synthesis, structural characterisation and cytotoxicity - nova
TRANSCRIPT

SYNTHESIS, STRUCTURAL CHARACTERISATION AND CYTOTOXICITY STUDY OF TIN(IV) COMPOUNDS CONTAINING ONS SCHIFF BASES
By
ENIS NADIA BINTI MD YUSOF
Thesis Submitted to the School of Graduate Studies, Universiti Putra Malaysia, Malaysia and The University of Newcastle, Australia in Fulfilment of the
Requirements for the Degree of Doctor of Philosophy
December 2019

All material contained within the thesis, including without limitation text, logos, icons, photographs and all other artwork, is copyright material of Universiti Putra Malaysia unless otherwise stated. Use may be made of any material contained within the thesis for non-commercial purposes from the copyright holder. Commercial use of material may only be made with the express, prior, written permission of Universiti Putra Malaysia. Copyright © Universiti Putra Malaysia

i
Abstract of thesis presented to the Senate of Universiti Putra Malaysia and The University of Newcastle in fulfilment of the requirement for the degree of Doctor of
Philosophy
SYNTHESIS, STRUCTURAL CHARACTERISATION AND CYTOTOXICITY STUDY OF TIN(IV) COMPOUNDS CONTAINING ONS SCHIFF BASES
By
ENIS NADIA BINTI MD YUSOF
December 2019
Chair : Associate Professor Thahira Begum, PhD (UPM) Associate Professor Alister J. Page, PhD (UON) Faculty : Science There is an urgent need for substantial investigation of non-platinum drugs with higher
activity and improved selectivity to address the problem associated with the use of
platinum-based compounds as therapeutic agents. In light of this, diphenyltin(IV),
dimethyltin(IV) and tin(IV) compounds were synthesised from the Schiff bases of three
series of dithiocarbazate (S-2-methylbenzyldithiocarbazate (S1), S-4-methylbenzyl
dithiocarbazate (S2), S-benzyldithiocarbazate (S3)) and two series of thiosemicarbazides
(4-methyl-3-thiosemicarbazide and 4-phenyl-3-thiosemicarbazide) with aldehydes, 2-
hydroxy-3-methoxybenzaldehyde (oVa) or 2,3-dihydroxybenzaldehyde (catechol). The
tin(IV) compounds formed were found to have a general formula of [R2Sn(ONS)] and
[Sn(ONS)2] (where R = Me and Ph). The compounds were fully characterised by physico-
chemical and spectroscopic methods. The spectroscopic results supported the coordination
geometry in which the Schiff bases behaved as tridentate ONS donor ligands coordinating
via azomethine nitrogen, thiolo sulphur and phenoxide oxygen atoms. A total of 11 crystal

ii
structures of the expected compounds were solved in this work. In order to verify the
experimental data, the compounds were optimised using the density functional theory
(DFT) method with the B3LYP hybrid exchange correlation functional with LanL2DZ
pseudopotential on tin and 6-311G(d,p) Pople basis set for all other atoms. Diphenyltin(IV)
compounds showed the most promising cytotoxicity with IC50 values ranging between
0.016 – 4.40 μM against a panel of twelve cancer cell lines (RT-112, EJ-28 (bladder), HT29
(colon), U87, SJ-G2, SMA (glioblastoma), MCF-7 (breast), A2780 (ovarian), H460 (lung),
A431 (skin), Du145 (prostate), BE2-C (neuroblastoma) and MIA (pancreatic)). The three
diphenyltin(IV) compounds of the oVa series were able to induce the production of
Reactive Oxygen Species (ROS) and acted as a cell apoptosis inducer. Good binding
interactions for all the diphenyltin(IV) compound series were observed and supported by
molecular docking analysis, where hydrogen, electrostatic and hydrophobic binding
interactions were observed. This highlights the important of two phenyl groups coordinated
directly to the tin ion to enhance the cytotoxicity by strong π-π stacking interactions to
biomacromolecules. Diphenyltin(IV) compounds could bring hope in the field of drug
development against various diseases including cancers.

iii
Abstrak tesis yang dikemukakan kepada Senat Universiti Putra Malaysia dan The University of Newcastle sebagai memenuhi keperluan untuk Ijazah Doktor Falsafah
SINTESIS, PENCIRIAN STRUKTUR DAN KAJIAN SITOTOSIK BAGI SEBATIAN TIN(IV) YANG MENGANDUNGI BES SCHIFF ONS
Oleh
ENIS NADIA BINTI MD YUSOF
Disember 2019
Pengerusi : Professor Madya Thahira Begum, PhD (UPM) Professor Madya Alister J. Page, PhD (UON) Fakulti : Sains Terdapat keperluan segera bagi penyiasatan penting ke atas ubat yang tidak mengandungi
platinum dengan aktitviti yang tinggi dan meningkatkan pemilihan untuk menyelesaikan
masalah berkaitan dengan penggunaan sebatian daripada platinum sebagai agen
terapeutik. Disebabkan keadaan ini, sebatian difenilstanum(IV), dimetilstanum(IV) dan
stanum(IV) telah disintesis daripada bes Schiff yang terdiri daripada tiga siri ditiokarbazat
(S-2-metilbenzilditiokarbazat (S1), S-4-metilbenzilditiokarbazat (S2), S-
benzilditiokarbazat (S3) dan dua siri tiosemikarbazid (4-metil-3-thiosemikarbazid dan 4-
fenil-3-tiosemikarbazid) dengan aldehid, 2-hidroksil-3-metoksibenzaldehid (oVa) atau
2,3-dihidroksibenzaldehid (katekol). Sebatian stanum(IV) yang dihasilkan telah ditemui
mempunyai formula umum [R2Sn(ONS)] dan [Sn(ONS)2] (di mana R = Me dan Ph).
Sebatian tersebut telah dicirikan sepenuhnya dengan kaedah fiziko-kimia dan
spektroskopi. Hasil spektroskopi menyokong geometri pengkoordinatan di mana bes
Schiff bertindak sebagai ligan penderma tridentat ONS berkoordinat melalui atom-atom
nitrogen azometina, sulfur tiolo, dan oksigen fenoksid. Sebanyak 11 struktur hablur

iv
sebatian yang dijangkakan telah diselesaikan dalam kajian ini. Bagi mengesahkan data
eksperimen, sebatian-sebatian itu telah dioptimum menggunakan kaedah teori berfungsi
ketumpatan (DFT) dengan fungsian korelasi pertukaran hibrid B3LYP dengan keupayaan
pseudo LanL2DZ ke atas stanum dan set asas Pople 6-311G(d,p) bagi semua atom-atom
yang lain. Sebatian difenilstanum(IV) menunjukkan kesitotoksikan yang paling
memberangsangkan dengan nilai IC50 diantara 0.016 – 4.40 μM terhadap satu panel
daripada dua belas siri sel kanser (RT-112, EJ-28 (pundi kencing), HT29 (usus besar),
U87, SJ-G2, SMA (glioblastoma), MCF-7 (payudara), A2780 (ovari), H460 (paru-paru),
A431 (kulit), Du145 (prostat), BE2-C (neuroblastoma) dan MIA (pankreas)). Tiga
sebatian difenilstanum(IV) daripada siri oVa berkebolehan untuk mendorong penghasilan
Spesis Oksigen Reaktif (ROS) dan bertindak sebagai pendorong sel apoptosis. Ikatan
interaksi yang baik bagi kesemua siri sebatian difenilstanum(IV) telah diperhatikan dan
disokong oleh analisis molekul docking, di mana ikatan interaksi hidrogen, elektrostatik
dan hidrofobik telah diperhatikan. Ini menunjukkan bahawa pentingnya dua kumpulan
fenil yang berkoordinat secara terus kepada ion stannum untuk meningkatkan sifat
sitotosik melalui interaksi π-π yang kuat kepada biomakromolekul. Difenilstannum(IV)
memberi harapan dalam bidang pembangunan dadah terhadap pelbagai penyakit
termasuklah kanser.

v
ACKNOWLEGEMENTS First of all, I would like thank to Allah for giving His blessing to complete my PhD
research project. I wish to express my deepest gratitude and appreciation to my principal
supervisors, Associate Professor Dr. Thahira Begum and Associate Professor Dr. Alister J.
Page as well as my co-supervisors, Professor Adam McCluskey, Dr. Mohamed Ibrahim
Mohamed Tahir, Professor Abhimanyu Veerakumarasivam and Dr. Muhammad Alif bin
Muhammad Latif, whose encouragement, guidance and support from the initial to the final
level. They spent countless hours clearing my doubts and problems with regards to my
research project. I would also like to extend my appreciation to Professor Karen Anne
Crouse for meaningful discussion, Dr. Michela Simone for her grateful NMR discussion,
Dr. Robert Burns for his kind suggestions about 119Sn NMR experiments, Dr. Jennette
Sakoff for anticancer screening and Professor Edward R. T. Tiekink for single crystal X-
ray structure analysis.
My special thanks to the wonderful computational chemistry group members, Ben,
Josh, Tilly, Kas, Krishna, Simone, Gareth, Izaac, Xinyu, Babu, Ryan, Thom, who had
guided me patiently and motivated me to get better in research. My special thanks also goes
to Inorganic Chemistry members, Nabihah, Nazhirah, Chee Keong, Lee Chin, Nabeel and
Ali for their kind assistance in helping me to complete my research. My sincere
appreciation goes to all lecturers and staff at Department of Chemistry, Faculty of Science,
Medical Genetic Laboratory, Faculty of Medicine and School of Chemistry, Faculty of
Environmental and Life Sciences and the Calvary Mater Hospital, Newcastle, Australia for
being helpful and cooperative throughout this research.
And finally, I would like to thank family members, especially my husband for his
endless love and encouragement, my parents, thank you for always loving, supporting and
wishing me the best for the whole of my life. To my siblings, thank you for always making

vi
me smile and releasing my tension during writing this thesis. Lastly, I offer my best wishes
to all of those who supported me in any aspect during the completion of this research.

vii
I certify that a Thesis Examination Committee has met on 2 December 2019 to conduct the final examination of Enis Nadia binti Md Yusof on her thesis entitled “Synthesis, Structural Characterisation and Cytotoxicity Study of Tin(IV) Compounds containing ONS Schiff Bases” in accordance with the Universities and University Colleges Act 1971 and the Constitution of the Universiti Putra Malaysia [P.U.(A) 106] 15 March 1998. The Committee recommends that the student be awarded the Doctoral of Philosophy.
Members of the Thesis Examination Committee were as follows: Lim Hong Ngee, PhD Associate Professor Faculty of Science Universiti Putra Malaysia (Chairman) Tan Kar Ban, PhD Associate Professor Faculty of Science Universiti Putra Malaysia (Internal Examiner) Christopher Scarlett, PhD Professor School of Environmental and Life Sciences The University of Newcastle Australia (Internal Examiner) Jagadese J. Vittal, PhD Professor Department of Chemistry National University of Singapore Singapore (External Examiner)
__________________________________ ZURIATI AHMAD ZUKARNAIN, PhD Professor and Deputy Dean School of Graduate Studies Universiti Putra Malaysia Date: 2 January 2020

viii
This thesis was submitted to the Senate of Universiti Putra Malaysia and has been accepted as fulfilment of the requirement for the degree of Doctor of Philosophy. The members of the Supervisory Committee were as follows:
Thahira Begum, PhD Associate Professor Faculty of Science Universiti Putra Malaysia (Chairman) Alister J. Page, PhD Associate Professor School of Environmental and Life Sciences The University of Newcastle, Australia (Member) Mohamed Ibrahim Mohamed Tahir, PhD Senior Lecturer Faculty of Science Universiti Putra Malaysia, Malaysia (Member) Abhimanyu Veerakumarasivam, PhD Professor Department of Biological Sciences School of Science and Technology Sunway University, Malaysia (Member) Muhammad Alif bin Muhammad Latif, PhD Senior Lecturer Centre of Foundation Studies for Agricultural Sciences Universiti Putra Malaysia, Malaysia (Member) Adam McCluskey, PhD Professor School of Environmental and Life Sciences The University of Newcastle, Australia (Member)
______________________________ ZALILAH MOHD SHARIFF, PhD Professor and Deputy Dean School of Graduate Studies Universiti Putra Malaysia Date:

ix
Declaration by graduate student
I hereby confirm that: this thesis is my original work; the thesis contains no material which has been accepted, or is being examined, for
the award of any other degree or diploma in any university or other tertiaryinstitution;
quotations, illustrations and citations have been duly acknowledged and appropriatecopyright permissions have been obtained for the content to be made availableelectronically;
ownership of intellectual property from the thesis is as stipulated in the Memorandumof Agreement (MoA), or as according to the Universiti Putra Malaysia (Research)Rules 2012, in the event where the MoA is absent;
permission from supervisor and the office of Deputy Vice-Chancellor (Research andInnovation) are required prior to publishing it (in the form of written, printed or inelectronic form) including books, journals, modules, proceedings, popular writings,seminar papers, manuscripts, posters, reports, lecture notes, learning modules or anyother materials as stated in the Universiti Putra Malaysia (Research) Rules 2012;
there is no plagiarism or data falsification/fabrication in the thesis, and scholarlyintegrity is upheld as according to the Universiti Putra Malaysia (Graduate Studies)Rules 2003 (Revision 2012-2013) and the Universiti Putra Malaysia (Research)Rules 2012 and the University of Newcastle Rules Governing Higher Degrees byResearch. The thesis has undergone plagiarism detection software;
i give consent to the final version of my thesis being made available worldwide whendeposited in the University’s Digital Repository, subject to the provisions of theCopyright Act 1968 and any approved embargo;
the thesis has been submitted to Universiti Putra Malaysia and the University ofNewcastle as part of a Jointly Awarded Doctoral Degree.
Signature: ___________________________ Date: 9/7/2019__________________
Name and Matric No.: Enis Nadia binti Md Yusof (GS44510 (UPM) and 3285394 (UON))

x
Declaration by Members of Supervisory Committee
This is to confirm that: the research conducted and the writing of this thesis was under our supervision; supervision responsibilities as stated in the Universiti Putra Malaysia (Graduate
Studies) Rules 2003 (Revision 2012-2013) and the University of Newcastle RulesGoverning Higher Degrees by Research and University of Newcastle Code ofPractice are adhered to.
Signature: Name of Chairman of Supervisory Committee: Thahira Begum
Signature: Name of Member of Supervisory Committee: Alister J. Page
Signature: Name of Member of Supervisory Committee: Mohamed Ibrahim Mohamed Tahir
Signature: Name of Member of Supervisory Committee: Abhi Veerakumarasivam
Signature: Name of Member of Supervisory Committee: Muhammad Alif bin Muhammad Latif
Signature: Name of Member of Supervisory Committee: Adam McCluskey

xi
TABLE OF CONTENTS Page ABSTRACT i ABSTRAK iii ACKNOWLEDGEMENTS v APPROVAL vii DECLARATION ix LIST OF TABLES xvi LIST OF FIGURES xviii LIST OF SCHEMES xxiii LIST OF ABBREVIATIONS xxiv CHAPTER
1 COORDINATION CHEMISTRY AND CYTOTOXICITY OF TIN(IV) COMPOUNDS
1
1.1 Introduction 1 1.2 Long-Standing Chemotherapeutic Agents 4 1.3 Non-Platinum-Based Preclinical Investigations for
Anticancer Drug Applications 7
1.3.1 Titanium Compound 8 1.3.2 Ruthenium Compounds 9 1.3.3 Gallium Compounds 10 1.3.4 Iron Compounds 12 1.3.5 Cobalt Compounds 13 1.3.6 Gold Compounds 14 1.4 Tin(IV) compounds as potential anticancer agents 15 1.4.1 Tin(IV) Compounds of Oxygen and/or Nitrogen
Donor Ligands 17
1.4.2 Tin(IV) Compounds of Sulphur Donor Ligands 23 1.5 Chemistry and Bioactivities of Schiff Bases and Their
Tin(IV) Compounds 27
1.5.1 Anticancer Activity of Dithiocarbazate Schiff Bases and Their Tin(IV) Compounds
28
1.5.2 Anticancer Activity of Thiosemicarbazone Schiff Bases and Their Tin(IV) Compounds
31
1.6 Mechanism of Action 36 1.6.1 DNA Binding 36 1.6.2 Apoptosis 38 1.7 Knowledge Gap 40 1.8 Project Aims 41 1.9 Thesis Outline 42 2 SYNTHESIS, STRUCTURAL EVALUATION AND
CYTOTOXICITY STUDIES OF O-VANILLIN DERIVED DITHIOCARBAZATE SCHIFF BASES
43
2.1 Introduction 43 2.2 Experimental and Computational Methods 44 2.2.1 Materials 44

xii
2.2.2 Synthesis 45 2.2.2.1 S-N-R-Benzyldithiocarbazates (N = 2,
3, R = Methyl) 45
2.2.2.2 S-2-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzyl methylene) dithiocarbazate (1)
46
2.2.2.3 S-4-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzyl methylene) dithiocarbazate (2)
47
2.2.2.4 S-Benzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene) dithiocarbazate (3)
47
2.2.3 Physical Measurements 48 2.2.4 Single Crystal X-ray Structure Determination 48 2.2.5 Density Functional Theory (DFT) Calculations 49 2.2.6 MTT Assays 50 2.3 Results and Discussion 52 2.3.1 Synthesis 52 2.3.2 IR Spectral Analysis 53 2.3.3 NMR Spectroscopic Analysis 54 2.3.4 Mass Spectral Analysis 55 2.3.5 UV-vis Absorption Spectroscopy 55 2.3.6 X-ray Crystallography 56 2.3.7 In vitro Cytotoxicity 58 2.4 Conclusions 61 3 ORGANOTIN(IV) COMPOUNDS OF O-VANILLIN
SCHIFF BASES: SYNTHESIS, STRUCTURAL CHARACTERISATION, IN-SILICO STUDIES AND CYTOTOXICITY
62
3.1 Introduction 62 3.2 Experimental 64 3.2.1 Materials 64 3.2.2 Synthesis 65 3.2.2.1 Diphenyltin(IV) Compounds 65 3.2.2.2 Dimethyltin(IV) Compounds 66 3.2.3 Physical Measurements 68 3.2.4 Single Crystal X-ray Structure Determination 68 3.2.5 Computational Methods 69 3.2.5.1 DFT Calculations 69 3.2.5.2 Molecular Docking Studies 70 3.2.6 Biological Assays 71 3.2.6.1 MTT Assays 71 3.2.6.2 Quantification of Apoptosis by
Annexin V 71
3.2.6.3 Measurement of Reactive Oxygen Species (ROS)
72
3.2.6.4 DNA Binding Studies 73 3.3 Results and Discussion 73 3.3.1 Synthesis 73

xiii
3.3.2 IR Spectral Analysis 74 3.3.3 NMR Spectroscopic Analysis 75 3.3.4 X-ray Crystallography 76 3.3.5 UV-vis Absorption Spectroscopy 85 3.3.6 Biological Assays 86 3.3.6.1 Cytotoxicity 86 3.3.6.2 Annexin V Assays of 4, 5 and 6-
Treated RT-112 Cells 88
3.3.6.3 Measurement of Reactive Oxygen Species (ROS) in RT-112 Cells Treated with 4 and 5
89
3.3.6.4 DNA Interaction Studies 92 3.3.7 Molecular Docking Studies 93 3.4 Conclusion 98 4 SELECTIVE CYTOTOXICITY OF ORGANOTIN(IV)
COMPOUNDS CONTAINING CATECHOL DITHIOCARBAZATE SCHIFF BASES
101
4.1 Introduction 101 4.2 Experimental 102 4.2.1 Materials and Physical Measurements 102 4.2.2 Synthesis 102 4.2.2.1 S-N-R-benzyldithiocarbazate
(N = 2, 3; R = methyl) 102
4.2.2.2 Schiff bases 102 4.2.2.3 Diphenyltin(IV) Compounds 104 4.2.2.4 Dimethyltin(IV) Compounds 106 4.2.3 X-ray Crystallographic Analysis 107 4.2.4 DFT Calculations and Molecular Docking
Studies 108
4.2.5 MTT Assay 108 4.3 Results and Discussion 108 4.3.1 Synthesis 108 4.3.2 IR Spectral Analysis 111 4.3.3 NMR Spectroscopic Analysis 112 4.3.4 Mass Spectral Analysis 114 4.3.5 UV-vis Absorption Spectroscopy 114 4.3.6 Structure Descriptions for 12, 14, 15, 17 and 18 115 4.3.7 In vitro Cytotoxicity 125 4.3.8 DNA Binding Analysis 126 4.3.9 Molecular Docking Analysis 127 4.4 Conclusions 134 5 HOMOLEPTIC TIN(IV) COMPOUNDS OF
DINEGATIVE ONS DITHIOCARBAZATE SCHIFF BASES: SYNTHESIS, X-RAY CRYSTALLOGRAPHY, DFT AND CYTOTOXICITY STUDIES
136
5.1 Introduction 136 5.2 Experimental 137 5.2.1 Materials and Physical Measurements 137

xiv
5.2.2 Synthesis 138 5.2.2.1 Synthesis of Schiff Bases 138 5.2.2.2 Synthesis of Tin(IV) Compounds (19-
24) 138
5.2.3 Single Crystal X-ray Structure Determination 141 5.2.4 DFT Calculations and Molecular Docking
Studies 141
5.2.5 MTT Assays 142 5.3 Results and Discussion 142 5.3.1 Synthesis 142 5.3.2 IR Spectral Analysis 143 5.3.3 NMR Spectroscopic Analysis 144 5.3.4 UV-vis Absorption Spectral Analysis 145 5.3.5 X-ray Crystallography 145 5.3.6 In vitro Cytotoxic Activity 151 5.3.7 Molecular Docking Analysis 151 5.4 Conclusions 156 6 TIN(IV) COMPOUNDS OF THIOSEMICARBAZONE
SCHIFF BASES: SYNTHESIS, STRUCTURAL CHARACTERISATION AND IN VITRO CYTOTOXICITY
158
6.1 Introduction 158 6.2 Experimental 159 6.2.1 Materials and Physical Measurements 159 6.2.2 Synthesis 160 6.2.2.1 Schiff Bases 160 6.2.2.2 Tin(IV) Compounds derived from 25
and 27 162
6.2.2.3 Tin(IV) Compounds derived from 26 165 6.2.2.4 Tin(IV) Compounds derived from 28 166 6.2.3 DFT Calculations and Molecular Docking
Simulations 168
6.2.4 MTT Assay 168 6.3 Results and Discussion 169 6.3.1 Synthesis 169 6.3.2 IR Spectral Analysis 170 6.3.3 NMR Spectroscopic Analysis 171 6.3.4 Mass Spectral Analysis 173 6.3.5 UV-vis Absorption Spectral Analysis 173 6.3.6 In vitro Cytotoxic Activity 174 6.3.7 DNA Binding Studies 176 6.3.8 Molecular Docking Analysis 177 6.4 Conclusions 182 7 CONCLUSIONS AND FUTURE RECOMMENDATIONS 184 7.1 Conclusions 184 7.2 Future Recommendations 187

xv
REFERENCES 189 APPENDICES 215 BIODATA OF STUDENT 344 LIST OF PUBLICATIONS 345

xvi
LIST OF TABLES Table
Page
1.1 Cytotoxic activity of tin(IV) compounds against a panel cancer cells and survival of MRC-5 cells, compared to cisplatin.
20
2.1 Physical data of synthesised S-substituted dithiocarbazate derivatives.
46
2.2 Crystal data and refinement details for Schiff base 3
50
2.3 Hydrogen-bond geometry (Å, °) of 3 obtained from single crystal X-ray diffraction analysis. The structure is shown in Figure 2.2.
57
2.4 Summary of the in vitro cytotoxicity of compounds 1-3 in several cell lines, determined by MTT assay and expressed as GI50 values with standard errors. (GI50 is the concentration at which cell growth is inhibited by 50% over 72 h).
60
3.1 Crystal data and refinement details for compounds 7, 8 and 9.
70
3.2 Selected geometric parameters (Å, °) for 7, 8 and 9.
82
3.3 Summary of the in vitro cytotoxicity of the Schiff bases and their organotin(IV) compounds.
90
3.4 Binding constants (Kb), hypochromism (%), bathochromic shifts (nm) and Gibbs free energy (kJ mol−1) values for the interaction of organotin(IV) compounds with calf thymus DNA (CT-DNA).
94
3.5 Molecular docking data for the organotin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
96
4.1 Crystallographic data and refinement details for 12, 14, 15, 17 and 18.
110
4.2 Selected geometric data (Å, °) characterising 12, 14, 15, 17 and 18.
120
4.3 Summary of the in vitro cytotoxicity of the Schiff bases (10-12) and their organotin(IV) compounds (13-18) in several cell lines, determined by MTT assay, expressed as GI50 values with standard errors. GI50 is the concentration at which cell growth was inhibited by 50% over 72 h.
129
4.4 Binding constants (Kb), hypochromism (%), bathochromic shifts (nm) and Gibbs free energy (kJ mol-1) values for the interaction of organotin(IV) compounds with CT-DNA.
131

xvii
4.5 Molecular docking data for the organotin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
133
5.1 Crystal data and refinement details for complexes 20 and 21.
142
5.2 Selected geometric parameters (Å, °) for 20 and 21.
147
5.3 Summary of the in vitro cytotoxicity of tin(IV) compounds in several cell lines, determined by the MTT assay and expressed as a GI50 value with standard error. GI50 is the concentration of tin(IV) compounds at which cell growth is inhibited at 50% over 72 hours.
153
5.4 Molecular docking data for the tin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
154
6.1 In vitro cytotoxicity of tin(IV) compounds (29-30, 32-40) derived thiosemicarbazone Schiff bases (25-28) in several cell lines, determined by the MTT assay and expressed as a GI50 value with standard error. GI50 is the concentration at which cell growth is inhibited by 50% over 72 hours.
178
6.2 Molecular docking data for 27, 32, 33 and 34 with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
181

xviii
LIST OF FIGURES
Figure
Page
1.1 Estimated number of new cancer cases and mortality in 2018, worldwide, all cancers, both sexes and all ages.
2
1.2 Structure of different platinum-based anticancer drugs.
5
1.3 Schematic diagram showing the cytotoxic pathway for cisplatin. After entering the tumour cells, cisplatin is equated, then binds to cellular DNA.
6
1.4 The new cancer drug allows for low-dose cancer chemotherapy and promises fewer side effects.
7
1.5 The titanium-based compounds, budotitane (left) and titanocene dichloride (right).
9
1.6 The ruthenium-based compounds, NAMI-A (left) and KP1019 (right).
10
1.7 Structure of gallium-based compound, KP46 which was designed and used in clinical trials.
11
1.8 Schematic representation of the mechanism of action of gallium-based compounds. Tf = transferrin; NDP = nucleoside diphosphate; dNDP = desoxynucleoside diphosphate; BAX = a proapoptotic protein.
12
1.9 The iron-based compounds, ferrocenium picrate (left) and ferrocenium trichloroacetate (right).
13
1.10 The cobalt-based compounds, hexacarbonyldicobalt complex of the propargylic ester of acetylsalicylic acid (Co-ASS).
14
1.11 The gold-based compounds, auranofin (left) and [Au(dppe)2]Cl (right).
15
1.12 The structure of tin(IV) compounds containing oxygen and/or nitrogen donor ligands.
21
1.13 The structures of tin(IV) compounds containing sulphur donor ligands.
25
1.14 Tautomerism in (a) DTC and (b) TSC Schiff bases.
28
1.15 Different substituents at position R in dithiocarbazate derivatives.
30

xix
1.16 The structure of tin(IV) compounds derived from dithiocarbazate Schiff bases.
31
1.17 Chemical structure of (a) Methisazone and (b) Triapine.
32
1.18 The structure of tin(IV) compounds derived from thiosemicarbazone Schiff bases.
35
1.19 Molecular docked model of diphenyltin(IV) complex with DNA dodecamer duplex of sequence d(CGCGAATTCGCG)2 (PDB ID: 1BNA). The image provides the side view of the docked model of complexes.
37
1.20 a) Molecular docking simulation of diethyltin(IV) complex-DNA complex (binding site of topoisomerase II) b) detailed molecular interactions of diethyltin(IV) complex with amino acid residues.
38
1.21 Mechanism of extrinsic and intrinsic apoptotic pathway.
40
2.1 The structures of (a) vanillin and (b) o-vanillin.
43
2.2 The (a) thione and (b) thiol tautomeric forms of the Schiff bases 1-3.
54
2.3 HOMO-LUMO of (a) 1, (b) 2 and (c) 3.
56
2.4 The molecular structure of 3, showing the atom- labelling scheme and displacement ellipsoids at the 50% probability level.
57
2.5 Molecular packing in 3: (a) a perspective view of the supramolecular layer sustained by thioamide-N-H…S(thione) and phenyl-C-H…O(hydroxy) interactions and, (b) a view of the unit-cell contents shown in projection down the b axis, highlighting one layer in space-filling mode. The N-H…S and C-H…O interactions are shown as blue and orange dashed lines, respectively. For (a), non-interacting H atoms have been omitted.
58
3.1 The structures of Schiff bases (1-3) and organotin(IV) compounds (4-9).
64
3.2 Molecular structures of the first independent molecules of (a) 7, (b) 8 and (c) 9 showing atom labelling schemes. (d) Overlay diagram.
79
3.3 Crystallographic diagrams for 7: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (red image) and inverted-b (green) drawn so the SnC2 atoms of the Me2Sn moiety are overlapped and (c) a view of the supramolecular layer in the ab-plane (left image) and a view of the unit cell contents in projection down the a-axis with one layer
80

xx
highlighted in space-filling mode (right image). The C‒H…O and C‒H…π interactions are shown as orange and purple dashed lines, respectively.
3.4 Crystallographic diagrams for 8: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (blue image) and inverted-b (pink) drawn so the SnC2 atoms of the Me2Sn moiety are overlapped and (c) a view of the supramolecular dimer sustained by C‒H…π (chelate) interactions (left image; non-participating hydrogen atoms have been omitted) and a view of the unit cell contents in projection down the a-axis. The C‒H…O and C‒H…π interactions are shown as orange and purple dashed lines, respectively.
80
3.5 Crystallographic diagrams for 9: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (yellow image) and b (aqua) drawn so the five-membered rings are overlapped and (c) supramolecular dimer sustained by edge-to-edge chelate ring…benzene interactions (upper image) between Sn1-containing molecules, supramolecular layer sustained by edge-to-edge π (chelate ring)…π (ethoxybenzene) and phenyl-C‒H…π (methoxybenzene) interactions occurring between Sn2-containing molecules and a view of the unit cell contents in projection down the b-axis. The edge-to-edge π (chelate ring)…π (ethoxybenzene) and C‒H…π interactions are shown as blue and purple dashed lines, respectively.
84
3.6 HOMO-LUMO of (a) 4, (b) 5, (c) 6, (d) 7, (e) 8 and (f) 9.
86
3.7 Apoptosis detection through fluorescence microscopy. Cells were treated for 24 h with 4 (0.31 µM), 5 (1.66 µM) and 6 (0.58 µM), and the negative control (DMSO) in complete media. After staining with Annexin V and PI, necrotic and apoptotic cells were detected by fluorescence microscopy (20×).
91
3.8 Percent reactive oxygen species (ROS) production in RT-112 cells treated with (a) 4 (0.31 μM) (b) 5 (1.66 μM) for 24 h and stained with 1 mM DCFH-DA for 60 minutes at 37 °C. DMSO and H2O2 acted as negative and positive controls, respectively.
92
3.9 (a) Electronic absorption spectra of (i) 4, (ii) 5 and (iii) 6; (b) Plot of [DNA]/εa − εf vs [DNA] for absorption titration of DNA with (i) 4, (ii) 5 and (iii) 6. The arrow indicates the change in absorbance in tandem with increasing DNA concentration.
95
3.10 (a) Schematic representation of organotin(IV) compounds that fit well in the grooves of the DNA strand obtained by docking simulations.
97

xxi
4.1 The structures of catechol dithiocarbazate Schiff bases (10-12) and
their organotin(IV) compounds (13-18).
102
4.2 (a) Thione and (b) thiol tautomerism of Schiff bases 10-12.
109
4.3 Frontier molecular orbitals LUMO and HOMO of the optimised (a) 10, (b) 11, (c) 12, (d) 13, (e) 14, (f) 15, (g) 16, (h) 17 and (i) 18 using B3LYP functional.
116
4.4 Molecular structure of 12 and atom labelling scheme.
117
4.5 ORTEP structures showing atom labelling scheme and overlay diagrams for (a) the two independent molecules of 14, (b) the two independent molecules of 15, (c) 17, (d) 18 and (e) overlay diagram for 17 and 18. For the overlay diagrams, molecules have been overlapped so that the CS2 residues are coincident.
119
4.6 A view of the linear supramolecular chain along [1 0 4] in the crystal of 12. The hydroxyl-O‒H…O(hydroxyl) and thioamide-N‒H…S(thione) hydrogen bonds are shown as orange and green dashed lines, respectively.
123
4.7 A view of the zig-zag chain along [0 0 1] in the crystal of 14. The hydroxyl-O‒H…O(hydroxyl) and thioamide-N‒H…S(thione) hydrogen bonds are shown as orange and green dashed lines, respectively. For reasons of clarity, the hydrogen atoms have been removed and only the ipso-carbon atoms of the tin-bound phenyl rings shown.
125
4.8 (a) Electronic absorption spectra of (i) 16, (ii) 17 and (iii) 18; (b) Plot of [DNA]/εa - εf vs [DNA] for absorption titration of DNA with (i) 16, (ii) 17 and (iii) 18. (The arrow indicates the change in absorbance in tandem with increasing DNA concentration).
130
4.9 (a) Schematic representation of organotin(IV) compounds that fit well in the grooves of the DNA strand obtained by docking simulations. The two double-stranded DNA comprised of the phosphate deoxyribose backbone with guanine (DG, red), cytosine (DC, blue), adenine (DA, pink) and thymine (DT, orange). (b) Molecular interactions of organotin(IV) compounds within the grooves of double stranded DNA residues.
132
5.1 The structures of homoleptic tin(IV) compounds (19-24).
137
5.2 Frontier MOs of (a) 19, (b) 20, (c) 21, (d) 22, (e) 23 and (f) 24.
146
5.3 Molecular structures of the molecules in (a) 20 (first independent molecule; the structure for the second independent molecule is
148

xxii
shown in Appendix Figure A5.1) and (b) 21, showing atom labelling schemes and 50% displacement ellipsoids. (c) Overlay diagram of the molecules in 20 (red image for the first independent molecule), 20a (green, inverted second molecule) and 21 (blue). Molecules have been overlapped so the Sn,S1,N2 chelate rings are coincident.
5.4 Homoleptic tin(IV) compounds that have been studied.
150
5.5 (a) Schematic representation of 19 (cyan), 21 (yellow), 22 (blue), 23 (grey) and 24 (green) that fit well in the grooves of the DNA strand obtained by docking simulations. The two double-stranded DNA comprised of the phosphate deoxyribose backbone (grey) with guanine (green), cytosine (purple), adenine (DA, red) and thymine (cyan). (b) Molecular interactions of 19, 21, 22, 23 and 24 within the grooves of double stranded DNA residues. Hydrogen bond, electrostatic and hydrophobic interactions are depicted by green, orange and pink dot lines, respectively.
155
5.6 The small modification (blue) of backbone of (a) dithiocarbazate Schiff bases to produce (b) thiosemicarbazone Schiff bases.
157
6.1 The structures of (a) dithiocarbazate and (b) thiosemicarbazone Schiff bases.
159
6.2 Frontier MOs (a) 25, (b) 26, (c) 27, (d) 28, (e) 29, (f) 30, (g) 31, (h) 32, (i) 33, (j) 34, (k) 35, (l) 36, (m) 37, (n) 38, (o) 39 and (p) 40.
174
6.3 Electronic absorption spectra of (i) 27, (ii) 32, (iii) 33 and (iv) 34. The arrow indicates the change in absorbance in tandem with increasing DNA concentration.
180
6.4 Molecular interactions of (a) 27, (b) 32, (c) 33 and (d) 34 within the grooves of double stranded DNA residues. Hydrogen bond, electrostatic and hydrophobic interactions are depicted by green, orange and pink dot lines, respectively.
180

xxiii
LIST OF SCHEMES
Scheme
Page
2.1 Synthetic pathway for the formation of 1-3.
53
3.1 Synthesis of organotin(IV) compounds (4-9).
74
4.1 Synthetic pathway for the formation of Schiff bases 10, 11 and 12.
109
4.2 Synthetic pathway for the synthesis of organotin compounds 13-18.
111
5.1 Synthetic pathway for the synthesis of 19-24.
143
6.1 Synthetic pathway of thiosemicarbazone Schiff bases 25-28.
169
6.2 Synthetic pathway of tin(IV) compounds (29-40) of thiosemicarbazone Schiff bases.
170

xxiv
LIST OF ABBREVIATIONS
3677 Human melanoma cell line A2780 Human ovarian carcinoma cancer cell line A431 Human skin cancer cell line A549 Human lung carcinoma line B16F10 Murine melanoma cell line B3LYP Berke's three-parameter exchange potential and the
Lee–Yang–Parr correlation functional Bcap37 Mammary tumour cell line BE2-C Neuroblastoma cancer cell line Bel-7402 Hepatocellular carcinoma line BGC-823 Gastric carcinoma cell line BSA Bovine serum albumins CDCl3 Deuterated chloroform CH3OH Methanol CHO Chinese hamster ovary cell line CIF Crystallographic Information File CML-T1 Chronic myeloid leukemic cell line CoLo205 Colon carcinoma cell line CT-DNA Calf thymus deoxyribonucleic acid DCF 2′,7′-Dichlorodihydrofluorescein DCF-DA Dichloro-dihydro-fluorescein diacetate DCM Dichloromethane DFT Density Functional Theory DMEM Dulbecco’s modified Eagle’s medium DMSO-d6 Deuterated dimethylfulfoxide DNA Deoxyribonucleic acid DTC Dithiocarbazate Du145 Human prostate carcinoma cancer cell line EJ-28 Muscle invasive bladder cancer cell line Et3N Triethylamine FT-IR Fourier transform infrared spectroscopy GI50 Concentration of the anti-cancer drug that inhibits the growth of
cancer cells by 50% relative to untreated control H2981 Human lung adenocarcinoma cell line H460 Human non-small lung carcinoma cell line HCT116 Human colorectal carcinoma cell line HCT-8 Colon carcinoma cell line HeLa Human cervical epithelioid carcinoma cell line HepG2 Human liver carcinoma cell line HL-60 Human acute myeloid leukemia cell line HT29 Human colon adenocarcinoma cell line IC50 Concentration of compound that inhibits a biological activity by
50% K562 Human myelogenous leukemia cell line KB Nasopharyngeal carcinoma cell line KP1019 Indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] KP46 Tris(8-quinolinolato)gallium(III)

xxv
LanL2DZ Los Alamos National Laboratory 2-double-z LMCT Ligand-metal charge transfer LMS Leiomysarcoma cell line LOX Lipoxygenase m/z Mass-to-charge ratio MCF-10A Normal breast cell line MCF-7 Human breast carcinoma with positive oestrogen receptor cell line MDA-MB-231 Human breast carcinoma with negative oestrogen receptor cell line Me2SnCl2 Dimethyltin(IV) dichloride MeOH Methanol MIA Pancreatic cancer cell line MKT4 Titanocene dichloride MRC-5 Normal foetal lung fibroblast cell line MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide NAMI-A Imidazolium trans-[tetrachloro(dimethylsulfoxide)(1H-
imidazole)ruthenate(III)] NCI National Cancer Institute NMR Nuclear magnetic resonance ONS Oxygen-nitrogen-sulphur oVa o-Vanillin; 2-hydroxy-3-methoxybenzaldehyde P388 Lymphocytic leukaemia cell line PDB Protein Data Bank Ph Phenyl Ph2SnCl2 Diphenyltin(IV) dichloride PI Propidium Iodide r.m.s Root-mean-square ROS Reactive Oxygen Species RR Ribonucleotide reductase RT-112 Minimum muscle invasive bladder cancer cell line S1 S-2-Methylbenzyldithiocarbazate S2 S-4-Methylbenzyldithiocarbazate S3 S-Benzyldithiocarbazate SJ-G2 Pediatric glioblastoma cell line SMMC-7721 Human hepatocellular carcinoma cell line TiO2 Titanium oxide Tris-HCl Tris(hydroxymethyl)aminomethane hydrochloride U2-OS Human osteosarcoma cell line U87 Human glioblastoma cell line UV-vis Ultraviolet-visible WiDr Human colon carcinoma cell line WRL-68 Normal liver cell ORTEP Oak Ridge thermal ellipsoid plot PBS Phosphate buffer saline PEG Polyethylene glycol


1
CHAPTER 1
COORDINATION CHEMISTRY AND CYTOTOXICITY OF TIN(IV) COMPOUNDS
1.1 Introduction
Cancer is the proliferation of abnormal cells which can occur in various body parts.
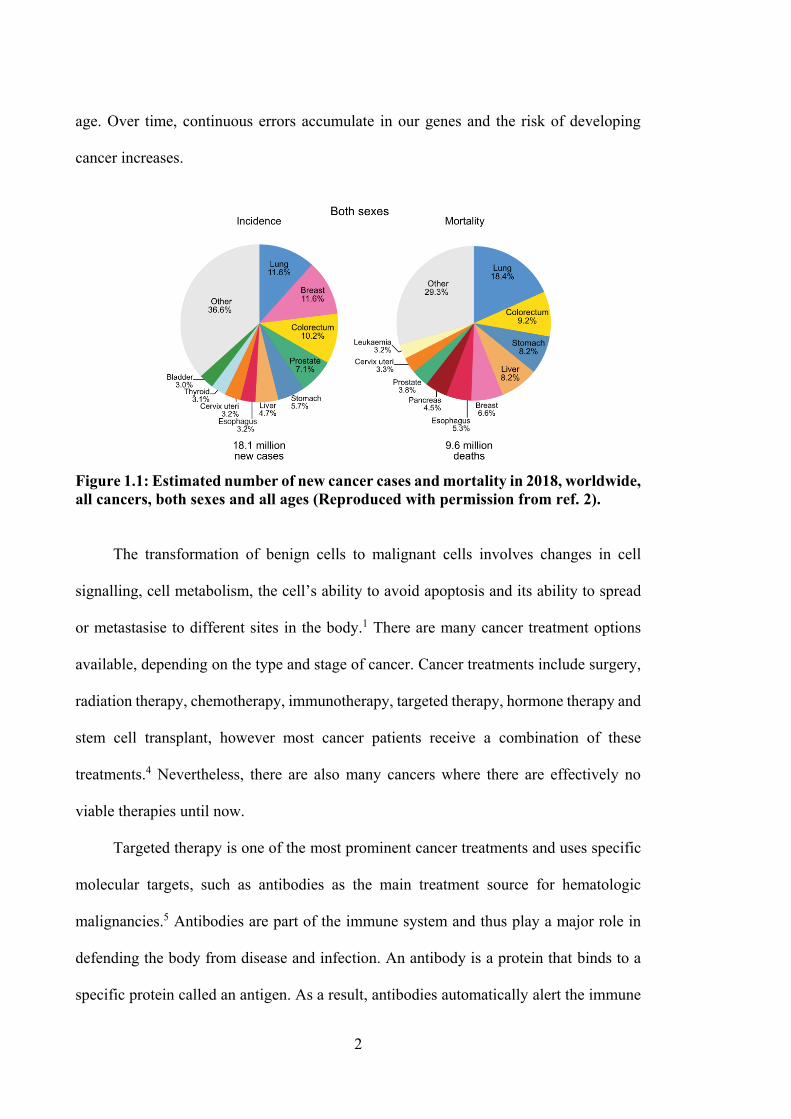
Globally, the International Agency for Research on Cancer has reported 18.1 million new
cancer cases (Figure 1.1) and 9.6 million deaths due to cancer in 2018.2 The types of
cancer with highest mortality rates are lung (1.76 million deaths), colorectum (880 792),
stomach (782 685), liver (781 631 deaths) and breast (626 679 deaths) cancer.2 Cancer
starts by certain changes to genes that control the cells function, especially on how the
cells grow and divide. Genes are essential and carry the instructions to make proteins in
cells. The changes or damages in genes called mutated genes, where they do not work
properly in giving instructions to the cells and ultimately the cells grow out of control,
which lead to cancer. Cancer cells are also immature and do not develop into mature cells
with their specific functions, able to avoid the immune system and capable to ignore
signals that tell them to stop dividing. Furthermore, they also can spread to other body
parts through circulatory and lymphatic systems.1 Cancer can be caused by person’s
genetic factors if the genetic changes present in germ cells, and as well as external agents
such as a high exposure rate to ultraviolet, ionising radiation and/or cigarette smoke,
particularly at an advanced age. Occupational exposure to chemical carcinogens is also a
factor, especially for those who are regularly exposed to aromatic amines such as beta-
naphthylamine, xenylamine and benzidine.3 Furthermore, infections from certain viruses,
bacteria and parasites also can initiate the development of cancer. Undoubtedly, ageing
is another factor for the development of cancer as the frequency of cancer increases with
2
age. Over time, continuous errors accumulate in our genes and the risk of developing
cancer increases.
Figure 1.1: Estimated number of new cancer cases and mortality in 2018, worldwide, all cancers, both sexes and all ages (Reproduced with permission from ref. 2). The transformation of benign cells to malignant cells involves changes in cell
signalling, cell metabolism, the cell’s ability to avoid apoptosis and its ability to spread
or metastasise to different sites in the body.1 There are many cancer treatment options
available, depending on the type and stage of cancer. Cancer treatments include surgery,
radiation therapy, chemotherapy, immunotherapy, targeted therapy, hormone therapy and
stem cell transplant, however most cancer patients receive a combination of these
treatments.4 Nevertheless, there are also many cancers where there are effectively no
viable therapies until now.
Targeted therapy is one of the most prominent cancer treatments and uses specific
molecular targets, such as antibodies as the main treatment source for hematologic
malignancies.5 Antibodies are part of the immune system and thus play a major role in
defending the body from disease and infection. An antibody is a protein that binds to a
specific protein called an antigen. As a result, antibodies automatically alert the immune

3
system to destroy cells containing antigens.6 To note here that this approach only works
if the targeted receptor is present on the cell surface and appropriately clustered. They are
also problematic as this approach shuts down a wide array of other signalling pathways.
Antibody-dependent cellular cytotoxicity is an effective treatment, under the right
conditions, but it is also a treatment with high numbers of level 5 adverse events which
are Type I immediate reactions (anaphylaxis, urticaria); Type II reactions (immune
thrombocytopenia, neutopenia, hemolytic anemia); Type III responses (vasculitis, serum
sickness; some pulmonary adverse events); and Type IV delayed mucocutaneous
reactions as well as infusion reactions/cytokine release syndrome, tumor lysis syndrome,
progressive multifocal leukoencephalopathy and cardiac events, especially the new
programmed cell death 1 (PD1) and anti-PD1 mAbs (nivolumab and pembrolizumab)
based approaches.7,8 Researchers have been successful in designing and developing many
copies of antibodies that specifically target cancer cell antigens, known as monoclonal
antibodies (mAbs).9 There are different types of mAbs used in cancer treatment, for
instance naked mAbs, bispecific mAbs and conjugated mAbs. Naked mAbs are
antibodies that work by themselves without the presence of drugs or radioactive materials
and are commonly used to treat cancer. However, antibodies can also bind to the antigen
of non-cancerous cells, as well as free-floating proteins. Alemtuzumab (Campath®) was
the first humanised mAb therapeutic, depleting lymphocytes, monocytes and dendritic
cells, where this antibody is very commonly used to treat chronic lymphocytic leukemia
patients.10 Bispecific mAbs work with two different proteins at the same time. For
example, blinatumomab (Blincyto) is used to treat some types of acute lymphocytic
leukemia, where the mAbs attached to the CD19 protein found on some leukemic and
lymphoma cells as well as to the CD3 protein found on immune cells (T cells).10
Conjugated mAbs consist of antibodies joined together with chemotherapy drugs or

4
radioactive particles. Even though new technologies have been developed to treat cancer,
these approaches do not guarantee a cure or remission, thus, chemotherapy remains of
much interest due to the effectiveness of the drugs that can work throughout the body.
1.2 Long-Standing Chemotherapeutic Agents
Chemotherapy is one of the most powerful tools used in cancer treatment. Ongoing
research continues to find new and effective low cost chemotherapy drugs with fewer side
effects as compared to existing ones. Chemotherapy can kill cancer cells that have spread
or metastasised to other parts of the body from their original primary tumour site.
Inorganic compounds offer potential advantages for chemotherapy over organic
compounds. The great bioactivity of inorganic compounds can be explained on the basis
of Tweedy’s chelation theory, where chelation reduces the polarity of the metal ion due
to partial sharing of its positive charge with donor atoms and also due to electron
delocalisation over the whole chelate rings. Such chelation increases the lipophilic
character of the inorganic compounds which favors the permeation of the compounds
through the lipid layer of the cell membrane.11–13 Importantly, inorganic drugs display a
wide range of coordination numbers, various geometries, oxidation states and
thermodynamic and kinetic characteristics of ligand substitution that offer the medicinal
chemist opportunities to explore metal-containing compounds with different strategies.14
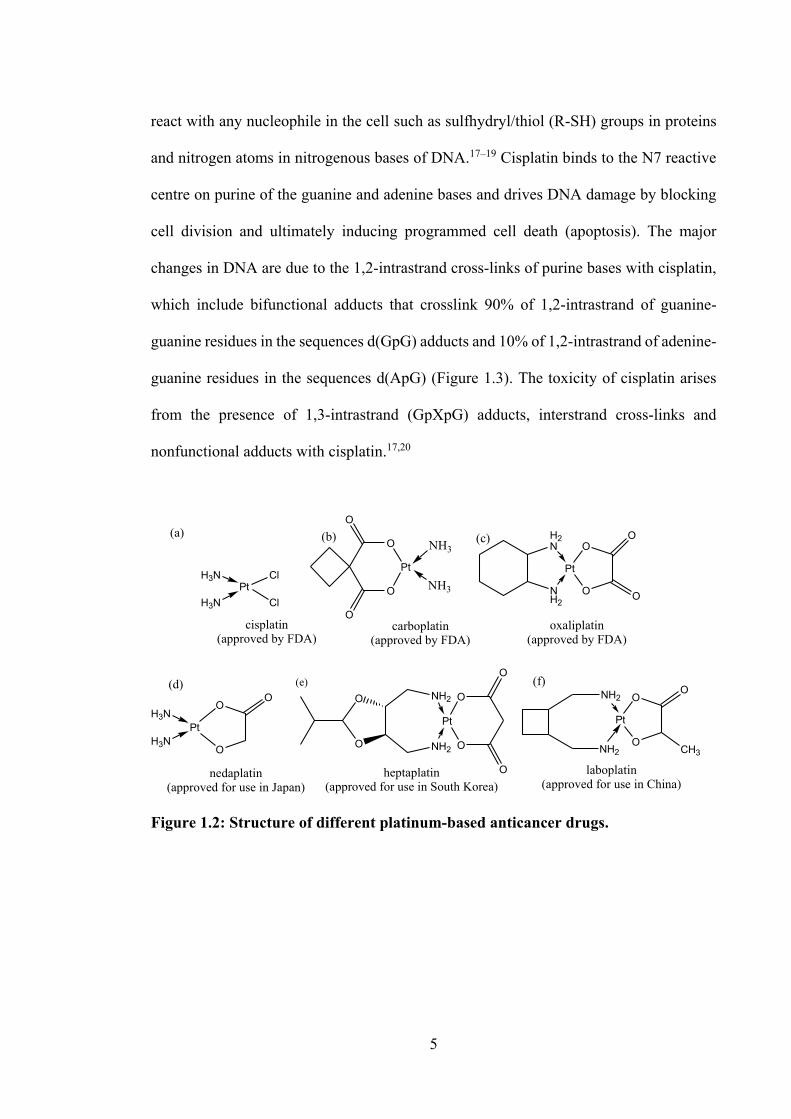
An effective and long-standing metallo-chemotherapy agent is cisplatin (Figure 1.2(a))
that falls into the class of deoxyribonucleic acid (DNA)-damaging agents. Cisplatin is
clinically proven to combat many cancers including testicular, ovarian, cervical, head and
neck, bladder, lung and colorectal cancers.15,16 Cisplatin becomes activated once it enters
the cell, where in the cytoplasm, the two Cl atoms are displaced by water molecules
through hydrolysis. The hydrolysed product formed behaves as an electrophile that can
5
react with any nucleophile in the cell such as sulfhydryl/thiol (R-SH) groups in proteins
and nitrogen atoms in nitrogenous bases of DNA.17–19 Cisplatin binds to the N7 reactive
centre on purine of the guanine and adenine bases and drives DNA damage by blocking
cell division and ultimately inducing programmed cell death (apoptosis). The major
changes in DNA are due to the 1,2-intrastrand cross-links of purine bases with cisplatin,
which include bifunctional adducts that crosslink 90% of 1,2-intrastrand of guanine-
guanine residues in the sequences d(GpG) adducts and 10% of 1,2-intrastrand of adenine-
guanine residues in the sequences d(ApG) (Figure 1.3). The toxicity of cisplatin arises
from the presence of 1,3-intrastrand (GpXpG) adducts, interstrand cross-links and
nonfunctional adducts with cisplatin.17,20
PtClH3N
H3N ClO
Pt
O
O
O
NH3
NH3
NH2
Pt
H2N
O
OO
O
cisplatin(approved by FDA)
carboplatin(approved by FDA)
oxaliplatin(approved by FDA)
PtH3N
H3NO
OO
nedaplatin(approved for use in Japan)
heptaplatin(approved for use in South Korea)
Pt
NH2
NH2O
OO
CH3
laboplatin(approved for use in China)
(a) (b) (c)
(d) (e) (f)O
Pt
O
O
O
NH2
NH2
O
O
Figure 1.2: Structure of different platinum-based anticancer drugs.

6
Figure 1.3: Schematic diagram showing the cytotoxic pathway for cisplatin. After entering the tumour cells, cisplatin is hydrolysed, then binds to cellular DNA (Reproduced with permission from ref. 21). Platinum-based compounds remain the most effective chemotherapeutic agents for
tumour therapy, such as carboplatin, oxaliplatin, nedaplatin, heptaplatin and laboplatin
(see Figure 1.2), except for all breast and colon cancers. However, such platinum drugs
are prone to side effects such as anemia, diarrhea, alopecia, petechia, fatigue
nephrotoxicity, emetogenesis, ototoxicity, neurotoxicity and also face increasing
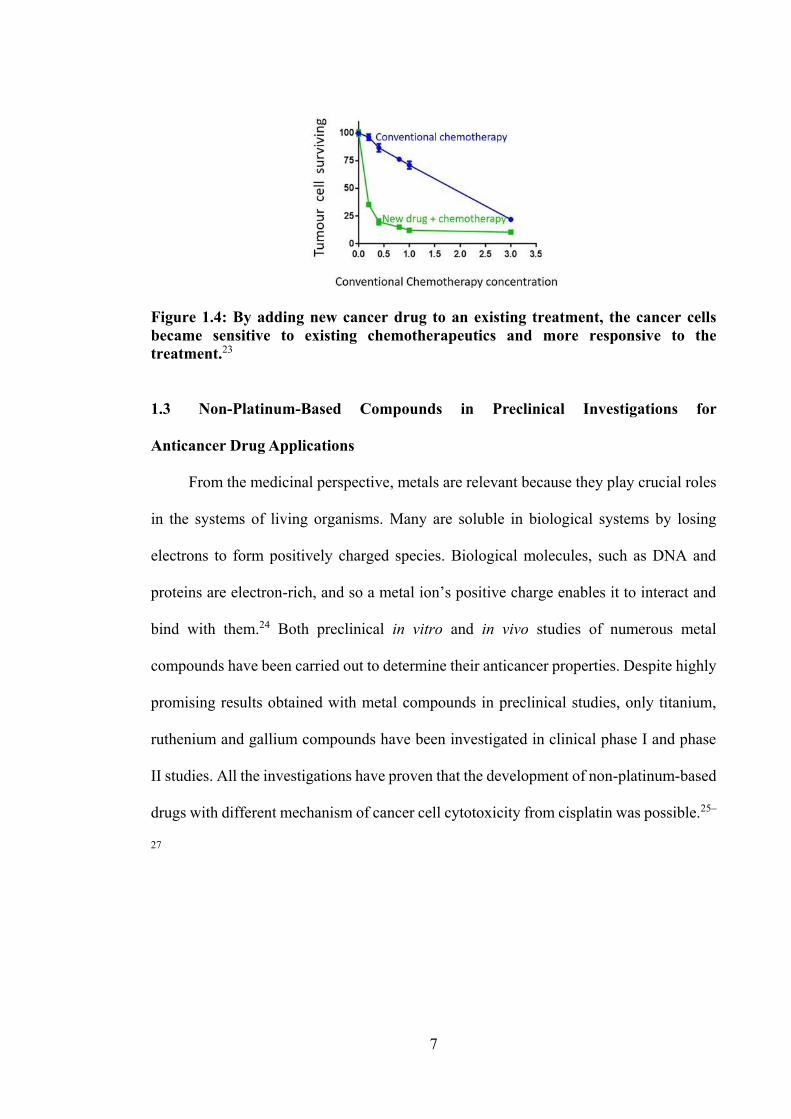
resistance from cancer cell lines.22 Figure 1.4 highlights the effectiveness of a
combination of new drugs and chemotherapy to prevent drug resistance in treating cancer
patients. It is therefore evident that the search for novel non-platinum-based compounds
will potentially open up new avenues in the development of chemotherapeutic drugs.
Furthermore, the discovery of new drugs with enhanced activity, selectivity and
bioavaibility, as well as fewer side effects than conventional drugs to treat cancer patients
is crucial.
7
Figure 1.4: By adding new cancer drug to an existing treatment, the cancer cells became sensitive to existing chemotherapeutics and more responsive to the treatment.23
1.3 Non-Platinum-Based Compounds in Preclinical Investigations for
Anticancer Drug Applications
From the medicinal perspective, metals are relevant because they play crucial roles
in the systems of living organisms. Many are soluble in biological systems by losing
electrons to form positively charged species. Biological molecules, such as DNA and
proteins are electron-rich, and so a metal ion’s positive charge enables it to interact and
bind with them.24 Both preclinical in vitro and in vivo studies of numerous metal
compounds have been carried out to determine their anticancer properties. Despite highly
promising results obtained with metal compounds in preclinical studies, only titanium,
ruthenium and gallium compounds have been investigated in clinical phase I and phase
II studies. All the investigations have proven that the development of non-platinum-based
drugs with different mechanism of cancer cell cytotoxicity from cisplatin was possible.25–
27

8
1.3.1 Titanium Compounds
Titanium in solution is dominated by a +4 oxidation state and has a strong tendency
to hydrolyse and form highly insoluble TiO2. However, this hydrolysis reaction can be
reduced by introducing appropriate ligands coordinated to the titanium centre, and this is
the common strategy for the development of titanium-based anticancer drugs. The
octahedral titanium β-diketone compound, budotitane (Figure 1.5, left), was the first non-
platinum-metal-based compound to enter clinical trials for the treatment of a broad
spectrum of cancers. The main target of the titanium-based compound was the inhibition
of DNA synthesis, triggering apoptosis and disrupting the topoisomerase II enzyme in
cancer cells, which led to the induction of the cell death pathway. Unfortunately, cancer
cells did not respond to budotitane after infusion twice in a week with a dose of 100
mg/m2 in an 18 patient in a Phase I clinical, which caused undesirable cardiac arrhythmia
in the patients. Unlike budotitane, titanocene dichloride (MKT4) (Figure 1.5, right)
showed good clinical outcome in Phase I trials. Testing on 40 patients with solid tumours
showed minor responses in bladder carcinoma and non-small cell lung cancer after
treatment with a lyophilised formulation of MKT4. The gastrointestinal side effects could
be reduced but were still dose-limiting because of possible accumulative nephrotoxicity.28
In clinical Phase II trials, no responses were noted in the 14 patients with metastatic renal-
cell carcinoma and 15 patients with metastatic breast cancer after administration of 270
mg/m2 of the complex every three weeks.29 Researchers also found that MKT4 inhibited
the synthesis of DNA by covalently binding to the DNA via the phosphate backbone and
as well as induced apoptosis. Binding studies suggested that the Ti(IV) ions were taken
up by the transport protein transferrin (which acts as a vehicle), where only this protein
had the capability of transporting other metal ions to the cells.29–31

9
Figure 1.5: The titanium-based compounds, budotitane (left) and titanocene dichloride (right) 27 1.3.2 Ruthenium Compounds
Ruthenium compounds have been investigated for their potential antitumour
activity since the 1970s. For example the octahedral imidazolium trans-
[tetrachloro(dimethylsulfoxide)(1H-imidazole)ruthenate(III)] (NAMI-A) and indazolium
trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019) complexes (Figure 1.6) both
entered clinical trials for the treatment of tumours. Both of them were significantly
different in their structures as compared to the typically square planar Pt(II)-based drugs.
KP1019 showed a wide range of growth inhibitory effects on in vitro tumour cells
including colorectal carcinomas and a variety of primary explanted human tumours.32 In
addition, NAMI-A had marked efficacy against the formation of metastases and had the
ability to control the angiogenic potential of tumours.33,34 The safe administration of
NAMI-A was a three hour infusion dose of 300 mg/m2/day for five days, over a duration
of three weeks. Only two patients showed common toxicity criteria with grade 3 and 4
hypersensitivity reaction.35 KP1019 had excellent uptake into the cells and hence the cells
underwent apoptosis by an intrinsic mitochondrial pathway. At this point, it caused
oxidative stress and DNA damage, which suggested that KP1019 could untwist and bend
DNA.36
OTi
O
OO O
O
TiCl
Cl

10
RuClCl
Ru
N
ClCl
OMe
Me
NH
RuCl
N
ClN
ClCl
NH
HN
Figure 1.6: The ruthenium-based compounds, NAMI-A (left) and KP1019 (right).27
1.3.3 Gallium Compounds
Among the p-block metals, gallium-based compounds have entered clinical trials
for the treatment of diverse disorders, categorised as (a) accelerated bone resorption, with
or without elevated plasma calcium; (b) autoimmune disease and allograft rejection; (c)
certain cancers; and (d) infectious diseases.37 Gallium nitrate, gallium chloride and
gallium maltolate were the gallium-based compounds which entered clinical trial Phase I
and II, but not all gallium species were effective for all types of malignancies. For
example, gallium nitrate was not successful in treating various malignancies such as
melanoma and breast cancer, but was effective in other types of cancer. This was because
gallium nitrate was readily hydrolysed in biological media to give non-soluble gallium
oxides which were responsible in blocking the absorption and membrane permeation of
the gallium ions thus reducing its effectivity in cancer treatments. During Phase II clinical
trials, gallium nitrate was administered as treatment for metastatic urothelial carcinomas
with a combination of two chemotherapy medication, vinblastine and ifosfamide and
returned a very good response rate (67%) on patients. However, patients suffered from
strong toxicity effects, especially granulocytopenia. Furthermore, gallium chloride was
administered in combination of paclitaxel, gemcitabine or vinorelbine at preclinical

11
stages. To improve the bioactivities of previous gallium-based compounds, researchers
developed the orally bioavailable gallium compound, [tris(8-quinolinolato)gallium(III)]
(KP46) (Figure 1.7) which were studied in phase I clinical trials. KP46 showed better
results than the combination of platinum compounds due to its lipophilic character of
ligands after oral administration.38,39 The mechanism of action of gallium-based
compounds is mainly by the binding of Ga3+ to the transferrin followed by accumulation
in endosomes. After Ga3+ was transported into the cytosol, Ga3+ ions were able to inhibit
the function of ribonucleotide reductase (RR), where Ga3+ ions played a role in catalysing
the conversion of ribonucleotides to deoxyribonucleotides. The coordination of Ga3+ with
the RR inhibitor activated the cell cycle arrest and ultimately the cells underwent
apoptosis through a mitochondrial pathway (Figure 1.8).40
N
Ga O
NO
N
O
Figure 1.7: Structure of gallium-based compound, KP46, which was designed and used in clinical trials.

12
Figure 1.8: Schematic representation of the mechanism of action of gallium-based compounds. Tf = transferrin; NDP = nucleoside diphosphate; dNDP = deoxynucleoside diphosphate; BAX = a proapoptotic protein (Reproduced with permission from ref. 41). 1.3.4 Iron Compounds
Iron is an essential element in biological systems and also an important nutrient
involved in cancer cells’ proliferation. The first antineoplastic iron complexes, which
were ferrocenium picrate and ferrocenium trichloroacetate (Figure 1.9) showed the best
cytotoxic effects with optimum cure rates of 100% against Ehrlich ascites tumour in CF1
mice.42 These two excellent ferrocene compounds underwent an oxidation process, where
ferrocene could be converted into ferrocenium ions inside the tumour cells. The
mechanism of ferrocenium salts was not via direct interaction with DNA, but instead via
the generation of reactive oxygen species (ROS) which led to protein and DNA damage,
and ultimately cell death. Moreover, the efficacy of ferrocene derivatives as antiestrogen
drugs depended on their proton-coupled electron transfer (redox potential) including
intramolecular interactions in the molecular π system.43 Other researchers claimed that
the stable iron complexes of pentadentate pyridyl ligands showed high cytotoxicity
against HeLa (human cervical epithelioid carcinoma cells) and HepG2 (human liver

13
carcinoma cells). They were also able to induce apoptosis by cleaving the supercoiled
plasmid DNA in vitro.44
Fe
NO2
NO2
O
O2N
Fe
Cl
Cl
Cl
O
O
Figure 1.9: The iron-based compounds, ferrocenium picrate (left) and ferrocenium trichloroacetate (right). 1.3.5 Cobalt Compounds
Cobalt alkyne complexes exhibit good potential as antitumour drugs and were first
reported in studies against murine leukemic cells. Further investigations and extensive
efforts have been carried out on the most active compound, the hexacarbonyldicobalt
complex of the propargylic ester of acetylsalicylic acid (Co-ASS) (Figure 1.10) against
3677 (human melanoma), H2981 (lung adenocarcinoma),45 MCF-7 (human breast
carcinoma with positive estrogen receptor) and MDA-MB-231 (human breast carcinoma
with negative estrogen receptor) cell lines.46 Studies of inhibitory potential revealed that
Co-ASS inhibits the cyclooxygenase enzyme (COX-1 and COX-2). A synergistic effect
was produced by the combination of Co-ASS with other antitumour drugs, for instance
tyrosine kinase inhibitor in HL-60 (human acute promyelocytic leukemia) and LAMA-
84 and CML-T1 (human chronic myeloid leukemic) cell lines in vitro.47

14
O
O
O
O
Co2(CO)6
Figure 1.10: The cobalt-based compounds, hexacarbonyldicobalt complex of the propargylic ester of acetylsalicylic acid (Co-ASS).
1.3.6 Gold Compounds
Early studies reported that gold-based compounds greatly inhibited the growth of
cultured cancer cells. Auranofin (Figure 1.11), gold-based compound was proven to
inhibit DNA, RNA and protein synthesis, where these inhibitions could affect the
propagation of cancer cells. Morphological changes, for example membrane blebbing and
pitting were observed under the exposure of cells to auranofin.48 The development of
gold-based compounds continued with bis(diphenylphosphine)ethane ligand,
[Au(dppe)2]Cl (Figure 1.11). This gold containing compound demonstrated good
cytotoxicity against cultured cancer cells, where it produced DNA-protein crosslinks and
DNA strand breaks in cells after [Au(dppe)2]Cl exposure. However, cardiotoxic effects
were observed which attributed to the disruption of mitochondrial function.49,50 The
successful development of chloro(triethylphosphine)gold(I), [Au(dppe)2]Cl and
auranofin as anticancer drugs by the inhibition of mitochondrial human glutathione
reductase and thioredoxin reductase enzyme was demonstrated, where this enzyme was
commonly involved in the process of cell division.51,52 As a result, the binding of gold
compounds with catalytic residues could cause alteration in the cellular process, finally
inducing apoptosis of the cells.

15
O SO
O
H3C
O O
O
H3C
O
H3C
O
AuP
CH3 CH3
H3C
CH3
O
P
P
P
P
Au
Figure 1.11: The gold-based compounds, auranofin (left) and [Au(dppe)2]Cl (right). 1.4 Tin(IV) compounds as potential anticancer agents
Among the non-platinum compounds, tin(IV) compounds have been a promising
new lead for the development of anticancer drugs. Tin, a post-transition metal in period
5 has two 5s and two 5p electrons in its outer shell orbital. Tin can lose two electrons
from the 5p orbital to form Sn2+, or share all four outer electrons with other atoms to
achieve a stable electron configuration. Tin(IV) compounds show a diverse range of
applications, from biological to industrial uses. In recent years, the interest in tin(IV)
compounds in medicinal chemistry has increased significantly, particularly with reference
to the development of anticancer drugs,53–55 which showed their effectiveness against a
number of tumours. The National Cancer Institute (NCI) has tested the largest number of
tin compounds compared to other metals, about 2000 tin-based compounds as compared
to platinum (1600), iron (900) and cobalt (800).56,57

16
Previous studies have reported that tin(IV) compounds may bind to the
glycoproteins or cellular proteins of living organisms, and also may cause cell death via
interaction with DNA.58–61 The general interaction mechanism between small
molecules/drugs and DNA mainly occurs via three non-covalent interactions:
intercalative binding, major/minor groove binding and electrostatic binding.
Spectroscopic and molecular modelling techniques are useful in order to investigate the
DNA binding interactions.62
The earliest studies on the cytotoxicity of tin(IV) compounds against mouse cancer
were carried out in 1929.63 Some 40 years later, a number of tin(IV) compounds
R2SnX2.Ln, (R = Me, Et, Pr, Bu, c-hexyl, and Ph groups; X = halogen; L = mono- (n=2)
or bidentate (n = 1) N/O-donor ligand) were investigated for their in vivo antitumour
activity against lymphocytic leukaemia (P388) in mice.64 It was reported that many
tin(IV) compounds showed promising activity against P388, but were found to be inactive
against some solid tumours tested, e.g. B16 melanocarcinoma, CD8F1 mammary tumour,
CX-1 colon xenograft, colon 38, L1210 lymphoid leukaemia, LX-l lung xenograft, Lewis
lung carcinoma and MX-1 breast xenograft.64 This study revealed that: (a) diethyl and/or
diphenyltin compounds possessed higher activities; (b) there was no real link between the
Lewis acidity of the parent tin(IV) halide and the P-388 inhibition activity; (c) a pre-
dissociation of the bidentate ligand could be the rate determining step in vivo; and (d)
several compounds showed poor activity against human cancer cell lines. These
discoveries triggered a number of tin(IV) compounds studies against several types of
cancer cells.
Tin-based compounds have been a particular focus due to their structural features
and cytotoxic properties, both of which are required for biocompatibility and DNA
cleavage. The structure-activity relationship of tin(IV) compounds was discussed in terms

17
of various factors such as the nature of metal, coordination number and geometry and the
effect of substitution on ligands.56,64 The possible structure-activity relationship was
summarised as follows: a) Diphenyltin(IV) compounds were more potent than
dimethyltin(IV) compounds in inhibiting cancer cell lines. This is due to the planarity of
the two phenyl groups present in the tin(IV) compounds, where it is favourable to interact
to the base pair of the DNA in cancer cell lines by π-interactions, thus causing cell death
by apoptosis.53,65,66 The tin(IV) compounds were also found to bind to the phosphodiester
backbone of DNA, thus disrupting DNA repair in the presence of the phosphodiesterase
enzyme.67 Moreover, tin(IV) compounds bind to glycoproteins and cellular proteins,
which act as cancer biomarkers.59–61
In this chapter, the review of anticancer activity of tin(IV) compounds is mainly
focused on phenyl- and methyltin(IV) compounds reported over the past ten years. The
cytotoxic activity of tin(IV) compounds is compared with the reference drugs used in that
particular study.
1.4.1 Tin(IV) Compounds of Oxygen and/or Nitrogen Donor Ligands
The tin(IV) compounds (1-5, see Figure 1.12) of 1-(4-chlorophenyl)-1-
cyclopentanecarboxylic acid were screened for their in vitro antitumor activities against
HL-60, hepatocellular carcinoma (Bel-7402), gastric carcinoma (BGC-823) and
nasopharyngeal carcinoma (KB) cell lines.68 However, only compound 3 was observed
to exhibit slightly higher antitumour activity than cisplatin, with the concentration of a
drug that inhibits a biological activity by 50% (IC50) values of 5.2 and 8.1 μM (Bel-7402),
and 4.9 and 6.5 μM (BGC-823), respectively.68 In contrast, findings reported by Hadi and
Rilyati, indicated that diphenyltin(IV) compounds (7) (IC50 = 17.89 µM) showed more
potency than dibutyltin(IV) (6) (IC50 = 41.25 µM).69 The cytotoxicity of octahedral

18
tin(IV) compounds with general formulae of R2SnL2 (R = Me (8), Et (9), Bu (10), Ph (11),
Bz (12) and L = 2-phenylmonomethylglutarate) were studied against KB cell line. 70
Among the tin(IV) compounds, compound 11 (IC50 = 0.30 μM) was equipotent cytotoxic
activity with cisplatin (IC50 = 0.37 μM).70
The cytotoxicity of dimethyl- (13, see Figure 1.12), dibutyl- (14, 16, see Figure
1.12) and diphenyl- (15, 17, see Figure 1.12) tin(IV) compounds containing an o-vanillin
(oVa) Schiff base were studied against three cisplatin resistant tumour cell lines, viz.
human lung (A549), HeLa and MCF-7.66 These tin(IV) compounds showed greater
cytotoxicity than cisplatin in all cancer cell lines tested. It was suggested that activity of
the tin(IV) compounds was related to the coordination geometry of the tin(IV)
compounds, since the octahedral tin(IV) compounds (13, 14 and 15) showed higher
cytotoxic activities than the tin(IV) compounds with trigonal bipyramidal geometry (16
and 17). The coordination of the organo groups complexed to the central tin also
influenced cytotoxicity, with cytotoxicity following the order n-Bu > Ph > Me for most
of the tumour cells. The cytotoxic mechanism for these reported compounds remains
unknown. However, protein binding studies with bovine serum albumins (BSA), show
that they bind to the albumin in the blood stream, resulting in a significant impact on the
distribution, free concentration, metabolism and toxicity of the drug. The binding constant
between the tin(IV) compounds and BSA suggested that all of the compounds could be
easily stored in the protein and also could be released in the affected areas.66
Four trigonal bipyramidal tin(IV) compounds, Me2SnL1 (18), Ph2SnL1 (19),
Me2SnL2 (20) and Ph2SnL2 (21) (H2L1 = 5-chlorosalicylaldehyde isonicotinoyl hydrazone
and H2L2 = 2-hydroxy-4-methoxybenzaldehyde isonicotinoyl hydrazone), see Figure
1.12, were synthesised and tested against A549 and HeLa cell lines, showing significant
activity with IC50 values ranging between 0.7 - 15.2 µM (as compared to their free Schiff

19
bases, IC50 > 100 µM and cisplatin, > 60 µM, respectively).53 The higher potency of the
diphenyltin(IV) compounds here were attributed to the presence of the phenyl groups,
which enabled intercalation of the compounds into the DNA base pairs via π-π stacking
interactions,53 confirmed via UV-vis spectroscopy. Drug-DNA binding constant values
of 1.58 x 104 M-1 (18), 2.02 x 104 M-1 (19), 1.47 x 104 M-1 (20) and 3.36 x 104 M-1 (21)
were reported, showing that 19 and 20 had significantly higher affinity to DNA.
The efficiency of dibutyl- and diphenyltin(IV) compounds (22-29, see Figure 1.12),
synthesised from 2-hydroxy-1-naphthaldehyde or 4-substituted-2-hydroxybenzaldehyde,
benzylhydrazine and tin(IV) oxide were tested against a murine melanoma (B16F10) cell
line at different doses response ranging from 10, 5, 2.5 and 1 μg mL-1 for 24 hours. These
compounds were stable for several months, except for compound 24. Compounds 22, 25
and 28 were identified as the most cytotoxic compounds, however, all diphenyl- and
dibutyltin(IV) compounds investigated in this study showed significant cytotoxic
activities against the B16F10 cell line.71
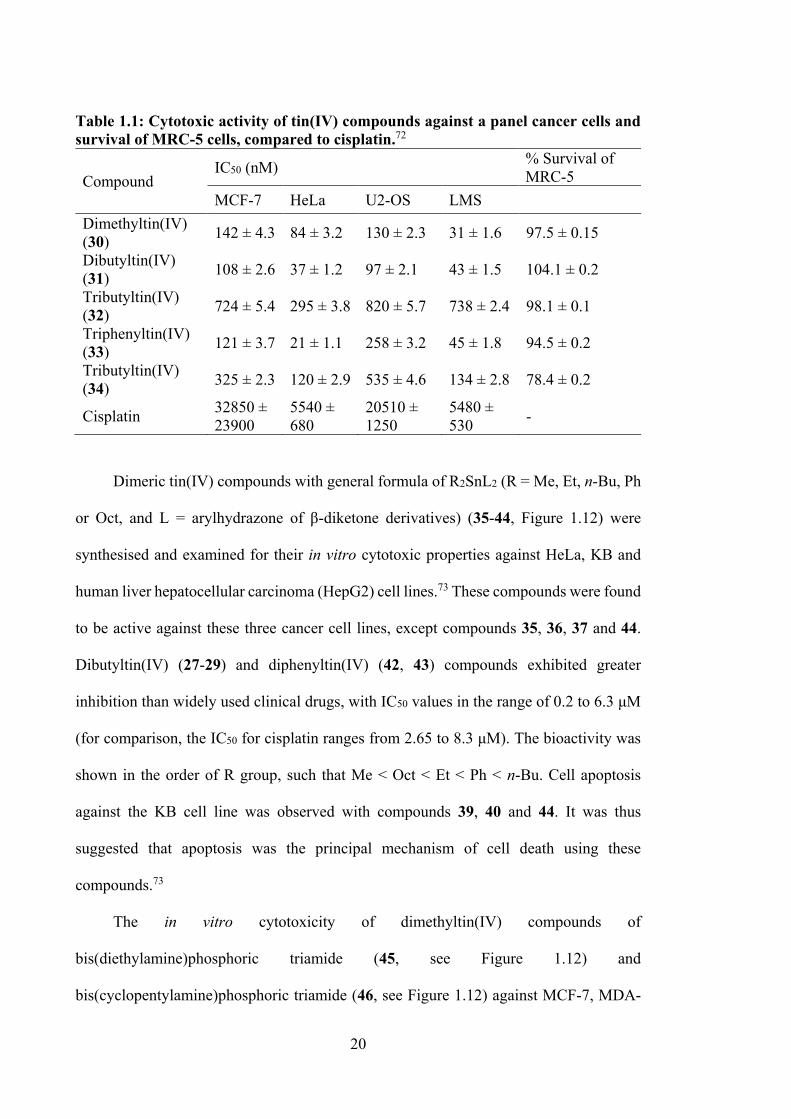
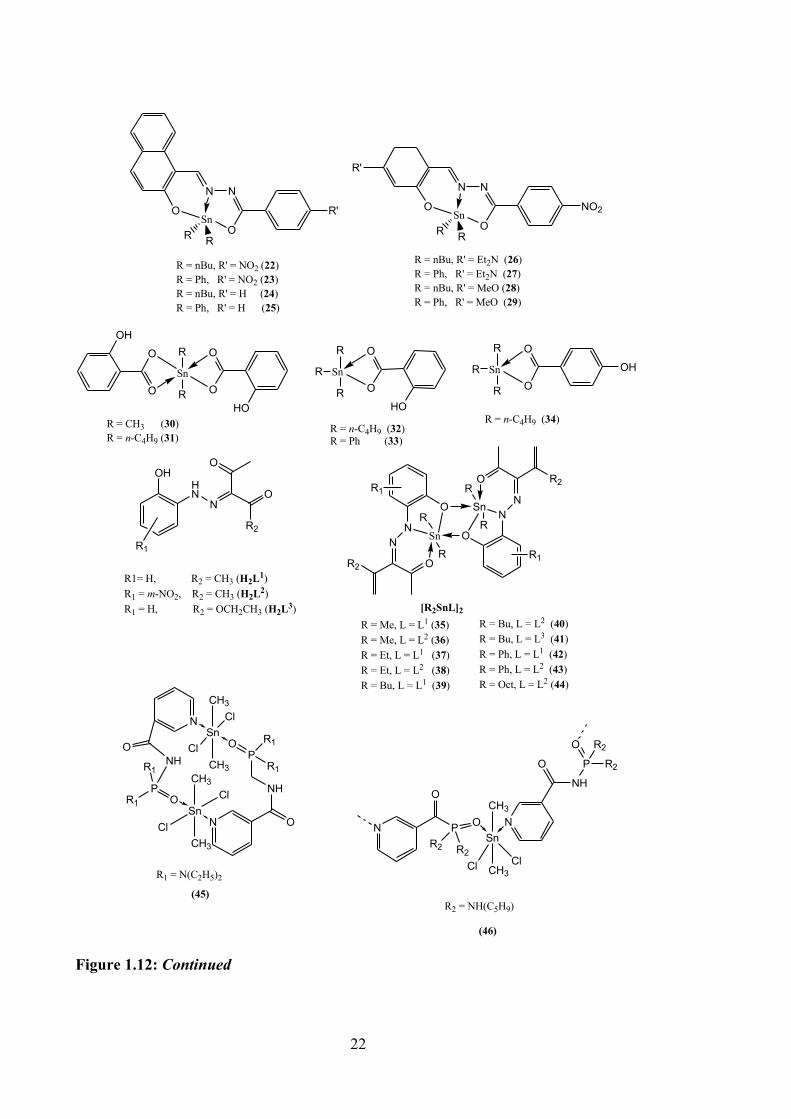
In vitro cytotoxicity properties were evaluated for five tin(IV) compounds with o-
hydroxy-benzoic or p-hydroxy-benzoic acids against four cancer cell lines, MCF-7,
HeLa, human osteosarcoma (U2-OS) and one normal cell line (normal foetal lung
fibroblast, MRC-5). The compounds demonstrated high potency against all cancer cell
lines tested (see Table 1.1). Compound 31 showed 304 and 210 times higher potency
compared to cisplatin for MCF-7 and U2-OS cell lines, respectively. Compounds 30 and
34 (see Figure 1.9) also exhibited higher cytotoxicity than 32, but slightly lower than 31.
Toxicity against MRC-5 cells showed no inhibitory activity, except for tributyltin.72
20
Table 1.1: Cytotoxic activity of tin(IV) compounds against a panel cancer cells and survival of MRC-5 cells, compared to cisplatin.72
Compound IC50 (nM) % Survival of
MRC-5 MCF-7 HeLa U2-OS LMS
Dimethyltin(IV) (30) 142 ± 4.3 84 ± 3.2 130 ± 2.3 31 ± 1.6 97.5 ± 0.15
Dibutyltin(IV) (31) 108 ± 2.6 37 ± 1.2 97 ± 2.1 43 ± 1.5 104.1 ± 0.2
Tributyltin(IV) (32) 724 ± 5.4 295 ± 3.8 820 ± 5.7 738 ± 2.4 98.1 ± 0.1
Triphenyltin(IV) (33) 121 ± 3.7 21 ± 1.1 258 ± 3.2 45 ± 1.8 94.5 ± 0.2
Tributyltin(IV) (34) 325 ± 2.3 120 ± 2.9 535 ± 4.6 134 ± 2.8 78.4 ± 0.2
Cisplatin 32850 ± 23900
5540 ± 680
20510 ± 1250
5480 ± 530 -
Dimeric tin(IV) compounds with general formula of R2SnL2 (R = Me, Et, n-Bu, Ph
or Oct, and L = arylhydrazone of β-diketone derivatives) (35-44, Figure 1.12) were
synthesised and examined for their in vitro cytotoxic properties against HeLa, KB and
human liver hepatocellular carcinoma (HepG2) cell lines.73 These compounds were found
to be active against these three cancer cell lines, except compounds 35, 36, 37 and 44.
Dibutyltin(IV) (27-29) and diphenyltin(IV) (42, 43) compounds exhibited greater
inhibition than widely used clinical drugs, with IC50 values in the range of 0.2 to 6.3 μM
(for comparison, the IC50 for cisplatin ranges from 2.65 to 8.3 μM). The bioactivity was
shown in the order of R group, such that Me < Oct < Et < Ph < n-Bu. Cell apoptosis
against the KB cell line was observed with compounds 39, 40 and 44. It was thus
suggested that apoptosis was the principal mechanism of cell death using these
compounds.73
The in vitro cytotoxicity of dimethyltin(IV) compounds of
bis(diethylamine)phosphoric triamide (45, see Figure 1.12) and
bis(cyclopentylamine)phosphoric triamide (46, see Figure 1.12) against MCF-7, MDA-
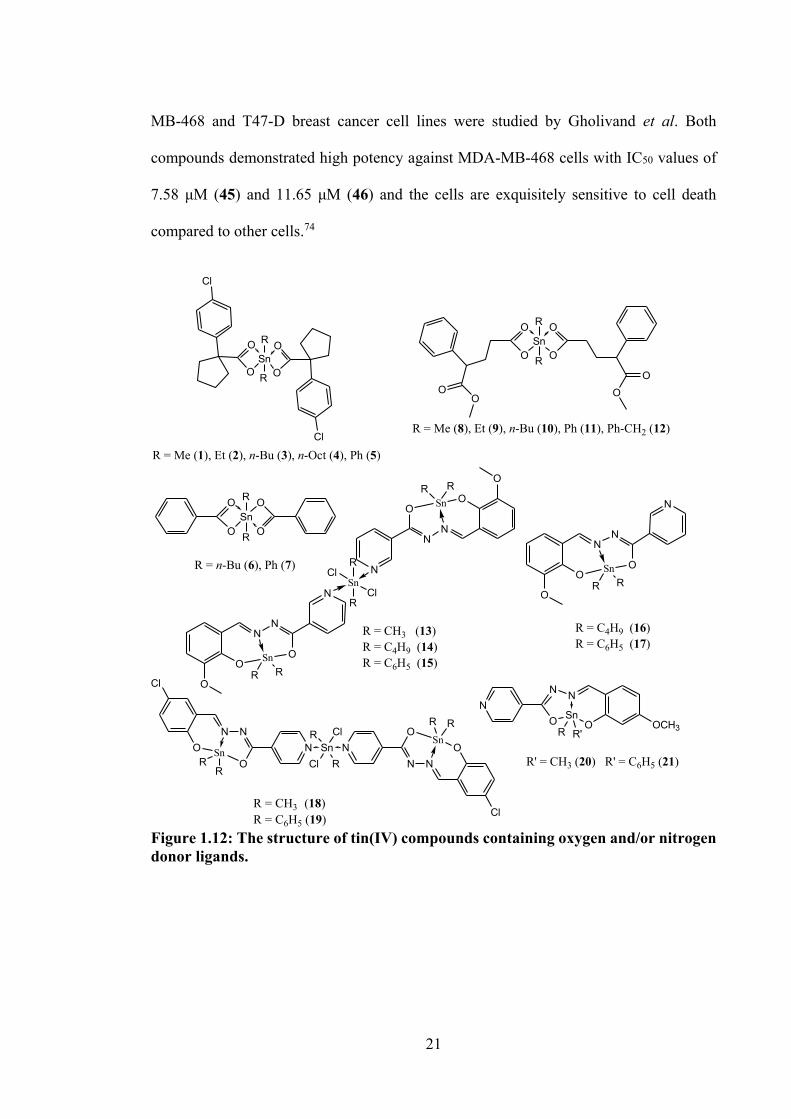
21
MB-468 and T47-D breast cancer cell lines were studied by Gholivand et al. Both
compounds demonstrated high potency against MDA-MB-468 cells with IC50 values of
7.58 μM (45) and 11.65 μM (46) and the cells are exquisitely sensitive to cell death
compared to other cells.74
SnO
O O
OR
R
Cl
Cl
R = Me (1), Et (2), n-Bu (3), n-Oct (4), Ph (5)
OSn
O
O
OR
R
R = n-Bu (6), Ph (7)
SnO
O O
OR
R
OO O
O
R = Me (8), Et (9), n-Bu (10), Ph (11), Ph-CH2 (12)
N
NN
OO
O
N
NN
OO
O
Sn
R
R
Cl
Cl
Sn
R R
Sn N
NN
OO
O
Sn
R R
R = CH3 (13)R = C4H9 (14)R = C6H5 (15)
R = C4H9 (16)R = C6H5 (17)
R R
N Sn NCl
Cl R
R O
N N
Cl
ONN
OO
SnSn
RR
RR
Cl
R = CH3 (18) R = C6H5 (19)
OCH3
N
SnO
N
ON
R R'
R' = CH3 (20) R' = C6H5 (21)
Figure 1.12: The structure of tin(IV) compounds containing oxygen and/or nitrogen donor ligands.
22
N
O
N
OSn
R R
R = nBu, R' = NO2 (22)R = Ph, R' = NO2 (23)R = nBu, R' = H (24)R = Ph, R' = H (25)
R'
N
O
N
OSn
R R
NO2
R'
R = nBu, R' = Et2N (26)R = Ph, R' = Et2N (27)R = nBu, R' = MeO (28)R = Ph, R' = MeO (29)
O
OSn
O
OOH
HO
R
R
R = CH3 (30)R = n-C4H9 (31)
SnO
O
HO
R
R
R = n-C4H9 (32)R = Ph (33)
R SnO
OR
R
R = n-C4H9 (34)
R OH
O
NSnN
R2 O
R1
R
RO
NSn N
R2O
R1
R
R
R1
OHHN
N
O
O
R2
R1= H, R2 = CH3 (H2L1)R1 = m-NO2, R2 = CH3 (H2L2)R1 = H, R2 = OCH2CH3 (H2L3) [R2SnL]2
R = Me, L = L1 (35)R = Me, L = L2 (36)R = Et, L = L1 (37)R = Et, L = L2 (38)R = Bu, L = L1 (39)
R = Bu, L = L2 (40)R = Bu, L = L3 (41)R = Ph, L = L1 (42)R = Ph, L = L2 (43)R = Oct, L = L2 (44)
SnClO
CH3
CH3
NCl
PR1
R1NH
NH
O
PR1
R1
OSn
CH3
CH3
Cl
Cl
N
O
R1 = N(C2H5)2
(45)
Sn
CH3
CH3
O N
Cl Cl
P
R2R2
N
O
O
NH
P R2
O R2
R2 = NH(C5H9)
(46)
Figure 1.12: Continued

23
1.4.2 Tin(IV) Compounds of Sulphur Donor Ligands
A five-coordinated anionic tin(IV) compound [C5H5NH][Ph2Sn(µ2-SCH2COO)Cl]
(47, see Figure 1.13) of mercaptoacetic acid with diphenyltin, was tested against A549
and colon carcinoma (HCT-8) cell lines with IC50 values of 1.9 μΜ and 0.7 μΜ,
respectively.75 R2Sn(IV) compounds (R = Me (48) and n-Bu (49), see Figure 1.13) of N-
acetyl-L-cysteine (H2NAC) were studied against hepatocellular carcinoma (HCC) and
non-tumour Chang liver cells.76 Cytotoxic data reported that 49 was effective in inhibition
of HCC cells as compared to H2NAC and 49 considered less toxic against non-tumour
Chang liver cells. This indicated that the coordination of anionic NAC2- was able to exert
modulatory activity. Moreover, the mechanism of death in HCC cells of 49 was reported
to relate to apoptotic pathway in inducing activation of caspase-3.76 Tin(IV) compounds
with heterocyclic thioamides, 2-mercapto-benzothiazole (Hmbzt), 5-chloro-2-mercapto-
benzothiazole (Hcmbzt) and 2-mercapto-benzoxazole (Hmbzo) of formulae
[(C6H5)3Sn(mbzt)] (50), [(C6H5)3Sn(mbzo)] (51), [(C6H5)3Sn(cmbzt)] (52) and
[(C6H5)2Sn(cmbzt)2] (53), [(n-C4H9)2Sn(cmbzt)2] (54) and [(CH3)2Sn(cmbzt)2] (55) were
tested for their cytotoxicity against leiomyosarcoma cells from the Wistar rat.77,78 Among
the triphenyltin(IV) compounds (50-52), 52 exhibited higher cytotoxic activity, while
among diorganotin(IV) compounds (53-55) compound 53 showed excellent cytotoxic
potency. Furthermore, compounds (50-55) were reported to strongly inhibit the
metalloenzyme of lipoxygenase (LOX) through a free radical mechanism in the same
manner.77,78
Milaeva and co-workers (2014) studied the mode of cytotoxic action of tin(IV)
compounds Me2Sn(SR)2 (56), Et2Sn(SR)2 (57), (n-Bu)2Sn(SR)2 (58), Ph2Sn(SR)2 (59),
Me3SnSR (60), Ph3SnSR (61) and R2SnCl2 (62) (R = 3,5-di-tert-butyl-4-hydroxyphenyl),
see Figure 1.13. All compounds were significantly active against MCF-7, HeLa and

24
MRC-5 cell lines.79 The cytotoxicity of dimethyl-, dibutyl- and diphenyltin(IV)
compounds (63-65, see Figure 1.13) of N-methyl-N-phenyldithiocarbamate and N-ethyl-
N-phenyldithiocarbamate were investigated against HeLa cell lines by Adeyemi et al.
The compounds exhibited better cytoselectivity (IC50 = 5 μM (dimethyltin(IV); 25 μM
(dibutyltin(IV); 12 μM (diphenyltin(IV)) compared to the standard drug, fluorouracil
(IC50 = 40 μM). It was demonstrated that 63 showed higher potency than 64 and 65, a
finding at odds with an earlier report showing that aryl and alkyl derivatives of organo
substituents with longer chains have better bioactivities.80 Nevertheless, the mechanism
of action of 64 remains unknown.
Coordinating the binuclear diphenyltin(IV) compounds (66-70, see Figure 1.13)
with 4,4’-bis(2-cyclohexylamino)acetamido) diphenylsulfone, 4,4’-bis(2-
isopropylamino) acetamido)diphenylsulfone, 4,4’-bis(n-butylamino)acetamido)
diphenylsulfone, 4,4’-bis(2-cyclohexylamino)acetamido)phenylene and 4,4’-bis(2-
isopropylamino)acetamido)phenylene increased cytotoxic activity against HepG2 cells
22-fold for 66 (IC50 = 3.32 ± 0.14 μM), 44‐fold for 67 (IC50 = 1.77 ± 0.11 μM), 37‐fold
for 68 (IC50 = 2.02 ± 0.47 μM), 8-fold for 69 (IC50 = 8.72 ± 0.19 μM) and 29‐fold for 70
(IC50 = 2.60 ± 0.67 μM), compared with the reference drug cisplatin (IC50 = 75.67 ± 0.51
μM). The toxicity studies demonstrated that the compounds had strong selectivity on
cancer cells over normal liver (WRL-68) cells. The shrinking of HepG2 cells were
observed after the treatment suggesting that the compounds caused apoptosis of the
cells.81
Two dinuclear phenyltin(IV) compounds (71 and 72, see Figure 1.13) of N,N’-
bis(2-hydroxybenzyl)-1,2-ethanebis(dithiocarbamate) ligands were investigated for their
cytotoxicity against colon carcinoma (CoLo205) and mammary tumour Bcap37) cell
lines.82 Both compounds (IC50 = 0.042 ± 0.001 μM (CoLo205), 0.058 ± 0.004 μM
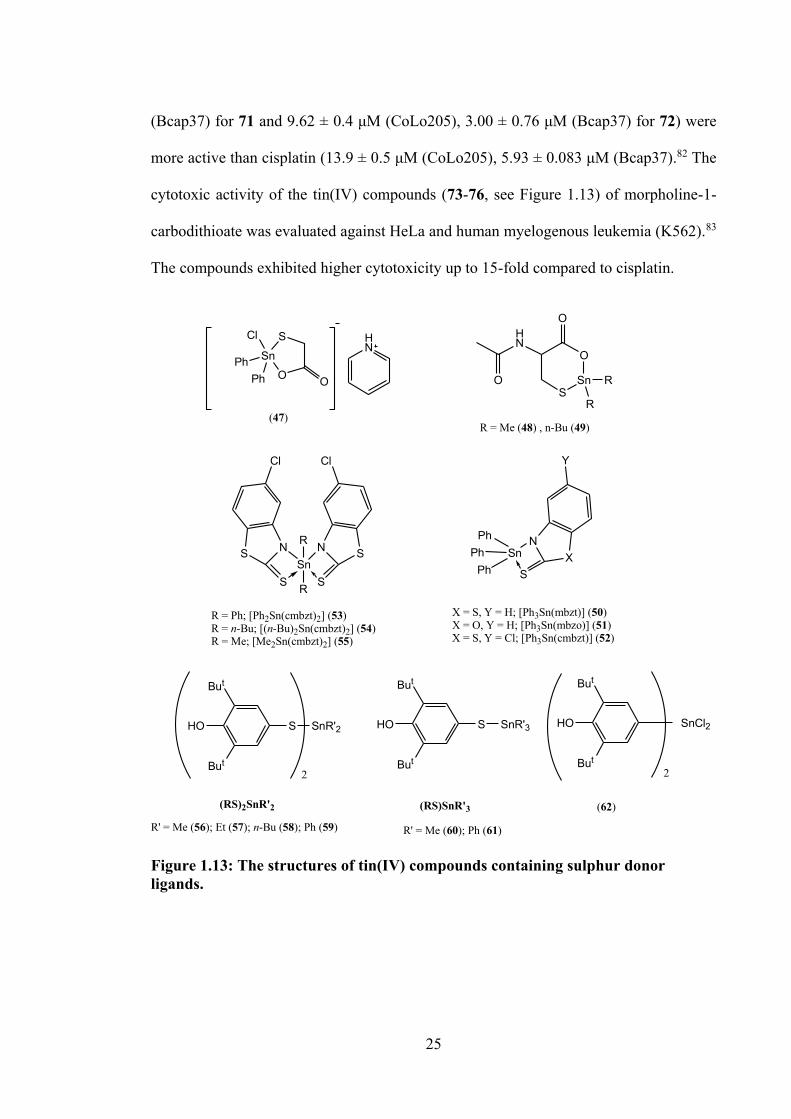
25
(Bcap37) for 71 and 9.62 ± 0.4 μM (CoLo205), 3.00 ± 0.76 μM (Bcap37) for 72) were
more active than cisplatin (13.9 ± 0.5 μM (CoLo205), 5.93 ± 0.083 μM (Bcap37).82 The
cytotoxic activity of the tin(IV) compounds (73-76, see Figure 1.13) of morpholine-1-
carbodithioate was evaluated against HeLa and human myelogenous leukemia (K562).83
The compounds exhibited higher cytotoxicity up to 15-fold compared to cisplatin.
HN
SSn
O
HN
O
O
R
R
R = Me (48) , n-Bu (49)(47)
NSn
SX
Y
PhPh
Ph
X = S, Y = H; [Ph3Sn(mbzt)] (50)X = O, Y = H; [Ph3Sn(mbzo)] (51)X = S, Y = Cl; [Ph3Sn(cmbzt)] (52)
SnN
S S
NR
R
S S
ClCl
R = Ph; [Ph2Sn(cmbzt)2] (53)R = n-Bu; [(n-Bu)2Sn(cmbzt)2] (54)R = Me; [Me2Sn(cmbzt)2] (55)
SSn
O O
Cl
PhPh
But
HO
But
S SnR'2
But
HO
But
S SnR'3
But
HO
But
SnCl2
2 2
R' = Me (56); Et (57); n-Bu (58); Ph (59) R' = Me (60); Ph (61)
(RS)2SnR'2 (RS)SnR'3 (62)
Figure 1.13: The structures of tin(IV) compounds containing sulphur donor ligands.
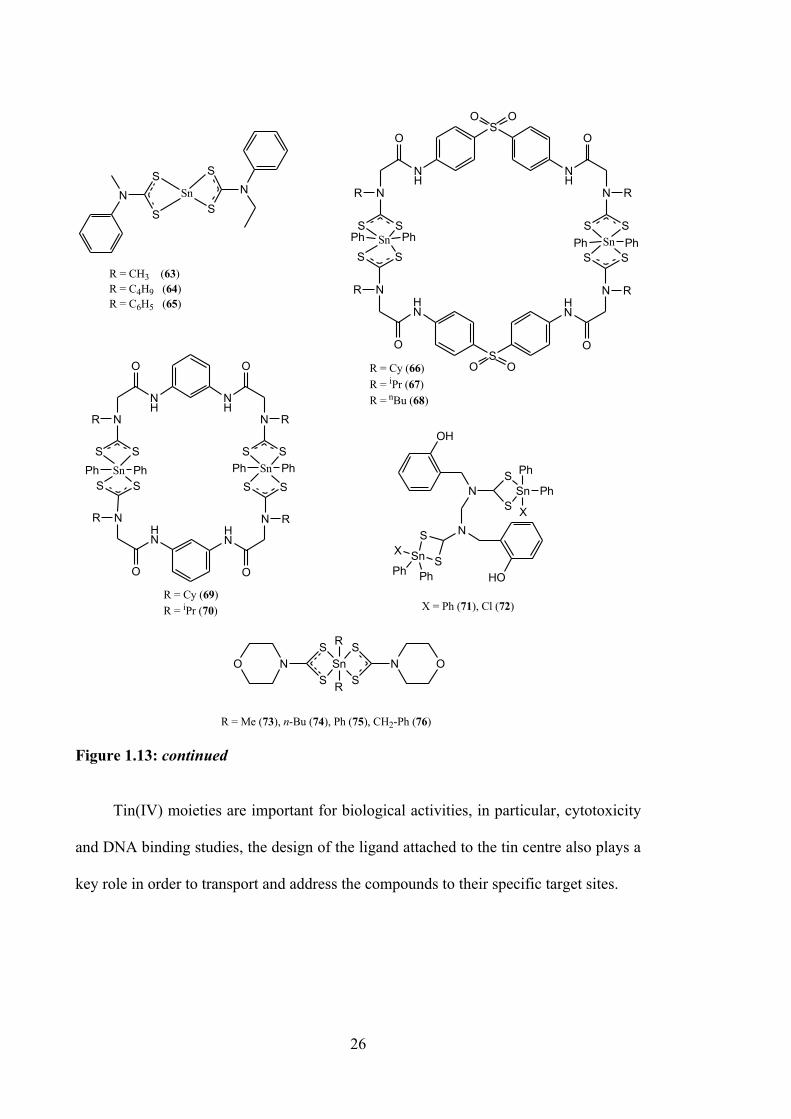
26
S
S
SnNS
SN
R = CH3 (63)R = C4H9 (64)R = C6H5 (65)
OH
NS
SnS Ph
Ph
XNS
Sn SX
Ph Ph HO
X = Ph (71), Cl (72)
NOS
SnS
S
SR
R
N O
R = Me (73), n-Bu (74), Ph (75), CH2-Ph (76)
SO O
NH
O
N
NH
O
NR
S S
R
S S
SOO
HN
O
NHN
O
N R
SS
R
SSSn Sn PhPhPhPh
R = Cy (66)R = iPr (67)R = nBu (68)N
H
O
NR
SSSn
NH
O
N R
S SSn
HN
O
N R
S S
HN
O
NR
SSPhPhPhPh
R = Cy (69)R = iPr (70)
Figure 1.13: continued Tin(IV) moieties are important for biological activities, in particular, cytotoxicity
and DNA binding studies, the design of the ligand attached to the tin centre also plays a
key role in order to transport and address the compounds to their specific target sites.

27
1.5 Chemistry and Bioactivities of Schiff Bases and Their Tin(IV) Compounds
Schiff bases are considered as “privileged ligands” and they are widely used as
pharmacophores for drug design due to their versatile synthesis and good solubility in
common organic solvents. The basic structure of the Schiff bases comprise stable imines
(C=N), where the basic nitrogen atom is able to stabilise metal ions in different oxidation
states. The nitrogen donor atom is also responsible for enhancing the biological activities,
by interacting via the nitrogen lone electron pair with the protein amino acid residues or
DNA nucleobases. Schiff bases are easily formed by condensation reaction of the
carbonyl group of aldehydes or ketones with primary amines, in particular dithiocarbazate
and thiosemicarbazide amine sources, which will be employed in this thesis.
Dithiocarbazate, NH2NHCS2, and thiosemicarbazide, NH2NHCSNH2, are di-
anionic chelating ligands, and can greatly modified by introducing different organic
substituents. Dithiocarbazates and thiosemicarbazides are capable of forming stable
compounds with a large number of metal elements, including tin, due to the presence of
the anionic nitrogen and sulphur moieties which enable a wide range of binding modes,
thus affording good coordination capacity. The interesting coordination modes and
stability of both dithiocarbazates and thiosemicarbazones with metals occurs where the
metal acts as a Lewis acid complex to react with the electron-rich dithiocarbazate/
thiosemicarbazone ligands according to the Hard Soft [Lewis] Acid Base principle.
Dithiocarbazate and thiosemicarbazone ligands have received significant attention
and they display diverse biological activities.66 However, from a synthetic perspective,
they can be easily derivatised with a range of organic substituents, and can coordinate to
metal ions by coordinating donors to yield complexes with a range of geometrical
structures and chemical/physical properties. They also can exist as thione and thiol
tautomers (Figure 1.14). In solid form, they usually exist in thione form, while in solution

28
they exist as an equilibrium mixture of thione-thiol tautomers (Ali and Livingstone,
1974). The tautomerism character of both these dithiocarbazate and thiosemicarbazone
ligands have been confirmed by FTIR and 1H NMR analysis.85–88,13,89–91
R1S N
H
N
S
R1S N
N
SH
R1NH
NH
N
S
R1NH
NN
SH
Thione form Thiol form
R1 indicate the presence of various different organic substituentsR2 indicate the presence of various different aldehydes/ketones
(a)
(b)
R2R2
R2 R2
Figure 1.14: Tautomerism in (a) DTC and (b) TSC Schiff bases. 1.5.1 Anticancer Activity of Dithiocarbazate Schiff Bases and Their Tin(IV)
Compounds
Dithiocarbazates and Schiff bases thereof have been shown to have promising and
wide-ranging biological activity over the past few decades. Much work has been done on
S-benzyldithiocarbazate (SBDTC) specifically, mostly focusing on SBDTC and S-
methyldithiocarabazate (SMBDTC) transition metal complexes.91–100 A number of
reports have developed dithiocarbazate analogues which specifically show selective
biological activity against certain cancer cell lines.98,101–106 For example, S-benzyl-β-N-
(benzoyl)dithiocarbazate exhibits IC50 = 3.32 µM against HL-60,107 S-2-
picolyldithiocarbazate with pyridine-2-carboxaldehyde exhibits CD50 (the concentrations
causing 50% of cell death) = 7.92 µM against CEM-SS (T-lymphoblastic leukemic cells)

29
and HT-29 (colon cancer cells),108 3-methylbenzyl-2-(6-methylpyridin-2-
ylmethylene)hydrazine carbodithioate exhibits CD50 = 0.95 and 6.36 µM against MCF-7
and MDA-MB-231, respectively,99 6-methyl-2-formylpyridine Schiff bases of SMBDTC
(IC50 = 4.43 µM), and 6-methyl-2-formylpyridine Schiff bases of SBDTC exhibit IC50 =
6.63 µM against Caov-3 (human ovarian cancer cells). The structure of these ligands can
be easily and widely modified via introducing different organic
substituents96,98,101,103,109,110 without substantially altering their coordination chemistry.
Dithiocarbazate derivatives can also be greatly modified, for instance by alkyl or aryl
substitutions at the sulphur atom (see Figure 1.15) forming S-allyl,111 isomeric S-2-/3-/4-
picolyl,108,112 isomeric S-2-/3-/4-methylbenzyl,97,99,103 S-napthylmethyl,107 S-quinolin-
2yl-methyl,107 S-4-nitrobenzyl,98 S-hexyl113,114 and S-4-chlorobenzyl115. Importantly, the
cytotoxic activity of tin(IV) compounds of dithiocarbazate Schiff base analogues remain
essentially unexplored.
The earliest tin(IV) compounds of dithiocarbazate Schiff bases were reported in
1985, where the compounds exhibited promising antibacterial activity against Gram
positive bacteria, Bacillus subtilis and Staphylococcus aureus. Since then, various tin(IV)
compounds were synthesised for various applications. However, the majority of the
studies mainly focused on structural aspects of tin(IV) compounds. The bioactivity of the
tin(IV) compounds remained less explored, particularly for anticancer
applications.85,91,93,116,117 The most recent studies focused on cytotoxicity and toxicity of
tin(IV) compounds SnL2 (73), SnLI2 (74) and SnLCl2 (75) (L = (2,2′-
(disulfanediylbis((ethylthio)methylene)) bis(hydrazin-2-yl-1-ylidene)bis(methanyl
ylidene)) diphenol (see Figure 1.13) against HeLa and MCF-7 cancer as well as Chinese
hamster ovary (CHO) normal cell lines.118 These compounds exhibited good cytotoxic
activity, with compounds L (IC50 = 0.1766 ± 0.022 μM) and 75 (IC50 = 0.05065 ± 0.0226 μM)
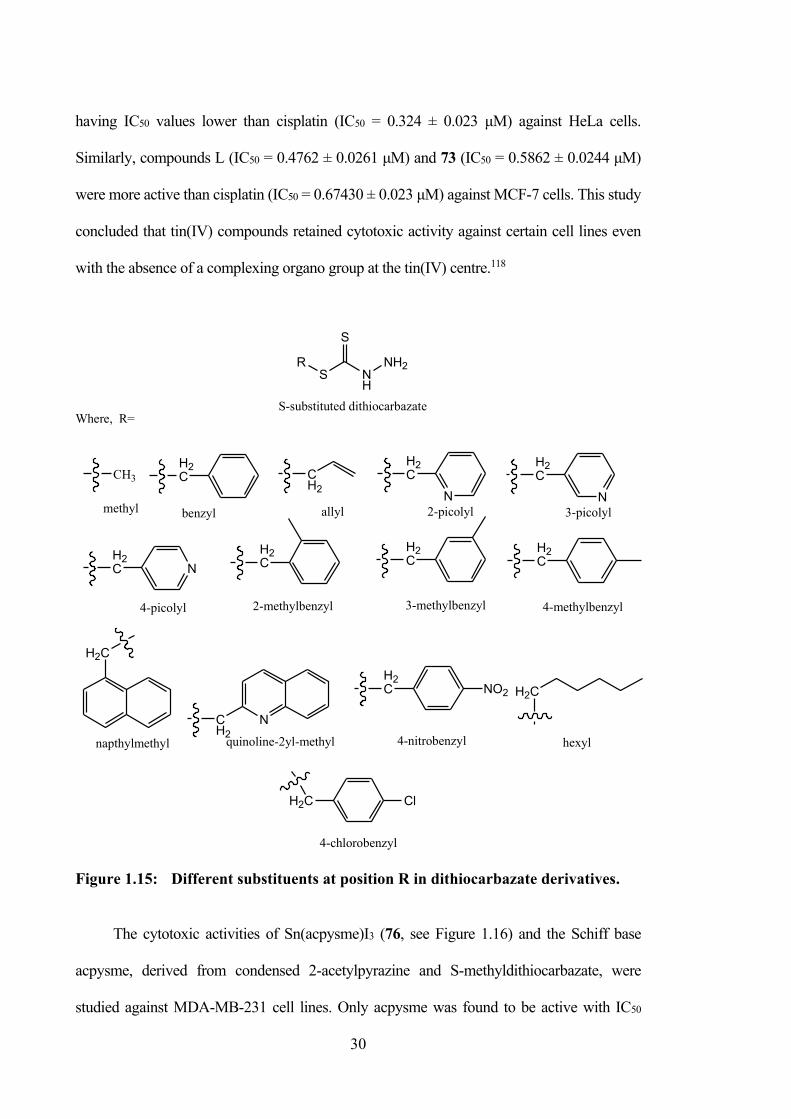
30
having IC50 values lower than cisplatin (IC50 = 0.324 ± 0.023 μM) against HeLa cells.
Similarly, compounds L (IC50 = 0.4762 ± 0.0261 μM) and 73 (IC50 = 0.5862 ± 0.0244 μM)
were more active than cisplatin (IC50 = 0.67430 ± 0.023 μM) against MCF-7 cells. This study
concluded that tin(IV) compounds retained cytotoxic activity against certain cell lines even
with the absence of a complexing organo group at the tin(IV) centre.118
S NH
S
NH2R
S-substituted dithiocarbazateWhere, R=
H2C
H2C
H2C
H2C
CH3
H2C
NCH2
N
H2C
H2C NO2
N
H2C
NH2C
H2C
ClH2C
CH2
methyl benzyl allyl 2-picolyl 3-picolyl
4-picolyl 2-methylbenzyl 3-methylbenzyl 4-methylbenzyl
napthylmethyl quinoline-2yl-methyl 4-nitrobenzyl hexyl
4-chlorobenzyl Figure 1.15: Different substituents at position R in dithiocarbazate derivatives. The cytotoxic activities of Sn(acpysme)I3 (76, see Figure 1.16) and the Schiff base
acpysme, derived from condensed 2-acetylpyrazine and S-methyldithiocarbazate, were
studied against MDA-MB-231 cell lines. Only acpysme was found to be active with IC50

31
values of 4.72 μM, while the tin(IV) compound 76 was found to be inactive.85 A hypothesis
that will be tested in this project is that the cytotoxicity of this compound can be improved via
manipulation of the S-substituent, aldehyde and organo groups.
S N
S
N
OSn
SN
S
N
O
S N
S
N
OSn
II
(73) (74)
(75)
S N
S
N
OSn
ClCl
S N
S
N N
NSn
I I
(76)
Figure 1.16: The structure of tin(IV) compounds derived from dithiocarbazate Schiff bases. 1.5.2 Anticancer Activity of Thiosemicarbazone Schiff Bases and Their Tin(IV)
Compounds
Thiosemicarbazones are an important class of compounds which have received
wide attention due to their remarkable biological and pharmacological properties, such as
antibacterial, antiviral, antineoplastic and antimalarial activities.119–122
Thiosemicarbazone Schiff bases are similar to dithiocarbazate and its derivatives, in that
complexation with a metal centre is achieved via the nitrogen and sulphur atoms
following deprotonation of the S-H and N-H groups.98,123–128 Thiosemicarbazones are an
interesting chemotherapeutic platform for the development of radiopharmaceuticals in the
treatment of cancer.129 Some synthetic thiosemicarbazone drugs are already available in

32
the market, such as methisazone (Figure 1.17(a)) used in the treatment of poxviruses, and
Triapine (Figure 1.17(b)), which is a promising anticancer agent following more than 30
clinical phase I/II trials in the treatment of leukaemia (principally advanced leukaemia),
lung, kidney, prostate and pancreatic cancers.130,131 The studies of Triapine in
combination with radiation or other cytotoxic agents have been promising candidates in
treating gynaecological tumours.132 It is proposed that these promising candidates interact
with intracellular iron and/or copper,133 which lead to the production of reactive oxygen
species (ROS). ROS is largely responsible for anti-proliferative activity in cancer cells,
which in turn effects ribonucleotide reductase (RR), the enzyme responsible for the
synthesis of DNA building blocks,134 by destroying the tyrosyl radical of the R2
subunit.135 The rapid growth of tumour cells are frequently characterised by RR
overexpression, thus this enzyme is a promising target for cancer therapy.136 In addition,
thiosemicarbazone compounds also exhibit other mechanisms of action, such as
promoting redox cycling reactions that lead to lysosomal membrane permeabilisation
which demonstrate the potency and selectivity against a range of tumours.137
N
H2N
NNH
S
H2NH2N
S
NH
N
NO
(a) (b)
Figure 1.17: Chemical structure of (a) Methisazone and (b) Triapine. Concerning the bioactivity of thiosemicarbazone Schiff base, in many cases
complexation with a metal ion enables higher activity than the parent thiosemicarbazone
Schiff bases, by lowering the concentration of drugs and essentially overcome some side

33
effects. The tin(IV) compounds (77, 78, 79, see Figure 1.18) of 3-methoxysalicylaldehyde
thiosemicarbazone (H2mstsc) were evaluated for their in vitro cytotoxicity against
immortalised line of human T lymphocyte cells, Jurkat cells. 138 It was observed that the
antitumour activity for dialkyltin(IV) compound increased with the length of the carbon
chain of the alkyl coordinated to the tin atom, while the most potent compound exhibited
an IC50 value of 110 µM. The activity explained the cytotoxicity order as dibutyl > dipehyl
> dimethyl = H2mstsc.138 The ability of 2-acetylpyridine N4-cyclohexyl
thiosemicarbazone Schiff base (L) and the distorted pentagonal bipyramidal tin(IV)
compound, [Ph2Sn(L)(OAc)].EtOH (80, see Figure 1.18) to inhibit tumour cell growth
against HepG2 cells were studied by Liu et al.139 Compound 80 and its ligand showed 3-
fold higher cytotoxic potency compared to the reference drug Mitoxantone (IC50 = 5.3 ±
2.38 µM), with IC50 values of 3.32 ± 0.52 µM IC50 = 10.10 ± 2.07 µM, respectively. The
better activity of 80 was attributed to chelation effects; the reduction in the polarity of the
central metal atom is due to partial sharing of its positive charge with the partial
negatively charged of Schiff base, as well as the nature R group attached directly to the
tin atom.139
In vitro cytotoxic activity of tin(IV) compounds (81-84, see Figure 1.18) of 2-
hydroxy-5-methoxybenzaldehyde and 4-methylthiosemicarbazide were studied against
human colorectal carcinoma (HCT116) cell line by Salam et al.,140 and significant activity
was observed in comparison to the standard drug 5-fluorouracil (IC50 = 7.6 µM). The
coordination of the Schiff base to the tin(IV) centre thus enhanced cytotoxic activity,
particularly for the compounds 83 and 84, where the reported IC50 values were 4.4 and
3.9 µM, which was better than 5-fluorouracil. The enhancement of cytotoxicity of both
compounds was attributed to the presence of phenyl groups coordinating the tin centre
and this phenyl group, which was suggested to interact with biomacromolecules in the

34
cells. A similar conclusion was reached by Li and co-workers,141 who studied
diphenyltin(IV) (86 and 88, see Figure 1.18) compounds of 2-benzoylpyridine N(4)-
phenylthiosemicarbazone and 2-acetylpyrazine N(4)-phenylthiosemicarbazone. These
compounds exhibited higher antitumour activities (against K562) with IC50 values of 5.8
μM and 14.8 μM, respectively, compared to the dimethyltin(IV) compounds (85 and 87)
and their free Schiff bases. Antiproliferative efficiencies of these compounds were found
to follow the order Ph2 > Ph > Me, implicating the structure of the organo group
coordinating the tin metal centre. An independent biological study suggested that the
diffusion, lipophilic character and steric effects associated with the ligand could also be
factors in determining cytotoxic activity in these compounds.140
Tin(IV) compounds of 2,3-dihydroxybenzaldehyde-N(4)-methyl
thiosemicarbazone (89-91, see Figure 1.18) and 2-hydroxy-5-methylbenzaldehyde-N(4)-
methylthiosemicarbazone (92-94, see Figure 1.18) showed significant antitumour activity
against MCF-7 cell line compared to 5-fluorouracil.65 The lowest IC50 values were 4.65
µM and 3.99 µM for diphenyltin(IV) compounds 91 and 94, due to the presence of the
bulky phenyl group chelating the centre tin, which affords π-π interactions with the cell
components of MCF-7 cells. The improvement of cytotoxic activity was also suggested
to be due to the presence of OH/NH groups, which enabled hydrogen bonding with DNA
base pairs.65 Similar studies of tin(IV) compounds of benzoylformic acid 3-hydroxy-2-
naphthoyl hydrazone142 reported that the diphenyltin(IV) compound (96, see Figure 1.18)
was more effective than its dimethyltin(IV) compound (95, see Figure 1.18) in inhibiting
the growth of A549, human hepatocellular carcinoma (SMMC-7721), P388 and HCT116
cell lines. Interestingly however, this dimethyltin(IV) compound exhibited promising
cytotoxicity against human colon carcinoma (WiDr) cell lines, which contradicted a
previous report53,128,143 that dimethyltin(IV) compounds have lower cytotoxic activities
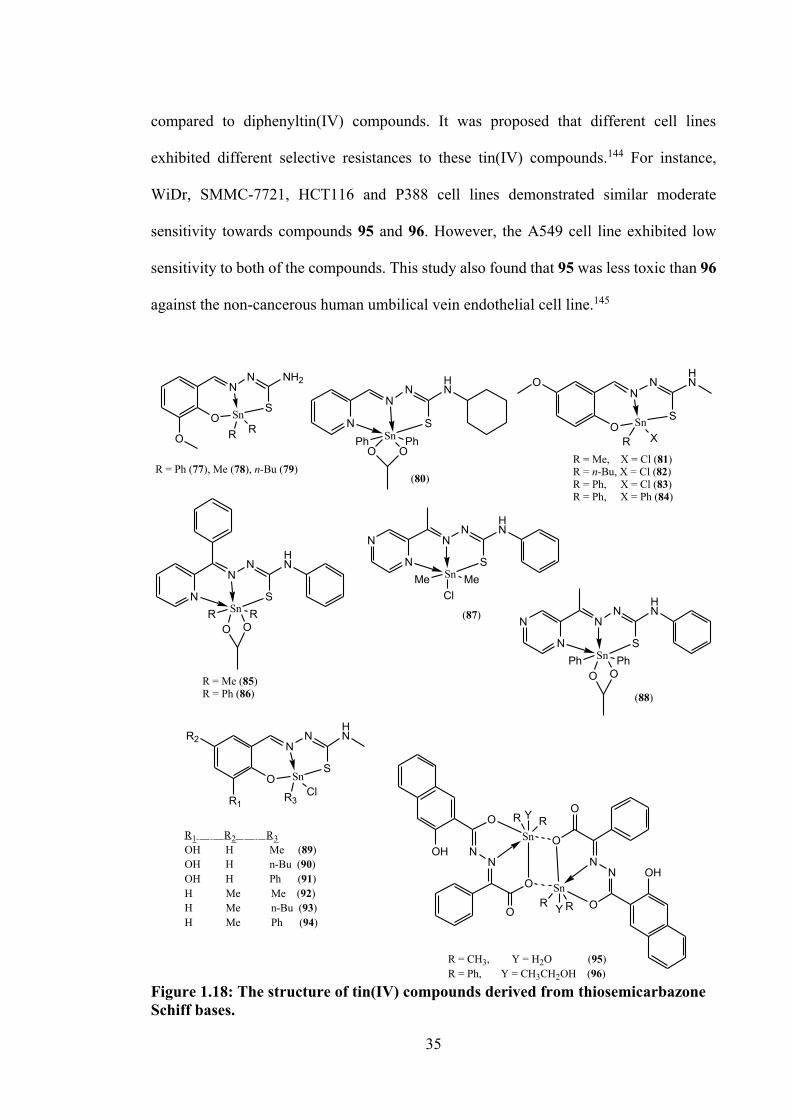
35
compared to diphenyltin(IV) compounds. It was proposed that different cell lines
exhibited different selective resistances to these tin(IV) compounds.144 For instance,
WiDr, SMMC-7721, HCT116 and P388 cell lines demonstrated similar moderate
sensitivity towards compounds 95 and 96. However, the A549 cell line exhibited low
sensitivity to both of the compounds. This study also found that 95 was less toxic than 96
against the non-cancerous human umbilical vein endothelial cell line.145
N
NN
HN
SnS
O OPh Ph
(80)
ON
NHN
SO Sn
R X
R = Me, X = Cl (81)R = n-Bu, X = Cl (82)R = Ph, X = Cl (83)R = Ph, X = Ph (84)
N
NN
HN
SSn
O ORR
R = Me (85)R = Ph (86)
N
N
NN
HN
SSn
O OPhPh
(88)
N
N
NN
HN
SSn MeMeCl
(87)
R2
R1
O
NN
HN
SSn
R3Cl
R1 R2 R3OH H Me (89)OH H n-Bu (90)OH H Ph (91)H Me Me (92)H Me n-Bu (93)H Me Ph (94)
O
O
NN
O
OHSn
R Y R
O
O
NN
O
OHSn
RYR
R = CH3, Y = H2O (95)R = Ph, Y = CH3CH2OH (96)
NN NH2
SO
O
Sn
R R
R = Ph (77), Me (78), n-Bu (79)
Figure 1.18: The structure of tin(IV) compounds derived from thiosemicarbazone Schiff bases.

36
1.6 Mechanism of Action
1.6.1 DNA Binding
Tin(IV) compounds potentially affect the production of energy by inhibiting
multimeric protein complexes (F1F0 ATP synthases), membrane-associated functions,
macromolecular synthesis, protein synthesis and DNA replication.146,147 Determining the
major site of inhibition is crucial to understanding the mechanism of a compound’s
activity. The binding ability of tin(IV) compounds towards DNA depends on the
coordination number and nature of groups bound to the tin centre. In solution, some
triorganotin(IV) compounds undergo spontaneous disproportionation into their
corresponding tetraorganotin(IV) and diorganotin(IV) compounds. While in vivo, the
spontaneous reaction into diorganotin(IV) compounds can occur by loss of the alkyl or
aryl group, via intervention of enzymes such as aromatase.148 Triorganotin(IV)
compounds are highly toxic, particularly those for which the tin centre is coordinated to
propyl, butyl, pentyl, phenyl and cyclohexyl groups.149 Therefore, diorganotin(IV)
compounds could be considered to be the ultimate cytotoxic agents; indeed, they have
been widely observed to display higher cytotoxicity than standard drugs tested against
several types of cancer cells,142,150,151 possibly related to pharmacokinetic effects. In
contrast, monoorganotin(IV) compounds are less potent.152
Tin(IV) compounds induce DNA damage by interaction with the phosphate
backbone, which leads to the contraction of the DNA and thus change in DNA
conformation. Phosphate oxygen and nitrogen atoms of DNA bases act as anchoring sites
for effective binding to tin(IV) compounds. In vitro DNA binding studies of dimethyltin
compounds of (R-/S-)2-amino-2-phenyl-ethanol and dibromoethane153 revealed strong
Sn-OPO3-2 bonding at the 5’-GMP phosphate group via outer-sphere coordination, and
negligible interaction between the tin(IV) compounds and purine nitrogen. UV-vis
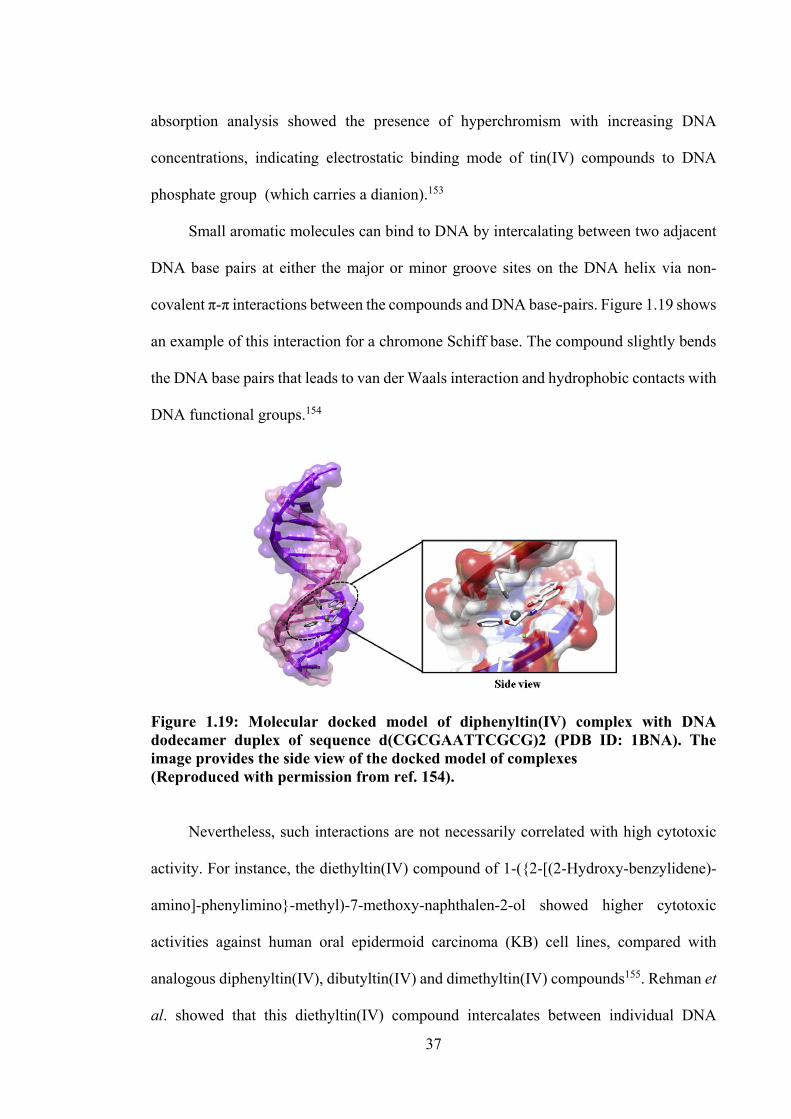
37
absorption analysis showed the presence of hyperchromism with increasing DNA
concentrations, indicating electrostatic binding mode of tin(IV) compounds to DNA
phosphate group (which carries a dianion).153
Small aromatic molecules can bind to DNA by intercalating between two adjacent
DNA base pairs at either the major or minor groove sites on the DNA helix via non-
covalent π-π interactions between the compounds and DNA base-pairs. Figure 1.19 shows
an example of this interaction for a chromone Schiff base. The compound slightly bends
the DNA base pairs that leads to van der Waals interaction and hydrophobic contacts with
DNA functional groups.154
Figure 1.19: Molecular docked model of diphenyltin(IV) complex with DNA dodecamer duplex of sequence d(CGCGAATTCGCG)2 (PDB ID: 1BNA). The image provides the side view of the docked model of complexes (Reproduced with permission from ref. 154). Nevertheless, such interactions are not necessarily correlated with high cytotoxic
activity. For instance, the diethyltin(IV) compound of 1-(2-[(2-Hydroxy-benzylidene)-
amino]-phenylimino-methyl)-7-methoxy-naphthalen-2-ol showed higher cytotoxic
activities against human oral epidermoid carcinoma (KB) cell lines, compared with
analogous diphenyltin(IV), dibutyltin(IV) and dimethyltin(IV) compounds155. Rehman et
al. showed that this diethyltin(IV) compound intercalates between individual DNA

38
strands and also binds with Topoisomerase II via hydrogen bonding at guanine (DG9)
and thymine (DT8) threonine (Thr911) and glutamic (Glu914, Glu831) residues (Figure
1.20), resulting in DNA damage and cell death.
Figure 1.20: a) Molecular docking simulation of diethyltin(IV) complex-DNA complex (binding site of topoisomerase II) b) detailed molecular interactions of diethyltin(IV) complex with amino acid residues (Reproduced with permission from ref. 155). 1.6.2 Apoptosis
Due to their lipophilicity, tin(IV) compounds can interact with the cellular
membrane and activate apoptosis. Apoptosis is a form of programmed cell death which
involves biochemical events, leading to morphological changes such as cell shrinkage,
membrane blebbing, chromatin cleavage, nuclear condensation and formation of
apoptotic bodies of condensed chromatin. Ultimately, the apoptotic bodies are rapidly
engulfed by the neighbouring cells. By contrast, necrosis is unprogrammed cell death in

39
living tissue by autolysis; necrotic cells exhibit loss of membrane integrity, cellular and
nuclear swelling and an associated inflammatory response.156 Unlike necrosis, apoptosis
produces cell fragments with two main death signals - extrinsic pathway via cell surface
receptors and intrinsic pathway though mitochondrial disturbance (Figure 2.14). Both of
these pathways involve p53 tumour suppressor proteins, TRAIL receptor, caspases and
Bcl-2 proteins.157 The p53 tumour suppressor gene is the most mutated gene in human
cancer cells, and the activation of p53 can lead to either cell cycle arrest, apoptosis or
DNA repair. Moreover, the increasing level of nuclear p53 gene in human neuroblastoma
cell line, SY5Y was activated by the trinuclear tin(IV) compound containing L-
tryptophan.57,158 The changes of p53 and histone phosphorylation, DNA damage markers,
which can be activated in response to DNA damage were studied by Esmail et al.159
Tin(IV) compound containing propyl gallate and 1,10‐phenanthroline reported to induce
MCF-7 cell p53-activated apoptosis through triggering DNA damage.159
Apoptosis seems to be the main type of cell death caused by tin(IV) compounds,
even though the path that leads to apoptosis may differ between them.160–162 Gennari and
co-workers61 reported that dibutyltin(IV) compounds initiate an increase of intracellular
Ca2+, which generates reactive oxygen species (ROS) and release of cytochrome C by
mitochondria. ROS include peroxides (R-O-O-R), superoxides (O2-), hydroxyl radicals
(•OH), singlet oxygen (1O2) and alpha-oxygen (α-O),163 and play key roles in cell
signalling and homeostasis.164 However, uncontrollable ROS generation results in
significant damage of cell structures. Similarly, cytochrome C has an intermediate role in
apoptosis.165 The release of uncontrollable ROS and cytochrome C results in activating
caspases, and thus cleaving defined target proteins and irreversible apoptotic damage of
the cells (Figure 1.21). The increase in intracellular Ca2+ may also be due to the disruption
of the cell membrane and cytoskeletal functioning.166 Tin(IV) compounds also could

40
accumulate in the Golgi apparatus and endoplasmic reticulum but not in the plasma
membrane and nucleus of the cells.167–169
Figure 1.21: Mechanism of extrinsic and intrinsic apoptotic pathway (Reproduced with permission from ref. 170). Apoptosis (22.4 ± 1.8%) and necrosis (2.1 ± 0.2%) of leiomysarcoma (LMS) cells
occurred following treatment with 50 nM of dimethyltin(IV) compound 30.72 However,
the percentage of cells that underwent apoptosis decreased to 21.1 ± 2.8% at higher
concentration (100 nM), while the rate of necrosis was essentially the same (1.6 ± 0.6%).
On the other hand, the rate of cell apoptosis increased with increasing concentration of
dibutyltin(IV) (31, 32 and 34, see Figure 1.12) and triphenyltin(IV) (33) compounds. The
characteristic DNA laddering pattern observed by electrophoresis gel indicated that the
LMS cells underwent apoptosis trigged by DNA damage and fragmentation.72
1.7 Knowledge Gap
Tin-based compounds were not considered as serious potential anticancer
candidates in the 1990s. They are now enjoying a resurgence and appear very promising

41
as anticancer drugs, particularly active against cisplatin-resistant cancers. Exciting
progress in medicinal tin(IV) chemistry has been reported in the past few years, allowing
the rational design of novel tin(IV) compounds with various types of substituted ligands.
The preceding literature review has established the potential of tin(IV) compounds in
inhibiting cancer cells over the common organic-based drugs via several mechanisms of
action. However, current literature demonstrates that the cytotoxicity of tin(IV) Schiff
base compounds derived from dithiocarbazate and thiosemicarbazone has not been
considered as good cytotoxic agents. Therefore, this project addresses this knowledge gap
by investigating cytotoxic activity of organotin(IV) and tin(IV) compounds of
dithiocarbazate and thiosemicarbazone Schiff bases. Dithiocarbazate and
thiosemicarbazone Schiff bases were selected as precursors for the development of
tin(IV) compounds and small modifications in their structure, by reacting o-vanillin (oVa)
or 2,3-dihydroxybenzaldehyde (catechol) with dithiocarbazate or thiosemicarbazide
ligands were investigated for their effect on cytotoxic activities. This project also
investigated DNA binding ability of the compounds as well as molecular modelling to
support the experimental data from the theoretical perspective.
1.8 Project Aims
Specific objectives of this project include:
1. Synthesis and structural characterisation of O, N, S tridentate Schiff bases and
their tin(IV) compounds.
2. Investigation of the cytotoxic activity of compounds formed in Aim 1 against a
panel of cancer cell lines
3. Experimental and theoretical investigation of the DNA binding interactions of
bioactive tin(IV) compounds obtained from Aim 2.

42
1.9 Thesis Outline
The synthesis and structural evaluation of oVa dithiocarbazate Schiff bases (1-3)
are presented and discussed in Chapter 2. Chapter 3 details the synthesis of diorganotin,
diphenyl- (4-6) and dimethyltin(IV) (7-9) compounds derived from oVa dithiocarbazate
Schiff bases (1-3), as well as their cytotoxicity. Chapter 3 also presents DNA binding
studies and molecular docking and molecular dynamic simulations for highly active
compounds that provide further insight regarding their mechanism of action. The
synthesis, characterisation and cytotoxicity of diphenyl- (13-15) and dimethyltin(IV) (16-
18) compounds of catechol dithiocarbazate Schiff bases (10-12) are presented in Chapter
4. The synthesis, structural study and biological evaluation of homoleptic tin(IV)
compounds (19-24) containing of two molecules of Schiff bases (1, 2, 3, 10, 11 and 12)
are reported in Chapter 5. Chapter 6 describes the synthesis, characterisation and
cytotoxicity of tin(IV) compounds (29-40) derived from thiosemicarbazone Schiff bases
(25-28) as well as their in-silico studies. Finally, the overall findings of this study are
summarised in Chapter 7 and recommendations for future study are suggested.

43
CHAPTER 2
SYNTHESIS, STRUCTURAL EVALUATION AND CYTOTOXICITY STUDIES OF O-VANILLIN DERIVED DITHIOCARBAZATE SCHIFF BASES
2.1 Introduction
As described in the Aims and Objectives section of Chapter 1, dithiocarbazate was
selected as precursor for the development of anticancer drug candidates. In
pharmaceuticals, vanillin (4-hydroxy-3-methoxybenzaldehyde) (Figure 2.1a) has been
used to synthesise trimethoprim (antibacterial drug), and papaverine, a drug that is
primarily used in the treatment of visceral spasms, vasospasms, and erectile
dysfunction.171 Ortho-vanillin (2-hydroxy-3-methoxybenzaldehyde) (oVa) (Figure 2.1b)
is a yellow crystalline material present in the extracts of essential oils of many plants. It
has distinctly different properties from its vanillin analogues. oVa-containing Schiff bases
are of immense interest in medicinal chemistry due to their potential biological properties
and a wide range of coordination modes. The azomethine connection between oVa and
the amine moieties (for example, dithiocarbazate) in Schiff bases, has been known to be
responsible for antitumour, antibacterial and antifungal activity.85,87,94,96,108,109,172–174
OH
OO
OO
OH(a) (b)
Figure 2.1: The structures of (a) vanillin and (b) o-vanillin In vitro cytotoxicity screening of two oVa Schiff bases derived from (R)-(+)-2-
amino-3-phenyl-1-propanol and 2-amino-2-ethyl-1,3-propanediol have recently been
evaluated against A549, HeLa, HL-60 and K-562 (chronic myelogenous leukemic) cell
lines.175 However, the concentrations needed to inhibit the cell lines were more than 50

44
μM, so they were considered as inactive compounds. Generally, most biological functions
of therapeutic agents are strongly dependent on the compound structure. Dithiocarbazate,
NH2NHC(=S)S-, and more specifically dithiocarbazate-substituted derivatives, have
attracted the attention of researchers for decades,84 since transition metal complexes of
these anions relate to potential biological activity.89,101,110,176 It is hypothesised, in this
project that the cytotoxic activity of dithiocarbazate Schiff base can be improved via
incorporation of oVa. This chapter investigates this hypothesis, and presents the
synthesis, structural characterisation and cytotoxic activities of three dithiocarbazate
Schiff bases derived from oVa.
This chapter contains material from published articles, adapted from reference177
with permission from the Elsevier and from reference178 with permission from MDPI.
Copyright permission attached in Appendices, page 222 and 223.
2.2 Experimental and Computational Methods
2.2.1 Materials
All solvents and reagents were of analytical reagent grade and used without further
purification. Chemicals: Hydrazine hydrate, 80% (Fluka, Buchs, Switzerland),
benzylchloride, ≥ 99% (Merck, New York, NY, USA), 2-methybenzyl chloride, 99%
(ACROS, Carson, CA, USA), 4-methylbenzyl chloride (ACROS, Carson, CA, USA),
potassium hydroxide (HmbG, Hamburg, Germany), carbon disulfide (BDH, Radnor, PA,
USA), 2-hydroxy-3-methoxybenzaldehyde (Merck, New York, NY, USA). Solvents:
Acetonitrile (Baker, Sanford, ME, USA), absolute ethanol, 99.8% (Scharlau, Barcelona,
Spain), ethanol, 95% (John Kollin Corporation, Midlothian, UK), methanol (Fisher,
Pittsburgh, PA, USA) and dimethylsulfoxide (Scharlau, Barcelona, Spain).

45
Chemicals used for biological assay were purchased as follows: RPMI-1640
medium with L-glutamine (without sodium bicarbonate) (Sigma Aldrich, St. Louis, MO,
USA), Penicillin/Streptomycin solution (Biowest, Riverside, MO, USA), Fetal Bovine
Serum (sterile filtered) (Biowest, Riverside, MO, USA), Trypsin 0.25%, EDTA in HSSS
without calcium, magnesium, with phenol red (Biowest, Riverside, MO, USA),
Phosphate Buffered Saline (PBS) 10× concentrate (Sigma Aldrich, St. Louis, MO, USA),
dimethyl sulfoxide (DMSO) (Fisher Scientific, Pittsburgh, PA, USA).
2.2.2 Synthesis
2.2.2.1 S-N-R-Benzyldithiocarbazates (N = 2, 3, R = Methyl)
The synthetic procedure used here was adapted from Tarafder and co-
workers.97,103,179 Potassium hydroxide (11.4 g, 0.2 mol) was dissolved in ethanol (70 cm3,
90%). Then, hydrazine hydrate (10 g, 0.2 mol) was added and the mixture was maintained
at 0 °C in an ice-salt bath. Carbon disulfide (15.2 g, 0.2 mol) was added dropwise with
vigorous stirring (750 rpm) over a period of 1 h. The two layers that formed were
separated and the light-brown lower layer was dissolved in 40% ethanol (60 cm3) below
5 °C. The mixture was kept in an ice-bath and 2-methylbenzyl chloride/4-methylbenzyl
chloride/benzyl chloride (0.2 mol) was added dropwise with vigorous stirring. The sticky
white product, which formed, S-2-methylbenzyldithiocarbazate (S1), S-4-
methylbenzyldithiocarbazate (S2) or S-benzyldithiocarbazate (S3), was filtered and left
to dry overnight in a desiccator over anhydrous silica gel. The products were then kept in
a freezer. The melting points of S-substituted dithiocarbazate derivatives are detailed in
Table 2.1.

46
Table 2.1: Physical data of synthesised S-substituted dithiocarbazate derivatives
Dithiocarbazates Yield (%) Melting point (°C) Related literature
S1 52 171.0-171.8 Lit. m.p: 171.2°C;103
S2 67 160.0-161.2 Lit. m.p: 86.0-89.0°C;97
S3 80 121.3-122.0 Lit. m.p: 125.0°C;179 2.2.2.2 S-2-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene)
dithiocarbazate (1)
The synthesis of 1 was adapted from literature.101,110 S1 (2.12 g, 10 mmol) was
dissolved in hot acetonitrile (100 cm3) and added to an equimolar amount of 2-hydroxy-
3-methoxybenzaldehyde (1.52 g, 10 mmol) in absolute ethanol (20 cm3). The mixture was
heated (80 °C) with continuous stirring for about 30 min and then allowed to stand
overnight at room temperature. The resultant product was recrystallised from
CH3CN/EtOH (1:1) to produce a light yellow crystalline solid that was filtered and
washed with cold absolute ethanol. Yield: 65%. M.p.: 180–183 °C. Elemental analysis:
calculated for C17H18N2O2S2: C, 58.93; H, 5.24; N, 8.09. Found: C, 59.75; H, 5.25; N,
7.81. FT-IR (ATR, cm−1): 3084, (N–H); 1600, (C=N); 1117, (N–N); 1026, (C=S). 1H
NMR (DMSO-d6) δ (ppm.): 13.34 (s,1H, NH), 9.57 (s, 1H, OH), 8.51 (s, 1H, CH), 6.75–
7.34 (multiplet, 7H, Ar–H), 4.40 (s, 2H, CH2), 3.76 (s, 3H, O–CH3), 2.30 (s,3H, Bz–CH3);
13C NMR (DMSO-d6) δ (ppm.): 196.1 (C=S), 148.6 (C=N); 148.6, 147.4, 144.9, 137.4,
134.4, 130.8, 130.7, 128.2, 126.7, 120.0, 118.8, 114.4 (aromatic-C), 56.4 (O–CH3), 36.9
(CH2), 19.4 (CH3). m/z calculated for C17H18N2O2S2: 346.47, found: 346.15.

47
2.2.2.3 S-4-Methybenzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene)
dithiocarbazate (2)
The synthesis of 2 was similar to 1, detailed above. S2 (2.12 g, 10 mmol) was
dissolved in hot acetonitrile (100 cm3) and added to an equimolar amount of 2-hydroxy-
3-methoxybenzaldehyde (1.52 g, 10 mmol) in absolute ethanol (20 cm3). The mixture was
heated (80 °C) with continuous stirring for about 30 min and then allowed to stand
overnight at room temperature. The resultant product was recrystallised from
CH3CN/EtOH (1:1) to produce a light yellow crystalline solid that was filtered and
washed with cold absolute ethanol. Yield: 68%. M.p.: 177–178 °C. Elemental analysis:
Calculated for C17H18N2O2S2: C, 58.93; H, 5.24; N, 8.09. Found: C, 58.48; H, 5.18; N,
8.76. FT-IR (ATR, cm−1): 3092, (N–H); 1598, (C=N); 1118, (N–N); 1030, (C=S). 1H
NMR (DMSO-d6) δ (ppm.): 13.32 (s,1H, NH), 9.61 (s, 1H, OH), 8.51 (s, 1H, CH), 6.97–
7.24 (multiplet, 7H, Ar–H), 4.39 (s, 2H, CH2), 3.76 (s, 3H, O-CH3), 2.23 (s,3H, Bz–CH3);
13C NMR (DMSO-d6) δ (ppm.): 196.2 (C=S), 148.6 (C=N); 148.6, 147.4, 145.0, 137.0,
134.0, 129.7, 129.6, 120.0, 118.8, 114.4 (aromatic-C), 56.4 (O–CH3), 38.0 (CH2), 21.2
(CH3). m/z calculated for C17H18N2O2S2: 346.47, found: 346.10.
2.2.2.4 S-Benzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene) dithiocarbazate (3)
The synthesis of 3 was similar to 1, detailed above. S3 (1.98 g, 10 mmol) was
dissolved in hot absolute ethanol (100 cm3) and added to an equimolar amount of 2-
hydroxy-3-methoxybenzaldehyde (1.52 g, 10 mmol) in absolute ethanol (20 cm3). The
mixture was heated (78 °C) with continuous stirring for about 30 min and then allowed
to stand overnight at room temperature. The resultant product was recrystallised from
ethanol to yield light yellow crystals that were filtered and washed with cold absolute
ethanol. Yield: 70%. M.p.: 173–174 °C. Elemental analysis: calculated for C16H16N2O2S2:

48
C, 57.81; H, 4.85; N, 8.43. Found: C, 58.21; H, 4.87; N, 8.28. FT-IR (ATR, cm−1): 3089,
(N–H); 1598, (C=N); 1125, (N–N); 1030, (C=S). 1H NMR (DMSO-d6) δ (ppm.): 13.34
(s,1H, NH), 9.58 (s, 1H, OH), 8.52 (s, 1H, CH), 6.77-7.37 (multiplet, 8H, Ar–H), 4.45 (s,
2H, CH2), 3.77 (s, 3H, O–CH3); 13C NMR (DMSO-d6) δ (ppm.): 196.1 (C=S), 148.6
(C=N); 148.6, 147.4, 144.9, 137.3, 129.8, 129.0, 127.8, 120.0, 118.8, 114.4 (aromatic-C),
56.4 (CH3), 38.1 (CH2). m/z calculated for C16H16N2O2S2: 332.44, found: 332.10.
2.2.3 Physical Measurements
Melting points were determined using an Electrothermal digital melting point
apparatus (Cole-Parmer, Staffordshire, UK). IR spectra were recorded using the Perkin
Elmer Spectrum 100 with Universal ATR Polarisation (PerkinElmer, Boston, MA, USA)
in the range 4000-280 cm−1. C, H and N elemental analyses were carried out using a
LECO CHNS-932 instrument (LECO, Saint Joseph, MI, USA). Electronic spectra were
recorded on a Shimadzu UV-1650 PC recording spectrophotometer (1000-200 nm)
(Shimadzu, Tokyo, Japan). 1H and 13C NMR spectra were recorded using an NMR JNM
ECA400 spectrometer (JEOL, Peabody, MA, USA) with tetramethylsilane (TMS) was
used as an internal standard. The mass spectra were recorded using a Shimadzu GC-MS
QP2010Plus mass spectrometer (Shimadzu, Tokyo, Japan).
2.2.4 Single Crystal X-ray Structure Determination
Crystals suitable for single-crystal X-ray diffraction studies were obtained for 3.
Unfortunately, suitable crystals for 1 and 2 were not able to be obtained despite repeated
crystal-growth attempts using various techniques and solvent systems. The intensity data
for 3 were measured at T = 100 K on an Oxford Diffraction Gemini E CCD diffractometer
(Oxford Diffraction Ltd., Agilent Technologies, Santa Clara, CA, USA) fitted with Mo

49
Kα radiation (λ = 0.71073 Å). Data reduction, including analytical absorption correction,
was accomplished with CrysAlisPro (Oxford Diffraction Ltd., UK).180 The structures
were solved by direct methods181 and refined (anisotropic displacement parameters, C-
bound H atoms in the riding model approximation) on F2. A weighting scheme w =
1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3).182 The molecular structure diagram
was generated with ORTEP for Windows183 with 50% displacement ellipsoids and the
packing diagrams were drawn with DIAMOND.184 Crystal data, data collection and
structure refinement details are summarized in Table 3.2. The carbon-bound H-atoms
were placed in calculated positions (C-H = 0.95–0.99 Å) and were included in the
refinement in the riding-model approximation, with Uiso(H) set to 1.2Ueq(C). The oxygen-
and nitrogen- bound H-atoms were located in a difference Fourier map but were refined
with distance restraints of O-H = 0.84±0.01 Å and N-H = 0.88±0.01 Å, and with Uiso(H)
set to 1.5Ueq(O) and 1.2Ueq(N).
2.2.5 Density Functional Theory (DFT) Calculations
All DFT calculations were performed using Gaussian09185 and Gaussview5186
software. The molecular structures and geometries of the Schiff bases were fully
optimised from the X-ray crystallographic structures using DFT method with the
B3LYP187,188 hybrid exchange correlation functional with a 6-311G(d,p) Pople basis set
for all atoms. Vibrational frequencies were calculated via the harmonic approximation to
ensure that all optimised structures corresponded to local minima of the potential energy
surface. All DFT vibrational frequencies were scaled using a scaling factor of 0.9682.189
The electronic stabilities of the optimised geometries were computed using the time-
dependent density functional theory (TD-DFT) formalism190,191 and included solvation

50
effects (DMSO) via the polarisable continuum method (PCM),192–194 using the same basis
set.
Table 2.2: Crystal data and refinement details for Schiff base 3 Formula C16H16N2O2S2
Molecular weight 332.43 Crystal system, space group Monoclinic, C2/c Temperature (K) 100 a, b, c (Å) 20.7896 (10), 4.6965 (2), 32.5217 (13) β (°) 95.004 (4) V (Å3) 3163.3 (2) Z 8 Radiation type Mo Kα μ (mm-1) 0.35 Crystal size (mm) 0.25x0.15x 0.07 Data collection Diffractometer Agilent Xcalibur Eos Gemini Absorption correction Multi-scan (CrysAlis PRO; Agilent, 2011) Tmin, Tmax 0.87, 0.98 No. of measured, independent and observed [I > 2σ(I)] reflection 6979, 3270, 2595 Rint 0.033 (sin θ/λ)max (Å-1) 0.628 Refinement, R[F2 > 2σ(F2)], wR(F2), S 0.041, 0.097, 1.06 No. of reflections 3270 No. of parameters 206 No. of restraints 2 H-atom treatment H atoms treated by a mixture of independent and constrained refinement ∆ ρmax, ∆ρmin (e Å-3) 0.33, -0.24
2.2.6 MTT Assays
The inhibitory effect of the synthesised compounds on the growth of bladder (EJ-
28 and RT-112) cancer cells was evaluated by MTT assay. The Schiff bases were
dissolved in DMSO and diluted in culture medium, where the final concentration of
DMSO is 0.05% (v/v) and was not toxic to the cancer cells. RT-112 and EJ-28, human
bladder cancer cell lines used in this study (ATCC, Virginia, USA) were cultured in

51
RPMI-1640 (High glucose) medium supplemented with 10% fetal bovine serum
containing 1% penicillin. The cells were cultured at 37 °C in a humidified atmosphere of
5% CO2 in air. Cells were seeded in 96-well plates at 6000 cells per well for 24 h, then
they were treated with different concentrations of compounds for 72 h. The medium was
subsequently removed and the wells were washed with 200 µL of phosphate buffer saline.
Aliquots of 20 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) was added to each well and incubated for 4 h at 37 °C. Aliquots of 200 µL of
media was then removed from each well and 200 µL of DMSO was added. Optical density
(OD) was measured using an ELISA plate reader at 570 nm. Control wells (100%
viability), containing media and DMSO, were included in all the experiments. All data
points represented an average of triplicate assays. Cytotoxicity was expressed as GI50,
i.e., the concentration that reduced the absorbance of treated cells by 50% with reference
to the control (untreated cells).195
For HT29 (colon), U87 and SJ-G2 (glioblastoma), MCF-7 (breast), A2780
(ovarian), H460 (lung), A431 (skin), Du145 (prostate), BE2-C (neuroblastoma), MIA
(pancreas) cell lines and one normal breast cell line, MCF-10A (normal breast), the MTT
assays were performed using the following methods:
Cell Culture and Stock Solutions. Stock solutions were prepared as follows and stored
at −20 °C: Trial compounds were stored as 10 mM solutions in DMSO. All cell lines were
cultured in a humidified atmosphere 5% CO2 at 37 °C. The cancer cell lines were
maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Trace Biosciences)
supplemented with 10% foetal bovine serum, 10 mM sodium bicarbonate, penicillin (100
IU/mL), streptomycin (100 μg/mL) and glutamine (4 mM). The normal cell line, MCF-
10A was cultured in DMEM:F12 (1:1) cell culture media, 5% heat inactivated horse
serum, supplemented with penicillin (50 IU/mL), streptomycin (50 μg/mL), 20mM

52
Hepes, L-glutamine (2 mM), epidermal growth factor (20 ng/mL), hydrocortisone (500
ng/mL), cholera toxin (100 ng/mL) and insulin (10 μg/mL).
In Vitro Growth Inhibition Assay. Cells in logarithmic growth were transferred to 96-
well plates. Cytotoxicity was determined by plating cells in duplicate in 100 μL medium
at a density of 2500–4000 cells/well. On day 0 (24 h after plating), when the cells were
in logarithmic growth, 100 μL medium, with or without the test agent, was added to each
well. After 72 h drug exposure, growth inhibitory effects were evaluated using the MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay and absorbance
read at 540 nm. Percentage growth inhibition was determined at a fixed drug
concentration of 25 μM. A value of 100% was indicative of complete cell growth
inhibition. Those analogues showing appreciable percentage growth inhibition underwent
further dose-response analysis allowing for the calculation of a GI50 value. This value is
the drug concentration at which cell growth is 50% inhibited based on the difference
between the optical density values on day 0 and those at the end of drug exposure.196,197
2.3 Results and Discussion
2.3.1 Synthesis
The Schiff bases, 1, 2 and 3, were prepared by condensation reaction of S-N-R-
benzyldithiocarbazates (N = 2, 3, R = methyl) and 2-hydroxy-3-methoxyenzaldehyde
(oVa) in equimolar ratios (Scheme 2.1). The Schiff bases were obtained in fairly good
yields of above 60 %. All the Schiff bases are non-hygroscopic, stable at room
temperature and soluble in common organic solvents especially dimethylsulfoxide
(DMSO) and dimethylformamide (DMF). The melting points of Schiff bases were sharp
(below 5 °C difference) indicating that the Schiff bases were free of impurities. All the
analytical data obtained were in good agreement with the proposed molecular structures.
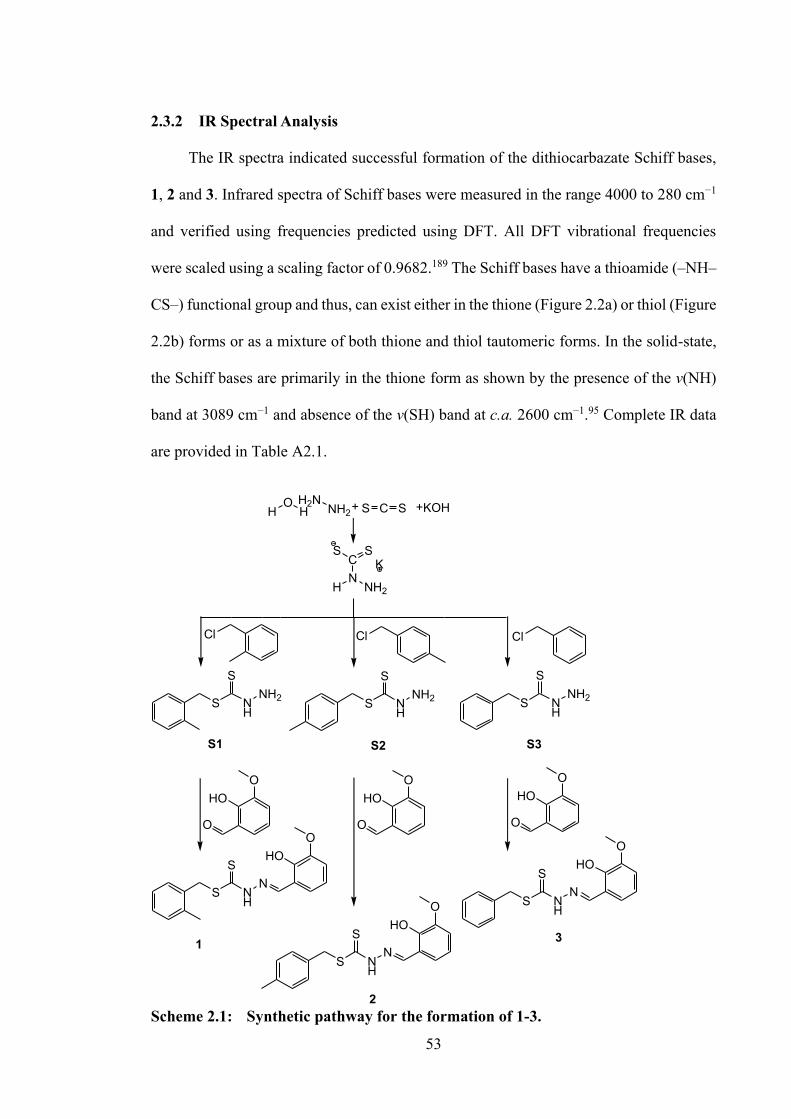
53
2.3.2 IR Spectral Analysis
The IR spectra indicated successful formation of the dithiocarbazate Schiff bases,
1, 2 and 3. Infrared spectra of Schiff bases were measured in the range 4000 to 280 cm−1
and verified using frequencies predicted using DFT. All DFT vibrational frequencies
were scaled using a scaling factor of 0.9682.189 The Schiff bases have a thioamide (–NH–
CS–) functional group and thus, can exist either in the thione (Figure 2.2a) or thiol (Figure
2.2b) forms or as a mixture of both thione and thiol tautomeric forms. In the solid-state,
the Schiff bases are primarily in the thione form as shown by the presence of the v(NH)
band at 3089 cm−1 and absence of the v(SH) band at c.a. 2600 cm−1.95 Complete IR data
are provided in Table A2.1.
Scheme 2.1: Synthetic pathway for the formation of 1-3.
C SS+ +KOH
HN
NH2
CSS
K
HO
HH2N
NH2
ClCl Cl
S
S
NH
N
HOO
S
S
NH
N
HOO
S
S
NH
N
HOO
HO
O
O
S
S
NH
NH2S
S
NH
NH2 S
S
NH
NH2
S3S1 S2
HO
O
OHO
O
O
1
2
3

54
NH
NS
SHO
O
(a)
NN
S
SHHO
O
(b)
R R
R = o-CH3 (1)R = p-CH3 (2)R = H (3)
Figure 2.2: The (a) thione and (b) thiol tautomeric forms of the Schiff bases 1-3.
2.3.3 NMR Spectroscopic Analysis
While IR spectra indicate that the dithiocarbazate Schiff bases 1, 2 and 3 exist as
thione tautomers, a previous study reported that the thione tautomers of similar Schiff
bases containing dithiocarbazate are relatively unstable in solution, and have a high
tendency to convert into thiol tautomers by enethiolisation.198 The solution-phase
structure of 1, 2 and 3 was therefore probed here via 1H and 13C NMR spectroscopy.
The absence of the exchangeable SH signal in the 1H NMR spectra of the Schiff
bases indicated the predominance of the thione tautomer in DMSO-d6.94 The spectra of
the Schiff bases also exhibited a downfield signal at ~13.32–13.34 ppm due to the NH
group. A singlet appeared in the downfield region of the 1H NMR spectra of the Schiff
bases, which corresponded to a sp2-C–OH proton. The presence of a methylene (-CH2)
proton between the sulphur atom and benzene ring was clearly observed at ~4 ppm,
slightly deshielded due to the close proximity to the electronegative sulphur atom. The
singlet peak was observed at ~8 ppm which corresponded to the –CH proton. The –CH
proton appeared at the deshielded region due to the electron repulsion from the
electronegative nitrogen atom. The singlet signal appearing at 2.30 and 2.23 ppm for 1
and 2, respectively, corresponding to a sp3 CH3 protons located at ortho and para position

55
of the benzene ring in the dithiocarbazate backbone. The methylene proton of the
methoxy group (O-CH3) was clearly observed at 3.76 (1 and 2) and 3.77 ppm (1). The
13C NMR spectra of the dithiocarbazate Schiff bases showed a downfield chemical shift
at ~196 ppm, indicating the existence of the C=S group and thus the predominance of the
thione tautomer in solution. The NMR details for 1, 2 and 3 are tabulated in Tables A2.2
and A2.3.
2.3.4 Mass Spectral Analysis
Mass spectral data for the Schiff bases 1-3 were recorded in DMSO and were found
to be consistent with the proposed formulation of the Schiff bases. Mass spectra displayed
prominent peaks at m/z 346.15 and 346.10 for 1 and 2, respectively, corresponding to
[C17H18N2O2S2]+, and m/z 332.10 for 3 corresponding to [C16H16N2O2S2]+. The mass
spectra for 1, 2 and 3 are supplied in Figure A2.1.
2.3.5 UV-vis Absorption Spectroscopy
The experimental UV-vis spectra of Schiff bases 1, 2 and 3 measured in DMSO
exhibited prominent absorption bands at 340–348 and 371–389 nm. The corresponding
peaks were predicted at 334 and 377–378 nm using B3LYP/6-311G(d,p). The highest
occupied molecular orbital and lowest unoccupied molecular orbital (HOMO-LUMO)
electron density distribution of the Schiff bases (see Figure 2.3) showed that the HOMO
was predominantly centred on the thiolate group and azomethine moieties of the
dithiocarbazate backbone in each of the compounds investigated here. Conversely, the
LUMO was consistently centred on the 2-hydroxy-3-methoxyphenyl ring, as well as on
the backbone of the dithiocarbazate moieties. Thus, the 334 nm absorption peak was
attributed to the n→π* transitions associated with the non-bonding electron pair of the

56
azomethine nitrogen and sulphur atoms. The π→π* transition was also observed related
to electron delocalisation in the aromatic rings. Complete experimental and calculated
UV-vis data are listed in Table A2.4.
Figure 2.3: HOMO-LUMO of (a) 1, (b) 2 and (c) 3.
2.3.6 X-ray Crystallography
Compound 3, Figure 2.4, comprises two almost planar regions, one being the
phenyl ring, the other being the remaining 14 heavy (non-hydrogen) atoms. The
maximum deviations from the least-squares plane through the latter plane, with a r.m.s.
deviation (RMSD) = 0.0410 Å, are 0.0715(15) Å for the O1 atom and -0.0796(18) for
atom C16. To a first approximation, the molecule can be described as having mirror
symmetry with the 1,4-atoms of the terminal ring being bisected by the plane. The
dihedral angle between the planes (82.72(5))°, substantiates this description, indicating a
very close to perpendicular relationship. The observed planarity in the larger fragment
may be ascribed, in part, to the presence of an intramolecular hydroxy-O-H…N(imine)
hydrogen bond (Table 2.3), which leads to the formation of an S(6) loop. The thione
C1=S2 bond length, 1.670(2) Å, is considerably shorter than the thiol C1-S1 and,
especially, C2-S1 bonds of 1.749(2) and 1.817(2) Å, respectively. The conformation
about the C N bond is E, and the amine-N-H atom is flanked on either side by the thione-

57
S and imine-H atoms. The detailed geometry for 3 is tabulated in the Appendix (Table
A2.5, A2.6, A2.7 and A2.8).
Figure 2.4: The molecular structure of 3, showing the atom- labelling scheme and displacement ellipsoids at the 50% probability level. The most prominent feature of the packing of compound 3 is the formation of
centrosymmetric, eight-membered …HNCS2 synthons through the agency of thio-
amide-N-H…S(thione) hydrogen bonds, Table 2.3. The dimeric aggregates are connected
by phenyl-C-H…O O(hydroxy) interactions to form a supramolecular layer in the bc-
plane, Figure 2.5a. The layers stack along the a axis with no directional interactions
between them, Figure 2.5b.
Table 2.3: Hydrogen-bond geometry (Å, °) of 3 obtained from single crystal X-ray diffraction analysis. The structure is shown in Figure 2.2. D-H…A D-H H…A D…A D-H…A O1-H1O…N2 0.84 (2) 1.90 (2) 2.639 (2) 146 (2) N1-H1N…S2i 0.88 (2) 2.47 (2) 3.3351 (18) 168 (2) C6-H6…O1ii 0.95 2.51 3.453 (3) 170

58
Figure 2.5: Molecular packing in 3: (a) a perspective view of the supramolecular layer sustained by thioamide-N-H…S(thione) and phenyl-C-H…O(hydroxy) interactions and, (b) a view of the unit-cell contents shown in projection down the b axis, highlighting one layer in space-filling mode. The N-H…S and C-H…O interactions are shown as blue and orange dashed lines, respectively. For (a), non-interacting H atoms have been omitted.
2.3.7 In Vitro Cytotoxicity
Compounds 1-3 were screened for in vitro cytotoxicity against a panel of twelve
cancer cell lines: EJ-28 (muscle invasive bladder), RT-112 (minimum-invasive bladder),
HT29 (colon), U87 and SJ-G2 (glioblastoma), MCF-7 (breast), A2780 (ovarian), H460
(lung), A431 (skin), Du145 (prostate), BE2-C (neuroblastoma), MIA (pancreatic) and one
normal breast cell line (MCF-10A) using the MTT metabolic assay. After 72 h of
incubation, a dose-dependent proliferative effect towards the cancer cell lines was
measured at very low micromolar (µM) concentrations of the compounds. The growth
inhibition concentration of the compounds required to inhibit 50% cell proliferation
relative to the cells that were treated with DMSO (control) are given in Table 2.4. The

59
stability of these compounds was assessed in DMSO as well as in a mixture of DMSO-
H2O. The absorbance obtained from UV-vis spectroscopy analysis was monitored for 72
h, and an unchanged pattern in the spectra was indicative that the compounds were stable
in both the solvent systems tested.
The GI50 values for the three Schiff bases showed a slight difference; the presence
of methyl group at ortho (1) and para (2) positions resulted in a slight reduction in
potency as compared to 1. This cytotoxicity difference was potentially due to the
hydrophobicity of the methyl group.199 2 exhibited the lowest potency against HT29 and
MIA with GI50 values of 8.0 ± 3.5 and 11 ± 2.3 µM, respectively. Taking cisplatin into
consideration, 1, 2 and 3 demonstrated equipotent cytotoxicity against EJ-28, RT-112 and
U87 cell lines. On both selective cell lines, HT29 and MIA, 1 and 3 displayed a twofold
increase in activity compared to cisplatin, indicating that the ortho methyl group yielded
better activity than the para methyl group. A twofold increase in activity was also
displayed in MCF-7 cell line treated with 1, 2 and 3. However, 1, 2 and 3 showed lower
activity against A2780, H460, A431, Du145, BE2-C and SJ-G2 cell lines compared to
cisplatin. A previous in vitro study on dithiocarbazate-containing compounds proposed
that these compounds induce apoptosis by DNA fragmentation and suppression of two
key genes, Epidermal Growth Factor Receptor (EGFR) and Mouse Double Minute 2
(MDM2) expression.200
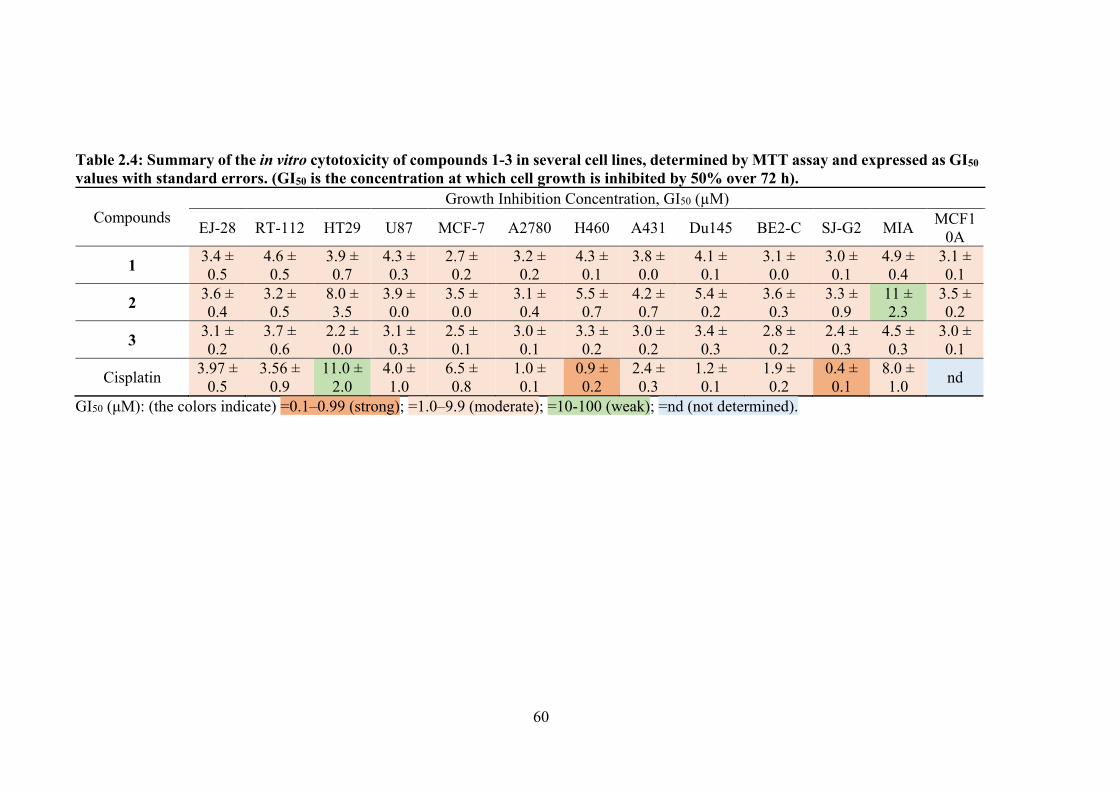
60
Table 2.4: Summary of the in vitro cytotoxicity of compounds 1-3 in several cell lines, determined by MTT assay and expressed as GI50 values with standard errors. (GI50 is the concentration at which cell growth is inhibited by 50% over 72 h).
Compounds Growth Inhibition Concentration, GI50 (µM)
EJ-28 RT-112 HT29 U87 MCF-7 A2780 H460 A431 Du145 BE2-C SJ-G2 MIA MCF10A
1 3.4 ± 0.5
4.6 ± 0.5
3.9 ± 0.7
4.3 ± 0.3
2.7 ± 0.2
3.2 ± 0.2
4.3 ± 0.1
3.8 ± 0.0
4.1 ± 0.1
3.1 ± 0.0
3.0 ± 0.1
4.9 ± 0.4
3.1 ± 0.1
2 3.6 ± 0.4
3.2 ± 0.5
8.0 ± 3.5
3.9 ± 0.0
3.5 ± 0.0
3.1 ± 0.4
5.5 ± 0.7
4.2 ± 0.7
5.4 ± 0.2
3.6 ± 0.3
3.3 ± 0.9
11 ± 2.3
3.5 ± 0.2
3 3.1 ± 0.2
3.7 ± 0.6
2.2 ± 0.0
3.1 ± 0.3
2.5 ± 0.1
3.0 ± 0.1
3.3 ± 0.2
3.0 ± 0.2
3.4 ± 0.3
2.8 ± 0.2
2.4 ± 0.3
4.5 ± 0.3
3.0 ± 0.1
Cisplatin 3.97 ± 0.5
3.56 ± 0.9
11.0 ± 2.0
4.0 ± 1.0
6.5 ± 0.8
1.0 ± 0.1
0.9 ± 0.2
2.4 ± 0.3
1.2 ± 0.1
1.9 ± 0.2
0.4 ± 0.1
8.0 ± 1.0 nd
GI50 (μM): (the colors indicate) =0.1–0.99 (strong); =1.0–9.9 (moderate); =10-100 (weak); =nd (not determined).

61
2.4 Conclusions
This chapter presented three dithiocarbazate Schiff bases derived from oVa which
were synthesised via condensation. The structures of synthesised Schiff bases were
confirmed by various spectroscopic techniques and DFT calculations. The Schiff bases
exist as thione tautomers even in solution which was proven by FT-IR, NMR and mass
spectroscopy analysis. The molecular structure of 3 was authenticated by single crystal
X-ray crystallography. The in vitro anticancer activity against a panel of cancer cell lines,
viz., EJ-28, RT-112, HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C,
MIA and one normal breast cell line (MCF10A) revealed that the oVa containing
dithiocarbazate Schiff bases afforded a modest activities, with slight enhancements
achieved by the introduction of the methyl groups at the ortho or para positions of the
Schiff base phenyl ring. The Schiff bases had no obvious cytotoxic cell line selectivity. It
is suggested that the presence of nitrogen, sulphur and oxygen donor atoms enabled
interaction with the donor atoms of nitrogenous bases of DNA, as well as proteins and
organelles of the cancer cells.
While this chapter demonstrated the synthesis and characterisation of three Schiff
bases with moderate cytotoxic activity, it is hypothesised that this activity could be
improved via their complexation with a metal such as tin. This hypothesis will be tested
in Chapter 3, which reports structural and cytotoxic studies of Schiff bases 1, 2 and 3 with
diphenyl- and dimethyltin, was further improved by complexation with diphenyl- and
dimethyltin. Since DNA is the primary pharmacological target of many chemotherapeutic
compounds, DNA-tin(IV) compounds interactions will also be discussed towards
establishing a mechanism of cancer cell inhibition in these compounds.

62
CHAPTER 3
ORGANOTIN(IV) COMPOUNDS OF O-VANILLIN SCHIFF BASES: SYNTHESIS, STRUCTURAL CHARACTERISATION, IN-SILICO STUDIES
AND CYTOTOXICITY
3.1 Introduction
In recent years, metal compounds with a stable d10 electronic configuration have
received considerable attention due to the wide variety of structures formed that depend
on the coordinating ligands and synthetic procedures.86,139,140,201–207 Tin, a group V metal,
can be found in various synthesis of inorganic and organometallic compounds with a
diverse range of applications. Organotin compounds were first discovered in 1894 by
Edward Frankland, with the first such compound being diethyltindiiodide ((C2H5)2SnI2).
The presence of one or more covalent C-Sn bonds in organotin compounds affects their
activity. In particular, the cytotoxicity of these compounds depends on the nature of alkyl
or aryl substituents coordinated to the tin center. Variation in the number and nature of
substituents in organotin(IV) compound has shown notable effects on their biological
activities, due to the unique characteristics of the compounds, such as structural diversity,
redox capacity and catalytic activity. Furthermore, such ligands play important roles in
transporting and addressing the organotin(IV) compound to specific sites in
biomacromolecules.
Organotin(IV) compounds derived from dithiocarbazate Schiff bases have received
attention due to their ability to stabilise specific stereochemistry. Many mechanistic
studies on organotin(IV) compounds have indicated that these compounds are able to
interact with DNA,151,155 one of the main targets in designing cytotoxic drugs. The
biological activity of organotin(IV) compounds is believed to arise from their tendency
in binding to or cleaving DNA.208,209 Wang et al. reported that organotin(IV) compounds

63
containing Schiff bases could interact with CT-DNA through intercalation of cytotoxic
drugs with serum albumin (which is involved in the transportation of compounds through
the blood stream).142 The nature and magnitude of these interactions could significantly
influence the pharmacokinetics.210,211 Organotin(IV) compounds have been found to
induce DNA damage by specifically binding to both nitrogenous DNA bases and the
phosphate backbone of DNA, thus leading to changes in DNA conformation.57,128,212
Organotin(IV) compounds have also been reported to bind to glycoprotein or cellular
proteins, hence interacting directly with DNA, causing cell death.
The activity of organotin(IV) compounds can be readily modified by the choice of
ligand coordinated to the central tin(IV) ion. Organotin(IV) centres coordinated to hetero-
donor atoms, especially oxygen, nitrogen and sulphur, have been investigated with a
particular focus on their structure-activity correlations.213–215 However, the cytotoxicity
of organotin(IV) compounds containing dithiocarbazate Schiff bases are essentially
unexplored in the literature. It is hypothesised here that complexing dithiocarbazate Schiff
bases with tin enhances the cytotoxicity of the compounds. This chapter investigates this
hypothesis, by reporting the synthesis, structural characterisation and cytotoxic activity
of six organotin(IV) compounds (4-9) of Schiff bases (1-3), see Figure 3.1.
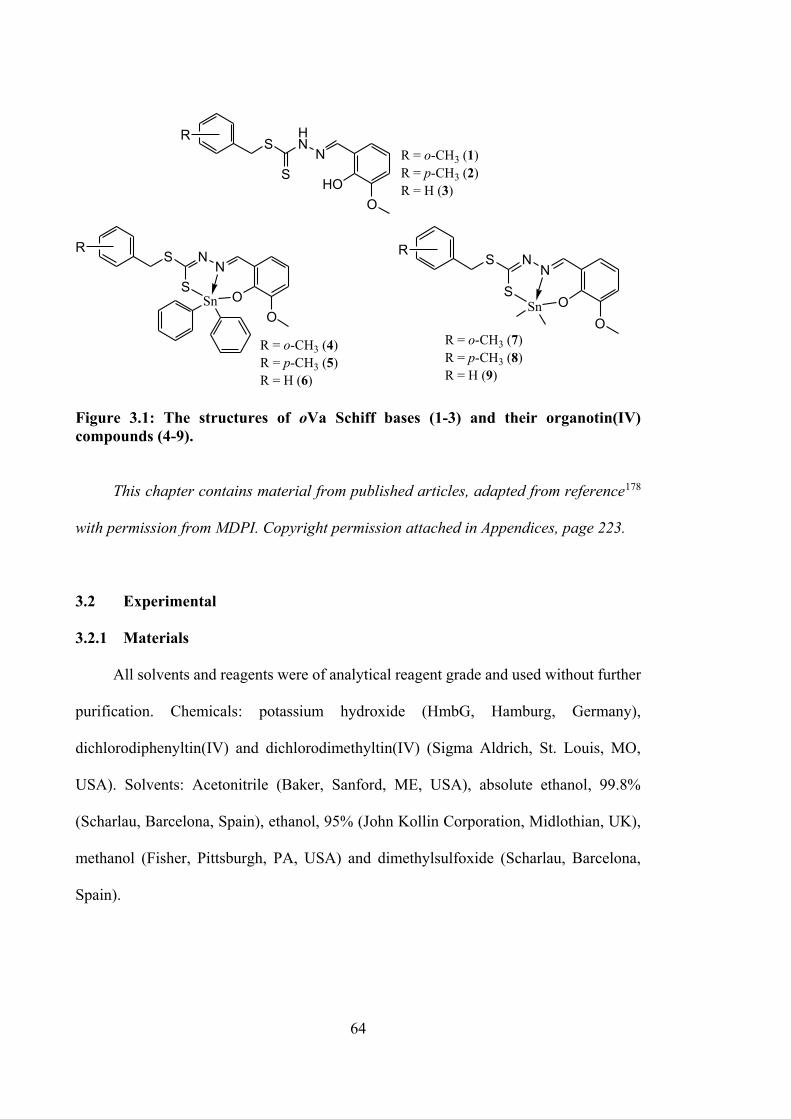
64
SHN
NS
HOO
S NN
SO
OSn
S NN
SO
OSn
R
R R
R = o-CH3 (4)R = p-CH3 (5)R = H (6)
R = o-CH3 (1) R = p-CH3 (2)R = H (3)
R = o-CH3 (7)R = p-CH3 (8)R = H (9)
Figure 3.1: The structures of oVa Schiff bases (1-3) and their organotin(IV) compounds (4-9).
This chapter contains material from published articles, adapted from reference178
with permission from MDPI. Copyright permission attached in Appendices, page 223.
3.2 Experimental
3.2.1 Materials
All solvents and reagents were of analytical reagent grade and used without further
purification. Chemicals: potassium hydroxide (HmbG, Hamburg, Germany),
dichlorodiphenyltin(IV) and dichlorodimethyltin(IV) (Sigma Aldrich, St. Louis, MO,
USA). Solvents: Acetonitrile (Baker, Sanford, ME, USA), absolute ethanol, 99.8%
(Scharlau, Barcelona, Spain), ethanol, 95% (John Kollin Corporation, Midlothian, UK),
methanol (Fisher, Pittsburgh, PA, USA) and dimethylsulfoxide (Scharlau, Barcelona,
Spain).

65
3.2.2 Synthesis
3.2.2.1 Diphenyltin(IV) Compounds
Each Schiff base (0.35 g (1, 2); 0.33 g (3), 1 mmol) from Chapter 2 was dissolved
in absolute ethanol (50 cm3) and mixed with an ethanolic solution (10 cm3) of Ph2SnCl2
(0.34 g, 1 mmol). The resultant yellow solution was refluxed for ca. 6 h. The mixture was
left overnight at room temperature and the reduction in the volume of the reaction mixture
resulted in the deposition of a yellow precipitate which was filtered off and recrystallised
from methanol.
3.2.2.1.1 Diphenyltin(IV) [S-2-Methybenzyl-β-N-(2-hydroxy-3-
methoxybenzylmethylene)dithiocarbazate] (4)
Yellow solid. Yield: 44%. M.p.: 149–150 °C. Elemental analysis: Calculated for
C29H34N2O6S2Sn: C, 56.42; H, 4.24; N, 4.54. Found: C, 56.52; H, 4.97; N, 4.06. FT-IR
(ATR, cm−1): 1588, (C=N); 1076, (N–N); 963, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.77
(s, 1H, CH), 4.47 (s, 2H, CH2), 2.43 (s, 3H, CH3), 3.97 (s, 3H, O–CH3), 6.69-7.94
(multiplet, 17H, Ar–H); 13C NMR (CDCl3) δ (ppm.): 171.9 (C–S), 166.1 (C=N); 159.3,
152.0, 142.0, 136.1, 130.6, 130.4, 130.2, 128.9, 127.9, 126.3, 126.1, 117.2, 116.2
(aromatic-C), 56.6 (O-CH3), 34.4 (CH2), 19.4 (Bz–CH3).119Sn NMR (CDCl3) δ (ppm.):
−236.5.
3.2.2.1.2 Diphenyltin(IV) [S-4-Methybenzyl-β-N-(2-hydroxy-3-methoxy
benzylmethylene)dithiocarbazate] (5)
Yellow solid. Yield: 72%. M.p.: 130–132 °C. Elemental analysis calculated for
C17H17N2O2S2: C, 54.82; H, 4.44; N, 4.41. Found: C, 54.42; H, 4.05; N, 4.18. FT-IR
(ATR, cm−1): 1589, (C=N); 1068, (N–N); 958, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.74

66
(s, 1H, CH), 4.41 (s, 2H, CH2), 2.32 (s, 3H, CH3), 3.94 (s,3H, O–CH3), 6.69-7.93
(multiplet, 17H, Ar–H); 13C NMR (CDCl3) δ (ppm.): 171.8 (C–S), 166.1 (C=N), 159.3,
152.0, 142.0, 137.2, 136.0, 135.8, 133.5, 130.2, 129.4, 129.2, 128.9, 126.1, 117.2, 116.3
(aromatic-C), 56.6 (O–CH3), 36.0 (CH2), 21.2 (Bz–CH3).119Sn NMR (CDCl3) δ (ppm.):
−236.3.
3.2.2.1.3 Diphenyltin(IV) [S-Benzyl-β-N-(2-hydroxy-3-methoxybenzyl methylene)
dithiocarbazate] (6)
Yellow crystals. Yield: 51%. M.p.: 131–133 °C. Elemental analysis calculated for
C28H24N2O2S2Sn: C, 55.74; H, 4.01; N, 4.64. Found: C, 56.35; H, 3.91; N, 4.98. FT-IR
(ATR, cm−1): 1579, (C=N); 1019, (N–N); 958, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.74
(s, 1H, CH), 4.45 (s, 2H, CH2), 3.96 (s, 3H, O-CH3), 6.69-7.93 (multiplet, 18H, Ar–H);
13C NMR (CDCl3) δ (ppm.): 171.6 (C–S), 166.2 (C=N), 152.0, 141.9, 136.7, 136.1, 130.3,
129.3, 128.9, 128.7, 127.5, 126.1, 117.2, 117.1, 116.2, 115.2, (aromatic-C), 56.6 (O–
CH3), 36.1 (CH2). 119Sn NMR (CDCl3) δ (ppm.): −236.3.
3.2.2.2 Dimethyltin(IV) Compounds
Each Schiff base (0.35 g (1, 2); 0.33 g (3), 1 mmol) from Chapter 2 was dissolved
separately in absolute ethanol (50 cm3) and dichloromethane (DCM) (20 cm3) and then
mixed with an ethanolic solution (10 cm3) of Me2SnCl2 (0.22 g, 1 mmol). The resultant
yellow solution was refluxed for ca. 6 h. The mixture was left overnight at room
temperature and kept for a few days yielding brown crystals that were suitable for single
crystal X-ray diffraction analysis.

67
3.2.2.2.1 Dimethyltin(IV) [S-2-Methybenzyl-β-N-(2-hydroxy-3-methoxy benzyl
methylene)dithiocarbazate] (7)
Yellow crystals. Yield: 78%. M.p.: 128–130 °C. Elemental analysis calculated for
C19H22N2O2S2Sn: C, 46.27; H, 4.50; N, 5.68. Found: C, 46.52; H, 4.51; N, 5.82. FT-IR
(ATR, cm−1): 1580, (C=N); 1076, (N–N); 959, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.76
(s, 1H, CH), 4.42 (s, 2H, CH2), 2.42 (s, 3H, CH3), 3.85 (s,3H, O–CH3), 6.69–7.34
(multiplet, 7H, Ar–H), 0.97 (Sn–CH3); 13C NMR (CDCl3) δ (ppm.): 173.9 (C–S), 166.3
(C=N), 158.5, 151.5, 137.2, 134.1, 130.6, 130.4, 127.9, 126.3, 126.1, 116.9, 116.5, 115.9
(aromatic-C), 56.3 (CH3), 34.6 (CH2), 19.4 (Bz–CH3), 7.1 (Sn–CH3).119Sn NMR (CDCl3)
δ (ppm.): −109.4.
3.2.2.2.2 Dimethyltin(IV) [S-4-Methybenzyl-β-N-(2-hydroxy-3-methoxy
benzylmethylene)dithiocarbazate] (8)
Yellow crystals. Yield: 55%. M.p.: 98–102 °C. Elemental analysis calculated for
C19H22N2O2S2Sn: C, 46.27; H, 4.50; N, 5.68. Found: C, 47.07; H, 4.66; N, 6.11. FT-IR
(ATR, cm−1): 1589, (C=N); 1071, (N–N); 958, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.73
(s, 1H, CH), 4.36 (s, 2H, CH2), 2.32 (s, 3H, CH3), 3.84 (s, 3H, O-CH3), 6.69–7.27
(multiplet, 7H, Ar–H), 0.95 (Sn–CH3); 13C NMR (CDCl3) δ (ppm.): 173.7 (C–S), 166.2
(C=N), 158.5, 151.5, 137.1, 133.6, 129.2, 126.1, 119.5, 116.8, 116.4, 115.9 (aromatic-C),
56.3 (CH3), 36.2 (CH2), 21.2 (Bz–CH3), 7.1 (Sn–CH3). 119Sn NMR (CDCl3) δ (ppm.):
−109.2.

68
3.2.2.2.3 Dimethyltin(IV) [S-Benzyl-β-N-(2-hydroxy-3-methoxybenzyl
methylene)dithiocarbazate] (9)
Yellow crystals. Yield: 48%. M.p.: 104–106 °C. Analysis calculated for
C18H20N2O2S2Sn: C, 45.11; H, 4.21; N, 5.85. Found: C, 45.36; H, 4.16; N, 6.02. FT-IR
(ATR, cm−1): 1581, (C=N); 1026, (N–N); 959, (C=S). 1H NMR (CDCl3) δ (ppm.): 8.73
(s, 1H, CH), 4.40 (s, 2H, CH2), 3.85 (s, 3H, O–CH3), 6.69-7.40 (multiplet, 8H, Ar–H),
0.95 (Sn–CH3); 13C NMR (CDCl3) δ (ppm.): 173.6 (C–S), 166.3 (C=N), 158.5, 151.5,
136.8, 129.3, 128.7, 127.4, 126.1, 116.9, 116.5, 115.9 (aromatic-C), 56.3 (O–CH3), 36.4
(CH2), 7.1 (Sn–CH3); 119Sn NMR (CDCl3) δ (ppm.): −109.0.
3.2.3 Physical Measurements
The melting point, CHNS analyser, FTIR, UV-vis and NMR (1H and 13C NMR)
instruments used in this chapter as the same as mentioned in Chapter 2 (§2.2.3). Molar
conductivities of 10−3 M solutions of the organotin(IV) compounds in DMSO were
measured at 27 °C using a Jenway 4310 conductivity meter fitted with a dip-type cell with
a platinised electrode (Cole-Parmer, Staffordshire, UK). 119Sn NMR were recorded using
a JOEL NMR spectrophotometer (JEOL, Peabody, MA, USA).
3.2.4 Single Crystal X-ray Structure Determination
Crystals suitable for single-crystal X-ray diffraction studies were obtained for 7, 8
and 9. Unfortunately, suitable crystals for the diphenyltin(IV) compounds were not
obtained despite repeated crystal-growth attempts using various techniques and solvent
systems. The intensity data for 7, 8 and 9 were measured at T = 150 K on an Oxford
Diffraction Gemini E CCD diffractometer (Oxford Diffraction Ltd., Agilent
Technologies, Santa Clara, CA, USA) fitted with Mo Kα radiation source (λ = 0.71073

69
Å) so that θmax was 29.4°. Data reduction, including analytical absorption correction, was
accomplished with CrysAlisPro.180 The structures were solved by direct methods181 and
refined (anisotropic displacement parameters, C-bound H atoms in the riding model
approximation) on F2. A weighting scheme w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2
+ 2Fc2)/3)182 was introduced in each case. The crystal of 7 was refined as a twin with the
fraction of the minor component being 0.1664(20); details are given in the deposited CIF.
In the final cycles of the refinement of 9, four reflections, i.e., (−3 4 0), (−1 −1 5), (0 6 0)
and (−3 4 4), were omitted owing to poor agreement. The molecular structure diagrams
was generated with ORTEP for Windows183 with 35, 50 and 70% displacement ellipsoids
for 7, 8 and 9, respectively, and the packing diagrams were drawn with DIAMOND
(Crystal Impact, GbR, Bonn, Germany).184 Additional data analysis was carried out with
PLATON.216 Crystal data and refinement details are given in Table 3.1.
3.2.5 Computational Methods
3.2.5.1 DFT Calculations
DFT calculations were performed as the same software and parameters mentioned
in Chapter 2. The molecular structures and geometries of 4, 5 and 6 were fully optimised
with the B3LYP187,188 hybrid exchange correlation functional in conjunction with the
LanL2DZ pseudopotential on Sn217–219 and 6-311G(d,p) Pople basis set for all other
atoms. The initial single crystal X-ray molecular structures and geometries of 7, 8 and 9
were used for DFT calculations using the same functional and basis set.

70
Table 3.1: Crystal data and refinement details for compounds 7, 8 and 9. Compound 7 8 9 Formula C19H22N2O2S2Sn C19H22N2O2S2Sn C18H20N2O2S2Sn Formula weight 493.19 493.19 479.19 Crystal colour light-yellow light-yellow light-yellow
Crystal size/mm3 0.03 × 0.05 × 0.10
0.05 × 0.13 × 0.20
0.03 × 0.05 × 0.10
Crystal system Monoclinic Triclinic Triclinic Space group Pn P1 P1 a/Å 11.3716(9) 10.5908(4) 8.9408(4) b/Å 12.0262(8) 13.9243(5) 10.4182(4) c/Å 16.3644(15) 14.3974(5) 22.6034(12) α/° 90 95.596(3) 97.713(4) β/° 109.285(9) 93.693(3) 94.600(4) γ/° 90 102.116(2) 111.634(4) V/Å3 2112.4(3) 2058.11(13) 1920.11(16) Z 4 4 4 Dc/g cm−3 1.551 1.592 1.658 F(000) 992 992 960 μ(MoKα)/mm−1 1.422 1.46 1.562 Measured data 6348 18090 15229 θ range/° 3.6–29.4 3.5–29.4 3.4–29.4 Unique data 6348 9402 8720 Observed data (I ≥ 2.0σ(I)) 5090 7605 6671
No. parameters 478 477 457 R, obs. data; all data 0.037; 0.064 0.034; 0.070 0.041; 0.083 a; b in weighting scheme 0.022; 0 0.026; 0.240 0.032; 0.781 Rw, obs. data; all data 0.055; 0.074 0.049; 0.078 0.062; 0.092 GoF 0.99 1.01 1.01 Range of residual electron
density peaks/eÅ−3 −0.56–0.66 −0.46–0.75 −1.03–0.63
3.2.5.2 Molecular Docking Studies
The coordinates of B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2
were obtained from the Protein Data Bank (http://www.rcsb.org/pdb). The coordinates of
the dimethyltin(IV) compounds, 7, 8 and 9 were taken from crystal structures, whereas
the coordinates for 4, 5 and 6 were obtained after DFT geometry optimisation. Molecular
docking studies were carried out using the AutoDock version 4.2.5.1 program (Molecular
Graphics Laboratory, La Jolla, CA, USA).220 Water molecules were removed, polar

71
hydrogen atoms and Kollman charges221 were added to the receptor (DNA sequences). In
the docking analysis, the binding site was assigned across all of the minor and major
grooves of the DNA molecule, which was enclosed in a 64 × 64 × 122 grid box with a
grid spacing of 0.375 Å. AutoDock was ran using the following parameters: 30 docking
trials, population size of 300, maximum number of energy evaluation ranges of 2,500,000,
maximum number of generations of 27,000, mutation rate of 0.02, cross-over rate of 0.8
and the default values were used for other parameters. The parameter file of Autodock
was modified to incorporate van der Waals interactions and other required parameters for
Sn obtained from the Autodock website.222 The lowest binding energy of the
conformation from the highest cluster was selected as the best binding conformation
between the ligand and receptor.
3.2.6 Biological Assays
3.2.6.1 MTT Assays
The protocol used for MTT assay followed that outlined in Chapter 2 (§2.2.6).
3.2.6.2 Quantification of Apoptosis by Annexin V
The apoptotic death of RT-112 cells was measured by fluorescence microscopy
using Annexin V/ PI double-staining. To observe the effect of the diphenyltin(IV)
compounds (4, 5 and 6) on cell apoptosis, RT-112 cells, which are minimum invasive
bladder cancer cells were seeded in 6-well plates at 1 × 106 cells per well. After growth
overnight, the inhibition concentration obtained from the GI50 analysis was used to treat
the cells. After 24 h, the cells were washed with phosphate buffer saline (PBS) and 100
µL of Annexin-V-FLUOS labelling solution containing the incubation buffer and then,
propidium iodide (PI) solution was added. The plate was then incubated for 10–15 min at

72
15–25 °C and analysed by fluorescence microscopy with a Fluorescence Inverted
Microscope (Olympus IX51, Waltham, USA). Green staining of the plasma membrane
indicated that the cells had bound to Annexin-V. The cells that showed red staining were
considered to have lost their membrane integrity. The cells stained green were concluded
to be apoptotic cells; the cells with both green and red staining were concluded as late
apoptotic cells whereas the cells that showed only red staining were considered as necrotic
cells.223
3.2.6.3 Measurement of Reactive Oxygen Species (ROS)
Measurements were performed using a fluorescence plate reader following the
procedure outlined in the ROS detection kit (OxiSelect™). RT-112 cells were cultured in
100 µL growth medium in a 96-well cell culture plate and incubated in an atmosphere of
5% CO2 at 37 °C for a period of 24 h. The growth medium was then removed, the cells
were loaded with an oxidation sensitive cell-permeable fluorescent probe, dichloro-
dihydro-fluorescein diacetate (DCFH-DA), and subsequently stabilised in the highly
reactive DCFH form. In this reactive state, ROS species in living cells were reacted with
DCFH, which is rapidly oxidised to the highly fluorescent 2′,7′-
dichlorodihydrofluorescein (DCF) The plates were then incubated for 1 h and the growth
medium was removed. The cells in each well were washed with PBS and the growth
medium containing 3.05 µM of 5, negative control (DMSO), or positive control (H2O2)
was added. The plates were incubated in an atmosphere of 5% CO2 at 37 °C for a period
of 24 h. After incubation, the cells were transferred to black 96-well plates with the
addition of 2× Cell Lysis Buffer. The cells were then read on a standard fluorescence
microplate reader (Infinite M200). The ROS or antioxidant content in unknown samples
was determined by comparison with the predetermined DCF standard curve.224

73
3.2.6.4 DNA Binding Studies
DNA binding experiments were carried out at 25°C. DNA concentration per
nucleotide was determined using the molar absorption coefficient (6600 M−1 cm−1) at 260
nm.53,225 Solutions of calf thymus (CT) DNA in Tris-HCl buffer containing (5 mM Tris,
pH 7.2, 50 mM NaCl)53 showed A260/A280 of 1.9, indicating that the DNA was sufficiently
free of proteins and contaminants.226 Absorption titration experiments were performed
maintaining the concentration of the organotin(IV) compounds solutions at 50 µM and
gradually increasing the concentration of CT-DNA. The organotin(IV) compounds were
dissolved in DMSO and diluted with Tris-HCI buffer at room temperature (25 °C). The
solutions were scanned over 230–600 nm. Absorbance values were recorded 10 min after
the addition of DNA solution. The binding constant, Kb was determined using equation
(1):
[DNA]
(εa − εf)=
[DNA]
(εb − εf)+
1
Kb(εb − εf) (1)
where [DNA] is the concentration of DNA in base pairs, εa corresponds to the apparent
molar extinction coefficient Aabs/[M], εf corresponds to the extinction coefficient for the
free organotin [M] and εb corresponds to the extinction coefficient for the fully bound
organotin compound.
3.3 Results and Discussion
3.3.1 Synthesis
Organotin(IV) compounds (4-9) were synthesised using the corresponding
diorganotin(IV) dichloride salts under reflux as shown in Scheme 3.1. The organotin(IV)
compounds are yellow, stable at room temperature and soluble in organic solvents
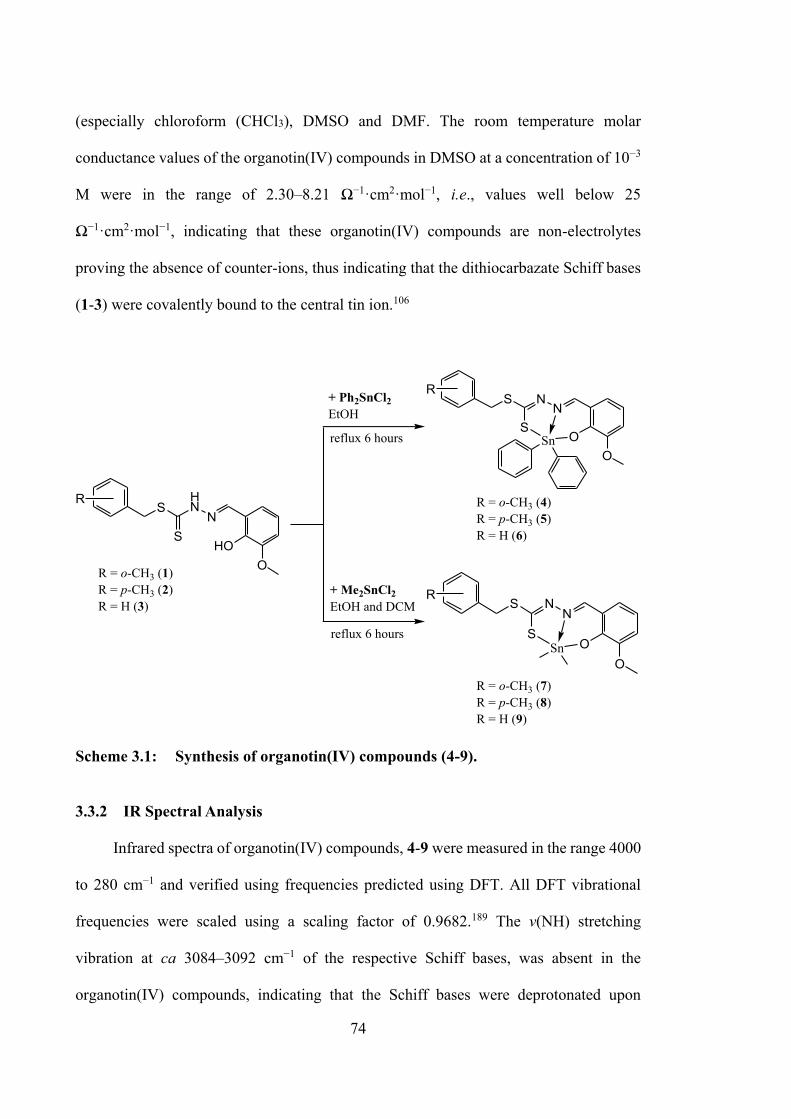
74
(especially chloroform (CHCl3), DMSO and DMF. The room temperature molar
conductance values of the organotin(IV) compounds in DMSO at a concentration of 10−3
M were in the range of 2.30–8.21 Ω−1·cm2·mol−1, i.e., values well below 25
Ω−1·cm2·mol−1, indicating that these organotin(IV) compounds are non-electrolytes
proving the absence of counter-ions, thus indicating that the dithiocarbazate Schiff bases
(1-3) were covalently bound to the central tin ion.106
SHN
NS
HOO
S NN
SO
OSn
S NN
SO
OSn
+ Ph2SnCl2EtOH
reflux 6 hours
+ Me2SnCl2EtOH and DCM
reflux 6 hours
R
R
R
R = o-CH3 (4)R = p-CH3 (5)R = H (6)
R = o-CH3 (1) R = p-CH3 (2)R = H (3)
R = o-CH3 (7)R = p-CH3 (8)R = H (9)
Scheme 3.1: Synthesis of organotin(IV) compounds (4-9).
3.3.2 IR Spectral Analysis
Infrared spectra of organotin(IV) compounds, 4-9 were measured in the range 4000
to 280 cm−1 and verified using frequencies predicted using DFT. All DFT vibrational
frequencies were scaled using a scaling factor of 0.9682.189 The v(NH) stretching
vibration at ca 3084–3092 cm−1 of the respective Schiff bases, was absent in the
organotin(IV) compounds, indicating that the Schiff bases were deprotonated upon

75
complexation. Sharp v(C=N) bands corresponding to the Schiff base azomethine group
were shifted upon complexation, indicating coordination via the azomethine nitrogen.227
The v(CSS) band splitting provides strong evidence of coordination of the thiolate sulphur
atom to the tin ion. The red shifted of the hydrazinic v(N–N) band in the organotin(IV)
compounds shows evidence of coordination via the azomethine nitrogen due to reduction
in the repulsion between lone pairs of electrons on the nitrogen atom.92,116,228 The
calculated frequencies correlate well with the experimental frequencies observed in the
FT-IR spectra, considering that DFT frequency calculations were performed in the gas-
phase. Only slight differences were observed in the experimental and calculated data for
v(C=N), v(N–N) and v(C=S). Complete IR data are provided in Appendix, Table A3.1.
3.3.3 NMR Spectroscopic Analysis
Complete 119Sn NMR data are provided in Experimental Section 3.2.3. The NMR
details for the organotin(IV) compounds are tabulated in Appendix, Tables A3.2 and
A3.3. NMR spectra of the compounds 4-9 were recorded in CDCl3. The data obtained
were in good agreement with the expected structures in solvents. For all compounds, the
1H NMR signal for the azomethine NH proton of Schiff bases (discussed in Chapter 2)
was absent due to the coordination with the metal ion, corroborating the absence of the
v(NH) FTIR band noted in §3.3.2. A singlet appeared in the downfield region of the 1H
NMR spectra of the Schiff bases, which corresponded to a sp2-C–OH proton. Proton
chemical shifts for both NH and OH disappeared in the spectra of the organotin(IV)
compounds suggesting double deprotonation of the Schiff bases and supporting
coordination of both the phenolic-oxygen and -nitrogen atoms of the Schiff base to the
tin ion.

