONDITIONING TO ALEMTUZUMAB/TBI CONDITIONING IMPROVES DONOR CHIMERISM IN MATCHED SIBLING DONOR STEM CELL TRANSPLANTATION IN ADULT SICKLE CELL DISEASE PATIENTS
Dovern, E1; Aydin, M1; Tang, M1; Suijk, L1; van Tuijn, C1; Zeerleder, S2; Hazenberg, M1; Biemond, B1; Nur, E1
1Amsterdam UMC, Amsterdam, NETHERLANDS; 2University Hospital Bern, Bern, SWITZERLAND
Background: Allogeneic hematopoietic stem cell transplantation (HSCT) is currently the only established curative treatment option for sickle cell disease (SCD). In adults, myeloablative conditioning is associated with significant toxicity, primarily due to cumulative organ damage. Matched sibling donor (MSD) transplantation with non-myeloablative condition-ing (alemtuzumab/3 Gy total body irradiation (TBI)) has shown prom-ising results in adult SCD patients.1,2 Patients treated with this regimen had their sickle cell phenotype corrected with only mild complications and no reports of graft-versus-host disease (GvHD). However, most of the described patients did not reach complete donor chimerism with graft failure rates of 13%. Furthermore, in almost 10% of patients, immunosuppressives could not be withdrawn because of too low T cell chimerism (<50%).2 We hypothesized that adding azathioprine and hydroxyurea as preconditioning to the alemtuzumab/TBI regimen might improve donor chimerism, reduce the risk of graft failure and improve successful withdrawal of immunosuppressives.Aims: In this study we prospectively investigate the effects of azathio-prine/hydroxyurea preconditioning on donor chimerism and graft fail-ure in patients receiving non-myeloablative MSD HSCT for SCD.Methods: Adult SCD patients who had an HLA-identical sibling donor were eligible for this treatment. After 3 months of azathioprine 150mg qd and hydroxyurea 25mg/kg qd, erythrocyte exchange transfusion was performed on day –10, aiming for HbS <30%. Conditioning with alemtuzumab/TBI was started on day –7. Graft versus host disease (GvHD) prophylaxis consisted of sirolimus.Results: As of October 2021, 13 SCD patients (median age 30.6 (range 19–49) years) were transplanted. Twelve patients (92.3%) engrafted successfully. After a median follow-up of 24 months, median donor myeloid and T cell chimerism of engrafted patients were 100% (IQR 84–100%) and 77% (IQR 70.5%-82.5%), respectively. These donor chimerism percentages are higher than previously reported with alemtuzumab/TBI only. All engrafted patients had a corrected SCD phenotype with normalized hemoglobin levels. Patients reaching 1-year post-transplantation were able to stop sirolimus without decreases in chimerism.Summary/Conclusion: Azathioprine/hydroxyurea preconditioning prior to alemtuzumab/TBI results in improved donor chimerism, poten-tially reducing the risk of graft failure after non-myeloablatieve MSD
transplantation in SCD patients. Importantly, all engrafted patients reached donor T cell chimerism >50% at 1-year post-transplantation and were able to stop immunosuppressives as scheduled.
References1. Hsieh et al, NEJM 2009; 361:242. Alzahrani et al, BJH 2021; 192:4
S106 LONG-TERM FOLLOW-UP OF BETA-THALASSEMIA PATIENTS TREATED WITH HEMATOPOIETIC STEM CELL GENE THERAPY
Marktel, S1; Scaramuzza, S1; Giglio, F2; Cicalese, M1; Lidonnici, M1; Rossi, C1; Calbi, V1; Masera, N3; D’Angelo, E4; Mirra, N4; Origa, R5; Tartaglione, I6; Perrotta, S6; Viarengo, G7; Santoleri, L8; Milani, R8; Gattillo, S8; Calabria, A1; Montini, E1; Graziadei, G9; Naldini, L1; Cappellini, M9; Aiuti, A1; Ciceri, F2; Ferrari, G1
1San Raffaele Telethon Institute for Gene Therapy, Milan, ITALY; 2Haematology and BMT Unit, IRCCS San Raffaele Scientific Institute, Milan, ITALY; 3Pediatric Department University of Milano-Bicocca, Monza, ITALY; 4Pediatric Clinic/DH Fondazione IRCCS Ca’ Granda, Milan, ITALY; 5Department of Biomedical Science and Biotechnology University of Cagliari, Cagliari, ITALY; 6Università degli studi della Campania “Luigi Vanvitelli”, Napoli, ITALY; 7Immunohematology and Transfusion Medicine Service Fondazione IRCCS Policlinico S. Matteo, Pavia, ITALY; 8Blood Transfusion Service, IRCCS San Raffaele Scientific Institute, Milan, ITALY; 9Rare Disease Center, Fondazione IRCCS Ca’ Granda, Milan, ITALY
Background: Transfusion-dependent ß-thalassemia (TDT) is a disor-der due to mutations in the gene encoding the ß-globin chain causing a reduced or absent production of haemoglobin A leading to severe anae-mia and lifelong transfusion dependence. Gene therapy has now been accepted as a possible alternative cure to allogeneic bone marrow (BM) transplantation.Aims: We developed a gene therapy approach based on autologous mobi-lized hematopoietic stem cell transduced by lentiviral vector, expressing human ß-globin gene, administered by intrabone injection, following a myeloablative conditioning (NCT02453477).Methods and Results: Nine patients with severe TDT with different gen-otypes have been treated with a drug product with a median cell dose of 19.5x106 CD34+ cells/kg, a transduction efficiency from 38 to 77% and a median vector copy number/genome (VCN) in bulk CD34+ cells of 0.9 (range: 0.7–1.5). Overall, gene therapy was generally well-tolerated with no adverse events related to the investigational product. No severe infectious-related adverse events were reported, except for those related to neutropenia as expected after myeloablative conditioning. Polyclonal vector integrations profiles with no evidence of clonal dominance have been detected in all patients with the expected genomic distribution for lentiviral vectors.Clinical outcome showed a reduction of transfusion requirement both in frequency and volume in adult patients up to more than 50%. Among the pediatric patients, 4 out of 6 discontinued transfusions shortly after gene therapy and are transfusion-independent at the last follow-up (up to 75 months).A robust and persistent engraftment was observed in 7 out of 9 patients, with a marking of BM progenitors that, in engrafted patients, ranged between 25.3 and 79.8% and with a median VCN in CD34+ cells of 0.53 (range: 0.34–2.21). As a relevant target for transgene expression, BM erythroid cells were stably marked (VCN range 0.3 - 2.5).Summary and Conclusions: A longer follow-up will provide further results on long-term clinical efficacy and safety of this approach.
References1. Marktel et al, Nat Med 2019; 25(2):234
S107 P-SELECTIN INHIBITOR INCLACUMAB REDUCES CELL ADHESION IN AN IN-VITRO ASSAYS SHOWING POTENTIAL FOR PREVENTION OF VASO-OCCLUSION EVENTS IN SICKLE CELL DISEASE
Tarasev, M; Herppich, A; Gao, X; Hines, P
Functional Fluidics Inc., Detroit, UNITED STATES
Introduction: Sickle cell disease (SCD) is a genetic disorder that leads to serious clinical complications including vaso-occlusive events (VOE). Development of VOEs had been linked to translocation of

ASCAT2022
4 | 2022; 6:S1
endothelial P-Selectin from Weibel–Palade bodies to the cell surface, a process promoting sickle Red Blood Cell (SS RBC) adhesion to vessel walls. Up-regulation of P-selectin contributes to cell–cell inter-actions, as activated platelets bind to neutrophils to form aggregates in a P-selectin–dependent manner. Previous studies have shown that anti-P-selectin agents decrease SS RBC adhesion in vivo, increase microvascular flow, and reduce adhesion of the leukocyte to endo-thelium, showing the potential of P-selectin as a therapeutic target. In November 2019, the FDA approved the first P-selectin inhibitor, crizanlizumab-tmca (Adakveo, Novartis), to reduce the frequency of vaso-occlusive crises (VOCs) in adults and pediatric patients with SCD. Different types of P-selectin inhibitors can offer physicians and patients new treatment options enhancing the arsenal of drugs to combat SCD. Inclacumab (Global Blood Therapeutics) is a fully human monoclonal antibody designed to bind to and selectively inhibit P-selectin. Differences in static cell adhesion to P-selectin substrate due to inclucumab were shown previously. The present study aimed to validate the dose-dependentability of inclacumab in blocking P-selectin cell adhesion in samples from SCD patients using in-vitro Whole Blood (WB) and Isolated White Blood Cell (I-WBC) assays.Methods: Five deidentified samples from SCD patients undergo-ing phlebotomy prior to blood exchange had been collected at the Children’s Hospital of Michigan with Flow Adhesion on P-selectin measured using proprietary assays in both Whole Blood (WB) and in Isolated WhiteBlood Cells (I-WBC), with and without Inclacumab after 5 minutes incubation at room temperature. Microfluidic channels were coated with P-selectin, and I-WBC (5x106/mL) suspension or WB sam-ple diluted 1:1 with phosphate buffer was passed through the channel for 6 (I-WBC) or 10 (WB) minutes at a shear rate of 1 dyne/cm and pulsatility of 1.67 Hz. The channel was washed for 5 minutes with the same buffer, and the number of cells adherring to the channel surface was quantified.Results: Incubation of either WB or I-WBC with inclacumab signifi-cantly reduced flow adhesion to P-Selectin with the overall inhibition being stronger when assessed on isolated white cells (ab. 35% inhi-bition on WB vs. ab. 55% on I-WBC after incubation with 40 mM Inclacumab as compared to baseline Flow Adhesion values, Fig. 1). Adhesion on P-selectin showed a pronounced dose response that followed logarithmic response curve, with changes in adhesion with I-WBC better correlated with inclacumab dose, than those measured using WB. Decrease in adhesion on P-selectin was reliably detected at 2 μg/mL treatment dose for 3 out of 5 samples, with dose-depemdent decrease in adhesion in all samples at 10 and 40 μg/mL treatment doses (for I-WBC; and similar for WB). For flow adhesion with both WB and I-WBC elevated adhesion at lower doses was observed in some samples.Conclusions: Both WB and I-WBC adhesion to P-Sselectin show a dose dependent inhibition by inclacumab, with clinically relevant doses likely demonstrating greater inhibition. P-selectin adhesion assays described in this study may serve as potential surrogate biomarkers of clinical response tp p-selectin inhibitors, like inclacumab.
References1. Schmitt et al, J Cardiovasc Pharmacol 2015; 65:611
Clinical and epidemiological studies
S108 A SYSTEMATIC POST-MARKETING (PM) REVIEW OF RARE INFUSION-RELATED REACTIONS (IRRS) PRESENTING AS PAIN EVENTS DURING OR AFTER CRIZANLIZUMAB INFUSION IN PATIENTS WITH SICKLE CELL DISEASE (SCD)
Kanter, J1; Shah, A2; Joshi, V3; Mehta, H3; Levine, M2; Arunagiri, U2; Paulose, J2; Donohue, B2; Scalera, A2; Manwani, D4
1University of Alabama, Birmingham, UNITED STATES; 2Novartis Pharmaceuticals Corporation, East Hanover, UNITED STATES; 3Novartis Healthcare Private Limited, Hyderabad, INDIA; 4Albert Einstein College of Medicine, The Children’s Hospital at Montefiore, New York, UNITED STATES
Background: Vaso-occlusive crises (VOCs) are the hallmark of SCD and can lead to complications and premature death. Crizanlizumab, an anti-P-selectin monoclonal antibody (mAb), is authorized to prevent/reduce VOCs in SCD patients aged ≥16 years.IRRs are defined as signs/symptoms experienced by patients during/within 24 hours of infusion of a pharmacologic/biologic agent.1 IRRs are quite common with mAbs (frequency 1.6–99%)2 and can present as

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 5
pain events, such as back pain, chest pain and myalgia. Pain events are known adverse drug reactions in the crizanlizumab label, however, pain events occurring during/within 24 hours of crizanlizumab infusion in SUSTAIN were not identified as potential IRRs.3 For SCD patients, IRR-related pain events may differ in location, severity and/or nature from their usual SCD/VOC pain.Aims: We reviewed PM data on IRRs presenting as pain events in SCD patients treated with crizanlizumab.Methods: A custom search of the Novartis safety database (comprising PM reports [spontaneous] and managed access/patient orientation program reports) was performed for reports received in November 2019–June 2021, using ~111 MedDRA terms associated with potential signs/symptoms of IRRs presenting as pain events. IRRs must have occurred during/within 24 hours of the most recent crizanlizumab infusion, and pain could differ from a patient’s usual SCD/VOC. As reports were not gathered via a uni-form data collection system, potential limitations include underreporting, incompletely documented cases, or bias towards reporting severe events.Results: IRRs presenting as pain events were experienced by 28 patients (Table), and most commonly presented as back pain, pain in extrem-ity, arthralgia, musculoskeletal chest pain and headache. Reporting rate was 1.67 cases per 100 patient-years (95% CI 1.11–2.42). Most patients (n=24) initially experienced IRR at the first or second infusion. IRR recurred on subsequent infusion(s) in six patients. Twenty patients (71%) were hospitalized for treatment, including analgesics, antihista-mines, IV fluids and/or steroids. Nine patients (32%) reported known complications of SCD following IRR (Table). Crizanlizumab was dis-continued in 23 patients (82%) after their most recent IRR, including all who experienced secondary SCD complications.All patients recovered (the majority within 3 days; one with sequelae), except one who died following SCD complications and refusal of blood transfusions for personal reasons. Resolution time was prolonged for patients who reported SCD complications following IRR. Causal anal-ysis of complications was confounded by the underlying disease and use of steroids to treat IRR (systemic corticosteroid exposure in SCD patients has been associated with pain, complications [including severe VOCs and hemorrhagic stroke] and death). We do not know whether the 28 patients had an active VOC or other SCD complications before receiving crizanlizumab.Summary – Conclusion: Although rare, based on review of PM data, healthcare professionals should be aware of the possibility of IRRs pre-senting as pain events during/after crizanlizumab infusion. Crizanlizumab labels have been/are being updated by Novartis to provide information on monitoring, management and prevention of IRRs, including a state-ment recommending caution when using corticosteroids. Given the limited data available regarding IRRs, Novartis is committed to further understanding these events.
References1. Kang & Saif. J Support Oncol 2007; 5:451–72. Rombouts et al, Anticancer Res 2020; 40:1201–183. Ataga et al, N Engl J Med 2017; 376:429–39
S109 ACTIVATION OF PYRUVATE KINASE-R WITH ETAVOPIVAT (FT-4202) IS WELL TOLERATED, IMPROVES ANEMIA, AND DECREASES INTRAVASCULAR HEMOLYSIS IN PATIENTS WITH SICKLE CELL DISEASE TREATED FOR UP TO 12 WEEKS
Telen, M1; Saraf, S2; Cruz, K3; Idowu, M4; Kalfa, T5; Osunkwo, I6; Hagar, R7; Geib, J8; Forsyth, S8; Schroeder, P8; Wu, E8; Kelly, P8; Brown, R9
1Duke University, Durham, UNITED STATES; 2University of Illinois, Chicago, UNITED STATES; 3Advanced Pharma, Miami, UNITED STATES; 4Memorial Hermann-Texas Medical Center, Houston, UNITED STATES; 5Cincinnati Children’s Hospital Medical Center, Cincinnati, UNITED STATES; 6Levine Cancer Institute, Atrium Health, Charlotte, UNITED STATES; 7UCSF Benioff Children’s Hospital San Francisco, Oakland, UNITED STATES; 8Forma Therapeutics, Inc, Watertown, UNITED STATES; 9Children’s Healthcare of Atlanta, Atlanta, UNITED STATES
Background: Etavopivat, an investigational, once daily (QD), selective, erythrocyte pyruvate kinase (PKR) activator, increases PKR activity, resulting in ↓2,3-DPG and ↑ATP in RBCs of healthy volunteers (HV) and patients (pts) with sickle cell disease (SCD) [1,2].Aims: Multiple-dose studies in pts with SCD (NCT03815695): 2-wk multiple ascending dose (MD) cohorts to identify the etavopivat QD dose providing maximum PD activity with an acceptable safety profile
and a 12-wk open-label (OL) study to characterize safety and clinical activity at the maximum pharmacodynamic (PD) dose.Methods: Completed MD cohorts: 20 pts with SCD were randomized 8:2 to etavopivat (300 mg, then 600 mg) or placebo (PBO) QD for 2 wks. Ongoing OL study: ≤20 pts will receive etavopivat 400 mg QD for 12 wks. Assessments: safety, PK, PD, and RBC health. All pts provided written informed consent.Results: MD cohorts (n=17 HbSS, n=2 HbSβ+thalassemia, n=1 HbSC) completed enrollment and data unblinded (300 and 600 mg etavopivat, n=8 each; PBO, n=4). OL cohort: As of July 13, 2021, 11 pts (HbSS/SC, n=10/1) were treated: median treatment (Trt) duration was 12 (range 1–12) wks; 6 pts completed 12 wks of Trt. In MD pts, etavopivat demon-strated dose-proportional PK with overlapping PD response (↓2,3-DPG; ↑ATP), confirming prior results in HVs that etavopivat 400 mg QD provides maximal PD activity. Etavopivat was well tolerated. AEs were reported in 1/4 (25%) MD PBO pts, most were grade (Gr) ≤3, with 1 Gr4 blood creatine phosphokinase (CPK) increase. AEs were reported in 13/16 (81%) etavopivat MD pts, most were Gr1/2 and commonly (n>2) included sickle cell pain (n=6[38%]), headache (n=5[31%]), and nausea (n=3[19%]). One pt had a serious AE (SAE) of Gr3 vaso-occlusive crisis (VOC, unrelated) after completing etavopivat. In the OL cohort, AEs were reported in 7/11 (64%) pts on etavopivat for ≥1 wk. AEs reported in >1 pt were headache and VOC (n=2[18%] each). Most AEs were Gr1/2; 1 pt had SAEs of Gr3 acute chest syndrome and VOC (unrelated), 1 pt had an SAE of Gr3 deep vein thrombosis (possibly related), and 1 pt had an AE of Gr4 transient blood CPK increase (unrelated). Hematologic and hemolytic parameters significantly improved at end of Trt (MD and OL cohorts); 11/15 (73%) evaluable MD pts had a hemoglobin (Hb) increase ≥1 g/dL over baseline (mean 1.1 g/dL, P<0.004) and markers of hemolysis decreased (Table). These initial findings were sustained in pts receiving ≤12 wks of etavopivat (Table, Fig.). Of 6 pts who completed 12 wks of etavopivat, 5 (83%) had >1 g/dL Hb increase over baseline (mean 1.39 g/dL). Markers of hemolysis decreased. Of 9 pts on etavopivat for ≥4 wks, 8 (89%) had an Hb increase >1 g/dL; highest mean Hb increase was 1.81 g/dL during Trt. Etavopivat-treated RBCs from MD pts (n=14) exhibited improved health, including point of sickling and deformability. Improved deformability persisted up to 1 wk after etavopivat in 36% of pts. Results in initial OL pts were similar. Data on additional pts will be presented.Summary: Etavopivat 400 mg QD for ≤12 wks was well tolerated, with a safety profile consistent with underlying SCD. Etavopivat increased sickle RBC lifespan and significantly improved severe anemia associ-ated with SCD. These early Phase 1 data show longer-living sickle RBCs have improved health, which may further reduce the risk of VOCs and end-organ damage, and support further evaluation of etavopivat in the Hibiscus Study (NCT04624659).
References1. Kalfa et al, Blood 2019;134(suppl 1):6162. Brown et al, Blood 2020;136(suppl 1):19

ASCAT2022
6 | 2022; 6:S1
S110 ACUTE AND CHRONIC PAIN MANAGEMENT IN SICKLE CELL DISEASE: OUTCOMES OF AN ENGLISH NATIONAL AUDIT.
Lugthart, S1; Kotsiopoulou, S2; Luqmani, A3; Eleftheriou, P4; Drasar, E5; Brown, R3; Webster, A6; Chakravorty, S7; Stuart-Smith, S7; Velangi, M8; Atoyebi, W9; Howard, J10; Telfer, P11
1University Hospitals of Bristol, Bristol, UNITED KINGDOM; 2Croydon Health Services NHS Trust, Croydon, UNITED KINGDOM; 3Imperial College Healthcare NHS Trust, London, UNITED KINGDOM; 4University College London Hospitals NHS Foundation Trust, London, UNITED KINGDOM; 5University College Hospitals London, London, UNITED KINGDOM; 6University Hospitals of Leicester NHS Trust, Leicester, UNITED KINGDOM; 7King’s College Hospital NHS Foundation Trust, London, UNITED KINGDOM; 8Birmingham Women’s and Children’s NHS Foundation Trust, Birmingham, UNITED KINGDOM; 9Oxford University Hospitals, Oxford, UNITED KINGDOM; 10Guy’s and St. Thomas’ NHS Foundation Trust, London, UNITED KINGDOM; 11Barts Health NHS Trust, London, UNITED KINGDOM
Background: Acute pain is the most common complication of sickle cell disease (SCD). Patients suffering severe pain often seek treatment in an acute hospital setting. Feedback from service users indicates a lack of satisfaction with quality of care. NHS England specialised commission-ing recommended forming a National Sickle Pain Group (NSPG) con-sisting of multi-professional stakeholders and patient representatives, to understand the range of practices and challenges in providing high-qual-ity hospital care.Aims: The objective was to develop national guidelines for acute and chronic pain management which will improve quality of care, patient experience and outcomes. The aim was to understand the variety of acute and chronic pain management policies and protocols used across England, identify aspects of care where there was unacceptable variation and examples of good care.Methods: A questionnaire was developed through discussion meetings with members of the NSPG. This was sent to haemoglobinopathy coor-dinating centre (HCC) leads, to be distributed to all specialist haemoglo-binopathy teams (SHTs) and local haemoglobinopathy centres (LHTs) in their network, inclusive of adult and paediatric services.Results: In total, 56 services (26 paediatric and 30 adult departments), in 39 centres completed the questionnaire (75% response rate). Of these, 51% were LHTs, 15% SHTs and 33% HCCs. The size of services varied between 0 and 808 patients for adult services and 0 and 439 patients for paediatric services. Hospital admissions with vaso-occlusive episode in the year April 2020 to March 2021 for adult services varied between 0 and 754. Admission rates in paediatric services are generally lower. For both adult and paediatric acute pain presentations, the majority of centres provided care in their hospital emergency department (ED), and only a small number offered direct access to a ward (5%) or an ambu-latory facility (12%). Ambulatory facilities’ opening days ranged from 5 – 7, with 64% only operating during standard hours. Access to an acute pain service (APS) was available in 83% and 65% for adult and paediatric departments respectively. Generic pain protocols were avail-able in 50 services. The protocols vary, but the most common analgesia prescribed in adults was morphine (oral or subcutaneous or). In children, morphine (oral or intranasal) was widely used. Individual pain proto-cols were used in 61% of responding centres. The NICE standard ‘<30 minutes time to first analgesia’ were not met in the majority of centres (range 30–60, outliers 80–128 min). Overall, the time to first analgesia was lower in services with ambulatory care facilities. The length of stay ranged between 3–5 days. Between 1–5% of the patients experienced a prolonged admission (>21 days). Frequent re-admissions occurred in 2–10% of the patient population (≥3 admissions/year). Education and teaching sessions were infrequently delivered for ED consultants, ED nurses, Acute Medicine consultants and pharmacists (30%, 40%, 21% and 5%).Summary: Hospital management of acute sickle pain is a significant challenge to NHS services and needs to be re-evaluated.Conclusion: The questionnaire results will inform the objectives and work plans of four working groups within the NSPG (acute pain, chronic pain, education and research). In order to develop national policies, it will be necessary to generate evidence through a more detailed audit of outcomes in scenarios of best practices identified here.No references, but a more detailed summarySummary: Hospital management of acute sickle pain is a significant chal-lenge to NHS services and needs to be re-evaluated at local and national level. Despite publication of NICE guidelines in 2012, few services in the NHS are able to consistently provide timely pain relief. The range of policies for analgesia management is surprisingly broad.
The availability of ambulatory care in some centres could function as an exemplar for national practice. There may be alternative models of care which could be effective. Patients with frequent attendance and prolonged hospital stay are present in most centres and, although rela-tively small as a proportion of the service, are especially challenging to manage. The management of these patients requires a multidisciplinary approach and the development of national guidance, as often this is out-side the expertise of haematologists. This may be helpful in improving outcomes in this patient cohort.The infrequent delivery of teaching reveals the need for regular local and national mandatory educational training of all ED staff providing care to SCD patients during acute presentations.
S111 BRAIN PERFUSION CHANGES IN BETA-THALASSEMIA
Manara, R1; Ponticorvo, S2; Tartaglione, I3; Canna, A3; Russo, A3; Fedele, M3; Rocco, M2; Cirillo, M3; Perrotta, S3; Esposito, F3
1UNIVERSITA’ DI PADOVA, padova, ITALY; 2Università di Salerno, Salerno, ITALY; 3Università degli studi della Campania “Luigi Vanvitelli”, Naples, ITALY
Background: Brain involvement in hereditary hemoglobinopathies (e.g. sickle cell disease, beta-thalassemia, spherocytosis) is commonly attributed to anemia-related relative hypoperfusion. Supratentorial and infratentorial vascular watershed regions seem to be especially vulnera-ble, but data are very scarce.Aims: We investigated a large beta-thalassemia sample with arterial spin labelling in order to characterize regional perfusion changes and their correlation with phenotype and anemia severity.Methods: We performed a multicenter single-scanner cross-sectional MRI study analyzing non-invasively in 71 beta-thalassemia patients and 56 healthy controls the brain perfusion changes. Clinical phenotype, age, hemoglobin levels, cognitive functioning and parenchymal lesions were also recorded.Results: Brain perfusion was globally increased in beta-thalassemia patients compared to healthy controls; using age and sex as covari-ates and scaling the perfusion maps for the global cerebral blood flow, beta-thalassemia patients showed: hyperperfusion in the white matter of the centrum semiovale bilaterally, located in the watershed regions between the vascular territories of the main cerebral arteries (Fig-1a) and in the cerebellar white matter corresponding to the cerebellar watershed regions. Subdividing patients according to anemia severity (hemoglobin level < or > 9.5g/dL), the hyperperfusion clusters persisted exclusively in the subgroup with lower hemoglobin levels.Summary and Conclusion: The relative hyperperfusion observed in vas-cular watershed territories does not support the previous hypothesis of a selective parenchymal hypoperfusion in the pathogenesis of brain injury in hereditary hemoglobinopathies. A careful management of ane-mia severity seems to be pivotal for preventing perfusion dysfunction, at least in beta-thalassemia.
Fig-1. Analysis of brain perfusion correcting for global rCBF. Upper row (panels a,b,c) shows t-maps of significantly globally increased perfusion in patients vs healthy controls (HC). Panel a. contains T-maps of the statistical contrast HC vs. thalassemia patients. Panel b. contains T-maps of the statistical contrast HC vs. transfusion dependent (TDT) thalas-semia patients. Panel c. contains T-maps of the statistical contrast HC vs. non-transfusion dependent (NTDT) thalassemia patients.
References1. Choi, Soyoung, Richard M. Leahy, and John C. Wood. 2020. “Lower
White Matter Volume in Beta-Thalassemia Associated with Anemia and Cognitive Performance.” American Journal of Hematology.
2. Choi, Soyoung, Sharon H. O’Neil, Anand A. Joshi, Jian Li, Adam M. Bush, Thomas D. Coates, Richard M. Leahy, and John C. Wood.

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 7
2019. “Anemia Predicts Lower White Matter Volume and Cognitive Performance in Sickle and Non-Sickle Cell Anemia Syndrome.” American Journal of Hematology 94 (10): 1055–65.
3. Manara R, Talenti G, Rampazzo P, Ermani M, Montanaro M, Baracchini C, Teso S, Basso G, Sainati L, Colombatti R. Longitudinal evaluation of cerebral white matter hyperintensities lesion volume in children with sickle cell disease. Br J Haematol. 2017 Feb;176(3):485–487. doi: 10.1111/bjh.13962. Epub 2016 Mar 27. PMID: 27018310.
S112 COMPARATIVE SCORE OF HOWELL-JOLLY BODIES IN SICKLE CELL DISEASE PATIENTS ACCORDING TO GENOTYPE, AGE, SPLENECTOMY OR STEM CELL TRANSPLANTATION HISTORY
Bernaudin, F1; Arnaud, C2; Kamdem, A2; Pondarré, C2; Hau, I3; Lelong, F4; Dalle, J5; Peffault de Latour, R6; Lezeau, H7
1Clinical Research Center, CHIC Hospital, Creteil, FRANCE; 2Referral Center for SCD, CHIC Hospital, Creteil, FRANCE; 3Pediatrics, CHIC Hospital, Creteil, FRANCE; 4Hematology, CHIC Hospital, Creteil, FRANCE; 5Hematology, Debré Hospital, Paris, FRANCE; 6Hematology, St-Louis Hospital, Paris, FRANCE; 7Pediatric surgery, Creteil, FRANCE
Background: Presence of Howell-Jolly bodies is associated with splenic dysfunction which progressively appears during aging or after splenec-tomy in patients with sickle cell disease. Improvement of splenic function has been found in several transplanted patients for SCD (Ferster 1993, Bernaudin 2007) encouraging our team to propose partial splenectomy to patients with hypersplenism and available matched-sibling donor.Aims: The goal of the present study was to evaluate the outcome of the splenic function from birth to adult age and the impact of splenectomy (partial or total) in SCD patients transplanted or not.Patients and Methods: In the pediatric Créteil SCD-cohort, including 907 SCD patients (111 SC, 47 Sb+, 725 SS, 21 Sb0 and 3 SDPunjab), Howell-Jolly bodies score (from 0 to 4) was prospectively assessed and recorded at each annual check-up. This study included 5013 annual check-ups (637 in SC/Sb+ and 4376 in SS/Sb0 patients) from Jan 1991 to July 2019.Results: Howell-Jolly bodies mean score increased during aging in SCD patients but was significantly lower in SC/Sb+ than in SS/Sb0 patients at each age of assessment (Figure 1). During the 2nd year of life, mean (SD Howell-Jolly bodies score was 0.34 (0.61) in SC/Sb+ vs 0.69 (0.82) in SS/Sb0 (P=0.01) while during the 18th year it was 0.61 (0.61) in SC/Sb+ vs 1.33 (1.1) in SS/Sb0 children (P<0.001).Among 749 SCA children, 86 were splenectomized at mean (SD) age of 6.4 years (3.9). As expected, mean (SD) Howell-Jolly bodies score was significantly higher at check-up in splenectomized than in non-splenec-tomized SCA-patients: 1.73 (1.2) vs 1.1 (1.0), p<0.001. However, the difference between both populations decreased during aging because of the progressive functional splenic dysfunction in non-splenectomized patients (Figure 2)Among SCA-children, 138 were transplanted with matched sibling donor at mean (SD) age of 9.0 years (4.9y). At each age of assessment, Howell-Jolly bodies score was strongly significantly (p<0.001) lower in transplanted than in non-transplanted children (Figure 3). At check-up per-formed during the 18th year of life, mean (SD) Howell-Jolly bodies score was 1.57 (1.0) in 241 non transplanted vs 0.54 (0.9) in 70 transplanted children (p<0.001) and the score in transplanted children was significantly positively correlated with the age at transplant (r=0.335, p=0.002). Seven children with splenic sequestration history or hypersplenism and available MSD-donor had partial splenectomy before transplant in order to try to preserve splenic function. Mean (SD) Howell-Jolly bodies score was 1.83 (1.2) before and was nul after transplant in all seven children.
Conclusion: This cohort study clearly shows that splenic dysfunction increases during aging in SCA children and that stem cell transplanta-tion allows to preserve splenic function especially since the transplant is performed early. In children with indication of splenectomy for recurrent
splenic sequestrations or hypersplenism who have an available matched sibling donor, the present study encourages to propose partial vs total splenectomy just before stem cell transplantation in order to preserve splenic function.
References1. Ferster et al, Blood 1993;81:11022. Bernaudin et al, Blood 2007;110:2749
S113 COST OF ILLNESS, EPIDEMIOLOGY AND HEALTHCARE RESOURCE UTILIZATION FOR SICKLE CELL DISEASE IN GULF COUNTRIES (CESCGU)
Alkindi, S1; Altooq, J2; Dewedar, H3; Soliman, D4; Elsawy, S5; Ibrahim, A6
1Sultan Qaboos University, Muscat, OMAN; 2Salmaniyah Medical Complex, Manama, BAHRAIN; 3Dubai Health Authority, Dubai, UNITED ARAB EMIRATES; 4Novartis, Dubai, UNITED ARAB EMIRATES; 5Accsight, Cairo, EGYPT; 6Accsight, Dubai, UNITED ARAB EMIRATES
Background: Sickle cell disease (SCD) is one of the most predominant form of haemoglobinopathy worldwide. In the Middle East, SCD is prevalent, with high impact on health care systems. Currently, there are insufficient information published on the economic burden of SCD in the Gulf Countries (GC).Aim: Estimate the economic burden incurred by payors in Oman (OMN), Bahrain (BHR) and United-Arab-Emirates (UAE).Methodology: A prevalence-based probabilistic model was used to esti-mate the direct medical costs (DMC) associated with SCD within differ-ent treatment sites across 3 countries in the gulf region. Data from three treatment sites across GC has been collected through meetings with one principle investigator from each site. Aggregate patients’ data input was gathered from hospital record databases of adult SCD patients managed in year 2019. Additionally, 50 Case Report Forms (CRFs) were filled from UAE site.Results: Aggregate data for 2,600 SCD patients in BHR, 2,000 in OMN and 350 in UAE were collected. The total DMC per patient per year was estimated at $15,300, $10,000 and $15,800 in BHR, OMN and UAE, respectively. The costs incurred by payors for “VOCs management” were identified as major cost driver.Conclusion: As far as our knowledge, this is the first COI study in SCD done in the GC. The economic burden of individuals affected by SCD in Gulf countries is rising. Severe SCD patients with ≥5 VOCs represent a significant burden on payors.
S114 FEASIBILITY OF HAPLOIDENTICAL ALLOGENEIC STEM CELL TRANSPLANTATION WITH POST-TRANSPLANT CYCLOPHOSPHAMIDE FOR PATIENTS WITH SEVERE SICKLE CELL DISEASE WITH END-STAGE RENAL DISEASE (ESRD) ON HEMODIALYSIS
Gomez Arteaga, A1; Orfali, N1; Pasciolla, M1; Baptiste, A2; Hsu, J1; Mayer, S1; Phillips, A1; Shore, T1; van Besien, K1
1Weill Cornell Medicine, NEW YORK, UNITED STATES; 2NewYork-Presbyterian Brooklyn Methodist Hospital, NEW YORK, UNITED STATES
Background: Adult patients with sickle cell disease (SCD) and ESRD have limited curative treatment options as many are deemed not to be candi-dates for an allogeneic transplant (SCT) or clinical trials. Furthermore,

