surface vibrational thermodynamics from ab initio calculations for fcc(100)
TRANSCRIPT

Surface Science 604 (2010) 308–317
Contents lists available at ScienceDirect
Surface Science
journal homepage: www.elsevier .com/ locate /susc
Surface vibrational thermodynamics from ab initio calculations for fcc(1 0 0)
Handan Yildirim a, Abdelkader Kara a,*, Talat S. Rahman a, Rolf Heid b, Klaus-Peter Bohnen b
a Department of Physics, University of Central Florida, Orlando, FL 32816, United Statesb Institut für Festkörperphysik, Forschungszentrum Karlsruhe, D-76021 Karlsruhe, Germany
a r t i c l e i n f o a b s t r a c t
Article history:Received 4 September 2009Accepted for publication 18 November 2009Available online 23 November 2009
Keywords:Density functional theoryVibrational dynamicsThermodynamicsMetal surfaces
0039-6028/$ - see front matter � 2009 Elsevier B.V. Adoi:10.1016/j.susc.2009.11.022
* Corresponding author. Tel.: +1 4078231527; fax:E-mail address: [email protected] (A. Kara).
We present vibrational dynamics and thermodynamics for the (1 0 0) surfaces of Cu, Ag, Pd, Pt and Auusing a real space approach. The force field for these systems is described by density functional theory.The changes in the vibrational dynamics and thermodynamics from those in bulk are confined mostly tothe first-layer. A substantial enhancement of the low-frequency end of the acoustic branch was found andis related to a loosening of the bond at the surface. The thermodynamics of the first-layer also show sig-nificant differences (higher heat capacity, lower free energy and higher mean vibrational square ampli-tudes) from what obtains in bulk. Comparing these results with those calculated using embedded-atom method potentials, we discovered that for Ag(1 0 0) and Cu(1 0 0), the two methods yield very sim-ilar results while for Pd(1 0 0), Pt(1 0 0) and Au(1 0 0) there are substantial differences.
� 2009 Elsevier B.V. All rights reserved.
1. Introduction
In considerations of relative stability of surfaces, nanostruc-tures, as well as bulk phases of different crystallographic orienta-tions, the quantity of interest is the free energy which includescontributions from the structural potential energy and the sys-tem’s vibrational and configurational entropy. While the latter isan important constituent for any system containing more thanone type of element (alloys), vibrational entropy is the quantityto be evaluated for surfaces and nanostructures of single elements.This is the factor that controls the temperature dependence of sur-face free energy, mean square vibrational amplitudes of surfaceatoms, the surface Debye temperature, and the surface heat capac-ity. It is also the quantity that may determine the equilibriumshape of the crystal surface and its propensity to undergo struc-tural transitions. Knowledge of surface free energy, together withthat of the step and kink free energy is also essential for consider-ations of surface faceting, bunching, and roughening. The extrac-tion of free energy from experimental data is, however, nontrivial[1]. The lattice contribution, which can be critical for determiningstructural transitions, is non zero, albeit a small fraction of thestructural energy. It is thus encouraging to see the flurry of activityin analyzing the contribution of vibrational entropy [2]. Several re-cent theoretical studies have been devoted to determining thevibrational contribution to the thermodynamic functions for sur-face systems [3–5]. These calculations have already provided aqualitative measure of the effect of vibrational entropy on surface
ll rights reserved.
+1 4078235112.
stability and structure. Durukanoglu et al. [4] and Kara andRahman [5] have also set the stage for a systematic evaluation ofthe local vibrational contribution to the free energy. Since thesecalculations were based on usage of many body interaction poten-tials [6], questions have been asked about their accuracy, particu-larly for 5d metals Pt, Ir and Au, for which these potentials arenot expected to work as well as they do for Ag, Cu, and Ni.
With the availability of ab initio electronic structure methodsbased on the density functional perturbation theory [7], surfacephonon dispersion curves can be drawn with remarkable accuracy[8]. These dispersion curves further lend themselves to the calcula-tion of vibrational density of states and thus of the vibrational con-tribution to surface free energy and entropy. Our aim in the presentpaper is to analyze the thermodynamic properties of the (1 0 0)surface of several metals and to compare the findings with thoseobtained earlier using potentials from the embedded-atom method(EAM) [4,5]. We carry out an analysis of the inter-atomic surfaceforce constants obtained from the ab initio methods, to examinethe range of the interaction and for comparison with those ob-tained from EAM. As is known, EAM potentials are relatively shortranged and do not extend to interactions beyond the fourth neigh-bor. These potentials are thought to be well suited for the six met-als Ag, Cu, Ni, Pd, Pt, Au, and although numerous efforts have beenmade to develop such potentials for a number of other metals andmetallic alloys [9]. Out of these six metals, we restrict ourselves inthis work to the five nonmagnetic systems. We are also aware thatAu(1 0 0) and Pt(1 0 0) undergo a hexagonal surface structuraltransition [10,11]. However, for the purpose of the present studywe use the results available for the surface phonons of the unre-constructed surface [8].

H. Yildirim et al. / Surface Science 604 (2010) 308–317 309
In Section 2 we present some details of the theoretical methodsused. In Section 3 we discuss our results. In Section 4 we comparethe vibrational dynamics, thermodynamics and the force constantscalculated using DFT with those calculated using EAM. In Section 5,we present our overall conclusions.
