st. anna kinderkrebsforschung
TRANSCRIPT

ST. ANNA KINDERKREBSFORSCHUNGFORSCHUNGSBERICHT 2016
ST. A
NN
A K
IND
ERK
REB
SFO
RSC
HU
NG
FO
RSC
HU
NG
SBER
ICH
T 20
16
kinderkrebsforschung.atscience.ccri.at

SCIENCE REPORTS 33Introduction 36
Non-genetic plasticity and variability as therapeutic and prognostic targets in Ewing sarcoma 38
Improving the routine enumeration of hematopoietic stem cells: Multi-color analysis of CD34 subtypes reveals unexpected differences between various stem cell sources 44
Characterization of a novel fusion gene in juvenile myelomonocytic leukemia associated with resistance to tyrosine kinase inhibitors 52
High resolution genomic and transcriptomic profiling of pediatric B-cell precursor acute lymphoblastic leukemia: implications for emergence of resistance and relapse 56
New insights in neuroblastoma biology – from spontaneous maturation to the relapse seeding clone 64
Chimeric antigen receptor (CAR)-based immunotherapy for treatment of reactivation of cytomegalovirus infection after stem cell transplantation 68
Large scale European trial demonstrates survival advantage for high risk neuroblastoma patients receiving high dose busulphan and melphalan treatment 72
EINLEITUNG 7Vorwort des Institutsleiters 8
Einleitung des wissenschaftlichen Direktors 10
Unseren Spendern sei Dank! 14
DATEN & FAKTEN 19Kompetitive Drittmittel 22
Zuweisung der Geldmittel 22
Finanzierung 22
Personelle Zusammensetzung 23
Nationen 23
Forschungsnetzwerke 24
Klinische Forschung 26
Anstieg der 2-Jahres-Überlebensraten 27
5-Jahres-Überleben von krebskranken Kindern 28
CAREER 79Working at CCRI 82
If you want to apply for a position 83
Scientific Staff 84
FINANZBERICHT 89Richtlinien zur Spendenverwendung 90
Mittelherkunft 94
Mittelverwendung 95
ANHANG 97Wissenschaftlicher Beirat 98
International fremdgeförderte Projekte 102
National fremdgeförderte Projekte 104
Danksagung 108
Diplom(Master)arbeiten /Dissertationen 2016 109
Publikationen 2016 112
Impressum 124

UNIV.-PROF. DR. WOLFGANG HOLTEREntwicklung zellulärer Therapie
EINLEITUNG

98
Einleitung
VORWORT DES INSTITUTSLEITERS
Die Erfolge meiner Kolleginnen und Kollegen machen es mir leicht, diesen Jahresbericht einzuleiten. Doch trotz aller persönlichen Genugtuung über den wissenschaftlichen Fort-schritt, trotz ermutigender Aner-
kennung unserer Arbeit auf internationaler Bühne, ist es doch immer der Zweck unserer Forschung, der uns antreibt. Es geht um das Überleben und Leben krebskranker Kinder und Jugendlicher und die Umsetzung unserer Erkenntnisse in die klinische Diagnostik und Therapie. Nichts – keine Auszeichnungen oder wissenschaftlichen Ehren – freut uns mehr als ein Beitrag zur Rettung der Betroffenen und zur Linderung von Leid und Schmerz bei deren Angehörigen.
Dass die Heilungsraten bei manchen Kinderkrebs-arten in lichte Höhen getrieben werden konnten, sodass wir nun die 100 % anpeilen können, hat die St. Anna Kinderkrebsforschung zu einem ge fragten Partner für staatenübergreifende Zusammen arbeit gemacht. So wurden wir für die Leitung eines drei-jährigen Pilot-Referenzprojektes ausgewählt, das die Aufgabe hat, die unter schiedlichen Überlebens-raten von Kindern mit Krebserkrankungen aufgrund ungleicher Leistungsfähigkeit der Gesundheitssys-teme an zugleichen und zu verbessern.
Neue, revolutionäre Möglichkeiten für die Diagnose des Neuroblastoms, einer besonders aggressiven Krebsart bei Kindern, hat unser Institut zur Leitung eines EU-weiten Forschungskonsortiums empfohlen. Die „Flüssige Biopsie“ wird in weiterführende klini-sche Studien eingebunden, um diese exakte mole-kulare Bestimmung des individuellen Tumor genoms voranzutreiben.
Eine weitere, vielversprechende Entdeckung der St. Anna Kinderkrebsforschung auf dem Gebiet der Immuntherapie gab grünes Licht zur Erforschung einer neuen Methode für die Behandlung lebens-bedrohlicher Virusinfektionen nach einer Stamm-zellentherapie, um nur einige Highlights aus der Forschung zu nennen.
Zu guter Letzt sollte nicht unerwähnt bleiben, dass wir im wissenschaftlichen Wett bewerb ebenfalls gepunktet haben. Ein Elise-Richter-Forschungs-stipendium, das hochkarätige Wissenschaftlerinnen auszeichnet, die eine Universitäts laufbahn an stre-ben, ging 2016 an eine Forscherin der St. Anna Kinderkrebsforschung.
Ich möchte mich herzlich bei den vielen Unter-stützerinnen und Unterstützern, den Mitgliedern des Ehrenkomitees, den Vorstandsmitgliedern, dem wissenschaftlichen Beirat und unseren vielen Förderern bedanken, die uns schon viele Jahre treu begleiten.
Ich hoffe, dass dieser Leistungsbericht zur weiteren und so dringend notwendigen Spendenbereitschaft motiviert, für die ich mich im Namen aller Mitarbei-terinnen und Mitarbeiter herzlich bedanken möchte.
Univ.-Prof. Dr. Wolfgang HolterInstitutsleiter

1110
Einleitung
EINLEITUNG DES WISSENSCHAFTLICHEN DIREKTORS
Mit dem rasanten technologischen Fortschritt in der biomedizini-schen Forschung eröffnen sich ungeahnte Dimensionen des Forschungs universums und ergeben sich immer neue,
überraschende Erkenntnisse. Im Bereich der Krebsforschung ist dies zweifellos die Einsicht, dass Tumore keine homogene Zellmasse darstellen, sondern sich aus einer Vielzahl verschiedener Zelltypen zusammensetzen, die, jeder für sich, ein hohes Maß an Heterogenität und Plastizität auf - weisen. Diese Erkenntnis trifft besonders auf Krebserkrankungen bei Kindern zu. Galt noch vor wenigen Jahren das Dogma, dass Krebs aus - schließlich eine Erkrankung der Gene ist, welche sich in irreversiblen, starren Mutationsmustern manifestiert, so zeigt sich heute mehr und mehr, dass Krebs bei Kindern in der Regel mit verhältnis-
mäßig wenigen genetischen Veränderungen einhergeht. Denn pädiatrische Krebserkrankungen müssen als Defekt in der normalen Entwicklungs-biologie unterschiedlicher Zelltypen gesehen werden. Um hier den Mechanismen als potentielle Therapie ansatzpunkte auf den Leib zu rücken, müssen wir die Regulationsmechanismen der Entwicklung verstehen. Die zunehmende Anzahl an Weiter entwicklungen neuer Technologien, wie etwa des Next Generation Sequencing (NGS), ermöglicht Einsichten in die normale und die krankhaft veränderte dreidimensionale Struktur des Chromatins, der Steuerzentrale der Genexpression, welches das Verhalten jeder Zelle bestimmt. Die hohe Sensitivität dieser Technologie erlaubt uns nicht nur genetische Zusammensetzung und Genexpression auf Einzelzellebene zu analysieren, sondern auch Tumorzellzerfallsprodukte in der Blutzirkulation nachzuweisen und zu bestimmen.
So erhalten wir Auskunft über die genetische und nicht-genetische Variation von Tumoren und deren Komponenten im Wechselspiel der verschiedenen Zelltypen und ihres Stoffwechsels. Diese Entwick-lungen spiegeln sich auch in den Forschungsaktivi-täten der St. Anna Kinderkrebsforschung wider. Wie in diesem Jahresbericht dargestellt, gelang es uns auch 2016 beachtliche Erfolge auf verschiedenen Gebieten der pädiatrisch-onkologischen Forschung zu erzielen, welche uns unserem Ziel einer für jeden Patienten maßgeschneiderten Therapie weiter näherbringen. Dies sei an einigen ausgewählten Beispielen aus den im Vorjahr veröffentlichten Forschungsergebnissen demonstriert.
Univ.-Prof. Dr. Heinrich Kovar Wissenschaftlicher Direktor

DR. MARTIN DISTELInnovative Krebsmodelle

Mag. Andrea PrantlLeiterin Spendenbüro
Einleitung
1514
UNSEREN SPENDERN SEI DANK!
Für einen wunderschönen Nachmittag mit vielen lachenden Gesichtern sorgte der Verein Öster-reichische Journalistenwerkstätte. Dieter Wally gelang es gemeinsam mit seinen engagierten Kolleginnen und Kollegen, die Österreichpremiere des Kinderfilms „Rettet Raffi“ zu einem speziel-len Benefizerlebnis zu machen. Die Abenteuer des mutigen Hamsters und seines jungen Besitzers konnten auch Patienten aus dem St. Anna Kinder-spital und deren Eltern genießen. Das Forscherteam freute sich, dank großzügiger Sponsoren, über einen hervorragenden Spendenbetrag.
Die Unterstützung der Bastelrunde Hirtenberg, unter der Leitung von Marianne Brandtner, hat bereits eine langjährige Tradition. Das ganze Jahr über wird dort für die St. Anna Kinderkrebs-forschung gebastelt, gemalt, gestrickt, genäht usw. Die Erlöse des jährlichen Oster- und Adventmarkts sind jedes Mal beeindruckend.
Auch das spannende Wettpaddeln in Scharndorf hat bereits Tradition! Viel Spaß für Jung und Alt ist beim Benefiz-Sautrogrennen mit Kultcharakter garantiert. Der gesamte Erlös wird von Jahr zu Jahr unglaublich gesteigert und wurde auch 2016 gespendet.
Sogar das derzeit wahrscheinlich hippste Duo der österreichischen Musik- und Unterhaltungsbranche schaute schon vor einem Tourstart im Spenden- büro der St. Anna Kinderkrebsforschung vorbei. Die Kunst-Kampagne „Horvathslos für eine gute Sache“ war die schöne Idee von „Mama Speer“. Der so gesammelte Betrag wurde von „Seiler und Speer“ verdoppelt und eine tolle Summe persönlich übergeben.
Jedes Jahr gestalten die Schülerinnen und Schüler der Schreibgruppe „Kreatives Schreiben“ am Gymnasium Schärding, gemeinsam mit der rührigen Initiatorin, Frau Prof. Mag. Thallinger, einen Lesezeichenkalender. Mit dem Reinerlös unter-stützen die jungen Fabulanten auch 2016 das Wiener Forschungsinstitut.
L ANGE NACHT DER (KINDERKREBS)FORSCHUNGForschung auf reizvolle Weise entdecken und Zukunft erleben! Dazu lädt die St. Anna Kinder-krebsforschung alljährlich ein. Im Rahmen der „Langen Nacht der Kinderkrebsforschung“ wird ein spielerisches Kennenlernen des Laborall-tages möglich und „Wissenschaft zum Anfassen“ greifbar. Interaktiv, spannend und leicht ver-ständlich wird Wissen über die Bausteine des Lebens und die molekularen Grundlagen der Krebsentstehung vermittelt. Vorführungen in den Labors und spannende wissenschaftliche Vorträge ver deutlichen, was unsere Forscherinnen und Forscher zur Verbesserung der Behandlungs-qualität und Er höhung der Heilungschancen an Krebs erkrankter Kinder und Jugendlicher leisten.
Unsere Spenderfamilie ist groß und großartig! Wir sind dankbar für die langjährige, treue Unter-stützung und wir freuen uns jedes Mal, ein neues Familienmitglied herzlich willkommen zu heißen! Das Faszinierende an der Arbeit im Spendenbüro der St. Anna Kinderkrebsforschung sind die Menschen, ihre Hilfsbereitschaft, ihre wunder baren Ideen und ihr großartiges Spendenengagement. Dank der konsequenten Forschung können heute bereits vier von fünf Kindern und Jugendlichen, die vor 40 Jahren noch als unheilbar galten, gerettet werden. Finanziert wird die St. Anna Kinderkrebsforschung, die seit 2002 das Österreichi-sche Spendengütesiegel führt und zum steuerlich begünstigten Empfängerkreis gehört, von Anfang an hauptsächlich durch Spenden. Dank gebührt daher unseren Spenderinnen und Spendern – denn sie alle schenken krebskranken Kindern eine Chance auf eine gesunde Zukunft.
BUNTE EINFÄLLE – SPANNENDE AKTIONEN – GROSSZÜGIGE SPENDENDas Spektrum der Spendenmöglichkeiten ist viel-fältig und die Unterstützung damit grenzenlos – wie einige Beispiele aus 2016 zeigen:Mit einer exklusiven Benefizgala im Goldenen Saal des Wiener Musikvereins feierte Aki Nuredini, prominenter und beliebter Patron des Wiener Nobel-Restaurants „Sole“, seinen 60. Geburtstag. Ein Teil des Reinerlöses des hochkarätig besetzten Konzerts kam der St. Anna Kinderkrebsforschung zugute. Die Besucher erlebten eine Musikgala der Extraklasse mit wunderbaren Interpreten, wie Ildikó Raimondi, Clemens Unterreiner, Ramón Vargas uvm. Sogar Starpianist Rudolf Buchbinder spielte ein Geburtstagsständchen.
Das ganze Jahr über sammelten die Mitglieder der Kinderkrebshilfe Haid / Ansfelden durch den Verkauf von Flohmarktware Spenden. Der Einsatz war enorm und die Einnahmen rekordverdächtig. Bereits zum 11. Mal wurden nicht nur Familien mit kleinen Krebspatienten unterstützt, sondern auch der St. Anna Kinderkrebsforschung wieder eine namhafte Spende überreicht.
Die Aschermittwochfeier der Künstlerinnen und Künstler in der Wiener Hofburgkapelle gilt als besondere Einstimmung auf die Fastenzeit. Die Wiener Hofmusikkapelle und alle Mitwirkenden agierten im Dienst der guten Sache und unter-stützten auch im Jahr 2016 mit den Einnahmen unsere Forschungs arbeit.

Einleitung
1716
2016 öffneten wir bereits zum 12. Mal unsere Pforten, aber erstmalig im Rahmen von Österreichs größtem Forschungsfest: der „Langen Nacht der Forschung“. Rund 1000 Besucherinnen und Besucher interessierten sich für unsere Arbeit, staunten über unsere Erfolge und nützten die Gelegenheit, sich zu informieren und einen Blick hinter die „Forschungs-kulissen“ zu werfen.
ST. ANNA KINDERKREBSFORSCHUNG-UNTERSTÜTZUNGSKOMITEESchon seit einiger Zeit begleitet ein prominent besetztes Ehrenkomitee die St. Anna Krebs-forschung. Die Mentoren, Personen aus Politik, Wirtschaft und Kultur, zeichnen sich durch eine besondere Position aus und sind ehrenamtlich tätig. Durch ihre Präsenz in der Öffentlichkeit können sie viel zugunsten der St. Anna Kinderkrebsforschung bewegen. Seit einigen Jahren unterstützt Eva Angyan, Gattin des Intendanten der Gesellschaft der Musikfreunde in Wien, Dr. Thomas Angyan, als Komiteepräsidentin das Forschungsinstitut. Die Unterstützung der Kinderkrebsforschung ist ihr ein Herzensanliegen und deshalb bittet Frau Angyan regelmäßig eine ausgesuchte Gästeschar zu einem Treffen, um gemeinsame Projekte zugunsten der St. Anna Kinderkrebsforschung zu planen.
Dr. Charlotte Rothensteiner, Mag. Maria Polsterer- Kattus, Erste Bank Vorstand Willibald Cernko, KR Karl Javurek, Isabella Kapsch, General direktorin Dr. Elisabeth Gürtler, Parlamentsabgeordnete KR Brigitte Jank, Prof. Erwin Ortner, Bäcker meister Senator Kurt Mann, sowie BM Dr. Michael Häupl, Meinungsforscher Prof. Rudolf Bretschneider, Interspot-Chefin Inge Klingohr, Psychologin Mag. Ulla Konrad, Maestro Franz Welser-Möst und Kapsch-Vorstand Ing. Mag. Thomas Schöpf sind Teil des Unterstützungskomitees und freuen sich, die St. Anna Kinderkrebsforschung als Mentorinnen und Mentoren zu begleiten und tat kräftig zu unterstützen.
Unsere Kuscheltiere: Kleine Lebensretter, die Freude schenkenBereits seit 24 Jahren sind die Kuscheltiere der St. Anna Kinderkrebsforschung bei Jung und Alt sehr beliebt und als Sammelobjekte auch heiß begehrt! Jedes Jahr im Oktober begrüßt die Maskottchen familie einen Neuzugang. 2016 war es „Gigi, der gutgelaunte Glückskäfer“. Die kleinen Lebensretter freuen sich auf ein nettes Zuhause und jede Menge Spielgefährten. Für 12,00 € schenkt man nicht nur krebskranken Kindern eine Chance, sondern auch sich selber oder seinen Lieben Freude. Bank Austria, Erste Bank und einige Sparkassen unterstützen die St. Anna Kinderkrebsforschung ebenso beim Vertrieb, wie die Raiffeisen Zentral-bank. Sogar an der Rezeption des renommierten Wiener Innenstadt-Hotels Imperial ist das jeweils aktuelle Maskottchen erhältlich. Welche Stofftiere angeboten und wie sie zu bestellen sind, steht auf unserer Internetseite.
WOLLEN SIE INFORMATIONEN, UNTERL AGEN ODER HABEN SIE FRAGEN?Das Spendenteam und ich freuen uns auf Ihre Kontaktaufnahme:
+43 (0)1 40 470 - [email protected]
Bank Austria IBAN: AT79 1200 0006 5616 6600 BIC: BKAUATWW
Erste Bank IBAN: AT66 2011 1000 0318 3777 BIC: GIBAATWW
Jede Spende hilft!Ob mit einer persönlichen Spende oder mit einer gemeinsamen Sammelaktion, ob per Zahlschein, Kreditkarte oder Onlinespende – jede finanzielle Zuwendung ermöglicht die Fortsetzung unserer Forschungsarbeit im Kampf gegen Kinderkrebs. Firmen spenden immer häufiger den für Kunden- Weihnachtsgeschenke vorgesehenen Betrag. Statt um Geschenke wird bei Geburtstagen, Jubiläen oder anderen Feiern gerne um Spenden gebeten. Auch der Verzicht auf Blumen und Kränze bei Begräbnissen, um stattdessen zu spenden, hilft krebskranken Kindern. Ein Testament oder ein Legat zugunsten der St. Anna Kinderkrebsforschung schenkt ebenfalls langfristig eine gesunde Zukunft.
Vielfältige Spendenmöglichkeiten:Onlinespende, Barspende, Spende mit Zahlschein, Spende mit Einziehungs- oder Dauerauftrag, Benefiz-veranstaltungen, Sammelaktionen, Spenden statt Geburtstags- oder Weihnachtsgeschenken, Erwerb unserer Kuscheltiere, Kranzablösespenden, Legat / Testament etc.
Das Team der St. Anna Kinderkrebsforschung ist dankbar für die langjährige spendenfreudige Unter-stützung und ich persönlich für die Begegnung mit so vielen großzügigen, warmherzigen und hilfs-bereiten Menschen. Herzlichen Dank!
Mag. Andrea PrantlLeiterin Spendenbüro
DATEN & FAKTEN