ASCAT2022
8 | 2022; 6:S1
many patients lack a matched donor. Haploidentical (haplo) SCT with post-transplant cyclophosphamide (Cy) has improved the access to SCT. Mitigating the risk of graft loss for SCD patients undergoing haplo-SCT, two groups have reported successful engraftment with modified condi-tioning regimens - Fludarabine (Flu), Cy with augmented total body irra-diation (TBI)(400 cGy) vs. Flu/Cy/TBI200cGy with additional thiotepa [1,2]. No patients with ESRD were included.Aims: To evaluate the feasibility of haplo-SCT with augmented TBI con-ditioning for a patient with SCD and ESRDMethods and Results: A 26-year-old male (B+, CMV-) with SCD and sickle nephropathy, on hemodialysis (HD), was referred for evaluation. The patient had a history of acute chest syndrome, priapism, and SVT. He had multiple hospitalizations in the 2 years prior to referral. His baseline Hb was 6 g/dl and he had mild liver iron overload. As there were no matched donors, his 19-yr-old, 8/12 sister (A+, CMV-), was evaluated. Due to a lack of data in ESRD, thiotepa was excluded and we opted for Flu/Cy/TBI400cGy conditioning. To avoid the risk of fludarabine neurotoxicity, dosing was decreased by 50% to 15 mg/m2 x 5 and HD was given more frequently. Rabbit-ATG was given. GvHD prophylaxis comprised of post-transplant Cy, MMF and sirolimus (fig 1). Prior to SCT, the patient underwent sperm cryopreservation, he received one dose of Rituximab for EBV prophylaxis, and he under-went exchange transfusion to target a Hb S <30%. Additional support-ive therapy included: levetiracetam; ursodiol; and infection prophylaxis with posaconazole, levofloxacin, and valacyclovir. A total of 5.41 x 106/kg CD34+ PBSC were infused. No G-CSF was planned but it was added on day +15 due to slow engraftment. Neutrophil engraftment occurred at day +27. Platelets were kept above 50k and engraftment occurred at day +48. Pre-engraftment adverse events included radiation-induced parotiditis, neutropenic fever, and medication-induced rhabdomyolysis with muscle pain attributed to posaconazole (CK peaked on day +13 with levels >7800; posaconazole was switched to micafungin with ade-quate resolution). Upon discharge pen-VK, Valtrex and fluconazol were continued and he started SMZ-TMP. He was re-admitted twice - once for SVT, and again for pneumonia. Chimerism studies revealed a 96% donor chimerism on day +30, with subsequent sustained 100% donor chimerism from day +100. Hb electrophoresis has been normal. CD4 count at day +120 was 1795 cells/ul. Our patient has had no GVHD. Tapering of immunosuppression started at 9 months and completed at 11 months post-SCT. His new baseline Hb is 14 g/dl, his last RBC transfusion was on day +33 and monthly therapeutic phlebotomies were started at 9 months. The patient is currently 1.4 yrs post-SCT. Renal transplant evaluation is underway.Conclusion: Haploidentical transplantation with post-transplant Cy is a feasible approach for patients with severe SCD and ESRD. Patients require very close monitoring for complications by a specialized mul-tidisciplinary team, but it can be a curative therapy with significant improvement in quality of life.
References1. Bolaños-Meade J et al. Lancet Haematol. 2019 Apr; 6(4) e183-e1932. de la Fuente J et al. Biol Blood Marrow Transplant. 2019; 25(6) 1197–1209
S115 LONG-TERM EFFICACY AND SAFETY OF DEFERIPRONE FOR PATIENTS WITH SICKLE CELL DISEASE OR OTHER ANEMIAS
Inusa, B1; Hamdy, M2; El-Beshlawy, A3; Ebeid, F4; Kwiatkowski, J5; Kanter, J6; Williams, S7; Lee, D8; Temin, N8; Fradette, C8; Tricta, F8; Elalfy, M4
1Evelina Children’s Hospital, Guy’s and St. Thomas’ NHS Foundation Trust, London, UNITED KINGDOM; 2Cairo University, Cairo, EGYPT; 3Pediatric Hospital of Cairo University, Cairo, EGYPT; 4Ain Shams University, Pediatric Hospital, Cairo, EGYPT; 5The Children’s Hospital of Philadelphia, Philadelphia, UNITED STATES; 6Medical University of South Carolina, Charleston, UNITED STATES; 7The Hospital for Sick Children, University of Toronto, Toronto, CANADA; 8Chiesi Canada Corporation, Toronto, CANADA
Background: FIRST-EXT was a prospective, multicenter, single-arm, open-label extension of the FIRST study in patients with sickle cell dis-ease (SCD) and other transfusion-dependent anemias. In FIRST, patients were randomly assigned 2:1 to receive oral deferiprone (DFP) or par-enteral deferoxamine (DFO) for 12 months. The noninferiority of DFP versus DFO was previously reported.1 Here we report the efficacy and safety of DFP in FIRST-EXT.
Aims: To evaluate the long-term efficacy and safety of DFP in iron-over-loaded patients with SCD or other anemias.Methods: Patients who completed FIRST could enter FIRST-EXT for up to 2 years. Patients previously treated with DFP continued on DFP (DFP-DFP), while those previously treated with DFO were switched to DFP (DFO-DFP). Baseline was defined as the start of FIRST for DFP-DFP patients, and the start of FIRST-EXT for DFO-DFP patients. Efficacy endpoints were yearly changes from baseline in liver iron concentration (LIC), cardiac MRI T2*, and serum ferritin (SF). We also report adverse drug reactions (ADRs), defined as adverse events at least possibly related to DFP. All patients provided informed consent or assent.Results: Patients (N=134: 89 DFP-DFP; 45 DFP-DFO) were 60.4% male, with a mean (SD) age of 16.2 (8.6) years. Most (85.8%) had SCD; 14.2% had other anemias. At baseline, all patients had elevated (≥1.8 mg/g dry weight [dw]) LIC; all but one had elevated (females >300 µg/L, males >400 µg/L) SF; and all had cardiac MRI T2* in the normal range (≥20 ms). A significant, progressive decline was seen in LIC, with mean (SD) changes from baseline to years 1, 2, and 3 of -2.64 (4.64), -3.91 (6.38), and -6.64 (7.72) mg/g dw, respectively (P<0.01 for all). A decline was also seen in SF, with mean (SD) changes from baseline to years 1, 2, and 3 of -1 (1986), -771 (2171), and -1016 (3617) µg/L, respectively (P<0.05 for years 2 and 3). Cardiac MRI T2* values changed little from baseline. The most frequent ADRs were neutropenia (9.0%), decreased neutrophil count (9.0%), and abdominal pain (7.5%); 2 patients (1.5%) experienced agranulocytosis. One patient withdrew due to ADRs of thrombocytopenia and neutropenia, which resolved. Another patient withdrew due to generalized edema and died for reasons unknown 17 days after withdrawal from the study.Conclusions: DFP long-term use (≤3 years) was effective in controlling body iron load in patients with SCD and other anemias. There were no new safety concerns.
References1. Kwiatkowski et al, Blood 2019; 134 (Supplement 1): 618.
S116 LONG-TERM EFFICACY AND SAFETY OF THE ORAL PYRUVATE KINASE ACTIVATOR MITAPIVAT IN ADULTS WITH NON—TRANSFUSION-DEPENDENT ALPHA- OR BETA-THALASSEMIA
Kuo, K1; Layton, D2; Lal, A3; Al-Samkari, H4; Bhatia, J5; Kosinski, P5; Tong, B5; Lynch, M5; Uhlig, K5; Vichinsky, E3
1Division of Hematology, University of Toronto, Toronto, ON, CANADA; 2Hammersmith Hospital, Imperial College Healthcare NHS Trust, London, UNITED KINGDOM; 3Division of Hematology, UCSF Benioff Children’s Hospital Oakland, Oakland, CA, UNITED STATES; 4Massachusetts General Hospital, Harvard Medical School, Boston, MA, UNITED STATES; 5Agios Pharmaceuticals, Inc., Cambridge, MA, UNITED STATES
Background: Thalassemia is characterized by ineffective erythropoie-sis and hemolysis that occur due to imbalanced production and pre-cipitation of globin chains.1,2 Thalassemic red blood cells (RBCs) have insufficient levels of ATP to meet increased energy demands associated with globin chain imbalance, protein degradation, and cellular oxidative stress responses.3,4 Mitapivat is an oral activator of RBC pyruvate kinase (PKR), a key glycolytic enzyme regulating ATP production.5 In a phase (ph) 2, open-label trial of mitapivat in adults with α- or β-non–transfu-sion-dependent (NTD) thalassemia (NCT03692052), 80% of patients (pts) met the primary endpoint of a hemoglobin (Hb) response (≥1.0 g/dL increase from baseline [BL] at ≥1 assessments between Weeks (Wks) 4–12, inclusive). Improvements in markers of hemolysis and ineffec-tive erythropoiesis were also observed and mitapivat was generally well tolerated.6
Aims: To report data from the ongoing long-term extension (LTE) period (up to Wk 72; data cutoff 27Mar2021).Methods: Pts aged ≥18 yrs with a known medical history of α- or β-thal-assemia, Hb concentration ≤10.0 g/dL, and ≤5 RBC units transfused in prior 24 wks and none in 8 wks prior to study drug were eligible. All pts started mitapivat 50 mg twice daily (BID), escalating to 100 mg BID based on individual safety and Hb assessments. After completion of the 24-wk core period, pts with a Hb response, or delayed Hb response (after Wk 12), with no ongoing study drug-related grade ≥3 treatment-emer-gent adverse events (AE) continued on mitapivat in the LTE at the same dose as the Wk 24 visit. Study visits occur every 12 wks for ≤10 yrs.Results: Of 19 pts who completed the core period, 17 entered the LTE (mitapivat 100 mg BID, n=16; 50 mg BID, n=1). As of data cutoff, 1 pt

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 9
discontinued (pt decision). Median duration of treatment for pts in the LTE was 70.9 wks (range 54.7, 105.6), with 8 pts receiving ≥72 wks of treatment as of data cutoff. Median pt age in the LTE was 44 yrs (range 29, 67). Mean BL (SD) Hb, total bilirubin and lactate dehydro-genase (LDH) was 8.1 (1.2) g/dL, 40.1 (26.2) μmol/L and 272.4 (121.7) U/L, respectively. Median BL erythropoietin (EPO) was 70.5 (range 15, 11191) IU/L. Hb improvements achieved in the core period were sustained in the LTE (Figure). Mean Hb (SD) increase from BL to Wk 60 (α-thalassemia, n=4; β-thalassemia, n=9) and Wk 72 (β-thalassemia, n=8) were 1.5 (0.4) and 1.7 (0.5) g/dL, respectively. Improvements in markers of hemolysis and ineffective erythropoiesis observed in the core period were maintained in the LTE up to Wk 72 (mean [SD] bilirubin and LDH, –15.8 [16.6] μmol/L and –63.6 [216.0] U/L, respectively; median [range] EPO, –33.0 [–72.0, –16.0] IU/L). The safety profile was consistent with that observed in the core period. AEs in ≥15% of pts were headache (5/17) and back pain (3/17), none were grade ≥3. No trends for decreases in bone mineral density were observed. No treat-ment-related serious AEs occurred.Conclusions: A favorable efficacy-safety profile was observed with long-term mitapivat in pts with α- or β-thalassemia. Data show sustained improvements in Hb, hemolysis and ineffective erythropoiesis despite globin genotypic heterogeneity, and no new safety findings. Mitapivat, through its unique mechanism of action, may represent a novel thera-peutic approach for this condition. Two ph 3 trials of mitapivat in α- and β-thalassemia, (NTD and transfusion-dependent pts), are enrolling.
References1. Taher et al. Lancet 2018; 391:155–67.2. Galanello et al. Ophanet J Rare Dis 2010; 5:11.3. Khandros et al. Blood 2012; 119:5265–75.4. Shaeffer. J Biol Chem 1988; 263:13663–9.5. Kung et al. Blood 2017; 130:1347–56.6. Kuo et al. HemaSphere 2021; Abstract S267.
S117 LONG-TERM FOLLOW UP OF DUTCH PATIENTS WITH SCD DIAGNOSED BY NEONATAL SCREENING -- EFFECT ON THE MORBIDITY AND MORTALITY IN THE NETHERLANDS.
Vuong, C; de Ligt, L; de Groot-Eckhardt, C; Heijboer, H; Fijnvandraat, K
Amsterdam UMC, location AMC, Amsterdam, NETHERLANDS
Background: Newborn screening for sickle cell disease (SCD) has been introduced in January 2007 in the Netherlands. The objective of this study is to assess the effect of this neonatal screening for SCD by describ-ing the residual risks of death and major disease-related events during the first fourteen years of life in children diagnosed with SCD at birth in the Netherlands.Aims: The objective of this study is to assess the effect of this neonatal screening for SCD by describing the residual risks of death and major disease-related events during the first fourteen years of life in children diagnosed with SCD at birth in the Netherlands.Methods: Here we report the first data of one center (Amsterdam UMC) of this prospective, national multicenter study. Following informed con-sent data were collected from medical files of all children born after 1 January 2007, diagnosed by neonatal screening. Descriptive data on
SCD genotype, occurrence of major disease-related events (hospitaliza-tion for vaso-occlusive crisis (VOC), acute chest syndrome/pneumonia, severe infections and neurological complications) are presented. Overall survival and survival without specific SCD-related complications were analyzed by Kaplan-Meier curves.Results: Up until now, 98 (56%) out of 174 eligible subjects from this institution were included, with a total follow-up of 805 patient-years. This concerns approximately 35% of the national number. The majority (55%) had the severe genotype (HbSS/ beta0-thalassemia), the remainder had the milder genotype (HbSC or HbS/beta+-thalassemia). Survival by the age of 14 was 98.9%, with 1 death at the age of 1 years due to sepsis. Seven patients (7.1%) had a severe infection (meningitis, sepsis, osteo-myelitis) caused by Streptococcus Pneumoniae in 3/7 cases. Two patients experienced a symptomatic cerebral infarction at the age of 11 months and 1.5 years. At the age of 10 years the survival without hospitalization for vaso-occlusive crisis was 27% (95% CI: 12.7 – 43.14%) and 51% (25.3 – 72.0%) for the SS/Sβ0 and SC/Sβ+ genotype respectively.Conclusion: In this cohort of neonatally screened patients with SCD, the SCD-related mortality and morbidity is still impressive with 1% mor-tality, 3 severe infections caused by Streptococcus Pneumoniae, and 2 patients with neurological complications. A final analysis of the effect of neonatal screening for SCD will follow after completion of data collec-tion in all participating centers in the Netherlands.
References1. Rettenbacher et al. J Pediatr Hematol Oncol 2021; 43:7.2. Lê et al. J Med Screen 2018. 25:2.3. Brousse et al. J Clin Med 2019. 8:10.
S118 LONG-TERM SAFETY AND EFFICACY OF VOXELOTOR FOR PATIENTS WITH SICKLE CELL DISEASE: RESULTS FROM AN OPEN-LABEL EXTENSION OF THE PHASE 3 HOPE TRIAL
Achebe, M1; Nduba, V2; Hassab, H3; Alkindi, S4; Brown, R5; Telfer, P6; Biemond, B7; Gordeuk, V8; Lipato, T9; Tonda, M10; Gray, S10; Howard, J11
1Brigham and Women’s Hospital, Harvard Medical School, Boston, Boston, UNITED STATES; 2Kenya Medical Research Institute, Kisumu, KENYA; 3Pediatric Department & Clinical research center, Faculty of Medicine, Alexandria University, Alexandria, EGYPT; 4Sultan Qaboos University, Muscat, OMAN; 5Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Altanta, UNITED STATES; 6Queen Mary, University of London, London, UNITED KINGDOM; 7Academic Medical Centre, University of Amsterdam, Amsterdam, NETHERLANDS; 8University of Illinois Hospital, Chicago, UNITED STATES; 9Virginia Commonwealth University Medical Center, Richmond, UNITED STATES; 10Global Blood Therapeutics, South San Francisco, UNITED STATES; 11Guy’s and St. Thomas’ NHS Foundation Trust, King’s College, London, UNITED KINGDOM
Background: Sickle cell disease (SCD), a lifelong, inherited blood disor-der, leads to sickle hemoglobin (HbS) formation. HbS polymerization causes red blood cell sickling, leading to hemolysis, chronic anemia, and vaso-occlusive crises (VOCs). Patients with SCD are at higher risk of end-organ damage, increased morbidity, and early mortality due to low hemoglobin (Hb) and increased hemolysis.1
Voxelotor, a HbS polymerization inhibitor, is approved in the US for SCD treatment in adults and adolescent patients aged ≥12 years.2 The randomized, placebo-controlled HOPE trial showed that significantly more patients on voxelotor 1500 mg had a >1 g/dL Hb increase than those on placebo at any time to week 72. These Hb increases were asso-ciated with reduced hemolysis markers.3 Here we report an interim anal-ysis of an ongoing open-label extension (OLE) of theMethods: Patients who completed the phase 3 HOPE trial were eligible to enroll in the multicenter OLE study and receive treatment as long as they continued to receive clinical benefit and/or until they had access to voxelotor through commercialization or a managed access program. All patients received voxelotor 1500 mg as ongoing treatment. Adverse event data were collected from the date of informed consent through 28 days after voxelotor discontinuation. Measurements of Hb and clinical markers of hemolysis are ongoing and summarized here for 48 weeks of the OLE. Data presented are based on an interim data cut (December 31, 2020).Results: Of the 199 patients who completed the HOPE trial, 178 (89.4%) were enrolled and dosed in the OLE. Median age at enrollment was 25 years (15.7% adolescents, 84.3% adults). At the cutoff date, the median voxelotor exposure duration in the OLE was 69.9 weeks (range:

ASCAT2022
10 | 2022; 6:S1
1.9–102.0 weeks), with 78 patients treated for ≥72 weeks, of whom 52 received voxelotor in the randomized part of the study, for a combined exposure duration ≥144 weeks. Among those who previously received placebo, the mean (SD) Hb change from baseline (ie, start of the OLE) to week 48 was 1.3 (1.51) g/dL, consistent with HOPE trial results. Hemolysis markers improved from baseline to week 48 in patients who received placebo in the HOPE trial (–39.5% indirect bilirubin; –28.6% reticulocytes). Patients who previously received voxelotor in the HOPE trial showed durability of response in Hb and clinical measures of hemolysis in the OLE. The annualized VOC incidence rate was 1.3 (95% CI: 1.1–1.4) events per year across all patients. 83.7% of patients (149/178) experienced a non-SCD-related treatment-emergent adverse event (TEAE), with the most commonly reported being arthralgia, head-ache, pain, nausea, and pain in extremity. Most non-SCD-related TEAEs were grade 1 or 2. Eleven patients (6.2%) had an adverse event that led to treatment discontinuation. No TEAEs consistent with lack of tissue oxygenation were observed.Conclusions: In this OLE study, treatment with voxelotor 1500 mg led to improvements in Hb and clinical measures of hemolysis at 48 weeks in patients who received placebo in the HOPE trial and showed durability of response in patients previously treated with voxelotor of any dosage in the HOPE trial. No new safety signals were identified with exposure through a combined 144 weeks of treatment. Per these results, long-term voxelotor treatment is safe, well tolerated, and effective in reducing anemia and hemolysis, with a low rate of VOCs, in patients with SCD.
References1. Kato GJ, Piel FB, Reid CD, et al. Nat Rev Dis Primers. 2018;4:18010.2. Oxbryta. Prescribing information. Global Blood Therapeutics; January
2021.3. Howard J, Ataga KI, Brown RC, et al. Lancet Haematol. 2021;8(5):E32
3-E333.
S119 PRELIMINARY RESULTS OF A PHASE 1 STUDY IN HEALTHY SUBJECTS ADMINISTERED INCLACUMAB, A FULLY HUMAN IGG4 ANTI-P-SELECTIN MONOCLONAL ANTIBODY IN DEVELOPMENT FOR TREATMENT OF SICKLE CELL DISEASE
Mayer, C1; Cooper, D2; Redfern, A3; Geng, X4; Shi, J4; Zutphen-van Geffen, M5; Kuan, I5; Koek, K5; Kastrissios, H5; Patel, K5; Davis, M4; Yue, P4
1Semivida Research, Dallas, UNITED STATES; 2Daniel S Cooper, MD, LLC, Palo Alto, UNITED STATES; 3Linear Clinical Research, Nedlands, AUSTRALIA; 4Global Blood Therapeutics, South San Francisco, UNITED STATES; 5Certara USA, Inc., Princeton, UNITED STATES
Background: Inclacumab, a fully human IgG4 anti-P-selectin monoclo-nal antibody, is being developed for the reduction of vaso-occlusive cri-ses (VOCs) in patients with sickle cell disease (SCD). P-selectin-mediated platelet-leukocyte aggregate (PLA) formation has been shown to con-tribute to vaso-occlusion. Safety and pharmacology of inclacumab have previously been well characterized in over 700 subjects (healthy volun-teers and patients with cardiovascular disease), at doses up to 20 mg/kg every 4 weeks for up to 9 months. The current Phase 1 study was initiated to evaluate the safety and pharmacology of inclacumab at doses of 20 mg/kg and 40 mg/kg in healthy subjects in support of a target Phase 3 dose of 30 mg/kg administered every 12 weeks to patients with SCD.Methods: Healthy adult subjects over 18 years of age without significant current or prior health conditions received a single intravenous (IV) dose of 20 mg/kg inclacumab infused over approximately one hour (Cohort 1). Following a review of safety, a second cohort received a single IV dose of 40 mg/kg infused over approximately one hour (Cohort 2). The total study duration and sample collection period was 29 weeks. Final safety and preliminary pharmacokinetics (PK), anti-drug antibody (ADA), and ex vivo thrombin receptor-activating peptide (TRAP)-activated PLA for-mation data are reported.Results: Fifteen subjects received a single dose of inclacumab 20 mg/kg (n=6) or 40 mg/kg (n=9). Median age was 42 years (range 22–52 years); median body weight was 73.6 kg (range 63.7–89.3 kg). Through the prespecified 72-hour post-infusion safety assessment period in both cohorts, no treatment-emergent adverse events (AEs) > grade 1 (mild) nor dose-limiting toxicities were reported. Across the duration of the study, there were no serious AEs, infusion-related reactions, or hypersen-sitivity reactions. Additionally, no clinically significant changes in vital signs, laboratory findings, or ECGs were observed. The most common
AEs were upper respiratory tract infections, headache, myalgia, back pain, and contact dermatitis. The only events assessed by the investiga-tor as potentially related to inclacumab were headache and dizziness, which were experienced by one subject (20 mg/kg) and occurred 4 hours following the end of infusion. In healthy subjects, inclacumab demon-strated dose-proportional PK over the dose range tested; PK parameter estimates were consistent with those reported for monoclonal antibod-ies. Geometric mean Cmax following single doses of 20 and 40 mg/kg were 402 and 970 µg/mL, respectively. Mean TRAP-activated pre-dose PLA formation was 33–39% across cohorts and decreased to 9–14% at 2 hours following end of infusion. PLA inhibition was sustained up to 23 weeks for both the 20 and 40 mg/kg doses. Two subjects in the 40 mg/kg cohort were ADA-positive on Week 12 and thereafter; a pre-liminary analysis demonstrated no apparent impact on PK or safety in these subjects.Conclusions: Inclacumab displayed a well-tolerated safety profile for up to 29 weeks following a single dose of 20 or 40 mg/kg in healthy subjects. Durable inhibition of TRAP-activated PLA formation was observed up to 23 weeks. Overall, the results support a Phase 3 dose of 30 mg/kg every 12 weeks in patients with SCD-related VOCs.Funding: This study was supported by Global Blood Therapeutics.
S120 RANDOMIZED CONTROLLED TRIAL OF THE EFFICACY AND SAFETY OF DEFERIPRONE: SUBGROUP ANALYSIS OF PEDIATRIC PATIENTS IN IRON-OVERLOADED PATIENTS WITH SICKLE CELL DISEASE AND OTHER ANEMIAS
Inusa, B1; Hamdy, M2; El-Beshlawy, A3; Ebeid, F4; Kwiatkowski, J5; Kanter, J6; Williams, S7; Lee, D8; Temin, N8; Fradette, C8; Tricta, F8; Elalfy, M4
1Paediatric Haematology, Evelina Children’s Hospital, Guy’s and St. Thomas’ NHS Foundation Trust, London, UNITED KINGDOM; 2Cairo University, Cairo, EGYPT; 3Pediatric Hospital of Cairo University, Cairo, EGYPT; 4Ain Shams University, Pediatric Hospital, Cairo, EGYPT; 5The Children’s Hospital of Philadelphia, Philadelphia, UNITED STATES; 6Medical University of South Carolina, Charleston, UNITED STATES; 7The Hospital for Sick Children, University of Toronto, Toronto, CANADA; 8Chiesi Canada Corporation, Toronto, CANADA
Background: Children with sickle cell disease (SCD) managed with blood transfusions often require iron chelation therapy to prevent iron overload.1 Deferoxamine (DFO) is an iron chelator approved for pedi-atrics that is often infused; however, adherence is a key challenge due to the burdensome route of administration.2 Deferiprone (DFP), an oral iron chelator, is a first-line treatment for transfusional iron overload in children and adults with SCD and other anemias.3 DFP is noninferior to DFO in SCD with iron overload (evaluated by liver iron concentra-tion [LIC]) and has an acceptable safety profile.4 This subgroup analysis of FIRST (NCT02041299) assessed whether efficacy and safety of DFP were comparable to DFO in children with SCD.Methods: In this phase 4 open-label study, patients were randomized 2:1 to DFP or DFO for 12 months. The subgroup analysis included children (2–16 years of age) with SCD or another rare anemia treated for trans-fusional iron overload. Children received either oral DFP three times a day or subcutaneous DFO infusion 5–7 days a week. Iron load was monitored and dosage adjustments were allowed. The primary endpoint was change in LIC from baseline to month 12. Data were analyzed for all patients with a baseline and a follow-up LIC assessment (efficacy population). Safety assessments were done for all patients who received at least 1 dose of study drug (safety population). All patients provided informed consent or assent.Results: Of 228 patients in the safety population, 128 (DFP, n=86; DFO, n=42) were children. Most children (DFP, 75.6%; DFO, 80.9%) had a primary diagnosis of SCD (HbS). Mean ages (SD) in the DFP and DFO groups were 9.9 (3.7) and 10.9 (3.0) years (P=0.09), respectively. There were no significant differences between the DFP and DFO groups in sex (males, 59.3% vs 57.1%; P=0.85), ethnicity (P=0.68), or race (P=0.34). 5 children withdrew due to AEs (all DFP) and 19 withdrew for other reasons (DFP, n=14; DFO, n=5). There was not a significant difference in number of withdraws between groups (P=0.23). Children treated with DFP or DFO showed no significant differences in overall incidence of AEs (P=0.77; including neutropenias [P=0.30]), severe AEs (P=0.10), serious AEs (P=0.16), or withdrawals due to AEs (P=0.17). A difference in overall incidence of nonserious AEs considered at least possibly related to DFP (59.3% vs 33.3%; P=0.01) was found. For AEs ≥5%: see Table 1. The only AE with a significantly higher rate with DFP
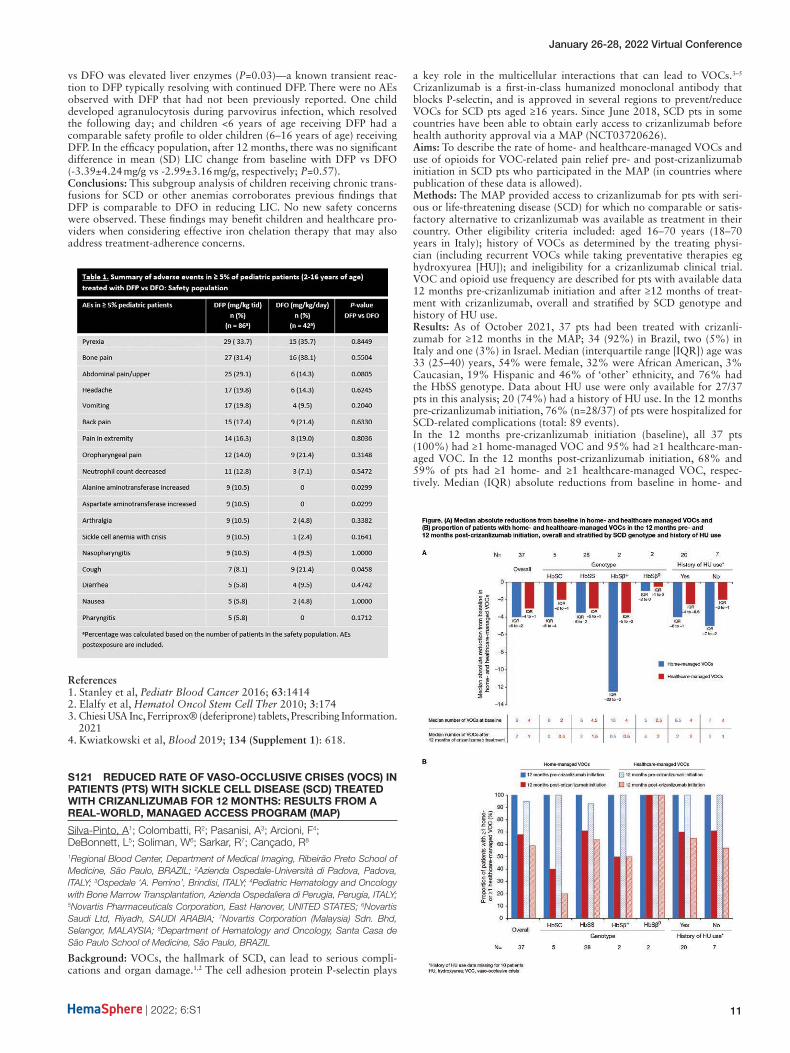
January 26-28, 2022 Virtual Conference
| 2022; 6:S1 11
vs DFO was elevated liver enzymes (P=0.03)—a known transient reac-tion to DFP typically resolving with continued DFP. There were no AEs observed with DFP that had not been previously reported. One child developed agranulocytosis during parvovirus infection, which resolved the following day; and children <6 years of age receiving DFP had a comparable safety profile to older children (6–16 years of age) receiving DFP. In the efficacy population, after 12 months, there was no significant difference in mean (SD) LIC change from baseline with DFP vs DFO (-3.39±4.24 mg/g vs -2.99±3.16 mg/g, respectively; P=0.57).Conclusions: This subgroup analysis of children receiving chronic trans-fusions for SCD or other anemias corroborates previous findings that DFP is comparable to DFO in reducing LIC. No new safety concerns were observed. These findings may benefit children and healthcare pro-viders when considering effective iron chelation therapy that may also address treatment-adherence concerns.
References1. Stanley et al, Pediatr Blood Cancer 2016; 63:14142. Elalfy et al, Hematol Oncol Stem Cell Ther 2010; 3:1743. Chiesi USA Inc, Ferriprox® (deferiprone) tablets, Prescribing Information.
20214. Kwiatkowski et al, Blood 2019; 134 (Supplement 1): 618.
S121 REDUCED RATE OF VASO-OCCLUSIVE CRISES (VOCS) IN PATIENTS (PTS) WITH SICKLE CELL DISEASE (SCD) TREATED WITH CRIZANLIZUMAB FOR 12 MONTHS: RESULTS FROM A REAL-WORLD, MANAGED ACCESS PROGRAM (MAP)
Silva-Pinto, A1; Colombatti, R2; Pasanisi, A3; Arcioni, F4; DeBonnett, L5; Soliman, W6; Sarkar, R7; Cançado, R8
1Regional Blood Center, Department of Medical Imaging, Ribeirão Preto School of Medicine, São Paulo, BRAZIL; 2Azienda Ospedale-Università di Padova, Padova, ITALY; 3Ospedale ‘A. Perrino’, Brindisi, ITALY; 4Pediatric Hematology and Oncology with Bone Marrow Transplantation, Azienda Ospedaliera di Perugia, Perugia, ITALY; 5Novartis Pharmaceuticals Corporation, East Hanover, UNITED STATES; 6Novartis Saudi Ltd, Riyadh, SAUDI ARABIA; 7Novartis Corporation (Malaysia) Sdn. Bhd, Selangor, MALAYSIA; 8Department of Hematology and Oncology, Santa Casa de São Paulo School of Medicine, São Paulo, BRAZIL
Background: VOCs, the hallmark of SCD, can lead to serious compli-cations and organ damage.1,2 The cell adhesion protein P-selectin plays
a key role in the multicellular interactions that can lead to VOCs.3–5 Crizanlizumab is a first-in-class humanized monoclonal antibody that blocks P-selectin, and is approved in several regions to prevent/reduce VOCs for SCD pts aged ≥16 years. Since June 2018, SCD pts in some countries have been able to obtain early access to crizanlizumab before health authority approval via a MAP (NCT03720626).Aims: To describe the rate of home- and healthcare-managed VOCs and use of opioids for VOC-related pain relief pre- and post-crizanlizumab initiation in SCD pts who participated in the MAP (in countries where publication of these data is allowed).Methods: The MAP provided access to crizanlizumab for pts with seri-ous or life-threatening disease (SCD) for which no comparable or satis-factory alternative to crizanlizumab was available as treatment in their country. Other eligibility criteria included: aged 16–70 years (18–70 years in Italy); history of VOCs as determined by the treating physi-cian (including recurrent VOCs while taking preventative therapies eg hydroxyurea [HU]); and ineligibility for a crizanlizumab clinical trial. VOC and opioid use frequency are described for pts with available data 12 months pre-crizanlizumab initiation and after ≥12 months of treat-ment with crizanlizumab, overall and stratified by SCD genotype and history of HU use.Results: As of October 2021, 37 pts had been treated with crizanli-zumab for ≥12 months in the MAP; 34 (92%) in Brazil, two (5%) in Italy and one (3%) in Israel. Median (interquartile range [IQR]) age was 33 (25–40) years, 54% were female, 32% were African American, 3% Caucasian, 19% Hispanic and 46% of ‘other’ ethnicity, and 76% had the HbSS genotype. Data about HU use were only available for 27/37 pts in this analysis; 20 (74%) had a history of HU use. In the 12 months pre-crizanlizumab initiation, 76% (n=28/37) of pts were hospitalized for SCD-related complications (total: 89 events).In the 12 months pre-crizanlizumab initiation (baseline), all 37 pts (100%) had ≥1 home-managed VOC and 95% had ≥1 healthcare-man-aged VOC. In the 12 months post-crizanlizumab initiation, 68% and 59% of pts had ≥1 home- and ≥1 healthcare-managed VOC, respec-tively. Median (IQR) absolute reductions from baseline in home- and
ASCAT2022
12 | 2022; 6:S1
healthcare-managed VOCs after ≥12 months of crizanlizumab treatment were –4.0 (–6.0 to –2.0) and –3.0 (–4.0 to –1.0), respectively. The lower rates of VOCs post- vs pre-crizanlizumab were also observed when strat-ifying the data by SCD genotype/prior HU use (Figure). Of note, a small number of pts are included in some groups when stratifying the data.Opioids were taken for VOC management by 95% of pts (n=35/37) in the 12 months before crizanlizumab initiation and by 68% (n=25/37) in the 12 months after; the most common opioid taken was morphine (74% [n=26/35] and 56% [n=14/25], respectively).Summary – Conclusion: Pts participating in the crizanlizumab MAP had a high burden of home- and healthcare-managed VOCs, as well as SCD-related complications at baseline, despite many pts reporting a his-tory of HU use. Most pts reported using opioids for VOC management. Crizanlizumab substantially reduced the median annualized rates of home- and healthcare-managed VOCs and use of opioids from baseline in this real-world setting, consistent with results from SUSTAIN.6
References1. Ballas & Lusardi. Am J Hematol 2005; 79:17–252. Herquelot et al, Value Health 2019; 22:S908; presented at ISPOR
20193. Zhang et al, Blood 2016; 127:801–94. Merle et al, JCI Insight 2018; 3:e969105. Kappelmayer & Nagy. Biomed Res Int 2017:61381456. Ataga et al, N Engl J Med 2017; 376:429–39
S122 SAFETY AND EFFICACY OF CRIZANLIZUMAB IN ADOLESCENTS WITH SICKLE CELL DISEASE (SCD): INITIAL DATA FROM THE PHASE II, MULTICENTER, OPEN-LABEL SOLACE-KIDS TRIAL
Heeney, M1; Rees, D2; de Montalembert, M3; Odame, I4; Brown, C5; Wali, Y6; Nguyen, T7; Lam, D8; Pfender, N9; Kanter, J10
1Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, UNITED STATES; 2Department of Paediatric Haematology, King’s College Hospital, London, UNITED KINGDOM; 3Hôpital Universitaire Necker-Enfants Malades, Paris, FRANCE; 4The Hospital for Sick Children (SickKids) and the University of Toronto, Toronto, CANADA; 5Children’s Healthcare of Atlanta, Atlanta, UNITED STATES; 6Sultan Qaboos University, Muscat, OMAN; 7Novartis Pharma S.A.S., Rueil-Malmaison, FRANCE; 8Novartis Pharmaceuticals Co., East Hanover, UNITED STATES; 9Novartis Pharma AG, Basel, SWITZERLAND; 10Division of Hematology-Oncology, University of Alabama-Birmingham, Birmingham, UNITED STATES
Background: Vaso-occlusive crises (VOCs) are the hallmark of SCD. P-selectin, a cell adhesion molecule, plays a key role in the multicellu-lar interactions leading to VOCs. In the SUSTAIN trial, crizanlizumab 5 mg/kg, a first-in-class humanized monoclonal antibody that blocks P-selectin, significantly reduced the annualized rate of VOCs versus pla-cebo and had a favorable safety profile in adults with SCD.Aims: To describe the initial safety and efficacy results for patients with SCD aged 12 to <18 years treated with crizanlizumab 5 mg/kg in the SOLACE-kids trial (ClinicalTrials.gov: NCT03474965; EudraCT: 2017-001747-12).Methods: SOLACE-kids is an ongoing Phase II study to establish and confirm appropriate dosing (Part A) and evaluate long-term safety and efficacy (Part B) of the confirmed dose of crizanlizumab, with or with-out hydroxyurea, in pediatric patients with SCD (any genotype) and ≥1 VOC leading to a healthcare visit within 12 months prior to screening. Patients (target enrollment N≥100) are stratified by age: Group 1 (12–<18 years), Group 2 (6–<12 years) and Group 3 (6 months–<6 years). Patients receive crizanlizumab on Day 1, Day 15, then every 4 weeks (up to 2 years). This analysis reports initial safety and efficacy results for Group 1 patients receiving crizanlizumab 5 mg/kg.Results: As of 28 August 2020, 50 patients have been enrolled in Group 1 (median [min–max] age 14.9 [12.0–17.9] years; 58% female; 88% HbSS genotype; 64% Black/African American; 84% receiving hydroxy-urea). Median (range) duration of crizanlizumab exposure was 36.6 (6–98) weeks; 88% of patients received treatment for ≥26 weeks.The most common adverse events (AEs) regardless of causality with study treatment were headache (n=14 [28%]), vomiting (n=12 [24%]) and back pain (n=9 [18%]). Treatment-related AEs occurred in 12 (24%) patients. Grade ≥3 AEs occurred in 13 (26%) patients, most com-monly anemia (n=3 [6%]) and back pain (n=2 [4%]). Grade ≥3 treat-ment-related AEs (back pain and pain in extremity) were reported in one (2%) patient. Eleven (22%) patients had serious AEs; none were
deemed treatment related. No AEs led to treatment discontinuation except one patient who died of suspected meningitis (considered unre-lated to treatment).The incidence of AEs of special interest (AESI) is shown in Table 1. No infusion-related reactions were serious, and all had resolved at data cut-off, except for one case of Grade 1 dizziness. There were no cases of anaphylaxis to crizanlizumab. No pain events were serious, and all had resolved or were resolving by data cut-off.The median (Q1–Q3) number of VOCs leading to a healthcare visit at baseline was 3.0 (1.0–5.0) versus 1.6 (0.0–3.7) while on treatment, a median absolute reduction from baseline of –1.0 (–3.0 to 0.5). At base-line, the median (Q1–Q3) rate of hospitalizations/emergency room visits was 4.0 (2.0–8.0) versus 1.5 (0.0–2.7) while on treatment, a median absolute reduction from baseline of –2.4 (–5.0 to 1.0). Eighteen (36%) patients did not experience a VOC leading to a healthcare visit during crizanlizumab treatment.Summary – Conclusion: In this initial analysis, crizanlizumab 5 mg/kg was safe and well tolerated in patients with SCD aged 12–<18 years, consistent with the established profile of crizanlizumab in adults. No new safety signals were identified. Crizanlizumab 5 mg/kg led to a clin-ically relevant reduction in VOCs in this patient population compared with baseline.
S123 SAFETY AND EFFICACY OF EARLY-START DEFERIPRONE IN INFANTS AND YOUNG CHILDREN WITH BETA-THALASSEMIA (START STUDY)
Elalfy, M1; El-Beshlawy, A2; Adly, A1; Ebeid, F1; Fradette, C3; Lee, D3; Temin, N3; Tricta, F3; Rozova, A3; Hamdy, M4
1Ain Shams University, Children’s Hospital, Cairo, EGYPT; 2Cairo University, Pediatric Hospital, Cairo, EGYPT; 3Chiesi Canada Corporation, Toronto, CANADA; 4Cairo University, Research Center, Cairo, EGYPT
Background: Patients with beta-thalassemia may need regular red blood cell transfusions in infancy.1 In the absence of iron chelation therapy, fre-quent transfusions cause iron to accumulate, which can lead to morbid-ity, organ damage, and death. However, in very young children, current practice is to delay chelation therapy until receipt of 10–20 transfusions, or until serum ferritin (SF) has reached 1000 μg/L, due to concerns over excessive iron depletion with deferoxamine.1,2 Unfortunately, this delay may increase the risk of iron accumulation in endocrine glands where toxicities could later manifest.3
Aims: START (NCT03591575) evaluated the safety and efficacy of the oral iron chelator deferiprone (DFP) in children with transfusion-depen-dent beta-thalassemia who did not yet meet the criteria for starting che-lation therapy per standard practice.Methods: Infants and children receiving regular blood transfusions (2 minimum) and whose SF level was between 200–600 μg/L were ran-domly assigned 1:1 to DFP oral solution or placebo until SF exceeded 1000 μg/L at 2 consecutive visits or 12 months of therapy. DFP was initiated at 25 mg/kg/d and increased to 50 mg/kg/d after 2 weeks; based on iron load, DFP was increased to 75 mg/kg/d. Efficacy was assessed by