2. Theoretical details
We have used density functional theory (DFT) [12,13] to calcu-late the structure within the mixed basis (MB) pseudopotentialrepresentation [14–18]. A density perturbation theory [19], asadapted for the MB representation [20], was used to evaluate theforce constants. The complete description of the calculation ofthe structure and force constants can be found in Ref. [8].
2.1. Structure and force constants
Our calculated relaxations using DFT and EAM for Ag, Cu, Pd, Ptand Au(1 0 0) are presented in Table 1 and compared with previouscalculations and experimental data when available [21–32]. Forboth Cu(1 0 0) and Ag(1 0 0), the calculated contraction betweenthe first- and second-layer is larger than the experimental observa-tion, except for the case of Cu(1 0 0) using EAM, for which aremarkable agreement is achieved with data. For Cu(1 0 0), the cor-rected effective medium theory calculations [28] show a largedeviation from the other DFT based values (Table 1). For Pd, Pt
Table 1The calculated % surface relaxation (Ddij) from DFT and EAM. Ddij = 100(dij � d0)/d0,where dij is the interlayer spacing between the atomic layers i and j. d0 is theinterlayer spacing in the unrelaxed surface.
Surfaces Dd12 DFT (EAM) present study Dd12 others
Ag(1 0 0) �2.1 (�1.9) �1.4 [21]�1.9 [22,27]�1.9 [23]0 ± 0.03 [24]a
Cu(1 0 0) �3.0 (�1.4) �2.9 [25]�1.4 [6]�3.0 [26]�3.5 [23]�6.0 [28]�1.1 ± 0.40 [30]a
Pd(1 0 0) �1.3 (�4.5) �0.6 [22]�1.2 [30]
Pt(1 0 0) �2.3 (�6.9) �2.2 [30]
Au(1 0 0) �1.6 (�6.0) �1.0 [27]�1.2 [31]
a Experimental data.
Fig. 1. The relative force constant strength vs. neighbors calculated using DFT for (a)
and Au(1 0 0), all DFT based calculations show a modest contrac-tion between the first- and second-layer while EAM potentials pre-dict rather larger values (by a factor of about four or larger). This isan indication that additional physical properties of these three sur-faces as calculated using DFT will show differences from those cal-culated using EAM.
Since we will be using, for the calculation of the vibrationaldynamics, a method that assumes a finite interaction range, a trun-cation on the number of neighbors is necessary. Because knowl-edge of the interaction range is not available a priori, one needsto investigate this effect for every element in order to achieve highaccuracy. For this purpose, we introduced a simple quantity toidentify the ‘‘strength” of the interaction between neighbors viaforce constants. This force constant strength (fcs) is evaluated inEq. (1) as the maximum strength for a given level of neighbors(the maximum is chosen over the average in order not to excludea possible case for which one particular nth neighbor has excep-tionally large strength). The force constant strength also helps usdecide how extensive a neighborhood we need to consider for aparticular system. The force constant strength is calculated be-tween the atoms (i, j) that are nth neighbors; where n can be any-thing between the 1st and 20th neighbor. For the nth neighbor, wedefine the force constant strength as:
ðfcsÞn ¼MaxXa;b
Ka;bi;j
� �2" #
over all nth neighbors
ð1Þ
where a and b are x, y or z. We normalize the force constantstrength by that of the 1st neighbors and designate the result as rel-ative strength (Rfcs). The DFT calculations for (1 0 0) surfaces pre-sented in Ref. [8] are the basis of our analysis. We present theRfcs for Rh, Ir, Pd, and Pt(1 0 0) in Fig. 1a, and for Ag, Au andCu(1 0 0) in Fig. 1b. As one may note from Fig. 1, the Rfcs decreasesvery rapidly with neighbors; hence we present their values in a logscale. In Fig. 1a, one notes that the second nearest neighbor contri-bution is less than 20%, while it is less than 10% for those repre-sented in Fig. 1b. For all cases, the contribution of the 5thneighbors is of the order of 2%. Contribution of yet higher-orderneighbors is less than 2% for all the cases with the exception ofthe 9th neighbor contribution for Rh and Ir(1 0 0) for which the Rfcsis about 10%. If we neglect the contribution of the neighbors withRfcs less than or equal to 2%, we can classify these systems intotwo categories: the first consists of Ag, Au, Cu, Pd and Pt(1 0 0)(up to 5th neighbors) and the second one including Ir andRh(1 0 0) (up to 5th neighbors plus the 9th). In this paper, we pres-ent a detailed study of the vibrational dynamics and thermodynam-ics of the elements in the first category.
Ir(1 0 0), Rh(1 0 0), Pd(1 0 0) and Pt(1 0 0); (b) Au(1 0 0), Ag(1 0 0) and Cu(1 0 0).

310 H. Yildirim et al. / Surface Science 604 (2010) 308–317
2.2. Vibrational densities of states (VDOS)
The vibrational modes and the density of vibrational states canbe obtained from lattice dynamics calculations [32] or moleculardynamics simulations [33]. Lattice dynamics methods can be fur-ther classified into those based on real space and those based onreciprocal space. Using a reciprocal space based method, Heidand Bohnen have calculated the dispersions of different modesfor the systems mentioned [8]. In the present paper, we use a realspace based method, more suitable for calculating directly the localvibrational densities of states (LVDOS) hence the corresponding lo-cal thermodynamics quantities. The force constants used in thepresent DFT part of the study were extracted from the results ofthe calculations by Heid and Bohnen [8], while the force constantsused in the EAM part are evaluated through the direct calculationof the partial second derivatives of the potential.