UNIV.-PROF. DR. RENATE PANZER-GRÜMAYERLeukämiebiologie
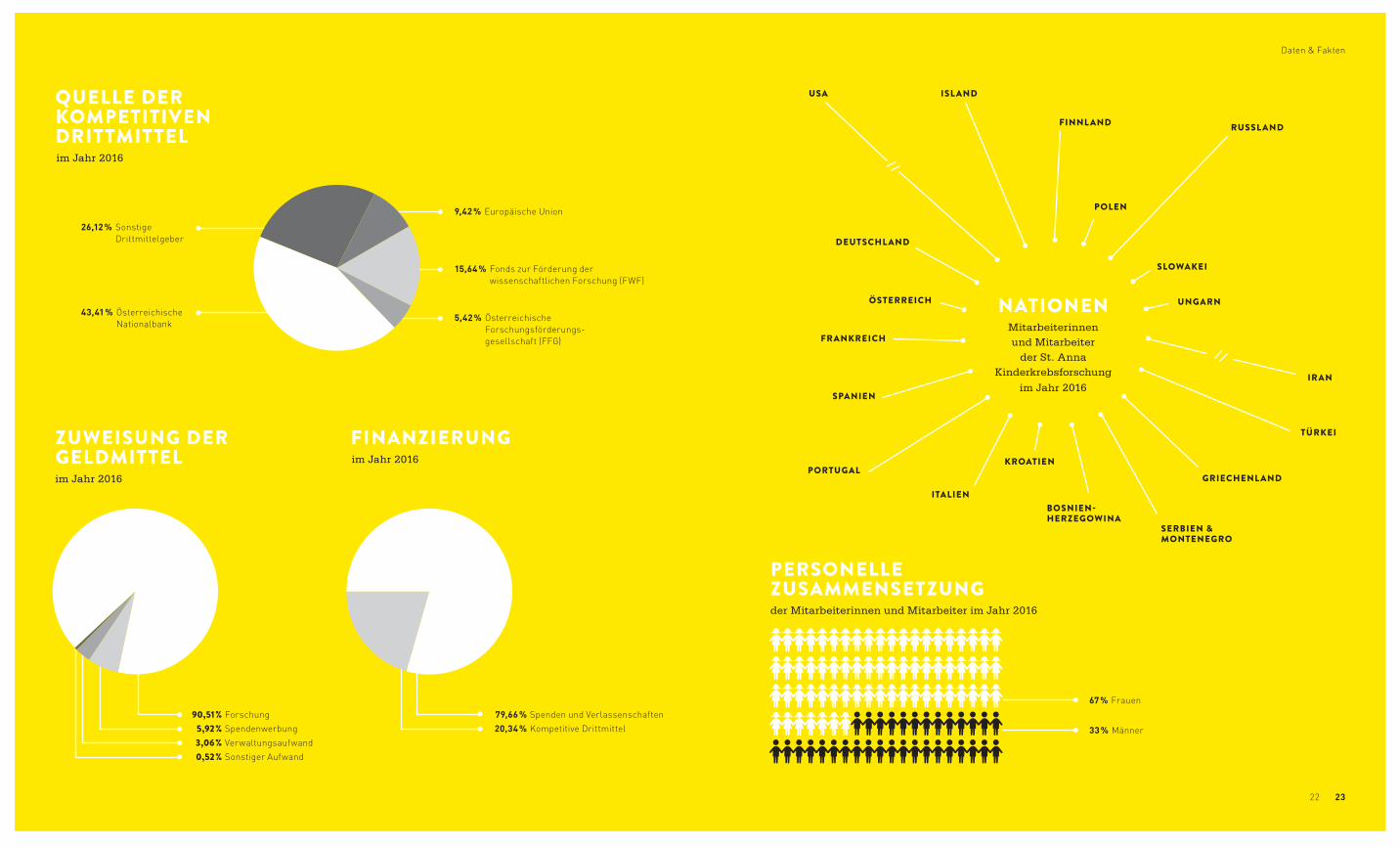
90,51 % Forschung 5,92 % Spendenwerbung 3,06 % Verwaltungsaufwand 0,52 % Sonstiger Aufwand
ZUWEISUNG DERGELDMITTELim Jahr 2016
FINANZIERUNGim Jahr 2016
79,66 % Spenden und Verlassenschaften 20,34 % Kompetitive Drittmittel
QUELLE DER KOMPETITIVENDRITTMITTELim Jahr 2016
9,42 % Europäische Union
26,12 % Sonstige Drittmittelgeber
43,41 % Österreichische Nationalbank
15,64 % Fonds zur Förderung der wissenschaftlichen Forschung (FWF)
5,42 % Österreichische Forschungsförderungs- gesellschaft (FFG)
2322
NATIONENMitarbeiterinnenund Mitarbeiter
der St. Anna Kinderkrebsforschung
im Jahr 2016
Daten & Fakten
PERSONELLEZUSAMMENSETZUNGder Mitarbeiterinnen und Mitarbeiter im Jahr 2016
ISL AND
DEUTSCHL AND
POLEN
RUSSL AND
TÜRKEI
IRAN
GRIECHENL AND
USA
KROATIEN
SERBIEN & MONTENEGRO
BOSNIEN- HERZEGOWINA
FINNL AND
PORTUGAL
SPANIEN
ÖSTERREICH
FRANKREICH
SLOWAKEI
UNGARN
ITALIEN
67 % Frauen
33 % Männer
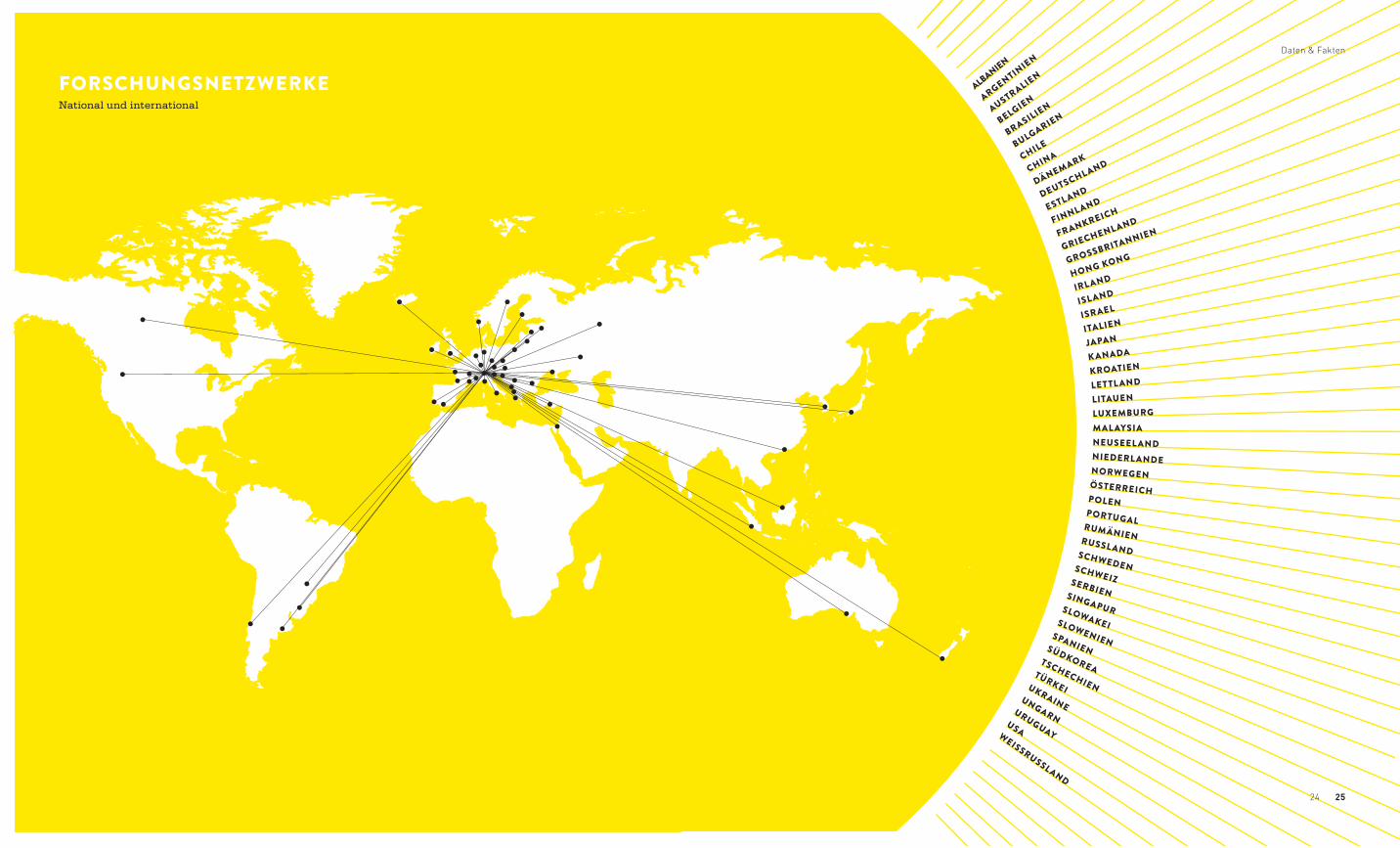
FORSCHUNGSNETZWERKENational und international
WEISSRUSSLAND
USA
URUGUAY
UNGARN
UKRAINE
TÜRKEI
TSCHECHIEN
SÜDKOREA
SPANIEN
SLOWENIEN
SLOWAKEI
SINGAPUR
SERBIEN
SCHWEIZ
SCHWEDEN
RUSSL AND
RUMÄNIEN
PORTUGAL
POLEN
ÖSTERREICH
NORWEGEN
NIEDERL ANDE
NEUSEEL ANDMAL AYSIALUXEMBURGLITAUENLETTL ANDKROATIENK ANADAJAPAN
ITALIENISRAEL
ISL ANDIRL ANDHONG KONGGROSSBRITANNIEN
GRIECHENL ANDFRANKREICH
FINNL ANDESTLANDDEUTSCHLAND
DÄNEMARKCHINACHILEBULGARIEN
BRASILIENBELGIEN
AUSTRALIEN
ARGENTINIEN
ALBANIEN
2524
Daten & Fakten
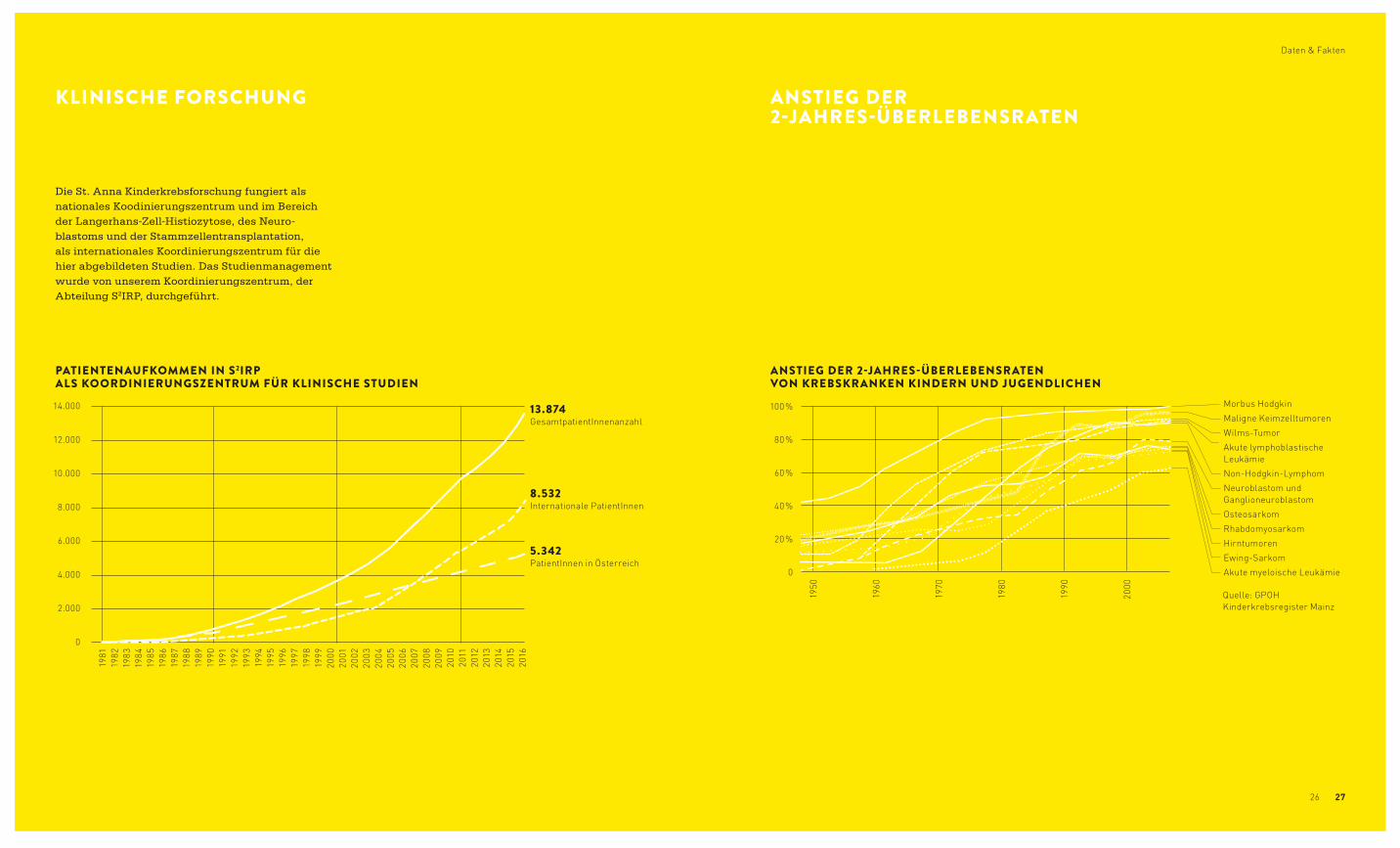
KLINISCHE FORSCHUNG ANSTIEG DER 2-JAHRES-ÜBERLEBENSRATEN
Die St. Anna Kinderkrebsforschung fungiert als nationales Koodinierungszentrum und im Bereich der Langerhans-Zell-Histiozytose, des Neuro-blastoms und der Stammzellentransplantation, als internationales Koordinierungszentrum für die hier abgebildeten Studien. Das Studienmanagement wurde von unserem Koordinierungszentrum, der Abteilung S2IRP, durchgeführt.
14.000
12.000
10.000
8.000
6.000
4.000
2.000
0
100 %
80 %
60 %
40 %
20 %
0
1981
1982
1983
1984
1985
1986
1987
1988
1989
1990
1991
1992
1993
1994
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
2010
2011
2012
2013
2014
2015
2016
PATIENTENAUFKOMMEN IN S2IRP ALS KOORDINIERUNGSZENTRUM FÜR KLINISCHE STUDIEN
ANSTIEG DER 2-JAHRES-ÜBERLEBENSRATEN VON KREBSKRANKEN KINDERN UND JUGENDLICHEN
13.874 GesamtpatientInnenanzahl
8.532 Internationale PatientInnen
5.342 PatientInnen in Österreich
Quelle: GPOH Kinderkrebsregister Mainz
1950
1960
1970
1980
1990
2000
Morbus HodgkinMaligne KeimzelltumorenWilms-TumorAkute lymphoblastische LeukämieNon-Hodgkin-LymphomNeuroblastom und GanglioneuroblastomOsteosarkomRhabdomyosarkomHirntumorenEwing-SarkomAkute myeloische Leukämie
2726
Daten & Fakten
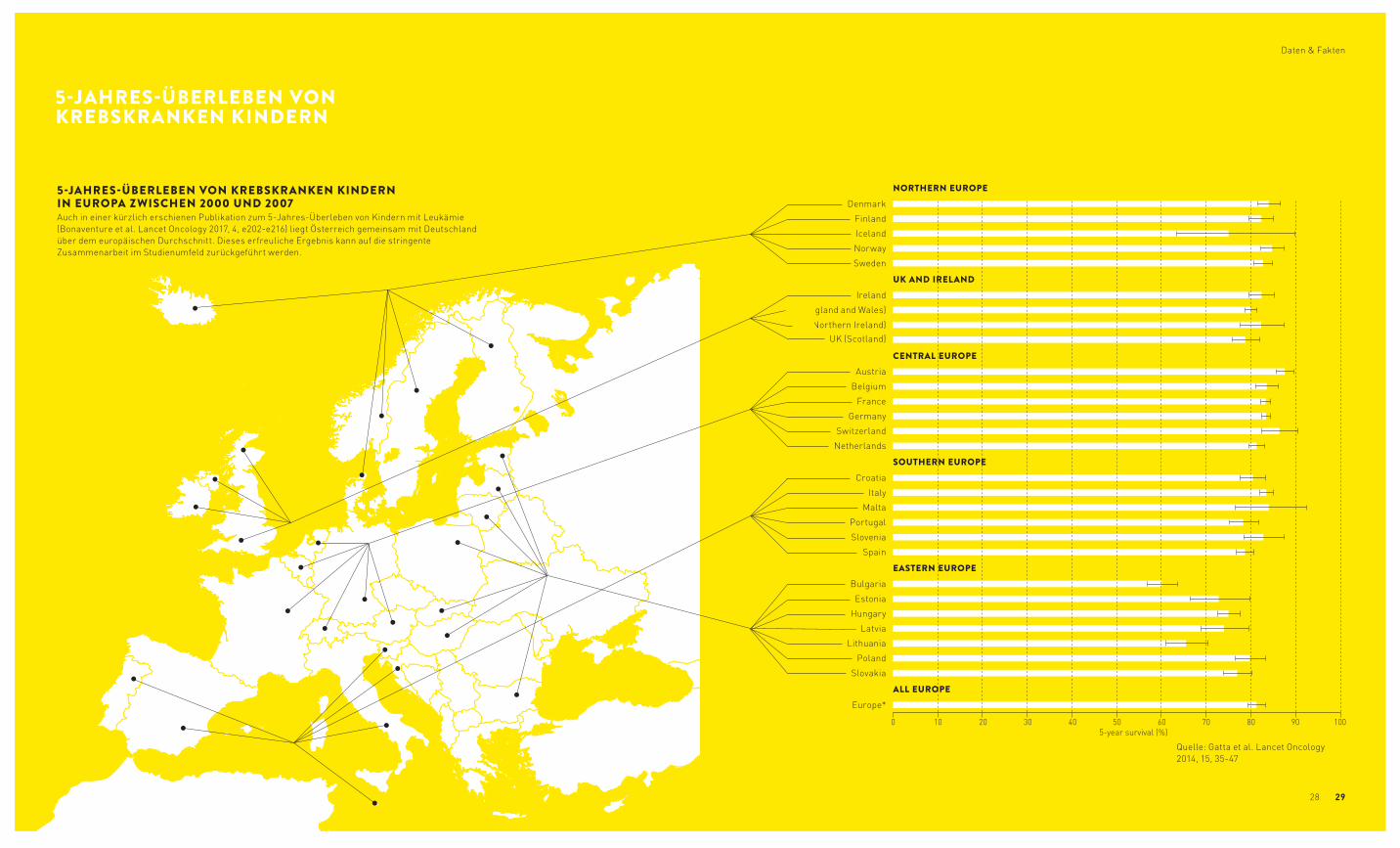
5-JAHRES-ÜBERLEBEN VON KREBSKRANKEN KINDERN IN EUROPA ZWISCHEN 2000 UND 2007Auch in einer kürzlich erschienen Publikation zum 5-Jahres-Überleben von Kindern mit Leukämie (Bonaventure et al. Lancet Oncology 2017, 4, e202-e216) liegt Österreich gemeinsam mit Deutschland über dem europäischen Durchschnitt. Dieses erfreuliche Ergebnis kann auf die stringente Zusammen arbeit im Studienumfeld zurückgeführt werden.
Quelle: Gatta et al. Lancet Oncology 2014, 15, 35-47
0 10 20 30 40 50 60 70 80 90 100
Europe*
Slovakia
Poland
Lithuania
Latvia
Hungary
Estonia
Bulgaria
Spain
Slovenia
Portugal
Malta
Italy
Croatia
Netherlands
Switzerland
Germany
France
Belgium
Austria
UK (Scotland) UK (Northern Ireland)
UK (England and Wales)
Ireland
Sweden
Norway
Iceland
Finland
Denmark
5-year survival (%)
CENTRAL EUROPE
UK AND IRELAND
NORTHERN EUROPE
SOUTHERN EUROPE
EASTERN EUROPE
ALL EUROPE
5-JAHRES-ÜBERLEBEN VON KREBSKRANKEN KINDERN
2928
Daten & Fakten

DR. ALEXANDER DOHNALTumorimmunologie
SCIENCE REPORTS

UNIV.-PROF. DR. HEINRICH KOVARMolekularbiologie

3736
Science Reports
circulation. This way, we are able to obtain infor-mation on the genetic and non-genetic variation of tumors and their components in their interplay with other cell types and metabolites. These develop-ments are also reflected in the activities of the Children´s Cancer Research Institute. As docu-mented by this annual report, we continued being successful in several areas of pediatric oncology research in 2016. These successes bring us a step closer to our goal of improved therapy tailored to the specific needs of individual patients. In support of this notion, this report includes several examples of research results published by the Children´s Cancer Research Institute in the past year.
Prof. Heinrich Kovar, PhD
INTRODUCTION
Rapid technological progress in biomed-ical research explores unimagined dimensions of the scientific universe and constantly produces new, unex-pected findings. In the field of cancer research thishas led to the insight
that tumors are not simply a homogenous mass of cells, but comprise a multitude of different cell types, each of which demonstrating a high degree of heterogeneity and plasticity. This finding espe-cially applies to pediatric cancers. Until recently, the dogma has been that cancer is a purely genetic disease, which manifests itself in irreversible, rigid mutation patterns. Now, however, it is becoming increasingly clear that childhood cancer is in fact associated with a relatively low number of genetic
alterations. As a result, pediatric cancers are to be considered a consequence of a perturbed develop-ment of different cell types. In order to exploit these aberrant mechanisms for therapeutic purposes, we need to understand the regulatory mechanisms of normal and malignant development that lie behind these aberrations. The increasing number of refined applications for novel technologies such as next generation sequencing (NGS) enables insights into the three-dimensional organization and aberrations of chromatin, the control center of gene expression, which determine the fate of each cell. The high sensitivity and specificity of this technology allows us not only to address the genetic composition and gene expression on the single cell level, but also to analyse tumor cell break-down products in the blood

Science Reports
3938
Figure 1
Levels of heterogeneity
Inter-cancer
Inter-individual
Intra-tumor
Figure 2 NON-GENETIC PLASTICITY AND VARIABILITY AS THERAPEUTIC AND PROGNOSTIC TARGETS IN EWING SARCOMA
Our group has been studying the molecular under-pinnings of Ewing sarcoma for many years. We try to understand how a single genetic aberration, the EWS-FLI1 gene fusion, can drive the pathogenesis of this disease. Since recent genome sequencing studies have confirmed an extremely low number of mutations and the absence of recurrent aberra-tions other than the EWS-ETS gene rearrangement and facultative whole chromosome copy number changes, we hypothesize that, if activated in the right developmental, epigenomic and genomic context, EWS-FLI1 is sufficient to induce and main-tain malignancy. We believe that a deep mecha-nistic understanding of the interaction between developmental cellular background, microenviron-ment, tumor epigenome and EWS-ETS activity on a systems level will allow us to better understand the biological and clinical variability of the disease, and ultimately lead to the identification of vulnerabilities with therapeutic potential.
Despite the low number of mutations, Ewing sarcoma shows significant biological, clinical, and immunophenotypic variation and plasticity, which have moved into our focus in the reporting period. To understand inter-tumor, inter-patient and intra-tumor variability in absence of significant genetic variation, we interrogated the epigenomes of more than 140 Ewing sarcomas in comparison to each other and to those of other tumors and normal tissues. As described below, this paradigmatic study revealed two dimensions of heterogeneity, providing unprecedented biological insights into the etiology of the disease.
Tumor cell plasticity is the basis for metastasis, the major threat to patients. Tumor progression is asso-ciated with different types of cellular stress imposed on the tumor cell by the microenvironment. We and others have previously demonstrated upregulation of gene products involved in handling of cellular stress, among them the metabolic sensor Sirtuin 1 (SIRT1) and the DNA repair enzyme poly(ADP-ribose) poly-merase 1 (PARP1). We hypothesize that interfering with the activity of these proteins may reduce the metastatic potential of Ewing sarcoma and poten-tially sensitize resistant tumor cells to a variety of chemotherapeutic drugs. As PARP1 and SIRT1 are responsive to changes in tumor and microenviron-mental metabolic states, we interrogated the role of the Ewing sarcoma metabolome as a potential vulnerability of the disease.
EPIGENETIC DIVERSIT Y IN EWING SARCOMAIn a collaborative study between the CCRI, the CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences in Vienna, and the Institut Curie in Paris, led by CCRI scientist Eleni Tomazou with contributions from several hospitals in Austria, Germany, France and Spain, we assessed genome-wide epigenomic patterns of more than 140 Ewing sarcomas. Using novel bio -informatic methods developed by Nathan Sheffield at CeMM, our team studied the tumors’ DNA meth-ylation patterns – one of the most important facets of the human epigenome. We applied a next-gen-eration-sequencing (NGS) based high-throughput method, RRBS (Reduced Representation Bisulfite Sequencing) to all samples, thus analyzing DNA methylation patterns of CpG-rich genome regions at single-nucleotide resolution. In addition, a subset of samples was analysed by WGBS (Whole Genome
Figure 1. DNA methylation profiling by RRBS identifies epigenetic diversity on different levels
Figure 2. RRBS profiling identifies a unique Ewing sarcoma specific DNA methylation pattern
CpGs
EwS
sam
ples
Oth
er ti
ssue
s
Reg
ion
sets
Odds ratio
Bisulfite Sequencing), and interrogated for open chromatin regions by ATAC-seq (Assay for Trans-posase-Accessible Chromatin with high throughput sequencing), and for histone modifications indic-ative of transcription-regulatory activity by ChIP-seq (Chromatin Immunoprecipitation sequencing). Comparing our data to the Ewing sarcoma reference epigenome that we had established previously
and to publicly accessible data banks for similarly obtained DNA methylation profiles of human cancers and normal tissues, we identified a diagnostic Ewing sarcoma specific DNA-methylation profile distinct from any other tumor and normal tissue, and predictive of activity of Ewing sarcoma specific transcriptional enhancers (Figure 2). In addition, we observed unexpected inter-individual diversity.
100%
0%
DN
Am
ethy
latio
n
Region set enrichment
ENCODE
CODEXDNase
LOLA Core database
UCSC
EwS-hypomethylated regionsLOLA
EwS-specific
Prostate
Science Reports
4140Vi
abili
ty(%
)
A673A673shA
150
150 150
150 150
100
100 100
100 100
50
50 50
50 50
0
0 0
0 0
0.1
0.1
0.10.1
0.1
0.1
0.150.46
2.651.06
5050
50
EWS-FLI1-highEWS-FLI1-low
50
0.1
1
1
11
1
1
1
10
10
1010
10
10
10
nmol/L
nmol/L nmol/L nmol/L
nmol/L nmol/L
nmol/L
nmol/L
nmol/Lnmol/L
nmol/L
nmol/L
nmol/L
0.01
0.01
0.010.01
0.01
0.1
ICIC
ICIC
0.1 0.1 0.1
0.1 0.1
1
1 1 1
1 1
10
10 10 10
10 10
100
100 100 100
100 100
0.01
0.01
STA-ET-2.2
STA-ET-11 RM-82
PC-3 HEK293 U2OS
MSCCLB-MAHeLa
SK-N-MC
TC32
TC32
Viab
ility
(%)
Viab
ility
(%)
Viab
ility
(%)
Viab
ility
(%)
B
100
100
100100
100
100
100
100
50
50
5050
50
50
50
50
0
0
00
0
0
0
0
0.1 1 10nmol/L
0.01
100
50
0
0.2550IC 0.58 0.2250 50IC IC
0.770.60 5050 ICIC
4.34 1.5050 50IC IC 4.4350IC
7.2450IC0.871.62 5050 ICIC
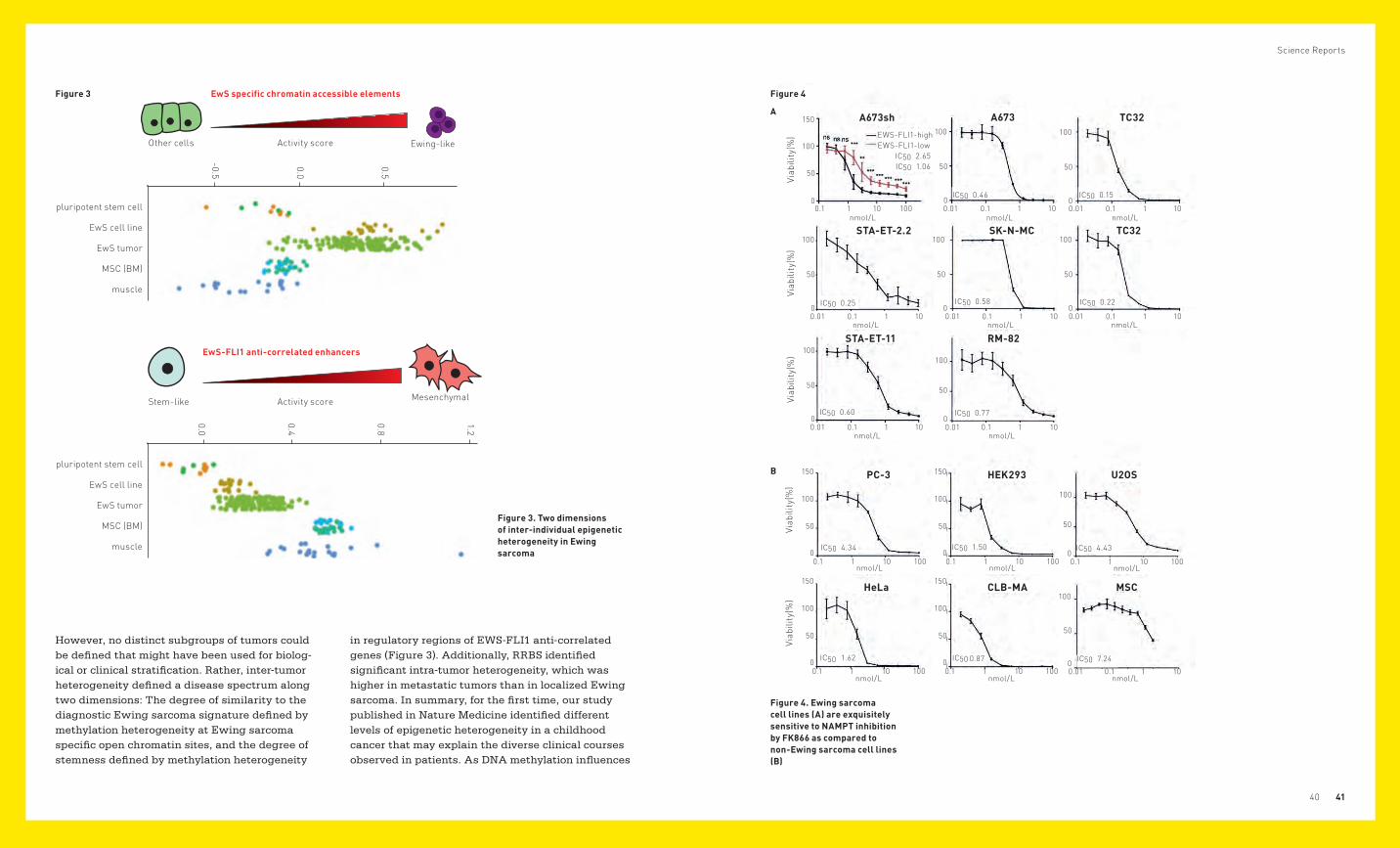
However, no distinct subgroups of tumors could be defined that might have been used for biolog-ical or clinical stratification. Rather, inter-tumor hetero geneity defined a disease spectrum along two dimensions: The degree of similarity to the diagnostic Ewing sarcoma signature defined by methylation heterogeneity at Ewing sarcoma specific open chromatin sites, and the degree of stemness defined by methylation heterogeneity
Figure 3 Figure 4
A
B
Figure 3. Two dimensions of inter-individual epigenetic heterogeneity in Ewing sarcoma
EwS specific chromatin accessible elements
Other cells
Stem-like
Activity score
Activity score
Ewing-like
Mesenchymal
Figure 4. Ewing sarcoma cell lines (A) are exquisitely sensitive to NAMPT inhibition by FK866 as compared to non-Ewing sarcoma cell lines (B)
in regulatory regions of EWS-FLI1 anti-correlated genes (Figure 3). Additionally, RRBS identified significant intra-tumor heterogeneity, which was higher in metastatic tumors than in localized Ewing sarcoma. In summary, for the first time, our study published in Nature Medicine identified different levels of epigenetic heterogeneity in a childhood cancer that may explain the diverse clinical courses observed in patients. As DNA methy lation influences
pluripotent stem cell
EwS cell line
EwS tumor
MSC (BM)
muscle
0.5
0.0
-0.5
1.2
0.8
0.4
0.0
pluripotent stem cell
EwS cell line
EwS tumor
MSC (BM)
muscle
EwS-FLI1 anti-correlated enhancers