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 13
monthly SF measurements to monitor iron load. The primary endpoint was the percentage of patients in each group with SF <1000 μg/L. All patients provided informed consent or assent.Results: The study enrolled 64 patients; 32 per group. The mean (SD) age was 3.03 (2.42) years in the DFP group and 2.63 (1.70) years in the placebo group; participants were 62.5% and 65.6% male in the DFP and placebo groups, respectively. There were no group statistical differences in the baseline demographics. The primary reason for with-drawal was SF levels that exceeded 1000 μg/L at 2 consecutive visits, which occurred in 25% of patients receiving DFP vs 63% of patients receiving placebo (P=0.0051). After completing 12 months of treatment, 65.6% of patients receiving DFP had a SF level <1000 mg/L vs 37.5% receiving placebo (P=0.0446). The percentage of patients who reached the 1000 mg/L SF threshold increased more rapidly in the placebo group vs the DFP group, and the difference in rates between the 2 groups was significant (P=0.0407). A summary of adverse events (AEs) is shown in Table 1. There were no significant group differences in the number of overall AEs (P=1.0000), serious AEs (P=0.4258), or the number of AEs that were considered to be at least possibly related to the study treatment (P=1.0000). Two patients receiving DFP withdrew from the study due to AEs: 1 patient experienced agranulocytosis and 1 patient experienced neutropenia of moderate severity and both patients recovered.Conclusions: Initiation of DFP chelation therapy at a lower threshold of SF values than currently recommended was safe and efficacious in pre-venting iron overload in most transfusion-dependent children. After 12 months of treatment, the number of patients with a SF level <1000 μg/L was significantly higher in the DFP group vs the placebo group. Moreover, there were significantly more patients who withdrew due to elevated SF levels in the placebo group (62.5%) vs the DFP group (25%). The safety and tolerability profile of DFP administered for up to 12 months in infants and young children was comparable to the profile established in older age groups and there were no instances of iron depletion.
References1. Thalassaemia International Federation, Guidelines for the management
of transfusion dependent thalassaemia (TDT) [internet]. 3rd ed: 2014.2. De Virgiliis et al, J Pediatr 1988; 113:6613. Berdoukas et al, Am J Hematol 2013; 88:E283
Infection, autoimmunity, nutritional deficiencies
S124 ADVERSE EVENTS FOLLOWING COVID-19 VACCINATION IN TRANSFUSION-DEPENDENT -THALASSEMIA PATIENTS
Delaporta, P1; Kyriakopoulou, D1; Ioakeimidou, N1; Solomou, E1; Binenbaum, I2; Toutoudaki, K1; Chatzielefteriou, M1; Papassotiriou, I3; Kattamis, A1
1First Department of Pediatrics, National and Kapodistrian University of Athens, ‘Aghia Sophia’ Children’s Hospital, Athens, GREECE; 2Pediatric Hematology Oncology Unit, First Department of Pediatrics, National and Kapodistrian University of Athens, Aghia Sophia, Athens, GREECE; 3Department of Clinical Biochemistry, Athens, GREECE
Background: Patients with transfusion-dependent-thalassemia (TDT) are considered as increased risk population for severe and/or morbid
COVID-19 infection. Timely vaccination is the main preventive method for severe COVID-19. Different adverse events and reactions following vaccination have been reported, with severe ones being extremely rare. TDT patients may have altered immunity due to chronic transfusions, iron overload and chelation therapy, and splenic dysfunction. The safety profile of vaccination in chronically transfused patients with thalassemia has not been reported.Aim: To evaluate the safety profile of COVID-19 vaccines in TDT patients.Patients and Methods: This is a single institution’s, retrospective analysis evaluating all TDT patients, older than 18 years old, who had completed the vaccination protocol at least 30 days before data cut-off time (July 20th 2021). Adverse events were reported by patients up to 30 days post each dose. Demographic data and hematological data, including mean hemoglobin levels before and up to 90 days after each dose, were recorded. T-test was performed to investigate changes in hematological profile and transfusion burden post vaccination.Results: 186 patients (median age:45; range:18–61 years old; male/female:87/99) were included for data analysis corresponding to 53% of all TDT patients followed in our Thalassemia Unit. Distribution of vaccine types were: Comirnaty-BNT162b2 (Pfizer Inc. and BioNTech)90.86% (n =168), Vaxzevria (previously COVID-19 vaccine, AstraZeneca) 1.61% (n=3), Moderna (Moderna TX Inc.) 6.99% (n =13) and JNJ-78436735 (Janssen Pharmaceuticals Companies of Johnson & Johnson) 0.54% (n =2). No patients had confirmed or suspected previ-ous COVID-19 infection.Adverse events were graded according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0. The incidence of adverse events after 1st and 2nd dose were 43.5% (81/186) and 54.8% (102/186), respec-tively. Adverse events after 1st dose included pain at injection site 26.3% (n=49), fatigue 9.7%(n=18), fever 5.4% (n=10),headaches 4.3% (n=8), arthralgia and myalgia 2.2% (n=4), and lymphadenopathy 0.5% (n=1). Adverse events after 2nd dose included fever 28.5% (n=53), fatigue 17.7% (n=33), pain at injection site 15.6%(n=29), arthralgia and myal-gia 11.3% (n=21), headaches 9.1% (n=17), lymphadenopathy 3.2% (n=6), dizziness 0,5% (n=1), tachycardia 0.5% (n=1), diarrhea/ vomiting 0.05% (n=1) and amaurosis fugax 0.5%: (n=1). No grade 4–5 events or anaphylaxis were observed. Two patients (both males, 51 years and 45 years old, respectively) presented with acute hemolytic crisis with hemoglobinuria in 3rd and 20th day after the second dose with Pfizer/BioNTech, respectively. They are receiving treatment with corticosteroids without partial response. Both patients had a history of acute hemolysis crisis within the last 3 years. A slight decrease in patients’ mean hemo-globin levels was observed three months after vaccination compared to the mean hemoglobin levels before vaccination (mean=9.9 /sd=0.63 g/dL vs mean=9.81 /sd=0.64 g/dl, respectively, p= 0.05).Conclusions: Compared to the vaccine trials, we observed some lower incidence of vaccine-related adverse events in our cohort of TDT patients. A temporary drop in hemoglobin levels may be noted. Of inter-est, two patients with previous history of alloimmunization, developed hemolysis. Close follow up is required in TDT patients post vaccination.
References1. Hu B et al. Nature Reviews Microbiology 2020; 19: 141–1542. Pollack FP et al, N Engl J Med 2020; 383:2603–26153. Lee JX et al. Front Med (Lausanne). 2021;13;8:757510
S125 THE ROLE OF THE MICROBIOME AS A SICKLE CELL DISEASE MODIFIER: AN EXPERT REVIEW
Oosterwyk, C1; Nguweneza, A1; Mnika, K2; Nembaware, V1; Wonkam, A1
1University of Cape Town, Cape Town, SOUTH AFRICA; 2University of Cape Town, Johanessburg, SOUTH AFRICA
Background: There is evidence that the microbiome is involved in numerous human systems’ pathobiology, however, its role in sickle cell disease (SCD) remains elusive.Methodology: An extensive systematic search was conducted by apply-ing the PRISMA guidelines (2009) and using three databases (Pubmed, Web of Science, and Scopus). The keywords were used separately and a combination of the following: “Microbiota” AND “Anemia” OR “Sickle Cell”. Articles included were confined to full-length articles written in English and focused on research and review articles on the contribu-tion of the microbiome in the development of sickle cell disease (SCD)

ASCAT2022
14 | 2022; 6:S1
severity. The main search was conducted, separately, by a MSc Student and a PhD student, both in Human Genetics and reviewed by a medical and human geneticist with expertise in SCD and a clinician-scientist with expertise in molecular biology and microbiology. A total of 229 arti-cles was compared and screened for duplications. The abstracts of the remaining articles were screened for suitability to the study. Articles were excluded based on its relevance to the scope of the review.Results: This systematic review examines available data on the role of the microbiome in SCD clinical events. A total of 13 published articles were selected; most were observational human studies (n =10), and five included SCD mouse models. Few studies performed microbiome 16S rRNA analysis, to identify/classify uncultured microbes (n = 8/13). Results showed that the microbiome influences inflammation, and vaso-occlusive crises (VOC) in SCD, and is disrupted by medication and diet. This review was able to provide insight on how immunity can be maintained by the microbiome through TLRs and Myd88 mechanisms. The exploration of bacterial translocation may be regulated by an alter-ing intestinal permeability and the microbial density, which is managed by the damage-associated molecular pattern (DAMPs). This may give insight into treating an altered intestinal microbiome in SCD patients. Whereas in SCD mouse models, neutrophil adhesion, Mac-1 activation, and heterotopic interaction in red blood cells were significantly influ-enced by microbiota.Conclusion: Data suggest that the microbiome can be disrupted by the SCD endophenotypes, specifically by triggering inflammation pathways, which could promote a cycle of VOC. This review emphasizes the need for investigating microbiome effects on SCD throughout the lifespan of patients.
References1. Liberati, A., Altman, D. G., Tetzlaff, J., Mulrow, C., Gøtzsche, P. C.,
Ioannidis, J. P. A., Clarke, M., Devereaux, P. J., Kleijnen, J. & Moher, D. 2009. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. PLOS Medicine, 6, e1000100.
2. Zhang, D., Chen, G., Manwani, D., Mortha, A., Xu, C., Faith, J. J., Burk, R. D., Kunisaki, Y., Jang, J.-E., Scheiermann, C., Merad, M. & Frenette, P. S. 2015. Neutrophil ageing is regulated by the microbiome. Nature, 525, 528–532.
3. Tavakoli, S. & Xiao, L. 2019. Depletion of Intestinal Microbiome Partially Rescues Bone Loss in Sickle Cell Disease Male Mice. Scientific Reports, 9, 8659.
4. Rana, K., Pantoja, K. & Xiao, L. 2018. Bone marrow neutrophil aging in sickle cell disease mice is associated with impaired osteoblast func-tions. Biochemistry and Biophysics Reports, 16, 110–114
5. Dutta, D., Methe, B., Amar, S., Morris, A. & Lim, S. H. 2019. Intestinal injury and gut permeability in sickle cell disease. Journal of Translational Medicine, 17, 183.
6. Xu, C., Lee, S. K., Zhang, D. & Frenette, P. S. 2020. The Gut Microbiome Regulates Psychological-Stress-Induced Inflammation. Immunity, 53, 417–428.e4.
7. Brim, H., Taylor, J., Abbas, M., Vilmenay, K., Daremipouran, M., Varma, S., Lee, E., Pace, B., Song-Naba, W. L., Gupta, K., Nekhai, S., O’neil, P. & Ashktorab, H. 2021. The gut microbiome in sickle cell disease: Characterization and potential implications. PLoS One, 16, e0255956
8. Rocha, L. C., Carvalho, M. O., Nascimento, V. M., Dos Santos, M. S., Barros, T. F., Adorno, E. V., Reis, J. N., Da Guarda, C. C., Santiago, R. P. & Goncalves, M. S. 2017. Nasopharyngeal and Oropharyngeal Colonization by Staphylococcus aureus and Streptococcus pneumo-niae and Prognostic Markers in Children with Sickle Cell Disease from the Northeast of Brazil. Front Microbiol, 8, 217.
9. Kimaro, F. D., Jumanne, S., Sindato, E. M., Kayange, N. & Chami, N. 2019. Prevalence and factors associated with renal dysfunction among children with sickle cell disease attending the sickle cell disease clinic at a tertiary hospital in Northwestern Tanzania. PLoS ONE, 14, 1–13.
10. De Matos, B. M., Ribeiro, Z. E. A., Balducci, I., Figueiredo, M. S., Back-Brito, G. N., Mota, A. J. D., Braga, J. A. P. & Koga-Ito, C. Y. 2014. Oral microbial colonization in children with sickle cell anae-mia under long-term prophylaxis with penicillin. Archives of Oral Biology, 59, 1042–1047.
11. Fonseca, P. B. B., Braga, J. A. P., Machado, A. M. D. O., Brandileone, M. C. C. & Farhat, C. K. 2005. Nasopharyngeal colonization by Streptococcus pneumoniae in children with sickle cell disease receiv-ing prophylactic penicillin. Jornal de Pediatria, 81, 149–154.
12. Mohandas, S., Soma, V. L., Tran, T. D. B., Sodergren, E., Ambooken, T., Goldman, D. L., Weinstock, G. & Herold, B. C. 2020. Differences in Gut Microbiome in Hospitalized Immunocompetent vs. Immunocompromised Children, Including Those With Sickle Cell Disease. Front Pediatr, 8, 583446.
13. Byeon, J., Blizinsky, K. D., Persaud, A., Findley, K., Lee, J. J., Buscetta, A. J., You, S., Bittinger, K., Minniti, C. P., Bonham, V. L. & Grice, E. A. 2021. Insights into the skin microbiome of sickle cell disease leg ulcers. Wound Repair Regen.
14. Costa, C. P. S., Alves, M. S., Lima-Neto, L. G., Valois, E. M., Monteiro-Neto, V. & Souza, S. F. C. 2021. Is there bacterial infection in the intact crowns of teeth with pulp necrosis of sickle cell anaemia patients? A case series study nested in a cohort. Int Endod J, 54, 817–825
Health services and outcomes research including psychology
S126 AN EU -- ARISE INITIATIVE GAP ANALYSIS APPROACH TO IMPROVING THE QUALITY OF LABORATORY SYSTEMS IN SUB-SAHARAN AFRICA.
Atoyebi, W1; Ruggieri, L2; Price, K3; Elion, J4; Bonifazi, D2; Inusa, B5
1Oxford University Hospitals NHS Foundation Trust, Oxford, UNITED KINGDOM; 2Benzi Foundation, Bari, ITALY; 3Royal College of Pathologists., London, UNITED KINGDOM; 4INSERM, Paris, FRANCE; 5Guys and St Thomas’s NHS Foundation Trust, London, UNITED KINGDOM
Background: Accurate and reliable laboratory results are critical to diag-nostics in sickle cell disease (SCD) newborn screening.In sub-Saharan Africa, due to neglect of some national health labo-ratories, there has been persistently high levels of laboratory error, lack of functioning quality management systems and laboratory accreditation.ARISE (African Research and Innovative Initiative for Sickle Cell Education) is a research project funded by the EU. It involves a sec-ondment plan across 8 work packages between institutions in EU (Italy, France, UK and Cyprus) and non-EU (Nigeria, Lebanon, Kenya and USA) countries, creating an interagency and multidisciplinary staff exchange programme to share best practices in newborn screen-ing, diagnosis and treatment of SCD, leading to improved disease outcomes.Secondments include medical, nursing, laboratory, administrative, aca-demic and research professionals.Led by the RCPath and INSERM, WP3 is tasked with improving labora-tory diagnostics, capacity, and quality assurance systems for population screening, diagnosis, treatment and monitoring of SCD through assess-ments, training and mentoring of laboratory health professionals. This is facilitated by baseline, interim and final gap assessmentsAims:
1. To compare a baseline gap analysis at the commencement of the project in 2019 with an interim gap assessment 2 years into the project, following secondments to UK laboratories from Nigeria
2. To identify areas of increased focus and support during planned secondments, workshops, and virtual lectures
Methods: 6 partner organisation laboratories participated in the base-line gap assessment in 2019, while 10 laboratories participated in the interim gap assessment in 2021, due to new institutions joining the ARISE initiative.An electronic online questionnaire (40 questions) was sent to each lab-oratory lead.Results: Survey results of the 6 laboratories that undertook both the baseline and interim gap assessment were analysed.A sample of comparative results between 2019 and 2021 include the following:
• Working towards accreditation - 2/6 versus 3/6: There is the need for each laboratory to progress towards an application for accreditation.
• SOP’s available for tests and processes - 38 versus 32: This might suggest standardisation and rebranding of documents previously labelled as SOP’s.

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 15
• No schedule of SOP revisions in 0/6 laboratories versus 4/6 labora-tories: This clearly demonstrates an understanding that SOP’s need to be updated via a document management system.
• Scientist certified competences - 22 versus 22.• Self -assessed improvement targets - 18 versus 15.• Laboratory lead for QA - 3/6 versus 6/6: The importance of
QA leadership for each laboratory identified prior to interim assessment.
Summary and Recommendations: Early evidence suggests that ARISE secondments have facilitated improved approaches to laboratory gover-nance procedures and QA.Mentoring by EU partner laboratories will further enhance this.ARISE related workshops and train - the- trainer programmes will sup-port capacity building and skills transfer.A final assessment will be undertaken at the end of the ARISE initiative.Conclusion: The ARISE initiative will build on the early success of improvement in laboratory systems, to further support service develop-ment and capacity building. The target for each partner laboratory will be a timeline of application to full accreditation
References1. Gershy-Damet GM et al, The World Health Organization African
region laboratory accreditation process: improving the quality of laboratory systems in the African region. Am J Clin Pathol. 2010 Sep;134(3):393–400
S127 CONSORTIUM ON NEWBORN SCREENING IN AFRICA FOR SICKLE CELL DISEASE
Nnodu, O1; Zapfel, A2
1University of Abuja Teaching Hospital, Abuja, NIGERIA; 2American Society of Hematology, Washington, DC, UNITED STATES
Background: Sickle cell disease (SCD) is an inherited blood disorder that causes abnormalities in the oxygen-carrying protein hemoglobin found in red blood cells, leading to severe long-term health effects. According to the World Health Organization, more than 300,000 babies are born each year with SCD globally with majority in sub-Saharan Africa fail-ing to reach the age of five. With early identification, low-cost treat-ments known to be effective in high-income settings for several years, can improve the health of children with SCD. Yet, SCD has not received much funding in many African countries as a health priority limiting available diagnostic and treatment services for it.The Consortium on Newborn Screening in Africa for SCD (CONSA), established in 2018, is a part of the American Society of Hematology’s broad initiative to strengthen the SCD response globally through advo-cacy and research generation.
CONSA Program Goals and Objectives
• Demonstrate the effectiveness of early identification and clinical interventions for newborns with SCD
• Create sustainable, expanded networks for newborn screening and clinical interventions
• Foster collaboration between African hematologists and public health services to develop an organized network of researchers for conducting quality studies and publishing results
• Increase hematology capacity throughout sub-Saharan Africa
CONSA introduces standard-of-care practices for screening and early intervention therapies (including antibiotics and malaria prophylaxis, folic acid supplements, family education and counseling, and immuni-zations) at participating clinical institutions, screening 10,000 – 16,000 babies per year in each country, and providing clinical follow-up for babies diagnosed with SCD. A shared registry captures data from CONSA institution sites, which will be used to estimate the prevalence of SCD in member countries and evaluate the effectiveness of the inter-ventions for the survival of newborns to five years of age.Currently, CONSA is working in seven countries, Ghana, Kenya, Liberia, Nigeria, Tanzania, Uganda, and Zambia. Hematologists and public health officials participating in the consortium have mobilized networks of screening laboratories, SCD or pediatric hematology clinics, teaching hospitals, regional referral hospitals, universities, and satellite clinics to screen newborns and provide clinical services protocol. Alongside the
research showcasing the health outcomes of newborns screened and delivered early interventions, the consortium is working to ensure the long-term sustainability of programs through government, corporate, and other partner support.All country sites launched screening in 2021. As of November 15, over 17,000 babies have been screened with over 150 found to be living with SCD. Despite challenges from the COVID-19 pandemic, including pop-ulation concerns of going to health clinics, need to protect SCD patients from transmission, and supply chain breakdowns, CONSA looks for-ward to continuing to expand newborn screening efforts for the next several years.Conclusion: The presentation will provide an overview of CONSA’s goals and current work to screen and provide care for newborns with SCD, despite challenges from the COVID-19 pandemic. The presentation will also include details from the Nigeria clinical sites, case studies of current babies with SCD, and recent work done to strengthen Nigeria’s national newborn screening and clinical eff
References1. World Health Organization, Sickle Cell Disease 2021.
S128 CONTINUOUS QUALITY IMPROVEMENT IN PAEDIATRIC SICKLE CELL DISEASE
Ahemad, E; Mitchell, R; Goridari, M; Wood, I; Mahmood, S; O’Driscoll, S; Cortes, A; Chakravorty, S
King’s College Hospital NHS foundation trust, London, UNITED KINGDOM
Background: Sickle cell disease (SCD) is characterised by severe episodic pain requiring prompt and effective analgesia. However, SCD patients frequently experience poor quality of care due to lack of awareness of the condition among non- specialist staff, pre-conceived biases and unfounded allegation of drug-seeking behaviour. Several reports and sur-veys using Patient Reported Experience Measure (PREM) tools indicate widespread prevalence of ineffective pain relief in emergency depart-ment(ED), poor access to psychological therapies and poor funding for service development.Aims: To use a validated PREM tool to survey patients or car-ers of sickle cell disease within the paediatric service of a Specialist Haemoglobinopathy Team. To use an established Continuous Quality Improvement (CQI) methodology (5-D’s-define, describe, design, deliver, digest) to ensure sustained improvement in patient- reported areas of service deficit.Methods: The PREM survey was conducted as part of a network- wide initiative in 2018. The survey responses were analysed and problem scores created for specialist care, emergency care, ward-based care, information and support. These problem areas were categorised into domains where further improvement action was needed. CQI tools were used to map the patient journey within Emergency Department (ED) when presenting with pain followed by thematic analysis to identify potential improvements in the patient journey.Results: The PREM survey analysis identified three domains for improvement:
• Domain A: To improve the effectiveness of pain relief and quality of empathic and informed care to paediatric sickle cell patients and their families in ED.
We developed multi-disciplinary teaching videos with patient involve-ment and patient information material based on ED/pain/sickle staff feedback.Excellent engagement from all staff groups obtained. Patient involve-ment in ED teaching video was well received. Thematic analysis of ED process mapping highlighted need for sustained staff education and re-writing ED pain protocol to align with current practice. Pre-conceived biases were identified among staff with respect to attributing drug-seek-ing behaviour to patients during severe sickle cell pain.
• Domain B: To improve the information presented to children regarding SCD and the care services available in our trust
We developed child friendly Information leaflets targeting primary school age and secondary school age readers
• Domain C: To improve the availability of, and access to, mental health services for paediatric SCD patients.