The LVDOS can be calculated using the continued fractionmethod (CF) based on the real space [34]. In this method, the sur-face matrix is treated as a perturbation to the bulk and the surfaceGreen’s function is obtained by projecting the bulk Green’s func-tion into the subspace defined by the perturbation matrix. If theinterest lies in gaining insights into the local contributions to thedynamics and thermodynamics of systems possessing site specificenvironments, a local approach in the real space is perhaps moreuseful than one based on k-space. The real space Green’s function(RSGF) method is one such method [35]. Instead of wave vectorsand Brillouin zones, one can focus on any ‘local’ region and analyzethe effect of the rest of the system on it, recursively. Since thismethod does not require the system to be periodic, it is particularlysuitable for studying local vibrational density of states of the sys-tems with defects and disorder. The only prerequisite is that the in-ter-atomic potential between the atoms in the system be of finite
Fig. 2. Layer-resolved vibrational densities of states
range, to enable writing the force constant matrix in a block-tridi-agonal form. There is also no a priori truncation in the system size,as would be the case for the matrix diagonalization methods basedon k-space. The real space Green’s function method also has anadvantage over the familiar ‘‘continued fraction” method [34] inthat it does not involve truncation schemes to determine the recur-sion coefficients, but applies a more general and simpler recursivescheme.
The first step in the application of RSGF method is to divide thesystem (with a finite interaction range) into regions such that theHamiltonian can be written in a block-tridiagonal matrix asfollows:
H ¼
. ..
ti;i�1 hi ti;iþ1
tiþ1;i hiþ1 tiþ1;iþ2
. ..
0BBBBB@
1CCCCCA ð2Þ
where the sub-matrices hi along the diagonals are (ni � ni) squarematrices and the sub-matrices ti;iþ1 along the ‘‘off-diagonals” arematrices of dimension 3ni � 3ni+1 with n being the number of par-ticles in the locality. The Green’s function associated with the ma-trix H is given by:
GðzÞ ¼ ðzI � HÞ�1 ð3Þ
where z = x2 + ie, e is the width of the Lorentzian representing thedelta function at x2, and I is a unit matrix of the same size as thatof H. The diagonal element of the Green’s function matrix corre-sponding to any chosen locality is given by:
for (a) Pd(1 0 0); (b) Pt(1 0 0) and (c) Au(1 0 0).

Fig. 3. The thermodynamic functions for Pd(1 0 0): (a) lattice heat capacity; (b) excess lattice heat capacity; (c) vibrational contribution to the free energy; (d) excessvibrational contribution to the free energy; (e) vibrational mean square amplitude.
H. Yildirim et al. / Surface Science 604 (2010) 308–317 311
Gii ¼ zI � hið Þ � ti;iþ1Dþiþ1tiþ1;i � ti;i�1D
�i�1ti�1;i
h i�1ð4Þ
(Dþi ) and (D�i ) are defined as forward and backward partial Green’sfunctions and described by:
Dþi ¼ zI � hi � ti;iþ1Dþi;iþ1tiþ1;i
� ��1ð5Þ
D�i ¼ zI � hi � ti;i�1D�i�1ti�1;i
� ��1 ð6Þ
The relation between the diagonal elements of the Green’s func-tion matrix G is [35]:
Gii ¼ D�i þ D�i ti;i�1Gi�1;i�1ti�1;iD�i ð7Þ
As seen in the above equations, in the method of resolvent ma-trix, the calculation of Green’s functions is reduced to the series ofinversion and multiplication of matrices whose dimensions areusually much smaller than the total number of degrees of freedomof the system under consideration. Another feature of the methodis that one can calculate the Green’s function corresponding to anyspecified locality in the system, a diagonal element of the Green’sfunction representing the entire system. This can be achievedthrough the forward and backward partial Green’s functions matri-ces D�i . In the present calculations, we apply this method layer-by-layer, viewing an infinite system with in-plane periodicity as two-dimensional atomic layers stacked one upon the other along theaxis normal to the planes. Hence, the elements of the sub-matricesin the block-tridiagonal matrix describe the force constants be-tween the particles within and between the associated localities

Fig. 4. The thermodynamic functions for Pt(1 0 0): (a) lattice heat capacity; (b) excess lattice heat capacity; (c) vibrational contribution to the free energy; (d) excessvibrational contribution to the free energy; (e) vibrational mean square amplitude.
312 H. Yildirim et al. / Surface Science 604 (2010) 308–317
containing certain number of two-dimensional atomic layers [36].The normalized vibrational density of states g(x2) and the fre-quency dependent vibrational density of states N(x) are:
gðx2Þ ¼ � 13np
lime!0
ImTr G x2 þ ie� �� �
ð8Þ
NðxÞ ¼ 2xg x2� �ð9Þ
2.3. Thermodynamic functions
Once the local vibrational density of states is calculated, we candetermine the local thermodynamic functions of the system, whichin the harmonic approximation, are given by:
Fvib ¼ 3kBTZ 1
0ln 2 sinh
�hx2kBT
� � NðxÞdx ð10Þ
CV ¼ 3kBTZ 1
0
�hx2kBT
� 2 1
sinh2
�hx2kBT
� NðxÞdx ð11Þ
u2a
�¼ �h
2M
Z 1
0
1x
coth 2�hxkBTð ÞNaðxÞdx ð12Þ
where Fvib, CV and hu2ai are the vibrational free energy, the lattice
heat capacity, and the vibrational mean square atomic displace-ments along the Cartesian direction a, and Na(x) is the a compo-nent of the vibrational density of states.