Science Reports
4342
We found that in the Ewing sarcoma model cell line A673 by activation of Tdo2 (tryptophan 2,3-dioxy-genase) knockdown of EWS-FLI1 led to tryptophan break-down and synthesis of the primary metab-olites kynurenine and kynurenic acid, which in an autocrine manner activated the aryl hydrocarbon receptor and its nuclear target genes. However, no further metabolization to NAD was observed and activation of TDO2 in response to low EWS-FLI1 levels was restricted to a subset of Ewing sarcoma cell lines only. Instead, we found the enzyme NAMPT (Nicotinamide Phosphoribosyltransferase) to be rate-limiting for NAD synthesis in Ewing sarcoma. We therefore investigated the conse-quences of NAMPT inhibition by the small molecule inhibitor FK866 on Ewing sarcoma growth. We observed that blocking NAMPT leads to exhaustive NAD depletion in Ewing sarcoma cells, followed by a metabolic collapse and cell death. Using conditional EWS-FLI1 knockdown by doxycycline-inducible shRNA revealed that EWS-FLI1 depletion signifi-cantly reduces the sensitivity of Ewing sarcoma cells to NAMPT inhibition. Consistent with this finding, a comparison of 7 Ewing sarcoma cell lines of differ-ent genotypes with 5 Non-Ewing sarcoma cell lines and mesenchymal stem cells revealed significantly higher FK866 sensitivity of EWS-ETS positive Ewing sarcoma cells, with IC50 values mostly below 1nM (Figure 4). Taken together, our data reveal evidence of the important role of the NAMPT-mediated NAD salvage pathway in the energy homeostasis of Ewing sarcoma cells and suggest NAMPT inhibi-tion as a potential new treatment approach in this disease. (Mutz C, et al., 2017, Oncotarget)
THE NAD METABOLOME AS THERAPEUTIC TARGET IN EWING SARCOMASeveral studies have previously indicated Ewing sarcoma specific overexpression of the DNA repair enzyme PARP1 and preclinical evidence for exqui-site sensitivity of the disease to PARP1 inhibi-tors. However, clinical trials have, so far, failed to confirm therapeutic efficacy of PARP1 inhibitors in monotherapy, and the question, which drugs to use in combination therapy sensitizing to PARP inhibition remains to be answered. PARP1 is a protein-modifying enzyme that requires NAD (Nico-tinamide Adenine Dinucleotide) as co-substrate for protein-ADP-ribosylation. NAD is a key metabolite of energy metabolism involved in cellular redox reactions, DNA repair, and in the maintenance of genomic stability. We have previously demonstrated overexpression of the metabolic sensor SIRT1 associ-ated particularly with Ewing sarcoma metastases. SIRT1 is also an enzyme competing for the co-sub-strate NAD to deacetylate acetylated proteins. In fact, PARP1 and SIRT1 are the major cellular consumers of NAD, and their high activity in Ewing sarcoma require continuous fuelling by the co-sub-strate. We therefore interrogated the major source of NAD in Ewing sarcoma and analysed the conse-quences of disrupting NAD supply. Analysing the transcriptional signature of EWS-FLI1, which drives Ewing sarcoma growth, we identified a number of genes in the NAD biosynthetic pathway to be dereg-ulated in expression. NAD can be metabolized from the amino acid tryptophan in the microenvironment.
gene activity, the combination of Ewing sarcoma specific and cell-of-origin specific patterns can lead to different outcomes. The epigenetic diversity also appears to correlate with the tumors’ aggressive-ness and metastatic state. These new insights into the biology of Ewing sarcoma provide the basis for developing epigenetic biomarkers that can reliably predict disease course and therapy response. Our findings in Ewing sarcoma also provide an inter-esting concept for other cancers with low genetic complexity.
Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schönegger A, Schuster M, Hadler J, Surdez D, Guillemot D, Lapouble E, Freneaux P, Champigneulle J, Bouvier R, Walder D, Ambros IM, Hutter C, Sorz E, Amaral AT, de Álava E, Schallmoser K, Strunk D, Rinner B, Liegl-Atzwanger B, Huppertz B, Leithner A, de Pinieux G, Terrier P, Laurence V, Michon J, Ladenstein R, Holter W, Windhager R, Dirksen U, Ambros PF, Delattre O, Kovar H, Bock C, Tomazou EM. (2017). DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med; 23:386-395.
Mutz CN, Schwentner R, Kauer MO, Katschnig AM, Kromp F, Aryee DN, Erhardt S, Goiny M, Alonso J, Fuchs D, Kovar H. (2016). EWS-FLI1 impairs aryl hydrocarbon receptor activation by blocking tryptophan breakdown via the kynurenine pathway. FEBS Lett, 590: 2063-2075.

CD45RA-
CD133 -/lowLympho-myeloid progenitors
Multi-lymphoidprogenitors
Erythro-myeloid progenitors
EoBP MEP
Megakaryocyteserythrocytes
Eosinophilsbasophils
Neutrophilsmonocytes
B-lymphoid progenitors
CD45RA +
CD133-
CD45RA +
CD133 +
CD45RA -
CD133+
CD38low
CD10-
MPP
LMPPCD38 low
CD10 - GMPCD38 +
CD10 -
MLPCD38 +
CD10 +
BLPCD38 ++
CD10 +
CD19 +
Late GMP
CD38 +
CD10 -
EMPCD38 +
CD10 -
Multi-potentprogenitors
Granulocytes and macrophages progenitors
B-lymphocytes
IMPROVING THE ROUTINE ENUMERATION OF HEMATOPOIETIC STEM CELLS: MULTI-COLOR ANALYSIS OF CD34 SUBTYPES REVEALS UNEXPECTED DIFFERENCES BETWEEN VARIOUS STEM CELL SOURCES
Science Reports
4544
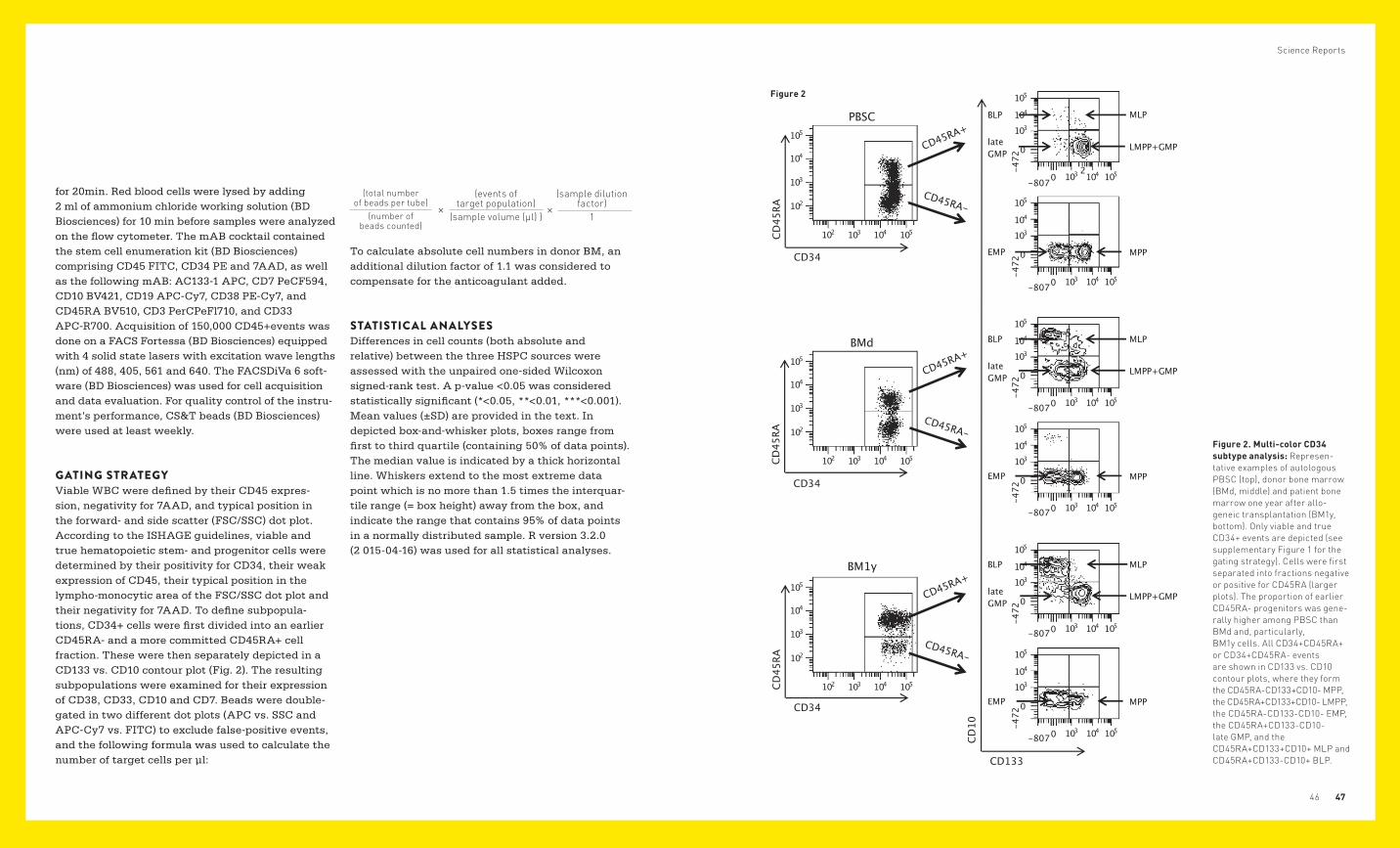
SUMMARYFlow cytometric analysis of CD34+ hematopoie-tic stem- and progenitor cells commenced in the early nineteen nineties and became a standardized 3-color procedure in 1998. In the clinical setting, this method has since been used worldwide, without any changes or improvements, to enumerate CD34+ cells as such, irrespective of their developmental stage or their hematopoietic potential. Based on the results of different research groups and on recent adjust-ments of the model of human hematopoiesis (Abb. 1, Görgens A. et al. 2013. Cell Rep.), we established a >10-color CD34 flow cytometric assay containing antibodies like CD133, CD45RA, CD10, CD19, CD38 and CD33. We showed that it enables phenotyping of at least 6 distinct CD34 subsets: The multipotent progenitors (MPP) are 133+/45RA-/10-/38low and represent the earliest of all CD34 subtypes. Differen-tiation of these MPP by asymmetrical division forms LMPP (lymphoid-primed multipotent progenitors) that acquired CD45RA (133+/45RA+/10-/38low), and EMP (erythro myeloid progenitors) that lost CD133 expression (133-/45RA-/10-/38+). LMPP represent progenitors of neutrophils and lymphoid blood cells while EMP can give rise to granulocytes other than neutrophils as well as to erythroid and megacaryocytic lineages. Differentiation of LMPP towards the more mature GMP (neutrophil gran-ulocyte and monocyte progenitors) goes along with a loss of CD133 expression (133-/45RA+/10-/38+), while gaining cell surface CD10 leads to the formation of MLP (myelo lymphoid progenitors; 133+/45RA+/10+/38+). Further maturation of MLP towards the progenitors of B lymphocytes (BLP) is accompanied by a loss of surface CD133 resulting in a 133-/45RA+/10+/38++/19+ phenotype.
Figure 1. Model of the human hematopoietic tree, adapted from the recent version by Görgens et al. 2013. Only CD34+ cell stages are depicted. The CD45RA-CD133+CD38lowCD10- multipotent progenitors (MPP, upper) either remain CD45RA- and down-regulate CD133 (middle right) to form cells of the erythro-myeloid lineage (EMP), or they acquire CD45RA and become CD133+ lympho-myeloid progenitors (LMPP, middle left) that comprise all lymphoid but also neutrophil and monocyte precursors (GMP). Upon down-regulation of CD133 (lower left), these form late GMP and, in case of CD10 acquisition, cells of the B-lymphoid lineage (BLP).
SPECIMEN COLLECTIONAll cell specimens used in this study were obtained for routine CD34 enumeration. Donor BM samples (BMd, n=31) were from healthy allogeneic BM donors aged from 2 to 48 (median 26) years. Patient BM specimens (BM1y, n=21) were from biopsies routinely drawn for clinical examination one year after allografting. Six of these were paired samples, i.e. we analyzed both BMd and BM1y from these patients. The BM recipients (11 males, 10 females) had been diagnosed with AML (4), ALL (6), MDS (4), CML (2), RCC (2), SCID (1), SAA (1) and Hyper IG E syndrome (1). Their median age was 10 years (range 0.8-18). Conditioning regimens were myelo-ablative (n=11) and reduced intensity (n=10). Fifteen BM recipients had received antithymocyte glob-ulin. PBSC samples (n=35) were from autologous stem cell collections from adult patients (22 males, 13 females) with Multiple Myeloma (n=19) and Non-Hodgkin´s lymphoma (n=16), with a median age of 54 years (range 22-73). The mobilization regimen comprised chemotherapy (CHT) and hematopoietic growth factors (hGF; n=13), CHT+hGF and plerixafor (n=6), hGF alone (n=5) or hGF and plerixafor (n=11). Informed consent was obtained from all patients and the extended staining experiments had been approved by the local ethical committee.
CELL PROCESSING, SINGLE PL ATFORM PROTOCOL AND FLOW CY TOMETRYBlood cell counts were obtained from a Sysmex KX-21N (Sysmex Corporation, Kobe, Japan). If neces-sary, cells were diluted with Dulbecco´s phosphate buffered saline (PBS, Carlsbad, CA, USA) and the white blood cells (WBC) adjusted to 5-15 x106 cells/ml prior to immune staining. All monoclonal anti-bodies (mAB) were used at pretested concentrations and after respective compensation. Isotype controls and fluorescence-minus-one analyses were used to define gating and compensation. One hundred µl of the cell sample were reverse-pipetted into a Trucount tube (BD Biosciences, San Jose, CA, USA). After adding the mAB cocktail, cells were mixed and incubated light-shielded at room temperature
Figure 1
Science Reports
4746
PBSC
BMd
BM1y
CD45
RA
CD45RA-
CD45RA+
CD45RA-
CD45RA+
CD45RA-
CD45RA+
CD34
CD45
RA
CD34
CD45
RA
CD34
CD133
CD10
1
1
2
2
5
late GMP
MLPBLP
LMPP+GMP
EMP MPP
late GMP
MLPBLP
LMPP+GMP
EMP MPP
late GMP
MLPBLP
LMPP+GMP
EMP MPP
102
102
103
103
104
1041030-807
-472
0104
105
105
105
104
103
1041030-807
-472
0
105
105
104
103
1041030-807
-472
0
105
105
104
103
1041030-807
-472
0
105
105
104
103
1041030-807
-472
0
105
105
104
103
1041030-807
-472
0
105
105
104
103
105
102
102
103
103
104
104
105
105
102
102
103
103
104
104
105
105
for 20min. Red blood cells were lysed by adding 2 ml of ammonium chloride working solution (BD Biosciences) for 10 min before samples were analyzed on the flow cytometer. The mAB cocktail contained the stem cell enumeration kit (BD Biosciences) comprising CD45 FITC, CD34 PE and 7AAD, as well as the following mAB: AC133-1 APC, CD7 PeCF594, CD10 BV421, CD19 APC-Cy7, CD38 PE-Cy7, and CD45RA BV510, CD3 PerCPeFl710, and CD33 APC-R700. Acquisition of 150,000 CD45+events was done on a FACS Fortessa (BD Biosciences) equipped with 4 solid state lasers with excitation wave lengths (nm) of 488, 405, 561 and 640. The FACSDiVa 6 soft-ware (BD Biosciences) was used for cell acquisition and data evaluation. For quality control of the instru-ment’s performance, CS&T beads (BD Biosciences) were used at least weekly.
GATING STRATEGYViable WBC were defined by their CD45 expres-sion, negativity for 7AAD, and typical position in the forward- and side scatter (FSC/SSC) dot plot. According to the ISHAGE guidelines, viable and true hematopoietic stem- and progenitor cells were determined by their positivity for CD34, their weak expression of CD45, their typical position in the lympho-monocytic area of the FSC/SSC dot plot and their negativity for 7AAD. To define subpopula-tions, CD34+ cells were first divided into an earlier CD45RA- and a more committed CD45RA+ cell fraction. These were then separately depicted in a CD133 vs. CD10 contour plot (Fig. 2). The resulting subpopulations were examined for their expression of CD38, CD33, CD10 and CD7. Beads were double-gated in two different dot plots (APC vs. SSC and APC-Cy7 vs. FITC) to exclude false-positive events, and the following formula was used to calculate the number of target cells per µl:
To calculate absolute cell numbers in donor BM, an additional dilution factor of 1.1 was considered to compensate for the anticoagulant added.
STATISTICAL ANALYSESDifferences in cell counts (both absolute and relative) between the three HSPC sources were assessed with the unpaired one-sided Wilcoxon signed-rank test. A p-value <0.05 was considered statistically significant (*<0.05, **<0.01, ***<0.001). Mean values (±SD) are provided in the text. In depicted box-and-whisker plots, boxes range from first to third quartile (containing 50% of data points). The median value is indicated by a thick horizontal line. Whiskers extend to the most extreme data point which is no more than 1.5 times the interquar-tile range (= box height) away from the box, and indicate the range that contains 95% of data points in a normally distributed sample. R version 3.2.0 (2 015-04-16) was used for all statistical analyses.
(total number of beads per tube)
(number of beads counted)
(events of target population)
(sample volume (µl) )
(sample dilution factor)
1× ×
Figure 2. Multi-color CD34 subtype analysis: Represen-tative examples of autologous PBSC (top), donor bone marrow (BMd, middle) and patient bone marrow one year after allo-geneic transplantation (BM1y, bottom). Only viable and true CD34+ events are depicted (see supplementary Figure 1 for the gating strategy). Cells were first separated into fractions negative or positive for CD45RA (larger plots). The proportion of earlier CD45RA- progenitors was gene-rally higher among PBSC than BMd and, particularly, BM1y cells. All CD34+CD45RA+ or CD34+CD45RA- events are shown in CD133 vs. CD10 contour plots, where they form the CD45RA-CD133+CD10- MPP, the CD45RA+CD133+CD10- LMPP, the CD45RA-CD133-CD10- EMP, the CD45RA+CD133-CD10- late GMP, and the CD45RA+CD133+CD10+ MLP and CD45RA+CD133-CD10+ BLP.
Figure 2
0
1000
2000
3000
4000
PBSC BMd BM1y
Total CD34 relative
% of
WBC
0
1
2
3
4
5
PBSC BMd BM1y
*****
**
0
500
1000
1500
2000
MPP EMP LMPP+GMP Late GMP MLP+BLP
0
20
40
60
80
MPP EMP LMPP+GMP Late GMP MLP+BLP
% of
CD
34+
*** ns
ns
**
*** *** ***
*** **
**
***
******** *** ********* * nsns
Total CD34 absolute
*** *** *** ***ns
*** *** *** ******
a
b
+
2000
2000
3000
4000
Total CD34 absolute Total CD34 relative
PBSC BMd
ns
ns
ns
BM1y
LMPP+GMPEMPMPP Late GMP MLP+BLP
LMPP+GMPEMPMPP Late GMP MLP+BLP
PBSC BMd BM1y
1500
1000
1000
500
0
0
Cells
/µI
%CD
34+
CD
34 c
ells
/µI
% o
f WB
C
b
a
0
20
40
60
80
5
4
3
2
1
0
nsns
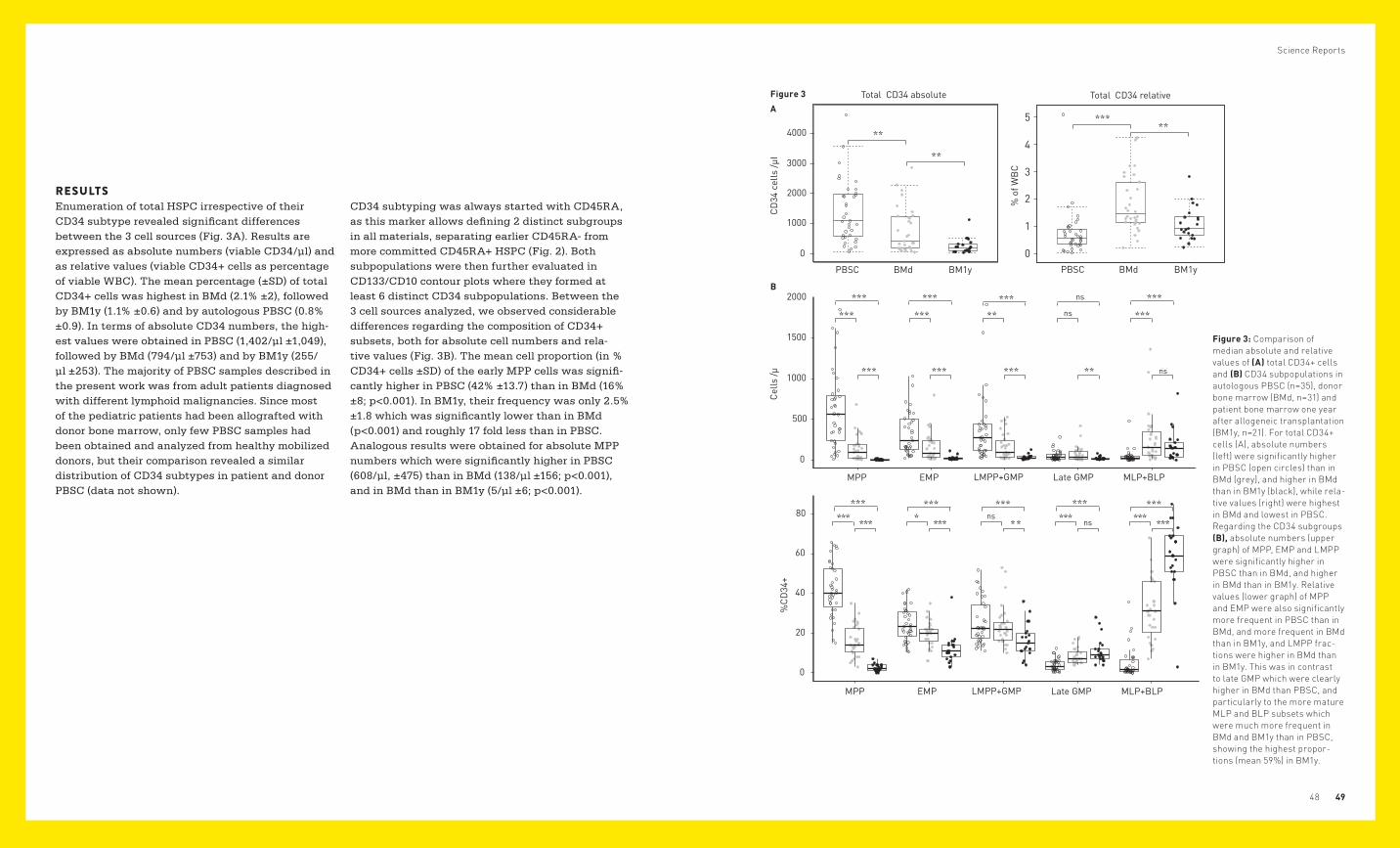
RESULTSEnumeration of total HSPC irrespective of their CD34 subtype revealed significant differences between the 3 cell sources (Fig. 3A). Results are expressed as absolute numbers (viable CD34/µl) and as relative values (viable CD34+ cells as percentage of viable WBC). The mean percentage (±SD) of total CD34+ cells was highest in BMd (2.1% ±2), followed by BM1y (1.1% ±0.6) and by autologous PBSC (0.8% ±0.9). In terms of absolute CD34 numbers, the high-est values were obtained in PBSC (1,402/µl ±1,049), followed by BMd (794/µl ±753) and by BM1y (255/µl ±253). The majority of PBSC samples described in the present work was from adult patients diagnosed with different lymphoid malignancies. Since most of the pediatric patients had been allografted with donor bone marrow, only few PBSC samples had been obtained and analyzed from healthy mobilized donors, but their comparison revealed a similar distribution of CD34 subtypes in patient and donor PBSC (data not shown).
CD34 subtyping was always started with CD45RA, as this marker allows defining 2 distinct subgroups in all materials, separating earlier CD45RA- from more committed CD45RA+ HSPC (Fig. 2). Both subpopulations were then further evaluated in CD133/CD10 contour plots where they formed at least 6 distinct CD34 subpopulations. Between the 3 cell sources analyzed, we observed considerable differences regarding the composition of CD34+ subsets, both for absolute cell numbers and rela-tive values (Fig. 3B). The mean cell proportion (in % CD34+ cells ±SD) of the early MPP cells was signifi-cantly higher in PBSC (42% ±13.7) than in BMd (16% ±8; p<0.001). In BM1y, their frequency was only 2.5% ±1.8 which was significantly lower than in BMd (p<0.001) and roughly 17 fold less than in PBSC. Analogous results were obtained for absolute MPP numbers which were significantly higher in PBSC (608/µl, ±475) than in BMd (138/µl ±156; p<0.001), and in BMd than in BM1y (5/µl ±6; p<0.001).
Science Reports
4948
Figure 3: Comparison of median absolute and relative values of (A) total CD34+ cells and (B) CD34 subpopulations in autologous PBSC (n=35), donor bone marrow (BMd, n=31) and patient bone marrow one year after allogeneic transplantation (BM1y, n=21). For total CD34+ cells (A), absolute numbers (left) were significantly higher in PBSC (open circles) than in BMd (grey), and higher in BMd than in BM1y (black), while rela-tive values (right) were highest in BMd and lowest in PBSC. Regarding the CD34 subgroups (B), absolute numbers (upper graph) of MPP, EMP and LMPP were significantly higher in PBSC than in BMd, and higher in BMd than in BM1y. Relative values (lower graph) of MPP and EMP were also significantly more frequent in PBSC than in BMd, and more frequent in BMd than in BM1y, and LMPP frac-tions were higher in BMd than in BM1y. This was in contrast to late GMP which were clearly higher in BMd than PBSC, and particularly to the more mature MLP and BLP subsets which were much more frequent in BMd and BM1y than in PBSC, showing the highest propor-tions (mean 59%) in BM1y.
A
Figure 3
B