76
The 13C NMR spectra of the dithiocarbazate Schiff bases showed a downfield
chemical shift at ~196 ppm, indicating the existence of the C=S group and thus the
predominance of the thione tautomer in solution. This signal was shifted upfield in the
organotin(IV) compounds (170–180 ppm), indicating a decrease in electron density at the
C=S carbon atom when the sulphur atom was complexed to the tin ion, consistent with
the structures proposed for other organotin(IV) compounds.229
The 119Sn NMR spectra of the organotin(IV) compounds were recorded at room
temperature in CDCl3. Theoretically, as the tin coordination number increases, chemical
shifts in 119Sn NMR move towards lower frequencies.230 119Sn NMR spectra are also very
sensitive to the type of donor atom (e.g., alkyl or aryl groups in the case of a C donor)
and the coordination environment (e.g., the bond angles around the tin center).231
However, shielding or deshielding of the tin nucleus does not have a significant effect on
the chemical shifts.232 Dimethyltin(IV) compounds showed less negative 119Sn chemical
shifts compared to diphenyltin(IV) compounds. The chemical shifts obtained were within
the range reported for other diphenyl- or dimethyl(IV) compounds with heterocyclic
ligands having penta-coordinated trigonal bipyramidal geometry.230,233
3.3.4 X-ray Crystallography
The crystal structures of 7, 8 and 9 each featured two independent molecules in the
crystallographic asymmetric unit cell, i.e., molecules a and b. Figure 3.2a–c shows the
molecular structures for the first independent molecule in each crystal and images for the
second independent molecules are shown in Figures 3.3a, 3.4a and 3.5a; selected
geometric parameters are collated in Table 3.2. The major conformational difference
between the molecules in each of 7, 8 and 9 is highlighted in the overlay diagram shown
in Figure 3.2d. Whereas in the tolyl species, the tolyl substituent is orientated away from

77
the coordinated S1 atom, i.e., can be considered anti to S1, in each molecule of 8, the
phenyl ring is syn to the S1 atom.
The tin atom in 7 is coordinated by the thiolate-S1, phenoxide-O1 and imine-N
atoms derived from the dinegative, [(3-methoxy-2-oxyphenyl)methyl] dithiocarbazate
ligand. The five-coordinate geometry is completed by a carbon atom from each of the two
methyl groups. The assignment of thiolate-S1 is readily made by comparing the length of
the C1‒S1 bond in 7, i.e., 1.726(9) and 1.717(8) Å, for the two independent molecules
comprising the asymmetric unit, with that in the uncoordinated Schiff base (which lacks
the 2-methyl group in the ester group) of 1.670(2) Å,234 i.e., the bond lengths in 7 are
significantly longer. The C1‒S2 bond lengths are experimentally equivalent at 1.744(8)
and 1.747(8) Å.234 However, the two C1=N1 imine bond lengths in 7 are 1.304(11) and
1.273(10) Å, which are significantly longer than the equivalent C1‒N1(H) bond of
1.338(2) Å in the respective free Schiff base. The coordination mode of the
dithiocarbazate ligand leads to the formation of five- and six-membered chelate rings,
with the chelate angle in the former being approximately 5° more acute than that in the
latter, see Table 3.2. The Sn, S1, N1, N2, C1 chelate ring in molecule is non-planar and
is twisted about the Sn‒S1 bond. On the other hand, the Sn, O1, N1, C10–C12 chelate
ring is best described as having an envelope conformation whereby the Sn atom lies
0.528(11) Å above the plane. However, the structure of the second independent molecule
in the asymmetric unit, molecule b, is slightly different, whereby the five-membered ring
is an envelope with the Sn atom lying 0.372(12) Å above the plane defined by the
remaining four atoms in this ring [RMSD = 0.008 Å]. The six-membered ring is also an
envelope with the Sn atom lying 0.528(11) Å out of the plane defined by the five
remaining atoms which present a RMSD = 0.022 Å. The dihedral angle between the best
planes through the chelate rings in the molecule a is 11.12(17)° while for molecule b, the

78
equivalent angle is 12.4(3)°. The benzyl ring is almost orthogonal to the five-membered
chelate ring for both independent molecules in the unit cell, forming dihedral angles of
88.3(3) and 86.8(3)°, respectively. Similarly, the dihedral angles between the outer rings
are 72.3(3) and 75.5(3)°, respectively, also indicating an approximate orthogonal
relationship. The similarity between the conformations found for the two independent
molecules in the asymmetric unit is highlighted in the overlay diagram shown in Figure
3.3. The five-coordinate geometry defined by the C2NOS donor set for molecule a, with
five-coordinate geometry descriptor τ = 0.48, is intermediate between ideal square-
pyramidal (τ = 0.0) and trigonal-bipyramidal geometries (τ = 1.0);235 the value of τ is 0.50
for molecule b. For each independent molecule, the widest angle corresponds to the S1‒
Sn‒O1 angle with the second widest angle being subtended by the tin-bound methyl
groups. The angles about the quaternary-C1 atom span nearly 20° with the widest angles
involving the doubly bonded N1 atom. Of these, the angle involving the S1 atom, being
the widest, is consistent with some double bond character in the C1‒S1 bond.
Concomitantly, the C1‒S1 bond lengths are systematically shorter than the C1‒S2 bonds
(see Table 3.2).
The isomeric compound, 8, having an ester 4-tolyl residue rather than 2-tolyl,
presents very similar characteristics to those exhibited in the structure of 7 (Figure 3.2b,
Figure 3.4 and Table 3.3). The conformations of the two chelate rings in the independent
molecules are approximately the same, and are based on an envelope conformation with
the tin atom being the flap in all cases. The envelope is more pronounced however, cf. 7,
in the five-membered rings with the tin atom lying 0.486(4) Å [0.490(4) Å for molecule
b] above the plane of the four remaining atoms which have a RMSD = 0.009 Å [0.012 Å]
but, is somewhat flattened in the six-membered rings with the tin atom lying 0.187(4) Å
[0.132(4) Å] out of the plane defined by the five remaining atoms with a RMSD = 0.013
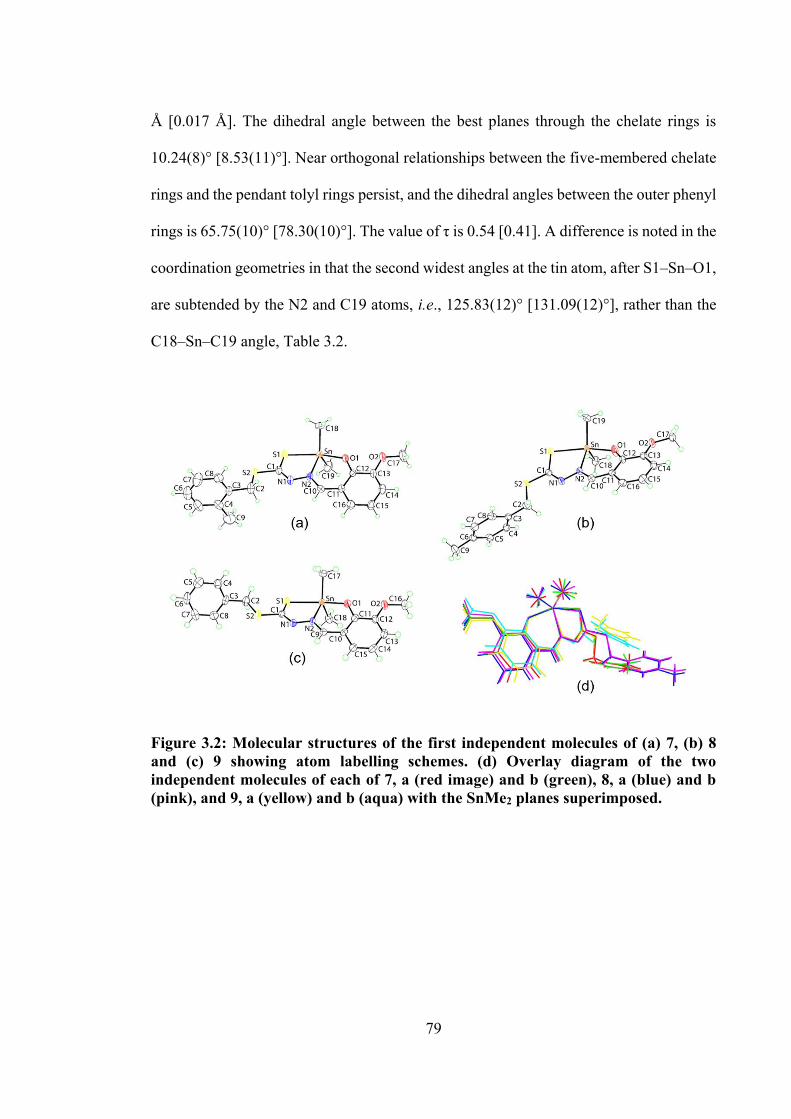
79
Å [0.017 Å]. The dihedral angle between the best planes through the chelate rings is
10.24(8)° [8.53(11)°]. Near orthogonal relationships between the five-membered chelate
rings and the pendant tolyl rings persist, and the dihedral angles between the outer phenyl
rings is 65.75(10)° [78.30(10)°]. The value of τ is 0.54 [0.41]. A difference is noted in the
coordination geometries in that the second widest angles at the tin atom, after S1‒Sn‒O1,
are subtended by the N2 and C19 atoms, i.e., 125.83(12)° [131.09(12)°], rather than the
C18‒Sn‒C19 angle, Table 3.2.
Figure 3.2: Molecular structures of the first independent molecules of (a) 7, (b) 8 and (c) 9 showing atom labelling schemes. (d) Overlay diagram of the two independent molecules of each of 7, a (red image) and b (green), 8, a (blue) and b (pink), and 9, a (yellow) and b (aqua) with the SnMe2 planes superimposed.
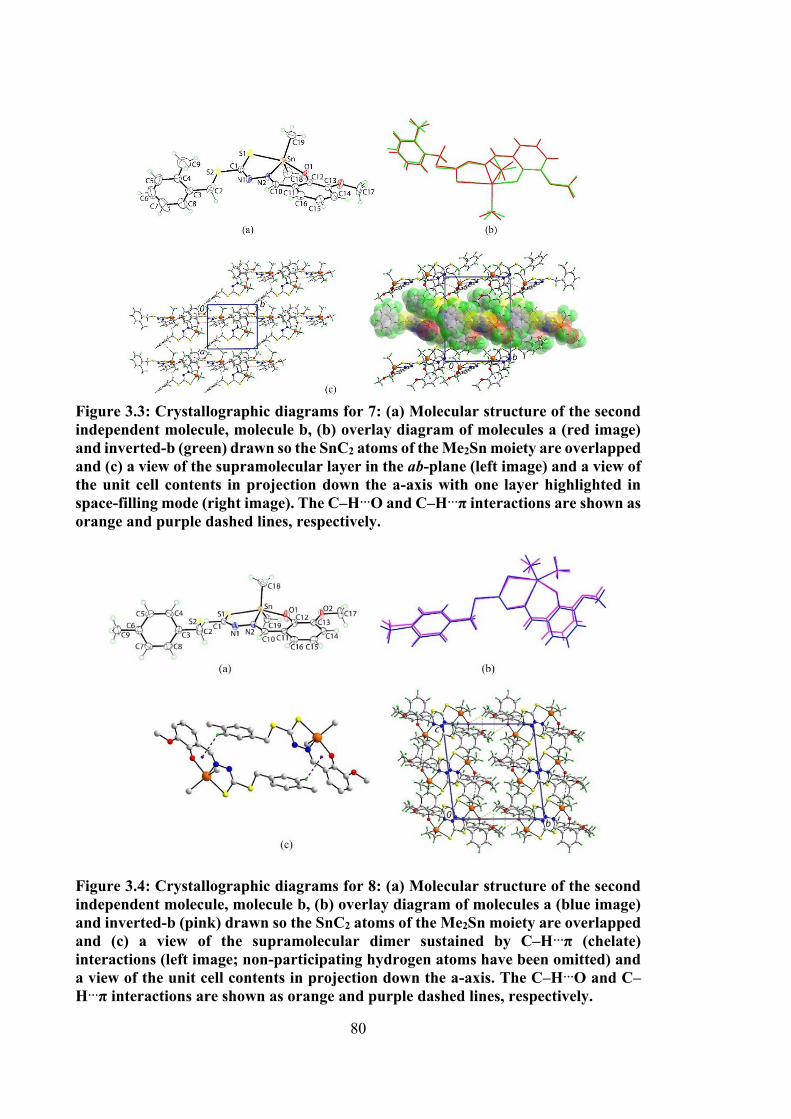
80
Figure 3.3: Crystallographic diagrams for 7: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (red image) and inverted-b (green) drawn so the SnC2 atoms of the Me2Sn moiety are overlapped and (c) a view of the supramolecular layer in the ab-plane (left image) and a view of the unit cell contents in projection down the a-axis with one layer highlighted in space-filling mode (right image). The C‒H…O and C‒H…π interactions are shown as orange and purple dashed lines, respectively.
Figure 3.4: Crystallographic diagrams for 8: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (blue image) and inverted-b (pink) drawn so the SnC2 atoms of the Me2Sn moiety are overlapped and (c) a view of the supramolecular dimer sustained by C‒H…π (chelate) interactions (left image; non-participating hydrogen atoms have been omitted) and a view of the unit cell contents in projection down the a-axis. The C‒H…O and C‒H…π interactions are shown as orange and purple dashed lines, respectively.

81
The unsubstituted analogue of the aforementioned isomeric structures, i.e., 9, is
similar to those just described (Figures 3.2c and 3.5, Table 3.2). A likely trend in the Sn‒
S1 bond lengths may be discerned, Table 3.2, in that these are longest in 9 compared to
the tolyl derivatives, 7 and 8, pointing to an electronic influence, i.e., activation of the
aromatic ring owing to the presence of electron-donating methyl groups; while there is
evidence for shorter Sn‒S1 bond lengths in 8 cf. 7, the standard uncertainty values in the
latter preclude a definitive statement on this although the trend for shorter Sn‒S1 bonds
in 8 is consistent with expectation. The conformational flexibility in the chelate ligands
formed by these tridentate ligands is reflected in the flattened envelope conformations for
the five-membered rings with the Sn atom lying 0.326(6) Å above the plane defined by
the four remaining atoms [RMSD = 0.021 Å; comparable parameters for molecule b are
0.287(6) and 0.024 Å, respectively]. By contrast, pronounced envelope conformations are
found for the six-membered rings [0.675(5) and 0.043 Å for molecule a; 0.706(5) and
0.048 Å for molecule b]. The dihedral angle between the best planes through the chelate
rings is 13.66(16)° [13.92(17)°]. The values of τ for the independent molecules compute
to 0.47 and 0.44, respectively. As for 8, after the S1‒Sn‒O1 angles, the widest angle
subtended at the tin atom in 9 for the second independent molecule involves the
coordinated imine-N2 and a tin-bound methyl-C17 atoms, i.e., 127.66(17)°. Of the three
new structures reported herein, the greatest agreement between the two independent
molecules comprising the asymmetric unit is found for 8, see Figure 3.5 and Table 3.2.
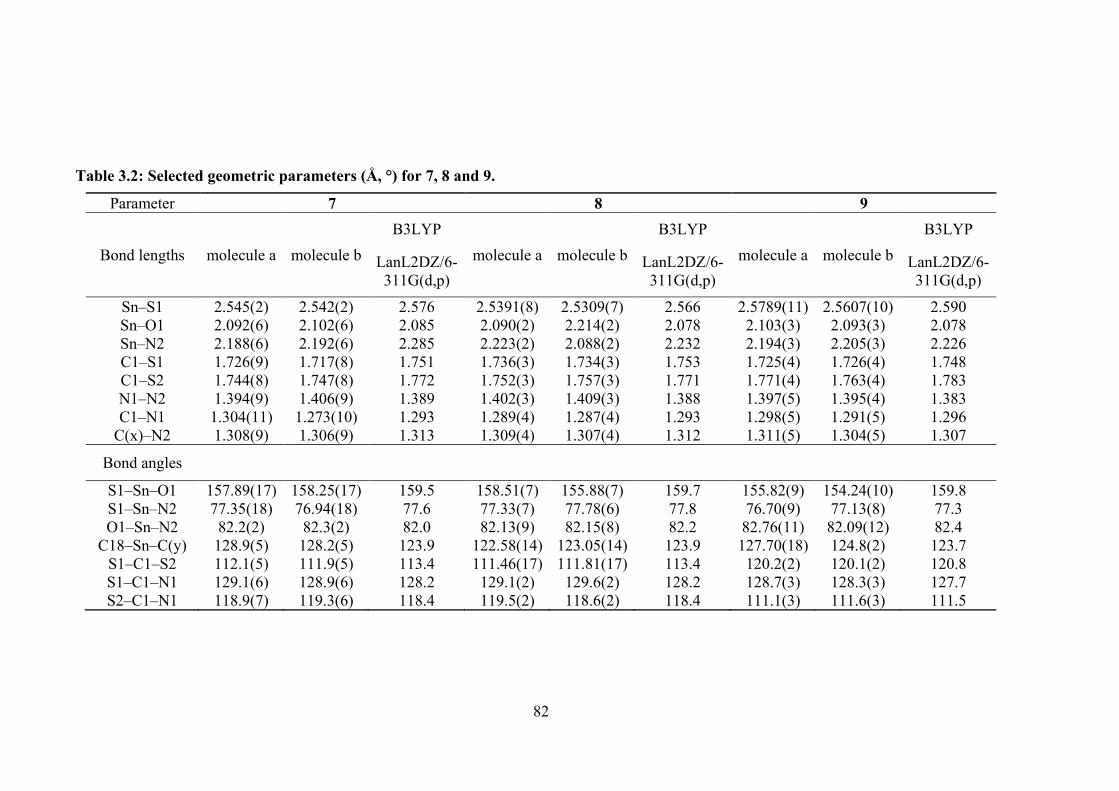
82
Table 3.2: Selected geometric parameters (Å, °) for 7, 8 and 9.
Parameter 7 8 9
Bond lengths molecule a molecule b B3LYP
molecule a molecule b B3LYP
molecule a molecule b B3LYP
LanL2DZ/6-311G(d,p)
LanL2DZ/6-311G(d,p)
LanL2DZ/6-311G(d,p)
Sn‒S1 2.545(2) 2.542(2) 2.576 2.5391(8) 2.5309(7) 2.566 2.5789(11) 2.5607(10) 2.590 Sn‒O1 2.092(6) 2.102(6) 2.085 2.090(2) 2.214(2) 2.078 2.103(3) 2.093(3) 2.078 Sn‒N2 2.188(6) 2.192(6) 2.285 2.223(2) 2.088(2) 2.232 2.194(3) 2.205(3) 2.226 C1‒S1 1.726(9) 1.717(8) 1.751 1.736(3) 1.734(3) 1.753 1.725(4) 1.726(4) 1.748 C1‒S2 1.744(8) 1.747(8) 1.772 1.752(3) 1.757(3) 1.771 1.771(4) 1.763(4) 1.783 N1‒N2 1.394(9) 1.406(9) 1.389 1.402(3) 1.409(3) 1.388 1.397(5) 1.395(4) 1.383 C1‒N1 1.304(11) 1.273(10) 1.293 1.289(4) 1.287(4) 1.293 1.298(5) 1.291(5) 1.296
C(x)‒N2 1.308(9) 1.306(9) 1.313 1.309(4) 1.307(4) 1.312 1.311(5) 1.304(5) 1.307
Bond angles
S1‒Sn‒O1 157.89(17) 158.25(17) 159.5 158.51(7) 155.88(7) 159.7 155.82(9) 154.24(10) 159.8 S1‒Sn‒N2 77.35(18) 76.94(18) 77.6 77.33(7) 77.78(6) 77.8 76.70(9) 77.13(8) 77.3 O1‒Sn‒N2 82.2(2) 82.3(2) 82.0 82.13(9) 82.15(8) 82.2 82.76(11) 82.09(12) 82.4
C18‒Sn‒C(y) 128.9(5) 128.2(5) 123.9 122.58(14) 123.05(14) 123.9 127.70(18) 124.8(2) 123.7 S1‒C1‒S2 112.1(5) 111.9(5) 113.4 111.46(17) 111.81(17) 113.4 120.2(2) 120.1(2) 120.8 S1‒C1‒N1 129.1(6) 128.9(6) 128.2 129.1(2) 129.6(2) 128.2 128.7(3) 128.3(3) 127.7 S2‒C1‒N1 118.9(7) 119.3(6) 118.4 119.5(2) 118.6(2) 118.4 111.1(3) 111.6(3) 111.5

83
In the absence of conventional hydrogen bonding interactions, the crystals of 7, 8
and 9 feature a range of weak non-covalent interactions. Packing diagrams are given in
Figures 3.3c, 3.4c and 3.5c and geometric details characterising the specified
intermolecular interactions are given in Appendix, Tables A3.4, A3.5 and A3.6. In the
molecular packing of 7, imine-C‒H…O(phenoxide), methylene-C‒
H…π(methoxybenzene), involving both methylene groups, and tin-bound-methyl-C‒H…π
(tolyl) interactions combine to stabilise supramolecular layers in the ab-plane; layers
stack without directional interactions between them. The crystal of 8 features additional
recognisable points of contact between molecules largely due to the participation of the
methoxy substituents. Thus, tolyl-C‒H…O (methoxy) and methoxy-methyl-C‒H…O
(methoxy, phenoxide) interactions are apparent as are tolyl-C5‒H…π(chelate) and tin-
bound-methyl-C‒H…π(tolyl) points of contact. These consolidate the three-dimensional
architecture, see Figure 3.3c. The presence of C‒H…π (chelate) interactions is of interest
and have only relatively recently been appreciated as being important in contributing to
the stability of coordination compounds.236–238 These interactions usually stabilise
(centrosymmetric) dimeric aggregates237 and that is the case in 8, see Figure 3.4c. In the
crystal of 9, both phenyl-C-H…π (methoxybenzene) and less common
π(chelate)…π(methoxybenzene) interactions are formed. The latter interactions have been
the subject of a recent comprehensive review.239 In the present case, Sn1-containing
molecules interact in this mode via association between the five-membered
(Sn/S1/N1/N2/C1) chelate ring and a methoxybenzene ring. However, these interactions
are best described as edge-to-edge interactions with the closest contact occurring between
a nitrogen atom of the chelate ring and the benzene-carbon, see Figure 3.5c. Similar edge-
to-edge contacts are found for the second independent molecule with the resulting dimeric
aggregates being connected into a supramolecular layer in the ab-plane by phenyl-C-H…π
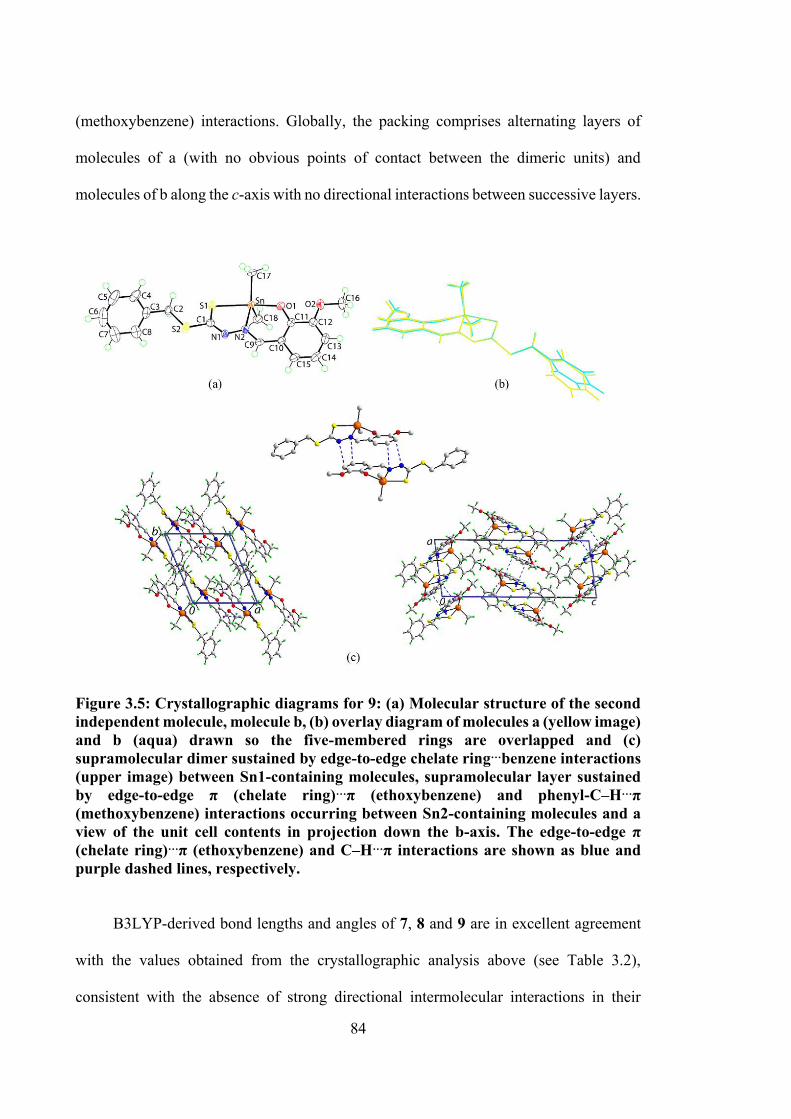
84
(methoxybenzene) interactions. Globally, the packing comprises alternating layers of
molecules of a (with no obvious points of contact between the dimeric units) and
molecules of b along the c-axis with no directional interactions between successive layers.
Figure 3.5: Crystallographic diagrams for 9: (a) Molecular structure of the second independent molecule, molecule b, (b) overlay diagram of molecules a (yellow image) and b (aqua) drawn so the five-membered rings are overlapped and (c) supramolecular dimer sustained by edge-to-edge chelate ring…benzene interactions (upper image) between Sn1-containing molecules, supramolecular layer sustained by edge-to-edge π (chelate ring)…π (ethoxybenzene) and phenyl-C‒H…π (methoxybenzene) interactions occurring between Sn2-containing molecules and a view of the unit cell contents in projection down the b-axis. The edge-to-edge π (chelate ring)…π (ethoxybenzene) and C‒H…π interactions are shown as blue and purple dashed lines, respectively.
B3LYP-derived bond lengths and angles of 7, 8 and 9 are in excellent agreement
with the values obtained from the crystallographic analysis above (see Table 3.2),
consistent with the absence of strong directional intermolecular interactions in their

85
crystal. For instance, the deviations in the selected bond lengths/angles of 7, 8 and 9
ranged from 0.002–0.144 Å/0–5.6°, whereby the maximum bond length deviation
corresponded to the Sn‒N2 bond for 8 and the maximum bond angle deviation
corresponded to the S1‒Sn‒O1 angle for 9.
3.3.5 UV-vis Absorption Spectral Analysis
The experimental UV-vis spectra of the organotin(IV) measured in DMSO
exhibited prominent absorption bands at 306-315, 364-373 and 433-447 nm. The
corresponding peaks were predicted at 304-320, 357-366 and 423-426 nm using B3LYP.
The highest occupied molecular orbital and lowest unoccupied molecular orbital
(HOMO-LUMO) electron density distribution of the organotin(IV) compounds (see
Figure 3.6) were comparable to those of the corresponding Schiff bases (1-3), i.e., the
HOMO was largely centered on the backbone of the Schiff bases and the LUMO mostly
covered the 2-hydroxy-3-methoxyphenyl ring as well as the backbone of the Schiff base
thus explaining the observed n→π* and π→π* transitions at 306–315 and 364–373 nm,
respectively. The additional, strong S→SnIV intra-ligand band at 433-447 nm indicated
coordination. The presence of the S→SnIV LMCT band was corroborated by IR evidence
which showed that the tin center was coordinated to the sulphur atom in the organotin(IV)
compounds.93 The HOMO and LUMO transitions were the same for all the organotin(IV)
compounds due to their similar structures, the difference being only the presence and
position of the methyl group attached to the phenyl ring. Complete experimental and
calculated UV-vis data are listed in Table A3.7.

86
Figure 3.6: HOMO-LUMO of (a) 4, (b) 5, (c) 6, (d) 7, (e) 8 and (f) 3.3.6 Biological Assays
3.3.6.1 Cytotoxicity
Compounds 4-9 were screened for in vitro cytotoxicity against a panel of 12 cancer
cell lines: EJ-28, RT-112, HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-
C, MIA and one normal breast cell line (MCF-10A) using the MTT metabolic assay. After
72 h of incubation, a dose-dependent proliferative effect towards the cancer cell lines was
measured at very low micromolar (µM) concentrations of the compounds. The growth
inhibition concentration of the organotin(IV) compounds required to inhibit 50% cell
proliferation relative to the cells that were treated with DMSO (control) are given in Table
3.3. The stability of these compounds was assessed in DMSO as well as in a mixture of

87
DMSO-H2O. The absorbance obtained from UV-vis spectroscopy analysis was monitored
for 72 h, and an unchanged pattern in the spectra indicated that the compounds were stable
in both the solvent systems tested.
It was also observed that the coordination of the Schiff bases with diphenyltin(IV)
induced potent selective growth inhibition in A2780, BE2-C and MIA cells, in which the
compounds exhibited potency higher than their respective Schiff bases, discussed in
Chapter 2. Cytotoxic selectivity was also observed for 5 and 6 against MCF-7 with GI50
values of 0.82 ± 0.14 and 0.90 ± 0.16 µM, respectively. This constituted a potency that
was almost three times higher than that of the corresponding Schiff bases, 2 and 3 which
had values of 2.5 ± 0.12 and 3.5 ± 0.00 µM, respectively. In addition, 5 exhibited a much
higher potency (GI50 value of 0.87 ± 0.22 µM) than 2 (GI50 value of 8.0 ± 3.5 µM) against
HT-29 cancer cells. A 3-fold greater activity against SJ-G2 was observed for 4 and 6 with
GI50 values of 0.84 ± 0.09 µM and 0.74 ± 0.15, respectively, than their respective Schiff
bases. It is noted that the diphenyltin(IV) compounds showed high selectivity against EJ-
28, RT-112, MCF-7, A2780, BE2-C, SJ-G2 and MIA cell lines in the range 1.4-5.0-fold
as compared to MCF10A.
In contrast, dimethyltin(IV) compounds showed almost similar cytotoxicity values
to their Schiff bases when assayed against the panel of cancer cell lines suggesting that
the presence of two phenyl groups coordinated directly to the tin ion improves the potency
of the Schiff bases. Electron delocalisation over the chelate rings, which increased the
lipophilicity of the molecules and favoured their permeation through the lipid layer of the
cancer cell membranes could also be a contributing factor to the enhancement of
cytotoxicity.240 A third contributing factor could be the ease of ability of the phenyl planes
to intercalate into the base pairs of DNA in the cancer cell lines.53 To better understand
the mode of biological action of the compounds that showed promising activity, apoptosis

88
assays using the minimally invasive bladder cancer RT-112 cells and DNA binding
studies were carried out to evaluate the compounds’ mechanisms of action.
3.3.6.2 Annexin V Assays of 4, 5 and 6-Treated RT-112 Cells
Cell death usually occurs by apoptosis or necrosis, the latter induces severe
inflammation. The apoptotic cells containing apoptotic bodies (small membrane-bound
vesicles), are engulfed by macrophages, and hence no inflammatory response is triggered.
On this account, as apoptotic cell induction is considered crucial in cytotoxic drug
development, investigations using the Annexin V assay were performed to study the
apoptosis effect on cancer cells of the organotin(IV) compounds. The three most highly
cytotoxic diphenyltin(IV) compounds at their specific GI50 concentrations were chosen
for this assay. The analysis was carried out using RT-112 minimum-invasive bladder
cancer cells with high levels of epidermal growth factor receptor expression. In the early
stages of apoptosis, changes occurred at the cell surface.241 The plasma membrane
alteration occurs due to the translocation of phosphatidyl-serine (PS) from the inner part
of the plasma membrane where it is normally located on the cytoplasmic surface, to the
outer layer. The PS exposed on the cell surface has high binding affinity to the Annexin
V. In the later stages of apoptosis, as well as necrosis, the cell membranes lose their
membrane integrity and allow propidium iodide (PI) to enter into the nucleus of the cells.
Annexin V/PI double staining was used to differentiate viable cells (Annexin V- and PI-)
in early apoptosis (Annexin V+ and PI−) or late apoptosis (Annexin V+ and PI+) and
necrosis (annexin V− and PI+) stages.242 Under fluorescence microscopy, the cells stained
by annexin V appear green, whereas the cells stained by PI appear red. After 24 h
exposure to the diphenyltin(IV) compounds, Figure 3.7 show that the cells undergoing

89
late apoptosis and necrosis features. Thus, this assay proved that the diphenyltin(IV)
compounds triggered the activation of apoptosis.243
3.3.6.3 Measurement of Reactive Oxygen Species (ROS) in RT-112 Cells Treated
with 4 and 5
Two compounds with the most promising bioactivity, 4 and 5 , were selected for
further cell apoptosis studies, in order to evaluate the primary damage to biological
macromolecules through redox reactions, particularly via formation of ROS.244 ROS are
highly reactive molecules/radicals (O2, O2•, H2O2, HO•) that are continuously generated
during aerobic metabolism, but when present in excessive amounts they can lead to
protein and DNA oxidation, protein cross-linking and cell death. Estimation of the levels
of ROS in cell culture is an important step towards better understanding the mechanisms
that lead to cell death or disease processes. Figure 3.8 shows that both 4 and 5 stimulated
ROS generation. ROS production was measured using the ROS-detecting fluorescent dye
DCFH-DA and DMSO was used as a control. Both compounds were able to induce ROS
production in RT-112 cell lines after 24 h. The significant increase in ROS generation
supports the findings that the compounds have high potential cytotoxicity and suggested
that the mechanism of cell death was via the simulation of mitochondrial-initiated
events.245
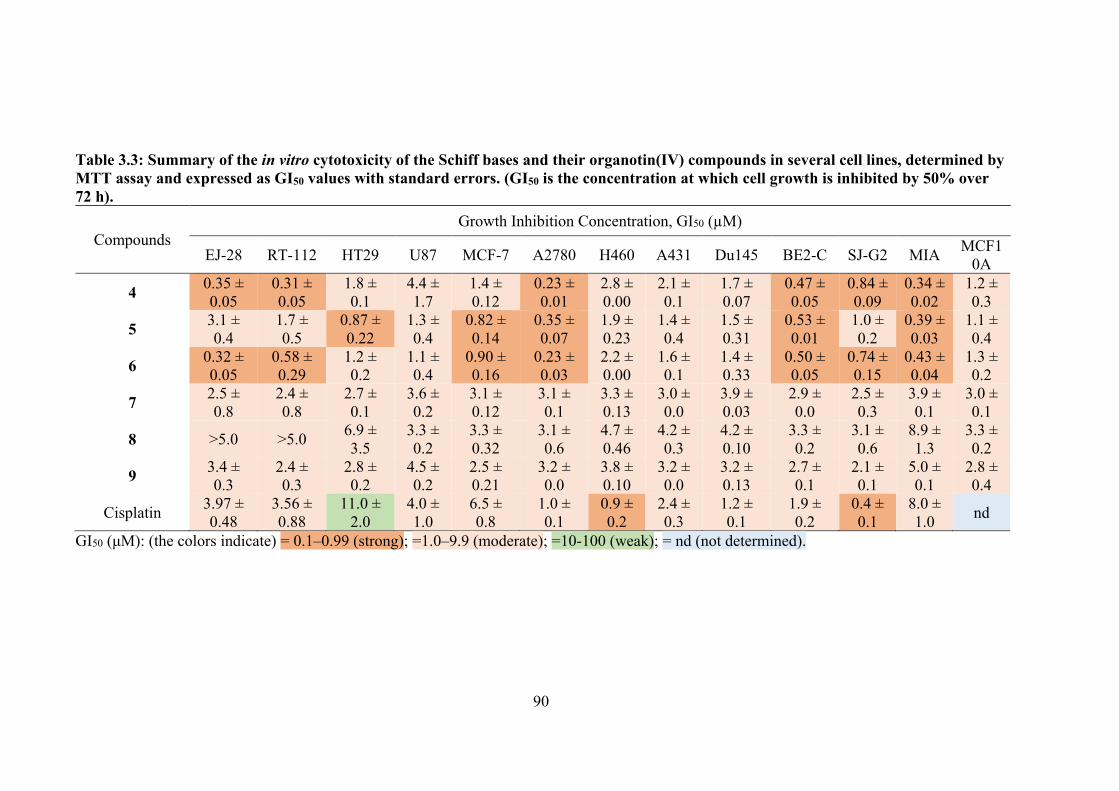
90
Table 3.3: Summary of the in vitro cytotoxicity of the Schiff bases and their organotin(IV) compounds in several cell lines, determined by MTT assay and expressed as GI50 values with standard errors. (GI50 is the concentration at which cell growth is inhibited by 50% over 72 h).
Compounds Growth Inhibition Concentration, GI50 (µM)
EJ-28 RT-112 HT29 U87 MCF-7 A2780 H460 A431 Du145 BE2-C SJ-G2 MIA MCF10A
4 0.35 ± 0.05
0.31 ± 0.05
1.8 ± 0.1
4.4 ± 1.7
1.4 ± 0.12
0.23 ± 0.01
2.8 ± 0.00
2.1 ± 0.1
1.7 ± 0.07
0.47 ± 0.05
0.84 ± 0.09
0.34 ± 0.02
1.2 ± 0.3
5 3.1 ± 0.4
1.7 ± 0.5
0.87 ± 0.22
1.3 ± 0.4
0.82 ± 0.14
0.35 ± 0.07
1.9 ± 0.23
1.4 ± 0.4
1.5 ± 0.31
0.53 ± 0.01
1.0 ± 0.2
0.39 ± 0.03
1.1 ± 0.4
6 0.32 ± 0.05
0.58 ± 0.29
1.2 ± 0.2
1.1 ± 0.4
0.90 ± 0.16
0.23 ± 0.03
2.2 ± 0.00
1.6 ± 0.1
1.4 ± 0.33
0.50 ± 0.05
0.74 ± 0.15
0.43 ± 0.04
1.3 ± 0.2
7 2.5 ± 0.8
2.4 ± 0.8
2.7 ± 0.1
3.6 ± 0.2
3.1 ± 0.12
3.1 ± 0.1
3.3 ± 0.13
3.0 ± 0.0
3.9 ± 0.03
2.9 ± 0.0
2.5 ± 0.3
3.9 ± 0.1
3.0 ± 0.1
8 >5.0 >5.0 6.9 ± 3.5
3.3 ± 0.2
3.3 ± 0.32
3.1 ± 0.6
4.7 ± 0.46
4.2 ± 0.3
4.2 ± 0.10
3.3 ± 0.2
3.1 ± 0.6
8.9 ± 1.3
3.3 ± 0.2
9 3.4 ± 0.3
2.4 ± 0.3
2.8 ± 0.2
4.5 ± 0.2
2.5 ± 0.21
3.2 ± 0.0
3.8 ± 0.10
3.2 ± 0.0
3.2 ± 0.13
2.7 ± 0.1
2.1 ± 0.1
5.0 ± 0.1
2.8 ± 0.4
Cisplatin 3.97 ± 0.48
3.56 ± 0.88
11.0 ± 2.0
4.0 ± 1.0
6.5 ± 0.8
1.0 ± 0.1
0.9 ± 0.2
2.4 ± 0.3
1.2 ± 0.1
1.9 ± 0.2
0.4 ± 0.1
8.0 ± 1.0 nd
GI50 (μM): (the colors indicate) = 0.1–0.99 (strong); =1.0–9.9 (moderate); =10-100 (weak); = nd (not determined).
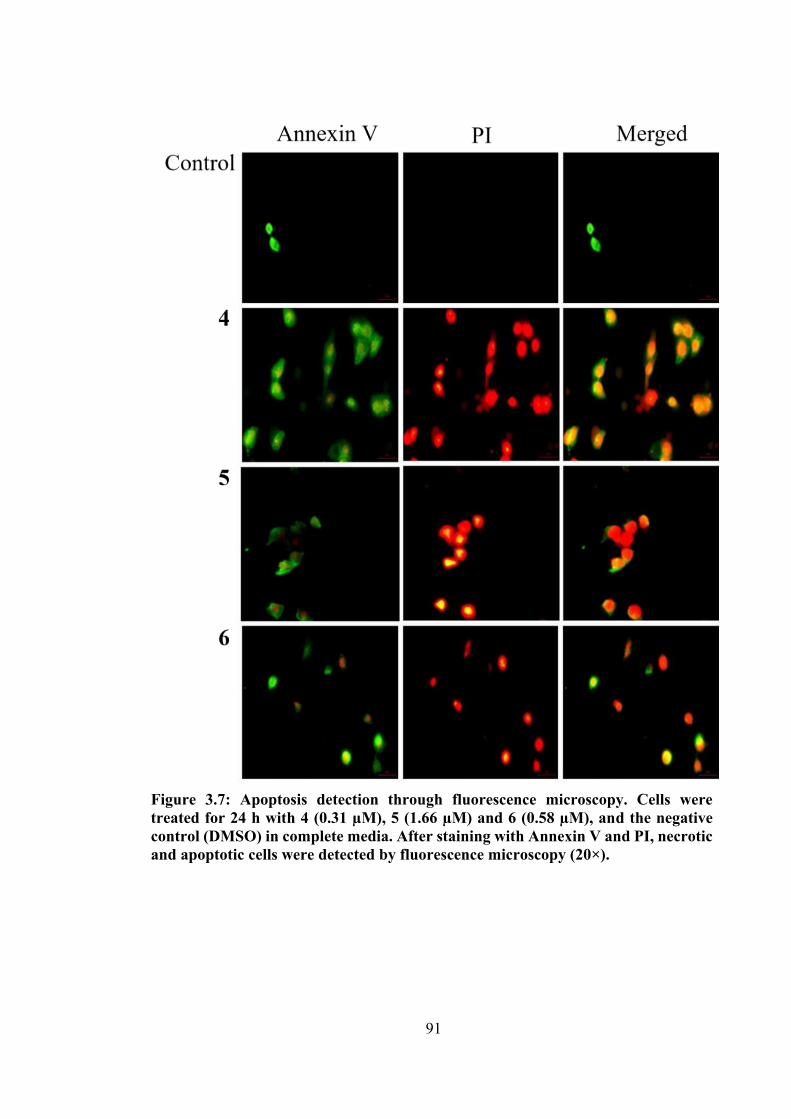
91
Figure 3.7: Apoptosis detection through fluorescence microscopy. Cells were treated for 24 h with 4 (0.31 µM), 5 (1.66 µM) and 6 (0.58 µM), and the negative control (DMSO) in complete media. After staining with Annexin V and PI, necrotic and apoptotic cells were detected by fluorescence microscopy (20×).
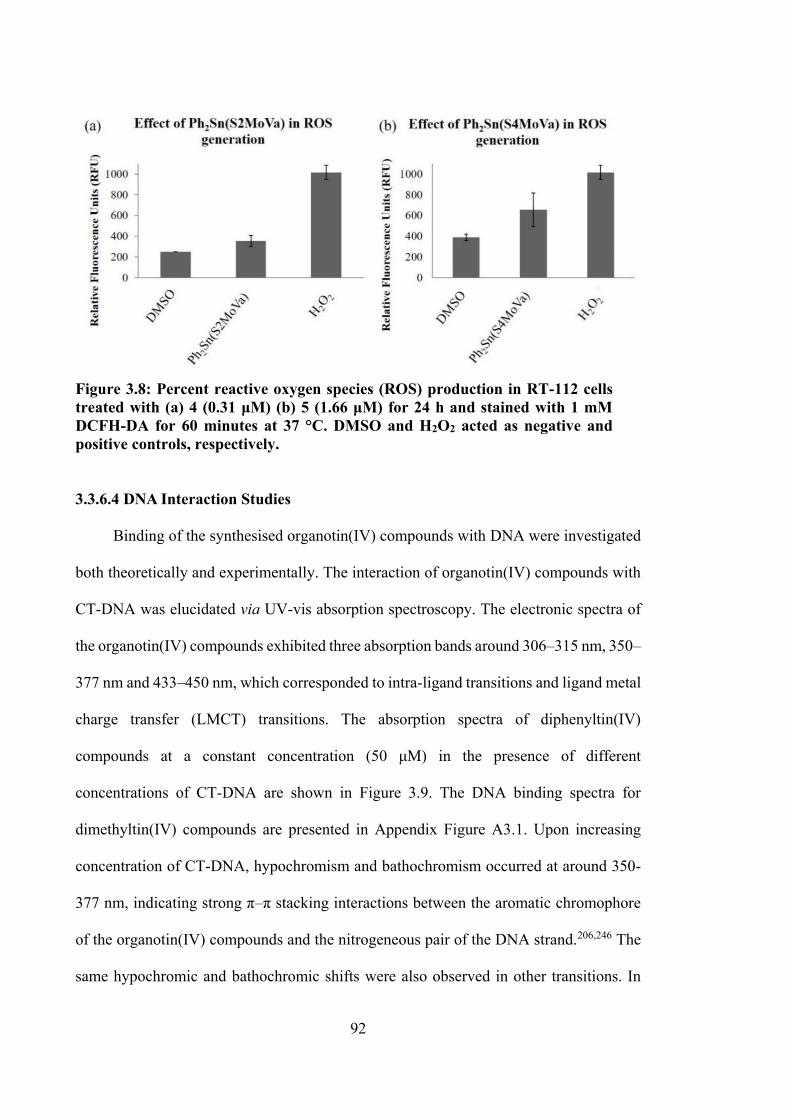
92
Figure 3.8: Percent reactive oxygen species (ROS) production in RT-112 cells treated with (a) 4 (0.31 μM) (b) 5 (1.66 μM) for 24 h and stained with 1 mM DCFH-DA for 60 minutes at 37 °C. DMSO and H2O2 acted as negative and positive controls, respectively.
3.3.6.4 DNA Interaction Studies
Binding of the synthesised organotin(IV) compounds with DNA were investigated
both theoretically and experimentally. The interaction of organotin(IV) compounds with
CT-DNA was elucidated via UV-vis absorption spectroscopy. The electronic spectra of
the organotin(IV) compounds exhibited three absorption bands around 306–315 nm, 350–
377 nm and 433–450 nm, which corresponded to intra-ligand transitions and ligand metal
charge transfer (LMCT) transitions. The absorption spectra of diphenyltin(IV)
compounds at a constant concentration (50 μM) in the presence of different
concentrations of CT-DNA are shown in Figure 3.9. The DNA binding spectra for
dimethyltin(IV) compounds are presented in Appendix Figure A3.1. Upon increasing
concentration of CT-DNA, hypochromism and bathochromism occurred at around 350-
377 nm, indicating strong π–π stacking interactions between the aromatic chromophore
of the organotin(IV) compounds and the nitrogeneous pair of the DNA strand.206,246 The
same hypochromic and bathochromic shifts were also observed in other transitions. In

93
order to compare the binding strength of the organotin(IV) compounds to CT-DNA, the
intrinsic binding constants (Kb) were quantitatively determined and are presented in Table
3.4. The Kb values show that these compounds have strong binding affinities with DNA,
and potentially prevent enzyme binding to the nitrogenous bases of DNA.
The Gibbs free energies of interaction between the organotin(IV) compounds and
DNA ranged from −31.0 to −37.6 kJ mol−1, meaning that the interaction of the
organotin(IV) compounds with DNA was spontaneous.247 To investigate and understand
the interactions between DNA and these compounds, molecular docking simulations were
carried out.
3.3.7 Molecular Docking Studies
Molecular docking analyses of the organotin(IV) compounds with the DNA duplex
of sequence d(CGCGAATTCGCG)2 dodecamer (PDB ID: 1BNA) were performed and
results are detailed in Table 3.5. These simulations demonstrated that the organotin(IV)
compounds considered here were commensurate with the dimensions of the grooves of
the DNA duplex sequence. Figure 3.10(a) revealed that these small organotin(IV)
compounds fitted well into the A-T rich region of the minor grooves of DNA. Generally,
the electronegativity of A-T sequences provided better van der Waals interactions due to
their narrower sequences compared to G-C regions. In groove regions, van der Waals and
hydrophobic interactions play important roles in the stabilisation of compounds.248 Their
interactions therefore, involved hydrogen bonding, hydrophobic and electrostatic
interactions yielding binding energies between −9.10 to −10.07 kcal mol−1, which is better
than cisplatin (−8.65 kcal mol−1).155 The interactions occurred either through the
phosphate-oxygen atom of the DNA or via DNA bases. 4 exhibited two hydrogen bonding
interactions through methoxy-oxygen atoms and methyl groups with phosphate-oxygen
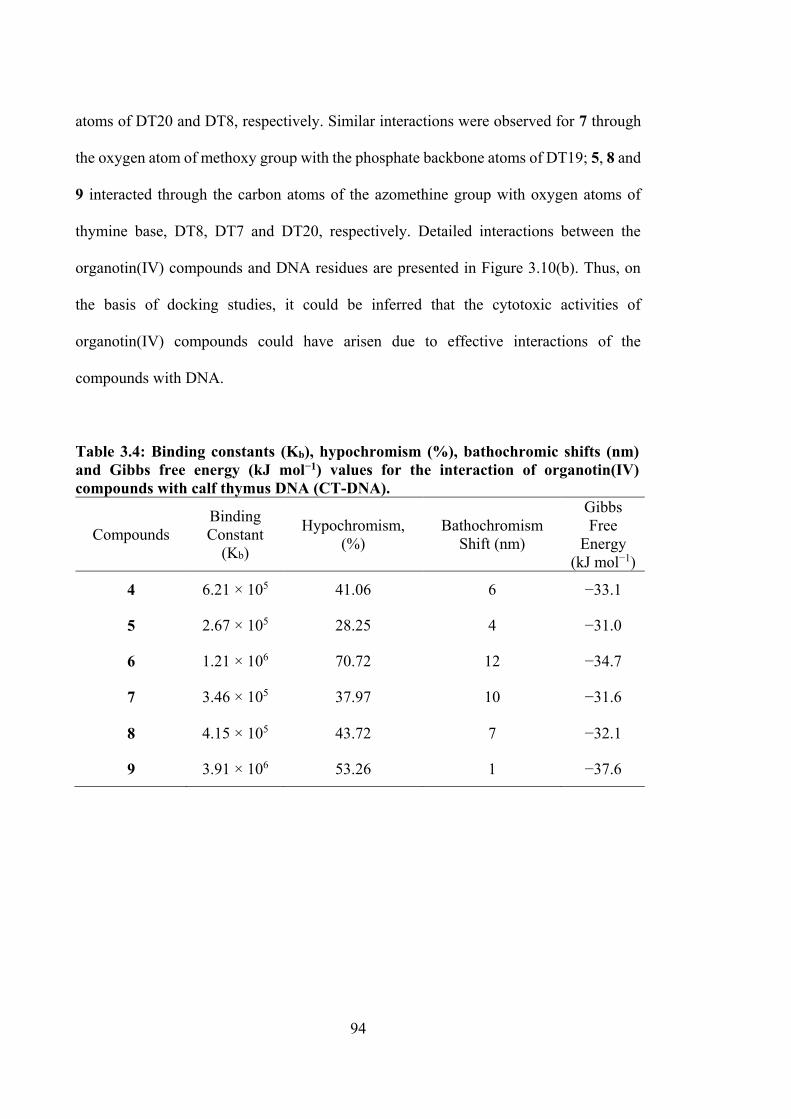
94
atoms of DT20 and DT8, respectively. Similar interactions were observed for 7 through
the oxygen atom of methoxy group with the phosphate backbone atoms of DT19; 5, 8 and
9 interacted through the carbon atoms of the azomethine group with oxygen atoms of
thymine base, DT8, DT7 and DT20, respectively. Detailed interactions between the
organotin(IV) compounds and DNA residues are presented in Figure 3.10(b). Thus, on
the basis of docking studies, it could be inferred that the cytotoxic activities of
organotin(IV) compounds could have arisen due to effective interactions of the
compounds with DNA.
Table 3.4: Binding constants (Kb), hypochromism (%), bathochromic shifts (nm) and Gibbs free energy (kJ mol−1) values for the interaction of organotin(IV) compounds with calf thymus DNA (CT-DNA).
Compounds Binding Constant
(Kb)
Hypochromism, (%)
Bathochromism Shift (nm)
Gibbs Free
Energy (kJ mol−1)
4 6.21 × 105 41.06 6 −33.1
5 2.67 × 105 28.25 4 −31.0
6 1.21 × 106 70.72 12 −34.7
7 3.46 × 105 37.97 10 −31.6
8 4.15 × 105 43.72 7 −32.1
9 3.91 × 106 53.26 1 −37.6
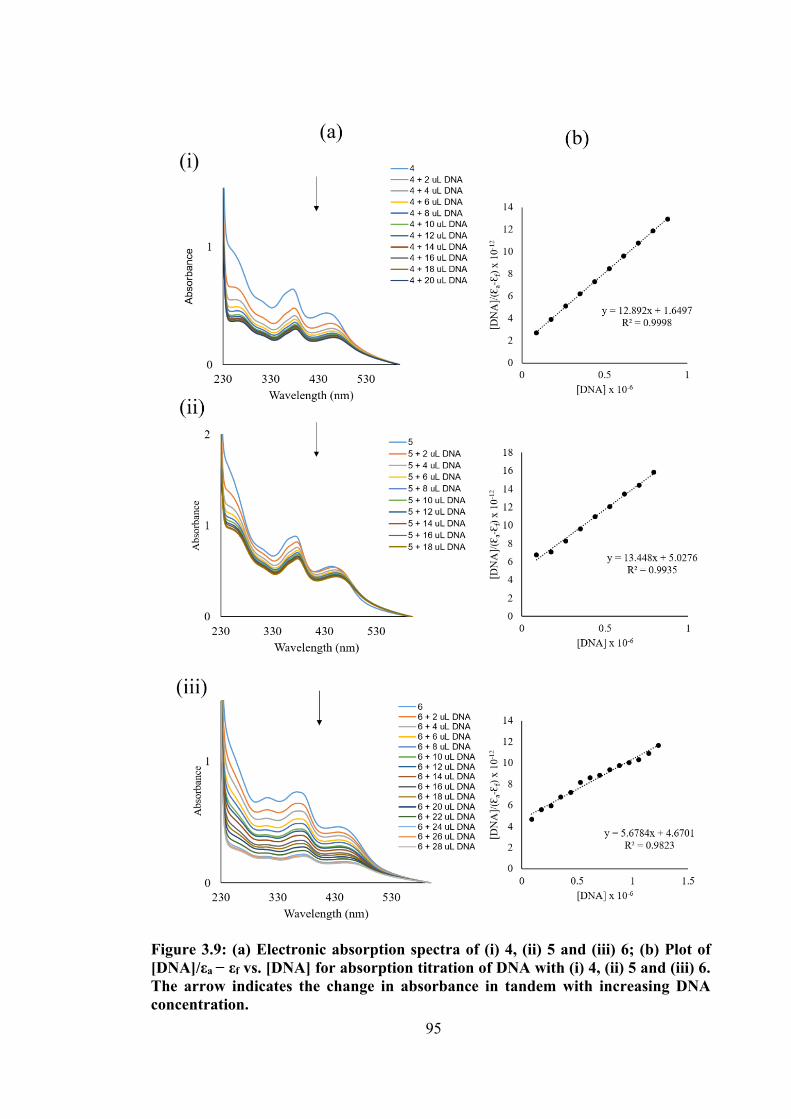
95
Figure 3.9: (a) Electronic absorption spectra of (i) 4, (ii) 5 and (iii) 6; (b) Plot of [DNA]/εa − εf vs. [DNA] for absorption titration of DNA with (i) 4, (ii) 5 and (iii) 6. The arrow indicates the change in absorbance in tandem with increasing DNA concentration.
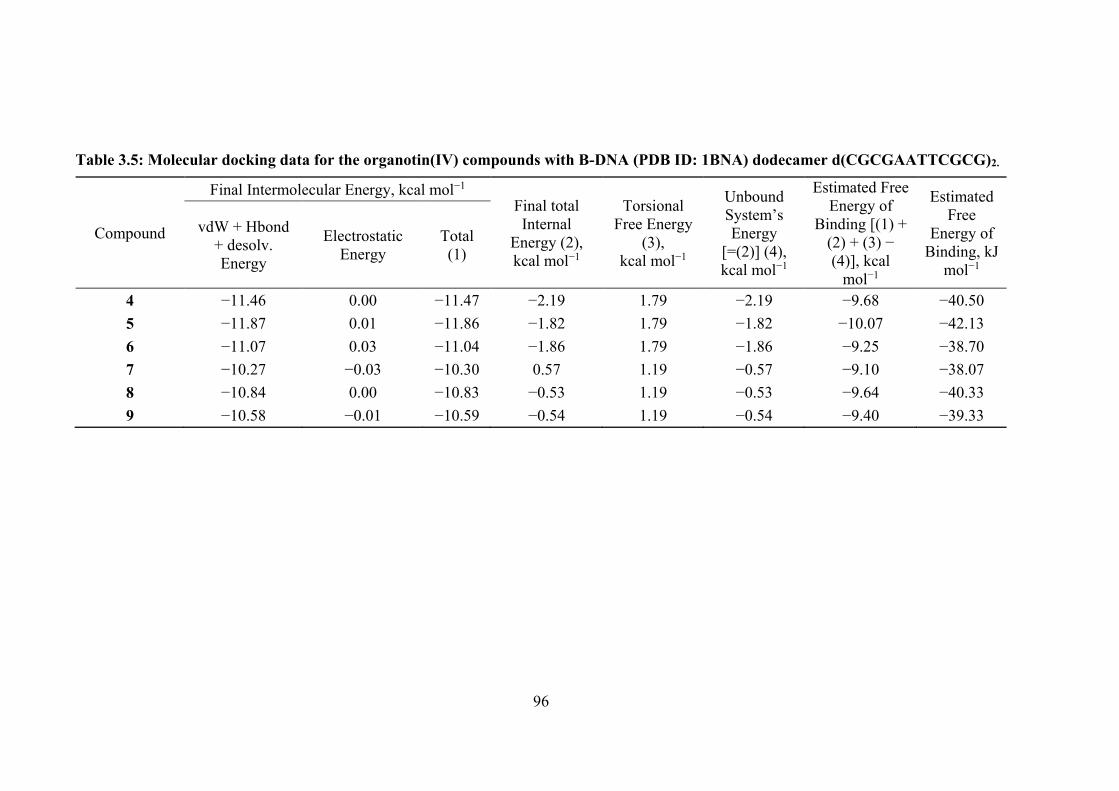
96
Table 3.5: Molecular docking data for the organotin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
Compound
Final Intermolecular Energy, kcal mol−1 Final total Internal
Energy (2), kcal mol−1
Torsional Free Energy
(3), kcal mol−1
Unbound System’s Energy
[=(2)] (4), kcal mol−1
Estimated Free Energy of
Binding [(1) + (2) + (3) − (4)], kcal
mol−1
Estimated Free
Energy of Binding, kJ
mol−1
vdW + Hbond + desolv. Energy
Electrostatic Energy
Total (1)
4 −11.46 0.00 −11.47 −2.19 1.79 −2.19 −9.68 −40.50 5 −11.87 0.01 −11.86 −1.82 1.79 −1.82 −10.07 −42.13 6 −11.07 0.03 −11.04 −1.86 1.79 −1.86 −9.25 −38.70 7 −10.27 −0.03 −10.30 0.57 1.19 −0.57 −9.10 −38.07 8 −10.84 0.00 −10.83 −0.53 1.19 −0.53 −9.64 −40.33 9 −10.58 −0.01 −10.59 −0.54 1.19 −0.54 −9.40 −39.33
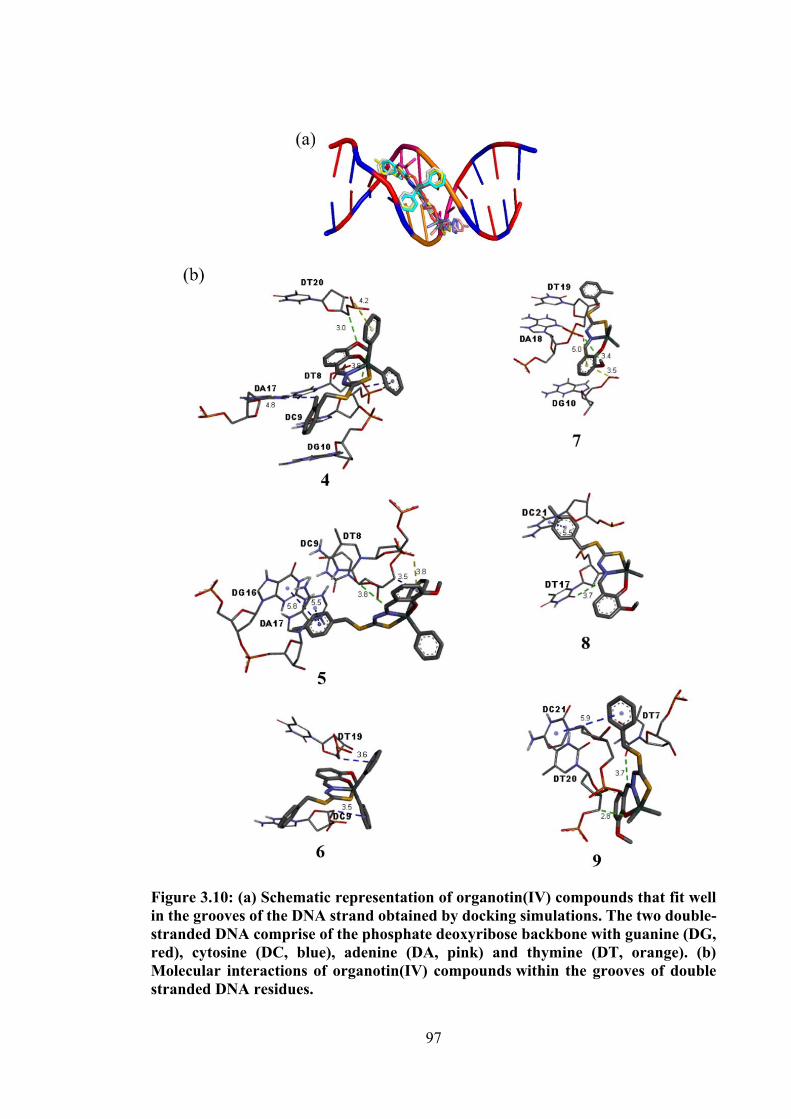
97
Figure 3.10: (a) Schematic representation of organotin(IV) compounds that fit well in the grooves of the DNA strand obtained by docking simulations. The two double-stranded DNA comprise of the phosphate deoxyribose backbone with guanine (DG, red), cytosine (DC, blue), adenine (DA, pink) and thymine (DT, orange). (b) Molecular interactions of organotin(IV) compounds within the grooves of double stranded DNA residues.

98
3.4 Conclusions
This chapter presented six organotin(IV) compounds derived from o-vanillin
dithiocarbazate Schiff bases (discussed in Chapter 2) characterised by elemental analysis
and various spectroscopic techniques (UV-visible, FTIR, 1H, 13C and 119Sn NMR). The
molecular structures of 7, 8 and 9 were elucidated by single crystal X-ray structure
analysis which indicated that the compounds had five-coordinate C2NOS geometries,
derived from thiolate-S1, phenoxide-O1 and imine-N atoms of a dinegative, tridentate
dithiocarbazate ligand along with two methyl-carbon atoms of the methyl groups, which
were intermediate between ideal trigonal-bipyramidal and square-pyramidal geometries.
The in vitro cytotoxicity against a panel of cancer cell lines viz., EJ-28, RT-112, HT29,
U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C and MIA cancer cell lines
revealed the remarkable cytotoxicity of the compounds 4, 5 and 6 against most of these
cell lines, which were more potent than cisplatin. In order to evaluate the mechanism of
death of the organotin(IV) compounds, Annexin-V and ROS assays were performed using
the RT-112 cell line, where the diphenyltin(IV) compounds were able to induce apoptosis
and ROS generation in the cells. The DNA binding of the organotin(IV) compounds were
investigated using UV-vis absorption and the diphenyltin(IV) compounds bound
reasonably well into the grooves of DNA with binding constants in the range 105–106
M−1. The mode of interaction of the organotin(IV) compounds with DNA was further
validated by molecular docking studies which were in accordance with the experimental
results, suggesting the strong binding of diphenyltin(IV) at the AT-rich region of the
minor groove of DNA. The observed trend in the cytotoxicity and binding interactions of
organotin(IV) compounds were attributed to the nature of the group bound to the tin metal
center where the compounds 4, 5 and 6 exhibited potent efficacy against cells tested.

99
The biologically active species formed from the cleavage of the tin-ligand bonds is
the major effect of the overall activity. The compounds with stronger Sn-O, Sn-N and Sn-
S bonds hence have difficulty forming could not easily form their active species, such as
[R2Sn(IV)]2+ (R = Ph and Me) species through the dissociation of these bonds. Such active
species reportedly cross the cell membrane by deeply affecting the structural arrangement
of membranes of biological systems.249,250 The moderate stability of the bonds results in
slow hydrolysis and gradual dissociation of the Sn-O, Sn-N and Sn-S bonds. It is
conceivable therefore that the gradual dissociation of these bonds could facilitate delivery
of these active species to their specific active site.251 For instance, the Sn-O (2.092-2.214
Å), Sn-N (2.088-2.223 Å) and Sn-S (2.542-2.5789 Å) bonds in the compounds discussed
in this chapter were shorter than that reported by Saxena and Huber,251 but still possessed
very good activity that is higher than the respective Schiff bases, particularly for the
diphenyltin(IV) compounds. It is concluded therefore that the bioactivity of tin(IV)
compounds cannot solely be attributed to the stability of the ligand-metal bonds.
While excellent cytotoxic activity of three oVa dithiocarbazate Schiff bases and
their diphenyl- and dimethyltin(IV) compounds were discussed in Chapter 2 and 3, it is
hypothesised that this activity could be improved via structural modification such as
introduction of hydroxyl group in compounds. The addition of hydroxyl group in
compounds were proven to enhance binding and activity due to their great ability of
forming hydrogen bond interactions with target macrobiomolecules such as DNA and
enzymes.252–254
This hypothesis will be tested in Chapter 4, which reports structural evaluation and
cytotoxic studies of catechol dithiocarbazate Schiff bases (10, 11, 12) with diphenyl- (13,
14, 15) and dimethyltin(IV) (16, 17, 18). The small modification of the structure was

100
explored by substituting a hydroxyl group at the position of the methoxy group of oVa
dithiocarbazate Schiff bases and their organotin(IV) compounds.

101
CHAPTER 4
SELECTIVE CYTOTOXICITY OF ORGANOTIN(IV) COMPOUNDS CONTAINING CATECHOL DITHIOCARBAZATE SCHIFF BASES
4.1 Introduction
A number of dihydroxybenzyl (catechol) containing drugs are currently available
on the market, such as L-dopa, which is used for treating Parkinson's disease, and
Protocatechualdehyde, which is used to treat human breast cancer255 and inhibit casein
kinase II activity in leukemic cells.256 While the discovery of catechol dithiocarbazate
Schiff base derived from S-methyldithiocarbazate was reported by Fayed et al.,257 the
cytotoxic activity of dithiocarbazate Schiff bases of catechol and their tin(IV) compounds
remains unexplored.
Building on results reported in Chapters 2 and 3, this chapter focuses on the
synthesis, structural characterisation and cytotoxic activity of catechol dithiocarbazate
Schiff bases (10-12) and their organotin(IV) compounds (13-18), see Figure 4.1.
Interactions of the synthesised organotin(IV) compounds with DNA as a function of the
coordination complex structure will also be discussed in the context of their structure
activity relationships.
This chapter contains material from submitted article for publication in Research
on Chemical Intermediates on 18 October 2019.
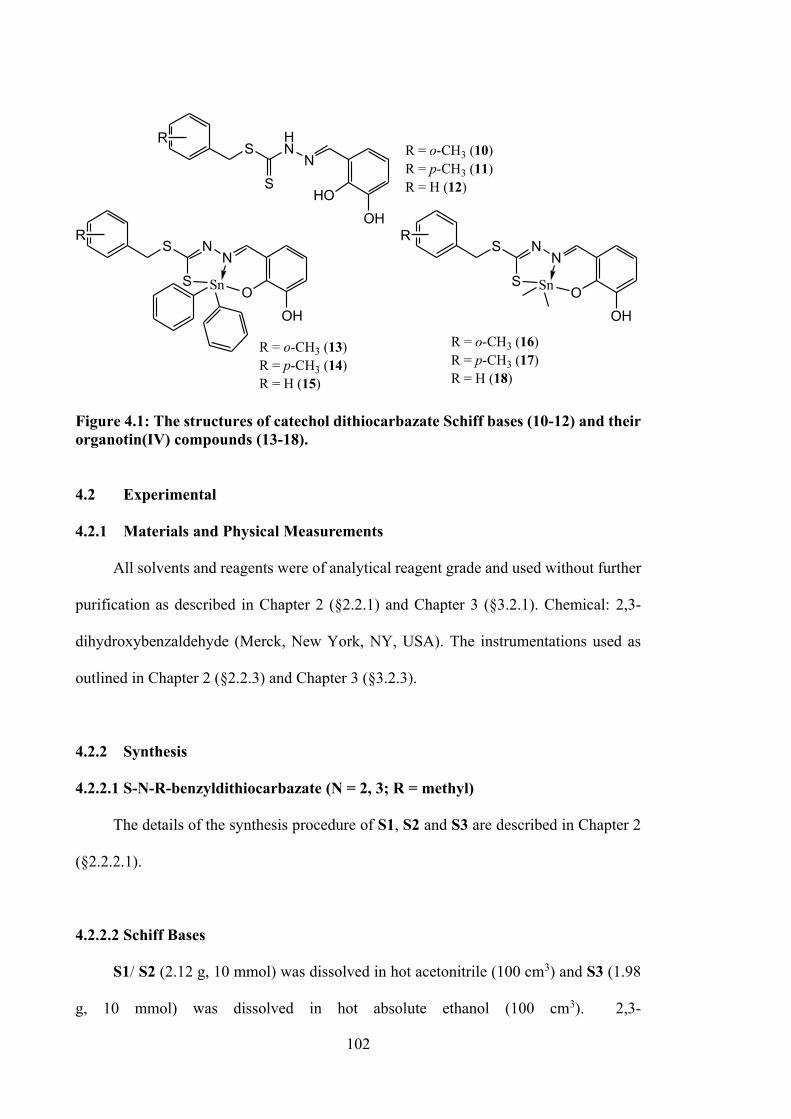
102
R = o-CH3 (13)R = p-CH3 (14)R = H (15)
R = o-CH3 (10) R = p-CH3 (11)R = H (12)
R = o-CH3 (16)R = p-CH3 (17)R = H (18)
SHN
SN
HOOH
R
S N
SN
OOH
R
Sn
S N
SN
OOH
R
Sn
Figure 4.1: The structures of catechol dithiocarbazate Schiff bases (10-12) and their organotin(IV) compounds (13-18).
4.2 Experimental
4.2.1 Materials and Physical Measurements
All solvents and reagents were of analytical reagent grade and used without further
purification as described in Chapter 2 (§2.2.1) and Chapter 3 (§3.2.1). Chemical: 2,3-
dihydroxybenzaldehyde (Merck, New York, NY, USA). The instrumentations used as
outlined in Chapter 2 (§2.2.3) and Chapter 3 (§3.2.3).
4.2.2 Synthesis
4.2.2.1 S-N-R-benzyldithiocarbazate (N = 2, 3; R = methyl)
The details of the synthesis procedure of S1, S2 and S3 are described in Chapter 2
(§2.2.2.1).
4.2.2.2 Schiff Bases
S1/ S2 (2.12 g, 10 mmol) was dissolved in hot acetonitrile (100 cm3) and S3 (1.98
g, 10 mmol) was dissolved in hot absolute ethanol (100 cm3). 2,3-

103
Dihydroxybenzaldehyde (1.38 g, 10 mmol) in ethanol (50 cm3) was then added to the
solution of S-substituted dithiocarbazate. The mixture was stirred and heated (78 °C) for
about 1 hour and then allowed to cool to room temperature. The pale-yellow Schiff bases
that formed were filtered and washed with absolute ethanol. The products were
recrystallised using absolute ethanol and dried over silica gel.101,110
4.2.2.2.1 S-2-methylbenzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate (10)
Pale yellow solid. Yield: 36%. M.p.: 150-152 °C. Analysis calculated for
C16H16N2O2S2: C, 57.81; H, 4.85; N, 8.43 %. Found: C, 56.98; H, 4.65; N, 8.21. FT-IR
(ATR, cm-1): 3496 (O-H), 3097 (N-H), 1608 (C=N), 1119 (N-N), 1013 (C=S). 1H NMR
(DMSO-d6) δ (ppm): 4.45 (s, 2H, CH2), 6.69-7.39 (multiplet, 7H, Ar-H), 8.51 (s, 1H,
CH), 9.51, 9.55 (s, 2H, OH), 13.41 (s,1H, NH). 13C NMR (DMSO-d6) δ (ppm): 19.3 (Ar-
CH3), 36.9 (CH2), 118.1, 118.5, 119.7, 120.0, 126.6, 128.2, 130.7, 130.8, 134.3, 137.3,
146.2, 146.2 (12 x aromatic-C), 146.5 (C=N), 195.6 (S-C=S). m/z calculated for
C16H16N2O2S2 332.44 g/mol, found 332.05.
4.2.2.2.2 S-4-methylbenzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate (11)
Pale yellow solid. Yield: 68%. M.p.: 203-205 °C. Analysis calculated for
C18H19N3O2S2: C, 57.81; H, 4.85; N, 8.43 %. Found: C, 57.55; H, 4.87; N, 8.60. FT-IR
(ATR, cm-1): 3501 (O-H), 3106 (N-H), 1607 (C=N), 1114 (N-N), 1012 (C=S). 1H NMR
(DMSO-d6) δ (ppm): 4.44 (s, 2H, CH2), 6.69-7.30 (multiplet, 7H, Ar-H), 8.51 (s, 1H,
CH), 9.53, 9.54 (s, 2H, OH), 13.39 (s,1H, NH). 13C NMR (DMSO-d6) δ (ppm): 21.2 (Ar-
CH3), 38.0 (CH2), 118.1, 118.4, 119.8, 119.9, 129.6, 129.7, 133.8, 137.0, 146.1, 146.2

104
(10 x aromatic-C), 146.5 (C=N), 195.8 (S-C=S). m/z calculated for C16H16N2O2S2 332.44
g/mol, found 332.05.
4.2.2.2.3 S-benzyl-β-N-(2,3-dihydroxybenzylmethylene)dithiocarbazate (12)
Pale yellow crystals. Yield: 77%. M.p.: 201-203 °C. Analysis calculated for
C15H14N2O2S2: C, 56.58; H, 4.43; N, 8.80 %. Found: C, 56.56; H, 4.67; N, 8.67. FT-IR
(ATR, cm-1): 3488 (O-H), 3097 (N-H), 1608 (C=N), 1119 (N-N), 1018 (C=S). 1H NMR
(DMSO-d6) δ (ppm): 4.47 (s, 2H, CH2), 6.67-7.39 (multiplet, 8H, Ar-H), 8.50 (s, 1H,
CH), 9.53, 9.56 (s, 2H, OH), 13.39 (s, 1H, NH). 13C NMR (DMSO-d6) δ (ppm): 38.06
(CH2), 118.1, 118.4, 119.7, 119.9, 127.7, 129.0, 129.7, 137.1, 146.1 (9 x aromatic-C);
146.5 (C=N); 195.6 (S-C=S). m/z calculated for C15H14N2O2S2 318.41 g/mol, found
318.05.
4.2.2.3 Diphenyltin(IV) Compounds
Each Schiff base (0.33 g (10 and 11); 0.32 g (12), 1 mmol) was dissolved separately
in absolute ethanol (50 cm3) and mixed with an ethanolic solution (10 cm3) of Ph2SnCl2
(0.34 g, 1 mmol). The yellow solution was refluxed for ca. 6 hours. The mixture was left
stirring overnight and the reduction in the volume (15 cm3) of the reaction mixture
resulted in the deposition of a yellow precipitate that was filtered off and recrystallised
from methanol.

105
4.2.2.3.1 Diphenyltin(IV)[S-2-methylbenzyl-β-N-(2,3-dihydroxybenzyl methylene)
dithiocarbazate] (13)
Yellow solid. Yield: 42%. M.p.: 112-113 °C. FT-IR (ATR, cm-1): 1610 (C=N),
1021 (N-N), 959 (C=S). 1H NMR (CDCl3) δ (ppm): 2.40 (s, 3H, CH3), 4.44 (s, 2H, CH2),
6.40-7.80 (multiplet, 17H, Ar-H), 8.81 (s, 1H, CH). 13C NMR (CDCl3) δ (ppm): 19.3
(CH3), 34.4 (CH2), 114.7, 118.3, 118.5, 125.1, 127.5, 128.6, 129.0, 129.2, 130.4, 135.6,
136.4, 141.8, 147.8, 154.9 (aromatic-C), 166.1 (C=N), 172.3 (S-C-S). 119Sn NMR
(CDCl3) δ (ppm.): -233.7.
4.2.2.3.2. Diphenyltin(IV)[S-4-methylbenzyl-β-N-(2,3-dihydroxybenzyl
methylene)dithiocarbazate] (14)
Light-orange crystals. Yield: 71%. M.p.: 137-138 °C. FT-IR (ATR, cm-1): 1611
(C=N), 1006 (N-N), 956 (C=S). 1H NMR (CDCl3) δ (ppm): 2.34 (s, 3H, CH3), 4.41 (s,
2H, CH2), 6.43-7.81 (multiplet, 17H, Ar-H), 8.80 (s, 1H, CH). 13C NMR (CDCl3) δ (ppm):
21.1 (CH3), 35.9 (CH2), 114.7, 118.2, 118.4, 125.1, 129.0, 129.1, 129.3, 130.4, 133.2,
135.6, 137.2, 141.8, 147.8, 154.9 (aromatic-C), 166.1 (C=N), 172.1 (S-C-S). 119Sn NMR
(CDCl3) δ (ppm.): -233.4.
4.2.2.3.3 Diphenyltin(IV)[S-benzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate] (15)
Light-yellow crystals. Yield: 41%. M.p.: 150-151 °C. FT-IR (ATR, cm-1): 1610
(C=N), 1016 (N-N), 960 (C=S). 1H NMR (CDCl3) δ (ppm): 4.44 (s, 2H, CH2), 6.43-7.80
(multiplet, 18H, Ar-H), 8.76 (s, 1H, CH). 13C NMR (CDCl3) δ (ppm): 36.0 (CH2), 114.7,
118.3, 118.5, 125.1, 127.5, 128.6, 129.0, 129.2, 130.4, 135.6, 136.4, 141.8, 147.8, 154.9
(aromatic-C), 166.2 (C=N), 172.0 (S-C-S). 119Sn NMR (CDCl3) δ (ppm.): -233.4.

106
4.2.2.4 Dimethyltin(IV) Compounds
Each Schiff base (0.33 g (10 and 11); 0.32 g (12), 1 mmol) was dissolved separately
in absolute ethanol (50 cm3) and dichloromethane (20 cm3) and was then mixed with an
ethanolic solution (10 mL) of Me2SnCl2 (0.22 g, 1 mmol). The yellow solution was
refluxed for ca. 6 hours. The mixture was left overnight and kept at room temperature for
a few days, yielding brown crystals that were suitable for single crystal X-ray diffraction
analysis.
4.2.2.4.1 Dimethyltin(IV)[S-2-methylbenzyl-β-N-(2,3-dihydroxybenzyl
methylene)dithiocarbazate] (16)
Yellow solid. Yield: 31%. M.p.: 184-185 °C. FT-IR (ATR, cm-1): 1597 (C=N),
1081 (N-N), 984 (C=S). 1H NMR (CDCl3) δ (ppm): 0.93 (Sn-CH3), 2.42 (s, 3H, CH3),
4.43 (s, 2H, CH2), 6.27-7.34 (multiplet, 7H, Ar-H), 8.79 (s, 1H, CH). 13C NMR (CDCl3)
δ (ppm): 6.9 (Sn-CH3), 19.5 (CH3), 34.6 (CH2), 114.5, 117.9, 119.0, 124.8, 126.1, 126.4,
128.0, 129.9, 130.4, 130.7, 137.2, 148.0 (aromatic-C), 166.1 (C=N), 173.3 (S-C-S). 119Sn
NMR (CDCl3) δ (ppm.): -99.9.
4.2.2.4.2 Dimethyltin(IV)[S-4-methylbenzyl-β-N-(2,3-dihydroxybenzyl
methylene)dithiocarbazate] (17)
Light-yellow crystals. Yield: 66%. M.p.: 180-182 °C. FT-IR (ATR, cm-1): 1613
(C=N), 1006 (N-N), 956 (C=S). 1H NMR (CDCl3) δ (ppm): 0.92 (Sn-CH3), 2.34 (s, 3H,
CH3), 4.37 (s, 2H, CH2), 6.24-7.27 (multiplet, 7H, Ar-H), 8.77 (s, 1H, CH). 13C NMR
(CDCl3) δ (ppm): 6.8 (Sn-CH3), 21.2 (CH3), 36.0 (CH2), 114.4, 117.7, 124.6, 129.1,
129.3, 133.4, 137.1, 147.8, 154.5 (aromatic-C), 166.1 (C=N), 173.3 (S-C-S). 119Sn NMR
(CDCl3) δ (ppm.): -99.8.

107
4.2.2.4.1 Dimethyltin(IV)[S-benzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate] (18)
Light-yellow crystals. Yield: 44%. M.p.: 110-112 °C. FT-IR (ATR, cm-1): 1612
(C=N), 1016 (N-N), 960 (C=S). 1H NMR (CDCl3) δ (ppm): 0.92 (s, 6H, Sn-CH3), 4.41
(s, 2H, CH2), 6.24-7.38 (multiplet, 8H, Ar-H), 8.76 (s, 1H, CH). 13C NMR (CDCl3) δ
(ppm): 6.8 (Sn-CH3), 36.2 (CH2), 114.3, 117.7, 124.6, 127.4, 128.6, 129.2, 136.6, 147.8,
154.2, 154.7 (aromatic-C), 166.2 (C=N), 173.1 (S-C-S). 119Sn NMR (CDCl3) δ (ppm.): -
99.5.
4.2.3 X-ray Crystallographic Analysis
X-ray intensity data for 12, 14, 15, 17 and 18 were measured at T = 150 K on an
Oxford Diffraction Gemini Eos CCD diffractometer equipped with Mo Kα radiation
source (λ = 0.71073 Å). Data reduction, including absorption correction, was
accomplished with CrysAlisPro.258 The structures were solved by direct-methods181 and
refined (anisotropic displacement parameters and C-bound H atoms in the riding model
approximation) on F2.182 When applicable, the O- and N-bound H atoms were refined
with O‒H and N‒H constrained to 0.84 ± 0.01 and 0.88 ± 0.01 Å, respectively, and with
Uiso(H) = 1.5Ueq(O) or 1.2Ueq(N). A weighting scheme of the form w = 1/[σ2(Fo2) + (aP)2
+ bP] where P = (Fo2 + 2Fc2)/3) was introduced in each case. The absolute structures of
17 and 18 were determined based on differences in Friedel pairs included in the respective
data sets. In 14, the maximum and minimum residual electron density peaks of 2.36 and
0.92 eÅ-3 were located 0.96 and 0.62 Å from the Sn1 atom. In 15, the residual peaks of
1.39 and -0.49 eÅ-3 were located 0.87 and 0.79 Å from the C9 and Sn1a atoms,
respectively. The molecular structure diagrams were generated with ORTEP for
Windows183 with 50% displacement ellipsoids, and the packing diagrams were drawn

108
with DIAMOND.184 Additional data analysis was made with PLATON.216 Crystal data
and refinement details are given in Table 4.1.
4.2.4 DFT Calculations and Molecular Docking Studies
DFT were performed using parameters detailed in Chapter 2 (§2.2.6) and Chapter
3 (§3.2.5.1), respectively. The coordinates of 14, 15, 17 and 18 were taken from their
crystal structures as a Crystallographic Information File (CIF) and converted to the
Protein Data Bank (PDB) format, whereas the coordinates for 13 and 16 were obtained
after minimisation of energy using DFT method. The parameters for molecular docking
studies described in Chapter 3 (§3.2.5.2).
4.2.5 MTT Assays
The protocol used for MTT assay followed that outlined in Chapter 2 (§2.2.6).
4.3 Results and Discussion
4.3.1 Synthesis
The general synthesis of the S-substituted Schiff bases (10-12) is outlined in
Scheme 4.1. The desired Schiff bases formed in good yields, except for 10, due to the
presence of an unexpected by-product (3-[(1Z)-2-[bis([(2-methylphenyl)methyl]
sulfanyl)methylidene]hydrazin-1-ylidenemethyl]benzene-1,2-diol) which was
manually isolated from the filtrate of 10.259 The Schiff bases were then synthesised via
condensation, by reacting the three S-substituted dithiocarbazates with 2,3-
dihydroxybenzaldehyde in an alcoholic medium.

109
RS N
H
SN
RS N
H
SNH2
HOOH
R = o-CH3 (10) R = p-CH3 (11) R = H (12)
O
HOOH
Scheme 4.1: Synthetic pathway for the formation of Schiff bases 10, 11 and 12.
Schiff bases containing the thioamide functional group [-NH-C(=S)] are capable of
undergoing thione-thiol tautomerism. They can exist either as thione (Figure 4.2a) or thiol
(Fig. 4.2b) tautomeric forms, or a mixture of both in solution. The three Schiff bases were
then each reacted with diphenyltin(IV) dichloride and dimethyltin(IV) dichloride
separately to produce six new organotin(IV) compounds (Scheme 4.2). The products were
isolated as yellow precipitates. Compounds 12, 14, 15, 17 and 18 produced crystalline
materials which were suitable for single crystal X-ray diffraction analysis. The Schiff
bases and their organotin(IV) compounds were non-hygroscopic and soluble in common
organic solvents, especially DMSO, CHCl3 and DMF. The organotin(IV) compounds
behaved as non-electrolytes in DMSO where the molar conductivity values were in the
range of 2.49-6.07 Ω−1cm2mol−1.106
RS N
H
SN
HOOH
RS N
SHN
HOOH
Where: R=o-CH3 (10) R=p-CH3 (11) R=H (12)
Figure 4.2: (a) Thione and (b) thiol tautomerism of Schiff bases 10-12.
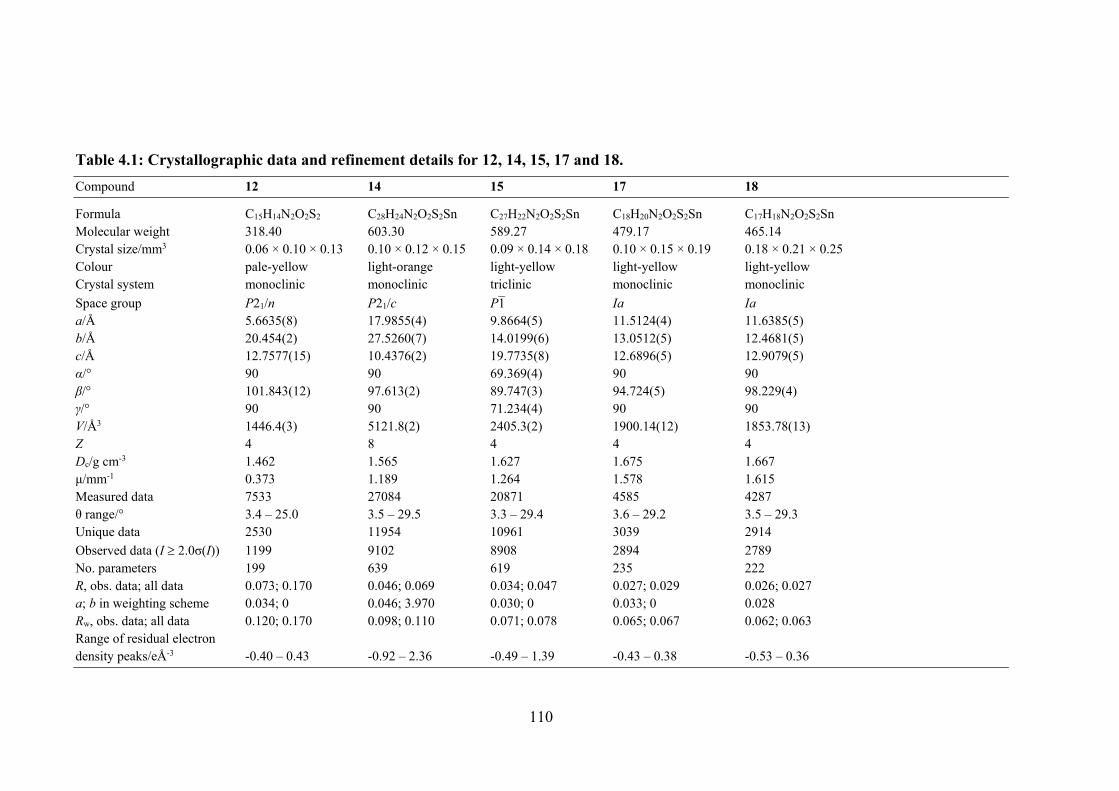
110
Table 4.1: Crystallographic data and refinement details for 12, 14, 15, 17 and 18. Compound 12 14 15 17 18
Formula C15H14N2O2S2 C28H24N2O2S2Sn C27H22N2O2S2Sn C18H20N2O2S2Sn C17H18N2O2S2Sn Molecular weight 318.40 603.30 589.27 479.17 465.14 Crystal size/mm3 0.06 × 0.10 × 0.13 0.10 × 0.12 × 0.15 0.09 × 0.14 × 0.18 0.10 × 0.15 × 0.19 0.18 × 0.21 × 0.25 Colour pale-yellow light-orange light-yellow light-yellow light-yellow Crystal system monoclinic monoclinic triclinic monoclinic monoclinic Space group P21/n P21/c P1 Ia Ia a/Å 5.6635(8) 17.9855(4) 9.8664(5) 11.5124(4) 11.6385(5) b/Å 20.454(2) 27.5260(7) 14.0199(6) 13.0512(5) 12.4681(5) c/Å 12.7577(15) 10.4376(2) 19.7735(8) 12.6896(5) 12.9079(5) α/° 90 90 69.369(4) 90 90 β/° 101.843(12) 97.613(2) 89.747(3) 94.724(5) 98.229(4) γ/° 90 90 71.234(4) 90 90 V/Å3 1446.4(3) 5121.8(2) 2405.3(2) 1900.14(12) 1853.78(13) Z 4 8 4 4 4 Dc/g cm-3 1.462 1.565 1.627 1.675 1.667 μ/mm-1 0.373 1.189 1.264 1.578 1.615 Measured data 7533 27084 20871 4585 4287 θ range/° 3.4 – 25.0 3.5 – 29.5 3.3 – 29.4 3.6 – 29.2 3.5 – 29.3 Unique data 2530 11954 10961 3039 2914 Observed data (I 2.0σ(I)) 1199 9102 8908 2894 2789 No. parameters 199 639 619 235 222 R, obs. data; all data 0.073; 0.170 0.046; 0.069 0.034; 0.047 0.027; 0.029 0.026; 0.027 a; b in weighting scheme 0.034; 0 0.046; 3.970 0.030; 0 0.033; 0 0.028 Rw, obs. data; all data 0.120; 0.170 0.098; 0.110 0.071; 0.078 0.065; 0.067 0.062; 0.063 Range of residual electron density peaks/eÅ-3 -0.40 – 0.43 -0.92 – 2.36 -0.49 – 1.39 -0.43 – 0.38 -0.53 – 0.36
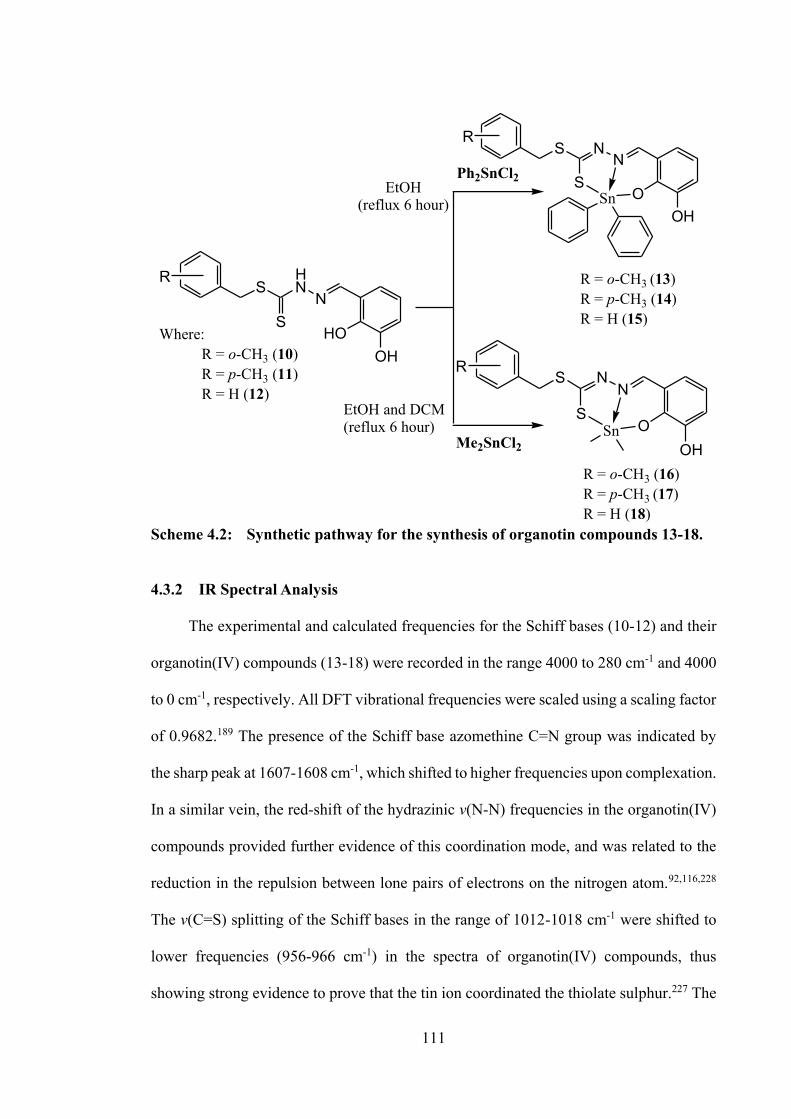
111
SHN
NS
HOOH
S NN
SO
OHSn
S NN
SO
OHSn
EtOH(reflux 6 hour)
EtOH and DCM(reflux 6 hour)
R
R
R
Me2SnCl2
Ph2SnCl2
Where: R = o-CH3 (10) R = p-CH3 (11) R = H (12)
R = o-CH3 (13)R = p-CH3 (14)R = H (15)
R = o-CH3 (16)R = p-CH3 (17)R = H (18)
Scheme 4.2: Synthetic pathway for the synthesis of organotin compounds 13-18.
4.3.2 IR Spectral Analysis
The experimental and calculated frequencies for the Schiff bases (10-12) and their
organotin(IV) compounds (13-18) were recorded in the range 4000 to 280 cm-1 and 4000
to 0 cm-1, respectively. All DFT vibrational frequencies were scaled using a scaling factor
of 0.9682.189 The presence of the Schiff base azomethine C=N group was indicated by
the sharp peak at 1607-1608 cm-1, which shifted to higher frequencies upon complexation.
In a similar vein, the red-shift of the hydrazinic v(N-N) frequencies in the organotin(IV)
compounds provided further evidence of this coordination mode, and was related to the
reduction in the repulsion between lone pairs of electrons on the nitrogen atom.92,116,228
The v(C=S) splitting of the Schiff bases in the range of 1012-1018 cm-1 were shifted to
lower frequencies (956-966 cm-1) in the spectra of organotin(IV) compounds, thus
showing strong evidence to prove that the tin ion coordinated the thiolate sulphur.227 The

112
spectra of the Schiff bases also exhibited v(N-H) at 3097-3196 cm-1 which was not present
in the spectra of the organotin(IV) compounds due to the protonation of the N-H group
upon complexation.95 Furthermore, the hydroxyl group attached at the ortho position of
the benzene ring participated in the coordination to the tin metal centre, which was
consistent with the disappearance of v(OH) upon complexation. The calculated
frequencies correlated well with the experimental frequencies, considering that the DFT
frequencies calculations were performed in the gas-phase. The N-H band in experimental
FT-IR spectra were at slightly lower frequencies due to intermolecular hydrogen
bonding260 interactions between the molecules. A similar pattern was observed for
hydroxyl stretching, where only one red-shifted hydroxyl band appeared in the spectra
which could be due to intermolecular and intramolecular hydrogen bonding as was
observed in similar reported organotin(IV) compounds.234 Complete IR data is provided
in Appendix, Table A4.1.
4.3.3 NMR Spectroscopic Analysis
The NMR spectra of the Schiff bases were recorded in DMSO-d6 and their
organotin(IV) compounds were recorded in CDCl3. The Schiff bases were present in the
thione tautomeric form in DMSO-d6 as indicated by the absence of an exchangeable SH
signal in the 1H NMR spectra.94 The spectra of the Schiff bases also exhibited a downfield
signal in the ~13.39-13.41 ppm range, which indicated the presence of the NH group. For
all the organotin(IV) compounds, the 1H NMR signal for the azomethine NH proton was
absent due to the coordination of the metal ion, in agreement with the absence of the
v(NH) FTIR peak mentioned above. Furthermore, a singlet signal appeared in the
downfield region of the spectrum, corresponding to a sp2-type OH proton. Both proton
chemical shifts for NH and OH disappeared in the spectra of the organotin(IV)

113
compounds, suggesting double deprotonation of the Schiff bases and hence confirming
the coordination to the tin(IV) ion through both phenolic oxygen and nitrogen atoms. The
complete 1H NMR data are collated in Appendix Table 4A.2.
The 13C NMR spectra of the Schiff bases showed a downfield chemical shift at ~196
ppm, indicating the presence of the C=S thione tautomer in solution. This signal was
shifted upfield in the organotin(IV) compounds in the range of 160-175 ppm, indicating
a decrease in electron density at the C-S carbon atom when the sulphur atom was
complexed to the tin atom.229 All other signals are tabulated in Appendix Table A4.3.
The tin chemical shift (119Sn) values of organotin(IV) compounds indicated the
metal’s coordination number, the substituent (phenyl, methyl, ethyl) group attached
directly to tin and the geometry of the organotin(IV) compounds: δ(119Sn) values of four-
coordinate complexes fall in the range +200 to −60 ppm; five-coordinate complexes fall
between −90 and −190 ppm, and six- and seven-coordinate complexes fall between −210
and −400 ppm.230 The high displacement of the 119Sn chemical shifts of the
diphenyltin(IV) compounds reported here (-233.4 to -233.7 ppm) is caused by the
electronegativity of the Schiff bases as well as the organo group attached to the tin
center.205 Dimethyltin(IV) compounds exhibited a higher chemical shift than
diphenyltin(IV) compounds in the range of -99.5 to -99.8 ppm, which was consistent with
their lower electron density and hence nuclear shielding. The 119Sn NMR values obtained
for both diphenyl- and dimethyltin(IV) compounds indicated that the compounds were in
a penta-coordinated trigonal bipyramidal geometry. This was corroborated by single
crystal XRD analysis.

114
4.3.4 Mass Spectral Analysis
The mass spectra were recorded for 10-12. The mass spectroscopic data revealed
that the molecular skeleton of 10-12 were obtained as predicted. The prominent molecular
ion peaks were observed at m/z 332.05 for 10 and 11, and m/z 318.05 for 12 respectively.
The mass spectra for 10, 11 and 12 are supplied in Appendix Figure A4.1.
4.3.5 UV-vis Absorption Spectroscopy
UV-vis spectra of compounds 10-18 were investigated experimentally and
theoretically in DMSO. B3LYP frontier orbitals of 10-12 are presented in Figure 4.2,
which shows that the HOMO was predominantly centred on the dithiocarbazate backbone
(S-(C=S)-NH-N), and the LUMO was largely centred on the 2,3-dihydroxyphenyl ring
and as well as dithiocarbazate backbone. Thus, the excitation of electrons observed in the
range 326-343 nm (experimental) and 335-345 nm (theoretical) was assigned as an n→π*
excitation, where nonbonding electrons of the azomethine nitrogen and sulphur atom in
the ground state were excited to the π* LUMO. The other excitations observed in the
range of 381-392 nm (experiment) and 372-386 nm (theoretical) indicated a π→π*
excitation involving the aromatic rings. In the spectra of the organotin(IV) compounds,
the HOMO was largely centred on the dithiocarbazate backbone and the 2,3-
dihydroxyphenyl ring moiety. Conversely, the LUMO was centred on the dithiocarbazate
backbone and 2,3-dihydroxyphenyl ring. The n→π* and π→π* transitions were observed
at 312-331 and 359-363 nm respectively, similar to the Schiff bases. The additional
intraligand band of the S→SnIV LMCT transition at 418-424 nm suggested coordination
of the Sn metal centre with sulphur atoms which was corroborated by evidence from
NMR and IR spectral analysis.93 All the transitions are summarised in Appendix Table
A4.4 and the HOMO-LUMO orbital diagrams are presented in Figure 4.3.

115
4.3.6 Structure Descriptions for 12, 14, 15, 17 and 18
The crystal and molecular structures of the hydrazinecarbodithioate, 12, a precursor
molecule for the structures of 15 and 18, and those of four diorganotin compounds, 14,
15, 17 and 18, were determined by X-ray crystallography. In 12, Figure 4.4 and Table
4.2, the central CN2S2 residue is planar with the RMSD of the fitted atoms being 0.011
Å; the maximum deviation from the least-squares plane is 0.018(4) Å for the N1 atom.
The dihedral angles formed between this central plane and the hydroxyl- and benzyl-rings
are 2.1(3) and 63.55(14)°, respectively, indicating co-planar and twisted relationships,
respectively; the dihedral angle between the rings is 61.63(17)°. The co-planarity in the
(phenylethylidene)hydrazine region of the molecule is due in part to the formation of
intramolecular hydroxyl-O‒H…O(hydroxyl) and hydroxyl-O‒H…N(imine) hydrogen
bonds to close S(5) and S(6) loops, respectively, details of which are given in Appendix
Figure A4.2.
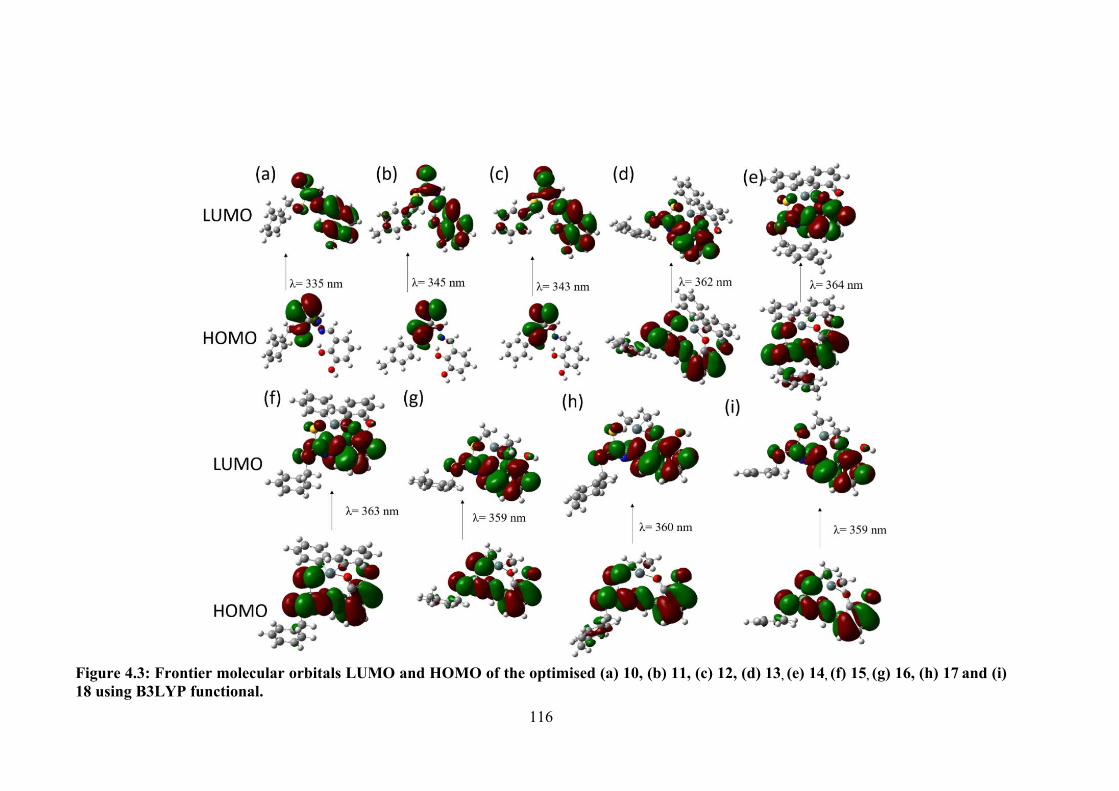
116
Figure 4.3: Frontier molecular orbitals LUMO and HOMO of the optimised (a) 10, (b) 11, (c) 12, (d) 13, (e) 14, (f) 15, (g) 16, (h) 17 and (i) 18 using B3LYP functional.

117
Figure 4.4: Molecular structure of 12 and atom labelling scheme.
The crystallographic asymmetric unit of 14 comprises two independent molecules,
as shown in Figure 4.5a. Selected geometric data for all diorganotin structures described
herein are collated in Table 4.2. The tin atom is coordinated by thiolate-sulphur, imine-
nitrogen and phenoxide-oxygen atoms derived from the di-negative tridentate ligand. The
five-coordinate geometry about the tin atom is completed by two ipso-carbon atoms
derived from two phenyl substituents. A measure of the distortion away from ideal
trigonal-bipyramidal and square-pyramidal coordination geometries is given by the
descriptor τ 235 for which these ideal geometries correspond to τ = 1.0 and 0.0,
respectively. In 14, τ is 0.58 and 0.52 for the two independent molecules, indicating
environments almost half-way between these extremes. The structure of 15 presents very
similar features, again with two molecules in the asymmetric unit, Figure 4.5b. In this
case, the values of τ for the two molecules are 0.41 and 0.46, indicating intermediate five-
coordinate geometries but, a greater tendency towards a square-pyramidal coordination
structure. The availability of the crystal structure of hydrazinecarbodithioate 12 allows
comparisons of bond length and angle data with the carbonodithiohydrazonate dianion of
15. From Table 4.2, obvious differences exist in key geometric parameters such as the
elongation of the C1‒S1 bonds for the molecules in 15 cf. that in 12 as well as the
elongation of the N1‒N2 and C2‒N2 bonds with a concomitant decrease in the C1‒N1

118
bond length; no significant change is noted for the C1‒S2 bond. These systematic
variations indicate the formation of a C1‒S1 thiolate ligand as well as a second imine C2‒
N2 bond in 15. Given the comparable geometric parameters are equivalent within
experimental error to those in 14, that same conclusion pertains as, indeed, for the
structures of 17 and 18, Table 4.2. The reorganisation in π-electron density upon
deprotonation of the ligand and coordination to tin results in systematic reductions in the
angles subtended at the N1 and N2 atoms. The decrease and increase of π-electron density
in the C1‒S1 and C1‒N1 bonds upon coordination results in a decrease in the S1‒C1‒S2
angles and accompanying increases in the S1‒C1‒N1 and S2‒C1‒N1 bond angles. The
structures of 17 and 18 are isostructural and present τ values of 0.55 and 0.50,
respectively, i.e. in the range of values established for 14 and 15. In the same way, the
C‒Sn‒C angles in 17 and 18 lie within the range of C‒Sn‒C angles seen in 14 and 15,
Table 4.2.
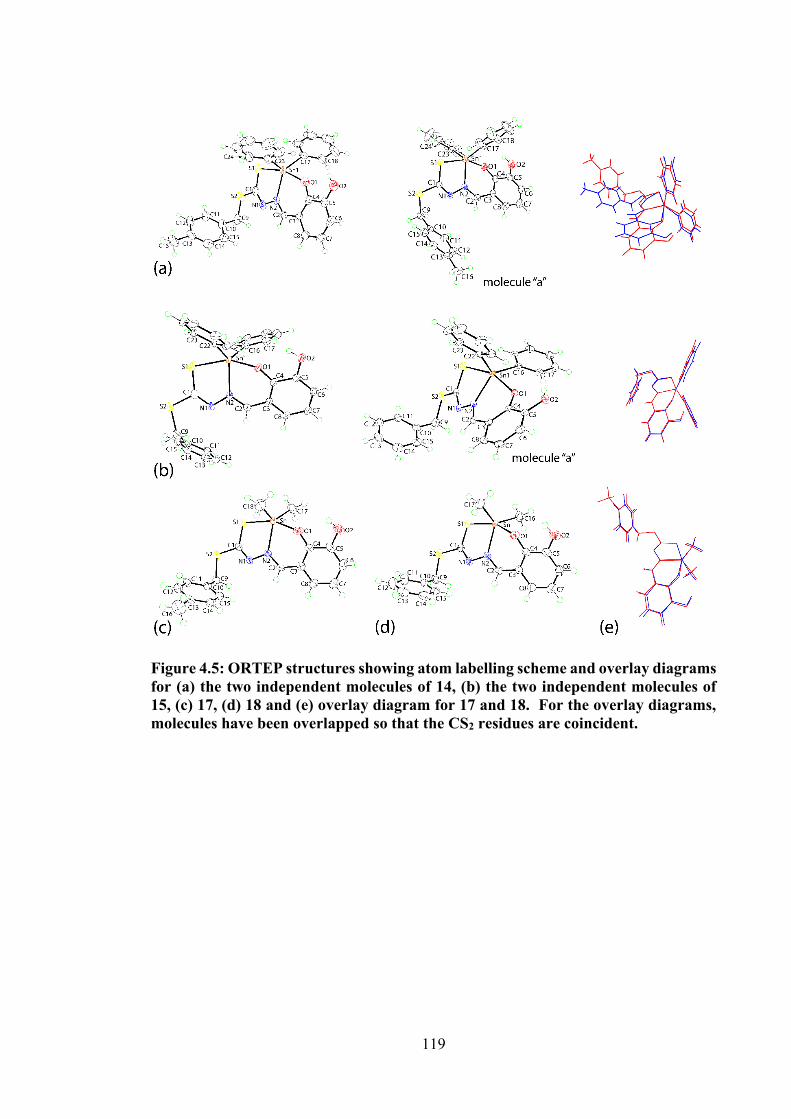
119
Figure 4.5: ORTEP structures showing atom labelling scheme and overlay diagrams for (a) the two independent molecules of 14, (b) the two independent molecules of 15, (c) 17, (d) 18 and (e) overlay diagram for 17 and 18. For the overlay diagrams, molecules have been overlapped so that the CS2 residues are coincident.
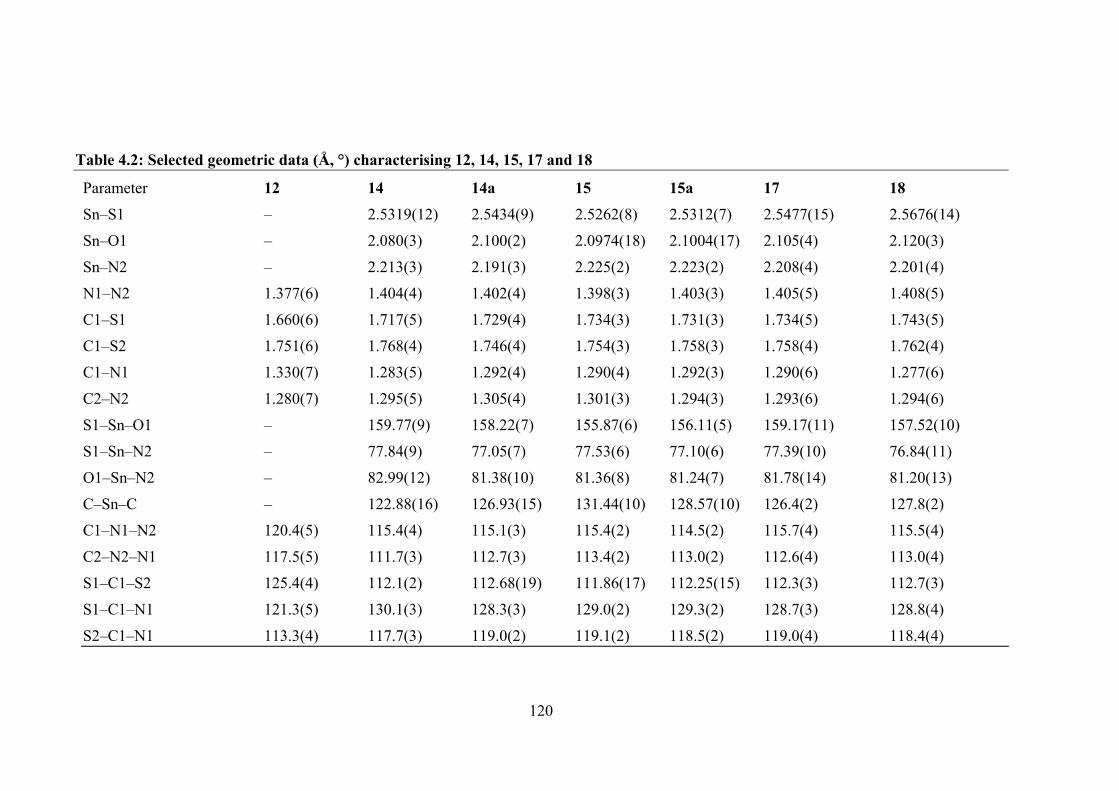
120
Table 4.2: Selected geometric data (Å, °) characterising 12, 14, 15, 17 and 18
Parameter 12 14 14a 15 15a 17 18 Sn‒S1 ‒ 2.5319(12) 2.5434(9) 2.5262(8) 2.5312(7) 2.5477(15) 2.5676(14) Sn‒O1 ‒ 2.080(3) 2.100(2) 2.0974(18) 2.1004(17) 2.105(4) 2.120(3) Sn‒N2 ‒ 2.213(3) 2.191(3) 2.225(2) 2.223(2) 2.208(4) 2.201(4) N1‒N2 1.377(6) 1.404(4) 1.402(4) 1.398(3) 1.403(3) 1.405(5) 1.408(5) C1‒S1 1.660(6) 1.717(5) 1.729(4) 1.734(3) 1.731(3) 1.734(5) 1.743(5) C1‒S2 1.751(6) 1.768(4) 1.746(4) 1.754(3) 1.758(3) 1.758(4) 1.762(4) C1‒N1 1.330(7) 1.283(5) 1.292(4) 1.290(4) 1.292(3) 1.290(6) 1.277(6) C2‒N2 1.280(7) 1.295(5) 1.305(4) 1.301(3) 1.294(3) 1.293(6) 1.294(6) S1‒Sn‒O1 ‒ 159.77(9) 158.22(7) 155.87(6) 156.11(5) 159.17(11) 157.52(10) S1‒Sn‒N2 ‒ 77.84(9) 77.05(7) 77.53(6) 77.10(6) 77.39(10) 76.84(11) O1‒Sn‒N2 ‒ 82.99(12) 81.38(10) 81.36(8) 81.24(7) 81.78(14) 81.20(13) C‒Sn‒C ‒ 122.88(16) 126.93(15) 131.44(10) 128.57(10) 126.4(2) 127.8(2) C1‒N1‒N2 120.4(5) 115.4(4) 115.1(3) 115.4(2) 114.5(2) 115.7(4) 115.5(4) C2‒N2‒N1 117.5(5) 111.7(3) 112.7(3) 113.4(2) 113.0(2) 112.6(4) 113.0(4) S1‒C1‒S2 125.4(4) 112.1(2) 112.68(19) 111.86(17) 112.25(15) 112.3(3) 112.7(3) S1‒C1‒N1 121.3(5) 130.1(3) 128.3(3) 129.0(2) 129.3(2) 128.7(3) 128.8(4) S2‒C1‒N1 113.3(4) 117.7(3) 119.0(2) 119.1(2) 118.5(2) 119.0(4) 118.4(4)

121
The tridentate mode of coordination of the carbonodithiohydrazonate leads to the
formation of five-membered SnSCN2 and six-membered SnOC3N chelate rings. In the
first independent molecule of 14, the five-membered ring is planar [RMSD = 0.007 Å]
and to a first approximation, so is the six-membered ring which presents a RMSD = 0.042
Å. However, a more apt description is one based on an envelope as the tin atom lies
0.162(5) Å above the plane of the five remaining atoms of the chelate ring which exhibit
a RMSD of 0.023 Å. The angle between chelate rings is 3.74(16)°, and the angle between
the six-membered ring and appended phenyl ring is 3.2(2)°, both consistent with a planar
ligand. Quite distinct conformations are adopted by the chelate rings in the second
independent molecule of 14. Thus, each is best described as having an envelope
conformation with the tin ion lying 0.544(5) Å above the plane defined by the four
remaining atoms of the five-membered chelate ring [RMSD = 0.008 Å], and with tin
being 0.761(4) Å above the plane of the five remaining atoms for the six-membered
chelate [RMSD = 0.038 Å]. The dihedral angle between the planar portions of the chelate
rings is 34.23(15)°; the angle between the five co-planar atoms of the larger ring and
appended phenyl ring is 2.3(2)°. This stark difference in coordination mode is clearly
evident from the overlay diagram in Figure 4.5a. As seen in the overlay diagram of Figure
4.5b, there is higher degree of concordance between the two independent molecules of
15. However, a key difference exists between 14 and 15 in that in the latter, each chelate
ring has the form of an envelope. Thus, for the five-membered ring, the tin atom lies
0.320(4) Å above the plane of the remaining atoms [RMSD = 0.015 Å; second molecule:
0.472(4) Å/0.020 Å]. The equivalent parameters for the six-membered ring are 0.750(3)
Å/0.047 Å [0.788(3) Å/0.052 Å]. The dihedral angle between the planar regions of the
chelate rings is 26.47(11) [31.20(11)°]. The overlay diagram in Figure 4.5e confirms the
similarity of the molecular structures of 17 and 18. The chelate rings in 17 and 18 adopt

122
conformations as for the first independent molecule of 14 and both molecules of 15, with
tin ion deviations from the planes through the lighter atoms being 0.452(7) Å/0.007Å and
0.620(6) Å/0.046 Å, respectively, for the five- and six-membered rings of 17; the
comparable values for 18 are 0.516(7) Å/0.009 Å and 0.857(5) Å/0.063 Å, respectively.
The dihedral angle between the chelate rings in 17 of 27.8(2)° is less than 35.8(2)° in 18.
With the exception of the very recently reported diorganotin derivatives bearing
ligands related to those in the present study, i.e. with a methoxy substituent adjacent to
the phenoxide-oxygen atom, and which present similar distorted five coordinate
geometries,178 there are only two other crystallographically-characterised tin structures
available in the Cambridge Structural Database.261 In each of these, the phenoxide ring
is unsubstituted and there is an ethyl group bound to the ester-sulphur atom, and similar
tridentate modes of coordination are noted. A distorted octahedral geometry based on a
N2O2S6 donor set with oxygen and sulphur atoms trans to each other is found in the
homoleptic species.118 In the other available structure, a distorted octahedral geometry is
also found with three positions occupied by the tridentate ligand, two by iodide atoms and
the sixth site by an oxygen atom derived from a dimethylformamide ligand.118
In the molecular packing of 12, centrosymmetric molecules assemble into dimeric
aggregates via hydroxyl-O‒H…O(hydroxyl) hydrogen bonds leading to 10-membered
…HOC2O2 synthons. These are connected into a linear supramolecular chain along [1
0 4] by thioamide-N‒H…S(thione) hydrogen bonds between centrosymmetrically related
molecules via eight-membered …HNCS2 synthons, Figure 4.6. The chains are
assembled into a three-dimensional architecture by hydroxyphenyl-C‒H…π(benzyl)
interactions; a view of the unit cell contents is shown Appendix Figure A4.3. Geometric
parameters characterising these interactions and those for the other structures discussed

123
herein are included in the respective figure captions of the Supplementary information, in
this case Appendix Figure A4.3.
Figure 4.6: A view of the linear supramolecular chain along [1 0 4] in the crystal of 12. The hydroxyl-O‒H…O(hydroxyl) and thioamide-N‒H…S(thione) hydrogen bonds are shown as orange and green dashed lines, respectively.
Along with the structures of 15, 17 and 18, there is an intramolecular charge-
assisted hydroxyl-O‒H…O(phenoxide) hydrogen bond in each molecule of 14 which
precludes the participation of this atom in hydrogen bonding in these crystals. An
interesting feature of the molecular packing of 14 is the presence of π…π interactions
involving the only planar chelate ring observed among the diorganotin species reported
herein (see above) and a symmetry-related hydroxyphenyl ring. The distance between
the ring centroids is 3.874(2) Å and the angle between rings is 0.95(15)° for symmetry
operation x, ½-y, -½+z; the slippage is 1.69 Å, indicating the usually-observed off-set
disposition of the rings.239 Such interactions are becoming increasingly apparent in the
supramolecular chemistry of heavy-element compounds and can impart energies of
stabilisation to their crystals equal to or greater than conventional hydrogen bonding
interactions.237,239 In the present structure, these interactions lead to the presence of
supramolecular chains with a zig-zag topology (glide symmetry) along the c-axis, Figure
4.7. The connections between the chains are tin-bound phenyl- and hydroxyphenyl-C‒
H…π(Sn-bound phenyl) and contribute to the stabilisation of a layer in the bc-plane; layers

124
stack along the a-axis without directional interactions between them, Appendix Figure
A4.3.
In the crystal of 15, distinct supramolecular motifs are observed containing one of
the independent molecules exclusively. Thus, a linear supramolecular chain containing
Sn1-molecules only is sustained by benzyl-phenyl-C‒H…O(hydroxyl) interactions,
Appendix Figure A4.4a, and a supramolecular dimer of Sn1a-molecules and sustained by
tin-bound-phenyl-C‒H…O(hydroxyl) interactions, Appendix Figure A4.4b, are noted.
The chains are connected into a three-dimensional architecture by a combination of
hydroxyphenyl-C‒H…π(tin-bound phenyl) and π(tin-bound phenyl)…π(tin-bound phenyl)
interactions, Appendix Figure A4.4c .
The crystals of 17 and 18 are isomorphous, as shown in Table 4.1. In the crystal of
17, a three-dimensional architecture is stabilised by tin-bound-methyl-C‒H...π(benzyl-
phenyl) and hydroxyphenyl-C‒H…O(hydroxyl) interactions, as detailed in Appendix
Figure A4.5. Analogous interactions are apparent in the crystal of 18, Appendix Figure
A4.6.
The gas phase B3LYP geometries of 12, 14, 15, 17 and 18 are in good agreement
with the values obtain from crystallographic analysis above (see Appendix Table A4.5),
consistent with the absence of strong directional of inter- and intramolecular interactions
in the solid form of the crystals. The deviation in selected bond lengths and bond angles
of 12, 14, 15, 17 and 18 are less than 0.038 Å and 8.0°, respectively, where the maximum
bond length deviation corresponded to the Sn-O1 bond and the maximum bond angle
deviation corresponded to the organo group attached to the tin, C-Sn-C angle.

125
Figure 4.7: A view of the zig-zag chain along [0 0 1] in the crystal of 14. The hydroxyl-O‒H…O(hydroxyl) and thioamide-N‒H…S(thione) hydrogen bonds are shown as orange and green dashed lines, respectively. For reasons of clarity, the hydrogen atoms have been removed and only the ipso-carbon atoms of the tin-bound phenyl rings shown.
4.3.7 In Vitro Cytotoxicity
Compounds 10-18 were tested for their cytotoxic activity against ten cancer cell
lines (HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, DU145, BE2-C, MIA) and one
normal breast cell line (MCF-10A) using the MTT metabolic assay. The growth inhibition
concentration of the Schiff bases (10-12) and their organotin(IV) compounds (13-18)
required to inhibit 50% cell proliferation relative to the cells that were treated with DMSO
(control) are tabulated in Table 4.3. The stability of these compounds was evaluated in
DMSO as well as in a mixture of DMSO-H2O by monitoring UV-vis absorbance. The
unchanged pattern in the spectra indicated that the compounds were stable in both
solvents.
Compound 10 showed selectivity against the BE2-C cell line and the highest
potency (GI50 = 0.38 ± 0.07 μM, refer Table 4.3) of the Schiff base analogues 11 and 12.
However, 10 exhibited the lowest cytotoxicity against HT29, U87, MCF-7, A431 and
Du145 cell lines. Moderate cytotoxic activity was observed for 11 with no significant
selectivity, with slightly lower activity than 12 in all cancer cell lines investigated here,
suggesting that the presence of the methyl group at the ortho and para positions did not

126
impact cytotoxicity significantly, an observation that correlated well with what was
reported in previous work.178 Complexation with diphenyltin(IV) dichloride improved the
activity of the Schiff bases, with diphenyltin(IV) compounds (13, 14 and 15)
demonstrating good cytotoxicity against A2780, BE2-C, SJ-G2 and MIA cells. The
highest level of selectivity of 13, 14 and 15 was observed for the A2780 cells, with GI50
values of 0.31 ± 0.05, 0.23 ± 0.02 and 0.19 ± 0.01 μM, respectively, which corresponded
to 6- and 10-fold higher potencies compared to 10, 11 and 12 (the latter had GI50 values
of 1.8 ± 1.2, 2.8 ± 0.20 and 2.2 ± 0.25 μM, respectively). Furthermore, 13 also showed
selectivity against HT29, MCF-7 and Du145 cell lines, with 10-fold, 15-fold and 57-fold
higher potency than its Schiff base, 10. A 3-fold greater activity against U87 was observed
for 15 with a GI50 value of 0.69 ± 0.03 μM as compared to its Schiff base, 12 (GI50 = 2.7
± 0.47 μM). The selectivity of 14 and 15 for A2780 was 8- and 5-fold higher, respectively,
over the normal breast cell line, MCF10A. There was a significant contrast in the
cytotoxic activity of dimethyltin(IV) compounds (16, 17 and 18) as compared to the
diphenyltin(IV) compounds, which suggested that the presence of the two phenyl groups
coordinating the tin(IV) centre enhanced the cytotoxicity of the compounds. Compounds
16, 17 and 18 exhibited no obvious enhancement in cytotoxicity as compared to their
respective Schiff bases, which was similar to that observed in previous work.178
4.3.8 DNA Binding Analysis
A number of studies66,142,152,262 have reported that cytotoxic agents can cleave or
bind to DNA. The interaction between organotin(IV) compounds (13-18) and CT-DNA
is therefore examined here via UV-vis absorption spectroscopy. Figure 4.8 shows the UV-
vis spectra observed where dimethyltin(IV) compounds (16, 17 and 18) in solutions of
varying DNA concentrations. These compounds had absorption peaks at 304 - 312, 358 -

127
362 and 426 - 434 nm, which indicated π-π* (due to aromatic group), n-π* (due to either
C=N or OH) and ligand-metal charge transfer excitations, respectively. By increasing the
concentrations of CT-DNA, significant hypochromism and red shifts were observed in
the spectra, see Table 4.4. This hypochromism was attributed to the strong interactions
between the organotin(IV) π electrons and DNA base pair π electrons. Therefore, the
energy level of the π→π* electron transition decreased and shifted to a higher
wavelength.263 The change in transitions were observed in the spectra of compounds 16,
17 and 18, but only slight changes were observed in diphenyltin(IV) compounds (13, 14
and 15) (Figure A4.7). It is therefore conceivable that the cytotoxic activities of
compounds 13, 14 and 15 could be partly due to the interaction between the compounds
and some proteins or organelles in cancer cells, not exclusively only via binding to
DNA.224 Nevertheless, negative Gibbs free energies (∆G) obtained from this
spectroscopic analysis indicate that the DNA - organotin(IV) compound interaction is
spontaneous.
4.3.9 Molecular Docking Analysis
In order to understand specific interactions between the six organotin(IV)
compounds (13-18) and CT-DNA, we turn to molecular docking simulations using a
model DNA duplex of sequence d(CGCGAATTCGCG)2. Results are detailed in Table
4.5. The organotin(IV) compounds mainly bound to the groove of the DNA helix via non-
covalent interactions, viz., hydrogen bonding, hydrophobic, van der Waals and
electrostatic interactions. These non-covalent interactions are usually stabilised by π-
interactions between aromatic groups of the compounds 13-18 and the DNA duplex, and
therefore provide a structural basis for the observed hypochromism detailed in the
previous section. Figure 4.9 shows the most favourable conformation of the docked pose

128
which exhibited the lowest binding energy, revealing that compounds 13-18 interact with
the AT-rich region of the minor groove. The DG10 and DT20 DNA residues played a
major role in the interaction with diphenyltin(IV) compounds (13-15), whereas DG10 and
DT8 of the DNA residues predominantly interacted with dimethyltin(IV) compounds (16-
18). The interactions mainly occurred through the phosphate backbone of the DNA, as
well as via nitrogenous bases of DNA.
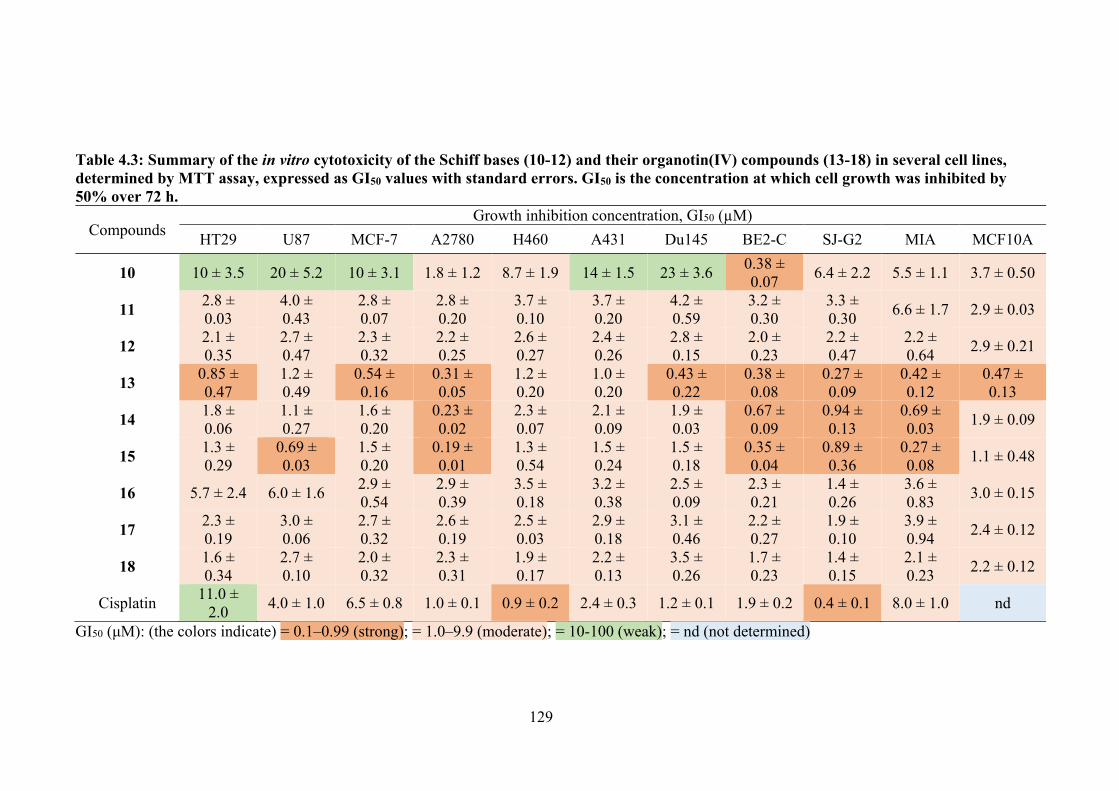
129
Table 4.3: Summary of the in vitro cytotoxicity of the Schiff bases (10-12) and their organotin(IV) compounds (13-18) in several cell lines, determined by MTT assay, expressed as GI50 values with standard errors. GI50 is the concentration at which cell growth was inhibited by 50% over 72 h.
Compounds Growth inhibition concentration, GI50 (µM)
HT29 U87 MCF-7 A2780 H460 A431 Du145 BE2-C SJ-G2 MIA MCF10A
10 10 ± 3.5 20 ± 5.2 10 ± 3.1 1.8 ± 1.2 8.7 ± 1.9 14 ± 1.5 23 ± 3.6 0.38 ± 0.07 6.4 ± 2.2 5.5 ± 1.1 3.7 ± 0.50
11 2.8 ± 0.03
4.0 ± 0.43
2.8 ± 0.07
2.8 ± 0.20
3.7 ± 0.10
3.7 ± 0.20
4.2 ± 0.59
3.2 ± 0.30
3.3 ± 0.30 6.6 ± 1.7 2.9 ± 0.03
12 2.1 ± 0.35
2.7 ± 0.47
2.3 ± 0.32
2.2 ± 0.25
2.6 ± 0.27
2.4 ± 0.26
2.8 ± 0.15
2.0 ± 0.23
2.2 ± 0.47
2.2 ± 0.64 2.9 ± 0.21
13 0.85 ± 0.47
1.2 ± 0.49
0.54 ± 0.16
0.31 ± 0.05
1.2 ± 0.20
1.0 ± 0.20
0.43 ± 0.22
0.38 ± 0.08
0.27 ± 0.09
0.42 ± 0.12
0.47 ± 0.13
14 1.8 ± 0.06
1.1 ± 0.27
1.6 ± 0.20
0.23 ± 0.02
2.3 ± 0.07
2.1 ± 0.09
1.9 ± 0.03
0.67 ± 0.09
0.94 ± 0.13
0.69 ± 0.03 1.9 ± 0.09
15 1.3 ± 0.29
0.69 ± 0.03
1.5 ± 0.20
0.19 ± 0.01
1.3 ± 0.54
1.5 ± 0.24
1.5 ± 0.18
0.35 ± 0.04
0.89 ± 0.36
0.27 ± 0.08 1.1 ± 0.48
16 5.7 ± 2.4 6.0 ± 1.6 2.9 ± 0.54
2.9 ± 0.39
3.5 ± 0.18
3.2 ± 0.38
2.5 ± 0.09
2.3 ± 0.21
1.4 ± 0.26
3.6 ± 0.83 3.0 ± 0.15
17 2.3 ± 0.19
3.0 ± 0.06
2.7 ± 0.32
2.6 ± 0.19
2.5 ± 0.03
2.9 ± 0.18
3.1 ± 0.46
2.2 ± 0.27
1.9 ± 0.10
3.9 ± 0.94 2.4 ± 0.12
18 1.6 ± 0.34
2.7 ± 0.10
2.0 ± 0.32
2.3 ± 0.31
1.9 ± 0.17
2.2 ± 0.13
3.5 ± 0.26
1.7 ± 0.23
1.4 ± 0.15
2.1 ± 0.23 2.2 ± 0.12
Cisplatin 11.0 ± 2.0 4.0 ± 1.0 6.5 ± 0.8 1.0 ± 0.1 0.9 ± 0.2 2.4 ± 0.3 1.2 ± 0.1 1.9 ± 0.2 0.4 ± 0.1 8.0 ± 1.0 nd
GI50 (μM): (the colors indicate) = 0.1–0.99 (strong); = 1.0–9.9 (moderate); = 10-100 (weak); = nd (not determined)
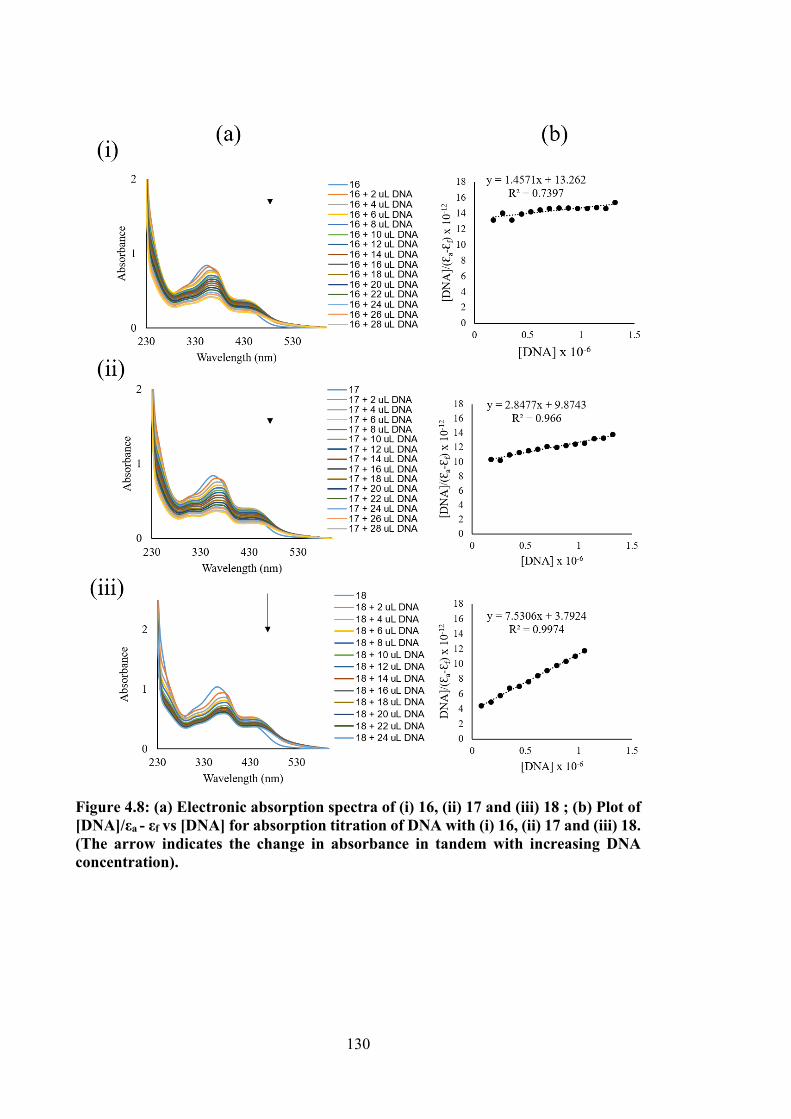
130
Figure 4.8: (a) Electronic absorption spectra of (i) 16, (ii) 17 and (iii) 18 ; (b) Plot of [DNA]/εa - εf vs [DNA] for absorption titration of DNA with (i) 16, (ii) 17 and (iii) 18. (The arrow indicates the change in absorbance in tandem with increasing DNA concentration).
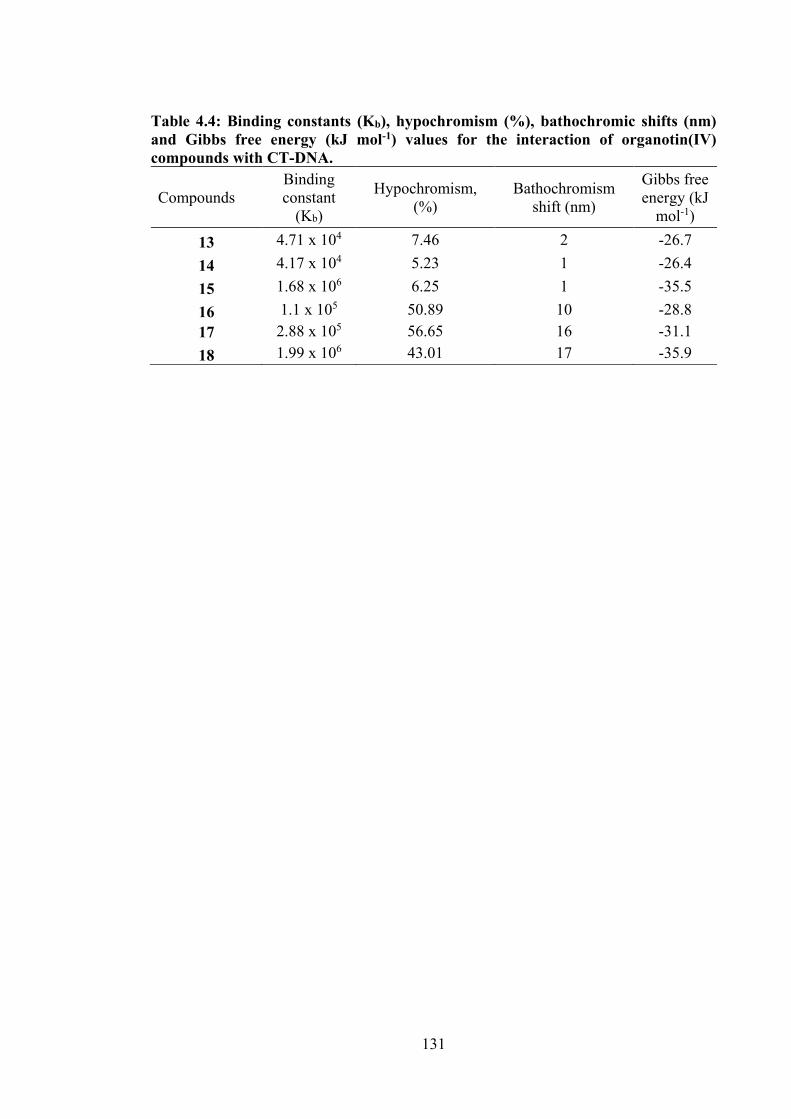
131
Table 4.4: Binding constants (Kb), hypochromism (%), bathochromic shifts (nm) and Gibbs free energy (kJ mol-1) values for the interaction of organotin(IV) compounds with CT-DNA.
Compounds Binding constant
(Kb)
Hypochromism, (%)
Bathochromism shift (nm)
Gibbs free energy (kJ
mol-1)
13 4.71 x 104 7.46 2 -26.7 14 4.17 x 104 5.23 1 -26.4 15 1.68 x 106 6.25 1 -35.5 16 1.1 x 105 50.89 10 -28.8 17 2.88 x 105 56.65 16 -31.1 18 1.99 x 106 43.01 17 -35.9
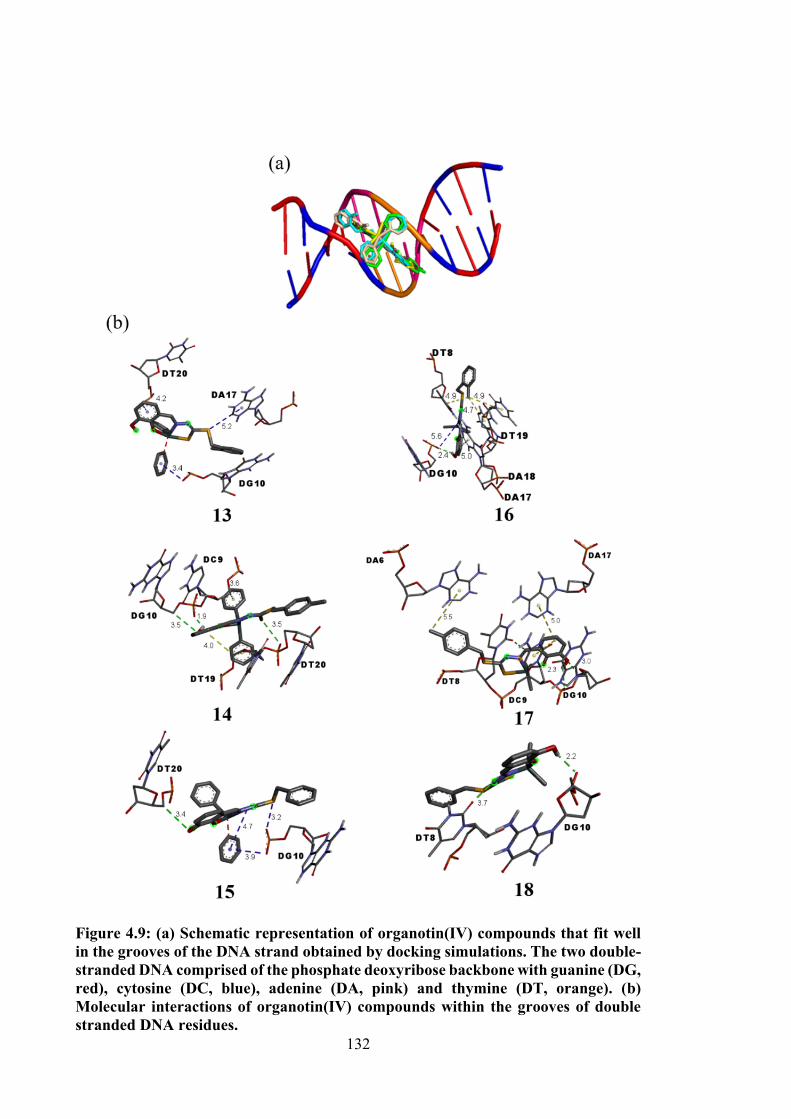
132
Figure 4.9: (a) Schematic representation of organotin(IV) compounds that fit well in the grooves of the DNA strand obtained by docking simulations. The two double-stranded DNA comprised of the phosphate deoxyribose backbone with guanine (DG, red), cytosine (DC, blue), adenine (DA, pink) and thymine (DT, orange). (b) Molecular interactions of organotin(IV) compounds within the grooves of double stranded DNA residues.
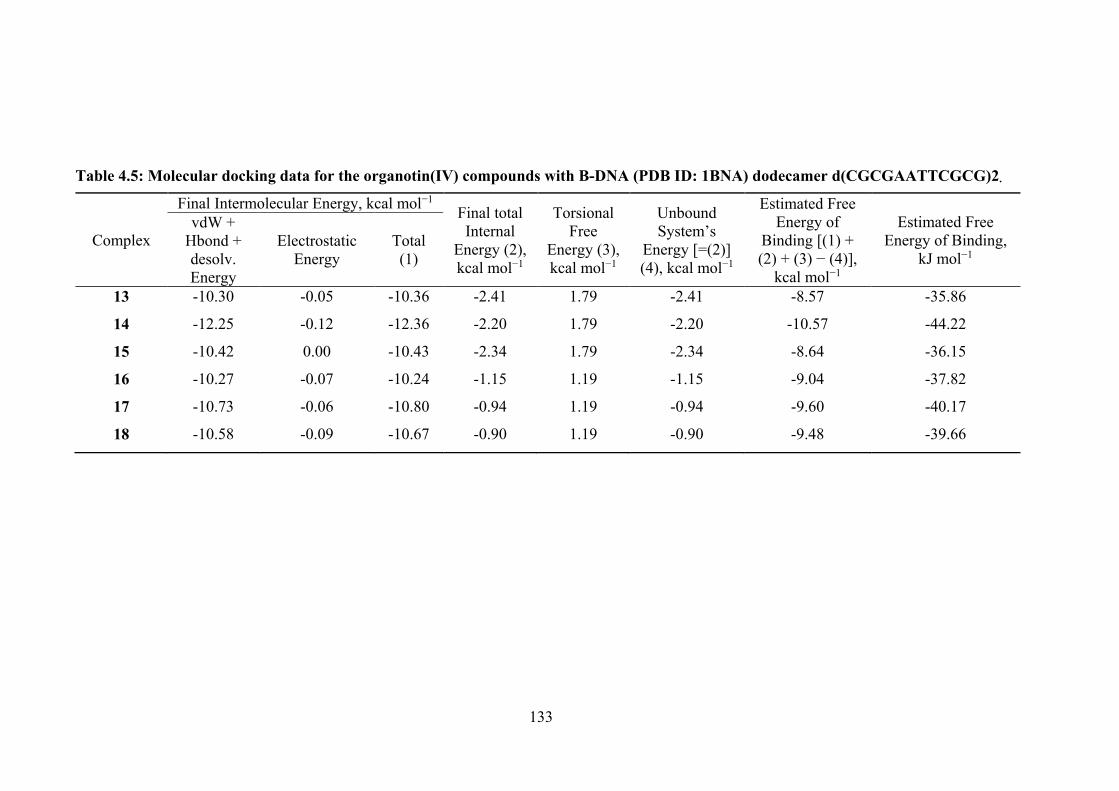
133
Table 4.5: Molecular docking data for the organotin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
Complex
Final Intermolecular Energy, kcal mol−1 Final total Internal
Energy (2), kcal mol−1
Torsional Free
Energy (3), kcal mol−1
Unbound System’s
Energy [=(2)] (4), kcal mol−1
Estimated Free Energy of
Binding [(1) + (2) + (3) − (4)],
kcal mol−1
Estimated Free Energy of Binding,
kJ mol−1
vdW + Hbond + desolv. Energy
Electrostatic Energy
Total (1)
13 -10.30 -0.05 -10.36 -2.41 1.79 -2.41 -8.57 -35.86
14 -12.25 -0.12 -12.36 -2.20 1.79 -2.20 -10.57 -44.22
15 -10.42 0.00 -10.43 -2.34 1.79 -2.34 -8.64 -36.15
16 -10.27 -0.07 -10.24 -1.15 1.19 -1.15 -9.04 -37.82
17 -10.73 -0.06 -10.80 -0.94 1.19 -0.94 -9.60 -40.17
18 -10.58 -0.09 -10.67 -0.90 1.19 -0.90 -9.48 -39.66

134
4.4 Conclusions
Diphenyltin(IV) (13-15) and dimethyltin(IV) (16-18) compounds containing ONS-
tridentate 2,3-dihydroxybenzyldithiocarbazate Schiff bases (10-12) were synthesised and
characterised by analytical, spectroscopic and single crystal X-ray diffraction studies. The
cytotoxic activities of these compounds against a panel of ten cancer cell lines were
evaluated. A good correlation was found between calculated geometries, electronic
absorptions and vibrational frequencies from experimental data, single crystal XRD, UV-
visible and FT-IR spectroscopy. X-ray crystallography established the anticipated
tridentate mode of coordination between the dinegative Schiff bases and the diorganotin
centres, resulting in highly distorted five-coordinate C2NOS geometries. In vitro
cytotoxic assays revealed remarkable cytotoxicity of diphenyltin(IV) compounds with
GI50 values in the range 0.19–2.3 μM for all cancer cell lines. This study has also
demonstrated that diphenyltin(IV) compounds exhibited selective potency against the
U87, A2780, BE2-C, SJ-G2 and MIA cell lines, with higher cytotoxicity than their Schiff
bases and dimethyltin(IV) compounds which were comparable with the diphenyltin
compounds discussed in Chapter 3. The DNA binding data obtained using UV-vis
spectroscopy showed that the dimethyltin(IV) compounds bound to the DNA via groove
binding, resulting in hypochromism and redshift in the principal UV-vis excitation. The
molecular docking results showed that DG10 and DT20 of the DNA residues were crucial
for the binding to diphenyltin(IV) compounds, whereas all dimethyltin(IV) compounds
were bound to DG10 and DT8 of the DNA residues.
Exciting cytotoxicity outcomes were observed for dithiocarbazate Schiff bases and
their organotin(IV) compounds which was previously discussed in this chapter and
previous chapters. It was also demonstrated that complexation enhanced the
antiproliferative effects against a panel of cancer cell lines tested, particularly

135
diphenyltin(IV) compounds suggesting that the presence of two phenyl groups attached
to the tin atom played a main role in inhibiting cancer cells, particularly by interacting
aromatic phenyl ring of compounds with nitrogenous bases of DNA of cancer cells.
Slightly higher cytotoxicity were also observed for dimethyltin(IV) compounds. Based
on this observation, further investigation required to study the absence of organo groups
attached to the tin atom. Recently, tin(IV) compound coordinated with two molecules of
dithiocarbazate Schiff base showed high potency against MCF-7 cell due to its ability to
bind to human serum albumin protein.118
Further investigations will be discussed in Chapter 5, which reports structural and
cytotoxicity studies of six homoleptic tin(IV) compounds coordinated with two molecules
of dithiocarbazate Schiff bases 1, 2, 3, 10, 11, 12 (discussed in Chapter 2 and 4). The
theoretical prediction of tin(IV) compound-DNA binding will also discussed in Chapter
5.

136
CHAPTER 5
HOMOLEPTIC TIN(IV) COMPOUNDS OF DINEGATIVE ONS DITHIOCARBAZATE SCHIFF BASES: SYNTHESIS, X-RAY
CRYSTALLOGRAPHY, DFT AND CYTOTOXICITY STUDIES
5.1 Introduction
Two decades ago, octahedral homoleptic tin(IV) compounds containing S-benzyl-
β-N-(2-hydroxyphenyl)methylene264, S-methyl-β-N-(2-hydroxyphenyl) methylene264, S-
methyl-β-N-(2-hydroxy-3-methoxy-phenyl)methylenedithiocar-bazate,265 S-methyl-β-N-
(2-hydroxy-1-naphthyl)methylenedithiocarbazate,265 S-benzyl-β-N-(2-hydroxy-3-
methoxyphenyl)methylenedithiocarbazate,266 S-benzyl-β-N-(2-hydroxy-1-naphthyl)
methylene dithiocarbazate266 and S-benzyl-β-N-(2-ferrocenoylacetone) methylene
dithiocarbazate267 were synthesised and structurally characterised. However, the
cytotoxic studies of these compounds remain undisclosed. Recently, the discovery of
homoleptic tin(IV) compounds coordinated with dithiocarbazate Schiff bases were
reported by Yekke-Ghasemi et al.118 They found that tin(IV) compound of
2,2’(disulfanediylbis((ethylthio) methylene)bis(hydrazin-2-yl-1-ylidene) bis(methanyl
ylidene))diphenol exhibited good cytotoxicity against MCF-7 cell compared to the
clinical drug, cisplatin. This compound was also observed to bind to the most abundant
protein in blood plasma, human serum albumin protein.118 However, the cytotoxicity
studies of tin(IV) compounds coordinated with two molecules of oVa or catechol
dithiocarbazate Schiff bases remains unexplored. It is hypothesised here that homoleptic
tin(IV) compounds containing Schiff bases would have good cytotoxic properties, and
are able to interact with biomacromolecules.
Following on the cytotoxicity results reported in Chapter 3 and 4, this chapter
investigates this hypothesis by focusing on the synthesis, structural evaluation and

137
cytotoxic studies of homoleptic tin(IV) compounds containing two molecules of oVa or
catechol dithiocarbazate Schiff bases with general formula, Sn(L)2 (L= Schiff bases 1, 2,
3, 10, 11 and 12), see Figure 5.1.
Figure 5.1: The structures of homoleptic tin(IV) compounds (19-24).
This chapter contains material from submitted article for publication in Journal of
Molecular Structure on 27 September 2019.
5.2 Experimental
5.2.1 Materials and Physical Measurements
All solvents and reagents were of analytical reagent grade and used without further
purification as described in Chapter 2 (§2.2.1) and Chapter 3 (§3.2.1). Tin(II) dichloride,
97% (Merck, New York, NY, USA) and triethylamine, > 99% (Sigma Aldrich, St. Louis,
RS N
SN
OR'
RSN
SN
OR' Sn
Where: R = o-CH3, R' = O-CH3 (19) R = p-CH3, R' = O-CH3 (20) R = H, R' = O-CH3 (21) R = o-CH3, R' = OH (22) R = p-CH3, R' = OH (23) R = H, R' = O-H (24)

138
MO, USA). The instrumentations used are as outlined in Chapter 2 (§2.2.3) and Chapter
3 (§3.2.3).
5.2.2 Synthesis
5.2.2.1 Synthesis of Schiff Bases
Schiff bases were synthesised following references 177,178. The details of this
synthesis procedure are described in Chapter 2 (§2.2.2).)
5.2.2.2 Synthesis of Tin(IV) Compounds (19-24)
The Schiff base (0.35 g (1, 2); 0.33 g (3, 10, 11); 0.32 g (12), 1 mmol) was dissolved
in absolute ethanol (50 cm3) and dichloromethane (20 cm3) and was mixed with an
ethanolic solution (2 mmol) of triethylamine. With continuous stirring, an ethanolic
solution of SnCl2 (0.20 g, 1 mmol) was added to the mixture. A cloudy solution formed
which was filtered. The yellow filtrate was then heated under reflux for ca. 6 h. The
mixture was left overnight and the reduction of the volume of the reaction mixture
resulted in an orange precipitate, which was filtered off. The precipitate was recrystallised
in dry ether to remove the remaining triethylamine hydrochloride salt from the mixture.
2.3.2.1. Tin(IV) [S-2-methybenzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene)
dithiocarbazate] (19)
Orange solid. Yield: 82 %. M.p.: >300 °C. Analysis calculated for
C34H32N4O4S4Sn: C, 50.56; H, 3.99; N, 6.94%. Found: C, 50.87; H, 3.58; N, 5.27%. FT-
IR (ATR, cm-1): 1587, v(C=N); 1088, v(N-N); 955, v(C-S). 1H NMR (CDCl3) δ (ppm.):
8.81, 8.44 (s, 2H, CH), 6.77-7.33 (multiplet, 14H, Ar-H), 4.47, 4.33 (s, 4H, CH2), 3.84,
3.57 (s, 3H, O-CH3), 2.40, 2.36 (s, 6H, CH3); 13C NMR (CDCl3) δ (ppm.): 177.2 (C-S),

139
171.0 (C=N); 115.1-171.0 (aromatic-C), 57.0, 55.1 (O-CH3), 19.4, 19.4 (CH3), 36.3, 33.9
(CH2).119Sn NMR (CDCl3) δ (ppm.): -446.3.
2.3.2.2. Tin(IV) [S-4-methybenzyl-β-N-(2-hydroxy-3-methoxybenzyl methylene)
dithiocarbazate] (20)
Orange crystals. Yield: 87 %. M.p.: 211-214 °C. Analysis calculated for
C34H32N4O4S4Sn: C, 51.56; H, 3.99; N, 6.94%. Found: C, 51.89; H, 4.22; N, 6.53%. FT-
IR (ATR, cm-1): 1593, v(C=N); 1083, v(N-N); 958, v(C=S). 1H NMR (CDCl3) δ (ppm.):
8.78, (s, 2H, CH), 6.76-7.36 (multiplet, 14H, Ar-H), 4.45 (s, 4H, CH2), 3.56 (s,3H, O-
CH3), 1.61 (s, 6H, CH3); 13C NMR (CDCl3) δ (ppm.): 170.8 (C-S), 165.4 (C=N); 117.2-
156.7 (aromatic-C), 56.9 (O-CH3), 21.2 (CH3), 35.5 (CH2).119Sn NMR (CDCl3) δ (ppm.):
-446.2.
2.3.2.3. Tin(IV) [S-benzyl-β-N-(2-hydroxy-3-methoxybenzylmethylene)
dithiocarbazate] (21)
Orange crystals. Yield: 53 %. M.p.: >300 °C. Analysis calculated for
C32H28N4O4S4Sn: C, 49.30; H, 3.62; N, 7.19%. Found: C, 49.26; H, 3.26; N, 6.02%. FT-
IR (ATR, cm-1): 1582, v(C=N); 1031, v(N-N); 958, v(C-S). 1H NMR (CDCl3) δ (ppm.):
8.78 (s, 2H, CH), 6.76-7.36 (multiplet, 16H, Ar-H), 4.45 (s, 2H, CH2), 3.56 (s, 3H, O-
CH3); 13C NMR (CDCl3) δ (ppm.): 170.7 (C-S), 165.5 (C=N); 117.2-156.7 (aromatic-C),
56.9 (CH3), 35.7 (CH2).119Sn NMR (CDCl3) δ (ppm.): -447.1.

140
2.3.2.4. Tin(IV)[S-2-methybenzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate] (22)
Orange solid. Yield: 40 %. M.p.: 188-192 °C. Analysis calculated for
C32H28N4O4S4Sn: C, 49.30; H, 3.62; N, 7.19%. Found: C, 49.14; H, 3.43; N, 7.50%. FT-
IR (ATR, cm-1): 1610, v(C=N); 1035, v(N-N); 964, v(C-S). 1H NMR (CDCl3) δ (ppm.):
8.90 (s, 2H, CH), 6.02-7.34 (multiplet, 14H, Ar-H), 4.48 (s, 4H, CH2), 2.42 (s, 6H, CH3);
13C NMR (CDCl3) δ (ppm.): 171.3 (C-S), 165.6 (C=N), 115.1, 119.0, 119.6, 125.4, 126.5,
128.3, 130.5, 130.8, 133.2, 137.4, 148.2, 152.1 (aromatic-C), 34.3 (CH2), 19.5
(CH3).119Sn NMR (DMSO-d6) δ (ppm.): -440.9.
2.3.2.5. Tin(IV) [S-4-methybenzyl-β-N-(2,3-dihydroxybenzylmethylene)
dithiocarbazate] (23)
Orange solid. Yield: 83 %. M.p.: 206-209°C. Analysis calculated for
C32H28N4O4S4Sn: C, 49.30; H, 3.62; N, 7.19%. Found: C, 49.02; H, 3.16; N, 7.85%. FT-
IR (ATR, cm-1): 1620, v(C=N); 1006, v(N-N); 965, v(C-S). 1H NMR (DMSO-d6) δ
(ppm.): 8.65, (s, 2H, CH), 6.50-7.35 (multiplet, 14H, Ar-H), 4.41 (s, 4H, CH2), 2.29 (s,
6H, CH3); 13C NMR (DMSO-d6) δ (ppm.): 150.8 (C-S), 145.2 (C=N); 118.4, 129.5, 129.6,
134.2, 136.8 (aromatic-C), 21.2 (CH3), 37.8 (CH2).119Sn NMR (DMSO-d6) δ (ppm.): -
443.1.
2.3.2.6. Tin(IV) [S-benzyl-β-N-(2,3-dihydroxy-benzylmethylene)
dithiocarbazate] (24)
Orange solid. Yield: 67 %. M.p.: 206-208 °C. Analysis calculated for
C30H24N4O4S4Sn: C, 47.95; H, 3.22; N, 6.46%. Found: C, 47.72; H, 2.93; N, 6.25 %. FT-
IR (ATR, cm-1): 1612, v(C=N); 1016, v(N-N); 960, v(C-S). 1H NMR (DMSO-d6) δ

141
(ppm.): 8.60, (s, 2H, CH), 6.27-7.41 (multiplet, 16H, Ar-H), 4.44 (s, 4H, CH2); 13C NMR
(DMSO-d6) δ (ppm.): 155.9 (C-S), 153.4 (C=N); 111.6, 114.3, 115.9, 116.9, 127.5, 128.9,
129.7, 137.6, 146.9 (aromatic-C), 38.1 (CH2).119Sn NMR (DMSO-d6) δ (ppm.): -461.9.
5.2.3 Single Crystal X-ray Structure Determination
An Oxford Diffraction Gemini Eos CCD diffractometer fitted with Mo Kα radiation
source (λ = 0.71073 Å) was employed to measure intensity data for the orange crystals of
20 and 21 at T = 150 K. The data reduction and analytical absorption corrections were
accomplished with CrysAlisPro180. The structures were solved by direct-methods181 and
refined (anisotropic displacement parameters, C-bound H atoms in the riding model
approximation) on F2 182. A weighting scheme w = 1/[σ 2(Fo2) + (aP)2 + bP] where P =
(Fo2 + 2Fc2)/3) was introduced in each case. In the final cycles of the refinement of 21, a
reflection, i.e. (-6 -4 8), was omitted owing to poor agreement. The molecular structure
diagrams were generated with ORTEP for Windows183 with 50% displacement ellipsoids
and the packing diagrams were drawn with DIAMOND184. Additional data analysis was
made with PLATON216. Crystal data and refinement details are given in Table 5.1.
5.2.4 DFT Calculations and Molecular Docking Studies
DFT and molecular docking simulations were performed using parameters detailed
in Chapter 2 (§2.2.6) and Chapter 3 (§3.2.5.1), respectively. The coordinates of 20 and
21 were taken from their respective crystal structures, whereas the coordinates for 19, 22,
23 and 24 were obtained after DFT optimisation. The parameters for molecular docking
simulations are described in Chapter 3 (§3.2.5.2).

142
Table 5.1: Crystal data and refinement details for complexes 20 and 21. Complex 20 21 Formula C34H32N4O4S4Sn C32H28N4O4S4Sn Formula weight 807.56 779.51 Crystal system Monoclinic Monoclinic Space group P21/n P21/c a/Å 12.8540(2) 12.8656(3) b/Å 19.4686(5) 8.4693(2) c/Å 27.2466(6) 29.8639(7) β/° 101.109(2) 96.634(2) V/Å3 6690.7(3) 3232.26(13) Z 8 4 Dc/g cm-3 1.603 1.602 F(000) 3280 1576 µ(MoKα)/mm-1 1.059 1.093 Measured data 44247 36728
range/° 3.3 – 25.2 3.4 – 25.2 Unique data 15972 7934 Observed data (I 2.0σ(I)) 10737 6324 No. parameters 855 408 R, obs. data; all data 0.041; 0.079 0.035; 0.072 a; b in weighting scheme 0.036; 1.389 0.032; 2.061 Rw, obs. data; all data 0.076; 0.093 0.052; 0.080 GoF 1.05 1.02 Range of residual electron density peaks/eÅ-3 -0.75 – 0.91 -0.57 – 0.50
5.2.5 MTT Assays
The protocol used for MTT assay followed that outlined in Chapter 2 (§2.2.6).
5.3 Results and Discussion
5.3.1 Synthesis
The six new tin(IV) compounds were synthesised by the condensation reaction of
S-substituted dithiocarbazate Schiff bases of 2-hydroxy-3-methoxybenzaldehyde or 2,3-
dihydroxybenzaldehyde with tin(II) chloride (Scheme 5.1), whereby the tin(II) precursor
underwent oxidation to tin(IV) after reflux step for 6 hours.159 The tin(IV) compounds
were stable at room temperature and soluble in most organic solvents, especially
dimethylsulfoxide (DMSO) and dimethylformamide (DMF). However, 20 was not
143
soluble in DMSO; consequently, cytotoxic activity of this compound was not determined.
The molar conductance of 10-3 M solutions of synthesised compounds in DMSO were in
the range 1.12-6.07 Ω-1 cm2 mol-1, indicating their non-electrolytic nature.106
Scheme 5.1: Synthetic pathway for the synthesis of 19-24.
5.3.2 IR Spectral Analysis
Complete experimental and calculated IR data are provided in Appendix, Table
A5.1. The FT-IR spectra of 19-24 were measured between 4000-280 cm−1 and verified
using DFT calculations. All DFT vibrational frequencies were scaled using a scaling
factor of 0.9682.189 For the uncomplexed Schiff bases, v(O-H), v(N-H), v(C=N), v(N-N)
and v(C=S) bands are observed at 3488-3501, 3084-3106, 1598-1608, 1114-1125 and
1012-1030 cm-1, respectively. However, v(O-H) of 1, 2 and 3 were not observed in the IR
spectra due to the intra- and inter-molecular interactions between the molecules, which
were reported in Chapter 2.178 The disappearance of the v(N-H) band for all Schiff bases
and the v(O-H) band for 10, 11 and 12 indicated the deprotonation of N-H and O-H groups
and its subsequent coordination to the central tin atom. The shifting of the v(C=N) band
RS N
H
SN
HOR'
RS N
SN
OR'
RSN
SN
OR' Sn
Et3N, EtOHreflux (6 hours)
Where: R = o-CH3, R' = O-CH3 (19) R = p-CH3, R' = O-CH3 (20) R = H, R' = O-CH3 (21) R = o-CH3, R' = OH (22) R = p-CH3, R' = OH (23) R = H, R' = O-H (24)
SnCl2

144
observed in the spectra of tin(IV) compounds further proves that the complexation
occurred through the azomethine nitrogen. The IR group assignments for the
experimental and theoretical calculation in gas phase appeared to be in a good agreement
with experimental data.
5.3.3 NMR Spectroscopic Analysis
All 1H NMR and 13C NMR spectra are tabulated in Appendix Tables A5.2 and A5.3,
respectively. The important signals observed in the range of 9.51-9.61 ppm and 13.32-
13.41 ppm were assigned to the OH and -NH protons in the Schiff base, respectively, as
reported previously in Chapter 2 and 4. Both of the signals disappeared in the 1H NMR
of the tin(IV) compounds indicating the deprotonation of the -OH and -NH groups upon
complexation. This confirmed that the coordination of the tin ion occurs via the oxygen
and nitrogen donor atom of the Schiff bases. Moreover, the signal of the HC=N proton
shifted to the downfield region due to the coordination of –NH with the tin centre, which
was a result of the formation of the C=N-N=C conjugated systems.66 The downfield shift
of the HC=N signal in the 13C NMR spectra, due to the electron density transfer from the
Schiff bases and the acceptor (tin atom), was consistent with that observed in earlier
reports.268,269 Furthermore, the signal attributable to the C-S moiety in the 13C NMR
spectra of the tin(IV) compounds was shifted upfield compared to their Schiff bases,
suggesting coordination of the sulphur atom to the tin centre. These observations
supported the FTIR analysis discussed above. 119Sn NMR spectroscopy provided further
validation of the geometries of 19-24. A sharp signal was observed in the range δ -441 to
-462 in the 119Sn NMR spectra of compounds 19-24, which strongly supported a six-
coordinated, distorted octahedral geometry around tin, comparable to reported
literature.100

145
5.3.4 UV-vis Absorption Spectral Analysis
Complete experimental and calculated UV-vis data are listed in Table A5.4.
Frontier MOs of 19-24 are presented in Figure 5.2. The experimental UV-vis spectra of
tin(IV) compounds showed a prominent absorption peak at 343-355 nm, in excellent
correlation with DFT calculations (334-348 nm, gas phase). The other experimental
absorption peak was observed at 415-427 nm, which indicated the presence of the
S→SnIV LMCT band. This was also verified by the excitations between 428-438 nm
observed from DFT calculations. The HOMOs in these compounds were primarily
located on the dithiocarbazate backbones, the phenyl ring of the aldehyde moieties and
the coordinating oxygen atoms. The LUMOs centred on the dithiocarbazate backbones,
and the phenyl rings of oVa or catechol. It is evident then that the UV-vis absorption
corresponds to a n→π* excitation, due to the presence of the electron lone pairs in the
azomethine nitrogen, thiolate sulphur and phenoxide oxygen atoms. The π→π*
transitions observed in all tin(IV) compounds corresponded to the electron delocalisation
around the aromatic rings.
5.3.5 X-ray Crystallography
Crystal structure determinations were achieved for the homoleptic compounds 20
and 21; selected geometric parameters are collected in Table 5.2. The crystallographic
asymmetric unit of 20 comprises two independent molecules, with the first shown in
Figure 5.3(a) and the second molecule shown in Appendix, Figure A5.1. For 21, one
molecule comprises the asymmetric unit, Figure 5.3(b). For the first independent
molecule of 20, the dinegative tridentate ligand coordinates the tin atom via thiolate-S,
phenoxide-O and imine-N atoms to establish five-membered Sn,S,C,N2 and six-
membered Sn,O,C3,N rings. Evidence for the presence of thiolate-sulphur atoms is found

146
in the elongation of the C1−S1 and C18−S3 bond lengths (Table 5.2) from 1.670(2) Å,
which is found in the most closely related acid molecule for which a crystallographic
analysis has been reported, i.e. the benzyl ester, rather than the 4-tolylCH2 ester.177
Concomitantly, the C1−N1 and C18−N3 bond lengths (Table 5.2) have also decreased
considerably from 1.338(2) Å in the acid.177 The conformation of the two tridentate
ligands is such that all like atoms are mutually trans in a distorted octahedral environment.
The major distortions from the ideal octahedral geometries are due to the acute chelate
angles in the five-membered rings, i.e. 78.88(7)° for S1−Sn−N2 and 78.94(7)° for the
S3−Sn−N4 angle.
Figure 5.2: Frontier MOs of (a) 19, (b) 20, (c) 21, (d) 22, (e) 23 and (f) 24.

147
The S1-five-membered chelate ring formed by the tridentate ligand is strictly planar
with the RMSD of the fitted atoms being 0.0036 Å. However, the S3-ring is less planar
with the RMSD being 0.0629 Å. A better description for the latter is an envelope in which
the tin atom lies 0.308(4) Å out of the least-squares plane defined by the remaining four
atoms (RMSD = 0.0060 Å). Envelope conformations also apply for the six-membered
chelate rings. For the O1-ring, the tin atom lies 0.579(4) Å out the plane through the five
remaining atoms. For the O3-ring the envelope is somewhat flattened with the tin atom
0.135(4) Å out of plane (RMSD = 0.0234 Å). The dihedral angle between the five-
membered chelate rings is 81.56(8)°.
Table 5.2: Selected geometric parameters (Å, °) for 20 and 21. Complex 20 20a 21 (x = 18, y = 19) (x = 18, y = 19) (x = 17, y = 18) Parameter Sn‒S1 2.4828(8) 2.4871(9) 2.4932(7) Sn‒S3 2.4961(9) 2.4898(8) 2.4873(7) Sn‒O1 2.025(2) 2.026(2) 2.0304(18) Sn‒O3 2.032(2) 2.032(2) 2.0242(18) Sn‒N2 2.198(2) 2.189(2) 2.169(2) Sn‒N4 2.189(2) 2.196(2) 2.173(2) N1‒N2 1.398(3) 1.399(3) 1.395(3) C1‒S1 1.753(3) 1.749(3) 1.744(3) C1‒S2 1.744(3) 1.743(3) 1.741(3) C1‒N1 1.288(4) 1.286(4) 1.289(3) C2‒N2 1.299(4) 1.309(4) 1.302(3) N3‒N4 1.393(3) 1.396(3) 1.397(3) C(x)‒S3 1.744(3) 1.754(3) 1.745(3) C(x)‒S4 1.743(3) 1.745(3) 1.751(2) C(x)‒N3 1.295(4) 1.289(4) 1.288(3) C(y)‒N4 1.307(4) 1.303(4) 1.304(3) S1‒Sn‒O1 162.46(6) 165.07(6) 164.51(5) S3‒Sn‒O3 164.42(6) 162.30(6) 165.46(5) N2‒Sn‒N4 170.18(10) 170.45(9) 177.76(8)
As seen from the overlay diagram in Figure 5.3(c), there are conformational
differences between the independent molecules of 20, at least with respect to the terminal
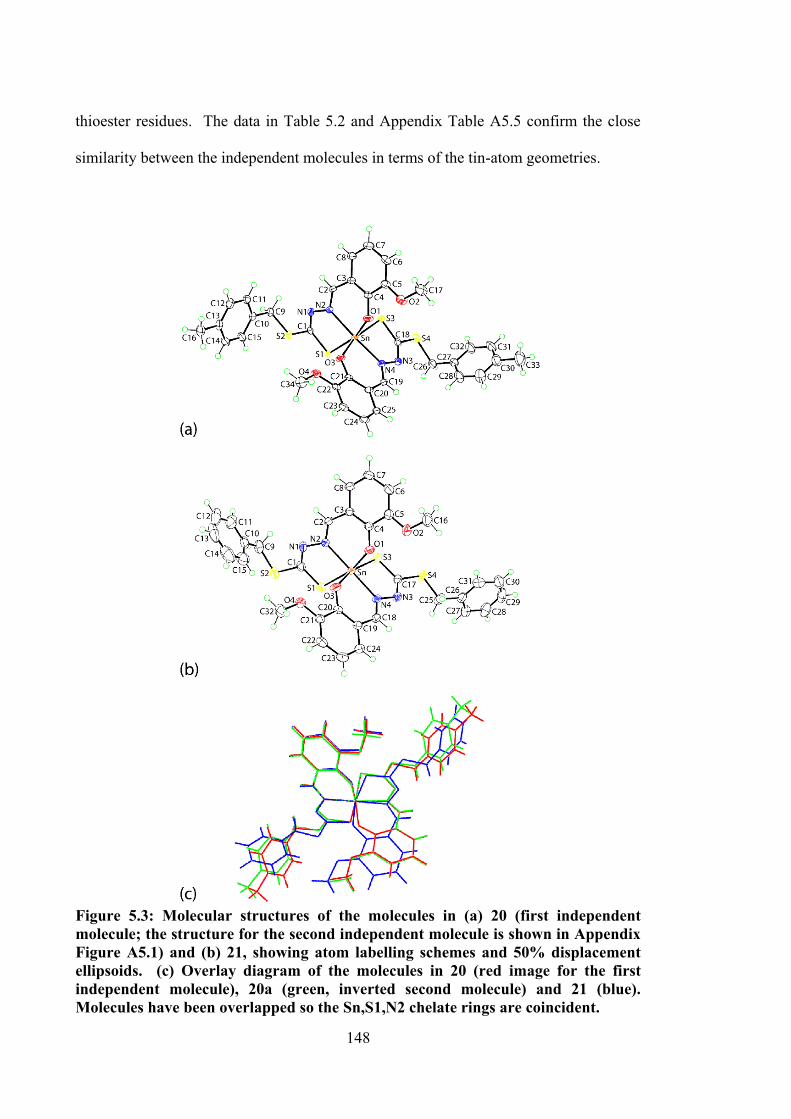
148
thioester residues. The data in Table 5.2 and Appendix Table A5.5 confirm the close
similarity between the independent molecules in terms of the tin-atom geometries.
Figure 5.3: Molecular structures of the molecules in (a) 20 (first independent molecule; the structure for the second independent molecule is shown in Appendix Figure A5.1) and (b) 21, showing atom labelling schemes and 50% displacement ellipsoids. (c) Overlay diagram of the molecules in 20 (red image for the first independent molecule), 20a (green, inverted second molecule) and 21 (blue). Molecules have been overlapped so the Sn,S1,N2 chelate rings are coincident.

149
The trends in the structure of 21 follow those established for 20, with the most
obvious difference relating to the coordination of the imine-N2 and N4 atoms. The angle
they subtend at the tin atom is wider by ca. 6-7°, and there is evidence to suggest the Sn–
N2, N4 bond lengths are marginally shorter than the equivalent bonds in the molecules of
20. Each of the chelate rings adopts an envelope conformation with data presented in
Appendix Table A5.5. The dihedral angle between the five-membered chelate rings in
21 is 81.77(5)°, i.e. within experimental error of the value computed for 20.
There are three related homoleptic tin(IV) compounds that have been structurally
characterised in the literature, namely the ethyl thioester and unsubstituted phenoxide
residue (Figure 5.4(a)),118 benzyl thioester, unsubstituted phenoxide residue and methyl
bound to the imine-carbon(Figure 5.4(b))270 and benzyl thioester with a ferrocenyl
substituent adjacent to the alkoxide-oxygen atom (Figure 5.4(c)).267 The contrasting and
curious feature of the literature structures is that the thiolate-sulphur atoms are mutually
cis, as are the alkoxide-oxygen atoms. The reasons for the different conformations
observed in 20 and 21, and those in the literature remain unclear.
In the absence of conventional hydrogen bonding, the crystals of 20 and 21 are
sustained by a variety of other non-covalent interactions; the geometric parameters
characterising these are included in the captions to the respective figures in the Appendix
Tables A5.6 and A5.7. In the molecular packing of 20, each of the independent molecules
form equivalent intermolecular contacts with the other independent molecule to sustain a
supramolecular layer in the ab-plane. These are imine-C2-H…S3(thiolate), imine-C19-
H…S1(thiolate), methoxybenzene-C7-H…O2(methoxy), methoxybenzene-C24-
H…O4(methoxy) and π(C3-C8)…π(C20-C25). The connections between layers along the
c-axis direction include methyl-C16a-H…O2a, methyl-C16-H…π(C10-C15) and
methoxy-C34-H…π(C10-C15) interactions. These occur between like-molecules and
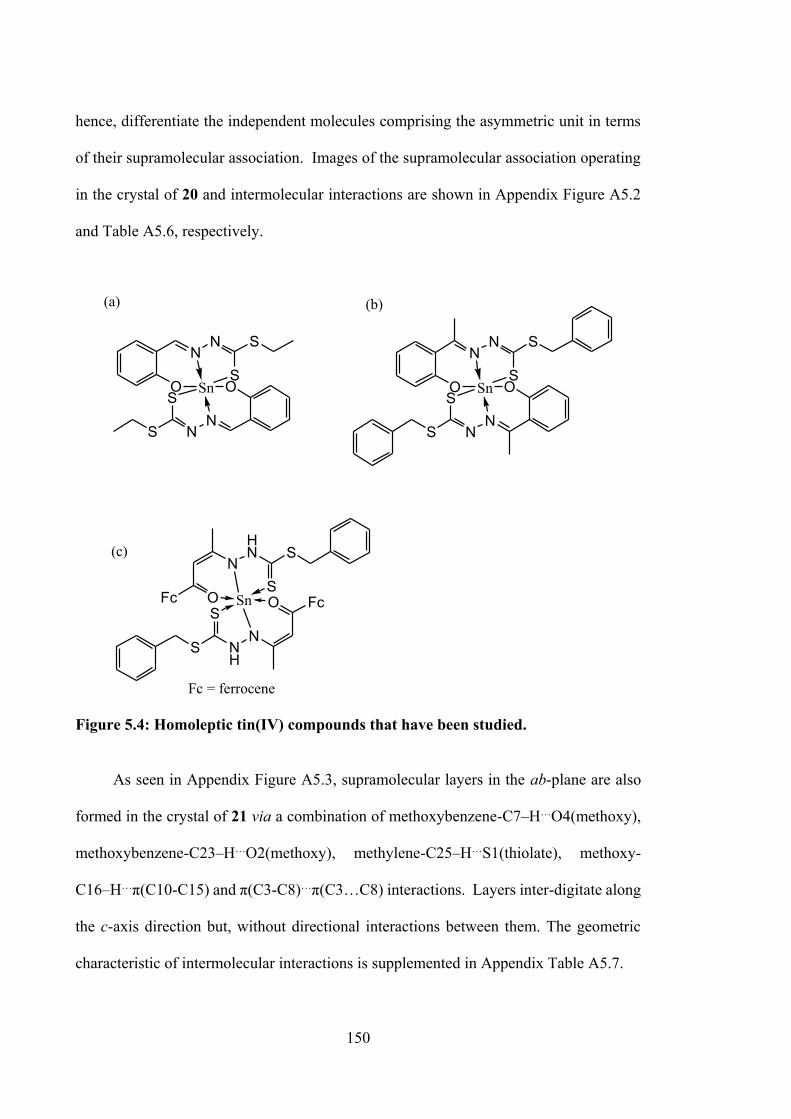
150
hence, differentiate the independent molecules comprising the asymmetric unit in terms
of their supramolecular association. Images of the supramolecular association operating
in the crystal of 20 and intermolecular interactions are shown in Appendix Figure A5.2
and Table A5.6, respectively.
S NN
SO
SNN
SO Sn
S NN
SO
SNN
SO Sn
S NH
NS
Sn O Fc
SHN
NS
OFc
Fc = ferrocene
(a) (b)
(c)
Figure 5.4: Homoleptic tin(IV) compounds that have been studied.
As seen in Appendix Figure A5.3, supramolecular layers in the ab-plane are also
formed in the crystal of 21 via a combination of methoxybenzene-C7–H…O4(methoxy),
methoxybenzene-C23–H…O2(methoxy), methylene-C25–H…S1(thiolate), methoxy-
C16–H…π(C10-C15) and π(C3-C8)…π(C3…C8) interactions. Layers inter-digitate along
the c-axis direction but, without directional interactions between them. The geometric
characteristic of intermolecular interactions is supplemented in Appendix Table A5.7.

151
5.3.6 In Vitro Cytotoxic Activity
The cytotoxic assays of 19, 21, 22, 23 and 24 were carried out against a panel ten
of cancer cells (HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C, MIA)
and one normal breast cell line (MCF-10A). Cytotoxicity for 20 could not be studied due
to its insolubility in 100% DMSO at 1 mM. Cisplatin was used as a positive control and
DMSO was used as a negative control to monitor the experiments. Table 5.3 shows that
the cytotoxicity of these tin(IV) compounds and their respective Schiff bases, 19, 21, 22,
23 and 24 exhibited moderate activity against all the tested cancer cell lines. The
cytotoxicities of these compounds against the HT29, MCF7 and MIA cell lines were
higher than cisplatin. Taking the Schiff bases into account, 19-24 were equipotent to their
corresponding Schiff bases, with the exception of 22. Compound 22 demonstrated higher
potency than their corresponding Schiff bases against the HT29, U87, MCF-7, A431 and
Du145 cell lines. The mechanism of action of 22 needs further in-depth investigation. In
general, for compounds 19, 20, 21, 23 and 24, it can be inferred that the presence of tin
does not influence cytotoxicity. This is possibly due to the bulkiness of the compound
and the possibility that only the coordinated Schiff bases playing a role in binding to the
biomacromolecules.271
5.3.7 Molecular Docking Analysis
Molecular docking analyses of compounds 19, 21, 22, 23 and 24 with the DNA
duplex sequence were performed, and the lowest binding energy conformation of the
docked structure are summarised in Table 5.4. From the docked structures obtained, it
was clear that 19, 21, 22, 23 and 24 considered here were fitted with the dimensions of
the grooves of the DNA duplex sequence. From the docked structures obtained here, it
was proposed that compounds 19-24 fit well into the cytosine and guanine region in the

152
minor groove of the targeted DNA (Figure 5.4(a)) with a relative binding energy of -35.82
(1), -37.15 (2), -33.56 (3), -31.42 (4), -32.07 (5) and -31.67 (6) kJ mol-1. These compounds
were predicted to interact with the DNA strand, suggesting that the planar aromatic ring
of the Schiff bases interacts with the nitrogenous bases of DNA via π-π stacking
interactions. These interactions are stabilised by other interactions, such as van der Waals,
hydrogen bonding, hydrophobic and electrostatic interactions. These interactions of the
energetically favourable conformation of the docked model are visualised in Figure
5.5(b). Is it worth noting that these interactions are consistent with the fact that the
cytotoxicity is the result of the Schiff base, and not necessarily the tin.
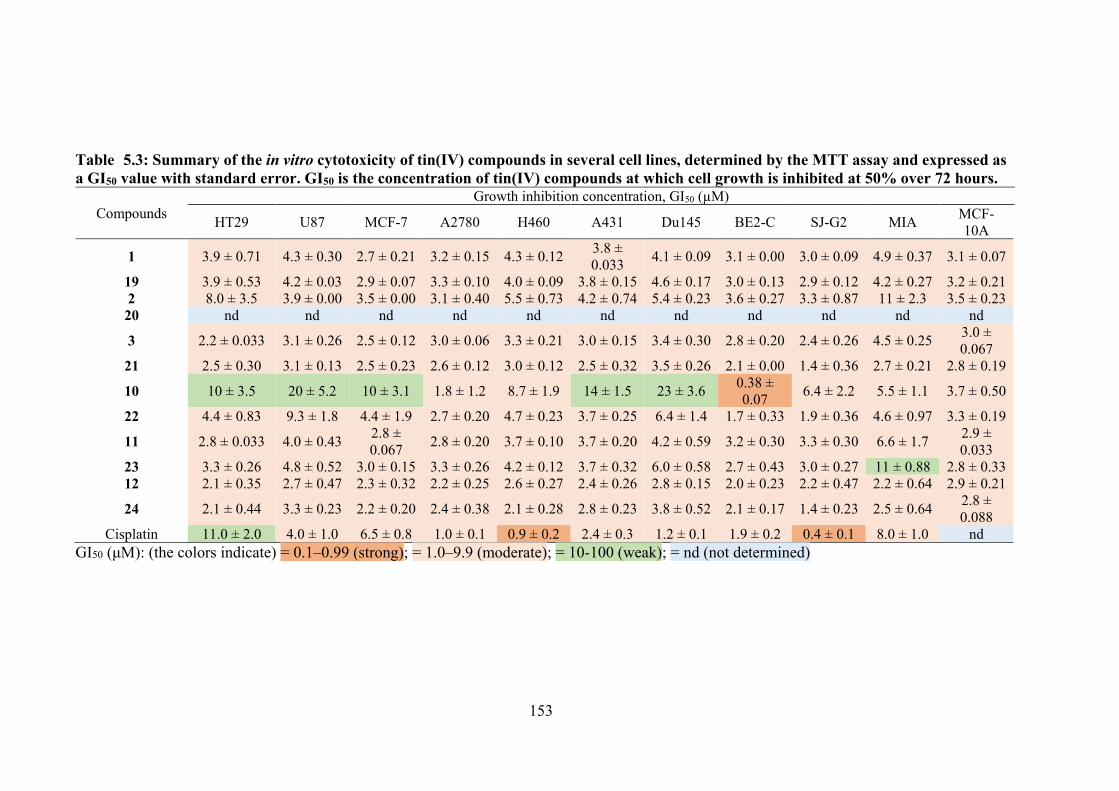
153
Table 5.3: Summary of the in vitro cytotoxicity of tin(IV) compounds in several cell lines, determined by the MTT assay and expressed as a GI50 value with standard error. GI50 is the concentration of tin(IV) compounds at which cell growth is inhibited at 50% over 72 hours.
Compounds Growth inhibition concentration, GI50 (µM)
HT29 U87 MCF-7 A2780 H460 A431 Du145 BE2-C SJ-G2 MIA MCF-10A
1 3.9 ± 0.71 4.3 ± 0.30 2.7 ± 0.21 3.2 ± 0.15 4.3 ± 0.12 3.8 ± 0.033 4.1 ± 0.09 3.1 ± 0.00 3.0 ± 0.09 4.9 ± 0.37 3.1 ± 0.07
19 3.9 ± 0.53 4.2 ± 0.03 2.9 ± 0.07 3.3 ± 0.10 4.0 ± 0.09 3.8 ± 0.15 4.6 ± 0.17 3.0 ± 0.13 2.9 ± 0.12 4.2 ± 0.27 3.2 ± 0.21 2 8.0 ± 3.5 3.9 ± 0.00 3.5 ± 0.00 3.1 ± 0.40 5.5 ± 0.73 4.2 ± 0.74 5.4 ± 0.23 3.6 ± 0.27 3.3 ± 0.87 11 ± 2.3 3.5 ± 0.23 20 nd nd nd nd nd nd nd nd nd nd nd
3 2.2 ± 0.033 3.1 ± 0.26 2.5 ± 0.12 3.0 ± 0.06 3.3 ± 0.21 3.0 ± 0.15 3.4 ± 0.30 2.8 ± 0.20 2.4 ± 0.26 4.5 ± 0.25 3.0 ± 0.067
21 2.5 ± 0.30 3.1 ± 0.13 2.5 ± 0.23 2.6 ± 0.12 3.0 ± 0.12 2.5 ± 0.32 3.5 ± 0.26 2.1 ± 0.00 1.4 ± 0.36 2.7 ± 0.21 2.8 ± 0.19
10 10 ± 3.5 20 ± 5.2 10 ± 3.1 1.8 ± 1.2 8.7 ± 1.9 14 ± 1.5 23 ± 3.6 0.38 ± 0.07 6.4 ± 2.2 5.5 ± 1.1 3.7 ± 0.50
22 4.4 ± 0.83 9.3 ± 1.8 4.4 ± 1.9 2.7 ± 0.20 4.7 ± 0.23 3.7 ± 0.25 6.4 ± 1.4 1.7 ± 0.33 1.9 ± 0.36 4.6 ± 0.97 3.3 ± 0.19
11 2.8 ± 0.033 4.0 ± 0.43 2.8 ± 0.067 2.8 ± 0.20 3.7 ± 0.10 3.7 ± 0.20 4.2 ± 0.59 3.2 ± 0.30 3.3 ± 0.30 6.6 ± 1.7 2.9 ±
0.033 23 3.3 ± 0.26 4.8 ± 0.52 3.0 ± 0.15 3.3 ± 0.26 4.2 ± 0.12 3.7 ± 0.32 6.0 ± 0.58 2.7 ± 0.43 3.0 ± 0.27 11 ± 0.88 2.8 ± 0.33 12 2.1 ± 0.35 2.7 ± 0.47 2.3 ± 0.32 2.2 ± 0.25 2.6 ± 0.27 2.4 ± 0.26 2.8 ± 0.15 2.0 ± 0.23 2.2 ± 0.47 2.2 ± 0.64 2.9 ± 0.21
24 2.1 ± 0.44 3.3 ± 0.23 2.2 ± 0.20 2.4 ± 0.38 2.1 ± 0.28 2.8 ± 0.23 3.8 ± 0.52 2.1 ± 0.17 1.4 ± 0.23 2.5 ± 0.64 2.8 ± 0.088
Cisplatin 11.0 ± 2.0 4.0 ± 1.0 6.5 ± 0.8 1.0 ± 0.1 0.9 ± 0.2 2.4 ± 0.3 1.2 ± 0.1 1.9 ± 0.2 0.4 ± 0.1 8.0 ± 1.0 nd GI50 (μM): (the colors indicate) = 0.1–0.99 (strong); = 1.0–9.9 (moderate); = 10-100 (weak); = nd (not determined)
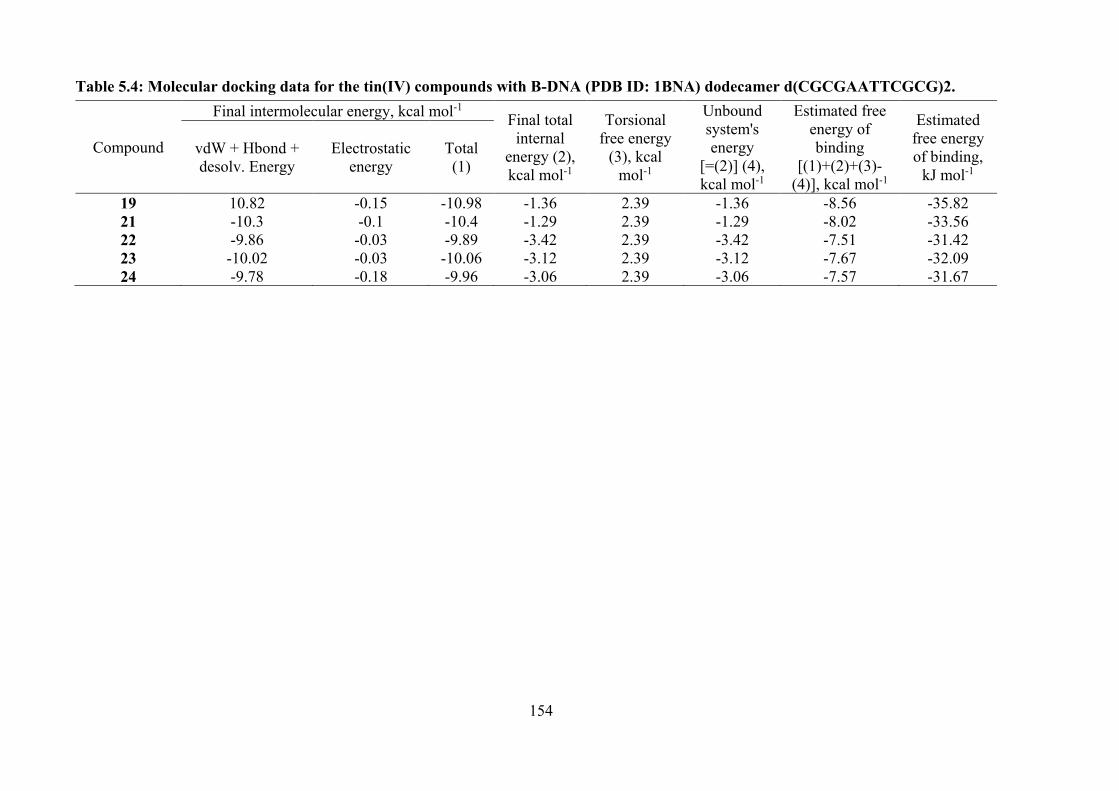
154
Table 5.4: Molecular docking data for the tin(IV) compounds with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
Compound
Final intermolecular energy, kcal mol-1 Final total internal
energy (2), kcal mol-1
Torsional free energy
(3), kcal mol-1
Unbound system's energy
[=(2)] (4), kcal mol-1
Estimated free energy of binding
[(1)+(2)+(3)-(4)], kcal mol-1
Estimated free energy of binding,
kJ mol-1
vdW + Hbond + desolv. Energy
Electrostatic energy
Total (1)
19 10.82 -0.15 -10.98 -1.36 2.39 -1.36 -8.56 -35.82 21 -10.3 -0.1 -10.4 -1.29 2.39 -1.29 -8.02 -33.56 22 -9.86 -0.03 -9.89 -3.42 2.39 -3.42 -7.51 -31.42 23 -10.02 -0.03 -10.06 -3.12 2.39 -3.12 -7.67 -32.09 24 -9.78 -0.18 -9.96 -3.06 2.39 -3.06 -7.57 -31.67
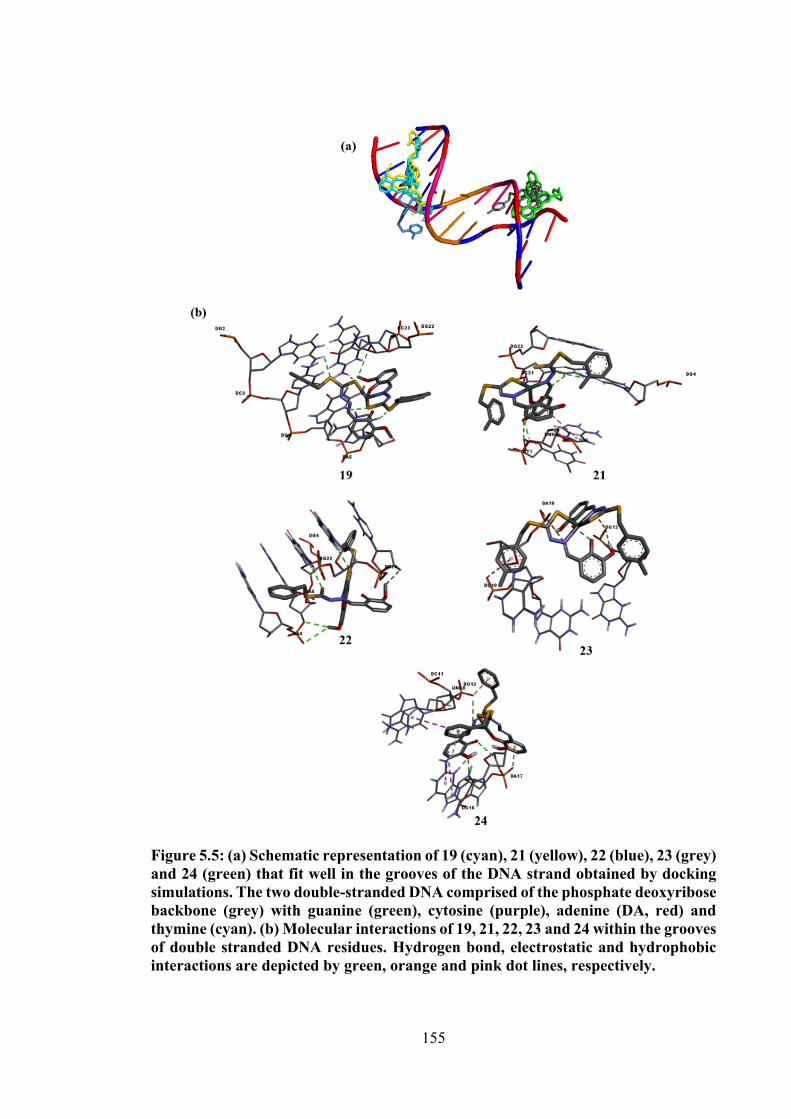
155
Figure 5.5: (a) Schematic representation of 19 (cyan), 21 (yellow), 22 (blue), 23 (grey) and 24 (green) that fit well in the grooves of the DNA strand obtained by docking simulations. The two double-stranded DNA comprised of the phosphate deoxyribose backbone (grey) with guanine (green), cytosine (purple), adenine (DA, red) and thymine (cyan). (b) Molecular interactions of 19, 21, 22, 23 and 24 within the grooves of double stranded DNA residues. Hydrogen bond, electrostatic and hydrophobic interactions are depicted by green, orange and pink dot lines, respectively.

156
5.4 Conclusions
In summary, six octahedral tin(IV) compounds were synthesised by the
condensation of tin(II) chloride with dithiocarbazate Schiff bases (1, 2, 3, 10, 11 and 12).
The oxidation of tin(II) to tin(IV) occurred as two dinegatively charge tridentate ONS
Schiff bases coordinated the tin centre with general formulae Sn(L)2 (L = 1, 2, 3, 10, 11
and 12). X-ray crystallography indicated that the dinegative tridentate ligands of 20 and
21 coordinated to the tin atoms via the thiolate-S, phenoxide-O and imine-N atoms,
leading to octahedral geometries in which the like-atoms were mutually trans. The in
vitro cytotoxicity against a panel of cancer cell lines viz., HT29, U87, SJ-G2, MCF-7,
A2780, H460, A431, Du145, BE2-C and MIA cancer cell lines revealed that compounds
19, 21, 22, 23 and 24 showed moderate cytotoxicity, similar to that of their respective
Schiff bases. However, compound 22 exhibited a higher potency against HT29, U87,
MCF-7, A431 and Du145 cells as compared to its Schiff base. Molecular docking predicts
that the tin(IV) compounds discussed in this chapter have a higher affinity wards guanine–
cytosine rich regions than the adenine–thymine rich regions of DNA due to their bulky
octahedral structures. The binding interactions of tin(IV) compounds-DNA were
stabilised by hydrogen, electrostatic and hydrophobic interactions.
Chapter 2, 3 and 4 demonstrated the synthesis, structural evaluation and
cytotoxicity studies of dithiocarbazate Schiff bases and their tin(IV) compounds, where
all the compounds showed good cytotoxic activity, particularly diphenyltin(IV)
compounds. From the cytotoxicity results obtained here, it is hypothesised that this
activity could be enhanced by modification of the backbone of dithiocarbazate (see Figure
5.6).

157
S NH
SN
RNH
NH
SN
HOR1
HOR1
R
R = o-CH3; R1 = OCH3 (1)R = p-CH3; R1 = OCH3 (2)R = H; R1 = OCH3 (3)R = o-CH3; R1 = OH (10)R = p-CH3; R1 = OH (11)R = H; R1 = OH (12)
R = CH3; R1 = OCH3 (25)R = C6H3; R1 = OCH3 (26)R = CH3; R1 = OH (27)R = C6H3; R1 = OH (28)
(a) (b)
4
Figure 5.6: The small modification (blue) of backbone of (a) dithiocarbazate Schiff bases to produce (b) thiosemicarbazone Schiff bases.
This hypothesis will be tested in Chapter 6, which reports the synthesis, structural
and cytotoxic studies of thiosemicarbazone Schiff bases (25-28) and their tin(IV)
compounds (29-40). Previous studies reported that the variation of the groups on the N(4)
atom (see Figure 5.6b) of the thiosemicarbazone Schiff bases affected the biological
action.272 This motivated further investigations on the relationship between
thiosemicarbazone Schiff base structures and cytotoxic activity, by changing the
substituents at N(4) position with methyl and phenyl groups, which will be discussed in
Chapter 6.

158
CHAPTER 6
TIN(IV) COMPOUNDS OF THIOSEMICARBAZONE SCHIFF BASES: SYNTHESIS, STRUCTURAL CHARACTERISATION AND IN VITRO
CYTOTOXICITY
6.1 Introduction
Thiosemicarbazones are an important class of compounds which have gained
attention owing to their remarkable biological and pharmacological properties.
Thiosemicarbazones differ from dithiocarbazates by replacing the sulphur atom with a
nitrogen atom attached to C=S (see Figure 6.1). One of the promising uses of
thiosemicarbazones is as anticancer agents. They play a key role in transporting and
directing molecules to the target site.98,119–121 In 1956, 2-formylpyridine
thiosemicarbazone was reported to exhibit a good effect on leukemic cells, but this
compound was found to be highly toxic.273 Almost a decade later, French and Blanz
formulated a hypotheses about the mode of action of tridentate α(N)-heterocyclic
thiosemicarbazones, postulating that the modification of the ring system while retaining
the ligand pattern could improve the cytotoxicity effect and decrease toxicity.274 The main
factors that contributed to the activity were electron densities, substituents and geometry
of the thiosemicarbazones. The anticancer activity of thiosemicarbazones also depend on
the topology of the cancer cells, which renders the selectivity of the target compounds.
Nevertheless, the presence of metal ions, and specifically tin, in this study almost
systematically increased the cytotoxicity and lowered the toxicity. Presently, the main
mechanism of action related to thiosemicarbazones is RR inhibition, which was discussed
in Chapter 1. Furthermore, thiosemicarbazones were also found to inhibit RNA-
dependent DNA polymerase and dihydrofolate reductase.275

159
S NH
SN
RNH
NH
SN
HOR1
HOR1
R
R = o-CH3; R1 = OCH3 (1)R = p-CH3; R1 = OCH3 (2)R = H; R1 = OCH3 (3)R = o-CH3; R1 = OH (10)R = p-CH3; R1 = OH (11)R = H; R1 = OH (12)
R = CH3; R1 = OCH3 (25)R = C6H3; R1 = OCH3 (26)R = CH3; R1 = OH (27)R = C6H3; R1 = OH (28)
(a) (b)
Figure 6.1: The structures of (a) dithiocarbazate and (b) thiosemicarbazone Schiff bases.
While the synthesis of thiosemicarbazone Schiff bases containing 4-methyl-3-
thiosemicarbazide or 4-phenyl-3-thiosemicarbazide with oVa or catechol have been
reported,276–280 the cytotoxic evaluation for these compounds and their tin(IV) compounds
remains largely unexplored. Following the discussion in previous chapters on the design
and synthesis of dithiocarbazate Schiff bases and their tin(IV) compounds, it was
hypothesised that their activity could be enhanced by modification of the dithiocarbazate
backbone. This hypothesis is investigated in this chapter, which presents the synthesis,
structural characterisation and cytotoxic activities of oVa and catechol thiosemicarbazone
Schiff bases and their tin(IV) compounds.
6.2 Experimental
6.2.1 Materials and Physical Measurements
All solvents and reagents were of analytical reagent grade and used without further
purification as described in Chapter 2 (§2.2.1), Chapter 3 (§3.2.1) and Chapter 4 (§4.2.1).
Chemicals: 4-methyl-3-thiosemicarbazide (Sigma Aldrich, St. Louis, MO, USA), 4-

160
phenyl-3-thiosemicarbazide (Sigma Aldrich, St. Louis, MO, USA). The instrumentation
used are as outlined in Chapter 2 (§2.2.3) and Chapter 3 (§3.2.3).
6.2.2 Synthesis
6.2.2.1 Schiff Bases
6.2.2.1.1 2-(2-hydroxy-3-methoxybenzylidene)-N-methylhydrazine
carbothioamide (25) and 2-(2-hydroxy-3-methoxybenzylidene)-N-phenylhydrazine
carbothioamide (26)
Compounds 25 and 26 were prepared according to the procedure described in the
literature,123,278 without reflux. 4-Methylthiosemicarbazide (1.05 g, 10 mmol)/ 4-
phenylthiosemicarbazide (1.67 g, 10 mmol) was dissolved in 40 cm3 of methanol with
stirring and heating over a period of 30 min. o-Vanillin (1.52 g, 10 mmol) in 10 cm3 of
methanol was added to the thiosemicarbazide solution and was stirred at room
temperature for 4 h. Upon cooling, a crystalline product began to separate and the product
was filtered, washed with cold methanol and dried in a desiccator over anhydrous silica
gel.
2-(2-hydroxy-3-methoxybenzylidene)-N-methylhydrazinecarbothioamide (25)
White crystalline solid. Yield: 92 %. Melting point: 242-243°C. Analysis calculated
for C10H13N3O2S: C, 50.19; H, 5.48; N, 17.56. Found: C, 49.93; H, 5.38; N, 17.22 %. FT-
IR (ATR, cm-1): 3337 v(OH), 3304 v(NH), 1610 v(C=N), 1109 v(N-N), 1037 v(C=S). 1H
NMR (DMSO-d6) δ (ppm.): 2.99 (d, 3H, CH3), 3.79 (s, 3H, O-CH3), 6.93-7.54 (multiplet,
3H, Ar-H), 8.37 (s, 1H, CH), 8.39 (q, 1H, C(=S)-NH), 9.18 (s, 1H, OH), 11.42 (s, 1H,
NH-N). 13C NMR (DMSO-d6) δ (ppm.): 30.8 (N-CH3), 56.4 (O-CH3), 113.1, 118.2,
119.2, 121.2, 139.6, 146.7 (6 x aromatic-C), 148.4 (C=N), 177.6 (C=S).

161
2-(2-hydroxy-3-methoxybenzylidene)-N-phenylhydrazinecarbothioamide (26)
White crystalline solid. Yield: 90 %. Melting point: 209-210°C. Analysis calculated
for C15H15N3O2S: C, 59.78; H, 5.02; N, 13.94. Found: C, 59.99; H, 5.15; N, 13.80 %. FT-
IR (ATR, cm-1): 3300 v(NH), 1609 v(C=N), 1103 v(N-N), 908 v(C=S). 1H NMR (DMSO-
d6) δ (ppm.): 3.80 (s, 3H, O-CH3), 6.77-7.69 (multiplet, 6H, Ar-H), 9.26 (s, 1H, CH), 8.50
(s, 1H, C(=S)-NH), 10.02 (s, 1H, OH), 11.78 (s, 1H, NH-N). 13C NMR (DMSO-d6) δ
(ppm.): 56.3 (O-CH3), 113.4, 118.8, 119.5, 121.2, 125.4, 126.4, 128.6, 139.7, 140.4, 146.6
(10 x aromatic-C), 148.6 (C=N), 176.1 (C=S).
6.2.2.1.2 2-(2,3-dihydroxybenzylidene)-N-methylhydrazinecarbothioamide
(27) and 2-(2,3-dihydroxybenzylidene)-N-phenylhydrazine carbothioamide (28)
Compounds 27 and 28 were prepared according to the procedure described in the
literature.280,281 A 25 cm3 ethanolic solution of 2,3-dihydroxybenzaldehyde (1.38 g, 10
mmol) was added to an equimolar ethanolic solution (10 cm3) of 4-methyl-3-
thiosemicarbazide (1.05 g, 10 mmol) / 4-phenyl-3-thiosemicarbazide (1.67 g, 10 mmol).
The mixture was heated (78°C) for 3 hours and the title compound was filtered. The title
compound was then recrystallised from methanol to remove all the impurities and kept in
a desiccator over anhydrous silica gel.
2-(2,3-dihydroxybenzylidene)-N-methylhydrazinecarbothioamide (27)
Pale yellow solid. Yield: 83 %. Melting point: 231-232°C. Analysis calculated for
C9H11N3O2S: C, 47.99; H, 4.92; N, 18.65. Found: C, 46.88; H, 4.85; N, 18.38 %. FT-IR
(ATR, cm-1): 3418 v(OH), 3140 v(NH), 1601 v(C=N), 1112 v(N-N), 1035 v(C=S). 1H
NMR (DMSO-d6) δ (ppm.): 3.00 (d, 3H, CH3), 6.67-7.38 (multiplet, 3H, Ar-H), 9.02 (s,
1H, CH), 8.37, 8.38 (s, 2H, OH), 9.49 (s, 1H, C(=S)-NH), 11.40 (s, 1H, NH-N). 13C NMR

162
(DMSO-d6) δ (ppm.): 31.3 (N-CH3), 116.7, 117.4, 119.4, 121.5, 140.1, 145.6 (6 x
aromatic-C), 146.0 (C=N), 178.0 (C=S).
2-(2,3-dihydroxybenzylidene)-N-phenylhydrazine carbothioamide (28)
Pale yellow solid. Yield: 70 %. Melting point: 215-216°C. Analysis calculated for
C14H13N3O2S: C, 58.52; H, 4.56; N, 14.62. Found: C, 57.61; H, 4.69; N, 14.67 %. FT-IR
(ATR, cm-1): 3443 v(OH), 3129 v(NH), 1597 v(C=N), 1047 v(N-N), 1029 v(C=S). 1H
NMR (DMSO-d6) δ (ppm.): 6.67-7.57 (multiplet, 8H, Ar-H), 8.49 (s, 1H, CH), 8.96, 9.52
(s, 2H, OH), 10.01 (s, 1H, C(=S)-NH), 11.75 (s, 1H, NH-N). 13C NMR (DMSO-d6) δ
(ppm.): 117.1, 117.9, 119.5, 121.3, 125.6, 126.0, 128.5, 139.6, 141.3, 145.9 (12 x
aromatic-C), 146.0 (C=N), 176.0 (C=S).
6.2.2.2 Tin(IV) Compounds derived from 25 and 27
To a solution of 25 (0.24 g, 1 mmol)/ 27 (0.23 g, 1 mmol) in 100 cm3 of methanol,
potassium hydroxide (0.11 g, 2 mmol) was added and the mixture was stirred and heated
for 30 minutes in methanol. 1 mmol of tin salt (Ph2SnCl2 (0.34 g)/ Me2SnCl2 (0.22 g)/
SnCl2 (0.19 g)) was added to the mixture and heated for 2 hours until the solution reduced
by half. The mixture was filtered while hot and then the filtrate was placed in the freezer
until bright yellow solids formed. The solid residue obtained was recrystallised from
methanol.
Diphenyltin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
methylhydrazinecarbothioamide (29)
Bright yellow solid. Yield: 74 %. Melting point: 138-139°C. Analysis calculated
for C22H21N3O2SSn: C, 51.79; H, 4.15; N, 8.24. Found: C, 51.03; H, 4.23; N, 8.16 %. FT-

163
IR (ATR, cm-1): 3299 v(N-H), 1596 v(C=N), 1066 v(N-N), 973 v(C=S). 1H NMR (CDCl3)
δ (ppm.): 3.01 (d, 3H, N-CH3), 3.96 (s, 3H, O-CH3), 7.96 (s, 1H, CH), 7.36-8.12 (m, 13H,
Ar-H), 8.59 (s, 1H, NH). 13C NMR (CDCl3) δ (ppm.): 29.8 (NH-CH3), 56.5 (O-CH3),
115.7, 116.7, 117.2, 125.1, 128.6, 129.9, 135.9, 142.5, 151.6 (aromatic-C), 157.1 (C=N),
160.3 (S-C-S). 119Sn NMR (CDCl3) δ (ppm.): -236.2.
Dimenyltin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
methylhydrazinecarbothioamide (30)
Bright yellow solid. Yield: 42 %. Melting point: 164-168°C. Analysis calculated
for C12H17N3O2SSn: C, 37.33; H, 4.44; N, 10.88. Found: C, 39.00; H, 4.76; N, 11.00 %.
FT-IR (ATR, cm-1): 3222 v(N-H), 1590 v(C=N), 1066 v(N-N), 973 v(C=S). 1H NMR
(CDCl3) δ (ppm.): 0.91 (s, 6H, Sn-CH3), 2.97 (d, 3H, N-CH3), 3.94 (s, 3H, O-CH3), 7.26
(s, 1H, CH), 6.66-6.87 (m, 3H, Ar-H), 8.55 (s, 1H, NH). 13C NMR (CDCl3) δ (ppm.): 8.7
(Sn-CH3), 31.3 (NH-CH3), 56.4 (O-CH3), 115.7, 115.8, 117.8, 118.5, 125.8, 151.3
(aromatic-C), 156.5 (C=N), 178.0 (S-C-S). 119Sn NMR (DMSO-d6) δ (ppm.): -154.4.
Tin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
methylhydrazinecarbothioamide (31)
Compound 31 was prepared following the same procedure as described for 29,
using 25 (0.48 g, 2 mmol). Bright yellow solid. Yield: 31 %. Melting point: 118-119°C.
Analysis calculated for C20H22N6O4S2Sn: C, 40.49; H, 3.74; N, 14.17. Found: C, 40.50;
H, 3.33; N, 14.17 %. FT-IR (ATR, cm-1): 3308 v(N-H), 1590 v(C=N), 1066 v(N-N), 973
v(C=S). 1H NMR (DMSO-d6) δ (ppm.): 2.36 (d, 6H, N-CH3), 3.82 (s, 6H, O-CH3), 7.60
(s, 2H, CH), 6.67-7.31 (m, 6H, Ar-H), 8.88 (s, 2H, NH). 13C NMR (DMSO-d6) δ (ppm.):
19.3 (NH-CH3), 56.7 (O-CH3), 105.4, 116.7, 117.4, 126.5, 128.6, 129.0, 130.7, 130.8,

164
134.9, 149.7, 151.8, 158.3 (aromatic-C), 163.7 (C=N), 170.6 (S-C-S). 119Sn NMR
(DMSO-d6) δ (ppm.): -354.2.
Diphenyltin(IV) compound containing 2-(2,3-dihydroxybenzylidene)-N-
methylhydrazinecarbothioamide (32)
Yellow solid. Yield: 49 %. Melting point: 186-192°C. Analysis calculated for
C21H19N3O2SSn: C, 50.83; H, 3.86; N, 8.47. Found: C, 48.06; H, 4.27; N, 8.02 %. FT-IR
(ATR, cm-1): 1593 v(C=N), 1006 v(N-N), 953 v(C=S). 1H NMR (DMSO-d6) δ (ppm.):
3.00 (s, 3H, NH-CH3), 8.57 (s, 1H, CH), 6.37-8.20 (m, 13H, Ar-H), 11.27 (s, 1H, NH).
13C NMR (DMSO-d6) δ (ppm.): 30.9 (NH-CH3), 112.8, 113.7, 115.7, 118.4, 128.3, 129.2,
136.0, 136.1, 141.1, 145.4, 153.3 (aromatic-C), 154.3 (C=N), 177.0 (S-C-S). 119Sn NMR
(DMSO-d6) δ (ppm.): -227.0.
Dimethyltin(IV) compound containing 2-(2,3-dihydroxybenzylidene)-N-
methylhydrazinecarbothioamide (33)
Yellow solid. Yield: 32 %. Melting point: 223-225°C. Analysis calculated for
C11H15N3O2SSn: C, 35.51; H, 4.06; N, 11.29. Found: C, 35.87; H, 3.86; N, 11.35 %. FT-
IR (ATR, cm-1): 1596 v(C=N), 1006 v(N-N), 951 v(C=S). 1H NMR (DMSO-d6) δ (ppm.):
0.62 (s, 6H, Sn-CH3), 3.00 (d, 3H, N-CH3), 6.32-7.10 (m, 3H, Ar-H), 8.38 (s, 1H, CH),
11.30 (s, 1H, NH). 13C NMR (DMSO-d6) δ (ppm.): 6.8 (Sn-CH3), 30.7 (NH-CH3), 112.6,
113.7, 115.6, 118.6, 140.7, 153.9 (aromatic-C), 155.0 (C=N), 177.2 (S-C-S). 119Sn NMR
(DMSO-d6) δ (ppm.): -122.9.
Tin(IV) compound containing 2-(2,3-dihydroxybenzylidene)-N-
methylhydrazinecarbothioamide (34)

165
Compound 34 was prepared following the same procedure as described for 29,
using 27 (0.46 g, 2 mmol). Orange solid. Yield: 79 %. Melting point: >300°C. Analysis
calculated for C18H18N6O4S2Sn: C, 38.25; H, 3.21; N, 14.87. Found: C, 36.62; H, 2.92;
N, 14.21 %. FT-IR (ATR, cm-1): 1585 v(C=N), 993 v(N-N), 951 v(C=S). 1H NMR
(DMSO-d6) δ (ppm.): 2.83 (d, 6H, N-CH3), 6.65-7.52 (m, 6H, Ar-H), 8.34 (s, 1H, OH),
8.77 (s, 2H, CH), 11.27 (s, 1H, NH). 13C NMR (DMSO-d6) δ (ppm.): 30.6 (NH-CH3),
113.8, 114.3, 116.7, 118.9, 140.0. 150.1 (aromatic-C), 150.6 (C=N), 177.5 (S-C-S). 119Sn
NMR (DMSO-d6) δ (ppm.): -519.4.
6.2.2.3 Tin(IV) Compounds derived from 26
Compound 26 (0.30 g, 1 mmol)/ 28 (0.29 g, 0.001 mol) was dissolved in methanol
(100 cm3) and triethylamine (Et3N) (0.28 cm3, 2 mmol) was added dropwise to the
solution of 26 or 28. The mixture was heated for about 2 hours until the solution was
reduced by half. Next 1 mmol of tin salt (Ph2SnCl2 (0.34 g)/ Me2SnCl2 (0.22 g)/ SnCl2
(0.19 g)) was added to the mixture. The mixture was heated (64°C) for about 2 hours and
filtered while hot to remove triethylamine salt and the filtrate was kept at room
temperature until the bright yellow product formed.
Diphenyltin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
phenylhydrazinecarbothioamide (35)
Yellow crystals. Yield: 73 %. Melting point: 205-207°C. Analysis calculated for
C27H23N3O2SSn: C, 56.67; H, 4.05; N, 7.34%. Found: C, 57.53; H, 4.26; N, 7.87%. FT-
IR (ATR, cm-1): 3331 v(N-H), 1586 v(C=N), 1075 v(N-N), 832 v(C=S). 1H NMR (CDCl3)
δ (ppm.): 3.96 (s, 3H, O-CH3), 7.99 (s, 1H, CH), 6.69-7.56 (m, 18H, Ar-H), 8.70 (s, 1H,
NH). 13C NMR (CDCl3) δ (ppm.): 56.7 (O-CH3), 115.2, 116.2, 116.9, 119.4, 120.7, 123.4,

166
124.1, 125.4, 128.7, 128.9, 130.0, 135.9, 139.3, 142.1, 148.3, 149.7, 151.7 (aromatic-C),
162.5 (C=N), 164.8 (S-C-S). 119Sn NMR (CDCl3) δ (ppm.): -241.6.
Dimethyltin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
phenylhydrazinecarbothioamide (36)
Yellow crystals. Yield: 64 %. Melting point: 176-179°C. Analysis calculated for
C17H19N3O2SSn: C, 45.56; H, 4.27; N, 9.38%. Found: C, 45.86; H, 4.40; N, 7.08%. FT-
IR (ATR, cm-1): 3294 v(N-H), 1577 v(C=N), 1059 v(N-N), 824 v(C=S). 1H NMR (CDCl3)
δ (ppm.): 3.85 (s, 3H, O-CH3), 7.53 (s, 1H, CH), 6.69-7.32 (m, 18H, Ar-H), 8.65 (s, 1H,
NH). 13C NMR (CDCl3) δ (ppm.): 6.5 (Sn-CH3), 56.2 (O-CH3), 115.3, 116.6, 116.7,
120.5, 123.3, 125.4, 128.9, 139.4, 151.3, 156.8 (aromatic-C), 162.5 (C=N), 163.9 (S-C-
S). 119Sn NMR (CDCl3) δ (ppm.): -114.8.
Tin(IV) compound containing 2-(2-hydroxy-3-methoxybenzylidene)-N-
phenylhydrazinecarbothioamide (37)
Compound 37 was prepared following the same procedure as described for 35,
using 26 (0.60 g, 2 mmol). Yellow solid. Yield: 50 %. Melting point: 293-294°C. Analysis
calculated for C30H26N6O4S2Sn: C, 50.23; H, 3.65; N, 11.71%. Found: C, 49.85; H, 3.73;
N, 11.60%. FT-IR (ATR, cm-1): 3303 v(N-H), 1581 v(C=N), 1063 v(N-N), 824 v(C=S).
1H NMR (DMSO-d6) δ (ppm.): 3.58 (s, 6H, O-CH3), 9.08 (s, 2H, CH), 6.80-7.73 (m, 16H,
Ar-H), 9.70 (s, 2H, NH). 13C NMR (DMSO-d6) δ (ppm.): 56.4 (O-CH3), 117.9, 118.2,
121.0, 123.4, 126.8, 129.2, 140.4, 151.4, 154.8 (aromatic-C), 160.3 (C=N), 162.2 (S-C-
S). 119Sn NMR (DMSO-d6) δ (ppm.): -451.1.

167
6.2.2.4 Tin(IV) Compounds derived from 28
Compound 28 (0.29 g, 0.001 mol) was dissolved in methanol (100 cm3) and
potassium hydroxide (0.11 g, 2 mmol) was added dropwise to the solution of 28. The
mixture was refluxed for about 30 minutes, where the colour changed from light yellow
to orange. Next 1 mmol of tin salt (Ph2SnCl2 (0.34 g)/ Me2SnCl2 (0.22 g)/ SnCl2 (0.19 g))
was added to the mixture. The mixture was refluxed for 6 hour and filtered while hot to
remove triethylamine salt and the filtrate was kept at room temperature until the product,
an orange precipitate, formed.
Diphenyltin(IV) compound containing 2-(2,3-dihydroxybenzylidene)-N-
phenylhydrazinecarbothioamide (38)
Orange solid. Yield: 71 %. Melting point: 133-137 °C. Analysis calculated for
C26H21N3O2SSn: C, 55.94; H, 3.79; N, 7.53%. Found: C, 56.30; H, 3.99; N, 7.42%. FT-
IR (ATR, cm-1): 1587 v(C=N), 998 v(N-N), 957 v(S-C-S). 1H NMR (DMSO-d6) δ (ppm.):
6.57-9.52 (m, 18H, Ar-H), 9.87 (s, 2H, CH), 11.69 (s, 2H, NH). 13C NMR (DMSO-d6) δ
(ppm.): 120.5, 120.7, 125.4, 125.5, 125.6, 125.7, 128.5, 128.6, 128.7, 128.8, 128.9, 129.0,
129.1, 129.5, 134.6, 135.2, 135.5, 136.3, 136.4, 136.7, 139.7 (aromatic-C), 148.8 (C=N),
175.2 (S-C-S). 119Sn NMR (DMSO-d6) δ (ppm.): -327.8.
Dimethyltin(IV) compound containing 2-(2,3-dihydroxybenzylidene)-N-
phenylhydrazinecarbothioamide (39)
Orange solid. Yield: 42 %. Melting point: 153-156°C. Analysis calculated for
C16H17N3O2SSn: C, 44.27; H, 3.95; N, 9.68. Found: C, 44.32; H, 3.72; N, 9.90%. FT-IR
(ATR, cm-1): 1575 v(C=N), 1001 v(N-N), 917 v(S-C-S). 1H NMR (DMSO-d6) δ (ppm.):
0.63 (s, 6H, CH3), 8.51 (s, 1H, CH), 6.33-7.75 (m, 8H, Ar-H), 9.84 (s, 1H, OH). 13C NMR

168
(DMSO-d6) δ (ppm.): 7.1 (Sn-CH3), 110.2, 114.7, 128.3, 128.4, 128.7, 128.8, 135.3,
152.1, 152.7, 153.6 (aromatic-C), 153.8 (C=N), 167.5 (S-C-S). 119Sn NMR (DMSO-d6) δ
(ppm.): -103.3.
Tin(IV) of 2-(2,3-dihydroxybenzylidene)-N-phenylhydrazinecarbothioamide (40)
Compound 40 was prepared following the same procedure as described for 38,
using 28 (0.58 g, 2 mmol). Orange solid. Yield: 48 %. Melting point: >300 °C. Analysis
calculated for C28H22N6O4S2Sn: C, 48.78; H, 3.22; N, 12.19%. Found: C, 48.30; H, 2.97;
N, 12.52 %. FT-IR (ATR, cm-1): 1588 v(C=N), 1001 v(N-N), 939 v(S-C-S). 1H NMR
(DMSO-d6) δ (ppm.): 6.66-7.55 (m, 16H, Ar-H), 8.48 (s, 2H), 10.00 (s, 2H, CH), 11.74
(s, 2H, NH). 13C NMR (DMSO-d6) δ (ppm.): 117.1, 119.5, 121.2, 125.6, 125.7, 125.9,
126.0, 128.4, 128.5, 128.5, 139.6 (aromatic-C), 145.9, 146.0 (C=N), 175.7, 176.1 (S-C-
S). 119Sn NMR (DMSO-d6) δ (ppm.): -540.7.
6.2.3 DFT Calculations and Molecular Docking Simulations
DFT were performed using parameters detailed in Chapter 2 (§2.2.6) and Chapter
3 (§3.2.5.1), respectively. The coordinates for all compounds (25-40) were obtained after
minimisation of energy using the DFT method. The parameters for molecular docking
studies are described in Chapter 3 (§3.2.5.2).
6.2.4 MTT Assay
The protocol used for MTT assay followed that outlined in Chapter 2 (§2.2.6).

169
6.3 Results and discussion
6.3.1 Synthesis
The synthetic pathway of Schiff bases 25-28 and their tin(IV) compounds (29-40)
are described in Schemes 6.1 and 6.2, respectively. The Schiff bases were synthesised by
the condensation reaction of oVa or catechol and the corresponding thiosemicarbazide
(4-methyl-3-thiosemicarbazide and 4-phenyl-3-thiosemicarbazide) in alcoholic
solution.278,282,283 The Schiff bases were then reacted with Ph2SnCl2, Me2SnCl2 and SnCl2
in the presence of potassium hydroxide (KOH) or triethyamine by conventional or reflux
methods (outlined in §6.2.2). The isolated tin(IV) compounds were in good yields with
yellow or orange colours. The resultant tin(IV) compounds were soluble in most organic
solvents especially DMSO and DMF, except 31 which was not soluble in DMSO and
DMF. Due to the insolubility of 31, cytotoxic activity was not determined. The molar
conductance values of the tin(IV) compounds ranged from 0.88-7.85 Ω-1 cm2 mol-1, which
were well below than 25 Ω-1 cm2 mol-1 indicating that all of them were non-electrolytes
in nature and no counter ions were present around the lattice of the compounds.106
NH
NH
R1
SN
HOR2
NH
NH
R1
SNH3 HO
R2
O
MeOH/EtOH
heat and stir (3-4 hours)
R1 = CH3, R2 = OCH3 (25)R1 = C6H5, R2 = OCH3 (26)R1 = CH3, R2 = OH (27)R1 = C6H5, R2 = OH (28)
Scheme 6.1: Synthetic pathway of thiosemicarbazone Schiff bases 25-28.
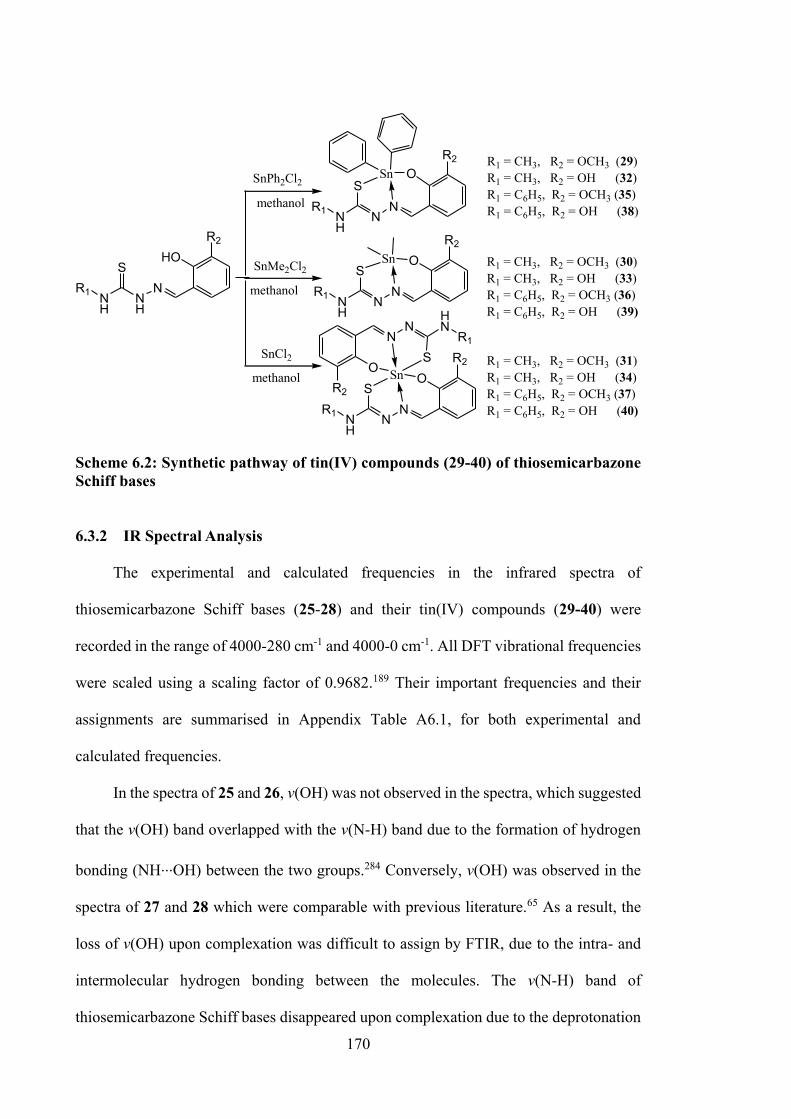
170
NH
NR1
SN
OR2
Sn
NH
NR1
SN
OR2
Sn
NH
NR1
SN
OR2
Sn
HNN
R1
SN
OR2
NH
NH
R1
SN
HOR2
methanol
methanol
methanol
SnPh2Cl2
SnMe2Cl2
SnCl2
R1 = CH3, R2 = OCH3 (29)R1 = CH3, R2 = OH (32)R1 = C6H5, R2 = OCH3 (35)R1 = C6H5, R2 = OH (38)
R1 = CH3, R2 = OCH3 (30)R1 = CH3, R2 = OH (33)R1 = C6H5, R2 = OCH3 (36)R1 = C6H5, R2 = OH (39)
R1 = CH3, R2 = OCH3 (31)R1 = CH3, R2 = OH (34)R1 = C6H5, R2 = OCH3 (37)R1 = C6H5, R2 = OH (40)
Scheme 6.2: Synthetic pathway of tin(IV) compounds (29-40) of thiosemicarbazone Schiff bases
6.3.2 IR Spectral Analysis
The experimental and calculated frequencies in the infrared spectra of
thiosemicarbazone Schiff bases (25-28) and their tin(IV) compounds (29-40) were
recorded in the range of 4000-280 cm-1 and 4000-0 cm-1. All DFT vibrational frequencies
were scaled using a scaling factor of 0.9682.189 Their important frequencies and their
assignments are summarised in Appendix Table A6.1, for both experimental and
calculated frequencies.
In the spectra of 25 and 26, v(OH) was not observed in the spectra, which suggested
that the v(OH) band overlapped with the v(N-H) band due to the formation of hydrogen
bonding (NH...OH) between the two groups.284 Conversely, v(OH) was observed in the
spectra of 27 and 28 which were comparable with previous literature.65 As a result, the
loss of v(OH) upon complexation was difficult to assign by FTIR, due to the intra- and
intermolecular hydrogen bonding between the molecules. The v(N-H) band of
thiosemicarbazone Schiff bases disappeared upon complexation due to the deprotonation

171
of the NH group, and the involvement of this group in the coordination to the tin centre.
Furthermore, the FTIR spectra of 25, 26, 27 and 28 exhibited an intense band due to the
presence of the v(C=N)azomethine stretch at 1610, 1609, 1601 and 1597 cm-1, respectively.
This band shifted to lower frequencies in the spectra of tin(IV) compounds suggesting
that the coordination with the tin centre occurred via the azomethine nitrogen atom. The
v(C=S) and v(N-N) were also shifted to lower frequencies upon complexation, indicating
the coordination via the thiolate sulphur and azomethine nitrogen atoms, forming a five-
membered chelate ring.
6.3.3 NMR Spectroscopic Analysis
The 1H NMR and 13C NMR spectra of thiosemicarbazone Schiff bases 25-28 were
recorded in DMSO-d6 solution, while spectra of tin(IV) compounds 29-40 were recorded
in DMSO-d6/CDCl3 solution at room temperature. The assignments of the relevant signals
are compiled in Appendix Tables A6.2 and A6.3.
In the 1H NMR spectra of 25, 26, 27 and 28, a singlet signal observed at 11.42,
11.48, 9.49 and 10.01 ppm respectively indicated the presence of NH protons. This NH
proton signal was not observed in the spectra of tin(IV) compounds indicating that the
Schiff bases were coordinated to the tin atoms via the nitrogen donor atom. A proton
signal appeared at 9.18 and 10.02 ppm for 25 and 26, corresponding to a hydroxyl proton,
which disappeared in the 1H NMR spectra of the tin(IV) compounds indicating that the
coordination through the hydroxyl group.229 By contrast, the two signals for the hydroxyl
group at 8.32, 8.38 ppm (27) and 11.75, 10.01 ppm (28) disappeared upon complexation,
which indicated the presence of intra- and intermolecular hydrogen bonding between the
molecules.65,285

172
The 13C NMR spectra of 25, 26, 27 and 28 showed carbon signals at 177.6, 176.1,
178.0 and 176.0 ppm respectively, at the downfield region attributed to -S-C(=S)N. The
position of these carbon signals proved that 25, 26, 27 and 28 predominated as the thione
tautomers, even in DMSO-d6 solution. This signal was shifted upfield in the spectra of
the respective tin(IV) compounds, which indicated a decrease of electron density at the
carbon atom of -S-C(-S)N when sulphur was chelated to the tin centre. The C=N signal
was observed at 148.4 ppm (25), 148.6 ppm (26), 146.0 ppm (27) and 146.0 ppm (28)
appeared downfield, as the carbon atom was attached an electronegative atom. However,
this C=N signal was shifted downfield in the spectra of tin(IV) compounds, due to the
increased electron density around the atom upon complexation. The methoxy -CH3-
signal appeared upfield, at 56.4 ppm (25) and 56.3 ppm (26). The same carbon signal was
observed in the spectra of tin(IV) compounds indicating that the methoxy group did not
coordinate the tin centre.
119Sn NMR was also used to assist in the structural elucidation of compounds 29-
40. The 119Sn chemical shift strongly depends on the alkyl/aryl group attached to the tin
atom and electronegativity of the ligand coordinated to the tin atom as well as temperature
employed in the experiments (room temperature here). Theoretically, as the coordination
number increases, the 119Sn chemical shift moves towards the shielding region.230 The
spectra of 29-40 each showed one sharp signal which strongly supported that the tin(IV)
compounds contain single species of the tin atom. The 119Sn NMR values of penta-
coordinated diphenyl- (29, 32, 35 and 38) and dimethyltin(IV) (30, 33, 36 and 39)
compounds fell in the range of -227 to -328 and -103 to -154 ppm, respectively. The 119Sn
NMR values of hexa-coordinated tin(IV) compounds (31, 34, 37 and 40) were observed
in the range of -354 to -541 ppm, which supported the hypothesis that by increasing the
coordination sphere, the chemical shift decreased.151

173
6.3.4 Mass Spectral Analysis
Mass spectral data for 25-28 were recorded in DMSO and were found to be
consistent with the proposed formulation of the Schiff bases. Mass spectra displayed
prominent peaks at m/z 239, 301, 225 and 287 for Schiff bases 25, 26, 27 and 28,
respectively, which corresponded to [C10H13N3O2S]+, [C15H15N3O2S]+, [C9H11N3O2S]+
and [C14H13N3O2S]+. The mass spectra for 25, 26, 27 and 28 are supplied in Appendix
Figure A6.1.
6.3.5 UV-vis Absorption Spectroscopy
The experimental and calculated electronic spectra of the thiosemicarbazone Schiff
bases and their tin(IV) compounds in DMSO are tabulated in Appendix Table A6.4. The
prominent experimental electronic absorptions for the thiosemicarbazone Schiff bases 25-
28 were observed at 328-334 nm, which were closely correlated with B3LYP excitations
at 317-330 nm. The frontier MOs of the thiosemicarbazone Schiff bases and their tin(IV)
compounds are depicted in Figure 6.2, which shows that the HOMOs in these compounds
are dominated by sulphur and nitrogen, whereas their LUMOs are largely centred on the
thiosemicarbazone backbones and the oVa or catechol groups. Thus, UV-vis absorption
maxima are assigned as n→π* and π→π* excitations for 25-28. For the tin(IV)
compounds 29-40, the HOMOs are largely centred on the thiosemicarbazone Schiff base,
whereas the LUMOs are centred on the entire thiosemicarbazone Schiff base, except the
coordinating sulphur atom. The overlapped n→π* and π→π* transitions were observed
at 328-360 nm (experimental) and 347-380 nm (DFT). The intraligand absorption
corresponded to S→SnIV LMCT transition was observed at 401-417 nm (experimental)
and 395-424 nm (DFT), consistent with coordination of tin by the sulphur atoms.

174
Figure 6.2: Frontier MOs (a) 25, (b) 26, (c) 27, (d) 28, (e) 29, (f) 30, (g) 31, (h) 32, (i) 33, (j) 34, (k) 35, (l) 36, (m) 37, (n) 38, (o) 39 and (p) 40.
6.3.6 In Vitro Cytotoxic Activity
The four thiosemicarbazone Schiff bases and twelve tin(IV) compounds reported in
this chapter were screened for their cytotoxicity against a panel of ten cancer cell lines,
HT29, U87 and SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C, MIA and one

175
normal cell line, MCF-10A (Table 6.1). The cytotoxicity of 31 was unable to be
determined due to its insolubility in 100% of DMSO at 1 mM concentration.
The cytotoxicity of oVa thiosemicarbazone Schiff bases (25 and 26) revealed an
increase in potency with the substituted methyl group at the α-nitrogen atom, where 25
exhibited 10 to 20 times higher antiproliferative activity than 26, 27 and 28 in all cancer
cell lines tested. Table 6.1 shows that the HT-29, A2780, A431, BE2-C and MIA cell
lines were more sensitive after treatment with 26. Compound 26 was approximately ~10-
100 times more potent than 3 (discussed in Chapter 2) against all cancer cell lines tested,
except for the Du145 cell line, which was more resistant towards 3. No obvious
cytotoxicity pattern was observed for the cytotoxicity of 26 which was similar to 3. In
contrast, 2,3-dihydroxybenzyl thiosemicarbazone Schiff bases (27 and 28) showed a
different pattern of cytotoxicity, which was attributed to the phenyl group attached to the
α-nitrogen. For instance, 28 was more active than the compound than 27 in all cancer cell
lines tested. Compound 27 showed poorer cytotoxicity at the 25 µM single point dose
evaluation pre-screening and was not selected for further GI50 determination, as it was
considered inactive.
The cytotoxicity studies of the tin(IV) compounds was comparable with the series
reported in Chapter 3 and 4.178 Diphenyltin(IV) compounds exhibited higher activities as
compared to their Schiff bases and other tin(IV) compounds, except 29, which exhibited
2.5-fold lower activity than its Schiff base (25) in MIA cells. Furthermore, 32 showed
promising cytotoxic activity with no specific selectivity against all cancer cell lines. The
cytotoxicity of dimethyltin(IV) compounds (30, 33, 36 and 39) and tin(IV) compounds
with the absence of organo group (31, 34, 37 and 40) exhibited no significant difference
as compared to their Schiff bases. It can be concluded that the presence of two phenyl
groups attached to the tin atom at the centre yields improved cytotoxicity against all

176
cancer cell lines considered here. Overall, the cytotoxicity results showed that HT29,
MCF-7, A2780, A431, BE2-C and MIA were more sensitive, whereas H460 and Du145
were less sensitive cancer cell lines to the Schiff bases and tin(IV) compounds in this
work.
6.3.7 DNA Binding Studies
In this chapter, DNA binding studies of compounds 27, 32, 33 and 34 were
investigated by absorption spectroscopy (Figure 6.3). These compounds were selected for
DNA binding evaluation in order to investigate the interactions of the inactive Schiff base
27 and its active tin(IV) compounds 32, 33 and 34 with DNA. The spectra were recorded
for a constant concentration of compounds (50 μM) with increasing concentrations of CT-
DNA. No significant changes were noted in the absorption spectra of DNA for 27 (Figure
6.3(i)) and 33 (Figure 6.3(iii)), which indicated that both compounds did not interact with
DNA. However, small changes were observed in the absorption spectra of DNA with
compounds 32 (Figure 6.3(ii)) and 34 (Figure 6.3(iv)), notably the decrease of the
absorption at λmax = 319 nm and 315 nm (i.e. hypochromism), respectively with absence
of any appreciable red shifts. Groove binding interactions is suggested as the most likely
mode of interaction, which was comparable to reported literature.286 The observed order
of hypochromism, viz 32 > 34 > 27 = 33, reflected a decrease in DNA binding affinity of
the tested compounds. This is consistent with the respective Kb values of 32 and 34, 4.62
x 104 and 7.51 x 103. The values obtained strongly supported that both compounds bound
to phosphate groups of the DNA surface.225 The DNA binding results correlated well with
cytotoxicity data, where compounds 32 and 34 showed more potency than compounds 27
and 33.

177
6.3.8 Molecular Docking Studies
As can be seen from the presented data in Table 6.2, there was an excellent
correlation between the growth inhibitions of cytotoxicity studies and free binding energy
of molecular docking simulations. A clear correlation was observed that 32 and 34 had
better activity than 27 and 33. Amongst the docked conformations, 32 showed the lowest
free binding energy (-31.42 kJ mol-1), which was in agreement with the DNA binding
studies discussed above. The compounds fitted well in the cytosine-guanine (G-C) rich
grooves (see Figure 6.4), and this region has a higher affinity than the adenine-thymine
(A-T) region. The G-C region is very important in terms of DNA stability, because the
guanine and cytosine are stabilised by three hydrogen bonds, and it has been suggested
that these regions could have a key role in anticancer activity.287 The compounds fitted in
the G-C region were stabilised by hydrogen, electrostatic and hydrophobic interactions.
Even though compounds 27 and 33 were not observed to have good DNA binding,
molecular docking simulations suggested that both compounds fitted at the grooves of
DNA. This can be explained by the fact that molecular docking simulations were
performed in rigid-rigid compounds-DNA conditions without explicit of water molecules
and other biological entities. Of note, significant potency was observed for compound 33,
where DNA binding studies revealed that 33 did not exclusively bind to the DNA. The
mechanism of action of 33 remains unknown and a subject for future research.
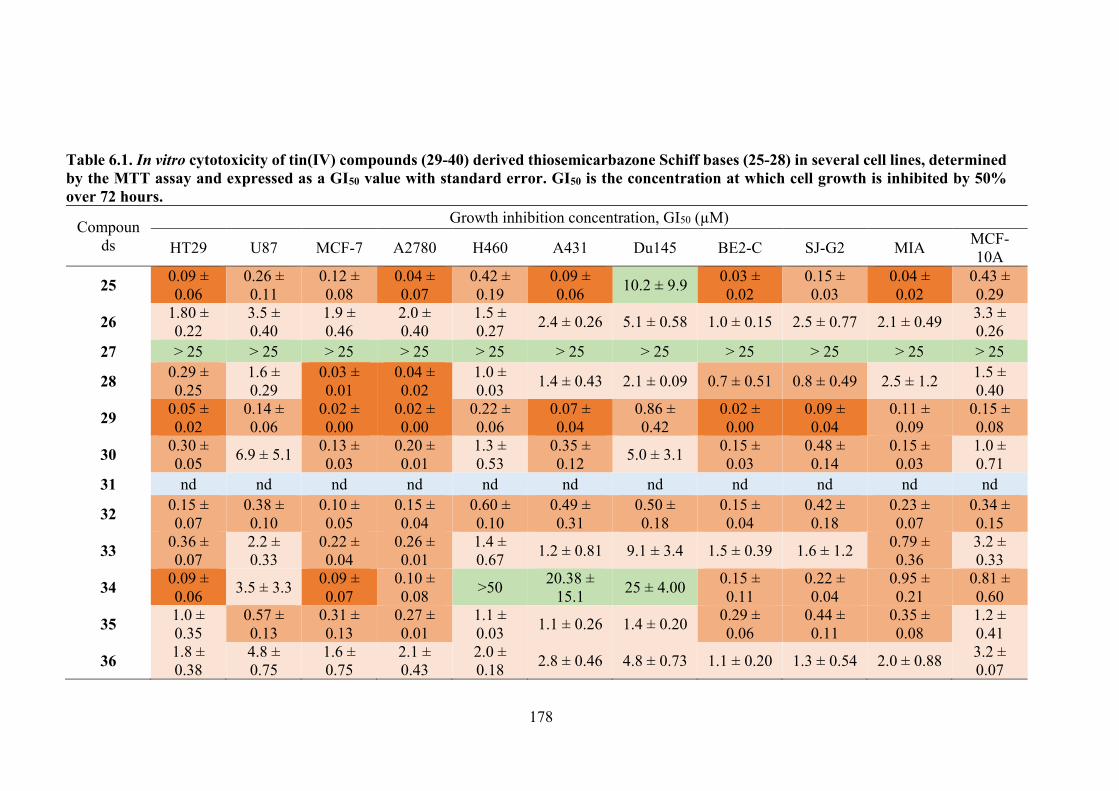
178
Table 6.1. In vitro cytotoxicity of tin(IV) compounds (29-40) derived thiosemicarbazone Schiff bases (25-28) in several cell lines, determined by the MTT assay and expressed as a GI50 value with standard error. GI50 is the concentration at which cell growth is inhibited by 50% over 72 hours.
Compounds
Growth inhibition concentration, GI50 (µM)
HT29 U87 MCF-7 A2780 H460 A431 Du145 BE2-C SJ-G2 MIA MCF-10A
25 0.09 ± 0.06
0.26 ± 0.11
0.12 ± 0.08
0.04 ± 0.07
0.42 ± 0.19
0.09 ± 0.06 10.2 ± 9.9 0.03 ±
0.02 0.15 ± 0.03
0.04 ± 0.02
0.43 ± 0.29
26 1.80 ± 0.22
3.5 ± 0.40
1.9 ± 0.46
2.0 ± 0.40
1.5 ± 0.27 2.4 ± 0.26 5.1 ± 0.58 1.0 ± 0.15 2.5 ± 0.77 2.1 ± 0.49 3.3 ±
0.26 27 > 25 > 25 > 25 > 25 > 25 > 25 > 25 > 25 > 25 > 25 > 25
28 0.29 ± 0.25
1.6 ± 0.29
0.03 ± 0.01
0.04 ± 0.02
1.0 ± 0.03 1.4 ± 0.43 2.1 ± 0.09 0.7 ± 0.51 0.8 ± 0.49 2.5 ± 1.2 1.5 ±
0.40
29 0.05 ± 0.02
0.14 ± 0.06
0.02 ± 0.00
0.02 ± 0.00
0.22 ± 0.06
0.07 ± 0.04
0.86 ± 0.42
0.02 ± 0.00
0.09 ± 0.04
0.11 ± 0.09
0.15 ± 0.08
30 0.30 ± 0.05 6.9 ± 5.1 0.13 ±
0.03 0.20 ± 0.01
1.3 ± 0.53
0.35 ± 0.12 5.0 ± 3.1 0.15 ±
0.03 0.48 ± 0.14
0.15 ± 0.03
1.0 ± 0.71
31 nd nd nd nd nd nd nd nd nd nd nd
32 0.15 ± 0.07
0.38 ± 0.10
0.10 ± 0.05
0.15 ± 0.04
0.60 ± 0.10
0.49 ± 0.31
0.50 ± 0.18
0.15 ± 0.04
0.42 ± 0.18
0.23 ± 0.07
0.34 ± 0.15
33 0.36 ± 0.07
2.2 ± 0.33
0.22 ± 0.04
0.26 ± 0.01
1.4 ± 0.67 1.2 ± 0.81 9.1 ± 3.4 1.5 ± 0.39 1.6 ± 1.2 0.79 ±
0.36 3.2 ± 0.33
34 0.09 ± 0.06 3.5 ± 3.3 0.09 ±
0.07 0.10 ± 0.08 >50 20.38 ±
15.1 25 ± 4.00 0.15 ± 0.11
0.22 ± 0.04
0.95 ± 0.21
0.81 ± 0.60
35 1.0 ± 0.35
0.57 ± 0.13
0.31 ± 0.13
0.27 ± 0.01
1.1 ± 0.03 1.1 ± 0.26 1.4 ± 0.20 0.29 ±
0.06 0.44 ± 0.11
0.35 ± 0.08
1.2 ± 0.41
36 1.8 ± 0.38
4.8 ± 0.75
1.6 ± 0.75
2.1 ± 0.43
2.0 ± 0.18 2.8 ± 0.46 4.8 ± 0.73 1.1 ± 0.20 1.3 ± 0.54 2.0 ± 0.88 3.2 ±
0.07
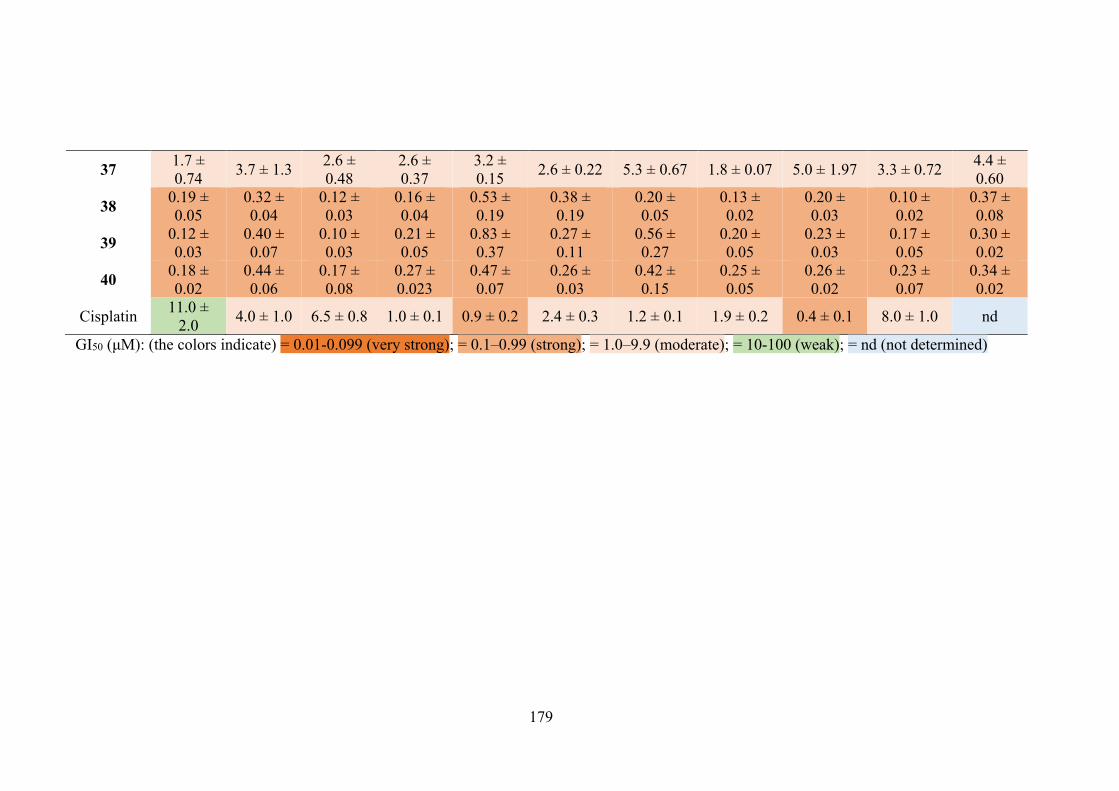
179
37 1.7 ± 0.74 3.7 ± 1.3 2.6 ±
0.48 2.6 ± 0.37
3.2 ± 0.15 2.6 ± 0.22 5.3 ± 0.67 1.8 ± 0.07 5.0 ± 1.97 3.3 ± 0.72 4.4 ±
0.60
38 0.19 ± 0.05
0.32 ± 0.04
0.12 ± 0.03
0.16 ± 0.04
0.53 ± 0.19
0.38 ± 0.19
0.20 ± 0.05
0.13 ± 0.02
0.20 ± 0.03
0.10 ± 0.02
0.37 ± 0.08
39 0.12 ± 0.03
0.40 ± 0.07
0.10 ± 0.03
0.21 ± 0.05
0.83 ± 0.37
0.27 ± 0.11
0.56 ± 0.27
0.20 ± 0.05
0.23 ± 0.03
0.17 ± 0.05
0.30 ± 0.02
40 0.18 ± 0.02
0.44 ± 0.06
0.17 ± 0.08
0.27 ± 0.023
0.47 ± 0.07
0.26 ± 0.03
0.42 ± 0.15
0.25 ± 0.05
0.26 ± 0.02
0.23 ± 0.07
0.34 ± 0.02
Cisplatin 11.0 ± 2.0 4.0 ± 1.0 6.5 ± 0.8 1.0 ± 0.1 0.9 ± 0.2 2.4 ± 0.3 1.2 ± 0.1 1.9 ± 0.2 0.4 ± 0.1 8.0 ± 1.0 nd
GI50 (μM): (the colors indicate) = 0.01-0.099 (very strong); = 0.1–0.99 (strong); = 1.0–9.9 (moderate); = 10-100 (weak); = nd (not determined)
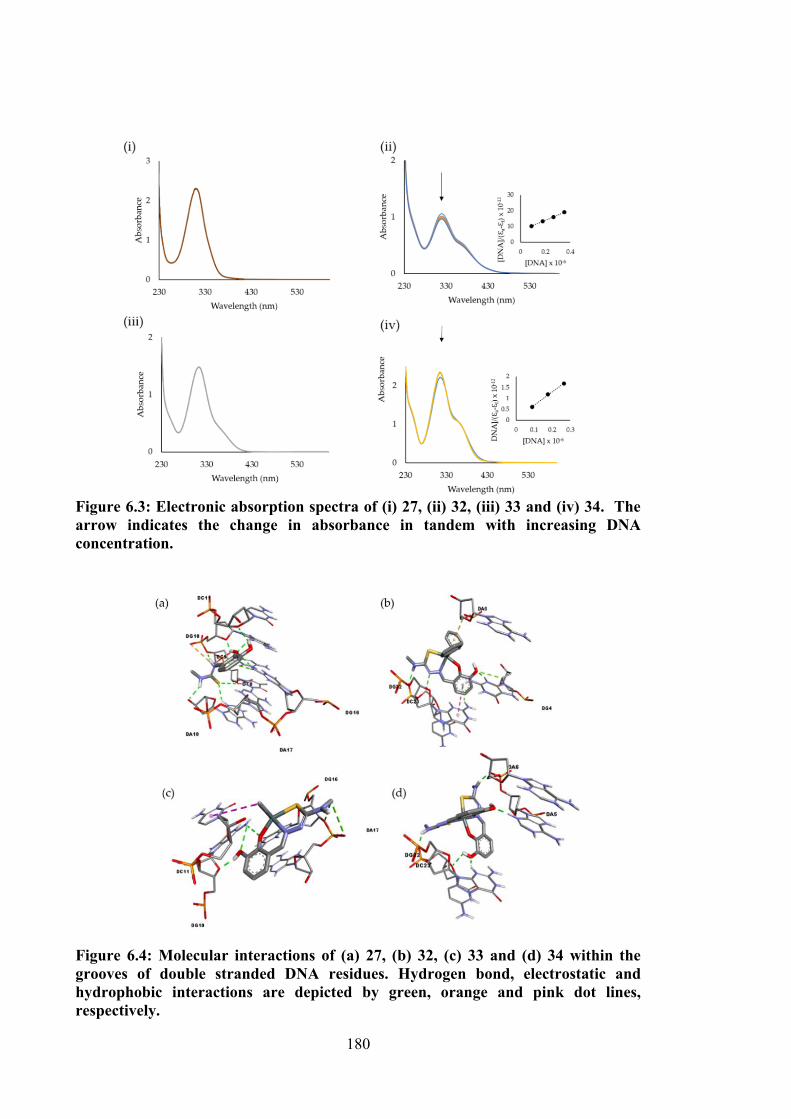
180
Figure 6.3: Electronic absorption spectra of (i) 27, (ii) 32, (iii) 33 and (iv) 34. The arrow indicates the change in absorbance in tandem with increasing DNA concentration.
Figure 6.4: Molecular interactions of (a) 27, (b) 32, (c) 33 and (d) 34 within the grooves of double stranded DNA residues. Hydrogen bond, electrostatic and hydrophobic interactions are depicted by green, orange and pink dot lines, respectively.
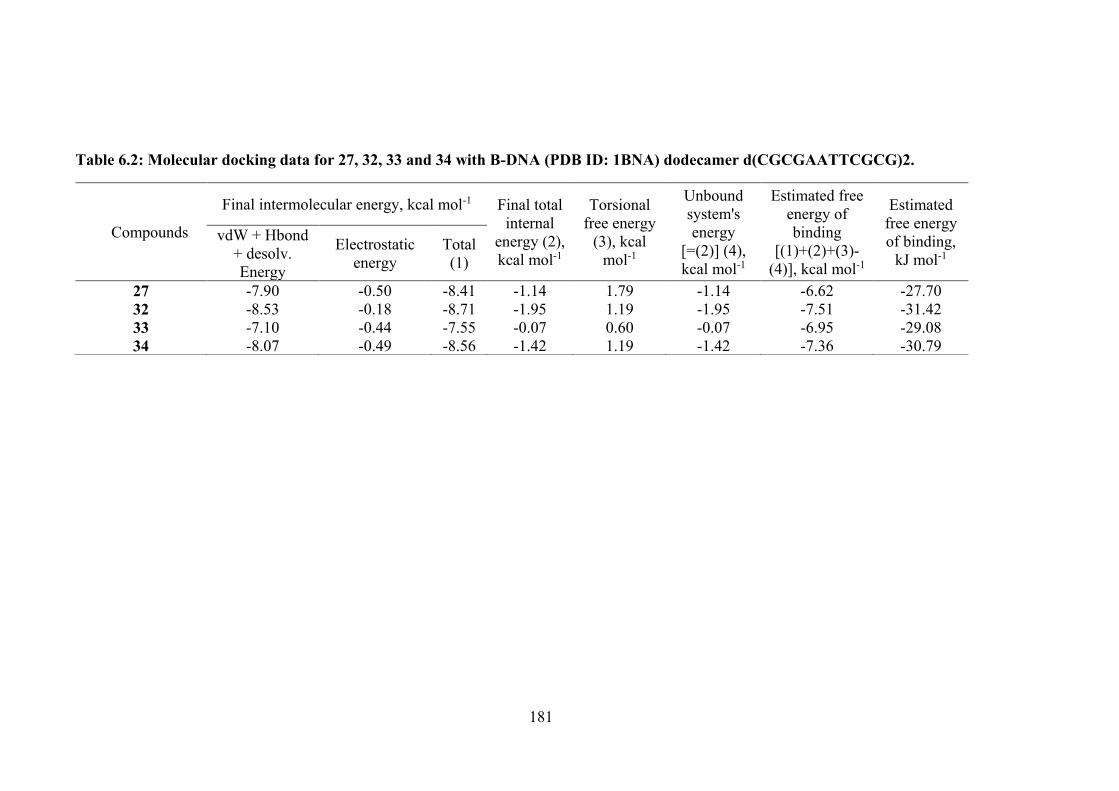
181
Table 6.2: Molecular docking data for 27, 32, 33 and 34 with B-DNA (PDB ID: 1BNA) dodecamer d(CGCGAATTCGCG)2.
Compounds
Final intermolecular energy, kcal mol-1 Final total internal
energy (2), kcal mol-1
Torsional free energy
(3), kcal mol-1
Unbound system's energy
[=(2)] (4), kcal mol-1
Estimated free energy of binding
[(1)+(2)+(3)-(4)], kcal mol-1
Estimated free energy of binding,
kJ mol-1 vdW + Hbond
+ desolv. Energy
Electrostatic energy
Total (1)
27 -7.90 -0.50 -8.41 -1.14 1.79 -1.14 -6.62 -27.70 32 -8.53 -0.18 -8.71 -1.95 1.19 -1.95 -7.51 -31.42 33 -7.10 -0.44 -7.55 -0.07 0.60 -0.07 -6.95 -29.08 34 -8.07 -0.49 -8.56 -1.42 1.19 -1.42 -7.36 -30.79

182
6.4 Conclusions
This chapter presents the synthesis and characterisation of four thiosemicarbazone
Schiff bases (25-28) containing 4-methyl-3-thiosemicarbazide or 4-phenyl-3-
thiosemicarbazide with oVa or catechol. Their penta-coordinated diphenyl- (29, 32, 35,
38) and dimethyltin(IV) (30, 33, 36, 39) and hexa-coordinated tin(IV) (31, 34, 37, 40)
compounds of 25, 26, 27 and 28 are also discussed in this chapter. The Schiff bases in
this work behaved as tridentate binegatively charged ONS which coordinated to the tin
ion via thiolate-S, phenoxide-O and imine-N atoms. The in vitro cytotoxicity of the
compounds were determined using a panel of cancer cell lines viz., HT29, U87, SJ-G2,
MCF-7, A2780, H460, A431, Du145, BE2-C and MIA and one normal breast cell line,
MCF-10A. Schiff bases 25 and 28 showed selective strong cytotoxicity against certain
cancer cells tested. 25 showed good selectivity against HT29, A2780, A431, BE2-C and
MIA, whereas 28 exhibited strong selectivity against MCF-7 and A2780. 26 displayed
moderate cytotoxicity with no selectivity. Very strong selective cytotoxicity against
HT29, MCF-7, A2780, A431, BE2-C and SJ-G2 were observed for 29, suggesting that
the complexation with diphenyltin(IV) enhanced the cytotoxicity. With the exception of
MIA, the same pattern was found here as that observed for the compounds discussed in
Chapter 3 and 4. Good cytotoxic activity with no selectivity was observed in tin(IV)
compounds 32, 36, 37, 38, 39 and 40. In general, H460, A431 and Du145 were more
resistant towards 25, 28, 30, 33, 35, 38, 39 and 40. DNA binding studies demonstrated
the existence of groove binding interactions between compounds 32 and 34 with DNA,
which suggested that binding with DNA was one of the mechanisms of action that
enhanced the cytotoxicity for both compounds. Molecular docking simulations predicted
that the compounds bound to the G-C rich groove of DNA and they were stabilised by
hydrogen, electrostatic and hydrophobic interactions.

183
The summary of the synthesis, structural evaluation and cytotoxicity studies for all
Schiff bases and tin(IV) compounds discussed in this research will be outlined in Chapter
7, which also contains of future direction of this research, such as to enhance solubility,
mechanism of action and toxicity evaluation and also molecular dynamic simulations in
order to strengthen the value of the compounds discussed in this research.

184
CHAPTER 7
CONCLUSIONS AND FUTURE RECOMMENDATIONS
7.1 Conclusions
The aim of this project was to synthesise, structurally characterise and evaluate the
cytotoxicity of tin(IV) compounds derived from six dithiocarbazate and four
thiosemicarbazone Schiff bases. The compounds were characterised by elemental
analysis, molar conductivity, various spectroscopic techniques and X-ray
crystallography. In vitro cytotoxicity was investigated against a panel of cancer cell lines
(EJ-28, RT-112, HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C, MIA)
and one normal breast cell (MCF-10A) using a MTT metabolic method based on their
mitochondrial dehydrogenase activity. The interactions between DNA and compounds
was also investigated via UV-vis absorption spectroscopy. Molecular docking simulation
was carried out in order to theoretically understand the potential types of interactions that
occurred. The compounds were classed in five libraries and discussed in Chapter 2-6.
Chapter 2 detailed the synthesis of three dithiocarbazate Schiff bases (1, 2 and 3)
derived from S1, S2 and S3 with oVa. The crystal structure of 3 comprised two almost
planar residues, i.e. the phenyl ring and the remaining 14 non-H atoms (r.m.s. deviation
= 0.0410 Å) which were orientated perpendicularly, forming a dihedral angle of
82.72(5)°. An analysis of the geometric parameters was consistent with the molecule
existing as the thione tautomer, and the conformation about the C=N bond was E. In vitro
cytotoxicity of 1, 2 and 3 showed moderate activity against all cancer cells (EJ-28, RT-
112, HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C, MIA) tested with
no specificity or selectivity. To enhance the cytotoxicity, these compounds were
complexed with tin ion and the findings were reported in Chapter 3.

185
Chapter 3 detailed the synthesis of nine organotin(IV) compounds (4-9) derived
from Schiff bases synthesised in Chapter 2. Three crystal structures of 7, 8 and 9 which
supported the coordination geometry of the compounds were obtained. Schiff bases
behaved as tridentate ONS donor ligands coordinating to the tin centre via azomethine
nitrogen, thiolo sulphur and oxygen atoms. Compounds 7, 8 and 9 adopted a five-
coordinated distorted trigonal bipyramidal environment, where one molecule of Schiff
base and two methyl groups were coordinated to the central tin. Organotin(IV)
compounds 4-9 were evaluated for their in vitro cytotoxicity against twelve cancer cell
lines (EJ-28, RT-112, HT29, U87, SJ-G2, MCF-7, A2780, H460, A431, Du145, BE2-C,
MIA) and one normal breast cell (MCF-10A). Selective strong cytotoxicity was observed
for MCF-7, A2780, BE2-C, SJ-G2 and MIA cell lines treated with diphenyltin
compounds (4, 5 and 6). Selected tin(IV) compounds, 4, 5 and 6 were evaluated for the
mechanism of death, ROS and Annexin V against RT-112 cell line, where these
compounds were found to induce ROS production and were marked as apoptosis inducer.
The significance of these target compounds was clearly shown by the high binding
interactions with DNA, where generally chemotherapeutic agents that are approved for
clinical use are compounds having the ability to damage DNA, block DNA synthesis by
inhibiting nucleic acid precursor biosynthesis or disrupting hormonal stimulation that
controls the cell growth.288 Theoretical understanding by molecular docking simulations
supported that the compounds bound to the groove of DNA strand via hydrogen bonding,
hydrophobic and electrostatic interactions.
Chapter 4 detailed the synthesis, characterisation of three dithiocarbazate Schiff
bases (10, 11 and 12) derived from catechol and their diphenyl- (13, 14 and 15) and
dimethyltin(IV) (16, 17 and 18) compounds. One crystal structure of Schiff base 12 and
four crystal structure data of five-coordinated distorted trigonal bipyramidal

186
organotin(IV) compounds 14, 15, 17 and 18 were obtained. All compounds were
investigated for their cytotoxicity against ten cancer cell lines (HT29, U87, SJ-G2, MCF-
7, A2780, H460, A431, Du145, BE2-C, MIA) and one normal breast cell (MCF-10A). A
similar selectivity pattern was observed for 13, 14 and 15, however, DNA was not one of
the cytotoxic targets, where the compounds had only weak DNA binding interactions.
Chapter 5 detailed the synthesis of homoleptic tin(IV) compounds (19, 20, 21, 22,
23 and 24) coordinated with two molecules of dithiocarbazate Schiff bases synthesised in
Chapter 2 and 4. Two crystal structure data of distorted octahedral 20 and 21 were
obtained. All compounds exhibited similar cytotoxic activity compared to their respective
Schiff bases, possibly due to the bulky structure of the compounds and hence the difficulty
of interaction with the biomacromolecules of the cancer cells. The mechanisms of action
of these compounds remain unknown and warrant further biochemical investigations.
Lastly, in Chapter 6, the synthesis of four thiosemicarbazone Schiff bases derived
from oVa and catechol and their tin(IV) compounds is reported. Amongst the Schiff
bases, thiosemicarbazone derived Schiff bases, 25 and 28 had the best cytotoxic potency
against HT29, A2780, A431, BE2-C and MIA cells. These compounds are useful lead
candidates for future organic drug design development to treat cancers. However, the
mechanism of action of these compounds have yet to be verified. Good cytotoxic activity
with no selectivity pattern was observed in tin(IV) compounds 32, 36, 37, 38, 39 and 40.
H460, A431 and Du145 were observed to be more resistant towards 25, 28, 30, 33, 35,
38, 39 and 40. DNA binding studies demonstrated that 32 and 34 had good binding ability
compared to 27 and 33, which suggested that DNA was one of the cytotoxic targets for
32 and 34.

187
7.2 Future Recommendations
A number of recommendations for future research have resulted from the detailed
investigations in this thesis. The poor water solubility of synthesised Schiff bases and
tin(IV) compounds is a major issue in the course of applications, particularly anticancer
activity in this work. Pre-dissolution in high concentration of DMSO was used to
overcome their low solubility in aqueous media for anticancer studies. Due to the toxicity
of final DMSO concentration is higher than 3% that has been shown to retard cancerous
cells at high concentrations, there is a need of highly water soluble compounds for future
use, where they will be able to enter preclinical and clinical investigations without the use
of any solubilisers. To overcome this problem, peptides as targeting vectors for the direct
transportation of Schiff bases and tin(IV) compounds can be utilised.289 The attachment
of synthesised compounds to one or more polyethylene glycol (PEG) chains might also
enhance the compounds aqueous solubility, concomitant with the increase in bioactivity.
PEG is a non-toxic, non-immunogenic and non-antigenic polymer that is highly water
soluble. The introduction of FDA-approved PEG into pharmaceuticals could enhance
their pharmacokinetics which “prolong residence in body, decrease degradation by
metabolic enzymes and reduce or eliminate protein immunogenicity”.290
In addition, the mechanism of action of Schiff bases and tin(IV) compounds has yet
to be verified. Cellular uptake will be the next crucial analysis in order to investigate the
compound uptake in the cells. Therefore, efforts to determine the concentration and the
mechanism of action of the compounds are important in the line of succeeding in vivo
studies and clinical trials. Given the exciting activity of synthesised Schiff bases and
tin(IV) compounds, future work could investigate the toxicity effect of the compounds
towards normal cells. In this case, MCF-10A is positive for telomerase reverse
transcriptase289 which is known to be up-regulated in many cancer cells as well. The

188
decrease in cell viability after treatment with synthesised compounds may be due to the
inactivation of this enzyme. The most relevant option to determine toxicity in vitro is by
using bone marrow or intestinal epithelial cells.291 Furthermore, the most accurate relative
toxicity can be measured by in vivo studies. Zebrafish is one of the model organisms used
for in vivo experiment, where the zebrafish have morphological and physiological
similarities to mammals. Zebrafish are grown rapidly on a large scale which is sufficient
for toxicity screening. Other than that, zebrafish promises to contribute to several other
aspects of the drug development process, including target identification, target validation,
morpholino oligonucleotide screens and structure-activity relationship studies.292–294
Hitherto, medicinal chemists face contemporary challenges in molecular docking
in order to evaluate the performance of synthesised compounds towards
biomacromolecules, such as DNA in this work. Even though software solutions for
molecular docking based on end point calculations (i.e. evaluation of binding energy and
interactions using a single, rigid structure of synthesised compounds and
biomacromolecules) have achieved performance that guarantees their great involvement
in drug discovery pipelines, several issues are to be addressed. High-accuracy of
molecular modelling is crucial in order to create the system mimics with biological
systems, and one of the prediction techniques is molecular dynamic (MD) simulations.
This approach could be an excellent method for both accuracy and cost-effective as
compared to the molecular docking method.

189
REFERENCES
[1] Cooper, G. M. The Cell: A Molecular Approach; 2nd ed.; Sinauer Associates Inc.: Massachusetts, United States, 2000.
[2] Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R. L.; Torre, L. A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424.
[3] Walker, B. J.; Gerber, A. Occupational Exposure to Aromatic Amines: Benzidine and Benzidine-Based Dyes. Natl. Cancer Inst. Monogr. 1982, 58, 11–13.
[4] Cancer treatment https://www.mayoclinic.org/tests-procedures/cancer-treatment/about/pac-20393344 (accessed May 2, 2017).
[5] Understanding Targeted Therapy https://www.cancer.net/navigating-cancer-care/how-cancer-treated/personalized-and-targeted-therapies/understanding-targeted-therapy (accessed Apr 3, 2018).
[6] Nicholson, L. B. The Immune System. Essays Biochem. 2016, 60, 275–301.
[7] Helissey, C.; Vicier, C.; Champiat, S. The Development of Immunotherapy in Older Adults: New Treatments, New Toxicities? J. Geriatr. Oncol. 2016, 7, 325–333.
[8] Baldo, B. A. Adverse Events to Monoclonal Antibodies Used for Cancer Therapy. Oncoimmunology 2013, 2, e26333.
[9] Scott, A. M.; Allison, J. P.; Wolchok, J. D. Monoclonal Antibodies in Cancer Therapy. Cancer Immun. 2012, 12, 1–8.
[10] Shepard, H. M.; Phillips, G. L.; Thanos, C. D.; Feldmann, M. Developments in Therapy with Monoclonal Antibodies and Related Proteins. Clin. Med. 2017, 17, 220–232.
[11] Akam, E. A.; Tomat, E. Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators. Bioconjug. Chem. 2016, 27, 1807–1812.
[12] Palma, E.; Mendes, F.; Morais, G. R.; Rodrigues, I.; Santos, I. C.; Campello, M. P. C.; Raposinho, P.; Correia, I.; Gama, S.; Belo, D.; Alves, V.; Abrunhosa, A. J.; Santos, I.; Paulo, A. Biophysical Characterization and Antineoplastic Activity of New Bis(thiosemicarbazonato) Cu(II) Complexes. J. Inorg. Biochem. 2017, 167, 68–79.
[13] Refat, M. S.; El-Deen, I. M.; Anwer, Z. M.; El-Ghol, S. Bivalent Transition Metal Complexes of Coumarin-3-yl Thiosemicarbazone Derivatives: Spectroscopic, Antibacterial Activity and Thermogravimetric Studies. J. Mol. Struct. 2009, 920, 149–162.
[14] Prasad, K. S.; Kumar, L. S.; Prasad, M.; Revanasiddappa, H. D. Novel

190
Organotin(IV)-Schiff Base Complexes: Synthesis, Characterization, Antimicrobial Activity, and DNA Interaction Studies. Bioinorg. Chem. Appl. 2010, 854514.
[15] Lebwohl, D.; Canetta, R. Clinical Development of Platinum Complexes in Cancer Therapy: An Historical Perspective and an Update. Eur. J. Cancer 1998, 34, 1522–1534.
[16] Galanski, M. Recent Developments in the Field of Anticancer Platinum Complexes. Recent Pat. Anticancer. Drug Discov. 2006, 1, 285–295.
[17] Alama, A.; Tasso, B.; Novelli, F.; Sparatore, F. Organometallic Compounds in Oncology: Implications of Novel Organotins as Antitumor Agents. Drug Discov. Today 2009, 14, 500–508.
[18] Fuertes, M. A.; Alonso, C.; Pérez, J. M. Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance. Chem. Rev. 2003, 103, 645–662.
[19] Jung, Y.; Lippard, S. J. Direct Cellular Responses to Platinum-Induced DNA Damage Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407.
[20] Dasari, S.; Bernard, T. P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378.
[21] Kim, J.; Pramanick, S.; Lee, D.; Park, H.; Kim, W. J. Polymeric Biomaterials for the Delivery of Platinum-Based Anticancer Drugs. Biomater. Sci. 2015, 3, 1002–1017.
[22] Subbaraj, P.; Ramu, A.; Raman, N.; Dharmaraja, J. Synthesis, Characterization, DNA Interaction and Pharmacological Studies of Substituted Benzophenone Derived Schiff Base Metal(II) Complexes. J. Saudi Chem. Soc. 2015, 19, 207–216.
[23] Drug resistance reversed in head and neck cancer https://www.labonline.com.au/content/life-scientist/news/drug-resistance-reversed-in-head-and-neck-cancer-1212457248 (accessed Feb 27, 2019).
[24] Pattan, S. R.; Pawar, S. B.; Vetal, S. S.; Gharate, U. D.; Bhawar, S. B. The Scope of Metal Complexes in Drug Design–a Review. Indian drugs 2012, 49, 5–12.
[25] Ott, I.; Gust, R. Non Platinum Metal Complexes as Anti-Cancer Drugs. Arch. Pharm. 2007, 340, 117–126.
[26] Ott, I.; Gust, R. Preclinical and Clinical Studies on the Use of Platinum Complexes for Breast Cancer Treatment. Anticancer. Agents Med. Chem. 2007, 7, 95–110.
[27] Ott, I.; Gust, R. Review Article Non Platinum Metal Complexes as Anti-Cancer Drugs. Arch Pharm Chem. Life Sci. 2007, 340, 117–126.
[28] Korfel, A.; Scheulen, M. E.; Hans-Joachim, S.; Grundel, O.; Harstrick, A.; Knoche, M.; Fels, L. M.; Skorzec, M.; Bach, F.; Baumgart, J.; Safi, G.; Seeber, S.; Thiel, E.; Benjamin, W. E. Phase I Clinical and Pharmacokinetic Dichloride in

191
Adults with Advanced Study of Titanocene Solid Tumors Agnieszka. Clin. Cancer Res. 1998, 4, 2701–2708.
[29] Kroger, N.; Kleeberg, U. R.; Mross, K.; Edler, L.; Sass, G.; Hossfeld, D. K. Phase II Clinical Trial of Titanocene Dichloride in Patients with Metastatic Breast Cancer. Onkologie 2000, 23, 60–62.
[30] Sun, H.; Li, H.; Weir, R. A.; Sadler, P. J. The First Specific Ti(IV)-Protein Complex: Potential Relevance to Anticancer Activity of Titanocenes. Angew. Chemie - Int. Ed. 1998, 37, 1577–1579.
[31] Guo, M.; Harvey, I.; Campopiano, D. J.; Sadler, P. J. Short Oxo-titanium(IV) Bond in Bacterial Transferrin: A Protein Target for Metalloantibiotics. Angew. Chemie - Int. Ed. 2006, 45, 2758–2761.
[32] Hartinger, C. G.; Jakupec, M. a; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P. J.; Keppler, B. K. KP1019, a New Redox-Active Anticancer Agent--Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Bodiversity 2008, 5, 2140–2155.
[33] Vacca, A.; Bruno, M.; Boccarelli, A.; Coluccia, M.; Ribatti, D.; Bergamo, A.; Garbisa, S.; Sartor, L.; Sava, G. Inhibition of Endothelial Cell Functions and of Angiogenesis by the Metastasis Inhibitor NAMI-A. Br. J. Cancer 2002, 86, 993–998.
[34] Morbidelli, L.; Donnini, S.; Filippi, S.; Messori, L.; Piccioli, F.; Orioli, P.; Sava, G.; Ziche, M. Antiangiogenic Properties of Selected ruthenium(III) Complexes That Are Nitric Oxide Scavengers. Br. J. Cancer 2003, 88, 1484–1491.
[35] Rademaker-Lakhai, J. M.; Bongard, D. Van Den; Pluim, D. A Phase I and Pharmacological Study with Imidazolium- Trans-DMSO-Imidazole-Tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727.
[36] Hartinger, C. G.; Zorbas-Seifried, S.; Jakupec, M. A.; Kynast, B.; Zorbas, H.; Keppler, B. K. From Bench to Bedside - Preclinical and Early Clinical Development of the Anticancer Agent Indazolium Trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904.
[37] Bernstein, L. R. Mechanisms of Therapeutic Activity for Gallium. Pharmacol. Rev. 1998, 50, 665–682.
[38] Senderowicz, A. M.; Reid, R.; Headlee, D.; Abornathy, T.; Horti, J.; Lush, R. M.; Reed, E.; Figg, W. D.; Sausville, E. A. A Phase II Trial of Gallium Nitrate in Patients with Androgen-Metastatic Prostate Cancer. Urol. Int. 1999, 63, 120–125.
[39] Galanski, M.; Arion, V. B.; Jakupec, M. A.; Keppler, B. K. Recent Developments in the Field of Tumor-Inhibiting Metal Complexes. Curr. Pharm. Des. 2003, 9, 2078–2089.

192
[40] Louie, A. Y.; Meade, T. J. Metal Complexes as Enzyme Inhibitors. Chem. Rev. 1999, 99, 2711–2734.
[41] Timerbaev, A. R. Advances in Developing tris(8-quinolinolato)gallium(III) as an Anticancer Drug: Critical Appraisal and Prospects. Metallomics 2009, 1, 193–198.
[42] Kopf-Maier, P.; Kopf, H.; Neuse, E. W. Ferricenium Complexes : A New Type of Water-Soluble Antitumor Agent. J. Cancer Res. Clin. Oncol. 1984, 108, 336–340.
[43] Hillard, E.; Vessières, A.; Thouin, L.; Jaouen, G.; Amatore, C. Ferrocene-Mediated Proton-Coupled Electron Transfer in a Series of Ferrocifen-Type Breast-Cancer Drug Candidates. Angew. Chemie - Int. Ed. 2006, 45, 285–290.
[44] Wong, E. L.-M.; Fang, G.-S.; Che, C.-M.; Zhu, N. Highly Cytotoxic Iron(II) Complexes with Pentadentate Pyridyl Ligands as a New Class of Anti-Tumor Agents. Chem. Commun. 2005, 36, 4578–4580.
[45] Jung, M.; Kerr, D. E.; Senter, P. D. Bioorganometallic Chemistry--Synthesis and Antitumor Activity of Cobalt Carbonyl Complexes. Arch. Pharm. (Weinheim). 1997, 330, 173–176.
[46] Ott, I.; Schmidt, K.; Kircher, B.; Schumacher, P.; Wiglenda, T.; Gust, R. Antitumor-Active Cobalt-Alkyne Complexes Derived from Acetylsalicylic Acid: Studies on the Mode of Drug Action. J. Med. Chem. 2005, 48, 622–629.
[47] Ott, I.; Abraham, A.; Schumacher, P.; Shorafa, H.; Gastl, G.; Gust, R.; Kircher, B. Synergistic and Additive Antiproliferative Effects on Human Leukemia Cell Lines Induced by Combining Acetylenehexacarbonyldicobalt Complexes with the Tyrosine Kinase Inhibitor Imatinib. J. Inorg. Biochem. 2006, 100, 1903–1906.
[48] Simon, T. M.; Kunishima, D. H.; Vibert, G. J.; Lorber, A. Inhibitory Effects of a New Oral Gold Compound on HELA Cells. Cancer 1979, 44, 1965–1975.
[49] Hoke, G. D.; Rush, G. F.; Mirabelli, C. K. The Mechanism of Acute Cytotoxicity of Triethylphosphine Gold(l) Complexes. Toxicol. Appl. Pharmacol. 1989, 99, 50–60.
[50] McKeage, M. J.; Maharaj, L.; Berners-Price, S. J. Mechanisms of Cytotoxicity and Antitumor Activity of Gold(I) Phosphine Complexes: The Possible Role of Mitochondria. Coord. Chem. Rev. 2002, 232, 127–135.
[51] Powis, G.; Kirkpatrick, D. L. Thioredoxin Signaling as a Target for Cancer Therapy. Curr. Opin. Pharmacol. 2007, 7, 392–397.
[52] Gromer, S.; Urig, S.; Becker, K. The Thioredoxin System - From Science to Clinic. Med. Res. Rev. 2004, 24, 40–89.
[53] Liu, K.; Yan, H.; Chang, G.; Li, Z.; Niu, M.; Hong, M. Organotin(IV) Complexes Derived from Hydrazone Schiff Base: Synthesis, Crystal Structure, In Vitro Cytotoxicity and DNA/BSA Interactions. Inorganica Chim. Acta 2017, 464, 137–146.

193
[54] Saeed, A.; Channar, P. A.; Larik, F. A.; Jabeen, F.; Muqadar, U.; Saeed, S.; Flörke, U.; Ismail, H.; Dilshad, E.; Mirza, B. Design, Synthesis, Molecular Docking Studies of Organotin-Drug Derivatives as Multi-Target Agents against Antibacterial, Antifungal, α-Amylase, α-Glucosidase and Butyrylcholinesterase. Inorganica Chim. Acta 2017, 464, 204–213.
[55] Wang, X.; Qiang, J.; Ni, X.; Jia, A.; Ma, X.; Tong, B.; Zhang, Q. Synthesis, Structure and DFT Calculation of a Facial Tris-Cyclometalated Phosphorescent Iridium(III) Complex Containing Substituted Phthalazine Ligands. Inorganica Chim. Acta 2017, 466, 343–348.
[56] Davies, A. G.; Gielen, M.; Pannell, K. H.; Tiekink, E. R. T. Tin Chemistry: Fundamentals, Fronteir, and Applications; Wiley: United Kingdom, 2008.
[57] Arjmand, F.; Parveen, S.; Tabassum, S.; Pettinari, C. Organo-Tin Antitumor Compounds : Their Present Status in Drug Development and Future Perspectives. Inorganica Chim. Acta 2014, 423, 26–37.
[58] Pellerito, L.; Nagy, L. Organotin(IV)n+ Complexes Formed with Biologically Active Ligands: Equilibrium and Structural Studies, and Some Biological Aspects. Coord. Chem. Rev. 2002, 224, 111–150.
[59] Pellerito, C.; Nagy, L.; Pellerito, L.; Szorcsik, A. Biological Activity Studies on organotin(IV)n+ Complexes and Parent Compounds. J. Organomet. Chem. 2006, 691, 1733–1747.
[60] Casas, J. S.; Castellano, E. E.; Couce, M. D.; Ellena, J.; Sanchez, A.; Sanchez, J. L.; Sordo, J.; Taboada, C. The Reaction of Dimethyltin(IV) Dichloride with Thiamine Diphosphate (H2TDP): Synthesis and Structure of [SnMe2(HTDP)(H2O]Cl·H2O, and Possibility of a Hitherto Unsuspected Role of the Metal Cofactor in the Mechanism of Vitamin-B 1 -Dependent Enzymes. Inorg. Chem. 2004, 43, 1957–1963.
[61] Gennari, A.; Viviani, B.; Galli, C. L.; Marinovich, M.; Pieters, R.; Corsini, E. Organotins Induce Apoptosis by Disturbance of [Ca2+]i and Mitochondrial Activity, Causing Oxidative Stress and Activation of Caspases in Rat Thymocytes. Toxicol. Appl. Pharmacol. 2000, 169, 185–190.
[62] Jaiswal, N. Organotin (IV) Complexes and DNA Interaction : A Promising Future for Tin Based Metallodrugs. Int. J. ChemTech Res. 2017, 10, 495–499.
[63] Collier, W. A. Zur Experimentellen Therapie Der Tumoren. H , Die Wirksamkeit Einiger Metallorganischen Blei- Uud Zinnverbindungen. Zeit. Hyg. Infektkr 1929, 10, 169–174.
[64] Crowe, A. J.; Smith, P. J. Investigations into the Antitumour Activity of Organotin Compounds. 2.* Diorganotin Dihalide and Di-Pseudohalide Complexes. Inorganica Chim. Acta 1984, 93, 179–184178.
[65] Haque, R. A.; Salam, M. A.; Arafath, M. A. New organotin(IV) Complexes with N (4)-Methylthiosemicarbazone Derivatives Prepared from 2,3-

194
Dihydroxybenzaldehyde and 2-Hydroxy-5-Methylbenzaldehyde: Synthesis, Characterization, and Cytotoxic Activity. J. Coord. Chem. 2015, 68, 2953–2967.
[66] Yang, Y.; Hong, M.; Xu, L.; Cui, J.; Chang, G.; Li, D.; Li, C. Organotin(IV) Complexes Derived from Schiff Base N’-[(1E)-(2-Hydroxy-3- methoxyphenyl)methylidene]pyridine-3-carbohydrazone: Synthesis, In Vitro Cytotoxicities and DNA/BSA Interaction. J. Organomet. Chem. 2016, 804, 48–58.
[67] Arakawa, Y. Invasion of Biofunctions by Organotins-Immune System, Brain Nervous System and Endocrine System. Biomed. Res. Trace Elem. 2000, 11, 269–286.
[68] Shang, X.; Meng, X.; Alegria, E. C. B. A.; Li, Q.; Guedes Da Silva, M. F. C.; Kuznetsov, M. L.; Pombeiro, A. J. L. Syntheses, Molecular Structures, Electrochemical Behavior, Theoretical Study, and Antitumor Activities of Organotin(IV) Complexes Containing 1-(4-Chlorophenyl)-1-Cyclopentanecarboxylato Ligands. Inorg. Chem. 2011, 50, 8158–8167.
[69] Hadi, S.; Rilyanti, M. Synthesis and in Vitro Anticancer Activity of Some Organotin(IV) Benzoate Compounds. Orient. J. Chem. 2010, 26, 775–779.
[70] Rehman, W.; Badshah, A.; Rahim, F.; Baloch, M. K.; Ullah, H.; Abid, O. U. R.; Nawaz, M.; Tauseef, I. Synthesis, Spectral Characterization, Antibacterial and Antitumor Studies of Some diorganotin(IV) Complexes Derived from 2-Phenylmonomethylglutarate. Inorganica Chim. Acta 2014, 423, 177–182.
[71] Jiménez-Pérez, V. M.; García-López, M. C.; Muñoz-Flores, B. M.; Chan-Navarro, R.; Berrones-Reyes, J. C.; Dias, H. V. R.; Moggio, I.; Arias, E.; Serrano-Mireles, J. A.; Chavez-Reyes, A. New Application of Fluorescent Organotin Compounds Derived from Schiff Bases: Synthesis, X-Ray Structures, Photophysical Properties, Cytotoxicity and Fluorescent Bioimaging. J. Mater. Chem. B 2015, 3, 5731–5745.
[72] Agiorgiti, M. S.; Evangelou, A.; Vezyraki, P.; Hadjikakou, S. K.; Kalfakakou, V.; Tsanaktsidis, I.; Batistatou, A.; Zelovitis, J.; Simos, Y. V.; Ragos, V.; Karkabounas, S.; Peschos, D. Cytotoxic Effect, Antitumour Activity and Toxicity of Organotin Derivatives with Ortho- or Para-Hydroxy-Benzoic Acids. Med. Chem. Res. 2018, 27, 1122–1130.
[73] Shang, X.; Zhao, B.; Xiang, G.; Guedes Da Silva, M. F. C.; Pombeiro, A. J. L. Dimeric Diorganotin(IV) Complexes with Arylhydrazones of β-Diketones: Synthesis, Structures, Cytotoxicity and Apoptosis Properties. RSC Adv. 2015, 5, 45053–45060.
[74] Gholivand, K.; Salami, R.; Shahsavari, Z.; Torabi, E. Novel Binuclear and Polymeric Diorganotin(IV) Complexes with N-Nicotinyl Phosphoramides: Synthesis, Characterization, Structural Studies and Anticancer Activity. J. Organomet. Chem. 2016, 819, 155–165.
[75] Zhong, G.-Y. Synthesis of [C5H5NH][Ph2Sn(μ2-SCH2COO)CI] with Good Biological Activity In Vitro Anti-Tumor. Adv. Mater. Res. 2013, 726–731, 222–224.

195
[76] Pellerito, L.; Prinzivalli, C.; Casella, G.; Fiore, T.; Pellerito, O.; Giuliano, M.; Scopelliti, M.; Pellerito, C. Diorganotin(IV) N-Acetyl-L-Cysteinate Complexes: Synthesis, Solid State, Solution Phase, DFT and Biological Investigations. J. Inorg. Biochem. 2010, 104, 750–758.
[77] Xanthopoulou, M. N.; Hadjikakou, S. K.; Hadjiliadis, N.; Kubicki, M.; Skoulika, S.; Bakas, T.; Baril, M.; Butler, I. S. Synthesis, Structural Characterization, and Biological Studies of Six- and Five-Coordinate Organotin(IV) Complexes with the Thioamides. Inorg. Chem. 2007, 46, 1187–1195.
[78] Xanthopoulou, M. N.; Hadjikakou, S. K.; Hadjiliadis, N.; Milaeva, E. R.; Gracheva, J. A.; Tyurin, V. Y.; Kourkoumelis, N.; Christoforidis, K. C.; Metsios, A. K.; Karkabounas, S.; Charalabopoulos, K. Biological Studies of New Organotin(IV) Complexes of Thioamide Ligands. Eur. J. Med. Chem. 2008, 43, 327–335.
[79] Milaeva, E. R.; Shpakovsky, D. B.; Gracheva, Y. A.; Antonenko, T. A.; Osolodkin, D. I.; Palyulin, V. A.; Shevtsov, P. N.; Neganova, M. E.; Vinogradova, D. V.; Shevtsova, E. F. Some Insight into the Mode of Cytotoxic Action of Organotin Compounds with Protective 2,6-Di- Tert -Butylphenol Fragments. J. Organomet. Chem. 2014, 782, 96–102.
[80] Adeyemi, J. O.; Onwudiwe, D. C.; Singh, M. Synthesis, Characterization, and Cytotoxicity Study of Organotin(IV) Complexes Involving Different Dithiocarbamate Groups. J. Mol. Struct. 2019, 1179, 366–375.
[81] Pillai, V.; Patel, S. K.; Buch, L.; Singh, V. K. Binuclear Diphenyltin(IV)dithiocarbamate Complexes Bearing Functionalized Linkers: Synthesis, Spectral Characterization, DFT and In Vitro Anticancer Activity. Appl. Organomet. Chem. 2018, 32, 1–12.
[82] Tian, L.; Zheng, X.; Zheng, X.; Sun, Y.; Yan, D.; Tu, L. Synthesis, Structure and Cytotoxic Activity of Binuclear Phenyltin(IV) Complexes with N,N-bis(2-Hydroxybenzyl)-1,2-ethanebis(dithiocarbamate) Ligand. Appl. Organomet. Chem. 2011, 25, 785–790.
[83] Yousefi, M.; Safari, M.; Torbati, M. B.; Kazemiha, V. M.; Sanati, H.; Amanzadeh, A. New Mononuclear Diorganotin(IV) Dithiocarboxylates: Synthesis, Characterization and Study of Their Cytotoxic Activities. Appl. Organomet. Chem. 2012, 26, 438–444.
[84] Ali, M. A.; Livingstone, S. E. Metal Complexes of Sulphur-Nitrogen Chelating Agents. Coord. Chem. Rev. 1974, 13, 101–132.
[85] Hamid, M. H. S. A.; Said, A. N. A. H.; Mirza, A. H.; Karim, M. R.; Arifuzzaman, M.; Akbar Ali, M.; Bernhardt, P. V. Synthesis, Structures and Spectroscopic Properties of Some Tin(IV) Complexes of the 2-Acetylpyrazine Schiff Bases of S-Methyl- and S-Benzyldithiocarbazates. Inorganica Chim. Acta 2016, 453, 742–750.
[86] Kumar, A.; Chaudhary, P.; Singh, R.; Kaushik, N. K. Organotin(IV) Complexes

196
of Thiohydrazones of Phenethylamine: Synthesis, Characterization, Biological and Thermal Study. Main Gr. Chem. 2016, 15, 163–178.
[87] Belwal, S.; Singh, R. V. Inventive Triorgano Tin(IV) Complexes of Biologically Potent Schiff Base Derivatives. Eur. J. Adv. Eng. Technol. 2015, 2, 34–42.
[88] Beshir, A. B.; Guchhait, S. K.; Gascón, J. A.; Fenteany, G. Synthesis and Structure-Activity Relationships of Metal-Ligand Complexes That Potently Inhibit Cell Migration. Bioorganic Med. Chem. Lett. 2008, 18, 498–504.
[89] Vijayan, P.; Viswanathamurthi, P.; Sugumar, P.; Ponnuswamy, M. N.; Balakumaran, M. D.; Kalaichelvan, P. T.; Velmurugan, K.; Nandhakumar, R.; Butcher, R. J. Unprecedented Formation of Organo-ruthenium(II) Complexes Containing 2-Hydroxy-1-Naphthaldehyde S-Benzyldithiocarbazate: Synthesis, X-ray Crystal Structure, DFT Study and Their Biological Activities In Vitro. Inorg. Chem. Front. 2015, 2, 620–639.
[90] Tian, L.; Yu, H.; Zheng, X.; Liu, X. Synthesis, Crystal Structure and Cytotoxic Activity of Tricyclohexyltin Complexes of Benzenedioxyacetic Acids. Appl. Organomet. Chem. 2015, 29, 725–729.
[91] Akbar Ali, M.; Huq Mirza, A.; Kok Wei, L.; Bernhardt, P. V.; Atchade, O.; Song, X.; Eng, G.; May, L. Synthesis and Characterization of Pentagonal Bipyramidal Organotin(IV) Complexes of 2,6-Diacetylpyridine Schiff Bases of S-Alkyl- and Aryldithiocarbazates. J. Coord. Chem. 2010, 63, 1194–1206.
[92] Ali, M. A.; Majumder, S. M. M.-H.; Butcher, R. J.; Jasinski, J. P.; Jasinski, J. M. The Preparation and Characterization of Bis-chelated Nickel(ll) Complexes of the 6-Methylpyridine-2-carboxaldehyde Schiff Bases of S-Alkyldithiocarbazates and the X-ray Crystal Structure of the BisS-methyl-β-N-(6-methylpyrid-2-yl)- methylenedithiocarbazatonickel(ll) Complex.. Polyhedron 1997, 16, 2749–2754.
[93] Akbar Ali, M.; Mirza, A. H.; Hamid, M. H. S. A.; Bernhardt, P. V. Diphenyltin(IV) Complexes of the 2-Quinolinecarboxaldehyde Schiff Bases of S-Methyl- and S-Benzyldithiocarbazate (Hqaldsme and Hqaldsbz): X-ray Crystal Structures of Hqaldsme and Two Conformers of Its Diphenyltin(IV) Complex. Polyhedron 2005, 24, 383–390.
[94] Ali, M. A.; Mirza, A. H.; Butcher, R. J.; Crouse, K. A. The Preparation, Characterization and Biological Activity of Palladium(II) and Platinum(II) Complexes of Tridentate NNS Ligands Derived from S-Methyl- and S-Benzyldithiocarbazates and the X-ray Crystal Structure of the [Pd(mpasme)Cl] Complex. Transit. Met. Chem. 2006, 31, 79–87.
[95] Elsayed, S. A.; Noufal, A. M.; El-Hendawy, A. M. Synthesis, Structural Characterization and Antioxidant Activity of Some Vanadium(IV), Mo(VI)/(IV) and Ru(II) Complexes of Pyridoxal Schiff Base Derivatives. J. Mol. Struct. 2017, 1144, 120–128.
[96] Low, M. L.; Maigre, L.; Tahir, M. I. M.; Tiekink, E. R. T.; Dorlet, P.; Guillot, R.; Ravoof, T. B.; Rosli, R.; Pages, J. M.; Policar, C.; Delsuc, N.; Crouse, K. A. New

197
Insight into the Structural, Electrochemical and Biological Aspects of Macroacyclic Cu(II) Complexes Derived from S-Substituted Dithiocarbazate Schiff Bases. Eur. J. Med. Chem. 2016, 120, 1–12.
[97] Omar, S. A.; Ravoof, T. B. S. A.; Tahir, M. I. M.; Crouse, K. A. Synthesis and Characterization of Mixed-Ligand copper(II) Saccharinate Complexes Containing Tridentate NNS Schiff Bases. X-Ray Crystallographic Analysis of the Free Ligands and One Complex. Transit. Met. Chem. 2013, 39, 119–126.
[98] Pavan, F. R.; Maia, P. I. da S.; Leite, S. R. A.; Deflon, V. M.; Batista, A. A.; Sato, D. N.; Franzblau, S. G.; Leite, C. Q. F. Thiosemicarbazones, Semicarbazones, Dithiocarbazates and Hydrazide/hydrazones: Anti - Mycobacterium Tuberculosis Activity and Cytotoxicity. Eur. J. Med. Chem. 2010, 45, 1898–1905.
[99] Ravoof, T. B. S. A.; Crouse, K. A.; Tahir, M. I. M.; How, F. N. F.; Rosli, R.; Watkins, D. J. Synthesis, Characterization and Biological Activities of 3-Methylbenzyl 2-(6-Methylpyridin-2-ylmethylene)hydrazine Carbodithioate and Its Transition Metal Complexes. Transit. Met. Chem. 2010, 35, 871–876.
[100] Singh, H. L.; Varshney, A. K. Synthetic, Structural, and Biochemical Studies of Organotin(IV) with Schiff Bases Having Nitrogen and Sulphur Donor Ligands. Bioinorg. Chem. Appl. 2006, 1–7.
[101] Yusof, E. N. M.; Ravoof, T. B. S. A.; Jamsari, J.; Tiekink, E. R. T.; Veerakumarasivam, A.; Crouse, K. A.; Tahir, M. I. M.; Ahmad, H. Synthesis, Characterization and Biological Studies of S-4-Methylbenzyl-β-N-(2-Furylmethylene)dithiocarbazate (S4MFuH) Its Zn2+, Cu2+, Cd2+ and Ni2+ Complexes. Inorganica Chim. Acta 2015, 438, 85–93.
[102] Shahzadi, S.; Ali, S.; Fettouhi, M. Synthesis, Spectroscopy, In Vitro Biological Activity and X-ray Structure of (4-Methylpiperidine-Dithiocarbamato-S,S′)triphenyltin(IV). J. Chem. Crystallogr. 2008, 38, 273–278.
[103] Ravoof, T. B. S. A.; Crouse, K. A.; Tahir, M. I. M.; Rosli, R.; Watkin, D. J.; How, F. N. F. Synthesis, Characterisation and Biological Activities of 2-Methylbenzyl 2-(Dipyridin-2-ylmethylene)hydrazinecarbodithioate. J. Chem. Crystallogr. 2011, 41, 491–495.
[104] Tarafder, M. T. H.; Ali, M. A.; Wee, D. J.; Azahari, K.; Silong, S.; Crouse, K. A. Complexes of a Tridentate ONS Schiff Base. Synthesis and Biological Properties. Transit. Met. Chem. 2000, 25, 456–460.
[105] Chew, K.-B.; Tarafder, M. T. .; Crouse, K. A.; Ali, A. .; Yamin, B. .; Fun, H.-K. Synthesis, Characterization and Bio-Activity of Metal Complexes of Bidentate N–S Isomeric Schiff Bases Derived from S-Methyldithiocarbazate (SMDTC) and the X-Ray Structure of the bis[S-Methyl-β-N-(2-Furyl-methylketone)dithiocarbazato]cadmium(II) Complex. Polyhedron 2004, 23, 1385–1392.
[106] Tarafder, M. T. H.; Chew, K.; Crouse, K. A.; Ali, A. M.; Yamin, B. M.; Fun, H. K. Synthesis and Characterization of Cu(II), Ni(II) and Zn(II) Metal Complexes of

198
Bidentate NS Isomeric Schiff Bases Derived from S-Methyldithiocarbazate (SMDTC): Bioactivity of the Bidentate NS Isomeric Schiff Bases , Some of Their Cu(II), Ni(II) and Zn(II). Polyhedron 2002, 21, 2683–2690.
[107] How, F. N.-F.; Crouse, K. A.; Tahir, M. I. M.; Tarafder, M. T. H.; Cowley, A. R. Synthesis, Characterization and Biological Studies of S-Benzyl-β-N-(Benzoyl) Dithiocarbazate and Its Metal Complexes. Polyhedron 2008, 27, 3325–3329.
[108] Crouse, K. A.; Chew, K. B.; Tarafder, M. T. H.; Kasbollah, A.; Ali, A. M.; Yamin, B. M.; Fun, H. K. Synthesis, Characterization and Bio-Activity of S-2-Picolyldithiocarbazate (S2PDTC), Some of Its Schiff Bases and Their Ni(II) Complexes and X-Ray Structure of S-2-Picolyl-β-N-(2-Acetylpyrrole)dithiocarbazate. Polyhedron 2004, 23, 161–168.
[109] Begum, M. S.; Zangrando, E.; Sheikh, M. C.; Miyatake, R.; Howlader, M. B. H.; Rahman, M. N.; Ghosh, A. Bischelated Complexes of a Dithiocarbazate N,S Schiff Base Ligand: Synthesis, Characterization and Antimicrobial Activities. Transit. Met. Chem. 2017, 42, 553–563.
[110] Yusof, E. N. M.; Ravoof, T. B. S. A.; Tiekink, E. R. T.; Veerakumarasivam, A.; Crouse, K. A.; Tahir, M. I. M.; Ahmad, H. Synthesis, Characterization and Biological Evaluation of Transition Metal Complexes derived from N, S Bidentate Ligands. Int. J. Mol. Sci. 2015, 16, 11034–11054.
[111] Islam, M. A. A. A. A.; Sheikh, M. C.; Alam, M. S.; Zangrando, E.; Alam, M. A.; Tarafder, M. T. H.; Miyatake, R. Synthesis, Characterization and Bio-Activity of a Bidentate NS Schiff Base of S-Allyldithiocarbazate and Its Divalent Metal Complexes: X-Ray Crystal Structures of the Free Ligand and Its nickel(II) Complex. Transit. Met. Chem. 2014, 39, 141–149.
[112] Khoo, T.-J.; Break, M. K. B.; Crouse, K. A.; Tahir, M. I. M.; Ali, A. M.; Cowley, A. R.; Watkin, D. J.; Tarafder, M. T. H. Synthesis, Characterization and Biological Activity of Two Schiff Base Ligands and Their Nickel(II), Copper(II), Zinc(II) and Cadmium(II) Complexes derived from S-4-Picolyldithiocarbazate and X-Ray Crystal Structure of Cadmium(II) Complex derived from Pyridine-2-carboxaldehyde. Inorganica Chim. Acta 2014, 413, 68–76.
[113] Zangrando, E.; Begum, M. S.; Sheikh, M. C.; Miyatake, R.; Hossain, M. M.; Alam, M. M.; Hasnat, M. A.; Halim, M. A.; Ahmed, S.; Rahman, M. N.; Gosh, A. Synthesis, Characterization, Density Functional Study and Antimicrobial Evaluation of a Series of Bischelated Complexes with a Dithiocarbazate Schiff Base Ligand. Arab. J. Chem. 2017, 10, 172–184..
[114] Zangrando, E.; Begum, M. S.; Miyatake, R.; Sheikh, M. C.; Hossain, M. M. Crystal Structure of bisS-Hexyl 3-[4-Dimethylamino)benzylidene]dithiocarbazato-κ 2 N 3,S -copper(II). Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, E71, 706–708.
[115] Li, H.-Q.; Luo, Y.; Li, D.-D.; Zhu, H.-L. (E)-4-Chlorobenzyl 3-(3-Nitrobenzylidene)dithiocarbazate. Acta Crystallogr. Sect. E Struct. Reports Online 2009, E65, o3101.

199
[116] Ali, M. A.; Mirza, A. H.; Tan, A. L.; Wei, L. K.; Bernhardt, P. V. The Preparation and Characterization of tin(IV) Complexes of 2-Quinolinecarboxaldehyde Schiff Bases of S-Methyl- and S-Benzyldithiocarbazates and the X-Ray Crystal and Molecular Structures of the 2-Quinolinecarboxaldehyde Schiff Base of S-Benzyldithiocarb. Polyhedron 2004, 23, 2405–2412.
[117] Ali, M. A.; Huq, A.; Haniti, M.; Hamid, S. A.; Bernhardt, P. V; Atchade, O.; Song, X.; Eng, G.; May, L. Synthesis , Spectroscopic and Structural Characterization of Diphenyltin(IV) Complexes of Acetone Schiff Bases of S -Alkyldithiocarbazates. Polyhedron 2008, 27, 977–984.
[118] Yekke-Ghasemi, Z.; Takjoo, R.; Ramezani, M.; Mague, J. T. Molecular Design and Synthesis of New Dithiocarbazate Complexes; Crystal Structure, Bioactivities and Nano Studies. RSC Adv. 2018, 8, 41795–41809.
[119] Scovill, J. P.; Klayman, D. L.; Lambros, C.; Childs, G. E.; Notsch, J. D. 2-Acetylpyridine Thiosemicarbazones. 9. Derivatives of 2-Acetylpyridine 1-Oxide as Potential Antimalarial Agents. J. Med. Chem. 1984, 27, 87–91.
[120] Hu, W.; Zhou, W.; Xia, C.; Wen, X. Synthesis and Anticancer Activity of Thiosemicarbazones. Bioorg. Med. Chem. Lett. 2006, 16, 2213–2218.
[121] Kovala-demerzi, D.; Domopoulou, A.; Demertzis, M. A.; Valle, G.; Papageorgiou, A. Palladium(II) Complexes of 2- Acetylpyridine N(4)-Methyl, N(4)-Ethyl and N(4)-Phenyl-Thiosemicarbazones. Crystal Structure of Chloro(2-Acetylpyridine N(4)-Methylthiosemicarbazonato) Palladium(II). Synthesis, Spectral Studies, In Vitro and In Vivo Antitumour. J. Inorg. Biocemistry 1997, 68, 147–155.
[122] Khan, S. A.; Yusuf, M. Synthesis, Spectral Studies and In Vitro Antibacterial Activity of Steroidal Thiosemicarbazone and Their Palladium (Pd(II)) Complexes. Eur. J. Med. Chem. 2009, 44, 2270–2274.
[123] Đilović, I.; Rubčić, M.; Vrdoljak, V.; Pavelić, S. K.; Kralj, M.; Piantanida, I.; Cindrić, M. Novel Thiosemicarbazone Derivatives as Potential Antitumor Agents: Synthesis, Physicochemical and Structural Properties, DNA Interactions and Antiproliferative Activity. Bioorg. Med. Chem. 2008, 16, 5189–5198.
[124] Palanimuthu, D.; Poon, R.; Sahni, S.; Anjum, R.; Hibbs, D.; Lin, H. Y.; Bernhardt, P. V.; Kalinowski, D. S.; Richardson, D. R. A Novel Class of Thiosemicarbazones Show Multi-Functional Activity for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2017, 139, 612–632.
[125] Parrilha, G. L.; Da Silva, J. G.; Gouveia, L. F.; Gasparoto, A. K.; Dias, R. P.; Rocha, W. R.; Santos, D. A.; Speziali, N. L.; Beraldo, H. Pyridine-Derived Thiosemicarbazones and Their Tin(IV) Complexes with Antifungal Activity against Candida Spp. Eur. J. Med. Chem. 2011, 46, 1473–1482.
[126] Satheesh, D.; Jayanthi, K. An In Vitro Antibacterial and Antifungal Activities of Copper(II) and Zinc(II) Complexes of N4-Methyl-3-Thiosemicarbazones. Int. J. Chem. Pharm. Anal. 2017, 1–7.