ASCAT2022
16 | 2022; 6:S1
We developed virtual parent support groups and psychological health screening for adolescent patients in clinics. We organised a ‘Tree of Life’ (ToL) patient empowerment workshop. The parent support groups were well attended. Plans are in place to continue ToL workshops and support groups on a regular basis.Future work: A rapid improvement workshop is being planned to engage a wider multi-disciplinary team to engender a culture of empathy in line with our trust values. Further input from workshop is expected to iden-tify and implement sustainable goals. We plan to undertake a repeat PREM survey following the end of this CQI project to understand the impact and the implemented improvements.
References1. Chakravorty, S, et al (2018) Archives of Disease in Childhood, 103
(12), pp. 1104–1109.2. All Party Parliamentary Group in sickle cell and thalassaemiareport:
No one’s listening(https://www.sicklecellsociety.org/wp-content/uploads/2021/11/No-Ones-Listening-PDF-Final.pdf)
3. Peer Review Overview Report 2020(https://haemoglobin.org.uk/wp-con-tent/uploads/2020/11/Adult-and-CYP-HD-Overview-Report-2020-V1.pdf)
S129 IMPLEMENTATION OF HYDROXYUREA THERAPY FOR SICKLE CELL DISEASE ON A LARGE SCALE IN GHANA
Ohene-Frempong, K1; Segbefia, C2; Spector, J3; Amoah, E1; Asubonteng, A1; Hammond-Addo, R4; Odame, I5; Odame, I5
1Sickle Cell Foundation of Ghana, Accra, GHANA; 2Department of Child Health, University of Ghana Medical School, Accra, Ghana, Accra, GHANA; 3Novartis Institutes for Biomedical Research, Cambridge, MA, UNITED STATES; 4Novartis Corporate Affairs & Global Health, Accra, GHANA; 5Departments of Medicine and Paediatrics, University of Toronto, Toronto, CANADA
Background: Hydroxyurea (hydroxycarbamide), has had a most pro-found and broad ameliorating impact on the clinical course of sickle cell disease (SCD.) Although relatively inexpensive, hydroxyurea (HU) has not been widely available to the large majority of people with SCD who happen to live in low-income countries.In 2018, in preparation for the establishment in 2019 of a broad-based Public Private Partnership (PPP) in SCD involving the Ghana govern-ment and Novartis, a group of parents Novartis to provide HU at a lower price for use in Ghana. Novartis produced 500mg capsule of HU and submitted it to Ghana Food and Drugs Authority registered the medicine for the specific indication of SCD in October 2018.The Sickle Cell Foundation of Ghana (SCFG), a partner in the PPP was tasked to develop the Ghana-Novartis Hydroxyurea-for-SCD Program (“Ahodwo [pr. A-ho-jo] Program”, meaning, “Program for Relief”.). The program was conceived as an implementation study to determine whether treatment with HU, specifically registered in Ghana for SCD, can be safely implemented and monitored on a large scale through an organized treatment program within the public health service in Ghana.Methods:1. Treatment Protocol: A team of Ghanaian SCD experts developed
an HU-for-SCD dose-escalating, maximum tolerated dose (MTD) Ahodwo Protocol adapted for Ghana. A unique feature of the pro-tocol is the selection of Hb level of 10g/dL as the primary goal of HU therapy with of a Therapeutic Dose (TD) defined as, “the dose at which Hb 10 g/dL or higher is achieved and maintained over a period of 12 weeks”.
2. Treatment Teams: Established SCD Treatment Centres (SCD TC) were surveyed for patient numbers, age groups, Hb Phenotypes, and available laboratory services. Doctor-nurse-pharmacist teams were recruited from 11 TC located in four Regions of Ghana in Phase 1 of the Program for training. A year later, 9 smaller SCD TC were added to the program, in Phase 2, extending it to two additional Regions.
3. Ahodwo Program App: In order to register and guide healthcare professionals (HCP) on the protocol, register all subjects, assist with dosing calculations, and monitor the entire program, a secure, smart-phone-based mobile application, Ahodwo Program App, was devel-oped, tested, and deployed to all HCP in the program. Recording toxicity and reporting all expected/unexpected adverse events were mandated and reportable through the App.
4. Steering Committee (ST): A ST comprising clinician leaders of the TC was established; the ST held bi-weekly online review meetings for the first year and monthly thereafter. All HCP teams met every quarter.
Results: Table, below, lists the number and characteristics of subjects registered in the Ahodwo Program.
Data Elements Phase 1 Phase 2 TOTAL
Number of SCD Treatment Centers (TC) 11 9 20Number of subjects on HU 3,357 291 3,648Average, No. of Subjects at TC 305 32 182Median, No. of Subjects at TC 256 26 88Range, No. of Subjects at TC (46, 694) (7, 94) (7, 694)Female, No. (%) 1,728 (47.4%)Male, No. (%) 1,920 (52.6%)Age < 10yr, No. (%) 1,561 (42.8%)Age 10-18yr, No. (%) 1,295 (35.5%)Age > 18yr, No. (%) 792 (21.7%)Presumed SCD-SS or S/beta-zero, No. (%) 3,526 (96.7%)SCD-SC, No. (%) 122 (3.3%)
On June 19th, 2021, World Sickle Cell Day, the government of Ghana announced the provision of HU for people with SCD through the National Health Insurance Scheme. Following preparatory meetings to establish the required regulatory standards, implementation of the national program is expectConclusion: Our experience supports the tenet that hydroxyurea can be safely and effectively administered at population scale in a low-income country. Long-term sustainability in this setting is likely to be dependent on a government-supported access programme. The pioneering efforts of the government of Ghana to provide HU to its citizens with SCD are laudable and serve as a model to guide similar efforts in other low-income countries.
S130 PROCESSING SPEED DECLINES OVER TIME IN 4--25-YEAR-OLDS WITH SICKLE CELL DISEASE
Walker, E1; Hood, A2; Kirkham, F1; Hogan, A1; Watkins, K3
1University College London, London, UNITED KINGDOM; 2University of Manchester, Manchester, UNITED KINGDOM; 3University of Oxford, Oxford, UNITED KINGDOM
Background: Alongside physiological symptoms, young people with sickle cell disease (SCD) may also experience cognitive difficulties, including poorer processing speed. Processing speed develops rapidly from birth to around mid-childhood, with steady improvements thereaf-ter into a person’s mid-twenties (Anderson, 2002). Nonetheless, little is known about the dynamic developmental trajectory of processing speed for young people with SCD.Aims: This study, we aimed to investigate if the change in processing speed index (PSI) over time is significantly different between younger participants (aged under 8.99 years at first assessment) and older partic-ipants (over 9 years at first assessment) with SCD.Methods: One hundred and five participants with SCD aged 4 – 18 (N < 8.99 = 47; N > 9 = 58) at recruitment consented to follow-up IQ assessments (WPPSI-R, WISC-III, WAIS-R or WAIS-III) and MRI scans
Figure 1. Processing speed index at timepoint 1 and timepoint 2 for par-ticipants under 9 (median age 6 years) and over 9 (median age 13 years) years by cerebral infarct status
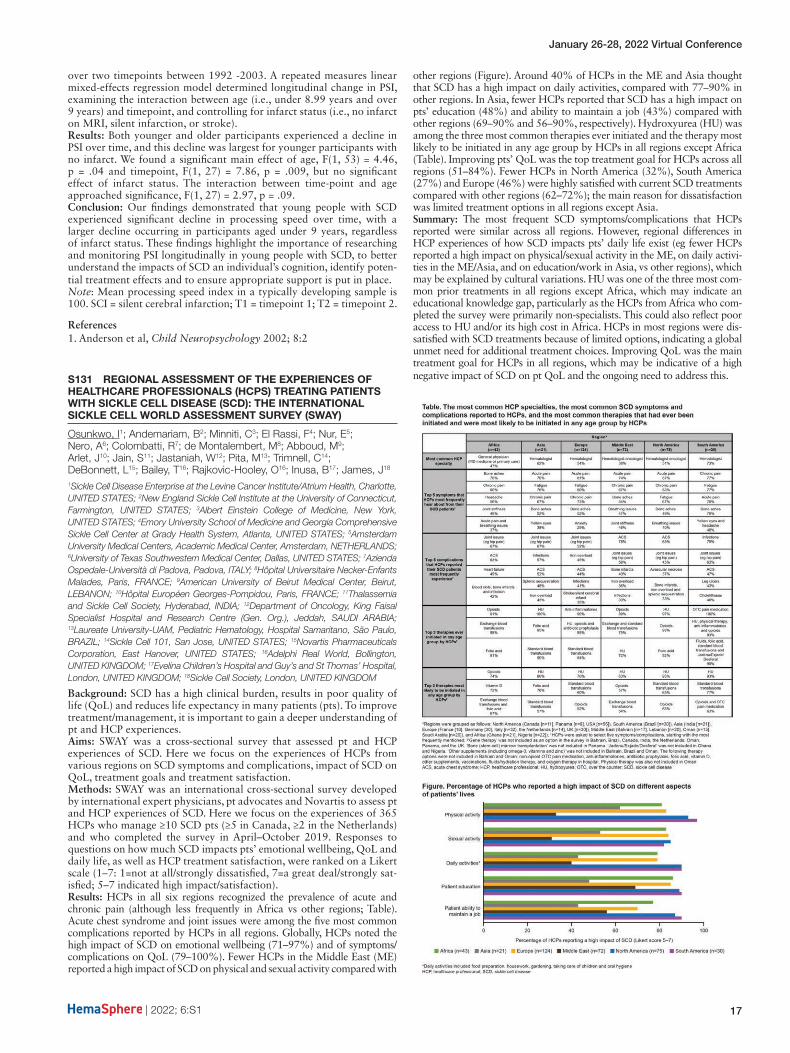
January 26-28, 2022 Virtual Conference
| 2022; 6:S1 17
over two timepoints between 1992 -2003. A repeated measures linear mixed-effects regression model determined longitudinal change in PSI, examining the interaction between age (i.e., under 8.99 years and over 9 years) and timepoint, and controlling for infarct status (i.e., no infarct on MRI, silent infarction, or stroke).Results: Both younger and older participants experienced a decline in PSI over time, and this decline was largest for younger participants with no infarct. We found a significant main effect of age, F(1, 53) = 4.46, p = .04 and timepoint, F(1, 27) = 7.86, p = .009, but no significant effect of infarct status. The interaction between time-point and age approached significance, F(1, 27) = 2.97, p = .09.Conclusion: Our findings demonstrated that young people with SCD experienced significant decline in processing speed over time, with a larger decline occurring in participants aged under 9 years, regardless of infarct status. These findings highlight the importance of researching and monitoring PSI longitudinally in young people with SCD, to better understand the impacts of SCD an individual’s cognition, identify poten-tial treatment effects and to ensure appropriate support is put in place.Note: Mean processing speed index in a typically developing sample is 100. SCI = silent cerebral infarction; T1 = timepoint 1; T2 = timepoint 2.
References1. Anderson et al, Child Neuropsychology 2002; 8:2
S131 REGIONAL ASSESSMENT OF THE EXPERIENCES OF HEALTHCARE PROFESSIONALS (HCPS) TREATING PATIENTS WITH SICKLE CELL DISEASE (SCD): THE INTERNATIONAL SICKLE CELL WORLD ASSESSMENT SURVEY (SWAY)
Osunkwo, I1; Andemariam, B2; Minniti, C3; El Rassi, F4; Nur, E5; Nero, A6; Colombatti, R7; de Montalembert, M8; Abboud, M9; Arlet, J10; Jain, S11; Jastaniah, W12; Pita, M13; Trimnell, C14; DeBonnett, L15; Bailey, T16; Rajkovic-Hooley, O16; Inusa, B17; James, J18
1Sickle Cell Disease Enterprise at the Levine Cancer Institute/Atrium Health, Charlotte, UNITED STATES; 2New England Sickle Cell Institute at the University of Connecticut, Farmington, UNITED STATES; 3Albert Einstein College of Medicine, New York, UNITED STATES; 4Emory University School of Medicine and Georgia Comprehensive Sickle Cell Center at Grady Health System, Atlanta, UNITED STATES; 5Amsterdam University Medical Centers, Academic Medical Center, Amsterdam, NETHERLANDS; 6University of Texas Southwestern Medical Center, Dallas, UNITED STATES; 7Azienda Ospedale-Università di Padova, Padova, ITALY; 8Hôpital Universitaire Necker-Enfants Malades, Paris, FRANCE; 9American University of Beirut Medical Center, Beirut, LEBANON; 10Hôpital Européen Georges-Pompidou, Paris, FRANCE; 11Thalassemia and Sickle Cell Society, Hyderabad, INDIA; 12Department of Oncology, King Faisal Specialist Hospital and Research Centre (Gen. Org.), Jeddah, SAUDI ARABIA; 13Laureate University-UAM, Pediatric Hematology, Hospital Samaritano, São Paulo, BRAZIL; 14Sickle Cell 101, San Jose, UNITED STATES; 15Novartis Pharmaceuticals Corporation, East Hanover, UNITED STATES; 16Adelphi Real World, Bollington, UNITED KINGDOM; 17Evelina Children’s Hospital and Guy’s and St Thomas’ Hospital, London, UNITED KINGDOM; 18Sickle Cell Society, London, UNITED KINGDOM
Background: SCD has a high clinical burden, results in poor quality of life (QoL) and reduces life expectancy in many patients (pts). To improve treatment/management, it is important to gain a deeper understanding of pt and HCP experiences.Aims: SWAY was a cross-sectional survey that assessed pt and HCP experiences of SCD. Here we focus on the experiences of HCPs from various regions on SCD symptoms and complications, impact of SCD on QoL, treatment goals and treatment satisfaction.Methods: SWAY was an international cross-sectional survey developed by international expert physicians, pt advocates and Novartis to assess pt and HCP experiences of SCD. Here we focus on the experiences of 365 HCPs who manage ≥10 SCD pts (≥5 in Canada, ≥2 in the Netherlands) and who completed the survey in April–October 2019. Responses to questions on how much SCD impacts pts’ emotional wellbeing, QoL and daily life, as well as HCP treatment satisfaction, were ranked on a Likert scale (1–7: 1=not at all/strongly dissatisfied, 7=a great deal/strongly sat-isfied; 5–7 indicated high impact/satisfaction).Results: HCPs in all six regions recognized the prevalence of acute and chronic pain (although less frequently in Africa vs other regions; Table). Acute chest syndrome and joint issues were among the five most common complications reported by HCPs in all regions. Globally, HCPs noted the high impact of SCD on emotional wellbeing (71–97%) and of symptoms/complications on QoL (79–100%). Fewer HCPs in the Middle East (ME) reported a high impact of SCD on physical and sexual activity compared with
other regions (Figure). Around 40% of HCPs in the ME and Asia thought that SCD has a high impact on daily activities, compared with 77–90% in other regions. In Asia, fewer HCPs reported that SCD has a high impact on pts’ education (48%) and ability to maintain a job (43%) compared with other regions (69–90% and 56–90%, respectively). Hydroxyurea (HU) was among the three most common therapies ever initiated and the therapy most likely to be initiated in any age group by HCPs in all regions except Africa (Table). Improving pts’ QoL was the top treatment goal for HCPs across all regions (51–84%). Fewer HCPs in North America (32%), South America (27%) and Europe (46%) were highly satisfied with current SCD treatments compared with other regions (62–72%); the main reason for dissatisfaction was limited treatment options in all regions except Asia.Summary: The most frequent SCD symptoms/complications that HCPs reported were similar across all regions. However, regional differences in HCP experiences of how SCD impacts pts’ daily life exist (eg fewer HCPs reported a high impact on physical/sexual activity in the ME, on daily activi-ties in the ME/Asia, and on education/work in Asia, vs other regions), which may be explained by cultural variations. HU was one of the three most com-mon prior treatments in all regions except Africa, which may indicate an educational knowledge gap, particularly as the HCPs from Africa who com-pleted the survey were primarily non-specialists. This could also reflect poor access to HU and/or its high cost in Africa. HCPs in most regions were dis-satisfied with SCD treatments because of limited options, indicating a global unmet need for additional treatment choices. Improving QoL was the main treatment goal for HCPs in all regions, which may be indicative of a high negative impact of SCD on pt QoL and the ongoing need to address this.

ASCAT2022
18 | 2022; 6:S1
POSTER PRESENTATIONS
Basic and translational
P101 GUT MICROBIOTA IMPACT ON ANGOLAN CHILDREN WITH SICKLE CELL DISEASE
Brito, M1; Delgadinho, M1; Ginete, C1; Mendes, J1; Vasconcelos, J2; Santos, B2
1H&TRC - Health & Technology Research Center, ESTeSL - Escola Superior de Tecnologia da Saúde, Instituto Politécnico de Lisboa, Lisbon, PORTUGAL; 2Centro de Investigação em Saúde de Angola, Luanda, ANGOLA
Background: Sickle cell disease (SCD) is one of the most prevalent genetic diseases, affecting between 20 and 25 million people worldwide. In the Sub-Saharan Africa, where it is more prevalent, it contributes to 50–80% of under-5 mortality. Clinical manifestations of SCD are very heterogeneous and the intestinal microbiome appears to be crucial in the modulation of inflammation, cell adhesion and induction of aged neutrophils, which are the main interveners of recurrent vaso-occlu-sive crisis. Enterocyte injury, increased permeability, altered microbial composition, and bacterial overgrowth have all been documented as microbial and pathophysiologic changes in the gut microbiome of SCD patients in recent research studies. Microbiota analysis in SCD popula-tions will be essential to demonstrate the importance of specific bacteria and their function in this disease and provide new insights for attenuat-ing symptoms and new drug targets.Aims: Given this, our aim is to sequence by NGS bacterial 16S RNA gene in order to characterize the gut microbiome of SCD children and healthy siblings, as a control. A written informed consent was presented and explained to all the guardian participants prior to the data collec-tion. A total of 72 stool samples were obtained from children between 3–14 years old.Results: Our preliminary results showed that the SCD and control samples exhibit some notable differences in microbiota relative abun-dance, at different levels of classification. Children with the disease have a higher number of the phylum Actinobacteria (p=0.013) with a mean of sequences of 5.47% (± 3.49), while the siblings have a mean of 3.25% (± 2.98). As for the genus level, only Clostridium cluster XI bac-teria was more prevalent in the SCD children, whereas the siblings had higher numbers of Blautia, Aestuariispira, Campylobacter, Helicobacter, Polaribacter and Anaerorhabdus.Conclusion: There is still much to learn before fully relying on the ther-apeutic approaches for gut modulation, which is why more research in this field is crucial to making this a reality.This works as been supported by FCT/Aga Khan (project nº330842553) and FCT/MCTES (UIDB/05608/2020 and UIDP/05608/2020) –H&TRC.
References1. Lim et al. J. Transl. Med. 14, 1–3 (2016).2. Lim et al. Am. J. Hematol. 93, E91–E93 (2018).3. Brim et al. Gastroenterology 152, S631 (2017).4. Dutta et al. Br. J. Haematol. 188, 488–493 (2020).
P102 PLASMA FREE HEMOGLOBIN AND REACTIVE OXYGEN SPECIES IN SICKLE CELL ANAEMIA PATIENTS UNDER HYDROXYUREA AND GLUTAMINE THERAPY
Giannaki, A1; Fortis, S1; Georgatzakou, H1; Xidaki, A2; Liosi, M2; Fountzoula, C3; Papageorgiou, E1; Antonelou, M4; Delicou, S2; Kriebardis, A1
1Laboratory of Reliability and Quality Control in Laboratory Hematology (HemQcR), Department of Biomedical Sciences, Egaleo, GREECE; 2Thalassemia and Sickle Cell Unit at Hippokration General Hospital Athens, Athens, GREECE; 3Laboratory of Chemistry, Biochemistry and Cosmetic Science (ChemBiochemCosm), Department of Biomedical Sciences, Egaleo, GREECE; 4Department of Biology, School of Science, National & Kapodistrian University of Athens (NKUA), Athens, GREECE
Background: Sickle Cell Anemia (SCA), a quite common hemoglobinop-athy in Greece, is a consequence of abnormal hemoglobin S production (HbS). When the oxygen tension is low, HbS polymerization occurs,
resulting in red blood cells (RBC) sickling that affects membrane integ-rity, cell deformability and rheological behavior. Thus, SCA provokes reduced RBCs survival, chronic hemolytic anemia, oxidative stress and microvascular occlusion. Currently, RBC transfusion and hydroxyurea supplementation are the major disease-modifying therapies available for SCD. In addition, L-glutamine oral therapy has been proposed as an anti-oxidative agent for SCA.Aims: The purpose of this study is to measure the oxidative status of RBCs and hemolysis in SCA patients under simultaneous hydroxyurea and L-glutamine therapy.Methods: Eighteen SCA patients (52±12 years old, HbS=68,02±9.40%, on average) as well as sixteen similar age-and gender-matched healthy volunteers (controls) participated in the present study. Six patients were under no therapy before glutamine intake, eight patients were under sin-gle hydroxyurea therapy and four patients were under blood transfusion therapy. After signing an informed consent, patients took a dose range of 10-30g glutamine (Glutamine DB EXTRA supplement) daily for a 4-month period. Blood sampling were taken before glutamine intake and during a period of 2–4 months after it. Intracellular RBC Reactive Oxygen Species (iROS) were measured by Flow Cytometry. Osmotic Fragility test (mean corpuscular fragility, MCF) and plasma free hemo-globin (p-F-Hb) were also determined. Statistical analysis was performed by GraphPad Prism 8.0.3 and SPSS v.27 software.Results: All patients had significantly increased iROS levels (632,30±44,83 AU) compared to controls (443.07±69.40 AU, p<0.01), while these under transfusion therapy had only slightly increased values (506.50±31.80 AU, p<0,05). The group of hydroxyurea treatment had lower iROS levels (584.00±87.00 AU) compared to the no therapy group (614.34±23.56 AU, p<0.05). Co-administration of hydroxyurea and glutamine resulted in a 22% decrease in iROS within a two-month period, which was pre-served until the end of the four-month period. All patients had decreased RBC MCF (MCF=0.337±0.040% NaCl) compared to controls (MCF=0.46±0.01% NaCl), a finding that was unaffected by hydroxy-urea intake (MCF=0.338±0.044% NaCl), a two (MCF=0,331±0,054% NaCl) and four-month (MCF=0.337±0.047% NaCl) glutamine intake period and transfusion therapy (MCF=0.326±0.031% NaCl) (p<0.01). Total bilirubin (T-BIL) was increased in the no therapy group (2.46mgr/dl±0.28) compared to controls (0.63mgr/dl±0.17), while p-F-Hb was increased in all patients (24.34mgr/dl±11.98 vs controls 7.12mgr/dl±2.90) (p<0.01). Patients under hydroxyurea treatment showed a 19.02% decrease in T-BIL levels but not significant different p-F-Hb levels. Finally, concomitant hydroxyurea and glutamine administra-tion seemed to cause a further decrease in T-BIL and p-F-Hb levels (T-BIL;23.24%, p-F-Hb;65.38% after a two-month period intake).Summary-Conclusion: RBCs of patients under simultaneous hydroxy-urea and glutamine supplementation scheme cope better with oxidative and hemolysis stresses. L-glutamine seems to be a candidate antioxidant supplement able to deal with basic symptoms of SCA.
References1. Ogu et al, Am J Hematol 2021; 96(1) E38-E402. Zaidi et al, Contemp Clin Trials 2021; 110
P103 THE ABNORMAL PRESENCE OF MITOCHONDRIA IN CIRCULATING SCD RED BLOOD CELLS ASSOCIATED WITH STRESS ERYTHROPOIESIS
Ramasamy, J1; Gallivan, A2; Hong, L1; Horneman, H3; Molokie, R1; Diamond, A1; Lavelle, D1; Rivers, A3
1University of Illinois at Chicago, Chicago, UNITED STATES; 2Berkley, Berkley, UNITED STATES; 3University of California, San Francisco, Oakland, UNITED STATES
Sickle Cell Disease (SCD) is an inherited blood disorder that affects millions of people worldwide, and it is caused by a mutation of the β-globin gene which results in the polymerization of HbS when deox-ygenated. Sickle cell patients experience severe pain, multi organ dam-age, and shortened life span. Changes arising from sickle hemoglobin (HbS) inside the red blood cell (RBC) leads to an imbalance of the oxi-dative reactions which increases reactive oxygen species (ROS) forma-tion. Consent was obtained from people with SCD and people with SCD. Our lab discovered that SCD patients have a significant fraction of RBCs retaining-mitochondria which were associated with excessive levels of ROS1. We also noticed and increase in immature mitochondria containing reticulocytes in peripheral blood of SCD patient compared to control subjects. The percentage of mitochondria retaining RBCs

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 19
directly correlated with absolute number of reticulocytes (n=18,r=0.69, P< 0.001). As would be expected we were able to link mitochondria-re-tention with hemolysis. In addition when we investigated energy metab-olism we made the novel finding that SCD RBC have increase oxygen consumption rate (OCR). Mitochondrial-RBC retention and markers of hemolysis (serum bilirubin, and LDH) were compared by using Pearson’s correlation coefficient analysis. The total serum bilirubin was directly associated with the percentage of mitochondria-contain-ing RBCs in SCD patients’ blood samples (r=0.31, n=41, p<0.04). A subgroup analysis showed a strong association between bilirubin and mitochondria-retaining RBCs in SCD patients who were not taking Hydroxyurea (r=0.55, n=17, P<0.02). There was a less significant asso-ciation of percentage of mitochondria-containing RBCs in SCD with serum LDH (r=0.02, n=40, P<0.2) when all patients were analyzed, however, a strong association was seen with serum LDH and mitochon-dria retaining RBCs in SCD patients who were not taking Hydroxyurea (r=0.67, n=17, P<0.002). Lactate dehydrogenase (LDH) is a known bio-marker for hemolysis-and associated disease complications and early mortality in patients with SCD. Percentage of mitochondria retaining RBCs also directly correlated with absolute number of reticulocytes (n=18,r=0.69, P< 0.001).We next investigated the mitochondrial RBCs energy metabolism in SCD as compared to controls in both human and mice. RBCs obtained from Townes SCD or control mice were seeded in an Agilent Seahorse XF 24-well plate using the Seahorse XF Base Medium with supplements and we monitored the OCR and extracellu-lar acidification rate (ECAR), in real time by Seahorse XFe-extracellular flux analyzer. OCR is significantly higher in sickle cell RBCs of both human and mice origin compared to control. OCR was reduced to nor-mal levels with oligomycin which is a known blocker of mitochondrial respiration. The mechanism responsible for mitochondrial retention in SCD is unknown. We further investigated that stress erythropoie-sis is responsible for abnormal mitochondrial retention in erythrocytes. Terminal erythrocyte precursors and reticulocyte from bone marrow and spleen and peripheral blood had ROS and mitochondrial content by FACS2. Phlebotomized showed similar increased in erythrocyte mitochondrial retention and similar pattern of precursor erythrocyte in bone marrow to SCD mice precursors.Conclusion: Our data suggest a novel pathophysiology of stress eryth-ropoiesis in SCD.
References1. Jagadeeswaran, R. et al. Pharmacological inhibition of LSD1 and mTOR
reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 50, 46–52 (2017).
2. Liu, J. et al. Quantitative analysis of murine terminal erythroid dif-ferentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood 121, e43–e49 (2013).
Novel therapies, gene therapies, bone marrow transplant and emerging diagnostics
P104 BASE EDITING REPAIRS THE HBE MUTATION RESTORING THE PRODUCTION OF NORMAL GLOBIN CHAINS IN SEVERE HBE/β-THALASSEMIA PATIENT HEMATOPOIETIC STEM AND ERYTHROID CELLS
Badat, M1; Hua, P2; Mettananda, S3; Fisher, C2; Roy, N4; Rice, S2; Roy, A2; Higgs, D2; Davies, J2
1Barts Health NHS Trust, London, UNITED KINGDOM; 2Weatherall Institute of Molecular Medicine, Oxford, UNITED KINGDOM; 3University of Kelaniya, Ragama, SRI LANKA; 4Dept Haematology, Oxford University Hospitals NHS Trust, Oxford, UNITED KINGDOM
Aims: HbE/β-thalassemia is the commonest form of severe β-thalas-semia, and comprises approximately 50% of all cases worldwide1. HbE/β-thalassemia is caused by the HbE codon 26 G>A mutation on one allele and any β0-thalassemia mutation on the other. There is a reduction in β-globin production, resulting in a relative excess in α-glo-bin chains that leads to ineffective erythropoiesis. Importantly, individ-uals with a mutation on one, but not two, alleles have β-thalassemia trait, a carrier state with a normal phenotype shared by 1.5% of the world’s population2. Recent gene therapy and gene editing approaches have been developed to treat β-thalassemia but do not directly repair the
causative mutation in-situ. Gene replacement approaches rely on lentivi-ral vector-based sequence insertion or homology directed repair (HDR). HbF induction strategies rely on non-homologous end joining targeting of enhancers in-trans. These approaches, whilst variably successful, are associated with potential safety concerns.Methods: Adenine base editors (ABEs) circumvent these problems by directly repairing pathogenic variants in-situ through deamination. ABEs catalyse A-T to G-C transitions. Conversion of the HbE codon to WT through base editing is an attractive strategy to recapitulate the phe-notypically normal β-thalassemia trait state without potentially harm-ful double-strand breaks or random vector insertions. ABEs are able to convert the HbE codon (AAG) to wild-type (GAG), but also to GGG or AGG (Fig A). GGG at codon 26 is found in a naturally occurring haemoglobin, Hb Aubenas3. Heterozygotes have normal red cell indices and are phenotypically normal. We electroporated the latest generation of ABE8 editors4 as mRNA into 3 different severe HbE/β-thalassemia donor HSPCs with sgRNAs targeting the HbE codon.Results: The mean conversion from the HbE codon to a normal or nor-mal variant in unselected cells was 86.2 (SD±8.1%, Fig B). The indel rate from inadvertent on-target Cas9 cleavage was below 0.5%. Edited cells did not show any perturbations in erythroid differentiation as assessed by Immunophenotyping and cellular morphology. In differentiated erythroid cells, RT-qPCR showed a mean fall in the α/β mRNA ratio to 0.65±0.08 (unedited patient cells normalised to 1, n=5, Figure C), indi-cating a reduction in the excess α-globin gene expression. Protein anal-ysis by CE-HPLC showed a 3.6-fold reduction in HbE levels (SD±1.3) and a 13.5-fold increase in HbA/Hb Aubenas (SD±2.4, Fig D & E).In serial NSG-murine xenotransplantation experiments, base edited cells were found to persist in secondary transplants, showing editing is possi-ble in long-term HSCs (mean editing efficiency 34.5%, Fig F). Potential off-target effects were assessed in-vitro by CIRCLE-seq5; most candidate sites were in intergenic and intronic regions (Fig G & H). The top 250 sites were sequenced using deep targeted NGS. Only 5 sites showed OT deaminations at low levels in patient cells (mean 1.5%). We developed a machine learning model to assess potential OT-effects on chromatin accessibility, at all candidate sites in 49 different blood cell types6. Only 17 potential edits were predicted to result in a significant change in chro-matin state (Fig I). 3 of these were in the top 250 sites sequenced previ-ously, and none showed deamination in-vivo.Conclusion: Together these data provide robust evidence for base editing being used as an effective and safe therapeutic strategy for HbE/β-thalassemia.
References1. Modell and Darlison, Bull World Health Organ (2008) 86(6):480–72. Origa, Genet Med (2017) 19(6):609–619

ASCAT2022
20 | 2022; 6:S1
3. Lacan, Hemoglobin (1996) 20(2):113–244. Richter, Nat Biotechnol (2020) 38(7):883–8915. Tsai, Nat Methods (2017) 14(6):607–6146. Schwessinger, Nat Methods (2020) 17(11):1118–1124
P105 BLOOD MICROVESICLES AS BIOMARKERS TO PREDICT VASO-OCCLUSIVE CRISIS IN SICKLE CELL ANEMIA
Chebbi, M1; Moumni, I1; Khalfaoui, K1; Safra, I1; Barmate, M1; Chaouechi, D1; Benkhaled, M2; Ouederni, M2; Mellouli, F2; Menif, S1
1Molecular and Cellular Laboratory, Pasteur Institute of Tunis, Tunis, TUNISIA; 2Department of pediatrics: Immunology, Hematology and Stem cell transplantation. Bone Marrow Transplant Center, Tunis, TUNISIA
Background/Aims: Sickle cell anemia (SCA) is a monogenetic disorder caused by a mutation that results an abnormal hemoglobin (HbS) with a susceptibility to polymerize and deform erythrocytes which leads to complications such as Haemolysis, chronic infections and vaso-occlu-sive and pain crises (Williams et Thein, 2018). The polymerization of HbS inside red blood cells leads to complex interactions with the cell membrane and other molecules which triggers the apoptosis of different blood cells and the release of microvesicles (MVs) (Mahfoudhi et al, 2012). MVs are phospholipid microparticles that are derived from the cytoplasmic membrane of cells submitted to stress conditions that result in apoptosis or activation. The generation of MVs in SCA could serve as a potential biomarker to predict serious cardiovascular complications (Olatunya et al, 2019). Therefore, our study suggests the research of MVs as potential cellular biomarkers and the implementation of a new strategy of innovative predictive diagnosis in order to avoid the serious complications of sickle cell anemia.Methods: Clinically diagnosed homozygous SCA patients from the Tunis national bone marrow transplant center and healthy donors were sampled for hematological and cellular assays. Flow cytometry was per-formed in to quantify apoptotic MVs derived from erythrocytes, plate-lets and endothelial cells using specific fluorescent antibodies and dyes.Results and Discussion: Our results showed a statistically significant increase in the number of apoptotic MVs which suggests a high apop-tosis rate in patients’ cells comparing to healthy donors. We also found that MVs derived from erythrocytes, platelets and endothelial cells were clearly elevated in SCA patients which suggests their potential contribu-tion in thrombotic risk and chronic hemolytic anemia. Our findings sug-gest then that microvesicles can be considered as hemolytic biomarkers for a predictive diagnosis so that disease complications could be avoided.
References1. Williams et Thein, Annual Review of Genomics and Human Genetics
2018;19:1132. Mahfoudhi et al, British Journal of Haematology 2012;156:5453. Olatunya et al, Annals of hematology 2019;98:2507
P106 EARLY PREVENTIVE DIAGNOSIS OF HEMOLYTIC ANEMIA IN SICKLE CELL PATIENTS BY DETECTING THE TRIGGERING OF ERYPTOSIS.
Khalfaoui, K1; Moumni, I1; Chebbi, M1; Safra, I1; Barmate, M1; Chaouechi, D1; Benkhaled, M2; Ouederni, M2; Mellouli, F2; Menif, S1
1Molecular and Cellular Laboratory,Pasteur Institue of Tunis, Tunis, TUNISIA; 2Department of pediatrics: Immunology, Hematology and Stem cell transplantation. Bone Marrow Transplant Center, Tunis, TUNISIA
Background/Aims: Sickle cell disease (SCD) also known as sickle cell anemia is one of the most worldwide-disseminated hereditary hemoglo-binopathies. It is caused by a single amino acid substitution at the sixth residue of β globin gene (Glu6Val), which results in an abnormal hemo-globin called hemoglobin S (HbS). The acceleration of HbS polymeriza-tion induces rigid and dysfunctional erythrocytes that play a central role in acute and chronic clinical manifestations of SCD(Conran et al.,2009). Vaso-occlusion and hemolytic anemia are the hallmarks of SCD.We aim to explore the cellular environment of red blood cells to explain the physiopathology of SCD. In fact, the life span of circulating eryth-rocytes in healthy individuals vary from 100 to 120 days. In SCD, red blood cells undergo a form of cell death, namely, eryptosis before they reach their full life span. Eryptosis is triggered by a wide variety of factors as hyperosmolarity, oxidative stress and energy depletion. It is characterized by the presence of membrane blebbing, cell shrinkage,
and phosphatidylserine (PS) exposure (Lang et al.,2012). In this study, we will explore the mechanism of triggering of eryptosis in Sickle cell disease.Methods: Following clinical diagnosis, 50 homozygous SCD patients and 30 healthy donors were identified for hematological and cellular assays. Flow cytometry was performed in order to determine the viability parameters of erythrocytes. The morphology of red blood cells and the externalization of phosphatidylserine was detected by labeling red blood cells with Annexine V. Moreover, we had identified intracellular calcium concentration and ceramide level by labeling erythrocytes with Fluo3-am and anti-ceramide antibodies. Finally, we had quantified reactive oxygen species (ROS) by labeling red blood cells with CM-H2DCFDA.Results and Discussion: Eryptosis in sickle cell patients is accelerated. In fact, PS (+) red blood cells are more present in patients than in healthy subjects. Therefore, eryptosis is triggered by oxidative stress, which stim-ulates the increase of calcium activity and subsequent externalization of PS and red blood cells shrinkage in sickle cell patients. However, the pathway of ceramide can also be considered a potential stimulating fac-tor of eryptosis in SCD. Eryptosis ensures healthy erythrocyte quantity in circulation, whereas excessive eryptosis is the cause of acute anemia and may contribute to vaso-occlusive crisis in SCD patients.
References1. Conran et al, Hemoglobin 2009;33:1–162. Lang et al, Transfusion Medicine and Hemotherapy 2012; 39: 308
Clinical and epidemiological studies
P107 A PHASE 2/3, RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED STUDY OF MITAPIVAT IN PATIENTS WITH SICKLE CELL DISEASE
Howard, J1; Kuo, K2; Oluyadi, A3; Shao, H3; Morris, S3; Zaidi, A3; Van Beers, E4; Thein, S5
1Department of Hematology, Guy’s and St Thomas’ NHS Foundation Trust, London, UNITED KINGDOM; 2Division of Hematology, University of Toronto, Toronto, ON, CANADA; 3Agios Pharmaceuticals, Inc., Cambridge, MA, UNITED STATES; 4Van Creveldkliniek, Department of Internal Medicine, University Medical Center Utrecht, Utrecht, NETHERLANDS; 5Sickle Cell Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, UNITED STATES
Background: The key pathology in sickle cell disease (SCD), a life-threat-ening, hereditary hemoglobin (Hb) disorder, is red blood cell (RBC) sickling due to polymerization of deoxygenated sickle Hb (HbS), which can be exacerbated by increased levels of the glycolytic metabolite 2,3-diphosphoglycerate (2,3-DPG), and decreased ATP.1–3 Sickled RBCs are rigid, not deformable, and fragile, resulting in vaso-occlusion trigger-ing pain and chronic hemolysis.4–6 SCD treatment options are limited, with an unmet need for safe and effective therapies to improve anemia and reduce pain. Mitapivat is an oral, activator of RBC pyruvate kinase (PKR), a key glycolytic enzyme. PKR activation decreases 2,3-DPG and increases ATP, which may reduce HbS polymerization, RBC sickling, and hemolysis in SCD.3,7–9 Data from the phase (ph) 1 National Institutes of Health multiple ascending dose study of up to 100 mg mitapivat twice daily (BID) in SCD (NCT04000165) showed that mitapivat was safe and tolerable, increased ATP and decreased 2,3-DPG in a dose-depen-dent manner, and improved anemia and hemolysis.8,10
Aims: To report the study design of RISE UP (NCT05031780, EudraCT: 2021-001674-34), a ph 2/3, double-blind, randomized, placebo-con-trolled, multicenter study evaluating the efficacy and safety of mitapivat in patients (pts) with SCD.Methods: Eligible: pts aged ≥16 yrs with documented SCD (HbSS, HbSC, HbSβ0/HbSβ+ thalassemia, other SCD variants), 2–10 sickle cell pain cri-ses (SCPCs; acute pain needing medical contact, acute chest syndrome, priapism, hepatic or splenic sequestration) in the prior 12 months, and Hb 5.5–10.5 g/dL. If taking hydroxyurea (HU), the dose must be stable for ≥90 days before starting study drug. Ineligible: pts receiving regularly scheduled blood transfusions, with severe kidney disease or hepatobili-ary disorders, currently receiving SCD therapies (excluding HU) or who have received gene therapy, bone marrow or stem cell transplantation.In the double-blind ph 2 part, 69 pts will be randomized (1:1:1) to 50 mg or 100 mg mitapivat, or placebo BID for 12 weeks (wks). The primary objective of ph 2 is to determine the recommended ph 3 mitapivat dose by evaluating anemia and safety vs placebo via the following endpoints: Hb response (≥1.0 g/dL increase in average Hb concentration over Wks

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 21
10–12 vs baseline [BL]), and type, severity, and relationship of adverse events and serious AEs.In the double-blind ph 3 part, 198 pts who did not participate in ph 2 will be randomized (2:1) to the selected ph 3 mitapivat dose or placebo BID for 52 wks, stratified by number of SCPCs in the prior yr (<5, ≥5) and HU use. The primary objectives of ph 3 are to determine the effect of mitapivat vs placebo on anemia, measured by Hb response (≥1.0 g/dL increase in average Hb concentration over Wks 24–52 vs BL), and the effect of mitapivat vs placebo on SCPC, measured by annualized rate of SCPCs. Key secondary endpoints include average change from BL in Hb concentration, indirect bilirubin, percent reticulocyte, and Pt-Reported Outcomes Measurement Information System® Fatigue 13a Short Form score over Wks 24–52; and annualized frequency of hospitalizations for SCPC.Pts who complete the double-blind period will be eligible to receive mitapivat in the open-label extension period in each ph.Results: Not yet available.Conclusion: This ph 2/3 study will investigate the efficacy and safety of mitapivat in pts aged ≥16 yrs with SCD. Enrollment is currently ongoing.
References1. Yawn et al. JAMA 2014; 312:1033–48.2. Ware et al. Lancet 2017; 390:311–23.3. Rab et al. Blood 2021; 137:2997–3001.4. Darbari et al. Eur J Haematol 2020; 105:237–46.5. Telen et al. Nat Rev Drug Discov 2019; 18:139–58.6. Nader et al. Front Immunol 2020; 11:454.7. Kung et al. Blood 2017; 130:1347–566.8. Xu et al. 42nd ASH 2020: Abstract 681.9. Eaton, Bunn. Blood 2017; 129:2719–26.10. Xu et al. 43rd ASH 2021: Abstract 10.
P108 ASSESSMENT OF HEALTHCARE WORKERS’ KNOWLEDGE AND RESOURCE AVAILABILITY FOR CARE OF SICKLE CELL DISEASE AT HEALTH FACILITIES IN DAR ES SALAAM, TANZANIA
Jonathan, A; Tutuba, H; Lloyd, W; Ndunguru, J; Makani, J; Ruggajo, P; Minja, I; Balandya, E
Muhimbili University of Health and Allied Science (MUHAS), Dar es Salaam, TANZANIA, UNITED REPUBLIC OF
Background: Sickle cell disease (SCD) is a global public health prior-ity due to its high morbidity and mortality. In Tanzania, SCD accounts for 7% of under-five mortality. Cost-effective interventions such as early diagnosis and linkage to care have been shown to prevent 70% of deaths but require knowledge among healthcare workers and availabil-ity of resources at healthcare facilities. In Tanzania, data on these critical determinants is currently lacking.Aim: To assess healthcare workers’ knowledge and resource availability for care of SCD at Health Facilities in Dar es Salaam, Tanzania.Methods: A facility-based cross-sectional study was conducted between December-2020 and February-2021 among 490 nurses and clinicians at Temeke, Amana, Mwananyamala and Muhimbili National Hospital in Dar es Salaam. Data was collected using structured questionnaire (knowledge) and inventory checklist (resources). Pearson’s Chi-square was used to determine association between level of knowledge and
demographic factors. Multivariate logistic regression was used to ascer-tain the strength of associations. P-values < 0.05 were considered statis-tically significant.Result: Of the 490 participants (28 years [IQR=26–35]), only 25.1% had good knowledge on SCD. The odds of good knowledge was 82% lower in nurses than clinicians (AOR= 0.177; 95% CI: 0.090, 0.349; p = 0.000); 95% lower in diploma than Master degree holders (AOR = 0.049; 95% CI: 0.008, 0.300; p = 0.001) and 4.6 times higher in those with 5 – 9 years than ≥ 10 years of experience (AOR=4.564; 95%CI: 1.341, 15.525; p=0.015). The regional-level hospitals lacked diagnostic tests and Hydroxyurea.Conclusion: There was a general lack of knowledge on SCD among healthcare workers and limited availability of critical resources for the diagnosis and care of SCD, especially at regional-level hospitals. Efforts are needed for their improvement in order to enhance care to patients, thus reducing the morbidity and mortality due to SCD in Tanzania.
References1. Adegoke et al, Trans R Soc Trop Med Hyg. 2018;112(2):81–7.2. Ambrose et al, Bull World Health Organ. 2020 Dec 1; 98(12):
859–868.3. Ansong et al, Mediterr J Hematol Infect Dis. 2013;5(1):1–6.4. Azonobi et al, BMC Pregnancy Childbirth. 2014;14(1):1–5.5. Chamba et al, Case Rep Hematol. 2018;2018:1–3.6. Chide et al, A Cross-sectional Survey. 2020;1–16.7. Cochran, W. G. 1977. Sampling Techniques, 3rd Ed., New York: John
Wiley and Sons, Inc.8. Diniz et al, Hematol Transfus Cell Ther. 2019;41(2):145–52.9. Diniz et al, Hematol Transfus Cell Ther. 2019;41(1):62–8.10. Drive et al, Adv Exp Med Biol. 2017; 1013: 59–8711. World Health Organisation, Agenda item 11.4. 59th World Health
Assembly, 27 May 2006. 2006.12. Piel et al, PLoS Med. 2013;10(7).13. Gomes et al, Mediterr J Hematol Infect Dis. 2015;7(1).14. Gomes et al, BMC Fam Pract. 2011;12.15. Ministry of Health et al Population Policy Compendium, 1–6.16. Hyochol et al, Physiol Behav. 2017;176(10):139–48.17. Jain et al, J Hematol Thromboembolic Dis. 2015;03(06).18. Jemima et al, Divers Equal Heal Care. 2008;241–54.19. Makani et al, PLoS One. 2011;6(2).20. Makani et al, BMC Hematol. 2018;18(1).21. Mcgann et al, Blood. 2018;129(2):155–62.22. McWalter et al, Am J Prev Med. 2011;41(6 SUPPL.4):384–9.23. Ministry of Health and Social Welfare. Minist Heal Soc Welf.
2015;(June):1–16.24. Mukinayi et al, Trop Med Infect Dis. 2020;5(3):127.25. Nkya et al, Int Health. 2019; 11(6): 589 – 595.26. Tanyi et al, J Am Acad Nurse Pract. 2003;15(9):389–97.27. Tluway et al, British Journal of Haematology. 2017;p. 919–29.28. Williams et al, BMC Med. 2015;13(1):1–3.
P109 CLINICAL CATEGORIZATION OF CHILDREN WITH SICKLE CELL DISEASE USING SEVERITY SCORE AT A TERTIARY CARE CENTER
Dipty Jain, L; Swaroop, K
Government medical college, nagpur, INDIA
Introduction and Aim: India has been identified as having the second largest number of births with sickle cell anaemia(SCA) in the world after Africa and exhibits wide clinical diversity (1). We therefore introduced a scoring system to evaluate the severity of SCA in India children using simple clinico-laboratory parameter.Objective and work plan: The main aim is to see if Indian score is dif-ferent from international scores -tweel et al.,(2) In India, the disease is largely undocumented. Thus, there is an urgent need to document the features of Indian disease so that locally appropriate models of care may be evolved across other countries too. We used cluster analysis of clini-cal parameters like avascular necrosis,stroke, hepatic or splenic seques-tration, acute chest syndrome, vaso-occlusive crisis, number of blood transfusions with combined clinical and laboratory parameters of inter-nationally validated score. The idea is to clinically categorise the children based on expert opinion into mild,moderate and severe using several clinical parameters and to match this with a validated international scor-ing system. The total severity score of 173 patients ranging from 0 to

ASCAT2022
22 | 2022; 6:S1
70 with mean ± standard deviation of 27.69 ± 24.51. Approximately 63.7% had mild disease, 13.2% moderate disease, and 35.7% severe disease, according to our severity score grading system. Comparison of international score with our score was done with a matching of 64.9%. Some minor modifications was done in a validated score due to local factors and clinical diversity.Present samples cohort (171) was divided randomly into two groups; Discovery group (N=100) and Validation group (N=71). Validation was done with overall matching of 81.5% after including parameters which was not present in an international score as per Indian patients. Score matching increased from 65% to 82% All weightings demonstrated a significant difference between the scores of mild, moderate, and severely affected patients, as classified by a sub-jective rating or with an existing index (P < 0.01).Conclusion: Routine evaluation of disease severity in children with SCA will help to prospectively identify children at higher risk for a turbulent clinical course who may need more active management and monitor-ing. Assessment of patients objectively after any type of intervention like counselling, treatment(pre and post hydroxyurea) can be done by score.
References1. Hockham et al, The spatial epidemiology of sickle-cell anemia in
india, Sci Rep. 2018;10.10382. W.van den Tweel et al, Develepment and validation of pediatric sever-
ity index for sickle cell patients, Am.J.hematology.85 (2010);746–751
P110 CLINICAL CHARACTERISTICS OF COVID-19 IN SICKLE CELL DISEASE (SCD) PATIENTS
Malhan, H; Iqbal, B; Ghazwani, Y; AlHakim, A; Ali, S; Omar, H; Dammag, E; Khalifah, H; Alqurbi, A; Ekpenyong, U;Subahi, N; Alqahiry, W; Bakkar, M
Prince Mohammad Bin Naser Hospital, Jazan, SAUDI ARABIA
Introduction SCA is a common genetic disorder in Saudi Arabia with estimate of 1.4 % of population(1,2). The prevalence of SCD in Jazan region is around 24 per 10,000 which consider the second region with high prevalence of SCD after eastern province. COVID 19 infection out-break has affected SCD with different outcome.Objectives: This research aims to determine the clinical picture of COVID-19 in patients with sickle cell disease SCD. The study repre-sented the risk factors for severe COVID-19 presentation and predictors for poor prognosis in this group of patients.Methods: This study is a retrospective, descriptive, observational study of SCD adult and pediatric patients in Prince Mohammed Bin Nasser Hospital, Jazan, Saudi Arabia, who were diagnosed with COVID-19 virus infection at the study center from March 2020 to September 2021.To describe clinical presentation of SCD patients of different severities, who got COVID-19 during the study, socio-demographic data, clin-ical presentations, laboratory parameters, medications use, as well as COVID-19 symptoms and severity were extracted and analyzed from the medical record files.Results: 43 medical records for adult and pediatric sickle cell patients with COVID-19 were collected, in order to determine the impact of COVID-19 on the clinical presentation of SCD patients. (53%) were females, (47%) were males with a mean age of 24 years (±1.9). (37%) of the sample suffered from major comorbidities, out of which 44% had ACS. and 11.6% have pulmonary embolism. The most prevalent clinical symptoms were fever (56%) and Shortness of Breath (37%). (16%) of included patients were admitted to the ICU with an average length of stay equals to 3.9 days (±0.6).CBC showed normal averages of PLT with a mean equal to 327 K/uL (±21.6), Basophils 0.05 K/uL (±0.01), Lymphocytes 3.6 K/uL (±0.4). High averages were found for WBC 13.8 K/uL (±1.1), Neutrophils 8.8 K/uL (± 0.9) and PT 14.2 seconds (±0.3). The only laboratory param-eter that showed low average reading was Hemoglobin, with a mean equal to 8 g/dl (±0.2). Out of the 41 patients who undergone CRP test, 85% were positive. The main CT chest finding were ground glass appearance in 30% of the patients 88% of the studied patients were on Hydroxyurea, 76% were on 1000 mg dose. For COVID-19 management, majority of patients were on Antibiotics 93%, 70% of patients started Anticoagulants, 51% of the patient has received antiviral treatment. 32.5% of the patient were treated with steroids. 70% of the patients have received blood transfusion. 5 patients 12% were managed at home. One patient had coinfection with falciparum malaria.Total deaths was 2 patients represented 4.6 % out of total SCD patients with COVID-19 who were included in the study.
Conclusion: After exploring the impact of COVID-19 on SCD patients, the main presenting symptom were fever and shortness of breath. The major complication were acute chest syndrome 44% and pulmonary embolism in 12%.The overall prognosis was excellent and most of the patient have recov-ered. The death was reported in 2 patients only. Outpatient treatments were feasible in 12% of the patients.
References1. AlHamdan NA, AlMazrou YY, AlSwaidi FM, Choudhry AJ. Premarital
screening for thalassemia and sickle cell disease in Saudi Arabia. Genet Med. 2007;9:3727.
2. Nasserullah Z, Alshammari A, Abbas MA, Abu-Khamseen Y, Qadri M, Jafer SA, et al. Regional experience with newborn screening for sickle cell disease, other hemoglobinopathies and G6PD deficiency. Ann Saudi Med. 2003;23:354–7
P111 COVID-19 IN PATIENTS WITH THALASSEMIA AND SICKLE CELL DISEASE: A SINGLE CENTER EXPERIENCE
Dimopoulou, M; Komninaka, V; Flevari, P; Bartzi, V; Repa, K; Voskaridou, E
Laikon General Hospital, Athens, GREECE
Background: Thalassemic patients present complications such as iron overload, cardiac disease or diabetes that are expected to make them more vulnerable to COVID-19. Acute complications of SCD triggered by a viral infection include painful vaso-occlusive episodes, acute chest syndrome (ACS) and venous thromboembolic disease (VTE). Published data report variable rates of severe COVID-19 in thalassemia whereas for SCD more robust data exist suggesting a strong association with severe disease and mortality.Aims: The aim of the present study was to report the severity and mor-tality of COVID-19 in thalassemic and SCD patients of a Greek Center and investigate possible risk factors for severe disease.Patients and Methods: The patient population of our Center includes 200 thalassemic patients and 320 SCD (of which 73 and 53 trans-fusion dependent, respectively). Baseline clinical and laboratory data and manifestations, clinical course, treatment and outcome of COVID-19 were collected from medical files for all the patients that had a PCR-documented COVID-19 infection since the pandemic ini-tiation. Association of characteristics with severe disease or hospital-ization was investigated with Fisher’s exact test.Results: Since August 2020 until November 2021 34 patients (21 thal and 13 SCD) were infected by SARS-CoV2 corresponding to 10.5% and 4% of our patient population, respectively. Their median age was 47 yrs and 14 of them were male. Baseline patients characteristics as well as COVID-19 manifestations and outcome are presented on Table 1. None of the patient had cardiac iron overload, 2 had medium and 1 severe hepatic iron overload. Five patients had commorbidities (pulmonary hypertension 2, heart failure 1, atrial fibrillation 3, diabetes mellitus 2, arterial hypertension 1, malignant disease 1). Five patients had been vac-cinated for COVID-19 before they presented with disease.The majority of patients (32/33) devepoped symptomatic disease of mild (26) or moderate (6) severity according to established criteria. Only one patient presented severe disease. Only 6/33 patients required hospitaliza-tion (4 thalassemic and 2 with SCD). In 4/6 hospitalized patients oxy-gen therapy with a nasal cannula was required and only one patient required a Venturi mask. None of the patients were intubated. One of the SCD patients developed an acute painful episode but there was no ACS post COVID-19. No increase in transfusion requirements was observed. There were no deaths in our patient population. All but one patients completely recovered whereas one SCD patient who was hospitalized is suffering from long-COVID.History of heart failure was associated with severe disease (p=0.03) and history of AF was associated with need for hospitalization (p=0.02). History of splenectomy, pulmonary hypertension or ACS and the degree of iron overload were not associated with disease severity or hospital-ization requirement.Conclusion: COVID-19 had mild to moderate severity in the majority of our patients and only a minority of the patients required hospitalization. No ICU admissions or deaths were observed, comparing favourably to published data. Factors predisposing for severe COVID-19 in the general population, especially cardiac disease, seem to play a role also in patients with hemoglobinopathy.

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 23
BASELINE PATIENTS CHARACTERISTICS
DIAGNOSIS TRANSFUSION DEPENDENT β-THALASSEMIA (TDT) 11NON-TRANSFUSION DEPENDENT β-THALASSEMIA (NTDT) 7HEMOGLOBINOPATHY H 2SCD (ON CHRONIC TRANFUSIONS) 13 (4)TOTAL 33AGE (YRS)* 47 (26–69)SEX (M/F) 14/17SPLENECTOMY/FUNCTIONAL ASPLENIA 11/4COMMORBIDITIES 5SERUM FERRITIN* (ng/μl) 738 (49–2900)HYDROXYUREA TREATMENT 6CHELATION THERAPY 14MONOTHERAPY 10COMBINATION 4ANTICOAGULANTS 3ANTI-PLATELETS 8HISTORY OF ACS/MULTIPLE RELAPSES 3/1HISTORY OF VTE 3LIC (mg/gr)* 4.7 (1.1–21.4)ANTI-COVID VACCINATION 5SEVERITY OF COVID INFECTION ASYMPTOMATIC 1MILD (NO DYSPNEA) 27MODERATE (DYSPNEA, SpO2 ≥94%) 5SEVERE DYSPNEA, SpO2 <94%) 1FEVER 23COUGH 20PAINFUL CRISIS 1PULMONARY INFILTRATES 3VTE 0HOSPITALIZATION 6TREATMENT ANTIBIOTICS 13STEROIDS 6REMDESIVIR 4LMWH 7anti-TNF 1OXYGEN THERAPY 5DEATHS 0*:MEDIAN (RANGE)
References1. Mucalo L et al, Blood Adv. 2021; 5(13):2717–2724.2. Clift AK, et al, Ann Intern Med. 2021; 174(10):1483–1487.3. Vilela TS, et al, Hematol Transfus Cell Ther. 2021;43(1):87–100.
P112 ENERGIZE AND ENERGIZE-T: TWO PHASE 3, RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED STUDIES OF MITAPIVAT IN ADULTS WITH NON--TRANSFUSION-DEPENDENT OR TRANSFUSION-DEPENDENT ALPHA- OR BETA-THALASSEMIA
Kuo, K1; Layton, D2; Al-Samkari, H3; Kattamis, A4; Sheth, S5; Taher, A6; Viprakasit, V7; Chamberlain, C8; Czapla, L8; Gheuens, S8; Lynch, M8; Tong, B8; Uhlig, K8; Cappellini, M9
1Division of Hematology, University of Toronto, Toronto, ON, CANADA; 2Hammersmith Hospital, Imperial College Healthcare NHS Trust, London, UNITED KINGDOM; 3Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, UNITED STATES; 4First Department of Pediatrics, National and Kapodistrian University of Athens BR, Athens, GREECE; 5Weill Cornell Medical College, New York, NY, UNITED STATES; 6American University of Beirut, Beirut, LEBANON; 7Siriraj-Thalassemia Center, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, THAILAND; 8Agios Pharmaceuticals, Inc., Cambridge, MA, UNITED STATES; 9University of Milan, Milan, ITALY
Background: Thalassemias are characterized by imbalanced globin-chain production resulting in excess α- or β-globin precipitation, hemolytic anemia, and ineffective erythropoiesis.1,2 ATP levels are reduced in
thalassemic red blood cells (RBCs), despite increased energy demands.3,4 Mitapivat is an oral activator of RBC pyruvate kinase (PKR), a glycolytic enzyme that regulates ATP production.5 In a phase 2 study of patients with α- or β-non–transfusion-dependent thalassemia (NTDT), twice-daily (BID) dosing with mitapivat increased hemoglobin (Hb) levels by ≥1.0 g/dL in 80% of patients,6 supporting the broadening of mitapivat’s development in thalassemia.Aims: To report the study designs of ENERGIZE (2021-000211-23) and ENERGIZE-T (2021-000212-34), two phase 3 trials to assess the efficacy and safety of mitapivat in adults with α- or β-NTDT or transfu-sion-dependent thalassemia (TDT), respectively.Methods: Both studies are phase 3, multicenter, randomized, dou-ble-blind, placebo-controlled trials (Figure). In ENERGIZE, approxi-mately 171 eligible adults with NTDT will be randomized (2:1) to receive 100 mg mitapivat BID or placebo for 24 weeks. Upon completion, eligible patients can transition to a 5-year, open-label extension. Key inclusion criteria: documented diagnosis of thalassemia (β-thalassemia ± α-globin mutations, Hb E β-thalassemia, or α-thalassemia [Hb H disease]), Hb concentration ≤10.0 g/dL, and NTDT defined as ≤5 RBC units during the 24-week period before randomization and no RBC transfusion ≤8 weeks prior. The primary endpoint is an Hb response defined as a ≥1.0 g/dL increase in average Hb concentration from Week 12 through 24 compared with baseline. Secondary endpoints include patient-reported outcomes, changes in Hb, markers of hemolysis and erythropoiesis, and safety. In ENERGIZE-T, approximately 240 eligible adults with TDT will be ran-domized (2:1) to receive 100 mg mitapivat BID or placebo for 48 weeks. Upon completion, eligible patients can transition to a 5-year, open-label extension. Key inclusion criteria: documented diagnosis of thalassemia (same genotypes as detailed for the ENERGIZE study), and TDT defined as 6–20 RBC units transfused and no transfusion-free period ≥6 weeks during the 24 weeks before randomization. The primary endpoint is a transfusion reduction response, defined as a ≥50% reduction in trans-fused RBC units with a reduction of ≥2 units of transfused RBCs in any consecutive 12-week period through Week 48 compared with baseline. Secondary endpoints include additional measures of transfusion burden, changes in iron markers, and safety.Results: Not yet available.Conclusions: ENERGIZE and ENERGIZE-T are the first pivotal studies to assess a potential treatment across a broad spectrum of patients with thalassemia (ie, patients with TDT and NTDT; α- and β-thalassemias). ENERGIZE and ENERGIZE-T will evaluate the efficacy and safety of mitapivat, a novel, first-in-class oral activator of PKR. Both studies are actively recruiting.
References1. Taher et al. Lancet 2018; 391:155–67.2. Cappellini et al. Blood Rev 2018; 32:300–11.

ASCAT2022
24 | 2022; 6:S1
3. Khandros et al. Blood 2012; 119:5265–75.4. Shaeffer. J Biol Chem 1988; 263:13663–95.5. Kung et al. Blood 2017; 130:1347–566.6. Kuo et al. HemaSphere 2021. Abstract S267.
P113 INCIDENCE OF HAEMOGLOBINOPATHY IN SLOVAKIA
Fabryova, V1; Vasiľová, A2; Kollárová, A3; Gbúrová, K2; Fabry, B4
1Hospital St. Michael, Bratislava, SLOVAKIA; 2L Pasteur University Hospital, Košice, SLOVAKIA; 3Faculty hospital, Nitra, SLOVAKIA; 4Comenius University, Faculty of Law and Ethics Comittee, Ministry of Health, Bratislava, SLOVAKIA
Background: The paper presents results of 27- year epidemiological study of screening and follow-up haemoglobinopathies in Slovakia.Aims: The incidence of haemoglobinopathies in Slovek RepublicMethods: Between 1993 – 2020, in two centres in Bratislava and in one centre in Kosice, carriers of beta-thalassaemic genes or other haemoglo-binopathies were searched for. Patients with a probability of having a haemoglobinopathy were sent to the research facilities. Diagnosis was performed by haematologists, whereby the family history was evaluated, together with overall clinical condition, blood count and blood smear, iron and haemolysis parameters, mutations of hereditary haemochroma-tosis, and haemoglobin electrophoresis testing.Results: A clinical suspicion of the heterozygous form of beta-thalas-saemia or other haemoglobinopathies was documented in 694 patients. Of them 25 (6.04%) patients were foreigners. 415 (59.85%) patients were genetically examined. In 385 (92.99%) of them heterozygote beta-thalassaemia was confirmed (in 98 families). Five patients (1.21%) were diagnosed for delta,beta-thalassaemia, 4 patients (0,97%) for del-ta,beta-gama1-thalasaemia or persistent hereditary fetal haemoglobin. In total we diagnosed 20 mutations of beta-globin gene. The most fre-quent mutations were IVS 1.110 (G-A), IVS II-1(G-A) and codon 39. Evidence of haemoglobin S (heterozygote sickle cell anaemia) was also notable in two non-relative children, whose fathers were of African ori-gin, in one patient of Ghana and in one patient from Nigeria. One female patient was followed up for haemoglobin Santa Ana (mutation de novo), one family for haemoglobin Bishopstown and one patient for mutation KLF1 gene.In our group were 14 patients (3.14%) diagnosed for alpha-thalassaemia.All patients were heterozygotes, only one female patient from Macedonia was a double heterozygote for beta-thalasaemie. Clinicaly all of the patients had a minor or intermedia form. In the years of 2012–2019 we observed 12 pregnant patients with beta-thalasaemie. One of them had multiple pregnancies, all deliveries were without haematological complications.Conclusions: The study showed that in the west and eastern Slovakia there is a higher number for thalassaemia and other haemoglobinopa-thies. Mutations are of historical origin or over the past years we have recorded an increase number of mutations from areas with high inci-dence of haemoglobinopathies. It is necessary to continue in search of pathological gene carriers to avoid serious forms of the disease.Key words: thalassaemia – sickle cell disease – prevention- epidemiolog-ical study – Slovakia
References1. Divoka et al, Hemoglobin 2016;40:156–1622. Engert et al, Haematologica 2016;101:115–2083. Fabryova et al, Centr Eur J Public Health 2017;25:67–714. Pinto et al, Internal and emergency medicine 2019;7:1051–645. Fabryova et al, J Hematol Transf 2020;7(1):10866. Vieves Corrons et al, Acta Biomed 2020;91:2
P114 NEWBORN SCREENING FOR HAEMOGLOBINOPATHIES IN BIDA, NORTH CENTRAL NIGERIA.
Folayan, O1; BELLO, A1; Ernest, S2
1Federal Medical Centre, Bida, NIGERIA; 2University of Ilorin Teaching Hospital, Ilorin, NIGERIA
Background: The global annual population of newborns with structural haemoglobin disorders is estimated at five million. Nigeria accounts for more than 30% of these in Sub-Saharan Africa with under-five mortality from haemoglobinopathies reaching 50–90%. Despite this huge burden and a 15-fold reduction in deaths from haemoglobinopathies in coun-tries that conduct newborn screening, most sub-Saharan Africa countries
do not have a screening program. Haemoglobin electrophoresis is also not sensitive for newborn screening.Objectives: This study was carried out to determine haemoglobin pheno-type patterns and frequency in neonates attending routine immunization clinics in Bida, and to identify factors associated with the occurrence of haemoglobinopathy.Methods: It was a descriptive cross-sectional study that recruited 254 neonates by multi-staged sampling technique from nine immunisation centres. Heel prick blood sample collected on Guthrie cards were tested using High-Performance Liquid Chromatography (HPLC). The relation-ship of various risk factors with the occurrence of an abnormal hae-moglobin variant was analysed with the Statistical Package for Social Sciences.Results: The Hb phenotypes found in this study were HbFA- 73.6% (187/254), HbFAS- 23.2% (59/254), HbFAC- 1.6% (4/254), HbFS- 1.2% (3/254), and HbFAD-0.4% (1/254). There was an almost equal abnormal haemoglobin occurrence in both genders. The majority (89%) of mothers did not know their Hb phenotype, one-quarter of these had a newborn with an abnormal phenotype and 20% married in consanguin-eous marriages. Wrong perception of sickle cell disease was common.Conclusion: Abnormal haemoglobin variants were present in more than one-quarter (26.4%) of the neonatal population studied in Bida. Most parents were not aware of their haemoglobin phenotype and had a wrong perception of sickle cell disease. Consanguinity though common in the population did not significantly affect the occurrence of an abnor-mal haemoglobin phenotype.
References1. Hsu L, Nnodu O. E, Brown B. J, Tluway F, King S, Dogara L. G et al. White Paper: Pathways to Progress in Newborn Screening for Sickle Cell Disease in Sub-Saharan Africa. J Trop Dis 2018;6(2):1–9.2. Weatherall DJ, Clegg J. B. Inherited haemoglobin disorders: An increas-ing global health problem. Bull World Health Organ 2001;79(8):704–12.3. Williams T. N, Weatherall D. J. World distribution, population genet-ics, and health burden of the hemoglobinopathies. Cold Spring Harb Perspect Med 2012;2(116):1–14.4. Upadhye DS. Neonatal screening & Natural history of sickle cell dis-ease. Immunohaematology Bull 2014;45(2):1–10.5. Akodu S, Diaku-Akinwumi I, Njokanma O. Age at Diagnosis of Sickle Cell Anaemia in Lagos, Nigeria. Mediterr J Hematol Infect Dis 2013;5(1):1–5.6. Fleming A. F, Storey J, Molineaux L, Iroko E. A, Attai E. D. Abnormal haemoglobins in the Sudan savanna of Nigeria I. Prevalence of haemo-globins and relationships between sickle cell trait, malaria and survival. Ann Trop Med Parasitol 1979;73(2):161–727. Odunvbun, M. E, Okolo A. A, Rahimy CM. Newborn screening for sickle cell disease in a Nigerian hospital. Public Health 2008;122(10):1111–116.8. Inusa B. P, Daniel Y, Lawson J. O, Dada J, Matthews C. E, Momi S et al. Sickle Cell Disease Screening in Northern Nigeria: The Co-Existence of Β- Thalassemia Inheritance. Pediat Ther 2015;5(3):1–4.9. Sam-Amoye TO. Prevalence of Sickle Cell Disorder and other Haemoglobinopathies among Newborn Infants Delivered at Government Hospitals in Lagos State. Faculty of Paediatrics. National Postgraduate Medical College. (Unpublished Dissertation). 2017.10. Ohene-Frempong K, Oduro J, Tetteh H, Nkrumah F. Newborn screen-ing for sickle cell disease in Ghana. Pediatrics 2008;121:120–121.
P115 REAL-WORLD DATA ON THE EFFICACY OF PHARMACEUTICAL-GRADE L-GLUTAMINE IN PREVENTING SICKLE CELL DISEASE-RELATED ACUTE COMPLICATIONS AND HEMOLYSIS IN PEDIATRIC AND ADULT PATIENTS.
Elenga, N1; Loko, G2; Etienne-Julan, M3; AlOkka, R4; Adel, A4; Yassin, M4
1Centre Hospitalier de Cayenne, Cayenne, FRENCH GUIANA; 2Centre de reference de la drepanocytose CHU de la Guadeloupe, Pointe-à Pitre, FRANCE; 3Centre de reference de la drepanocytose CH de Fort de France, Fort de France, FRANCE; 4Hamad Medical Corporation, Doha, QATAR
Background: Oxidative stress is a key contributors to the pathophysiol-ogy of sickle cell disease (SCD) and related complications including acute pain (vaso-occlusive crisis VOC) and acute chest syndrome (ACS) [1, 2]. L-glutamine (L-Gln), a precursor of nicotinamide adenine dinucleotide (NAD), has been shown to play a key role in the regulation of oxida-tive stress[3]. In a pivotal Phase 3 trial, L-Gln demonstrated significant

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 25
reduction in VOCs, hospitalizations, and ACS events compared to pla-cebo in patients with SCD, with or without hydroxyurea (HU) use, over a 48-week period [4]. In September 2021, a re-analysis of the trial data revealed that L-Gln decreased the number of VOCs by 45% [5].Aims: To confirm the efficacy of pharmaceutical-grade L-glutamine in pediatric and adult patients with SCD at follow-up time points of 24, 48 and 72 weeks.Methods: In a retrospective study conducted from October 2019 to April 2020, 19 patients (4 patients from Qatar and 15 patients from French Guyana) were treated orally with L-Gln (0.3mg/kg) twice daily. Laboratory parameters (hemoglobin levels (Hg), hematocrit proportion (Ht), WBC counts, reticulocyte counts, and LDH levels) were measured at baseline and follow-up time points. Clinical parameters (number of VOCs, hospitalizations, days hospitalized, ACS events, and blood transfusions) were documented for the year prior to treatment initiation as baseline values. These parameters were also collected at 24, 48, and 72 weeks from treatment initiation. Adverse events (AEs) were also collected during the treatment period. The data values at 24, 48, and 72 weeks have been annu-alized. Statistical analysis was performed using MedCalc Version 20.015.Results: 19 patients (HbSS, age range, 8–54 years; 53% patients <18 years) were retrospectively analyzed. 63% of the patients were receiving HU at baseline and 47% received concomitant HU during the study. Compared to baseline, patients had significantly fewer VOCs at 24, 48, and 72 weeks following L-Gln therapy (median change from 3.0 to 0; P<0.00001). Compared to baseline, there were fewer hospitalizations (median change from 3.0 to 0; P<0.00001) and patients spent fewer days in hospital (median change from 15.0 to 0; P<0.00001). Moreover, at 24, 48, and 72 weeks, the number of blood transfusions was considerably lower than at baseline (median, from 3.0 to 0; P<0.00001). In the year prior to treatment initiation, 2 patients reported a single ACS event, but, no such events were observed during therapy. Following treatment with L-Gln, the mean Hg level increased significantly from baseline to 72 weeks (8.2 to 8.8 g/dL; P<0.001) with peak mean increase from baseline of 11.2% at 48 weeks. A similar increasing trend was observed for Ht from baseline to 72 weeks (24% to 27%; P<0.001) with highest mean improvement from baseline of 15.5% at 48 weeks. Conversely, mean reticulocyte counts and LDH levels were significantly reduced at follow-up time points compared to baseline (P=0.003 and P<0.001, respectively). Only few AEs occurred and were mild. No compliance issue was reported.Conclusions: This study demonstrated that L-Gln therapy in SCD patients from Qatar and French Guyana resulted in significant and sus-tained improvements in clinical outcomes (number of VOCs, number and duration of hospitalizations, and number of blood transfusions) and an increase in Hg and a reduction of hemolysis. 2 patients with a history of ACS did not experience any of those events during L-Gln therapy.
References1. Klings ES, Farber HW (2001) Role of free radicals in the pathogenesis
of acute chest syndrome in sickle cell disease. Respir Res 2(5):280–5. https://doi.org/10.1186/rr70.
2. Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, et al. (2008) Erythrocyte glutamine depletion, altered redox envi-ronment, and pulmonary hypertension in sickle cell disease. Blood 111(1):402–10. https://doi.org/10.1182/blood-2007-04-081703.
3. Niihara Y, Zerez CR, Akiyama DS, Tanaka KR (1998) Oral L-glutamine therapy for sickle cell anemia: I. Subjective clin-ical improvement and favorable change in red cell NAD redox potential. Am J Hematol 58(2):117–21. https://doi.org/10.1002/(sici)1096–8652(199806)58:2<117::aid-ajh5>3.0.co;2-v.
4. Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. (2018) A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med 379(3):226–35. https://doi.org/10.1056/NEJMoa1715971.
5. Zaidi AU, Estepp J, Shah N, Alkindi S, Ezzat H, Lam H, et al. (2021) A reanalysis of pain crises data from the pivotal l-glutamine in sickle cell disease trial. Contemp Clin Trials 110:106546. https://doi.org/10.1016/j.cct.2021.106546.
P116 THE IMPACT OF COVID19 PANDEMIC ON SICKLE CELL MANAGEMENT: EXPERIENCE OF A SINGLE PEDIATRIC CENTER
Maria Vousvouki, M1; Aikaterini Teli, A1; Alkistis Adramerina, A1; Stamatia Theodoridou, S2; Marina Economou, M1
11st Pediatric Department, Aristotle University of Thessaloniki, Thessaloniki, GREECE; 2Blood Bank, Hippokration General Hospital of Thessaloniki, Thessaloniki, GREECE
Background: COVID19 pandemic has put an incomparable pressure on all health services – including those offered to hemoglobinopathy patients. Sickle cell disease management, although not routinely requir-ing inpatient facilities, is dependent upon hospital based services on a great extent.Aims: Aim of the present study was to evaluate the impact of COVID19 pandemic on medical management of sickle cell disease patients fol-lowed at a single pediatric center in Northern Greece.Methods: Patient records were reviewed in order to assess changes reflecting limited access to specialized care, i.e. number of disease related complications, number of hospital routine visits and number of disease related hospitalizations, during the 18month pandemic in Greece.Results: The study included 23 patients, 17 female (74%) and 6 male (26%). Age range was 2 to 19 years. Eighteen (18) patients (78.3%) were double heterozygotes for sickle cell and beta thalassemia, 4 (17.4%) were sickle cell homozygotes and 1 patient (4.3%) was double heterozygote for sickle cell disease and hemoglobinopathy D. Out of 23 patients, 4 were on regular blood transfusions due disease related issues (primary prophylaxis for cerebrovascular disease or hypersplenism in 2 cases, severe anemia or repeated pain crises in 2 other cases). No sig-nificant difference was recorded in number of hospital visits during the 18month period before and during the pandemic (5.45 visits/year and 6 visits/year, respectively – p = 0.49), reflecting a stable course for patients not receiving regular transfusions and an unaffected by blood shortage or limited hospital access course for regularly transfused patients. To that end, no significant changes were recorded in non-COVID related hospitalizations between the two groups (0.57/year before the pandemic and 0.39/year during the pandemic, p = 0.32). In addition, no difference was found between the reported number of pain crises (0.63 episodes per year vs 0.63 episodes per year, p = 1.0). An otherwise unremark-able overall clinical and laboratory course was reported for all patients during the two time periods compared. No changes in regular monitor-ing was noted and no changes in drug prescription or drug availability for patients on hydroxyurea was recorded.Summary-Conclusions: Although sickle cell patients require close mon-itoring, mostly dependent upon hospital based services, the COVID19 pandemic does not seem to have limited access to recommended care in this patient group.
References1. Orphanet J Rare Dis 2021 Mar 1;16(1):114
P117 THE INTERNATIONAL HAEMOGLOBINOPATHY RESEARCH NETWORK (INHERENT): AN INTERNATIONAL INITIATIVE TO STUDY THE ROLE OF GENETIC MODIFIERS IN HAEMOGLOBINOPATHIES
Kountouris, P1; Stephanou, C1; Archer, N2; Bonifazi, F3; Giannuzzi, V3; Kuo, K4; Maggio, A5; Makani, J6; Mañú Pereira, M7; Michailidou, K1; Nkya, S6; Nnodu, O8; Trompeter, S9; Tshilolo, L10; Wonkam, A11; Zilfalil, B12; Inusa, B13; Kleanthous, M1
1The Cyprus Institute of Neurology and Genetics, Nicosia, CYPRUS; 2Dana-Farber/Boston Children’s Cancer and Blood Disorder Centre, Boston, UNITED STATES; 3Fondazione per la ricerca farmacologica Gianni Benzi onlus, Bari, ITALY; 4University of Toronto, Toronto, CANADA; 5Azienda Ospedaliera Ospedali Riuniti Villa Sofia, Palermo, ITALY; 6Muhimbili University of Health and Allied Sciences, Dar es Salaam, TANZANIA, UNITED REPUBLIC OF; 7Vall d’Hebron Institut de Recerca, Barcelona, SPAIN; 8University of Abuja, Abuja, NIGERIA; 9University College London Hospitals NHS Foundation Trust, London, UNITED KINGDOM; 10Centre Hospitalier Monkole, Kinshasa, CONGO, THE DEMOCRATIC REPUBLIC OF THE; 11University of Capetown, Cape Town, SOUTH AFRICA; 12Universiti Sains Malaysia - Kampus Kesihatan, Kubang Kerian, MALAYSIA; 13Guy’s and St Thomas’ NHS Trust, London, UNITED KINGDOM
Background: Haemoglobinopathies, including sickle cell disease (SCD) and thalassaemia syndromes, represent the commonest monogenic dis-eases in the world. Although their pathogenicity is well established, the diverse clinical manifestations and the varying degree of severity are less understood and are thought to be governed, in part, by genetic modifiers. Despite the identification and characterisation of a few genetic modifiers by previous studies, these are as yet insufficient to guide treatment rec-ommendations or stratify patients reliably. Larger, multi-ethnic studies are needed to identify and validate further disease modifiers that can be used for patient stratification and personalised treatment. There is a growing need for deeper insight with the availability of novel targeted therapies and potentially curative options like gene therapy in both SCD and thalaessemia.
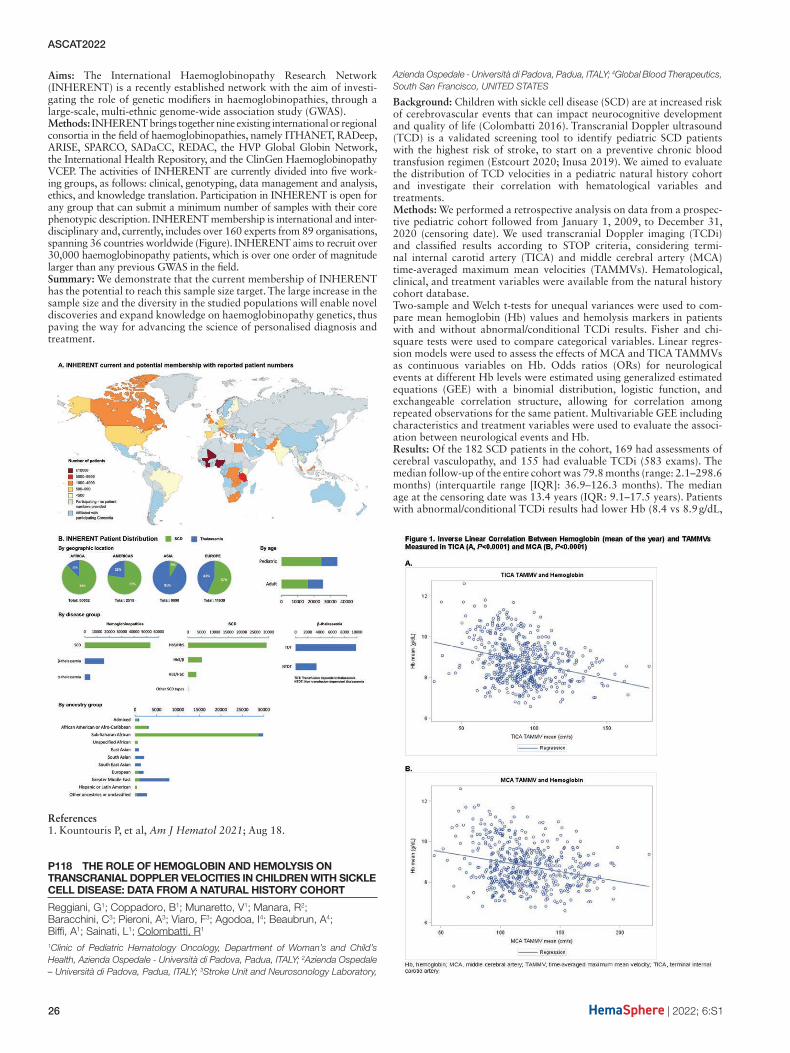
ASCAT2022
26 | 2022; 6:S1
Aims: The International Haemoglobinopathy Research Network (INHERENT) is a recently established network with the aim of investi-gating the role of genetic modifiers in haemoglobinopathies, through a large-scale, multi-ethnic genome-wide association study (GWAS).Methods: INHERENT brings together nine existing international or regional consortia in the field of haemoglobinopathies, namely ITHANET, RADeep, ARISE, SPARCO, SADaCC, REDAC, the HVP Global Globin Network, the International Health Repository, and the ClinGen Haemoglobinopathy VCEP. The activities of INHERENT are currently divided into five work-ing groups, as follows: clinical, genotyping, data management and analysis, ethics, and knowledge translation. Participation in INHERENT is open for any group that can submit a minimum number of samples with their core phenotypic description. INHERENT membership is international and inter-disciplinary and, currently, includes over 160 experts from 89 organisations, spanning 36 countries worldwide (Figure). INHERENT aims to recruit over 30,000 haemoglobinopathy patients, which is over one order of magnitude larger than any previous GWAS in the field.Summary: We demonstrate that the current membership of INHERENT has the potential to reach this sample size target. The large increase in the sample size and the diversity in the studied populations will enable novel discoveries and expand knowledge on haemoglobinopathy genetics, thus paving the way for advancing the science of personalised diagnosis and treatment.
References1. Kountouris P, et al, Am J Hematol 2021; Aug 18.
P118 THE ROLE OF HEMOGLOBIN AND HEMOLYSIS ON TRANSCRANIAL DOPPLER VELOCITIES IN CHILDREN WITH SICKLE CELL DISEASE: DATA FROM A NATURAL HISTORY COHORT
Reggiani, G1; Coppadoro, B1; Munaretto, V1; Manara, R2; Baracchini, C3; Pieroni, A3; Viaro, F3; Agodoa, I4; Beaubrun, A4; Biffi, A1; Sainati, L1; Colombatti, R1
1Clinic of Pediatric Hematology Oncology, Department of Woman’s and Child’s Health, Azienda Ospedale - Università di Padova, Padua, ITALY; 2Azienda Ospedale – Università di Padova, Padua, ITALY; 3Stroke Unit and Neurosonology Laboratory,
Azienda Ospedale - Università di Padova, Padua, ITALY; 4Global Blood Therapeutics, South San Francisco, UNITED STATES
Background: Children with sickle cell disease (SCD) are at increased risk of cerebrovascular events that can impact neurocognitive development and quality of life (Colombatti 2016). Transcranial Doppler ultrasound (TCD) is a validated screening tool to identify pediatric SCD patients with the highest risk of stroke, to start on a preventive chronic blood transfusion regimen (Estcourt 2020; Inusa 2019). We aimed to evaluate the distribution of TCD velocities in a pediatric natural history cohort and investigate their correlation with hematological variables and treatments.Methods: We performed a retrospective analysis on data from a prospec-tive pediatric cohort followed from January 1, 2009, to December 31, 2020 (censoring date). We used transcranial Doppler imaging (TCDi) and classified results according to STOP criteria, considering termi-nal internal carotid artery (TICA) and middle cerebral artery (MCA) time-averaged maximum mean velocities (TAMMVs). Hematological, clinical, and treatment variables were available from the natural history cohort database.Two-sample and Welch t-tests for unequal variances were used to com-pare mean hemoglobin (Hb) values and hemolysis markers in patients with and without abnormal/conditional TCDi results. Fisher and chi-square tests were used to compare categorical variables. Linear regres-sion models were used to assess the effects of MCA and TICA TAMMVs as continuous variables on Hb. Odds ratios (ORs) for neurological events at different Hb levels were estimated using generalized estimated equations (GEE) with a binomial distribution, logistic function, and exchangeable correlation structure, allowing for correlation among repeated observations for the same patient. Multivariable GEE including characteristics and treatment variables were used to evaluate the associ-ation between neurological events and Hb.Results: Of the 182 SCD patients in the cohort, 169 had assessments of cerebral vasculopathy, and 155 had evaluable TCDi (583 exams). The median follow-up of the entire cohort was 79.8 months (range: 2.1–298.6 months) (interquartile range [IQR]: 36.9–126.3 months). The median age at the censoring date was 13.4 years (IQR: 9.1–17.5 years). Patients with abnormal/conditional TCDi results had lower Hb (8.4 vs 8.9 g/dL,

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 27
P≤0.0001) and higher reticulocyte counts (317,766 vs 262,750/mm3, P≤0.0001), lactate dehydrogenase (843 vs 690 U/L, P=0.0012), and aspartate aminotransferase (61 vs 54 U/L, P=0.0007) compared with patients with normal/low results. We detected an inverse linear cor-relation between TICA/MCA TAMMVs and Hb (Figure 1A and 1B). Univariate analysis showed significant inverse correlation between abnormal/conditional TCDi results and Hb considered as a continuous variable (OR: 0.484, P<0.001). In the multivariate analysis, the correla-tion between TCDi results and Hb remained significant; moreover, the risk of presenting abnormal/conditional TCDi results decreased with age (OR: 0.833, P<0.0064).Conclusions: This analysis from our natural history cohort shows a sig-nificant inverse correlation between Hb and MCA and TICA velocities, supporting the beneficial effect of higher Hb levels in reducing TAMMV.
References1. Colombatti R, et al. PLoS One. 2016;11(6):e-157090.2. Estcourt LJ, et al. Cochrane Database Syst Rev. 2020;4(4):CD012389.3. Inusa BPD, et al. J Clin Med. 2019;9(1):44.
P119 TRIAL IN PROGRESS: THE RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED PHASE 1B CROSSWALK-A TRIAL EVALUATING THE SAFETY OF CROVALIMAB FOR THE MANAGEMENT OF ACUTE UNCOMPLICATED VASO-OCCLUSIVE EPISODES (VOES) IN PATIENTS (PTS) WITH SICKLE CELL DISEASE (SCD)
Bartolucci, P1; Ataga, K2; Callaghan, M3; De Franceschi, L4; Minniti, C5; Alexandrou, A6; Imbs, D7; Fox, R8; Patel, H9; Sostelly, A10; Schimmel, J11
1University Paris-East Créteil, Sickle Cell Referral Center - UMGGR & IMRB, Laboratory of Excellence LABEX GRex, Créteil, FRANCE; 2UTHSC Center for Sickle Cell Disease, Memphis, TN, UNITED STATES; 3Division of Pediatric Hematology/Oncology, Central Michigan University SOM, Children’s Hospital of Michigan, Detroit, MI, UNITED STATES; 4Department of Medicine, University of Verona and AOUI Verona, Verona, ITALY; 5Albert Einstein College of Medicine, Bronx, New York, NY, UNITED STATES; 6Roche Products Limited, Welwyn Garden City, UNITED KINGDOM; 7Certara, Inc., Paris, FRANCE; 8Parexel International, Uxbridge, UNITED KINGDOM; 9Genentech, Inc., South San Francisco, CA, UNITED STATES; 10F. Hoffmann-La Roche Ltd, Basel, SWITZERLAND; 11Icahn School of Medicine at Mount Sinai, Department of Emergency Medicine, New York. NY, UNITED STATES
Background: SCD is a group of autosomal recessive red blood cell (RBC) disorders caused by a single point mutation in the β-globin gene, result-ing in the production of hemoglobin S (HbS). HbS polymerizes within RBCs under certain conditions, leading to the distortion of the RBC membrane and generation of dense and sickle RBCs. These pathologic RBCs contribute to microvascular occlusions, which may present as acute, painful episodes called VOEs. Although most VOEs are managed at home, >70% of emergency department visits and >90% of hospital-izations for SCD are VOE-related.1,2 Additionally, acute chest syndrome is one of the most severe complications of VOEs and is a leading cause of mortality.3,4 As there are no targeted therapies for the management of VOEs, treatment is currently limited to pain management, blood exchange transfusion (with the risk of complications), and other sup-portive care, which represents a significant unmet medical need. Overall, accumulating nonclinical data suggest a multimodal role for comple-ment dysregulation in the pathophysiology of SCD including vaso-occlu-sion, hemolysis, inflammation, thrombogenicity, endothelial activation, and end-organ damage.5 Complement pathway activation has been described in pts with SCD at baseline, in acute pain crises, and in pts with delayed hemolytic transfusion reaction. The complement pathway can be targeted with crovalimab, which is a novel, engineered, anti-com-plement C5 monoclonal antibody. In a Phase 1/2 paroxysmal nocturnal hemoglobinuria (a complement-mediated disorder) study, crovalimab showed rapid and sustained complement inhibition with promising effi-cacy and safety.6
Aims: CROSSWALK-a (NCT04912869) is a randomized, double-blind, placebo-controlled, Phase 1b study evaluating the safety of crovalimab in managing acute uncomplicated VOEs in pts with SCD.Methods: Pts aged 12 to 55 years, weighing ≥40 kg, and with a con-firmed diagnosis of SCD homozygous hemoglobin S (HbSS) or sickle cell β0 thalassemia (HbSβ0) will be enrolled (Figure). Pts must present with an acute uncomplicated VOE, requiring hospitalization and treat-ment with parenteral opioid analgesics. Vaccinations against Neisseria
meningitidis, Hemophilus influenzae type B, and Streptococcus pneumo-niae must be current. Eligible pts will be randomized 2:1 to receive either a single intravenous weight-based tiered dose of crovalimab or placebo. All pts will continue with pain management and other supportive care for their VOE, and may continue concurrent SCD-directed therapies. Pts will be followed during hospitalization until discharge and will also be followed post-discharge during an observational period. The maximum total study duration for an individual pt, including the hospitalization and observational periods, will be 12 weeks. The primary objective is to evaluate the incidence and severity of adverse events according to National Cancer Institute Common Terminology Criteria for Adverse Events, Version 5.0, incidence and severity of infusion-related reactions and hypersensitivity, and change from baseline in targeted vital signs and clinical laboratory test results. Efficacy, pharmacokinetics, pharmacody-namics, immunogenicity, and exploratory biomarker endpoints will also be evaluated.Results: CROSSWALK-a is scheduled to be completed in September 2023.Summary: CROSSWALK-a is enrolling pts with SCD in 5 countries.
References1. Ballas et al, Am J Hematol 2005; 79:172. Lanzkron et al, Am J Hematol 2010; 85:7973. Vichinsky et al, N Engl J Med 2000; 342:18554. Bartolucci et al, eBioMedicine 2016; 10:3055. Roumenina et al, Am J Hematol 2020; 95:4566. Röth et al, Blood 2020; 135:912
P120 TRIAL IN PROGRESS: THE RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED PHASE 2A CROSSWALK-C TRIAL EVALUATING THE EFFICACY OF CROVALIMAB AS ADJUNCT TREATMENT IN THE PREVENTION OF VASO-OCCLUSIVE EPISODES (VOES) IN PATIENTS (PTS) WITH SICKLE CELL DISEASE (SCD)
Callaghan, M1; Ataga, K2; De Franceschi, L3; Minniti, C4; Balachandran, N5; Imbs, D6; Perretti, T7; Ramos, J8; Sostelly, A7; Bartolucci, P9
1Division of Pediatric Hematology/Oncology, Central Michigan University SOM, Children’s Hospital of Michigan, Detroit, MI, UNITED STATES; 2UTHSC Center for Sickle Cell Disease, Memphis, TN, UNITED STATES; 3Department of Medicine, University of Verona and AOUI Verona, Verona, ITALY; 4Albert Einstein College of Medicine, Bronx, New York, NY, UNITED STATES; 5Roche Products Limited, Welwyn Garden City, UNITED KINGDOM; 6Certara, Inc., Paris, FRANCE; 7F. Hoffmann-La Roche Ltd, Basel, SWITZERLAND; 8Genentech Inc., South San Francisco, CA, UNITED STATES; 9University Paris-East Créteil, Sickle Cell Referral Center - UMGGR & IMRB, Laboratory of Excellence LABEX GRex, Créteil, FRANCE
Background: SCD is a group of autosomal recessive red blood cell (RBC) disorders caused by a single point mutation in the β-globin gene, with either homozygous inheritance or heterozygous co-inheritance with other pathogenic variants of the β-globin gene. Hemoglobin S, produced as a result of this point mutation, polymerizes within RBCs under cer-tain conditions, distorting them and generating dense and sickle RBCs. These pathologic RBCs contribute to microvascular occlusions, which may present as acute painful episodes called VOEs. Pts with SCD may also have severe chronic anemia, chronic pain, immune dysfunction, and progressive multi-organ damage. Current therapies for SCD include hydroxyurea, as well as newer treatments such as L-glutamine, crizan-lizumab, and voxelotor. Despite these treatments, considerable morbid-ity and mortality among pts with SCD represents a significant unmet medical need. Complement pathway activation has been reported in pts with SCD at baseline, in acute pain crises, and in delayed hemolytic

ASCAT2022
28 | 2022; 6:S1
transfusion reaction. Accumulating nonclinical data suggest the poten-tial multimodal role for complement dysregulation in the pathophys-iology of SCD, including vaso-occlusion, hemolysis, inflammation, thrombogenicity, endothelial activation, and end-organ damage.1 The complement pathway can be targeted with crovalimab, a novel anti-C5 monoclonal antibody that allows for small-volume subcutaneous (SC) self-injection. In a Phase 1/2 paroxysmal nocturnal hemoglobinuria (a complement-mediated disorder) study, crovalimab showed rapid and sustained complement inhibition with promising efficacy and safety.2
Aim: CROSSWALK-c (NCT05075824) is a randomized, double-blind, placebo-controlled, Phase 2a study evaluating the efficacy and safety of crovalimab as adjunct therapy in preventing VOEs in pts with SCD.Methods: Pts aged 12 to 55 years, weighing ≥40 kg, with a confirmed diagnosis of SCD, homozygous hemoglobin S (HbSS) or sickle cell β0 thalassemia (HbSβ0), and presenting with 2 to 10 VOEs are eligi-ble, including pts on concurrent SCD-directed therapies. Vaccinations against Neisseria meningitidis, Haemophilus influenzae type B, and Streptococcus pneumoniae are required. Pts with a history of hema-topoietic stem cell transplant will be excluded. Eligible pts will be randomized 1:1 to the crovalimab or placebo arms (Figure). An ini-tial intravenous loading dose of crovalimab or placebo will be given on Week 1, Day 1, followed by SC dose on Week 1, Day 2, and then weekly SC doses on Weeks 2–4. Starting Week 5, a maintenance dose will be given every 4 weeks for 48 weeks. All study treatment will be given according to a weight-based tiered dosing schedule (pts weighing ≥40 kg to <100 kg and pts >100 kg). The primary objective is to evaluate the efficacy of crovalimab vs placebo based on the annualized rate of medical facility VOEs. Key secondary efficacy objectives are the annu-alized rate of acute chest syndrome, the annualized rate of home VOE, and change in urinary albumin-creatinine ratio, tricuspid regurgitant jet velocity, and Patient-Reported Outcomes Measurement Information System (PROMIS)-Fatigue score in adults, from baseline to Week 49. Safety, pharmacokinetics, immunogenicity, and exploratory biomarker objectives will also be evaluated.Results: Primary results for CROSSWALK-c are expected in July 2024.Summary: CROSSWALK-c is enrolling pts with SCD and chronic VOEs in 6 countries.
References1. Roumenina et al, Am J Hematol 2020; 95:4562. Röth et al, Blood 2020; 135:912
Infection, autoimmunity, nutritional deficiencies
P121 PREVALENCE OF VERTEBRAL COMPRESSION FRACTURE AND HYPOVITAMINOSIS D IN CHILDREN WITH THALASSEMIA MAJOR
Rahmartani, L1; Sari, T1; Bermanshah, E2; Wulandari, H2; Wahidiyat, P1
1Pediatric Hematology-Oncology Division, Department of Child Health, Universitas Indonesia - Cipto Mangunkusumo National Hospital, Jakarta, INDONESIA; 2Pediatric Imaging Division, Department of Child Health, Universitas Indonesia - Cipto Mangunkusumo National Hospital, Jakarta, INDONESIA
Background: Thalassemia major has various complications that can increase morbidity and mortality. Osteoporosis is a common com-plication in thalassemia major and has a multifactorial pathogenesis. Osteoporosis can cause vertebral compression fractures and lead to dis-ability. In TIF recommendation, a DEXA-scan is used to measure bone density in thalassemia patients. However, most patients in Indonesia could not afford the DEXA-scan examination because it is not covered by insurance. The 2019 ISCD Pediatric Positions Task Forces stated that the diagnosis of pediatric osteoporosis included the presence of a nontraumatic vertebral compression fracture (VF), without the need for
BMD criteria. In Indonesia, there has been no study for the prevalence of hypovitaminosis D and vertebral compression fractures in children with thalassemia major and its correlation.Objective: To describe the prevalence of VF and hypovitaminosis D in children with thalassemia major.Methods: A cross-sectional study design was conducted in children with thalassemia major aged 7–18 years at the Thalassemia Center at Cipto Mangunkusumo National Hospital. Detection and evaluation techniques for vertebral fractures were using plain radiograph from Siemens Healthineers – Ysio Max – X-ray. The classification is based on Global Consensus Recommendations on Prevention and Management of Nutritional Rickets, whereas <30 nmol/L or <12 ng/mL are deficient, 30–50 nmol/L or 12–20 ng/mL are insufficient.Results: This study obtained 165 subjects (50,1% boys, 66% homozy-gous beta-thalassemia). Fifty-two subjects (31,5%) have VF, and three of them have both thoracal and lumbar fractures. The most common sites of VF were lumbar five and thoracic 11–12. The youngest subject who has VF is seven years old. Osteopenia was found in 67,3% of patients. There were no subjects with fractures who had complaints of pain or a history of previous trauma. Vitamin D levels [25(OH)D] were measured in 122 subjects, and hypovitaminosis D occurred in 77,9% of the subject (32,8% deficiency, 45,1% insufficiency). Our study found no significant relationship between vitamin D levels on the incidence of vertebral com-pression fractures (p>0,05).Conclusion: To our knowledge, this is the highest prevalence of VF among various studies that have been conducted. Vertebral X-ray is beneficial to detect bone density from osteopenia to fracture in children with thalassemia major. Hypovitaminosis D is common in thalassemia, requiring regular vitamin D administration. Further research is needed to consider the other contributing factors.
References1. Gaudio A, et al. J Clin Res Pediatr Endocrinol. 2019;11:110–7.2. Steffey. Pediatric osteoporosis. Peds in Rev. 2019;40;259.3. Piga A. Hematology Am Soc Hematol Educ Program. 2017(1):272–277.4. Bishop N, et al. J Clin Densitom. 2014;17:275–80.5. Munns CF, et al. J Clin Endocrinol Metab. 2016;101:394–415.6. Soliman A, Det al. Mediterr J Hematol Infect Dis. 2013;5(1):e2013057.7. Pulungan A, et al. Ann Pediatr Endocrinol Metab. 2021;26:92–8.8. Vogiatzi MG, et al. J Bone Miner Res. 2009;24(3):543–557.9. Dhale S, et al. J Med Sci Clin Res. 2009;07:254–7.10. Agrawal A, et al. Pediatric Rev: Intl J Ped Res. 2016;9:652–6.
Ageing and end organ damage
P122 DEEP LEARNING-BASED ALGORITHM FOR AUTOMATIC SPLEEN LENGTH MEASUREMENT
Yuan, Z1; King, A1; Inusa, B2
1King’s College London, London, UNITED KINGDOM; 2Evalina Children’s Hospital, London, London, UNITED KINGDOM
Background: In Sickle Cell Disease (SCD), spleen enlargement hap-pens frequently in children. Acute splenic sequestration occurs when the red pulp of the spleen filtering action is exaggerated and the red blood cells are trapped, which causes the spleen sudden enlargement. If left untreated, this condition can be life-threatening. The spleen length is typically measured in the clinic, including manual palpation and fol-lowing ultrasound examination. However, the common clinical work-flow is non-quantitative (manual palpation) and suffers from inter- and intra- experts variability (ultrasound examination). In the developing area, where there is a high prevalence of SCD, there is often a lack of experienced sonographers to conduct the length measurement while ultrasound examination. To address this problem, we set out to use a deep learning-based strategy to develop a full pipeline, which consists of a quality control system and an automated length measurement for spleen assessment.Methods: We investigated the use of deep learning to automate the pro-cess of spleen length measurement. A deep learning model based on the ResNet was developed to classify whether the input ultrasound image could be used to measure the length. A U-net based network was trained to automatically segment the spleen. The principal components analy-sis was then applied to the segmentation to find the longest axis of the spleen, and the splenic length was computed based on the projection of the points within the segmentation to the axis.