MPP differentiate to either LMPP (CD45RA+CD133+) or EMP (CD45RA-CD133-/low). Both subsets showed a higher CD38 expression than MPP supporting their higher differentiation (Fig. 4). As shown in Fig. 3B, the mean frequency of LMPP was similar between PBSC and BMd (25.6% ±11.1, and 23.7% ±10.1; ns), but significantly lower in BM1y (16.3% ±8.3) than in BMd (p<0.01). Absolute LMPP cell numbers differed significantly between PBSC (383/µl ±412) and BMd (170/µl ±158; p<0.01), and between BMd and BM1y (33/µl ±28; p<0.001). This may suggest a higher proportion of neutrophil progenitors in favor of B-lymphoid progenitors in the LMPP subfraction of PBSC compared to BMd. EMP which give rise to erythrocytes, megakaryocytes and granulocytes other than neutrophils, showed results comparable to those obtained for LMPP. Their mean frequency was similar in PBSC (24.5% ±8.7) and BMd (19.5% ±6.5; p<0.05) but differed significantly between BMd and BM1y (11.9% ±7.3; p<0.001). Absolute EMP numbers differed clearly and were 331/µl ±261 for PBSC vs. 162/µl ±176 for BMd (p<0.001), and 24/µl ±25 for BM1y vs. BMd (p<0.001). In terms of LMPP and EMP, BM1y thus mediates an impression of exhaustion when compared with BMd. Late myeloid progenitors (late GMP) with a CD45RA+CD133-CD33+CD10- phenotype differed mainly with regard to relative values, which were significantly lower in PBSC (4% ±3.1) than in BMd (8.5% ±4.1; p<0.001), but similar between BMd and BM1y (11.1% ±6.5; ns). In terms of absolute values, they were 51/µl ±62 in PBSC vs. 72/µl ±91 in BMd (ns), and slightly lower in BM1y (23/µl ±20) than in BMd (p<0.01). Due to the low frequency of the CD133+ MLP, this CD34 subset was evaluated together with the CD133- BLP. These cells were hardly detectable in PBSC (5% ±7.9) but clearly present in BMd (31.9% ±15.1; p<0.001). In BM1y, they represented the largest CD34 subfraction (58.5% ±17.6) which was significantly higher than in BMd (p<0.001).
Despite the rather low proportions of the CD133dim MLP in the BM samples, it has to be noted that this CD34 subset, when expressed as percentage of the MLP/BLP fraction, represented a clearly lower median cell proportion (p=1.66e-09; one-sided unpaired Wilcoxon rank sum test) in BM1y (3.6%) than in BMd (11.9%). A representative example is depicted in Fig. 2. In terms of absolute CD10+ stem cell numbers, significant differences were observed between PBSC (43/µl ±83) and BMd (255/µl ±302; p<0.001), whereas the values were similar between BMd and BM1y (171/µl ±191; ns). Out of the 31 BMd and 21 BM1y specimens analyzed, 6 were paired samples, i.e. we examined BMd and BM1y pairs from the same patients. The results were virtually identical to those obtained from the whole groups: The median proportions of the CD34 subsets for BMd/BM1y were 12.1%/1.8% (MPP), 22%/17.4% (LMPP), 20.1%/10% (EMP), 5.5%/9.1% (late GMP) and 34%/65.2% (MLP and BLP).
The expression intensity of distinct markers often correlates with differentiation as shown for CD38 (see below). Such differences were also observed for CD33 and CD133. Expression of CD133 on MPP and LMPP was generally higher in PBSC than in BMd and BM1y (not depicted), suggesting that these cell stages are more differentiated in BM than in PBSC. Nevertheless, it was possible to distinguish the different CD133+/- subpopulations in all cell samples (Fig. 2). The myeloid marker CD33 was expressed in all CD34 subfractions, although it was weaker on MPP in PBSC with high CD133 expression than on MPP in BM with weaker CD133 expression (not depicted). Distinct CD10+ (and CD19+) HSPC subsets were detectable among both the CD45RA+ and the CD133- progenitors, and the CD33 expres-sion was clearly higher in BM1y than PBSC (not shown). In all cell sources, a potential coexpression
of CD7 as a marker of T- and NK-cell progenitors was only seen on 0% to 2.8% of CD34+ cells). As non-spe-cific staining could not be excluded, this subtype was not pursued any further.
We used the PE-Cy7-labelled H7 clone of CD38 in all experiments performed. Virtually all CD34+ cells were positive for this antibody, albeit at clearly different intensity (Fig. 4). MPP showed the lowest expression intensity, followed by LMPP and EMP. The intensity was higher among CD45RA+ late myeloid precursors, and highest among the CD45RA+CD10+CD19+ BLP. This differential CD38 expression was generally observed in all specimens examined although the differences were not always as clear as depicted in the BM1y sample shown in Fig. 4. Due to the considerable overlap between the different CD34 subgroups, CD38 was never used as first-line antibody for subgroup distinction.
CONCLUSIONWe conclude that the presented analysis can identify and enumerate distinct CD34 subfractions in any conventional CD34 cell source. This approach may provide a solid basis for future studies to determine the impact of different CD34 subsets in the graft as well as in post-transplant bone marrow on engraft-ment kinetics and immune reconstitution. Whether or not the analysis will allow predicting engraftment kinetics on a routine basis remains to be examined.
Dmytrus J, Matthes-Martin S, Pichler H, Worel N, Geyeregger R, Frank N, Frech C, Fritsch G. (2016). Multi-color immune-pheno-typing of CD34 subsets reveals unexpected differences between various stem cell sources. Bone Marrow Transplant, 51: 1093-1100
Science Reports
5150
CD38
CD34 subset
MPP 18.191
LMPP 46.974
EMP 66.165
Late GMP
BLP 96.670
71.427
MFI
Figure 4. Rise of CD38 mean fluorescence intensity (MFI) values on HSPC with increa-sing differentiation of CD34+ subsets. Representative bone marrow sample from a patient one year after allogeneic transplantation (BM1y) depic-ting CD45RA-CD133+CD10- multi-potent progenitors (MPP), CD45RA+CD133+CD10- lymphoid-primed multi-po-tent progenitors (LMPP), CD45RA-CD133-CD10- erythro-myeloid precursors (EMP), CD45RA+CD133-CD10- late granulocyte monocyte progenitors (late GMP) and CD45RA+CD133-CD10+ B-lym-phoid progenitors (BLP).
Figure 4

B
A
HPDGFRb
H657 D850
R853
E946 E946
H853
H857
D850
D850
R853
+ -
D850
K657
4.1A3.0A
2.9AE850
H657
5.1A2.7A
+3
A L M S E L K I M S H L G K I C D F G L A R D I M R D S N YK I C D F G L A R D I M H D S N YK I C D F G L A R D I M S D S N YK I C D F G L A R D I M N D S N YK I C D F G L A R D I K N D S N Y
K I C D F G L A R D I M N D S N Y
A L M S E L K I M S H L GA L M S E L K V L S Y L G
A L M S E L K I M T H L G
A L M S E L K I M S H L G
A L M S E L K M MT O L GPDGFRa
FLT3CSF1R
c-Kit
100%
0%
Consensus
αC-helix A-loop
Conservation
G
E
F
DC
B
A
HPDGFRb
H657 D850
R853
E946 E946
H853
H857
D850
D850
R853
+ -
D850
K657
4.1A3.0A
2.9AE850
H657
5.1A2.7A
+3
A L M S E L K I M S H L G K I C D F G L A R D I M R D S N YK I C D F G L A R D I M H D S N YK I C D F G L A R D I M S D S N YK I C D F G L A R D I M N D S N YK I C D F G L A R D I K N D S N Y
K I C D F G L A R D I M N D S N Y
A L M S E L K I M S H L GA L M S E L K V L S Y L G
A L M S E L K I M T H L G
A L M S E L K I M S H L G
A L M S E L K M MT O L GPDGFRa
FLT3CSF1R
c-Kit
100%
0%
Consensus
αC-helix A-loop
Conservation
G
E
F
DC
Science Reports
5352
Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative neoplasia occur-ring in young children, characterized by excessive proliferation of monocytic and granulocytic cells infiltrating different organs. About 90% of JMML patients have mutations in NF1, K-RAS, N-RAS, CBL, or PTPN11, all implicated in activation of the RAS-RAF-MAPK pathway. Despite recent efforts exploiting whole exome sequencing, the genetic alterations underlying the disease in the remain-ing 10% of patients remain elusive. Hematopoietic stem cell transplantation (HSCT) is currently the only curative therapy for most JMML patients, but advances in the understanding of the underlying molecular mechanisms in JMML have permitted the introduction of different therapeutic agents such as the DNA-hypomethylating substance azacitidine. We have identified a JMML case with a chromo-somal translocation, t(5;17)(q33;p11.2), resulting in the fusion of the platelet-derived growth factor receptor β (PDGFRB) gene to a novel partner, the nuclear distribution protein nudE-like 1 (NDEL1), which has not been implicated in any translocation event to date. In contrast to earlier data on fusion genes involving PDGRFB in myeloid malignancies, which were generally responsive to treatment with imatinib, the patient became refractory to both imatinib and nilotinib. This observation represented the first clinical finding of a PDGFRB gene fusion resistant to therapy with tyrosine kinase inhibitors (TKIs), and our work focused on elucidating the hith-erto unknown mechanism of TKI resistance.
IDENTIFICATION OF A NOVEL FUSION GENE INVOLVING PDGFRB AND MUTATIONAL ANALYSISSequencing of 5’-RACE-PCR products revealed an in-frame fusion between NDEL1 and PDGFRB. Screening of archived diagnostic specimens from 40 JMML patients provided no evidence for the occurrence of the NDEL1-PDGFRB fusion gene in other individuals. The observed development of resistance to two different TKIs including imati-nib and nilotinib prompted screening of the entire tyrosine kinase domain (TKD) of PDGFRB for the presence of mutations. Sanger sequencing revealed the missense point mutation C2550G in the acti-vation loop of the TKD converting the aspartate residue at position 850 into glutamate (D850E). This mutation was identified at the time of both relapses but not in the diagnostic PB or BM samples.
STRUCTURAL MODELLING OF THE PDGFRβ T YROSINE KINASE DOMAINIn order to elucidate the structural effects mediated by the D850E mutation in the PDGFRβ TKD, we have generated protein models of the kinase domain both in active and inactive conformations. Since the structure of PDGFRβ TKD is not available, we have modelled the kinase domain on the basis of estab-lished structures of highly homologous proteins from the PDGFR family including c-KIT, CSF1R, and VEGFR2. All structural models indicated that the observed TKI type-II resistance of cells expressing NDEL1-PDGFRβD850E was conceivably related to stabilization of the activation loop (A-loop) in the active conformation. In this conformation, three crit-ical amino acids, D844, F845, and G846 (DFG) serv-ing as a hypomochlion for the activation loop, adopt the so-called DFG-in position. The modelled struc-ture of the inactive DFG-out conformation revealed
the typical auto-inhibitory interaction between D850 and the amino acid at the +3 position, R853, which is commonly observed in inactive kinase domains of other receptor tyrosine kinases (RTKs) from the PDGFR family, and is believed to stabilize the A-loop in the inactive conformation (fig.1A, orange). However, modelling of the mutant PDGFRβ TKD in the inactive conformation could not explain the enhanced kinase activity and resistance to type-II TKIs, because the negatively charged glutamate at position 850 is also able to form a salt bridge with the positively charged side chain of R853 (fig.1B). Moreover, the DFG-out model of the TKD did not display any indication of weakened electrostatic interaction between E850 and R853 which would destabilize the inactive conformation of the kinase domain. However, the DFG-in model suggested the occurrence of two intriguing amino acid inter-actions upon transition of the A-loop from inactive to the active state (fig.1A, green). One interaction implicated the negatively charged D850 and the positively charged, conserved H657 in the αC-helix
(fig. 1C), which is expected to stabilize the A-loop in the active conformation. This interaction can be further enhanced by the D850E mutation, because the longer side chain of glutamate in comparison to aspartate brings the negatively charged carboxylic group 1.1 Å closer to the positively charged histi-dine residue, thus increasing the stability of the activation loop in the active conformation (fig. 1D). The other interaction involved R853 and E946 in the C-lobe of the TKD (fig. 1F). The +3 position to D850 is one of the least conserved positions in the activation loop of receptor tyrosine kinases (RTKs) from the PDGFR family (fig. 1H), and the arginine at this position in PDGFRβ (R853) has the longest side chain among all members. The DFG-in model suggested that the positively charged side chain of R853 can reach a distance of approximately 2.5 Å to the negatively charged carboxyl group of E946, which may facilitate electrostatic bonds and provide additional stabilization of the DFG-in conformation of the PDGFRβ TKD (fig. 1G). The structural model therefore suggested resistance of
CHARACTERIZATION OF A NOVEL FUSION GENE IN JUVENILE MYELOMONOCYTIC LEUKEMIA ASSOCIATED WITH RESISTANCE TO TYROSINE KINASE INHIBITORS
Figure 1. Structural model of PDGFRβ TKD
Figure 1