3. Results and discussion
We first present our DFT results on the LVDOS (the locality herebeing an atom in a given layer) for each surface and analyze mainlythe differences between the first- and the fourth-layer LVDOS,
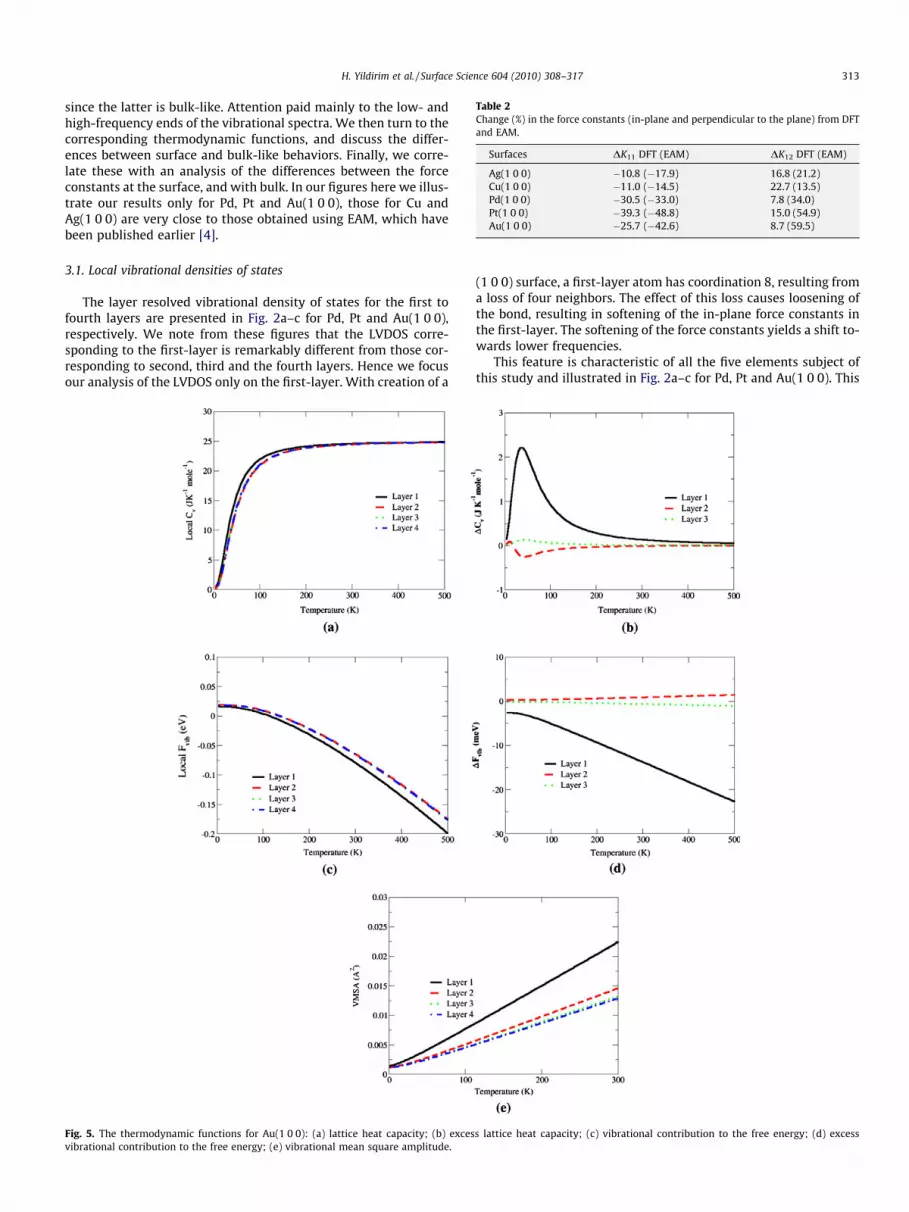
Table 2Change (%) in the force constants (in-plane and perpendicular to the plane) from DFTand EAM.
Surfaces DK11 DFT (EAM) DK12 DFT (EAM)
Ag(1 0 0) �10.8 (�17.9) 16.8 (21.2)Cu(1 0 0) �11.0 (�14.5) 22.7 (13.5)Pd(1 0 0) �30.5 (�33.0) 7.8 (34.0)Pt(1 0 0) �39.3 (�48.8) 15.0 (54.9)Au(1 0 0) �25.7 (�42.6) 8.7 (59.5)
H. Yildirim et al. / Surface Science 604 (2010) 308–317 313
since the latter is bulk-like. Attention paid mainly to the low- andhigh-frequency ends of the vibrational spectra. We then turn to thecorresponding thermodynamic functions, and discuss the differ-ences between surface and bulk-like behaviors. Finally, we corre-late these with an analysis of the differences between the forceconstants at the surface, and with bulk. In our figures here we illus-trate our results only for Pd, Pt and Au(1 0 0), those for Cu andAg(1 0 0) are very close to those obtained using EAM, which havebeen published earlier [4].
3.1. Local vibrational densities of states
The layer resolved vibrational density of states for the first tofourth layers are presented in Fig. 2a–c for Pd, Pt and Au(1 0 0),respectively. We note from these figures that the LVDOS corre-sponding to the first-layer is remarkably different from those cor-responding to second, third and the fourth layers. Hence we focusour analysis of the LVDOS only on the first-layer. With creation of a
Fig. 5. The thermodynamic functions for Au(1 0 0): (a) lattice heat capacity; (b) excesvibrational contribution to the free energy; (e) vibrational mean square amplitude.
(1 0 0) surface, a first-layer atom has coordination 8, resulting froma loss of four neighbors. The effect of this loss causes loosening ofthe bond, resulting in softening of the in-plane force constants inthe first-layer. The softening of the force constants yields a shift to-wards lower frequencies.
This feature is characteristic of all the five elements subject ofthis study and illustrated in Fig. 2a–c for Pd, Pt and Au(1 0 0). This
s lattice heat capacity; (c) vibrational contribution to the free energy; (d) excess
314 H. Yildirim et al. / Surface Science 604 (2010) 308–317
is not a global shift of the density of states but rather a relativelyimportant depletion of the high-frequency band accompanyingan enhancement of the low-frequency band. As the figures makeplain, these effects are marginal for the second-layer atoms andare absent for the third and fourth layers. We have found thatthe LVDOS for Cu and Ag(1 0 0) show the same trend as exhibitedby the three surfaces just discussed.
3.2. Thermodynamic functions
As a consequence of the features in the LVDOS discussed in theprevious sub-section, we expect deviations in the thermodynamicsfrom the bulk values to be localized at the top layer atoms. This isin line with findings reported in earlier publications on the vibra-tional dynamics and thermodynamics of vicinal and kinked fcc me-tal surfaces using EAM potentials [4,5,37]. The quantities ofinterest here are the lattice heat capacity (local and excess), thecontribution of the vibrational dynamics to the free energy (localand excess) and the mean square displacement. We present in Figs.3–5 the local and excess thermodynamical functions for Pd, Pt andAu(1 0 0), respectively. As shown in Figs. 3a, 4a and 5a, the locallattice heat capacity (CV) of the first-layer atoms differs from thatof the other atoms in the system and it is temperature dependent.These differences are better described by the local excess from thebulk as illustrated in Figs. 3b, 4b, and 5b. Indeed, for Pd andPt(1 0 0), the maximum deviation was found to be 3.4 and2.8 J K�1 mol�1, respectively, both occurring at a temperature of50 K. However, the first-layer atoms of Au(1 0 0) behave differ-ently; with a maximum deviation of only 2.2 J K�1 mol�1 occurringat a lower temperature (30 K).
Let us now turn our attention to the local and excess vibrationalfree energy. The results are presented in Figs. 3c,d, 4c,d, 5c and d,
Fig. 6. Comparison of the LVDOS from EAM and DFT
for Pd, Pt and Au(1 0 0), respectively. Here again, only the first-layer atoms show differences from the atoms in the other layers.As shown in the figures, the local contributions to vibrational freeenergies for the layers beyond the second converge to the corre-sponding bulk values. For the atoms in the first-layer, the local con-tribution to the vibrational free energy decreases with temperatureand reaching �48, �64 and �79 meV/atom for Pd, Pt and Au(1 0 0),respectively at 300 K. The excess vibrational free energies are sig-nificantly different for the first-layer atoms as can be seen in Figs.3d, 4d, and 5d, with the contribution amounting 19, 17 and14 meV/atom at 300 K. Our DFT calculations for Cu and Ag(1 0 0)show 15 meV/atom, which is in good agreement with the EAM re-sults (about 18 meV/atom for both surfaces at 300 K).
In the harmonic approximation, the vibrational mean squareamplitude (VMSA) is expected to vary linearly with temperatures.Note that at 0 K, the VMSA does not go to zero due to the zero pointmotion. In Figs. 3e, 4e and 5e, we present our DFT results for theVMSA of Pd, Pt and Au(1 0 0), respectively. The VMSA of the atomsin the third and fourth layers are bulk- like (0.008,0.0075, and 0.0130 Å2 at 300 K for Pd, Pt and Au(1 0 0), respec-tively). Through VMSA of the second-layer atoms is close to the bulkvalues, that corresponding to the first-layer atoms shows largedeviations. The ratio between the first-layer and the bulk VMSA is2.06, 1.73, and 1.73 for Pd, Pt and Au(1 0 0), respectively. This devi-ation is due to the decrease of coordination at the surface as com-pared to the bulk. Note that the ratio is the same for Pt andAu(1 0 0) and the highest for Pd(1 0 0), reflecting the mass effect.
4. Comparison between DFT and EAM results
The physical properties of interest in this study depend inti-mately on the local details of the interactions that describe the sys-
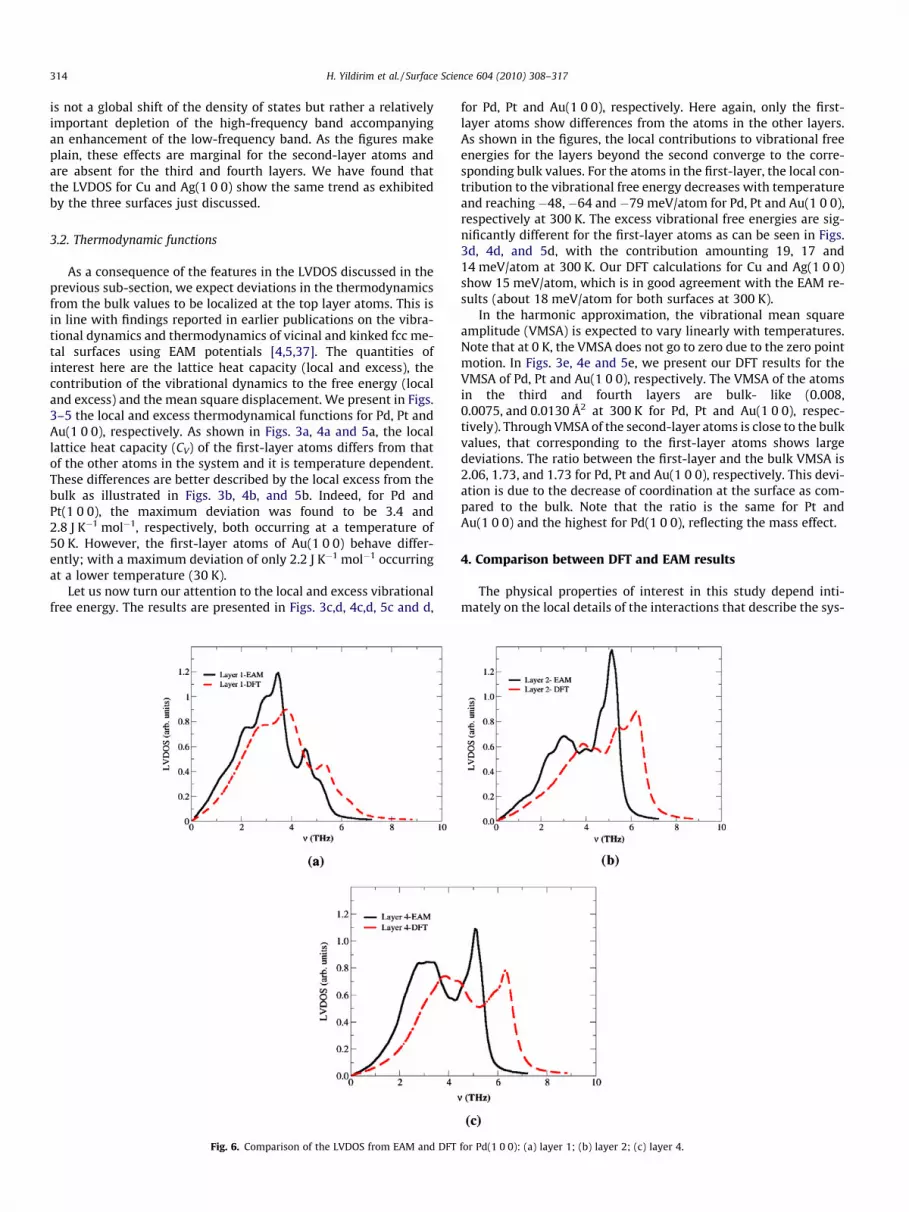
for Pd(1 0 0): (a) layer 1; (b) layer 2; (c) layer 4.

H. Yildirim et al. / Surface Science 604 (2010) 308–317 315
tem at hand. Indeed, the vibrational dynamics, hence the corre-sponding thermodynamics, emerge from treatments of the partialsecond derivatives of the potential. Comparison between twopotentials at this level is expected to reveal important differencesinheritent to the different classes of potentials (first principles vs.semi-empirical). We start our comparison by presenting changesin some key force constants and correlate these changes with dif-ferences in the LVDOS and corresponding thermodynamics.
4.1. Force constants
A complete analysis of the changes in the force constants foreach system is possible but not necessary. We choose to presentthe changes (with respect to bulk) in the two most affected forceconstants (matrix elements), namely the ones connecting twoneighbors that are in the first-layer (K11, corresponding to the ma-trix element Kaa where a (x or y) can be chosen to host both surfaceatoms) and two neighbors in the first- and the second-layer (K12,corresponding to the matrix element Kzz, where z is the directionperpendicular to the surface). Note that a positive value reflectsstiffening, while a negative reflects softening of the force constants.
In Table 2, we summarize these force constants changes for thefive surfaces studied. Let us first analyze the changes as calculatedusing DFT and highlighting the elemental effect. The softening inthe in-plane force constants (K11) is caused by the loss of fourneighbors experienced by the first-layer atoms and, as one notes,it is very large for Pd, Pt and Au(1 0 0). For K12, we note that thelargest changes are found for Cu, Ag and Pt(1 0 0), owing to the rel-atively large contraction (shortening of the bond between atoms inthe first and second-layers) of d12 (see Table 1). Let us now com-
Fig. 7. Comparison of the LVDOS from EAM and DFT
pare the changes in the force constants as calculated under DFTand EAM. As one may have already noted from Table 1, the inter-layer contractions calculated using DFT differ notably from thosecalculated using EAM for Pd, Pt and Au(1 0 0). This in turn reflectsthe large discrepancies in DK12 for which the ratio between theEAM and DFT values is 4.5, 3.7, and 6.8 for Pd, Pt and Au(1 0 0),respectively. On the other hand, because the change in K11 is dueto the loss of neighbors (four), which is independent of the choiceof the potential, we expect differences between DFT and EAM to beless dramatic than for K12 Indeed, these differences stand at a ratioless than two (Table 2).
