targeting reactive astrogliosis by novel biotechnological strategies
TRANSCRIPT

Biotechnology Advances 30 (2012) 261–271
Contents lists available at ScienceDirect
Biotechnology Advances
j ourna l homepage: www.e lsev ie r.com/ locate /b iotechadv
Research review paper
Targeting reactive astrogliosis by novel biotechnological strategies
Anna Maria Colangelo b,⁎, Giovanni Cirillo a, Maria Luisa Lavitrano c, Lilia Alberghina b, Michele Papa a,⁎⁎a Laboratory of Morfology of Neural Networks, Department of Medicina Pubblica Clinica e Preventiva, Second University of Napoli, 80138 Napoli, Italyb Laboratory of Neuroscience “R. Levi-Montalcini”, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Milano, Italyc Department of Surgical Sciences and Intensive Therapy, University of Milano-Bicocca, 20052 Monza, Italy
Abbreviations: CNS, Central Nervous System; GFAP,EAAT1, excitatory amino acid transporter 1; GLAST, gluGLT, glutamate transporter; GSH, glutathione; i.t., intFactor; BB14®, Nerve Growth Factor-like peptide; Mchronic constriction injury; SNI, Spared Nerve Injury.⁎ Correspondence to: A.M. Colangelo, Department of B
Laboratory of Neuroscience “R. Levi-Montalcini”, Univedella Scienza 4, 20126 Milano, Italy. Tel.: +39 02 64483⁎⁎ Correspondence to: M. Papa, Department of MPreventiva, Institute of Human Anatomy, Second UniveItaly. Tel./fax: +39 081 296636.
E-mail addresses: [email protected] ([email protected] (M. Papa).
0734-9750/$ – see front matter © 2011 Elsevier Inc. Aldoi:10.1016/j.biotechadv.2011.06.016
a b s t r a c t
a r t i c l e i n f oAvailable online 5 July 2011
Keywords:Neuro-glial networkNerve injuryNGF-like peptideReactive astrocytosisSynaptic homeostasis
Neuroglial cells are fundamental for control of brain homeostasis and synaptic plasticity. Decades of pathologicaland physiological studies have focused on neurons in neurodegenerative disorders, but it is becomingincreasingly evident that glial cells play an irreplaceable part in brain homeostasis and synaptic plasticity. Animalmodels of brain injury and neurodegenerative diseases have largely contributed to current understanding ofastrocyte-specific mechanisms participating in brain function and neurodegeneration. Specifically, gliotransmis-sion (presence of glial neurotransmitters, and their receptors and active transporters), trophic support (release,maturation and degradation of neurotrophins) andmetabolism (production of lactate and GSH components) arerelevant aspects of astrocyte function in neuronal metabolism, synaptic plasticity and neuroprotection.Morpho-functional changes of astrocytes andmicroglial cells after traumatic or toxic insults to the central nervous system(namely, reactive gliosis) disrupt the complex neuro-glial networks underlying homeostasis and connectivitywithin brain circuits. Thus, neurodegenerative diseases might be primarily regarded as gliodegenerativeprocesses, in which profound alterations of glial activation have a clear impact on progression and outcomes ofneuropathological processes. This reviewprovides anoverviewof current knowledgeof astrocyte functions in thebrain andhow targeting glial-specific pathwaysmight ultimately impact the development of therapies for clinicalmanagement of neurodegenerative disorders.
Glial Fibrillary Acidic Protein;tamate-aspartate transporter;rathecal; NGF, Nerve GrowthMPs, metalloproteinases; CCI,
iotechnology and Biosciences,rsity of Milano-Bicocca, piazza536; fax: +39 0264483519.edicina Pubblica Clinica ersity of Napoli, 80100 Napoli,
.M. Colangelo),
l rights reserved.
© 2011 Elsevier Inc. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2612. Astrocytes in synapse formation and plasticity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2623. Glial activation and neurodegenerative pathologies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2634. Neuroglia, neuroinflammation and neurodegeneration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2645. Neurotrophins, astrogliosis and neurodegenerative diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2666. Neurotrophins and reactive gliosis in ophthalmic pathologies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2687. Neurotrophin-derived drug candidates for neuroprotection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2698. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 269Acknowledgments. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 269References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 269
1. Introduction
The relevance of glial cells to the central nervous system (CNS)function and plasticity started to be noticed relatively recently, about20 years ago. Until then, in fact, astrocytes and other cells of the gliallineage, such as oligondendrocytes and microglia, were believed to bestructural, electrically silent elements, lacking transmitter receptorsand transporters, with the main function of holding neurons together(“brain glue”). This view of “embedded elements among neurons”was challenged by a series of in vitro and in vivo studies that clearly

262 A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
demonstrated that astrocytes and neurons share almost the same setof ion channels, receptors and transporters, allowing to conclude thatglial cells sense and respond to neuronal activity.
The complexity of cellular circuitries in the CNS, an unicum in livingsystems, resides in the dynamic changes of neural connections (i.e.synaptic plasticity). This function, through a highly developed blood–brain barrier, is entrusted to glial cells. In particular, astrocytes providefor the micro architecture of the gray matter by forming relativelyindependent structural domains. Within these domains, each astrocytecovers synaptic contacts (tripartite synapse) and establishes connec-tions with neuronal membranes and blood vessels. Through gapjunctions, astrocytes of distinct domains create an astroglial syncitium,thus providing a glial information-transfer system, a pathway for rapidintercellular diffusion and long-range signaling.
It is now clear that glial cells are fundamental for control of brainhomeostasis and constitute an intrinsic brain defense system. In fact,they possess an evolutionary conserved program of activation inresponse to brain damage. Microglial cells represent the residentmacrophages of the CNS. Like astrocytes, microglial cells disseminatethroughout the brain and occupy well-defined territorial domains,which do not overlap with neighboring microglia. Resident microgliahave small somatas and multiple fine processes constantly moving andscanning the microenvironment of their domains (Davalos et al., 2005;Nimmerjahn et al., 2005), thus providing the first line of defense assensors of nervous system injury (Hanisch and Kettenmann, 2007;Ransohoff and Perry, 2009). Microglia and astrocytes become activated(reactive gliosis) in response to several CNS insults. Astrocytic activationoccurs throughmechanisms involving specific structural and functionalalterations, such as hypertrophy and increased expression of glialfibrillary acidic protein (GFAP) (Pekny and Nilsson, 2005). Phenotypicchanges affecting reactive astrocytes impair neuronal network functionby producing or boosting neuronal degeneration as “non cell-autono-mous diseases” (Lobsiger and Cleveland, 2007).
Decades of pathological and physiological studies have focused onneuronal abnormalities in brain disorders, but it is becomingincreasingly evident that astrocytes are also important players. Ourunderstanding of the normative biology of astrocytes has beenfostered by the development of animal models in which astrocyte-specific proteins and pathways have been manipulated. Models ofneurodegenerative diseases have also led to current knowledge ofglial function in neurodegenerative pathologies. Thus, a comprehen-sive understanding of mechanisms contributed by astrocytes appearsto be relevant for the development of targeted therapies for clinicalmanagement of neurodegenerative disorders.
2. Astrocytes in synapse formation and plasticity
Several studies support the key role for astrocytes from synapticformation to metabolic support and neurotransmitter release.Astrocytes represent the key elements in synaptogenesis. It hasbeen reported that addition of astrocytes to in vitro neuronal culturestriggers a significant increase in synapse formation (Pfrieger andBarres, 1996): through production of cholesterol (Nieweg et al., 2009)and release of trophic factors, astrocytes are crucial for synapsematuration and maintenance (Pfrieger, 2009); through productionand release of thrombospondins 1 and 2, they promote synaptogen-esis suggesting their crucial role in post-lesion synaptic plasticity,remodeling and regeneration.
Astrocytes are also essential for neuronal energy metabolism andglutathione synthesis. Astrocytic endfeet contact neighboring capil-laries through perivascular processes, thus forming a functional linkbetween neurons and blood vessels. An increase in neural activitywithin an astrocytic domain results in the release of vasoactivesubstances (arachidonic acid metabolites) (Hirase, 2005) thatpromote dilation in nearby arterioles (Takano et al., 2006). Metabolicsupport to neurons is achieved through the astrocyte–neuron lactate
shuttle. Astrocytes convert glucose to lactic acid, which is then takenup into neurons and converted to pyruvate for energy metabolism(Danbolt, 2001). Thus, astrocytes play a central role in couplingsynaptic plasticity and glucose metabolism (neurometabolic cou-pling) through mechanisms involving the sodium-coupled glutamatere-uptake, which stimulates aerobic glycolysis and production/releaseof lactate (Magistretti, 2006). The metabolic function of astrocytes issupported by a large number of studies. More recently, astrocyticglycogen breakdown and astrocyte-neuron lactate transport havebeen demonstrated to be essential for long-term memory formation,and for the maintenance of long-term potentiation (LTP) of synapticstrength (Suzuki et al., 2011).