Science Reports
5554
NIL,100 nM
1 2 3 4 5 6 7 8 9 10 11 12
pY751-PDGFRβ
pY857-PDGFRβ
PDGFRβ
pErk
Erk
Gapdh
WT
IMA
TYPE
IITY
PE I
80
40
4090
20
20
50
25
2530
30
30
10
10
10
10
10
10
15
15
15
15
15
15
2525
12
12
12
12
2
2
N/A
N/A
N/A N/A
1
8
7
4
1312
TKI WT R853H
>5000 >5000
2200
2100
2100
1900
DR H657K HR WT D842ED850EFIP1L1-PDGFRαNDEL1-PDGFRβ
NIL
SOR
DAS
MID
PAC
RB
A
H HR D DR
NIL,100 nM
1 2 3 4 5 6 7 8 9 10 11 12
pY751-PDGFRβ
pY857-PDGFRβ
PDGFRβ
pErk
Erk
Gapdh
WT
IMA
TYPE
IITY
PE I
80
40
4090
20
20
50
25
2530
30
30
10
10
10
10
10
10
15
15
15
15
15
15
2525
12
12
12
12
2
2
N/A
N/A
N/A N/A
1
8
7
4
1312
TKI WT R853H
>5000 >5000
2200
2100
2100
1900
DR H657K HR WT D842ED850EFIP1L1-PDGFRαNDEL1-PDGFRβ
NIL
SOR
DAS
MID
PAC
RB
A
H HR D DR
NDEL1-PDGFRBD850E mutation to type-II TKIs, which can only bind to the inactive conformation of the PDGFRβ TKD, but indicated sensitivity to type-I TKIs binding to the active conformation. To address the predictions provided by the protein model, we have introduced several mutations affecting the aforementioned interactions and tested the sensitiv-ity of generated constructs against a panel of TKIs.
TRANSFORMING ACTIVIT Y AND TKI-SENSITIVIT Y OF THE WILDT YPE AND MUTANT NDEL1-PDGFRB FUSION GENESTo assess the oncogenic potential of the newly identified fusion gene, the murine cell line Ba/F3 was stably transduced with wildtype or mutant NDEL1-PDGFRB constructs by employing a trans-poson-based system (Byrgazov K et al. Oncotarget.
2016). In addition to the clinically identified D850E mutant, we have made a construct carrying H657K mutation, which, according to our structural model, would strengthen the electrostatic interaction between D850 and αC-helix thus stabilizing the DFG-in conformation of the PDGFRβ TKD (fig.1E). In order to probe the influence of R853 on the kinase activity and TKI-sensitivity of PDGRFβ TKD, we have also generated the constructs carrying the R853H mutation. Ba/F3 cells expressing the mutants H657K and D850E versions of NDEL1-PDG-FRB displayed a slightly higher proliferation rate, possibly reflecting an elevated kinase activity of the mutants, whereas the R853H mutation leveled the proliferation rate of these mutants to wildtype level. In line with this observation, the level of auto-phos-phorylation of PDGFRβ and its target, ERK1/2, was higher in Ba/F3 cells carrying the H657K and D850E mutation (fig. 2B, lanes 5 and 9) in comparison to the wildtype and R853H-carrying versions of the fusion gene (fig. 2B, lanes 1, 3, 7, and 11).
The in vitro responsiveness of Ba/F3 cells trans-duced with wildtype or mutant NDEL1-PDGFRB constructs to different TKIs of type-I (dasatinib
(DAS), midostaurin (MID) and pacritinib (PAC)) and type-II (imatinib (IMA), nilotinib (NIL) and sorafenib (SOR)) was determined by in vitro cell survival (MTT) assays. Oncogene-addicted proliferation of Ba/F3 cells carrying NDEL1-PDGFRBD850E could only be inhibited by the type-I TKIs dasatinib, midostau-rin, and pacritinib at sub-micromolar concentrations, in line with predictions by the protein model.
DIFFERENTIAL EFFECTS OF CORRESPONDING MUTATIONS IN PDGFRβ AND PDGFRαThe intriguing observation of TKI resistance appar-ently induced by the D850E mutation in the kinase domain of PDGFRβ, which was in contrast to the same amino acid exchange at the corresponding site in PDGFRα (D842E), raised questions regarding important structural differences between the two highly homologous RTKs. While PDGFRβ displays an arginine in the +3 position to the mutation site (R853), PDGFRα has the much shorter and less basic histidine in the corresponding position (H845) (fig.1H). It appeared conceivable therefore that inter-action between the side chains of R853 and E946 in the mutant PDGFRβ TKD could stabilize the active conformation in the mutants H657K and D850E, thus mediating resistance to type-II TKIs. To address this notion, we introduced a mutation into NDEL1-PDG-FRB constructs replacing arginine at position 853 by histidine (R853H), thus mimicking the sequence of the activation loop in the PDGFRα TKD (fig. 1G). This change reduced the NDEL1-PDGFRB-driven proliferation of Ba/F3 cells, and restored the sensitiv-ity of cells carrying the H657K and D850E mutant of NDEL1-PDGFRβ to type-II TKIs, in line with the prop-erties of the D842E mutant of the FIP1L1-PDGFRα fusion (fig.2A). The restored sensitivity to nilotinib was also confirmed by western blot analysis (fig. 2B).
Our data provide first evidence for the occurrence of a point mutation in the activation loop of PDGFRB mediating resistance to type-II TKIs, and for a major alteration of responsiveness to TKI treatment mediated by an exchange between two negatively charged amino acids in a tyrosine kinase. The protein model indicated sensitivity of cells carry-ing the mutant NDEL1-PDGFRB to type-I TKIs, which was confirmed by sensitivity testing in vitro. Availability of the model at the time of relapse after failure of imatinib or nilotinib could have assisted in selecting potentially effective treatment options. The observations therefore provide new insights into specific amino acid interactions in mutant RTKs which are of clinical relevance for improved selection of appropriate TKI treatment (Preuner S et al. Int J Mol Sci 2016). In addition, it provides a new molec-ular insight into activation of PDGFRβ which could serve for de novo design of specific inhibitors of this versatile RTK.
Byrgazov K, Lucini CB, Berkowitsch B, Koenig M, Haas OA, Hoermann G, Valent P, Lion T. (2016). Transposon-mediated generation of BCR-ABL1-expressing transgenic cell lines for unbiased sensitivity testing of tyrosine kinase inhibitors. Oncotarget, 7: 78083-78094
Preuner S, Barna A, Frommlet F, Czurda S, Konstantin B, Alikian M, Machova Polakova K, Sacha T, Richter J, Lion T (corresp.author), Gabriel C. (2016). Quantitative Analysis of Mutant Subclones in Chronic Myeloid Leukemia: Comparison of Different Methodo-logical Approaches. Int J Mol Sci, 17: 642
Byrgazov K, Kastner R, Gorna M, Hoermann G, Koenig M, Lucini CB, Ulreich R, Benesch M, Strenger V, Lackner H, Schwinger W, Sovinz P, Haas OA, van den Heuvel-Eibrink M, Niemeyer CM, Hantschel O, Valent P, Superti-Furga G, Urban C, Dworzak MN, Lion T. (2017, Epub 2016 Oct 7). NDEL1-PDGFRB fusion gene in a myeloid malignancy with eosinophilia associated with resistance to tyrosine kinase inhibitors. Leukemia, 31: 237-240
Figure 2 Figure 2. (A) Displayed are IC50 values of different TKIs against Ba/F3 cells expressing wildtype (wt) or mutant NDEL1-PDGFR? fusion proteins. The corres-ponding IC50 values for Ba/F3 expressing FIP1L1-PDGFR? WT and D842E are given for compa-rison. (B) Western blot analysis of Ba/F3 cells transduced with wildtype or mutant (R = R853H, H = H657K, HR = H657K/R853H, D = D850E, and DR = D850E/R853H) NDEL1-PDGFRB genes. The phosphorylation levels of NDEL1-PDGFR? at Y751 and Y857, and Erk are displayed. Shown are also the total expression levels of NDEL1-PDGFR?, Erk, and the control gene Gapdh upon mock treat-ment with DMSO (indicated by “-“) or with 100 nM of nilotinib (indicated by “+”) for 4 h.
A
B
Science Reports
5756
HIGH RESOLUTION GENOMIC AND TRANSCRIPTOMIC PROFILING OF PEDIATRIC B-CELL PRECURSOR ACUTE LYMPHOBLASTIC LEUKEMIA: IMPLICATIONS FOR EMERGENCE OF RESISTANCE AND RELAPSEAcute lymphoblastic leukemia (ALL) is the most frequent malignancy in childhood and adolescence. ALL comprises various disease entities character-ized by chromosomal translocations, aneuploidy, structural variants, and sequence mutations that interfere with critical cellular pathways affecting lymphoid development, tumor suppression, cell cycle control, homing, as well as kinase and cytokine receptor signaling. With contemporary treatment protocols, up to 90% of the patients remain in long-term remission. Still, relapses are one of the leading causes of death in children and young people.
The goal of our basic and translational research is, therefore, to explore genomic and transcriptomic alterations in specific ALL subgroups to better understand the evolution of leukemia, the emer-gence of relapse, and the nature of the resistant clone. Thereby, not only new insight into the mech-anisms of relapse development is gained, but also the biological impact of genomic alterations, their potential role in resistance mechanisms, as well as their applicability as biomarkers and drug targets can be inferred.
IMPLICATIONS OF GLUCOCORTICOID SIGNALING ALTERATIONS IN CHILDREN WITH REL APSED ETV6/RUNX1-POSITIVE LEUKEMIAThe ETV6/RUNX1 (E/R) gene fusion is the genetic hallmark of the largest subgroup of childhood B cell precursor acute lymphoblastic leukemia, which also has the overall most favorable prognostic outlook. Yet despite its low risk features and rapid response to current treatment regimens, up to 15% of cases still relapse. These disease recurrences are more difficult to treat and therefore also responsible for a dismal outcome in a considerable proportion of affected children.
Previous studies from others and our group have shown that deletions of genes involved in the glucocorticoid (GC) mediated signaling pathway prevail at relapse (Kuster et al). This finding was therefore taken as an indication that these altera-tions could render the affected cells resistant to GC – an integral component of all major childhood ALL treatment protocols – and would then constitute a significant precondition for disease recurrence.
Frequency of genetic deletions in E/R-positive relapses Similar to previous studies, we classified deletions into those that affect genes involved in the GC sign-aling pathway, B cell development and cell cycle. The overall incidence of deletions in the GC signa-ling gene components BTG1, NR3C1, NR3C2, BMF, MSH1 and MSH6 was, with 58% (18/31 cases), simi-lar to the one in our previous report (Kuster et al.). The most common of all remaining recurrent dele-tions concerned the tumor-suppressor gene ETV6 (61%), followed by BCL2L14, a gene that encodes a mainly pro-apoptotic BH3-only family member, and CDKN1B, which generates the cyclin kinase inhib-itor p27. Since both latter genes flank ETV6, they were co-deleted in 38% and 35% cases, respectively. Other common deletions affected genes that encode cell cycle regulators, such as CDKN2A, CDKN2B and RB1 in 35%, 29% and 10% cases, respectively.
Association of genetic alterations with clinical characteristics and outcome Of all GC signaling pathway-associated gene deletions only those in NR3C1, which generates the glucocorticoid receptor (GR), were associated with a subsequent relapse (50 vs. 8%, p<0.04) and tended to occur more frequently in cases with a poor MRD response to relapse treatment (25 vs. 7%; p<0.04). ETV6 gene deletions prevailed among MRD poorly
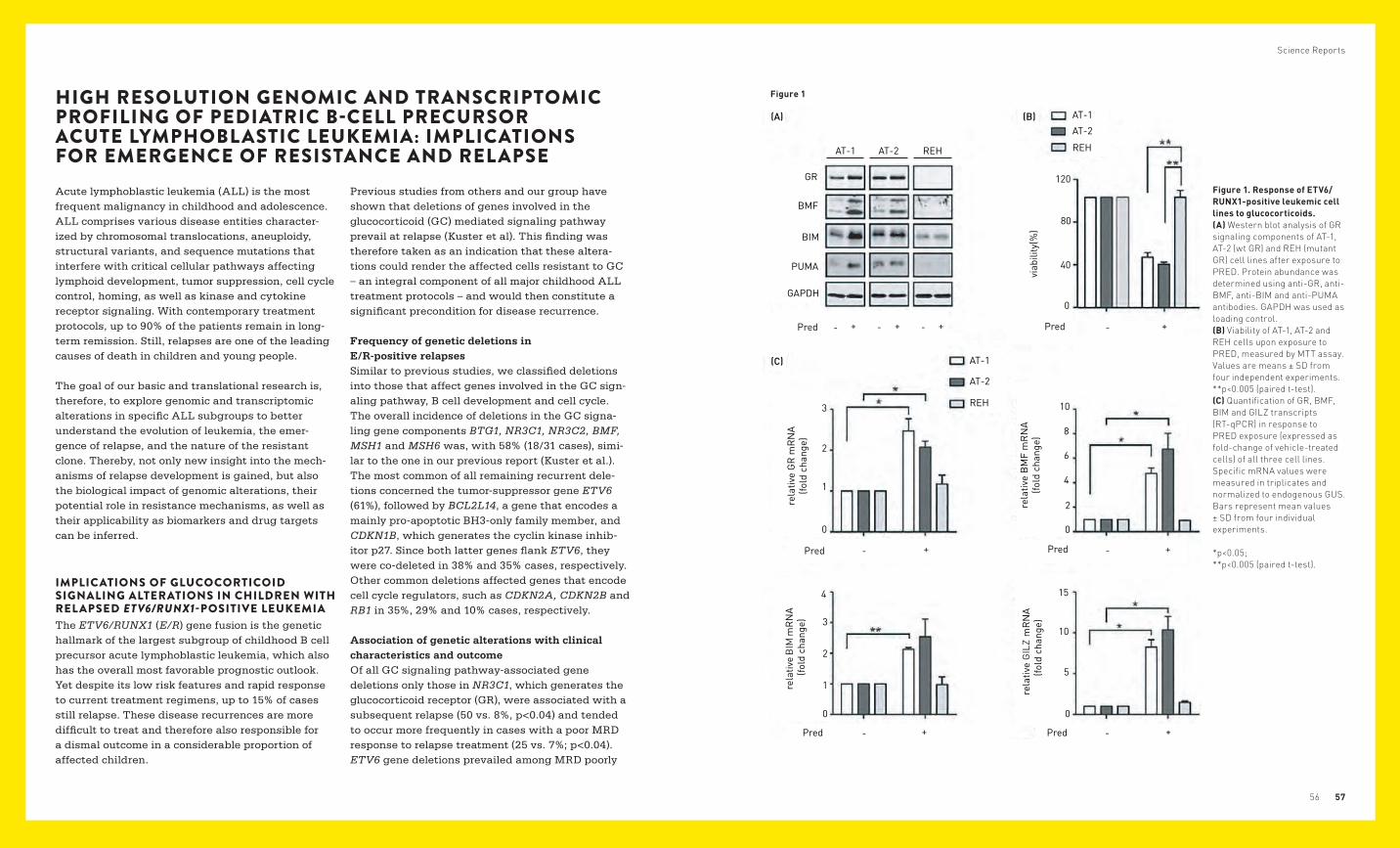
Figure 1. Response of ETV6/RUNX1-positive leukemic cell lines to glucocorticoids. (A) Western blot analysis of GR signaling components of AT-1, AT-2 (wt GR) and REH (mutant GR) cell lines after exposure to PRED. Protein abundance was determined using anti-GR, anti-BMF, anti-BIM and anti-PUMA antibodies. GAPDH was used as loading control. (B) Viability of AT-1, AT-2 and REH cells upon exposure to PRED, measured by MTT assay. Values are means ± SD from four independent experiments. **p<0.005 (paired t-test). (C) Quantification of GR, BMF, BIM and GILZ transcripts (RT-qPCR) in response to PRED exposure (expressed as fold-change of vehicle-treated cells) of all three cell lines. Specific mRNA values were measured in triplicates and normalized to endogenous GUS. Bars represent mean values ± SD from four individual experiments. *p<0.05; **p<0.005 (paired t-test).
Figure 1

Science Reports
5958
responding cases (81 vs. 40%, p<0.05). However, since ETV6 deletions are similarly frequent at diag-nosis and at relapse and the gene is not expressed in the remaining, non-deleted cases, it is difficult to imagine that they can play a major role in the development of drug resistance. The concomitant deletions of the BCL2L14 and/or CDKN1B genes are in fact much better candidates, because even a partial loss of their function might affect apoptosis and drug response in a substantial way.
The glucocorticoid receptor determines the response to GC in vitro We used GC-resistant (REH) and GC-sensitive (AT-1, AT-2) E/R-harboring leukemic cell lines to model and assess the consequences of a functional loss of the GR in E/R-positive leukemias. Consistent with the lack of a functional GR, REH cells were resistant to prednisolone (PRED) when exposed to clinically meaningful concentrations, as indicated by their unchanged viability as well as the inability to induce the GR downstream targets BCL2 modifying factor (BMF), the BCL2-like gene BIM, glucocorti-coid-induced leucine zipper (GILZ) and BCL2 binding component 3 (PUMA) at the transcript and the protein levels [Fig. 1A–C]. By contrast, both GC- sen-sitive cell lines showed a reduced viability upon exposure to the same PRED concentration and the concomitant up-regulation of GR, as well as of the downstream targets at both transcript and protein levels [Fig. 1. A–C].
Overall, our findings, together with previous ones, corroborate the notion that the functional impair-ment of many GC signaling pathway elements is involved in the emergence of GC-resistant E/R-pos-itive cell populations. Many of the affected genes are not only engaged in specific GC signaling alone but also in a variety of other pathways that are,
for instance, coordinating the cell cycle and cell survival. GC resistance at relapse can therefore not be viewed in isolation but must always be seen as part of a more global system of drug resistance. In such a context, a distinct form of GC resistance will lose its relevance as soon as effective drugs, such as obatoclax, become available for clinical use.
Grausenburger R, Bastelberger S, Eckert C, Kauer M, Stanulla M, Frech C, Bauer E, Stoiber D, von Stackelberg A, Attarbaschi A, Haas OA, Panzer-Grümayer R. (2016). Genetic alterations in glucocorticoid signaling pathway components are associated with adverse prognosis in children with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia. Leuk Lymphoma, 57: 1163-1173
GENOMIC AND TRANSCRIPTIONAL L ANDSCAPE OF P2RY8-CRLF2-POSITIVE CHILDHOOD ACUTE LYMPHOBL ASTIC LEUKEMIARecently, a novel subgroup of childhood ALL has been described whose defining characteristic is the deregulated expression of the cytokine receptor- like factor 2 (CRLF2) gene, which is located in the pseudoautosomal region 1 on the short arm of the X and Y chromosome. The two most common causative genetic defects are a small interstitial deletion that fuses the first non-coding exon P2RY8 to the entire coding region of CRLF2 and occurs in 5–8% of childhood B-cell precursor (BCP) ALL cases and a more rare translocation that places CRLF2 under the control of the IGH enhancer. P2RY8-CRLF2 fusion harboring leukemias often carry additional alterations in JAK/STAT path-way genes and they may cooperatively activate downstream pathways. They are associated with a significantly increased relapse risk in AIEOP/BFM protocols, which is independent of the size of the P2RY8-CRLF2-positive clone (Morak et al.). Respective cases are primarily classified as non-high risk by clinical and molecular response criteria and relapses occur predominantly late.
IKZF1 encodes the lymphoid transcription factor IKAROS, which is a key regulator in early lympho-cyte development and prevails in poorly respond-ing cases in major treatment protocols. IKZF1 deletions were also reported in small cohorts of P2RY8-CRLF2-positive leukemia cases recruited to various treatment protocols. It seems, therefore, likely that their presence contributes to relapse development in this particular subgroup, a notion that has, so far, not been systematically investigated in large and well-characterized cohorts. Therefore, we performed whole exome sequencing (WES) and transcriptional profiling (RNA-seg) in 41 relapsing and non-relapsing major clone P2RY8-CRLF2-posi-tive cases that were treated primarily according to BFM protocols.
Clonal heterogeneity and instability of kinase-activating pathway alterations At initial diagnosis, the overall frequency of JAK/STAT pathway mutations was 51% and equally distributed between relapsing and non-relapsing cases. They were subclonal in 47% of cases. Irre-spective of their original size, JAK/STAT pathway mutation carrying clones were lost at relapse in 60% of the cases (Figure 2A). Mutations in RTK/Ras pathway genes were found in 29% of the cases at diagnosis, lost in almost half of the cases, and increased to 68% in relapses (P = 0.06). P2RY8-CRLF2 was also lost in 32% of relapses.

Science Reports
6160
INITIAL LEUKEMIA RELAPSE
Normal cellFounder PAX5PAR1CDKN2A/BIKZF1JAK/STATRAS
IKZF1 alterations predict relapse in P2RY8-CRLF2-positive ALL and display distinctive transcriptional signaturesExcept for IKZF1, the frequency of alterations in genes implicated in lymphoid development and in tumor suppression/cell cycle regulation did not significantly differ between relapsing and non-re-lapsing cases. IKZF1 alterations prevailed in the relapsing cohort with a frequency of 41% versus 10% in non-relapsing cases (P = 0.001).
Therefore, we profiled 22 leukemia samples by RNA-Seq according to their IKZF1 status. Tran-scriptional profiling revealed specific regulations in the IK6 deletions and biallelic alterations group and, albeit to a much lesser degree, in the group with larger deletions compared with the wtIKZF1 one (Figure 3). Gene set enrichment analysis revealed a highly significant correlation of differentially expressed genes with various human hematopoie-tic and lymphoid stem cells sets, and concordantly for gene sets specifically expressed in immature B cells. This suggests that IKZF1 alterations lead to impaired B-cell differentiation and the acquisition of stem cell-like features. We also found enrichment in gene sets that are upregulated in the context of microenvironment, focal adhesion kinase and inte-grin pathways, as well as of genes that are higher expressed in response to hypoxia, downstream VEGF/VEGFR signaling and to EPO signaling.
IKZF1 alterations are associated with dismal outcome Except for the genomic IKZF1 status and white blood cell count, which were not associated with each other (P = 0.66), we found no other biological or clinical parameters such as other genetic alter-ations, age at diagnosis, clinical risk group assign-ment or morphological and molecular response to treatment to be correlated with the occurrence of relapses. There was no difference between Down Syndrome (DS) and non-DS cases. Of note, only one of eight IKZF1-altered relapsing cases initially had a poor MRD response and was, therefore, assigned to high-risk treatment. Yet, IKZF1-mutated cases had a significantly poorer outcome than their IKZF1 wild-type counterparts as evidenced by an adverse pEFS (P = 0.026) and pOS (P = 0.051).
The P2RY8-CRLF2 fusion was lost in one-third of the relapses. Together with previous observations – that, for instance, these fusions frequently affect subclones that never evolve into major relapse clones – this indicates that it is primarily a second-ary change that may potentially supply the respec-tive cells with a certain proliferative but certainly not with an evolutionary advantage ( Morak et al.). In line with other types of childhood BCP ALL, B-cell differentiation genes are also commonly deleted in cases with a P2RY8-CRLF2 fusion. These abnormalities are usually preserved in the corre-sponding relapses. Consistent with their role in drug resistance, IKZF1 alterations prevail already in relapse-prone cases at diagnosis but become even more abundant in relapses. IKZF1- deleted cases also seem to profit from other associated B-cell development and cell cycle gene defects, particu-larly those of PAX5 and/or CDKN2A/B, which are found in half of the cases, but also from mutations in specific proliferation-promoting pathway genes.
Figure 2
A
B
Figure 2. Clonal composi-tion and stability of genomic alterations. (A) Clonal composition of JAK/STAT (blue) and RTK/Ras pathway (red) signaling gene mutations according to individual genes (symbol code). Dots represent the adjusted allelic frequency (adj. AF) of mutations. Black dots mark conserved mutations at diagnosis and relapse. (B) Model for the evolution of leukemia and selection of relapse clones. Leukemia-in-itiating (founder) alterations occur in a hematopoietic stem/progenitor cell, while the ensuing RAG-mediated micro-deletions evolve later during early B cell differentiation. Microdeletions affect genes critical for B cell differentia-tion and tumor suppression (color code at the bottom of the graph). JAK/STAT or RTK/Ras pathway activating alterations continuously emerge but do not outcompete each other at initial presentation of leukemia. Chemotherapy then selects for resistant clones, which represent frequently only minor subclones at initial diagnosis, but they may vary regarding their proliferation driving mutation.