4.2. Local vibrational densities of states
In Figs. 6–8, we present the comparison of the LVDOS for Pd, Ptand Au(1 0 0), for the first, second and the fourth layers as calcu-lated from DFT and EAM. As is discussed above, the LVDOS featuresfor the layers above the second represent bulk-like behavior. Wefind the calculated LVDOS under DFT for Cu and Ag(1 0 0) to bevery close to those produced by EAM. For these systems, we findthe top of the bulk band to be 0.5 THz higher for DFT than forEAM. This shows that the vibrational dynamics for copper and sil-ver can be well described using EAM.
However, for Pd, Pt and Au(1 0 0), as shown in Figs. 6c, 7c, and8c, the top of the band for the fourth layer (hence for the bulkatoms) is substantially higher for the spectrum calculated usingDFT than for that using EAM. For Pd(1 0 0) (Fig. 6c), the top ofthe DFT-band is higher (1.2 THz) than that of the EAM-band. Wealso note that the ratio (relative intensity) of the top of the high-frequency band to the top of the low-frequency band is 1.08 and
for Pt(1 0 0): (a) layer 1; (b) layer 2; (c) layer 4.

Fig. 8. Comparison of the LVDOS from EAM and DFT for Au(1 0 0): (a) layer 1; (b) layer 2; (c) layer 4.
Table 3Vibrational contribution to the free energy (meV) for layer 1 and layer 2 at 5, 100 and300 K.
Surfaces Temperature (K) DFT (EAM) 1st layer DFT (EAM) 2nd layer
Ag(1 0 0) 5 0.018 (0.018) 0.022 (0.022)100 0.008 (0.008) 0.015 (0.015)300 �0.066 (�0.067) �0.050 (0.048)
Cu(1 0 0) 5 0.027 (0.025) 0.032 (0.031)100 0.022 (0.019) 0.029 (0.027)300 �0.033 (�0.039) �0.017 (�0.020)
Pd(1 0 0) 5 0.022 (0.019) 0.029 (0.024)100 0.015 (0.010) 0.024 (0.018)300 �0.048 (0.062) �0.027 (�0.043)
Pt(1 0 0) 5 0.018 (0.015) 0.024 (0.018)100 0.009 (0.002) 0.017 (0.009)300 �0.064 (�0.083) �0.044 (�0.064)
Au(1 0 0) 5 0.015 (0.013) 0.018 (0.016)100 �0.003 (0.002) 0.009 (0.004)300 �0.079 (�0.095) �0.065 (�0.077)
316 H. Yildirim et al. / Surface Science 604 (2010) 308–317
1.29 from DFT and EAM, respectively. We can detect the sametrend for both Pt and Au(1 0 0) as is shown in Figs. 7c and 8c,respectively. The results calculated using DFT show that the topof the band are 1.2 and 1.0 THz higher than those derived fromEAM for Pt and Au(1 0 0), respectively. We find the ratio betweenthe top of the two frequency bands to be 1.09 and 1.07 (fromDFT) and 1.52 and 1.31 (from EAM) for Pt and Au(1 0 0). Similarto the LVDOS features for the fourth layer, we find for the sec-ond-layer that the top of the frequency band calculated usingDFT is about 1 THz higher than the EAM results, for each surface(Figs. 6b, 7b, and 8b). Interestingly, the comparison of the LVDOSresults from DFT and EAM for the first-layer shows less pro-nounced deviations from each other as compared to the rest ofthe layers mentioned above.
4.3. Free energies
In Table 3, we present comparison between the DFT and EAMresults for the vibrational contribution to the free energy at 5,100 and 300 K for the first and the second-layers. From the table,it is clear that for both Cu and Ag(1 0 0), the calculated contributionto the free energies from DFT and EAM are in good agreement. Wefind at most 6 meV/atom deviation (for copper, for the first-layer)between the EAM and DFT results at 300 K. The agreement be-tween the EAM and DFT results for the thermodynamical functionscan be traced to the excellent agreement in the vibrational dynam-ics (both for bulk and at surfaces), as discussed above. However, wefind the vibrational contribution to the free energy (first-layer) at
300 K for Pd, Pt and Au(1 0 0) to deviate by more than 15 meV/atom for each surface, the highest being for Pt(1 0 0). The contribu-tion is found to be larger for EAM than for DFT. The vibrational con-tribution to the free energy (at relatively low temperature) iscontrolled by the low-frequency end of the vibrational spectrumhence future analysis of this region may explain the large devia-tions between the EAM and DFT results. Figs. 6a, 7a, and 8a showa more pronounced enhancement of the low-frequency band for

H. Yildirim et al. / Surface Science 604 (2010) 308–317 317
EAM than for DFT results. Of the three elements, the deviation be-tween the EAM and DFT results (first-layer) at the low-frequencyend of the spectrum is the largest for Pt(1 0 0).
5. Conclusions
We have calculated the force constants between neighbors upto the 20th order using density functional perturbation theory.We then used these force constants to calculate the vibrationaldynamics and thermodynamics of five surfaces Ag, Cu, Pd, Pt andAu(1 0 0). The changes, from the bulk, in the local properties ofthese surfaces were found to be confined mainly to the first-layeratoms. The vibrational dynamics of all these surfaces (Ag, Cu, Pd,Pt and Au(1 0 0)) show analogous qualitative behaviors with a sub-stantial reshuffling of the densities of states. The low-frequencyend of the spectrum is enhanced at the expense of the high-fre-quency range, resulting in a lower contribution to the vibrationalfree energy as compared with that of the bulk. The vibrationalmean square amplitudes for atoms in the first-layer were foundto be as much as twice those in the bulk. When comparing resultsusing density functional theory with those using embedded-atommethod potentials, we found that the two methods yield very closeresults for silver and copper. However, for palladium, platinum andgold, we found that the embedded-atom method describes verypoorly the interactions in these systems and thus delivers mislead-ing picture of their vibrational dynamics and thermodynamics.