200
[127] Singh, H. L.; Singh, J. B.; Bhanuka, S. Synthesis, Spectroscopic Characterization, Biological Screening, and Theoretical Studies of Organotin(IV) Complexes of Semicarbazone and Thiosemicarbazones Derived from (2-Hydroxyphenyl)(pyrrolidin-1-yl)methanone. Res. Chem. Intermed. 2016, 42, 997–1015.
[128] Wiecek, J.; Dokorou, V.; Ciunik, Z.; Kovala-Demertzi, D. Organotin Complexes of Pyruvic Acid Thiosemicarbazone: Synthesis, Crystal Structures and Antiproliferative Activity of Neutral and Cationic Diorganotin Complexes. Polyhedron 2009, 28, 3298–3304.
[129] Oliveira, A. A.; Franco, L. L.; dos Santos, R. G.; Perdigão, G. M. C.; da Silva, J. G.; Souza-Fagundes, E. M.; Beraldo, H. Neutron Activation of In(III) Complexes with Thiosemicarbazones Leads to the Production of Potential Radiopharmaceuticals for the Treatment of Breast Cancer. New J. Chem. 2017, 41, 9041–9050.
[130] Giles, F. J.; Fracasso, P. M.; Kantarjian, H. M.; Cortes, J. E.; Brown, R. A.; Verstovsek, S.; Alvarado, Y.; Thomas, D. A.; Faderl, S.; Garcia-Manero, G.; Wright, L. P.; Samson, T.; Cahill, A.; Lambert, P.; Plunkett, W.; Sznol, M.; DiPersio, J. F.; Gandhi, V. Phase I and Pharmacodynamic Study of Triapine®, a Novel Ribonucleotide Reductase Inhibitor, in Patients with Advanced Leukemia. Leuk. Res. 2003, 27, 1077–1083.
[131] Karp, J. E.; Giles, F. J.; Gojo, I.; Morris, L.; Greer, J.; Johnson, B.; Thein, M.; Sznol, M.; Low, J. A Phase I Study of the Novel Ribonucleotide Reductase Inhibitor 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP, Triapine®) in Combination with the Nucleoside Analog Fludarabine for Patients with Refractory Acute Leukemias and Aggressive Myelopro. Leuk. Res. 2008, 32, 71–77.
[132] Kunos, C. A.; Radivoyevitch, T.; Waggoner, S.; Debernardo, R.; Zanotti, K.; Resnick, K.; Fusco, N.; Adams, R.; Redline, R.; Faulhaber, P.; Dowlati, A. Radiochemotherapy plus 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone (3-AP, NSC #663249) in Advanced-Stage Cervical and Vaginal Cancers. Gynecol. Oncol. 2013, 130, 75–80.
[133] Jansson, P. J.; Sharpe, P. C.; Bernhardt, P. V.; Richardson, D. R. Novel Thiosemicarbazones of the ApT and DpT Series and Their Copper Complexes: Identification of Pronounced Redox Activity and Characterization of Their Antitumor Activity. J. Med. Chem. 2010, 53, 5759–5769.
[134] Kolberg, M.; Strand, K. R.; Graff, P.; Andersson, K. K. Structure, Function, and Mechanism of Ribonucleotide Reductases. Biochim. Biophys. Acta 2004, 1699, 1–34.
[135] Aye, Y.; Long, M. J. C.; Stubbe, J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase In Vitro and in Mammalian Cells: Tyrosyl Radical Quenching Not Involving Reactive Oxygen Species. J. Biol. Chem. 2012, 287, 35768–35778.

201
[136] Lane, D. J. R.; Mills, T. M.; Shafie, N. H.; Merlot, A. M.; Saleh Moussa, R.; Kalinowski, D. S.; Kovacevic, Z.; Richardson, D. R. Expanding Horizons in Iron Chelation and the Treatment of Cancer: Role of Iron in the Regulation of ER Stress and the Epithelial-Mesenchymal Transition. Biochim. Biophys. Acta - Rev. Cancer 2014, 1845, 166–181.
[137] Yuan, J.; Lovejoy, D. B.; Richardson, D. R. Novel Di-2-Pyridyl-Derived Iron Chelators with Marked and Selective Antitumor Activity: In Vitro and In Vivo Assessment. Blood 2004, 104, 1450–1458.
[138] Khandani, M.; Sedaghat, T.; Erfani, N.; Haghshenas, M. R.; Khavasi, H. R. Synthesis, Spectroscopic Characterization, Structural Studies and Antibacterial and Antitumor Activities of Diorganotin Complexes with 3-Methoxysalicylaldehyde Thiosemicarbazone. J. Mol. Struct. 2013, 1037, 136–143.
[139] Liu, T.; Sun, J.; Tai, Y.; Qian, H.; Li, M. Synthesis, Spectroscopic Characterization, Crystal Structure, and Biological Evaluation of a Diorganotin(IV) Complex with 2-Acetylpyridine N 4-Cyclohexylthiosemicarbazone. Inorg. Nano-Metal Chem. 2017, 47, 813–817.
[140] Salam, M. A.; Hussein, M. A.; Ramli, I.; Islam, S. Synthesis, Structural Characterization and Evaluation of Biological Activity of Organotin(IV) Complexes with 2-Hydroxy-5-methoxybenzaldehyde-N(4)-methylthiosemicarbazone. J. Organomet. Chem. 2016, 813, 71–77.
[141] Li, M. X.; Zhang, D.; Zhang, L. Z.; Niu, J. Y.; Ji, B. S. Diorganotin(IV) Complexes with 2-Benzoylpyridine and 2-Acetylpyrazine N(4)-Phenylthiosemicarbazones: Synthesis, Crystal Structures and Biological Activities. J. Organomet. Chem. 2011, 696, 852–858.
[142] Wang, F.; Yin, H.; Cui, J.; Zhang, Y.; Geng, H.; Hong, M. Synthesis, Structural Characterization, In Vitro Cytotoxicities, DNA-Binding and BSA Interaction of Diorganotin(IV) Complexes Derived from Hydrazone Schiff Base. J. Organomet. Chem. 2014, 759, 83–91.
[143] Rehman, W.; Badshah, A.; Khan, S.; Tuyet, L. T. A. Synthesis, Characterization, Antimicrobial and Antitumor Screening of Some Diorganotin(IV) Complexes of 2-[(9H-Purin-6-ylimino)]-phenol. Eur. J. Med. Chem. 2009, 44, 3981–3985.
[144] Vicente-Dueñas, C.; Romero-Camarero, I.; Cobaleda, C.; Sánchez-García, I. Function of Oncogenes in Cancer Development: A Changing Paradigm. EMBO J. 2013, 32, 1502–1513.
[145] Shawish, H. B.; Wong, W. Y.; Wong, Y. L.; Loh, S. W.; Looi, C. Y.; Hassandarvish, P.; Phan, A. Y. L.; Wong, W. F.; Wang, H.; Paterson, I. C.; Ea, C. K.; Mustafa, M. R.; Maah, M. J. Nickel(II) Complex of Polyhydroxybenzaldehyde N4-Thiosemicarbazone Exhibits Anti-Inflammatory Activity by Inhibiting NF-κB Transactivation. PLoS One 2014, 9, 1–13.
[146] Koch, B.; Baul, T. S. B.; Chatterjee, A. Cell Proliferation Inhibition and Antitumor Activity of Novel Alkyl Series of Diorganotin(IV) Compounds. J. Appl. Toxicol.

202
2008, 28, 430–438.
[147] Marinovich, M.; Viviani, B.; Galli, C. L. Reversibility of Tributyltin-Chloride-Induced Protein Synthesis Inhibition after ATP Recovery in HEL-30 Cells. Toxicol. Lett. 1990, 52, 311–317.
[148] Lee, R. F. Metabolism of Tributyltin Oxide by Crabs, Oysters and Fish. Mar. Environ. Res. 1985, 17, 145–148.
[149] Kimmel, E. C.; Fish, R. H.; Casida, J. E. Bioorganotin Chemistry. Metabolism of Organotin Compounds in Microsomal Monooxygenase Systems and in Mammals. J. Agric. Food Chem. 1977, 25, 1–9.
[150] Basu Baul, T. S.; Paul, A.; Pellerito, L.; Scopelliti, M.; Singh, P.; Verma, P.; Duthie, A.; de Vos, D.; Tiekink, E. R. T. Dibutyltin(IV) Complexes Containing Arylazobenzoate Ligands: Chemistry, In Vitro Cytotoxic Effects on Human Tumor Cell Lines and Mode of Interaction with Some Enzymes. Invest. New Drugs 2011, 29, 285–299.
[151] Sirajuddin, M.; Ali, S.; Tahir, M. N. Pharmacological Investigation of Mono-, Di- and Tri-organotin(IV) Derivatives of Carbodithioates: Design, Spectroscopic Characterization, Interaction with SS-DNA and POM Analyses. Inorganica Chim. Acta 2016, 439, 145–158.
[152] Zeglis, B. M.; Pierre, V. C.; Barton, J. K.; Zeglis, B. M.; Pierre, V. C.; Barton, J. K. Metallo-Intercalators and Metallo-Insertors. Chem. Commun. 2007, 44, 4549–4696.
[153] Arjmand, F.; Sharma, G. C.; Sayeed, F.; Muddassir, M.; Tabassum, S. De Novo Design of Chiral Organotin Cancer Drug Candidates : Validation of Enantiopreferential Binding to Molecular Target DNA and 5’-GMP by UV–visible, Fluorescence, 1H and 31P NMR. J. Photochem. Photobiol. B Biol. 2011, 105, 167–174.
[154] Arjmand, F.; Yousuf, I. Synthesis, Characterization and In Vitro DNA Binding of Chromone Schiff Base Organotin(IV) Complexes. J. Organomet. Chem. 2013, 743, 55–62.
[155] Rehman, W.; Yasmeen, R.; Rahim, F.; Waseem, M.; Guo, C. Y.; Hassan, Z.; Rashid, U.; Ayub, K. Synthesis Biological Screening and Molecular Docking Studies of Some Tin(IV) Schiff Base Adducts. J. Photochem. Photobiol. B Biol. 2016, 164, 65–72.
[156] Doonan, F.; Cotter, T. G. Morphological Assessment of Apoptosis. Methods 2008, 44, 200–204.
[157] Chen, F.; Vallyathan, V.; Castranova, V.; Shi, X. Cell Apoptosis Induced by Carcinogenic Metals. Mol. Cell. Biochem. 2001, 222, 183–188.
[158] Chauhan, M.; Banerjee, K.; Arjmand, F. DNA Binding Studies of Novel Copper(II) Complexes Containing L-Tryptophan as Chiral Auxiliary: In Vitro

203
Antitumor Activity of Cu-Sn2 Complex in Human Neuroblastoma Cells. Inorg. Chem. 2007, 46, 3072–3082.
[159] Esmail, S. A. A.; Shamsi, M.; Chen, T.; Al-asbahy, W. M. Design, Synthesis and Characterization of Tin-Based Cancer Chemotherapy Drug Entity: In Vitro DNA Binding, Cleavage, Induction of Cancer Cell Apoptosis by Triggering DNA Damage-Mediated p53 Phosphorylation and Molecular Docking. Appl. Organomet. Chem. 2019, 33, 1–14.
[160] Nakatsu, Y.; Kotake, Y.; Ohta, S. Concentration Dependence of the Mechanisms of Tributyltin-Induced Apoptosis. Toxicol. Sci. 2007, 97, 438–447.
[161] Grondin, M.; Marion, M.; Denizeau, F.; Averill-bates, D. A. Tributyltin Induces Apoptotic Signaling in Hepatocytes through Pathways Involving the Endoplasmic Reticulum and Mitochondria. Toxicol. Appl. Pharmacol. 2007, 222, 57–68.
[162] Tada-oikawa, S.; Kato, T.; Kuribayashi, K.; Nishino, K.; Murata, M.; Kawanishi, S. Critical Role of Hydrogen Peroxide in the Differential Susceptibility of Th1 and Th2 Cells to Tributyltin-Induced Apoptosis. Biochem. Pharmacol. 2008, 75, 552–561.
[163] Hayyan, M.; Hashim, M. A.; Alnashef, I. M. Superoxide Ion : Generation and Chemical Implications. Chem. Rev. 2016, 116, 3026–3085.
[164] Devasagayam, T.; Tilak, J.; Boloor, K.; Sane, K.; Ghaskadbi, S.; Lele, R. Free Radicals and Antioxidants in Human Health: Current Status and Future Prospects. J. Assoc. Physicians India 2004, 52, 794–804.
[165] Liu, X.; Kim, C. N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 1996, 86, 147–157.
[166] Chow, S. C.; Orrenius, S. Rapid Cytoskeleton Modification in Thymocytes Induced by the Immunotoxicant Tributyltin. Toxicol. Appl. Pharmacol. 1994, 127, 19–26.
[167] Smith, P. J. Chemistry of Tin; Blackie Academic & Professionall: Glasgow, U.K, 1998.
[168] Das, V. G. K.; Gielen, M. Chemistry and Technology of Silicon and Tin; Oxford University Press: Oxford, 1992.
[169] Das, V. G. K. Main Group Elements and Their Compounds; Narosa Publishing House: New Delhi, India, 1996.
[170] Reed, J. C. Mechanisms of Apoptosis. Am. J. Pathol. 2000, 157, 1415–1430.
[171] Sasmal, A.; Garribba, E.; Ugone, V.; Rizzoli, C.; Mitra, S. Synthesis, Crystal Structures, EPR and DFT Studies of First Row Transition Metal Complexes of Lignin Model Compound Ethylvanillin. Polyhedron 2016, 121, 107–114.
[172] Singh, H. L.; Varshney, A. K. Synthesis and Characterization of Coordination

204
Compounds of Organotin(IV) with Nitrogen and Sulfur Donor Ligands. Appl. Organomet. Chem. 2001, 15, 762–768.
[173] Singh, R. V.; Chaudhary, P.; Chauhan, S.; Swami, M. Microwave-Assisted Synthesis, Characterization and Biological Activities of Organotin(IV) Complexes with Some Thio Schiff Bases. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 72, 260–268.
[174] Manan, M. A. F. A.; Tahir, M. I. M.; Crouse, K. a.; How, F. N.-F.; Watkin, D. J. Synthesis, Characterization and Antibacterial Activity of Schiff Base Derived from S-Methyldithiocarbazate and Methylisatin. J. Chem. Crystallogr. 2012, 42, 173–179.
[175] Niu, M. J.; Li, Z.; Chang, G. L.; Kong, X. J.; Hong, M.; Zhang, Q. Crystal Structure, Cytotoxicity and Interaction with DNA of Zinc(II) Complexes with O-Vanillin Schiff Base Ligands. PLoS One 2015, 10, 1–14.
[176] Basha, M. T.; Chartres, J. D.; Pantarat, N.; Ali, M. A.; HuqMirza, A.; Kalinowski, D. S.; Richardson, D. R.; Bernhardt, P. V. Heterocyclic Dithiocarbazate Iron Chelators: Fe Coordination Chemistry and Biological Activity. Dalt. Trans. 2012, 41, 6536–6548.
[177] Yusof, E. N. M.; Jotani, M. M.; Tiekink, E. R. T.; Ravoof, T. B. S. A. 2-[(1 E)-([(Benzylsulfanyl)methanethioyl]aminoimino)methyl]-6-Methoxyphenol: Crystal Structure and Hirshfeld Surface Analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 516–521.
[178] Yusof, E. N. M.; Latif, M. A. M.; Tahir, M. I. M.; Sakoff, J. A.; Simone, M. I.; Page, A. J.; Veerakumarasivam, A.; Tiekink, E. R. T.; Ravoof, T. B. S. A. O-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-Silico Studies and Cytotoxicity. Int. J. Mol. Sci. 2019, 20, 854.
[179] Ali, M. A.; Tarafder, M. T. . Metal Complexes of Sulphur and Nitrogen-Containing Ligands: Complexes of S-Benzyldithiocarbazate and a Schiff Base Formed by Its Condensation with Pyridine-2-carboxaldehyde. J. Inorg. Nucl. Chem. 1977, 39, 1785–1791.
[180] Agilent. CrysAlis PRO. Agilent Technologies: Yarnton, England 2011.
[181] Sheldrick, G. M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, A64, 112–122.
[182] Sheldrick, G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8.
[183] Farrugia, L. J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854.
[184] Brandenburg, K. DIAMOND, Crystal Impact GbR. Bonn, Germany 2006.

205
[185] Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; R., J. C.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Caricato, M., Li, Xiaosong, Hratchian, H. P., Izmaylov, Artur F., Bloino, Julien, Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, François, Bearpark, Michael J., Heyd, Jochen, Brothers, E. N., Kudin, K. N., Staroverov, V. N., Kobayashi, Rika, Normand, J., Raghavachari, Krishnan, Rendell, Alistair P., Burant, J. C., Iyengar, S. S., Tomasi, Jacopo, Cossi, M., Rega, N., Millam, N. J., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ödön, Foresman, J. B., Ortiz, J. V., Cioslowski, J., Fox, Douglas J. Gaussian 09, Revision D.01. Wallingford CT. 2013.
[186] Roy, D.; Todd, K.; John, M. GaussView, Ver 5.0.9. Semichem Inc: Shawnee Mission, KS, USA, 2009.
[187] Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789.
[188] Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652.
[189] Jeffrey, P. M.; Damian, M.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700.
[190] Chen, K.-Y.; Tsai, H.-Y. Synthesis, X-Ray Structure, Spectroscopic Properties and DFT Studies of a Novel Schiff Base. Int. J. Mol. Sci. 2014, 15, 18706–18724.
[191] Scalmani, G.; Frisch, M. J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and Properties of Excited States in the Gas Phase and in Solution: Theory and Application of a Time-Dependent Density Functional Theory Polarizable Continuum Model. J. Chem. Phys. 2006, 124, 94107.
[192] Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041.
[193] Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094.
[194] Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093.
[195] Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63.

206
[196] Sakoff, J. A.; Ackland, S. P. Thymidylate Synthase Inhibition Induces S-Phase Arrest, Biphasic Mitochondrial Alterations and Caspase-Dependent Apoptosis in Leukaemia Cells. Cancer Chemother. Pharmacol. 2000, 46, 477–487.
[197] Bergman, A. M.; van Haperen, V. W. T. R.; Veerman, G.; Kuiper, C. M.; Peters, G. J. Synergistic Interaction between Cisplatin and Gemcitabine In Vitro. Clin. Cancer Res. 1996, 2, 521–530.
[198] Ali, M. A.; Butcher, R. J.; Bryan, J. C. Synthetic, Spectroscopic and X-Ray Crystallographic Structural Studies on Some Copper(II) Complexes of the 6-Methylpyridine-2-carboxaldehyde Schiff Base of S-Methyldithiocarbazate. Inorganica Chim. Acta 1999, 287, 8–13.
[199] Anto, R. J.; Sukumaran, K.; Kuttan, G.; Rao, M. N. A.; Subbaraju, V.; Kuttan, R. Anticancer and Antioxidant Activity of Synthetic Chalcones and Related Compounds. Cancer Lett. 1995, 97, 33–37.
[200] Awidat, S. A. K. Biological Activities and Molecular Analysis of Novel Dithiocarbazate Complex Compounds on Glioma Cell Lines, Ph.D. Dissertation, Universiti Putra Malaysia, Malaysia, 2005.
[201] Bacchi, A.; Bonardi, A.; Carcelli, M.; Mazza, P.; Pelagatti, P.; Pelizzi, C.; Pelizzi, G.; Solinas, C.; Zani, F. Organotin Complexes with Pyrrole-2,5-Dicarboxaldehyde Bis(Acylhydrazones). Synthesis, Structure, Antimicrobial Activity and Genotoxicity. J. Inorg. Biochem. 1998, 69, 101–112.
[202] Jain, M.; Gaur, S.; Singh, V. P.; Singh, R. V. Organosilicon(IV) and Organotin(IV) Complexes as Biocides and Nematicides : Synthetic, Spectroscopic and Biological Studies of N∩N Donor Sulfonamide Imine and Its Chelates. Main Gr. Met. Compd. 2004, 18, 73–82.
[203] Malhotra, R.; Ravesh, A.; Singh, V. Synthesis, Characterization, Antimicrobial Activities and QSAR Studies of Organotin(IV) Complexes. Phosphorus. Sulfur. Silicon Relat. Elem. 2017, 192, 73–80.
[204] Singh, R.; Kaushik, N. K. Spectral and Thermal Studies with Anti-Fungal Aspects of Some Organotin(IV) Complexes with Nitrogen and Sulphur Donor Ligands Derived from 2-Phenylethylamine. Spectrochim. Acta - Part A Mol. Biomol. Spectrosc. 2008, 71, 669–675.
[205] Sirajuddin, M.; Ali, S.; Mckee, V.; Sohail, M.; Pasha, H. Potentially Bioactive Organotin(IV) Compounds: Synthesis, Characterization, In Vitro Bioactivities and Interaction with SS-DNA. Eur. J. Med. Chem. 2014, 84, 343–363.
[206] Tariq, M.; Ali, S.; Muhammad, N.; Shah, N. A.; Sirajuddin, M.; Tahir, M. N.; Khalid, N.; Khan, M. R. Biological Screening, DNA Interaction Studies, and Catalytic Activity of Organotin(IV) 2-(4-Ethylbenzylidene) Butanoic Acid Derivatives: Synthesis, Spectroscopic Characterization and X-ray Structure. J. Coord. Chem. 2014, 67, 323–340.
[207] Win, Y. F.; Choong, C. S.; Dang, J. C.; Iqbal, M. A.; Quah, C. K.; Majid, A. M. S.

207
A.; Teoh, S. G. Polymeric Seven-Coordinated Organotin(IV) Complexes Derived from 5-Amino-2-Chlorobenzoic Acid and In Vitro Anti-Cancer Studies. J. Coord. Chem. 2014, 67, 3401–3413.
[208] Basu Baul, T. S.; Masharing, C.; Ruisi, G.; Jirásko, R.; Holčapek, M.; de Vos, D.; Wolstenholme, D.; Linden, A. Self-Assembly of Extended Schiff Base Amino Acetate Skeletons, 2-[(2Z)-(3-Hydroxy-1-Methyl-2-Butenylidene)]aminophenylpropionate and 2-[(E)-1-(2-Hydroxyaryl)alkylidene]aminophenylpropionate Skeletons Incorporating Organotin(IV) Moieties: Synthesis, Sp. J. Organomet. Chem. 2007, 692, 4849–4862.
[209] Katsoulakou, E.; Tiliakos, M.; Papaefstathiou, G.; Terzis, A.; Raptopoulou, C.; Geromichalos, G.; Papazisis, K.; Papi, R.; Pantazaki, A.; Kyriakidis, D.; Cordopatis, P.; Manessi-Zoupa, E. Diorganotin(IV) Complexes of Dipeptides Containing the α-Aminoisobutyryl Residue (Aib): Preparation, Structural Characterization, Antibacterial and Antiproliferative Activities of [(N-Bu)2Sn(H-1L)] (LH = H-Aib-L-Leu-OH, H-Aib-L-Ala-OH). J. Inorg. Biochem. 2008, 102, 1397–1405.
[210] Hou, H.-N.; Qi, Z.-D.; Ouyang, Y.-W.; Liao, F.-L.; Zhang, Y.; Liu, Y. Studies on Interaction between Vitamin B12 and Human Serum Albumin. J. Pharm. Biomed. Anal. 2008, 47, 134–139.
[211] Sielecki, T. M.; Boylan, J. F.; Benfield, P. A.; Trainor, G. L. Cyclin-Dependent Kinase Inhibitors: Useful Targets in Cell Cycle Regulation. J. Med. Chem. 2000, 43, 1–18.
[212] Rudorf, W. Reactions of Carbon Disulfide with C- Nucleophiles. Sulfur Reports 1991, 11, 51–141.
[213] Chandrasekhar, V.; Nagendran, S.; Baskar, V. Organotin Assemblies Containing Sn-O Bonds. Coord. Chem. Rev. 2002, 235, 1–52.
[214] Nath, M.; Pokharia, S.; Yadav, R. Organotin(IV) Complexes of Amino Acids and Peptides. Coord. Chem. Rev. 2001, 215, 99–149.
[215] Song, X.; Zapata, A.; Eng, G. Organotins and Quantitative-Structure Activity/property Relationships. J. Organomet. Chem. 2006, 691, 1756–1760.
[216] Spek, A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, D65, 148–155.
[217] Hay, P. J.; Wadt, W. R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283.
[218] Hay, P. J.; Wadt, W. R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310.

208
[219] Wadt, W. R.; Hay, P. J. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298.
[220] Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J. Autodock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexiblity. J. Comput. Chem. 2009, 16, 2785–2791.
[221] Besler, B. H.; Merz, K. M.; Kollman, P. A. Atomic Charges Derived from Semiempirical Methods. J. Comput. Chem. 1990, 11, 431–439.
[222] ADL: Parameters for docking with metal ions in receptor http://autodock.1369657.n2.nabble.com/ADL-Parameters-fordocking-with-metal-ions-in-receptor-td2505649.html (accessed Sep 7, 2016).
[223] Sudan, S.; Rupasinghe, H. V. Antiproliferative Activity of Long Chain Acylated Esters of Quercetin-3-O-Glucoside in Hepatocellular Carcinoma HepG2 Cells. Exp. Biol. Med. 2015, 240, 1452–1464.
[224] Hong, M.; Chang, G.; Li, R.; Niu, M. Anti-Proliferative Activity and DNA/BSA Interactions of Five Mono- or Di-organotin(IV) Compounds derived from 2-Hydroxy-N’-[(2-Hydroxy-3-Methoxyphenyl)methylidene]-Benzohydrazone. New J. Chem. 2016, 40, 7889–7900.
[225] Ganeshpandian, M.; Loganathan, R.; Suresh, E.; Riyasdeen, A.; Akbarsha, M. A.; Palaniandavar, M. New Ruthenium(II) Arene Complexes of Anthracenyl-Appended Diazacycloalkanes: Effect of Ligand Intercalation and Hydrophobicity on DNA and Protein Binding and Cleavage and Cytotoxicity. Dalt. Trans. 2014, 43, 1203–1219.
[226] Loganathan, R.; Ramakrishnan, S.; Suresh, E.; Riyasdeen, A.; Akbarsha, M. A.; Palaniandavar, M. Mixed Ligand Copper(II) Complexes of N, N-Bis(benzimidazol-2-ylmethyl)amine (BBA) with Diimine Co-Ligands: Efficient Chemical Nuclease and Protease Activities and Cytotoxicity. Inorg. Chem. 2012, 51, 5512–5532.
[227] Malhotra, R.; Singh, J. P.; Dudeja, M.; Dhindsa, K. S. Ligational Behavior of N-Substituted Acid Hydrazides towards Transition Metals and Potentiation of Their Microbiocidal Activity. J. Inorg. Biochem. 1992, 46, 119–127.
[228] Ali, M. A.; Mirza, A. H.; Butcher, R. J. Synthesis and Characterization of Copper(II) Complexes of the Methylpyruvate Schiff Base of S-Methyldithiocarbazate (Hmpsme) and the X-Crystal Structures of Hmpsme and [Cu(mpsme)Cl]. Polyhedron 2001, 20, 1037–1043.
[229] Naqeebullah; Farina, Y.; Chan, K. M.; Mun, L. K.; Rajab, N. F.; Ooi, T. C. Diorganotin(IV) Derivatives of N-Methyl P-fluorobenzo-hydroxamic Acid: Preparation, Spectral Characterization, X-Ray Diffraction Studies and Antitumor Activity. Molecules 2013, 18, 8696–8711.
[230] Holeček, J.; Nádvorník, M.; Handlíř, K.; Lyčka, A. 13C and 119Sn NMR Spectra of

209
Di-N-butyltin(IV) Compounds. J. Organomet. Chem. 1986, 315, 299–308.
[231] Zhang, Y.-Y.; Zhang, R.-F.; Zhang, S.-L.; Cheng, S.; Li, Q.-L.; Ma, C.-L. Syntheses, Structures and Anti-Tumor Activity of Four New Organotin(IV) Carboxylates Based on 2-Thienylselenoacetic Acid. Dalt. Trans. 2016, 45, 8412–8421.
[232] Barreiro, S.; Durán-Carril, M. L.; Viqueira, J.; Sousa-Pedrares, A.; García-Vázquez, J. A.; Romero, J. Structural Studies and Bioactivity of Diorganotin(IV) Complexes of Pyridin-2-thionato Derivatives. J. Organomet. Chem. 2015, 791, 155–162.
[233] Ma, C.; Zhang, J.; Tian, G.; Zhang, R. Syntheses, Crystal Structures and Coordination Modes of Tri- and Di-Organotin Derivatives with 2-Mercapto-4-Methylpyrimidine. J. Organomet. Chem. 2005, 690, 519–533.
[234] Yusof, E. N. M.; Jotani, M. M.; Tiekink, E. R. T.; Ravoof, T. B. S. A. 2-[(1E)-([(Benzylsulfanyl)methanethioyl]amino-imino)methyl]-6-methoxyphenol: Crystal Structure and Hirshfeld Surface Analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 516–521.
[235] Addison, A. W.; Rao, T. N. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen-Sulphur Donor Ligands ; the Crystal and Molecular Structure of Aqua[l,7-bis(N-Methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper(II) Perchlorate. J. Chem. Soc. Dalt. Trans. 1984, 7, 1349–1356.
[236] Milčič, M. K.; Medakovič, V. B.; Sredojevič, D. N.; Juranič, N. O.; Zarič, S. D. Electron Delocalization Mediates the Metal-Dependent Capacity for CH/π Interactions of Acetylacetonato Chelates. Inorg. Chem. 2006, 45, 4755–4763.
[237] Tiekink, E. R. T. Supramolecular Assembly Based on “emerging” Intermolecular Interactions of Particular Interest to Coordination Chemists. Coord. Chem. Rev. 2017, 345, 209–228.
[238] Chen, D.; Lai, C. S.; Tiekink, E. R. T. Crystal Structures of 2,2’-bipyridine Adducts of Two Cadmium O-Alkyl Dithiocarbonates: Rationalisation of Disparate Coordination Geometries Based on Different Crystal Packing Environments. Zeitschrift fur Krist. 2002, 217, 747–752.
[239] Malenov, D. P.; Janjić, G. V.; Medaković, V. B.; Hall, M. B.; Zarić, S. D. Noncovalent Bonding: Stacking Interactions of Chelate Rings of Transition Metal Complexes. Coord. Chem. Rev. 2017, 345, 318–341.
[240] González-garcía, C.; Mata, A.; Zani, F.; Mendiola, M. A.; López-torres, E. Synthesis and Antimicrobial Activity of Tetradentate Ligands Bearing Hydrazone and / or Thiosemicarbazone Motifs and Their Diorganotin (IV) Complexes. J. Inorg. Biochem. 2016, 163, 118–130.
[241] Andree, H. A. M.; Reutelingsperger, C. P. M.; Hauptmann, R.; Hemker, H. C.; Hermens, W. T.; Willems, G. M. Binding of Vascular Anticoagulant Alpha (VAC

210
Alpha) to Planar Phospholipid Bilayers. J. Biol. Chem. 1990, 265 (9), 4923–4928.
[242] Gea, R.; Ma, W.-H.; Li, Y.-L.; Li, Q.-S. Apoptosis Induced Neurotoxicity of Di-N-butyl-di-(4-chlorobenzohydroxamato) Tin(IV) via Mitochondria-Mediated Pathway in PC12 Cells. Toxicol. Vitr. 2013, 27, 92–102.
[243] Poon, I. K. H.; Hulett, M. D.; Parish, C. R. Molecular Mechanisms of Late Apoptotic/necrotic Cell Clearance. Cell Death Differ. 2010, 17, 381–397.
[244] Prosser, K. E.; Chang, S. W.; Saraci, F.; Le, P. H.; Walsby, C. J. Anticancer Copper Pyridine Benzimidazole Complexes: ROS Generation, Biomolecule Interactions and Cytotoxicity. J. Inorg. Biochem. 2017, 167, 89–99.
[245] Fani, S.; Kamalidehghan, B.; Lo, K. M.; Hashim, N. M.; Chow, K. M.; Ahmadipour, F. Synthesis, Structural Characterization, and Anticancer Activity of a Monobenzyltin Compound against MCF-7 Breast Cancer Cells. Drug Des. Devel. Ther. 2015, 9, 6191–6201.
[246] Hussain, S.; Bukhari, I. H.; Ali, S.; Shahzadi, S.; Shahid, M.; Munawar, K. S. Synthesis and Spectroscopic and Thermogravimetric Characterization of Heterobimetallic Complexes with Sn(IV) and Pd(II); DNA Binding, Alkaline Phosphatase Inhibition and Biological Activity Studies. J. Coord. Chem. 2015, 68, 662–677.
[247] Hussain, S.; Ali, S.; Shahzadi, S.; Tahir, M. N.; Shahid, M. Synthesis, Characterization, Biological Activities, Crystal Structure and DNA Binding of Organotin(IV) 5-Chlorosalicylates. J. Coord. Chem. 2015, 68, 2369–2387.
[248] Yadav, S.; Yousuf, I.; Usman, M.; Ahmad, M.; Arjmand, F.; Tabassum, S. Synthesis and Spectroscopic Characterization of Diorganotin(IV) Complexes of N′-(4-Hydroxypent-3-en-2-ylidene)isonicotinohydrazide: Chemotherapeutic Potential Validation by In Vitro Interaction Studies with DNA/HSA, DFT, Molecular Docking and Cytotoxic Ac. RSC Adv. 2015, 5, 50673–50690.
[249] Aldridge, W. H. The Influence of Organotin Compounds on Mitochondrial Functions. In Organotin Compounds: New Chemistry and Applications; Zuckerman, J. J., Ed.; American Chemical Society: Washington, DC, 1976; pp 186–196.
[250] Ventrella, V.; Nesci, S.; Trombetti, F.; Bandiera, P.; Pirini, M.; Borgatti, A. R.; Pagliarani, A. Tributyltin Inhibits the Oligomycin-Sensitive Mg-ATPase Activity in Mytilus Galloprovincialis Digestive Gland Mitochondria. Comp. Biochem. Physiol. Part C 2011, 153, 75–81.
[251] Saxena, A. K.; Huber, F. Organotin Compounds and Cancer Chemotheraphy. Coord. Chem. Rev. 1989, 95, 109–123.
[252] Murdock, K. C.; Child, R. G.; Fabio, P. F.; Angier, R. B.; Wallace, R. E.; Durr, F. E.; Citarella, R. V. Antitumor Agents. 1. 1,4-Bis[(aminoalkyl)amino]-9,10-Anthracenediones. J. Med. Chem. 1979, 22, 1024–1030.

211
[253] Pereañez, J. A.; Núñez, V.; Patiño, A. C.; Londoño, M.; Quintana, J. C. Inhibitory Effects of Plant Phenolic Compounds on Enzymatic and Cytotoxic Activities Induced By a Snake Venom Phospholipase A2. Vitae 2011, 19, 295–304.
[254] Johnson, M. G.; Kiyokawa, H.; Tani, S.; Koyama, J.; Morris-natschke, S. L.; Bowers-daines, A. M. M. M.; Lange, C.; Lee, K. Antitumor Agents-CLXVII. Synthesis and Structure-Activity Correlations of the Cytotoxic Anthraquinone 1,4-Bis-(2,3-Epoxypropylamino)-9,10-Anthracenedione, and of Related Compounds. Bioorg. Med. Chem. 1997, 5, 1469–1479.
[255] Kim, K. J.; Kim, M. A.; Jung, J. H. Antitumor and Antioxidant Activity of Protocatechualdehyde Produced from Streptomyces Lincolnensis M-20. Arch. Pharm. Res. 2008, 31, 1572–1577.
[256] Lee, B. H.; Yoon, S. H.; Kim, Y. S.; Kim, S. K.; Moon, B. J.; Bae, Y. S. Apoptotic Cell Death through Inhibition of Protein Kinase CKII Activity by 3,4-Dihydroxybenzaldehyde Purified from Xanthium Strumarium. Nat. Prod. Res. 2008, 22, 1441–1450.
[257] Fayed, A. M.; Elsayed, S. A.; El-Hendawy, A. M.; Mostafa, M. R. Complexes of Cis-dioxomolybdenum(VI) and Oxovanadium(IV) with a Tridentate ONS Donor Ligand: Synthesis, Spectroscopic Properties, X-Ray Crystal Structure and Catalytic Activity. Spectrochim. Acta - Part A Mol. Biomol. Spectrosc. 2014, 129, 293–302.
[258] Rigaku Oxford Diffraction. CrysAlis PRO. Agilent Technologies Inc. Santa Clara, CA, USA p 2015.
[259] Yusof, E. N. M.; Tiekink, E. R. T.; Jotani, M. M.; Simone, M. I.; Page, A. J.; Ravoof, T. B. S. A. Synthesis, Characterisation and Structure Determination of 3-[(1Z)-2-ylidenemethyl]benzene-1,2-diol. J. Mol. Struct. 2018, 1171, 650–657.
[260] Mıhçıokur, Ö.; Özpozan, T. Molecular Structure, Vibrational Spectroscopic Analysis (IR & Raman), HOMO-LUMO and NBO Analysis of Anti-Cancer Drug Sunitinib Using DFT Method. J. Mol. Struct. 2017, 1149, 27–41.
[261] Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179.
[262] Frija, L. M. T.; Pombeiro, A. J. L.; Kopylovich, M. N. Coordination Chemistry of Thiazoles, Isothiazoles and Thiadiazoles. Coord. Chem. Rev. 2016, 308, 32–55.
[263] Javed, F.; Ali, S.; Shahzadi, S.; Sharma, S. K.; Qanungo, K.; Munawar, K. S.; Khan, I. Synthesis, Characterization, and Biological Activity of Organotin(IV) Complexes with 4-Oxo-4-[3-(Trifluoromethyl)phenylamino]butanoic Acid. Russ. J. Gen. Chem. 2017, 87, 2409–2420.
[264] Saxena, A.; Tandon, J. P.; Molloy, K. C.; Zuckerman, J. J. Tin(IV) Complexes of Tridentate Schiff Bases Having ONS Donor Systems. Inorganica Chim. Acta 1982, 63, 71–74.

212
[265] Degaonkar, M. P.; Gopinathan, S.; Gopinathan, C. Preparation and Characterisation of Some New Dithioca Bazate Schiff Base Complexes of Titanium(IV), Tin(IV) and Lead(IV). Indian J. Chem. 1989, 28, 678–682.
[266] Degaonkar, M. P.; Gopinathan, S.; Gopinathan, C. Synthesis and Characterisation of Titanium(IV), Tin(IV) and Lead(IV) Complexes of Schiff Bases Derived from S-Benzyldithiocarbazate. Synth. React. Inorg. Met. Chem. 1989, 19, 613–625.
[267] Shi, Y. C.; Yang, H. M.; Song, H. Bin; Yan, C. G.; Hu, X. Y. Syntheses and Crystal Structures of the Potential Tridentate Ligand Formed from Condensation of Ferrocenoylacetone and S-Benzyldithiocarbazate and Its Bivalent Metal Complexes. Polyhedron 2004, 23, 567–573.
[268] Yin, H. D.; Hong, M.; Li, G.; Wang, D. Q. Synthesis, Characterization and Structural Studies of Diorganotin(IV) Complexes with Schiff Base Ligand Salicylaldehyde Isonicotinylhydrazone. J. Organomet. Chem. 2005, 690, 3714–3719.
[269] Wrackmeyer, B. l19Sn-NMR Parameters. Annu. Reports NMR Spectrosc. 1985, 16, 73–186.
[270] Sousa, G. F. de; Gatto, C. C.; Ellena, J.; Ardisson, J. D. Synthesis and Crystal Structures of Octahedral Sn(IV) Complexes Prepared from SnCl2·2H2O and 2-Hydroxyacetophenone (S-Benzydithiocarbazate) Ligand (H2L). J. Chem. Crystallogr. 2011, 41, 838–842.
[271] Nakanishi, T.; Masuda, A.; Suwa, M.; Akiyama, Y.; Hoshino-Abe, N.; Suzuki, M. Synthesis of Derivatives of NK109, 7-OH Benzo[c]phenanthridine Alkaloid, and Evaluation of Their Cytotoxicities and Reduction-Resistant Properties. Bioorganic Med. Chem. Lett. 2000, 10, 2321–2323.
[272] Maia, P. I. da S.; Pavan, F. R.; Leite, C. Q. F.; Lemos, S. S.; de Sousa, G. F.; Batista, A. A.; Nascimento, O. R.; Ellena, J.; Castellano, E. E.; Niquet, E.; Deflona, V. M. Vanadium Complexes with Thiosemicarbazones: Synthesis, Characterization, Crystal Structures and Anti-Mycobacterium Tuberculosis Activity. Polyhedron 2009, 28, 398–406.
[273] Wallace, R.; Richard Thomson, J.; Bell, M. J.; Skipper, H. E. Observations on the Antileukemic Activity of Pyridine- 2-Carboxaldehyde Thiosemicarbazone and Thiocarbohydrazone. Cancer Res. 1956, 16, 167–170.
[274] French, F. A.; Blanz, E. J. The Carcinostatic Activity of α-(N) Heterocyclic Carboxaldehyde Thiosemicarbazones. Cancer Res. 1965, 25, 1454–1458.
[275] West, D. X.; Liberta, A. E.; Padhye, S. B.; Chikate, R. C.; Sonawane, P. B.; Kumbhar, A. S.; Yerande, R. G. Thiosemicarbazone Complexes of Copper(II): Structural and Biological Studies. Coord. Chem. Rev. 1993, 123, 49–71.
[276] Shanmugapriya, A.; Jain, R.; Sabarinathan, D.; Kalaiarasi, G.; Dallemer, F.; Prabhakaran, R. Structurally Different Mono-, Bi- and Trinuclear Pd(II) Complexes and Their DNA/protein Interaction, DNA Cleavage, and Anti-Oxidant,

213
Anti-Microbial and Cytotoxic Studies. New J. Chem. 2017, 41, 10324–10338.
[277] Kalaiarasi, G.; Jain, R.; Shanmugapriya, A.; Puschman, H.; Kalaivani, P.; Prabhakaran, R. New Binuclear Ni(II) Metallates as Potent Antiproliferative Agents against MCF-7 and HeLa Cells. Inorganica Chim. Acta 2017, 462, 174–187.
[278] Kalaivani, P.; Prabhakaran, R.; Ramachandran, E.; Dallemer, F.; Paramaguru, G.; Renganathan, R.; Poornima, P.; Vijaya Padma, V.; Natarajan, K. Influence of Terminal Substitution on Structural, DNA, Protein Binding, Anticancer and Antibacterial Activities of palladium(II) Complexes Containing 3-Methoxy Salicylaldehyde-4(N) Substituted Thiosemicarbazones. Dalt. Trans. 2012, 41, 2486.
[279] Kalaiarasi, G.; Jain, R.; Puschman, H.; Poorna Chandrika, S.; Preethi, K.; Prabhakaran, R. New Binuclear Ni(II) Metallates Containing ONS Chelators: Synthesis, Characterisation, DNA Binding, DNA Cleavage, Protein Binding, Antioxidant Activity, Antimicrobial and In Vitro Cytotoxicity. New J. Chem. 2017, 41, 2543–2560.
[280] Swesi, A. T.; Farina, Y.; Baba, I. Synthesis and Characterization of Some diorganotin(IV) Complexes of 2,3-Dihydroxybenzaldehyde N(4)-Substituted Thiosemicarbazone. Sains Malaysiana 2007, 36, 21–26.
[281] Swesi, A. T.; Farina, Y.; Kassim, M.; Ng, S. W. 2,3-Dihydroxybenzaldehyde Thiosemicarbazone Hemihydrate. Acta Crystallogr. Sect. E Struct. Reports Online 2006, E62, o5457–o5458.
[282] Vrdoljak, V.; Cindrić, M.; Milić, D.; Matković-Čalogović, D.; Novak, P.; Kamenar, B. Synthesis of Five New Molybdenum(VI) Thiosemicarbazonato Complexes. Crystal Structures of Salicylaldehyde and 3-Methoxy-Salicylaldehyde 4-Methylthiosemicarbazones and Their molybdenum(VI) Complexes. Polyhedron 2005, 24, 1717–1726.
[283] Rocha, F. V.; Barra, C. V.; Mauro, A. E.; Carlos, I. Z.; Nauton, L.; El Ghozzi, M.; Gautier, A.; Morel, L.; Netto, A. V. G. Synthesis, Characterization, X-Ray Structure, DNA Cleavage and Cytotoxic Activities of Palladium(II) Complexes of 4-Phenyl-3-Thiosemicarbazide and Triphenylphosphane. Eur. J. Inorg. Chem. 2013, 25, 4499–4505.
[284] Matsuda, Y.; Ebata, T.; Mikami, N. Vibrational Spectroscopy of 2-Pyridone and Its Clusters in Supersonic Jets: Structures of the Clusters as Revealed by Characteristic Shifts of the NH and C=O Bands. J. Chem. Phys. 1999, 110, 8397.
[285] Shawish, H. B.; Paydar, M.; Looi, C. Y.; Wong, Y. L.; Movahed, E.; Halim, S. N. A.; Wong, W. F.; Mustafa, M. R.; Maah, M. J. Nickel(II) Complexes of Polyhydroxybenzaldehyde N4-Thiosemicarbazones: Synthesis, Structural Characterization and Antimicrobial Activities. Transit. Met. Chem. 2014, 39, 81–94.
[286] Shaheen, F.; Sirajuddin, M.; Ali, S.; Zia-ur-Rehman; Dyson, P. J.; Shah, N. A.;

214
Tahir, M. N. Organotin(IV) 4-(benzo[d][1,3]dioxol-5-ylmethyl)piperazine-1-carbodithioates: Synthesis, Characterization and Biological Activities. J. Organomet. Chem. 2018, 856, 13–22.
[287] Miskolci, C.; Labádi, I.; Kurihara, T.; Motohashi, N.; Molnár, J. Guanine–cytosine Rich Regions of Plasmid DNA Can Be the Target in Anti-Plasmid Effect of Phenothiazines. Int. J. Antimicrob. Agents 2002, 14, 243–247.
[288] Foye, W. O. Cancer Chemotherapeutic Agents; American Chemical Society: Washington, DC, 1995.
[289] Sishc, B. J.; Nelson, C. B.; Mckenna, M. J.; Battaglia, C. L. R.; Tanzarella, C. Telomeres and Telomerase in the Radiation Response: Implications for Instability, Reprograming, and Carcinogenesis. Front. Oncol. 2015, 5, 1–19.
[290] Veronese, F. M.; Pasut, G. PEGylation, Successful Approach to Drug Delivery. Drug Discov. Today 2005, 10, 1451–1458.
[291] Jong, J. H. De; Rodermond, H. M.; Zimberlin, C. D.; Lascano, V.; Melo, F. D. S. E.; Richel, D. J.; Medema, J. P.; Vermeulen, L. Fusion of Intestinal Epithelial Cells with Bone Marrow Derived Cells Is Dispensable for Tissue Homeostasis. Sci. Rep. 2012, 2, 271.
[292] Szakacs, G.; Varadi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME–Tox). Drug Discov. Today 2008, 13, 379–393.
[293] Hill, A. J.; Teraoka, H.; Heideman, W.; Peterson, R. E. Zebrafish as a Model Vertebrate for Investigating Chemical Toxicity. Toxicol. Sci. 2005, 86, 6–19.
[294] Zon, L. I.; Peterson, R. T. In Vivo Drug Discovery in the Zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35.
[295] Garberoglio, G. OBGMX: A Web-Based Generator of GROMACS Topologies for Molecular and Periodic Systems Using the Universal Force Field. J. Comput. Chem. 2012, 33, 2204–2208.
[296] Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J. L.; Dror, R. O.; Shaw, D. E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct. Funct. Bioinforma. 2010, 78, 1950–1958.
[297] Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–936.

215
APPENDICES
Copyright Permission for Figure 1.1:

216
Copyright Permission for Figure 1.3:

217

218
Copyright Permission for Figure 1.8:

219
Copyright Permission for Figure 1.19:

220
Copyright Permission for Figure 1.20:

221
Copyright Permission for Figure 1.21:
222
Copyright Permission Contents Presented in Chapter 2:

223
Copyright Permission Contents Presented in Chapter 2 and 3:

224
APPENDIX - ANALYSES
Melting Points
Melting points were determined using an Electrothermal digital melting point apparatus
at Department of Chemistry, Universiti Putra Malaysia.
Elemental (CHN) Analyses
CHNS was determined using a LECO CHNS-932 instrument at Department of
Chemistry, Universiti Putra Malaysia and Thermo Flash EA110 elemental analyser at
Department of Chemistry, Universiti Teknologi Mara, Malaysia. The samples were
weighed to around 1.8-2.1 mg into the tin capsule, crimped and then combusted under
excess of oxygen. Sulphamethazine was used as a calibration standard before analyses.
The presented results obtained are based on weight percent composition.
Molar conductivity Analyses
The tin(IV) compounds were diluted into 1x10-3 concentrations in DMSO. Molar
conductance was determined by immersing a dip-type cell with platinised electrode into
the tin(IV) compounds solution at 28°C using a 4310 Jenway Conductivity Meter at
Department of Chemistry, Universiti Putra Malaysia. The molar conductivity values were
determined by using the following equation:
Molar conductance, ΛM = 1000R/C[Λsolution - Λsolvent] x 10-6
Where; R = cell constant = 1 C = Concentration (0.001 M) Λsolution = Conductivity of solution Λsolvent = Conductivity of solvent

225
Fourier Transformed Infrared (FTIR) Spectroscopic Analyses
Infrared spectra were recorded using the Perkin Elmer Spectrum 100 with Universal ATR
Polarization in the range 4000-280 cm-1 at Department of Chemistry, Universiti Putra
Malaysia. All the spectra were recorded at room temperature.
Ultraviolet-visible (UV-vis) Spectral Analyses
The tin(IV) compounds were weighed accurately and dissolved in DMSO as a solvent.
The compounds were prepared in three different concentration (10-3 M, 10-4 M, and 10-5
M). Electronic spectra were recorded on a Shimadzu UV-1650 PC recording
spectrophotometer (1000–200 nm) at Department of Chemistry, Universiti Putra
Malaysia.
Mass spectroscopic (MS) Analyses
The mass spectra were recorded using a Shimadzu GC-MS QP2010Plus mass
spectrometer at Department of Chemistry, Universiti Putra Malaysia.
Multinuclear (1H, 13C and 119Sn) Nuclear Magnetic Resonance (NMR) spectroscopic
analyses
1H and 13C NMR spectra were recorded using NMR JNM ECA400 spectrometer at
Universiti Putra Malaysia and tetramethylsilane (TMS) was used as an internal standard.
At room temperature, 5-20 mg of compounds were weighed and dissolved in DMSO-d6
or CDCl3. 119Sn NMR of compounds 4-9 and 19-21 were recorded using a JOEL NMR
spectrophotometer at Universiti Malaya, Malaysia and 119Sn NMR of compounds 13-18
and 22-40 were measured using Bruker BioSpin Avance III (600MHz) spectrometer at
The University of Newcastle, Australia.

226
Cytotoxic Assay
The cytotoxic assay against bladder cancer cell lines (EJ-28 and RT-112) was evaluated
at the Molecular Genetics Laboratory, Department of Biomedical Sciences, Universiti
Putra Malaysia. For HT29 (colon), U87 and SJ-G2 (glioblastoma), MCF-7 (breast),
A2780 (ovarian), H460 (lung), A431 (skin), Du145 (prostate), BE2-C (neuroblastoma),
MIA (pancreas) cell lines and one normal breast cell line, MCF-10A (normal breast), the
MTT assays were performed at Department of Medical Oncology, Calvary Mater
Newcastle Hospital, Australia.
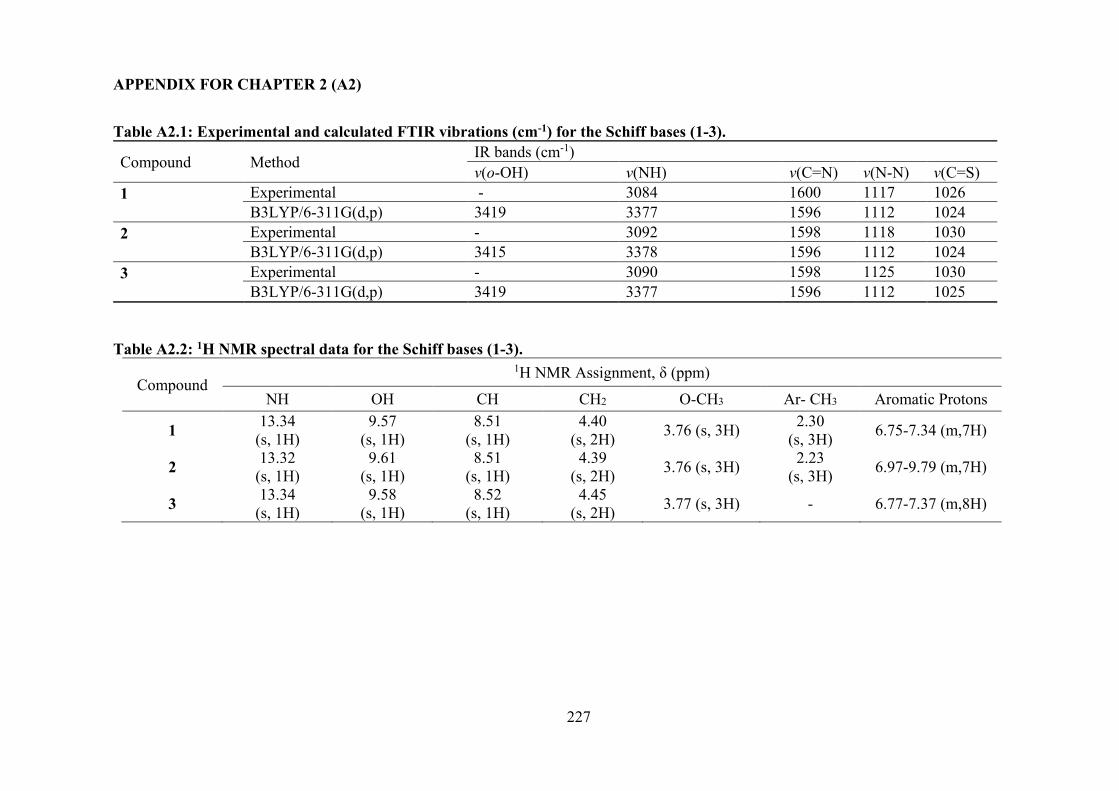
227
APPENDIX FOR CHAPTER 2 (A2)
Table A2.1: Experimental and calculated FTIR vibrations (cm-1) for the Schiff bases (1-3).
Compound Method IR bands (cm-1) v(o-OH) v(NH) v(C=N) v(N-N) v(C=S)
1
Experimental - 3084 1600 1117 1026 B3LYP/6-311G(d,p) 3419 3377 1596 1112 1024
2
Experimental - 3092 1598 1118 1030 B3LYP/6-311G(d,p) 3415 3378 1596 1112 1024
3
Experimental - 3090 1598 1125 1030 B3LYP/6-311G(d,p) 3419 3377 1596 1112 1025
Table A2.2: 1H NMR spectral data for the Schiff bases (1-3).
Compound 1H NMR Assignment, δ (ppm)
NH OH CH CH2 O-CH3 Ar- CH3 Aromatic Protons
1 13.34 (s, 1H)
9.57 (s, 1H)
8.51 (s, 1H)
4.40 (s, 2H) 3.76 (s, 3H) 2.30
(s, 3H) 6.75-7.34 (m,7H)
2 13.32 (s, 1H)
9.61 (s, 1H)
8.51 (s, 1H)
4.39 (s, 2H) 3.76 (s, 3H) 2.23
(s, 3H) 6.97-9.79 (m,7H)
3 13.34 (s, 1H)
9.58 (s, 1H)
8.52 (s, 1H)
4.45 (s, 2H) 3.77 (s, 3H) - 6.77-7.37 (m,8H)
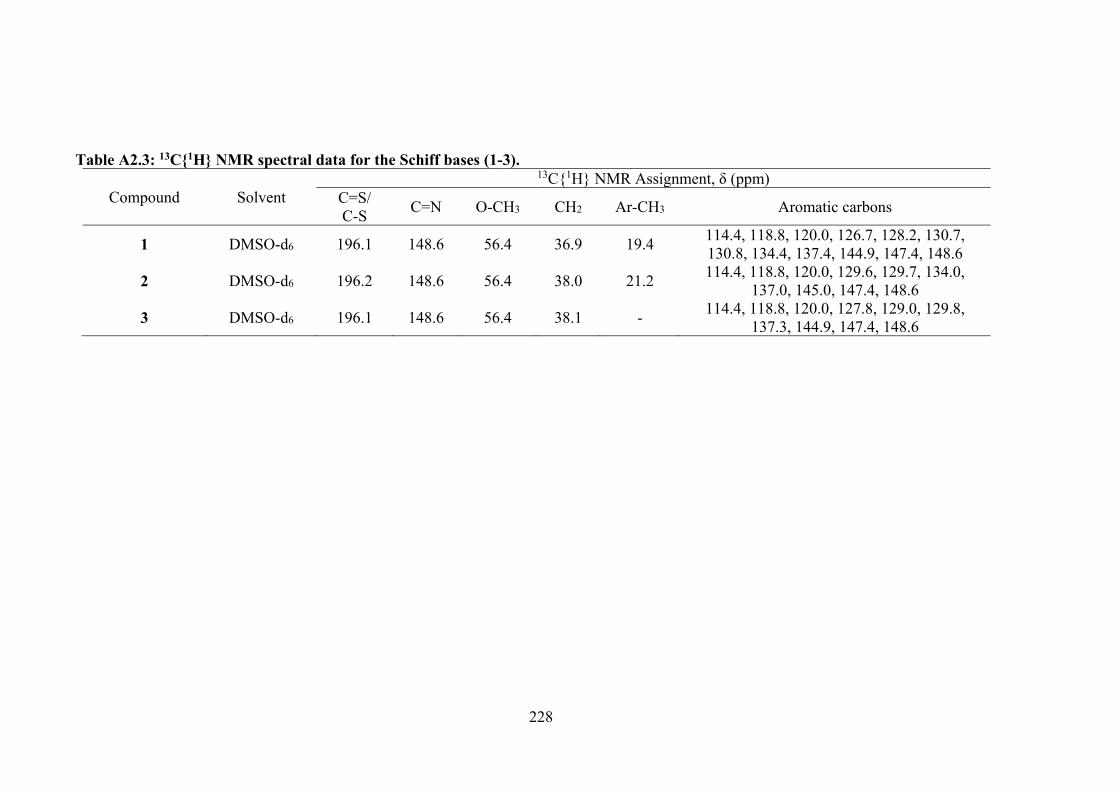
228
Table A2.3: 13C1H NMR spectral data for the Schiff bases (1-3).
Compound Solvent 13C1H NMR Assignment, δ (ppm)
C=S/ C-S C=N O-CH3 CH2 Ar-CH3 Aromatic carbons
1 DMSO-d6 196.1 148.6 56.4 36.9 19.4 114.4, 118.8, 120.0, 126.7, 128.2, 130.7, 130.8, 134.4, 137.4, 144.9, 147.4, 148.6
2 DMSO-d6 196.2 148.6 56.4 38.0 21.2 114.4, 118.8, 120.0, 129.6, 129.7, 134.0, 137.0, 145.0, 147.4, 148.6
3 DMSO-d6 196.1 148.6 56.4 38.1 - 114.4, 118.8, 120.0, 127.8, 129.0, 129.8, 137.3, 144.9, 147.4, 148.6
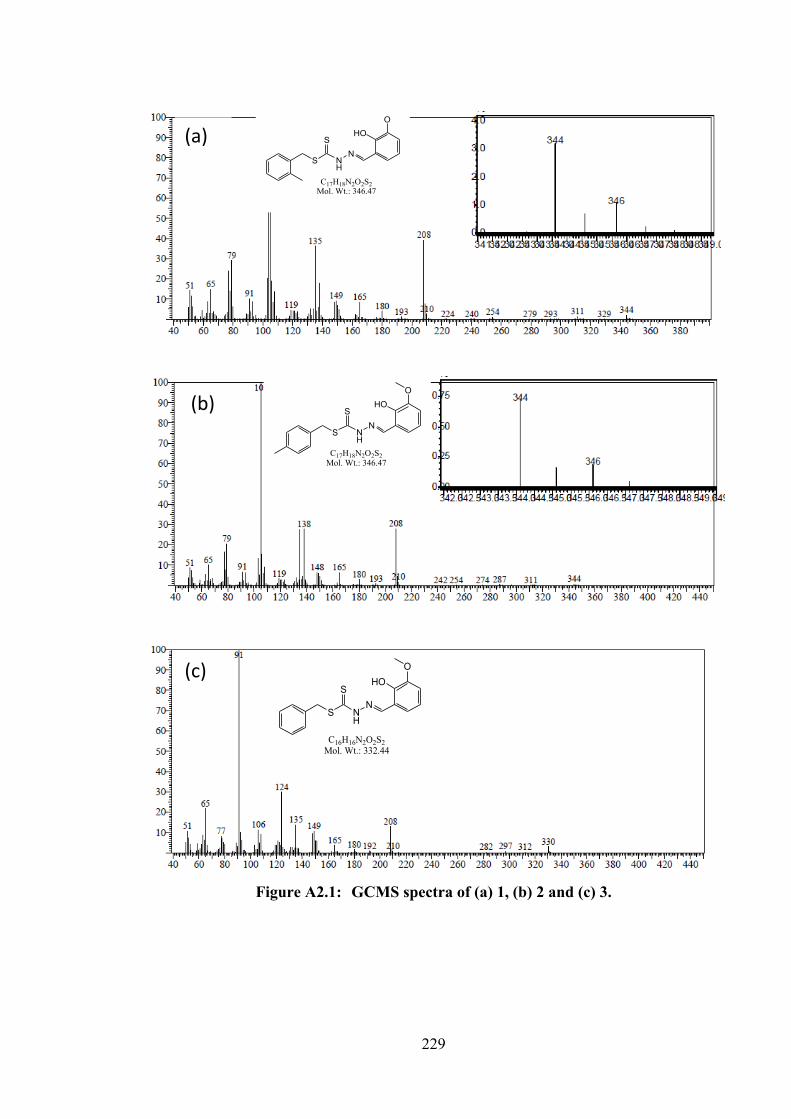
229
Figure A2.1: GCMS spectra of (a) 1, (b) 2 and (c) 3.
S NH
NS
HOO
C16H16N2O2S2Mol. Wt.: 332.44
(c)
S NH
NS
HOO
C17H18N2O2S2Mol. Wt.: 346.47
S NH
NS
HOO
C17H18N2O2S2Mol. Wt.: 346.47
(a)
(b)
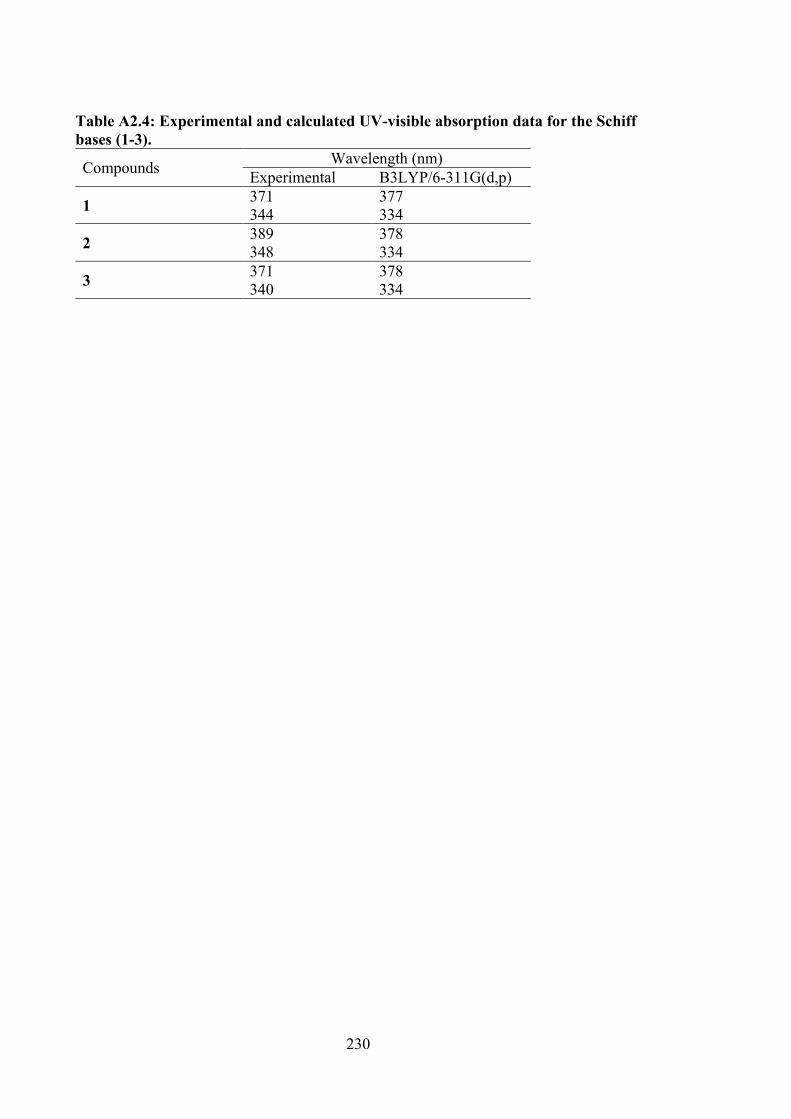
230
Table A2.4: Experimental and calculated UV-visible absorption data for the Schiff bases (1-3).
Compounds Wavelength (nm) Experimental B3LYP/6-311G(d,p)
1 371 377 344 334
2 389 378 348 334
3 371 378 340 334
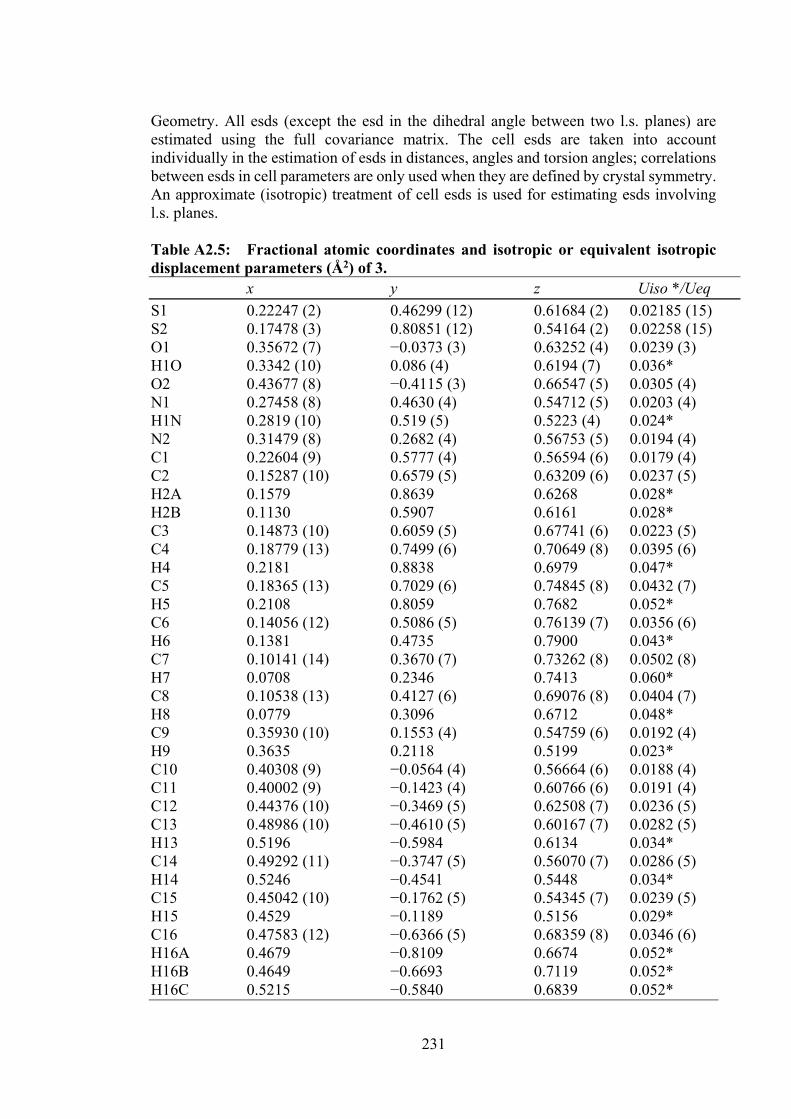
231
Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. Table A2.5: Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) of 3. x y z Uiso */Ueq S1 0.22247 (2) 0.46299 (12) 0.61684 (2) 0.02185 (15) S2 0.17478 (3) 0.80851 (12) 0.54164 (2) 0.02258 (15) O1 0.35672 (7) −0.0373 (3) 0.63252 (4) 0.0239 (3) H1O 0.3342 (10) 0.086 (4) 0.6194 (7) 0.036* O2 0.43677 (8) −0.4115 (3) 0.66547 (5) 0.0305 (4) N1 0.27458 (8) 0.4630 (4) 0.54712 (5) 0.0203 (4) H1N 0.2819 (10) 0.519 (5) 0.5223 (4) 0.024* N2 0.31479 (8) 0.2682 (4) 0.56753 (5) 0.0194 (4) C1 0.22604 (9) 0.5777 (4) 0.56594 (6) 0.0179 (4) C2 0.15287 (10) 0.6579 (5) 0.63209 (6) 0.0237 (5) H2A 0.1579 0.8639 0.6268 0.028* H2B 0.1130 0.5907 0.6161 0.028* C3 0.14873 (10) 0.6059 (5) 0.67741 (6) 0.0223 (5) C4 0.18779 (13) 0.7499 (6) 0.70649 (8) 0.0395 (6) H4 0.2181 0.8838 0.6979 0.047* C5 0.18365 (13) 0.7029 (6) 0.74845 (8) 0.0432 (7) H5 0.2108 0.8059 0.7682 0.052* C6 0.14056 (12) 0.5086 (5) 0.76139 (7) 0.0356 (6) H6 0.1381 0.4735 0.7900 0.043* C7 0.10141 (14) 0.3670 (7) 0.73262 (8) 0.0502 (8) H7 0.0708 0.2346 0.7413 0.060* C8 0.10538 (13) 0.4127 (6) 0.69076 (8) 0.0404 (7) H8 0.0779 0.3096 0.6712 0.048* C9 0.35930 (10) 0.1553 (4) 0.54759 (6) 0.0192 (4) H9 0.3635 0.2118 0.5199 0.023* C10 0.40308 (9) −0.0564 (4) 0.56664 (6) 0.0188 (4) C11 0.40002 (9) −0.1423 (4) 0.60766 (6) 0.0191 (4) C12 0.44376 (10) −0.3469 (5) 0.62508 (7) 0.0236 (5) C13 0.48986 (10) −0.4610 (5) 0.60167 (7) 0.0282 (5) H13 0.5196 −0.5984 0.6134 0.034* C14 0.49292 (11) −0.3747 (5) 0.56070 (7) 0.0286 (5) H14 0.5246 −0.4541 0.5448 0.034* C15 0.45042 (10) −0.1762 (5) 0.54345 (7) 0.0239 (5) H15 0.4529 −0.1189 0.5156 0.029* C16 0.47583 (12) −0.6366 (5) 0.68359 (8) 0.0346 (6) H16A 0.4679 −0.8109 0.6674 0.052* H16B 0.4649 −0.6693 0.7119 0.052* H16C 0.5215 −0.5840 0.6839 0.052*
232
Table A2.6: Atomic displacement parameters (Å2) of 3. U11 U22 U33 U12 U13 U23 S1 S2 O1 O2 N1 N2 C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 C11 C12 C13 C14 C15 C16
0.0221 (3) 0.0225 (3) 0.0258 (8) 0.0399 (9) 0.0206 (9) 0.0180 (8) 0.0181 (10) 0.0239 (11) 0.0234 (11) 0.0430 (15) 0.0523 (17) 0.0411 (14) 0.0549 (18) 0.0493 (16) 0.0225 (10) 0.0184 (10) 0.0185 (10) 0.0264 (11) 0.0239 (11) 0.0228 (11) 0.0223 (11) 0.0411 (14)
0.0271 (3) 0.0273 (3) 0.0281 (9) 0.0269 (9) 0.0253 (10) 0.0208 (9) 0.0179 (10) 0.0257 (12) 0.0231 (12) 0.0457 (16) 0.0504 (17) 0.0470 (16) 0.066 (2) 0.0480 (16) 0.0203 (11) 0.0170 (10) 0.0189 (11) 0.0194 (11) 0.0238 (12) 0.0301 (13) 0.0269 (12) 0.0237 (13)
0.0169 (3)0.0180 (3) 0.0183 (7) 0.0238 (8) 0.0154 (8) 0.0194 (9) 0.0176 (10) 0.0222 (11) 0.0212 (11) 0.0293 (13) 0.0258 (13) 0.0200 (11) 0.0310 (14) 0.0244 (12) 0.0150 (10) 0.0210 (10) 0.0197 (10) 0.0239 (11) 0.0355 (13) 0.0331 (13) 0.0228 (11) 0.0363 (14)
0.0071 (2) 0.0075 (2) 0.0072 (7) 0.0075 (7) 0.0044 (8) 0.0039 (7) −0.0018 (8) 0.0080 (9) 0.0093 (9) −0.0093 (13) −0.0002 (14) −0.0188 (16) −0.0172 (13) 0.0152 (12) −0.0017 (9) −0.0013 (9) −0.0013 (8) −0.0004 (9) 0.0068 (10) 0.0063 (10) 0.0012 (9) −0.0008 (11)
0.0051 (2) 0.0024 (2) 0.0044 (6) −0.0035 (7) 0.0040 (7) 0.0024 (7) 0.0016 (8) 0.0061 (9) 0.0059 (9) 0.0010 (12) −0.0034 (12) 0.0103 (11) 0.0104 (13) 0.0063 (12) 0.0028 (9) 0.0015 (8) 0.0004 (9) −0.0046 (9) −0.0067 (10) 0.0035 (10) 0.0041 (9) −0.0129 (12)
0.0040 (2) 0.0047 (2) 0.0034 (7) 0.0064 (7) 0.0058 (8) 0.0026 (8) 0.0000 (9) 0.0038 (10) 0.0005 (9) 0.0040 (12) −0.0029 (13) 0.0032 (12) 0.0078 (14) −0.0001 (12) 0.0010 (9) −0.0030 (9) −0.0039 (9) −0.0011 (9) −0.0046 (11) −0.0077 (11) −0.0030 (10) 0.0070 (11)
233
Table A2.7: Geometric parameters (Å, º) of 3. S1—C1 S1—C2 S2—C1 O1—C11 O1—H1O O2—C12 O2—C16 N1—C1 N1—N2 N1—H1N N2—C9 C2—C3 C2—H2A C2—H2B C3—C4 C3—C8 C4—C5 C4—H4 C5—C6 C5—H5 C1—S1—C2 C11—O1—H1O C12—O2—C16 C1—N1—N2 C1—N1—H1N N2—N1—H1N C9—N2—N1 N1—C1—S2 N1—C1—S1
1.749 (2) 1.817 (2) 1.670 (2) 1.354 (2) 0.836 (10) 1.368 (3) 1.429 (3) 1.338 (2) 1.371 (2) 0.874 (9) 1.290 (2) 1.504 (3) 0.9900 0.9900 1.370 (3) 1.376 (3) 1.392 (3) 0.9500 1.370 (4) 0.9500 101.75 (10) 108.6 (16) 117.13 (17) 119.96 (16) 120.0 (15) 120.0 (15) 117.70 (17) 121.37 (15) 113.89 (15)
C6—C7 C6—H6 C7—C8 C7—H7 C8—H8 C9—C10 C9—H9 C10—C11 C10—C15 C11—C12 C12—C13 C13—C14 C13—H13 C14—C15 C14—H14 C15—H15 C16—H16A C16—H16B C16—H16C C3—C8—C7 C3—C8—H8 C7—C8—H8 N2—C9—C10 N2—C9—H9 C10—C9—H9 C11—C10—C15 C11—C10—C9 C15—C10—C9 O1—C11—C10
1.359 (4) 0.9500 1.388 (3) 0.9500 0.9500 1.450 (3) 0.9500 1.401 (3) 1.408 (3) 1.408 (3) 1.383 (3) 1.400 (3) 0.9500 1.370 (3) 0.9500 0.9500 0.9800 0.9800 0.9800 120.5 (2) 119.7 119.7 121.21 (18) 119.4 119.4 119.1 (2) 121.72 (18) 119.15 (18) 123.47 (19)
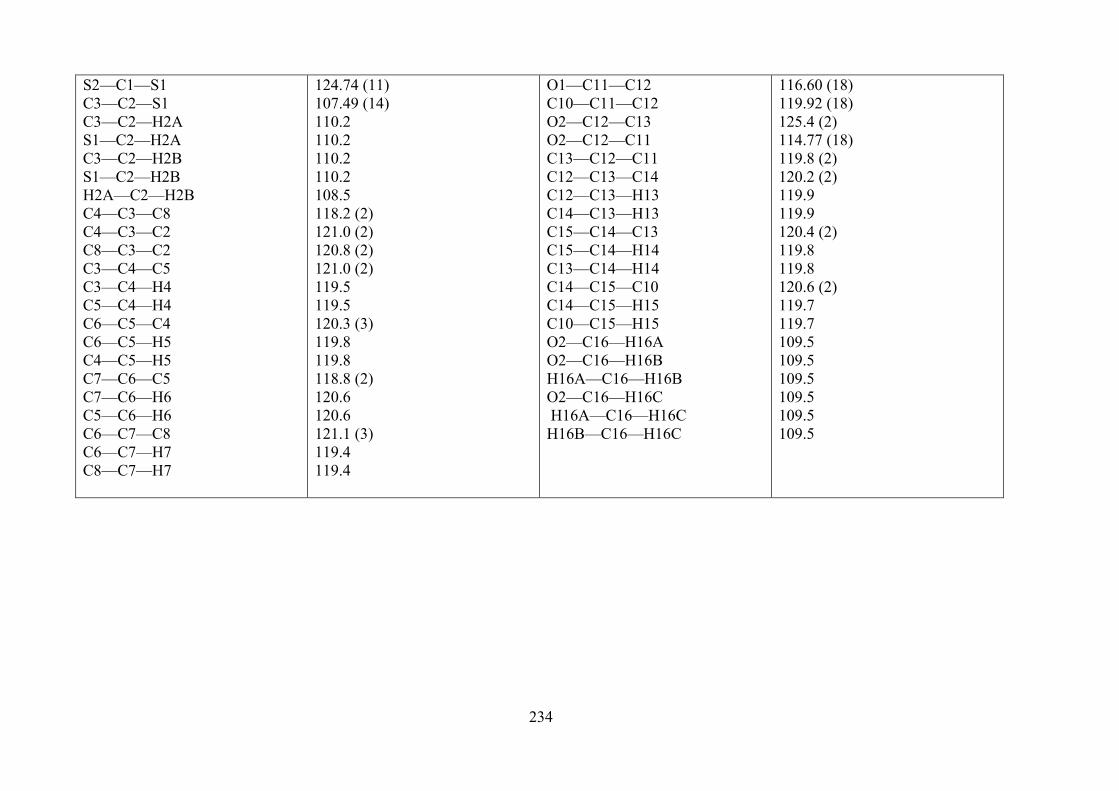
234
S2—C1—S1 C3—C2—S1 C3—C2—H2A S1—C2—H2A C3—C2—H2B S1—C2—H2B H2A—C2—H2B C4—C3—C8 C4—C3—C2 C8—C3—C2 C3—C4—C5 C3—C4—H4 C5—C4—H4 C6—C5—C4 C6—C5—H5 C4—C5—H5 C7—C6—C5 C7—C6—H6 C5—C6—H6 C6—C7—C8 C6—C7—H7 C8—C7—H7
124.74 (11) 107.49 (14) 110.2 110.2 110.2 110.2 108.5 118.2 (2) 121.0 (2) 120.8 (2) 121.0 (2) 119.5 119.5 120.3 (3) 119.8 119.8 118.8 (2) 120.6 120.6 121.1 (3) 119.4 119.4
O1—C11—C12 C10—C11—C12 O2—C12—C13 O2—C12—C11 C13—C12—C11 C12—C13—C14 C12—C13—H13 C14—C13—H13 C15—C14—C13 C15—C14—H14 C13—C14—H14 C14—C15—C10 C14—C15—H15 C10—C15—H15 O2—C16—H16A O2—C16—H16B H16A—C16—H16B O2—C16—H16C H16A—C16—H16C H16B—C16—H16C
116.60 (18) 119.92 (18) 125.4 (2) 114.77 (18) 119.8 (2) 120.2 (2) 119.9 119.9 120.4 (2) 119.8 119.8 120.6 (2) 119.7 119.7 109.5 109.5 109.5 109.5 109.5 109.5
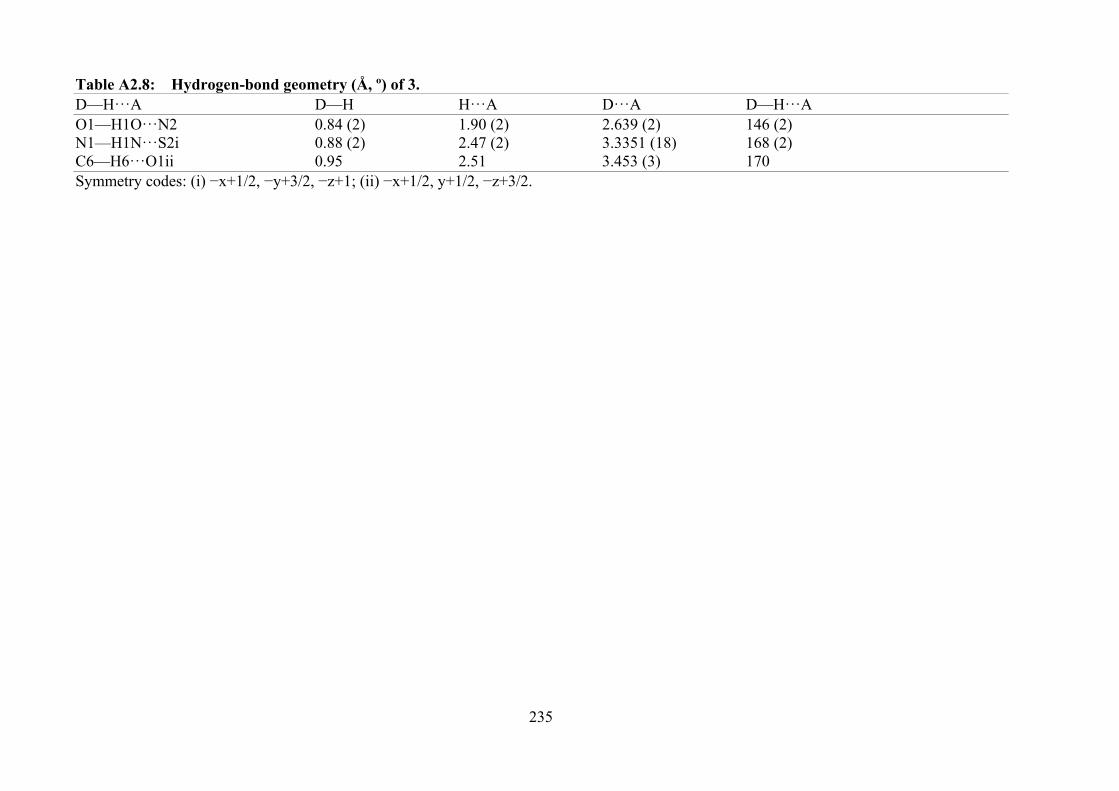
235
Table A2.8: Hydrogen-bond geometry (Å, º) of 3. D—H···A D—H H···A D···A D—H···A O1—H1O···N2 0.84 (2) 1.90 (2) 2.639 (2) 146 (2) N1—H1N···S2i 0.88 (2) 2.47 (2) 3.3351 (18) 168 (2) C6—H6···O1ii 0.95 2.51 3.453 (3) 170 Symmetry codes: (i) −x+1/2, −y+3/2, −z+1; (ii) −x+1/2, y+1/2, −z+3/2.
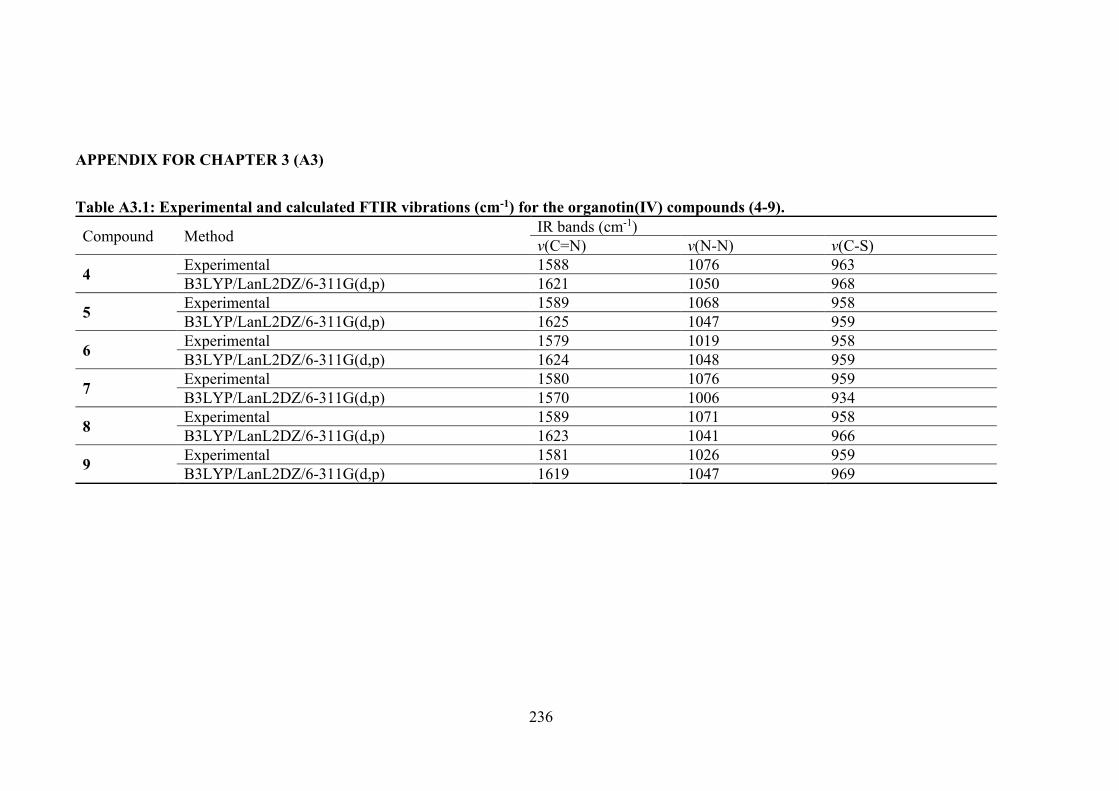
236
APPENDIX FOR CHAPTER 3 (A3)
Table A3.1: Experimental and calculated FTIR vibrations (cm-1) for the organotin(IV) compounds (4-9).
Compound Method IR bands (cm-1) v(C=N) v(N-N) v(C-S)
4 Experimental 1588 1076 963 B3LYP/LanL2DZ/6-311G(d,p) 1621 1050 968
5 Experimental 1589 1068 958 B3LYP/LanL2DZ/6-311G(d,p) 1625 1047 959
6 Experimental 1579 1019 958 B3LYP/LanL2DZ/6-311G(d,p) 1624 1048 959
7 Experimental 1580 1076 959 B3LYP/LanL2DZ/6-311G(d,p) 1570 1006 934
8 Experimental 1589 1071 958 B3LYP/LanL2DZ/6-311G(d,p) 1623 1041 966
9 Experimental 1581 1026 959 B3LYP/LanL2DZ/6-311G(d,p) 1619 1047 969
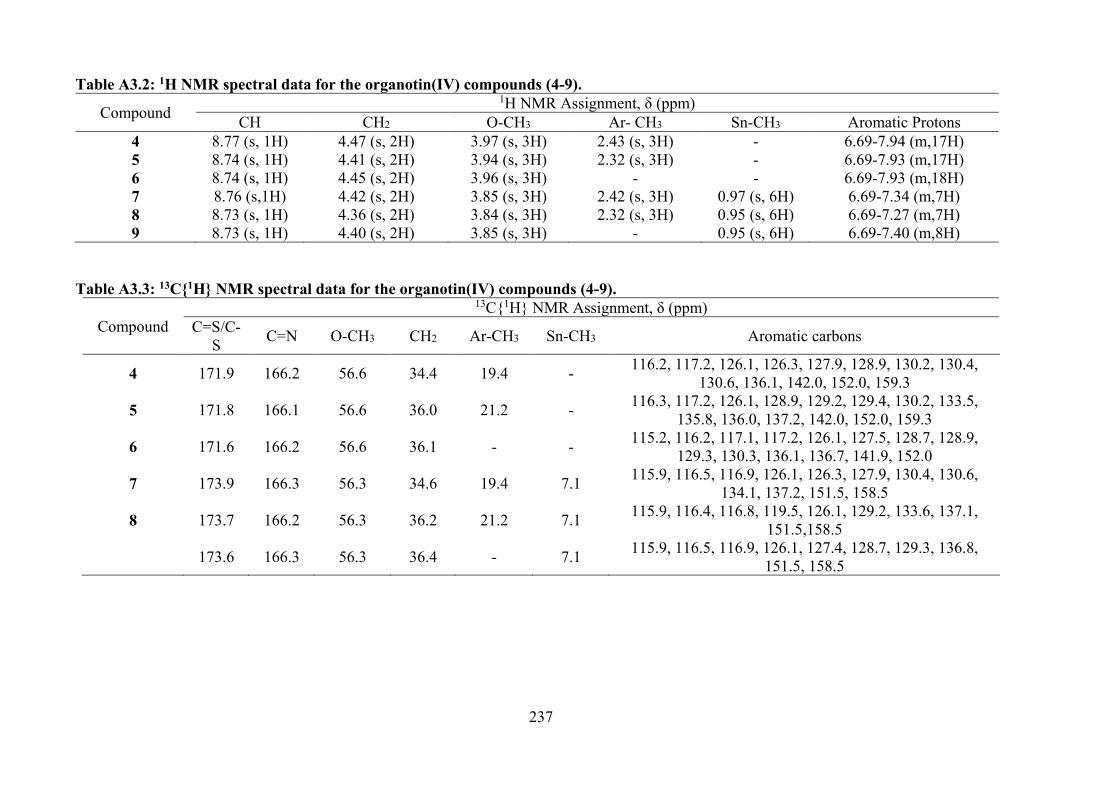
237
Table A3.2: 1H NMR spectral data for the organotin(IV) compounds (4-9).
Compound 1H NMR Assignment, δ (ppm)
CH CH2 O-CH3 Ar- CH3 Sn-CH3 Aromatic Protons 4 8.77 (s, 1H) 4.47 (s, 2H) 3.97 (s, 3H) 2.43 (s, 3H) - 6.69-7.94 (m,17H) 5 8.74 (s, 1H) 4.41 (s, 2H) 3.94 (s, 3H) 2.32 (s, 3H) - 6.69-7.93 (m,17H) 6 8.74 (s, 1H) 4.45 (s, 2H) 3.96 (s, 3H) - - 6.69-7.93 (m,18H) 7 8.76 (s,1H) 4.42 (s, 2H) 3.85 (s, 3H) 2.42 (s, 3H) 0.97 (s, 6H) 6.69-7.34 (m,7H) 8 8.73 (s, 1H) 4.36 (s, 2H) 3.84 (s, 3H) 2.32 (s, 3H) 0.95 (s, 6H) 6.69-7.27 (m,7H) 9 8.73 (s, 1H) 4.40 (s, 2H) 3.85 (s, 3H) - 0.95 (s, 6H) 6.69-7.40 (m,8H)
Table A3.3: 13C1H NMR spectral data for the organotin(IV) compounds (4-9).
Compound 13C1H NMR Assignment, δ (ppm)
C=S/C-S C=N O-CH3 CH2 Ar-CH3 Sn-CH3 Aromatic carbons
4 171.9 166.2 56.6 34.4 19.4 - 116.2, 117.2, 126.1, 126.3, 127.9, 128.9, 130.2, 130.4, 130.6, 136.1, 142.0, 152.0, 159.3
5 171.8 166.1 56.6 36.0 21.2 - 116.3, 117.2, 126.1, 128.9, 129.2, 129.4, 130.2, 133.5, 135.8, 136.0, 137.2, 142.0, 152.0, 159.3
6 171.6 166.2 56.6 36.1 - - 115.2, 116.2, 117.1, 117.2, 126.1, 127.5, 128.7, 128.9, 129.3, 130.3, 136.1, 136.7, 141.9, 152.0
7 173.9 166.3 56.3 34.6 19.4 7.1 115.9, 116.5, 116.9, 126.1, 126.3, 127.9, 130.4, 130.6, 134.1, 137.2, 151.5, 158.5
8 173.7 166.2 56.3 36.2 21.2 7.1 115.9, 116.4, 116.8, 119.5, 126.1, 129.2, 133.6, 137.1, 151.5,158.5
173.6 166.3 56.3 36.4 - 7.1 115.9, 116.5, 116.9, 126.1, 127.4, 128.7, 129.3, 136.8, 151.5, 158.5
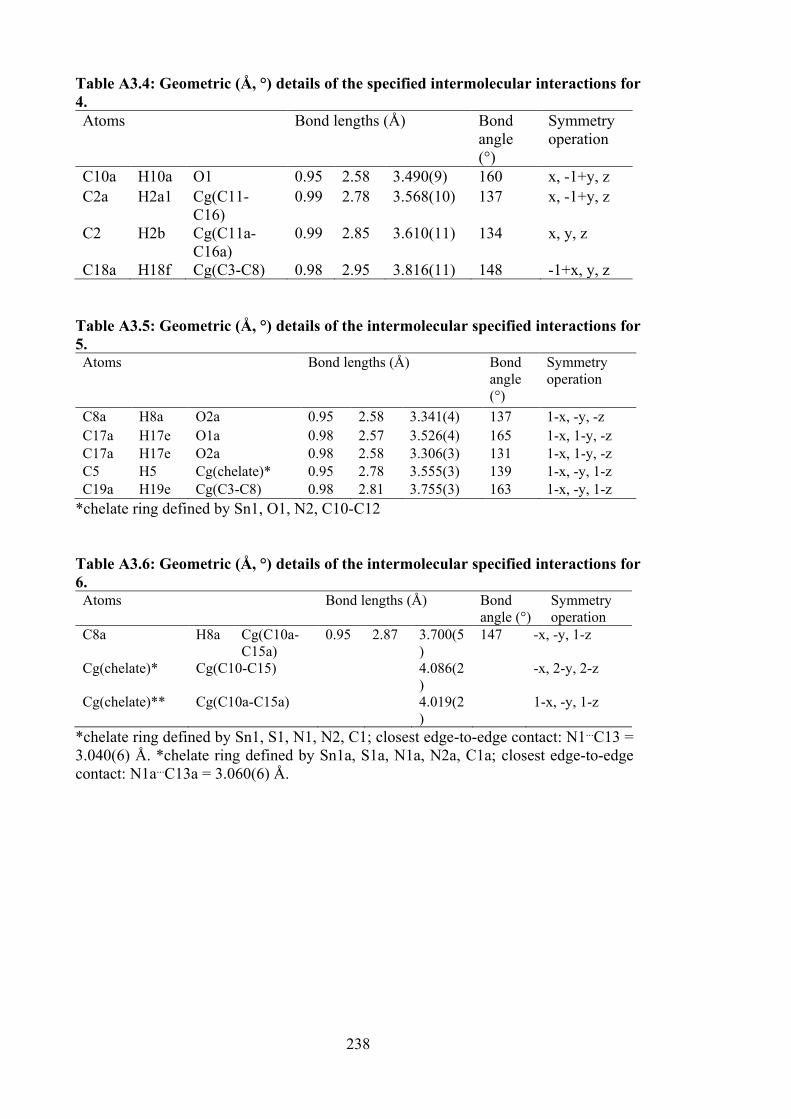
238
Table A3.4: Geometric (Å, °) details of the specified intermolecular interactions for 4. Atoms Bond lengths (Å) Bond
angle (°)
Symmetry operation
C10a H10a O1 0.95 2.58 3.490(9) 160 x, -1+y, z C2a H2a1 Cg(C11-
C16) 0.99 2.78 3.568(10) 137 x, -1+y, z
C2 H2b Cg(C11a-C16a)
0.99 2.85 3.610(11) 134 x, y, z
C18a H18f Cg(C3-C8) 0.98 2.95 3.816(11) 148 -1+x, y, z Table A3.5: Geometric (Å, °) details of the intermolecular specified interactions for 5. Atoms Bond lengths (Å) Bond
angle (°)
Symmetry operation
C8a H8a O2a 0.95 2.58 3.341(4) 137 1-x, -y, -z C17a H17e O1a 0.98 2.57 3.526(4) 165 1-x, 1-y, -z C17a H17e O2a 0.98 2.58 3.306(3) 131 1-x, 1-y, -z C5 H5 Cg(chelate)* 0.95 2.78 3.555(3) 139 1-x, -y, 1-z C19a H19e Cg(C3-C8) 0.98 2.81 3.755(3) 163 1-x, -y, 1-z
*chelate ring defined by Sn1, O1, N2, C10-C12 Table A3.6: Geometric (Å, °) details of the intermolecular specified interactions for 6. Atoms Bond lengths (Å) Bond
angle (°) Symmetry operation
C8a H8a Cg(C10a-C15a)
0.95 2.87 3.700(5)
147 -x, -y, 1-z
Cg(chelate)* Cg(C10-C15) 4.086(2)
-x, 2-y, 2-z
Cg(chelate)** Cg(C10a-C15a) 4.019(2)
1-x, -y, 1-z
*chelate ring defined by Sn1, S1, N1, N2, C1; closest edge-to-edge contact: N1...C13 = 3.040(6) Å. *chelate ring defined by Sn1a, S1a, N1a, N2a, C1a; closest edge-to-edge contact: N1a...C13a = 3.060(6) Å.
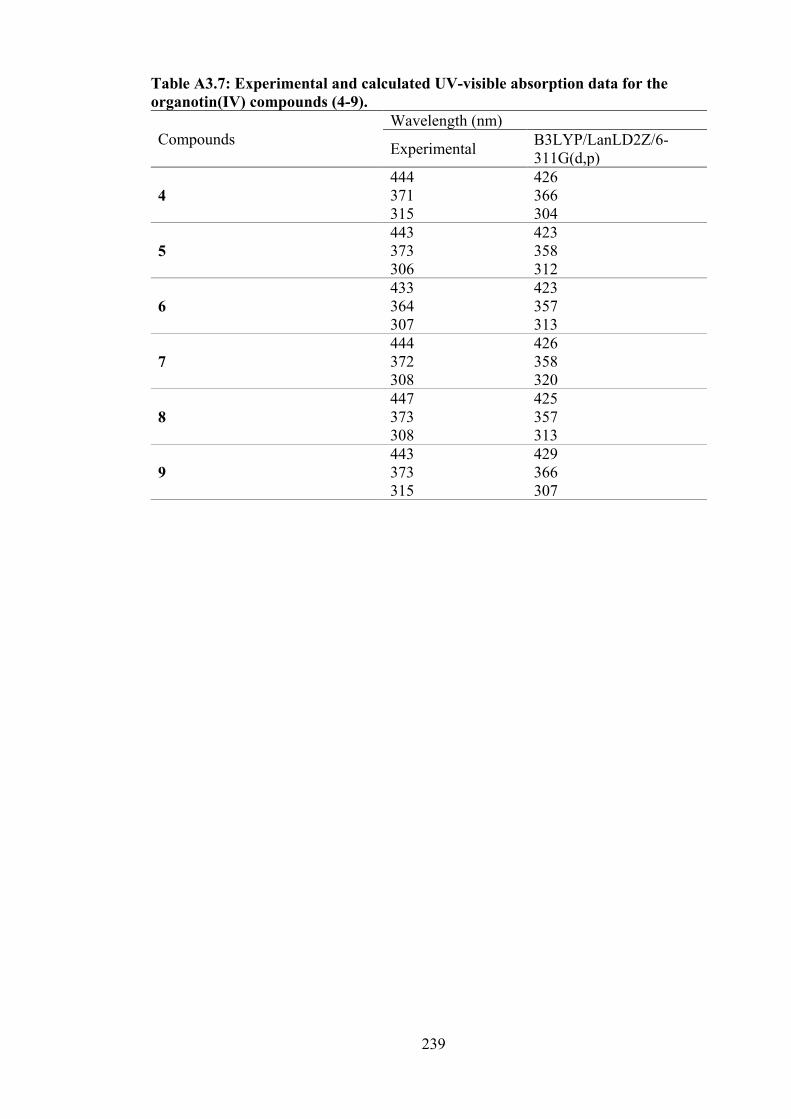
239
Table A3.7: Experimental and calculated UV-visible absorption data for the organotin(IV) compounds (4-9).
Compounds Wavelength (nm)
Experimental B3LYP/LanLD2Z/6-311G(d,p)
4 444 426 371 366 315 304
5 443 423 373 358 306 312
6 433 423 364 357 307 313
7
444 426 372 358 308 320
8 447 425 373 357 308 313
9
443 429 373 366 315 307
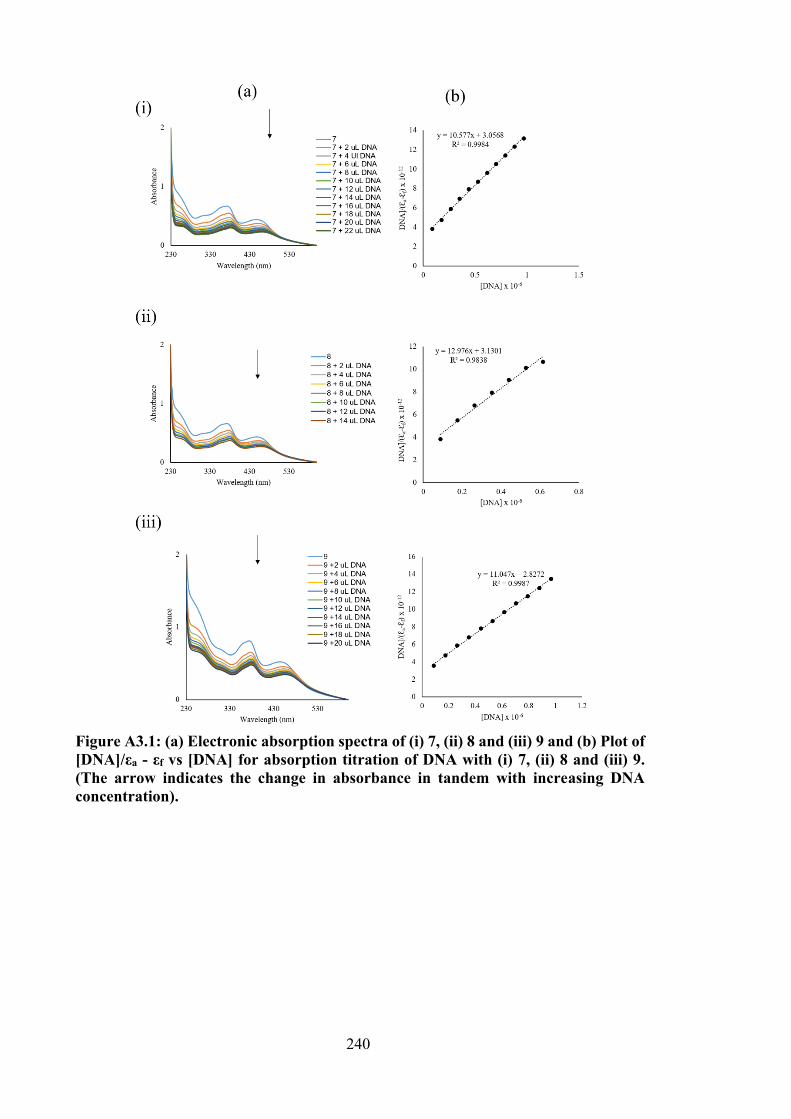
240
Figure A3.1: (a) Electronic absorption spectra of (i) 7, (ii) 8 and (iii) 9 and (b) Plot of [DNA]/εa - εf vs [DNA] for absorption titration of DNA with (i) 7, (ii) 8 and (iii) 9. (The arrow indicates the change in absorbance in tandem with increasing DNA concentration).
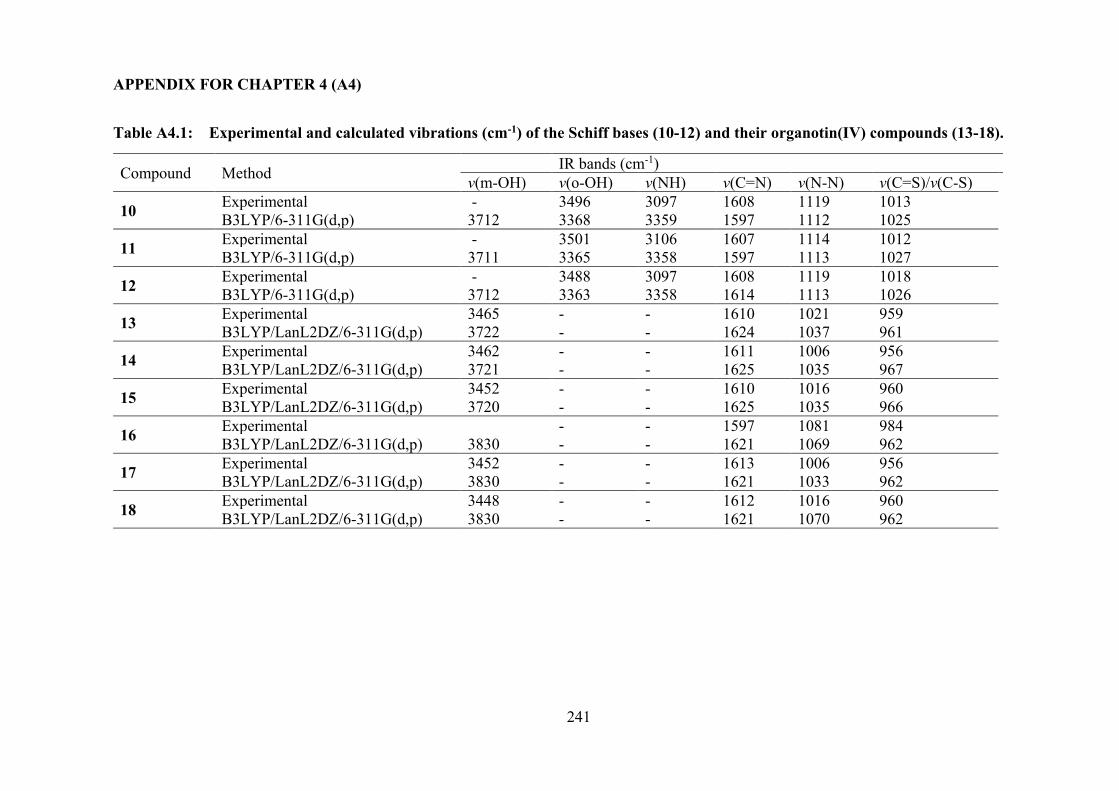
241
APPENDIX FOR CHAPTER 4 (A4)
Table A4.1: Experimental and calculated vibrations (cm-1) of the Schiff bases (10-12) and their organotin(IV) compounds (13-18).
Compound Method IR bands (cm-1) v(m-OH) v(o-OH) v(NH) v(C=N) v(N-N) v(C=S)/v(C-S)
10 Experimental - 3496 3097 1608 1119 1013 B3LYP/6-311G(d,p) 3712 3368 3359 1597 1112 1025
11 Experimental - 3501 3106 1607 1114 1012 B3LYP/6-311G(d,p) 3711 3365 3358 1597 1113 1027
12 Experimental - 3488 3097 1608 1119 1018 B3LYP/6-311G(d,p) 3712 3363 3358 1614 1113 1026
13 Experimental 3465 - - 1610 1021 959 B3LYP/LanL2DZ/6-311G(d,p) 3722 - - 1624 1037 961
14 Experimental 3462 - - 1611 1006 956 B3LYP/LanL2DZ/6-311G(d,p) 3721 - - 1625 1035 967
15 Experimental 3452 - - 1610 1016 960 B3LYP/LanL2DZ/6-311G(d,p) 3720 - - 1625 1035 966
16 Experimental - - 1597 1081 984 B3LYP/LanL2DZ/6-311G(d,p) 3830 - - 1621 1069 962
17 Experimental 3452 - - 1613 1006 956 B3LYP/LanL2DZ/6-311G(d,p) 3830 - - 1621 1033 962
18 Experimental 3448 - - 1612 1016 960 B3LYP/LanL2DZ/6-311G(d,p) 3830 - - 1621 1070 962
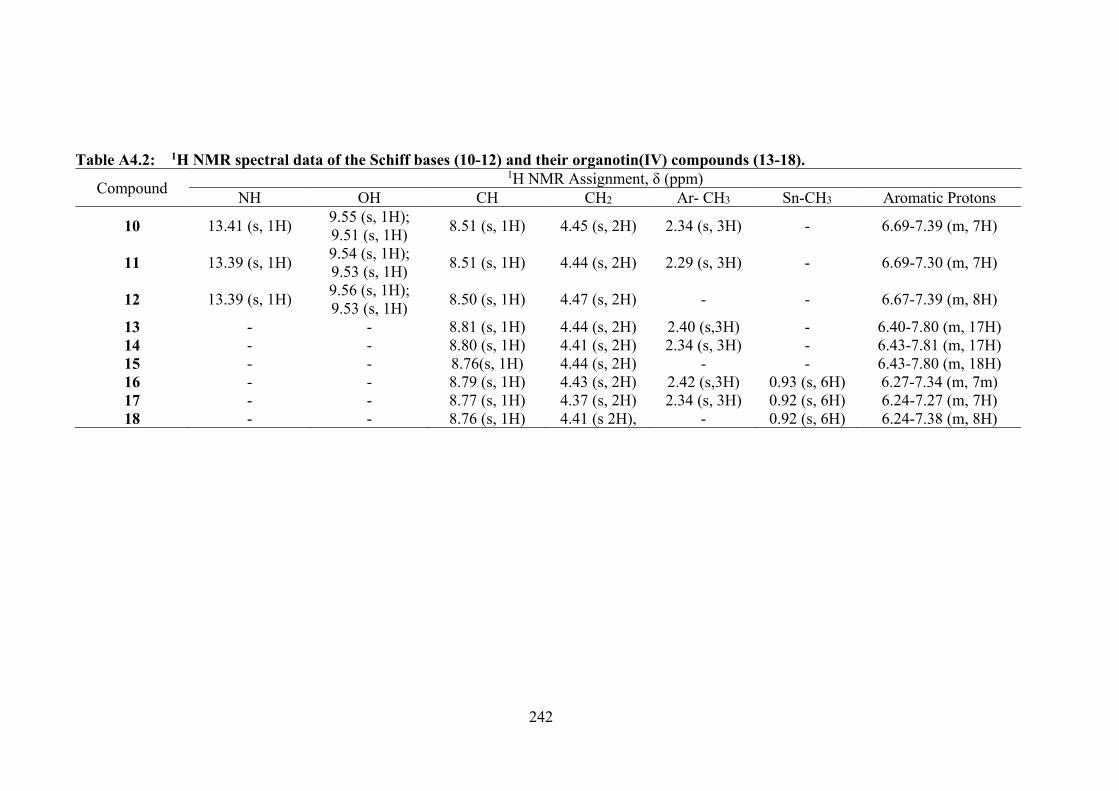
242
Table A4.2: 1H NMR spectral data of the Schiff bases (10-12) and their organotin(IV) compounds (13-18).
Compound 1H NMR Assignment, δ (ppm)
NH OH CH CH2 Ar- CH3 Sn-CH3 Aromatic Protons
10 13.41 (s, 1H) 9.55 (s, 1H); 9.51 (s, 1H) 8.51 (s, 1H) 4.45 (s, 2H) 2.34 (s, 3H) - 6.69-7.39 (m, 7H)
11 13.39 (s, 1H) 9.54 (s, 1H); 9.53 (s, 1H) 8.51 (s, 1H) 4.44 (s, 2H) 2.29 (s, 3H) - 6.69-7.30 (m, 7H)
12 13.39 (s, 1H) 9.56 (s, 1H); 9.53 (s, 1H) 8.50 (s, 1H) 4.47 (s, 2H) - - 6.67-7.39 (m, 8H)
13 - - 8.81 (s, 1H) 4.44 (s, 2H) 2.40 (s,3H) - 6.40-7.80 (m, 17H) 14 - - 8.80 (s, 1H) 4.41 (s, 2H) 2.34 (s, 3H) - 6.43-7.81 (m, 17H) 15 - - 8.76(s, 1H) 4.44 (s, 2H) - - 6.43-7.80 (m, 18H) 16 - - 8.79 (s, 1H) 4.43 (s, 2H) 2.42 (s,3H) 0.93 (s, 6H) 6.27-7.34 (m, 7m) 17 - - 8.77 (s, 1H) 4.37 (s, 2H) 2.34 (s, 3H) 0.92 (s, 6H) 6.24-7.27 (m, 7H) 18 - - 8.76 (s, 1H) 4.41 (s 2H), - 0.92 (s, 6H) 6.24-7.38 (m, 8H)
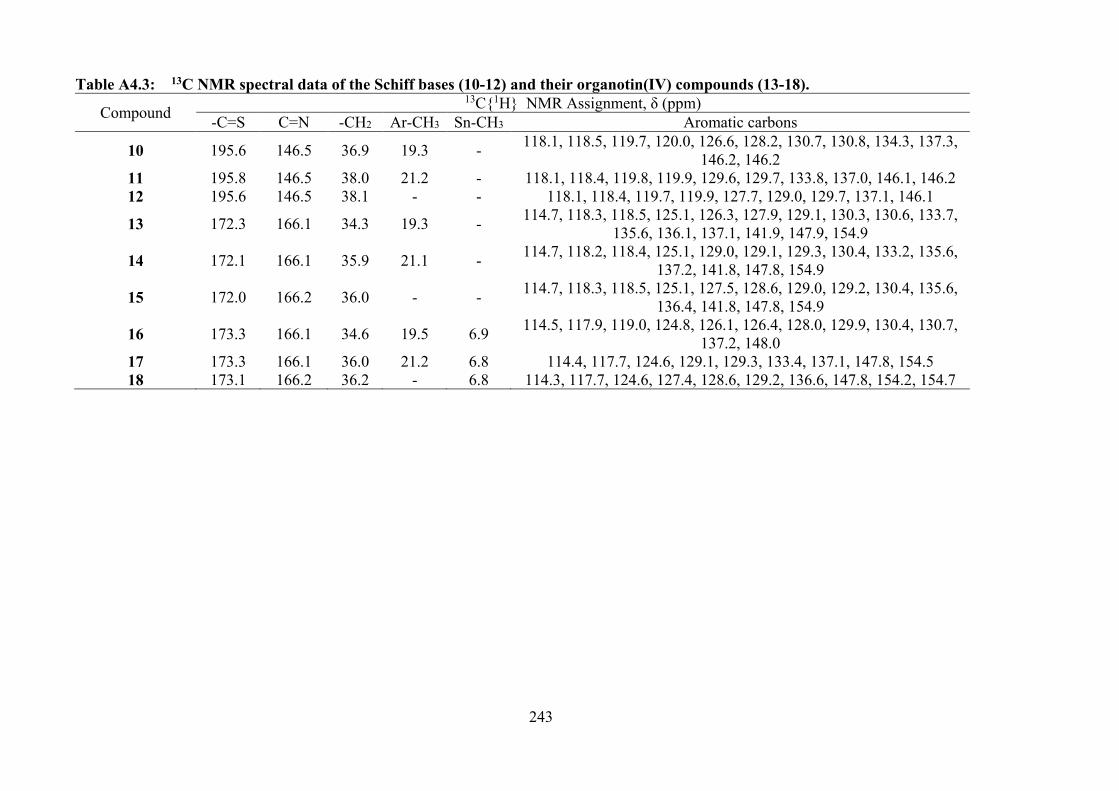
243
Table A4.3: 13C NMR spectral data of the Schiff bases (10-12) and their organotin(IV) compounds (13-18).
Compound 13C1H NMR Assignment, δ (ppm)
-C=S C=N -CH2 Ar-CH3 Sn-CH3 Aromatic carbons
10 195.6 146.5 36.9 19.3 - 118.1, 118.5, 119.7, 120.0, 126.6, 128.2, 130.7, 130.8, 134.3, 137.3, 146.2, 146.2
11 195.8 146.5 38.0 21.2 - 118.1, 118.4, 119.8, 119.9, 129.6, 129.7, 133.8, 137.0, 146.1, 146.2 12 195.6 146.5 38.1 - - 118.1, 118.4, 119.7, 119.9, 127.7, 129.0, 129.7, 137.1, 146.1
13 172.3 166.1 34.3 19.3 - 114.7, 118.3, 118.5, 125.1, 126.3, 127.9, 129.1, 130.3, 130.6, 133.7, 135.6, 136.1, 137.1, 141.9, 147.9, 154.9
14 172.1 166.1 35.9 21.1 - 114.7, 118.2, 118.4, 125.1, 129.0, 129.1, 129.3, 130.4, 133.2, 135.6, 137.2, 141.8, 147.8, 154.9
15 172.0 166.2 36.0 - - 114.7, 118.3, 118.5, 125.1, 127.5, 128.6, 129.0, 129.2, 130.4, 135.6, 136.4, 141.8, 147.8, 154.9
16 173.3 166.1 34.6 19.5 6.9 114.5, 117.9, 119.0, 124.8, 126.1, 126.4, 128.0, 129.9, 130.4, 130.7, 137.2, 148.0
17 173.3 166.1 36.0 21.2 6.8 114.4, 117.7, 124.6, 129.1, 129.3, 133.4, 137.1, 147.8, 154.5 18 173.1 166.2 36.2 - 6.8 114.3, 117.7, 124.6, 127.4, 128.6, 129.2, 136.6, 147.8, 154.2, 154.7
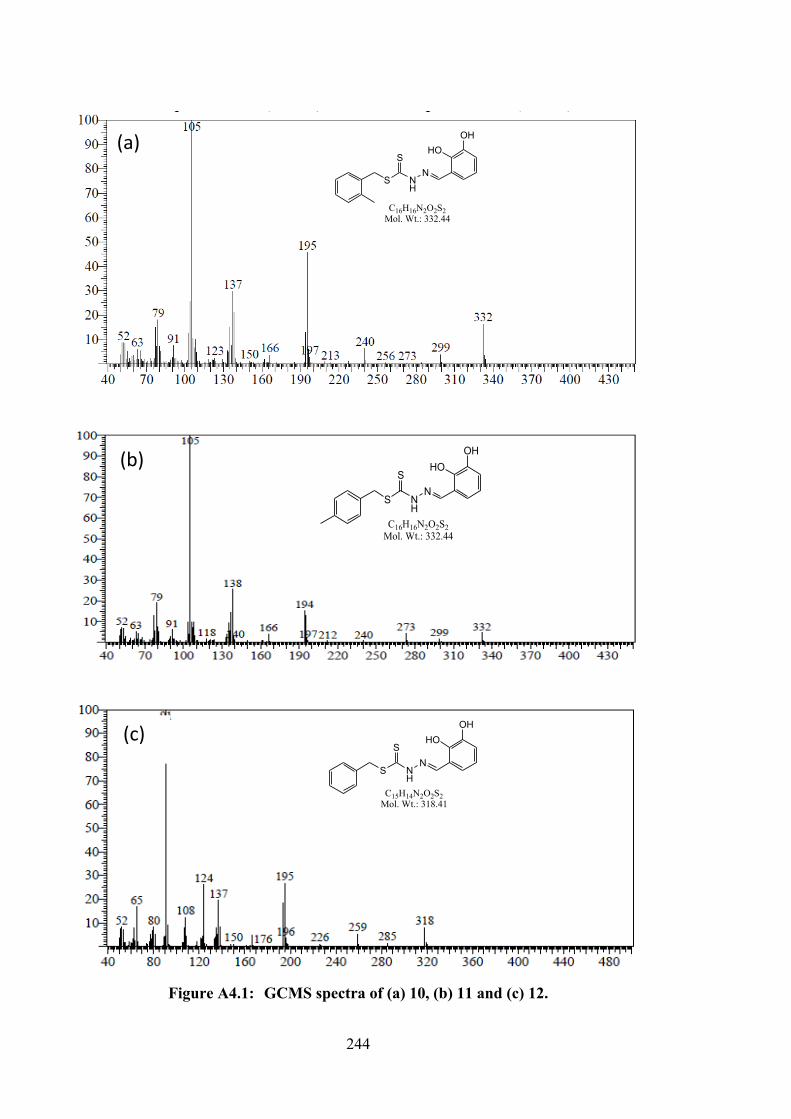
244
Figure A4.1: GCMS spectra of (a) 10, (b) 11 and (c) 12.
S NH
SN
HOOH
C16H16N2O2S2Mol. Wt.: 332.44
(b)
(a)
S NH
SN
HOOH
C16H16N2O2S2Mol. Wt.: 332.44
S NH
SN
HOOH
C15H14N2O2S2Mol. Wt.: 318.41
(c)
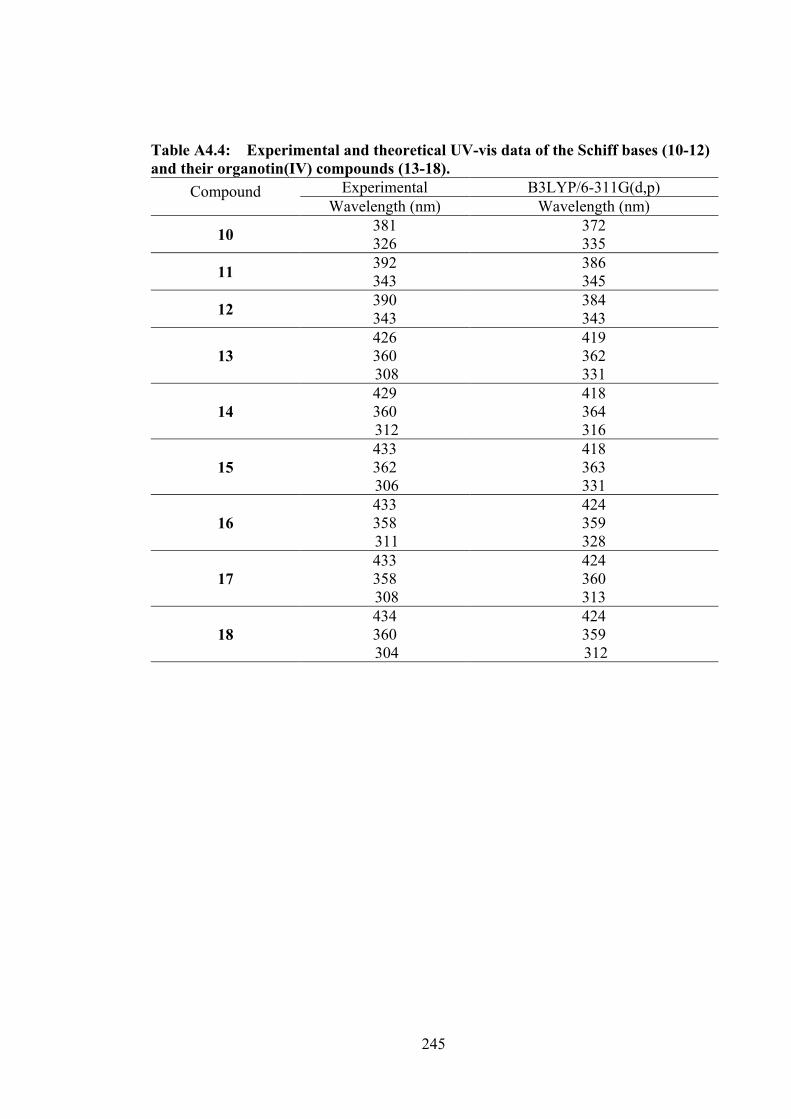
245
Table A4.4: Experimental and theoretical UV-vis data of the Schiff bases (10-12) and their organotin(IV) compounds (13-18).
Compound Experimental B3LYP/6-311G(d,p) Wavelength (nm) Wavelength (nm)
10 381 372 326 335
11 392 386 343 345
12 390 384 343 343
13 426 419 360 362 308 331
14 429 418 360 364 312 316
15 433 418 362 363 306 331
16 433 424 358 359 311 328
17 433 424 358 360 308 313
18 434 424 360 359 304 312

246
Figure A4.2: Molecular packing in 12: a view of the unit cell contents in projection down the a-axis. The hydroxyl-O‒H…O(hydroxyl) and thioamide-N‒H…S(thione) hydrogen bonds are shown as orange and green dashed lines, respectively. The hydroxyphenyl-C‒H…π(benzyl-phenyl) interactions are shown as purple dashed lines. Intramolecular hydrogen bonding O1 H1o N2 0.84(6) 1.87(5) 2.635(6) 151(6) x, y, z O2 H2o O1 0.84(5) 2.31(6) 2.703(7) 109(5) x, y, z Intermolecular interactions N1 H1n S1 0.88(4) 2.53(4) 3.390(5) 169(5) 2-x, 2-y, 1-z O2 H2o O1 0.84(5) 2.07(6) 2.814(6) 148(5) 1-x, 2-y, 2-z C6 H6 Cg(C10-C15) 0.95 2.66 3.500(7) 147 1-x, 2-y, 2-z C6 H6 Cg(C10-C15) 0.95 2.76 3.550(7) 141 1½-x, ½+y, 1½-z
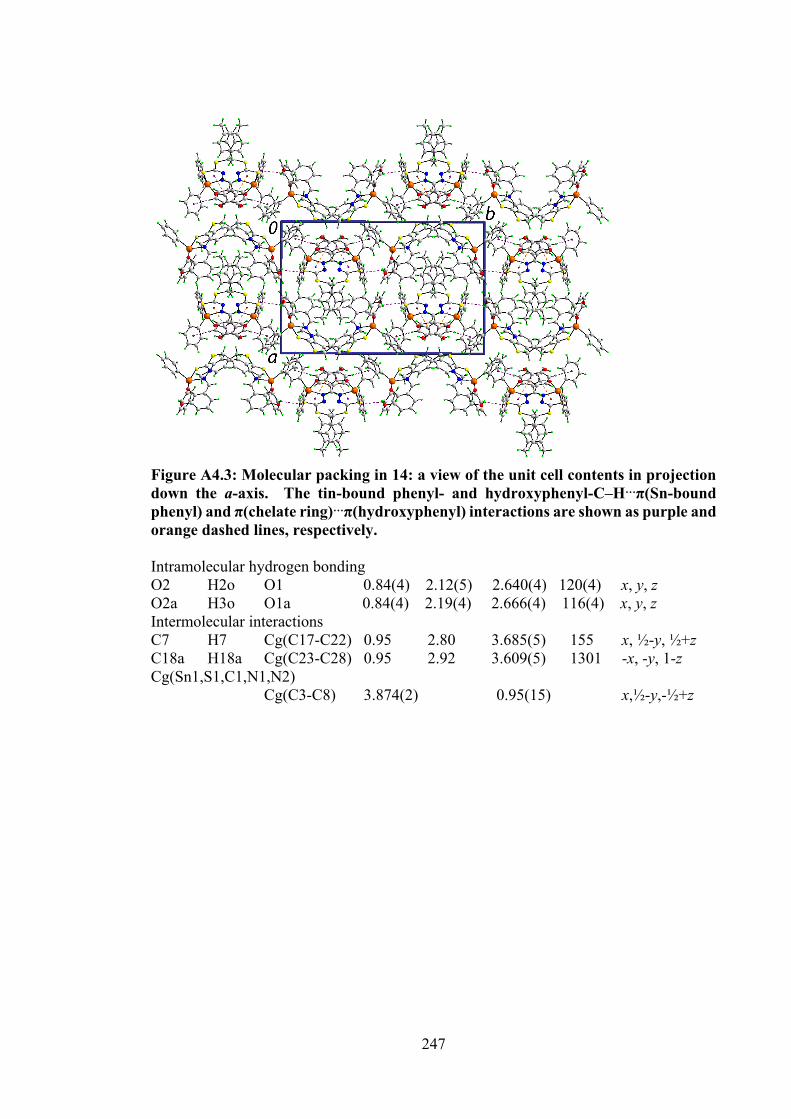
247
Figure A4.3: Molecular packing in 14: a view of the unit cell contents in projection down the a-axis. The tin-bound phenyl- and hydroxyphenyl-C‒H…π(Sn-bound phenyl) and π(chelate ring)…π(hydroxyphenyl) interactions are shown as purple and orange dashed lines, respectively. Intramolecular hydrogen bonding O2 H2o O1 0.84(4) 2.12(5) 2.640(4) 120(4) x, y, z O2a H3o O1a 0.84(4) 2.19(4) 2.666(4) 116(4) x, y, z Intermolecular interactions C7 H7 Cg(C17-C22) 0.95 2.80 3.685(5) 155 x, ½-y, ½+z C18a H18a Cg(C23-C28) 0.95 2.92 3.609(5) 130 1 -x, -y, 1-z Cg(Sn1,S1,C1,N1,N2) Cg(C3-C8) 3.874(2) 0.95(15) x,½-y,-½+z
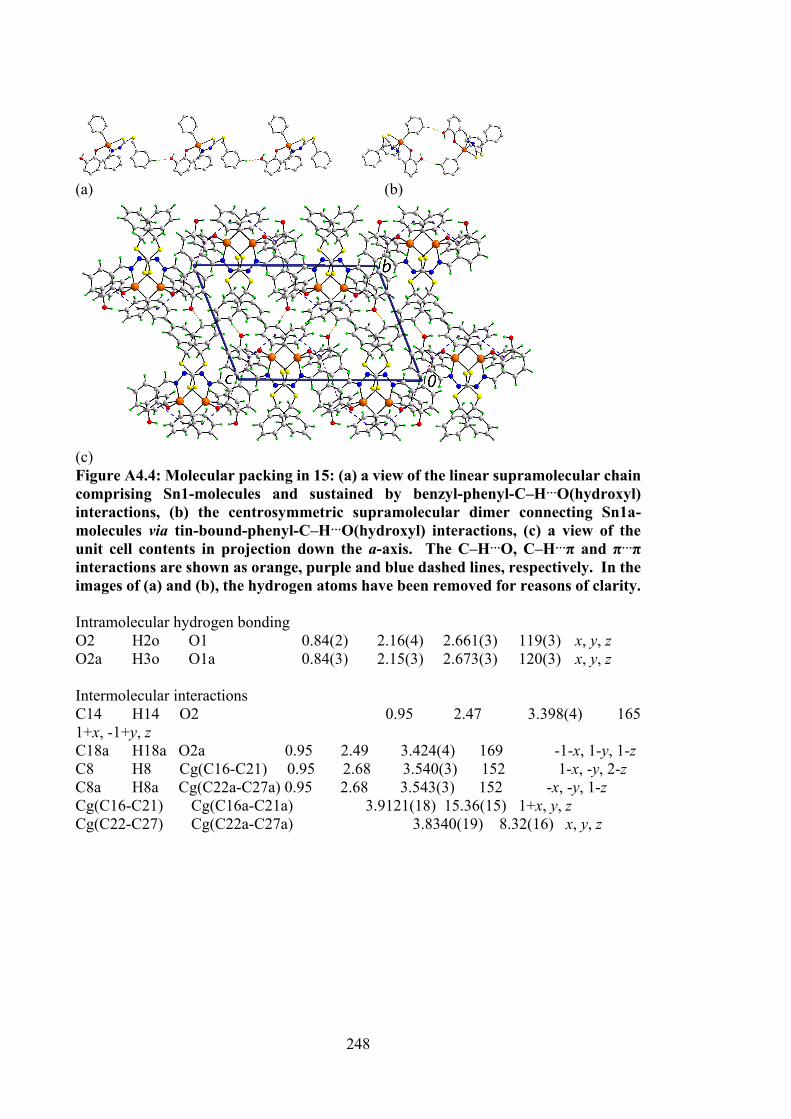
248
(a) (b)
(c) Figure A4.4: Molecular packing in 15: (a) a view of the linear supramolecular chain comprising Sn1-molecules and sustained by benzyl-phenyl-C‒H…O(hydroxyl) interactions, (b) the centrosymmetric supramolecular dimer connecting Sn1a- molecules via tin-bound-phenyl-C‒H…O(hydroxyl) interactions, (c) a view of the unit cell contents in projection down the a-axis. The C‒H…O, C‒H…π and π…π interactions are shown as orange, purple and blue dashed lines, respectively. In the images of (a) and (b), the hydrogen atoms have been removed for reasons of clarity. Intramolecular hydrogen bonding O2 H2o O1 0.84(2) 2.16(4) 2.661(3) 119(3) x, y, z O2a H3o O1a 0.84(3) 2.15(3) 2.673(3) 120(3) x, y, z Intermolecular interactions C14 H14 O2 0.95 2.47 3.398(4) 165 1+x, -1+y, z C18a H18a O2a 0.95 2.49 3.424(4) 169 -1-x, 1-y, 1-z C8 H8 Cg(C16-C21) 0.95 2.68 3.540(3) 152 1-x, -y, 2-z C8a H8a Cg(C22a-C27a) 0.95 2.68 3.543(3) 152 -x, -y, 1-z Cg(C16-C21) Cg(C16a-C21a) 3.9121(18) 15.36(15) 1+x, y, z Cg(C22-C27) Cg(C22a-C27a) 3.8340(19) 8.32(16) x, y, z
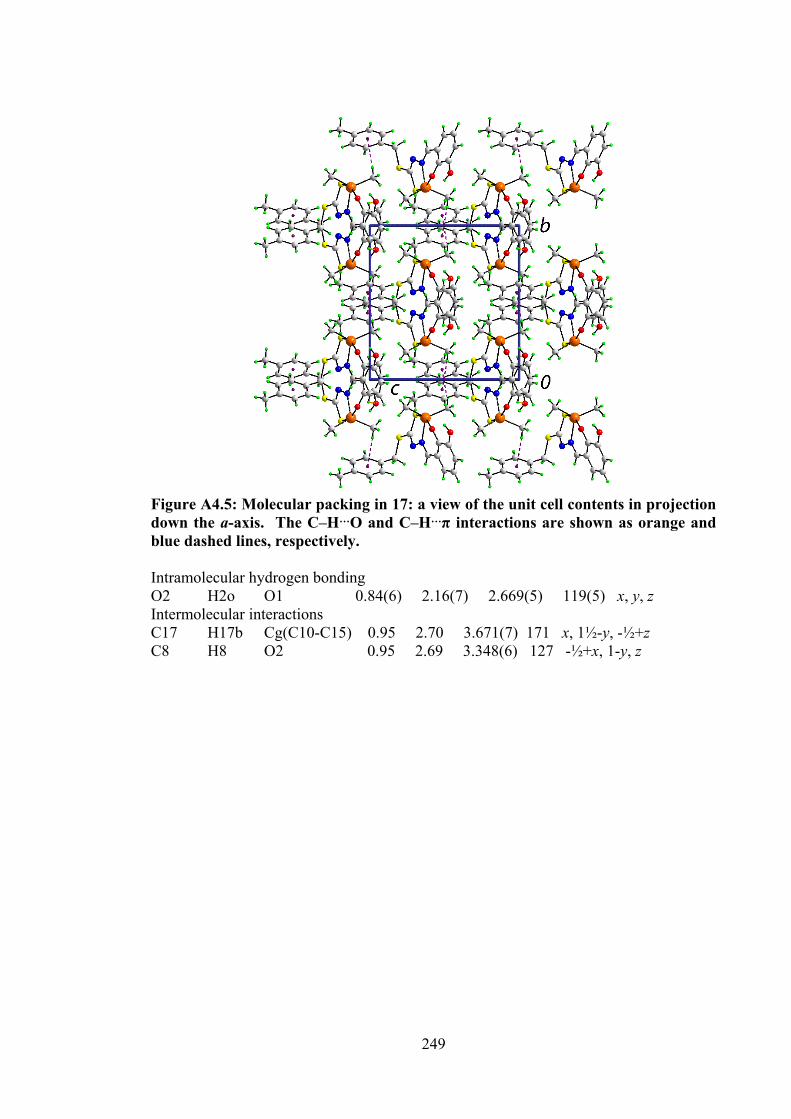
249
Figure A4.5: Molecular packing in 17: a view of the unit cell contents in projection down the a-axis. The C‒H…O and C‒H…π interactions are shown as orange and blue dashed lines, respectively. Intramolecular hydrogen bonding O2 H2o O1 0.84(6) 2.16(7) 2.669(5) 119(5) x, y, z Intermolecular interactions C17 H17b Cg(C10-C15) 0.95 2.70 3.671(7) 171 x, 1½-y, -½+z C8 H8 O2 0.95 2.69 3.348(6) 127 -½+x, 1-y, z
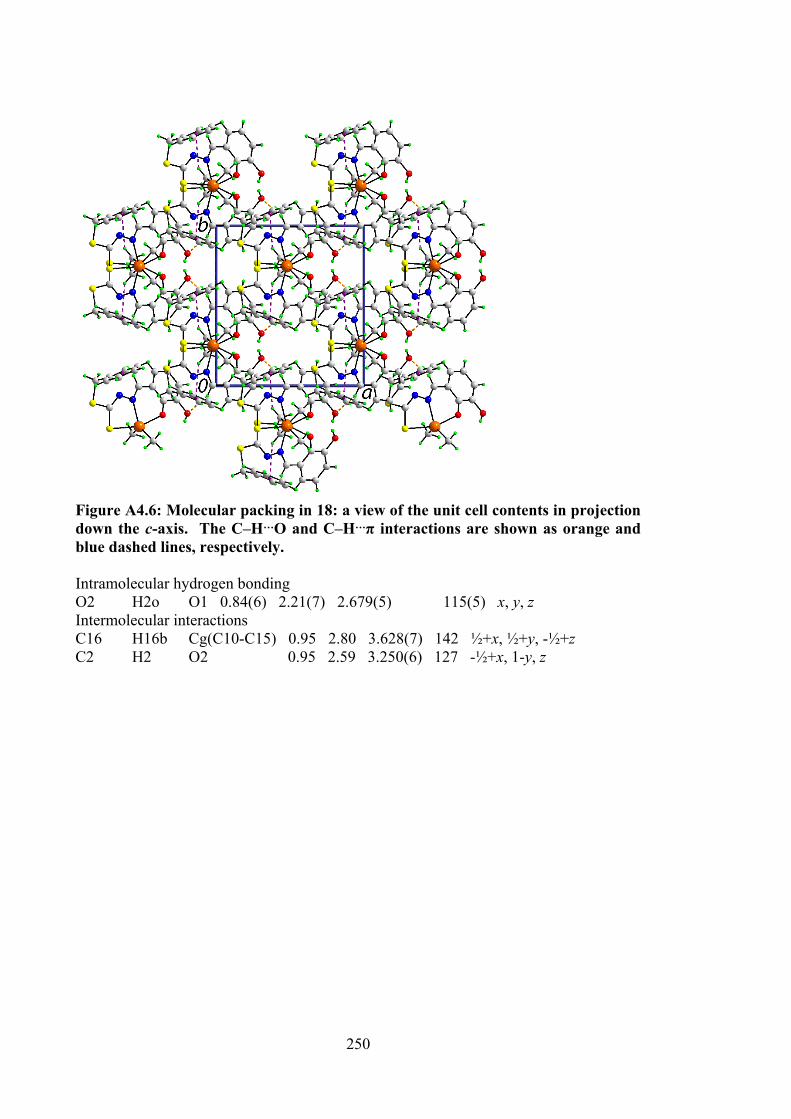
250
Figure A4.6: Molecular packing in 18: a view of the unit cell contents in projection down the c-axis. The C‒H…O and C‒H…π interactions are shown as orange and blue dashed lines, respectively. Intramolecular hydrogen bonding O2 H2o O1 0.84(6) 2.21(7) 2.679(5) 115(5) x, y, z Intermolecular interactions C16 H16b Cg(C10-C15) 0.95 2.80 3.628(7) 142 ½+x, ½+y, -½+z C2 H2 O2 0.95 2.59 3.250(6) 127 -½+x, 1-y, z
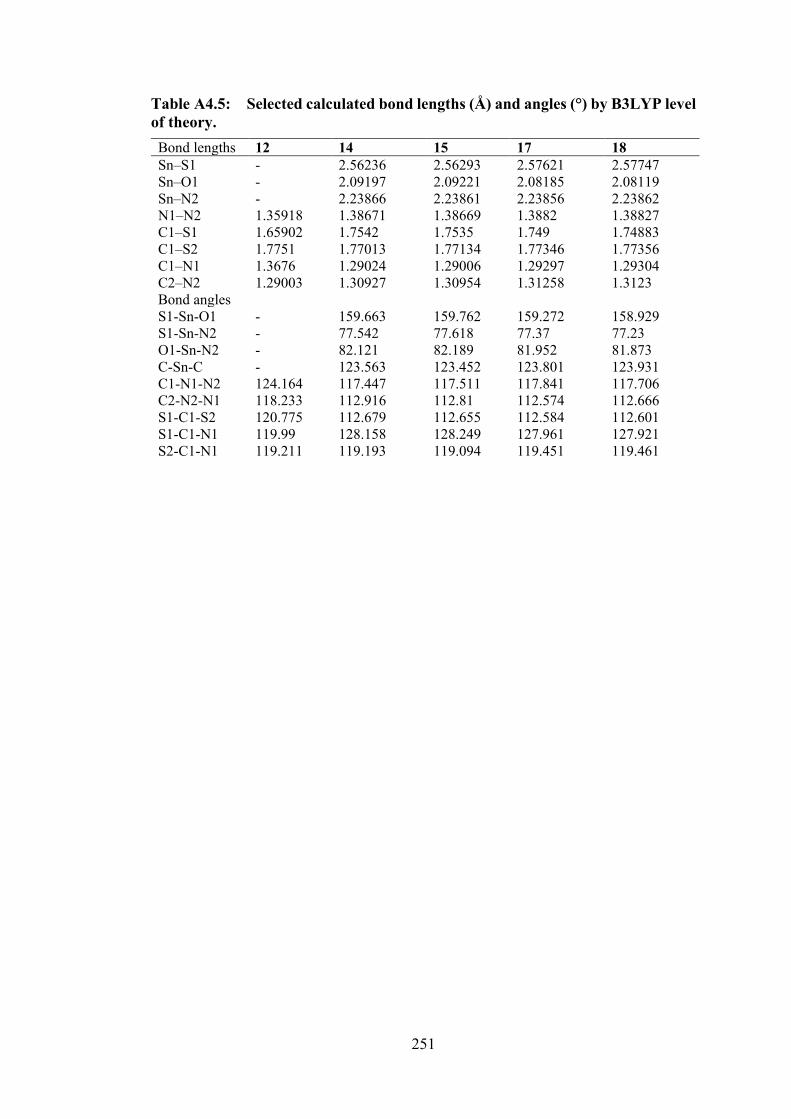
251
Table A4.5: Selected calculated bond lengths (Å) and angles (°) by B3LYP level of theory.
Bond lengths 12 14 15 17 18 Sn‒S1 - 2.56236 2.56293 2.57621 2.57747 Sn‒O1 - 2.09197 2.09221 2.08185 2.08119 Sn‒N2 - 2.23866 2.23861 2.23856 2.23862 N1‒N2 1.35918 1.38671 1.38669 1.3882 1.38827 C1‒S1 1.65902 1.7542 1.7535 1.749 1.74883 C1‒S2 1.7751 1.77013 1.77134 1.77346 1.77356 C1‒N1 1.3676 1.29024 1.29006 1.29297 1.29304 C2‒N2 1.29003 1.30927 1.30954 1.31258 1.3123 Bond angles S1-Sn-O1 - 159.663 159.762 159.272 158.929 S1-Sn-N2 - 77.542 77.618 77.37 77.23 O1-Sn-N2 - 82.121 82.189 81.952 81.873 C-Sn-C - 123.563 123.452 123.801 123.931 C1-N1-N2 124.164 117.447 117.511 117.841 117.706 C2-N2-N1 118.233 112.916 112.81 112.574 112.666 S1-C1-S2 120.775 112.679 112.655 112.584 112.601 S1-C1-N1 119.99 128.158 128.249 127.961 127.921 S2-C1-N1 119.211 119.193 119.094 119.451 119.461
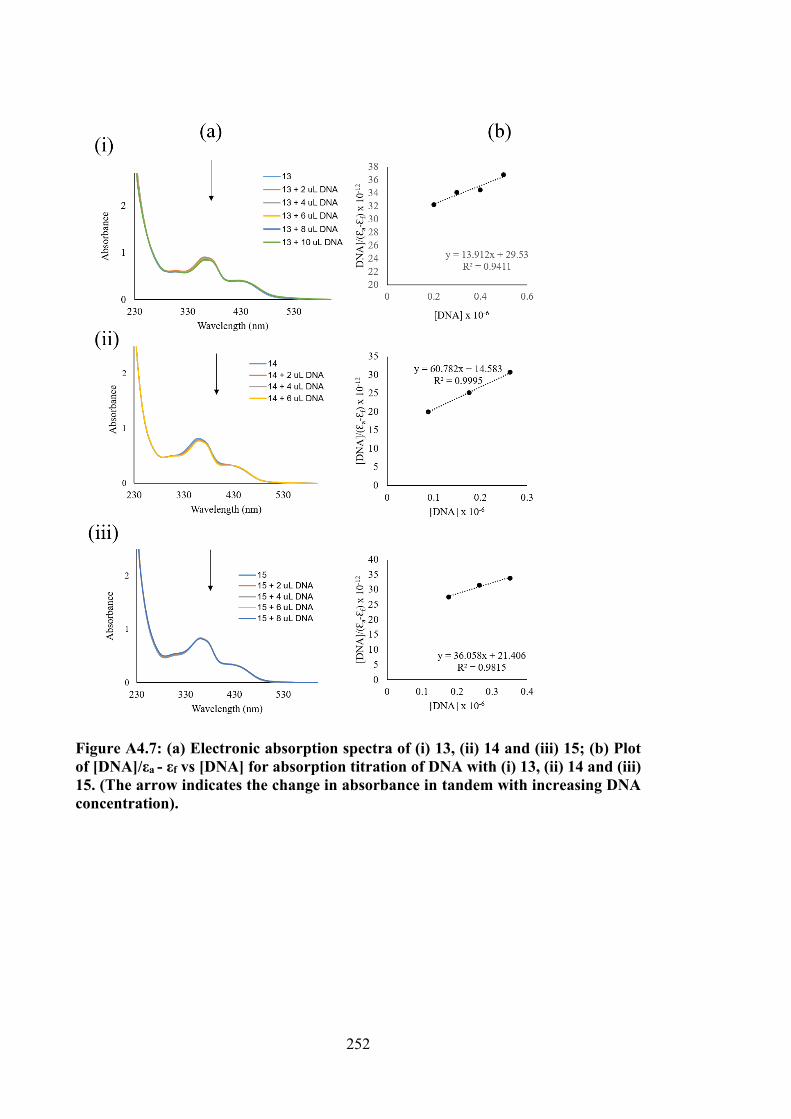
252
Figure A4.7: (a) Electronic absorption spectra of (i) 13, (ii) 14 and (iii) 15; (b) Plot of [DNA]/εa - εf vs [DNA] for absorption titration of DNA with (i) 13, (ii) 14 and (iii) 15. (The arrow indicates the change in absorbance in tandem with increasing DNA concentration).
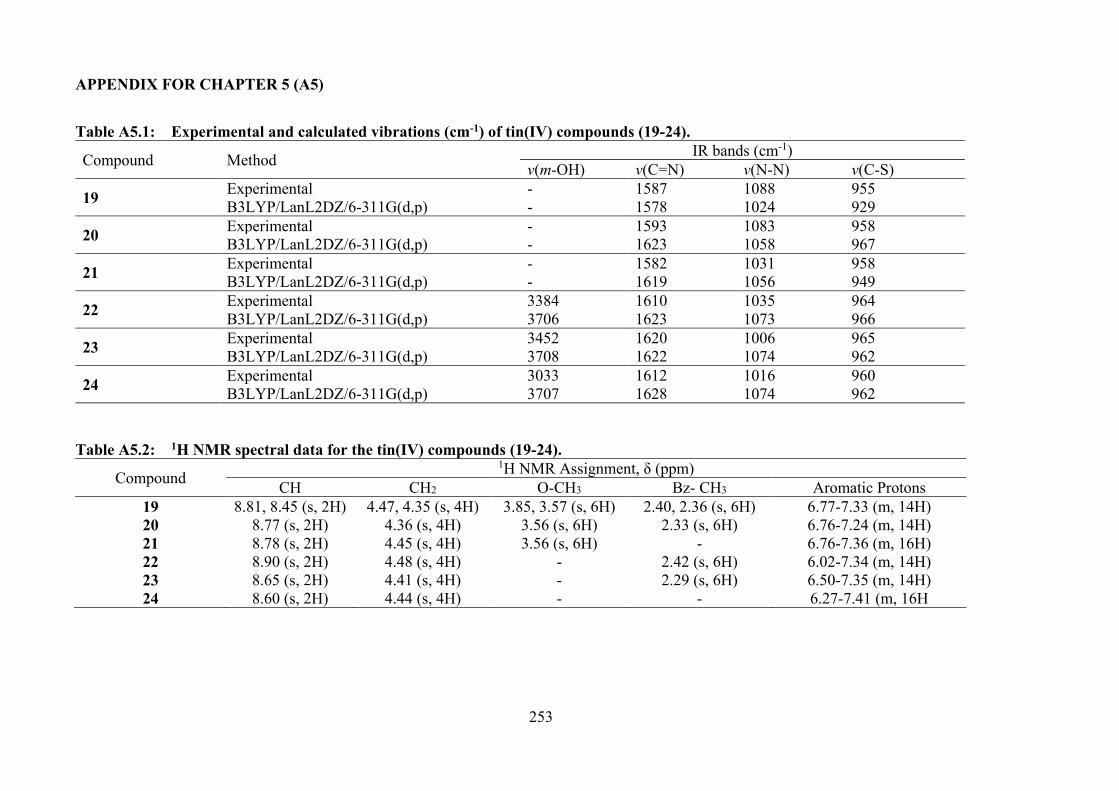
253
APPENDIX FOR CHAPTER 5 (A5)
Table A5.1: Experimental and calculated vibrations (cm-1) of tin(IV) compounds (19-24).
Compound Method IR bands (cm-1) v(m-OH) v(C=N) v(N-N) v(C-S)
19 Experimental - 1587 1088 955 B3LYP/LanL2DZ/6-311G(d,p) - 1578 1024 929
20 Experimental - 1593 1083 958 B3LYP/LanL2DZ/6-311G(d,p) - 1623 1058 967
21 Experimental - 1582 1031 958 B3LYP/LanL2DZ/6-311G(d,p) - 1619 1056 949
22 Experimental 3384 1610 1035 964 B3LYP/LanL2DZ/6-311G(d,p) 3706 1623 1073 966
23 Experimental 3452 1620 1006 965 B3LYP/LanL2DZ/6-311G(d,p) 3708 1622 1074 962
24 Experimental 3033 1612 1016 960 B3LYP/LanL2DZ/6-311G(d,p) 3707 1628 1074 962
Table A5.2: 1H NMR spectral data for the tin(IV) compounds (19-24).
Compound 1H NMR Assignment, δ (ppm)
CH CH2 O-CH3 Bz- CH3 Aromatic Protons 19 8.81, 8.45 (s, 2H) 4.47, 4.35 (s, 4H) 3.85, 3.57 (s, 6H) 2.40, 2.36 (s, 6H) 6.77-7.33 (m, 14H) 20 8.77 (s, 2H) 4.36 (s, 4H) 3.56 (s, 6H) 2.33 (s, 6H) 6.76-7.24 (m, 14H) 21 8.78 (s, 2H) 4.45 (s, 4H) 3.56 (s, 6H) - 6.76-7.36 (m, 16H) 22 8.90 (s, 2H) 4.48 (s, 4H) - 2.42 (s, 6H) 6.02-7.34 (m, 14H) 23 8.65 (s, 2H) 4.41 (s, 4H) - 2.29 (s, 6H) 6.50-7.35 (m, 14H) 24 8.60 (s, 2H) 4.44 (s, 4H) - - 6.27-7.41 (m, 16H
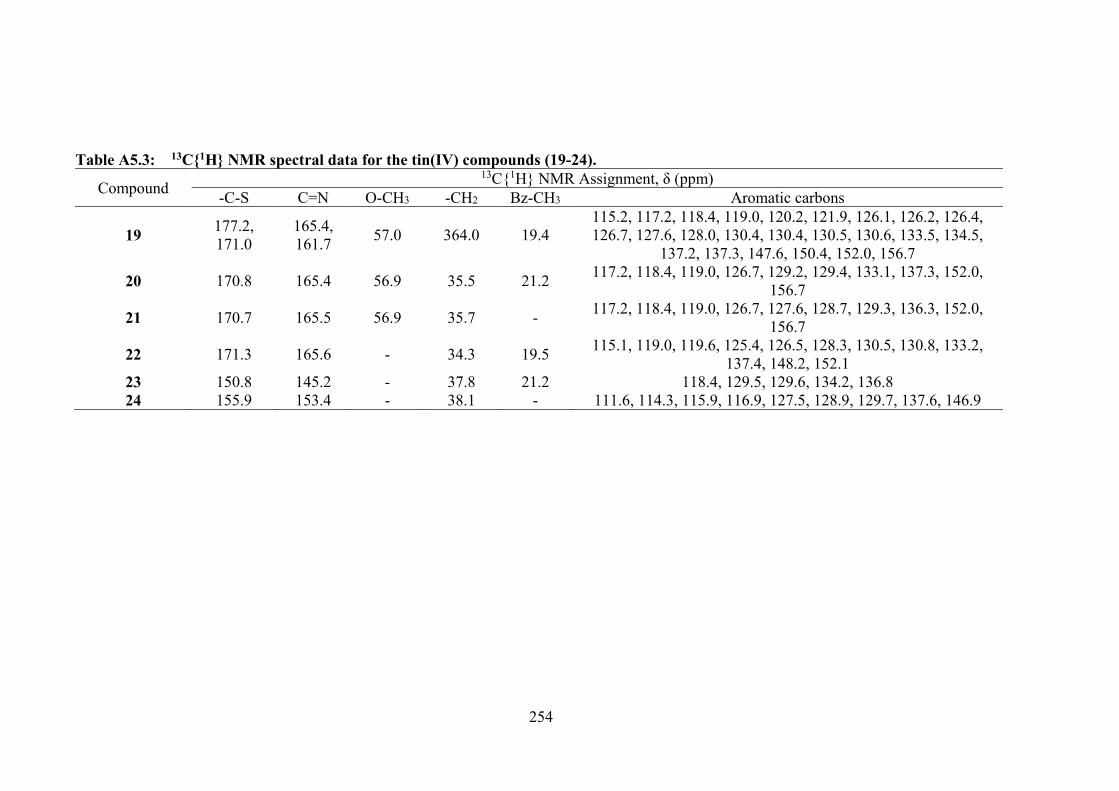
254
Table A5.3: 13C1H NMR spectral data for the tin(IV) compounds (19-24).
Compound 13C1H NMR Assignment, δ (ppm)
-C-S C=N O-CH3 -CH2 Bz-CH3 Aromatic carbons
19 177.2, 171.0
165.4, 161.7 57.0 364.0 19.4
115.2, 117.2, 118.4, 119.0, 120.2, 121.9, 126.1, 126.2, 126.4, 126.7, 127.6, 128.0, 130.4, 130.4, 130.5, 130.6, 133.5, 134.5,
137.2, 137.3, 147.6, 150.4, 152.0, 156.7
20 170.8 165.4 56.9 35.5 21.2 117.2, 118.4, 119.0, 126.7, 129.2, 129.4, 133.1, 137.3, 152.0, 156.7
21 170.7 165.5 56.9 35.7 - 117.2, 118.4, 119.0, 126.7, 127.6, 128.7, 129.3, 136.3, 152.0, 156.7
22 171.3 165.6 - 34.3 19.5 115.1, 119.0, 119.6, 125.4, 126.5, 128.3, 130.5, 130.8, 133.2, 137.4, 148.2, 152.1
23 150.8 145.2 - 37.8 21.2 118.4, 129.5, 129.6, 134.2, 136.8 24 155.9 153.4 - 38.1 - 111.6, 114.3, 115.9, 116.9, 127.5, 128.9, 129.7, 137.6, 146.9
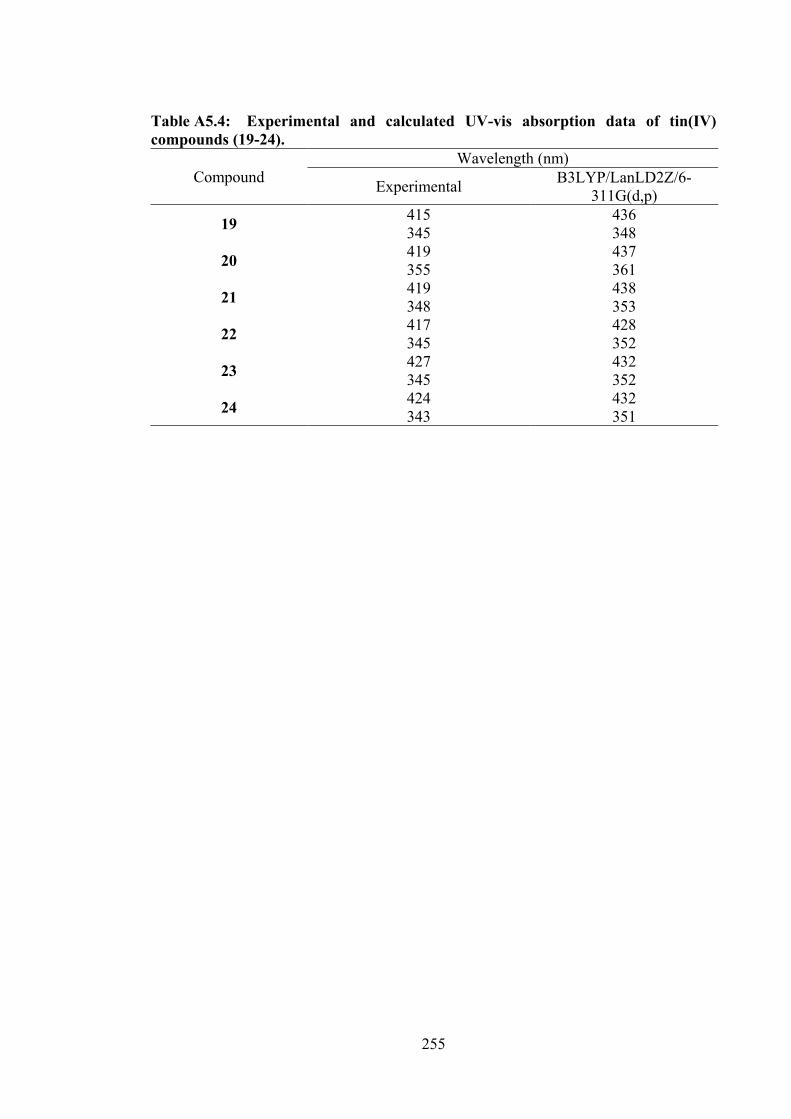
255
Table A5.4: Experimental and calculated UV-vis absorption data of tin(IV) compounds (19-24).
Compound Wavelength (nm)
Experimental B3LYP/LanLD2Z/6-311G(d,p)
19 415 436 345 348
20 419 437 355 361
21 419 438 348 353
22 417 428 345 352
23 427 432 345 352
24 424 432 343 351

256
Table A5.5: Least-squares plane data for 20a and 21. 20a S1a-chelate ring: envelope with Sn1a lying 0.307(4) Å above the remaining atoms (r.m.s. deviation = 0.0060 Å) S3a-chelate ring: planar with r.m.s. deviation = 0.0057 Å O1a-chelate ring: envelope with Sn1a lying 0.579(4) Å above the remaining atoms (r.m.s. deviation = 0.0279 Å) O3a-chelate ring: envelope with Sn1a lying 0.135(4) Å above the remaining atoms (r.m.s. deviation = 0.0234 Å) 21 S1-chelate ring: envelope with Sn1a lying 0.392(4) Å above the remaining atoms (r.m.s. deviation = 0.0066 Å) S3-chelate ring: envelope with Sn1a lying 0.309(4) Å above the remaining atoms (r.m.s. deviation = 0.0124 Å) O1-chelate ring: envelope with Sn1a lying 0.463(4) Å above the remaining atoms (r.m.s. deviation = 0.0392 Å) O3-chelate ring: envelope with Sn1a lying 0.271(4) Å above the remaining atoms (r.m.s. deviation = 0.0343 Å)
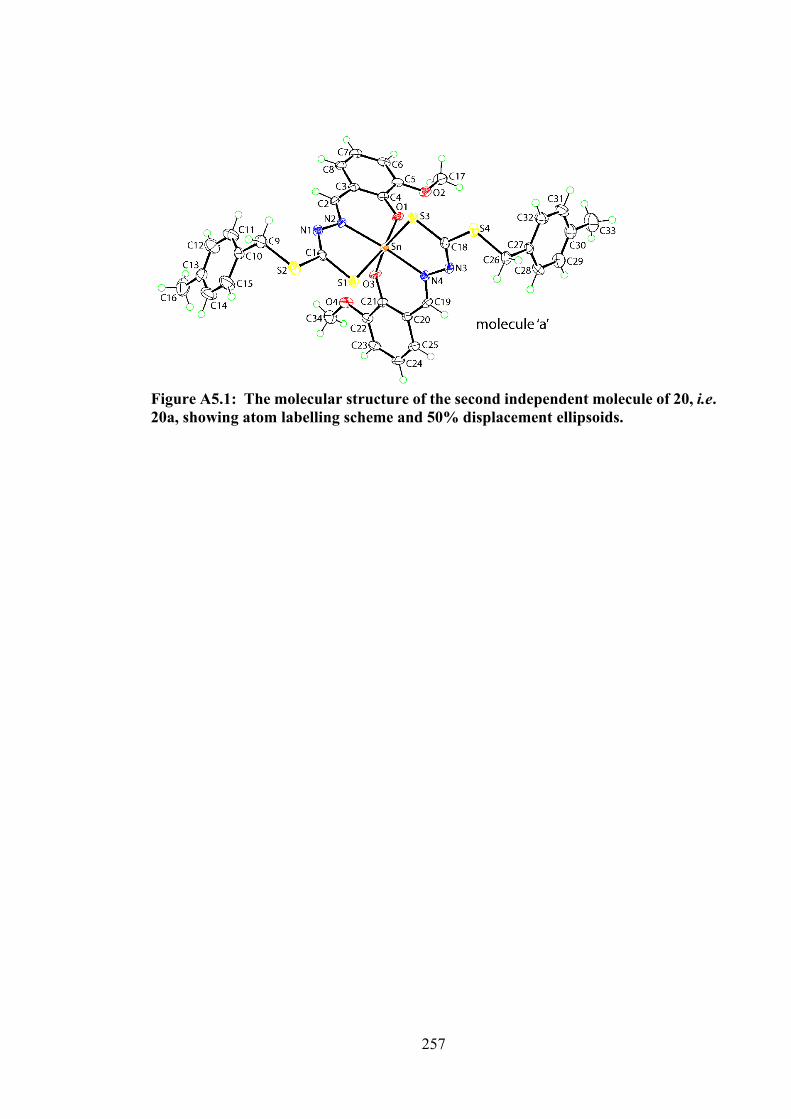
257
Figure A5.1: The molecular structure of the second independent molecule of 20, i.e. 20a, showing atom labelling scheme and 50% displacement ellipsoids.
258
Figure A5.2: Molecular packing in 20: (a) a side-on view of the supramolecular layer in the ab-plane, (b) plane view of the supramolecular layer (non-participating hydrogen atoms have been removed for clarity) and (c) unit cell contents in projection down the a-axis. The C–H…O, C–H…S, C–H…π and π…π interactions are shown as orange, green, purple and blue dashed lines, respectively.
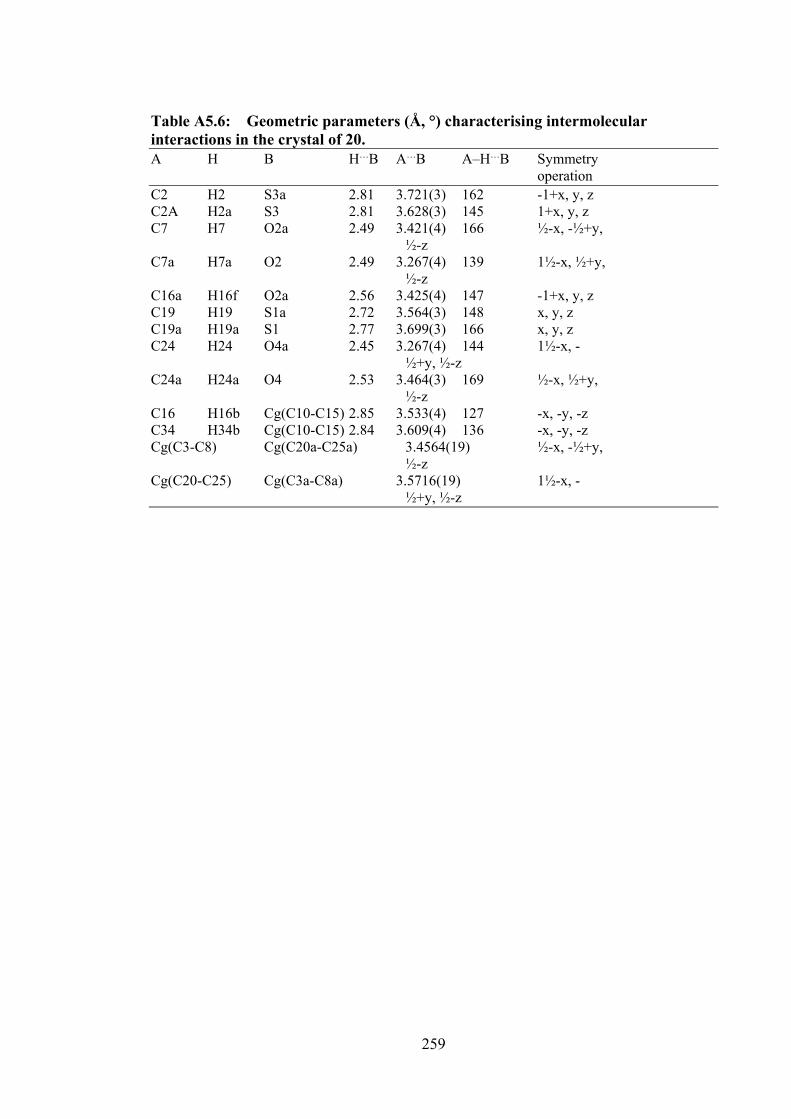
259
Table A5.6: Geometric parameters (Å, °) characterising intermolecular interactions in the crystal of 20. A H B H…B A…B A–H…B Symmetry operation C2 H2 S3a 2.81 3.721(3) 162 -1+x, y, z C2A H2a S3 2.81 3.628(3) 145 1+x, y, z C7 H7 O2a 2.49 3.421(4) 166 ½-x, -½+y, ½-z C7a H7a O2 2.49 3.267(4) 139 1½-x, ½+y, ½-z C16a H16f O2a 2.56 3.425(4) 147 -1+x, y, z C19 H19 S1a 2.72 3.564(3) 148 x, y, z C19a H19a S1 2.77 3.699(3) 166 x, y, z C24 H24 O4a 2.45 3.267(4) 144 1½-x, - ½+y, ½-z C24a H24a O4 2.53 3.464(3) 169 ½-x, ½+y, ½-z C16 H16b Cg(C10-C15) 2.85 3.533(4) 127 -x, -y, -z C34 H34b Cg(C10-C15) 2.84 3.609(4) 136 -x, -y, -z Cg(C3-C8) Cg(C20a-C25a) 3.4564(19) ½-x, -½+y, ½-z Cg(C20-C25) Cg(C3a-C8a) 3.5716(19) 1½-x, - ½+y, ½-z
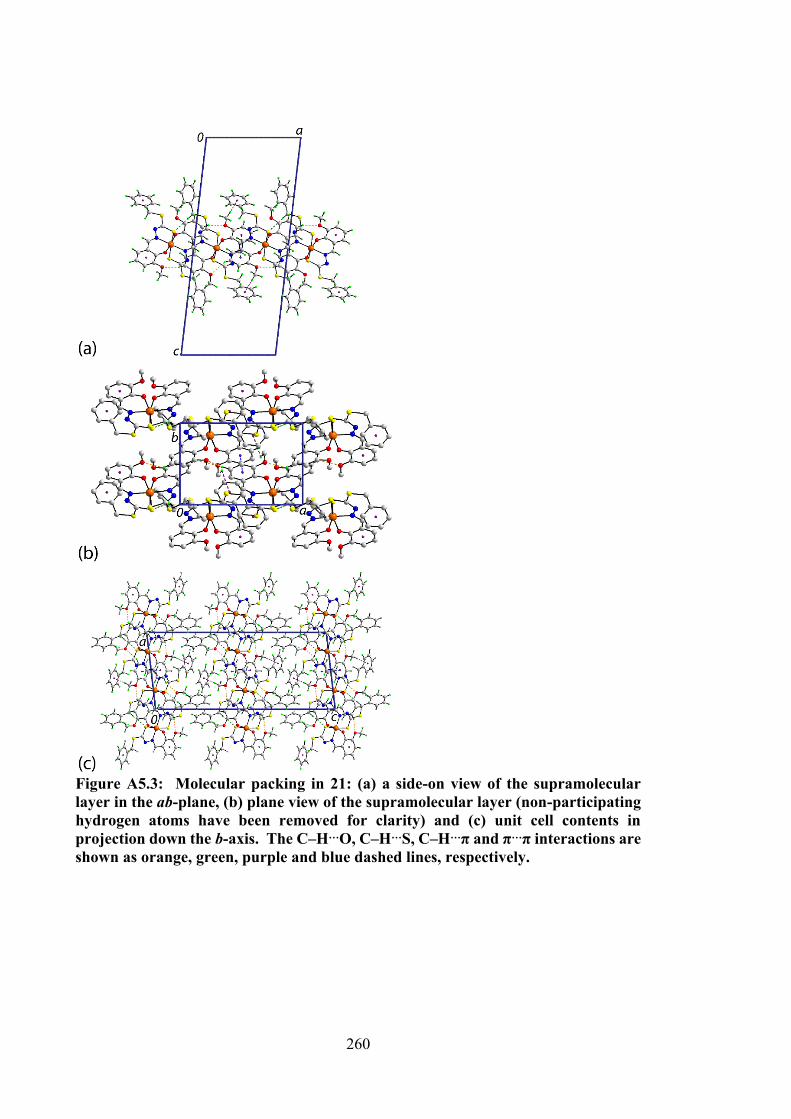
260
Figure A5.3: Molecular packing in 21: (a) a side-on view of the supramolecular layer in the ab-plane, (b) plane view of the supramolecular layer (non-participating hydrogen atoms have been removed for clarity) and (c) unit cell contents in projection down the b-axis. The C–H…O, C–H…S, C–H…π and π…π interactions are shown as orange, green, purple and blue dashed lines, respectively.
261
Table A5.7: Geometric parameters (Å, °) characterising intermolecular interactions in the crystal of 21. A H B H…B A…B A–H…B Symmetry operation C7 H7 O4 2.60 3.487(3) 156 1-x, -y, 2-z C23 H23 O2 2.37 3.140(3) 138 2-x, -y, 2-z C25 H25b S1 2.79 3.645(3) 145 2-x, 1-y, 2-z C16 H16a Cg(C10-C15) 2.95 3.354(4) 106 1-x, -y, 2-z Cg(C3-C8) Cg(C3-C8) 3.5623(15) 1-x, -y, 2-z
262
APPENDIX FOR CHAPTER 6 (A6) Table A6.1: Experimental and calculated frequencies (cm-1) of the thiosemicarbazone Schiff bases (25-28) and tin(IV) compounds (29-40).
Compound Method IR bands (cm-1)
v(m-OH) v(o-OH) v(NH) v(NH) v(C=N) v(N-N) v(C=S)/ v(C-S)
25 Experimental - 3337 3304 1610 1109 1037 B3LYP/6-311G(d,p) - 3647 3472 3410 1611 1109 1034
26 Experimental - - - 3300 1609 1103 908 B3LYP/6-311G(d,p) - 3646 3407 3399 1611 1099 942
27 Experimental 3418 3140 1601 1112 1035 B3LYP/6-311G(d,p) 3712 3354 3511 3407 1601 1131 1016
28 Experimental 3443 - 3129 1597 1047 1029 B3LYP/6-311G(d,p) 3712 3401 3480 3367 1601 1138 1020
29 Experimental - - 3299 - 1596 1066 973 B3LYP/LanL2DZ/6-311G(d,p) - - 3665 - 1629 1075 1001
30 Experimental - - 3222 - 1590 1066 973 B3LYP/LanL2DZ/6-311G(d,p) - - 3548 - 1575 1039 971
31 Experimental - - 3308 - 1590 1066 973 B3LYP/LanL2DZ/6-311G(d,p) - - 3665 - 1629 1085 1002
32 Experimental - - 1593 1006 953 B3LYP/LanL2DZ/6-311G(d,p) 3720 - 3668 - 1629 1062 822
33 Experimental - - 1596 1006 951 B3LYP/LanL2DZ/6-311G(d,p) 3706 - 3666 - 1598 1064 821
34 Experimental - - 1585 993 951 B3LYP/LanL2DZ/6-311G(d,p) 3702 - 3664 - 1570 1087 819
35 Experimental - - 3331 - 1586 1075 832 B3LYP/LanL2DZ/6-311G(d,p) - - 3619 - 1626 1081 836
263
36 Experimental - - 3294 - 1577 1059 824 B3LYP/LanL2DZ/6-311G(d,p) - - 3505 - 1572 1045 811
37 Experimental - - 3303 - 1581 1063 824 B3LYP/LanL2DZ/6-311G(d,p) - - 3503 - 1573 1055 809
38 Experimental - - 1587 998 957 B3LYP/LanL2DZ/6-311G(d,p) 3708 - 3616 - 1624 1090 852
39 Experimental - - 1575 1001 917 B3LYP/LanL2DZ/6-311G(d,p) 3707 - 3619 - 1623 1075 839
40 Experimental - - 1588 1001 939 B3LYP/LanL2DZ/6-311G(d,p) 3708 - 3616 - 1628 1084 852
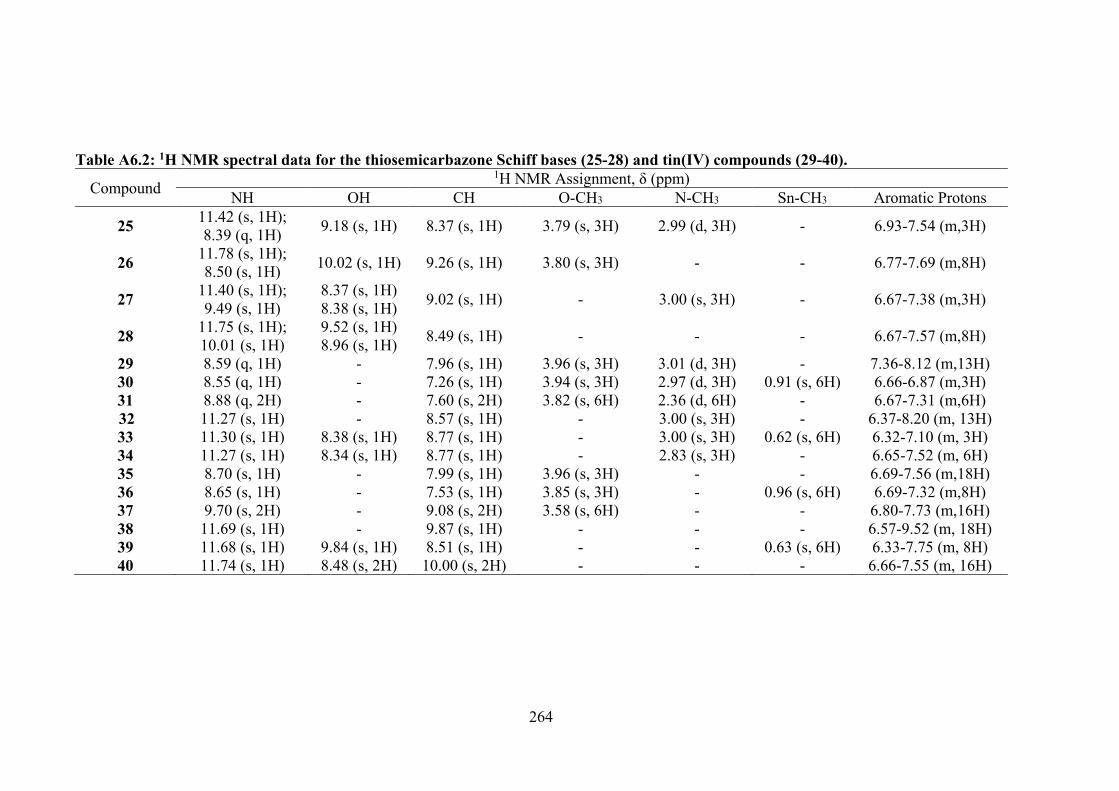
264
Table A6.2: 1H NMR spectral data for the thiosemicarbazone Schiff bases (25-28) and tin(IV) compounds (29-40).
Compound 1H NMR Assignment, δ (ppm)
NH OH CH O-CH3 N-CH3 Sn-CH3 Aromatic Protons
25 11.42 (s, 1H); 8.39 (q, 1H) 9.18 (s, 1H) 8.37 (s, 1H) 3.79 (s, 3H) 2.99 (d, 3H) - 6.93-7.54 (m,3H)
26 11.78 (s, 1H); 8.50 (s, 1H) 10.02 (s, 1H) 9.26 (s, 1H) 3.80 (s, 3H) - - 6.77-7.69 (m,8H)
27 11.40 (s, 1H); 9.49 (s, 1H)
8.37 (s, 1H) 8.38 (s, 1H) 9.02 (s, 1H) - 3.00 (s, 3H) - 6.67-7.38 (m,3H)
28 11.75 (s, 1H); 10.01 (s, 1H)
9.52 (s, 1H) 8.96 (s, 1H) 8.49 (s, 1H) - - - 6.67-7.57 (m,8H)
29 8.59 (q, 1H) - 7.96 (s, 1H) 3.96 (s, 3H) 3.01 (d, 3H) - 7.36-8.12 (m,13H) 30 8.55 (q, 1H) - 7.26 (s, 1H) 3.94 (s, 3H) 2.97 (d, 3H) 0.91 (s, 6H) 6.66-6.87 (m,3H) 31 8.88 (q, 2H) - 7.60 (s, 2H) 3.82 (s, 6H) 2.36 (d, 6H) - 6.67-7.31 (m,6H) 32 11.27 (s, 1H) - 8.57 (s, 1H) - 3.00 (s, 3H) - 6.37-8.20 (m, 13H) 33 11.30 (s, 1H) 8.38 (s, 1H) 8.77 (s, 1H) - 3.00 (s, 3H) 0.62 (s, 6H) 6.32-7.10 (m, 3H) 34 11.27 (s, 1H) 8.34 (s, 1H) 8.77 (s, 1H) - 2.83 (s, 3H) - 6.65-7.52 (m, 6H) 35 8.70 (s, 1H) - 7.99 (s, 1H) 3.96 (s, 3H) - - 6.69-7.56 (m,18H) 36 8.65 (s, 1H) - 7.53 (s, 1H) 3.85 (s, 3H) - 0.96 (s, 6H) 6.69-7.32 (m,8H) 37 9.70 (s, 2H) - 9.08 (s, 2H) 3.58 (s, 6H) - - 6.80-7.73 (m,16H) 38 11.69 (s, 1H) - 9.87 (s, 1H) - - - 6.57-9.52 (m, 18H) 39 11.68 (s, 1H) 9.84 (s, 1H) 8.51 (s, 1H) - - 0.63 (s, 6H) 6.33-7.75 (m, 8H) 40 11.74 (s, 1H) 8.48 (s, 2H) 10.00 (s, 2H) - - - 6.66-7.55 (m, 16H)
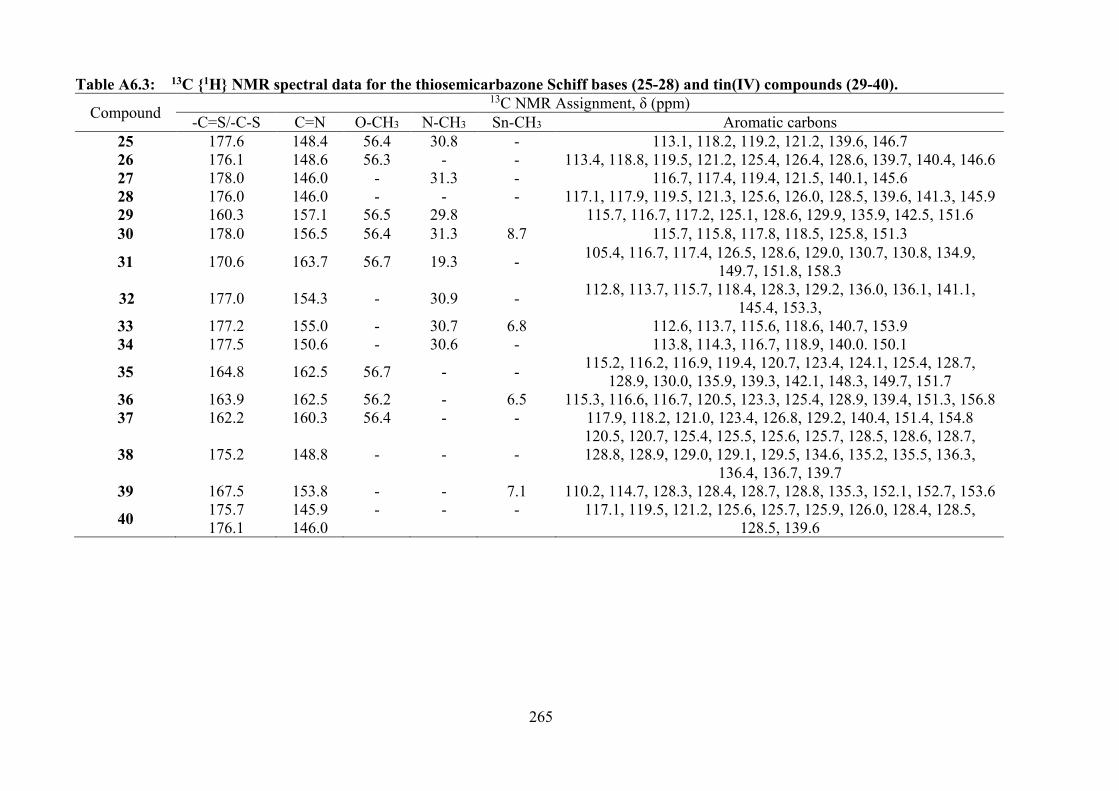
265
Table A6.3: 13C 1H NMR spectral data for the thiosemicarbazone Schiff bases (25-28) and tin(IV) compounds (29-40).
Compound 13C NMR Assignment, δ (ppm)
-C=S/-C-S C=N O-CH3 N-CH3 Sn-CH3 Aromatic carbons 25 177.6 148.4 56.4 30.8 - 113.1, 118.2, 119.2, 121.2, 139.6, 146.7 26 176.1 148.6 56.3 - - 113.4, 118.8, 119.5, 121.2, 125.4, 126.4, 128.6, 139.7, 140.4, 146.6 27 178.0 146.0 - 31.3 - 116.7, 117.4, 119.4, 121.5, 140.1, 145.6 28 176.0 146.0 - - - 117.1, 117.9, 119.5, 121.3, 125.6, 126.0, 128.5, 139.6, 141.3, 145.9 29 160.3 157.1 56.5 29.8 115.7, 116.7, 117.2, 125.1, 128.6, 129.9, 135.9, 142.5, 151.6 30 178.0 156.5 56.4 31.3 8.7 115.7, 115.8, 117.8, 118.5, 125.8, 151.3
31 170.6 163.7 56.7 19.3 - 105.4, 116.7, 117.4, 126.5, 128.6, 129.0, 130.7, 130.8, 134.9, 149.7, 151.8, 158.3
32 177.0 154.3 - 30.9 - 112.8, 113.7, 115.7, 118.4, 128.3, 129.2, 136.0, 136.1, 141.1, 145.4, 153.3,
33 177.2 155.0 - 30.7 6.8 112.6, 113.7, 115.6, 118.6, 140.7, 153.9 34 177.5 150.6 - 30.6 - 113.8, 114.3, 116.7, 118.9, 140.0. 150.1
35 164.8 162.5 56.7 - - 115.2, 116.2, 116.9, 119.4, 120.7, 123.4, 124.1, 125.4, 128.7, 128.9, 130.0, 135.9, 139.3, 142.1, 148.3, 149.7, 151.7
36 163.9 162.5 56.2 - 6.5 115.3, 116.6, 116.7, 120.5, 123.3, 125.4, 128.9, 139.4, 151.3, 156.8 37 162.2 160.3 56.4 - - 117.9, 118.2, 121.0, 123.4, 126.8, 129.2, 140.4, 151.4, 154.8
38 175.2 148.8 - - - 120.5, 120.7, 125.4, 125.5, 125.6, 125.7, 128.5, 128.6, 128.7, 128.8, 128.9, 129.0, 129.1, 129.5, 134.6, 135.2, 135.5, 136.3,
136.4, 136.7, 139.7 39 167.5 153.8 - - 7.1 110.2, 114.7, 128.3, 128.4, 128.7, 128.8, 135.3, 152.1, 152.7, 153.6
40 175.7 176.1
145.9 146.0
- - - 117.1, 119.5, 121.2, 125.6, 125.7, 125.9, 126.0, 128.4, 128.5, 128.5, 139.6
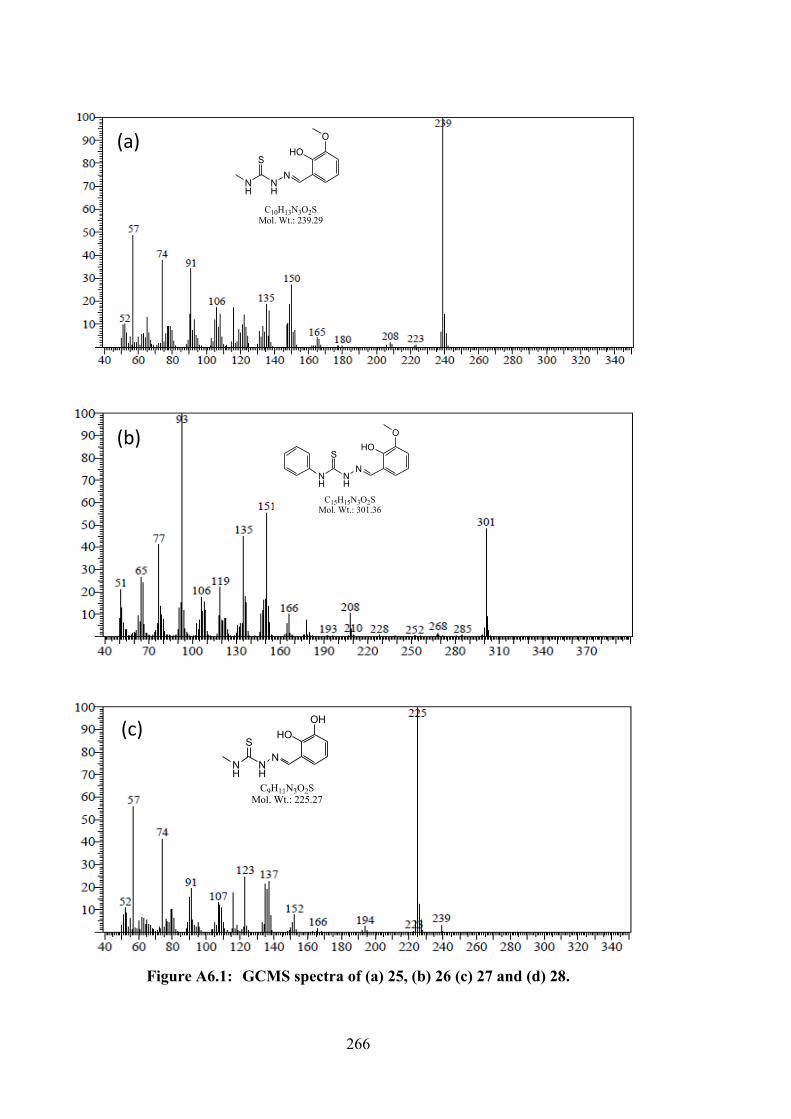
266
Figure A6.1: GCMS spectra of (a) 25, (b) 26 (c) 27 and (d) 28.
NH
NH
SN
HOOH
C9H11N3O2SMol. Wt.: 225.27
(c)
NH
NH
SN
HOO
C10H13N3O2SMol. Wt.: 239.29
NH
NH
SN
HOO
C15H15N3O2SMol. Wt.: 301.36
(a)
(b)
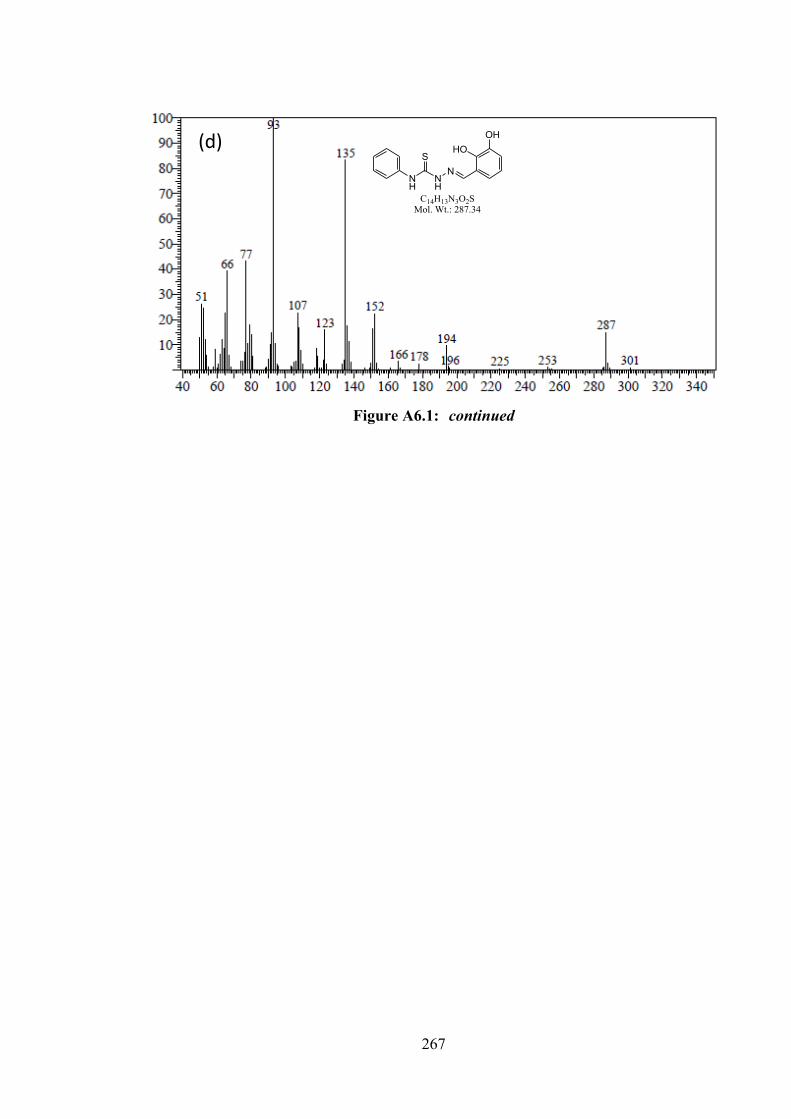
267
Figure A6.1: continued
NH
NH
SN
HOOH
C14H13N3O2SMol. Wt.: 287.34
(d)
268
Table A6.4: Experimental and calculated absorption data for the thiosemicarbazone Schiff bases (25-28) and tin(IV) compounds (29-40). Compounds
Wavelength (nm) Experimental B3LYP/6-311G(d,p) or
B3LYP/LanLD2Z/6-311G(d,p)
25 353 335 328 318
26 374 348 334 330
27 368 348 328 337
28 375 361 330 326
29 406 403 343 348
30 412 405 333 348
31 406 410 345 356
32 408 395 328 353
33 401 402 328 353
34 406 408 330 347
35 413 414 350 365
36 419 420 354 368
37
417 428 360 376
38 414 414 352 370
39 414 414 352 373
40 413 424 339 380
269
APPENDIX B - FTIR SPECTRA OF SCHIFF BASES AND TIN(IV) COMPOUNDS
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
1 4 7 19
Figure B1: FTIR spectra of 1, 4, 7 and 19.
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
2 5 8 20
Figure B2: FTIR spectra of 2, 5, 8 and 20.
270
3600 3000 2400 1800 1200 600
wavenumber (cm-1)
3 6 9 21
Figure B3: FTIR spectra of 3, 6, 9 and 21.
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
10 13 16 22
Figure B4: FTIR spectra of 10, 13, 16 and 22.
271
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
11 14 17 23
Figure B5: FTIR spectra of 11, 14, 17 and 23.
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
12 15 18 24
Figure B6: FTIR spectra of 12, 15, 18 and 24.
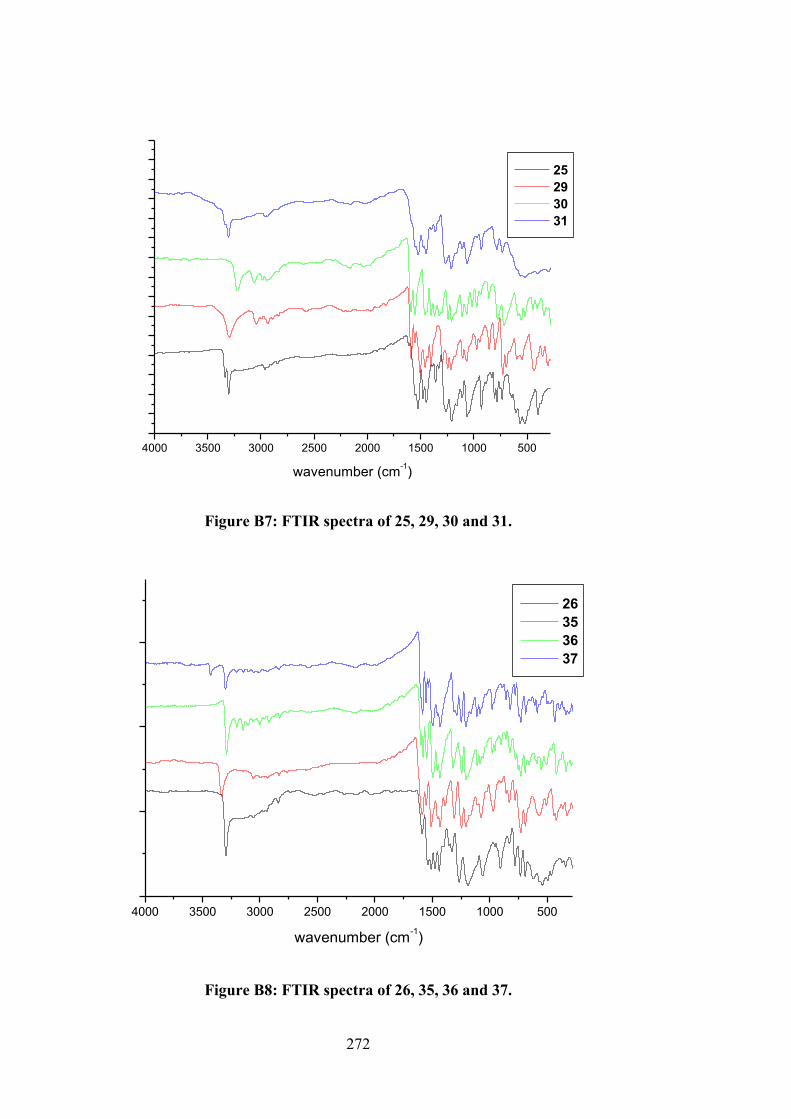
272
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
25 29 30 31
Figure B7: FTIR spectra of 25, 29, 30 and 31.
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
26 35 36 37
Figure B8: FTIR spectra of 26, 35, 36 and 37.
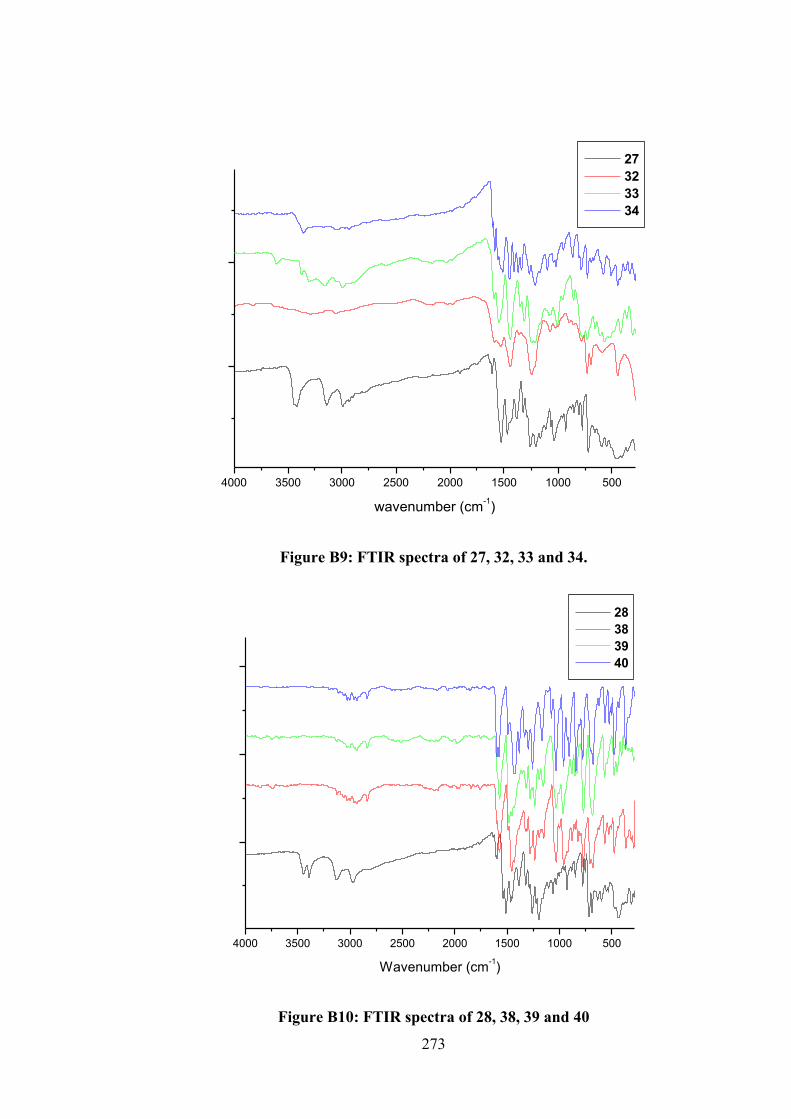
273
4000 3500 3000 2500 2000 1500 1000 500
wavenumber (cm-1)
27 32 33 34
Figure B9: FTIR spectra of 27, 32, 33 and 34.
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber (cm-1)
28 38 39 40
Figure B10: FTIR spectra of 28, 38, 39 and 40
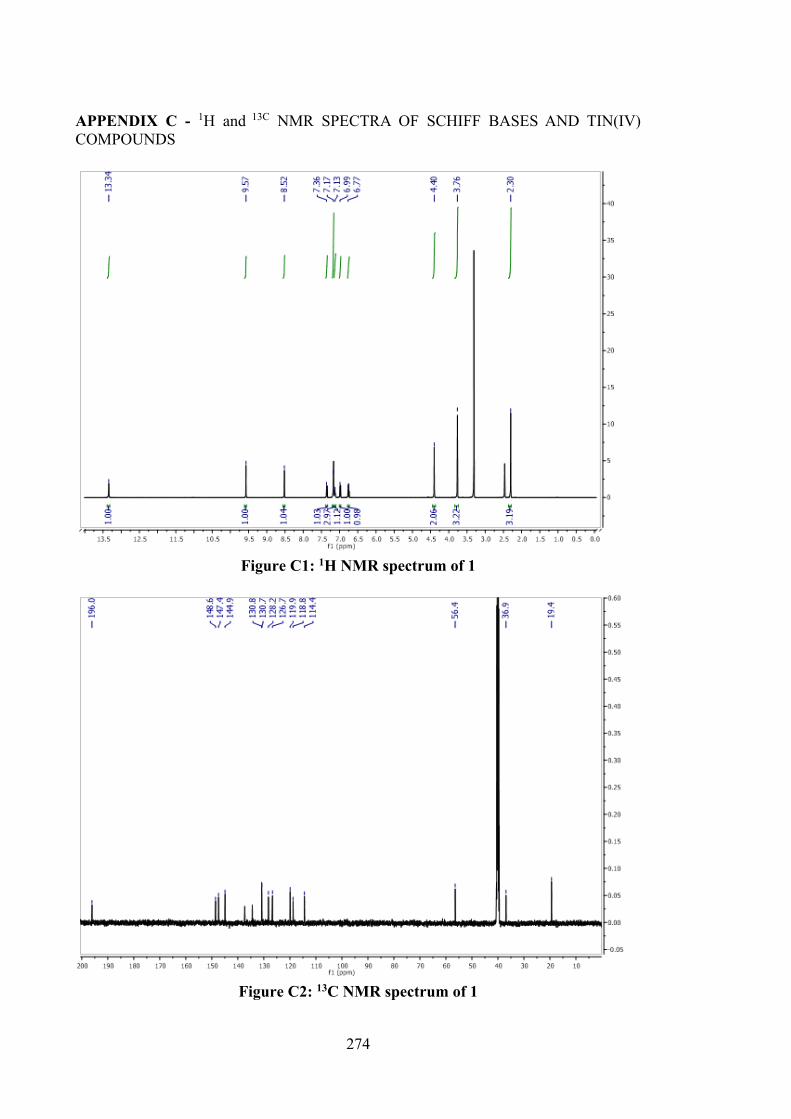
274
APPENDIX C - 1H and 13C NMR SPECTRA OF SCHIFF BASES AND TIN(IV) COMPOUNDS
Figure C1: 1H NMR spectrum of 1
Figure C2: 13C NMR spectrum of 1
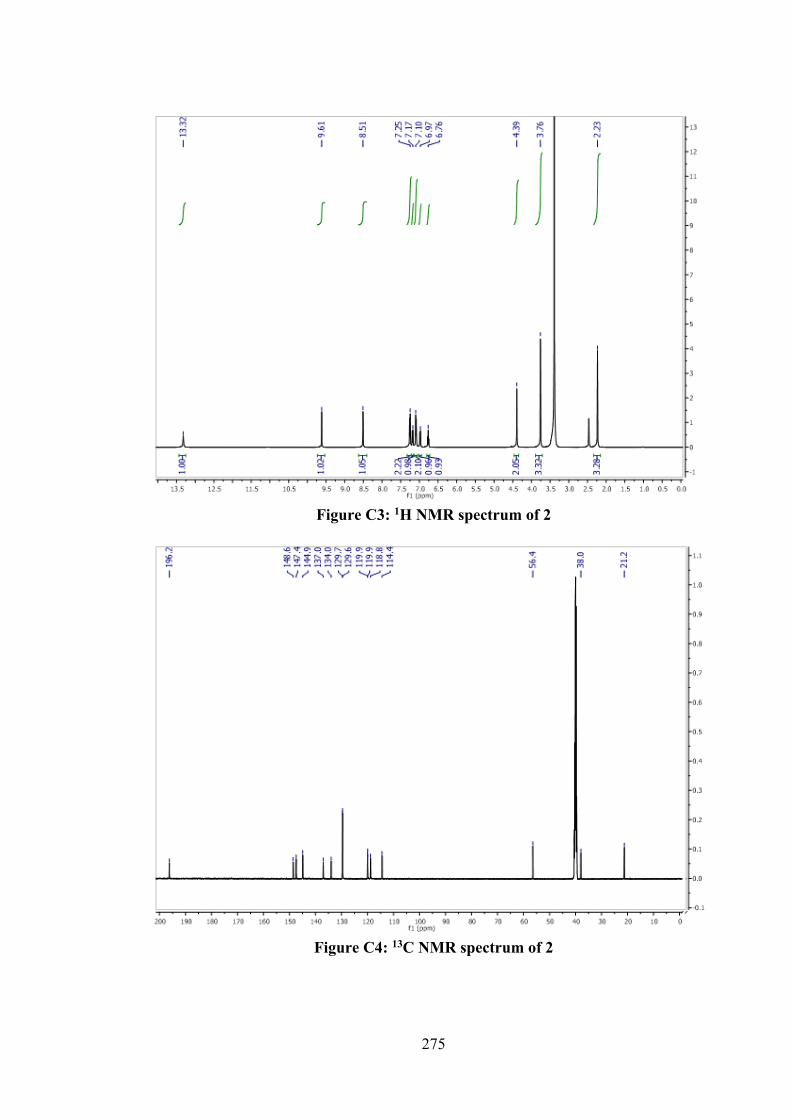
275
Figure C3: 1H NMR spectrum of 2
Figure C4: 13C NMR spectrum of 2
276
Figure C5: 1H NMR spectrum of 3
Figure C6: 13C NMR spectrum of 3
277
Figure C7: 1H NMR spectrum of 4
Figure C8: 13C NMR spectrum of 4
278
Figure C9: 1H NMR spectrum of 5
Figure C10: 13C NMR spectrum of 5
279
Figure C11: 1H NMR spectrum of 6
Figure C12: 13C NMR spectrum of 6
280
Figure C13: 1H NMR spectrum of 7
Figure C14: 13C NMR spectrum of 7
281
Figure C15: 1H NMR spectrum of 8
Figure C16: 13C NMR spectrum of 8
282
Figure C17: 1H NMR spectrum of 9
Figure C18: 13C NMR spectrum of 9
283
Figure C19: 1H NMR spectrum of 10
Figure C20: 13C NMR spectrum of 10
284
Figure C21: 1H NMR spectrum of 11
Figure C22: 13C NMR spectrum of 11
285
Figure C23: 1H NMR spectrum of 12
Figure C24: 13C NMR spectrum of 12
286
Figure C25: 1H NMR spectrum of 13
Figure C26: 13C NMR spectrum of 13
287
Figure C27: 1H NMR spectrum of 14
Figure C28: 13C NMR spectrum of 14
288
Figure C29: 1H NMR spectrum of 15
Figure C30: 13C NMR spectrum of 15
289
Figure C31: 1H NMR spectrum of 16
Figure C32: 13C NMR spectrum of 16
290
Figure C33: 1H NMR spectrum of 17
Figure C34: 13C NMR spectrum of 17
291
Figure C35: 1H NMR spectrum of 18
Figure C36: 13C NMR spectrum of 18
292
Figure C37: 1H NMR spectrum of 19
Figure C38: 13C NMR spectrum of 19
293
Figure C39: 1H NMR spectrum of 20
Figure C40: 13C NMR spectrum of 20
294
Figure C41: 1H NMR spectrum of 21
Figure C42: 13C NMR spectrum of 21
295
Figure C43: 1H NMR spectrum of 22
Figure C44: 13C NMR spectrum of 22
296
Figure C45: 1H NMR spectrum of 23
Figure C46: 13C NMR spectrum of 23
297
Figure C47: 1H NMR spectrum of 24
Figure C48: 13C NMR spectrum of 24
298
Figure C49: 1H NMR spectrum of 25
Figure C50: 13C NMR spectrum of 25
299
Figure C51: 1H NMR spectrum of 26
Figure C52: 13C NMR spectrum of 26
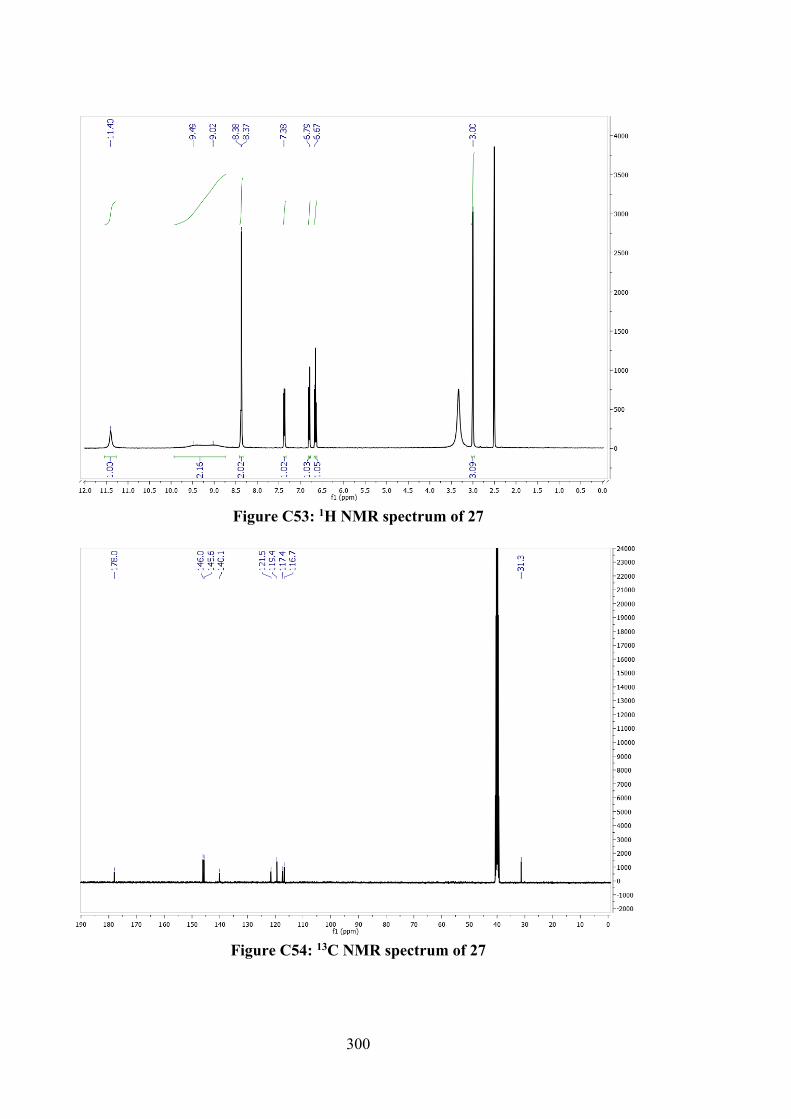
300
Figure C53: 1H NMR spectrum of 27
Figure C54: 13C NMR spectrum of 27
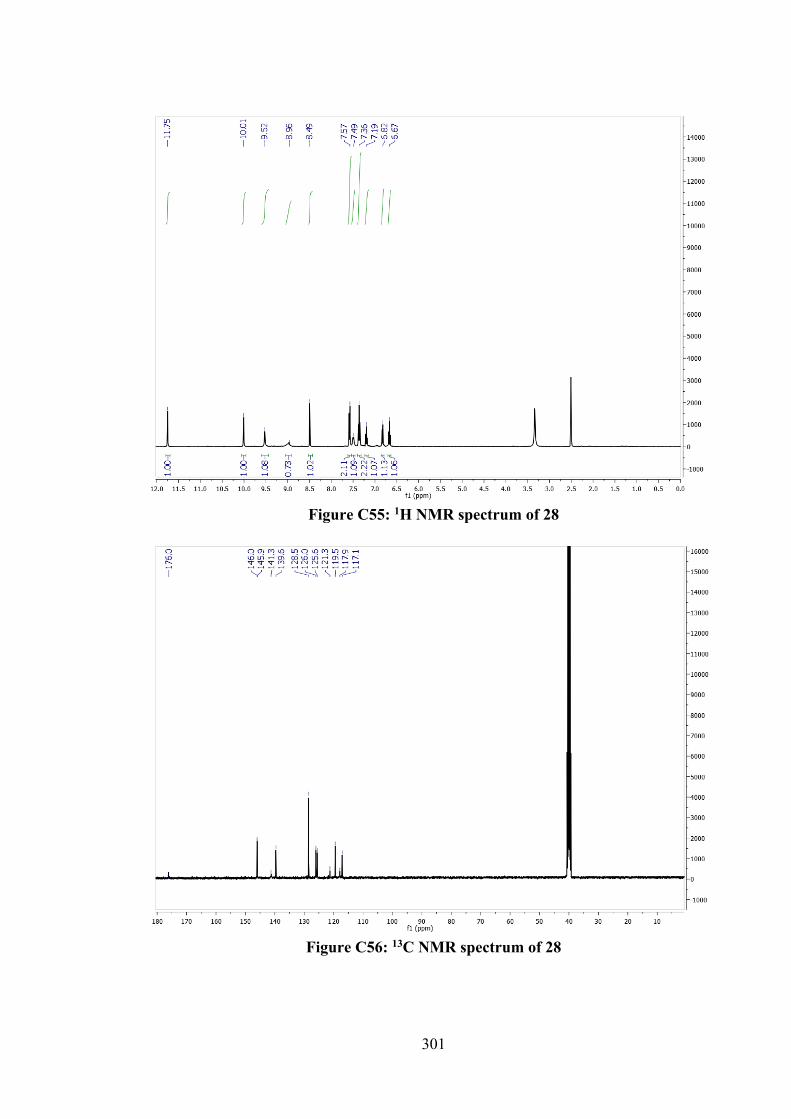
301
Figure C55: 1H NMR spectrum of 28
Figure C56: 13C NMR spectrum of 28
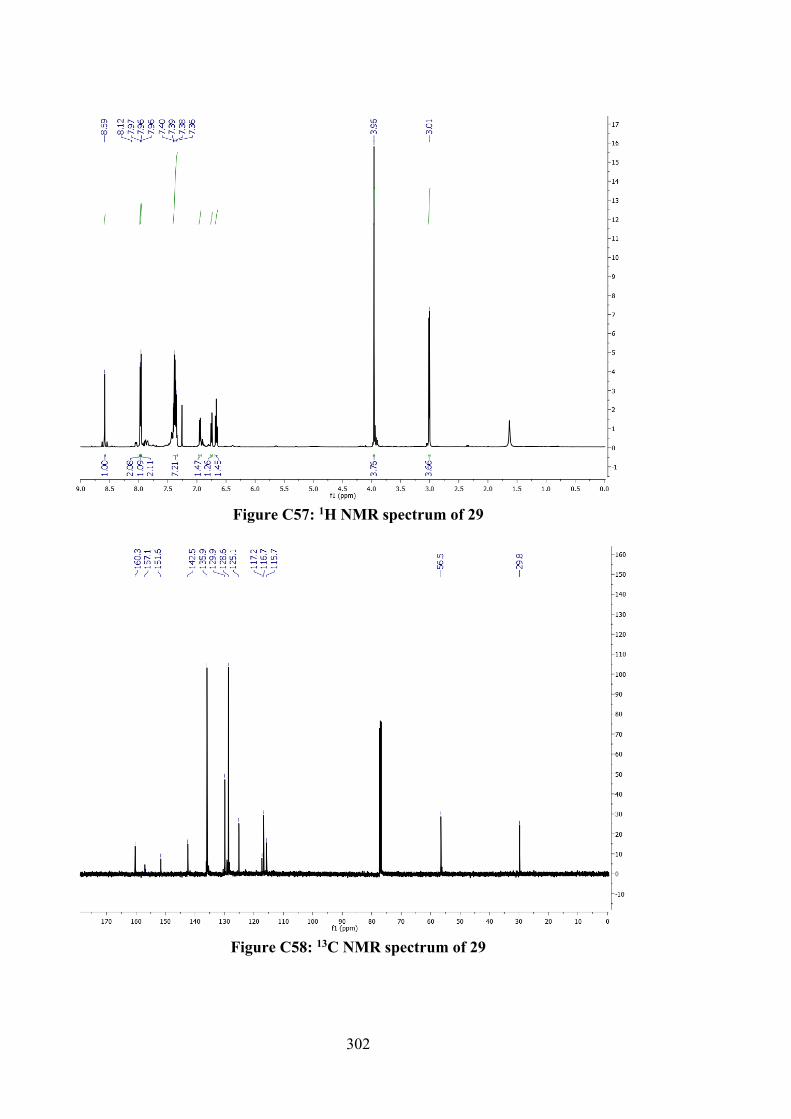
302
Figure C57: 1H NMR spectrum of 29
Figure C58: 13C NMR spectrum of 29
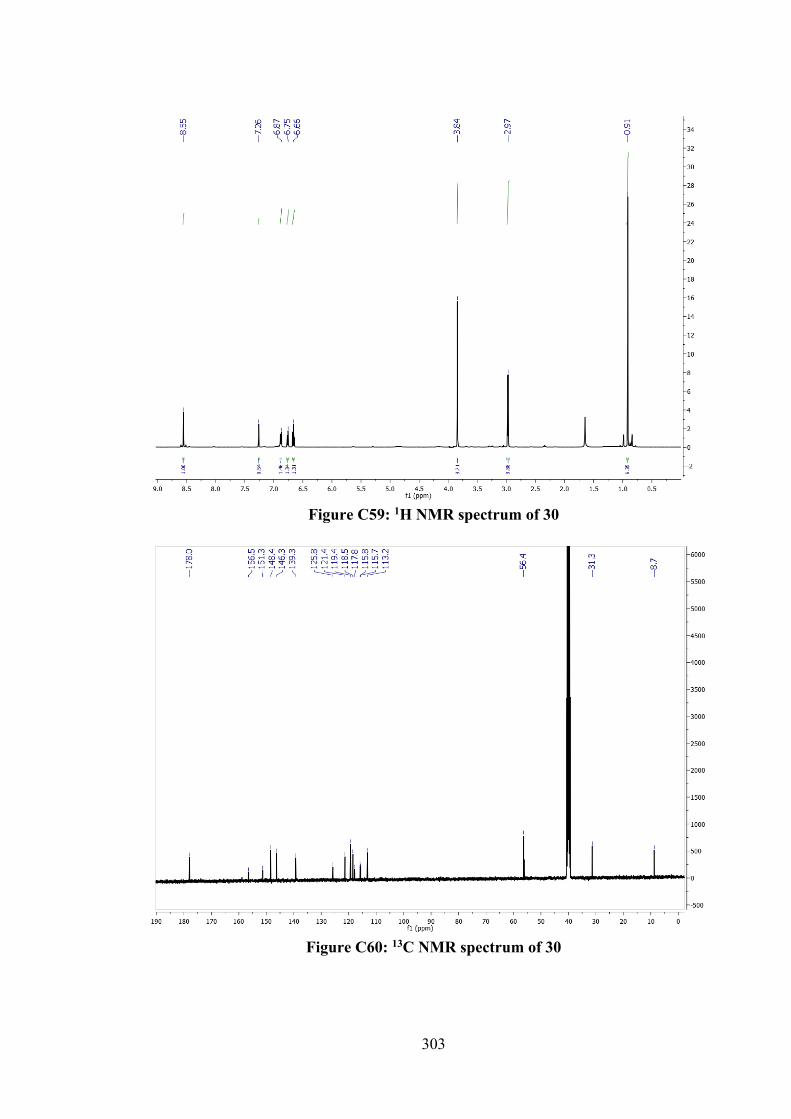
303
Figure C59: 1H NMR spectrum of 30
Figure C60: 13C NMR spectrum of 30
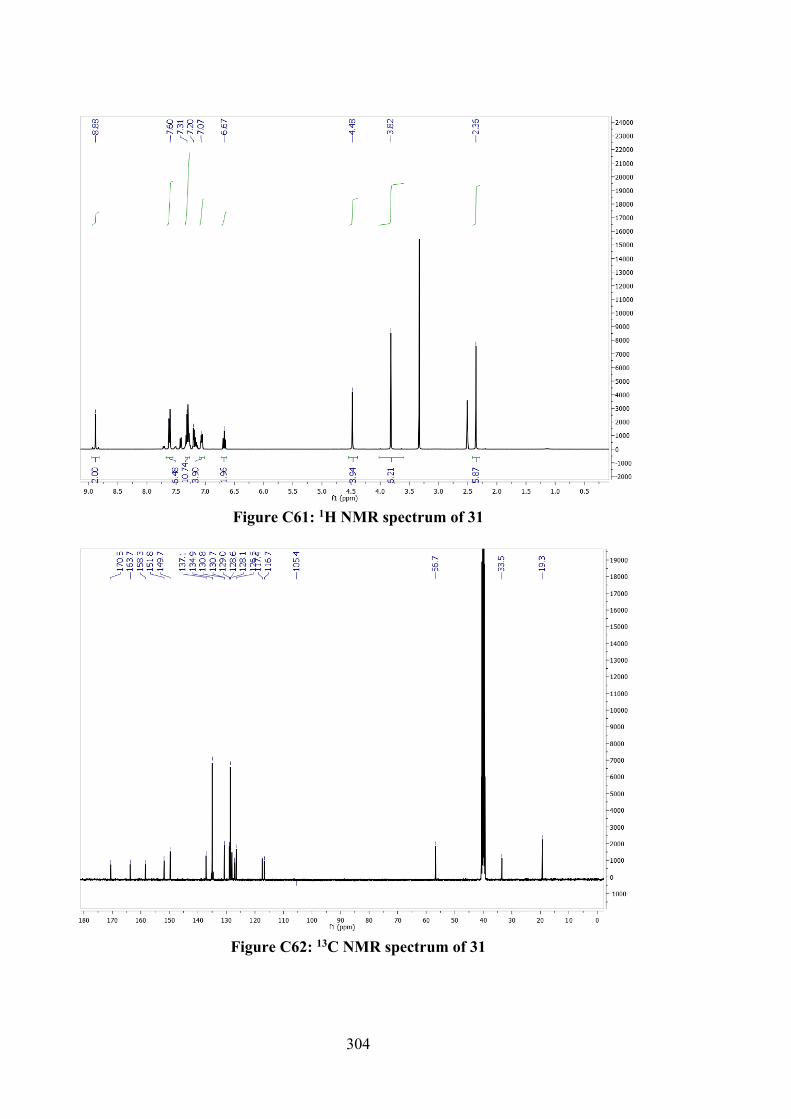
304
Figure C61: 1H NMR spectrum of 31
Figure C62: 13C NMR spectrum of 31
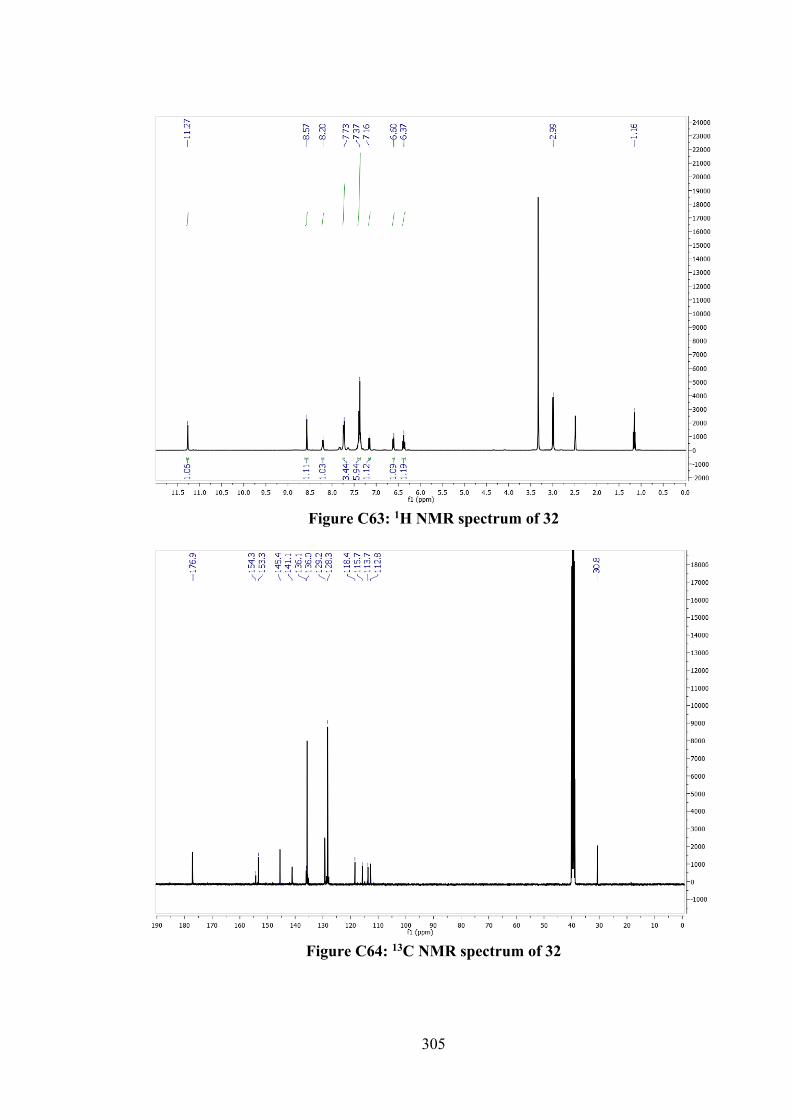
305
Figure C63: 1H NMR spectrum of 32
Figure C64: 13C NMR spectrum of 32
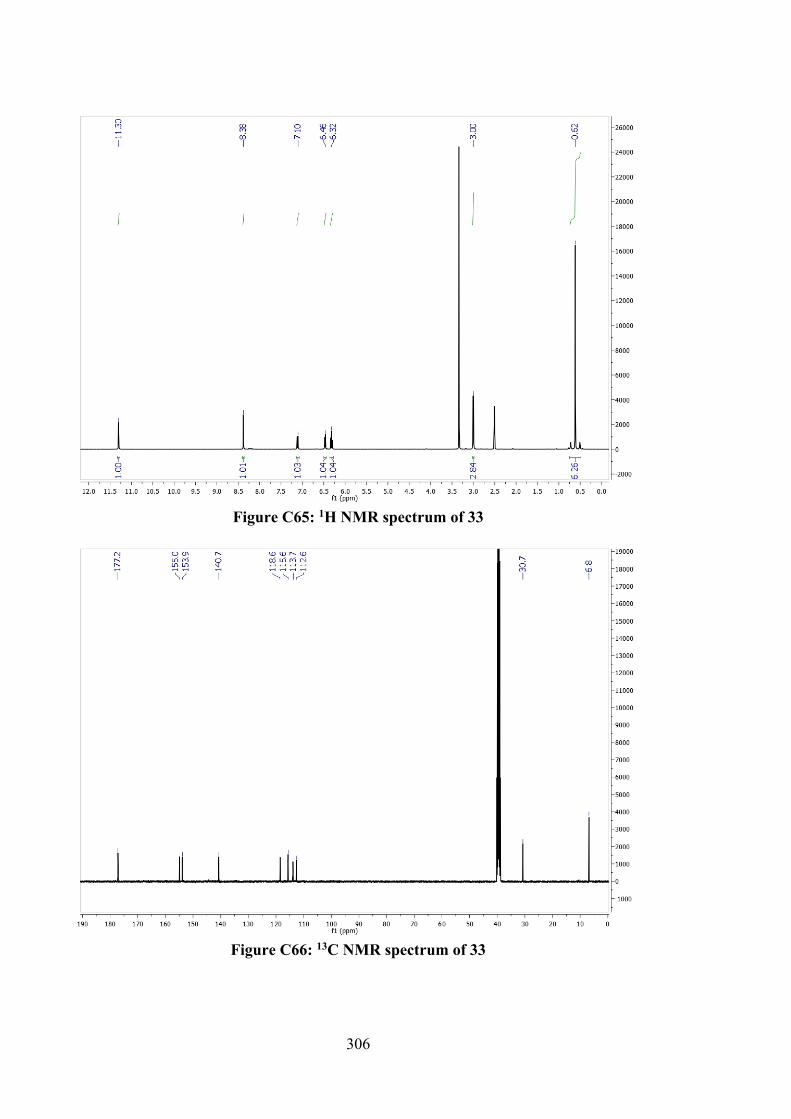
306
Figure C65: 1H NMR spectrum of 33
Figure C66: 13C NMR spectrum of 33
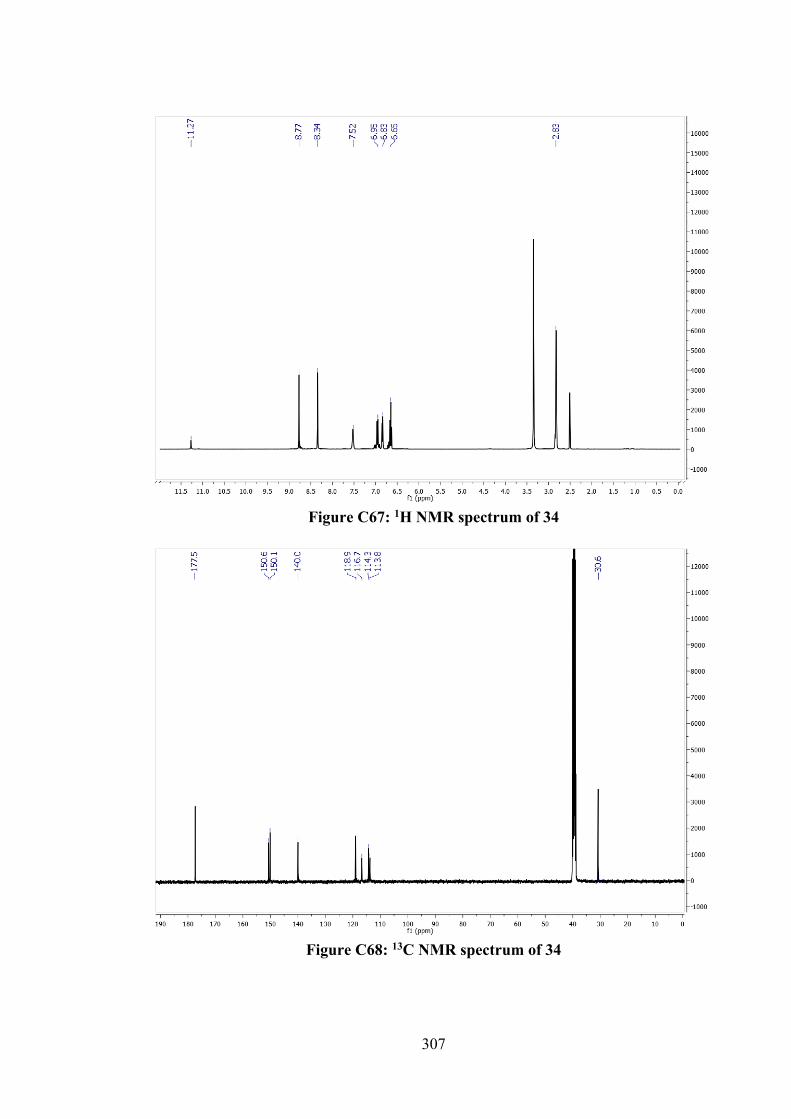
307
Figure C67: 1H NMR spectrum of 34
Figure C68: 13C NMR spectrum of 34
308
Figure C69: 1H NMR spectrum of 35
Figure C70: 13C NMR spectrum of 35
309
Figure C71: 1H NMR spectrum of 36
Figure C72: 13C NMR spectrum of 36
310
Figure C73: 1H NMR spectrum of 37
Figure C74: 13C NMR spectrum of 37
311
Figure C75: 1H NMR spectrum of 38
Figure C76: 13C NMR spectrum of 38
312
Figure C77: 1H NMR spectrum of 39
Figure C78: 13C NMR spectrum of 39
313
Figure C79: 1H NMR spectrum of 40
Figure C80: 13C NMR spectrum of 40
314
APPENDIX D - 119Sn NMR SPECTRA OF TIN(IV) COMPOUNDS
Figure D1: 119Sn NMR spectrum of 4
Figure D2: 119Sn NMR spectrum of 5
315
Figure D3: 119Sn NMR spectrum of 6
Figure D4: 119Sn NMR spectrum of 7
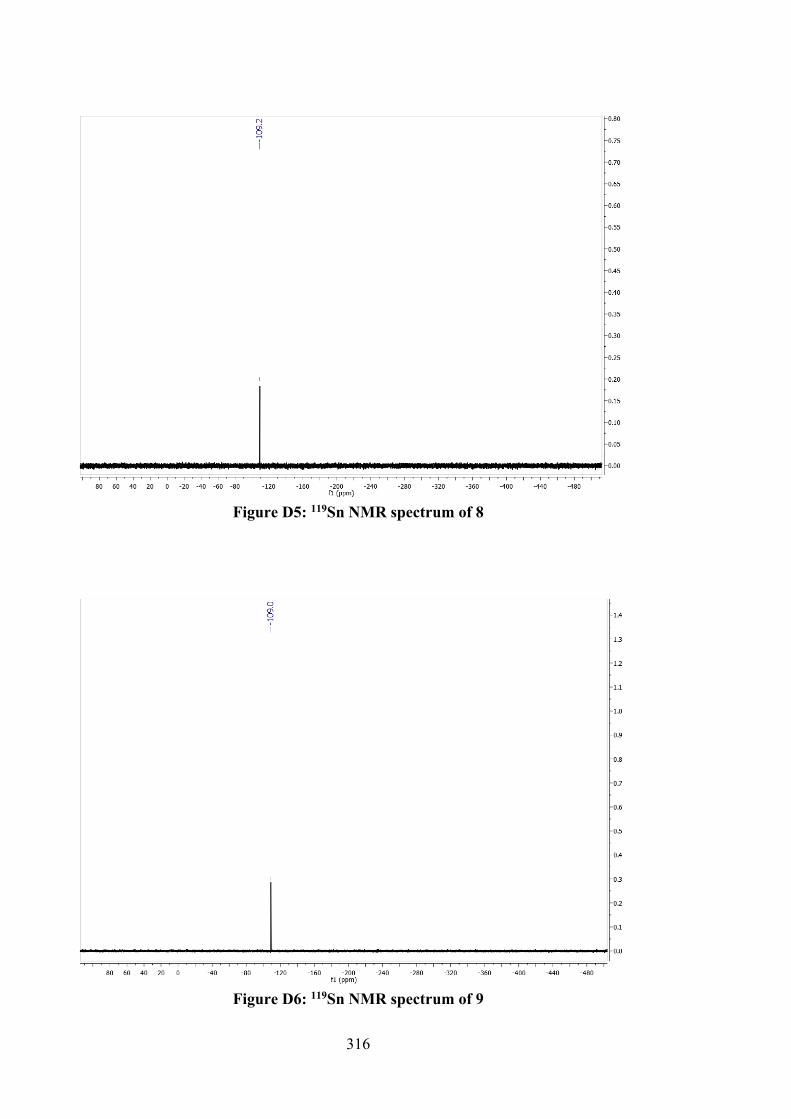
316
Figure D5: 119Sn NMR spectrum of 8
Figure D6: 119Sn NMR spectrum of 9
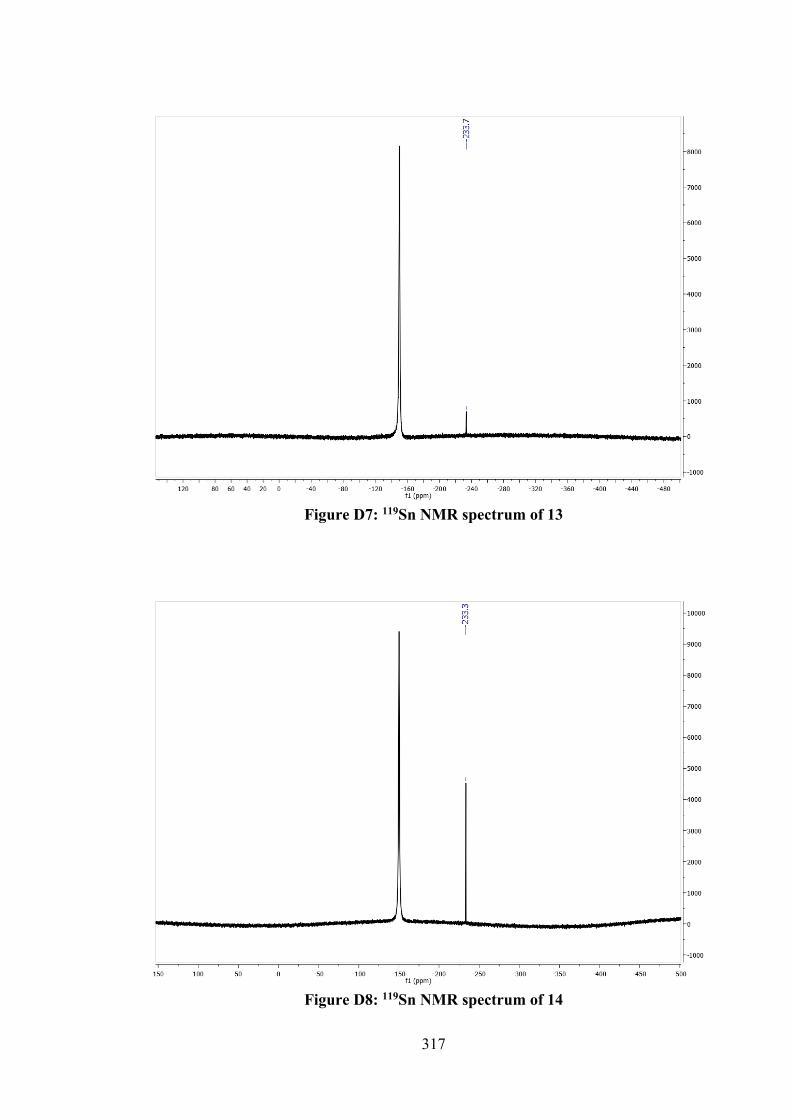
317
Figure D7: 119Sn NMR spectrum of 13
Figure D8: 119Sn NMR spectrum of 14
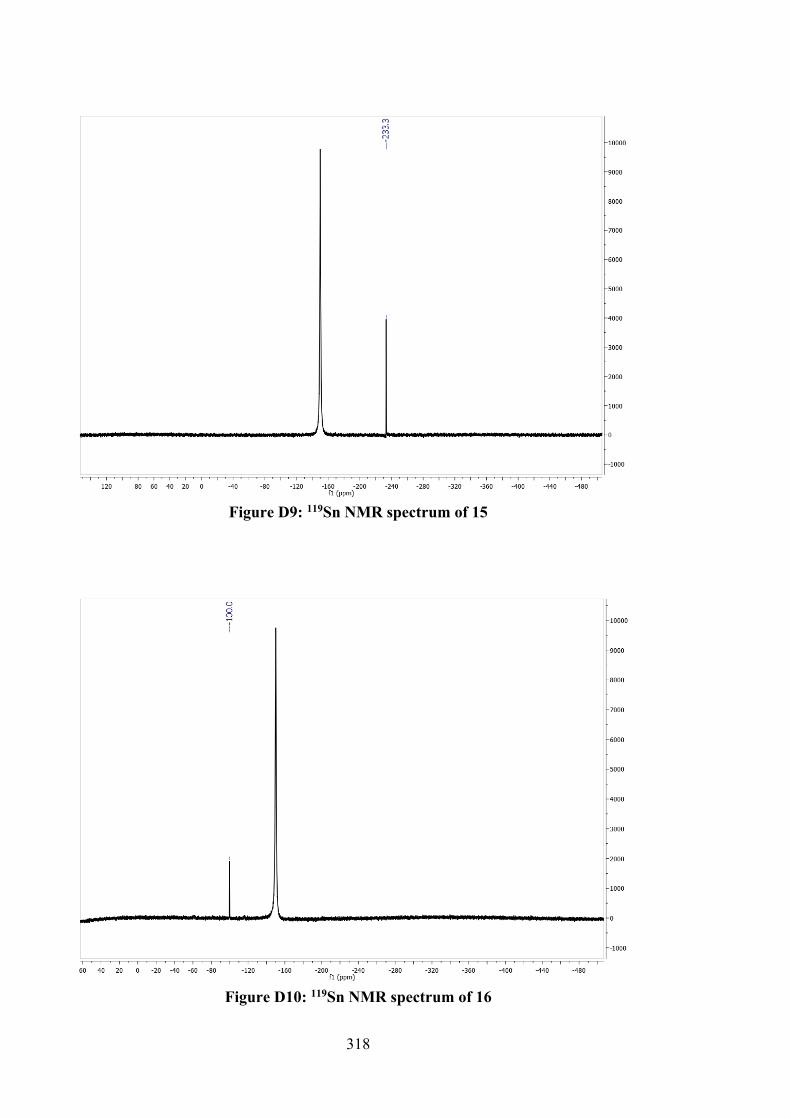
318
Figure D9: 119Sn NMR spectrum of 15
Figure D10: 119Sn NMR spectrum of 16
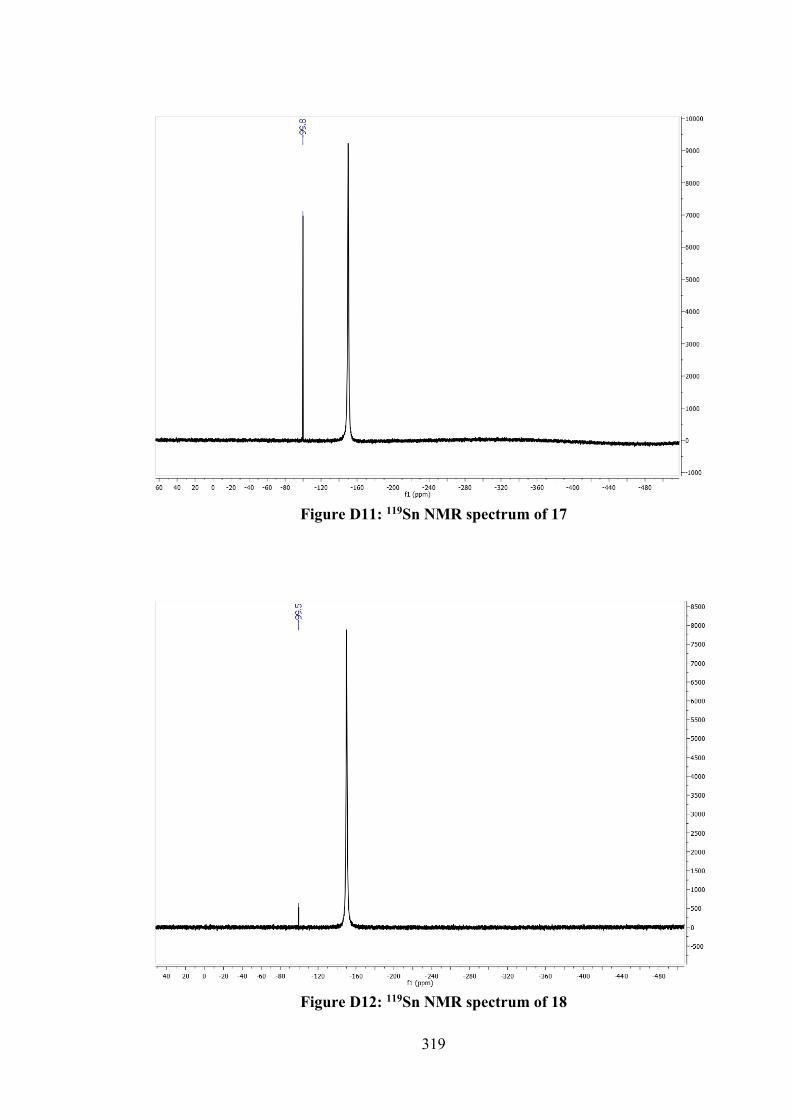
319
Figure D11: 119Sn NMR spectrum of 17
Figure D12: 119Sn NMR spectrum of 18
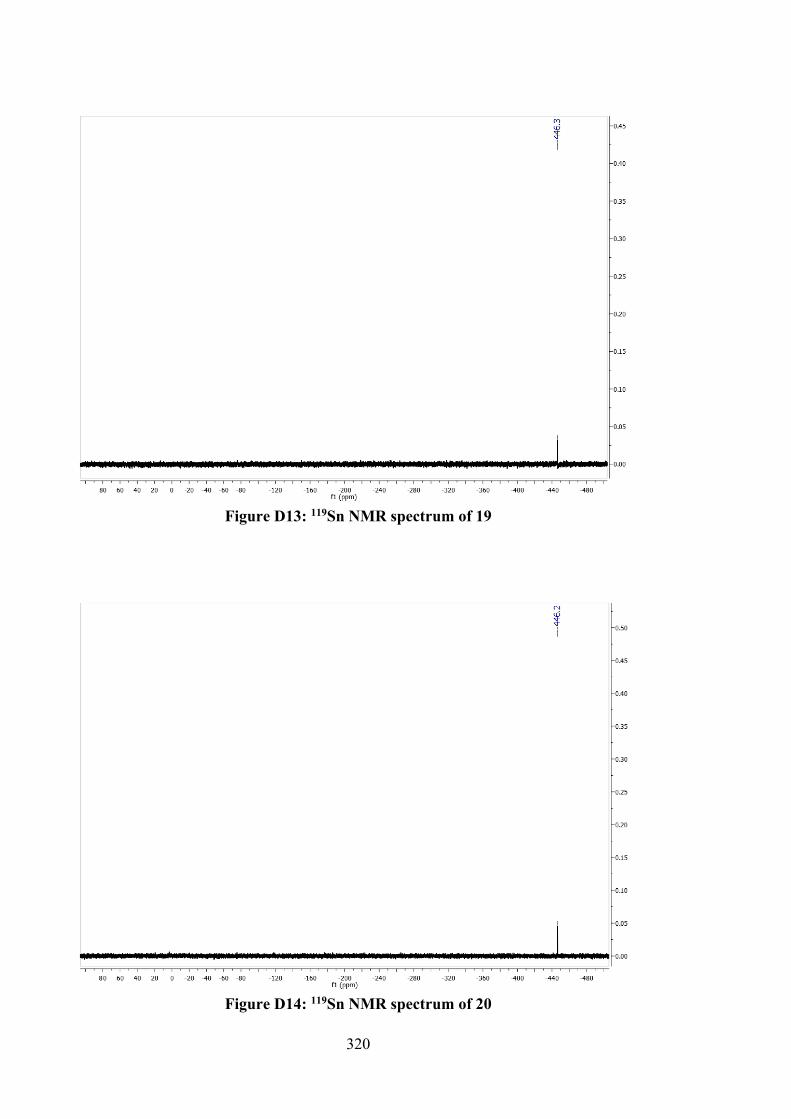
320
Figure D13: 119Sn NMR spectrum of 19
Figure D14: 119Sn NMR spectrum of 20
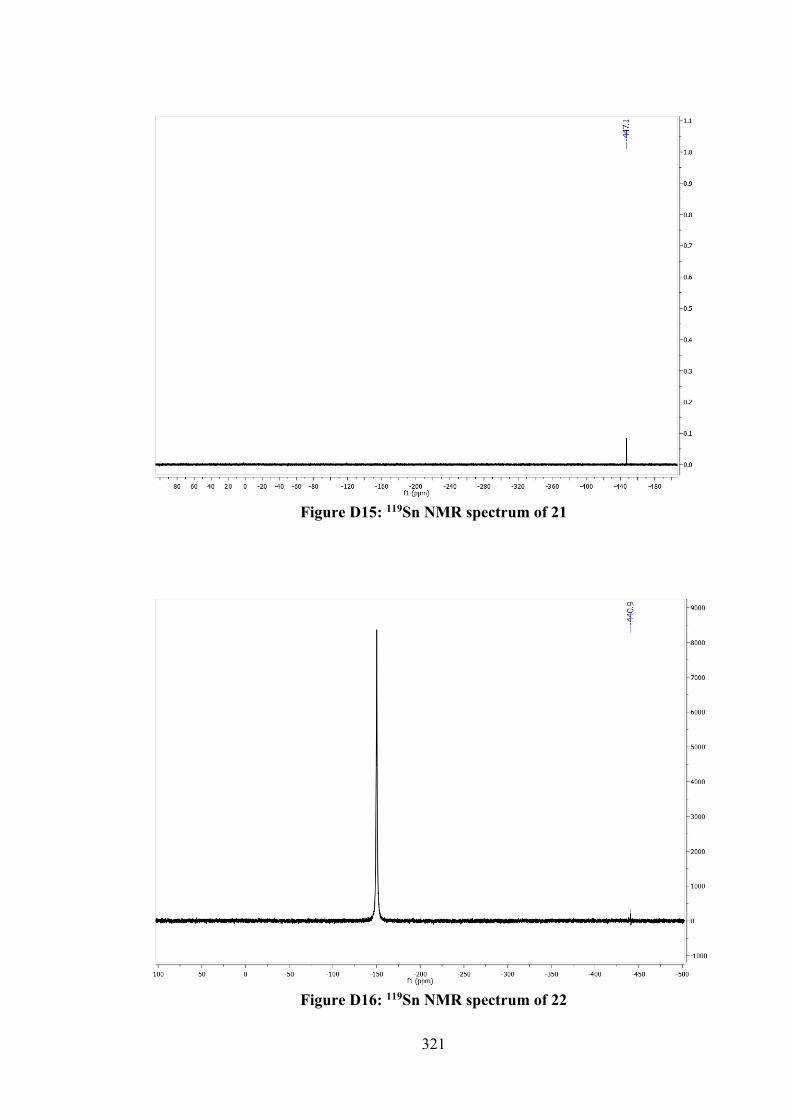
321
Figure D15: 119Sn NMR spectrum of 21
Figure D16: 119Sn NMR spectrum of 22
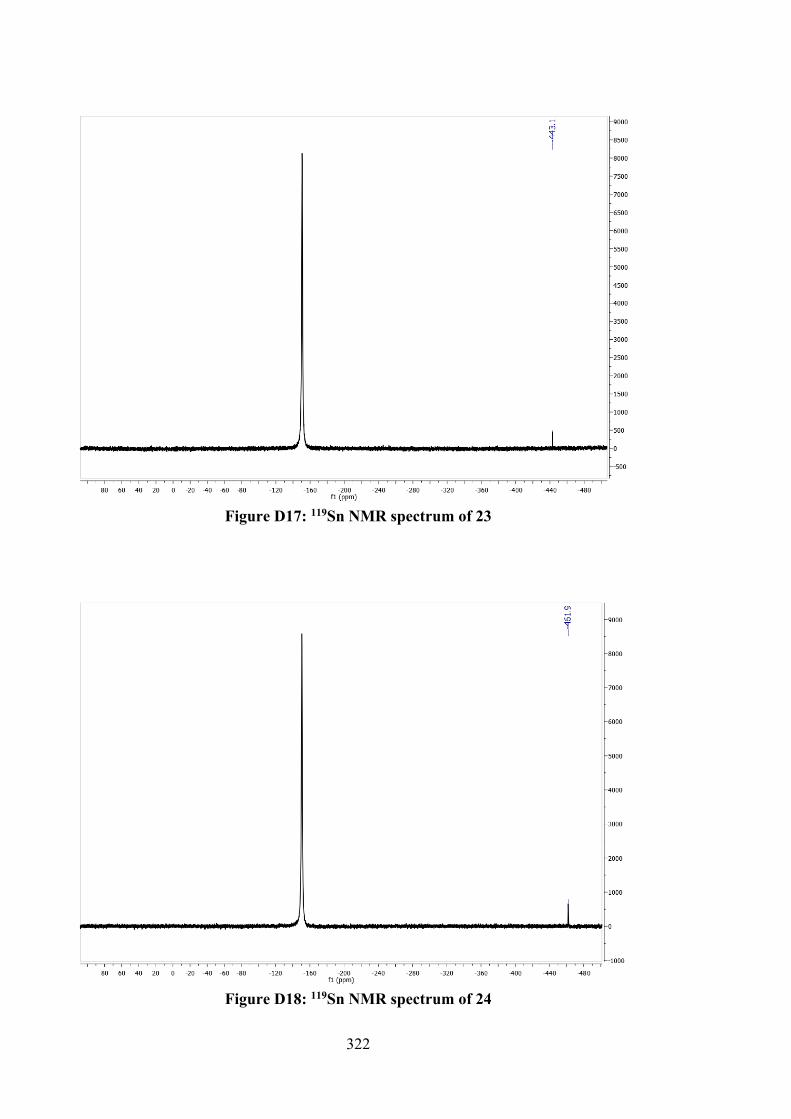
322
Figure D17: 119Sn NMR spectrum of 23
Figure D18: 119Sn NMR spectrum of 24
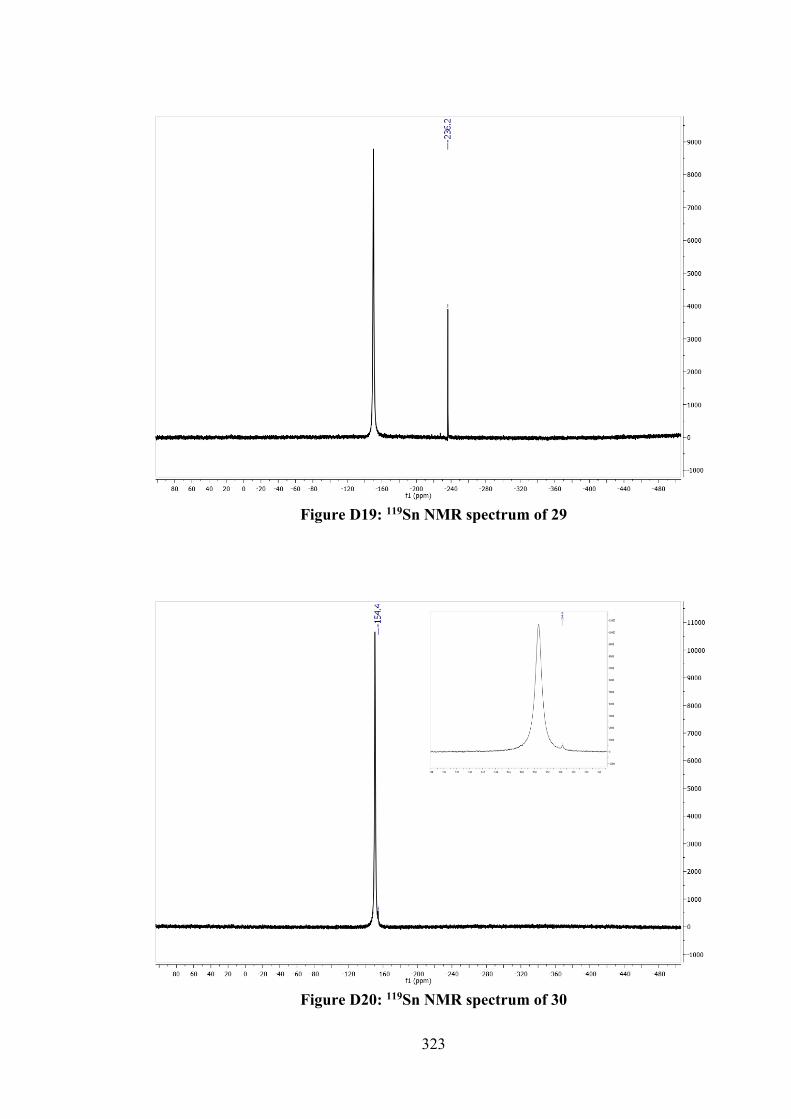
323
Figure D19: 119Sn NMR spectrum of 29
Figure D20: 119Sn NMR spectrum of 30
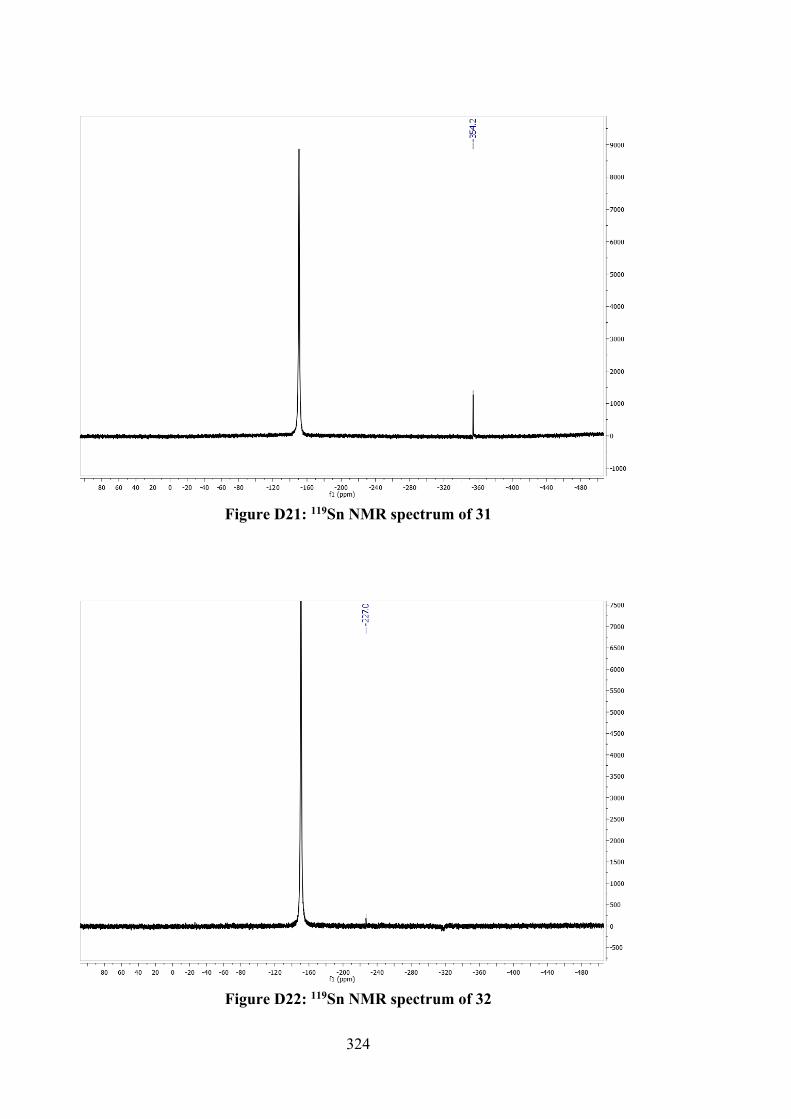
324
Figure D21: 119Sn NMR spectrum of 31
Figure D22: 119Sn NMR spectrum of 32
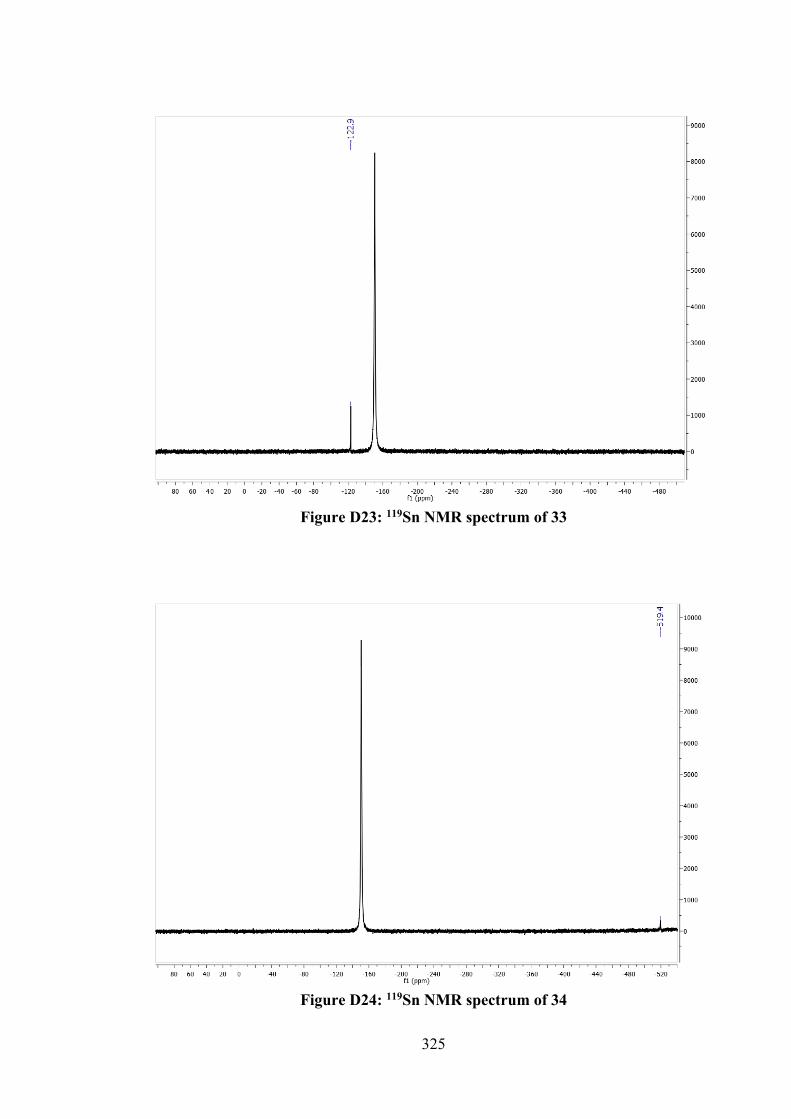
325
Figure D23: 119Sn NMR spectrum of 33
Figure D24: 119Sn NMR spectrum of 34
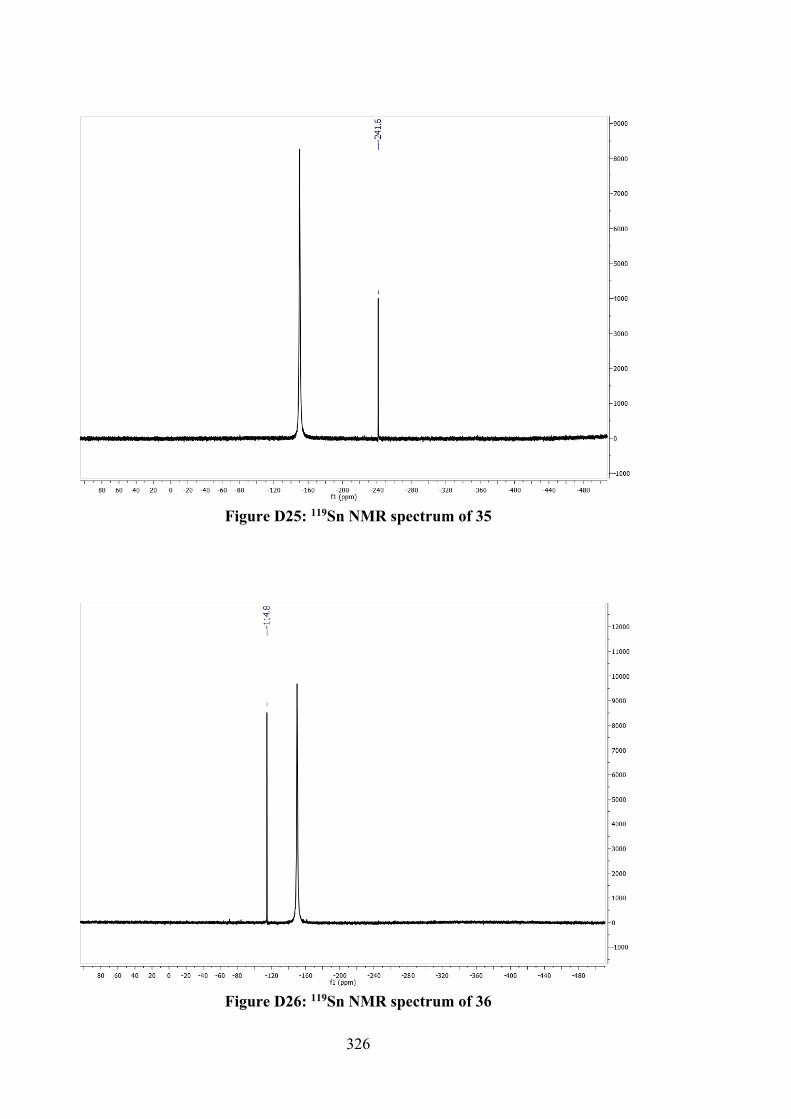
326
Figure D25: 119Sn NMR spectrum of 35
Figure D26: 119Sn NMR spectrum of 36
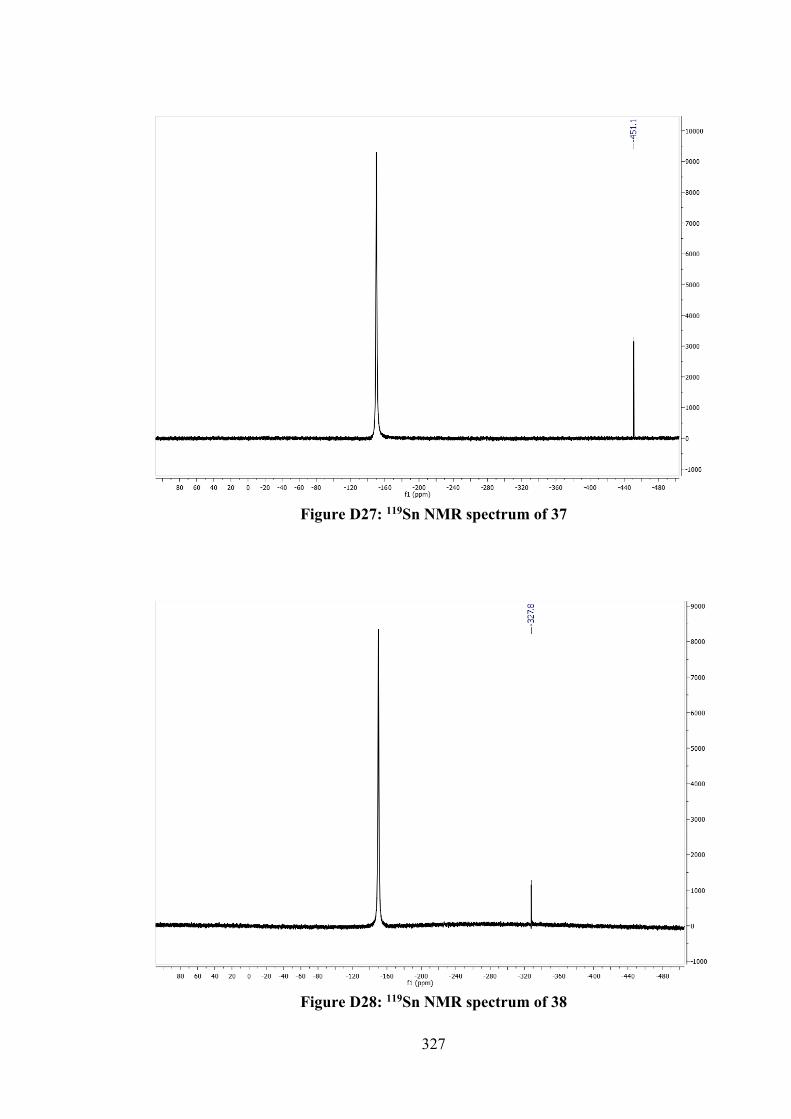
327
Figure D27: 119Sn NMR spectrum of 37
Figure D28: 119Sn NMR spectrum of 38
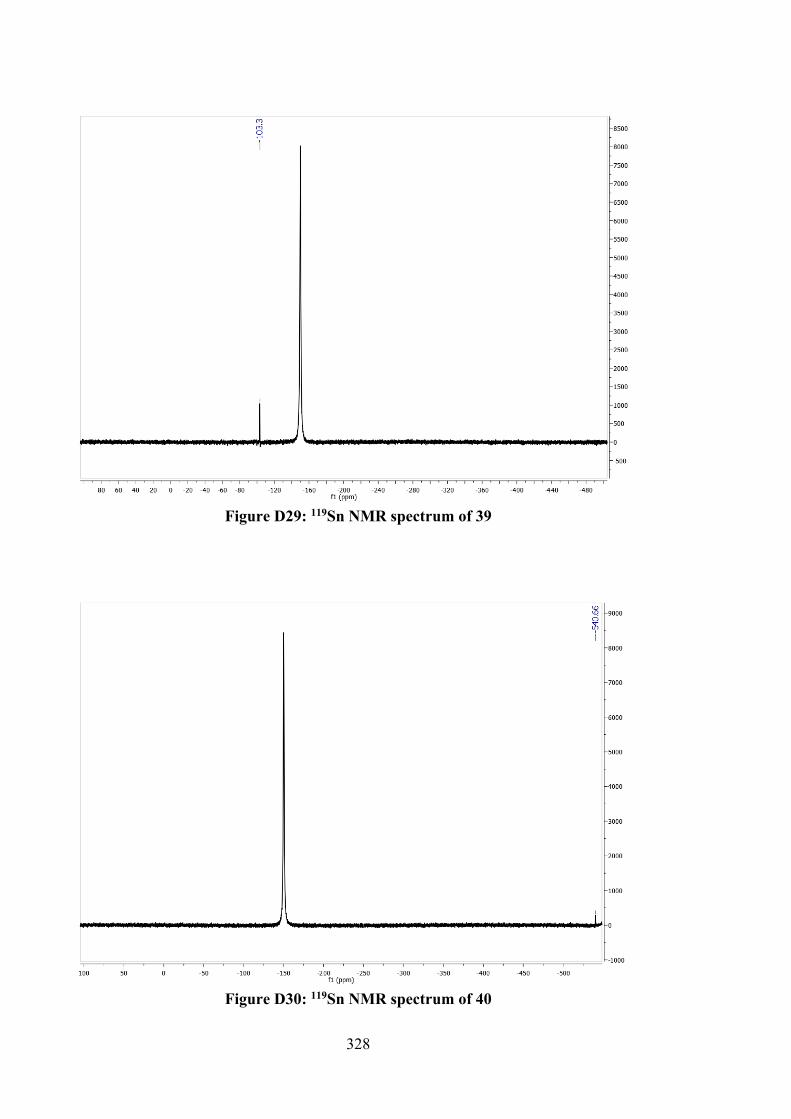
328
Figure D29: 119Sn NMR spectrum of 39
Figure D30: 119Sn NMR spectrum of 40
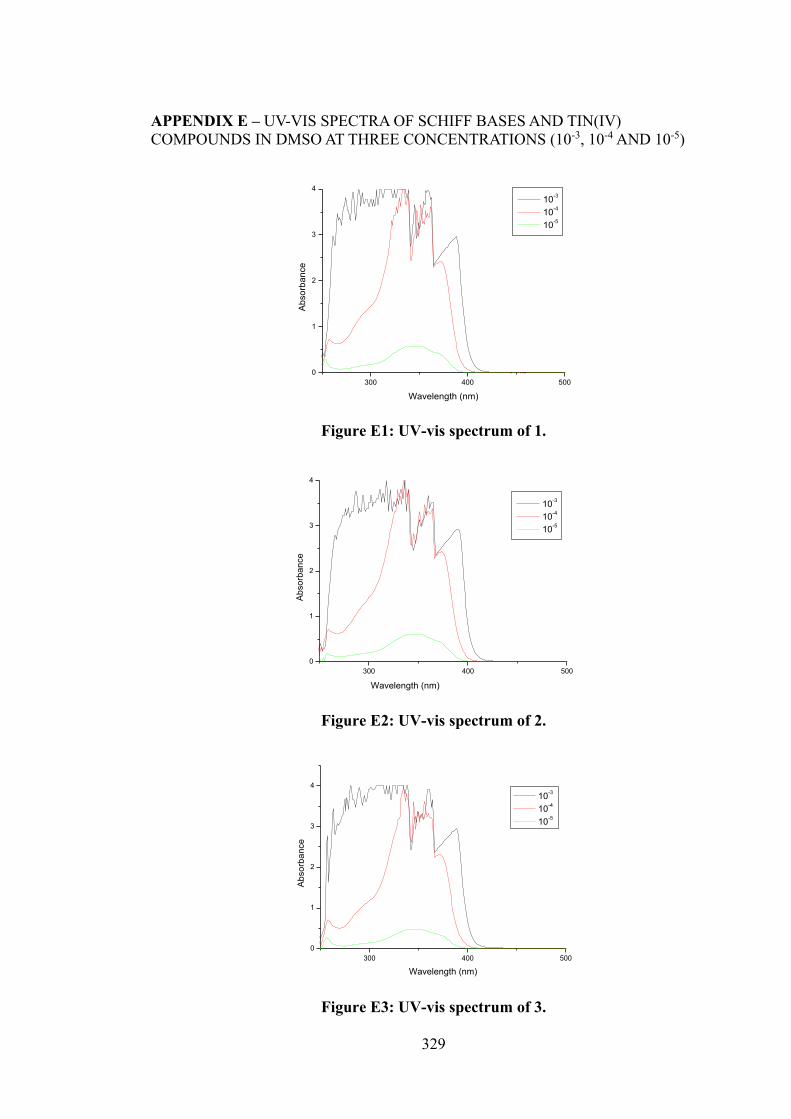
329
APPENDIX E – UV-VIS SPECTRA OF SCHIFF BASES AND TIN(IV) COMPOUNDS IN DMSO AT THREE CONCENTRATIONS (10-3, 10-4 AND 10-5)
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E1: UV-vis spectrum of 1.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E2: UV-vis spectrum of 2.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E3: UV-vis spectrum of 3.
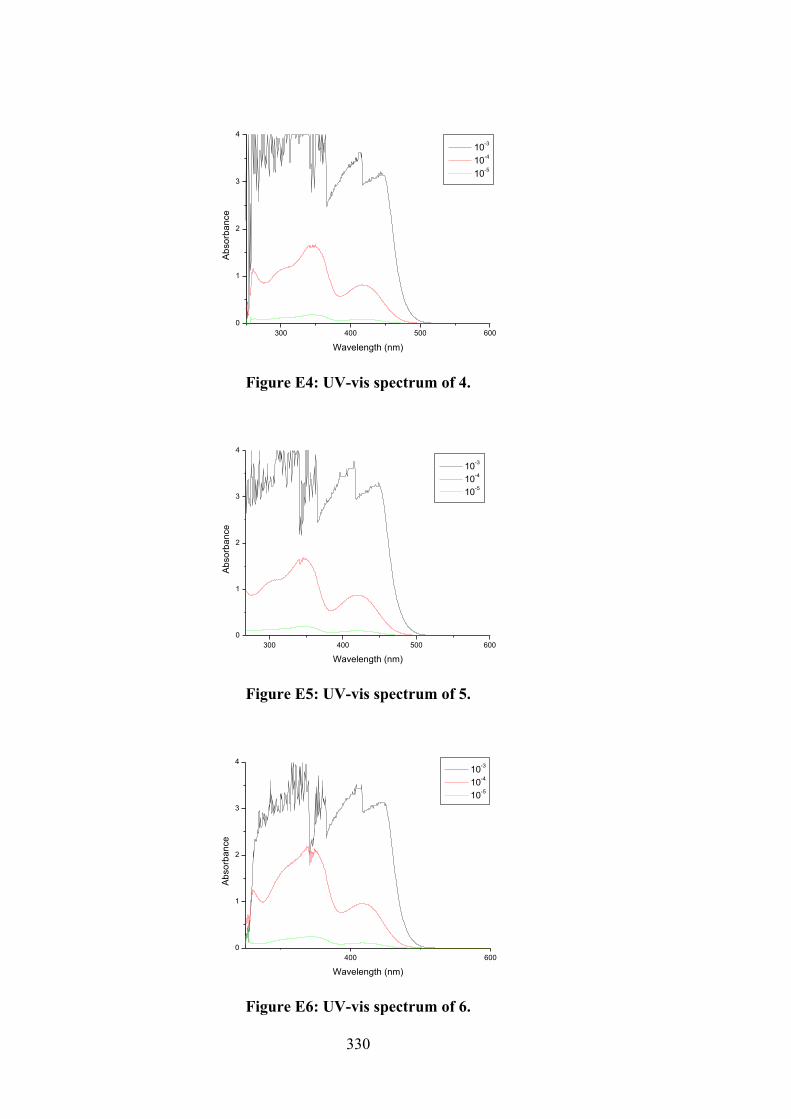
330
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E4: UV-vis spectrum of 4.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E5: UV-vis spectrum of 5.
400 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E6: UV-vis spectrum of 6.
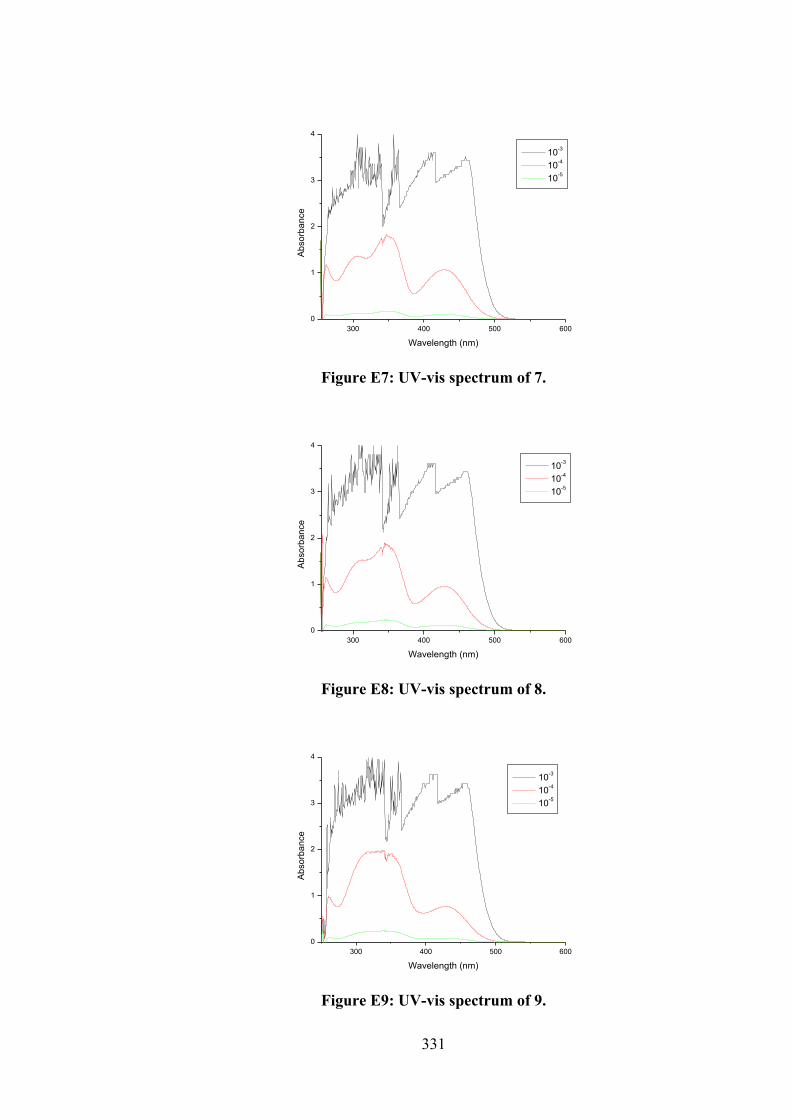
331
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E7: UV-vis spectrum of 7.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E8: UV-vis spectrum of 8.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E9: UV-vis spectrum of 9.
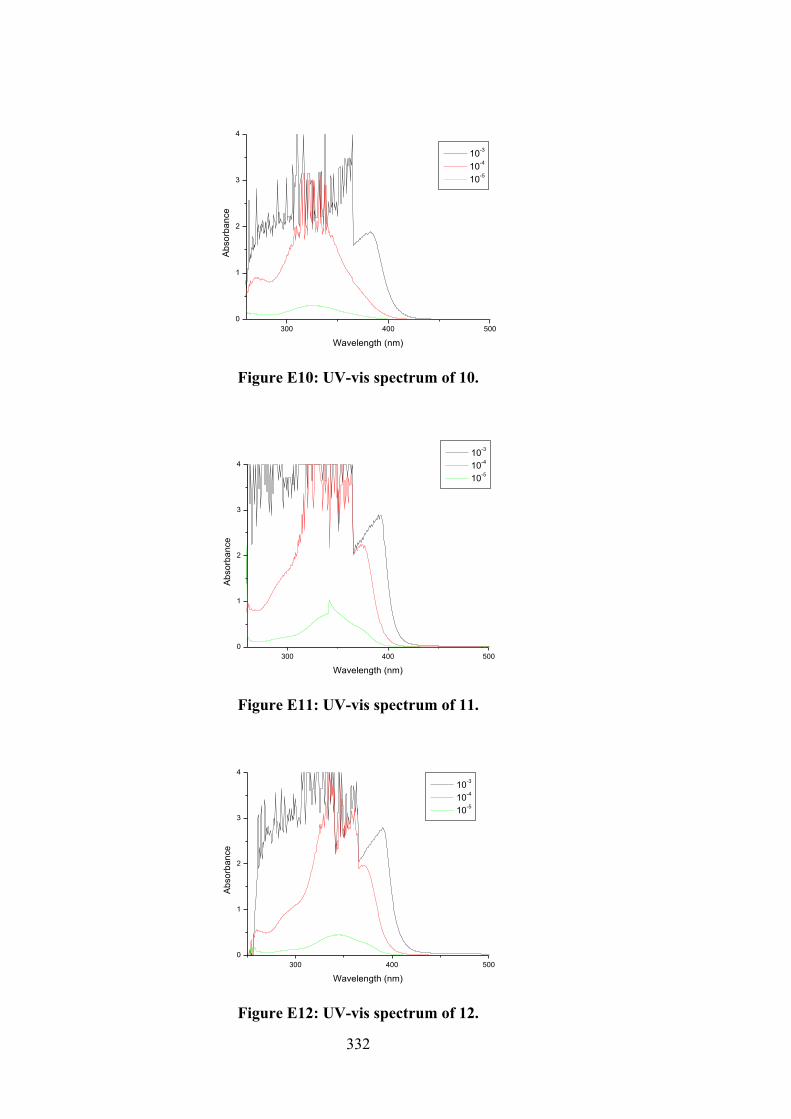
332
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E10: UV-vis spectrum of 10.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E11: UV-vis spectrum of 11.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E12: UV-vis spectrum of 12.
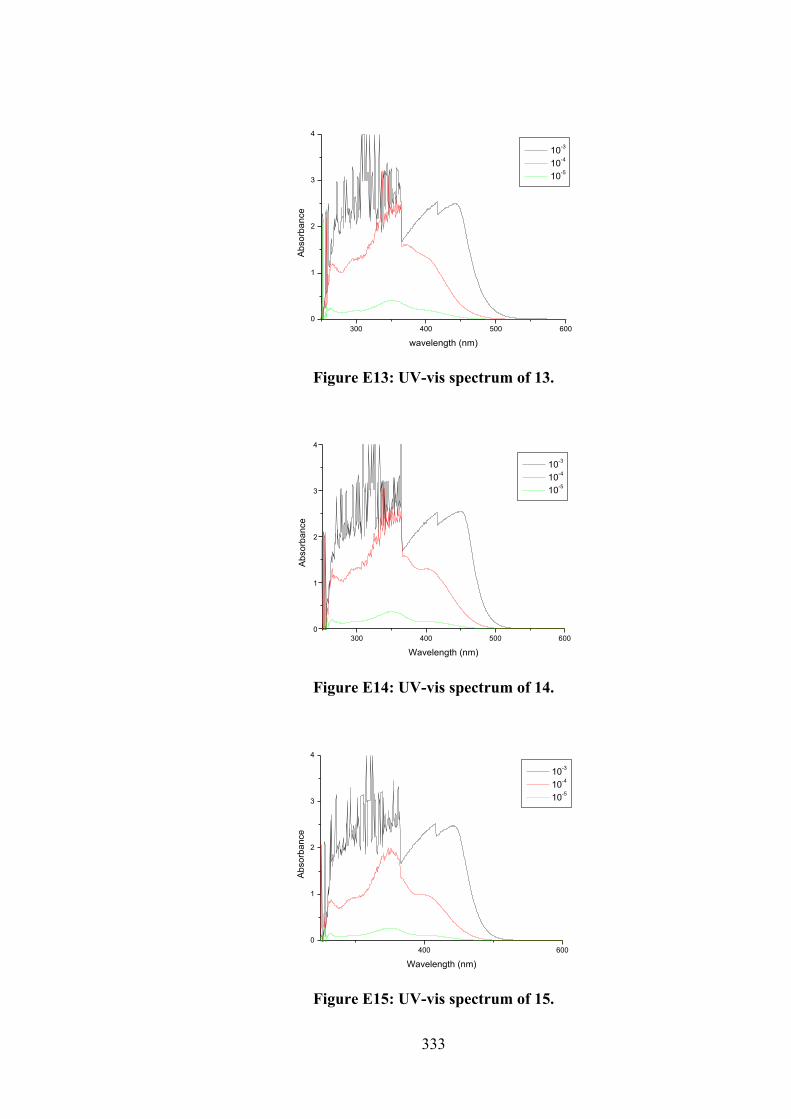
333
300 400 500 6000
1
2
3
4
Abso
rban
ce
wavelength (nm)
10-3
10-4
10-5
Figure E13: UV-vis spectrum of 13.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E14: UV-vis spectrum of 14.
400 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E15: UV-vis spectrum of 15.
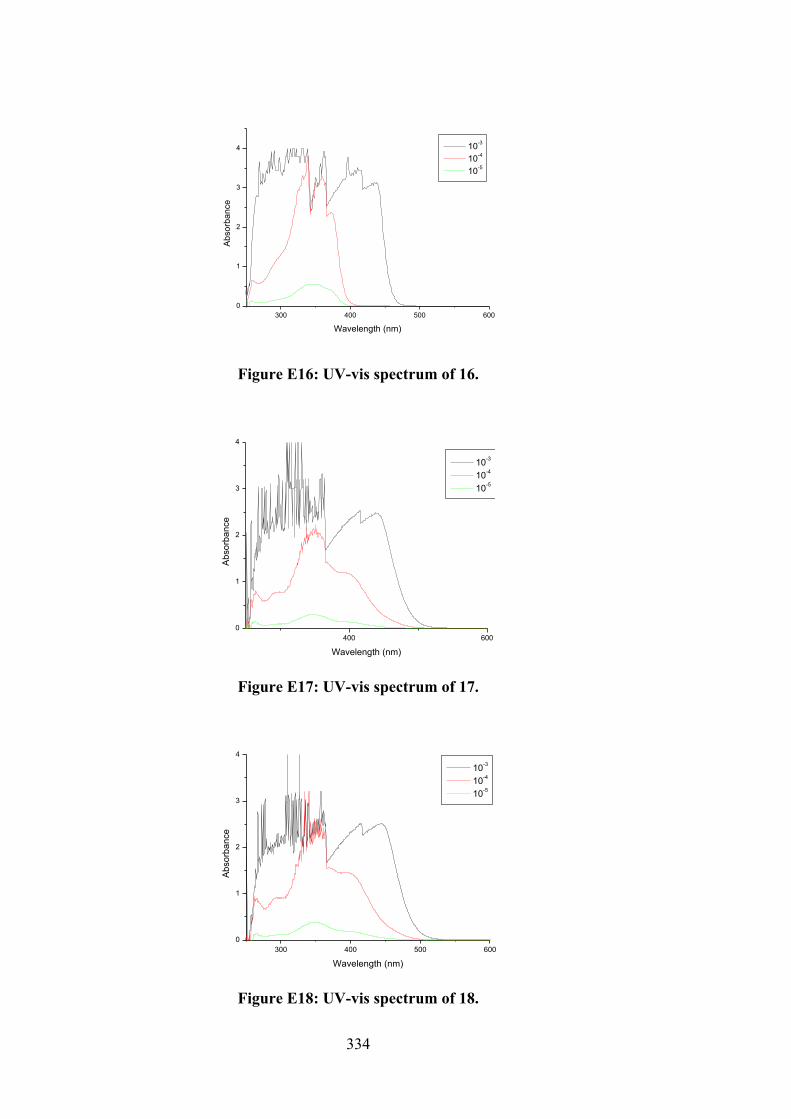
334
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E16: UV-vis spectrum of 16.
400 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E17: UV-vis spectrum of 17.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E18: UV-vis spectrum of 18.
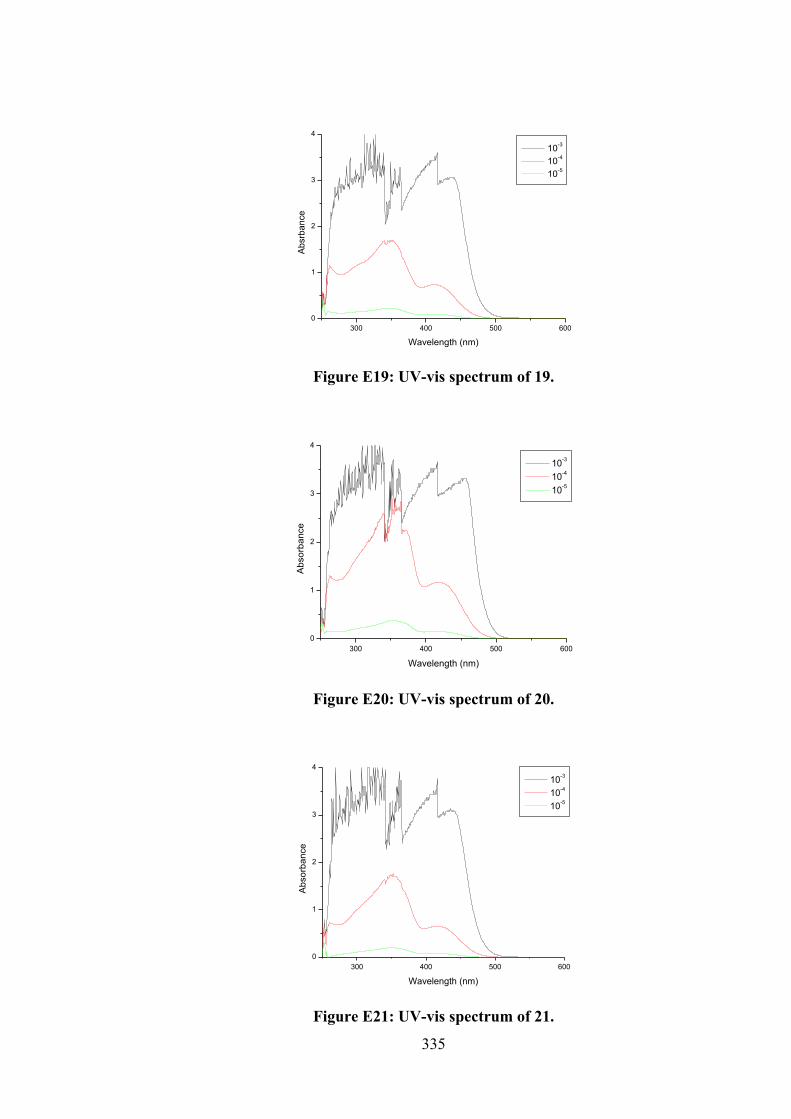
335
300 400 500 6000
1
2
3
4
Absr
banc
e
Wavelength (nm)
10-3
10-4
10-5
Figure E19: UV-vis spectrum of 19.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E20: UV-vis spectrum of 20.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E21: UV-vis spectrum of 21.
336
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E22: UV-vis spectrum of 22.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E23: UV-vis spectrum of 23.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E24: UV-vis spectrum of 24.
337
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength(nm)
10-3
10-4
10-5
Figure E25: UV-vis spectrum of 25.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength(nm)
10-3
10-4
10-5
Figure E26: UV-vis spectrum of 26.
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E27: UV-vis spectrum of 27.
338
300 400 5000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E28: UV-vis spectrum of 28.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E29: UV-vis spectrum of 29.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E30: UV-vis spectrum of 30.
339
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E31: UV-vis spectrum of 31.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E32: UV-vis spectrum of 32.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E33: UV-vis spectrum of 33.
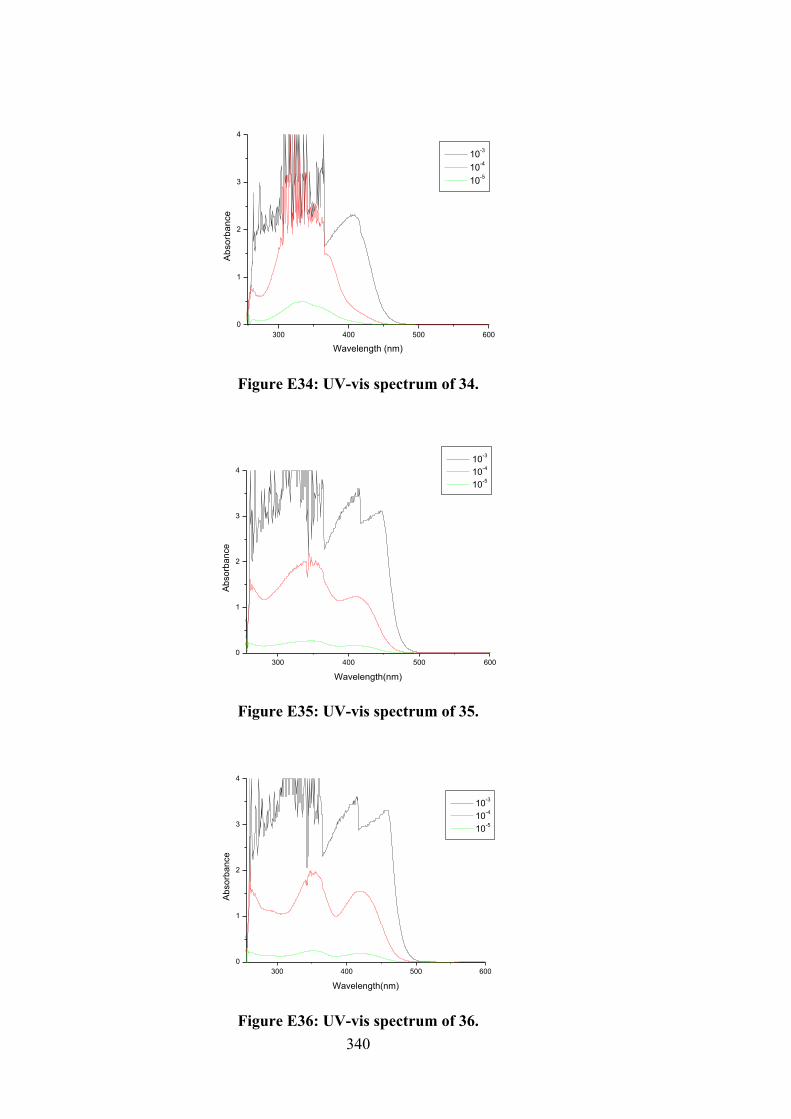
340
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E34: UV-vis spectrum of 34.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength(nm)
10-3
10-4
10-5
Figure E35: UV-vis spectrum of 35.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength(nm)
10-3
10-4
10-5
Figure E36: UV-vis spectrum of 36.
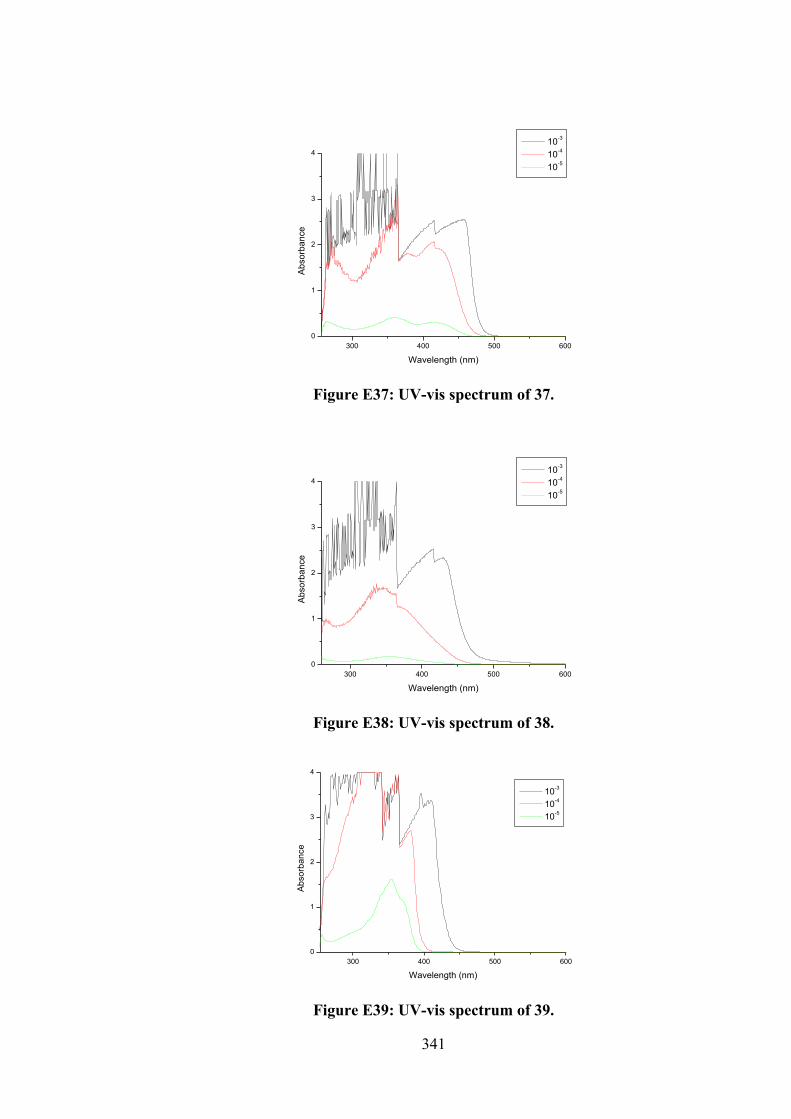
341
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E37: UV-vis spectrum of 37.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E38: UV-vis spectrum of 38.
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E39: UV-vis spectrum of 39.
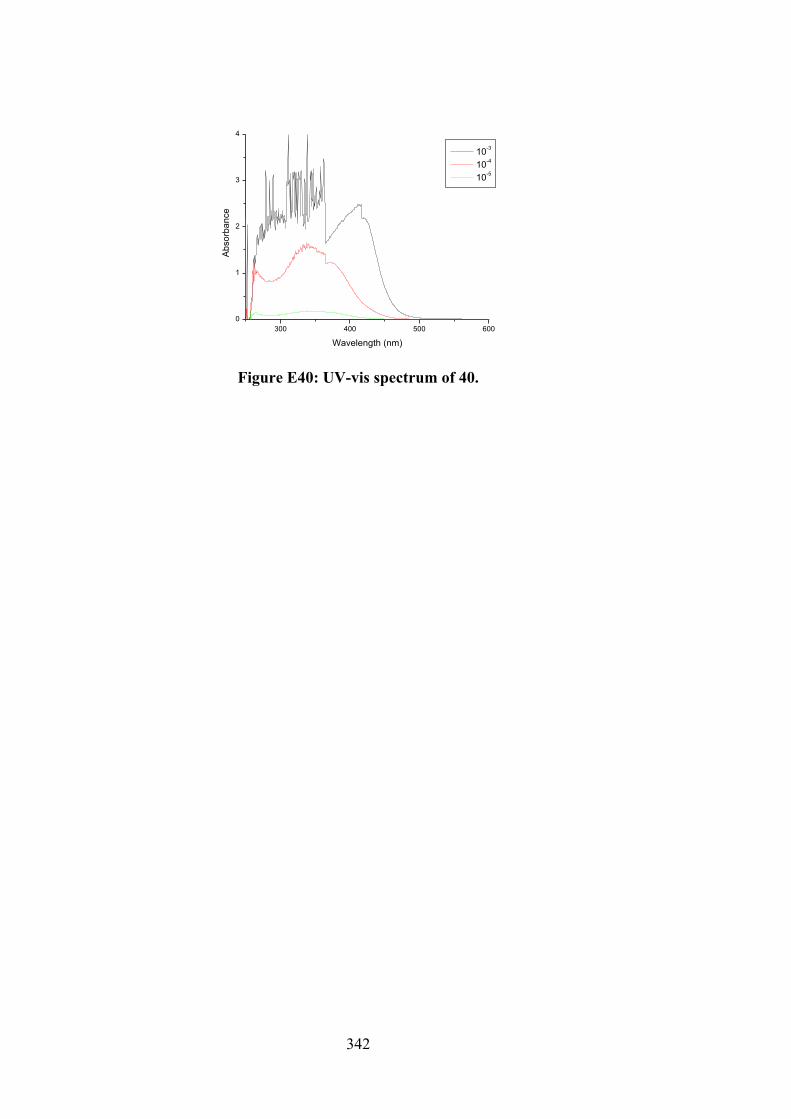
342
300 400 500 6000
1
2
3
4
Abso
rban
ce
Wavelength (nm)
10-3
10-4
10-5
Figure E40: UV-vis spectrum of 40.

343
Acknowledgement of Authorship
I hereby certify that the work embodied in this thesis contains scholarly work of which I am a joint author. I have included a written declaration below endorsed in writing by my supervisor, attesting to my contribution to the scholarly work.
By signing below I, Associate Professor Dr. Alister J. Page, confirm that Enis Nadia Md Yusof contributed to the following scholarly works:
1. 2-[(1E )-([(Benzylsulfanyl)methanethioyl]aminoimino)methyl]-6-methoxyphenol: Crystal Structure and Hirshfeld Surface Analysis.Yusof, E. N. M.; Jotani, M. M.; Tiekink, E. R. T.; Ravoof, T. B. S. A.Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 516–521.
As leading author, Enis Nadia Md Yusof; Carried out experiments presented in the publication Performed data analysis and figure formatting for publication Wrote initial manuscript draft Collaborated with co-authors on manuscript revision, submission and reviewer
comments Provided corrections for proofs prior to publication
2. o-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis,Structural Characterisation, In-Silico Studies and CytotoxicityYusof, E. N. M.; Latif, M. A. M.; Tahir, M. I. M.; Sakoff, J. A.; Simone, M. I.;Page, A. J.; Veerakumarasivam, A.; Tiekink, E. R. T.; Ravoof, T. B. S. A.Int. J. Mol. Sci. 2019, 20, 854
As leading author, Enis Nadia Md Yusof; Carried out experiments presented in the publication Performed data analysis and figure formatting for publication Wrote initial manuscript draft Collaborated with co-authors on manuscript revision, submission and reviewer
comments Provided corrections for proofs prior to publication
Signed,
Alister J. Page (Date: 15/7/2019)

344
BIODATA OF STUDENT
Enis Nadia binti Md Yusof was born on 22nd October 1989 in Terengganu, Malaysia. She
received her primary education at Sekolah Kebangsaan Kuala Pilah from 1995-1996 and
changed to Sekolah Kebangsaan Cherating, Kuantan from 1997-2001. She continued her
secondary education at Sekolah Menengah Kebangsaan Chukai, Terengganu from 2002-
2006. She had completed her matriculation program in biological science at Malacca
Matriculation College in 2008. In 2011, she was awarded her first degree, Bachelor of
Science (Hons.) majoring in Chemistry Resource and minoring in Biotechnology from
Universiti Malaysia Sarawak, Malaysia. In 2015, she was awarded Master of Science
(Inorganic Synthesis) from Universiti Putra Malaysia, Malaysia under the supervision of
Dr. Thahira Begum. In the same year 2015, she started her Doctor of Philosophy in
Inorganic Synthesis at Universiti Putra Malaysia, Malaysia under supervision of
Associate Professor Dr. Thahira Begum, supported by the Ministry of Education under
MyBrain15 Program, MyPhD. In September 2017, she joined Joint Awarded Doctoral
Degree programme with The University of Newcastle, Australia under the supervision
Associate Professor Dr. Alister J. Page and Professor Adam McCluskey. During her study
in Australia, she was granted the University of Newcastle International Postgraduate
Research Scholarship (UNIPRS) and the University of Newcastle Research Scholarship
Central (UNRSC) for 20 months.

345
LIST OF PUBLICATIONS
1. Yusof, E. N. M.; Jotani, M. M.; Tiekink, E. R. T.; Ravoof, T. B. S. A. 2-[(1E)-([(Benzylsulfanyl)methanethioyl]aminoimino)methyl]-6-methoxyphenol: Crystal Structure and Hirshfeld Surface Analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 516–521.
2. Yusof, E. N. M.; Ravoof, T. B. S. A.; Tahir, M. I. M.; Jotani, M. M.; Tiekink, E. R. T. Bis4-methylbenzyl 2-[4-(propan-2-yl)benzyl-idene]hydrazine carbodithioato-κ2N2,Snickel(II): Crystal Structure and Hirshfeld Surface Analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, E73, 397–402.
3. Yusof, E. N. M; Tahir, M. I. M.; Ravoof, T. B. S. A.; Loon, S. A. Cinnamaldehyde
Schiff Base of S-(4-methylbenzyl)dithiocarbazate: Crystal Structure, Hirshfeld Surface Analysis and Computational Study. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, E73, 543–549.
4. Mokhtaruddin, N. S. M.; Yusof, E. N. M.; Ravoof, T. B. S. A.; Tiekink, E. R. T.;
Veerakumarasivam, A.; Tahir, M. I. M. Unusual Saccharin-N,O (Carbonyl) Coordination in Mixed-Ligand copper(II) Complexes: Synthesis, X-Ray Crystallography and Biological Activity. J. Mol. Struct. 2017, 1139, 1–9.
5. Ravoof, T. B. S. A.; Crouse, K. A.; Tiekink, E. R. T.; Tahir, M. I. M.; Yusof, E. N. M.;
Rosli, R. Synthesis, Characterisation and Biological Activities of S-2- or S-4-methylbenzyl-β-N-(di-2-pyridyl)methylenedithiocarbazate and Cu(II), Ni(II), Zn(II) and Cd(II) Complexes. Polyhedron 2017, 133, 383–392.
6. Yusof, E. N. M.; Tiekink, E. R. T.; Jotani, M. M.; Simone, M. I.; Page, A. J.; Ravoof, T. B. S. A. Synthesis, Characterisation and Structure Determination of 3-[(1Z)-2-ylidenemethyl]benzene-1,2-diol. J. Mol. Struct. 2018, 1171, 650–657.
7. Yusof, E. N. M.; Latif, M. A. M.; Tahir, M. I. M.; Sakoff, J. A.; Simone, M. I.; Page, A. J.; Veerakumarasivam, A.; Tiekink, E. R. T.; Ravoof, T. B. S. A. O-Vanillin Derived Schiff Bases and Their Organotin(IV) Compounds: Synthesis, Structural Characterisation, In-silico Studies and Cytotoxicity. Int. J. Mol. Sci. 2019, 20, 854.
8. Yusof, E. N. M.; Nasri, N. M.; Ravoof, T. B. S. A.; Tiekink, E. R. T. A Ternary
Nickel(II) Schiff Base Complex Containing Di-Anionic and Neutral Forms of a Dithiocarbazate Schiff Base. Molbank 2019, M1057, 1–7.
9. Yusof, E. N. M.; Nasri, N. M.; Ravoof, T. B. S. A.; Jotani, M. M.; Tiekink, E. R. T.
Bis[S-benzyl 3-(furan-2-ylmethylidene)dithiocarbazato-K2N3,S]copper(II): crystal structure and Hirshfeld surface analysis. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, E75, 794-799.
10. Yusof, E. N. M., Latif, M. A. M., Tahir, M. I. M., Sakoff, J. A., Abhi Veerakumarasivam, Page, A. J., Tiekink, E. R. T., Ravoof, T. B. S. A. Homoleptic tin(IV) compounds of dinegative ONS dithiocarbazate Schiff bases: Synthesis, X-ray crystallography, DFT and cytotoxicity studies. J. Mol. Struct. 2020, 1205, 127635.

346
11. Yusof, E. N. M., Ishak, N. M., Latif, M. A. M., Tahir, M. I. M., Sakoff, J. A., Page, A. J., Tiekink, E. R. T., Ravoof, T. B. S. A.. Selective cytotoxicity of organotin(IV) compounds with 2,3-dihydroxybenzyl dithiocarbazate Schiff bases. Res. Chem. Intermediat. 2020, 1-29.

347
LIST OF CONFERENCES/ WORKSHOP ATTENDED
E. N. M. Yusof, A. J. Page, M. I. Simone and T. B. S. A. Ravoof. Cytotoxicity of organotin(IV) complexes containing dithiocarbazate Schiff bases. ASEAN Emerging Researchers Conference 2019, Sunway University, Malaysia (9-10 December 2019). Poster Presentation.
E. N. M. Yusof, A. J. Page, M. I. Simone and T. B. S. A. Ravoof. Cytotoxicity of organotin(IV) complexes containing dithiocarbazate Schiff bases. Medicinal Chemistry and Chemical Biology (MCCB) 2018, Brisbane Convention & Exhibition Centre, Brisbane, Australia (18-21 November 2018). Poster Presentation.
E. N. M. Yusof and T. B. S. A. Ravoof. Synthesis, structural characterisation and
biological activities of Sn(IV) complexes derived from S-2-methylbenzyl-β-N-(2-hydroxy-3-methoxybenzylene) dithiocarbazate. 6th International Conference of Young Chemists (ICYC), St. Giles The Wembley Hotel, Penang, Malaysia (16-18 August 2017). Poster presentation.
E. N. M. Yusof and T. B. S. A. Ravoof. Sn(II) and Diphenyltin(IV) complexes of the
o-vanillin Schiff bases derived from S-benzyl- and S-4-methylbenzyldithiocarbazate: Synthesis, spectroscopic and structural characterization. Fundamental Science Congress (FSC), Universiti Putra Malaysia, Malaysia (9-10 August 2016). Oral presentation.
16th BCA/CCG Intensive Teaching School in X-Ray Structure Analysis, Trevelyan College, Durham, UK (25th March - 2nd April 2017), As Participant.

348
UNIVERSITI PUTRA MALAYSIA
STATUS CONFIRMATION FOR THESIS / PROJECT REPORT AND COPYRIGHT
ACADEMIC SESSION : FIRST SEMESTER 2019/2020
TITLE OF THESIS / PROJECT REPORT :
SYNTHESIS, STRUCTURAL CHARACTERISATION AND CYTOTOXICITY
STUDY OF TIN(IV) COMPOUNDS CONTAINING ONS SCHIFF BASES
NAME OF STUDENT: ENIS NADIA BINTI MD YUSOF
I acknowledge that the copyright and other intellectual property in the thesis/project report belonged to Universiti Putra Malaysia and I agree to allow this thesis/project report to be placed at the library under the following terms:
1. This thesis/project report is the property of Universiti Putra Malaysia.
2. The library of Universiti Putra Malaysia has the right to make copies for educational purposes only.
3. The library of Universiti Putra Malaysia is allowed to make copies of this thesis for academic exchange.
I declare that this thesis is classified as:
*Please tick (√ )
CONFIDENTIAL (Contain confidential information under Official Secret Act 1972).
RESTRICTED (Contains restricted information as specified by the
organization/institution where research was done).
OPEN ACCESS I agree that my thesis/project report to be published as hard
copy or online open access.
√

349
This thesis is submitted for:
PATENT Embargo from ____________ until ______________ (date) (date)
Approved by: ________________________ ________________________ (Signature of Student) (Signature of Chairman IC No/ Passport No.: of Supervisory Committee) 891022115274 / A38956205 Name: Thahira Begum / Alister J. Page Date : Date : [Note : If the thesis is CONFIDENTIAL or RESTRICTED, please attach with the letter from the organization/institution with period and reasons for confidentially or restricted. ]