Science Reports
6362
Thus, we consider the P2RY8-CRLF2 fusion as one of the many secondary proliferative driver altera-tions that - in line with those activating JAK/STAT and RTK/Ras pathways – are highly instable at relapse, albeit to a lesser degree if initially present in the major clone, and not as a bona fide primary genetic alteration that is always stable at relapse and critical for the maintenance of the leukemia.
Taken together and as schematically depicted in Figure 2B, we envision the following scenario of relapse evolution in P2RY8-CRLF2-positive leuke-mias. Predisposing constitutional or acquired genomic alterations – as those affecting chromo-some 21 – facilitate the expansion of a pre/leukemic B cell precursor clone. Its regular development is
then obstructed by defects in genes whose products are required for normal B cell differentiation. Cells that carry specific combinations of abnormalities may gain a competitive and selective advantage and eventually predominate in the pre-leukemic cell population. Parallel to these alterations and increas-ingly during later stages of leukemia evolution, mutations in specific genes activate signaling path-ways that enable their unrestrained proliferation.
Figure 3. Transcriptional signature of leukemias accor-ding to IKZF1 status. Cluster heatmap of the top 50 up- and downregulated genes in both IKZF1-altered groups („IKN“ denotes IKZF1 altera-tions leading to a dominant-ne-gative effect and biallelic alterations and „IKD“ designa-tes IKZF1 alterations resulting in haploinsufficiency) according to fold-change (P≤1E−8 for IKN versus IKZF1 wt (IKC), P≤2E−3 for IKD versus IKC); IKN cases are indicated in red, IKD ones in blue and IKC in gray at the top of the map.
Since chemotherapy primarily eliminates the bulk of rapidly proliferating cells, it spares those less active resistant stem-cell-like ones that eventually gener-ate relapses. In this scenario, the P2RY8-CRLF2 fusion is only one of several proliferation activating alterations that merely serves as a common marker for an otherwise genetically heterogeneous group, whose other and probably more relevant features are IKZF1 alterations. These alterations are well-known disease drivers in many types of drug-resistant leukemias, such as BCR/ABL1-positive ones, which all share a similar gene expression signature. The transcriptional profile of IKZF1-altered cases reflects their strong homing preference to the bone marrow niche as well as their high repopulation capacity, attributes that also become apparent in mouse models in which IKZF1-/- pre-B cells acquire stem cell and adhesion properties including activation of the focal adhesion kinase pathway.
Besides these biological insights, our findings also provide some clues that may become relevant in future treatment decisions. Apart from their prog-nostic implications, IKZF1 alterations may even-tually serve as markers for specific therapeutic interventions. It appears reasonable to try to restore IKAROS signaling especially in those IKZF1-altered cases that still have retained a functional wild-type allele. For the other 20% of cases with biallelic IKZF1 alterations, inhibition of the activated focal adhesion kinase pathway may become a viable treatment option. Such approaches might perhaps be combined with a cocktail of other signaling inhibitors given the availability of various JAK/STAT, Ras/MEK/ERK and PI3K/mTOR pathway inhibitors.
Figure 3
Vesely C, Frech C, Eckert C, Cario G, Mecklenbrauker A, zur Stadt U, Nebral K, Kraler F, Fischer S, Attarbaschi A, Schuster M, Bock C, Cavé H, von Stackelberg A, Schrappe M, Horstmann MA, Mann G, Haas OA, Panzer-Grümayer R. (2017). Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia, Epub 2017 Jan 6

6564
Science Reports
NEW INSIGHTS IN NEUROBLASTOMA BIOLOGY – FROM SPONTANEOUS MATURATION TO THE RELAPSE SEEDING CLONE
Neuroblastoma is the most common extracranial solid tumor in childhood and accounts for around 15% of all pediatric oncology deaths. This embryonal tumor arises from sympathetic neuronal precursor cells and shows a unique biological and clinical spectrum, encompassing spontaneous regression, spontaneous maturation, or malignant progression. While most patients whose tumors undergo sponta-neous regression or maturation (ganglioneuroblasto-mas, ganglioneuromas) have an excellent outcome, not all children with aggressive tumors can be cured (Figure 1). Thus, a better understanding of the biol-ogy of both tumor types, spontaneously regressing/maturing and aggressive ones is of high interest to develop novel treatment approaches. Thus, our recent research has focused on two distinct aspects of neuroblastoma biology – spontaneous maturation and relapse formation.
MEDIATORS OF SPONTANEOUS MATURATION IN NEUROBL ASTOMANeuroblastomas show a so far unique feature in oncology research, i.e., their ability to mature spon-taneously into a benign tumor. Maturing neuroblas-tomas are composed of two cell populations 1) tumor cells differentiating into a mitotically quiescent, benign state and 2) Schwann cells forming a dense tumor stroma. Already in 1996, we have shown that these stromal Schwann cells are not of tumor origin and postulated a prominent role of Schwann cells in the maturation process of this favorable neuroblastoma subtype (Ambros et al. 1996. NEJM). Schwann cells are known to closely interact with neurons regulating axon integrity in the adult as well as axon differentiation during development. Furthermore, it was demonstrated that Schwann cells are able to transform into dedicated repair cells with distinct functions essential to promote
axonal re-growth after nerve injury. Based on these findings, we hypothesized that Schwann cells exert their physiological functions, i.e., the regulation of neuronal differentiation during development and nerve regeneration, on neuroblastoma cells facilitat-ing spontaneous tumor maturation in vivo. Subse-quent studies further demonstrated that Schwann cells are also able to impair the growth of aggressive neuroblastoma cell lines, obtained from tumors lacking a spontaneous maturation capacity in vitro, indicating a therapeutic potential of Schwann cells or the factors they express.
To gain insights into the role of Schwann cells in the neuroblastoma maturation process, we tested our hypothesis on the similarities of physiological interactions between nerve regeneration and neuro-blastoma maturation. In a first step, we analyzed the proteome and transcriptome and cellular processes active in repair Schwann cells after nerve injury using high-resolution mass spectrometry and RNA-sequencing of highly enriched human Schwann cells and injured nerve tissue (Figure 2). Our results revealed that cultured Schwann cells and injured nerves share a similar repair Schwann cell-associ-ated expression signature including previously not described molecules involved in axonal differenti-ation and two novel repair Schwann cell functions, i.e., debris clearance via phagocytosis and a type II immune-regulation (Weiss et al. 2016). These find-ings extended the functional spectrum of Schwann cells in regenerative processes after nerve injury and strengthened our hypothesis on their vital role during neuroblastoma maturation. Hence, two-fol-low up projects now explore the signaling events between Schwann cells and neuroblastoma and/or immune cells in detail. Our preliminary results using a FACS based read-out system and immunofluores-cence, demonstrated growth-inhibitory, differenti-
neuroblastoma
blood vessel
SCs in periphery
MSCs SC progenitors
fully matures ganglioneuroma
ganglioniccells
time
benignly behaving
aggressive
unfavorable genetics
any ploidy
±MNA
SCAs
mutations
favorable genetics
aneuploidy
no MNA
mostly no SCAs
Schwann cell recruitment
differentiatingneurblastoma
un- or poorlydifferentiated
neuroblastoma
progressingneuroblastoma
SC stroma
spontaneous maturation
malignant progression
Figure 1. Clinical and biological heterogeneity of neuroblastoma.
Figure 2. Schwann cells in spontaneous neuroblastoma maturation. Schwann cells adopt new functions upon nerve repair and as stromal cells in the tumor microenviron-ment. High-resolution mass spectrometry (proteomics) and functional tests revealed novel neuritogenic/neurotro-phic factors, phagocytosis and MHCII up-regulation.
Nerve (control)
3 (8%)
2 (16%)
HLA-ABC HLA-DRα1
SCs
SC
NB
EGFL8FLRT3GFRA1MDGA1NRP2SEM3BSHOT1TEN3
FBs coun
ts
NB
cel
l/p1
SC
co-c
ultu
re
1 (52%)
Nerve (injured)Schwann cellFibroblast
MHCII upregulation
Neurotrophic/NeuritogenichrMS Proteomics
Phagocytosis
Figure 1
Figure 2

Science Reports
6766
ation- and apoptosis-inducing effects of Schwann cells on co-cultivated aggressive neuroblastoma cells. Secretome analysis and RNA-sequencing of repair- and tumor-associated Schwann cells, neuro-blastoma cell lines and primary tumors will aid to identify the signaling factors and receptors partic-ipating in this cross-talk. All in all, understanding the Schwann cell biology in neuronal differentiation is of utmost interest for new approaches in regenera-tive medicine as well as neuroblastoma therapy.
TUMOR HETEROGENEIT Y AND IDENTIFICATION OF THE REL APSE SEEDING CLONE IN METASTATIC NEUROBL ASTOMAAlthough there has been substantial improvement in the outcome of patients with certain subsets of neuroblastoma, long-term survival of high-risk neuroblastoma patients is still less than 40% and effective relapse therapies are lacking. To improve the outcome of these patients, efforts are currently focusing on understanding fundamental genomic alterations driving neuroblastoma progression, therapy resistance and relapse development. Tumor relapse in patients with metastatic disease (stage M) is the main cause of mortality in these patients. Therefore, early and reliable detection and charac-terization of the relapse-seeding clone/s will help to monitor disease and to choose an appropriate treat-ment. However, due to intra-tumor heterogeneity, tumor biopsies may fail to identify the most aggres-sive tumor cell clone(s). In order to identify the most appropriate tissue for detecting the relapse-seeding clones, we recently studied the genomic evolution of neuroblastoma tumors and bone marrow-derived disseminated tumor cells by analyzing geographi-cally and temporally separated samples of stage M neuroblastoma patients (Abbasi et al. 2017. Clin. Cancer Research). In a single, well-characterized
stage M patient, we found a unique aberration, deletion 1q, in the relapse samples besides a high number of concordant genomic aberrations present in all analyzed samples. Interestingly, this aberra-tion was present in the disseminated tumor cells at diagnosis but not in the primary tumor, despite analyzing seven different tumor pieces (Figure 3). In a cohort of 154 patients, the highest incidence of this aberration was found in relapse samples and occurred in DTCs at diagnosis nearly twice as frequently as compared to primary tumors. Our results indicate that analysis of bone marrow-de-rived disseminated tumor cells at diagnosis besides the tumor biopsies may increase the probability for detecting the relapse-seeding clone and thus allow a more precise diagnosis of the most aggressive ‘drivers’ of tumor progression.
Weiss T*, Taschner-Mandl S*, Bileck A, Slany A, Kromp F, Rifat begovic F, Frech C, Windhager R, Kitzinger H, Tzou CH and others. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Gila. 2016 Dec; 64(12):2133-2153. doi: 10.1002/glia.23045.
* Contributed equaly
Figure 3. Tumor evolution in neuroblastoma. Graphical representation of clonal expan-sion in a stage 4 neuroblastoma patient. Each quadrant repre-sents the clonal architecture of a tissue/time point. Each color represents a group of chromo-somal aberrations and the size of each colored area represents the pro portion of cells with these aberrations within the analyzed samples. A 1q terminal deletion (W) which was present in the diagnostic DTCs and also in both, DTCs and metastatic tumor, at relapse was not found in seven pieces of the primary tumor.
Figure 3
Tumor at diagnosis
DTCs at diagnosis DTCs at relapse
Tumor at relapse
B
A
Fibroblasts +/- HCMV-infection
293T cells +/- gB-transfection
isotype
Crough et al. 2011
gB
CAR
gB
HCMV-infected cell
MembranegB
gH
NucleocapsidTegument
Crough et al. 2011
gB
CAR
gB
HCMV-infected cell
MembranegB
gH
NucleocapsidTegument
Cytomegalovirus T cell
Science Reports
6968
Figure 1. Schematics of Cytomegalovirus (A) and of the CAR-T cell approach (B).
Figure 2. CAR-T cells do not lyse HCMV-infected cells. (A) The histograms show the expression of gB in fibroblasts (HFF, infected or non-infected) and in 293T cells transduced or non-transduced with a gB-en-coding vector. (B) Shown is the lytic activity of αCD3/αCD28-expanded T cells transfected with either the gB-specific CAR or an irrelevant CAR specific for the carcinoembroynic antigen (CEA) by electroporation of CAR-encoding mRNA. Lytic activity of these CAR-T cells (effector:target ratio 25:1) was determined one day after elec-troporation using HFF (non-in-fected or 4 days after infection with AD169, MOI 5) and 293T cells transduced or non-trans-duced with gB (filled symbols: infected HFF or gB-transduced 293T cells; empty symbols: non-infected HFF or non-trans-duced 293T; three donors).
Figure 1
A BCHIMERIC ANTIGEN RECEPTOR (CAR)-BASED IMMUNOTHERAPY FOR TREATMENT OF REACTIVATION OF CYTOMEGALOVIRUS INFECTION AFTER STEM CELL TRANSPLANTATIONReactivation of cytomegalovirus (HCMV) infection after hematopoietic stem cell transplantation is still associated with substantial morbidity and mortal-ity. Prophylactically and preemptively adminis-tered antiviral chemotherapy after transplantation frequently results in toxicity and selection of resist-ant virus variants. Adoptive transfer of HCMV-spe-cific memory T cells has been successfully applied, however, generation of specific T cells from seron-egative donors is difficult. We hypothesized that HCMV could be targeted by adoptive transfer of CAR-T cells in an HLA-independent manner, because HCMV-infected cells display intact viral proteins such as glycoprotein B (gB) on their surface.
HCMV is a complex virus with possibly 750 trans-lational products encoded by its genome. Since many of these products are highly immunogenic, HCMV triggers immune responses from all arms of the immune system and leads to a high frequency of responding HCMV-specific CD8pos and CD4pos T cells (up to 40% of the whole T cell repertoire). Despite this strong T cell reaction HCMV persists and establishes lifelong latency, possibly explained by the fact that a large proportion of its genome encodes for RNAs and proteins interfering with the antiviral immune response. One of the most import and most intensively studied defense mechanism of HCMV is the prevention of recognition of infected cells by T and NK cells and by antibodies. Like many other viruses, HCMV escapes recognition by T cells through interfering with antigen processing and inhibiting antigen presentation by MHC class I and II molecules on the surface of infected cells.
RESISTANCE OF HCMV-INFECTED CELLS TO T CELL CY TOTOXICIT Y DESPITE HL A-INDEPENDENT TARGETING
In collaboration with our partner Armin Ensser (Institute of Virology, Universitätsklinikum Erlan-gen, Germany) we have thus previously constructed a CAR for targeting a conserved region in gB (Full F et al. 2010), which is abundantly expressed on the surface of HCMV-infected cells and conserved among different viral strains. Targeting of HCMV-in-fected cells by CAR expressing T cells in an HLA-in-dependent manner is attractive, because it obviates the need for enriching antigen-specific memory T cells and circumvents immune evasion by impaired antigen presentation. Originally, this approach has been proposed for the treatment of HIV and was tested in clinical phase II trials finally. Recent appli-cations of a CAR-T cell approach also for fighting Hepatitis B and C have shown promising results both in vitro and in vivo in a preclinical model.
Interestingly and unexpectedly, however, when we tested polyclonally activated T cells expressing the gB-specific CAR, we found that these T cells could not directly eliminate cells infected with HCMV (Figure 2). We have observed this with two well-characterized HCMV laboratory strains Ad169 and Towne and also after prolonged co-culture of infected and effector cells. In order to exclude that the observed lack of lysis of HCMV-infected fibro-blasts was due to inhibition of T cell activation, we determined the level of T cell degranulation and cytokine production. However, we clearly found that the CAR triggers degranulation as well as release of cytokines IFN-g and TNF in the T cells. In order to further exclude that any possible defective function of our gB-specific CAR was responsible for the observed lack of lysis, we switched to a
A
B
Figure 2
non-infected HFF
coun
tssp
ecifi
c ly
sis
[%]
gB102
0 00
50
50
50
100
100
100
150
150
150
250
250
350
450
40025
0
200
200
200
300
300
102 102 102103 103 103 103
specific antibody
HCMV-infected HFF 293T
isotype
gB-CAR
80 80
70 70
60 60
50 50
40 40
30 30
20 20
10 10
-10 -10
0 0
gB-CAR
Fibroblasts +/- HCMV-infection
293T cells +/- gB-transfection
CEA-CAR CEA-CAR
293T-gB
105 105 105 105104 104 104 104
050
100
150
250
350
200
300
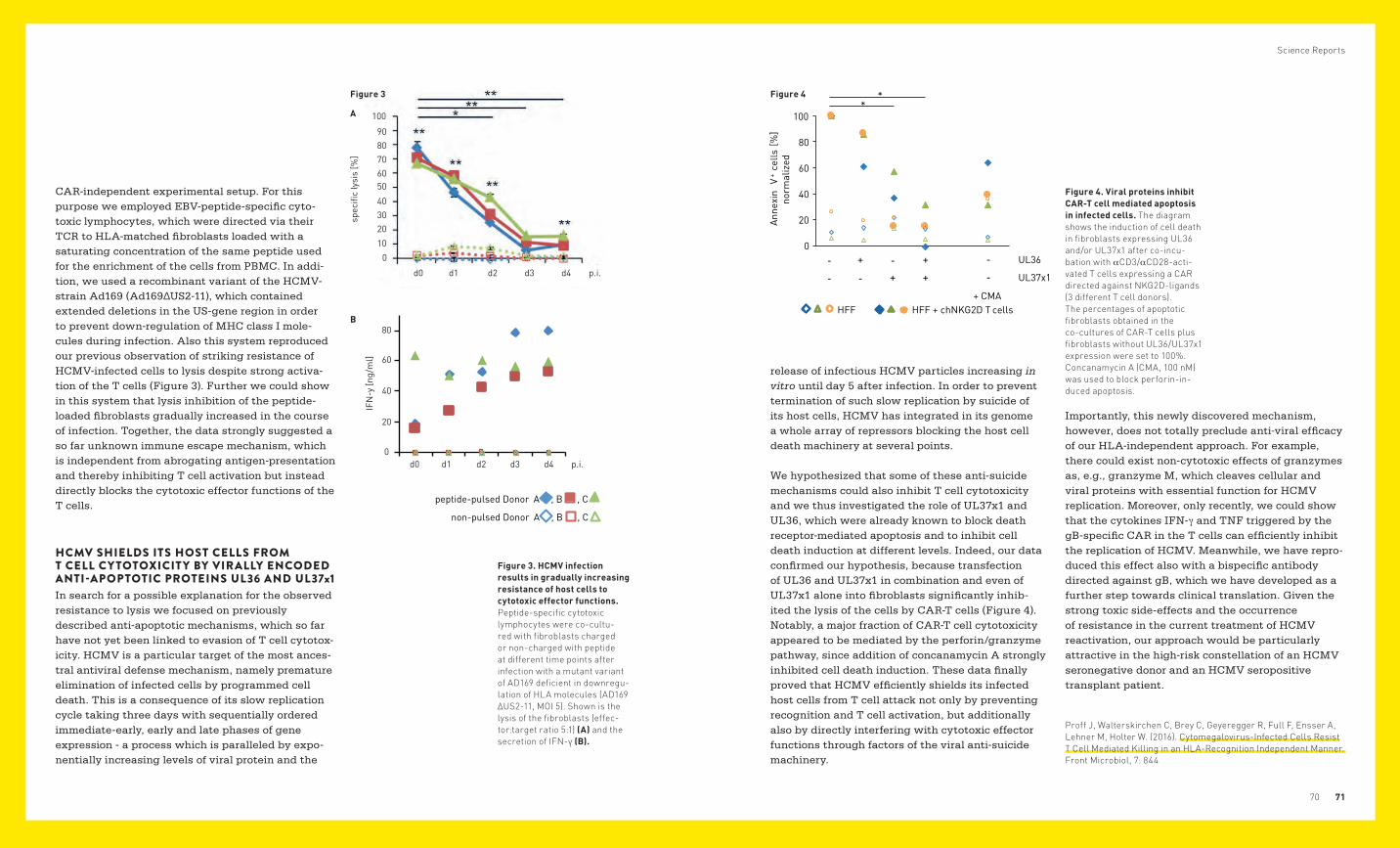
B
A
%IF
N-y
ng/
ml
d0 d1 d2 d3 d4 p.i.
d0 d1 d2 d3 d4 p.i.
0
0
20
20
40
40
60
60
80
80 70
50
30
10
90 100
peptide-pulsed Donor A , B , C
non-pulsed Donor A , B , C
B
A
%IF
N-y
ng/
ml
d0 d1 d2 d3 d4 p.i.
d0 d1 d2 d3 d4 p.i.
0
0
20
20
40
40
60
60
80
80 70
50
30
10
90 100
peptide-pulsed Donor A , B , C
non-pulsed Donor A , B , C
HFF HFF + chNKG2D T cells
Anne
xin
V+
cells
[%]
norm
aliz
ed
**
-
-
+
-
-
+
+
+
UL36
UL37x1
+ CMA
-
-
0
20
40
60
80
100
Science Reports
7170
release of infectious HCMV particles increasing in vitro until day 5 after infection. In order to prevent termination of such slow replication by suicide of its host cells, HCMV has integrated in its genome a whole array of repressors blocking the host cell death machinery at several points.
We hypothesized that some of these anti-suicide mechanisms could also inhibit T cell cytotoxicity and we thus investigated the role of UL37x1 and UL36, which were already known to block death receptor-mediated apoptosis and to inhibit cell death induction at different levels. Indeed, our data confirmed our hypothesis, because transfection of UL36 and UL37x1 in combination and even of UL37x1 alone into fibroblasts significantly inhib-ited the lysis of the cells by CAR-T cells (Figure 4). Notably, a major fraction of CAR-T cell cytotoxicity appeared to be mediated by the perforin/granzyme pathway, since addition of concanamycin A strongly inhibited cell death induction. These data finally proved that HCMV efficiently shields its infected host cells from T cell attack not only by preventing recognition and T cell activation, but additionally also by directly interfering with cytotoxic effector functions through factors of the viral anti-suicide machinery.
CAR-independent experimental setup. For this purpose we employed EBV-peptide-specific cyto-toxic lymphocytes, which were directed via their TCR to HLA-matched fibroblasts loaded with a saturating concentration of the same peptide used for the enrichment of the cells from PBMC. In addi-tion, we used a recombinant variant of the HCMV-strain Ad169 (Ad169∆US2-11), which contained extended deletions in the US-gene region in order to prevent down-regulation of MHC class I mole-cules during infection. Also this system reproduced our previous observation of striking resistance of HCMV-infected cells to lysis despite strong activa-tion of the T cells (Figure 3). Further we could show in this system that lysis inhibition of the peptide-loaded fibroblasts gradually increased in the course of infection. Together, the data strongly suggested a so far unknown immune escape mechanism, which is independent from abrogating antigen-presentation and thereby inhibiting T cell activation but instead directly blocks the cytotoxic effector functions of the T cells.
HCMV SHIELDS ITS HOST CELLS FROM T CELL CY TOTOXICIT Y BY VIRALLY ENCODED ANTI-APOPTOTIC PROTEINS UL36 AND UL37x1In search for a possible explanation for the observed resistance to lysis we focused on previously described anti-apoptotic mechanisms, which so far have not yet been linked to evasion of T cell cytotox-icity. HCMV is a particular target of the most ances-tral antiviral defense mechanism, namely premature elimination of infected cells by programmed cell death. This is a consequence of its slow replication cycle taking three days with sequentially ordered immediate-early, early and late phases of gene expression - a process which is paralleled by expo-nentially increasing levels of viral protein and the
Importantly, this newly discovered mechanism, however, does not totally preclude anti-viral efficacy of our HLA-independent approach. For example, there could exist non-cytotoxic effects of granzymes as, e.g., granzyme M, which cleaves cellular and viral proteins with essential function for HCMV replication. Moreover, only recently, we could show that the cytokines IFN-γ and TNF triggered by the gB-specific CAR in the T cells can efficiently inhibit the replication of HCMV. Meanwhile, we have repro-duced this effect also with a bispecific antibody directed against gB, which we have developed as a further step towards clinical translation. Given the strong toxic side-effects and the occurrence of resistance in the current treatment of HCMV reactivation, our approach would be particularly attractive in the high-risk constellation of an HCMV seronegative donor and an HCMV seropositive transplant patient.
Proff J, Walterskirchen C, Brey C, Geyeregger R, Full F, Ensser A, Lehner M, Holter W. (2016). Cytomegalovirus-Infected Cells Resist T Cell Mediated Killing in an HLA-Recognition Independent Manner. Front Microbiol, 7: 844
Figure 3. HCMV infection results in gradually increasing resistance of host cells to cytotoxic effector functions. Peptide-specific cytotoxic lymphocytes were co-cultu-red with fibroblasts charged or non-charged with peptide at different time points after infection with a mutant variant of AD169 deficient in downregu-lation of HLA molecules (AD169 ΔUS2-11, MOI 5). Shown is the lysis of the fibroblasts (effec-tor:target ratio 5:1) (A) and the secretion of IFN-γ (B).
Figure 4. Viral proteins inhibit CAR-T cell mediated apoptosis in infected cells. The diagram shows the induction of cell death in fibroblasts expressing UL36 and/or UL37x1 after co-incu-bation with αCD3/αCD28-acti-vated T cells expressing a CAR directed against NKG2D-ligands (3 different T cell donors). The percentages of apoptotic fibroblasts obtained in the co-cultures of CAR-T cells plus fibroblasts without UL36/UL37x1 expression were set to 100%. Concanamycin A (CMA, 100 nM) was used to block perforin-in-duced apoptosis.
Figure 3 Figure 4
A
B
0
0
d0 d1 d2 d3 d4 p.i.
20
2010
30
40
40
60
6050
70
80
8090
100
d0 d1 d2 d3 d4 p.i.
spec
ific
lysi
s [%
]
IFN
-y [n
g/m
l]

Science Reports
7372
LARGE SCALE EUROPEAN TRIAL DEMONSTRATES SURVIVAL ADVANTAGE FOR HIGH RISK NEUROBLASTOMA PATIENTS RECEIVING HIGH DOSE BUSULPHAN AND MELPHALAN TREATMENT (A RANDOMISED PHASE 3 TRIAL OF THE SIOP EUROPE NEUROBLASTOMA GROUP (SIOPEN)).
Neuroblastoma, the commonest paediatric extra-cra-nial solid tumour, is responsible for a large propor-tion of deaths from cancer in childhood. High-risk neuroblastoma, defined by metastatic disease over the age of 12 or 18 months and MYCN amplification (MNA) at any age remains associated with poor long-term survival rates. High-dose chemotherapy with haematopoietic stem cell rescue (HDT/SCR) improves event free survival (EFS) of patients with high-risk neuroblastoma (HR-NBL); however which regimen has the greatest patient benefit is under investigation.
The high-risk neuroblastoma trial (HR-NBL1/SIOPEN) opened in 2002 and has tested, a number of hypo theses (Ladenstein et al., J Clin Oncol 2010, 28(21):3516–24 / Ladenstein et al., MAbs. 2013; 5(5): 801–9). Here we report results of the hypothesis that HDT with busulphan and melphalan (BuMel) results in a superior EFS than HDT with carboplatin, etoposide and melphalan (CEM).
METHODSPatients were enrolled and randomised by 128 SIOPEN member institutions/hospitals in 18 countries using the SIOPEN-R-NET web based, online remote data entry system (https://www.siopen-r-net.org/). Randomisation was carried out just prior to HDT by minimisation balancing age at diagnosis, stage, MYCN amplification and national group between the arms. Tumour evaluations were carried out at the time of trial enrolment, after completion of induction, before HDT, following HDT and at the end of therapy.
Eligibility and response was evaluated by 123I-mIBG scintigraphy assessing the primary tumour and metastatic sites. In the case of mIBG nega-tive tumour, bone scintigraphy with 99mTc-hy-droxy-methylenediphosphanate (MDP) scintigraphy was carried out; CTor MRI scan of primary tumour and radiological visualisation of any other evaluable disease; examination of bone marrow aspirates and trephines at two sites and measurement of urinary catecholamines. Patients were evaluated according to the National Cancer Institute Common Toxicity Criteria (CTC, Version 2) and by Bearman toxic-ity grades for pulmonary toxicity, veno-occclusive disease (VOD) and haemorrhagic cystitis. Monitor-ing for adverse event monitoring was continuous.
The trial was approved as required by national regulatory authorities and by national as well as institutional ethical committees/review boards in participating countries. Three year EFS from randomisation was the primary endpoint. Analyses were done by intention to treat, but a per-protocol approach was used for the safety analysis. The trial was registered with ClinicalTrials.gov (number NCT01704716) and EudraCT (number 2006-001489-17).
Trial efficacy results remained masked until release by the independent Data Monitoring and Safety Committee (DMSC) and only the DMSC and the study statistician were aware of interim efficacy monitoring results. At the pre-planned interim analysis of October 2010 the DMSC recommended stopping randomisation as the Peto rule on EFS was met (p-value <0.001 on the primary endpoint).
Figure 1
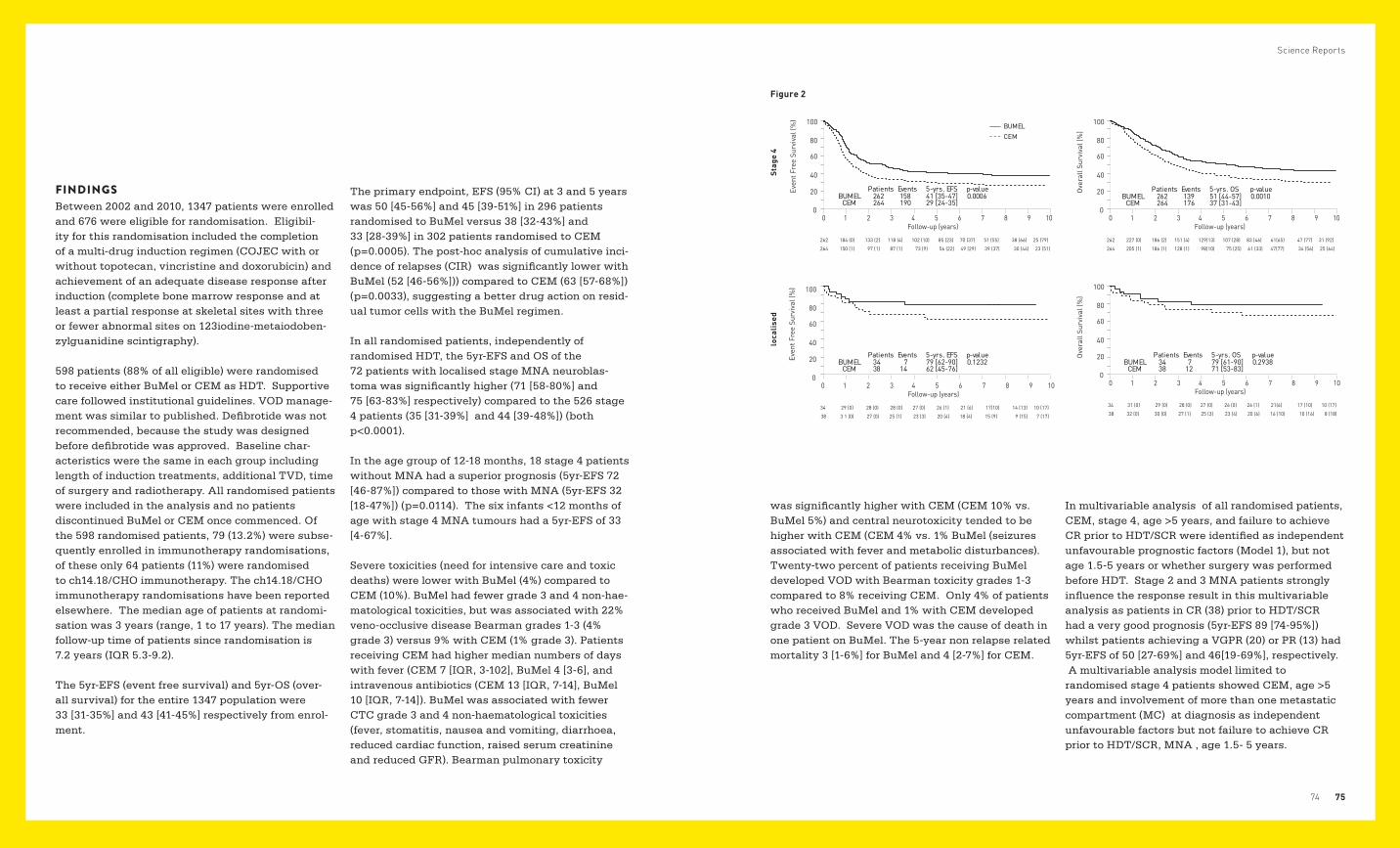
Science Reports
7574
FINDINGSBetween 2002 and 2010, 1347 patients were enrolled and 676 were eligible for randomisation. Eligibil-ity for this randomisation included the completion of a multi-drug induction regimen (COJEC with or without topotecan, vincristine and doxorubicin) and achievement of an adequate disease response after induction (complete bone marrow response and at least a partial response at skeletal sites with three or fewer abnormal sites on 123iodine-metaiodoben-zylguanidine scintigraphy).
598 patients (88% of all eligible) were randomised to receive either BuMel or CEM as HDT. Supportive care followed institutional guidelines. VOD manage-ment was similar to published. Defibrotide was not recommended, because the study was designed before defibrotide was approved. Baseline char-acteristics were the same in each group including length of induction treatments, additional TVD, time of surgery and radiotherapy. All randomised patients were included in the analysis and no patients discontinued BuMel or CEM once commenced. Of the 598 randomised patients, 79 (13.2%) were subse-quently enrolled in immunotherapy randomisations, of these only 64 patients (11%) were randomised to ch14.18/CHO immunotherapy. The ch14.18/CHO immunotherapy randomisations have been reported elsewhere. The median age of patients at randomi-sation was 3 years (range, 1 to 17 years). The median follow-up time of patients since randomisation is 7.2 years (IQR 5.3-9.2).
The 5yr-EFS (event free survival) and 5yr-OS (over-all survival) for the entire 1347 population were 33 [31-35%] and 43 [41-45%] respectively from enrol-ment.
The primary endpoint, EFS (95% CI) at 3 and 5 years was 50 [45-56%] and 45 [39-51%] in 296 patients randomised to BuMel versus 38 [32-43%] and 33 [28-39%] in 302 patients randomised to CEM (p=0.0005). The post-hoc analysis of cumulative inci-dence of relapses (CIR) was significantly lower with BuMel (52 [46-56%])) compared to CEM (63 [57-68%]) (p=0.0033), suggesting a better drug action on resid-ual tumor cells with the BuMel regimen.
In all randomised patients, independently of randomised HDT, the 5yr-EFS and OS of the 72 patients with localised stage MNA neuroblas-toma was significantly higher (71 [58-80%] and 75 [63-83%] respectively) compared to the 526 stage 4 patients (35 [31-39%] and 44 [39-48%]) (both p<0.0001).
In the age group of 12-18 months, 18 stage 4 patients without MNA had a superior prognosis (5yr-EFS 72 [46-87%]) compared to those with MNA (5yr-EFS 32 [18-47%]) (p=0.0114). The six infants <12 months of age with stage 4 MNA tumours had a 5yr-EFS of 33 [4-67%].
Severe toxicities (need for intensive care and toxic deaths) were lower with BuMel (4%) compared to CEM (10%). BuMel had fewer grade 3 and 4 non-hae-matological toxicities, but was associated with 22% veno-occlusive disease Bearman grades 1-3 (4% grade 3) versus 9% with CEM (1% grade 3). Patients receiving CEM had higher median numbers of days with fever (CEM 7 [IQR, 3-102], BuMel 4 [3-6], and intravenous antibiotics (CEM 13 [IQR, 7-14], BuMel 10 [IQR, 7-14]). BuMel was associated with fewer CTC grade 3 and 4 non-haematological toxicities (fever, stomatitis, nausea and vomiting, diarrhoea, reduced cardiac function, raised serum creatinine and reduced GFR). Bearman pulmonary toxicity
was significantly higher with CEM (CEM 10% vs. BuMel 5%) and central neurotoxicity tended to be higher with CEM (CEM 4% vs. 1% BuMel (seizures associated with fever and metabolic disturbances). Twenty-two percent of patients receiving BuMel developed VOD with Bearman toxicity grades 1-3 compared to 8% receiving CEM. Only 4% of patients who received BuMel and 1% with CEM developed grade 3 VOD. Severe VOD was the cause of death in one patient on BuMel. The 5-year non relapse related mortality 3 [1-6%] for BuMel and 4 [2-7%] for CEM.
In multivariable analysis of all randomised patients, CEM, stage 4, age >5 years, and failure to achieve CR prior to HDT/SCR were identified as independent unfavourable prognostic factors (Model 1), but not age 1.5-5 years or whether surgery was performed before HDT. Stage 2 and 3 MNA patients strongly influence the response result in this multivariable analysis as patients in CR (38) prior to HDT/SCR had a very good prognosis (5yr-EFS 89 [74-95%]) whilst patients achieving a VGPR (20) or PR (13) had 5yr-EFS of 50 [27-69%] and 46[19-69%], respectively. A multivariable analysis model limited to randomised stage 4 patients showed CEM, age >5 years and involvement of more than one metastatic compartment (MC) at diagnosis as independent unfavourable factors but not failure to achieve CR prior to HDT/SCR, MNA , age 1.5- 5 years.
Figure 2
Stag
e 4
loca
lise
dSt
age
4lo
calis
ed
Even
t Fre
e Su
rviv
al (%
)
0
20
60
40
80
100 BUMEL CEM
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
262 184 (0) 133 (2) 118 (4) 102 (10) 85 (23) 70 (37) 51 (55) 38 (66) 25 (79)
264 150 (1) 97 (1) 87 (1) 73 (9) 56 (22) 49 (29) 39 (37) 30 (44) 23 (51)
Ove
rall
Surv
ival
(%)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
262 227 (0) 186 (2) 151 (4) 129(13) 107 (28) 83 (46) 61(65) 47 (77) 31 (92)
264 205 (1) 186 (1) 128 (1) 98(10) 75 (25) 61 (33) 47(77) 34 (56) 25 (64)
Even
t Fre
e Su
rviv
al (%
)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
34 29 (0) 28 (0) 28 (0) 27 (0) 26 (1) 21 (6) 17(10) 14 (13) 10 (17)
38 3 1 (0) 27 (0) 25 (1) 23 (3) 20 (4) 18 (4) 15 (9) 9 (15) 7 (17)
Ove
rall
Surv
ival
(%)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
34 31 (0) 29 (0) 28 (0) 27 (0) 26 (0) 26 (1) 21(6) 17 (10) 10 (17)
38 32 (0) 30 (0) 27 (1) 25 (3) 23 (4) 20 (6) 16 (10) 10 (16) 8 (18)
Patients Events 5-yrs. EFS p-valueBUMEL 262 158 41 [35-47] 0.0006
CEM 264 190 29 [24-35]
Patients Events 5-yrs. OS p-valueBUMEL 262 139 51 [44-57] 0.0010
CEM 264 176 37 [31-43]
Patients Events 5-yrs. EFS p-valueBUMEL 34 7 79 [62-90] 0.1232
CEM 38 14 62 [45-76]
Patients Events 5-yrs. OS p-valueBUMEL 34 7 79 [61-90] 0.2938
CEM 38 12 71 [53-83]
Stag
e 4
loca
lised
Even
t Fre
e Su
rviv
al (%
)
0
20
60
40
80
100 BUMEL CEM
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
262 184 (0) 133 (2) 118 (4) 102 (10) 85 (23) 70 (37) 51 (55) 38 (66) 25 (79)
264 150 (1) 97 (1) 87 (1) 73 (9) 56 (22) 49 (29) 39 (37) 30 (44) 23 (51)
Ove
rall
Surv
ival
(%)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
262 227 (0) 186 (2) 151 (4) 129(13) 107 (28) 83 (46) 61(65) 47 (77) 31 (92)
264 205 (1) 186 (1) 128 (1) 98(10) 75 (25) 61 (33) 47(77) 34 (56) 25 (64)
Even
t Fre
e Su
rviv
al (%
)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
34 29 (0) 28 (0) 28 (0) 27 (0) 26 (1) 21 (6) 17(10) 14 (13) 10 (17)
38 3 1 (0) 27 (0) 25 (1) 23 (3) 20 (4) 18 (4) 15 (9) 9 (15) 7 (17)
Ove
rall
Surv
ival
(%)
0
20
60
40
80
100
0 1 2 3 4 5 6 7 8 9 10 Follow-up (years)
34 31 (0) 29 (0) 28 (0) 27 (0) 26 (0) 26 (1) 21(6) 17 (10) 10 (17)
38 32 (0) 30 (0) 27 (1) 25 (3) 23 (4) 20 (6) 16 (10) 10 (16) 8 (18)
Patients Events 5-yrs. EFS p-valueBUMEL 262 158 41 [35-47] 0.0006
CEM 264 190 29 [24-35]
Patients Events 5-yrs. OS p-valueBUMEL 262 139 51 [44-57] 0.0010
CEM 264 176 37 [31-43]
Patients Events 5-yrs. EFS p-valueBUMEL 34 7 79 [62-90] 0.1232
CEM 38 14 62 [45-76]
Patients Events 5-yrs. OS p-valueBUMEL 34 7 79 [61-90] 0.2938
CEM 38 12 71 [53-83]

Science Reports
7776
INTERPRETATION This randomised trial has evaluated a critical component of high risk neuroblastoma therapy and compared BuMel with CEM. After COJEC induction, BuMel significantly improved EFS in children with HR-NBL with an adequate response to induction therapy and induced less severe toxicities. BuMel showed an advantage in patients with stage 4 metastatic disease and less than 5 years. The improved survival of patients receiving BuMel was supported by the post-hoc analysis demonstrating that the cumulative incidence of relapse (CIR) was significantly lower with BuMel. The outcome of patients with stage 2 and 3 MNA appeared to be improved (79% 5yr-EFS) compared to approaches without HDT/SCR.
BuMel was found tolerable in spite of more frequent VOD and spared important other life-threatening toxicities observed with CEM. Both in COG and SIOPEN further randomised studies are evaluating additional intensification of consolidation including two HDT procedures in tandem.
BuMel is now considered standard HDT/SCR for SIOPEN and ongoing randomised SIOPEN studies will continue to optimise therapy for high-risk neuro-blastoma.
FUNDING SUPPORTEuropean Commission 5th Frame Work Grant (SIOPEN-R-NET EC grant No. QLRI-CT-2002-01768, www.siopen-r-net.org), Pierre Fabre Médicament providing Busilvex® and the St. Anna Kinder-krebsforschung. Neither the European Commission nor Pierre Fabre had a role in study design, data collection, data analysis, data interpretation or writing of the report. All authors participated in the report and had full access to all the data in the study. The corresponding author had final responsi-bility for the decision to submit for publication.
PUBLICATION ON THE HR-NBL1/SIOPEN TRIAL:Ladenstein R, Pötschger U, Pearson ADJ, et al. For the SIOP Europe Neuroblastoma Group (SIOPEN). (2017). Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemo-therapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lanct Oncology, Volume 18, No. 4
Ladenstein R, Poetschger U, Gray J, et al. (2016). Toxicity and outcome of anti-GD2 antibody ch14.18/CHO in front-line, high-risk patients with neuro-blastoma: Final results of the phase III immuno-therapy randomisation (HR-NBL1/SIOPEN trial). ASCO Annual Meeting Abstracts. ASCO Annual Meeting, J Clin Oncol 34, (suppl; abstr 10500.)
CAREER

UNIV.-PROF. DR. RUTH L ADENSTEINS2IRP: Studien und Statistik

8382
Career
A SHARED MISSION MAKES A DIFFERENCE IN RESEARCH!The Children’s Cancer Research Institute (CCRI) is an internationally renowned biomedical research institute, dedicated to the advancement of diagno-sis, prognosis and treatment of childhood cancer. Science at the CCRI addresses a broad range of topics, covered by 12 research groups whose members work closely together and in cooperation with clinicians from St. Anna Children’s Hospital. We are at the forefront of international pediatric cancer research and scientists at CCRI are striving to make the most innovative and effective therapies available to young patients.
At CCRI, we are an international team with employees from all over the world, offering students and postdocs an ideal combination of freedom and guidance to foster their personal and intellectual development while at the same time introducing them to the world of science and academic competition.
The CCRI offers an intellectually stimulating environ ment with lots of opportunities for scientific exchange and discussions in regular seminars, retreats, and national and international conference attendances. The quality of our work is periodically reviewed by a panel of top international experts guaranteeing consistently high standards of CCRI research. PhD students are accompanied by PhD-committees to guide them through their thesis and postdocs have the option to exercise their ability to teach by mentoring and training younger colleagues.
WORKING AT CCRI IF YOU WANT TO APPLY FOR A POSITION
please proceed according to the details given on our web site. http://science.ccri.at

8584
Career
CLINICAL RESEARCHHelga Björk ArnardóttirSaelde BaumgartnerTijana FrankEvgenia GlogovaCorinne GraflIngeborg HirschDasa JanousekSusanne KarlhuberMonica KiesewetterBarbara KristufekNora MühleggerMarek NykielUlrike PötschgerIngrid PribillMarion SebekEva SorzElfriede Thiem
PHD STUDENTSCharlotte BreyBarbara DillingerFriedrich ErhartTeresa GerberClara HechenbergerAnna-Maria HusaAnna Maria KatschnigFlorian KrompCornelia MutzLeonel PereiraKatarzyna PietrzykowskaFikret RifatbegovicBenjamin SalzerKlara SoukupTamara WeissMartin Zeppetzauer
DIPLOMA STUDENTSJakob BernerBernadette BlauensteinerClemens BrunnerHelena DodigKristin FischerKatharina MartinMagdalena ReiterMaria Regina StroblJakob Winkler
TECHNICIANS IN RESEARCH &DIAGNOSTICSBettina BerkowitschMaria BernederBettina Brunner- HerglotzHelga DaxbergerGudrun DivokyUlrike EngelSusanna FischerMichaela FortscheggerNelli FrankBrigitte GrimmAngela HalfmannSabrina HaslingerChristine Hoffmann- FreimüllerStefanie HosinerAndrea InthalJovana JovanovicGunhild JugDragana JugovicMargit KönigSusanna KoskelaFiona Anna-Maria KralerIsabella KricklErika MartonAstrid MecklenbräukerGerda ModarresKarin MühlbacherNadine NirtlBettina NockerSusana PascoalMaya-Marisol PlankMichaela PregesbauerSandra Preuner-StixDieter Printz
Christina SatkeDaniela ScharnerAngela SchumichManuela StadlerJulia StembergerSusanne SuhendraDijana TrbojevicEva WinklerSven WohlmacherMarion ZeginiggAndrea ZieglerElke Zipperer
RESEARCH SUPPORTOFFICENuno-Miguel Andrade GomesCaterina BarresiBarbara BrunmairZoltán DobaiMelanie Brunhofer
SCIENTIFIC STAFF
GROUP LEADERPeter AmbrosMartin DistelAlexander DohnalMichael DworzakGerhard FritschOskar HaasWolfgang HolterHeinrich KovarRuth LadensteinThomas LionRenate Panzer-GrümayerSabine Strehl
POSTDOCTORAL FELLOWS & STAFF SCIENTISTSReza Abbasi M.Sarah Ahmadi-ErberIngeborg AmbrosDave AryeeJozef BanDominik BogenKonstantin ByrgazovSara Colomer LahigueraStefan CzurdaFilomena De Almeida NogueiraMarkus DiemKlaus FortscheggerChristian FrechRené GeyereggerZvenyslava HusakMaximilian Otto KauerMarkus Abraham Kernbauer-HölzlStefanie KirchbergerKarin KosulinZsuzsanna LehnerManfred LehnerChantal LuciniKarin NebralMichael ReiterDagmar SchinnerlRaphaela SchwentnerCaterina SturtzelSabine Taschner-MandlEleni Tomazou-BockMurat TugrulCornelia Vesely
AFFILIATED CLINICIANSAndishe AttarbaschiHeidrun BoztugKaan BoztugChristofer DiakosBernhard FahrnerCaroline HutterLeo KagerUlrike KastnerAnita LawitschkaGeorg MannSusanne Matthes- LeodolterMilen MinkovChristina PetersHerbert PichlerChristian PoschStefan RieglerAndreas VécseiVolker Witt

UNIV.-DOZ. DR. MICHAEL DWORZAKImmunologische Diagnostik
FINANZBERICHT

9190
Finanzbericht
QUALITÄTSSICHERUNG DER WISSENSCHAFTLICHEN ARBEITDas Forschungsinstitut verfügt über ein Scientific Advisory Board – ein Gremium aus externen Exper-ten – mit der Aufgabe der laufenden Evalu ierung der wissenschaftlichen Arbeiten und Beratung der Institutsleitung. Darüber hinaus werden regelmäßig neue wissenschaftliche Projekte bei renommierten forschungsfördernden nationalen und internatio-nalen Stellen eingereicht und Forschungsergebnisse in international anerkannten, wissenschaftlichen Journalen publiziert. In regelmäßigen Abständen findet zusätzlich eine objektive Beurteilung der wissenschaftlichen Leistung durch ausgewiesene externe Fachleute auf dem Gebiet statt.
Die St. Anna Kinderkrebsforschung wird zum über wiegenden Teil durch private Spenden finanziert. Für den Betrieb des Forschungs-institutes werden jährlich mehr als sieben Millionen Euro benötigt, der
Verein verfügt jedoch über keine Basisfinanzie-rung durch die öffentliche Hand. Zusätzliche Mittel werden im Rahmen von kompetitiv ausgeschriebe-nen Projektförderungen von anerkannten nationalen und internationalen Stellen akquiriert.
Wir fühlen uns unseren Spenderinnen und Spen-dern gegenüber zu einer sparsamen und effizienten Verwendung der uns anvertrauten Gelder ver - pflichtet. Aus diesem Grund verwenden wir weniger als 10 % für die Verwaltung und das Fundraising, das bedeutet: mehr als 90 % der Spenden fließen direkt in die Forschung.
Der Jahresabschluss wird gemäß den Bestimmun-gen des Vereinsgesetzes für große Vereine erstellt, wobei die gleichen Richtlinien wie für Kapitalge-sellschaften gelten. Die Finanzgebarung und der Jahresabschluss des Vereins werden jährlich von einem beeideten Wirtschaftsprüfer kontrolliert und mit einem uneingeschränkten Bestätigungsvermerk versehen. Damit wird der sach- und zweckgemäße Umgang mit den erhaltenen Spenden sichergestellt und bestätigt.
SPENDENGÜTESIEGEL UND STEUERLICHE ABSETZBARKEITSeit dem Jahr 2002 trägt die St. Anna Kinderkrebs-forschung als eine der ersten Organisationen Öster reichs das Spendengütesiegel der Kammer der Wirtschaftstreuhänder. Für die jährliche Neu-verleihung führt ein Wirtschaftsprüfer zusätzlich eine Prüfung der transparenten und ordnungs ge-mäßen Verwendung der Mittel gemäß den strengen Richt linien des Spendengütesiegels durch.
Auf Grundlage eines von der Finanzlandes- direk tion für Wien erlassenen Bescheides zählt die St. Anna Kinderkrebsforschung zum begünstig-ten Empfänger kreis, sodass Spenden sowohl von der Lohnsteuer als Sonderausgabe, als auch von der Einkommensteuer als Betriebsausgabe steuerlich absetzbar sind.
RICHTLINIEN ZUR SPENDENVERWENDUNG

UNIV.-DOZ. DR. GERHARD FRITSCH Klinische Zellbiologie

9594
Finanzbericht
MITTELHERKUNFT MITTELVERWENDUNG
2015 2016
I. Spenden a) ungewidmete € 0,00 € 0,00
b) gewidmete € 13.202.063,51 € 7.831.440,93
II. Mitgliedsbeiträge € 0,00 € 0,00
III. Betriebliche Einnahmen a) betriebliche Einnahmen aus öffentlichen Mitteln € 1.606.752,78 € 1.480.409,97 b) sonstige betriebliche Einnahmen € 2.740.734,54 € 2.567.045,14
IV. Subventionen und Zuschüsse der öffentlichen Hand € 0,00 € 0,00
V. Sonstige Einnahmen a) Vermögensverwaltung € 0,00 € 0,00 b) sonstige andere Einnahmen sofern nicht
in Punkt I bis IV festgehalten€ 441.545,44 € 557.065,46
VI. Auflösung von Passivposten für noch nicht widmungsgemäß verwendete Spenden bzw. Subventionen
€ 0,00 € 0,00
VII. Auflösung von Rücklagen € 0,00 € 0,00
VIII. Jahresverlust € 0,00 € 0,00
TOTAL € 17.991.096,27 € 12.435.961,50
2015 2016
I. Leistungen für die statutarisch festgelegten Zwecke € 10.950.509,20 € 10.645.229,50
II. Spendenwerbung € 598.537,96 € 695.899,94
III. Verwaltungsaufwand € 346.652,69 € 359.480,99
IV. Sonstiger Aufwand sofern nicht unter Punkt I bis III festgehalten
€ 2.639,13 € 60.763,57
V. Zuführung zu Passivposten für noch nicht widmungsgemäß verwendete Spenden bzw. Subventionen
€ 6.092.757,29 € 674.587,50
VI. Zuführung zu Rücklagen € 0,00 € 0,00
VII. Jahresüberschuss € 0,00 € 0,00
TOTAL € 17.991.096,27 € 12.435.961,50
JAHRESERGEBNIS € 0,00 € 0,00
ANHANG

9998
Anhang
WISSENSCHAFTLICHER BEIRAT
PROF. DR. KL AUS-MICHAEL DEBATINUniversitätsklinik fürKinder- und Jugendmedizin89075 Ulm, Germany
PROF. JAMES R. DOWNING, MDScientific DirectorSt. Jude Children’s Research Hospital Memphis, TN 38105-3678, USA
PROF. LEE J. HELMAN, MDScientific Director for Clinical ResearchCenter for Cancer Research National Cancer Institute, National Institutes of Health Bethesda, MD 20892-2440, USA
PROF. STEPHAN L ADISCH, MDBosworth Chair for Cancer Biology Center for Cancer and Immunology Research Children’s National Medical Center Washington, DC 20010, USA
PROF. MEGAN SYKES, MDColumbia University Medical CenterNew York, NY 10032, USA
PROF. JOSEF VORMOOR, MDNewcastle Cancer Centre at the Northern Institute for Cancer Research Newcastle University, Paul O’Gorman Building, Medical School, Framlington PlaceNewcastle upon Tyne, NE2 4HH

UNIV.-PROF. DDR. THOMAS LIONMolekulare Mikrobiologie

103102
Anhang
Molecular mechanisms of human fungal pathogen host interaction (ImResFun) CCRI partner: Thomas Lion Coordinator: Karl Kuchler (Medical University of Vienna, Austria) European Commission Grant – FP7 Initial Training Network (Marie Curie Actions) N°: 606786 | Duration: 01/10/2013 to 30/09/2017
Expert paediatric oncology reference network for diagnostics and treatment (ExPO-r-Net) Coordinator: Ruth Ladenstein (CCRI) Grant from the European Consumers, Health, Agriculture and Food Executive Agency (CHAFEA) N°: 20131207 | Duration: 01/03/2014 to 31/08/2017
Targeted modulation of immune-system responses in cell therapies (MODICELL) Coordinator: Originally Andreas Heitger (5 May 2014), presently Wolfgang Holter European Commission Grant – FP7 Industry- Academia Partnerships and Pathways (Marie Curie Actions) N°: 285875 | Duration: 01/01/2013 to 31/12/2016
International study for treatment of childhood relapsed ALL 2010 (IntReALL) CCRI partners: Georg Mann, Andishe Attarbaschi and Ruth Ladenstein Coordinator: Jeremy Whelan (University College London, UK) European Commission Grant – FP7 Cooperation Project N°: 278514 | Duration: 01/10/2011 to 30/09/2017
INTERNATIONAL FREMDGEFÖRDERTE PROJEKTE
The interplay of NOTCH and MAPK pathway in LCH Principal investigator: Raphaela Schwentner Grant from the Histiocytosis Association Duration: 01/01/2016 to 31/12/2016
Optimized diagnostics for improved treatment stratification in invasive fungal diseases (FUNGITECT) Coordinator: Thomas Lion (CCRI/Labdia) European Commission Grant – FP7 Cooperation Project N°: 602125 | Duration: 01/02/2014 to 31/01/2019
Automation of flow cytometric analysis for quality-assured follow-up assessment to guide curative therapy for acute lymphoblastic leukaemia in children (AutoFLOW) Labdia/CCRI partner: Michael Dworzak Coordinator: Martin Kampel (Technical University of Vienna, Austria) European Commission Grant – FP7 Industry-Academia Partnerships and Pathways (Marie Curie Actions) N°: 610872 | Duration: 01/02/2014 to 31/01/2018
EURO EWING Consortium – International clinical trials to improve survival from Ewing sarcoma (EEC) CCRI partner: Heinrich Kovar Coordinator: Jeremy Whelan (University College London, UK) European Commission Grant – FP7 Cooperation Project N°: 602586 | Duration: 01/10/2013 to 31/09/2018
Analysing and Striking the Sensitivities of Embryonal Tumours (ASSET) CCRI partner: Heinrich Kovar Coordinator: Walter Koch (University College Dublin, Ireland) European Commission Grant – FP7 Cooperation Project N°: 259348 | Duration: 01/11/2010 to 30/04/2016
PanCare childhood and adolescent cancer survivor care and follow-up studies (PanCareSurFup) CCRI/St Anna Spital partners: Eva Frey Coordinator: Lars Hjorth (Lund University, Sweden) European Commission Grant – FP7 Cooperation Project N°: 257505 | Duration: 01/02/2011 to 31/01/2017

Einleitung
105104
Liquid biopsy in neuroblastoma: chance for diagnosis, prognosis and disease monitoring CCRI responsible principal investigator: Peter Ambros Grant from the Austrian National Bank (OeNB), Jubiläumsfonds N°: OeNB 16611 | Duration: 01/01/2016 to 31/12/2017
Permanent consequences in childhood Langerhans cell histiocytosis CCRI responsible principal investigator: Milen Minkov Grant from the Austrian National Bank (OeNB), Jubiläumsfonds N°: 16431 | Duration: 01/09/2015 to 31/08/2017
Regulation of the MYCN oncogene by Nuclear Lamina Proteins upon therapy-induced senescence in aggressive neuroblastoma CCRI responsible principal investigator: Sabine Taschner-Mandl Grant from the Herzfelder‘sche Familienstiftung Duration: 01/05/2015 to 31/10/2016
Verbesserte Patientenkommunikation in der Onkologie mittels INTERACCT App(OCCURSUS) CCRI responsible principal investigator: Anita Lawitschka Grant from the „Österreichische Gesellschaft für Hämatologie und Medizinische Onkologie“ Duration: 01/05/2016 to 31/07/2017
Integrating entertainment and reaction assessment into child cancer therapy (INTERACCT) CCRI/St Anna Spital partner: Anita Lawitschka Coordinator: Helmut Hlavacs (University of Vienna, Austria) Grant from the Austrian Research Promotion Agency (FFG), Call Bridge (Brückenschlagprogramm) N°: 838594 | Duration: 01/05/2013 to 30/04/2016
Prä-klinische Entwicklung einer Off-the-Shelf individualisierten Krebsimmuntherapie (IN SITU DC-CIT) CCRI partner: Alexander Dohnal Coordinator: Wolfgang Schöfberger (University of Linz, Austria) Grant from the Austrian Research Promotion Agency (FFG), Call Bridge (Brückenschlagprogramm) N°: 836532 | Duration: 01/10/2012 to 30/09/2016
Virus-specific immunotherapy (VISIT) Labdia/CCRI: coordinator René Geyeregger (Labdia), partner: Matthes-Leodolter (CCRI) Grant from the Wirtschaftsagentur Wien, Call Life Sciences 2014 N°: 1207846 | Duration: 01/04/2015 to 31/03/2018
Automated MRD-assessment in AML (flowCLUSTER) Labdia coordinator: Michael Dworzak Grant from the Wirtschaftsagentur Wien, Call Life Sciences 2014 N°: 1207843 | Duration: 01/03/2015 to 28/02/2018
Myeloproliferative neoplasms CCRI partner: Thomas Lion Coordinator: Peter Valent (Medical University of Vienna, Austria) Grant from the Austrian Science Fund (FWF), Special Research Programme (SFB) N°: F4705-B20 | Duration: 01/03/2013 to 28/02/2017
Single molecule array platform for sensitive diagnostics (SmardScout) CCRI partner: Thomas Lion Coordinator: Jan Hesse (Center for Advanced Bioanalysis GmbH, Linz, Austria) Grant from the Austrian Research Promotion Agency (FFG), Research Studios Austria, 4th Call N°: 844738 | Duration: 01/09/2014 to 22/11/2016
Verfahren zur hochautomatisierten Bewertung und Klassifikation von Zellen in Gewebeschnitten anhand räumlicher Markerprofile (TisQuant) LABDIA responsible principal investigator: Peter Ambros Grant from the Austrian Research Promotion Agency (FFG), ERA-SME N°: 844198 | Duration: 01/06/2014 to 31/05/2017
Directed in vitro differentiation of induced pluripotent stem cells towards the B lymphoid lineage (B-different) CCRI coordinator: Klaus Fortschegger Grant from the Austrian Research Promotion Agency (FFG), Call Bridge (Brückenschlagprogramm) N°: 843456 | Duration: 01/06/2014 to 22/12/2017
Overcoming Neuroblastoma Tumour HETerogeneity, Resistance and RecurrAnCe_ (ONTHETRRAC) CCRI responsible principal investigator: Peter Ambros Grant from the Austrian Science Fund (FWF), ERA-Net Transcan N°: 2799 B28 | Duration: 01/01/2016 to 30/06/2018
Ewing sarcoma – an enhancer disease? CCRI responsible principal investigator: Eleni Tomazou Grant from the Austrian Science Fund (FWF), Elise Richter Programme N°: V 506 B28 | Duration: 01/04/2016 to 31/01/2021
TRANSCALL (Translational research in childhood acute lymphoblastic leukemia) CCRI responsible principal investigator: Renate Panzer-Grümayer Grant from the Austrian Science Fund (FWF), ERA-Net Transcan N°: I1226-B19 | Duration: 01/07/2013 to 30/06/2017
PROVABES (prospective validation of biomarkers in Ewing sarcoma for personalized treatment) CCRI responsible principal investigator: Heinrich Kovar Grant from the Austrian Science Fund (FWF), ERA-Net Transcan N°: I1225-B19 | Duration: 01/04/2013 to 31/03/2017
NATIONAL FREMDGEFÖRDERTE PROJEKTE

DR. SABINE STREHLLeukämiegenetik

109108
Anhang
Wir möchten uns an dieser Stelle gerne bei unseren zahlreichen privaten Spenderinnen und Spendern bedanken, die uns seit vielen Jahren treu unter-stützen. Des Weiteren bedanken wir uns bei den folgenden nationalen und internationalen Förder-gebern und Organisationen:
DANKSAGUNG
• 7. Forschungsrahmenprogramm der Europäischen Kommission (FP7)
• 3rd Health Programme of the European Commission
• Bundesministerium für Wissenschaft, Forschung und Wirtschaft (BMWFW)
• Fonds zur Förderung der wissenschaftlichen Forschung (FWF)
• Österreichische Forschungsförderungsgesellschaft (FFG)
• Österreichische Nationalbank (OeNB)
• Wirtschaftsagentur Wien
• Histiocytosis Association
• Herzfelder’sche Familienstiftung
• Gigax Privatstiftung FL
• Kapsch AG
• Österreichische Gesellschaft für Hämatologie & Medizinische Onkologie (ÖGHO)
• Österreichischen Gesellschaft für Kinder- und Jugendheilkunde (ÖGKJ)
• Dachverband der Österreichischen Kinder-Krebs-Hilfe
• Medac Gesellschaft für klinische Spezialpräparate mbH, Deutschland
• Verein für Dermatologie, Wien
DIPLOM(MASTER)ARBEITEN / DISSERTATIONEN 2016
REZA ABBASIGenomic Analysis of Bone Marrow-Derived Disseminated Neuroblastoma CellsSupervised by Assoc.-Prof. Peter F. Ambros, PhDPhD Thesis
STEFANIE ANDERLThe role of PAX5 fusion genes in the pathogenesis of childhood B-cell precursor acute lymphoblastic leukemia. Supervised by Sabine Strehl, PhD and Klaus Fortschegger, PhD.PhD Thesis
L ARISSA KOLLERGenerierung von BCR-ABL1-exprimierenden Zelllinien zur Untersuchung der Ph+ Leukämie mutierten Subklon-EvolutionSupervised by Dr. Konstantin Byrgazov, PhD and Univ.-Prof. Thomas Lion, MD, PhDBachelor Thesis
VANESSA MAYRDynamic in vivo MAP kinase reporters to study development and disease in zebrafishSupervised by Martin Distel, PhD
CORNELIA MUTZInvestigating the NAD metabolome in Ewing sarcomaSupervised by Univ.-Prof. Heinrich Kovar, PhDPhD thesis
MAGDALENA REITERFunction of ETV6/RUNX1 in leukaemiaSupervised by Univ.-Prof. Dr. Renate Panzer-Grümayer, MDMaster Thesis

UNIV.-DOZ. DR. PETER F. AMBROSTumorbiologie

113112
Anhang
PUBLIKATIONEN 2016
Byrgazov K, Kastner R, Gorna M, Hoermann G, Koenig M, Lucini CB, Ulreich R, Benesch M, Strenger V, Lackner H, Schwinger W, Sovinz P, Haas OA, van den Heuvel-Eibrink M, Niemeyer CM, Hantschel O, Valent P, Superti-Furga G, Urban C, Dworzak MN, Lion T. (2017, Epub 2016 Oct 7). NDEL1-PDGFRB fusion gene in a myeloid malignancy with eosino-philia associated with resistance to tyrosine kinase inhibitors. Leukemia, 31: 237-240
Byrgazov K, Lucini CB, Berkowitsch B, Koenig M, Haas OA, Hoermann G, Valent P, Lion T. (2016). Transposon-mediated generation of BCR-ABL1- expressing transgenic cell lines for unbiased sensitivity testing of tyrosine kinase inhibitors. Oncotarget, 7: 78083-78094
Chagtai T, Zill C, Dainese L, Wegert J, Savola S, Popov S, Mifsud W, Vujanic G, Sebire N, Le Bouc Y, Ambros PF, Kager L, O'Sullivan MJ, Blaise A, Bergeron C, Mengelbier LH, Gisselsson D, Kool M, Tytgat GA, van den Heuvel-Eibrink MM, Graf N, van Tinteren H, Coulomb A, Gessler M, Williams RD, Pritchard-Jones K. (2016). Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol, 34: 3195-3203
Colomer-Lahiguera S, Strehl S. (2016). Complexity of NOTCH1 juxtamembrane insertion mutations in T-cell acute lymphoblastic leukemia. Leuk Lymphoma, 57: 1219-1222
Crazzolara R, Kropshofer G, Haas OA, Matthes- Martin S, Kager L. (2017, Epub 2016 Dec 7). Reduced-intensity conditioning and stem cell trans-plantation in infants with Diamond Blackfan anemia. Haematologica, 102: e73-e75
Creutzig U, Rössig C, Dworzak M, Stary J, von Stack-elberg A, Wössmann W, Zimmermann M, Reinhardt D. (2016). Exchange Transfusion and Leukapheresis in Pediatric Patients with AML With High Risk of Early Death by Bleeding and Leukostasis. Pediatr Blood Cancer, 63: 640-645
Creutzig U, Zimmermann M, Reinhardt D, Rasche M, von Neuhoff C, Alpermann T, Dworzak M, Perglerova K, Zemanova Z, Tchinda J, Bradtke J, Thiede C, Haferlach C. (2016). Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer, 122: 3821-3830 Czurda S, Lion T. (2017, Epub 2016 Nov 12). Broad-Spectrum Molecular Detection of Fungal Nucleic Acids by PCR-Based Amplification Tech-niques. Methods Mol Biol, 1508: 257-266
Czurda S, Lion T. (2017, Epub 2016 Nov 12). Prerequisites for Control of Contamination in Fungal Diagnosis. Methods Mol Biol, 1508: 249-255
Czurda S, Smelik S, Preuner-Stix S, Nogueira F, Lion T. (2016). Occurrence of Fungal DNA Contami-nation in PCR Reagents: Approaches to Control and Decontamination. J Clin Microbiol, 54: 148-152
Araki A, Chocholous M, Gojo J, Dorfer C, Czech T, Heinzl H, Dieckmann K, Ambros IM, Ambros PF, Slavc I, Haberler C. (2016). Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol Commun, 4: 88
Berbegall AP, Villamon E, Piqueras M, Tadeo I, Djos A, Ambros PF, Martinsson T, Ambros IM, Canete A, Castel V, Navarro S, Noguera R. (2016). Comparative genetic study of intratumoral hetero-genous MYCN amplified neuroblastoma versus aggressive genetic profile neuroblastic tumors. Oncogene, 35: 1423-1432
Bileck A, Mayer RL, Kreutz D, Weiss T, Taschner- Mandl S, Meier SM, Slany A, Gerner C. (2017, Epub 2016 Nov 13). Evaluation of inflammation-related signaling events covering phosphorylation and nuclear translocation of proteins based on mass spectrometry data. J Proteomics, 152: 161-171
Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, Kuiper RP, Hoogerbrugge P, Horstmann M, Zaliova M, Palmi C, Trka J, Fronkova E, Emerenciano M, do Socorro Pombo-de-Oliveira M, Mlynarski W, Szczepanski T, Nebral K, Attarbaschi A, Venn N, Sutton R, Schwab CJ, Enshaei A, Vora A, Stanulla M, Schrappe M, Cazzaniga G, Conter V, Zimmermann M, Moorman AV, Pieters R, den Boer ML. (2016). Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia, 30: 32-38
Bogen D, Brunner C, Walder D, Ziegler A, Abbasi R, Ladenstein RL, Noguera R, Martinsson T, Amann G, Schilling FH, Ussowicz M, Benesch M, Ambros PF, Ambros IM. (2016). The genetic tumor background is an important determinant for heterogeneous MYCN-amplified neuroblastoma. Int J Cancer, 139: 153-163
Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, Pickl W, Förster-Waldl E, Boztug K. (2016). NF-kappaB1 Haploinsufficiency Causing Immunodeficiency and EBV-Driven Lympho-proliferation. J Clin Immunol, 36: 533-540
Boztug H, Mühlegger N, Pötschger U, Attarbaschi A, Peters C, Mann G, Dworzak M. (2017, Epub 2016 Oct 4). Antibiotic prophylaxis with teicoplanin on alternate days reduces rate of viridans sepsis and febrile neutropenia in pediatric patients with acute myeloid leukemia. Ann Hematol, 96: 99-106
Brunner C, Brunner-Herglotz B, Ziegler A, Frech C, Amann G, Ladenstein R, Ambros IM, Ambros PF. (2016). Tumor Touch Imprints as Source for Whole Genome Analysis of Neuroblastoma Tumors. PLoS One, 11:8
Burchill SA, Beiske K, Shimada H, Ambros PF, Seeger R, Tytgat GA, Brock PR, Haber M, Park JR, Berthold F. (2017, Epub 2016 Dec 16). Recommenda-tions for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma; on behalf of the International Neuro-blastoma Response Criteria Bone Marrow Working Group. Cancer, 123: 1095-1105

115114
Anhang
Dittrich C, Kosty M, Jezdic S, Pyle D, Berardi R, Bergh J, El Saghir N, Lotz JP, Österlund P, Pavlidis N, Purkalne G, Awada A, Banerjee S, Bhatia S, Bogaerts J, Buckner J, Cardoso F, Casali P, Chu E, Close JL, Coiffier B, Connolly R, Coupland S, De Petris L, De Santis M, de Vries EG, Dizon DS, Duff J, Duska LR, Eniu A, Ernstoff M, Felip E, Fey MF, Gilbert J, Girard N, Glaudemans AW, Gopalan PK, Grothey A, Hahn SM, Hanna D, Herold C, Herrstedt J, Homicsko K, Jones DV Jr, Jost L, Keilholz U, Khan S, Kiss A, Köhne CH, Kunstfeld R, Lenz HJ, Lichtman S, Licitra L, Lion T, Litière S, Liu L, Loehrer PJ, Markham MJ, Markman B, Mayerhoefer M, Meran JG, Michielin O, Moser EC, Mountzios G, Moynihan T, Nielsen T, Ohe Y, Öberg K, Palumbo A, Peccatori FA, Pfeilstöcker M, Raut C, Remick SC, Robson M, Rutkowski P, Salgado R, Schapira L, Schernhammer E, Schlum-berger M, Schmoll HJ, Schnipper L, Sessa C, Shapiro CL, Steele J, Sternberg CN, Stiefel F, Strasser F, Stupp R, Sullivan R, Tabernero J, Travado L, Verheij M, Voest E, Vokes E, Von Roenn J, Weber JS, Wildiers H, Yarden Y. (2016). ESMO/ASCO recommendations for a Global Curriculum (GC) in medical oncology-edition 2016. Ann Oncol, 27: 1378-1381
Dittrich C, Kosty M, Jezdic S, Pyle D, Berardi R, Bergh J, El-Saghir N, Lotz JP, Österlund P, Pavlidis N, Purkalne G, Awada A, Banerjee S, Bhatia S, Bogaerts J, Buckner J, Cardoso F, Casali P, Chu E, Close JL, Coiffier B, Connolly R, Coupland S, De Petris L, De Santis M, de Vries EG, Dizon DS, Duff J, Duska LR, Eniu A, Ernstoff M, Felip E, Fey MF, Gilbert J, Girard N, Glaudemans AW, Gopalan PK, Grothey A, Hahn SM, Hanna D, Herold C, Herrstedt J, Homicsko K, Jones DV Jr, Jost L, Keilholz U, Khan S, Kiss A, Köhne CH, Kunstfeld R, Lenz HJ, Licht-man S, Licitra L, Lion T, Litière S, Liu L, Loehrer PJ, Markham MJ, Markman B, Mayerhoefer M, Meran JG, Michielin O, Moser EC, Mountzios G, Moynihan T, Nielsen T, Ohe Y, Öberg K, Palumbo A, Peccatori FA, Pfeilstöcker M, