synthesis and anticancer screening of combinatorial
TRANSCRIPT

Synthesis and Anticancer Screening of
Combinatorial Libraries of Cyclic Peptides
and Other Compounds
Thesis Submitted for the Partial Fulfillment of the Degree of
DOCTOR OF PHILOSOPHY
By
SAMREEN ASHRAF
H. E. J. Research Institute of Chemistry
International Center for Chemical and Biological Sciences,
University of Karachi, Karachi-75270, Pakistan
(2017)


i
Certificate
To Whom It May Concern
This is to certify that the thesis entitled, “Synthesis and Anticancer Screening of
Combinatorial Libraries of Cyclic Peptide and Other Compounds”, has been
submitted by Ms. Samreen Ashraf to the Board of Advance Studies and Research,
University of Karachi, for the award of the degree of Ph.D. in Chemistry. This thesis, in
full on in parts, has not been submitted to any other institute or university for the award of
any degree or diploma.
Prof. Dr. Farzana Shaheen
Research Supervisor

Contents
ACKNOWLEDGMENTS ...................................................................................................... i
PERSONAL INTRODUCTION............................................................................................ ii
SUMMARY ..........................................................................................................................iii
Urdu Khulasa...………………………………………………………………………….…vi
List of Figures ...................................................................................................................... vii
List of Tables ........................................................................................................................ ix
List of Schemes ...................................................................................................................... x
CHAPTER-1
INTRODUCTION
1.1. Drug Discovery and Development ................................................................................. 1
1.1.1. The Lead Discovery Process ............................................................................ 1
1.2. Peptides: An Attractive Source of New Lead Molecules ............................................... 3
1.2.1. Peptide Hormones as Drugs ............................................................................. 5
1.2.2. Peptide as Radionuclide Carrier....................................................................... 6
1.2.3. Peptide as Vaccines.......................................................................................... 8
1.2.4. Peptides in Targeted Drug Delivery ................................................................ 9
1.2.5. Anti-Tumor Peptides ...................................................................................... 11
1.2.6. Cell Penetrating Peptides ............................................................................... 11
1.2.7. Anti-inflammatory Peptides ........................................................................... 12
1.2.7.1. Duanbanhuains (A–C) .................................................................... 13
1.2.7.2. Cyclomarins .................................................................................... 13
1.2.7.3. Cyclosquamosin .............................................................................. 14
1.2.7.4. Anti-inflammatory Peptides from Marine Sponges ........................ 15
1.3. Cyclic Peptides ............................................................................................................. 16
1.3.1. Stability of Cyclic Peptides ............................................................................ 17
1.3.2. Strategies to Develop Peptide Libraries ......................................................... 18
1.3.2.1. Combinatorial Library Approach .................................................... 19
1.3.2.2. Phage-Display Peptide Library Method .......................................... 19

1.3.2.3. Parallel Library Method .................................................................. 20
1.3.2.4. Combinatorial Library Methods Requiring Deconvolution ............ 20
1.3.2.5. Affinity Selection Method .............................................................. 21
1.3.2.6. Self-Assembled PNA-Encoded Chemical Microarrays .................. 21
1.3.2.7. OBOC Combinatorial Library Method ........................................... 21
1.3.3. Cyclization Strategies for Synthetic Cyclic Peptides ..................................... 22
1.3.4. Analysis of Cyclic Peptide through Mass Spectrometry ............................... 25
1.3.5. Conformational Analysis of Cyclic Peptides ................................................. 27
1.4. Importance of Cationic Cyclic Peptides ....................................................................... 28
1.5. Cancer Targeting Strategies .......................................................................................... 29
1.6. Inflammation ................................................................................................................. 33
1.6.1. Mediators of Inflammation ............................................................................ 36
1.6.1.1. Role of Interlukin-1 (IL-1) .............................................................. 36
1.6.1.2. Role of Interlukin-2 (IL-2) .............................................................. 37
1.6.1.3. Role of Interlukin-6 (IL-6) .............................................................. 37
1.6.1.4. Role of tumor necrosis factor-alpha (TNF-α) ................................. 38
1.6.1.5. Role of Nitric Oxide (NO.) ............................................................. 38
1.6.1.6. Role of Reactive Oxygen Species ................................................... 39
1.7. Solid-phase Peptide Synthesis ...................................................................................... 39
1.7.1. Solid Supports for Peptide Synthesis ............................................................. 40
1.7.2. Linkers used in Solid-phase Peptide Synthesis (SPPS) ................................. 41
1.7.2.1. Kenner’s Safety-catch Linker ......................................................... 41
1.7.2.2. Wang Linker ................................................................................... 42
1.7.2.3. Rink-amide Linker .......................................................................... 42
1.7.3. Protecting Groups Used in Solid-phase Peptide Synthesis ............................ 43
1.7.4. Coupling Reagents ......................................................................................... 44
1.8. Side-reactions in Peptide Bond Formation ................................................................... 45
1.8.1. Racemization: ................................................................................................ 45
1.8.2. Synthesis of Diketopiperazine During Peptide Chain Elongation ................. 45
1.9. Targets of Current Study ............................................................................................... 46

Chapter - 2
Result and Discussions
2.1. Design, Synthesis and Screening of Cyclic Peptide Library ........................................ 47
2.2. Structural Studies of Anticancer Peptides from Library 1 ............................................ 50
2.2.1. Cyclic Peptide 2 ............................................................................................. 50
2.2.2. Cyclic Peptide 14 ........................................................................................... 57
2.2.3. Cyclic Peptide 15 ........................................................................................... 62
2.2.4. Cyclic Peptide 16 ........................................................................................... 68
2.2.5. Anticancer Activities of Cyclic Peptides. ...................................................... 73
2.3. Studies on Stylissatin A Analogues .............................................................................. 81
2.3.1. Cyclic Peptide 26 ........................................................................................... 84
2.3.2. Cyclic Peptide 27 ........................................................................................... 90
2.3.3. Cyclic Peptide 28 ........................................................................................... 97
2.3.4. Cyclic Peptide 29 ......................................................................................... 105
2.3.5. Cyclic Peptide 30 ......................................................................................... 112
2.3.6. Cyclic Peptide 31 ......................................................................................... 118
2.4. Immunomodulatory Activities of Stylissatin A Analogues ........................................ 125
2.5. Conclusion .................................................................................................................. 126
Chapter - 3
EXPERIMENTAL PROCEDURES
3.1. General Experimental Details ..................................................................................... 128
3.2. General Synthesis Procedure for Cyclic Peptide Library I ......................................... 128
3.3. Synthesis of Biologically Active Compounds ............................................................ 129
3.3.1. Synthesis of Cyclic Peptide 2 ...................................................................... 129
3.3.1.1. Characterization Data of Peptide 2 ............................................... 130
3.3.2. Synthesis of Cyclic Peptide 14 .................................................................... 130
3.3.2.1. Characterization Data of Peptide 14 ............................................. 131
3.3.3. Synthesis of Cyclic Peptide 15 .................................................................... 131
3.3.3.1. Characterization of Peptide 15 ...................................................... 132

3.3.4. Synthesis of Cyclic Peptide 16 .................................................................... 132
3.3.4.1. Characterization of Peptide 16 ..................................................... 133
3.4. Synthesis Procedure for Stylissatin A Analogues ....................................................... 133
3.4.1. Synthesis of Cyclic Peptide 26 .................................................................... 133
3.4.1.1. Characterization Data of Peptide 26 ............................................. 134
3.4.2. Synthesis of Cyclic Peptide 27 .................................................................... 135
3.4.2.1. Characterization Data of Peptide 27 ............................................ 135
3.4.3 Synthesis of Cyclic Peptide 28. .................................................................... 136
3.4.3.1. Characterization Data of Peptide 28 ............................................. 136
3.4.4. Synthesis of Cyclic Peptide 29 .................................................................... 137
3.4.4.1. Characterization Data of Peptide 29 ............................................ 137
3.4.5. Synthesis of Cyclic Peptide 30 .................................................................... 138
3.4.5.1. Characterization Data of Peptide 30 ............................................ 138
3.4.6. Synthesis of Cyclic Peptide 31 .................................................................... 139
3.4.6.1. Characterization Data of Peptide 31 ............................................ 139
3.5. Screening Protocol ...................................................................................................... 140
3.5.1. MTT (3- (4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide)
Assay……………………………………………………………………………..140
3.5.2. Chemiluminescence Assay .......................................................................... 140
3.5.3. Nitric Oxide Screening Protocol: ................................................................. 140
3.5.4. IL-2 Production and Quantification: ............................................................ 141
References .......................................................................................................................... 142
Glossary ............................................................................................................................. 168
Abbreviations……………………………………..……………………………………………..172

i
ACKNOWLEDGEMENTS
First of all I present my sincere gratitude to the Allah (SWT), who is the most beneficent and merciful
and Hazrat Muhammad (PBUH), whose teachings enlighten our souls and minds.
I present my honor to the pioneer of this prestigious institute Prof. Dr. Salim-uz-Zaman Siddiqui FRS
whose dedication makes this institute the home of great research. I also present a note of thanks to
revolutionary scientist Prof. Dr. Atta-ur-Rahman FRS. His dynamic leadership and guidance has driven
this country towards prosperity and development. I am also thankful to honorable Prof. Dr.
Muhammad Iqbal Choudhary H.I. S.I. T.I., Director of ICCBS. He is always a source of inspiration
throughout my Ph. D. studies. I wish to express my deepest gratitude for his expert guidance, sincere
advice and continuous support through the period of this research study.
I expressed my profound gratitude to my research supervisor Prof. Dr. Farzana Shaheen. Her endless
efforts, encouragements and attention allowed me to finally complete my Ph.D. work. I am highly
indebted to her for the contribution of experience and knowledge being shared and conveyed to me.
I am very thankful to Dr. Almas Jabeen for the biological evaluation of compounds reported in this
thesis.
I cordially express my thanks to all faculty members especially Dr. Shabana Simjee for their guidance,
cooperation and assistance and thanks are to all technical and non-technical staff of the institute who
have helped me in the completion of my work in so many ways.
It cheers me up to remember all my lab colleagues, Dr. Zafar Ali Shah, Comail Abbas, Asad Ziaee,
Muhammad Nadeem-ul-Haque, Salma Nazir, Attiya Hameed, Anila Bashir and Hira Shehzed and Lab.
attendant Syed Muhammad Nasar.
I also want to acknowledge the financial support for this study from Pakistan Science Foundation
(PSF) project PSF/NSLP(290).
I am also pleased to present my utmost gratitude to my family, parents, my husband, and my mother-in-
law, their patience, trust and prayers kept me going ahead widening the path and making ways to
achieve my goal.
Samreen Ashraf
Karachi, 2017

ii
PERSONAL INTRODUCTION:
On 17th of June, 1985, it was a hot afternoon of summer when ALLAH
bestowed HIS blessing on my parents and I open my eyes in this world. I
have been very fortune to be in the “City of Lights” Karachi, Pakistan, where
number of educational institutes is enlightening the lives of people with
knowledge and skills. After schooling I completed my higher secondary
education from Govt. Degree Science College Malir Cantt. I pursued my
Master’s degree from University of Karachi with organic chemistry as major
subject.
The thirst of knowledge was still not satisfied, and on March 2010, I joined the H.E.J. Research
Institute of Chemistry, International Center for Chemical and Biological Sciences, University of
Karachi. I have been very fortunate to carry out my research studies at one of the best institutions of
the region. I worked on the “Synthesis and Anticancer Screening of Combinatorial Libraries of Cyclic
Peptide and Other Compounds” for my Ph.D. studies under the kind and able supervision of Prof. Dr.
Farzana Shaheen.
At H.E.J. Institute, I received generous guidance by highly experienced and dedicated faculty members
who always helped me to broaden my scientific knowledge and skills. I learnt different ways of tackling
complex situations, which not only enhanced my self-confidence but also improved my capacity of
critical thinking. In the area of research my focus remains the peptide synthesis. In years to come, I
would like to take part in the development of theoretical chemistry and to correlate it with the peptide
synthesis for the development of biologically active molecules against diseases like cancer, diabetes,
tuberculosis, etc. I have a dream of serving the humanity through relevant science all over the world
generally, and Pakistan particularly by contributing in the development of field laboratories to promote
the scientific knowledge, and ultimately contribute towards national development.

iii
SUMMARY
The structural features of cyclic peptides make them good drug candidates, and up till now,
several cyclic peptides have been developed for clinical use. Many structurally important
cyclic peptides have been developed with different synthetic approaches. In recent years,
there has been an increasing interest in the synthesis of biologically active cyclic peptides.
The current study was focused on the synthesis and identification of novel biologically
active compounds from libraries based on cyclic peptide scaffold. During the synthesis of
cyclic peptides, different cyclization strategies were adopted to develop diverse forms of
cyclic peptides. These cyclic peptides were designed to contain some unusual building
blocks, such as phenyl glycine, naphthyl alanine, ornithine, 2-amino benzoic acid, D amino
acids as well as cationic residues at different positions. The synthesis of all cyclic peptides
was accomplished by macrocyclization strategy using standard Fmoc-peptide synthesis
protocol. The cyclic peptide libraries were screened against four human cancer cell lines
MCF-7, NCI-H460, and DoHH2 and HeLa cell lines to identify active compounds.
Detailed structural studies were performed on anticancer peptides discovered from the
peptide libraries.
Compound 16 was found very potent anticancer peptide during the screening of peptide
library against DoHH2 (IC50 4.4 µM) and MCF-7 (IC50 1.1 µM) cell lines, while
compounds 2 and 15 were found active against NCI-H460 (IC50 62.6 µM and IC50 41.9
µM) respectively. Compound 15 also showed some activity against HeLa cell lines (IC50
92.4 µM). Compound 14 was found active against MCF-7 with IC50 29.6 µM. All
compounds were found non-cytotoxic to 3T3 normal cell lines.
In the second part of this study, a small library was synthesized around a naturally
occurring cyclic peptide stylissatin A. stylissatin A is reported as a natural inhibitor of
nitric oxide. The solid- phase total synthesis of this natural product was also accomplished
by our research group. The current study was focused on the synthesis, structure
elucidation, and SAR study of a small library of stylissatin A. All compounds from this
library were tested against different cancer cell lines but all of them are found inactive.

iv
These all compounds were tested for immunomodulatory activity. The Synthetic analogue
27 inhibited ROS on whole blood, as well as neutrophils. Related analogues of stylissatin
A were identified as more potent inhibitors of interleukin 2 than peptide stylissatin A.
Potent Anti-cancer Cyclic Peptide 16
MCF-7 (IC50 = 1.1 ± 0.07 uM)
DoHH2 (IC50 = 4.4 ± 0.06 uM)

v
SAR Studies of Stylissatin A

vi
خالصہ
حلقئ پیپٹائیڈز اپنی ساختی خصوصیات کی بنیاد پر بہترین طبی کارکردگی کے حامل ہیں اور اب
تک کئی حلقئ پیپٹائیڈز طبی استعمال کیلئے منظور کئے جا چکے ہیں۔
کئی ساختی اعتبار سے اہم حلقئ پیپٹائیڈز مختلف ترکیبی طریقہ کار کے تحت تیار کئے جا چکے
وں میں حیاتیاتی سرگرمی رکھنے والے حلقئ پیپٹائیڈز کی تالیف پر بہت زیادہ ہیں۔ گذشتہ چند سال
توجہ اور تحقیق کی گئی ہے۔
زیر مطالعہ تحقیقی مقالے میں حیاتیاتی سرگرمی کے حامل نادر مرکبات کی پہچان اور تالیفی
ہ کار کو تجربات پر بحث کی گئی ہے۔ ان حلقئ مرکبات کی تالیف کے دوران مختلف حلقوی طریق
استعمال کیا گیا ہے۔ یہ حلقئ پیپٹائیڈز کچھ غیر معمولی لحمی ترشوں کو بنیادی اکائی کے طور پر
Fmocاستعمال کرتے ہوئے بنائے گئے ہیں۔تمام پیپٹائیڈزکو ایک بڑے حلقے میں ڈھالنے کیلئے۔
پیپٹائیڈ تالیفی طریقہ کار استعمال کیا گیا ہے۔
یری کوچار مختلف کینسرسیل الئینزکے خالف ضد سرطانی سرگرمی ان حلقوی پیپٹائیڈز کی الئبر
پیپٹائیڈ الئبریری سے دریافت ہونے والے سب سے زیادہ ضد معلوم کرنے کیلئے جانچا گیا ہے۔
۔سرطانی صالحیت رکھنے والے مرکبات کا تفصیلی ساختی مطالعہ کیا گیاہے
DoHH2 سے زیادہ کینسر مدافعتی نے سب 16سیل الئینز کی جانچ کے دوران مرکب
سیل الئینز کے خالف بالترتیب )آئی NCI-H460 15اور 2سرگرمی ظاہر کی ہے۔ جبکہ مرکب
ملی مولر(کے مطابق سرگرم پائے گئے ہیں۔ 41.9 50ملی مولر(اور )آئی۔سی 62.6 50۔سی
HELAملی مولر( جبکہ مرکب 29.6 50سیل الئینز کے خالف )آئی۔سی MCF-7 14مرکب
پر غیر خلیہ (3T3)سیل الئینز کے خالف بھی سرگرم پایا گیا ہے۔ یہ تمام مرکبات عام خلیوں 15
پاش پائے گئے ہیں۔ اس تحقیقی مقالے کے دوسرے حصے میں قدرتی طور پر پائے جانے والے
کے مماثالت کی ایک چھوٹی الئبریری کی تالیف پر بحث کی گئی Stylissatin Aحلقئ پیپٹائیڈ
کی پیداوار کی مزاحمت کرتا ہے۔ زیر بحث مقالے (NO)ہ قدرتی پیپٹائیڈ نائٹریک اکسائیڈ ی ہے۔
میں اسکے مماثالت کی تالیف، ساختی جانچ اور ساختی وسرگرمی کے باہمی تعلقات کا مظاہرہ
کے خالف زیادہ سرگرم پائے گئے ہیں جبکہ تالیفی Inteeleukin-2کیا گیا ہے۔ یہ تمام مماثالت
کے خالف سب سے زیادہ سرگرم پایا گیا ہے۔ ROS 27 مماثل

vii
List of Figures
Figure No Title Page No.
Figure-1: Lead Discovery Process ................................................................................... 2
Figure-2: Role of Peptides in Different Possible Ways of Cancer Treatment ................. 5
Figure-3: Binding and killing mechanism of radio-labeled peptide ................................ 8
Figure-4: Schematic representation of the four possible ways for peptide
macrocyclization. ........................................................................................................... 23
Figure-5: Peptide Fragmentation Notation Roepstorrff and Fohlman ........................... 27
Figure-6: Peptidyl-Prolyl conformation of Cis/Trans Isomers. ..................................... 28
Figure-7: Process of Inflammation ................................................................................ 35
Figure-8: The General Scheme for Solid-Phase Peptide Synthesis ............................... 40
Figure-9: Resin for SPPS ............................................................................................... 41
Figure-10: Wang Linker ................................................................................................ 42
Figure-11: Rink-amide Linker ....................................................................................... 43
Figure-12: Protecting Groups used in SPPS .................................................................. 43
Figure-13: Coupling Reagents used in SPPS ................................................................. 44
Figure-14: Diketopiperazine Formation ........................................................................ 46
Figure-15: Analytical HPLC Profile of Cyclic Peptide 2 .............................................. 52
Figure-16: Key HMBC Interaction of Cyclic Peptide 2 ................................................ 53
Figure-17: Key COSY Correlations of Cyclic Peptide 2 ............................................... 54
Figure-18: Key NOESY Correlation of Cyclic Peptide 2 .............................................. 54
Figure-19: Key HMBC and COSY Correlations of Cyclic Peptide 14 ......................... 60
Figure-20: Key HMBC Interactions of Cyclic Peptide 15............................................. 65
Figure-21: Key COSY Correlations of Cyclic Peptide 15 ............................................. 65
Figure-22: Key HMBC Interactions of Cyclic Peptide 16............................................. 72
Figure-23: Analytical HPLC Profile of Cyclic Peptide 26 ............................................ 86
Figure-24: Key HMBC Interactions of Cyclic Peptide 26............................................. 87

viii
Figure-25: Key COSY Correlations of Cyclic Peptide 26 ............................................. 88
Figure-26: Key COSY Correlations of Cyclic Peptide 26 ............................................. 89
Figure-27: Analytical HPLC Profile of Cyclic Peptide 27 ............................................ 93
Figure-28: Key HMBC Interactions of Cyclic Peptide 27............................................. 94
Figure-29: Key COSY Correlations of Cyclic Peptide 27 ............................................. 95
Figure-30: Key NOESY correlations of Cyclic Peptide 27 ........................................... 96
Figure-31: Analytical HPLC Profile of Cyclic Peptide 28 .......................................... 100
Figure-32: Key HMBC Correlation of Cyclic Peptide 28 ........................................... 101
Figure-33: Key COSY Correlation of Cyclic Peptide 28 ............................................ 102
Figure-34: Key NOESY Correlation of Cyclic Peptide 28 .......................................... 103
Figure-35: Analytical HPLC Profile of Cyclic Peptide 29 .......................................... 107
Figure-36: Key HMBC Interactions of Cyclic Peptide 29........................................... 108
Figure-37: Key COSY Correlations of Cyclic Peptide 29 ........................................... 109
Figure-38: Key NOESY Correlation of Cyclic Peptide 29 .......................................... 110
Figure-39: Analytical HPLC Profile of Cyclic Peptide 30 .......................................... 114
Figure-40: Key HMBC Interactions of Cyclic Peptide 30........................................... 115
Figure-41: Key COSY Correlation of Cyclic Peptide 30 ............................................ 116
Figure-42: Key NOESY Correlations of Cyclic Peptide 30 ........................................ 117
Figure-43: HPLC Profile of Cyclic Peptide 31 ............................................................ 121
Figure-44: Key HMBC Interactions of Cyclic Peptide 31........................................... 122
Figure-45: Key COSY Correlations of Cyclic Peptide 31 ........................................... 123
Figure-46: Key NOESY Correlations of Cyclic Peptide 31 ........................................ 123

ix
List of Tables
Table No. Title Page No.
1(a) Cyclic Peptide Library containing Unusual and Cationic
Residues.
48
1(b) Cyclic Peptide Library Based on Stylissatin A 49
2 NMR Data of Cyclic Peptide 2 55
3 NMR Data of Cyclic Peptide 14 61
4 NMR Data of Cyclic Peptide 15 67
5 NMR Data of Cyclic Peptide 16 74
6 Cytotoxicity of New Cyclic Peptide 81
7 Alanine Substituted Analogues of Stylissatin A 83
8 NMR Spectroscopic Data of Cyclic Peptide 26 91
9 NMR Spectroscopic Data of Cyclic Peptide 27 98
10 NMR Spectroscopic Data of Cyclic Peptide 28 105
11 NMR Spectroscopic Data of Cyclic Peptide 29 112
12 NMR Spectroscopic Data of Cyclic Peptide 30 119
13 NMR Spectroscopic Data of Cyclic Peptide 31 125
14 Mass Spectroscopic Analysis and [α]D of Peptides 26-31. 126
15 Effect of Peptides 26-31 on Oxidative Burst, Nitric Oxide
(NO.) and IL-2 Production.
126

x
List of Schemes
Scheme No. Caption Page No.
1 Kenner’s Safety-catch Principle 42
2 Racemization during Peptide Bond Formation in SPPS 45
3 Synthesis of Cyclic Peptide 2 51
4 Synthesis of Cyclic Peptide 14 58
5 Synthesis of Cyclic Peptide 15 64
6 Synthesis of Cyclic Peptide 16 70
7 Solid-Phase Synthesis of Cyclic Peptides (26-31) 84
8 Synthesis of Cyclic Peptide 26 86
9 Synthesis of Cyclic Peptide 27 93
10 Synthesis of Cyclic Peptide 28 100
11 Synthesis of Cyclic Peptide 29 107
12 Synthesis of Cyclic Peptide 30 114
13 Synthesis of Cyclic Peptide 31 121

CHAPTER-1
INTRODUCTION

1
1.1. Drug Discovery and Development:
The drug discovery process is initiated from the search of new lead molecule either from
natural sources or by high-throughput screening of synthetic libraries. The new lead
molecules can be further developed as new drug for the requisite therapeutic effect
(Panchagnula & Thomas, 2000). Drug discovery is a time consuming and laborious
process which comprises of different phases such as disease target identification, target
validation, high-throughput identification of hits and leads, lead optimization, preclinical
and clinical evaluation (Gombar, Silver, & Zhao, 2003). From high throughput screening
or from the primary screening of the natural molecules, thousands of compounds may be
potential candidates for development as a new drug, but most of them are filtered out at
next stages of development and only a small number of compounds successfully go
through development stages for new drug.
In development process, several experiments are conducted to evaluate the efficacy along
with safety/ toxicity of new candidate molecules. The efficient and safest (least toxic or
non-toxic) molecule is further tested for the absorbency, distribution, metabolism and
excretion in the body. Figure-1represents the stages of drug discovery and development
process, this whole process requires a huge investment of money and time. It is estimated
that out of ten candidate molecule, only one enters into clinical development, which costs
approximately 10 – 15 years in time domain and US $ 500-800 million from pre-clinical
stage to marketing (Pastan, Hassan, FitzGerald, & Kreitman, 2006).
1.1.1. The Lead Discovery Process:
The molecule which exhibit desired and reproducible activity against disease in a
compound screening is called the “Lead” or “Hit” compound. A variety of screening
procedures exist to identify hit molecules. There are several procedures that can provide
potential candidate to become the lead compound, such as, isolation of compounds from
the natural sources like plant, lower terrestrial or marine organisms, or from synthesis of
libraries of compounds by using computer aided drug design and/or combinatorial
synthesis (Panchagnula & Thomas, 2000). High throughput screening or in vitro screening
assays are used to identify “Hit” molecule from different sources.

2
When hits are identified, their optimization is the next step. Empirical and semi empirical
structure-activity relationship (SAR) studies are used to modify the structure in order to
improve the effectiveness and lessen the toxicity of molecule. However, good in vitro
activity does not guarantee a good in vivo activity; it depends on good bioavailability of
drug and a desirable duration of action.
When “Hit” compound passes through in vivo screening, optimization of ADMET
(Absorption, Distribution, Metabolism, Excretion and Toxicity) is also performed to
improve the degree of potency, which has already been achieved (Y. Yang, Adelstein, &
Kassis, 2009), (Hughes, Rees, Kalindjian, & Philpott, 2011), (Caldwell, Yan, Tang,
Dasgupta, & Hasting, 2009). In vivo screening involves testing on a suitable animal model
to evaluate its pharmacokinetic and toxico-kinetic profile. This whole process is a part of
pre-clinical studies in drug discovery and development. Once the candidate drug molecule
passes this phase, an investigation IND (Investigational New Drug) is filed to the drug
regulating authorities. After IND application approval, the clinical phase starts which
involves testing in human subjects. Although the small group of these therapies has
significant curative preclinical results for cancer, very fewer have progressed toward
commercialization (Wolinsky, Colson, & Grinstaff, 2012).
Figure-1: Lead Discovery Process

3
1.2. Peptides: An Attractive Source of New Lead Molecules
Due to recent advancement in the peptide-based lead discovery process, they are
considered as fascinating therapeutic agents. Peptides have acquired an important place in
the field of medicine and are being used individually or in conjugation with drugs or nano-
materials of all reported compositions. Over the years, peptides have been reported for
treatment of diabetes, cancer, congestive heart diseases and inflammation. Nearly, 60
peptide drugs have been marketed and results in an annual sale of more than $13 billion
(Thayer, 2011). Three out of four marketed peptide drugs are being used in the direct
treatment of cancer or in the episodes of treatment that is associated with certain tumors
(leuprolide, goserelin, and octreotide) and are generating global sales of more than $1
billion.
From 2000 till present, hundreds of peptide candidate entered in the clinic and pre-clinic
development, out of which mostly are used for the cancer detection and therapy (18%) and
metabolic disorders (17%) (Borghouts, Kunz, & Groner, 2005), (Reichert, Pechon, Tartat,
& Dunn, 2010).
Some anti-microbial peptides like, a few cathelicidin-derived AMPs, such as LL-37,
indolicidin (IN), bactenecin, BMAP-27, and β-defensin, can potentially inhibit LPS-
induced cellular cytokine and/or NO. release by binding directly to LPS or by blocking the
binding of LPS to LBP (Bowdish, Davidson, Scott, & Hancock, 2005), (Zughaier, Shafer,
& Stephens, 2005).

4
Peptide hybrids also can be a good choice for the inflammation therapy. Yi-Fan Liu et al
studied the effect of hybrid peptide and found that hybrid peptides have a potential to
inhibit the expression of LPS-induced pro-inflammatory cytokines and chemokines (Y.
Liu, Xia, Xu, & Wang, 2013).
At present, many anti-inflammatory peptides have been reported from various natural
sources and successfully synthesized. Cyclomarin A isolated from a culture of Streptomyce
showed cytotoxicity towards cancer cells but its anti-inflammatory properties were also
significant (Renner et al., 1999). Solomonamide A, a peptide, with a unique chemical
structure was isolated from the marine sponge Theonella swinhoei. It showed significant
reduction of inflammation in the carrageenan-induced paw edema model in rats. These two
compounds showed promising results as potential non-steroidal anti-inflammatory drugs.

5
Peptides can be used for the treatment of cancer in several different ways, including tumor
targeting agents that deliver cytotoxic drugs specifically to the tumor site, peptides
angiogenesis inhibitors, radionuclides, hormones, and vaccines (Aina, Sroka, Chen, &
Lam, 2002), (Meng et al., 2012), (X.-X. Zhang, Eden, & Chen, 2012).Figure-3 describes
the various plausible ways of treatment for cancer using peptides. Peptides have excellent
binding affinity with various receptors and are involved in several biochemical pathways
this is the reason that peptides are being used in diagnosis and as biomarkers in cancer
progression. There is continues progress in various other fields, such as development of
peptide-vaccine and peptide angiogenesis inhibitors. Several clinical trials are in progress
which will provide better options to millions of cancer patients (Lee et al., 2008).
Figure-2: Role of Peptides in Different Possible Ways of Cancer Treatment (Thundimadathil,
2012)
1.2.1. Peptide Hormones as Drugs:
For the first time, peptides were used for cancer therapy by Schally et al. He used LHRH
(luteinizing hormone-releasing hormone) agonists as prostate cancer therapy. A major
increase in different formulations for the LHRH agonists has been observed. The
mechanism of action of LHRH agonists is the down-regulation of the LHRH receptors in
the pituitary glands which results in the obstacle in release of follicle stimulating hormone

6
(FSH) and LH. Reduction in concentration of these hormones also reduces the associated
testosterone production. Ultimately, it helps in suggesting a new therapy of androgen
deficiency in prostate cancer patients.
There are other effective LHRH antagonists which are in clinical use nowadays. The first
LHRH antagonist available in market is cetrorelix (Debruyne, Bhat, & Garnick, 2006).
Many other potent LHRH antagonists such as abarelix and degarelix are now approved for
human use (Broqua et al., 2002), (D. Kwekkeboom, Krenning, & de Jong, 2000).
1.2.2. Peptide as Radionuclide Carrier:
Cancer diagnosis is equally important as its treatment. In the diagnosis of cancer, apart
from several other diagnostic tools, peptides are also implicated at this step. Specifically,
peptides associated with radionuclide can work as a diagnostic and as well as radio-
therapeutic agents. For instance, somatostatin hormone, which consists of fourteen amino
acid residues and present in delta (δ) cells of the pancreas, hypothalamic and other

7
gastrointestinal cells. It is normally employed in Peptide Receptor Radionuclide Therapy
(PRRT). In this therapy, radio-peptides or radio-labeled somatostatin analogues were
formed by the combination of a radionuclide and somatostatin analogue which finally
forms highly specialized molecules (D. J. Kwekkeboom et al., 2005), (Krenning et al.,
1999), (Esser et al., 2006), (Nicolas, Giovacchini, Müller-Brand, & Forrer, 2011),
(Grozinsky-Glasberg, Shimon, Korbonits, & Grossman, 2008).
This radio-labeled somatostatin analogue connects to tumor cells containing receptors for
somatostatin. Now the site of radioactivity or tumor cells connected with radio-labeled
somatostatin analogue can be detected using a radiation-measuring device ultimately clear
the sites of tumor cells in the body. Among many effective analogues of somatostatin,
[111In-DTPA]-octreotide (Octreoscan) is the first available radio-labeled analogue that is
used in the diagnosis of sst-positive NETs (Somatostatin positive Neuro-Endocrine
Tumors) (Virgolini et al., 2002), (Strowski & Blake, 2008). After binding with the tumor
cells, radiations are emitted from the radio-peptides which kill the tumor cells. On the
other hand, 111In-coupled peptides have not shown efficiency against PRRT; because after
emission, Auger electrons travelled very short distance. Hence, for effective tumoricidal
behavior of 111In, decay of the nuclei has to take place close to the tumor cells (Krenning et
al., 1999), (D. J. Kwekkeboom et al., 2005), (Saltz et al., 1993). At this time, five sub-
types of somatostatin receptor are identified (sst1-5) (Van de Wiele et al., 2008). Their
density is much higher in comparison to non-tumor tissues. Hence, radio-labeled
somatostatin analogues are commonly used for supply of radioactive somatostatin. In

8
recent years, few other receptors like gastrin-releasing peptide/bombesin (GRP) and
cholecystokinin (CCK) have been involved in tumor imaging and PRRT (H. Zhang et al.,
2004), (Ginj et al., 2006). Radio-labeled receptor antagonists are also developing as
substitutes in this field (Wild et al., 2011), (Henderson, Mossman, Nairn, & Cheever,
2005).
Figure-3: Binding and killing mechanism of radio-labeled peptide.(Thundimadathil, 2012)
1.2.3. Peptide as Vaccines:
Like many other diseases, cancer could also be treated by active immunization. Many
studies have been conducted on immune cells related to tumor or immune molecule.
(Berzofsky, Ahlers, & Belyakov, 2001), (Hareuveni et al., 1990). The host immune system
(T-cells) can easily recognized Tumor-associated antigens (TAAs) which are expressed by
tumor cell (Coulie, Hanagiri, & Takenoyama, 2001), (Eisenbach, Bar-Haim, & El-Shami,
2000). These TAAs can work through the procedure known as active immunotherapy or
vaccination. In this process the host’s immune system is either activated de novo or re-
stimulated to mount an effective, tumor-specific immune reaction that may ultimately lead

9
to tumor regression. The TAAs is injected into the patient’s body which induces a systemic
immune response that eradicates cancer cells from the body tissues. Tumor antigen can be
protein or peptide in nature that has abnormal structure due to mutation of the concerned
gene (Parmiani et al., 2002), (Beck et al., 2007), (Knutson, Schiffman, & Disis, 2001).
Several peptide vaccines have cleared phase I/II/III clinical trials and exhibited excellent
results in immunological as well as clinical responses. These peptide based vaccines
include HER-2 / new immune dominant peptide tested for breast, lung, or ovarian tumor
(Sole, 2006), (Hueman et al., 2005), (Ramanathan et al., 2005), mucin-1 peptide (MUC-1,
stimuvax), (breast or colon cancer) (YAMAMOTO et al., 2005), (J. Wang et al., 2012),
carcino-embryonic antigen (colorectal, gastric, breast, pancreatic and non-small-cell lung
cancers) (Weber et al., 2011), (Grunnet & Sorensen, 2012), prostate-specific membrane
antigen (prostate cancer) (Garetto et al., 2009), (Akhtar, Pail, Saran, Tyrell, & Tagawa,
2011), (Muderspach et al., 2000), HPV-16 E7 peptide (cervical cancer) (Khleif et al.,
1999), rasonco protein peptide (colorectal and pancreatic carcinomas) (Gjertsen &
Gaudernack, 1998), (Abrams, Hand, Tsang, & Schlom, 1996), (Kaufman, 2012), and
melanoma antigens (Melanoma) (Markowicz et al., 2012), (J. C. Yang, 2011). GV-1001,
an injectable form of promiscuous MHC class II peptide, which is in phase III trial for
adenocarcinomas (Nava-Parada & Emens, 2007), (Reubi, 2007).
The use of peptide as vaccine is synthetically economical and does not have problem of
batch to batch variation, having definite structure that is why easy to manipulate.
Drawback of peptide vaccine is their low immunogenicity. Different strategies are being
employed to improve the immunogenicity and efficacy of peptide vaccines (YAMAMOTO
et al., 2005), (J. Wang et al., 2012), (Garetto et al., 2009).
1.2.4. Peptides in Targeted Drug Delivery
In traditional drug delivery system, drug releases instantaneously, that result in the peak
concentration at the start of the dose administration that is also toxic to the normal tissues.
Particularly in cancer, drug distribution is not so specific and can cause severe side effects
to the other tissue than the tumor. Matte et al first published the delivery of ligand-directed

10
drug to leukemic cells. Since then many successful targeting systems have been studied
that effectively directed drugs to the specific site of action.
Peptides are also a better option for transferring drug to the targets. Peptide-drug
conjugates can be easily prepared and can carry the drug to the specific receptors. Several
peptides; known as cell targeting peptides have the ability to target cells expressing their
receptors. Studies showed that many peptide receptors can be excellent targets in cancer
therapy (Van de Wiele et al., 2008), (Wild et al., 2011), (Reubi, 2007), (Gotthardt et al.,
2006).
A number of researches have been conducted on the cancer treatment through peptides.
During recent years, several targeting peptides are discovered through phage display
technique that selectively binds to various diseased tissues. The RGD and NGR motif are
the first generation of targeting peptides. Studies demonstrate that these peptides bind to
the tumor, independent of its type, which indicates the up-regulation of receptors of these
peptides during angiogenesis (Zitzmann, Ehemann, & Schwab, 2002).
A model study indicates that RGD peptide not only identifies angiogenic vessels in general
but also has good affinity towards the αv integrin receptors in the angiogenic vasculature.
The NGR peptide was first recognized as a cell adhesion motif by an in vivo screening on
human breast carcinoma xenografts and specifically target angiogenic blood vessels
(Temming, Schiffelers, Molema, & Kok, 2005), (Burg, Pasqualini, Arap, Ruoslahti, &
Stallcup, 1999), (Porkka, Laakkonen, Hoffman, Bernasconi, & Ruoslahti, 2002). Arap et
al. reported in vivo effects of neo-vasculature targeting peptides RGD-4C and NGR
conjugated with doxorubicin in a mouse xenograft (Arap, Pasqualini, & Ruoslahti, 1998).
Ellerby et al. reported that hybrid peptides composed of D-amino acid pro-apoptotic
peptide, NGR motif and doxorubicin selectively target and destroy angiogenic endothelial
cells in mice (Ellerby et al., 1999). Chen et. al. reported tumor retardation by RGD-peptide
coupled to tachyplesin (Chen et al., 2001). Various chemotherapeutic drugs were also
studied in combination with TNF-NGR peptide and it was found that targeted delivery of
low doses of NGR-TNF-α to tumor vasculature increased the efficacy of various drugs
acting via different mechanisms (Curnis et al., 2000), (Curnis et al., 2002).

11
1.2.5. Anti-Tumor Peptides:
A large number of peptides having anti-tumor activity are reported to date. These peptides
work through a variety of mechanisms to eliminate or restrict the tumor, such as by
angiogenesis inhibition, protein-protein interactions, signal transduction pathways, or gene
expression (V Rosca et al., 2011), (Karagiannis & Popel, 2008), (Kritzer, Stephens,
Guarracino, Reznik, & Schepartz, 2005), (Mochly-Rosen & Qvit, 2009). Peptide
antagonists also behave as antitumor peptides and preferentially bind to a known receptor
(Cornelio, Roesler, & Schwartsmann, 2007), (Sotomayor et al., 2010). “Pro-apoptotic”
peptides induce apoptosis in tumors (Ellerby et al., 1999), (Smolarczyk et al., 2005),
(Walensky et al., 2004). Angiogenesis is the process which involves growth, migration and
differentiation of endothelial cells of blood vessels. A number of studies are conducted to
discover peptide based angiogenesis inhibitors for the safest treatment of cancer (V Rosca
et al., 2011). Recently it is reported that angiotensin-(1–7) inhibit growth of bronchogenic
carcinoma in mice by restricting blood vessel development (Soto-Pantoja, Menon,
Gallagher, & Tallant, 2009). An RGD peptide derivative cilengitide is found selective for
αv integrins and behaves as an anti-angiogenic agent (Alghisi, Ponsonnet, & Rüegg, 2009),
(Hariharan et al., 2007), (Reardon et al., 2008), (K. Park et al., 2008). This derivative is
now in phase II clinical study for the therapy of glioblastoma and refractory brain tumors
in children.
1.2.6. Cell Penetrating Peptide:
Efficient passage through the cellular plasma membrane remains a major hurdle for some
drugs, particularly molecules that are large, ionized or highly bound to plasma protein
(Peck & Hill, 2014). Prochiantz et al. proposed the concept of cell penetrating peptide
(CPP) which can go across the cellular boundary and facilitates drug internalization
(Derossi, Joliot, Chassaing, & Prochiantz, 1994). These peptides deliver various high
molecular weight cargos across plasma membrane (Schwarze & Dowdy, 2000). Although
the general mechanism of CPP uptake is not clearly understood but there is consensus that
CPP interact with plasma membrane through electrostatic interaction with proteoglycans.
The mechanism of interaction and deliverance not only depends on structure of the CPP

12
but nature of cargo and its concentration also play a key role (El-Andaloussi, Holm, &
Langel, 2005).
Endosomal escape is being considered now a days as efficient pathway for CPP-cargo
conjugates internalization (Wadia, Stan, & Dowdy, 2004), (Magzoub, Pramanik, &
Gräslund, 2005). CPP can conjugate with cargo through two main strategies: through
covalent linkage with cargo or by forming non-covalent stable complexes. CPP conjugated
to nano carriers or cargos have been extensively studied, this method appeared very
advantageous in in vivo testing. This procedure is reproducible, rationalized and
stoichiometry of CPP-cargo can be controlled. (Torchilin, 2008), (Mäe & Langel, 2006),
(Gupta, Levchenko, & Torchilin, 2005). However, alteration in the chemical activity of the
cargo is limited as it can alter the biological response of cargo (Juliano, Alam, Dixit, &
Kang, 2008).
Short amphiphilic peptide carriers that carries both hydrophobic and hydrophilic
characteristics usually works through non-covalent strategy. Primary or secondary
structure of these short peptides gives them an amphiphilic character (Deshayes, Morris,
Divita, & Heitz, 2005). The hydrophobic domain forms complex with hydrophobic cargos
and used for membrane anchoring, while the hydrophilic domain forms complex with
hydrophilic negatively charged molecules. Mostly the amphiphilic peptide folds to α
helical structure and the hydrophilic and hydrophobic domains arranges on one side of α
helix. The non-covalent strategy was initially designed for gene delivery. Peptide of HA2
subunit of influenza hemaglutinin (Lear & DeGrado, 1987), (Parente, Nadasdi, Subbarao,
& Szoka Jr, 1990) and histidine-rich peptides (Kichler, Mason, & Bechinger, 2006),
(Midoux, Kichler, Boutin, Maurizot, & Monsigny, 1998), (Kichler, Leborgne, März,
Danos, & Bechinger, 2003) have been described as potent gene delivery systems
1.2.7. Anti-inflammatory Peptides:
Peptides always appear as a valuable solution for several disease treatments. Cyclic
peptides have an extra advantage of fixed conformation compared to linear peptides.
Several cyclic peptides are being used as drug for different diseases. Different classes of

13
cyclic peptides are reported to behave as potential anti-inflammatory agents. Some of them
are discussed below.
1.2.7.1. Duanbanhuains (A–C):
Brachystemma calycinum, is a plant used as folk medicine in China to treat diseases related
to inflammatory conditions. Its roots contain three anti-inflammatory cyclic peptides called
duanbanhuains (A–C). These peptides showed inhibition of MCP-1, IL-6, collagen IV and
reactive oxygen species (ROS) against high-glucose-stimulated cells and was non-toxic.
Duanbanhuain A and B were more potent against the production of IL-6, collagen IV and
ROS at the concentration of 10 µM. while, duanbanhuain A showed inhibition against
collagen IV 1 µM (Cheng, Zhou, Yan, Chen, & Hou, 2011).
1.2.7.2. Cyclomarins:
Cyclomarins (A-C) were isolated from cultured marine actinomycetes. Among the three,
cyclomarin A, was found as major metabolite and found as potent anti-inflammatory
compound in the phorbol ester (PMA)-induced mouse ear edema assay. Structurally, this
compound is attractive due to the presence of four unique amino acids which are 5-
hydroxyleucin, β-methoxyphenylalanine, tert-prenylated β-hydroxytryptophan and 2-
amino-3,5-dimethyl-4-hexenoic acid. This compound is also effective against
Mycobacterium tuberculosis and Plasmodium falciparum (Renner et al., 1999), (Schmitt et
al., 2011).

14
1.2.7.3. Cyclosquamosin:
Cyclosquamosin is the family of cyclic peptide obtained from the seeds of Annona
squamosa. After the isolation of first peptide annosquamosin A, a number of cyclic peptide
such as cyclosquamosin A and B, cyclosquamosin H and I, squamin A and B,
cyclosquamosin D and E, and cherimolacyclopeptide B have been isolated from this plant.
Cyclosquamosin D appears as a major cyclic peptide constituent among all the cyclic
peptides isolated from the plant. It behaves as inhibitor of TNFα and IL-6 production in a
dose dependent manner (Y.-l. Yang et al., 2007). The first total synthesis of
cyclosquamosin D and parallel synthesis of its analogues was reported by Dellai et. al. It
was found that its analogues effectively suppressed IL-6 secretion but had no effect on
TNFα (Dellai et al., 2010).

15
1.2.7.4. Anti-inflammatory Peptides from Marine Sponges:
Marine organisms are the most important source of anti-inflammatory cyclic peptide.
Many peptides have been isolated from different marine sponges like, perthamide C and
perthamide D of the genus Theonella swinhoei (Festa et al., 2009). Solomonamides A and
B are effective anti-inflammatory cyclic peptides isolated from the same genus.
Stylissa is another genus of the marine sponges that is a source of various bioactive cyclic
peptides. To-date, there have been 13 cyclic peptides reported from sponges in this genus.
These include: stylissamides A - H, isolated from S. caribica (X. Wang, Morinaka, &
Molinski, 2014), (Schmidt, Grube, & Köck, 2007), (Cychon & Ko ck, 2010), stylissamide
X from Stylissasp (Arai, Yamano, Fujita, Setiawan, & Kobayashi, 2012), stylisins 1 and 2
from S. caribica (Mohammed, Peng, Kelly, & Hamann, 2006), phakellistatin 13 from S.

16
caribica (Mohammed et al., 2006) and stylissatin A from S. massa (Kita, Gise, Kawamura,
& Kigoshi, 2013). Interestingly, all of the peptides isolated are heptapeptide except
stylissamide X which is an octapeptide.
Stylissatin A, a heptapeptide was isolated from the Papua New Guinean marine sponge
Stylissa massa. Stylissatin A exhibited inhibitory effect on nitric oxide production in LPS-
stimulated murine macrophage RAW264.7 cells with an IC50 value of 87 µM (Kita et al.,
2013). Total synthesis of this peptide was also reported earlier by the combination of
solution and solid-phase synthesis (Akindele, Gise, Sunaba, Kita, & Kigoshi, 2015). Its
first total solid-phase synthesis was also reported by our research group (Shaheen et al.,
2016).
1.3. Cyclic Peptides:
Cyclic peptides represent a large privileged and yet under exploited category of molecules
for drug discovery. These macrocycles forms a chemical space where the biological
expertise collaborates with chemical approaches in search of leads against different targets.
Cyclic peptides are well known for broad spectrum of biological activities. Structurally,
polypeptides chain adopts cyclic form by connecting one terminal of the peptide with
another through amide bond or any other chemically stable bond such as lactone, thioether,
ether, and disulfide and so on. Many biologically active cyclic peptides are formed by
head-to-tail cyclization. The group of cyclic peptide is continuously growing and
thousands of cyclic peptide have been isolated or synthesized and many of them are used
medicinally. For example tyrocidine, gramicidin, vancomycin, polymixin and colistin are
used as antibiotics, cyclosporin A is an immunosuppressive agent, and octreotide is used in
radio therapy, calcitonin in hypercalcemia and osteoporosis, nisin as food preservatives.
Peptide cyclization has many advantages; peptide molecule attains fixed geometry by
cyclization which helps them to bind with their targets more efficiently. Even if the
receptors have several subtypes; it is believed that definite conformation of these peptides
will be selective to particular receptor subtypes (Roxin & Zheng, 2012). Cyclic peptides
can adopt limited molecular conformation in solution this is the reason; when these
molecules are used as ligands to target disease biomarkers, their limited conformations

17
allow effective binding to the different isoforms or subtypes of specific receptors (Deem &
Bader, 1996).
Cyclic peptides attain receptor selectivity and enhanced binding by losing entropy when
they interact with the target which results in the reduction of free energy of target-ligand
complex. Cyclic peptides lack both free carboxyl and amino terminal which makes them
less susceptible to hydrolysis by exo as well as endo-peptidases (Liskamp, Rijkers, &
Bakker, 2008).
The main goal of cyclization is to induce structural constrain in the peptide chain but the
site of cyclization greatly affect the binding affinity. Kumar et al. investigated a linear
sequence Ac-CIYKYY and reported the comparison of 20 linear peptides in which side
chain of amino acid has been modified and 11 cyclic analogues of original sequence (A.
Kumar et al., 2006).
The cyclization was performed by using different strategies such as head to tail cyclization,
head to side chain cyclization and side chain to side chain cyclization. The results showed
that the head to tail cyclized peptide was 62.5 times more effective than the parent linear
peptide. This suggests that mode of cyclization and the orientation of the cyclized amide
bond greatly alter the biological response.
Cyclic peptides have structural features that allow the easy passage of molecule across the
plasma membrane. For example, presence of intramolecular hydrogen bonding keeps the
hydrophobic groups on the surface of the molecule and facilitates its transport across the
membrane (Mandal, Nasrolahi Shirazi, & Parang, 2011).
1.3.1. Stability of Cyclic Peptide:
In solution, small linear peptides exist in fast equilibrium of easily interconvertible
conformations. From these conformations very few adopts the orientation that matches the
receptors active site while some conformers also fit in the active site of the proteolytic
enzymes that cause their degradation. Cyclization was performed to minimize the number
of possible conformations that enhance the receptors selectivity and avoid the
susceptibility of degradation by proteolytic enzymes (Gilon, Halle, Chorev, Selincer, &

18
Byk, 1991). Generally cyclization increases stability of peptides also that directly prolong
their biological activity by resisting the enzymatic degradation (Liskamp et al., 2008).
Bogdanowich-Knipp et. al. studied the cyclo-(1,6)-Ac-CRGDF-Pen-NH2 (Pen = 8,8-
dimethylcysteine) peptide and its linear analogue NH2-RGDF-OH and found that the cyclic
form is 30 time more stable than linear in the solution of pH from 3 to 7. The disulfide
bond is more prone to reduction at high pH that ultimately facilitates the degradation of
cyclic RGD (Gilon et al., 1991). This result suggests that the disulfide bond has key role in
the stabilization of peptide.
Besser et al. studied the comparison between cyclization through disulfide bond and
cyclization through amide bond. This study was conducted on SRIF derivatives
Somatostatin-14, an endogenous disulfide-cyclized peptide with the sequence AG
(CKNFFWKTFTSC) and nine cyclized derivatives of somatostatin receptor-binding
octapeptide with the sequence cyclo (fFYwKV)FT-NH2 in which cyclization was done
through D-phenylalanine and valine residues but the orientation of cyclized bond is
different and extended linkers were also used (L. Yang et al., 1998). It is found that the
amide cyclized peptide was stable for 15 hours while the disulfide-cyclized peptide
hydrolyzed within 1 hour. This study showed that receptor binding affinity might improve
by disulfide cyclization but due to enzymatic reduction its use is limited. Both targets
stability and selectivity might achieve by amide bond cyclization (Besser et al., 2000).
1.3.2. Strategies to Develop Peptide Libraries.
Peptides are considered as attractive drug lead molecules due to several reasons, that is
why scientists are making continuous efforts to develop medicinally important compound
based on peptides structure. Peptides can be synthesized by two methods, Genetic or
Synthetic. In genetic method, peptide generation is limited to 20 ribosomal amino acids but
the sequence determination is straight forward. Synthetic method is more versatile, many
natural and unnatural amino acids can be incorporated into the cyclic peptide chain. Some
of the common methods that are used for peptide generation are described below.

19
1.3.2.1. Combinatorial Library Approach:
Combinatorial library synthesis is the simple, economical and versatile method for the
synthesis of peptide libraries. These libraries can be synthesized through biological or
chemical synthesis approaches. Biological method is generally limited to naturally
occurring amino acids while in synthetic approach; versatility is achieved by incorporating
D-amino acids, unnatural amino acids, specific secondary structure or scaffold that can
enhance the biological activity as well as small organic molecules and biological building
blocks such as monosaccharides. These combinatorial peptide libraries can be screened
against a variety of biological targets for the rapid discovery of ligands and substrate and
inhibitors (Pandeya & Thakkar, 2005). Since its discovery the combinatorial chemistry has
become a powerful tool for the drug discovery and molecular recognition. There are six
general procedures for the synthesis and screening of combinatorial libraries:
(i) The biological peptide library method (phage-display peptide library, bacterial peptide
display library system (Salmon et al., 1996), FliTrx, polysome library, (Jayawickreme et
al., 1999), (Silen et al., 1998) or plasmid peptide library (Aina et al., 2007).
(ii) The spatially addressable parallel library method (Frank, 1992).
(iii) Combinatorial library methods requiring deconvolution (Houghten et al., 1991),
(Brown, Wagner, & Geysen, 1995)
(iv) Affinity selection method (Songyang et al., 1995)
(v) One-bead-one-Compound (OBOC) combinatorial library method (Lam et al., 1991),
(Lam, Lebl, & Krchnák, 1997)
(vi) Self-assembled peptide nucleic acid (PNA) encoded chemical microarrays (Winssinger
et al., 2004).
1.3.2.2. Phage-Display Peptide Library Method:
Smith et al introduced phage display in 1985. In this method, each phage particle displays
unique peptide on its surface and hit molecules was decided on the basis of binding with
the target. Usually, peptides are displayed on the N-terminus, middle, or C-terminus of coat
proteins, and DNA sequence of the phage directs the peptide sequence this allows the easy
determination of sequence. Repeated screening called bio-pinning can be done until the

20
DNA molecules are preserved. Cyclic peptides having even number of cysteine units can
be synthesized (Koivunen et al., 1993). The phage particles are exposed to oxygen rich
periplasmic space of bacteria, which forced the two Cys units to form disulfide bond. 1 ×
108 to 1 × 109 different phages are routinely generated through this method. The first main
drawback is, only 20 natural amino acids can be incorporated in the library; secondly
complex bicyclic, branched structures, or molecules with special chemistry of cyclization
cannot be synthesized; and only binding study and some functional assays can be achieved.
1.3.2.3. Parallel Library Method:
Parallel peptide libraries are generated through solid- phase peptide synthesis by using
manual or automated high-throughput synthetic methods. The amino acid sequence of each
of these compounds are known (STRYER, t AMY, & SOLAS, 1991). This method forms
the basis of many other techniques such as SPOT synthesis, (Frank, 1992) multipin
technology, NanoKan, peptide microarray, multi-syringe system, and the 96 dee p-well
plate system. Screening of parallel peptide library can be done by direct solid-phase
binding assay or by a solution-phase releasable assay. The main disadvantage of this
method is that limited number of peptides can synthesized and therefore the library size is
rather small (Lam, 1997), (R. Liu & Lam, 2008).
1.3.2.4. Combinatorial Library Methods Requiring Deconvolution:
This strategy is based on the synthesis of mixture of compounds and their biological
screening. This synthetic approach is the basis of many other methods such as the iterative
method (Houghten et al., 1991), (Geysen, Rodda, & Mason, 1986) positional scanning
method (Dooley & Houghten, 1993), orthogonal partition method (Deprez et al., 1995),
recursive deconvolution method (Erb, Janda, & Brenner, 1994)and the dual recursive
deconvolution method. A large peptide library (1 × 106 to 1 ×108) can be generated and
screening can be done through many existing biological assays including functional assays.
This method is very suitable if target protein has only one predominant motif. Targets with
multiple binding motifs likely lead to scramble and un-interpretable results.

21
1.3.2.5. Affinity Selection Method:
This method is based on the affinity of ligand with receptors. Peptides from solution
mixture library are passed through affinity column with immobilized receptors that
separates ligands from library. This mixture library is usually synthesized by split-pool
method (Furka, SEBESTYÉN, ASGEDOM, & DIBÓ, 1991), (Lam et al., 1991),
(Houghten et al., 1991) The bounded ligands elute by extensive washing of column and
their structure is elucidated. This method has been successfully used for the synthesis of
combinatorial libraries of oligo deoxynucleotide. Libraries greater than 10000 peptides are
difficult to generate. Main concerns about this approach are nonspecific binding to
receptors and un-interpretable results are obtained if more than one predominant motif is
present in the mixture.
1.3.2.6. Self-Assembled PNA-Encoded Chemical Microarrays:
In this technique, split-pool method is used to generate the peptide library. This library is
cleaved from resin and an encoded solution phase library is formed in which each
compound is the PNA-encoded peptide or small molecule, the library compound is linked
to a PNA code via a hydrophilic linker (Winssinger et al., 2004). The library is then mixed
with the target protein and later exposed to a planar oligonucleotide microarray of
predetermined sequences. The identity of the positive library compound that interacts with
the target protein can be determined by knowing the nucleic acid sequences of the
oligonucleotide microarrays. Like the OBOC method, a whole cell binding assay can also
be applied to the encoded planar chemical microarrays.
1.3.2.7. OBOC Combinatorial Library Method:
One-bead-one-compound (OBOC) library is synthesized by using split pool method
through solid-phase-peptide synthesis. In this library, every single 80–100 μm bead
contains approximately 100 pmol of single chemical compound(Lam et al., 1991), (Lam et
al., 1997), this OBOC library is then subjected to high throughput screening. On bead
binding assays as well as releasable solution phase assays can be used(Lam, Liu,

22
Miyamoto, Lehman, & Tuscano, 2003).On-bead binding assays includes standard enzyme-
linked colorimetric assays, fluorescent assays, or radionuclide assays. Color probe can also
be used for the screening. The positive beads are identified by color change. OBOC
method has given very good outcome in whole-cell binding assay and successfully
employed in the identification of cell surface binding ligands. The procedure includes the
incubation of library beads with living cells; then the beads with monolayer of cells are
considered as positive beads, these are picked and characterized. OBOC library method has
also been useful in the identification of protease substrate and inhibitors (Meldal,
Svendsen, Breddam, & Auzanneau, 1994), (Meldal, 2002), (Olivos, Bachhawat‐Sikder, &
Kodadek, 2003), (Juskowiak, Stachel, Tivitmahaisoon, & Van Vranken, 2004). For this
assay highly porous resin were used for the peptide library synthesis so that enzyme can
access to the bead interior. OBOC library can also be screened by solution-phase assay. In
situ releasable approach is used in which library beads are immobilized on a thin layer of
agar the compound from the beads is released in the vicinity of bead in the semi-solid
matrix (Salmon et al., 1996), (Jayawickreme et al., 1999), (Silen et al., 1998). The OBOC
method is advantageous because a large number (1 × 106 to 1 × 108) of compounds can be
rapidly synthesized and screened. Secondly it does not require deconvolution and multiple
peptide ligands with completely different motifs can often be identified in a single screen.
Usually the library compound is linked to the solid support via linker that may cause steric
hindrance between library compound and cellular receptor, but mostly linkers play a role
of convenient handle that links the ligand to the therapeutic payload.
1.3.3. Cyclization Strategies for Synthetic Cyclic Peptides:
Cyclic peptides are a very valuable class in the modern drug development and a large
number of bioactive cyclic peptides are reported but there are some major challenges re-
main for the efficient synthesis of cyclic peptides. The peptide can be cyclized through
lactamization (Parenty, Moreau, & Campagne, 2006), lactonization (Lundquist IV &
Pelletier, 2002) or disulfide bridge formation (X.-Y. Wang, Wang, Huang, Wang, & Yu,
2006). Synthesis of some cyclic peptides are difficult and the success of the synthetic
approach often largely depends on the amino acid constituents of the cyclic peptide, the

23
specific site of ring-closure, the ring-closing strategy and the desired ring-size (Roxin &
Zheng, 2012). These issues have stimulated the development of novel efficient methods for
peptide cyclization. Various strategies have been reported for peptide cyclization,
depending on its functional groups, on the application of the cyclic peptides and/ or on the
desired technique for conjugation to other molecules, there are four different methods for
peptide cyclization: head-to-tail (C-terminus to N-terminus), head-to-side chain, side
chain-to-tail or side chain-to-side chain. Cyclization can take place via classical amide-
bond formation reactions (lactamization), disulfide-formation, or via the use of orthogonal
ligation methods (Horton, Bourne, & Smythe, 2000)
Figure-4: Schematic representation of the four possible ways for peptide macrocyclization.
Peptide macrocyclization reactions can be carried out in solution or on the solid support
and especially the field of solid-supported macrocyclizations has been actively explored in
recent years. The macrocyclizations on solid-supported is advantageous because the
standard washing and filtration procedures used in solid phase peptide synthesis are often
enough for purification. Macrocyclizations in solution are usually best performed in very
dilute conditions to minimize unwanted intermolecular reactions. Although these
conditions increase the selectivity of the reaction, they generally slow down the reaction
speed, thereby increasing reaction times and the risk on side product-formation. In
addition, macrocyclization in solution is usually performed on partially protected peptides,
and the solubility of these peptides is unpredictable and often poor (White & Yudin, 2011).

24
The interactions between protein binding sites often do not involve backbone-interactions
between large peptide- or protein-fragments, but mostly rely on certain contact residues
that are present in the epitopes. When cyclic peptides are used for mimicry of these
epitopes, proper positioning of the main contact residues is of great importance, and
peptide conformation is a crucial feature with respect to its biological activity. Therefore,
the site and method for macrocyclization must be carefully selected, since these factors can
strongly influence the peptide conformation
In addition, alternative approaches have been developed that make use of orthogonal
ligation methods. An important advantage of the use of orthogonal ligation methods over
amide-bond formation for macrocyclization reactions is that cyclization can be performed
on side chain-unprotected peptides to improve the solubility of peptides (Kimmerlin &
Seebach, 2005).
The demand for a detailed understanding of complex biological processes has stimulated
the development of highly advanced chemical tools, in order to improve the efficiency of
peptide cyclization. Ideally, peptide cyclization should be sequence independent, the
cyclization strategy should be applicable on both short and longer peptides and the strategy
should only require easily accessible functionalities or chemical moieties. A variety of
chemo-selective ligation methods have been described for this purpose.
Kent and co-workers first reported the chemo-selective ligation methods, like the thiol-
mediated intermolecular native chemical ligation (NCL) of peptide segments have been
heavily exploited for both existing and novel purposes (Shao, Lu, & Kent, 1998). The
feasibility of this method for peptide cyclization via intramolecular trans thio esterification
and ring contraction was first demonstrated by Tam and Zhang. This method for the
synthesis of head-to-tail cyclized peptides has also been extended to a solid-phase based
approach. An obvious limitation of NCL is the necessity of a cysteine residue at the site of
cyclisation. In answer to that, several research efforts have focused on circumventing this
requirement, resulting in a number of methods that has been reported for peptide
cyclization mediated through removable sulfur-containing auxiliaries (L. Zhang & Tam,
1997).

25
Another powerful method for the introduction of ring-shaped constraints is ring-closing
metathesis (RCM). The chemistry of RCM, relying on the action of ruthenium-based
catalysts, has been applied for the first time to rigidify amino acids and peptides by Grubbs
and co-workers (Miller, Blackwell, & Grubbs, 1996).
The copper catalyzed azide-alkyne cycloaddition as developed by Sharpless has effectively
been translated to the macrocyclization of peptides. In addition to the high efficiency and
regioselectivity of this reaction, this triazole-forming reaction appeared to be of special
value in the synthesis of the small cyclic peptides that are usually difficult to synthesize.
The conformational restrictions imposed by the resulting triazole-ring can positively
influence the formation of these small macrocycles (Rostovtsev, Green, Fokin, &
Sharpless, 2002).
Whereas above-mentioned methods for the synthesis of cyclic peptides make use of main
chain or (modified) side chain functionalities of linear peptide precursors or involve
removable auxiliaries, peptide cyclization can also be achieved using scaffold molecules.
A successful example of the use of a synthetic scaffold for conformational fixation of
peptides is CLIPS-technology (Chemical Linkage of Peptides onto Scaffolds).A first
example of this approach involves the immobilization of a dicysteine-containing linear
peptide to an α,α’-dibromoxylene-scaffold, resulting in a scaffolded cyclic peptide. After
the introduction of CLIPS-technology for the generation of single peptide loops, the
technology has been extended for the synthesis of multicyclic peptides towards the
generation of mimics of more complex protein binding sites(Horton et al., 2000).
1.3.4. Analysis of Cyclic Peptide through Mass Spectrometry:
Mass spectrometry has gained signification attention and much development has been done
since last three decades. Mass spectrometry is also considered as an effective tool for the
identification and characterization of biological macromolecules. With the use of different
ionization methods such as field desorption (FD) (Beckey, 2016), plasma desorption (PD)
(Tsarbopoulos et al., 1994) and fast atomic bombardment (FAB) mass spectrometers can
detect ~40kDa. Recently matrix assisted laser desorption ionization technique (MALDI)
has emerged as a promising tool for the fast and accurate determination of number of

26
biomolecules (Yergey et al., 2002). This technique has been employed for the detection
and characterization of several biopolymers such as protein, polysaccharides, peptides and
many organic macromolecules like dendrimers and other synthetic polymers.
Peptide sequencing through mass spectrometry is not that much easy task as it may seems,
cyclic peptides are even more difficult. The reason is during CID multiple ring opening
reactions occurs that make the interpretation difficult. Some amino acids direct the
pathway of fragmentation and generate specific fragmentation patterns that make the
identification easy. For example, proline generates a specific fragmentation pattern that
occurs at the peptidyl-prolyl (Xaa-Pro) amide bond, leading to a linear peptide C- ended by
an acylium ion (bn), which upon further fragmentation gives a series of acylium ions which
leads to the structure determination.
Peptides can be fragmented through the dissociation of three types of bond; CO-NH,CO-
CH and CH-NH, as a result of each dissociation one neutral and one charged specie is
formed but only positively charged species are detected (Figure-5). For each amino acid
six fragment ions are possible that can be divided into two categories. If the N- terminus
retained the charged the ions formed will be labeled as a, b and c ions and if the charge
retain on the C-terminus the x, y and z ions will be formed. The CO-NH bond is the most
labile bond in the peptide chain and gives rise to “b” ions with the formation of ammonium
ion and “y” ion with the formation of acylium ion. These ions give characteristic peaks and
their difference indicates the loss of particular amino acid residue from the peptide chain.
The peptide identification can be made simpler by MS/MS scan. Hence the mass
spectrometry is a valuable tool for sequencing all synthetic and natural peptides including
both cyclic and linear peptide.

27
Figure-5: Peptide Fragmentation Notation Roepstorrff and Fohlman (1984)
1.3.5. Conformational Analysis of Cyclic Peptide:
The structural features of proteins have been extensively studied for the better
understanding of their functions. Conformation of proteins is important structural feature
that play a key role in their binding to different receptors. For the conformational studies of
proteins cyclic peptides are often used as model (Takahashi, Hayano, & Suzuki, 1989).
Cyclic peptides can be both rigid and flexible and can adopt many stable conformations.
Different physical and chemical factors such as polarity of solvent, intermolecular forces
and the nature of the side chain of amino acids can effect these conformations. Extensive
studies have been done on the small and large cyclic peptide to determine their
conformational preferences.
Proline has a significant position in peptide conformation due to its cyclic structure. The
free energy barrier between cis/trans conformers is very small that cause rapid inter-
conversion of these isomers. These changes affect the whole protein or peptide
conformations that ultimately alter their function. The real mechanism of this cis/trans
isomerism is still under investigation, several theoretical studies have been conducted to
understand the phenomenon. Recently a study has been conducted that uses the empirical
energy function and ab-initio molecular orbital calculations. Molecular dynamics
simulation techniques have also been used to demonstrate proline isomerism.

28
Figure-6: Peptidyl-Prolyl conformation of Cis/Trans Isomers.
1.4. Importance of Cationic Cyclic Peptide:
Structurally diverse libraries of cyclic peptides are important sources of new medicinal
agents. Therefore, the development of diverse forms of new and novel compounds is an
important area of research in medicinal chemistry. Cationic Peptides (CPs) (also called as
amphiphilic peptides or antimicrobial peptides) have been discovered in nearly all species
from micro-organism to human being (Zasloff, 2002), (Lehrer, 2004), (Giuliani, Pirri, &
Nicoletto, 2007). Despite of having extensive variety in their sequences and structures,
these peptides share some common characteristics. The chain length of naturally occurring
CPs generally includes 12-50 amino acids having a net positive charge, as basic residues
especially lysine and arginine and sometime histidine are in excess as compared to acidic
residues and having around 50% hydrophobicity (Zasloff, 2002), (Hancock, 2001).
Most of naturally occurring cationic peptides contain two main types of cationic amino
acids, lysine and arginine, while most synthetic cationic peptides are lysine (K) rich
peptides. Very few synthetic arginine rich (R) peptides are reported except the RW-rich
peptides (Papo, Shahar, Eisenbach, & Shai, 2003), (Li et al., 2011).
Morris et al reported that, Pep-1; a Lys rich peptide consists of three domains: a
tryptophan-rich hydrophobic region, a flexible linker and a lysine-rich domain. In addition,
the ends of the peptide are capped by acetyl and cysteamide groups (Morris, Deshayes,
Heitz, & Divita, 2008). Pep-1 is considered as cell penetrating peptide and believed to
work by first forming a complex with its cargo through electrostatic and/or hydrophobic

29
interactions and delivers it directly into the cytoplasm, even at 4 °C, where energy-
dependent endocytosis. However, endocytosis is the predominant transport mode at 37 °C.
Pep-1 induces pore-like defects in bilayers that are clearly visible in ionic current
measurements (Lein, Sgolastra, Tew, & Holden, 2015). Anjaneya.et al reported lysine rich
cyclotides isolated from Australasian violaceae, Malicytus chathamicus and M. latifolius.
Cyclotides are plant-derived disulfide-rich mini-proteins ranging from 28 to 37 amino
acids in size (Ravipati et al., 2015), (Craik, Daly, Bond, & Waine, 1999). Structurally these
are consists of head-to-tail cyclic backbone as part of their characteristic cyclic cystine
knot (CCK) that is formed by three disulfide bonds. This unique structural motif provokes
cyclotides with exceptional resistance to thermal, chemical, or enzymatic degradation
(Craik, Daly, & Waine, 2001), (Colgrave & Craik, 2004).These lysine rich cyclotides are
able to bind with the lipid bilayer, but showed very low binding affinity to neutral 1-
palmitoyl-2-oleoylphosphatidylcholine (POPC) and 1-palmitoyl-2-oleoylphosphatidyl
glycerol (POPG) bilayer compared to the negatively charged POPC/POPE/POPG
membrane. 1-palmitoyl-2-oleoylphosphatidylethanolamine (POPE) improves the affinity
of lysine-rich cyclotides to the membrane. Binding of cationic peptides to lipid bilayer is
greatly affected by overall hydrophobicity, net charge, and location of charges (Henriques
et al., 2012), (Ravipati et al., 2015). The current study was focused on the synthesis of
diverse forms of cyclic peptide libraries containing various cationic, anionic residues and
unusual building blocks and structural motif of naturally occurring bioactive peptides to
identify new lead molecules against cancer and inflammation.
1.5. Cancer Targeting Strategies:
Cancer is not a disease it is rather a group of diseases characterized by the uncontrolled
growth and proliferation of abnormal cells from the site of origin to the distant organs that
may ultimately lead to death. Several factors are involved in the progression of cancer,
such as exposure to UV radiation, pollution smoking habits, Viruses, and an unhealthy
diet. Moreover many chronic infections like hepatitis B and C (liver cancer), human
papilloma (cervical and ano-genital cancers) and Helicobacter pylori (stomach cancer) also
convert into cancerous situation. It may also be caused by genetic mutations, hormones,

30
and immunological disorder etc. One or all of these factors may involve in the progression
of disease. Several therapies including surgery, hormone therapy, radiation, immune
therapy, chemotherapy and targeted therapy (administration of the drugs that specifically
inhibit cancer cell growth) are being used. Cancer that is caused by unhealthy life style,
overweight or obesity, physical inactivity, poor nutrition, by smoking and/or excess
alcohol consumption could be completely obstructed by abandon smoking or alcohol
intake (Petti, 2009).
Cancer can be detected earlier befor the appearance of symptoms which usually results in
less extensive treatment and better outcomes (Anand et al.). One of the common and major
approaches for the cancer is chemotherapy. This treatment is done by delivering cytotoxic
drugs to the site of cancer. This is mostly administered to the intermediate and late stages
cancers patients and can be provided with or without radiation and before or after surgical
resection. The drugs that are administered during chemotherapy are not targeted and
selective towards cancer cells. These drugs also accumulate in normal tissues and distrub
their function. For example, some unwanted effects such as hypersensitivity reactions,
myelo-suppression, and neurotoxicity has been reported against paclitaxel (PTX) a drug
which is used against lung, ovarian, and breast cancers (S. Kumar et al., 2010). Another
drug doxorubicin (DOX) that is being used in chemotherapy has also been described to
have cardio toxic side effects (Prados et al., 2012). Low aqueous solubility is another issue
associated with most of chemotherapeutic drugs that prevents intravenous administration.
Those drugs are then chemically modified as pro-drugs (e.g. Irinotecan) or formulated in a
surfactant containing solution. These strategies enhance the aqueous solubility but the
bioavailability is compromised and also leads to sensitization reaction and other secondary
side effects (Kakde, Jain, Shrivastava, Kakde, & Patil, 2011), (Gelderblom, Verweij,
Nooter, & Sparreboom, 2001).

31
The limitation of intravenous systemic chemotherapy is, it is not specifically targeted to
the tumor, not only it is difficult to achieve therapeutic levels of drug within or adjacent to
the tumor, significant concentrations of drug also accumulate in normal tissue, that cause
dose-limiting toxicity and severe side effects. Moreover, acquired chemo resistance is also
a limitation of chemotherapy (Thayer, 2011).
The treatment of cancer is expensive because either radiation therapy or chemotherapy or
both, has high material costs and require specialist personnel for delivery of therapy,
appropriate monitoring, and treatment of side effects. These treatments are time consuming
for both patients and personnel, requiring frequent visits over months of therapy.
To cut the long story short, drug resistance, bio-transformation, altered bio-distribution,
drug clearance and non-specific drug delivery collectively urge the search of new localized
anti-tumor drug lead or ligand molecule to improve the efficacy of the therapy.
The main goal of cancer therapy is to abolish cancer cells, while sparing normal tissues.
This can achieve only by the selective targeting of cancer cells at the site of malignancy.

32
Certain tumor can be distinguished by their specific gene expression patterns as numerous
genes are over expressed in tumors comparative to normal tissue and expressions of
membrane proteins produced specifically by tumor cells (Alizadeh, Ross, Perou, & Van De
Rijn, 2001), (Nielsen & Marks, 2000). Due to this over expression these proteins or genes
appeared as extremely useful markers for targeting cancer (Shadidi & Sioud, 2003).
Previously, number of active anti-tumor agents and methods has been employed for the
targeted drug delivery. Macromolecules such as protein ligands and monoclonal antibodies
(mAbs) have also been tested clinically as therapeutics for targeted delivery (Allen, 2002),
(Pastan et al., 2006), (Thorpe, 2004), but there is still two major limitations: due to their
large size these are not specifically delivered to tumors and passive diffusion across
endothelial cell membranes in capillaries is also difficult; and dose-limiting toxicity to the
liver and bone marrow due to nonspecific uptake by the liver and the reticuloendothelial
system (RES) (Aina et al., 2002), (Mori, 2004), (Reff, Hariharan, & Braslawsky, 2002).
Another approach is the polymer-based drug delivery depots. This method has been
studied to improve the systemic morbidities and unspecified tumor targeted treatments.
These exist in various forms to facilitate the drug delivery to the effected site for the long
period and in controlled manner. According to their mechanism of action and mode of
administration polymer based approach can be classify into two groups;
1) Systemic administration, 2) Controlled release drug delivery system.
Systemic administration includes polymer liposomes, nanoparticles and dendrimers.
Localization of these nano-meterial is difficult due to uptake of these by the
reticuloendothelial system. The controlled release drug delivery system includes various
form- factors such as drug-eluting films, rods, wafers, gels and particles that usually
implanted intra-tumorally or adjacent to the effected tissues. The polymer used in this
technique can be natural or synthetic. Natural polymer including polysaccharides (X. Liu,
Heng, Li, & Chan, 2006), (Abe et al., 2008), (Bouhadir, Alsberg, & Mooney, 2001),
dextran (Al-Ghananeem et al., 2009) and chitosan (Gerber et al., 2011), (Li et al., 2011),
(Ampollini et al., 2010), hyaluronic acid (Vassileva, Grant, De Souza, Allen, & Piquette-
Miller, 2007), polypeptides including albumin (Davidson et al., 1995), (Almond et al.,

33
2003), collagen (Kakinoki & Taguchi, 2007),elastin (McDaniel, Callahan, & Chilkoti,
2010),and gelatin (Konishi et al., 2003), (Stuart, Stokes, Jenkins, Trey, & Clouse, 1993).
1.6. Inflammation:
A vigilant defence system against attack and injury is vital for the survival of living
organisms. The innate immune system continuously surveys the body for the presence of
invaders. When tissue damage by an invasion of pathogenic organism is encountered, it
voluntarily sets in motion a discrete, localized inflammatory response to thwart most
pathogenic threat (K. Tracey, 2002). Inflammation is a normal host defence mechanism
that is initiated by a complex series of interactions involving several chemical mediators.
This defence response not only protects the host from infection and other insults but also
restores hemostasis at damaged or infected areas (Barrientos-Salcedo, Rico-Rosillo,
Giménez-Scherer, & Soriano-Correa, 2009). The magnitude of inflammatory response is
critical: insufficient responses are caused by immunodeficiency that leads to serious
infections; excessive responses also causes diseases such as Crohn’s disease, rheumatoid
arthritis, diabetes and many others, but generally it is well regulated, self-limiting and
resolves rapidly. This self-regulation involves the activation of negative feedback
mechanisms such as the secretion of anti-inflammatory cytokines, inhibition of pro-
inflammatory signaling cascades, shedding of receptors for inflammatory mediators, and
activation of regulatory cells (K. J. Tracey, 2002), (Calder, 2009).
Inflammation is classified into two categories: (1) acute and (2) chronic ones (Lawrence,
Willoughby, & Gilroy, 2002), (Ahmed, 2011). Acute inflammation is a body’s short-term
responses that result mostly in healing and repairing damaged tissues and cells. Chronic
inflammation, on the other hand, is a prolonged inflammatory response that can cause
damage to the tissues. The prolonged and persistent inflammation is uncontrolled and it is
associated with many chronic human diseases such as cancer, multiple sclerosis, arthritis,
and allergy. An uncontrolled inflammatory response can be harmful to the host either in an
acute setting, such as during an infection, or in a chronic context, such as arthritis
(Mantovani, Allavena, Sica, & Balkwill, 2008).

34
Inflammation includes a series of complex reactions that involves cytokines, enzymes,
growth factors, and nitric oxide (NO) produced by macrophage cells. The onset of
infections and inflammation leads to an increased level of enzymes and signal proteins in
the affected area. This biological response, triggered and promoted by cytokines, is a
protective mechanism where the injurious stimuli, pathogens, irritants, and infections will
be removed via phagocytosis to protect the cells or tissues and initiate the healing process
(Huang et al., 2013). During inflammation, cell signaling proteins known as cytokines play
an important role by controlling the host’s responses to infections and injuries as
inflammatory or anti-inflammatory reactions. Some cytokines promote or make infections
and diseases to escalate and are referred as pro-inflammatory cytokines. Other cytokines
control and reduce the level of inflammation and promote healing processes, and are
known as anti-inflammatory cytokines (Dinarello, 2000), (Opal & DePalo, 2000).
Almost all gram-negative bacteria have lipopolysaccharide (LPS; endotoxin) as the major
outer surface membrane component which acts as an extremely strong stimulator of
mononuclear phagocytes (monocytes and macrophages), which are part of the innate
immune system of various eukaryotic species ranging from insects to humans (P. Wang et
al., 2010). The activation mechanism of macrophages by LPS starts when LPS binds with
the LPS-binding protein (LBP), accelerating the binding of CD14, which is the primary
receptor for LPS and is expressed mainly on macrophages (Rosenfeld, Papo, & Shai,
2006).The LPS–LBP–CD14 complex initiates intracellular signaling by interacting with
the trans-membrane protein Toll-like receptor-4 (TLR-4), and triggers, activation of
nuclear factor kappa B (NF-κB). The interactions of the receptors results in transmitting
signals to nucleus where the activation of a selective set of genes takes place through
transcriptional and posttranscriptional mechanisms. For example, the activated NF-κB will
be detached from the inhibitory protein kappa B (IκB) and released into the cell nucleus
and binds onto the deoxyribonucleic acid (DNA). The attachment of NF-κB to the DNA
will induce enzymes such as inducible nitric oxide synthase (iNOS) andcyclooxygenase 2
(COX-2) to synthesize prostaglandin, thromboxane and nitric oxide, respectively as an
inflammatory response (S. Y. Park et al., 2011), (Tabas & Glass, 2013). During the onset
of inflammatory responses, cells also release pro-inflammatory cytokines to help the

35
process which sometimes results in chronic condition causing cell death and tissue
damages. To control this condition, the immune system will try to balance the
inflammation process by releasing anti-inflammatory cytokines that regulate and lower
inflammation level where the infections are cured and cells are repaired (Figure-7). After
the onset of inflammation, whether it is chronic or acute, cytokines play an important role.
For example, tumor necrosis factor α (TNF- α) and interleukin 1 (IL-1) are pro-
inflammatory cytokines. They cause inflammation, fever, tissue destruction, and
sometimes results in shock and death.
Figure-7: Process of Inflammation
On the other hand, IL-4, IL-10, and IL-13 are potent anti-inflammatory cytokines. Anti-
inflammatory cytokines have the ability to suppress and inhibit the synthesis of pro-
inflammatory cytokines resulting in tissue recovery and healing. Over the years, a lot of
research has been carried out to better understand and cure the diseases associated with
inflammation. Though inflammation is very critical for survival, over production of
inflammatory cytokines and mediators can cause infection and diseases. Many of the anti-
inflammatory drugs inhibit the synthesis of inflammatory mediators such as eicosanoids.
Eicosanoids are signaling molecules that play a vital role in an inflammatory response,
which are produced as a result of oxidation of arachidonic, eicosapentanoic, and dihomo-
gamma-linolenic acids (fatty acids containing 20 carbons). The phospholipase A2 (PLA2)

36
catalyzes the conversion of glycerol phospholipids from the cell membrane to arachidonic
acid, and their inhibition could potentially block the synthesis of all eicosanoids (Rosenfeld
et al., 2006). It is very important to identify anti-inflammatory drug candidates that could
selectively inhibit the activation and synthesis of enzymes and signaling molecules that
promote inflammation.
1.6.1. Mediators of Inflammation:
Usually, the clinically relevant drug targets for the treatment of inflammation are the
cytokines and interleukins. There are two types of cytokines, 1) pro-inflammatory
cytokines, which increases inflammation, e.g., IL-1, IL-2, IL-6, TNF-α, 2) anti-
inflammatory cytokines, which decrease inflammation and increase healing process, e.g.,
IL-3 and IL-4 (Sugano et al., 1991).
1.6.1.1. Role of Interlukin-1 (IL-1):
Interleukin-1(IL-1) is a proinflammatory, multifunctional cytokine, that regulates the
expression of more than 150 genes associated with inflammation and immunity(Fitzgerald
& Luke, 2000).It was first focused for its ability to cause fever, but later it was found to
perform various biological functions including neutrophil recruitment, lymphocyte
activation, induction of inflammatory mediators, induction of hepatic acute-phase proteins,
and up-regulated prostanoid synthesis(Mills & Dunne, 2009).
IL-1 is a member of the IL-F or sometimes called IL-1 family. At present 11 members of
this family are known, but the most prominent are the two forms of IL-1 i-e IL-1α and IL-
1β(Geddes, Magalhães, & Girardin, 2009). These two forms of the IL-1 are encoded by
distinct genes and both are translated as 31kDa molecules present in the cytoplasm and do
not have signal peptide. At the amino acid level, these both forms are only 23%
homologous but the tertiary structure of both forms consists of barrel like structure and 12
β-pleated sheets.The physiologically active form is IL-1α but the precursor form of IL-1β
is activated by intracellular cysteine protease caspase-1 (Murphy, Robert, & Kupper,
2000), (Mizutani, Black, & Kupper, 1991). IL-1α is an intracellular cytokine which
performs both as a cytokine and as a transcription factor while IL-1β performs a part in the

37
fundamental inflammatory response(Dinarello, 2006). Excessive production of IL-1β
caused the repeated episodes of fever and systemic inflammation in autoinflammatory
diseases such as familial cold autoinflammatory syndrome (FCAS), Muckle-Wells
syndrome (MWS), and gout (Masters, Simon, Aksentijevich, & Kastner, 2009).
1.6.1.2. Role of Interlukin-2 (IL-2):
IL-2 was originally known as T cell growth factor (TCGF). It is a 15 kDa glycoprotein. It
is mainly produced by activated T helper cells and acts as a growth factor/activator for T
cells, B cells and NK cells. It has a critical role in regulation of cellular and hormonal
chronic inflammatory responses. Binding of IL-2 to the IL-2 receptor on T lymphocytes
leads to cell proliferation, increased lymphokine secretion (IFN-γ, lymphotoxin, IL-4, IL-3,
IL-5, GM-CSF), and increases the expression of class II MHC molecules (Feghali &
Wright, 1997).
1.6.1.3. Role of Interlukin-6 (IL-6):
Interleukin-6 (IL-6) is a phosphorylated four-helical glycoprotein comprises of 184 amino
acids and a molecular weight of 26 kDa (Ataie-Kachoie, Pourgholami, & Morris, 2013). It
is a T-cell derived β cell stimulating factor. The cDNA of IL-6 was cloned in 1986 and it
belongs to a large cytokines family, sharing the four-helical protein topology (Rose-John,
2012).It is a pro inflammatory cytokine, mainly involved in inflammation by controlling
differentiation, migration, proliferation, and apoptosis of target cells. This is also involved
in regulation of immune system and plays role in multiple processes such as metabolism,
liver regeneration, embryonic development and memory consolidation. Increased levels of
IL-6 of malfunctioning of complex regulatory cytokine network may cause autoimmune or
chronic inflammatory diseases or neoplastic (Scheller, Garbers, & Rose-John, 2014),
(Alonzi et al., 1998), (Ohshima et al., 1998). Therefore, suppression or inhibition of IL-6
seems an attractive therapy for wide range of diseases (Heikkilä, Ebrahim, & Lawlor,
2008).

38
1.6.1.4. Role of tumor necrosis factor-alpha (TNF-α):
Tumour necrosis factor (TNF-α), a cytokine with a relative molecular mass of 17,000 (17
KDa), is produced by activated macrophages in response to pathogens and other injurious
stimuli, and is necessary mediator of local and systemic inflammation (K. J. Tracey et al.,
1986), (H. Wang et al., 1999). It is essential for the complete expression of inflammation
during invasion, and self-limiting inflammation is normally characterized by decreasing
TNF-α activity. Low amounts of TNF-α can contribute to host defence by limiting the
spread of pathogenic organisms into the blood stream, initiating coagulation to localize the
invader, and stimulating the growth of damaged tissues. The duration and magnitude of
TNF-α release is limited, its beneficial and protective activities predominate, and it is not
released systemically (K. Tracey, Vlassara, & Cerami, 1989).
1.6.1.5. Role of Nitric Oxide (NO.):
Nitric oxide is a short lived, gaseous free radical that acts as a signaling molecule and
regulates various physiological and pathophysiological responses in the human body that
includes circulation and blood pressure, platelet function, host defense, and
neurotransmission in central nervous system and in peripheral nerves (Korhonen, Lahti,
Kankaanranta, & Moilanen, 2005). During inflammation process NO. is the mediator of a
number of regulatory responses such as infection control, regulation of signaling cascades
and transcription factors, regulation of vascular responses, and regulation of leukocyte
rolling, migration, cytokine production, proliferation and apoptosis (Knowles & Moncada,
1994), (Marletta, 1994), (Alderton, Cooper, & Knowles, 2001).
Large amounts of "inflammatory NO." from myeloid cells are usually generated side by
side with large amounts of superoxide anion (O2-). These two can form peroxynitrite
(ONOO-) which mediates the cytotoxic effects of NO, such as DNA damage, LDL
oxidation, isoprostane formation, tyrosine nitration, inhibition of aconitase and
mitochondrial respiration (Channon & Guzik, 2002), (Guzik, West, Pillai, Taggart, &
Channon, 2002), (Ischiropoulos & Al-Mehdi, 1995). Nitric oxide is also involved in the
regulation of the release of hormones which have been shown to control inflammatory

39
process centrally, For example, inhibition of CRH-induced ACTH secretion and inhibition
of corticosterone secretion (Givalois, Li, & Pelletier, 2002). Nitric oxide may also regulate
mast cell functions that are considered to be a key player in the initiation of inflammatory
responses (Nathan, 2002). Nitric oxide may act like a double edged sword, on one hand it
may act as a mediator of inflammatory responses to mast cell derived histamine, on the
other hand NO was shown to inhibit mast cell activation, mediate inhibitory effects of IFN-
g, or inhibit vascular adhesion molecule expression thus limiting allergic inflammation.
1.6.1.6. Role of Reactive Oxygen Species:
Reactive oxygen species (ROS) production plays an important role in the modulation of
inflammatory reactions. Major ROS produced within the cell are superoxide anion,
hydrogen peroxide and hydroxyl radical (Salvemini, Ischiropoulos, & Cuzzocrea, 2003).
Extracellular release of large amounts of superoxide, produced as respiratory burst in
leukocytes, is an important mechanism of pathogen killing and also leads to endothelial
damage resulting in an increased vascular permeability as well as cell death (Tiidus, 1998).
These ROS production plays an important role in modulation or release of other mediators
of inflammation. They also increase chemokine and cytokine expression (Kimura et al.,
2003), (Brzozowski et al., 2003).
1.7. Solid-Phase Peptide Synthesis:
The solution-phase methodology of peptide synthesis was firstly introduced by Emil
Fischer but he encountered the major issue of carboxyl and amino group protection of the
two amino acids so that they undergo side reactions (Fischer & Fourneau, 1901). Then
carbobenzoxy group was introduced as a protecting agent (Bergmann & Zervas, 1932).
The mode of peptide synthesis was laborious and time consuming because each step
requires purification of the product.
R. B. Merrifield revolutionized the peptide synthesis in 1963 by giving the concept of solid
phase synthesis. In this technique, the peptide chain was tethered on a solid support and
amino acids were linked until the required sequence was obtained (Merrifield, 1963)
(Figure--6).

40
Figure-8: The General Scheme for Solid-Phase Peptide Synthesis
In solid-phase-synthesis, usually high equivalents of reagents are used to force the reaction
to completion. The excess reagents are just washed off in between the steps of synthesis
and the final cleaved product is carefully purified and characterized. The success of SPS is
incomplete without the advancement in purification and characterization techniques such
as HPLC, capillary electrophoresis, mass spectrometry, NMR spectrometry and automated
amino acid analysis.
1.7.1. Solid Supports for Peptide Synthesis:
Chemically, the solid support is an insoluble polymer having sufficient mechanical
stability desirable physiochemical properties. These physiochemical properties includes
constant shape and size, chemically inert to reaction conditions, according to desired
peptide sequence, suitable functionality should be present in the polymer (Miranda &
Alewood, 2000). The most commonly used solid supports in SPPS are resins. These are
small polymeric beads cross linked with polystyrene (Figure--22), polyacrylamide and
polyethylene glycol (PEG).

41
Figure-9: Resin for SPPS
1.7.2. Linkers used in Solid-phase Peptide Synthesis (SPPS):
Linker acts as a connecting medium between peptide and resin bead. Chemically linkers
should be stable against the reaction condition and make the attachment of the peptide with
resin easy. Some commonly used linkers are discussed below.
1.7.2.1. Kenner’s Safety-catch Linker
Safety-catch linker usually inert toward the reaction condition used for the chain
elongation until it is chemically activated for the cleavage. The use of this linker was
introduced by Kenner in 1971. He employed N- acylsulfonamide linkage for the
preparation of carboxylic acids and primary amides(Kenner, McDermott, & Sheppard,
1971). Utilization of this linker is associated with some major concerns that are poor
reactivity, racemization and poor loading efficiency. A large stoichiometric excess of
reagent have to use to overcome these issues (Backes, Virgilio, & Ellman, 1996).
Consequently more reactive linker was developed by replacing diazomethane with halo
acetonitrile (Backes & Ellman, 1999) (Scheme-5).

42
Scheme-1: Kenner’s Safety-catch Principle
1.7.2.2. Wang Linker:
The mostly used linker for solid phase peptide synthesis is the Wang linker (S.-S. Wang,
1973). This linker is used in the synthesis of acids, alcohols, phenols and peptide. Peptide
synthesis was performed by using the Fmoc / t-Bu protection strategy (Guillier, Orain, &
Bradley, 2000). The Wang linker (4-(hydroxymethyl) phenoxymethyl), is attached to a
resin through an aryl benzyl ether bond (Figure--8).
Figure-10: Wang Linker
1.7.2.3. Rink-Amide Linker:
A variety of supports are available which have rink amide linker (4-(2,4-dimethoxyphenyl)
(Fmoc-amino) methyl) phenoxy methyl) in both forms protected with Fmoc or
unprotected. This resin is used for the synthesis of peptide carboxamide (Atherton, Clive,
& Sheppard, 1975); (Albericio et al., 1990). These linkers are acid labile and release

43
peptides on treatment with 95 % trifluoroacetic acid (TFA). The exposure to high
concentration of TFA sometime results in the formation of side product in the last cleavage
step.
Figure-11: Rink-amide Linker
1.7.3. Protecting Groups Used in Solid Phase Peptide Synthesis:
There are two types of protecting groups that can be used in the synthesis i.e., temporary
and permanent (Figure--12). The “temporary” protecting group is used for the amine
functionality of amino acid. These protecting groups are removed after linking the amino
acid with the linker or with another amino acid. The side chain protection of tri-functional
amino acid is done with the permanent protecting groups. These groups are removed only
when the whole synthesis is complete.
Figure-12: Protecting Groups used in SPPS

44
1.7.4. Coupling Reagents:
Coupling agents are used in the synthesis for two reasons; firstly, in N terminal to C
terminal amino acid synthesis, it is necessary to activate the carboxylic group of amino
acid that further reacts with the amine part of the another amino acid residue. Coupling
agents does this job by converting the carboxylic acid in more reactive state. Secondly,
there are some side-reactions (such as racemization or Diketopiperazine formation)
occurring during the amide bond formation or sometimes incomplete coupling takes place.
Coupling agents are used to avoid the formation of such side products and to ensure the
complete coupling.
Different types of coupling reagents (Figure-13), are used, e.g., benzotriazol-1-yl-
oxytripyrrolidinophosphonium hexafluorophosphate (PyBop), ethyl 2-cyano-2-
(hydroxyimino) acetate (Oxymapure), (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyl-
uroniumhexafluorophosphate (HBTU), hydroxybenzotriazole (HOBT),
diisopropylcarbodiimide (DIC) and dicyclohexylcarbodiimide (DCC) etc. Oxymapure
showed clear superiority to HOBt / DIC or carbodiimide alone in terms of purity and yield
(El-Faham, Al Marhoon, Abdel-Megeed, & Albericio, 2013; STEINAUER, CHEN, &
LEO BENOITON, 1989).
Figure-13: Coupling Reagents used in SPPS

45
1.8.Side-reactions in Peptide Bond Formation:
1.8.1. Racemization:
Racemization is one of the main issues usually encountered during peptide synthesis. This
reaction results in the formation of aza-lactone (Scheme-2). This reaction can be suppress
by incorporation of urethane-type blocking groups, coupling agents that also suppress
racemization (such as oxyma pure) and auxiliary nucleophiles that minimize the chances of
racemization during peptide chain elongation (STEINAUER et al., 1989).
Scheme-2: Racemization during Peptide Bond Formation in SPPS
1.8.2. Synthesis of Diketopiperazine during Peptide Chain Elongation:
Diketopiperazine (DKP), a side product which results in the poor yield of final product.
Due to its formation during the cleavage process hydroxyl sites are generated on the
polymer which results in undesired side reactions (Giralt, Eritja, & Pedroso, 1981).

46
Figure-14: Diketopiperazine Formation
1.9.Targets of Current Study:
This study is designed by considering the emerging importance of peptides in cancer and
inflammatory diseases. We designed and synthesized diverse cyclic peptide library and
screened the compounds against different cancer cell lines which has led to discovery of
four cyclic peptides which were active against different cell lines.
It is very important to develop new anti-inflammatory agents that can meet the growing
need of effective anti-inflammatory drug. In the current study, we synthesized a series of
closely related analogues of anti-inflammatory natural product, stylissatin A. The
immunomodulatory effect of all analogues was studied and compared with the natural
stylissatin A in order to find the more potent and least toxic anti- inflammatory compound.

Chapter # 2
RESULTS
AND
DISCUSSION

47
2.1. Design, Synthesis and Screening of Cyclic Peptide Library:
The current study was focused on the identification of new and novel biologically active
compounds from randomly developed as well as focused libraries based on cyclic peptide
scaffold. A 25-member cyclic peptide library was synthesized using Fmoc-solid phase
chemistry to get collection of exceedingly diverse forms of cyclic peptides. In the synthesis
of diverse cyclic peptide library, some unusual building blocks such as phenyl glycine,
naphthyl alanine, 3-nitro tyrosine, 2-amino benzoic, D-phenylalanine, D-alanine residues
as well as cationic and anionic residues were used. The cyclic peptide library was screened
against different cancer cell lines, DoHH2, MCF-7, NCI-H460, HeLa and 3T3 cell lines
using MTT assay. From this library, four cyclic peptides 2, 14, 15, 16, were identified as
active compounds against different cancer cell lines. The detailed structural and biological
studies of 2, 14, 15 and 16 are presented in the following section 2.2.
In the second part of current study, a focused library around naturally occurring cyclic
peptide stylissatin A was synthesized. Stylissatin A is reported as a natural inhibitor of
nitric oxide. Its first solid phase total synthesis was also carried out by our research group.
The current study focused on the design, synthesis, structure elucidation and SAR study of
six member library of stylissatin A which has been discussed in this dissertation in section
2.3.

48
Table-1(a): Cyclic Peptide Library containing Unusual and Cationic Residues.

49
Table-1(b): Cyclic Peptide Library Based on Stylissatin A

50
2.2. Structural Studies of Active Peptides from Library I:
2.2.1. Cyclic Peptide 2:
The cyclic peptide 2 was designed to contain cationic residues like Lysine, Ornithine and
D-amino acid residues. Peptide 2 was synthesized on Ring amide AM resin by on-resin
macrocyclization strategy using standard Fmoc-peptide synthesis protocol. The first amino
acid phe (D- phenylalanine) was loaded onto the Rink amide resin by using the coupling
agent ethyl hydroxyiminocyanoacetate (oxyma pure) and diisopropyl carbodiimide (DIC).
The next residues Ile and phe were sequentially coupled followed by coupling with Fmoc-
Glu-Allyl-OH under standard Fmoc peptide synthesis conditions. After the removal of
Fmoc group of glutamic acid, the sequence of cyclic part of peptide 2 was constructed by
using two consecutive coupling with Fmoc-Lys (Boc)-OH followed by coupling with
Fmoc-Orn(Boc)-OH, Fmoc-phe-OH, and Fmoc-Lys(Alloc)-OH. Finally, palladium
catalyst was used to remove the protecting groups (allyl group of Glu and alloc group of
Lys residues) to allow head-to-side chain cyclization between -acid group of Glu and
amino group of Lys residue of peptide 2. TFA cocktail was used to cleave the peptide
from Rink resin (Scheme-3). The crude peptide product was purified by RP-HPLC using
the isocratic solvent system 60% ACN in water H2O. The pure peptide 2 was obtained as a
yellow solid material with an overall yield of 40% and [α]D25 +9.97 (c 0.00341, 60% ACN:
H2O). The high resolution electron spray ionization (HR-ESI) technique exhibited

51
molecular ion (M+H)+ at 1020.6325 (calc. 1020.6314) corresponding to molecular formula
C51H82 N13O9. IR spectrum on KBr disc showed absorptions at 3422.6, 3085.8, 2930.8,
1671.1, 1198.7 and 1133.3 cm-1.
Scheme-3: Synthesis of Cyclic Peptide 2

52
Figure-15: Analytical HPLC Profile of Cyclic Peptide 2
Conditions: C4 RP-analytical column/ 60% ACN: H2O
Deuterated solvent d6-DMSO was used for recording NMR spectra because it allowed the
clear detection of NH protons of peptide. All 1HNMR assignments were confirmed by 2D
NMR experiments. The phe1 residue exhibited clear signals for α proton (δH 4.41 / δC 54.1)
and CH2 group (δH 2.69, 3.08 / δC 37.2). The other prominent signals of phe1 were
observed for aromatic protons (δH 7.15 to δH 7.23), carbonyl carbon (δC172.2) and NH
proton (δH 8.40). The terminal free NH2 protons (δH7.42) were also observed. Val2 residue
exhibited signals for α proton, (δH 3.96 / δC 58.4), β CH (δH 1.68, δC 29.9), the gem-
dimethyl group (δH 0.47 / δC 18.9), (δH 0.57 / δC 18.2) and NH proton (δH 7.86). α and γ
acidic carbonyl groups of Glu3, exhibited signals at δC 171.0 and δC 171.1 respectively.
While, β CH2and γ CH2 groups (δH 1.49, 1.33, δC 26.7), (δH 2.58, 2.72, δC 38.0) and NH
proton (δH 8.15) of Glu3 were also observed. The signals for Lys4, Lys7 and Lys8 were
overlapped. The overlapping signals of CH proton of Lys4, Lys7 and Lys8 were observed
in the range of δH 4.20-4.23. For Orn5 residue, α CH (δH 4.41 / δC 54.1), CH2 groups (δH
1.51 / δC 30.5), (δH 1.49, 1.59 / δC 23.4), (δH 2.58, 2.73 / δC 38.8) and NH (δH 8.41) were
assigned. The aromatic residue phe6 exhibited signals for α proton (δH 4.71 / δC 54.1), β
CH2 (δH 2.64. 3.04 / δC 38.7) aromatic protons (δH 7.15- 7.23), NH proton (δH 8.31) and
carbonyl carbon (δC 173.3). All NMR assignments of peptide 2 were established by 2D
NMR techniques including HSQC, HMBC, COSY and NOESY interactions. The HMBC
showed very prominent interaction of α proton (δH 4.41) of phe1 with β CH2 (δC 37.2), C-1
(δC 138.2) and carbonyl carbon (δC 172.2). The terminal free NH2 protons (δH 7.42)

53
showed interaction with CO group of phe1, while the NH proton (δH 8.31) was found to
interact with α carbon (δC 54.1) and β carbon (δC 37.2) of phe1. α proton (δH 3.96) of Val2
residue exhibited interactions with β CH (δC 29.9), γ CH3 (δC 18.2), and carbonyl carbon
(δC 171.1), while NH proton showed interaction with α CH (δC 58.4), β CH (δC 29.9) of
Val2 and carbonyl carbon (δC 171.1) of the next amino acid i.e. Glu3. α proton of Glu3 (δH
4.23) showed the signals with β CH2 (δC 26.7) and carbonyl carbon (δC 171.1) while
interaction of γ protons (δH 2.58, 2.72) with β carbon was also observed. Similarly, α
proton (δH 4.41) of Orn5 showed correlation with β CH2 (δC 30.5), γ CH2 (δC 23.4), and NH
proton (δH 8.4) had interaction with α CH (δC 54.1), β CH2 (δC 30.5) and carbonyl carbon
(δC 171.0) of Orn5. The CH proton (δH 4.71) of phe6 was correlated with β CH2 (δC 38.7),
C-1’ (δC 137.7) of the phenyl ring and the carbonyl carbon (δC 173.3), while the correlation
of NH proton (δH 8.31) was observed with α CH (δC 53.5) and β CH2 (δC 38.7).
Figure-16: Key HMBC Interaction of Cyclic Peptide 2
The COSY spectrum showed the cross peaks among CH α of all residues with adjacent NH
protons. Thus α CH proton (δH 4.41) of phe1 was correlated to the β CH2 (δH 2.69, 3.08)
and NH (δH 8.32). α proton (δH 3.96) of Val2 exhibited the correlation with NH proton (δH
7.86), while β CH proton (δH 1.68) showed the cross peaks with gem-dimethyl (δH 0.47,
0.57). α proton (δH 4.23) of Glu3 was correlated with NH proton (δH 8.15), while β CH2
protons (δH 1.49, 1.33) were correlated with the γ CH2 protons (δH 2.58, 2.72). The NH
protons (δH 8.32) of the Lys4 and (δH 8.15) of Lys7 was correlated with their adjacent α

54
protons (δH 4.23), while, α proton (δH 4.41) of Orn5 showed interaction with NH proton (δH
8.40). The CH α proton (δH 4.71) of phe6 showed cross peaks with β CH2 (δH 2.68, 3.08)
and NH proton (δH 8.31).
Figure-17: Key COSY Correlations of Cyclic Peptide 2
The NOESY spectrum of cyclic peptide 2 showed the NH/NH interaction among residues,
Phe1/ Val2, Glu3/ Lys8, Lys4/ Orn5.
Figure-18: Key NOESY Correlation of Cyclic Peptide 2

55
Table-2: NMR Data of Peptide 2 (d6 DMSO, 600 MHz for 1H and 150 MHz for 13C)

56

57
2.2.2. Cyclic Peptide 14:
The cyclic peptide 14 was synthesized on Ring amide AM resin by linking the side chain
of Fmoc-Glu-O-Allyl to Rink linker by using standard coupling procedure. After
successful loading of first residue glutamic acid, all amino acid were linked in a sequence
containing tyr, three units of Arg, Lys followed by the deprotection of both Glu α allyl and
alloc group of Lys side chain to produce a cyclic peptide 14 by head-to-side chain
cyclization on solid-phase (Scheme-4). The unusual amino acid Nal was linked to cyclic
structure through terminal amino acid Lys followed by its acylation with p-
methoxybenzoic acid. TFA cocktail was used to cleave the crude peptide from resin. The
crude peptide product was purified by RP-HPLC using the isocratic solvent system 60%
ACN in water. The pure peptide 14 was obtained as a yellow viscous material with an
overall yield of 41.9 %. [α]D25 + 5.85 (c 0.0022, 60% ACN: H2O). The high resolution
electron spray ionization (HR-ESI) technique exhibited molecular ion [M+2H]+2 at
610.3445 corresponding to molecular formula C59H84N18O11. IR spectrum on KBr disc
showed absorptions at 3348.8, 2932.6, 1662.7, 1543.1, 1444.6, 1253.0, 1179.2 and 1106.6
cm-1. Deuterated solvent d6-DMSO was used for recording NMR spectra because it
allowed the detection of NH protons of all six amino acid residues of peptide 14.

58
Scheme-4: Synthesis of Cyclic Peptide 14
The 1HNMR showed the presence of α proton ranging between δH 4.01- 4.84. The Gln1
residue was recognized by signal of two methylene groups (δH 1.65, 1.82 / δC 27.4), (δH
2.79, 2.61 / δC 36.6), γ acidic carbonyl (δC 173.7) and NH proton (δH 8.10). Signals for tyr2
were prominent and identified for overlapping signals of H-2 / H-6 (δH 6.96 / δC 130.1) and

59
H-3 / H-5 (δH 6.62 / δC 114.9), the downfield resonance of hydroxyl proton (δH 9.18) along
with βCH2 (δH 2.92, 3.15 / δC 38.1), NH (δH 8.53) and CO (δC 172.1) groups. The 1HNMR
spectrum showed the overlapping signals of methylene groups of Arg3, Arg4, Arg5 and
Lys6 in the range of δH 1.42 to δH 3.12. However the NAL7 residue was fully characterized
by downfield signals of CH2 (δH 3.56, 3.66 / δC 34.0), while signals for naphthyl protons,
H-2ʹ (δH 7.73 / δC 126.9), H-3ʹ (δH 7.35 / δC 125.3), H-4ʹ (δH 7.49 / δC 127.2), H-5ʹ (δH 8.24 /
δC 123.7), H -6ʹ (δH 7.57 / δC 126.0), H-7ʹ (δH 7.85 / δC128.6), H-8ʹ (δH 7.49 / δC125.5) were
identified. While quaternary carbons C-9ʹ and C-10ʹ were found at δC134.3 and δC 131.6
respectively. The carbonyl carbon of Nal7 residue appeared at δC171.7, while amide proton
appeared at δH 8.60. p-Methoxy benzoic acid residue was identified by the sharp singlet of
methoxy group (δH 3.76 / δC 55.3) and aromatic protons (δH 7.71 / δC 129.2) H-2” / H-6”,
(δH 6.92 / δC 113.4) H-3” / H-5”, C-1” (δC 161.6) and C-4” (δC 156.9). The structure was
further confirmed by the HMBC (Figure-19).
The HMBC interactions of Arg3, Arg4, Arg5 and Lys6 residues of compound 14 were not
very clear due to extensive overlapping signals of these residues. The H-2 / H-6 (δH 6.96)
of tyr2 exhibited the interaction with β CH2 (δC 38.1) and C-4 (δC 155.7) of the ring. The
overlapping signals of H-3 and H-5 (δH 6.62) were correlated with the C-1 (δC 127.7) and
C-4 (δC 155.7) of the ring, while hydroxyl proton (δH 9.18) showed correlation with C-4
(δC 155.7) C-3 and C-5 (δC 114.9) of the phenyl ring. In case of NAL7 residue, the H-2’ (δH
7.73) and H-3’ (δH 7.35) exhibited the correlation with C-1’ (δC 127.7), C-9’ (δC 134.3) and
C-10’ (δC 131.6). While H-4’ (δH 7.49) was correlated with C-5’ (δC 123.7) and C-9’ (δH
134.3). H-5’ (δH 8.24) was correlated with C-4’ (δC 127.2), C-9’ (δH 134.3) and C-10’ (δC
131.6). H-6’ (δH 7.57) showed correlation with C-7’ (δC 128.6) and C-10’ (δC 131.6). H-7’
(δH 7.87) displayed correlation with C-9’ (δC 134.3) and C-10’ (δC 131.6). H-8 (δH 7.49)
was correlated to the β CH2 (δC 34.0) and with C-9’ (δC 134.3). The correlations for the H-
2” / H-6” (δH 7.71) of p-methoxybenzoic acid was observed with C-1” (δC 161.6) and
carbonyl carbon (δC 165.8) of the acid. H-3” and H-5” displayed the correlation with C-1”
(δC 161.6) and C-4” (δC 156.9), while the methoxy protons (δH 3.76) showed the
correlation with C-4” (δC 156.9) of the ring.

60
The clear COSY cross peaks were observed between α proton (δH 4.01) and NH proton
(δH8.10) of Gln1 residue. The CH proton in position (δH4.10) of tyr2 was correlated with
the β CH2 proton (δH 2.92, 3.15) and NH proton (δH 8.53), while aromatic protons H-2 / H-
6 (δH 6.96) were correlated with H-3 / H-5 (δH 6.62). The α proton of Arg3, (δH 4.04), Arg4
(δH 4.24) and Arg5 (δH 4.15) were correlated with the adjacent NH protons (δH 8.10), (δH
8.08) and (δH 7.94) respectively. The α proton (δH 4.21) of Lys6 was found to have
correlation with vicinal NH proton (δH 8.48). Nal7 had clear COSY interaction of α proton
(δH 4.84) with vicinal NH proton (δH 8.63). The aromatic protons H-2’ (δH 7.73), H-3’ (δH
7.35) and H-4’ (δH 7.49) of Nal7 residue were correlated with each other. H-5’ (δH 8.24),
H-6’ (δH 7.57), H-7’ (δH 7.49) and H-8’ (δH 7.87) were correlated with each other in the
same spin system. Protons of p-methoxybenzoic group, H-2”, H-6” (δH 7.71) and H-3”, H-
5” (δH 6.92) were correlated with each other.
Figure-19: Key HMBC and COSY Correlations of Cyclic Peptide 14

61
Table-3: NMR Data for Peptide 14 (d6 DMSO, 600 MHz for 1H, 150 MHz for 13C)

62
2.2.3. Cyclic Peptide 15:
The cyclic peptide 15 was synthesized on Ring amide AM resin by linking the side chain
of glutamic acid residue to the Rink linker under standard coupling procedure. After
successful loading of glutamic acid on the resin, all amino acid were sequentially linked by
Asn, Lys, Arg, Lys, Lys, followed by the deprotection of α-allyl and alloc group of Glu
and Lys residues to allow cyclic peptide 15 formation by head-to-side chain cyclization
under cyclization coupling conditions (Scheme-5). After the completion of cyclic sequence
of peptide 15, tyr and Ile were linked to the N-terminal after the removing Fmoc of the
terminal Lys residue. TFA cocktail was used to cleave the crude peptide from resin. The
crude peptide product was purified by RP-HPLC using the isocratic solvent system 60%
ACN: H2O. The pure peptide 15 was obtained as a yellow viscous material with an overall
yield of 45 %. [α]D25 +4.25 (c 0.00282, 60% ACN: H2O). The high resolution electron
spray ionization (HR-ESI) technique exhibited molecular ion (M+H)1+ at m/z 1059.6481
corresponding to molecular formula C48H83N16O11. IR spectrum on KBr disc showed
absorptions at 3421.9 cm-1, 2925.6 cm-1, 1672.9 cm-1, 1629.8 cm-1, 1439.2 cm-1, 1207.2
cm-1, 1134.1 cm-1. Deuterated solvent d6-DMSO was used for recording NMR spectra
because it allowed the detection of NH protons of all eight amino acid residues of peptide
15. The α protons of all residues showed resonance ranging from δH 3.59 - δH 4.57. The
Gln1residue was identified by the presence of two CH2 (δH 1.68, 1.52 / δC 29.1) (δH 2.22,
2.19 / δC 39.6) and γ CO (δC 174.5) was identified through HMBC correlation with γ CH2.

63
Scheme-5: Synthesis of Cyclic Peptide 15
The downfield signal of CH2 (δH 2.49, 2.51 / δC 37.4) was assigned to Asn2. The signals for
Lys3, Lys5, Lys6 methylene groups were overlapped and found in the range of δH 1.42 –
2.90. The β CH2 (δH 1.21, 1.30 / δC 28.9), γ CH2 (δH 1.45 / δC 25.0), δ (δH 3.08 / δC 40.2)
and CO (δC 174.0) were assign to Arg4. The aromatic residue tyr7 was fully characterized

64
by resonances of H-2 / H-6 (δH 7.03 / δC 130.2), H-3 / H-5 (δH 6.62 / δC 115.4) and OH
proton (δH 9.20). The carbon shifts (δC 127.2), (δC 155.9), (δC 171.3) were assigned to
quaternary carbon C-1, C-4 and CO respectively. The proton shift (δH 8.19) was given to
NH proton. Ile8 was characterized by β CH (δH1.77 / 36.8), γ CH2 (δH 1.03, 1.05 / 22.3), γ’
CH3 (δH 0.84 / 14.8), δ CH3 (δH 0.78 / δC11.2), carbonyl carbon (δC 167.9) and NH2
protons (δH 1.50).
The evaluation of HMBC interactions of compound 15 was difficult due to overlapping
methylene groups of Lys3, Lys5 and Lys6 residues. The amide protons exhibited interaction
with next carbonyl functionality of each residue (Figure-20). The NH proton (δH 8.05) of
Gln1 showed interaction with α CO (δC 171.5) of adjacent Asn2 residue. The NH proton (δH
8.01) of Asn2 showed interaction with CO group (δC 171.5) of Lys3. Similarly the amide
proton (δH 8.25) of Lys3, Lys5 and Lys6 showed the HMBC correlation with their adjacent
carbonyl groups of Arg4 (δC 174.0), Lys6 (δC 171.3) and tyr7 (δC 171.3) respectively. The H-
2 / H-6 (δH 7.03) of tyr7 exhibited the interaction with β CH2 (δC 38.2), C-3/C-5 (δC 114.9)
and C-4 (δC 155.9) of the ring. The β methylene protons (δH 2.73, 2.81) showed interaction
with the C-2 / C-6 (δC 130.1). The overlapping signal of H-3 and H-5 (δH 6.62) was
correlated with the C-1 (δC 127.2) and C-4 (δC 155.9) of the ring, while hydroxyl proton
(δH 9.18) showed correlation with C-4 (δC 155.9) C-3 and C-5 (δC 114.9) of the phenyl
ring. The β proton (δH 1.77) of Ile8 showed interaction with carbonyl carbon (δC 167.9), the
γ’ protons (δH 0.84) exhibited the correlation with α-carbon (δC 56.2) and γ carbon (δC 22.3)
while δ protons (δH 0.78) of Ile8 were correlated with β carbon (δC 36.8) and γ carbon (δC
22.3).
The clear COSY cross peaks were observed between α proton (δH 3.62) and NH proton (δH
8.05) of Glu1 residue. While α proton (δH 4.15) of Asn2 was correlated with the β CH2
proton (δH 2.41, 2.50). α proton of Lys3 (δH 4.57) was correlated with vicinal NH proton
(δH 8.25). While α proton of Arg4, (δH 4.05), was correlated with adjacent NH protons (δH
8.1) and β methylene protons (δH 1.21, 1.30).The α proton (δH 4.20) of Lys5 was found to
have correlation with vicinal NH proton (δH 8.0).
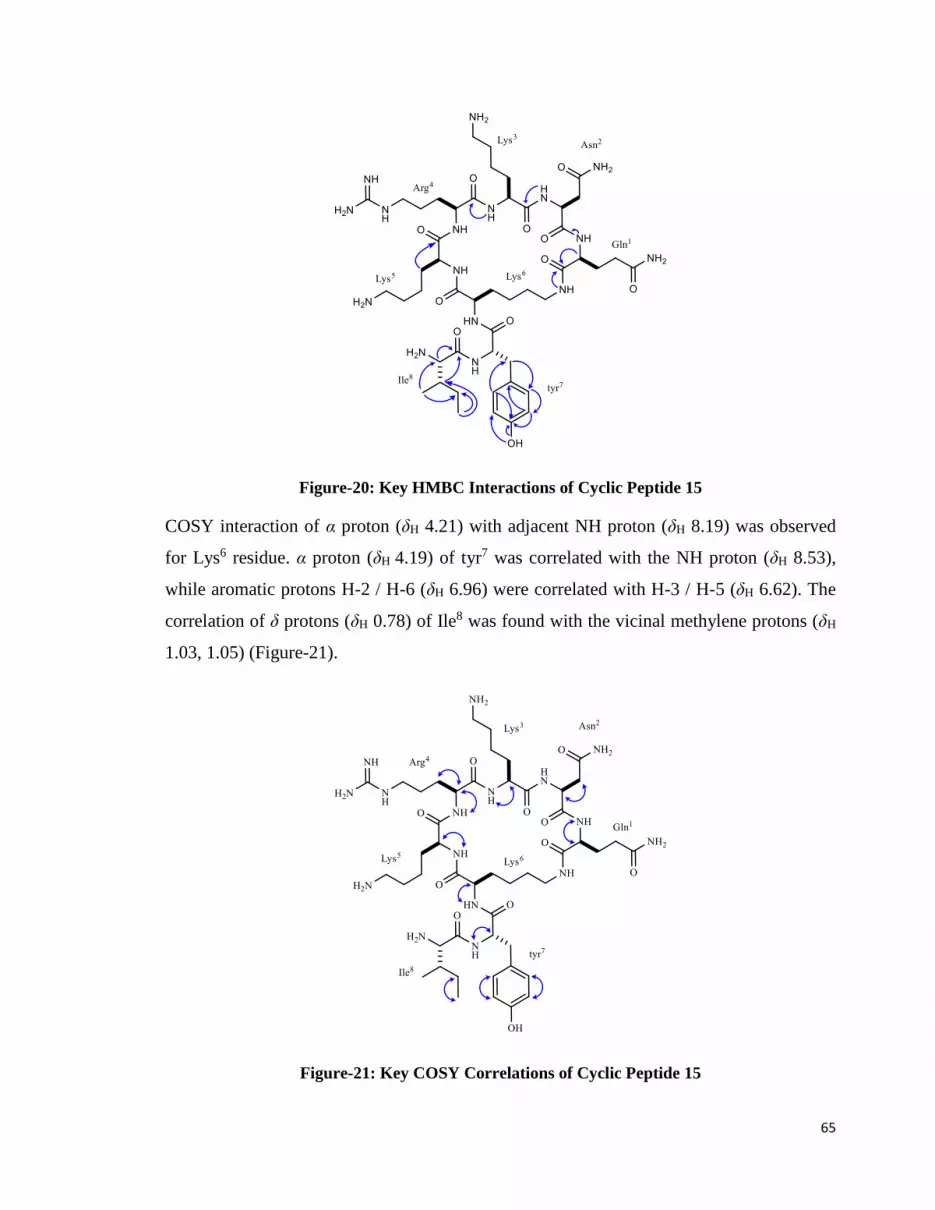
65
Figure-20: Key HMBC Interactions of Cyclic Peptide 15
COSY interaction of α proton (δH 4.21) with adjacent NH proton (δH 8.19) was observed
for Lys6 residue. α proton (δH 4.19) of tyr7 was correlated with the NH proton (δH 8.53),
while aromatic protons H-2 / H-6 (δH 6.96) were correlated with H-3 / H-5 (δH 6.62). The
correlation of δ protons (δH 0.78) of Ile8 was found with the vicinal methylene protons (δH
1.03, 1.05) (Figure-21).
Figure-21: Key COSY Correlations of Cyclic Peptide 15

66
Table-4: NMR Data for Peptide 15 (d6 DMSO, 600 MHz for 1H, 150 MHz for 13C)

67

68
2.2.4. Cyclic Peptide 16:
The novel cyclic peptide 16 was a cyclic derivative of an anticancer linear peptide
temporin ICEa (C. Wang, Li, Li, Tian, & Shang, 2012) (Scheme-6). It was synthesized on
Ring amide AM resin by linking the side chain of Glu residue to Rink linker by using
standard coupling procedure. After successful coupling of glutamic acid, all amino acid
were sequentially linked by using Asn, Lys, Arg, Lys Lys followed by the deprotection of
allyl group of Glu residue and alloc group of Lys to produce a cyclic peptide by head-to-
side chain cyclization. The two residues tyr and Ile were linked to cyclic structure after
removal of Fmoc group of Ile. TFA cocktail was used to cleave the crude peptide from
resin. The crude peptide product was purified by RP-HPLC using the isocratic solvent
system 60% ACN: H2O. The pure peptide 16 was obtained as a whitish yellow solid
material with an overall yield of 35%. The high resolution matrix assisted laser desorption
ionization (MALDI) technique exhibited molecular ion (M+H)+ at 2775.6401
corresponding to molecular formula C131H215N35O31. Deuterated solvent d6-DMSO was
used for recording NMR spectra because it allowed the detection of NH protons of all
amino acid residues. The α protons of all residues appeared in the range of δH 3.59 - δH
4.57. The Glu1residue was identified by two CH2 (δH 1.50, 1.68 / δC 26.7) (δH 2.44, 2.49 /
δC 39.6), γ CO (δC 172.3), α CO (δC172.1), NH proton (δH 8.15) and terminal NH2 (δH
7.41).

69
Scheme-6: Synthesis of Cyclic Peptide 16
The downfield signals of CH2 (δH 2.45, 2.52, δC 38.4), γ CO (δC 171.32), γ NH2 (δH 7.94),
α CO (δC 170.67), α NH (δH 6.96) were assigned to Asn2 residue. The overlapped signals
for Lys3,Lys5, Lys6, Lys19, Lys18 methylene groups were found in the range of δH 1.40 –
2.75. The aromatic amino acid tyr7 was fully characterized by protons signals H-2 / H-6
(δH 6.96 / δC 130.2), H-3 / H-5 (δH 6.62 / δC 115.3) and OH proton (δH 9.21). The carbon
shifts δC 133.8, 156.2, 169.2, were assigned to quaternary carbon C-1, C-4 and CO,
respectively, while the proton shift at δH 7.01 was given to NH proton. Ile8was identified

70
by β CH (δH 1.74 / 36.5), γ CH2 (δH 1.37, 1.50 / 24.2), γ’ CH3 (δH 0.64 / 15.3), δ CH3 (δH
0.66 / δC 11.1) and carbonyl carbon (δC 171.0). The Phe9 residue has signals for aromatic
protons H-2’ / H-6’ (δH 7.31 / δC 128.3), H-3’ / H-5’ (δH 7.19 / δC 129.9) and H-4’ (δH 7.14
/ δC 126.2). Signals for β CH2 (δH 2.93, 3.04 / δC 37.1) C-1 (δC 134.7), and CO (δC 169.2)
were also observed. Ile10, Ile13, Ile14, Ile17 showed overlapping signals of β CH from δH
1.58 – 1.74, γ CH2 δH 1.36 – 1.52, γ’ CH3 and δ CH3 δH 0.57 – 0.81. The most downfield
methylene signal of CH2 (δH 3.49, 4.41 / δC 61.7) were assigned to Ser11 residue. The NMR
spectrum also showed the overlapping downfield signals of β CH2 (δH 2.66 – 2.78) of
Asn12 and Asn15. Signals for α-carbonyl carbon (δC 170.6), γ CO (δC 171.0), α-NH (δH
6.96) were also observed. Ala16 was recognized by its CH3 signal (δH 1.17 / δC 18.26)
along with NH signal (δH 8.55) and CO signal (δC 171.9). The NMR shifts for β CH2 (δH
2.38, 2.41 / δC 40.87), γ CH (δH 1.36 / δC 24.1), δ CH3 (δH 0.78 / δC 21.4), δ’CH3 (δH 0.82 /
δC 23.34), NH proton (δH 7.21) and carbonyl group (δC 170.9) were assign to Leu20. The
Asp21 were assigned β CH2 shift (δH 2.67, 2.71 / δC 38.8), NH proton (δH 8.42) and α-CO
(δC 170.1) and γ-CO of acid functionality (δC 170.4). The signals for gem dimethyl (δH 0.82
/ δC 18.4), (δH 0.84 / δC 19.1) CH (δH 1.90 / δC 31.3), NH (δH 8.10) and CO (δC 167.8) were
assign to Val22. The Phe23 were characterized by aromatic protons H-2” / H-6” (δH 7.18 /
δC 129.9), H-3” / H-5” (δH 7.21 / δC 128.4), H-4” (δH 7.24 / δC 127.3) and carbonyl carbon
(δC 169.2).
The assignment of NMR shifts was most arduous due to overlapping signals of methyl
groups common in various amino acid residues. However, HMBC correlation helped us to
correctly assign the resonance to different residues. α proton of Asn2 (δH 4.58) exhibited
interaction with β CH2 (δC 38.4), while β protons (δH 2.45, 2.52) showed correlation with α
carbon (δC 49.4) and γ carbonyl group (δC 171.3). The γ protons (δH 1.42- 1.74) of Lys3,
Lys5, Lys6, Lys18, Lys19 were giving interaction with δ carbon (δC 26.7), while the δ protons
(δH 1.50) showed interaction with γ carbon (δC 23.6) and ϵ carbon (δC 40.0). α proton (δH
4.41) of tyr7 showed correlation with C-2 (δC 130.2), C-3 (δC 115.3), while β protons (δH
2.91, 3.03) were correlated with carbonyl carbon (δC 167.8). The overlapping signals of H-
2 / H-6 (δH 6.96) showed interaction with β carbon (δC 37.1), C-3 / C-5 (δC 115.3) and C-4
(δC156.2). H-3 / H-5 exhibited interaction with C-4 (δC 156.2) and OH proton (δH 9.21)
showed interaction with C-4 (δC 156.2). In case of residue at position 8, α proton (δH 4.13)

71
of Ile8 showed correlation with γ carbon (δC 24.2), while δ CH3 (δH 0.66) were correlated
with α carbon (δC 57.2), β carbon (δC 36.5), γ carbon (δC 24.2). The CH α proton (δH 4.12)
of Phe9 exhibited interaction with β carbon (δC 37.1), while β methylene protons (δH 2.93,
3.04) showed correlation with carbonyl carbon (δC 167.8), H-2’ / H-6’ (δH 7.31) showed
correlation with C-3’ / C-5’ (δC 129.9), H-3’ / H-5’ (δH 7.19) was correlated with C-2’ / C-
6’ (δC 128.3) and C-4’ (δC 126.4). The distinct α proton (δH 4.52) of Asn12 and Asn15
showed correlation with β carbon (δC 38.8), while β protons (δH 2.66, 2.72) were interacted
with α carbon (δC 49.4) and γ carbonyl carbon (δC 171.0). The CH3 protons (δH 1.17) of
Ala16 exhibited the interaction with α carbon (δC 48.2) and carbonyl carbon (δC 171.9). δ
CH3 protons (δH 0.78) of Leu20 were correlated with α carbon (δC 56.9), β carbon (δC 40.8)
and δ’ carbon (δC 23.3). α proton (δH 4.55) of Asp21showed interaction with β carbon (δC
38.8), while, CH2 β protons (δH 2.67, 2.69) showed interaction with α carbon (δC 49.8) and
γ-acidic group (δC 170.4). α proton (δH 4.24) of Val22 was interacted with β carbon (δC
31.3) and carbonyl carbon (δC 167.8), while β protons (δC 171.9) were interacted with α
carbon (δC 57.3), γ carbon (δC 18.4) and γ’carbon (δC 19.1). γ CH3 protons (δH 0.82)
showed interaction with γ’ carbon (δC 19.1). α proton (δH 4.16) of Phe23 exhibited
interaction with β carbon (δC 37.1), β methylene protons (δH 2.94, 3.05) showed interaction
with carbonyl carbon (δC 167.8), H-2” / H-6” (δH 7.18) exhibited correlation with C-1” (δC
137.2) and C-4” (δC 127.3), H-3” / H-5” (δH 7.21) were correlated with C-1” (δC 137.2).

72
Figure-22: Key HMBC Interactions of Cyclic Peptide 16

73
Table 5: NMR Data for Peptide 16 (d6 DMSO, 600 MHz for 1H, 150 MHz for 13C)

74

75

76

77

78
2.2.5. Anticancer Activities of Cyclic Peptides.
Cancer is a group of diseases characterized by the uncontrolled growth and proliferation of
abnormal cells from the site of origin to the distant organs that may ultimately lead to
death. Several factors are involved in the progression of cancer, such as exposure to UV
radiation, pollution smoking habits, viruses, and an unhealthy diet. Moreover many chronic
infections like hepatitis B and C (liver cancer), human papilloma (cervical and ano-genital
cancers) and Helicobacter pylori (stomach cancer) also convert into cancerous situation. It
may also be caused by genetic mutations, hormones, and immunological disorder etc.
Several therapies including surgery, hormone therapy, radiation, immune therapy,
chemotherapy, and targeted therapy (administration of the drugs that specifically inhibit
cancer cell growth) are being used (Morris et al., 2008).
Breast cancer is about one third of the all malignancies found in females. This arises from
breast epithelial elements and can be grouped into in situ carcinomas and invasive
carcinomas. In situ carcinomas confined in one place and may arise from ductal or lobular
epithelium, while invasive carcinomas extend from ductal or lobular epithelium from
basement membrane of epithelial border. Radiation therapy was suggested but with care
taken to avoid damage to the heart and lungs. Systemic adjuvant chemotherapy is never
recommended for non-invasive cancer. For invasive cancer standard adjuvant
chemotherapy includes adriamycin, cyclophosphamide, taxol, methotrexate, fluorouracil,
epirubicin drugs. Hormonal therapy is also used to inhibit estrogen that is believed to
stimulate cancer cell growth. Tamoxifen is most commonly used in hormonal therapy.
Peptide therapeutics is considered promising for the advancement of breast cancer drug
target development. Several peptides based vaccines have been developed and studied
against this disease (Mittal, Kaur, Gautam, & Mantha, 2017). Chemist and biologist are
making continuous efforts to develop peptide based therapy to fight against breast cancer

79
effectively. Keeping the same in mind the cyclic peptide library 1 was screened against
MCF-7 cancer cell line. The results of the screening are summarized in table-6
Lung cancer is considered as the most common human cancer. It is reported that non-small
cell lung cancer (NSCLC) accounts for 80 % of all lung cancer cases and five-year survival
rate of this cancer is less than 15% despite recent advancement made in surgery,
radiotherapy and chemotherapy. In chemotherapy, the cisplatin is widely used
chemotherapeutic agents for NSCLC treatment. This is considered unsatisfactory due to
drug resistance and severe side effects. Thus, identification of new and more effective
medicinal agents is utmost requirement for treatment of this disease. For the screening of
cyclic peptide library, NC-H460 cell line was used to find new anticancer compound
against non-small cell lung cancer (NSCLC) (Xu et al., 2013).
Non-Hodgkin lymphomas are the most frequent human cancers (Siegel et al, 2014).
Despite the noticeable progress in their treatment, they still face reversion after a first
complete remission (Dreyling et al, 2013; Ghielmini et al, 2013), thus demand new
therapeutic agents for the complete cure of disease. In the current study, cyclic peptide
library was also screened against non-Hodgkin lymphoma cell line DOHH2.
In the current study, anticancer screening results showed that the novel peptide 16 was a
strong inhibitor of MCF-7 and DOHH2 cell lines. This peptide 16 was designed as a novel
cyclic analogue of an anticancer linear peptide temporin-1CEa. Temporin-1CEa is an
antimicrobial peptide reported from the skin secretions of the Chinese brown frog (Rana
chensinensis). It is reported that temporin-1CEa-induced rapid cytotoxicity on MCF-7
(IC50 54.9 µM). In the current study, peptide 16 more potently inhibited MCF-7 cancer cell
line (IC50 1.1µM) compared to temporin-1CEa and also strongly inhibited the DOHH2 cell
line (IC50 4.4 µM). The peptides 15 exhibited inhibitions against NCI-H460 and HeLa cell
lines with IC50 41.9 µM, and 92.4 M, respectively, While the peptide, 2 was active against
NCI-H460 (IC50 62.6 µM) and compound 14 was active against MCF-7 cell line (IC50 29.6
µM). All four compounds were found inactive against normal 3T3 cell lines till 100 M.

80
Table-6: Cytotoxicity of New Cyclic Peptides.
*Not tested

81
2.3. Studies on Stylissatin A Analogues:
The cyclic heptapeptide stylissatin A has been isolated from the Papua New Guinean
marine sponge Stylissa massa. The natural peptide had an inhibitory effect on nitric oxide
production in LPS-stimulated murine macrophage RAW264.7 cells with an IC50 value of
87 µM (Kita et al., 2013). The completed total synthesis of natural product was reported by
our research group. Here we report the complete solid-phase synthesis, structural studies
and immunomodulatory activities of different related analogues of stylissatin A (26-31).
Inflammation is an immediate cellular response of body against harmful foreign elements
and tissue injury. Upon first exposure to pathogens or other stimuli phagocytes, mainly
neutrophils and macrophages, releases various mediators including reactive oxygen (ROS)
and nitrogen species (RNS), chemokines and cytokines. The release of these mediators
progresses the processes of inflammation and activation of cell mediated immune
responses (Jadhav, Singh, & Bhutani, 2003).
Reactive oxygen species (ROS) and nitric oxide (NO.) released by inflammatory cells are
involved directly and indirectly in the advancement of oxidative damage (Valko et al.,
2007). Nitric oxide (NO.) has various physiological roles including maintenance of vessel
homeostasis, however the excessive production of NO. in inflammatory condition has
damaging effect on cells and organs.
T lymphocytes are the main cells involve in the generation of adaptive immune response.
The activation and proliferation of various immune cells depends on the cytokines secreted
by T cells. IL-2 is an important immunomodulatory cytokine, secreted by T cells. Along
with the effect on production of other cytokines, it is known to activate T cells in an
autocrine manner. Activated T cells have high affinity receptors for IL-2 on their
surface(Cantrell, Collins, & Crumpton, 1988). Inhibition of IL-2 provides strong
immunosuppressive approach by preventing the activation and proliferation of T cells in
transplantation rejection and other autoimmune diseases (Waldmann, 1993).
Different strategies for the synthesis of cyclic peptides are reported. Among them, the on-
resin cyclization approach has been successfully employed in the synthesis of several

82
biologically active natural peptides in good yields with minimum side products (Kumarn,
Chimnoi, & Ruchirawat, 2013), (Shaheen et al., 2012).
Six analogues 26-31 of stylissatin A were synthesized on Wang resin via on-resin
cyclization approach. Linear precursors of peptide analogues were anchored to the Wang
resin through the side chain of glutamic acid by Fmoc-peptide synthesis. Peptide sequences
of 26-31 were made different by Ala substitution of amino acids residues of original
peptide (Table-7). After the synthesis of linear chain, the allyl group of the side chain of
the Glu was removed by using palladium catalyst (Scheme 7).
Table-7: Alanine-Substituted Analogues (26-31) of Stylissatin A
The Fmoc group of terminal amino acid residue was removed by 4-methylpiperidine in
DMF. On-resin cyclization was carried out by using oxymapure / DIC. TFA cocktail was
used to cleave final product from resin. Structures of analogues were confirmed by
MALDI (Table-7) and NMR studies. Purification of all peptides was achieved by using
preparative recycling HPLC.

83
Scheme 7: Solid-Phase Synthesis of Cyclic Peptides (26-31).

84
2.3.1. Cyclic Peptide 26:
The cyclic peptide 26 contain 7 amino acid residues, all having “L” configuration. The
peptide 26 was synthesized by substitution of Pro4 and Pro6 residues of original peptide
sequence of stylissatin A with Ala4 and Glu6 respectively as shown in scheme-8.
The pure peptide 26 was obtained as a yellowish white solid. UV spectra were recorded in
DMSO and showed λmax 229 nm. IR (KBr disc) showed absorptions at 3400, 3019, 2400,
1706, 1219, 791, 628 cm-1. [α]D25was −21.8 (c 0.055, MeOH). The melting point was
determined to be 155 0C. The high resolution matrix assisted laser desorption ionization
(MALDI) technique exhibited pseudo-molecular ion (M+Na)+1 at 906.4372 corresponding
to molecular composition C47H61N7O10Na. Deuterated solvent d6-DMSO was used for
recording NMR spectra which allowed the detection of all seven NH protons of analogue
26. The 1HNMR showed seven amide protons, resonating at δ 8.06 (1H, Phe3-NHCO), δ
7.99 (1H, NHCO), δ 7.90 (1H, Ala4-NHCO), δ 7.85 (1H, Tyr1-NHCO), δ 7.71 (1H,
NHCO), δ 7.1 (1H, Ile5-NHCO), δ 7.10 (1H, Glu6-NHCO). NMR shifts for α proton of
constituent amino acids of 26 appeared in the range of (δH 3.80 - 4.51). Tyr1 has prominent
signals for its aromatic protons δH 6.95 (H-2, H-6) and δH 6.57 (H-3, H-5). Ile2 and Ile5
exhibited the characteristic resonances for methine, methylene and two groups of gem-
dimethyl (Table-8).

85
Scheme-8: Synthesis of Cyclic Peptide 26

86
Figure-23: Analytical HPLC Profile of Cyclic Peptide 26
Conditions: C4 RP-analytical column/ 60% ACN: H2O
The CH2 groups of Phe3 and Phe7 appeared at (δH 2.74, 2.93, δC 36.5) and (δH 2.82, 3.15,
δC 36.9) respectively. Ala4 residue signals were appeared at (δH 1.13, δC 18.2) and CH2
groups of Glu6 were observed at (δH 2.21, 2.32, δC 30.1) and (δH 1.90, 1.70, δC 28.5). The
HMBC spectrum showed the correlation of α methine proton of Tyr1 (δH 4.42) with β
methylene carbon (δC 37.4), while β CH2 protons were further correlated with α CH (δC
53.5) of Tyr1, as well as with C-1 (δC 137.9), C-2 and C-6 (δC 129.9) of aromatic ring of
Tyr1 residue. The HMBC correlation of the H-2 and H-6 (δH 6.95) with the aromatic
carbons (δC 114.8, 155.7) of Tyr1 residue were observed. The hydroxyl proton (δH 9.1) of
Tyr1 residue showed the HMBC interaction with the aromatic carbon C-3 and C-5 (δC
114.8) and NH proton (δH 7.85) showed interaction with the carbonyl carbon (δC 170.45)
of the next residue Ile2.
HMBC correlation of α CH of Ile2 (δH 4.12) was observed with the β CH (δC 36.1), γ CH2
carbon (δC 23.9) and carbonyl carbon (δC 170.4). The β-methine proton (δH 1.59) was
correlated with methylene carbon (δC 23.9). The NH proton (δH 7.76) of Ile2 showed
correlation with the carbonyl carbon (δC 171.9) of next residue Phe3. The β CH2 protons of
Phe3 (δH 2.71, 2.80) exhibited HMBC correlation with α carbon (δC 53.5) and with C-1ʹ of
the phenyl ring (δC 137.6). The H-3ʹ, H-5ʹ (δH 7.15) of the phenyl ring were correlated with
C-1ʹ (δC 137.6), C-2ʹ, C-6ʹ (δC 127.9) and C-4ʹ(δC 126.11) of the ring. The amide proton (δH
8.06) of Phe3was correlated with the carbonyl carbon (δC 171.5) of the next residue i.e.,
Ala4. The HMBC correlation was observed for α proton (δH 4.25) with methyl carbon (δC
18.2) and carbonyl carbon (δC 172.6) of Ala4 unit. The NH proton (δH 7.90) of Ala4 was

87
further correlated with the carbonyl carbon (δC 170.4) of Ile5 residue. HMBC correlation of
α proton (δH 3.84) of Ile5 residue was seen with the β methine carbon (δC 36.6), γ
methylene carbon (δC 24.3) and with carbonyl carbon (δC 170.4). The NH proton (δH 7.1)
of Ile5 was correlated with the carbonyl carbon (δC 166.2) of the Glu6 residue. The CH β
proton (δH 2.21, 2.32) of Glu6 residue showed HMBC interaction with the carbonyl carbon
(δC 173.8), and with γ carbon (δC 28.5). The NH proton of Glu6 residue showed correlation
with the carbonyl carbon (δC 166.2) of the Phe7. The HMBC correlation of α proton of
Phe7 residue (δH 4.49) was observed with the β CH2 (δC 36.9) and with carbonyl carbon (δC
166.2) of Phe7. β CH2 of Phe7 (δH 2.82, 2.99) showed correlation with α carbon (δC 53.1)
and with C-1ʹʹ (δC 137.5) of the phenyl ring. The correlation of the H-4ʹʹ (δH 7.12) with C-
1ʹʹ (δC 137.5), C-3ʹʹ, C-5ʹʹ (δC 129.99) and C-2ʹʹ, C-6ʹʹ (δH 126.22) of aromatic ring was also
observed. Furthermore, the NH proton (δH 7.87) of the Phe7 showed HMBC correlation
with the carbonyl carbon (δC 170.75) of the adjacent Tyr1 residue.
Figure-24: Key HMBC Interactions of Cyclic Peptide 26
In the COSY spectrum of Tyr1 residue, α proton (δH 4.42) was correlated with the β
methylene protons (δH 2.65, 2.82) and amide proton (δH 7.85). The aromatic protons were
also found to have correlation with each other. COSY correlations of Ile2 residue were
clearly observed between α proton and NH proton (δH 7.71) and between β methine proton

88
(δH 1.60) and γ’ methyl protons (δH 0.65). Prominent cross peaks were also observed
between γ methylene protons (δH 1.03, 1.35) and σ methyl group (δH 0.76).
COSY correlations were observed for α proton (δH 4.51) of Phe3 residue with the NH
proton (δH8.06). The correlation was also seen between α proton resonance (δH 4.25) and
methyl protons (δH 1.13) and the NH group (δH 7.90) of Ala4 unit. α proton of Ile5showed
cross peaks with the NH proton (δH 7.1). The β methine proton (δH 1.65) is correlated with
that of γ’ methyl protons (δH 0.75). COSY correlation of γ methylene protons (δH 1.26,
1.30) was seen with the δ methyl (δH 0.74). COSY correlation of α proton of Phe7 was
observed with β methylene protons (δH 2.82, 3.15) and NH proton (δH 7.99).
Figure-25: Key COSY Correlations of Cyclic Peptide 26
The key NOESY interaction observed in the cyclic peptide 26 were between Tyr1 Hα /
Phe7 NH, Ile2 Hα / Tyr1 NH, Ile2 Hα / Phe3 NH, Ala4 NH / Ile5 Hα, Glu NH / Phe7 Hα.

89
Figure-26: Key COSY Correlations of Cyclic Peptide 26

90
Table-8: NMR Data of Peptide 26 (d6 DMSO, 500 MHz for 1H and 150 MHz for 13C)

91
2.3.2. Cyclic Peptide 27:
The cyclic peptide 27 was composed of seven amino acids building blocks all having “L”
configuration. The peptide 27 was synthesized by substitution of Pro4 and Pro6 residues of
stylissatin A with Glu4 and Ala6 respectively as shown in scheme-9.
The pure peptide was obtained as a yellow solid. UV spectra were recorded in DMSO and
showed λmax 229 nm. IR (KBr disc) showed absorptions at 3283, 3074, 2400, 1706, 1219,
791, 628 cm-1. [α]D25 − 47.60(c 0.055, MeOH). Melting point was found to be 175.2 0C.
The molecular formula of analogue 27 was found to be C47H61N7O10Na on the basis of
pseudo-molecular ion (M+Na)+1 at 906.43 in the high-resolution matrix assisted laser
desorption ionization (MALDI) technique. Deuterated solvent d6-DMSO was used for
recording NMR spectra which facilitated the detection of all seven NH protons of peptide
27. The 1HNMR showed of all seven NH protons resonating at δ 8.15 (1H, Ala6-NHCO), δ
8.07 (1H, Phe7-NHCO), δ 7.91 (1H, Ile2-NHCO), δ 7.9 (1H, Ile5-NHCO), δ 7.89 (1H, Tyr1-
NHCO), δ 7.85 (1H, Glu4-NHCO), δ 7.78 (1H, Phe3-NHCO). The signals of α proton for
amino acid residues of peptide 27 were present in the range of δH 4.14 – 4.52. The
aromatic moiety of Tyr1 residue was characterized by the overlapped signals of H-2 / H-6
(δH 6.99, δC 130.0) and H-3 / H5 (δH 6.62, δC 114.7). The presence of two signals for each
of β CH proton (δH 1.58, δC 36.8) (δH 1.61, δC 36.9), γ CH2 (δH 0.95, 1.28, δC 24.0) (δH
1.27, 1.32, δC 24.2) and presence of four sets of gem-dimethyl (Table-9.) confirmed the
presence of two isoleucine residues.

92
Scheme-9: Synthesis of Cyclic Peptide 27

93
Figure-27: Analytical HPLC Profile of Cyclic Peptide 27
Conditions: C4 RP-analytical column/ 60% ACN: H2O
The methylene protons (β CH2) of Tyr1, Phe3 and Phe7 were seen in the range of δH 2.59 –
2.79. The NMR signals for β protons (δH 2.60, 2.81, δC 29.8) of Glu4 were also observed.
Ala6 unit was easily identified by α proton shift (δH 4.34, δC 48.0) and methyl shift (δH 1.13,
δC 18.1). The 13C resonances were assigned to all the amino acid residues with the help of
broad band and DEPT-135 spectra. The structure was further confirmed by HMBC and
COSY correlation. The HMBC correlation of α proton of Tyr1 residue (δH 4.44) was
observed with the β CH2 protons (δH 2.59, 2.79), which were further correlated with the α
carbon (δC 53.8), C-1 (δC 135.9) and C-2, C-6 (δC 130.0) of the aromatic ring. H-2, and H-6
(δH 6.94) showed correlation with the β CH2 (δC 37.2). While H-3, H-5 (δH 6.56) of
Tyr1were correlated with C-2, C-6 and C-4. The NH proton (δH 7.89) showed interaction
with the carbonyl carbon (δC 170.4) of Ile2.
The CH α of Ile2 (δH 4.14) showed the HMBC correlation with the β carbon (δC 36.8), γ
carbon (δC 24.0) and carbonyl carbon (δC 170.54) of Ile2 itself. The γ’ methyl protons (δH
0.70) were related with α CH (δC 56.7), β CH (δC 36.8) and with γ CH2 (δC 24.0). The NH
proton (δH 7.91) signal was correlated with the carbonyl carbon (δC 170.9) of Phe3.
The α CH proton (δH 4.18) of Phe3residue exhibited interaction with the β CH2 (δC 36.5),
which was further correlated with C-1ʹ (δC 137.5) and C-2ʹ, C-6ʹ (δC 128.2) of the ring. The
H-2ʹ, H-6ʹ protons (δH 7.21) were correlated with the C-3ʹ, C-5ʹ (δC 127.8), with C-1ʹ (δC
137.5) and C-4ʹ (δC 127.5) of the ring. The NH proton (δH 7.78) showed interaction with
the carbonyl carbon (δC 173.0) of Glu4. The correlation of β proton (δH 2.60, 2.81) of Glu4
residue with α CH (δC 52.5) and with carbonyl carbon of acid group (δC 174) were

94
identified. For Ile5 residue, α proton (δH 4.16) was correlated with β CH (δC 36.0). The γ’
methyl protons (δH 0.67) showed the cross peaks with α CH (δC 56.7), β CH (δC 36.0), and
γ CH2 (δC 24.2). Furthermore HMBC interaction of the σ methyl protons (δH 0.75) are
observed with γ CH2 (δC 24.2). The NH proton (δH 8.0) showed interaction with the
carbonyl carbon (δC 172.5) of Ala6. The Ala6 unit was identified from the HMBC
correlation between α CH (δH 4.34), methyl (δC 18.1) and carbonyl carbon (δC 172.5) of
Ala6. The NH proton (δH 8.15) was correlated with the carbonyl carbon of the Phe7.
In COSY spectrum, clear cross peaks of α CH of Phe7 (δH 4.54) were observed with β CH2
(δH 2.79, 2.99) and NH proton (δH 8.07). The β methylene protons displayed the HMBC
correlation with α carbon (δC 53.4), with C-1ʹʹ(δC 138.4) and C-2ʹʹ, C-6ʹʹ (δC 129.0) of the
ring. H-2ʹʹ, H-6ʹʹ (δH 7.22) were observed with the C-1ʹʹ (δC 138.4), C-3ʹʹ, C-5ʹʹ (δC 129.18)
and C-4ʹʹ (δC 128.29).
Figure-28: Key HMBC Interactions of Cyclic Peptide 27
The COSY correlations α CH proton of Tyr1 was observed with β CH2 protons (δH 2.59,
2.79) and amide proton (δH 7.88). H-2, H-6 (δH 6.94) were correlated with the H-3, H-5 (δH
6.56). α proton of Ile2 (δH 4.14) was correlated with the NH proton (δH 7.91), while β CH
proton (δH 1.55) showed interaction with the γ’ CH3 (δH 0.66). The CH2 γ (δH 0.95, 1.28)
showed interaction with the σ CH3 (δH 0.78). COSY correlation of α proton of Phe3 with
the β proton (δH 2.99, 2.78) and amide proton (δH 7.78) was observed. On the other hand,
CH α proton of Glu4 (δH 4.4) was correlated with the amide proton (δH 7.78) and β
methylene protons (δH 2.6, 2.8). CH α proton of Ile5 (δH 4.16) was correlated with the

95
amide proton (δH 7.9) and β CH proton was correlated with the γ’ methyl (δH 0.67). The
COSY correlation observed for CH resonance (δH 4.34) with the methyl (δH 1.18) and
NH group (δH 8.15) identified the Ala6 unit, While α CH proton of Phe7 displayed the
correlation with the β CH2 protons (δH 2.99, 2.78) and amide proton (δH 7.78).
Figure-29: Key COSY Correlations of Cyclic Peptide 27
The NOESY spectrum showed the cross peaks of Tyr1 Hα / Phe7 NH, Ile2 Hα / Tyr1 NH,
Phe3 Hα / Ile2 NH, Ala6 NH / Phe7 Hα.

96
Figure-30: Key NOESY correlations of Cyclic Peptide 27

97
Table-9: NMR Data of Peptide 27 (d6-DMSO, 500 MHz for 1H and 150 MHz for 13C)

98
2.3.3. Cyclic Peptide 28:
The peptide 28 was synthesized by substitution of Pro6and Phe7 residues with Glu4 and
Ala6 respectively, as shown in scheme-10.
The pure peptide was obtained as a white solid. UV spectra were recorded in DMSO and
showed λmax 230 nm. IR (KBr disc) showed absorptions at 3207, 3074, 2400, 1729, 1219,
791, 628 cm-1. [α]D25 −77.3 (c 0.052, MeOH). Melting point was found to be 171.7 0C. The
molecular formula of analogue 28 was found to be C43H59N7O10Na on the basis of high
resolution matrix assisted laser desorption ionization (MALDI) technique, which gave the
pseudo-molecular ion (M+Na)+1 856.4216. Deuterated solvent d6-DMSO was used for
recording 1HNMR which helped in clear detection of seven amide protons of peptide 28.
According to 1HNMR, the NH protons were present at δ 8.16 (1H, Tyr1-NHCO), δ 8.16
(1H, Glu6-NHCO), δ 8.14 (1H, Ala7-NHCO), δ 8.01 (1H, Phe3-NHCO), δ 7.88 (1H, Ile2-
NHCO), δ 7.72 (1H, Ile5-NHCO), The Tyr1 residue was identified by aromatic moiety
resonances of (δH 6.99, δC 129.1,C-1, C-6) and (δH 6.58, δC 114.7,C-3, C-5). The methyl
group of Ala2 residue appeared at (δH 1.09, δC 18.2).The presence of two CH2 in the shift
range (δH 2.78 – 3.01, δC 37.30, 37.97) confirmed the two phenylalanine residues which
were present at 3 and 7 positions. The presence of CH2 signals at (δH 1.68, 1.83, δC 29.0)
(δH 1.48, 1.49, δC 24.1) and CH2 at (δH 3.49, 3.68, δC 47.1) characterized the Pro4 residue.

99
The Ile5 was confirmed by the CH signal at (δH 1.70, δC 36.0), CH3 signals (δH 0.82, δC
14.9) and (δH 0.78, δC 10.8).
Scheme-10: Synthesis of Cyclic Peptide 28

100
Figure-31: Analytical HPLC Profile of Cyclic Peptide 28
Conditions: C4 RP-analytical column/ 60% ACN: H2O
The structure was confirmed by HMBC and COSY correlation. In the HMBC spectrum, α
CH of Tyr1 residue (δH 4.42) displayed the correlation with the β CH2 (δC 35.5). While β
CH2 protons (δH 2.78, 2.94) were correlated with C-1 (δC 137.6) and C-2, C-6 (δC 129.1) of
the ring. The aromatic protons H-2 / H-6 were correlated with the C-3, C-5 (δC 114.8) and
C-4 (δC 115.8). The hydroxyl proton (δH 9.18) showed the relation with C-2, C-4, and C-6.
The HMBC correlation of NH proton (δH 8.16) of Phe6 residue with the carbonyl carbon of
Tyr1 (δC 172.8) further supported their linkage. In case of Ile2 residue, α CH (δH 4.10)
exhibited the correlation with the β CH (δC 36.3), γ CH2 (δC 24.0), γ’ CH3 (δC 10.82) and
carbonyl carbon (δC 170.5). The γ’ methyl protons (δH 0.70) were correlated with α carbon
(δC 56.6) and NH proton (δH 7.88) was correlated with the carbonyl carbon (δC 167.69) of
next Phe3 residue.
The correlation of α CH proton (δH 4.33) with the β CH2 (δC 28.5) and β protons with C-1’
(δC 137.6), C-2’, C-6’, HMBC relation of the H-2’, H-6’ (δH 7.19) with the C-3’, C-5’ (δC
127.9) and C-4’ (δC 126.1) and correlation of NH proton with the carbonyl carbon (δC
170.6) of Pro4 provided the full characterization of Phe3 residue. For the Pro4 residue, the
correlation of α proton (δH 4.26) was observed with β carbon (δC 29.2) and carbonyl carbon
(δC 170.6). Another correlation of CH2 γ protons (δH 3.69, 3.44) with α carbon (δC 51.3)
was also present.
For Ile5 residue, CH α proton (δH 4.26) was correlated to β CH2 (δC 36.0), γ CH2 protons
(δH 1.79, 1.93) and carbonyl carbon (δC 169.0). Further, γ’ CH3 protons (δH 0.75) and σ
CH3 (δH 0.71) showed correlation with β CH (δC 36.0) and γ CH2 (δC 23.8). The NH proton

101
(δH 7.72) was correlated with the carbonyl carbon of the next Glu6 residue. For the Glu6
residue, α CH proton resonating at (δH 4.32) exhibit correlation with adjacent carbonyl
carbon (δC 167.69). Furthermore, β CH2 protons (δH 2.37, 2.38) were correlated with the γ
CH2 (δC 27.7). The NH proton was correlated with the CO group of the next Ala7 residue.
The correlation observed between the CH α proton at δH 3.80 with the methyl group at δC
47.2 and carbonyl carbon (δC 172.0) identified the Ala7 unit. The cis and trans
configuration around the Pro4 residue was assign on the basis of 13C chemical shift
difference as the 13C chemical shifts for Pro residue in cis and trans configurations vary
significantly both in small peptides and proteins. The signals of the β-C and γ-C atoms are
more separated in cis-Pro than trans-Pro peptide bond. If the 13C NMR chemical shift
difference between β-C and γ-C of proline is small then the Pro-peptide bond would be
trans and larger 13C chemical shifts difference of β-C / γ-C indicates the cis geometry. The
cyclic peptide 28 has proline residue at position 4 which was found to have trans Ile5-
Pro4 peptide bonds, respectively, as revealed by 13CNMR chemical shift differences of
Pro2 Cβ − Cγ i.e., (Pro4, Δδ Cβ (23.7) − Cγ (29.2) = 5.5. this trans geometry was further
confirmed by NOESY interaction (Figure-34).
Figure-32: Key HMBC Correlation of Cyclic Peptide 28
COSY correlations were observed for α proton (δH 4.42) with the β CH2 protons (δH 2.78,
2.94) and amide proton (δH7.95) of Tyr1 residue. In case of Ile2 residue, α proton (δH 4.10)

102
was correlated with the β methine proton (δH 1.62) and NH proton (δH 7.72). β CH proton
was further correlated with the adjacent methyl protons (δH 0.70). Phe3 residue had α
proton (δH 4.33) which was correlated with amide proton (δH 8.01). In case of Pro4 residue,
COSY correlation were observed between α CH proton (δH 4.26) and β CH2 protons (δH
1.03, 1.46), γ protons (δH 1.96, 1.86) and protons (δH 3.69, 3.44). Ile5 residue exhibited
correlation between α CH proton (δH 4.26) and β CH2 proton (δH 1.65), and γ’ CH3 proton
(δH 0.75), γ CH2 proton (δH 1.79, 1.93) and σ CH3 protons (δH 0.71). The COSY
correlations of Glu6α proton (δH 4.32) were observed with the amide proton (δH 7.72), and
β CH2 protons (δH 2.73, 2.38). The COSY correlations were also observed among CH
proton (δH 3.85) with the methyl group (δH 1.25) and NH group at (δH 8.14).
Figure-33: Key COSY Correlation of Cyclic Peptide 28
The NOESY spectrum showed correlation between Tyr1 Hα / Ile2 Hα, Tyr1 NH / Ile2 NH,
Ile2 Hα / Try1 NH, Pro4 Hδ / Ile5 Hα, Glu6 NH / Ala7 Hα. The presence ofcross peak
between Pro4 Hδ / Ile2 Hα confirmed the trans geometry of Pro4 / Ile5 peptide bond.

103
Figure-34: Key NOESY Correlation of Cyclic Peptide 28

104
Table 10: NMR Data of Peptide 28 (d6-DMSO, 500 MHz for 1H and 150 MHz for 13C)

105
2.3.4.Cyclic Peptide 29:
The cyclic peptide 29 had “7” L-amino acids linked by peptide bond in a cyclic form. The
peptide 29 was synthesized by substitution of Phe3 and Pro6 residues of stylissatin A with
Ala3 and Glu6 respectively, as shown in scheme-11.
The pure peptide was obtained as a white solid. IR (KBr disc) showed absorptions at 3297,
3063, 2400, 1731, 1219, 791, 628 cm-1. [α]D25 −49.5 (c 0.047, MeOH). Melting point was
found to be 185.2 0C. The molecular formula of cyclic peptide 29 was found to be
C43H59N7O10Na by means of high resolution matrix assisted laser desorption ionization
(MALDI) technique, which gave the pseudo-molecular ion [M+Na]+ at 856.4216 (calcd.
833.43) Deuterated solvent d6-DMSO was used for recording NMR spectra which revealed
the clear shifts of seven amide protons of peptide 29. The 1HNMR showed the protons
resonating at δ 8.18 (1H, Tyr1-NHCO), δ 8.07 (1H, Ile5-NHCO), δ 8.01 (1H, Ala3-NHCO),
δ 8.0 (1H, Phe7-NHCO), δ 7.80 (1H, Glu6-NHCO), δ 7.6 (1H, Ile2-NHCO). Tyr1 unit was
identified by aromatic signals (H-2 / H-6, δH 6.92, δC 129.1) (H-3 / H-5, δH 6.56, δC 114.7),
β CH2 (δH 2.82, 3.10, δC 37.9). The presence of two Ile residues were evident from CH
signals (δH 1.59, δC 36.5) (δH 1.73, δC 36.1) and CH2 (δH 0.91, 1.23, δC 24.0) (δH 1.50, 1.78,
δC 24.2) along with 4 CH3 signals (δH 0.62, δC 10.8) (δH 0.70, δC 15.1) (δH 0.81, δC 11.0) (δH
0.88, δC 14.9). Ala3 residue was identified by the β CH3 signal (δH 1.13, δC 17.5).

106
Scheme-11: Synthesis of Cyclic Peptide 29

107
Figure-35: Analytical HPLC Profile of Cyclic Peptide 29
Conditions: C4 RP-analytical column/ 60% ACN: H2O
Pro4 residue was characterized by δ proton (δH 3.52, 3.72, δC 47.1). β and γ CH2 methylene
groups (δH 1.03,1.23 δC 28.6) and (δH 1.79, 1.82, δC 24.3) respectively. The β CH2 (δH 2.22,
2.26 δC 30.2) and γ CH2 (δH 1.68, 1.98, δC 28.5) of Glu6 were also observed. Phe7 residue
showed the signals of aromatic moiety (H-2’ / H-6’, δH7.12, C-2’/ C-6’, δC 130.4), (H-3’ /
H-5’, δH 7.23, C-3’ / C-5’, δC 128.3) and (H-4’ δH 7.17, δC 126.7).
The structure was further confirmed by HMBC and COSY correlation. In HMBC spectra,
α proton of Tyr1 residue (δH 4.18) showed the correlation with the β methylene carbon (δC
37.9), C-1 (δC 135.9) and carbonyl carbon (δC 167.0). The H-2 / H-6 protons (δH 6.92) were
correlated with β carbon (δC 37.9), C-3 / C-5 (δC 114.7) and C-4 (δC 155.7). The hydroxyl
proton (δH 9.10) showed signal with the C-3, C-5 and C-4 of the ring. The NH proton (δH
8.18) showed correlation with the carbonyl carbon (δC 170.54) of the adjacent Ile2. α
proton of Ile2 residue (δH 4.02) showed correlation with β CH (δC 36.9), γ CH2 (δC 24.0), σ
CH3 (δC 15.1) and carbonyl carbon (δC 170.54) of its own. The δ methyl proton (δH 0.62)
were related with α CH (δC 56.2), β CH2 (δC 36.9) and γ CH2 (δC 24.0). The γ’ methyl (δH
0.70) also showed signal with β CH2 and γ CH2. The NH proton (δH 7.6) had correlation
with the carbonyl carbon (δC 172.0) of the adjacent Ala3.
The HMBC correlation observed for α proton (δH 4.18) with methyl group (δC 17.5) and
carbonyl carbon (δC 172.0) supported the Ala3 unit. While the amide proton (δH 8.01)
showed correlation with the carbonyl carbon (δC 166.3) of the next residue i-e Pro4.
The HMBC correlation supported the assignment of Pro4 residue. HMBC correlations were
observed from α proton (δH 3.65) with β carbon (δC 28.4) and carbonyl carbon (δC 166.3). γ

108
CH2 protons (δH 1.79, 1.82) exhibited correlation with β CH2 (δC 28.4) and α CH (δC 52.8).
While CH2 protons (δH 3.52, 3.72) showed correlation with β carbon (δC 28.47). The
HMBC correlation further supported the assignments of Ile5 residue. α proton (δH 4.28)
exhibited interaction with β CH (δC 36.1) and carbonyl carbon (δC 170.0). γ’ CH3 (δH 0.81)
showed correlation with β CH (δC 36.1) and γ CH2 (δC 24.2). The correlation of methyl
with α CH (δC 54.9), β CH (δC 36.1), and γ CH2 as well as correlation of NH proton (δH
8.07) with carbonyl carbon (δC 172.0) supported the Ile5 residue. For the Glu6 residue,
HMBC correlation of NH proton with carbonyl carbon (δC 172.0) was observed. The
HMBC correlation of α CH proton of Phe7 (δH 4.51) with β CH2 (δC 37.9) were observed.
The β CH2 (δH 2.81, 3.15) also showed the correlation with the C-1’ (δC 135.9) and C-2’ /
C-6’ (δC 130.2) of the phenyl ring. HMBC correlation of the H-2’/ H-6’ (δH 7.12) with the
β CH2, C-3’/ C-5’ (δC 127.9) and C-4’ (δC 126.7), while H-4’ (δH 7.16) interaction with C-
2’/ C-6’ (δC 130.2) was also observed. The peptide 29 also has proline residue at position
4 so the 13CNMR chemical shift differences of Pro4 Cβ − Cγ i.e., (Pro4, Δδ Cβ (28.9) −
Cγ (24.3) = 4.6 indicated the trans peptide bond between Ile5- Pro4.
Figure-36: Key HMBC Interactions of Cyclic Peptide 29
The COSY interaction of α CH proton (δH 4.18) with β CH2 protons (δH 2.82, 3.10) and
amide protons (δH 8.18) along with the interaction of H-2 / H-6 (δH 6.92) with H-3 / H-5
(δH 6.56) supported Tyr1 residue. α proton (δH 4.08) of Ile2 displayed the COSY correlation
with the CH proton (δH 1.59) and also with amide proton (δH 7.65). The methine proton

109
further had correlation with γ’ methyl (δH 0.62). The COSY correlation observed for the
proton resonance (δH 4.20) with both the methyl group (δH 1.13) and the NH group at (δH
8.19) supported the Ala3 unit. α proton (δH 3.65) of Pro4 was correlated with the β CH2
protons (δH 1.03, 1.23). While α proton (δH 4.31) of Ile5was correlated with the β CH2
proton (δH 1.73). The methine proton further showed correlation with γ’ methyl (δH 0.81).
α proton showed correlation with amide proton (δH 8.07). The correlation between γ
protons (δH 1.50, 1.78) and δ CH3 protons (δH 0.88) were also observed. The COSY
correlations for Glu6 residue between α CH proton (δH 4.37) and amide proton (δH 7.80)
were also observed. α proton of Phe7 showed the correlation with the β CH2 protons (δH
2.81, 3.12) and amide proton (δH 8.0).
Figure-37: Key COSY Correlations of Cyclic Peptide 29
The trans geometry of Ile5-Pro4 amide bond was further confirmed by NOESY correlation
spectrum that showed prominent cross peaks of Pro4 Hδ / Ile5 Hα. Other NOESY
correlations were observed among Tyr1 Hα / Phe7 NH, Ile2 NH / Ala3 Hα, Ile5 Hα / Glu6
Hα, Glu6 NH / Phe7 NH, Tyr1 Hα / Ile2 NH, Ile5 NH / Glu6 NH.

110
Figure-38: Key NOESY Correlation of Cyclic Peptide 29

111
Table 11: NMR Data of Peptide 29 (d6-DMSO, 500 MHz for 1H and 150 MHz for 13C)

112
2.3.5. Cyclic Peptide 30:
The cyclic peptide 30 had “7” amino acids all having “L” configuration. . The peptide 30
was synthesized by substitution of Tyr1 and Pro6 residues of stylissatin A with Ala1 and
Glu6 respectively as shown in scheme-12.
The pure peptide was obtained as a white solid. IR (KBr disc) showed absorptions at 3303,
3067, 2400, 1706, 1219, 791, 628 cm-1. [α]D25 −71.1 (c 0.06, MeOH). Melting point was
found to be 171.5 0C. The molecular formula was found to be C43H59N7O9Na by high
resolution matrix assisted laser desorption ionization (MALDI) technique, which gave the
pseudo-molecular ion [M+Na]+ at 840.4266. Deuterated solvent d6-DMSO was used for
recording NMR spectra because the amide protons signals appeared very clear in DMSO.
1HNMR showed seven amide protons, resonating at δ 7.99 (1H, overlap, Ala1-NHCO), δ
7.80 (1H, overlap, Ile2-NHCO), δ 7.89 (1H, m, Phe3-NHCO), δ 8.01 (1H, b, Ile5-NHCO), δ
8.01 (1H, b, Glu6-NHCO), δ 7.89 (1H, m, Phe7-NHCO). α protons of all constituent amino
acid appeared between δH 4.12 and δH 4.48. In analogue 30, Ala1 was used to substitute
Tyr1 of original sequence of stylissatin A, and it was evident by the appearance of CH3
signal at δH1.12, δC 18.1.

113
Scheme-12: Synthesis of Cyclic Peptide 30

114
Figure-39: Analytical HPLC Profile of Cyclic Peptide 30
Conditions: C4 RP-analytical column/ 60% ACN: H2O
The two CH signals (δH 1.65, δC 36.1) and (δH 1.68, δC 36.1) along with the two CH2
signals (δH 1.35, 1.01, δC 24.1), (δH 1.71, 1,68, δC 24.1) and four methyl signals (δH 0.73, δC
14.9), (δH 0.72, δC 10.8), (δH 0.81, δC 15.1), (δH 0.84, δC 10.9) confirmed the presence of
two isoleucine residues. The presence of two phenyl alanine residues was evident by the
aromatic proton signals, for Phe3, H-2 / H-6 (δH 7.12, δC 130.3), H-3 / H-5 (δH 7.19, δC
129.1) H-4 (δH 7.14, δC 126.1), β CH2 (δH 2.77, 2.83 δC 37.2), while for Phe7 residue the
signals of aromatic protons observed at (δH 7.14, δC 127.9) for H-2’ / H-6’, (δH 7.19, δC
129.2) for H-3’ / H-5’ and (δH 7.18, δC 126.1) for H-4’. For Glu6 the CH2 resonances (δH
2.22, 2.25, δC 29.7) (δH 1.72, 1.85, δC 28.6) were observed. The structure was further
confirmed by the HMBC and COSY correlation. The correlation of α proton (δH 4.29) of
Ala1 residue to the β methyl carbon (δC 18.1) and carbonyl carbon (δC 172.8) was observed.
The correlation of amide proton (δH 7.99) with carbonyl carbon (δC 170.6) of next Ile2 was
also present. For the Ile2 residue, α proton (δH 4.12) displayed the correlation with the β
CH (δC36.1), γ CH2 (δC 24.1) and γ’ CH3 (δC 14.9). The γ CH2 protons (δH 1.35, 1.01)
showed the correlation with the δ CH3 (δC 10.8). The γ’ CH3 protons (δH 0.73) was
correlated to β CH (δC 36.1) and α carbon (δC 56.6). The NH proton of Ile2 is correlated
with carbonyl carbon (δC 171.8) of adjacent Phe3 residue.
In the Phe3 residue, the correlation of α proton (δH 4.48) with β- methylene carbon (δC
37.2) and with the carbonyl carbon (δC 171.8) was detected. The β methylene protons
showed the correlation with α carbon (δC 53.5), C-1 (δC 137.5) and C-2 / C-6 (δC 130.3) of
the phenyl ring. The NH proton is correlated with the carbonyl carbon (δC 170.3) of Pro4.

115
For the Pro4 residue, correlation of α proton (δH 4.30) is seen with the β CH2 (δC 28.5) and
γ CH2 (δC 24.2). β proton (δH 1.77, 1.92) and γ protons (δH 1.48, 1.13) were correlated to
the δ carbon (δC 47.12). For the Ile5 residue, the correlation of α proton (δH 4.30) with the β
CH (δC 36.1), γ CH2 (δC 24.1) and with the carbonyl carbon (δC 170.3) of Ile5 was
observed. Correlation of amide proton (δH 8.01) is observed with the carbonyl carbon (δC
170.3) of the adjacent amino acid. For the Glu6 residue, α proton (δH 4.34) had correlation
with carbonyl carbon (δC 170.7). The correlation of β protons (δH 2.22, 2.25) was observed
with the carbonyl carbon (δC 173.8) of acidic group and γ carbons (δC 28.6), while the NH
proton (δH 8.01) showed the correlation with the carbonyl carbon (δC 171.3) of adjacent
Phe7. For Phe7 residue, the HMBC correlation of α proton was observed with the β
methylene carbon (δC 37.9) and carbonyl carbon (δC 171.3). The correlation of β protons
with the C-1’ (δC 137.5), with C-2’ / C-4’ (δC 127.9) and with α carbon (δC 4.48) were also
present and the NH proton (δH 7.89) showed the signals with the carbonyl carbon of the
Ala1 (δC 172.8). The peptide 30 has proline residue at position 4 and the 13CNMR chemical
shift differences of Pro4 Cβ − Cγ i.e., (Pro4, Δδ Cβ (28.5) − Cγ (24.2) = 4.3 which indicated
its trans geometry around the Ile5-Pro4 amide bond.
Figure-40: Key HMBC Interactions of Cyclic Peptide 30

116
In COSY spectrum α proton (δH 4.29) of Tyr1 showed the cross peaks with β proton (δH
1.12) and NH proton (δH 7.99). For Ile2 residue, α proton had the correlation with the β CH
(δH 1.65) and NH proton (δH 7.80). β CH further correlated with the γ’ CH3 (δH 0.72). γ
CH2 exhibited the relation with the δ CH3 (δH 0.75). For Phe3 residue, α proton (δH 4.48)
showed the correlation with β protons (δH 2.77, 2.83) and with the NH proton (δH 7.89).
For Pro4, α proton (δH 4.34) was correlated with the β protons (δH 1.77, 1.92). For Ile5
residue, α proton (δH 4.30) was correlated with the β proton (δH 1.68) and NH proton (δH
8.04). β proton is further correlated with the γ’ CH3 (δH 0.81). The γ methylene protons (δH
1.73, 1.84) are further correlated to the δ methyl protons (δH 0.84). For Glu6 COSY
correlation of α proton (δH 4.30) with the NH (δH 8.01), β protons (δH 2.22, 2.25) with the γ
methylene protons (δH 1.72, 1.85) was observed. For Phe7, α proton (δH 4.48) showed the
relation with the β protons (δH 2.78, 2.96) and NH proton (δH 7.89).
Figure-41: Key COSY Correlation of Cyclic Peptide 30

117
The NOESY spectrum indicated the key correlation of Ala1 NH / lle2 Hα, Ile2 Hα / Phe3
NH, Ile2 NH / Phe3 Hα, Phe3 NH / Pro4 Hα, Pro4 Hδ / Ile5 Hα, Glu6 NH / Phe7 Hα, Phe7 Hα
/ Ala1 NH.
Figure-42: Key NOESY Correlations of Cyclic Peptide 30

118
Table-12: NMR Data of Peptide 30 (d6-DMSO, 500 MHz for 1H and 150 MHz for 13C)

119
2.3.6. Cyclic Peptide 31:
The cyclic peptide consists of 7 amino acids building blocks all having “L” configuration.
The peptide 31 was synthesized by substitution of Ile2 and Pro6 residues with Ala2 and Glu6
respectively, as shown in scheme-13.
The pure peptide was obtained as a yellow solid. IR (KBr disc) showed absorptions at
3302, 3062, 2400, 1732, 1245, 752, 628 cm-1. [α]D25 −60.36 (c 0.055, MeOH). Melting
point was found to be 172.6 0C. The molecular formula of analogue 31 was found to be
C46H57N7O10Na by high resolution matrix assisted laser desorption ionization (MALDI)
technique, which exhibited the pseudo-molecular ion [M+Na]+ at m/z 890.4059 (calcd.
866.4214) Deuterated solvent d6-DMSO was used for recording NMR spectra. 1HNMR
showed seven amide protons, resonating at δH 8.02 (1H, overlap, Tyr1-NHCO), δH 7.90
(1H, overlap, Ala2-NHCO), δH 7.88 (1H, m, Phe3-NHCO), δH 8.07 (1H, b, Ile5-NHCO), δ
8.07 (1H, b, Glu6-NHCO), δ 7.8 (1H, m, Phe7-NHCO), Pro4 does not contain any amide
proton. The α proton of all constituent amino acid appears between (δH 4.17 and δH 4.51).
Tyr1 residue had the β CH2 at (δH 2.62, 2.85 δC 37.32) and aromatic protons, H-2 / H-6 at
(δH 6.92, δC 130.03), H-3 / H-5 at (δH 6.58, δC 114.78).

120
Scheme-13: Synthesis of Cyclic Peptide 31

121
Figure-43: HPLC Profile of Cyclic Peptide 31
Conditions: C4 RP-analytical column/ 60% ACN: H2O
Ala2 was confirmed by CH3 signal (δH 1.09, δC 18.2). The two phenyl alanine present at
position “3” and “7” were confirmed by the aromatic proton signals for Phe3, H-2ʹ / H-6ʹ
(δH 7.10, δC 130.2), H-3ʹ / H-5ʹ (δH 7.25, δC 129.2) H-4ʹ (δH 7.28, δC 128.0), CH2 (δH 2.78,
2.99 δC 37.3), while for Phe7 the signals of aromatic protons located at (δH 7.14, δC 130.0)
for H-2ʹʹ / H-6ʹʹ, (δH 7.07, δC 128.5) for H-3ʹʹ / H-5ʹʹ and (δH 7.15, δC 127.9) for H-4ʹʹ and
CH2 was present at (δH 2.83, 3.01, δC 37.97). The CH signal at (δH 1.70, δC 36.0) along with
CH2 signal at (δH 1.05, 1.48, δC 24.3) and two methyl signals at (δH 0.78, δC 10.8), (δH 0.82,
δC 14.9) confirmed the presence of isoleucine residue at position “5”. Glu6, had two CH2
(δH 2.26, 2.28, δC 29.8) and (δH 1.75, 1.86, δC 28.2). This structure was further confirmed
by the HMBC and COSY interactions.
The HMBC correlation of α proton (δH 4.34) of Tyr1 residue was observed with the β
methylene (δC 37.32), and with its carbonyl carbon (δC 170.4). While β methylene protons
were related with C-1 (δC 135.9) and C-2 / C-6 (δC 129.19) of the phenyl ring. H-2 / H-6
(δH 6.92) showed correlation with C-3 / C-5 (δC 114.7), and C-4 (δC 155.7). The hydroxyl
proton was related with C-4 and C-3 / C-5 of ring. Ala2 unit was identified by HMBC
correlation of α proton (δH 4.17, δC 48.6) with both the methyl group (δH 1.09, δC 18.2) and
with the carbonyl carbon (δC 172.8). For Phe3 residue, α proton (δH 4.38) was correlated
with the β CH2 (δC 37.3) and with its carbonyl carbon (δC 169.9). The β protons (δH 2.76,
2.99) were further related with α carbon (δC 54.0), with the C-1ʹ of the phenyl ring (δC
135.9) and with the C-2ʹ / C-6ʹ (δC 130.2). For Pro4, no signal is observed. For Ile5 residue,
signal of α proton (δH 4.29) was correlated with the β methine (δC 36.0), γ methylene (δC

122
24.3), γ’ methyl (δC 14.9) and with its carbonyl carbon (δC 170.6), β CH was also
correlated with the γ CH2. For Phe7 interaction of α proton (δH 4.51) was observed with the
β CH2 (δC 37.97) and with its carbonyl carbon (δC 170.8). The β CH2 of phe7 (δH 2.83, 3.01,
δC 37.97) was correlated with α carbon (δC 53.4) and with the C-1 of the phenyl ring (δC
135.9). The peptide 31 has proline residue at position 4 and the 13CNMR chemical shift
differences of Pro4 Cβ − Cγ i.e., (Pro4, Δδ Cβ (29.0) − Cγ (24.1) = 4.9 which indicates the
trans orientation of the Pro4-Ile5 peptide bond.
Figure-44: Key HMBC Interactions of Cyclic Peptide 31
For Tyr1, the COSY correlation of α proton (δH 4.34) was observed with the β protons (δH
2.62, 2.83) and NH proton (δH 8.02). The correlations between H-2 / H-6 (δH 6.92) and H-3
/ H-5 (δH 6.58) were also observed. α proton (δH 4.17) of Ala2 was correlated with both the
methyl (δH 1.09) and NH proton (δH 7.9). α proton (δH 4.38) of Phe3 was observed with
that of NH proton (δH 7.88). α proton of Ile5 exhibited relation with the methine proton (δH
1.70) and NH proton (δH 8.07), while CH2 protons (δH 1.05, 1.48) showed interaction with
δ CH3 protons (δH 0.78). For glu6 residue, COSY correlation between β protons (δH 2.26,
2.28) and γ protons (δH 1.75, 1.86) was present. For Phe7, residue COSY correlation of α
proton (δH 4.51) was observed with NH proton (δH 7.8).

123
Figure-45: Key COSY Correlations of Cyclic Peptide 31
The NOESY spectrum showed cross peaks between Tyr1 Hα / Ile2 Hα, Tyr1 NH / Ile2 NH,
Tyr1 NH / Ile2 Hα, Pro4 Hδ / Ile5 Hα and Glu6 NH / Ala7 Hα.
Figure-46: Key NOESY Correlations of Cyclic Peptide 31

124
Table 13: NMR Data of Peptide 31 (d6-DMSO, 500 MHz for 1H and 150 MHz for 13C)

125
Table-14: Mass Spectroscopic Analysis and [α]D of Cyclic Peptides 26-31.
Table-15: Effect of Peptides on Oxidative Burst, Nitric Oxide (NO.) and IL-2
Production.
2.4. Immunomodulatory Activities of Stylissatin A analogues:
To evaluate the structure activity relationship, six analogs 26–31 were synthesized
(scheme-7) and evaluated for their anti-inflammatory potential by investigating different
immune parameters including effects on production of NO, intracellular ROS, and IL-2

126
cytokine (Table-15). The natural product was good inhibitor of NO (IC50 63.0 ± 5.4 μM) as
compared to the standard drug (IC50 97.5 ± 3.2 μM) (Kita et al., 2013).
The analogues 26-31 showed weak inhibitory activity of NO. and more potent inhibition of
interleukin 2 production compared to parent peptide. It also appeared that by substitution
of both proline residues (Pro4 and Pro6) of natural peptide stylissatin A, with Ala and Glu
respectively, the resulting peptide analogue 26 was found to be a strong inhibitor of ROS
with no inhibition of NO.. It potently inhibited ROS from neutrophils as well as strongly
inhibited the release of cytokine interleukin 2 (IC50 13.4 ± 0.1 μM). All analogues were
found more potent inhibitor of IL-2 production compared to the synthetic natural product.
This indicated that both proline residues are involved in the mechanism of NO. inhibition.
The peptide analogue 31, in which Ile2 and Pro6 were replaced by Ala and Glu,
respectively, was found to be the most potent inhibitor of interleukin 2 (IC50 6.0 ± 0.9 μM)
releases among the peptides 26-31. The results obtained from current studies showed
significant anti-inflammatory potential of analogues 26-31. All peptides were also
evaluated against different cell lines including MCF-7, NCI-H460, DoHH2, HeLa as well
as 3T3 cell lines. It was releaved that the stylissatin A analogues 26 – 31 were non
cytotoxic to all standard cell lines
2.5. Conclusion:
The synthesis and anticancer screening of cyclic peptide libraries has led to discovery of
many hit compounds including anticancer compounds 2, 14, 15, 16 and anti-inflammatory
peptides 26-31. All these peptides were found non cytotoxic to normal cell lines (3T3
normal mouse cell line), thus they have potential as more safe new medicinal agents
against cancer and inflammation. gfggfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfgfff

Chapter # 3
EXPERIMENTAL
PROCEDURES

128
3.1. General Experimental Details:
The protected amino acids, resins, coupling reagents and solvents were purchased from
Sigma Aldrich, Novabiochem and Chem- impex. Chromatographic solvents were synthetic
grade and were filtered through 0.45 µm and degassed by sonication prior to use. RP-
HPLC was done on (LC-900 Japan). C-18 Column Jaigel ODS-MAT 80 with a flow rate
of 4 ml / min using ACN: H2O (60: 40). IR spectra were recorded on shimadzu® Japan,
FTIR-8900 Fourier Transform infrared spectrophotometer. Optical rotation was
determined on P-2000 polarimeter JASCO® Japan. UV-Visible spectra were recorded at
Thermoscientific® Evolution 300 UV-visible spectrophotometer. NMR spectra were
recorded at Bruker® and Avance AV-600 Nuclear Magnetic Resonance Spectrometers were
used for recording 1H (600 MHz) and 13C NMR spectra (150 MHz), operating NMR
solvent was d6- DMSO. Chemical shifts were reported in parts per million (ppm). Matrix-
assisted laser desorption ionization (MALDI) mass spectra were recorded on Ultraflex III
TOF/TOF (Bruker Daltonics, Bremen, Germany).
3.2. General Synthesis Procedure for Cyclic Peptide Library I:
Peptide 1 to 25 was synthesized by solid-phase chemistry by using Fmoc protocol. The
sequences of all peptides are shown in Table 1 (a). The resin was soaked in synthetic grade
DCM for 2 hours for thorough exposure of reaction sites. The first amino acid was loaded
by using the oxyma pure and DIC as the coupling agents. The reaction mixture was kept
under gentle shaking for two hours at room temperature. The coupling was confirmed by
the ninhydrin test. The Fmoc deprotection was achieved by treating peptide-bound resin
with 20% 4-methylpiperidine in DMF for 30 min. Peptide chain was further elongated by
using Fmoc protected amino acids. Each cyclic peptide was composed of eight residues.
As the linear sequence was synthesized, the acid terminal having allyl protecting group and
amine terminal having alloc group were deprotected by a mixture of palladium catalyst
[tetrakistripehnylphosphine palladium (0)] in DCM with acetic acid and N-methyl
morpholine (37: 2: 1) respectively. The macrocyclization was carried out between free
acid and amino groups by using oxyma pure and DIC. After the deprotection of terminal
Fmoc group by 20% 4-methylpiperidine, the resin was treated with 90% TFA cocktail
(TFA: Phenol: H2O: TIPS, 90: 4: 4: 2) for simultaneous cleavage of peptide from resin and

129
removal of side chain protecting groups. Extensive washing was carried out after each
coupling and deprotection step by DMF, DCM, MeOH, and DCM, respectively. The crude
reaction mixture was treated with cold ether and peptide was obtained in the form of
precipitate.
3.3. Synthesis of Biologically Active Compounds:
3.3.1. Synthesis of Cyclic Peptide-2:
Compound 2 was synthesized by solid-phase chemistry using Fmoc protocol on Rink
amide AM resin (130 µm, 2.5 g, 0.51 mmol / g). The resin was washed and soaked in
solvent as described in the general procedure. Fmoc-D-phe-OH (100.7 mg, 0.25 mmol)
was loaded by using the oxyma pure (36.9 mg, 0.25 mmol) and DIC (0.037 ml, 0.26
mmol). The reaction mixture was kept on shaking for two hours and reaction progress was
monitored by the ninhydrin test. The Fmoc deprotection was done by passing a solution of
20% 4-methylpiperidine in DMF (30 min) into the reaction vessel containing peptide-
bound resin. Peptide chain was further elongated with Fmoc-Val-OH (88.2 mg, 0.25
mmol), Fmoc-Glu-α-allyl-OH (106.4 mg, 0.25 mmol), Fmoc-Lys(Boc)-OH (121.8 mg,
0.25mmol), Fmoc-Orn(Boc)-OH (118.1mg, 0.26 mmol) Fmoc-D-phe-OH (100.7 mg, 0.25
mmol), Fmoc-Lys(Boc)-OH (121.8mg , 0.25 mmol) and the peptide sequence was
completed with Fmoc-Lys (Alloc)-OH (117.6 mg, 0.25 mmol). The side chain NH2 group
of lysine and α acidic group of glutamic acid was deprotected by palladium (0) catalyst in
DCM with acetic acid and N-methylmorpholine (37: 2: 1). Cyclization was achieved
between acid group of glutamic acid and amino group of lysine side chain by using oxyma
pure and DIC. The resin was extensively washed with DMF, DCM, MeOH, and DCM
respectively, after each coupling / deprotection step. The Fmoc group of terminal residue
Lys was removed by 20% 4-methylpiperidine and resin bound peptide was treated with
90% TFA cocktail (TFA: phenol: H2O: TIPS, 90: 4: 4: 2) for two hours for simultaneous
cleavage of side chain protecting groups and release of crude peptide from the resin. The
peptide was precipitated out with cold ether and further purified by HPLC using isocratic
solvent system of 60% acetonitrile: water. The structure of purified product 2 was
elucidated by interpretation of spectral data.

130
3.3.1.1. Characterization Data of Peptide 2:
3.3.2. Synthesis of Cyclic Peptide 14
Compound 14 was prepared by solid-phase synthesis by using Fmoc chemistry on Rink
amide AM resin (130 µm, 2.5 g, 0.51 mmol / g). After soaking of resin in DCM, the first
amino acid Fmoc-Glu-α-allyl-OH (106.4 mg, 0.25 mmol), was loaded using oxyma pure
(36.9 mg, 0.25 mmol) and DIC (0.037 ml, 0.26 mmol) as coupling agents. The reaction
was gently shaked for two hours. The coupling reactions were driven to completion and
confirmed by the ninhydrin test. The Fmoc deprotection was carried out by passing a
solution of 20% 4-methylpiperidine in DMF (30 min) into the reaction vessels containing
peptide-bound resin. Peptide chain was further elongated by coupling with Fmoc-D-tyr
(OtBu)-OH (119.4 mg, 0.25 mmol), and three consecutive couplings of Fmoc-Arg(Pbf)-
OH (168.68 mg, 0.25 mmol for each coupling), followed by coupling with Fmoc-
Lys(Alloc)-OH (117.65 mg, 0.25 mmol). Alloc group of lysine and allyl group of glutamic
acid were removed by using palladium (0) catalyst in the presence of DCM, acetic acid
and N-methylmorpholine (37: 2: 1). Cyclization was carried out between acid group of
glutamic acid and amino group of lysine side chain by using oxyma pure and DIC. The
resin was thoroughly washed after each coupling / deprotection with DMF, DCM, MeOH,
and DCM. Finally, Fmoc group of Lys was removed by 20% 4-methylpiperidine and resin
bound peptide was treated with 90 % TFA mixture (TFA: phenol: H2O: TIPS, 90: 4: 4: 2)
for two hours for simultaneous deprotection of side chain protecting groups and release of
crude cyclic peptide from the resin. The peptide was precipitated out with cold ether and

131
further purified by HPLC using isocratic solvent system of 60% acetonitrile: water. The
structure of purified product 14 was elucidated by interpretation of spectral data.
3.3.2.1. Characterization Data of Peptide 14:
3.3.3. Synthesis of Cyclic Peptide 15:
Compound 15 was prepared by solid-phase synthesis by using Fmoc chemistry on Rink
amide AM resin (130 µm, 2.5 g, 0.51 mmol / g). After soaking of resin in DCM, the first
amino acid Fmoc-Glu-α-allyl-OH (106.4 mg, 0.25 mmol), was loaded using oxyma pure
(36.94 mg, 0.25 mmol) and DIC (0.037 ml, 0.26 mmol). The reaction was gently shaked
for two hours. The coupling reaction was driven to completion and confirmed by the
ninhydrin test. The Fmoc deprotection was achieved by passing a solution of 20% 4-
methylpiperidine in DMF into the reaction vessels containing peptide-bound resin. Peptide
chain was further extended with Fmoc-Asn-OH (155.14 mg, 0.25 mmol), Fmoc-Lys(Boc)-
OH (121.3 mg, 0.25 mmol), Fmoc-Arg(Pbf)-OH (168.6 mg, 0.25 mmol), Fmoc-Lys(Boc)-
OH (121.3 mg, 0.25 mmol), Fmoc-Lys(Alloc)-OH (117.6 mg, 0.25 mmol), Fmoc-D-
tyr(tBu)-OH (119.47 mg, 0.25 mmol) and Fmoc-D-ile-OH (91.88 mg, 0.25 mmol). Allyl
protecting groups of lysine and glutamic acid were deprotected by palladium (0) catalyst in
DCM with acetic acid and N-methylmorpholine (37: 2: 1). Cyclization was carried out
between -acidic functionality of glutamic acid and ϵ amino group of lysine side chain by
using oxyma pure and DIC. The resin was washed after each coupling / deprotection step.
The terminal NH2 group of D- ile was deprotected by 20% 4-methylpiperidine and resin

132
bound peptide was treated with 90% TFA cocktail (TFA: phenol: H2O: TIPS, 90: 4: 4: 2)
for two hours for the simultaneous cleavage of peptide from the resin and side chain
deprotection. The peptide was precipitated out with cold ether and further purified by
HPLC using isocratic solvent system of 60% acetonitrile in water. The structure of purified
product 15 was elucidated by interpretation of spectral data.
3.3.3.1. Characterization of Peptide 15:
3.3.4. Synthesis of Cyclic Peptide 16:
Compound 16 was prepared by solid-phase synthesis by using Fmoc chemistry on Rink
amide AM resin (130 µm, 2.5 g, 0.51 mmol / g). After soaking of resin in DCM, the first
amino acid Fmoc-Glu-α-allyl-OH (106.4 mg, 0.25 mmol), was loaded by using PyBop
(benzotriazol-1-yl-oxy-tris(pyrrolidino)phosphoniumhexafluoro-phosphate) (81.1 g, 0.156
mmol) and N,N-diisiopropylethylamine (DIEA, 44.5 µl, 0.26 mmol. The reaction was
gently shaked for two hours. The coupling of amino acid was confirmed by ninhydrin test.
The Fmoc deprotection was performed by passing a solution of 20% 4-methylpiperidine in
DMF for 30 min into the reaction vessels containing peptide-bound resin. Peptide chain
was further extended by coupling with Fmoc-Asn-OH (2.37g, 3.9 mmol), Fmoc-Lys(Boc)-
OH (1.86 g, 3.9 mmol) Fmoc-Arg(Pbf)-OH (2.58 g, 3.9 mmol), Fmoc-Lys(Boc)-OH (1.86
g, 3.9 mmol), Fmoc-Lys(Alloc)-OH (1.80 g, 3.9 mmol), Fmoc-D-tyr(tBu)-OH (1.82g, 3.9
mmol), and Fmoc-d-ile-OH (1.40g, 3.9 mmol). ϵ NH2 group of lysine and α acid group of
glutamic acid was deprotected by palladium (0) catalyst in DCM with acetic acid and N-

133
methylmorpholine (37: 2: 1). Cyclization occurs between acid group of glutamic acid and
NH2 group of lysine side chain using oxyma pure and DIC. The Fmoc group of D-ile was
removed by 20% 4-methylpiperidine and peptide conjugation with temporin ICea peptide
was initiated by coupling with Fmoc-Phe-OH (1.80 g, 3.9 mmol), Fmoc-Ile-OH( 1.40 g,
3.9 mmol), Fmoc-Ser(tBu)-OH (1.52 g, 3.9 mmol), Fmoc-Asn(trt)-OH (2.37 g, 3.9 mmol),
Fmoc-Ile-OH (1.40 g, 3.9 mmol), Fmoc-Ile-OH (1.40 g, 3.9 mmol), Fmoc-Asn(trt)-OH
(2.37 g, 3.9 mmol), Fmoc-Ala-OH (1.23 g, 3.9 mmol), Fmoc-Ile-OH (1.40 g, 3.9 mmol),
Fmoc-Lys(Boc)-OH (1.86 g, 3.9 mmol), Fmoc-Lys(Boc)-OH (1.86 g, 3.9 mmol), Fmoc-
Leu-OH (1.40 g, 3.9 mmol), Fmoc-Asp(tBu)-OH (1.63 g, 3.9 mmol), Fmoc-Val-OH (1.35
g, 3.9 mmol), Fmoc-Phe-OH (1.80 g, 3.9 mmol). The NH2 group of Phe residue was
deprotected and the resin was treated with 90% TFA mixture (TFA: EDT: H2O: TIPS, 94:
2.5: 2.5: 1) for the simultaneous cleavage of peptide from resin and removal of all side
chain protecting groups. The cold diethyl ether was used for precipitation of peptide and
further purified by HPLC using isocratic solvent system of 60% acetonitrile in water. The
structure of purified product 16 was elucidated by interpretation of spectral data.
3.3.4.1. Characterization of Peptide 16:
3.4. Synthesis Procedure for Library 2 (stylissatin A Analogues):
Peptide 26 to 31 was synthesized by solid-phase chemistry by using Fmoc protocol on 4-
benzyloxybenzyl alcohol resin (Wang resin). The detail synthetic procedure was discussed
below.
3.4.1. Synthesis of Cyclic Peptide 26:
Peptide analogue 26 was synthesized by using Fmoc solid-phase peptide synthesis protocol
on 4-benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g).

134
(Chem-impex Int. INC. USA). The properly soaked resin was loaded with first amino acid
Fmoc-Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g,
13.4 mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9
mg, 0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling
was confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Phe-OH (2.0 g, 5.39 mmol), Fmoc-
Tyr(tBu)-OH (2.6 g, 5.4 mmol), Fmoc-Ile-OH (1.9 g, 5.4 mmol), Fmoc-Phe-OH (2.0 g,
5.39 mmol), Fmoc-Ala-OH (1.6 g, 5.1 mmol), Fmoc-Ile-OH (1.9 mg, 5.4 mmol) were
sequentially attached to the first residue Glu. The deprotection of allyl group of glutamic
acid was carried out by using palladium (0) in DCM, acetic acid and N-methylmorpholine
(37: 2: 1) for 24 hours. The terminal NH2 group of Ile residue was deprotected by 20 % 4-
methylpiperidine and cyclization was allowed between α acidic group of glutamic acid and
amino group of Ile by using oxyma pure and DIC. The resin was thoroughly washed after
each coupling / deprotection step. The cyclized product was cleaved from resin by 95%
TFA in DCM / TIS (few drops). The cleaved product was precipitated out by cold diethyl
ether and precipitate was further washed with ether and purified by Recycling HPLC using
isocratic solvent system of acetonitrile/ water (ACN: H2O 6: 4) 0.1% TFA. The structure
of pure peptide 26 was elucidated by interpretation of spectral data.
3.4.1.1. Characterization Data of Peptide 26:

135
3.4.2. Synthesis of Cyclic Peptide 27:
Peptide analogue 27 was synthesized by using Fmoc solid-phase peptide synthesis protocol
on 4-benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g).
(Chem-impex Int. INC. USA). The properly soaked resin was loaded with first amino acid
Fmoc-Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g,
13.4 mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9
mg, 0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling
was confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Ile-OH (1.9 g, 5.4 mmol), Fmoc-
Ala-OH (1.6 g, 5.1 mmol), Fmoc-Phe-OH (2.0 g, 5.39 mmol), Fmoc-Tyr(tBu)-OH (2.6 g,
5.4 mmol), Fmoc-Ile-OH (1.9 mg, 5.4 mmol) and Fmoc-Phe-OH (2.0 g, 5.39 mmol). Acid
group of glutamic acid was deprotected by using palladium (0) in DCM, acetic acid and N-
methylmorpholine (37: 2: 1) for 24 hours. The terminal NH2 group of Phe residue was
deprotected by 20% 4-methylpiperidine and cyclization was allowed between α acidic
group of glutamic acid and amino group of Phe using oxymapure and DIC. The resin was
thoroughly washed after each coupling / deprotection step. The cyclized product was
cleaved from resin by 95% TFA in DCM with TIS (few drops). The cleaved product was
precipitated out by cold diethyl ether and precipitate was further washed with ether and
purified by Recycling HPLC using isocratic solvent system of acetonitrile/ water (ACN:
H2O 6: 4) 0.1% TFA. The structure of pure peptide 27 was elucidated by interpretation of
spectral data.
3.4.2.1. Characterization Data of Peptide 27:

136
3.4.3 Synthesis of Cyclic Peptide 28.
Peptide 28 was synthesized by using Fmoc solid-phase peptide synthesis protocol on 4-
benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g). (Chem-
impex Int. INC. USA). The properly soaked resin was loaded with first amino acid Fmoc-
Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g, 13.4
mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9 mg,
0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling was
confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Ala-OH (1.6 g, 5.1 mmol), Fmoc-
Tyr(tBu)-OH (2.6 g, 5.4 mmol), Fmoc-Ile-OH (1.9 g, 5.4 mmol), Fmoc-Phe-OH (2.0 g,
5.39 mmol), Fmoc-Pro-OH (1.8 g, 5.3 mmol) and Fmoc-Ile-OH (1.9 g, 5.4 mmol). Acid
group of glutamic acid was deprotected by using palladium (0) in DCM, acetic acid and N-
methylmorpholine (37: 2: 1) for 24 hours. The terminal NH2 group of Ile residue was
deprotected by 20% 4-methylpiperidine and cyclization was allowed between α acidic
group of glutamic acid and amino group of Ile using oxymapure and DIC. The resin was
thoroughly washed after each coupling / deprotection step. The cyclized product was
cleaved from resin by 95% TFA in DCM with TIS (few drops). The cleaved product was
precipitated out by cold diethyl ether and precipitate was further washed with ether and
purified by Recycling HPLC using isocratic solvent system of acetonitrile/ water (ACN:
H2O 6: 4) 0.1% TFA. The structure of pure peptide 28 was elucidated by interpretation of
spectral data.
3.4.3.1. Characterization Data of Peptide 28:

137
3.4.4. Synthesis of Cyclic Peptide 29
Peptide 29 was synthesized by using Fmoc solid-phase peptide synthesis protocol on 4-
benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g). (Chem-
impex Int. INC. USA). The properly soaked resin was loaded with first amino acid Fmoc-
Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g, 13.4
mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9 mg,
0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling was
confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Phe-OH (2.0 g, 5.39 mmol), Fmoc-
Tyr(tBu)-OH (2.6 g, 5.4 mmol), Fmoc-Ile-OH (1.9 g, 5.4 mmol), Fmoc-Ala-OH (1.6 g, 5.1
mmol), Fmoc-Pro-OH (1.8 g, 5.3 mmol) and Fmoc-Ile-OH (1.9 g, 5.4 mmol). Acid group
of glutamic acid was deprotected by using palladium (0) in DCM, acetic acid and N-
methylmorpholine (37: 2: 1) for 24 hours. The terminal NH2 group of Ile residue was
deprotected by 20% 4-methylpiperidine and cyclization was allowed between α acidic
group of glutamic acid and amino group of Ile using oxyma pure and DIC. The resin was
thoroughly washed after each coupling / deprotection step. The cyclized product was
cleaved from resin by 95% TFA in DCM with TIS (few drops). The cleaved product was
precipitated out by cold diethyl ether and precipitate was further washed with ether and
purified by Recycling HPLC using isocratic solvent system of acetonitrile/ water (ACN:
H2O 6: 4) 0.1% TFA. The structure of pure peptide 29 was elucidated by interpretation of
spectral data.
3.4.4.1. Characterization Data of Peptide 29

138
3.4.5. Synthesis of Cyclic Peptide 30
Peptide 30 was synthesized by using Fmoc solid-phase peptide synthesis protocol on 4-
benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g). (Chem-
impex Int. INC. USA). The properly soaked resin was loaded with first amino acid Fmoc-
Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g, 13.4
mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9 mg,
0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling was
confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Phe-OH (2.0 g, 5.39 mmol),Fmoc-
Ala-OH (1.6 g, 5.1 mmol), Fmoc-Ile-OH (1.9 g, 5.4 mmol), Fmoc-Phe-OH (2.0 g, 5.39
mmol), Fmoc-Pro-OH (1.8 g, 5.3 mmol) and Fmoc-Ile-OH (1.9 g, 5.4 mmol). Acid group
of glutamic acid was deprotected by using palladium (0) in DCM, acetic acid and N-
methylmorpholine (37: 2: 1) for 24 hours. The terminal NH2 group of Ile residue was
deprotected by 20% 4-methylpiperidine and cyclization was allowed between α acidic
group of glutamic acid and amino group of Ile using oxyma pure and DIC. The resin was
thoroughly washed after each coupling / deprotection step. The cyclized product was
cleaved from resin by 95% TFA in DCM with TIS (few drops). The cleaved product was
precipitated out by cold diethyl ether and precipitate was further washed with ether and
purified by Recycling HPLC using isocratic solvent system of acetonitrile/ water (ACN:
H2O 6: 4) 0.1% TFA. The structure of pure peptide 30 was elucidated by interpretation of
spectral data.
3.4.5.1. Characterization Data of Peptide 30:

139
3.4.6. Synthesis of Cyclic Peptide 31
Cyclic peptide 31 was synthesized by using Fmoc solid-phase peptide synthesis protocol
on 4-benzyloxybenzyl alcohol resin (Wang resin, 100 – 200 µm, 3 g, 1.2 mmol / g).
(Chem-impex Int. INC. USA). The properly soaked resin was loaded with first amino acid
Fmoc-Glu-α-allyl (3.3 g, 8.0 mol) in the presence of coupling agents, oxyma pure (1.91g,
13.4 mmol) and DIC (2.113 ml, 13.5 mmol) along with 4-dimethylamino pyridine (32.9
mg, 0.269 mmol) as a catalyst. The reaction was gently shaked for two hours and coupling
was confirmed by ninhydrin test. After Fmoc deprotection with a solution of 20% 4-
methylpiperidine in DMF, the next amino acids, Fmoc-Phe-OH (2.0 g, 5.39 mmol), Fmoc-
Tyr(tBu)-OH (2.6 g, 5.4 mmol), Fmoc-Ala-OH (1.6 g, 5.1 mmol), Fmoc-Phe-OH (2.0 g,
5.39 mmol), Fmoc-Pro-OH (1.8 g) (5.3 mmol) and Fmoc-Ile-OH (1.9 g) (5.4 mmol). Acid
group of glutamic acid was deprotected by using palladium (0) in DCM, acetic acid and N-
methylmorpholine (37: 2: 1) for 24 hours. The terminal NH2 group of Ile residue was
deprotected by 20% 4-methylpiperidine and cyclization was allowed between α acidic
group of glutamic acid and amino group of Ile using oxyma pure and DIC. The resin was
thoroughly washed after each coupling / deprotection step. The cyclized product was
cleaved from resin by 95% TFA in DCM with TIS (few drops). The cleaved product was
precipitated out by cold diethyl ether and precipitate was further washed with ether and
purified by Recycling HPLC using isocratic solvent system of acetonitrile/ water (ACN:
H2O 6: 4) 0.1% TFA. The structure of pure peptide 31 was elucidated by interpretation of
spectral data.
3.4.6.1. Characterization Data of Peptide 31:

140
3.5. Screening Protocol:
3.5.1. MTT (3- (4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide) Assay
Cytotoxicity of compounds was determined using (3- (4, 5-Dimethylthiazol-2-yl)-2, 5-
Diphenyltetrazolium Bromide) reagent (Hansen et al., 1989). Cell lines were purchased
from ATCC (USA) and used in the present study were NCI-H460 (lung cancer cells), and
MCF-7 (breast cancer cells). The NCI-H460 cell were cultured at the concentrations of 2 x
104 /ml, and MCF-7 were cultured at 3 x 104 /ml. Plates were incubated overnight at 37°C
with 5% CO2. Following incubation, cells were treated for 48 hrs. with 25, 50, 100 and 200
µM with the test compounds. Since the test compounds were soluble in DMSO (0.5%)
therefore a vehicle control was also set. Following 48 hr. treatment, wells were emptied
and MTT dye (0.5 mg / mL final concentration in each well) along with the fresh medium
was added. Plates were allowed to stand for 4 hours in sterile environment. Then,
incubation medium was removed and formazan crystals were dissolved in DMSO with
gentle shaking. Plates were read at 550 nm filter of photometer (Thermofisher Scientific)
and percent inhibition was calculated using the following formula:
3.5.2 Chemiluminescence Assay
Luminol or lucigenin enhanced chemiluminescence assays were performed to study the
effect of compounds on ROS from phagocytes. This assay was performed as described by
Mesaik et al (Mesaik et al., 2012).
3.5.3. Nitric Oxide Screening Protocol:
Mouse macrophage cell lines (J774.2) (European Collection of Cell Cultures, UK) was
used to carry out the nitric oxide assay. The sample peptide was used in three different
concentrations (2, 10, and 50μM) and the protocol was used as described in Shaheen et al
(Shaheen et al., 2014), (Shaheen et al., 2016).
% inhibition = [(OD of the treated well- OD of untreated well) / OD of the untreated well] x 100

141
3.5.4. IL-2 Production and Quantification:
The IL-2 production and quantification was performed on Jurkat (human T lymphocyte
leukemia) cells. The protocol used was reported in Shaheen et al (Shaheen et al., 2016) and
according to manufacturer’s instructions (Manger, Hardy, Weiss, & Stobo, 1986).
3.4.12. T-cell Proliferation Assay:
Cell proliferation assay performed by standard thymidine, incorporation assay (Tabudravu,
Morris, Kettenes-van den Bosch, & Jaspars, 2002). The complete procedure was reported
in Shaheen et al (Shaheen et al., 2016).

142
References:
Abe, T., Sakane, M., Ikoma, T., Kobayashi, M., Nakamura, S., & Ochiai, N. (2008).
Intraosseous delivery of paclitaxel-loaded hydroxyapatitealginate composite beads
delaying paralysis caused by metastatic spine cancer in rats: Laboratory
investigation. Journal of Neurosurgery: Spine, 9(5), 502-510.
Abrams, S. I., Hand, P. H., Tsang, K. Y., & Schlom, J. (1996). Mutant ras epitopes as
targets for cancer vaccines. Paper presented at the Seminars in oncology.
Ahmed, A. U. (2011). An overview of inflammation: mechanism and consequences.
Frontiers in Biology, 6(4), 274-281.
Aina, O. H., Liu, R., Sutcliffe, J. L., Marik, J., Pan, C.-X., & Lam, K. S. (2007). From
combinatorial chemistry to cancer-targeting peptides. Molecular pharmaceutics,
4(5), 631-651.
Aina, O. H., Sroka, T. C., Chen, M. L., & Lam, K. S. (2002). Therapeutic cancer targeting
peptides. Peptide Science, 66(3), 184-199.
Akhtar, N. H., Pail, O., Saran, A., Tyrell, L., & Tagawa, S. T. (2011). Prostate-specific
membrane antigen-based therapeutics. Advances in urology, 2012.
Akindele, T., Gise, B., Sunaba, T., Kita, M., & Kigoshi, H. (2015). Total Synthesis of
Stylissatin A, A Cyclic Peptide That Inhibits Nitric Oxide Production. Bulletin of
the Chemical Society of Japan(0).
Al-Ghananeem, A. M., Malkawi, A. H., Muammer, Y. M., Balko, J. M., Black, E. P.,
Mourad, W., & Romond, E. (2009). Intratumoral delivery of paclitaxel in solid
tumor from biodegradable hyaluronan nanoparticle formulations. Aaps
Pharmscitech, 10(2), 410-417.
Albericio, F., Kneib-Cordonier, N., Biancalana, S., Gera, L., Masada, R. I., Hudson, D., &
Barany, G. (1990). Preparation and application of the 5-(4-(9-
fluorenylmethyloxycarbonyl) aminomethyl-3, 5-dimethoxyphenoxy)-valeric acid
(PAL) handle for the solid-phase synthesis of C-terminal peptide amides under
mild conditions. The Journal of Organic Chemistry, 55(12), 3730-3743.
Alderton, W. K., Cooper, C. E., & Knowles, R. G. (2001). Nitric oxide synthases:
structure, function and inhibition. Biochemical Journal, 357(3), 593-615.

143
Alghisi, G. C., Ponsonnet, L., & Rüegg, C. (2009). The integrin antagonist cilengitide
activates αVβ3, disrupts VE-cadherin localization at cell junctions and enhances
permeability in endothelial cells. PLoS One, 4(2), e4449.
Alizadeh, A. A., Ross, D. T., Perou, C. M., & Van De Rijn, M. (2001). Towards a novel
classification of human malignancies based on gene expression patterns. The
Journal of pathology, 195(1), 41-52.
Allen, T. M. (2002). Ligand-targeted therapeutics in anticancer therapy. Nature Reviews
Cancer, 2(10), 750-763.
Almond, B. A., Hadba, A. R., Freeman, S. T., Cuevas, B. J., York, A. M., Detrisac, C. J.,
& Goldberg, E. P. (2003). Efficacy of mitoxantrone-loaded albumin microspheres
for intratumoral chemotherapy of breast cancer. Journal of controlled release,
91(1), 147-155.
Alonzi, T., Fattori, E., Lazzaro, D., Costa, P., Probert, L., Kollias, G., . . . Ciliberto, G.
(1998). Interleukin 6 is required for the development of collagen-induced arthritis.
The Journal of experimental medicine, 187(4), 461-468.
Ampollini, L., Sonvico, F., Barocelli, E., Cavazzoni, A., Bilancia, R., Mucchino, C., . . .
Carbognani, P. (2010). Intrapleural polymeric films containing cisplatin for
malignant pleural mesothelioma in a rat tumour model: a preliminary study.
European Journal of Cardio-Thoracic Surgery, 37(3), 557-565.
Anand, P., Kunnumakkara, A. B., Kunnumakara, A. B., Sundaram, C., Harikumar, K. B.,
Tharakan, S. T., . . . Aggarwal, B. B. Pharmaceutical research Volume: 25 ISSN:
0724-8741 ISO Abbreviation: Pharm. Res. Publication Date: 2008 Sep. Detail:.
Arai, M., Yamano, Y., Fujita, M., Setiawan, A., & Kobayashi, M. (2012). Stylissamide X,
a new proline-rich cyclic octapeptide as an inhibitor of cell migration, from an
Indonesian marine sponge of Stylissa sp. Bioorganic & medicinal chemistry letters,
22(4), 1818-1821.
Arap, W., Pasqualini, R., & Ruoslahti, E. (1998). Cancer treatment by targeted drug
delivery to tumor vasculature in a mouse model. Science, 279(5349), 377-380.
Ataie-Kachoie, P., Pourgholami, M. H., & Morris, D. L. (2013). Inhibition of the IL-6
signaling pathway: a strategy to combat chronic inflammatory diseases and cancer.
Cytokine & growth factor reviews, 24(2), 163-173.

144
Atherton, E., Clive, D., & Sheppard, R. (1975). Polyamide supports for polypeptide
synthesis. Journal of the American Chemical Society, 97(22), 6584-6585.
Backes, B. J., & Ellman, J. A. (1999). An alkanesulfonamide “safety-catch” linker for
solid-phase synthesis. The Journal of Organic Chemistry, 64(7), 2322-2330.
Backes, B. J., Virgilio, A. A., & Ellman, J. A. (1996). Activation Method to Prepare a
Highly Reactive Acylsulfonamide “Safety-Catch” Linker for Solid-Phase
Synthesis1. Journal of the American Chemical Society, 118(12), 3055-3056.
Barrientos-Salcedo, C., Rico-Rosillo, G., Giménez-Scherer, J. A., & Soriano-Correa, C.
(2009). Computational study of the electronic structure characterization of a
novelanti-inflammatory tripeptide derived from monocyte locomotion
inhibitoryfactor (MLIF)-pentapeptide. European journal of medicinal chemistry,
44(8), 3114-3119.
Beck, A., Klinguer‐Hamour, C., Bussat, M. C., Champion, T., Haeuw, J. F., Goetsch, L., . .
. Van Dorsselaer, A. (2007). Peptides as tools and drugs for immunotherapies.
Journal of Peptide Science, 13(9), 588-602.
Beckey, H.-D. (2016). Principles of Field Ionization and Field Desorption Mass
Spectrometry: International Series in Analytical Chemistry: Elsevier.
Bergmann, M., & Zervas, L. (1932). Über ein allgemeines Verfahren der Peptid‐Synthese.
Berichte der deutschen chemischen Gesellschaft (A and B Series), 65(7), 1192-
1201.
Berzofsky, J. A., Ahlers, J. D., & Belyakov, I. M. (2001). Strategies for designing and
optimizing new generation vaccines. Nature Reviews Immunology, 1(3), 209-219.
Besser, D., Müller, B., Kleinwächter, P., Greiner, G., Seyfarth, L., Steinmetzer, T., . . .
Reissmann, S. (2000). Synthesis and characterization of octapeptide somatostatin
analogues with backbone cyclization: comparison of different strategies, biological
activities and enzymatic stabilities. Journal für praktische Chemie, 342(6), 537-
545.
Borghouts, C., Kunz, C., & Groner, B. (2005). Current strategies for the development of
peptide‐based anti‐cancer therapeutics. Journal of Peptide Science, 11(11), 713-
726.

145
Bouhadir, K. H., Alsberg, E., & Mooney, D. J. (2001). Hydrogels for combination delivery
of antineoplastic agents. Biomaterials, 22(19), 2625-2633.
Bowdish, D. M., Davidson, D. J., Scott, M. G., & Hancock, R. E. (2005).
Immunomodulatory activities of small host defense peptides. Antimicrobial agents
and chemotherapy, 49(5), 1727-1732.
Broqua, P., Riviere, P. J.-M., Conn, P. M., Rivier, J. E., Aubert, M. L., & Junien, J.-L.
(2002). Pharmacological profile of a new, potent, and long-acting gonadotropin-
releasing hormone antagonist: degarelix. Journal of Pharmacology and
Experimental Therapeutics, 301(1), 95-102.
Brown, B. B., Wagner, D. S., & Geysen, H. M. (1995). A single-bead decode strategy
using electrospray ionization mass spectrometry and a new photolabile linker: 3-
amino-3-(2-nitrophenyl) propionic acid. Molecular diversity, 1(1), 4-12.
Brzozowski, T., Konturek, P. C., Konturek, S. J., Kwiecień, S., Sliwowski, Z., Pajdo, R., . .
. Hahn, E. G. (2003). Implications of reactive oxygen species and cytokines in
gastroprotection against stress-induced gastric damage by nitric oxide releasing
aspirin. International journal of colorectal disease, 18(4), 320-329.
Burg, M. A., Pasqualini, R., Arap, W., Ruoslahti, E., & Stallcup, W. B. (1999). NG2
proteoglycan-binding peptides target tumor neovasculature. Cancer research,
59(12), 2869-2874.
Calder, P. C. (2009). Polyunsaturated fatty acids and inflammatory processes: new twists
in an old tale. Biochimie, 91(6), 791-795.
Caldwell, G. W., Yan, Z., Tang, W., Dasgupta, M., & Hasting, B. (2009). ADME
optimization and toxicity assessment in early-and late-phase drug discovery.
Current topics in medicinal chemistry, 9(11), 965-980.
Cantrell, D., Collins, M., & Crumpton, M. (1988). Autocrine regulation of T-lymphocyte
proliferation: differential induction of IL-2 and IL-2 receptor. Immunology, 65(3),
343.
Channon, K., & Guzik, T. (2002). MECHANISMS OF SUPEROXIDE PRODUCTION IN
HUMAN BLOOD. Journal of physiology and pharmacology, 53(4), 515-524.
Chen, Y., Xu, X., Hong, S., Chen, J., Liu, N., Underhill, C. B., . . . Zhang, L. (2001).
RGD-Tachyplesin inhibits tumor growth. Cancer research, 61(6), 2434-2438.

146
Cheng, Y.-X., Zhou, L.-L., Yan, Y.-M., Chen, K.-X., & Hou, F.-F. (2011). Diabetic
nephropathy-related active cyclic peptides from the roots of Brachystemma
calycinum. Bioorganic & medicinal chemistry letters, 21(24), 7434-7439.
Colgrave, M. L., & Craik, D. J. (2004). Thermal, chemical, and enzymatic stability of the
cyclotide kalata B1: the importance of the cyclic cystine knot. Biochemistry,
43(20), 5965-5975.
Cornelio, D., Roesler, R., & Schwartsmann, G. (2007). Gastrin-releasing peptide receptor
as a molecular target in experimental anticancer therapy. Annals of Oncology.
Coulie, P. G., Hanagiri, T., & Takenoyama, M. (2001). From tumor antigens to
immunotherapy. International journal of clinical oncology, 6(4), 163-170.
Craik, D. J., Daly, N. L., Bond, T., & Waine, C. (1999). Plant cyclotides: a unique family
of cyclic and knotted proteins that defines the cyclic cystine knot structural motif.
Journal of molecular biology, 294(5), 1327-1336.
Craik, D. J., Daly, N. L., & Waine, C. (2001). The cystine knot motif in toxins and
implications for drug design. Toxicon, 39(1), 43-60.
Curnis, F., Arrigoni, G., Sacchi, A., Fischetti, L., Arap, W., Pasqualini, R., & Corti, A.
(2002). Differential binding of drugs containing the NGR motif to CD13 isoforms
in tumor vessels, epithelia, and myeloid cells. Cancer research, 62(3), 867-874.
Curnis, F., Sacchi, A., Borgna, L., Magni, F., Gasparri, A., & Corti, A. (2000).
Enhancement of tumor necrosis factor α antitumor immunotherapeutic properties
by targeted delivery to aminopeptidase N (CD13). Nature biotechnology, 18(11),
1185-1190.
Cychon, C., & Kock, M. (2010). Stylissamides E and F, cyclic heptapeptides from the
Caribbean sponge Stylissa caribica. Journal of natural products, 73(4), 738-742.
Davidson, B. S., Izzo, F., Cromeens, D. M., Stephens, L. C., Siddik, Z. H., & Curley, S. A.
(1995). Collagen matrix cisplatin prevents local tumor growth after margin-positive
resection. Journal of Surgical Research, 58(6), 618-624.
Debruyne, F., Bhat, G., & Garnick, M. B. (2006). Abarelix for injectable suspension: first-
in-class gonadotropin-releasing hormone antagonist for prostate cancer. Future
Oncology, 2(6), 677-696.

147
Deem, M. W., & Bader, J. S. (1996). A configurational bias Monte Carlo method for linear
and cyclic peptides. Molecular Physics, 87(6), 1245-1260.
Dellai, A., Maricic, I., Kumar, V., Arutyunyan, S., Bouraoui, A., & Nefzi, A. (2010).
Parallel synthesis and anti-inflammatory activity of cyclic peptides cyclosquamosin
D and Met-cherimolacyclopeptide B and their analogs. Bioorganic & medicinal
chemistry letters, 20(19), 5653-5657.
Deprez, B., Williard, X., Bourel, L., Coste, H., Hyafil, F., & Tartar, A. (1995). Self-
deciphering, orthogonal combinatorial libraries of soluble organic compounds:
Discovery of a potent V2 vasopressin antagonist. Paper presented at the
PEPTIDES-EUROPEAN SYMPOSIUM-.
Derossi, D., Joliot, A. H., Chassaing, G., & Prochiantz, A. (1994). The third helix of the
Antennapedia homeodomain translocates through biological membranes. Journal
of Biological Chemistry, 269(14), 10444-10450.
Deshayes, S., Morris, M., Divita, G., & Heitz, F. (2005). Cell-penetrating peptides: tools
for intracellular delivery of therapeutics. Cellular and Molecular Life Sciences
CMLS, 62(16), 1839-1849.
Dinarello, C. A. (2000). Proinflammatory cytokines. Chest Journal, 118(2), 503-508.
Dinarello, C. A. (2006). Interleukin 1 and interleukin 18 as mediators of inflammation and
the aging process. The American journal of clinical nutrition, 83(2), 447S-455S.
Dooley, C. T., & Houghten, R. A. (1993). The use of positional scanning synthetic peptide
combinatorial libraries for the rapid determination of opioid receptor ligands. Life
sciences, 52(18), 1509-1517.
Eisenbach, L., Bar-Haim, E., & El-Shami, K. (2000). Antitumor vaccination using peptide
based vaccines. Immunology letters, 74(1), 27-34.
El-Andaloussi, S., Holm, T., & Langel, U. (2005). Cell-penetrating peptides: mechanisms
and applications. Current pharmaceutical design, 11(28), 3597-3611.
El-Faham, A., Al Marhoon, Z., Abdel-Megeed, A., & Albericio, F. (2013).
OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-
(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester
Derivatives. Molecules, 18(12), 14747-14759.

148
Ellerby, H. M., Arap, W., Ellerby, L. M., Kain, R., Andrusiak, R., Del Rio, G., . . .
Ruoslahti, E. (1999). Anti-cancer activity of targeted pro-apoptotic peptides.
Nature medicine, 5(9), 1032-1038.
Erb, E., Janda, K. D., & Brenner, S. (1994). Recursive deconvolution of combinatorial
chemical libraries. Proceedings of the National Academy of Sciences, 91(24),
11422-11426.
Esser, J.-P., Krenning, E., Teunissen, J., Kooij, P., Van Gameren, A., Bakker, W., &
Kwekkeboom, D. J. (2006). Comparison of [177Lu-DOTA0, Tyr3] octreotate and
[177Lu-DOTA0, Tyr3] octreotide: which peptide is preferable for PRRT?
European journal of nuclear medicine and molecular imaging, 33(11), 1346-1351.
Feghali, C. A., & Wright, T. M. (1997). Cytokines in acute and chronic inflammation.
Front Biosci, 2(1), d12-d26.
Festa, C., De Marino, S., Sepe, V., Monti, M. C., Luciano, P., D'Auria, M. V., . . .
Zampella, A. (2009). Perthamides C and D, two new potent anti-inflammatory
cyclopeptides from a Solomon Lithistid sponge Theonella swinhoei. Tetrahedron,
65(50), 10424-10429.
Fischer, E., & Fourneau, E. (1901). Ueber einige derivate des glykocolls. Berichte der
deutschen chemischen Gesellschaft, 34(2), 2868-2877.
Fitzgerald, K. A., & Luke, A. (2000). The role of the interleukin-1/Toll-like receptor
superfamily in inflammation and host defence. Microbes and infection, 2(8), 933-
943.
Frank, R. (1992). Spot-synthesis: an easy technique for the positionally addressable,
parallel chemical synthesis on a membrane support. Tetrahedron, 48(42), 9217-
9232.
Furka, A., SEBESTYÉN, F., ASGEDOM, M., & DIBÓ, G. (1991). General method for
rapid synthesis of multicomponent peptide mixtures. International journal of
peptide and protein research, 37(6), 487-493.
Garetto, S., Sizzano, F., Brusa, D., Tizzani, A., Malavasi, F., & Matera, L. (2009). Binding
of prostate-specific membrane antigen to dendritic cells: a critical step in vaccine
preparation. Cytotherapy, 11(8), 1090-1100.

149
Geddes, K., Magalhães, J. G., & Girardin, S. E. (2009). Unleashing the therapeutic
potential of NOD-like receptors. Nature reviews Drug discovery, 8(6), 465-479.
Gelderblom, H., Verweij, J., Nooter, K., & Sparreboom, A. (2001). Cremophor EL: the
drawbacks and advantages of vehicle selection for drug formulation. European
Journal of Cancer, 37(13), 1590-1598.
Gerber, D. E., Gallia, G. L., Tyler, B. M., Eberhart, C. G., Royer, G., & Grossman, S. A.
(2011). A novel polymer gel for the delivery of local therapies to intracranial
tumors: In vivo safety evaluation. Journal of Biomedical Materials Research Part
A, 99(3), 479-484.
Geysen, H. M., Rodda, S. J., & Mason, T. J. (1986). A priori delineation of a peptide
which mimics a discontinuous antigenic determinant. Molecular immunology,
23(7), 709-715.
Gilon, C., Halle, D., Chorev, M., Selincer, Z., & Byk, G. (1991). Backbone cyclization: a
new method for conferring conformational constraint on peptides. Biopolymers,
31(6), 745-750.
Ginj, M., Zhang, H., Waser, B., Cescato, R., Wild, D., Wang, X., . . . Reubi, J. C. (2006).
Radiolabeled somatostatin receptor antagonists are preferable to agonists for in
vivo peptide receptor targeting of tumors. Proceedings of the National Academy of
Sciences, 103(44), 16436-16441.
Giralt, E., Eritja, R., & Pedroso, E. (1981). Diketopiperazine formation in acetamido-and
nitrobenzamido-bridgedpolymeric supports. Tetrahedron Letters, 22(38), 3779-
3782.
Giuliani, A., Pirri, G., & Nicoletto, S. F. (2007). Antimicrobial peptides: an overview of a
promising class of therapeutics. Central European Journal of Biology, 2(1), 1-33.
doi: 10.2478/s11535-007-0010-5
Givalois, L., Li, S., & Pelletier, G. (2002). Central nitric oxide regulation of the
hypothalamic–pituitary–adrenocortical axis in adult male rats. Molecular brain
research, 102(1), 1-8.
Gjertsen, M., & Gaudernack, G. (1998). Mutated Ras peptides as vaccines in
immunotherapy of cancer. Vox sanguinis, 74(S2), 489-495.

150
Gombar, V. K., Silver, I. S., & Zhao, Z. (2003). Role of ADME characteristics in drug
discovery and their in silico evaluation: in silico screening of chemicals for their
metabolic stability. Current topics in medicinal chemistry, 3(11), 1205-1225.
Gotthardt, M., Béhé, M. P., Grass, J., Bauhofer, A., Rinke, A., Schipper, M. L., . . . Behr,
T. M. (2006). Added value of gastrin receptor scintigraphy in comparison to
somatostatin receptor scintigraphy in patients with carcinoids and other
neuroendocrine tumours. Endocrine-related cancer, 13(4), 1203-1211.
Grozinsky-Glasberg, S., Shimon, I., Korbonits, M., & Grossman, A. B. (2008).
Somatostatin analogues in the control of neuroendocrine tumours: efficacy and
mechanisms. Endocrine-related cancer, 15(3), 701-720.
Grunnet, M., & Sorensen, J. (2012). Carcinoembryonic antigen (CEA) as tumor marker in
lung cancer. Lung Cancer, 76(2), 138-143.
Guillier, F., Orain, D., & Bradley, M. (2000). Linkers and cleavage strategies in solid-
phase organic synthesis and combinatorial chemistry. Chemical reviews, 100(6),
2091-2158.
Gupta, B., Levchenko, T. S., & Torchilin, V. P. (2005). Intracellular delivery of large
molecules and small particles by cell-penetrating proteins and peptides. Advanced
drug delivery reviews, 57(4), 637-651.
Guzik, T. J., West, N. E., Pillai, R., Taggart, D. P., & Channon, K. M. (2002). Nitric oxide
modulates superoxide release and peroxynitrite formation in human blood vessels.
Hypertension, 39(6), 1088-1094.
Hancock, R. E. (2001). Cationic peptides: effectors in innate immunity and novel
antimicrobials. The Lancet infectious diseases, 1(3), 156-164.
Hareuveni, M., Gautier, C., Kieny, M.-P., Wreschner, D., Chambon, P., & Lathe, R.
(1990). Vaccination against tumor cells expressing breast cancer epithelial tumor
antigen. Proceedings of the National Academy of Sciences, 87(23), 9498-9502.
Hariharan, S., Gustafson, D., Holden, S., McConkey, D., Davis, D., Morrow, M., . . .
O'Bryant, C. (2007). Assessment of the biological and pharmacological effects of
the ανβ3 and ανβ5 integrin receptor antagonist, cilengitide (EMD 121974), in
patients with advanced solid tumors. Annals of Oncology, 18(8), 1400-1407.

151
Heikkilä, K., Ebrahim, S., & Lawlor, D. A. (2008). Systematic review of the association
between circulating interleukin-6 (IL-6) and cancer. European journal of cancer,
44(7), 937-945.
Henderson, R. A., Mossman, S., Nairn, N., & Cheever, M. A. (2005). Cancer vaccines and
immunotherapies: emerging perspectives. Vaccine, 23(17), 2359-2362.
Henriques, S. T., Huang, Y.-H., Castanho, M. A., Bagatolli, L. A., Sonza, S., Tachedjian,
G., . . . Craik, D. J. (2012). Phosphatidylethanolamine binding is a conserved
feature of cyclotide-membrane interactions. Journal of Biological Chemistry,
287(40), 33629-33643.
Horton, D. A., Bourne, G. T., & Smythe, M. L. (2000). Exploring privileged structures: the
combinatorial synthesis of cyclic peptides. Molecular diversity, 5(4), 289-304.
Houghten, R. A., Pinilla, C., Blondelle, S. E., Appel, J. R., Dooley, C. T., & Cuervo, J. H.
(1991). Generation and use of synthetic peptide combinatorial libraries for basic
research and drug discovery. Nature, 354(6348), 84-86.
Huang, G.-J., Deng, J.-S., Huang, S.-S., Wang, S.-Y., Chang, Y.-S., & Kuo, Y.-H. (2013).
Bioassay guided isolation and identification of anti-inflammatory active
compounds from the root of Ficus formosana. Journal of agricultural and food
chemistry, 61(46), 11008-11015.
Hueman, M. T., Dehqanzada, Z. A., Novak, T. E., Gurney, J. M., Woll, M. M., Ryan, G.
B., . . . Ioannides, C. G. (2005). Phase I clinical trial of a HER-2/neu peptide (E75)
vaccine for the prevention of prostate-specific antigen recurrence in high-risk
prostate cancer patients. Clinical Cancer Research, 11(20), 7470-7479.
Hughes, J., Rees, S., Kalindjian, S., & Philpott, K. (2011). Principles of early drug
discovery. British journal of pharmacology, 162(6), 1239-1249.
Ischiropoulos, H., & Al-Mehdi, A. B. (1995). Peroxynitrite‐mediated oxidative protein
modifications. FEBS letters, 364(3), 279-282.
Jadhav, H., Singh, A., & Bhutani, K. (2003). Rationale for immunomodulatory and anti-
inflammatory effects of Ocimum sanctum: radical scavenging potential and effect
on nitric oxide production. Paper presented at the III WOCMAP Congress on
Medicinal and Aromatic Plants-Volume 4: Targeted Screening of Medicinal and
Aromatic Plants, Economics 678.

152
Jayawickreme, C. K., Sauls, H., Bolio, N., Ruan, J., Moyer, M., Burkhart, W., . . . Shaffer,
J. (1999). Use of a cell-based, lawn format assay to rapidly screen a 442,368 bead-
based peptide library. Journal of pharmacological and toxicological methods,
42(4), 189-197.
Juliano, R., Alam, M. R., Dixit, V., & Kang, H. (2008). Mechanisms and strategies for
effective delivery of antisense and siRNA oligonucleotides. Nucleic acids research,
36(12), 4158-4171.
Juskowiak, G. L., Stachel, S. J., Tivitmahaisoon, P., & Van Vranken, D. L. (2004).
Fluorogenic peptide sequences-transformation of short peptides into fluorophores
under ambient photooxidative conditions. Journal of the American Chemical
Society, 126(2), 550-556.
Kakde, D., Jain, D., Shrivastava, V., Kakde, R., & Patil, A. (2011). Cancer therapeutics-
opportunities, challenges and advances in drug delivery.
Kakinoki, S., & Taguchi, T. (2007). Antitumor effect of an injectable in-situ forming drug
delivery system composed of a novel tissue adhesive containing doxorubicin
hydrochloride. European Journal of Pharmaceutics and Biopharmaceutics, 67(3),
676-681.
Karagiannis, E. D., & Popel, A. S. (2008). Novel anti‐angiogenic peptides derived From
ELR‐containing CXC chemokines. Journal of cellular biochemistry, 104(4), 1356-
1363.
Kaufman, H. L. (2012). Vaccines for melanoma and renal cell carcinoma. Paper presented
at the Seminars in oncology.
Kenner, G., McDermott, J., & Sheppard, R. (1971). The safety catch principle in solid
phase peptide synthesis. Journal of the Chemical Society D: Chemical
Communications(12), 636-637.
Khleif, S. N., Abrams, S. I., Hamilton, J. M., Bergmann-Leitner, E., Chen, A., Bastian, A.,
. . . Schlom, J. (1999). A phase I vaccine trial with peptides reflecting ras oncogene
mutations of solid tumors. Journal of Immunotherapy, 22(2), 155-165.
Kichler, A., Leborgne, C., März, J., Danos, O., & Bechinger, B. (2003). Histidine-rich
amphipathic peptide antibiotics promote efficient delivery of DNA into mammalian
cells. Proceedings of the National Academy of Sciences, 100(4), 1564-1568.

153
Kichler, A., Mason, A. J., & Bechinger, B. (2006). Cationic amphipathic histidine-rich
peptides for gene delivery. Biochimica et Biophysica Acta (BBA)-Biomembranes,
1758(3), 301-307.
Kimmerlin, T., & Seebach, D. (2005). ‘100 years of peptide synthesis’: ligation methods
for peptide and protein synthesis with applications to β‐peptide assemblies. The
Journal of peptide research, 65(2), 229-260.
Kimura, T., Iwase, M., Kondo, G., Watanabe, H., Ohashi, M., Ito, D., & Nagumo, M.
(2003). Suppressive effect of selective cyclooxygenase-2 inhibitor on cytokine
release in human neutrophils. International immunopharmacology, 3(10), 1519-
1528.
Kita, M., Gise, B., Kawamura, A., & Kigoshi, H. (2013). Stylissatin A, a cyclic peptide
that inhibits nitric oxide production from the marine sponge Stylissa massa.
Tetrahedron letters, 54(50), 6826-6828.
Knowles, R. G., & Moncada, S. (1994). Nitric oxide synthases in mammals. Biochemical
Journal, 298(Pt 2), 249.
Knutson, K. L., Schiffman, K., & Disis, M. L. (2001). Immunization with a HER-2/neu
helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer
patients. The Journal of clinical investigation, 107(4), 477-484.
Konishi, M., Tabata, Y., Kariya, M., Suzuki, A., Mandai, M., Nanbu, K., . . . Fujii, S.
(2003). In vivo anti-tumor effect through the controlled release of cisplatin from
biodegradable gelatin hydrogel. Journal of controlled release, 92(3), 301-313.
Korhonen, R., Lahti, A., Kankaanranta, H., & Moilanen, E. (2005). Nitric oxide production
and signaling in inflammation. Current Drug Targets-Inflammation & Allergy,
4(4), 471-479.
Krenning, E., De Jong, M., Kooij, P., Breeman, W., Bakker, W., De Herder, W., . . .
Pauwels, S. (1999). Radiolabelled somatostatin analogue (s) for peptide receptor
scintigraphy and radionuclide therapy. Annals of Oncology, 10(suppl 2), S23-S29.
Kritzer, J. A., Stephens, O. M., Guarracino, D. A., Reznik, S. K., & Schepartz, A. (2005).
β-peptides as inhibitors of protein–protein interactions. Bioorganic & medicinal
chemistry, 13(1), 11-16.

154
Kumar, A., Ye, G., Wang, Y., Lin, X., Sun, G., & Parang, K. (2006). Synthesis and
structure-activity relationships of linear and conformationally constrained peptide
analogues of CIYKYY as Src tyrosine kinase inhibitors. Journal of medicinal
chemistry, 49(11), 3395-3401.
Kumar, S., Mahdi, H., Bryant, C., Shah, J. P., Garg, G., & Munkarah, A. (2010). Clinical
trials and progress with paclitaxel in ovarian cancer. Int J Womens Health, 2, 411-
427.
Kumarn, S., Chimnoi, N., & Ruchirawat, S. (2013). Synthesis of integerrimide A by an on-
resin tandem Fmoc-deprotection–macrocyclisation approach. Organic &
biomolecular chemistry, 11(44), 7760-7767.
Kwekkeboom, D., Krenning, E. P., & de Jong, M. (2000). Peptide receptor imaging and
therapy. Journal of Nuclear Medicine, 41(10), 1704-1713.
Kwekkeboom, D. J., Teunissen, J. J., Bakker, W. H., Kooij, P. P., de Herder, W. W.,
Feelders, R. A., . . . Krenning, E. P. (2005). Radiolabeled somatostatin analog
[177Lu-DOTA0, Tyr3] octreotate in patients with endocrine gastroenteropancreatic
tumors. Journal of Clinical Oncology, 23(12), 2754-2762.
Lam, K. S. (1997). Mini-review. Application of combinatorial library methods in cancer
research and drug discovery. Anti-cancer drug design, 12(3), 145-167.
Lam, K. S., Lebl, M., & Krchnák, V. (1997). The “one-bead-one-compound”
combinatorial library method. Chemical reviews, 97(2), 411-448.
Lam, K. S., Liu, R., Miyamoto, S., Lehman, A. L., & Tuscano, J. M. (2003). Applications
of one-bead one-compound combinatorial libraries and chemical microarrays in
signal transduction research. Accounts of chemical research, 36(6), 370-377.
Lam, K. S., Salmon, S. E., Hersh, E. M., Hruby, V. J., Kazmierski, W. M., & Knapp, R. J.
(1991). A new type of synthetic peptide library for identifying ligand-binding
activity. Nature, 354(6348), 82-84.
Lawrence, T., Willoughby, D. A., & Gilroy, D. W. (2002). Anti-inflammatory lipid
mediators and insights into the resolution of inflammation. Nature Reviews
Immunology, 2(10), 787-795.

155
Lear, J. D., & DeGrado, W. F. (1987). Membrane binding and conformational properties of
peptides representing the NH2 terminus of influenza HA-2. Journal of Biological
Chemistry, 262(14), 6500-6505.
Lee, T.-H., Lin, Y.-H., Seow, K.-M., Hwang, J.-L., Tzeng, C.-R., & Yang, Y.-S. (2008).
Effectiveness of cetrorelix for the prevention of premature luteinizing hormone
surge during controlled ovarian stimulation using letrozole and gonadotropins: a
randomized trial. Fertility and sterility, 90(1), 113-120.
Lehrer, R. I. (2004). Primate defensins. Nat Rev Micro, 2(9), 727-738. doi:
http://www.nature.com/nrmicro/journal/v2/n9/suppinfo/nrmicro976_S1.html
Lein, M., Sgolastra, F., Tew, G. N., & Holden, M. A. (2015). Protein transport across
membranes: Comparison between lysine and guanidinium-rich carriers. Biochimica
et Biophysica Acta (BBA)-Biomembranes, 1848(11), 2980-2984.
Li, X., Kong, X., Zhang, J., Wang, Y., Wang, Y., Shi, S., . . . Wei, Y. (2011). A novel
composite hydrogel based on chitosan and inorganic phosphate for local drug
delivery of camptothecin nanocolloids. Journal of pharmaceutical sciences, 100(1),
232-241.
Liskamp, R. M., Rijkers, D. T., & Bakker, S. E. (2008). Bioactive macrocyclic peptides
and peptide mimics Modern Supramolecular Chemistry: Strategies for Macrocycle
Synthesis (pp. 1-27): Wiley-VCH.
Liu, R., & Lam, K. S. (2008). Peptide Combinatorial Libraries. Wiley Encyclopedia of
Chemical Biology.
Liu, X., Heng, W. S., Li, Q., & Chan, L. W. (2006). Novel polymeric microspheres
containing norcantharidin for chemoembolization. Journal of controlled release,
116(1), 35-41.
Liu, Y., Xia, X., Xu, L., & Wang, Y. (2013). Design of hybrid β-hairpin peptides with
enhanced cell specificity and potent anti-inflammatory activity. Biomaterials,
34(1), 237-250.
Lundquist IV, J. T., & Pelletier, J. C. (2002). A new tri-orthogonal strategy for peptide
cyclization. Organic letters, 4(19), 3219-3221.
Mäe, M., & Langel, Ü. (2006). Cell-penetrating peptides as vectors for peptide, protein and
oligonucleotide delivery. Current opinion in pharmacology, 6(5), 509-514.

156
Magzoub, M., Pramanik, A., & Gräslund, A. (2005). Modeling the endosomal escape of
cell-penetrating peptides: transmembrane pH gradient driven translocation across
phospholipid bilayers. Biochemistry, 44(45), 14890-14897.
Mandal, D., Nasrolahi Shirazi, A., & Parang, K. (2011). Cell‐Penetrating Homochiral
Cyclic Peptides as Nuclear‐Targeting Molecular Transporters. Angewandte Chemie
International Edition, 50(41), 9633-9637.
Manger, B., Hardy, K. J., Weiss, A., & Stobo, J. D. (1986). Differential effect of
cyclosporin A on activation signaling in human T cell lines. Journal of Clinical
Investigation, 77(5), 1501.
Mantovani, A., Allavena, P., Sica, A., & Balkwill, F. (2008). Cancer-related inflammation.
Nature, 454(7203), 436-444.
Markowicz, S., Nowecki, Z. I., Rutkowski, P., Lipkowski, A. W., Biernacka, M.,
Jakubowska-Mucka, A., . . . Polowniak-Pracka, H. (2012). Adjuvant vaccination
with melanoma antigen-pulsed dendritic cells in stage III melanoma patients.
Medical Oncology, 29(4), 2966-2977.
Marletta, M. A. (1994). Nitric oxide synthase: aspects concerning structure and catalysis.
Cell, 78(6), 927-930.
Masters, S. L., Simon, A., Aksentijevich, I., & Kastner, D. L. (2009). Horror
autoinflammaticus: the molecular pathophysiology of autoinflammatory disease.
Annual review of immunology, 27, 621.
McDaniel, J. R., Callahan, D. J., & Chilkoti, A. (2010). Drug delivery to solid tumors by
elastin-like polypeptides. Advanced drug delivery reviews, 62(15), 1456-1467.
Meldal, M. (2002). The one‐bead two‐compound assay for solid phase screening of
combinatorial libraries. Peptide Science, 66(2), 93-100.
Meldal, M., Svendsen, I., Breddam, K., & Auzanneau, F.-I. (1994). Portion-mixing peptide
libraries of quenched fluorogenic substrates for complete subsite mapping of
endoprotease specificity. Proceedings of the National Academy of Sciences, 91(8),
3314-3318.
Meng, L., Yang, L., Zhao, X., Zhang, L., Zhu, H., Liu, C., & Tan, W. (2012). Targeted
delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One,
7(4), e33434.

157
Merrifield, R. B. (1963). Solid phase peptide synthesis. I. The synthesis of a tetrapeptide.
Journal of the American Chemical Society, 85(14), 2149-2154.
Mesaik, M. A., Jabeen, A., Halim, S. A., Begum, A., Khalid, A. S., Asif, M., . . .
Choudhary, M. I. (2012). In silico and in vitro immunomodulatory studies on
compounds of Lindelofia stylosa. Chemical biology & drug design, 79(3), 290-299.
Midoux, P., Kichler, A., Boutin, V., Maurizot, J.-C., & Monsigny, M. (1998). Membrane
permeabilization and efficient gene transfer by a peptide containing several
histidines. Bioconjugate chemistry, 9(2), 260-267.
Miller, S. J., Blackwell, H. E., & Grubbs, R. H. (1996). Application of ring-closing
metathesis to the synthesis of rigidified amino acids and peptides. Journal of the
American Chemical Society, 118(40), 9606-9614.
Mills, K. H., & Dunne, A. (2009). Immune modulation: IL-1, master mediator or initiator
of inflammation. Nature medicine, 15(12), 1363-1364.
Miranda, L. P., & Alewood, P. F. (2000). Challenges for protein chemical synthesis in the
21st century: bridging genomics and proteomics. Peptide Science, 55(3), 217-226.
Mittal, S., Kaur, H., Gautam, N., & Mantha, A. K. (2017). Biosensors for breast cancer
diagnosis: A review of bioreceptors, biotransducers and signal amplification
strategies. Biosensors and Bioelectronics, 88, 217-231.
Mizutani, H., Black, R., & Kupper, T. (1991). Human keratinocytes produce but do not
process pro-interleukin-1 (IL-1) beta. Different strategies of IL-1 production and
processing in monocytes and keratinocytes. Journal of Clinical Investigation,
87(3), 1066.
Mochly-Rosen, D., & Qvit, N. (2009). Peptide inhibitors of protein-protein interactions.
Trends Endocrinol Metab, 20, 25-33.
Mohammed, R., Peng, J., Kelly, M., & Hamann, M. T. (2006). Cyclic heptapeptides from
the Jamaican sponge Stylissa caribica. Journal of natural products, 69(12), 1739-
1744.
Mori, T. (2004). Cancer-specific ligands identified from screening of peptide-display
libraries. Current pharmaceutical design, 10(19), 2335-2343.
Morris, M. C., Deshayes, S., Heitz, F., & Divita, G. (2008). Cell‐penetrating peptides:
from molecular mechanisms to therapeutics. Biology of the Cell, 100(4), 201-217.

158
Muderspach, L., Wilczynski, S., Roman, L., Bade, L., Felix, J., Small, L., . . . Weber, J.
(2000). A phase I trial of a human papillomavirus (HPV) peptide vaccine for
women with high-grade cervical and vulvar intraepithelial neoplasia who are HPV
16 positive. Clinical Cancer Research, 6(9), 3406-3416.
Murphy, J.-E., Robert, C., & Kupper, T. S. (2000). Interleukin-1 and cutaneous
inflammation: a crucial link between innate and acquired immunity. Journal of
investigative dermatology, 114(3), 602-608.
Nathan, C. (2002). Points of control in inflammation. Nature, 420(6917), 846-852.
Nava-Parada, P., & Emens, L. A. (2007). GV-1001, an injectable telomerase peptide
vaccine for the treatment of solid cancers. Current opinion in molecular
therapeutics, 9(5), 490.
Nicolas, G., Giovacchini, G., Müller-Brand, J., & Forrer, F. (2011). Targeted radiotherapy
with radiolabeled somatostatin analogs. Endocrinology and metabolism clinics of
North America, 40(1), 187-204.
Nielsen, U. B., & Marks, J. D. (2000). Internalizing antibodies and targeted cancer therapy:
direct selection from phage display libraries. Pharmaceutical science & technology
today, 3(8), 282-291.
Ohshima, S., Saeki, Y., Mima, T., Sasai, M., Nishioka, K., Nomura, S., . . . Suemura, M.
(1998). Interleukin 6 plays a key role in the development of antigen-induced
arthritis. Proceedings of the National Academy of Sciences, 95(14), 8222-8226.
Olivos, H. J., Bachhawat‐Sikder, K., & Kodadek, T. (2003). Quantum dots as a visual aid
for screening bead‐bound combinatorial libraries. ChemBioChem, 4(11), 1242-
1245.
Opal, S. M., & DePalo, V. A. (2000). Anti-inflammatory cytokines. Chest Journal, 117(4),
1162-1172.
Panchagnula, R., & Thomas, N. S. (2000). Biopharmaceutics and pharmacokinetics in drug
research. International journal of pharmaceutics, 201(2), 131-150.
Pandeya, S., & Thakkar, D. (2005). Combinatorial Chemistry: A novel method in drug
discovery and its application. Indian Journal of Chemistry, 44, 335-348.

159
Papo, N., Shahar, M., Eisenbach, L., & Shai, Y. (2003). A novel lytic peptide composed of
DL-amino acids selectively kills cancer cells in culture and in mice. Journal of
Biological Chemistry, 278(23), 21018-21023.
Parente, R. A., Nadasdi, L., Subbarao, N. K., & Szoka Jr, F. C. (1990). Association of a
pH-sensitive peptide with membrane vesicles: role of amino acid sequence.
Biochemistry, 29(37), 8713-8719.
Parenty, A., Moreau, X., & Campagne, J.-M. (2006). Macrolactonizations in the total
synthesis of natural products. Chemical reviews, 106(3), 911-939.
Park, K., Kim, Y.-S., Lee, G. Y., Park, R.-W., Kim, I.-S., Kim, S. Y., & Byun, Y. (2008).
Tumor endothelial cell targeted cyclic RGD-modified heparin derivative: inhibition
of angiogenesis and tumor growth. Pharmaceutical research, 25(12), 2786-2798.
Park, S. Y., Kim, Y. H., Kim, Y., Kim, S. G., Shon, K. J., Choi, Y.-W., & Lee, S.-J.
(2011). Upregulation of heme oxygenase-1 via PI3K/Akt and Nrf-2 signaling
pathways mediates the anti-inflammatory activity of Schisandrin in Porphyromonas
gingivalis LPS-stimulated macrophages. Immunology letters, 139(1), 93-101.
Parmiani, G., Castelli, C., Dalerba, P., Mortarini, R., Rivoltini, L., Marincola, F. M., &
Anichini, A. (2002). Cancer immunotherapy with peptide-based vaccines: what
have we achieved? Where are we going? Journal of the National Cancer Institute,
94(11), 805-818.
Pastan, I., Hassan, R., FitzGerald, D. J., & Kreitman, R. J. (2006). Immunotoxin therapy of
cancer. Nature Reviews Cancer, 6(7), 559-565.
Peck, T. E., & Hill, S. A. (2014). Pharmacology for anaesthesia and intensive care:
Cambridge University Press.
Petti, S. (2009). Lifestyle risk factors for oral cancer. Oral oncology, 45(4), 340-350.
Porkka, K., Laakkonen, P., Hoffman, J. A., Bernasconi, M., & Ruoslahti, E. (2002). A
fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor
endothelial cells in vivo. Proceedings of the National Academy of Sciences, 99(11),
7444-7449.
Prados, J., Melguizo, C., Ortiz, R., Velez, C., J Alvarez, P., L Arias, J., . . . Aranega, A.
(2012). Doxorubicin-loaded nanoparticles: new advances in breast cancer therapy.

160
Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal
Chemistry-Anti-Cancer Agents), 12(9), 1058-1070.
Ramanathan, R. K., Lee, K. M., McKolanis, J., Hitbold, E., Schraut, W., Moser, A. J., . . .
Kim, H. (2005). Phase I study of a MUC1 vaccine composed of different doses of
MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic
cancer. Cancer Immunology, Immunotherapy, 54(3), 254-264.
Ravipati, A. S., Henriques, S. T., Poth, A. G., Kaas, Q., Wang, C. K., Colgrave, M. L., &
Craik, D. J. (2015). Lysine-rich cyclotides: a new subclass of circular knotted
proteins from Violaceae. ACS chemical biology, 10(11), 2491-2500.
Reardon, D. A., Fink, K. L., Mikkelsen, T., Cloughesy, T. F., O'Neill, A., Plotkin, S., . . .
Rich, K. M. (2008). Randomized phase II study of cilengitide, an integrin-targeting
arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme.
Journal of Clinical Oncology, 26(34), 5610-5617.
Reff, M. E., Hariharan, K., & Braslawsky, G. (2002). Future of monoclonal antibodies in
the treatment of hematologic malignancies. Cancer Control, 9(2), 152-168.
Reichert, J., Pechon, P., Tartat, A., & Dunn, M. (2010). Development trends for peptide
therapeutics: A comprehensive quantitative analysis of peptide therapeutics in
clinical development. Peptide Therapeutics Foundation, 1-11.
Renner, M. K., Shen, Y.-C., Cheng, X.-C., Jensen, P. R., Frankmoelle, W., Kauffman, C.
A., . . . Clardy, J. (1999). Cyclomarins AC, new antiinflammatory cyclic peptides
produced by a marine bacterium (Streptomyces sp.). Journal of the American
Chemical Society, 121(49), 11273-11276.
Reubi, J. C. (2007). Targeting CCK receptors in human cancers. Current topics in
medicinal chemistry, 7(12), 1239-1242.
Rose-John, S. (2012). IL-6 trans-signaling via the soluble IL-6 receptor: importance for the
pro-inflammatory activities of IL-6. Int J Biol Sci, 8(9), 1237-1247.
Rosenfeld, Y., Papo, N., & Shai, Y. (2006). Endotoxin (Lipopolysaccharide)
Neutralization by Innate Immunity Host-Defense Peptides PEPTIDE
PROPERTIES AND PLAUSIBLE MODES OF ACTION. Journal of Biological
Chemistry, 281(3), 1636-1643.

161
Rostovtsev, V. V., Green, L. G., Fokin, V. V., & Sharpless, K. B. (2002). A stepwise
huisgen cycloaddition process: copper (I)‐catalyzed regioselective “ligation” of
azides and terminal alkynes. Angewandte Chemie, 114(14), 2708-2711.
Roxin, Á., & Zheng, G. (2012). Flexible or fixed: a comparative review of linear and
cyclic cancer-targeting peptides. Future medicinal chemistry, 4(12), 1601-1618.
Salmon, S. E., Liu-Stevens, R. H., Zhao, Y., Lebl, M., Krchñák, V., Wertman, K., . . .
Lam, K. S. (1996). High-volume cellular screening for anticancer agents with
combinatorial chemical libraries: a new methodology. Molecular diversity, 2(1-2),
57-63.
Saltz, L., Trochanowski, B., Buckley, M., Heffernan, B., Niedzwiecki, D., Tao, Y., &
Kelsen, D. (1993). Octreotide as an antineoplastic agent in the treatment of
functional and nonfunctional neuroendocrine tumors. Cancer, 72(1), 244-248.
Salvemini, D., Ischiropoulos, H., & Cuzzocrea, S. (2003). Roles of nitric oxide and
superoxide in inflammation. Inflammation Protocols, 291-303.
Scheller, J., Garbers, C., & Rose-John, S. (2014). Interleukin-6: from basic biology to
selective blockade of pro-inflammatory activities. Paper presented at the Seminars
in immunology.
Schmidt, G., Grube, A., & Köck, M. (2007). Stylissamides A–D–New Proline‐Containing
Cyclic Heptapeptides from the Marine Sponge Stylissa caribica. European journal
of organic chemistry, 2007(24), 4103-4110.
Schmitt, E. K., Riwanto, M., Sambandamurthy, V., Roggo, S., Miault, C., Zwingelstein,
C., . . . Rao, S. P. (2011). The natural product cyclomarin kills Mycobacterium
tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease.
Angewandte Chemie International Edition, 50(26), 5889-5891.
Schwarze, S. S. R., & Dowdy, S. S. F. (2000). In vivo protein transduction: intracellular
delivery of biologically active proteins, compounds and DNA. Trends in
Pharmacological Sciences, 21(2), 45-48.
Shadidi, M., & Sioud, M. (2003). Selective targeting of cancer cells using synthetic
peptides. Drug Resistance Updates, 6(6), 363-371.

162
Shaheen, F., Jabeen, A., Ashraf, S., Nadeem‐ul‐Haque, M., Shah, Z. A., Ziaee, M. A., . . .
Ganesan, A. (2016). Total synthesis, structural, and biological evaluation of
stylissatin A and related analogs. Journal of Peptide Science, 22(9), 607-617.
Shaheen, F., Rasoola, S., Shah, Z. A., Soomro, S., Jabeen, A., Mesaik, M. A., &
Choudhary, M. I. (2014). Chemical constituents of Marrubium vulgare as potential
inhibitors of nitric oxide and respiratory burst. Natural product communications,
9(7), 903-906.
Shaheen, F., Rizvi, T. S., Musharraf, S. G., Ganesan, A., Xiao, K., Townsend, J. B., . . .
Choudhary, M. I. (2012). Solid-phase total synthesis of cherimolacyclopeptide E
and discovery of more potent analogues by alanine screening. Journal of natural
products, 75(11), 1882-1887.
Shao, Y., Lu, W., & Kent, S. B. (1998). A novel method to synthesize cyclic peptides.
Tetrahedron letters, 39(23), 3911-3914.
Silen, J. L., Lu, A. T., Solas, D. W., Gore, M. A., Maclean, D., Shah, N. H., . . . Tsutsui, K.
T. (1998). Screening for novel antimicrobials from encoded combinatorial libraries
by using a two-dimensional agar format. Antimicrobial agents and chemotherapy,
42(6), 1447-1453.
Smolarczyk, R., Cichoń, T., Graja, K., Hucz, J., Sochanik, A., & Szala, S. (2005).
Antitumor effect of RGD-4C-GG-D (KLAKLAK) 2 peptide in mouse B16 (F10)
melanoma model. Acta Biochimica Polonica, 53(4), 801-805.
Sole, K. (2006). HER-2/neu peptide vaccine for the prevention of prostate cancer
recurrence. Nature Clinical Practice Urology, 3(1), 6.
Songyang, Z., Carraway 3rd, K., Eck, M. J., Harrison, S. C., Feldman, R. A., Mohammadi,
M., . . . Eng, C. (1995). Catalytic specificity of protein-tyrosine kinases is critical
for selective signalling. Nature, 373(6514), 536-539.
Soto-Pantoja, D. R., Menon, J., Gallagher, P. E., & Tallant, E. A. (2009). Angiotensin-(1-
7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in
vascular endothelial growth factor. Molecular cancer therapeutics, 8(6), 1676-
1683.
Sotomayor, S., Muñoz‐Moreno, L., Carmena, M. J., Schally, A. V., Sánchez‐Chapado, M.,
Prieto, J. C., & Bajo, A. M. (2010). Regulation of HER expression and

163
transactivation in human prostate cancer cells by a targeted cytotoxic bombesin
analog (AN‐215) and a bombesin antagonist (RC‐3095). International Journal of
Cancer, 127(8), 1813-1822.
STEINAUER, R., CHEN, F. M., & LEO BENOITON, N. (1989). Studies on racemization
associated with the use of benzotriazol‐1‐yl‐tris (dimethylamino) phosphonium
hexafluorophosphate (BOP). International journal of peptide and protein research,
34(4), 295-298.
Strowski, M. Z., & Blake, A. D. (2008). Function and expression of somatostatin receptors
of the endocrine pancreas. Molecular and cellular endocrinology, 286(1), 169-179.
STRYER, L., t AMY, T. L., & SOLAS, D. (1991). Light-directed, spatially addressable
parallel.
Stuart, K., Stokes, K., Jenkins, R., Trey, C., & Clouse, M. (1993). Treatment of
hepatocellular carcinoma using doxorubicin/ethiodized oil/gelatin powder
chemoembolization. Cancer, 72(11), 3202-3209.
Sugano, M., Sato, A., Iijima, Y., Oshima, T., Furuya, K., Kuwano, H., . . . Hanzawa, H.
(1991). Phomactin A; a novel PAF antagonist from a marine fungus Phoma sp.
Journal of the American Chemical Society, 113(14), 5463-5464.
Tabas, I., & Glass, C. K. (2013). Anti-inflammatory therapy in chronic disease: challenges
and opportunities. Science, 339(6116), 166-172.
Tabudravu, J. N., Morris, L. A., Kettenes-van den Bosch, J. J., & Jaspars, M. (2002).
Axinellin C, a proline-rich cyclic octapeptide isolated from the Fijian marine
sponge Stylotella aurantium. Tetrahedron, 58(39), 7863-7868.
Takahashi, N., Hayano, T., & Suzuki, M. (1989). Peptidyl-prolyl cis-trans isomerase is the
cyclosporin A-binding protein cyclophilin. Nature, 337(6206), 473-475.
Temming, K., Schiffelers, R. M., Molema, G., & Kok, R. J. (2005). RGD-based strategies
for selective delivery of therapeutics and imaging agents to the tumour vasculature.
Drug Resistance Updates, 8(6), 381-402.
Thayer, A. M. (2011). Improving peptides. Chemical & Engineering News, 89(22), 13-+.
Thorpe, P. E. (2004). Vascular targeting agents as cancer therapeutics. Clinical Cancer
Research, 10(2), 415-427.

164
Thundimadathil, J. (2012). Cancer treatment using peptides: current therapies and future
prospects. Journal of amino acids, 2012.
Tiidus, P. M. (1998). Radical species in inflammation and overtraining. Canadian journal
of physiology and pharmacology, 76(5), 533-538.
Torchilin, V. P. (2008). Cell penetrating peptide‐modified pharmaceutical nanocarriers for
intracellular drug and gene delivery. Peptide Science, 90(5), 604-610.
Tracey, K. (2002). The inflammatory reflex Nature 420: 853–859. Find this article online.
Tracey, K., Vlassara, H., & Cerami, A. (1989). Peptide regulatory factors:
cachectin/tumour necrosis factor. The Lancet, 333(8647), 1122-1126.
Tracey, K. J. (2002). The inflammatory reflex. Nature, 420(6917), 853-859.
Tracey, K. J., Beutler, B., Lowry, S. F., Merryweather, J., Wolpe, S., Milsark, I. W., . . .
Albert, J. D. (1986). Shock and tissue injury induced by recombinant human
cachectin. Science, 234(4775), 470-474.
Tsarbopoulos, A., Karas, M., Strupat, K., Pramanik, B. N., Nagabhushan, T. L., &
Hillenkamp, F. (1994). Comparative mapping of recombinant proteins and
glycoproteins by plasma desorption and matrix-assisted laser desorption/ionization
mass spectrometry. Analytical chemistry, 66(13), 2062-2070.
V Rosca, E., E Koskimaki, J., G Rivera, C., B Pandey, N., P Tamiz, A., & S Popel, A.
(2011). Anti-angiogenic peptides for cancer therapeutics. Current pharmaceutical
biotechnology, 12(8), 1101-1116.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., & Telser, J. (2007). Free
radicals and antioxidants in normal physiological functions and human disease. The
international journal of biochemistry & cell biology, 39(1), 44-84.
Van de Wiele, C., Phonteyne, P., Pauwels, P., Goethals, I., Van den Broecke, R., Cocquyt,
V., & Dierckx, R. A. (2008). Gastrin-releasing peptide receptor imaging in human
breast carcinoma versus immunohistochemistry. Journal of Nuclear Medicine,
49(2), 260-264.
Vassileva, V., Grant, J., De Souza, R., Allen, C., & Piquette-Miller, M. (2007). Novel
biocompatible intraperitoneal drug delivery system increases tolerability and
therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model.
Cancer chemotherapy and pharmacology, 60(6), 907-914.

165
Virgolini, I., Traub, T., Novotny, C., Leimer, M., Fuger, B., Li, S., . . . Raderer, M. (2002).
Experience with indium-111 and yttrium-90-labeled somatostatin analogs. Current
pharmaceutical design, 8(20), 1781-1807.
Wadia, J. S., Stan, R. V., & Dowdy, S. F. (2004). Transducible TAT-HA fusogenic peptide
enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nature
medicine, 10(3), 310-315.
Waldmann, T. A. (1993). The IL-2/IL-2 receptor system: a target for rational immune
intervention. Immunology today, 14(6), 264-270.
Walensky, L. D., Kung, A. L., Escher, I., Malia, T. J., Barbuto, S., Wright, R. D., . . .
Korsmeyer, S. J. (2004). Activation of apoptosis in vivo by a hydrocarbon-stapled
BH3 helix. Science, 305(5689), 1466-1470.
Wang, C., Li, H.-B., Li, S., Tian, L.-L., & Shang, D.-J. (2012). Antitumor effects and cell
selectivity of temporin-1CEa, an antimicrobial peptide from the skin secretions of
the Chinese brown frog (Rana chensinensis). Biochimie, 94(2), 434-441.
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., . . .
Borovikova, L. (1999). HMG-1 as a late mediator of endotoxin lethality in mice.
Science, 285(5425), 248-251.
Wang, J., Ma, Y., Zhu, Z.-H., Situ, D.-R., Hu, Y., & Rong, T.-H. (2012). Expression and
prognostic relevance of tumor carcinoembryonic antigen in stage IB non-small cell
lung cancer. Journal of thoracic disease, 4(5), 490-496.
Wang, P., Nan, Y. H., Yang, S.-T., Kang, S. W., Kim, Y., Park, I.-S., . . . Shin, S. Y.
(2010). Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich α-helical
model antimicrobial peptide and its diastereomeric peptides. Peptides, 31(7), 1251-
1261.
Wang, S.-S. (1973). p-Alkoxybenzyl alcohol resin and p-
alkoxybenzyloxycarbonylhydrazide resin for solid phase synthesis of protected
peptide fragments. Journal of the American Chemical Society, 95(4), 1328-1333.
Wang, X.-Y., Wang, Q., Huang, X.-Y., Wang, T., & Yu, X.-Q. (2006). Synthesis of small
cyclic peptides containing the disulfide bond. Arkivoc, 11, 1-7.

166
Wang, X., Morinaka, B. I., & Molinski, T. F. (2014). Structures and solution
conformational dynamics of stylissamides G and H from the Bahamian sponge
Stylissa caribica. Journal of natural products, 77(3), 625-630.
Weber, J. S., Vogelzang, N. J., Ernstoff, M. S., Goodman, O. B., Cranmer, L. D., Marshall,
J. L., . . . Qiu, Z. (2011). A phase 1 study of a vaccine targeting preferentially
expressed antigen in melanoma and prostate-specific membrane antigen in patients
with advanced solid tumors. Journal of immunotherapy (Hagerstown, Md.: 1997),
34(7), 556.
White, C. J., & Yudin, A. K. (2011). Contemporary strategies for peptide
macrocyclization. Nature chemistry, 3(7), 509-524.
Wild, D., Fani, M., Behe, M., Brink, I., Rivier, J. E., Reubi, J. C., . . . Weber, W. A.
(2011). First clinical evidence that imaging with somatostatin receptor antagonists
is feasible. Journal of Nuclear Medicine, 52(9), 1412-1417.
Winssinger, N., Damoiseaux, R., Tully, D. C., Geierstanger, B. H., Burdick, K., & Harris,
J. L. (2004). PNA-encoded protease substrate microarrays. Chemistry & biology,
11(10), 1351-1360.
Wolinsky, J. B., Colson, Y. L., & Grinstaff, M. W. (2012). Local drug delivery strategies
for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. Journal
of controlled release, 159(1), 14-26.
Xu, X.-M., Zhang, Y., Qu, D., Liu, H.-B., Gu, X., Jiao, G.-Y., & Zhao, L. (2013).
Combined anticancer activity of osthole and cisplatin in NCI-H460 lung cancer
cells in vitro. Experimental and therapeutic medicine, 5(3), 707-710.
YAMAMOTO, K., UENO, T., KAWAOKA, T., HAZAMA, S., FUKUI, M., SUEHIRO,
Y., . . . OKA, M. (2005). MUC1 peptide vaccination in patients with advanced
pancreas or biliary tract cancer. Anticancer Research, 25(5), 3575-3579.
Yang, J. C. (2011). Melanoma vaccines. The Cancer Journal, 17(5), 277-282.
Yang, L., Berk, S. C., Rohrer, S. P., Mosley, R. T., Guo, L., Underwood, D. J., . . . Mitra,
S. W. (1998). Synthesis and biological activities of potent peptidomimetics
selective for somatostatin receptor subtype 2. Proceedings of the National Academy
of Sciences, 95(18), 10836-10841.

167
Yang, Y.-l., Hua, K.-f., Chuang, P.-H., Wu, S.-h., Wu, K.-y., Chang, F.-R., & Wu, Y.-c.
(2007). New cyclic peptides from the seeds of Annona squamosa L. and their anti-
inflammatory activities. Journal of agricultural and food chemistry, 56(2), 386-
392.
Yang, Y., Adelstein, S. J., & Kassis, A. I. (2009). Target discovery from data mining
approaches. Drug discovery today, 14(3), 147-154.
Yergey, A. L., Coorssen, J. R., Backlund, P. S., Blank, P. S., Humphrey, G. A.,
Zimmerberg, J., . . . Vestal, M. L. (2002). De novo sequencing of peptides using
MALDI/TOF-TOF. Journal of the American Society for Mass Spectrometry, 13(7),
784-791.
Zasloff, M. (2002). Antimicrobial peptides of multicellular organisms. nature, 415(6870),
389-395.
Zhang, H., Chen, J., Waldherr, C., Hinni, K., Waser, B., Reubi, J. C., & Maecke, H. R.
(2004). Synthesis and evaluation of bombesin derivatives on the basis of pan-
bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for
targeting bombesin receptor-expressing tumors. Cancer research, 64(18), 6707-
6715.
Zhang, L., & Tam, J. P. (1997). Metal ion-assisted peptide cyclization. Tetrahedron letters,
38(25), 4375-4378.
Zhang, X.-X., Eden, H. S., & Chen, X. (2012). Peptides in cancer nanomedicine: drug
carriers, targeting ligands and protease substrates. Journal of controlled release,
159(1), 2-13.
Zitzmann, S., Ehemann, V., & Schwab, M. (2002). Arginine-glycine-aspartic acid (RGD)-
peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer research,
62(18), 5139-5143.
Zughaier, S. M., Shafer, W. M., & Stephens, D. S. (2005). Antimicrobial peptides and
endotoxin inhibit cytokine and nitric oxide release but amplify respiratory burst
response in human and murine macrophages. Cellular microbiology, 7(9), 1251-
1262.

168
Glossary
Anticancer/ Antitumor Agents
Agents that inhibit the formation of tumor or to stop the irregular cell division are known
as anticancer or antitumor agents.
Bioassay
The comparison of test and standard compound in-vitro and in-vivo analysis is known as
bioassay.
Broad-Band Decoupled 13C-NMR Spectrum
It is a fully decoupled 13C-NMR spectrum, which gives information about the number of
carbon atoms in a molecule and their electronic environment.
Chemical Shifts (δ)
The difference between the precession frequencies of a nucleus to standard (TMS) is called
chemical shift value. It is expressed in ppm (Parts per million).
Chromatography
It is a separation technique in which the constituents of the mixture are separated between
the two phases (Stationary and mobile phases).
COSY-45° Spectrum
It is a homonuclear two-dimensional NMR spectroscopic technique, used for the
determination of vicinal and geminal 1H-1H couplings.
Coupling Constant (J)
The magnitude of splitting of NMR signal is known as coupling constant. It is a constant
number which is employed to determine the scalar coupling between adjacent protons and
other nuclei. It also determines the strength of interaction of a proton with a non-equivalent
proton on same carbon (geminal coupling or 1, 2 coupling) or on adjacent carbon (vicinal
coupling or 1,3 coupling). It is expressed in cycle/second or Hz.

169
Distortionless Enhancement by Polarization Transfer (DEPT)
A one-dimensional 13C-NMR spectroscopic technique, employed to differentiate the
multiplicity among CH3, CH2, and CH. The DEPT spectra is recoded at different pulse
angles,i.e. = 45°, 90°, and 135°.
Electrospray Ionization Mass Spectrometry (ESI-MS)
The low resolution mass spectrum in which the ionization and fragmentation of the
compound is brought about by the bombardment of electron beam with 70 ev energy. This
gives an important clue about the structure of molecule through fragmentation pattern.
Elute
The process in which the solute molecules is carrying away by solvent in a
chromatographic operation is called elute.
Eluent
Mobile phase which is used in chromatographic technique for the separation of compounds
from mixture is called eluent.
Elution
The process by which the dissolved constituents in column were carried by mobile phase is
called elution.
Flow Rate
The time required for mobile phase to pass through the column is called flow rate.
Gradient Elution
The process in which the polarity of mobile phase changed with time is called gradient
elution. It is used to decrease the separation time by increasing the polarity of mobile phase
over time in chromatography. It is also known solvent programming system. Gradient
elution may be continuous or stepwise. In routine, HPLC based separation technique,
binary, tertiary and quaternary gradients solvent are used.

170
Heteronuclear Multiple Bond Connectivity (HMBC)
It is a heteronuclear two-dimensional NMR technique, which determines the long-range
couplings between carbons and hydrogens (1J, 2J, 3J and 4J in conjugated system).
Heteronuclear Single Bond Coherence (HSQC)
It is an inverse heteronuclear two-dimensional NMR spectroscopy, which is used to
determinethe 1H/13C one bond correlations.
High-resolution Electron Impact Mass Spectrum (HREI-MS)
It is a mass spectrometry technique which is used directly for the determination of
elemental composition of a compound through exact mass of the molecule. The HREI-MS
is recorded by a double focusing mass spectrometer.
Heteronuclear Multiple Bond Connectivity (HMBC)
It is an inverse two-dimensional heteronuclear NMR experiment, which determines the
long-range coupling between proton and carbon (1H/13C, 1J, 2J, 3J and 4J in conjugation).
Heteronuclear Single Quantum Coherence (HSQC)
It is an inverse two-dimensional NMR technique used to deduce the direct linkage of
proton to carbon (1H/13C) through one-bond connectivity.
Inflammation
The body’s adaptive and immediate to tissue and cell damage by pathogens stimuli, or
physical injuries is called Inflammation.
Pharmacokinetic
The process by which a drug is absorbed, distributed, metabolized and eliminated by the
body.

171
Infrared Spectroscopy
It is a spectroscopic technique, which is used for the determination of functionalities in
molecule. The infrared spectra are generated by the absorption of infrared radiations in
region ranging from 320-4000 cm-1 by a substance.
Isocratic solvent system
The process in which the polarity of mobile phase remains same throughout the operation
in column chromatography is known as isocratic solvent system.
Mobile Phase
The solvent (liquid, gas) used to elute the solute in column chromatography is called
mobile phase.
Optical Rotation
It is a physical property of a molecule that rotates the plane polarized light either towards
the right or towards the left. Its presence in a molecule indicates the chiral nature of the
molecule.
Proton-NMR Spectrum (1H-NMR)
A one-dimensional NMR technique which gives information about the electronic
environment of a proton in a molecule.
TOCSY
TOCSY is a total correlation spectroscopy, which is also called HOHAHA (Homonuclear
Hartmann Hahn). It provides couplings of all the protons in an isolated spin system.

172
Abbreviation
ACPs Anticancer peptides
Boc tert-Butoxycarbonyl
Cbz Benzyloxycarbonyl
DCC Dicyclohexylcarbodiimide
DIC Diisopropylcarbodiimide
DIPEA Diisopropylethylamine (Hünig’s base)
DMF Dimethylformamide
Fmoc Fluoren-9-ylmethyloxycarbonyl
PyBOP Benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphoniumhexafluoro
phosphate
HATU N-[(Dimethylamino)-1H-1, 2, 3-triazole [4, 5-b]pyridin-1- yl methy
lene]-N-methylmethanaminiumhexafluorophosphate N-oxide
HBTU N-[(1H-Benzotriazol-1-yl)(dimethylamino)methylene]-N-methyl
methan-aminium hexafluorophosphate N-oxide
HOAt 1-Hydroxy-7-azabenzotriazole
HOBt 1-Hydroxybenzotriazole
HPLC High performance liquid chromatography
IC50 Inhibitor concentration giving 50 % inhibition
MALDI – TOF Matrix Assisted Laser Desorption/Ionization Time-of-Flight
NMM N-Methylmorpholine

173
NMP N-Methyl-2-pyrrolidone
NMR Nuclear magnetic resonance (spectroscopy)
Pbf 2, 2, 4, 6, 7-Pentamethyldihydrobenzofuran-5-sulfonyle
SAR Structure–activity relationship
TBTU O-(Benzotriazol-1-yl)-N, N, N', N'-tetra methyl uranium tetra fluoro
borate
TFA Trifluoroacetic acid
TIS Triisopropylsilane
Ala (L) Alanine
ala (D) Alanine
BNZ 2-Amino-1- methyl benzimidazole
Gly Glycine
iPr Isopropyl
Ileu Isoleucine
Lys Lysine
2-Nal 2-Naphthyl alanine
Thr Threonine
Trp Triptophane
Tyr Tyrosine