The different metabolic requirements of astrocytes and neurons(high and low glycolytic, respectively) is based upon their differentenzymatic assets. Neurons have very low activity of the glycolysis-promoting enzyme PFKFB3 (6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase, isoform-3), which is constantly degraded by the E3ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C)–Cdh1. This mechanism appears to be ultimately linked to the neuronalneed to redirect glucose metabolism to the pentose-phosphate (PPP)pathway to generate NADPH(H+), a necessary cofactor in theregeneration of reduced glutathione (GSH) (Herrero-Mendez et al.,2009; Bolaños et al., 2010). Neurons are particularly vulnerable toreactive oxygen (ROS) and nitrogen species (RNS), and also have lowactivity of γ-glutamyl cysteine synthetase, the limiting enzyme forGSH synthesis. Therefore, neuronal downregulation of glycolysis andglucose metabolism through the PPP pathway seem to be required tomaintain their antioxidant status (Herrero-Mendez et al., 2009).
Control of extracellular ion concentrations and water homeostasisis also a well recognized function of astrocytes. Extracellular K+
accumulated from neural activity (Kofuji and Newman, 2004) islowered by inward rectifying K+ channels (Kir-channels) controllingion concentration; water homeostasis occurs through aquaporin-4type expression on perivascular endfeet and perisynaptic processes.
Another key function of astrocytes is, moreover, the removal ofneurotransmitters that are released by active neurons, in particularglutamate (Fig. 1A). Glutamate, themain excitatory neurotransmitter inCNS, is also the most powerful neurotoxin when it accumulates in theextracellular space. From the bulk of glutamate released during synaptictransmission, about 20% is re-captured by postsynaptic neurons, theremaining 80% is taken up by perisynaptic astrocytes (Rothstein et al.,1996). Astrocytes selectively express two glutamate transporters,EAAT1 and EAAT2 (in rodents known as GLAST and GLT1, respectively)(Rothsteinet al., 1994; Chaudhry et al., 1995; Lehre et al., 1995;Miltonetal., 1997). Besides their role in glutamate uptake, astrocytes are alsocrucial for the recovery of glutamate in presynaptic terminals throughthe glutamate–glutamine shuttle system. Astrocytic cytosolic glutamateis converted by the astrocytic-specific glutamine synthetase into thenon toxic glutamine, that is released in the extracellular space and afterentering the neuronal compartment is converted into glutamate toreconstitute the neurotransmitter pool.
The cystine–glutamate antiporter system (xCT) is also functionallyrelated to the role of astrocytes in regulating extracellular glutamateand providing intracellular cystine for neuronal GSH homeostasis.Compelling evidence suggests that altered glutamate uptake byastrocytes is directly involved in the pathogenesis of some neurolog-ical disorders. We also reported a decrease of glial glutamatetransporters after peripheral nerve injury, together with a decreaseof GSH levels and a functional block of the xCT system by extracellularglutamate accumulation (Cirillo et al., 2011). These data are inagreement with the increase of xCT under conditions of glutamateoxidative stress and GSH depletion (Dun et al., 2006; Lewerenz et al.,2006).
The ability of astrocytes to release chemical transmitters(gliotransmitters) is also fundamental for their involvement inneuro-glial networks. Among gliotransmitters, astrocytes can release

Fig. 1. Schematic representation of a tripartite synapse in “normal” (A) and under conditions of reactive astrocytosis (B). Excitatory aminoacids (EAs) (glutamate and glycine) arereleased in synaptic clefts and retrieved by neuronal and mainly glial glutamate and glycine transporters (GTs). The xCT transporter, through the cystine–glutamate antiporterexchange, supplies the cysteine for GSH synthesis. In normal conditions, the turn-over of intermediate filaments, in particular GFAP, and GTs occurs through the activity of calpain I, aCa2+-dependent protease. B) Reactive astrocytosis determines an increase of calpain I activity and degradation of GTs, and failure of glutamate uptake. Extracellular glutamatespillover to perisynaptic spaces contributes to NMDA receptors sensitization and changes of synaptic efficacy. The increase of extracellular glutamate also contributes toexcitotoxicity through a functional block of the xCT system and failure of GSH production.
263A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
glutamate (Haydon, 2001), ATP, D-serine, GABA and other moleculesthrough a Ca2+-dependent exocytosis or diffusion through eitherlarge pore channels [e.g., P2X7 receptors or hemichannels (Ye et al.,2003)], transporters (glutamate transporters), or the cystine–glutamate antiporter system. This function strengthens the doublerole of astrocytes in the tripartite synapse: astrocytes sense neuronalactivity and neurotransmitter release through the expression ofneurotransmitters receptor on astrocytic membrane; on the otherhand, they modulate the efficacy of the synapse by releasinggliotransmitters that in turn might modulate the strength ofinhibitory or excitatory synaptic transmission through activation ofneuronal receptors. Moreover, each astrocyte can reach thousands ofsynapses simultaneously, and the release of gliotransmitters mightlead to the synchronization of neuronal firing patterns (Allegriniet al., 2009). Propagation of glutamate transmission through theastrocytic syncitium occurs through two Ca2+-mediated systems:one involving gap junctions (via connexin-43), the other involvingthe paracrine release of ATP (Simard and Nedergaard, 2004).
Given the complexity of neuro-glial networks in CNS homeostasisand function, a systems biology approach of neurodegeneration(Alberghina and Colangelo, 2006) might be crucial to a comprehen-sive understanding of the glia–neuron interplay in synaptic function,and how changes of metabolic fluxes might influence the complexinteractions between neuronal and glial compartments and lead tothe structural/functional modifications underlying neuro-glial rear-rangements during the degeneration process.
3. Glial activation and neurodegenerative pathologies
Glial cells play a crucial role in neurological diseases bydetermining the progression and outcome of the neuropathologicalprocess. Insults to the nervous system trigger a complex and multi-stage activation of microglia which results in both phenotypic andfunctional changes (activated microglia). Under pathological situa-tions, these cells migrate to and surround damaged or dead cells,clearing cellular debris from the area, similarly to the macrophages ofthe peripheral immune system (Fig. 2). Activated microglia undergofundamental morphological changes from a ramified phenotype toactivated amoeboid cells, and up-regulate a variety of surfacereceptors (includingMHC and complement receptors) and the releaseof pro-inflammatory factors (cytokines, reactive oxygen species —
ROS, nitric oxide — NO), which contribute to neuronal dysfunctionand cell death, ultimately creating a vicious cycle.
Current knowledge indicates that early stages of neurodegenera-tive processes are associated with neuroinflammation, involvingmicroglial cells and subsequent activation of astrocytes (Fig. 3).Astrocytes seem to be involved in several mental disorders (Narita etal., 2006; Machado-Vieira et al., 2009; Musholt et al., 2009), as well asin neurodegenerative diseases (Maragakis and Rothstein, 2006;Lobsiger and Cleveland, 2007) including Alzheimer's disease (AD)(Verkhratsky et al., 2010), amyotrophic lateral sclerosis (ALS)(Sheldon and Robinson, 2007), spinocerebellar ataxia type 1 (SCA1)(Giovannoni et al., 2007), Parkinson's disease (PD) (Saijo et al., 2009)
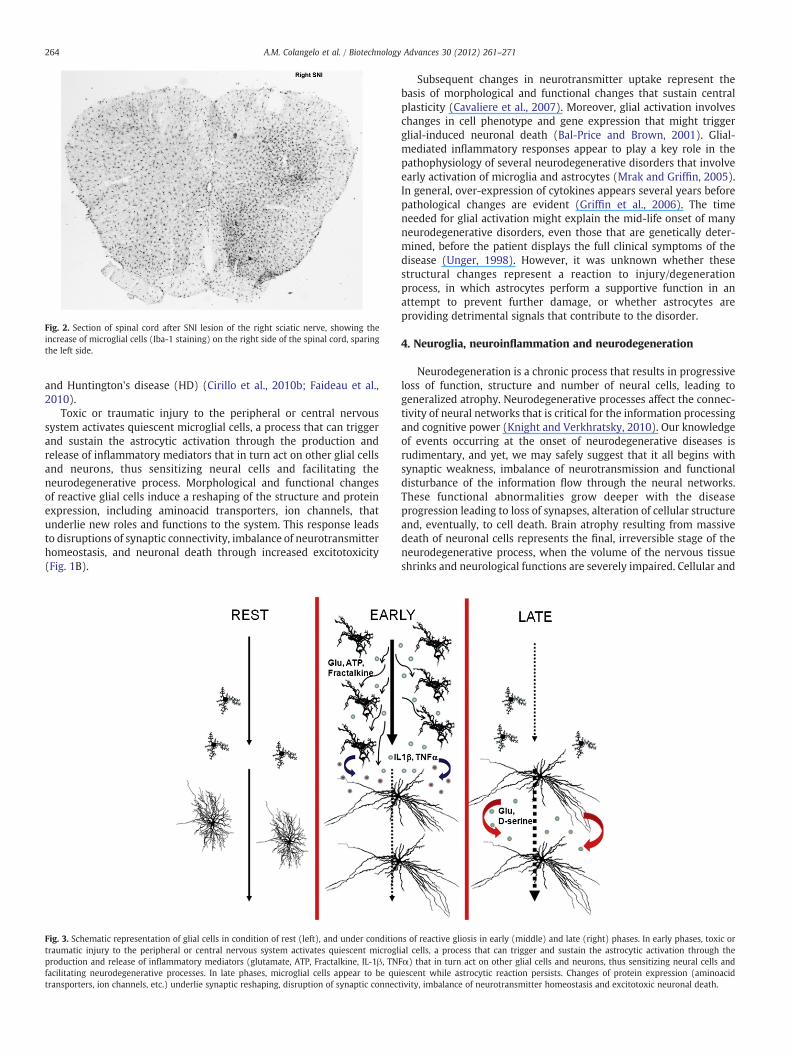
Fig. 2. Section of spinal cord after SNI lesion of the right sciatic nerve, showing theincrease of microglial cells (Iba-1 staining) on the right side of the spinal cord, sparingthe left side.
264 A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
and Huntington's disease (HD) (Cirillo et al., 2010b; Faideau et al.,2010).
Toxic or traumatic injury to the peripheral or central nervoussystem activates quiescent microglial cells, a process that can triggerand sustain the astrocytic activation through the production andrelease of inflammatory mediators that in turn act on other glial cellsand neurons, thus sensitizing neural cells and facilitating theneurodegenerative process. Morphological and functional changesof reactive glial cells induce a reshaping of the structure and proteinexpression, including aminoacid transporters, ion channels, thatunderlie new roles and functions to the system. This response leadsto disruptions of synaptic connectivity, imbalance of neurotransmitterhomeostasis, and neuronal death through increased excitotoxicity(Fig. 1B).
Fig. 3. Schematic representation of glial cells in condition of rest (left), and under conditiotraumatic injury to the peripheral or central nervous system activates quiescent microglproduction and release of inflammatory mediators (glutamate, ATP, Fractalkine, IL-1β, TNfacilitating neurodegenerative processes. In late phases, microglial cells appear to be qutransporters, ion channels, etc.) underlie synaptic reshaping, disruption of synaptic connec
Subsequent changes in neurotransmitter uptake represent thebasis of morphological and functional changes that sustain centralplasticity (Cavaliere et al., 2007). Moreover, glial activation involveschanges in cell phenotype and gene expression that might triggerglial-induced neuronal death (Bal-Price and Brown, 2001). Glial-mediated inflammatory responses appear to play a key role in thepathophysiology of several neurodegenerative disorders that involveearly activation of microglia and astrocytes (Mrak and Griffin, 2005).In general, over-expression of cytokines appears several years beforepathological changes are evident (Griffin et al., 2006). The timeneeded for glial activation might explain the mid-life onset of manyneurodegenerative disorders, even those that are genetically deter-mined, before the patient displays the full clinical symptoms of thedisease (Unger, 1998). However, it was unknown whether thesestructural changes represent a reaction to injury/degenerationprocess, in which astrocytes perform a supportive function in anattempt to prevent further damage, or whether astrocytes areproviding detrimental signals that contribute to the disorder.
4. Neuroglia, neuroinflammation and neurodegeneration
Neurodegeneration is a chronic process that results in progressiveloss of function, structure and number of neural cells, leading togeneralized atrophy. Neurodegenerative processes affect the connec-tivity of neural networks that is critical for the information processingand cognitive power (Knight and Verkhratsky, 2010). Our knowledgeof events occurring at the onset of neurodegenerative diseases isrudimentary, and yet, we may safely suggest that it all begins withsynaptic weakness, imbalance of neurotransmission and functionaldisturbance of the information flow through the neural networks.These functional abnormalities grow deeper with the diseaseprogression leading to loss of synapses, alteration of cellular structureand, eventually, to cell death. Brain atrophy resulting from massivedeath of neuronal cells represents the final, irreversible stage of theneurodegenerative process, when the volume of the nervous tissueshrinks and neurological functions are severely impaired. Cellular and
ns of reactive gliosis in early (middle) and late (right) phases. In early phases, toxic orial cells, a process that can trigger and sustain the astrocytic activation through theFα) that in turn act on other glial cells and neurons, thus sensitizing neural cells andiescent while astrocytic reaction persists. Changes of protein expression (aminoacidtivity, imbalance of neurotransmitter homeostasis and excitotoxic neuronal death.

265A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
molecular mechanisms involved in the development of neurodegen-erative processes are many. Conceptually, the neurodegeneration canbe viewed as a chronic and progressive failure of the brainhomeostasis that finally assumes toxic proportions leading to massivecell demise.
For many years, neurodegenerative diseases were considered to bea specific pathology of neurons. Recent years, however, havechallenged the neurocentric views and evidence demonstrating thepathological potential of neuroglia began to accumulate (Rossi andVolterra, 2009; Heneka et al., 2010). Astroglial changes in theprogression of neurodegenerative diseases are complex. It wasgenerally believed that neurodegeneration triggers reactive micro-and macro-gliosis; reactive astrocytes in turn contribute to neuroin-flammation, which is considered to be an important component ofneurodegenerative diseases (Lobsiger and Cleveland, 2007). More-over, morphological and functional changes in astroglia occurring atthe early stages of neurodegenerative processes, may result in fadingastroglial support that can affect synaptic transmission.
Neurodegenerative diseases represent major unmet challenges fortherapeutic interventions. Characterization and targeting of theprocesses that initiate specific disease pathologies and act primarilyat the level of neurons are clearly important areas for continuedinvestigation. The emerging evidence for both protective andpathogenic roles of microglia and astrocytes, and the activation ofcommon inflammation pathways in these cells in several neurode-generative diseases, supports the concept that glia-induced inflam-mation is an amplifier of the pathology (Glass et al., 2010; Bajramovic,2011). Although inhibition of neuroinflammation may not alter theunderlying cause of disease, it may reduce the production of factorsthat contribute to neurotoxicity, thereby resulting in clinical benefit.Further knowledge of the inducers, sensors, transducers, and effectorsof neuroinflammation can make possible the attainment of this goal.
Virchow's seminal descriptions of activated glial cells anticipatedby more than a century the current interest in roles of the innate andadaptive immune systems in diverse forms of neurodegenerativedisease. Direct evidence for inflammatory responses in AD wasdescribed nearly 20 years ago and subsequent studies have docu-mented inflammatory components in PD (Tansey and Goldberg, 2010;Chung et al., 2010), ALS, multiple sclerosis (MS) (Glass et al., 2010;van Noort et al., 2011), as well as in a growing number of otherpathologies of the nervous system. Although inflammation may nottypically represent an initiating factor in neurodegenerative disease,there is emerging evidence in animal models that sustainedinflammatory responses involving microglia and astrocytes contrib-ute to disease progression. A major unresolved question is whetherinhibition of these responses will be a safe and effective means forreversing or slowing the course of disease. To effectively address thisquestion, it will be necessary to learn more about how inflammatoryresponses are induced within the CNS and the mechanisms by whichthese responses ultimately contribute to the pathology.
Recently, our groups have focused on different experimentalmodels of reactive gliosis in rodents to better understand earlymorphological and functional glial changes underlying the responseof the CNS to injury and neurodegeneration. We have evidence ofmicroglial and astrocytic activation as early signs of the pathology in amousemodel of SCA1, in a rat model of toxic-induced HD, as well as inthe central response to peripheral nerve injury.
SCA1 is a dominantly inherited, progressive, neurodegenerativedisorder clinically characterized by ataxia and cranial motor neurop-athies due to expansion of a CAG repeat in the coding region of theSCA1 gene (Schols et al., 2004; Orr and Zoghbi, 2007), encoding forataxin-1 protein. Ataxin-1 is expressed throughout the brain, inPurkinje cells in cerebellum but also in non-neuronal cells, mainlyradial Bergmann glia (Servadio et al., 1995). We generated aconditional mouse model using the tetracycline-responsive gene(TET) system to control the expression of ataxin-1 (Giovannoni et al.,
2007) using a brain specific promoter to induce ataxin-1 throughoutbrain tissues. Our results indicate that expression of mutated ataxin-1was associated with the onset and progression of cerebellar motordeficits, together with a peculiar clustering of the glutamatetransporter in the cerebellar cortex and reactive astrocytosis (GFAPupregulation). These phenotypic and morpho-molecular changesappeared to be related to cerebellar dysfunction of both Purkinjecells and Bergmann glia. In this regard, neuronal degeneration mightbe the ultimate step of the neuropathology, when rescue from thedisease is no longer possible. Our findings provide evidence that thismouse model exhibits features of inflammatory pathology and that itmight be useful for studying the role of inflammatory pathways inCAG triplet diseases.
As observed in the cerebellum of SCA1mouse, astrocytic activation inthe striatum is a common reaction after toxic, ischemic or degenerativeprocesses and seems toplay a role in thedevelopmentofHD(Hickeyet al.,2008).HD is aneurodegenerative autosomicdominant inheriteddisorder,characterized, in humans, by involuntary choreiform movements andcognitive impairment caused by a CAG expansion in the coding region ofthe gene huntingtin (Landles and Bates, 2004). HD is characterized bymarkedstriatal atrophywithneuronal loss, astrocytic activationand intra-neuronal protein aggregates (Paulson and Fischbeck, 1996; Paulsen et al.,2006). Mechanisms underlying the selective neuronal death of theGABAergic medium spiny neurons (MSN) in the striatum still remainsunknown. One hypothesis suggests that the genetic defect may cause animpairment of energy metabolism that subsequently makes theseneurons more vulnerable to excitotoxic degeneration (Brouillet et al.,2005). 3-Nitropropionic acid (3NP), a suicide inhibitor of mitochondrialcomplex II succinate dehydrogenase has been shown to cause a HD-likesyndrome in rodents (Borlongan et al., 1997; Cirillo et al., 2010b) andprimates (Brouillet et al., 2005). We administered 3NP intra-peritoneallyon a subchronic schedule and after treatment we found an intensereactive astrocytosis in the striatum, and altered expression of the glialglutamate transporters, without clear sign of neuronal death. Our resultssupport the hypothesis that the onset and progression of motor deficitsare linked to the reactive astrocytosis and changes of glial glutamatetransporter expression, thus altering the glutamate uptake system andexposing neurons to glutamate excitotoxicity. These could represent earlyevents leading to neuronal death, which occurs only in the late phase ofneurodegenerative diseases.