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 29
Results: 475 ultrasound images of the spleen were used in this study. 475 images were used to train the quality control network (420 good quality images and 55 bad quality images), and 420 images (good qual-ity images) were used in developing the automated spleen length mea-surement system. These images were obtained from SCD patients and were acquired as part of routine examinations at the Evelina Children’s Hospital, London. 108 images were manually measured by three expe-rienced sonographers to computer the inter-expert variability as a com-parison to our methods.The mean percentage error of our length measurement has reached 4.58% on good quality images which was within the human expert error (5.78%). Our quality control system has reached 0.964 sensitivity and 0.636 specificity. After incorporating bad quality images and the quality control system, the full pipeline reached 4.88% percentage length error, which is still within the human expert variability.Conclusion: We have demonstrated that deep learning can be used for automating the length measurement for the spleen from ultra-sound images. The performance of the developed pipeline has reached the human expert level. This can be potentially used in a real clinical scenario.
References1. Chakravorty, T.N. Williams, Sickle cell disease: A neglected chronic
disease of increasing global health importance, Arch. Dis. Child. 100 (2015). https://doi.org/10.1136/archdischild-2013–303773.
2. B. Piel, A.P. Patil, R.E. Howes, O.A. Nyangiri, P.W. Gething, M. Dewi, W.H. Temperley, T.N. Williams, D.J. Weatherall, S.I. Hay, Global epi-demiology of Sickle haemoglobin in neonates: A contemporary geo-statistical model-based map and population estimates, Lancet. 381 (2013). https://doi.org/10.1016/S0140-6736(12)61229-X.
3. D. Grosse, I. Odame, H.K. Atrash, D.D. Amendah, F.B. Piel, T.N. Williams, Sickle cell disease in Africa: A neglected cause of early child-hood mortality, Am. J. Prev. Med. 41 (2011). https://doi.org/10.1016/j.amepre.2011.09.013.
4. He, X. Zhang, S. Ren, J. Sun, Deep residual learning for image rec-ognition, in: Proc. IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit., 2016. https://doi.org/10.1109/CVPR.2016.90.
5. Ronneberger, P. Fischer, T. Brox, U-net: Convolutional networks for biomedical image segmentation, in: Lect. Notes Comput. Sci. (Including Subser. Lect. Notes Artif. Intell. Lect. Notes Bioinformatics), 2015. https://doi.org/10.1007/978-3-319-24574-4_28.
Health services and outcomes research including psychology
P123 A SYSTEMATIC REVIEW OF THE BARRIERS TO UPTAKE AND ADHERENCE TO HYDROXYCARBAMIDE FROM THE PERSPECTIVE OF CHILDREN AND ADULTS WITH SICKLE CELL DISEASE, PARENTS AND CLINICIANS.
Clark, C
Cambridge University Hospital, Cambridge, UNITED KINGDOM
Aim: The aim of this study was to identify the reasons for the poor uptake and adherence to hydroxycarbamide from the perspective of peo-ple with sickle cell disease, parents of children with sickle cell disease and clinicians of people with sickle cell disease.Background: Children and adults with sickle cell disease have health related complications which impact on their health, quality of life and life expectancy. Hydroxycarbamide has been demonstrated to offer clinical benefits with minimal toxicities but uptake and adherence to hydroxycarbamide remains poor.Method: This study was a systematic review. A search strategy cov-ered several databases including Academic Search Complete, Medline, CINAHL Complete, SocIndex, PubMed, Psych Info, British Nursing Index, Cochrane and Science Direct. An inclusion and exclusion criteria was applied and 9 studies were then critically appraised using validated appraisal tools. Data was extracted, themes identified and results and findings synthesised.Results: Four key themes and further subthemes were identified.
• Safety of hydroxycarbamide, subthemes: side effects, long term side effects, still experimental
• Knowledge and Understanding, subthemes: knowledge of hydroxycarbamide, knowledge of sickle cell disease, perceived
effectiveness of hydroxycarbamide, perception of disease severity, perception of therapeutic effect in the community
• Clinician/healthcare, subthemes: clinician choice, clinician rela-tionship trust in medical system
• Burden of taking, subthemes: remembering to take, aversion to taking pills, clinic reviews (missed work, school), number of blood tests, obtaining prescriptions/refills, costs, transport
Conclusions: Whilst scientific evidence suggests that the benefits of hydroxycarbamide far outweigh the risks, the participants in the nine included studies report barriers that effect its utilisation. Whilst many of the concerns have been reported previously this systematic review confirms that these barriers still exist. Clinicians and healthcare profes-sionals must explore barriers with their colleagues, patients and families and need to make provisions to help reduce the burden of treatment with hydroxycarbamide. Continuing discussions and education through-out childhood, transition and into adulthood will enable patients and families to make informed decisions. Further qualitative studies would enhance the findings of this research.
References of Included Studies 1. Haywood, C., Beach, M.C., Bediako, S., Caroll, C.P., Lattimer, L.,
Jarrett, D. and Lanzkron, S, American Journal Haematology 2011; 68:12. Oyeku, S.O., Driscoll, C., Cohen, H.W., Pashankar, F., Mullen, C.,
Giardina, P.J., Velazco, N., Racine, A.D. and Green, N.S, Pediatric Blood Cancer, 2013; 60:4
3. Bekele, E., Thornburg, C.D., Brandow, A.M., Sharma, M., Smaldone, A.M., Jin, Z. and Green, N.S, 2014: 61:9
4. Creary, S., Zickmund, S., Ross, D., Krishnamurti, L. and Bogen, D.L, Biomedical Research Notes, Pediatric Blood Cancer, 2015; 8:372
5. Vaidyanathan, A. Dissertation.University of Pittsburgh, http://d-schol-arship.pitt.edu/26082/, 2015;
6. Badawy, S.M., Thompson, A.A., Lais, J.S. and Liem, R.I, European Journal of Haematology, 2017;98:6
7. Bakshi, N., Sinha, C.B., Ross, D., Khemani, K., Loewenstein, G. and Krishnamurti, L, PLOS ONE, 2017; doi: 10.1371/journal.pone.0178413
8. Cevher, F., Ünal, S., Küҫükkavruk, Ş.P. and Tunҫtan, B, Turkiye Klinikleri Journal of Medical Science, 2018; 38:1
9. Sinha, C.B., Bakshi, N., Ross, D. and Krishnamurti, L, PLOS ONE, 2018; doi:10.1371/journal.pone.0199375
Key References 1. Hankins, J.S., Ware, R.E., Rogers, Z.R., Wynn, L.W., Lane, P.A., Scott,
P and Wang, W.C, Blood, 2005; 1062. Hankins, J.S., Aygun, B., Nottage, K., Thornburg, C., Smeltzer, M.P.,
Ware, R.E., and Wang, W.C, Medicine, 2014; 93: 283. Nevitt, S.J., Jones, A.P., and Howard, J. Hydroxyurea (hydroxy-
carbamide) for sickle cell disease. (Review) Cochrane Database of Systematic Reviews, 2017; 4
4. Public Health England (PHE) SCD in childhood; standards and guide-lines for clinical care, 2010; 2
5. Qureshi, A., Kaya, B., Pancham, S., Keenan, R., Anderson, J., Akanni, M. and Howard, J. (on behalf of British Society of Haematology - BSH), British Journal of Haematology, 2018; 181
6. Wang, W.C., Ware, R.E., Miller, S.T., Iyer, R.V., Casella, J.F., Minniti, C.P., Rana, S., Thornburg, C.D., Rogers, Z.R., Kalpatthi, R.V., Barredo, J.C., Brown, R.C., Sarnaik, S.A., Howard, T.H., Wynn, L.W., Kutlar, A., Armstrong, F.D., Files, B.A., Goldsmith, J.C., Waclawiw, M.A., Myron, A., Huang, X. and Thompson, B.W, The Lancet, 2011; 377:9778)
P124 EFFICACY OF NON-PHARMACOLOGICAL INTERVENTIONS TO REDUCE PAIN IN CHILDREN WITH SICKLE CELL DISEASE: A SYSTEMATIC REVIEW
Vuong, C; van Veelen, S; Gerritsma, J; de Groot-Eckhardt, C; Verbeek, S; Peters, M; Fijnvandraat, K
Amsterdam UMC, location AMC, Amsterdam, NETHERLANDS
Background: Pain is the clinical hallmark of sickle cell disease (SCD) and leads to frequent emergency department visits, psychological sequelae and a decreased health-related quality of life. Although pharmacolog-ical treatments to target and prevent pain are evolving, children still experience frequent pain. Although pharmacological treatments are effective, they are accompanied by side effects. Therefore, complemen-tary non-pharmacological pain strategies are needed. The literature on

ASCAT2022
30 | 2022; 6:S1
this topic is heterogeneous and we urgently require an evaluation of the evidence supporting efficacy of non-pharmacological interventions in reducing the frequency and intensity of sickle cell related pain in chil-dren with SCD.Aims: The aim of this systematic review is to evaluate the evidence on the efficacy of non-pharmacological interventions in reducing the frequency and intensity of sickle cell related pain in children with SCD.Methods: We performed a literature search through October 2021 using the terms ‘sickle cell’, ‘pain’, ‘psychotherapy’, ‘analgesic’, and ‘health ser-vice’. We included randomized controlled trials and quasi-experimental studies that investigated the efficacy of non-pharmacological interven-tions on (1) pain frequency and intensity, and (2) analgesic and health service use in children with SCD.Results: Eleven articles including 441 participants were included. Nine studies performed in the outpatient clinic investigated the efficacy of cognitive behavioral therapy, biofeedback, and massage. Two studies performed during hospital admission included virtual reality and yoga. Five studies reported significant reductions in the frequency and/or intensity of pain. One study reported a significantly reduced analgesic use. The remaining six studies did not report a reduction in pain related outcomes.Conclusion: The evidence for non-pharmacological interventions to reduce pain in children with SCD seems promising, but due to heteroge-neity of the included/published studies no firm conclusions can be drawn. To target SCD related pain in children, a multidisciplinary approach focusing on the physical, psychological and social aspects seems most appropriate, but the clinical implementation of non-pharmacological interventions first needs further exploration.
References1. Brandow et al, Blood advances 2020; 4:12.2. Williams et al, Journal of Pain and Symptom Management 2016; 51:2.3. Anie et al, The Cochrane database of systematic reviews 2015; 5:5.
P125 EMPOWERING YOUNG ADOLESCENTS ON THE TRANSITION BY THE EDUCATION METHOD ORBIS SENSUALIUM PICTUS - THEIR OWN IMPROVING BLOOD CELL MORPHOLOGY AND MEDICAL RESULTS ON ACTIVE TREATMENT
Kotoucek, P; Caples, R; Culpin, R; Abdussalam, A
Broomfield Hospital, Chelmsford, UNITED KINGDOM
Background: The transition from childhood to adulthood is an import-ant part of a journey of people with inherited haemoglobinopathy and their families.The most difficult part is to find the way of motivation, so children and young adolescents can comply with taking their medications and attend-ing their appointments even without the support of their families to achieve desirable targets and to maintain their good health.Method: We started regular education of young adolescents on the transition in adult haematology department by an efficient and pow-erful method ORBIS SENSUALIUM PICTUS developed by Jan Amos Komensky, Czech teacher, living in the Netherlands in 17th century - teaching with images. We educate and motivate our clients by mor-phology of their own blood cells before the commencement of treat-ment and during it with showing them their results of full blood count, biochemistry, HbS, HbF levels, reticulocytes and others in the same time.Visualising their own blood cells under microscope and comparison of previous unhealthy and now healthy blood cells on hydroxyurea or after regular transfusions empower young adolescents to take ownership of their own health and can improve their self esteem and confidence.We have included into the transition also regular visits in Haematology laboratory, Adult Day therapy unit and Adult haematology department.Results: We are presenting improvement in compliance of the atten-dance and taking regular medication of young adolescents empowered by imaginary education in our transition clinic.Summary: Empowering young adolescents by visualising technique of reviewing their own blood cells under microscope and understanding the purpose of each medication and medical result is very powerful method of motivation. It can help them to engage better in regular consultantions and to find the new meaning on their journey with inherited haemoglo-binopathy. It can open the way of understanding their own disease with consideration of therapeutical options including hydroxyurea, voxelotor, crizanlizumab and gene therapy and allogeneic transplant.
References1. Komensky, Orbis Sensualium Pictus, Nurember 1658, (Sydney University
Press, 1968)2. Kotoucek et al, The Morphology Ward Rounds improve haematology
care and training, BJHaem 20183. Frankl, Man’s searching of meaning, Rider 20044. NICE guideline NG43, Transition from children’s to adult’s services
for young people, 2016
P126 HEALTH LITERACY AND DISEASE KNOWLEDGE IN YOUNG PERSONS WITH SICKLE CELL DISEASE
Ikediashi, B; Roser, K; Michel, G
University of Lucerne, Lucerne, SWITZERLAND
Background: People living with Sickle Cell Disease (SCD) often suf-fer excruciating painful episodes, which can be triggered by infection, extreme weather conditions or stress, and lead to frequent hospital admissions (1,2). Having the requisite knowledge about the disease and adequate health literacy may be beneficial for early recognition of symp-toms, avoidance of triggers of these painful episodes and overall man-agement of the condition (3). This is a pilot study in preparation for a larger study to examine relationships between health literacy, disease knowledge and health outcomes in young persons with SCD.Aim: This pilot study aimed to test the use of a French version of The Health Literacy Measure for Adolescents (HELMA) questionnaire and a sickle cell disease-specific knowledge questionnaire to assess the health literacy and disease knowledge levels of young people with SCD in a clinical setting in West Africa.Methods: Young patients (aged 15 to 24 years) attending a routine Hematology clinic of The University Hospital (CNHU) in Benin and who could speak French were invited to participate in a questionnaire study. An SCD knowledge questionnaire was developed by administer-ing a survey to 7 medical doctors to gather questions they considered relevant for young patients living with SCD. These responses formed the basis of the questionnaire with 18 questions. Of these, 17 questions were given a score of 1 point while 1 question was given a score of 6 points giving a total of 23 points, with a score greater than 16 indicating high knowledge. The HELMA questionnaire was translated into French. It consists of 44 items transferred to a score from 0 to 100. A score of over 66 is considered an adequate health literacy level.Results: A total of 27 patients were recruited for the study (17 males, 63%). The mean age of the patients was 20 years (range: 15–24 years). Most of the patients in this sample (n=17, 63%) were attending secondary schools, 33% (n=9) were in tertiary institutions while 4% (n=1) had completed pri-mary education. The average time used to complete the SCD questionnaire was 6 minutes (range: 4–8 minutes) while most of the patients completed the health literacy questionnaire after 30 minutes (range: 25–35 minutes). Few patients (n=3; 11%) scored less than 16 points on the SCD knowledge questionnaire indicating a high SCD knowledge in this group, while most of the patients (n=20, 73%) had a score of less than 66 on the HELMA questionnaire indicating inadequate health literacy levels.Summary/Conclusions: This study suggests that it may be challenging to use the HELMA in a clinical setting due to the long duration needed for the administration. A shorter version of the HELMA for use in a clinical setting might be helpful. However, the sickle cell disease-specific ques-tionnaire is suitable to assess patients’ knowledge about their condition in a clinical setting.
References1. Ballas et al, Am J Hematol 2005;79:17–252. Roth-Isigkeit et al, Pediatrics 2005: e152-e162.3. Carden et al, Pediatr Hemat Oncol 2016: 121–133.
P127 HEALTHCARE RESOURCE UTILIZATION PATTERNS AND RELATED COSTS IN PATIENTS WITH SICKLE CELL DISEASE IN NORTH LEBANON- A REAL-WORLD EXPERIENCE
Adlette Inati, A1; Maha Sabbagh, M2; Chadi Al Alam, C3; Ammar Al Kheir, A4; Ziad Massouh, Z5; Rawan Demachkie, R6; Cristel Ojaimy, C5; Nidhi Dani, N7; Asif Siddiqui, A7; Rishi Rajat Adhikary, R8; Brendan Lynch, B9; Abhishek Srivastava, A8
1Lebanese American University, Byblos and NINI Hospital, Tripoli, LEBANON; 2Novartis Pharma Services, Beirut, LEBANON; 3American Center for Psychiatry and Neurology,
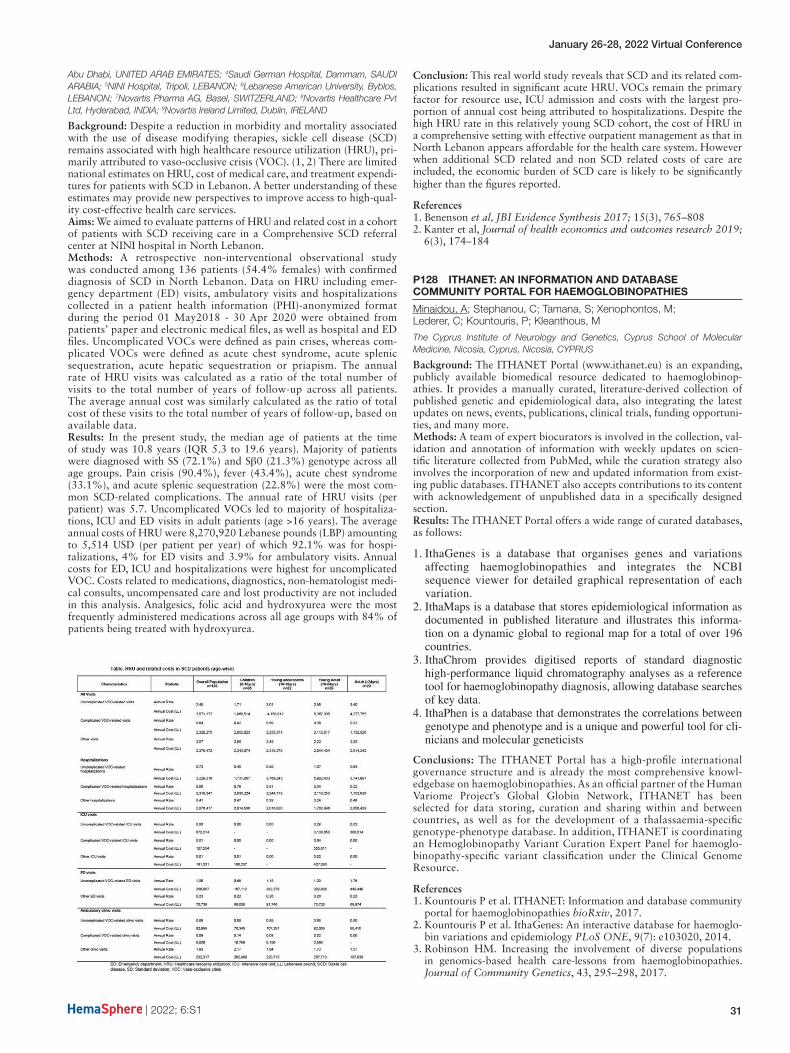
January 26-28, 2022 Virtual Conference
| 2022; 6:S1 31
Abu Dhabi, UNITED ARAB EMIRATES; 4Saudi German Hospital, Dammam, SAUDI ARABIA; 5NINI Hospital, Tripoli, LEBANON; 6Lebanese American University, Byblos, LEBANON; 7Novartis Pharma AG, Basel, SWITZERLAND; 8Novartis Healthcare Pvt Ltd, Hyderabad, INDIA; 9Novartis Ireland Limited, Dublin, IRELAND
Background: Despite a reduction in morbidity and mortality associated with the use of disease modifying therapies, sickle cell disease (SCD) remains associated with high healthcare resource utilization (HRU), pri-marily attributed to vaso-occlusive crisis (VOC). (1, 2) There are limited national estimates on HRU, cost of medical care, and treatment expendi-tures for patients with SCD in Lebanon. A better understanding of these estimates may provide new perspectives to improve access to high-qual-ity cost-effective health care services.Aims: We aimed to evaluate patterns of HRU and related cost in a cohort of patients with SCD receiving care in a Comprehensive SCD referral center at NINI hospital in North Lebanon.Methods: A retrospective non-interventional observational study was conducted among 136 patients (54.4% females) with confirmed diagnosis of SCD in North Lebanon. Data on HRU including emer-gency department (ED) visits, ambulatory visits and hospitalizations collected in a patient health information (PHI)-anonymized format during the period 01 May2018 - 30 Apr 2020 were obtained from patients’ paper and electronic medical files, as well as hospital and ED files. Uncomplicated VOCs were defined as pain crises, whereas com-plicated VOCs were defined as acute chest syndrome, acute splenic sequestration, acute hepatic sequestration or priapism. The annual rate of HRU visits was calculated as a ratio of the total number of visits to the total number of years of follow-up across all patients. The average annual cost was similarly calculated as the ratio of total cost of these visits to the total number of years of follow-up, based on available data.Results: In the present study, the median age of patients at the time of study was 10.8 years (IQR 5.3 to 19.6 years). Majority of patients were diagnosed with SS (72.1%) and Sβ0 (21.3%) genotype across all age groups. Pain crisis (90.4%), fever (43.4%), acute chest syndrome (33.1%), and acute splenic sequestration (22.8%) were the most com-mon SCD-related complications. The annual rate of HRU visits (per patient) was 5.7. Uncomplicated VOCs led to majority of hospitaliza-tions, ICU and ED visits in adult patients (age >16 years). The average annual costs of HRU were 8,270,920 Lebanese pounds (LBP) amounting to 5,514 USD (per patient per year) of which 92.1% was for hospi-talizations, 4% for ED visits and 3.9% for ambulatory visits. Annual costs for ED, ICU and hospitalizations were highest for uncomplicated VOC. Costs related to medications, diagnostics, non-hematologist medi-cal consults, uncompensated care and lost productivity are not included in this analysis. Analgesics, folic acid and hydroxyurea were the most frequently administered medications across all age groups with 84% of patients being treated with hydroxyurea.
Conclusion: This real world study reveals that SCD and its related com-plications resulted in significant acute HRU. VOCs remain the primary factor for resource use, ICU admission and costs with the largest pro-portion of annual cost being attributed to hospitalizations. Despite the high HRU rate in this relatively young SCD cohort, the cost of HRU in a comprehensive setting with effective outpatient management as that in North Lebanon appears affordable for the health care system. However when additional SCD related and non SCD related costs of care are included, the economic burden of SCD care is likely to be significantly higher than the figures reported.
References1. Benenson et al, JBI Evidence Synthesis 2017; 15(3), 765–8082. Kanter et al, Journal of health economics and outcomes research 2019;
6(3), 174–184
P128 ITHANET: AN INFORMATION AND DATABASE COMMUNITY PORTAL FOR HAEMOGLOBINOPATHIES
Minaidou, A; Stephanou, C; Tamana, S; Xenophontos, M; Lederer, C; Kountouris, P; Kleanthous, M
The Cyprus Institute of Neurology and Genetics, Cyprus School of Molecular Medicine, Nicosia, Cyprus, Nicosia, CYPRUS
Background: The ITHANET Portal (www.ithanet.eu) is an expanding, publicly available biomedical resource dedicated to haemoglobinop-athies. It provides a manually curated, literature-derived collection of published genetic and epidemiological data, also integrating the latest updates on news, events, publications, clinical trials, funding opportuni-ties, and many more.Methods: A team of expert biocurators is involved in the collection, val-idation and annotation of information with weekly updates on scien-tific literature collected from PubMed, while the curation strategy also involves the incorporation of new and updated information from exist-ing public databases. ITHANET also accepts contributions to its content with acknowledgement of unpublished data in a specifically designed section.Results: The ITHANET Portal offers a wide range of curated databases, as follows:
1. IthaGenes is a database that organises genes and variations affecting haemoglobinopathies and integrates the NCBI sequence viewer for detailed graphical representation of each variation.
2. IthaMaps is a database that stores epidemiological information as documented in published literature and illustrates this informa-tion on a dynamic global to regional map for a total of over 196 countries.
3. IthaChrom provides digitised reports of standard diagnostic high-performance liquid chromatography analyses as a reference tool for haemoglobinopathy diagnosis, allowing database searches of key data.
4. IthaPhen is a database that demonstrates the correlations between genotype and phenotype and is a unique and powerful tool for cli-nicians and molecular geneticists
Conclusions: The ITHANET Portal has a high-profile international governance structure and is already the most comprehensive knowl-edgebase on haemoglobinopathies. As an official partner of the Human Variome Project’s Global Globin Network, ITHANET has been selected for data storing, curation and sharing within and between countries, as well as for the development of a thalassaemia-specific genotype-phenotype database. In addition, ITHANET is coordinating an Hemoglobinopathy Variant Curation Expert Panel for haemoglo-binopathy-specific variant classification under the Clinical Genome Resource.
References1. Kountouris P et al. ITHANET: Information and database community
portal for haemoglobinopathies bioRxiv, 2017.2. Kountouris P et al. IthaGenes: An interactive database for haemoglo-
bin variations and epidemiology PLoS ONE, 9(7): e103020, 2014.3. Robinson HM. Increasing the involvement of diverse populations
in genomics-based health care-lessons from haemoglobinopathies. Journal of Community Genetics, 43, 295–298, 2017.

ASCAT2022
32 | 2022; 6:S1
P129 NARRATING SICKLE CELL DISEASE: THE EXPERIENCES OF PATIENTS AND CARGIVERS
De Franceschi, L1; Galvani, C1; Ruffo, G2; Colombatti, R3; Graziadei, G4; Palazzi, G5; Venturelli, D6; Forni, G7; Quota, A8; Scopinaro, A9; Vindigni, R10; Zini, M11; Chesi, P12; Reale, L12
1Department of Medicine, Section of Internal Medicine, University of Verona, Policlinico GB Rossi, AOUI, Verona, ITALY; 2U.O. Ematologia con Talassemia ARNAS Civico Di Cristina Benfratelli, Palermo, ITALY; 3Clinic of Pediatric Hematology Oncology, Department of Child and Maternal Health, Azienda Ospedaliera - University of Padova, Padova, ITALY; 4Department of Internal Medicine, Rare Disease Center, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, ITALY; 5Dipartimento Integrato Materno Infantile U. O. Complessa di Pediatria Università degli Studi di Modena e Reggio Emilia, Modena, ITALY; 6Transfusion Medicine Department University Hospital Modena, Modena, ITALY; 7Centro della Microcitemia e Anemie Congenite e del Dismetabolismo del Ferro, Ospedale Galliera, Genova, ITALY; 8U.O.S talassemia PO Vittorio Emanuele, Gela, ITALY; 9UNIAMO Federazione Italiana Malattie Rare APS ETS, Roma, ITALY; 10UNITED – Federazione Nazionale delle Associazioni Talassemia, Drepanocitosi e Anemia Rare, Ferrara, ITALY; 11Novartis Pharma, Origgio, ITALY; 12Istud, healthcare area, Milano, ITALY
Aims: SCAN (Sickle Cell Anemia Narrations) is a project aimed to col-lect narratives of patients with sickle cell disease (SCD) and their family members, to gain insight on living with SCD. Italian SCD population is multicultural, melting endemic patients of Caucasian descent and sub-jects of African descent as first or second-generation Italian citizen.Methods: The research was carried in Italy between January-September 2021 and involved two patients’ associations and seven comprehensive sickle cell centers. Illness plots for patients with SCD and family mem-bers, together with a sociodemographic survey, were collected online through the project’s website. In-depth interviews were also conducted. Narratives were analyzed through narrative-based medicine classifica-tions, NVivo software, and interpretative coding.Results: Twenty-two patients with SCD (53% Caucasian, 33% African, 14% other) and ten family members participated in the survey. The patient’s care pathway in 44% of the cases was described as diffi-cult at the beginning but in the present was considered satisfactory. Misdiagnose of SCD was reported in 33% of patients’ narratives, and their caregivers remarked the overall lack of empathy in communication of the diagnosis. Transitional care was described as positive in 17% of the cases. Although in 41% of the cases family members were described by patients as supportive, in the other cases difficult family situations were reported. Caregivers confirmed the difficulties encountered in man-aging the care pathway and the lack of coordination between compre-hensive SCD centers and general practitioners. Lack of knowledge about SCD, even among health professionals, was remarked by 56% of the respondents. Weakness (53%), and pain in the limbs and hips (41%) impact patients’ quality of life. Acceptance of SCD was the main coping element (79%). Patients’ absence from job showed an average of 39 days per year due to the illness related to SCD. When stimulated about future expectations, patients indicated less pain (30%) and more efficient cures (20%) as still unmet needs.Conclusion: SCAN is the first project on narratives from patients with SCD. Our findings highlight the SCD disease burden for both patients and caregivers. We also identify the need to increase disease awareness and to activate/improve services such as psychological/anthropologic support and transitional care.
References1. SLF Freitas et al, Quality of life in adults with sickle cell disease: an
integrative review of the literature Rev Bras Enferm. 2018 Jan-Feb; 71(1):195–205
2. Pelandri F et al, Life for patients with myelofibrosis: the physical, emo-tional and financial impact, collected using narrative medicine-Results from the Italian ‘Back to Life’ project Qual Life Res 2018; (6):1545–1554
3. Kato GJ et al, Sickle Cell disease Nat Rev dis Primers 2018; Mar 15;4:180104. Ragusa L et al, Caring and living with Prader-Willi syndrome in Italy:
integrating children, adults and parents’ experiences through a mul-ticentre narrative medicine research Bmj Open 2020; 10(8):e036502
5. Charon R, The patient-physician relationship. Narrative Medicine: a model for empathy, reflection, profession, and trust JAMA 2001; 286(15):1897–1902
6. Fioretti C et al, Research studies on patients’ illness experience using the narrative medicine approach: a systematic review BMJ Open 2016; 6(7):e011220
7. Greenhalgh T et al, Narrative based medicine: why study narrative? BMJ 1999; 318(7175):48–50
8. Marini MG. Languages of Care in Narrative Medicine. Words, Space and Time in the Healthcare Ecosystem. Springer International Publishing; 2019.
9. Midena E et al, Real-life patient journey in neovascular age-related macular degeneration: a narrative medicine analysis in the Italian set-ting. Eye 2021 doi:10.1038/s41433-021-01470-9
10. Tonini MC et al, Narrative medicine to integrate patients’, caregiv-ers’ and clinicians’ migraine experiences: the DRONE multicentre project. Neurol Sci. 2021; 15:1–12.
P130 PROVIDING ADEQUATE HEALTHCARE TO PEOPLE WITH HEMOGLOBINOPATHIES DURING THE PANDEMIC (COVID-19)
Sophia Delicou, S1; Aikaterini Xydaki, A1; Konstantinos Manganas, K1; Lucia Evliati, L2; Chrysoula Kalkana, C2; Michael Diamantidis, M3; Achilles Manafas, A3; Marianna Katsatou, M4; Leonidas Roumpatis, L4; Theodoros Aforozis, T1
1Hippokratio General Hospital Athens, ATHENS, GREECE; 2General Hospital Evaggelismos, ATHENS, GREECE; 3General Hospital of Larissa, LARISSA, GREECE; 4Corfu General Hospital, CORFU, GREECE
Background: Countries around the world were dealing with an increase in demand for COVID-19 healthcare, which was exacerbated by fear, confusion, and mobility restrictions, all of which impacted the provision of health care for all conditions. Patients with severe diseases, such as hemoglobinopathies, seem to pose additional challenges.Aim-Methods: An anonymous questionnaire was developed and given to 130 patients from four Thalassemia and Sickle Cell Disease Units of the National Health System of Greece Hospitals using stratified ran-dom sampling technique. To perform statistical analysis on the question-naires, the MedCalc 2018 application was used.Results: There were 130 participants (51 % women, 49 % males) with transfusion-dependent thalassemia (84%), transfused sickle cell dis-ease (15%), and other conditions (1%). During the pandemic, patients’ main concern was a lack of blood for transfusions (64 percent). The consistency of scheduled transfusions was not impaired (72 %) during the lock-down, while they were occasionally delayed (21 %) or did not appear at all (7 %). Similarly, when it came to systemic iron chelation therapy, 82 % were consistent, whereas 6 % stopped taking the drug on a regular basis because of worry of not having enough. The pandemic and lockdown had an impact on the annual follow-up of basic disease comorbidities, with 42% postponing the standard cardiac evaluation, 30% postponing magnetic resonance imaging of the liver-heart, and 23% cancelling major assessments or treatments such as biopsies or in vitro fertilisation (IVF) therapeutic interventions. Finally, 6% of sched-uled surgeries had to be rescheduled.Conclusion: Patients with hemoglobinopathies received their scheduled transfusions without delay during the pandemic. A small percentage of patients modified their home medications because they were concerned about not being adequate during the pandemic.The significant consequence of the pandemic on our patients was the postponement of scheduled assessments and medical procedures required for chronic complications of their underlying condition.
References1. World Health Organization. (2020). COVID-19: Operational guid-
ance for maintaining essential health services during an outbreak.
P131 REAL-TIME VACCINATION IMPACTS IN SICKLE CELL DISEASE: A REAL-WORLD PATIENT CASE STUDY FOR INFLUENZA AND COVID-19 VACCINATION
Agrippa, O1; Summers, K1; Anie, K2; Telfer, P3; James, J4; Low, E5
1Eleven Health, London, UNITED KINGDOM; 2London North West University Healthcare NHS Trust, London, UNITED KINGDOM; 3Barts Health NHS Trust, London, UNITED KINGDOM; 4The Sickle Cell Society, London, UNITED KINGDOM; 5Eric Low Consulting, Edinburgh, UNITED KINGDOM
Background: Sickle Cell Disease (SCD) comprises a group of red blood cell (RBC) disorders, with a diagnosed global population of ~20 million1. It is a chronic disease characterised by morphological RBC abnormali-ties in low oxygen due to β-globin mutation, causing vascular obstruc-tion and complications including pain crisis and stroke2-4.The pandemic has seen patients with SCD face a higher risk of severe forms of COVID-19 infection and mortality5. Despite the approval of
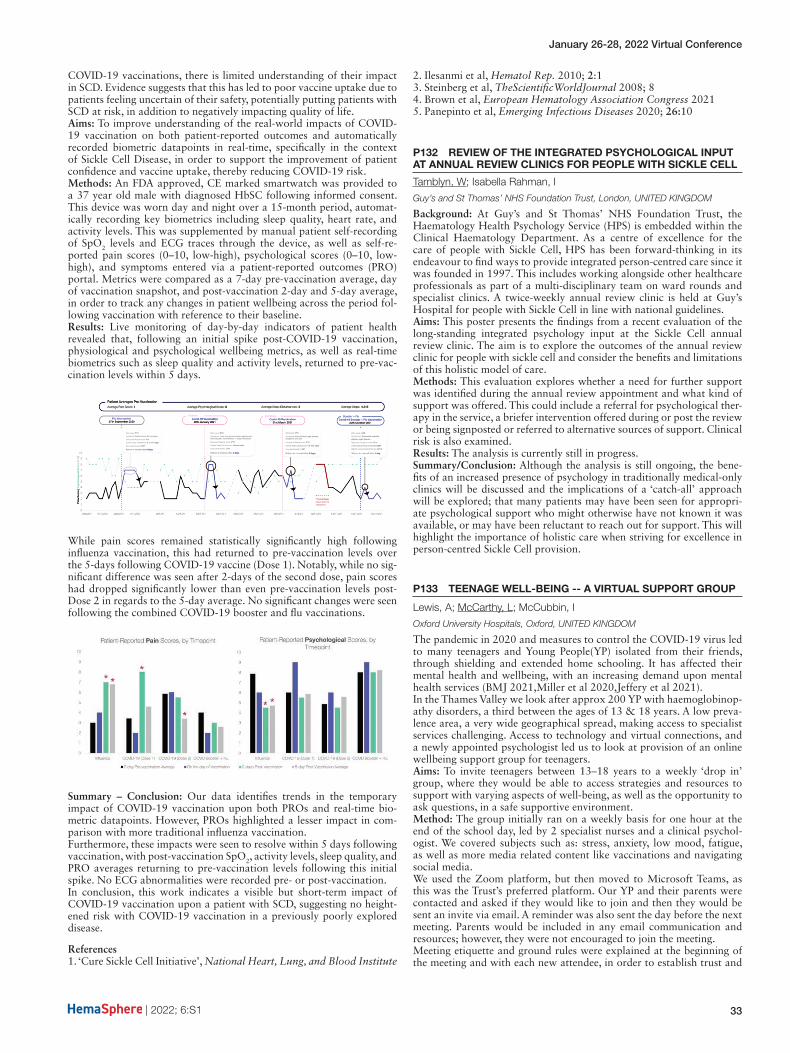
January 26-28, 2022 Virtual Conference
| 2022; 6:S1 33
COVID-19 vaccinations, there is limited understanding of their impact in SCD. Evidence suggests that this has led to poor vaccine uptake due to patients feeling uncertain of their safety, potentially putting patients with SCD at risk, in addition to negatively impacting quality of life.Aims: To improve understanding of the real-world impacts of COVID-19 vaccination on both patient-reported outcomes and automatically recorded biometric datapoints in real-time, specifically in the context of Sickle Cell Disease, in order to support the improvement of patient confidence and vaccine uptake, thereby reducing COVID-19 risk.Methods: An FDA approved, CE marked smartwatch was provided to a 37 year old male with diagnosed HbSC following informed consent. This device was worn day and night over a 15-month period, automat-ically recording key biometrics including sleep quality, heart rate, and activity levels. This was supplemented by manual patient self-recording of SpO2 levels and ECG traces through the device, as well as self-re-ported pain scores (0–10, low-high), psychological scores (0–10, low-high), and symptoms entered via a patient-reported outcomes (PRO) portal. Metrics were compared as a 7-day pre-vaccination average, day of vaccination snapshot, and post-vaccination 2-day and 5-day average, in order to track any changes in patient wellbeing across the period fol-lowing vaccination with reference to their baseline.Results: Live monitoring of day-by-day indicators of patient health revealed that, following an initial spike post-COVID-19 vaccination, physiological and psychological wellbeing metrics, as well as real-time biometrics such as sleep quality and activity levels, returned to pre-vac-cination levels within 5 days.
While pain scores remained statistically significantly high following influenza vaccination, this had returned to pre-vaccination levels over the 5-days following COVID-19 vaccine (Dose 1). Notably, while no sig-nificant difference was seen after 2-days of the second dose, pain scores had dropped significantly lower than even pre-vaccination levels post-Dose 2 in regards to the 5-day average. No significant changes were seen following the combined COVID-19 booster and flu vaccinations.
Summary – Conclusion: Our data identifies trends in the temporary impact of COVID-19 vaccination upon both PROs and real-time bio-metric datapoints. However, PROs highlighted a lesser impact in com-parison with more traditional influenza vaccination.Furthermore, these impacts were seen to resolve within 5 days following vaccination, with post-vaccination SpO2, activity levels, sleep quality, and PRO averages returning to pre-vaccination levels following this initial spike. No ECG abnormalities were recorded pre- or post-vaccination.In conclusion, this work indicates a visible but short-term impact of COVID-19 vaccination upon a patient with SCD, suggesting no height-ened risk with COVID-19 vaccination in a previously poorly explored disease.
References1. ‘Cure Sickle Cell Initiative’, National Heart, Lung, and Blood Institute
2. Ilesanmi et al, Hematol Rep. 2010; 2:13. Steinberg et al, TheScientificWorldJournal 2008; 84. Brown et al, European Hematology Association Congress 20215. Panepinto et al, Emerging Infectious Diseases 2020; 26:10
P132 REVIEW OF THE INTEGRATED PSYCHOLOGICAL INPUT AT ANNUAL REVIEW CLINICS FOR PEOPLE WITH SICKLE CELL
Tamblyn, W; Isabella Rahman, I
Guy’s and St Thomas’ NHS Foundation Trust, London, UNITED KINGDOM
Background: At Guy’s and St Thomas’ NHS Foundation Trust, the Haematology Health Psychology Service (HPS) is embedded within the Clinical Haematology Department. As a centre of excellence for the care of people with Sickle Cell, HPS has been forward-thinking in its endeavour to find ways to provide integrated person-centred care since it was founded in 1997. This includes working alongside other healthcare professionals as part of a multi-disciplinary team on ward rounds and specialist clinics. A twice-weekly annual review clinic is held at Guy’s Hospital for people with Sickle Cell in line with national guidelines.Aims: This poster presents the findings from a recent evaluation of the long-standing integrated psychology input at the Sickle Cell annual review clinic. The aim is to explore the outcomes of the annual review clinic for people with sickle cell and consider the benefits and limitations of this holistic model of care.Methods: This evaluation explores whether a need for further support was identified during the annual review appointment and what kind of support was offered. This could include a referral for psychological ther-apy in the service, a briefer intervention offered during or post the review or being signposted or referred to alternative sources of support. Clinical risk is also examined.Results: The analysis is currently still in progress.Summary/Conclusion: Although the analysis is still ongoing, the bene-fits of an increased presence of psychology in traditionally medical-only clinics will be discussed and the implications of a ‘catch-all’ approach will be explored; that many patients may have been seen for appropri-ate psychological support who might otherwise have not known it was available, or may have been reluctant to reach out for support. This will highlight the importance of holistic care when striving for excellence in person-centred Sickle Cell provision.
P133 TEENAGE WELL-BEING -- A VIRTUAL SUPPORT GROUP
Lewis, A; McCarthy, L; McCubbin, I
Oxford University Hospitals, Oxford, UNITED KINGDOM
The pandemic in 2020 and measures to control the COVID-19 virus led to many teenagers and Young People(YP) isolated from their friends, through shielding and extended home schooling. It has affected their mental health and wellbeing, with an increasing demand upon mental health services (BMJ 2021,Miller et al 2020,Jeffery et al 2021).In the Thames Valley we look after approx 200 YP with haemoglobinop-athy disorders, a third between the ages of 13 & 18 years. A low preva-lence area, a very wide geographical spread, making access to specialist services challenging. Access to technology and virtual connections, and a newly appointed psychologist led us to look at provision of an online wellbeing support group for teenagers.Aims: To invite teenagers between 13–18 years to a weekly ‘drop in’ group, where they would be able to access strategies and resources to support with varying aspects of well-being, as well as the opportunity to ask questions, in a safe supportive environment.Method: The group initially ran on a weekly basis for one hour at the end of the school day, led by 2 specialist nurses and a clinical psychol-ogist. We covered subjects such as: stress, anxiety, low mood, fatigue, as well as more media related content like vaccinations and navigating social media.We used the Zoom platform, but then moved to Microsoft Teams, as this was the Trust’s preferred platform. Our YP and their parents were contacted and asked if they would like to join and then they would be sent an invite via email. A reminder was also sent the day before the next meeting. Parents would be included in any email communication and resources; however, they were not encouraged to join the meeting.Meeting etiquette and ground rules were explained at the beginning of the meeting and with each new attendee, in order to establish trust and

ASCAT2022
34 | 2022; 6:S1
inclusion. Following the meeting, a summary and any material used was circulated to the whole group.Results: 13 meetings January- November 2021 (Initially weekly, how-ever we changed to monthly over the summer as demand dropped off).Subjects covered include: COVID-19 & dealing with anxiety, stress, Low mood/dealing with sadness, Vaccine safety, fatigue, returning to school, Paintalking to people about your diagnosis.Attendance was variable – with the maximum being 6 young people. A short survey was circulated to try and identify what was working, what needed to change and to have input from the YP on content. As a result of this feedback, we moved to a different day of the week and a later session time.Summary: small numbers YP would regularly attend, parents commented they looked forward to this group, which we took to be a sign of success. Other attendees, joined sporadically on 1 or more occasions. We found the YP to be engaged. Most were happy to say hello and introduce them-selves on camera but would then prefer the camera off. We didn’t have anyone comment on difficulties with access, many used their smartphone devices. Session preapration was time consuming, but as we progressed, we became more efficient at re-using material and needing less ‘team brief’ time as we understood how the sessions ran. It is an expensive service, professional time wise. However, we haven’t compared to using a clinic environment or education room on site, which would take more organising and possibly less availability. There are no transport cost or travel time, so equitable for allConclusion: This has been a valuable service for a small cohort
References1. BMJ 2021;372:n614 Mental health of children and young people
during pandemic https://doi.org/10.1136/bmj.n6142. Millar, R. Quinn, N. Cameron, J. Colson, A. (2020). Considering the
evidence of the impacts of lockdown on the mental health and wellbe-ing of children and young people within the context of the individual, the family, and education. Glasgow: Mental Health Foundation
3. Jeffery, M., Lereya, T., Edbrooke-Childs, J., Deighton, J., Tait, N. & Cortina, M. A. (2021). Emerging evidence (Issue 8): coronavirus and children and young people’s mental health. Evidence Based Practice Unit, London.
P134 THE AREAL PROJECT: A VIRTUAL REALITY APPLICATION TO SUPPORT THE THERAPEUTIC EXPERIENCE AND THERAPY EDUCATION OF ADULT PATIENTS WITH THALASSEMIA AND SICKLE CELL DISEASE
Megale, V1; Casale, M2; Colombatti, R3; Pinto, V4; Baido, I5; Brentan, C4; Forni, G4; Forte, M2; Grieco, F2; Munaretto, V5; Perrotta, S2; Putti, M5; Robello, G4
1Softcare Studios, Rome, ITALY; 2Università degli Studi della Campania Luigi Vanvitelli, Napoli, ITALY; 3Azienda Ospedale-Università di Padova, Padova, ITALY; 4E.O. Ospedali Galliera, Genova, Genova, ITALY; 5Azienda Ospedaliera-Università di Padova, Padova, ITALY
Background: The chronic transfusion regimen consists of procedures that take time and that significantly impact the quality of life of patients with thalassemia or sickle cell disease; sometimes these procedures are carried out in uncomfortable environments where the patient tries with his own means to get distracted or “estranged”.Therefore, the need to pay attention not only to the strictly physical needs but also to the psychological, emotional and social needs of patients emerges with the aim of recovering valuable time “monopo-lized” by therapy by enriching the therapeutic path with activities capa-ble of involving more the person.Aims: From this need and the opportunities offered by the digital tech-nologies available today, the AREAL project was born, developed by the Italian startup Softcare Studios in partnership with the pharma company Novartis. AREAL consists of a gaming experience enjoyed in virtual reality thanks to the use of suitable VR viewers, designed in collabora-tion with medical staff to enhance the time spent in the hospital during the transfusion routine of young adult and adult patients.Methods: AREAL consists of three gaming activities developed to immerse the patient in a sensorially stimulating scenario different from the hospital one in which the user is forced to spend his time, and designed to stimulate cognitive skills (memory, reflexes, attention, auditory processing and visual, logic and association, spatial naviga-tion), providing added value to the patient’s engagement / entertainment and compensating for the near physical immobility required during the
transfusion / medical procedure (Figure 1). AREAL also integrates ele-ments of patient education in therapy and recommended lifestyle habits to promote their well-being, and is equipped with a social functionality thanks to which patients can play together with other patients in the same hospital or between hospitals. different, laying the foundations for building a delocalized patient community.Results: AREAL is currently in use in 3 pilot hospital centers in Italy, Padova, Genova, Napoli, where, over a period of 4 months, evidence will be collected on its impact on the therapeutic experience of patients, usability will be assessed and the integration in the staff work routine will be refined. doctor, with the ultimate goal of extending the concept of “care” not only to clinical but also emotional and psychological aspects during all the time spent in the hospital.
Figure 1. Example of different scenarios of the virtual reality
References1. Colombatti R, Casale M, Russo G.Disease burden and quality of life
of in children with sickle cell disease in Italy: time to be considered a priority.Ital J Pediatr. 2021 Jul 29;47(1):163.
2. Current challenges in the management of patients with sickle cell disease - A report of the Italian experience.Russo G, De Franceschi L, Colombatti R, Rigano P, Perrotta S, Voi V, Palazzi G, Fidone C, Quota A, Graziadei G, Pietrangelo A, Pinto V, Ruffo GB, Sorrentino F, Venturelli D, Casale M, Ferrara F, Sainati L, Cappellini MD, Piga A, Maggio A, Forni GL. Orphanet J Rare Dis. 2019 May 30;14(1):120.
3. Pinto VM, Poggi M, Russo R, Giusti A, Forni GL. Management of the aging beta-thalassemia transfusion-dependent population - The Italian experience. Blood Rev. 2019 Nov;38:100594.
P135 THE CLINGEN HEMOGLOBINOPATHY VARIANT CURATION EXPERT PANEL
Coralea Stephanou, C1; Petros Kountouris, P1; Carsten W Lederer, C1; Celeste Bento, C2; Cornelis L Hartveld, C3; Jan Traeger-Synodinos, J4; John S Waye, J5; Zhiyu Peng, Z6; Irene Fylaktou, I4; Hashim Halim-Fikri, H7; Tamara T. Koopmann, T3; Landry Nfonsam, L5; Jun Sun, J6; Franck Nzengu-Lukusa, F8; Michael Angastiniotis, M9; Catherine Badens, C10; Bertha Ibarra Cortes, B11; Johan T. den Dunnen, J3; Jacques Elion, J12; Suthat Fucharoen, S13; Kyriaki Michailidou, K1; Thessalia Papasavva, T1; Antonio Piga, A14; Raj Ramesar, R15; Swee Lay Thein, S16; Léon Tshilolo, L8; Zilfalil Bin Alwi, Z7; Marina Kleanthous, M1
1The Cyprus Institute of Neurology and Genetics, Cyprus School of Molecular Medicine, Nicosia, CYPRUS; 2Centro Hospitalar e Universitário de Coimbra, Coimbra, PORTUGAL; 3Leiden University Medical Center, Leiden, NETHERLANDS; 4National and Kapodistrian University of Athens, Athens, GREECE; 5Hamilton Health Sciences, McMaster University, Hamilton, CANADA; 6BGI Genomics, Shenzhen, Guangdong, CHINA; 7Universiti Sains Malaysia, Penang, MALAYSIA; 8Centre Hospitalier Monkole, Kinshasa, DRC; 9Thalassaemia International Federation, Nicosia, CYPRUS; 10Aix-Marseille Université, Marseille, FRANCE; 11Universidad de Guadalajara, Guadalajara, MEXICO; 12Université Paris Diderot, Paris, FRANCE; 13Mahidol University, Nakhon Pathom, THAILAND; 14Università degli Studi di Torino, Turin, ITALY; 15University of Cape Town, Cape Town, SOUTH AFRICA; 16National Heart, Lung and Blood Institute (NHLBI), Bethesda, UNITED STATES
Background: Accurate and consistent interpretation of sequence vari-ants is integral to the delivery of safe and reliable diagnostic genetic ser-vices. To standardize the interpretation process, in 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published a joint guideline based on different lines of evidence for the classification of sequence variants in Mendelian diseases. The generality of this guideline necessitates the application of expert judgment when evaluating and weighing evidence for variant interpretation. The Clinical Genome Resource (ClinGen) assembles Variant Curation Expert Panels (VCEPs) to perform gene- and

January 26-28, 2022 Virtual Conference
| 2022; 6:S1 35
disease-specific modifications of the ACMG/AMP framework. The ClinGen Hemoglobinopathy VCEP was created collaboratively between the ITHANET portal and the Global Globin Network of the Human Variome Project towards comprehensive annotation of all variants related to hemoglobinopathies.Aim: The adaptation of the ACMG/AMP variant interpretation guide-lines of use in hemoglobinopathies.Methods: The Hemoglobinopathy VCEP focuses on the review and anno-tation of variants located in the globin gene clusters, namely α-globin locus (NG_000006), which includes genes HBA1, HBA2 and HBZ, and β-globin locus (NG_000007) which includes genes HBB, HBD, HBG1, HBG2 and HBE and the regulatory element LCRB. Using a consensus approach and guidance by the ClinGen Sequence Variant Interpretation Working Group, the Hemoglobinopathy VCEP has prepared a pre-final version of the specified ACMG/AMP criteria for hemoglobinopathies.Results: The Hemoglobinopathy VCEP developed disease-specific rules for sequence variant classification based on evidence criteria that assess variant frequency, variant types and disease causality, protein domains and mutational hotspots implicated in disease, clinical manifestations, segregation, in silico predictions and functional evidence.Conclusions: For the first time, the Hemoglobinopathy VCEP will pro-vide a standardised classification of the pathogenicity of variants related to hemoglobinopathies. The Hemoglobinopathy VCEP specifications were approved by ClinGen in April 2021 (Step 2 approval), which initi-ated the process of further validation and adaptation with known globin gene variants in a pilot study (toward Step 3 approval).
References1. Kountouris P et al, Human Mutation 2021, doi: 10.1002/humu.24280
P136 THE UPTAKE OF VACCINATIONS IN ADULTS WITH SICKLE CELL DISEASE RECEIVING CARE AT THE CAMBRIDGE UNIVERSITY HOSPITALS NHS FOUNDATION TRUST
Kucharczak, J1; Garcia, V2; Jolley, R2; Mahajan, S2; Auturally, R2; Thomas, S3; Besser, M2
1University of Cambridge, School of Clinical Medicine, Cambridge, UNITED KINGDOM; 2Department of Haematology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UNITED KINGDOM; 3East of England Health Protection, UK Health Security Agency, Cambridge, UNITED KINGDOM
Those affected by sickle cell disease have an increased susceptibility to infection by encapsulated bacteria and hepatitis B virus due to reduced splenic function and increased likelihood of receiving blood transfu-sions. Sickle cell disease patients are also more likely to suffer from complications, such as vaso-occlusive crises or acute chest syndrome, following infection with influenza or COVID-19. Standards for Clinical Care of Adults with Sickle Cell Disease in the UK (2018) outline that that those with sickle cell disease are recommended to be vaccinated against invasive pneumococcal disease, Haemophilus influenza type B, Neisseria meningitis types ACWY and type B, hepatitis B, and influenza. These patients are also recommended to have their hepatitis B immunity reviewed annually and to receive a hepatitis B vaccination booster if hepatitis B surface antibody (HBsAb) levels are less than 100mIU/ml. According to the Standards, hospital staff is advised to remind and check with the patients’ primary care teams whether these vaccinations have been administered.In this audit, we examined the records of 64 patients with sickle cell disease who receive regular care at the Cambridge University Hospitals NHS Foundation Trust. We collected data on the uptake of the pneu-mococcal conjugate vaccine (PCV13), pneumococcal polysaccharide vaccine (PPV23 or Pneumovax) within 5 years, two doses of Meningitis B vaccine, Meningitis ACWY vaccine (MenACWY), Haemophilus influenzae type b vaccine (Hib/MenC), influenza vaccine within one year, hepatitis B vaccine (HepB), whether HBsAb levels have been reviewed within one year, HepB booster if HBsAb levels were less than 100mIU/ml, and two doses of COVID-19 vaccine. These records were obtained from electrical medical records provided by patients’ general practitioners. Data collection took place from 23rd September to 4th November 2021.The uptake of vaccinations was 67.4% for PCV13, 61.0% for PPV23 or Pneumovax within 5 years, 75.0% for Hib/MenC, 45.3% for MenACWY, 42.2% for the first dose of MenB and 29.3% for the sec-ond dose of MenB, 54.7% for influenza within one year, 75.0% for HepB, 71.9% for the first dose of the COVID-19 vaccine, and 68.3%
for the second dose of the COVID-19 vaccine. 43.8% had their HbsAB reviewed and 20.0% received a HepB booster following HBsAb levels of less than 100mIU/ml.The uptake levels for the recommended vaccinations are lower than expected in our hospital trust. The COVID-19 pandemic has highlighted the effect of health inequalities and the uptake of the vaccination pro-gramme by patients of different ethnicities. During our patient support group, patients identified the Tuskegee syphilis experiment as one of the reasons why there is still distrust of the medical profession by those with Afro-Caribbean heritage. Beyond directed patient education, more com-munication is needed with the primary care teams to raise awareness of which vaccinations are required for sickle cell patients. Certain vacci-nations, such as MenACWY and MenB were only introduced in 2015, meaning that some general practitioners may be still unaware of their necessity in adults with sickle cell disease.
References1. Standards for Clinical Care of Adults with Sickle Cell Disease in the
UK, 2nd Edition (2018)
P137 WHAT’S IN A NAME? PATIENTS WANT US TO RETHINK ‘SICKLE CELL DISEASE’
Otasowie, C1; Pearse, A2; Gilmour-Hamilton, C2; Dasaolu, F2; Fordwor, K2; Sinclair, M3; Igbineweka, N4; Njoku, S5; Hayes, S2; Atoyebi, W2; Roy, N2
1University of Oxford, Oxford, UNITED KINGDOM; 2Oxford University Hospitals NHS Foundation Trust and University of Oxford, John Radcliffe Hospital, Oxford, UNITED KINGDOM; 3Milly Sinclair Associates Ltd, Stroud, UNITED KINGDOM; 4Imperial College London, London, UNITED KINGDOM; 5SOAS University of London, London, UNITED KINGDOM
Racism impacts all aspects of patient care, not least for those with Sickle Cell Disease (SCD). Minimal research exists in this area, despite being long identified as a compelling issue for patients [1], and highlighted as a “Top 10” Priority in a workshop during ASCAT 2019. In response, we conducted a series of workshops to explore how race and racism impacts the quality of care in the SCD community and to identify action-able areas of change. This comprised three 90 minute sessions held over Zoom in May, June and July 2021. Attendees included people living with SCD, doctors, nurses, medical students, and members of the local community.Outcomes broadly centred around the themes of medical education; public education; and ways of better supporting the SCD patient com-munity. However, we were struck by a rich discussion on how the name ‘Sickle Cell Disease’ may, in itself, influence the mindset of caregivers and perpetuate stigma in both clinical and social settings.The term ‘Sickle Cell Anaemia’ traditionally refers to HbSS only, though OMIM uses it to cover all genotypes [2]. Meanwhile the ICD-10 classi-fication code D57 refers to ‘Sickle Cell Disorders’, with sub-categories using ‘Disease’ and divided by genotype [3].From the workshops, the patient group shared a strong consensus view that Sickle Cell Disorder, rather than ‘Disease’, would be preferable as a more neutral term of reference.To verify the results of the workshop in an independent sample, a questionnaire was circulated to a small SCD patient support group (12 individuals) to assess their preference for one or other label. 9/12 (75%) preferred ‘Disorder’, one preferred ‘Disease’ and two expressed no preference. Which label was used by patients themselves, their fam-ilies and friends, and their medical teams was important to patients, with 67% expressing a wish that this issue be pursued further. Interestingly, the acceptability of Disorder/Disease showed some vari-ation by the patient’s country of birth. These findings point towards a pressing need for increased sensitivity around the language used to talk about illness, particularly where the condition has been histori-cally stigmatised.
References1. Igbineweka et al. Medical science must address health disparities
amongst different ethnic groups. Nat Hum Behav. 2021.2. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins
University, Baltimore, MD. MIM Number: 603903: 03/08/2021:. World Wide Web URL: https://omim.org/
3. World Health Organization (WHO). The ICD-10 Classification of Mental and Behavioural Disorders. World Health Organization. 1993.

ASCAT2022
36 | 2022; 6:S1
P138 WHITE PAPER: ASSESSMENT OF INTERNATIONAL SICKLE CELL DISEASE GUIDELINES IN HIGH-, MIDDLE-, AND LOW-INCOME COUNTRIES
Suthar, N1; Halpern, T2; Hsu, L3
1University of Illinois at Chicago College of Medicine, Chicago, UNITED STATES; 2University of Illinois at Chicago School of Public Health, Chicago, UNITED STATES; 3University of Illinois at Chicago Department of Pediatric Hematology, Chicago, UNITED STATES
Sickle cell disease (SCD) is a common genetic condition defined by abnor-malities in hemoglobin, an oxygen carrying protein in red blood cells. About 5% of the world’s population carries genes for hemoglobin disor-ders, with the highest prevalence in countries in Subsaharan Africa (SSA). SCD causes significant morbidity, mortality, and economic burden in the countries and populations affected. Various countries have established national guidelines for SCD management, which serve to standardize care and improve patient outcomes. Ten such guidelines were identified for compilation and analysis in this paper as of summer 2021. Qualitative analysis of the guidelines has identified the priorities of each government and medical institutions’ stance on best practices across high-, middle-, and low-income countries. The analysis identified that care for acute complications of SCD is more frequently endorsed in the guidelines of
low-and-middle income countries (LMIC), which also have higher disease burden, over high-income countries (HIC). Chronic preventative care for SCD disease, which is shown to be an evidence-based effective approach to improve patient outcomes, is more frequently recommended in guide-lines of HIC over LMIC. Furthermore, national newborn screening guide-lines are scarce in the countries with high prevalence rates and adolescent mortality. While most countries recommend screening programs, LMIC have limited recommendations for newborn preventative measures. Incorporation and prioritization of preventive care guidelines in LMIC may serve as a path to inform sustainable nationwide interventions to improve patient outcomes for SCD. Implementation of these guidelines would contribute to preventing acute complications and improving mor-bidity and mortality for individuals with SCD. In addition, prioritization and inclusion of preventive and chronic care measures in healthcare ser-vices may mitigate the economic burden of SCD in LMIC.
References1. Yawn et al. JAMA 2014. 312;10.2. Piel et al, PLOS Medicine 2013; 10:73. Kato et al, Nat Rev Dis Primers 2018; 15:4.4. Ryan et al. Br J Haematol 2010; 149;1.5. Alli et al. S Afr Med J 2014; 104;11.