Acknowledgements
This work is supported partially by US DOE under Grant No. DE-FG02-07ER46354. We thank Lyman Baker for his help with themanuscript.
References
[1] C. Bombis, A. Emundts, M. Nowicki, H.P. Bonzel, Surf. Sci. 511 (2002) 83.
[2] H.P. Bonzel, A. Emundts, Phys. Rev. Lett. 84 (2000) 5804.[3] J.W.M. Frenken, P. Stoltze, Phys. Rev. Lett. 82 (1999) 3500.[4] S. Durukanoglu, A. Kara, T.S. Rahman, Phys. Rev. B 67 (2003) 235405.[5] A. Kara, T.S. Rahman, Surf. Sci. Rep. 56 (2005) 159.[6] S.M. Foiles, M.I. Baskes, M.S. Daw, Phys. Rev. B 33 (1986) 7983.[7] S. Baroni, P. Giannozzi, A. Testa, Phys. Rev. Lett. 58 (1987) 1861;
P. Giannozzi, S. de Gironcoli, P. Pavone, S. Baroni, Phys. Rev. B 43 (1991) 7231.[8] R. Heid, K.-P. Bohnen, Phys. Rep. 387 (2003) 151.[9] R. Zope, Y. Mishin, Phys. Rev. B 68 (2003) 024102;
Y. Mishin, M.J. Mehl, D.A. Papaconstantopoulos, Phys. Rev. B 65 (2002) 224114.[10] Xiao-Qian Wang, Phys. Rev. Lett. 67 (1991) 3547.[11] G. Ritz, M. Schmid, P. Varga, A. Borg, M. Rønning, Phys. Rev. B 56 (1997) 10518.[12] P. Hohenberg, W. Kohn, Phys. Rev. B 136 (1964) 864.[13] W. Kohn, L.J. Sham, Phys. Rev. A 140 (1965) 1133.[14] S.G. Louie, K.-M. Ho, M.L. Cohen, Phys. Rev. B 19 (1979) 1774.[15] C.-L. Fu, K.M. Ho, Phys. Rev. B 28 (1983) 5480.[16] C. Elsasser, Ph.D. Thesis, Universittat Stuttgart, 1990.[17] B. Meyer, Ph.D. Thesis, Universittat Stuttgart, 1998.[18] B. Meyer, C. Elsasser, M. Fahnle, FORTRAN 90 Program for Mixed-basis
Pseudopotential Calculations for Crystals, Max-Planck-Institut furMetallforschung, Stuttgart, unpublished.
[19] S. Baroni, S. de Gironcoli, A. Dal Corso, P. Giannozzi, Rev. Mod. Phys. 73 (2001)515.
[20] R. Heid, K.-P. Bohnen, Phys. Rev. B 60 (1999) R3709.[21] B.D. Yu, M. Scheffler, Phys. Rev. Lett. 77 (1996) 1095.[22] M. Methfessel, D. Henning, M. Scheffler, Phys. Rev. B 46 (1992) 4816.[23] G. Boisvert, L.J. Lewis, Phys. Rev. B 56 (1997) 7643.[24] H. Li, J. Quinn, Y.S. Li, D. Tian, F. Jona, P.M. Marcus, Phys. Rev. B 43 (1991) 7305.[25] J.L.F. Silva, K. Schroeder, S. Blügel, Phys. Rev. B 69 (2004) 245411.[26] Th. Rodach, K.-P. Bohnen, K.M. Ho, Surf. Sci. 286 (1993) 66.[27] G. Boisvert, L.J. Lewis, M.J. Puska, R.M. Nieminen, Phys. Rev. B 52 (1995) 9078.[28] S.B. Sinnott, M.S. Stave, T.J. Raeker, A.E. DePristo, Phys. Rev. B 44 (1991) 8927.[29] H.L. Davis, J.R. Noonan, Surf. Sci. 126 (1983) 245.[30] P.J. Feibelman, R. Stump, Phys. Rev. B 59 (1999) 5892.[31] B.D. Yu, M. Scheffler, Phys. Rev. B 56 (1997) R15569.[32] R.E. Allen, G.P. Alldredge, F.W. de Wette, Phys. Rev. B 4 (1971) 1648.[33] R.E. Allen, F.W. de Wette, A. Rahman, Phys. Rev. 179 (1969) 887.[34] R. Haydok, V. Heine, M.J. Kelly, J. Phys. C: Solid State Phys. 5 (1972) 2845.[35] S.Y. Wu, J. Cocks, C.S. Jayanthi, Phys. Rev. B 49 (1994) 7957.[36] A. Kara, C.S. Jayanthi, S.Y. Wu, F. Ercolessi, Phys. Rev. Lett. 49 (1994) 2223;
A. Kara, C.S. Jayanthi, S.Y. Wu, F. Ercolessi, Phys. Rev. B 51 (1995) 17046.[37] T.S. Rahman, A. Kara, S. Durukanoglu, J. Phys, J. Phys.: Condens. Matter 15
(2003) S3197.