Our recent studies also indicate that glial activation afterperipheral nerve injury is an important component of the neuropathicpain-like syndrome characterized by allodynia and thermal hyper-algesia (Cavaliere et al., 2007; Cirillo et al., 2010a). After peripheralnerve injury, a rapid glial response occurs within the CNS, as revealedby microglial activation and increased expression of GFAP inastrocytes in the spinal cord (Fig. 4) (Watkins et al., 2001;Raghavendra et al., 2002; Wieseler-Frank et al., 2004). Activation ofquiescent microglial cells in the spinal cord sustains the astrocyticactivation through the release of pro-inflammatory mediators(Watkins and Maier, 2003; Zhuang et al., 2005), such as interleukin-1β (IL-1 β) (Hashizume et al., 2000) and cyclooxygenase 2 (COX-2)(McMahon et al., 2005), which sensitize dorsal horn neurons (Ji andStrichartz, 2004) and facilitate pain transmission (Watkins and Maier,2003; Fellin and Carmignoto, 2004).
Themassive cytoskeletal rearrangement due to reactive astrocytosisseems to be sustainedby calpain I-dependentprocesses (Lee et al., 2000;Gray et al., 2006), a Ca2+-dependent protease that is highly activefollowing nerve injury and has several substrates, including membraneglutamate and glycine transporters (Li et al., 1996; Serbest et al., 2007)(Fig. 1). Since glial cells through glutamate/aspartate transporter EAAT1(also knownasGLAST) andEAAT2 (also knownasGLT1) are responsiblefor the majority of extracellular glutamate uptake, we investigatedchanges of glial glutamate transporters (GTs) expression followingreactive astrocytosis-induced nerve injury. Both neuronal and glialaminoacid transporter systems actively participate to synaptic plasticity
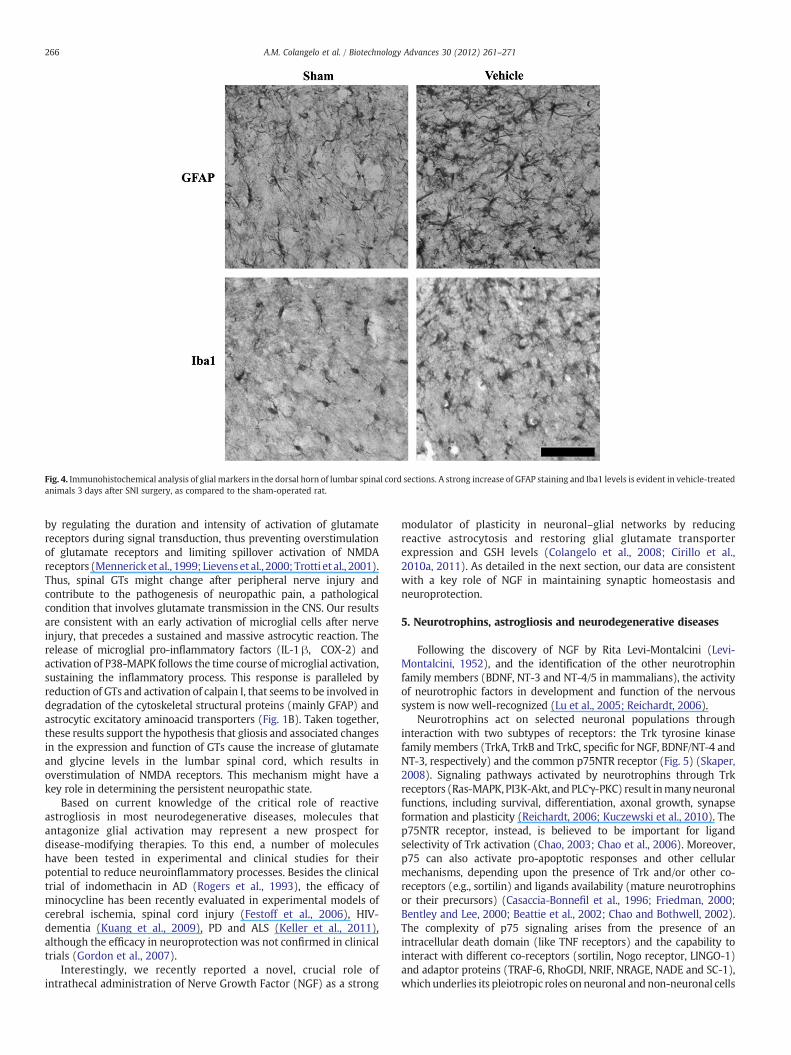
Fig. 4. Immunohistochemical analysis of glial markers in the dorsal horn of lumbar spinal cord sections. A strong increase of GFAP staining and Iba1 levels is evident in vehicle-treatedanimals 3 days after SNI surgery, as compared to the sham-operated rat.
266 A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
by regulating the duration and intensity of activation of glutamatereceptors during signal transduction, thus preventing overstimulationof glutamate receptors and limiting spillover activation of NMDAreceptors (Mennerick et al., 1999; Lievenset al., 2000; Trotti et al., 2001).Thus, spinal GTs might change after peripheral nerve injury andcontribute to the pathogenesis of neuropathic pain, a pathologicalcondition that involves glutamate transmission in the CNS. Our resultsare consistent with an early activation of microglial cells after nerveinjury, that precedes a sustained and massive astrocytic reaction. Therelease of microglial pro-inflammatory factors (IL-1 β, COX-2) andactivation of P38-MAPK follows the time course ofmicroglial activation,sustaining the inflammatory process. This response is paralleled byreduction of GTs and activation of calpain I, that seems to be involved indegradation of the cytoskeletal structural proteins (mainly GFAP) andastrocytic excitatory aminoacid transporters (Fig. 1B). Taken together,these results support the hypothesis that gliosis and associated changesin the expression and function of GTs cause the increase of glutamateand glycine levels in the lumbar spinal cord, which results inoverstimulation of NMDA receptors. This mechanism might have akey role in determining the persistent neuropathic state.
Based on current knowledge of the critical role of reactiveastrogliosis in most neurodegenerative diseases, molecules thatantagonize glial activation may represent a new prospect fordisease-modifying therapies. To this end, a number of moleculeshave been tested in experimental and clinical studies for theirpotential to reduce neuroinflammatory processes. Besides the clinicaltrial of indomethacin in AD (Rogers et al., 1993), the efficacy ofminocycline has been recently evaluated in experimental models ofcerebral ischemia, spinal cord injury (Festoff et al., 2006), HIV-dementia (Kuang et al., 2009), PD and ALS (Keller et al., 2011),although the efficacy in neuroprotection was not confirmed in clinicaltrials (Gordon et al., 2007).
Interestingly, we recently reported a novel, crucial role ofintrathecal administration of Nerve Growth Factor (NGF) as a strong
modulator of plasticity in neuronal–glial networks by reducingreactive astrocytosis and restoring glial glutamate transporterexpression and GSH levels (Colangelo et al., 2008; Cirillo et al.,2010a, 2011). As detailed in the next section, our data are consistentwith a key role of NGF in maintaining synaptic homeostasis andneuroprotection.
5. Neurotrophins, astrogliosis and neurodegenerative diseases
Following the discovery of NGF by Rita Levi-Montalcini (Levi-Montalcini, 1952), and the identification of the other neurotrophinfamily members (BDNF, NT-3 and NT-4/5 in mammalians), the activityof neurotrophic factors in development and function of the nervoussystem is now well-recognized (Lu et al., 2005; Reichardt, 2006).
Neurotrophins act on selected neuronal populations throughinteraction with two subtypes of receptors: the Trk tyrosine kinasefamily members (TrkA, TrkB and TrkC, specific for NGF, BDNF/NT-4 andNT-3, respectively) and the common p75NTR receptor (Fig. 5) (Skaper,2008). Signaling pathways activated by neurotrophins through Trkreceptors (Ras-MAPK, PI3K-Akt, and PLCγ-PKC) result inmanyneuronalfunctions, including survival, differentiation, axonal growth, synapseformation and plasticity (Reichardt, 2006; Kuczewski et al., 2010). Thep75NTR receptor, instead, is believed to be important for ligandselectivity of Trk activation (Chao, 2003; Chao et al., 2006). Moreover,p75 can also activate pro-apoptotic responses and other cellularmechanisms, depending upon the presence of Trk and/or other co-receptors (e.g., sortilin) and ligands availability (mature neurotrophinsor their precursors) (Casaccia-Bonnefil et al., 1996; Friedman, 2000;Bentley and Lee, 2000; Beattie et al., 2002; Chao and Bothwell, 2002).The complexity of p75 signaling arises from the presence of anintracellular death domain (like TNF receptors) and the capability tointeract with different co-receptors (sortilin, Nogo receptor, LINGO-1)and adaptor proteins (TRAF-6, RhoGDI, NRIF, NRAGE, NADE and SC-1),whichunderlies its pleiotropic roles onneuronal and non-neuronal cells

Fig. 5. Schematic diagram of neurotrophins, receptors, key signaling pathways and activities.
Fig. 6. Scheme of the tPA/plasminogen/plasmin/metalloproteinases (MMPs) system onNGF maturation and degradation. The tPA/plasmin system promotes maturation ofproNGF to mature NGF, as well as cleavage of proMMPs to their mature active forms(MMPs). NGF degradation by MMPs can be blocked by GM6001, a generic MMPsinhibitor.
267A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
(Wong et al., 2002; Chao, 2003; Domeniconi et al., 2007; Jansen et al.,2007; Cragnolini and Friedman, 2008). Besides its activity on glial cells(astrocytes, microglia, Schwann cells and oligodendrocytes) and its rolein myelination, migration and proliferation (Bentley and Lee, 2000;Cosgaya et al., 2002; Vilar et al., 2006; Cragnolini et al., 2009),particularly relevant to the pathophysiology of neurodegenerativeconditions is its role in pro-neurotrophin-induced neuronal death (Leeet al., 2001; Harrington et al., 2004; Teng et al., 2005; Domeniconi et al.,2007; Yano et al., 2009).
Neurotrophins are secreted as pro-neurotrophins (pro-NGF, pro-BDNF, pro-NT-3) and proteolytically cleaved to the mature proteins inthe extracellular space (Fahnestock et al., 2001; Bruno and Cuello,2006). Therefore, the neurotrophic activity of NGF is currentlybelieved to be the result of a dynamic balance between conversionof pro-NGF to mature NGF and its degradation by the tissueplasminogen activator (tPA)–plasmin–matrix metalloproteinase-9(MMP-9) system (Fig. 6) (Bruno and Cuello, 2006; Cuello andBruno, 2007). Pro-neurotrophins are high-affinity ligands of p75NTRin complex with its co-receptor sortilin (Lee et al., 2001; Teng et al.,2005; Domeniconi et al., 2007; Yano et al., 2009). Thus, the biologicalfunctions of these receptors in the adult brain appear far morecomplex, as differential activation of p75 by mature neurotrophins ortheir precursors can have distinct and sometimes opposing actions inregulating cell death/survival and in modulating synaptic plasticity(Chao and Bothwell, 2002).
Alteration of NGF maturation/degradation processes and increaseof proNGF levels in the brains of AD patients are currently believed tounderlie the vulnerability and atrophy of NGF-dependent cholinergicneurons in AD, as well as in PD and age-related neurodegeneration(Fahnestock et al., 2001; Cuello and Bruno, 2007; Al-Shawi et al.,2007; Chen et al., 2008). Similar mechanisms of cell death have beendescribed for motor and sympathetic neurons in response to proNGF,proBDNF and proNT-3, and suggested to play a crucial role in severalneurodegenerative pathologies, such as ALS (Teng et al., 2005;Domeniconi et al., 2007; Yano et al., 2009). This hypothesis is inagreement with the evidence of increased p75NTR expression inseveral brain disorders (AD and motor diseases), as well as followingCNS injury (Masoudi et al., 2009).
These findings provide a challenging interpretation of thecomplex interplay between neuronal survival and neurotrophinprocessing/signaling in health and under pathological conditions andappear strictly linked to mechanisms of reactive gliosis (Fig. 7).
As extensively described above, astrocytes play a crucial role inmaintaining neuronal homeostasis during development and inadulthood (Heales et al., 2004; Benarroch, 2005). Glial function inneuroprotection is known to be also due to their role in productionand secretion of neurotrophic factors for neuronal survival. Inparticular, astrocytes have been shown to up-regulate neurotrophinproduction and secretion in response to glial stimulation byinflammatory mediators and pro-inflammatory cytokines (IL-1β,TNF-α, INFγ, LPS), as well as by neurotransmitters (glutamate and
beta-adrenergic stimulation) and nitric oxide donors (Friedman et al.,1996; Galve-Roperh et al., 1997; Colangelo et al., 1998; Mocchetti andColangelo, 2001; Domeniconi et al., 2007). Increased production ofpro-NGF and pro-BDNF by activatedmicroglia and astrocytes has beenshown to occur following CNS and spinal cord injury and trigger p75-mediated death of cortical and motor neurons (Harrington et al.,2004; Pehar et al., 2004; Domeniconi et al., 2007). We also found thatfollowing peripheral nerve lesion by chronic constriction injury (CCI)or spared nerve injury (SNI), glial activation and changes of synaptichomeostasis were paralleled by i) increase of MMPs activity, ii)decrease of mature NGF levels, iii) and increase of both pro-NGF andp75 receptor expression (Cirillo et al., 2010a, 2011). All theseresponses were restored by i.t. administration of NGF or GM6001, ageneric MMP inhibitor (Cirillo et al., 2011). These findings areconsistent with a model in which NGF is essential in maintainingsynaptic homeostasis (through modulation of glutamatergic andGABAergic components) and neuroprotection (through control ofsynaptic glutamate levels and providing neurons with GSH) (Cirillo

Fig. 7. Schematic representation of neuro-glial dysfunction linked to decreased NGF activity. Reactive gliosis determines the reduction of glial glutamate transporters and glutamateuptake. Glutamate spillover sensitizes NMDA and non-NMDA (mGlu, AMPA) glutamate receptors. The increase of extracellular glutamate also impairs the cystine–glutamateantiporter system, thus decreasing GSH synthesis and neuroprotection. These changes are prevented by i.t. administration of NGF or by increasing endogenous NGF levels by MMPsinhibition.
268 A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
et al., 2011). Thus, by establishing a strict correlation between glialdysfunction and aberrant NGF metabolisms and signaling, our dataprovide additional evidence of the detrimental role of pro-NGFfollowing abnormal NGF metabolism and the relevance of NGF-mediated function of astrocytes in neuroprotection.
6. Neurotrophins and reactive gliosis in ophthalmic pathologies
The activity of neurotrophins, in particular NGF, and thedetrimental effect of altered NGF metabolism following glial activa-tion may be also relevant for pathologies of the visual system. Theneural retina contains Muller (radial) glial cells, which span throughthe entire retina. Muller cells interact with all neurons and constitutethe functional link between retinal neurons and the other compart-ments (blood vessels, vitreous chamber and subretinal space) for theexchange of trophic substances and metabolic waste products. Mullerglia also plays a critical role in the regulation of extracellular spacevolume, ion and water homeostasis in the retina, as well as in themaintenance of the blood–retinal barrier and the retinal blood flow.
Reactive gliosis plays a critical role also in ophthalmic neurode-generative pathologies (de Melo Reis et al., 2008; Bringmann et al.,2009). Besides their role in trophic support and metabolism, Mullerglia modulates neuronal excitability and transmission. Like astrocytes,Muller cells participate in neurotransmission through: i) release ofgliotransmitters; ii) uptake and metabolism of aminoacid neurotrans-mitters (glutamate, GABA and glycine, as well as the purinergicreceptor agonists ATP and adenosine); and iii) release of precursors ofneurotransmitters to the neurons (de Melo Reis et al., 2008; Loiolaand Ventura, 2011). Glutamate is the most prominent excitatoryneurotransmitter in the retina, and is used in retinal transmission ofvisual signals by photoreceptors, bipolar cells and ganglion cells. Theremoval of glutamate from extracellular sites involves all theexcitatory amino acid transporters (EAATs): Muller cells and
astrocytes express EAAT1-5 transporters. Glutamate is metabolizedto glutamine by Muller cells, released and taken up by neurons as aprecursor for the synthesis of glutamate and GABA (glutamate–glutamine cycle). The metabolism of glutamate in Muller cells alsoproduces various substrates for the oxidative metabolism of photo-receptors (glutamine, lactate, alanine, and α-ketoglutarate). Mullercells (but not neurons) of the rat retina also express a chloride-dependent cystine–glutamate antiporter (system Xc-) that mediatesthe uptake of cystine in exchange with glutamate, both used for theproduction of glutathione (Mysona et al., 2009).
Alterations of Muller glia function and elevated levels ofextracellular glutamate have been described in the pathophysiologyof neuronal loss in ophthalmic disorders such as glaucoma. Mechan-ical stimulation of retinal ganglion axons due to elevation of theintraocular pressure is the major risk for glaucoma and causesreduced GLAST activity, decreased glutamate uptake and excitotoxicdamage to the retina. Disruption of glutamate transport inMuller cellsfollowing the increase in the intraocular pressure has been shown tobe also caused by inner retinal hypoxia (due to compression of bloodvessels), increased formation of free radicals in the mitochondria andlipid peroxidation (Mysona et al., 2009).
Glial dysfunction linked to ophthalmic diseases has been found tobe paralleled by aberrant production and activity of pro-NGF. Underphysiological conditions, TrkA is expressed in retinal ganglion cells(RGC), whereas p75NTR is mainly present on glial Muller cells.Alteration of NGF and NGF receptors has been shown to occur inseveral ophthalmic pathologies, such as glaucoma and retinal injury(Sposato et al., 2008; Colafrancesco et al., 2011). Induction of reactivegliosis in these pathological conditions seems to involve up-regulationof p75 and proNGF which causes RGC death through an indirectmechanism: activation of a p75NTR/sortilin/NRAGE-dependent path-way and production of TNFα by Müller glial cells (Bai et al., 2010;Lebrun-Julien et al., 2010).

269A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
7. Neurotrophin-derived drug candidates for neuroprotection
Based on the extensive literature regarding the several functions ofneurotrophins in the nervous system, it is generally recognized thatdecreased neurotrophin availability and signaling play a crucial role inthe pathophysiology of many neurological and psychiatric disorders(Weickert et al., 2005; Calissano et al., 2010). Moreover, the functionallink between all the components of reactive gliosis (excitotoxicity,decreased neuronalmetabolism and antioxidant properties, alterationof neurotrophins metabolism/signaling, and impaired synaptic plas-ticity), is all indicative of the impact that alterations of this complexneuro-glial network have on disease progression both during braininjury and in neurodegenerative diseases and age-related disorders.
Because of their crucial activity in modulating multiple pathwaysunderlying synaptic homeostasis and function in the nervous system,neurotrophins are the most likely drug candidates for neuroprotec-tion in many neurological diseases. For instance, the therapeuticpotential of NGF in restoring the cholinergic function in AD has beenlargely demonstrated in several experimental models (Williams et al.,1986; Koliatsos et al., 1991) and confirmed by small clinical trials(Tuszynski et al., 2005). Experimental and clinical findings are alsoavailable about the therapeutic activity of topic application of murineNGF (mNGF) in several ophthalmic pathologies (neurotrophickeratites, dry eye, glaucoma, etc.) (Aloe et al., 2008; Lambiase et al.,2010; Bagnis et al., 2011), as well as on cutaneous ulcers (Aloe et al.,2008; Sun et al., 2010) and peripheral neuropathies (Apfel, 2002).Neuroprotection by other neurotrophins, such as BDNF, in AD andother neurodegenerative conditions has also been largely demon-strated (Monteggia, 2011; Nagahara et al., 2009).
However, the development of neurotrophin proteins as drugsholds a number of intrinsic drawbacks, including: i) difficulties toobtain large amount of correctly folded recombinant proteins; ii) poorpharmacokinetic properties due to their in vivo instability (as target ofproteinases) and low permeability through the blood–brain barrier,thus raising delivery problems; and iii) pleiotropic actions due to theirinteraction with receptors and co-receptors (p75 and sortilin) thatactivate unwanted effects (p75-mediated neuronal death).
More recently, however, a valid alternative for neurotrophin-based therapies is the construction of small molecules that caninteract and activate specific receptor(s). This strategy has beenemployed to construct small functional mimetics of NGF, BDNF andNT-3 with activity of agonists or antagonists (Zaccaro et al., 2005;Peleshok and Saragovi, 2006; Massa et al., 2006, 2010; Molina-Holgado et al., 2008). This approach might be even more suitable forboth pharmaceutical development and therapeutic properties of themolecules. Advantages also include: i) specific receptor targeting, ii)lower molecular weight, and better pharmacological properties, iii)stability to proteinases, iv) lack of immunogenicity, and v) lower costof production. For instance, given the role of p75 in mediating pro-neurotrophin-death signals, NGF peptidomimetics endowed withagonist activity for TrkA or antagonism for p75 might be betterneurotherapeutics by targeting more specifically Trk-dependentsurvival and neuronal function, or blocking the p75-mediated deathsignaling. Some of these molecules have been found to display someefficacy both in a model of cholinergic dysfunction (Bruno et al., 2004)and in animal models of glaucoma (Shi et al., 2007). More recently, aTrkB antagonist also showed to be useful in reducing anxiety anddepression-related behaviors (Cazorla et al., 2011).
Our group recently developed a NGF-like peptide, BB14, thatbehaved as a strong TrkA agonist both in vitro, as demonstrated by itsneurotrophic activity on DRG and PC12 cell differentiation throughTrkA phosphorylation, and in a rat model of peripheral nerve injury byCCI where BB14 showed to be effective in reducing reactive gliosis andneuropathic behavior (Colangelo et al., 2008). Based on the impact ofreactive astrogliosis in most neurodegenerative pathologies, thisnovel anti-gliosis function displayed by BB14 might render this
molecule very suitable for therapeutic applications in a wide numberof neurological conditions, as well as in ophthalmic pathologies likeglaucoma, neurotrophic keratites and dry eye, which so far lackeffective therapeutic interventions.
8. Conclusions
Molecular dissection of neurodegenerative pathologies stronglyindicates that reactive astrogliosis is a complex process that involves anumber of changes ranging from alterations in gene expression andmorphology to modification of synaptic circuitry. The relevance ofneurotrophins in neuronal survival and function, and the strictcorrelation between glial dysfunction and aberrant NGF metabolismsand signaling, all suggest that the glia-neurotrophin system might bean effective therapeutic target in neurodegenerative diseases. Indeed,the positive effects of recombinant neurotrophins or peptidomimeticsin animal models and small clinical trials also provide compellingevidence of the impact that novel biotechnological approaches mighthave in the development of novel and effective molecules forneuroprotection. It is expected that small neurotrophin moleculesendowedwith better selectivity and potency, and void of the potentialcomplications of systemic neurotrophin stimulation, might be thenext generation therapeutics for neurodegenerative diseases.
Acknowledgments
Thisworkwas supported by grants fromRegione Campania (L.R. N.5Bando 2003 to M.P.), the Italian Minister of Research and University(PRIN2007 to M.P. and to A.M.C.), Regione Campania (Prog. Spec art 12E.F. 2000 to M.P.), the CNR (Neurobiotecnologie 2003 to M.P.), FIRB-ITALBIONET to L.A., PRIMM srl, Blueprint Biotech and Associazione Levi-Montalcini.
References
Alberghina L, Colangelo AM. The modular systems biology approach to investigate thecontrol of apoptosis in Alzheimer's disease neurodegeneration. BMC Neurosci2006;7(Suppl 1):S2. Oct 30.
Allegrini P, Fronzoni L, Pirino D. The influence of the astrocyte field on neuronaldynamics and synchronization. J Biol Phys 2009;35:413–23.
Aloe L, Tirassa P, Lambiase A. The topical application of nerve growth factor as apharmacological tool for human corneal and skin ulcers. Pharmacol Res 2008;57:253–8.
Al-Shawi R, Hafner A, Chun S, Raza S, Crutcher K, Thrasivoulou C, et al. ProNGF, sortilin,and age-related neurodegeneration. Ann NY Acad Sci 2007;1119:208–15.
Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: what wentwrong, what went right, and what does the future hold? Int Rev Neurobiol2002;50:393–413.
Bagnis A, Papadia M, Scotto R, Traverso CE. Current and emerging medical therapies inthe treatment of glaucoma. Expert Opin Emerg Drugs 2011;16:293–307.
Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC, et al. Chronic and acute models ofretinal neurodegeneration TrkA activity are neuroprotective whereas p75NTRactivity is neurotoxic through a paracrine mechanism. J Biol Chem 2010;285:39392–400.
Bajramovic JJ. Regulation of innate immune responses in the central nervous system.CNS Neurol Disord Drug Targets 2011;10:4–24.
Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide fromactivated glia-inhibiting neuronal respiration, causing glutamate release andexcitotoxicity. J Neurosci 2001;21:6480–91.
Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, et al. ProNGF inducesp75-mediated death of oligodendrocytes following spinal cord injury. Neuron2002;36:375–86.
Benarroch EE. Neuron–astrocyte interactions: partnership for normal function anddisease in the central nervous system. Mayo Clin Proc 2005;80:1326–38.
Bentley CA, Lee KF. p75 is important for axon growth and Schwann cell migrationduring development. J Neurosci 2000;20:7706–15.
Bolaños JP, Almeida A, Moncada S. Glycolysis: a bioenergetic or a survival pathway?Trends Biochem Sci 2010;35:145–9.
Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model andHuntington's disease. Neurosci Biobehav Rev 1997;21:289–93.
Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, et al. Cellularsignaling and factors involved in Müller cell gliosis: neuroprotective anddetrimental effects. Prog Retin Eye Res 2009;28:423–51.
Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin touncover physiopathological mechanisms underlying striatal degeneration inHuntington's disease. J Neurochem 2005;95:1521–40.

270 A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor,conversion to mature nerve growth factor, and its degradation by a proteasecascade. Proc Natl Acad Sci USA 2006;103:6735–40.
Bruno MA, Clarke PB, Seltzer A, Quirion R, Burgess K, Cuello AC, et al. Long-lastingrescue of age-associated deficits in cognition and the CNS cholinergic phenotype bya partial agonist peptidomimetic ligand of TrkA. J Neurosci 2004;24:8009–18.
Calissano P, Matrone C, Amadoro G. Nerve growth factor as a paradigm ofneurotrophins related to Alzheimer's disease. Dev Neurobiol 2010;70:372–83.
Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytesmediated by the interaction of nerve growth factor with its receptor p75. Nature1996;383:716–9.
Cavaliere C, Cirillo G, Bianco MR, Rossi F, De Novellis V, Maione S, et al. Gliosis altersexpression and uptake of spinal glial amino acid transporters in a mouseneuropathic pain model. NGB 2007;2:141–53.
Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of alow-molecular weight TrkB antagonist with anxiolytic and antidepressant activityin mice. J Clin Invest 2011;121:1846–57.
Chao MV. Neurotrophins and their receptors: a convergence point for many signallingpathways. Nat Rev Neurosci 2003;4:299–309.
ChaoMV, Bothwell M. Neurotrophins: to cleave or not to cleave. Neuron 2002;33:9–12.Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci
2006;110:167–73.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-
Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiatedlocalizations revealed by quantitative ultrastructural immunocytochemistry. Neuron1995;15:711–20.
Chen LW, Yung KKL, Chan YS, Shum DKY, Bolam JP. The proNGF–p75NTR–sortilinsignalling complex as new target for the therapeutic treatment of Parkinson'sdisease. CNS Neurol Disord Drug Targets 2008;7:512–23.
Chung YC, Ko HW, Bok E, Park ES, Huh SH, Nam JH, et al. The role of neuroinflammationon the pathogenesis of Parkinson's disease. BMB Rep 2010;43:225–32.
Cirillo G, Cavaliere C, Bianco MR, De Simone A, Colangelo AM, Sellitti S, et al. IntrathecalNGF administration reduces reactive astrocytosis and changes neurotrophinreceptors expression pattern in a rat model of neuropathic pain. Cell Mol Neurobiol2010a;30:51–62.
Cirillo G, Maggio N, Bianco MR, Vollono C, Sellitti S, Papa M. Discriminative behavioralassessment unveils remarkable reactive astrocytosis and early molecular correlatesin basal ganglia of 3-nitropropionic acid subchronic treated rats. Neurochem Int2010b;56:152–60.
Cirillo G, Bianco MR, Colangelo AM, Cavaliere C, Daniele de L, Zaccaro L, et al. Reactiveastrocytosis-induced perturbation of synaptic homeostasis is restored by nervegrowth factor. Neurobiol Dis 2011;41:630–9.
Colafrancesco V, Parisi V, Sposato V, Rossi S, Russo MA, Coassin M, et al. Ocularapplication of nerve growth factor protects degenerating retinal ganglion cells in arat model of glaucoma. J Glaucoma 2011;20:100–8.
Colangelo AM, Johnson PF, Mocchetti I. Beta-adrenergic receptor-induced activationof nerve growth factor gene transcription in rat cerebral cortex involvesCCAAT/enhancer-binding protein delta. Proc Natl Acad Sci USA 1998;95:10920–5.
Colangelo AM, Bianco MR, Vitagliano L, Cavaliere C, Cirillo G, De Gioia L, et al. A newnerve growth factor-mimetic peptide active on neuropathic pain in rats. J Neurosci2008;28:2698–709.
Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positivemodulator of myelination. Science 2002;298:1245–8.
Cragnolini AB, Friedman WJ. The function of p75NTR in glia. Trends Neurosci 2008;31:99–104.
Cragnolini AB, Huang Y, Gokina P, Friedman WJ. Nerve growth factor attenuatesproliferation of astrocytes via the p75 neurotrophin receptor. Glia 2009;57:1386–92.
Cuello AC, Bruno MA. The failure in NGF maturation and its increased degradation asthe probable cause for the vulnerability of cholinergic neurons in Alzheimer'sdisease. Neurochem Res 2007;32:1041–5.
Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1–105.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid
microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752–8.de Melo Reis RA, Ventura AL, Schitine CS, de Mello MC, de Mello FG. Müller glia as an
active compartment modulating nervous activity in the vertebrate retina:neurotransmitters and trophic factors. Neurochem Res 2008;33:1466–74.
Domeniconi M, Hempstead BL, Chao MV. Pro-NGF secreted by astrocytes promotesmotor neuron cell death. Mol Cell Neurosci 2007;34:271–9.
Dun Y, Mysona B, Van Ells T, Amarnath L, Ola MS, Ganapathy V, et al. Expression of thecystine–glutamate exchanger (xc-) in retinal ganglion cells and regulation by nitricoxide and oxidative stress. Cell Tissue Res 2006;324:189–202.
Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factoris the predominant form of nerve growth factor in brain and is increased inAlzheimer's disease. Mol Cell Neurosci 2001;18:210–20.
Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, et al. In vivo expression ofpolyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamatetransport: a correlation with Huntington's disease subjects. HumMol Genet 2010;19:3053–67.
Fellin T, Carmignoto G. Neurone-to-astrocyte signalling in the brain represents a distinctmultifunctional unit. J Physiol 2004;559:3–15.
Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS, Citron BA. Minocyclineneuroprotects, reduces microgliosis, and inhibits caspase protease expression earlyafter spinal cord injury. J Neurochem 2006;97:1314–26.
Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75receptor. J Neurosci 2000;20:6340–6.
FriedmanWJ, Thakur S, Seidman L, Rabson AB. Regulation of nerve growth factor mRNAby interleukin-1 in rat hippocampal astrocytes is mediated by NFkappaB. J BiolChem 1996;271:31115–20.
Galve-Roperh I, Malpartida JM, Haro A, Brachet P, Díaz-Laviada I. Regulation of nervegrowth factor secretion and mRNA expression by bacterial lipopolysaccharide inprimary cultures of rat astrocytes. J Neurosci Res 1997;49:569–75.
Giovannoni R, Maggio N, Bianco MR, Cavaliere C, Cirillo G, Lavitrano M, et al. Reactiveastrocytosis and glial glutamate transporter clustering are early changes in aspinocerebellar ataxia type 1 transgenic mouse model. Neuron Glia Biol 2007;3:335–51.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlyinginflammation in neurodegeneration. Cell 2010;140:918–34.
Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Studygroup. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: aphase III randomised trial. Lancet Neurol 2007;6:1045–53.
Gray BC, Skipp P, O'Connor VM, Perry VH. Increased expression of glial fibrillary acidicprotein fragments and mu-calpain activation within the hippocampus of prion-infected mice. Biochem Soc Trans 2006;34:51–4.
GriffinWS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewybody pathologies. J Neuroinflammation 2006;3:5.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in thenormal and pathologic brain. Nat Neurosci 2007;10:1387–94.
Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Mörl K, et al. SecretedproNGF is a pathophysiological death-inducing ligand after adult CNS injury. ProcNatl Acad Sci USA 2004;16:6226–30.
Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokineexpression after lumbar root injury in the rat. Spine 2000;25:1206–17.
Haydon PG. Glia: listening and talking to the synapse. Nat Rev Neurosci 2001;2:185–93.Heales SJ, Lam AA, Duncan AJ, Land JM. Neurodegeneration or neuroprotection: the
pivotal role of astrocytes. Neurochem Res 2004;29:513–9.HenekaMT, Rodriguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Res Rev
2010;63:189–211.Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The
bioenergetic and antioxidant status of neurons is controlled by continuousdegradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 2009;11:747–52.
Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, et al. Extensive earlymotor and non-motor behavioral deficits are followed by striatal neuronal loss inknock-in Huntington's disease mice. Neuroscience 2008;157:280–95.
Hirase H. A multi-photon window onto neuronalglial-vascular communication. TrendsNeurosci 2005;28:217–9.
Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O, Sjoegaard SS, et al. Roles for thepro-neurotrophin receptor sortilin in neuronal development, aging and braininjury. Nat Neurosci 2007;10:1449–57.
Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Science STKE2004:reE14.
Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset altersastrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol2011;228(1):69–79.
Knight RA, Verkhratsky A. Neurodegenerative diseases: failures in brain connectivity?Cell Death Differ 2010;17:1069–70.
Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience2004;129:1045–56.
Koliatsos VE, Clatterbuck RE, Nauta HJW, Knusel B, Burton LE, Hefti FF, et al. HumanNerve Growth Factor prevents degeneration of basal forebrain cholinergic neuronsin primates. Ann Neurol 1991;30:831–40.
Kuang X, Scofield VL, Yan M, Stoica G, Liu N, Wong PK. Attenuation of oxidative stress,inflammation and apoptosis by minocycline prevents retrovirus-induced neuro-degeneration in mice. Brain Res 2009;1286:174–84.
Kuczewski N, Porcher C, Gaiarsa JL. Activity-dependent dendritic secretion of brain-derived neurotrophic factor modulates synaptic plasticity. Eur J Neurosci 2010;32:1239–44.
Lambiase A, Mantelli F, Bonini S. Nerve growth factor eye drops to treat glaucoma. DrugNews Perspect 2010;23:361–7.
Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington'sdisease. Fourth in molecular medicine review series. EMBO Rep 2004;5:958–63.
Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR, Di Polo A, et al.ProNGF induces TNFalpha-dependent death of retinal ganglion cells through ap75NTR non-cell-autonomous signaling pathway. Proc Natl Acad Sci USA2010;107:3817–22.
Lee YB, Du S, Rhim H, Lee EB, Markelonis GJ, Oh TH. Rapid increase in immunoreactivityto GFAP in astrocytes in vitro induced by acidic pH is mediated by calcium influxand calpain I. Brain Res 2000;864:220–9.
Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secretedproneurotrophins. Science 2001;294:1945–58.
Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expressionof two glial glutamate transporters in the rat brain: quantitative and immunocy-tochemical observations. J Neurosci 1995;15:1835–53.
Levi-Montalcini R. Effects of mouse tumor transplantation on the nervous system. AnnNY Acad Sci 1952;55:330–44.
Lewerenz J, Klein M, Methner A. Cooperative action of glutamate transporters andcystine/glutamate antiporter system Xc- protects from oxidative glutamatetoxicity. J Neurochem 2006;98:916–25.
Li Z, Hogan EL, Banik NL. Role of calpain in spinal cord injury: increased calpainimmunoreactivity in rat spinal cord after impact trauma. Neurochem Res 1996;21:441–8.

271A.M. Colangelo et al. / Biotechnology Advances 30 (2012) 261–271
Lievens JC, Salin P, Had-Aissouni L, Mahy N, Kerkerian-Le GL. Differential effects ofcorticostriatal and thalamostriatal deafferentation on expression of the glutamatetransporter GLT1 in the rat striatum. J Neurochem 2000;74:909–19.
Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomousneurodegenerative disease. Nat Neurosci 2007;10:1355–60.
Loiola EC, Ventura AL. Release of ATP from avian Müller glia cells in culture. NeurochemInt 2011;58:414–22.
Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci2005;6:603–14.
Machado-Vieira R, Manji HK, Zarate CA. The role of the tripartite glutamatergic synapsein the pathophysiology and therapeutics of mood disorders. Neuroscientist2009;15:525–39.
Magistretti PJ. Neuron–glia metabolic coupling and plasticity. J Exp Biol 2006;209:2304–11.
Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerativedisease. Nat Clin Pract Neurol 2006;2:679–89.
Masoudi R, Ioannou MS, Coughlin MD, Pagadala P, Neet KE, Clewes O, et al. Biologicalactivity of Nerve Growth Factor precursor is dependent upon relative levels of itsreceptors. J Biol Chem 2009;284:18424–33.
Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, et al. Small, nonpeptidep75NTR ligands induce survival signaling and inhibit proNGF-induced death. JNeurosci 2006;26:5288–300.
Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, et al. Small molecule BDNF mimeticsactivate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest2010;120:1774–85.
McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediatorsand modulators. Exp Neurol 2005;192:444–62.
Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, et al. Substrate turnoverby transporters curtails synaptic glutamate transients. J Neurosci 1999;19:9242–51.
Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, et al. Expression of theglial glutamate transporter EAAT2 in the human CNS: an immunohistochemicalstudy. Brain Res Mol Brain Res 1997;52:17–31.
Mocchetti I, Colangelo AM. Transcriptional regulation of NGF in the central nervoussystem. In: Mocchetti I, editor. Neurobiology of the neurotrophins. F.P. GrahamPublishing Co.; 2001. p. 631–54.
Molina-Holgado F, Doherty P, Williams G. Tandem repeat peptide strategy for thedesign of neurotrophic factor mimetics. CNS Neurol Disord Drug Targets 2008;7:110–9.
Monteggia LM. Toward neurotrophin-based therapeutics. Am J Psychiatry 2011;168:114–6.
Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration.Neurobiol Aging 2005;26:349–54.
Musholt K, Cirillo G, Cavaliere C, Bianco MR, Bock J, Helmeke C, et al. Neonatal separationstress reduces glial fibrillary acidic protein and S100beta-immunoreactive astrocytesin the rat medial precentral cortex. Dev Neurobiol 2009;69:203–11.
Mysona B, Dun Y, Duplantier J, Ganapathy V, Smith SB. Effects of hyperglycemia andoxidative stress on the glutamate transporters GLAST and system xc- in mouseretinal Müller glial cells. Cell Tissue Res 2009;335:477–88.
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al.Neuroprotective effects of brain-derived neurotrophic factor in rodent and primatemodels of Alzheimer's disease. Nat Med 2009;15:331–7.
Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, et al. Directevidence of astrocytic modulation in the development of rewarding effects inducedby drugs of abuse. Neuropsychopharmacology 2006;31:2476–88.
Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis betweenneurons and glial cells from postnatal rats. J Neurochem 2009;109:125–34.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamicsurveillants of brain parenchyma in vivo. Science 2005;308:1314–8.
Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007;30:575–621.
Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, et al. Brainstructure in preclinical Huntington's disease. Biol Psychiatry 2006;59:57–63.
Paulson HL, Fischbeck KH. Trinucleotide repeats in neurogenetic disorders. Annu RevNeurosci 1996;19:79–107.
Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, et al. Astrocyticproduction of nerve growth factor in motor neuron apoptosis: implications foramyotrophic lateral sclerosis. J Neurochem 2004;89:464–73.
Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia 2005;50:427–34.Peleshok J, Saragovi HU. Functional mimetics of neurotrophins and their receptors.
Biochem Soc Trans 2006;34:612–7.Pfrieger FW. Roles of glial cells in synapse development. Cell Mol Life Sci 2009;66:
2037–47.Pfrieger FW, Barres BA. New views on synapse–glia interactions. Curr Opin Neurobiol
1996;6:615–21.Raghavendra V, Rutkowski MD, De Leo JA. The role of spinal neuroimmune activation in
morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. JNeurosci 2002;22:9980–9.
Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses.Annu Rev Immunol 2009;27:119–45.
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond BBiol Sci 2006;361:1545–64.
Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, et al. Clinical trialof indomethacin in Alzheimer's disease. Neurology 1993;43:1609–11.
Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration.Brain Res Bull 2009;80:224–32.
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, et al. Localization ofneuronal and glial glutamate transporters. Neuron 1994;13:713–25.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout ofglutamate transporters reveals a major role for astroglial transport in excitotoxicityand clearance of glutamate. Neuron 1996;16:675–86.
Saijo K, Winner B, Carson TC, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoRESTpathway in microglia and astrocytes protects dopaminergic neurons frominflammation-induced death. Cell 2009;137:47–59.
Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias:clinical features, genetics, and pathogenesis. Lancet Neurol 2004;3:291–304.
Serbest G, Burkhardt MF, Siman R, Raghupathi R, Saatman KE. Temporal profiles ofcytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res2007;32:2006–14.
Servadio A, Koshy B, Armstrong D, Antalffy B, Orr HT, Zoghbi HY. Expression analysis ofthe ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1individuals. Nat Genet 1995;10:94–8.
Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerativediseases and potential opportunities for intervention. Neurochem Int 2007;51:333–55.
Shi Z, Birman E, Saragovi HU. Neurotrophic rationale in glaucoma: a TrkA agonist, butnot NGF or a p75 antagonist, protects retinal ganglion cells in vivo. Dev Neurobiol2007;67:884–94.
Simard M, Nedergaard M. The neurobiology of glia in the context of water and ionhomeostasis. Neuroscience 2004;129:877–96.
Skaper SD. The biology of neurotrophins, signalling pathways, and functional peptidemimetics of neurotrophins and their receptors. CNS Neurol Disord Drug Targets2008;7:46–62.
Sposato V, Bucci MG, Coassin M, Russo MA, Lambiase A, Aloe L. Reduced NGF level andTrkA protein and TrkA gene expression in the optic nerve of rats withexperimentally induced glaucoma. Neurosci Lett 2008;446:20–4.
Sun W, Lin H, Chen B, Zhao W, Zhao Y, Xiao Z, et al. Collagen scaffolds loaded withcollagen-binding NGF-beta accelerate ulcer healing. J BiomedMater Res A 2010;92:887–95.
Suzuki A, Stern SA, Bozdagi O, Huntley GW,Walker RH,Magistretti PJ, et al. Astrocyte-neuronlactate transport is required for long-term memory formation. Cell 2011;144:810–23.
Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, et al. Astrocyte-mediated controlof cerebral blood flow. Nat Neurosci 2006;9:260–7.
Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role inneuronal death and implications for therapeutic intervention. Neurobiol Dis2010;37:510–8.
Teng HK, Teng KK, Lee R,Wright S, Tevar S, Almeida RD, et al. ProBDNF induces neuronalapoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci2005;25:5455–63.
Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown Jr RH, et al. Amyotrophiclateral sclerosis-linked glutamate transporter mutant has impaired glutamateclearance capacity. J Biol Chem 2001;276:576–82.
Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, et al. A phase 1 clinical trial ofnerve growth factor gene therapy for Alzheimer disease. Nat Med 2005;11:551–5.
Unger JW. Glial reaction in aging and Alzheimer's disease. Microsc Res Tech 1998;43:24–8.
van Noort JM, van den Elsen PJ, van Horssen J, Geurts JJ, van der Valk P, Amor S. Preactivemultiple sclerosis lesions offer novel clues for neuroprotective therapeutic strategies.CNS Neurol Disord Drug Targets 2011;10:68–81.
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes inAlzheimer's disease. Neurotherapeutics 2010;7:399–412.
Vilar M, Murillo-Carretero M, Mira H, Magnusson K, Besset V, Ibáñez CF. Bex1, a novelinteractor of the p75 neurotrophin receptor, links neurotrophin signaling to the cellcycle. EMBO J 2006;25:1219–30.
Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev DrugDiscov 2003;2:973–85.
Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain.Trends Neurosci 2001;24:450–5.
Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM, et al.Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patientswith schizophrenia. Mol Psychiatry 2005;10:637–50.
Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain.Neurochem Int 2004;45:389–95.
Williams LR, Varon S, Peterson GM,Wictorin K, Fisher W, Bjorklund A, et al. Continuousinfusion of nerve growth factor prevents basal forebrain neuronal death afterfimbria fornix transection. Proc Natl Acad Sci USA 1986;83:9231–5.
Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. A p75(NTR) andNogo receptor complex mediates repulsive signaling by myelin-associatedglycoprotein. Nat Neurosci 2002;5:1302–8.
Yano H, Torkin R, Martin LA, Chao MV, Teng KK. Proneurotrophin-3 is a neuronalapoptotic ligand: evidence for retrograde-directed cell killing. J Neurosci 2009;29:14790–802.
Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes:a novel mechanism of glutamate release. J Neurosci 2003;23:3588–96.
Zaccaro MC, Lee HB, Pattarawarapan M, Xia Z, Caron A, L'Heureux PJ, et al. Selectivesmall molecule peptidomimetic ligands of TrkC and TrkA receptors afford discreteor complete neurotrophic activities. Chem Biol 2005;12:1015–28.
Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia,and astrocytes by spinal nerve ligation and contributes to mechanical allodynia inthis neuropathic pain model. Pain 2005;114:149–59.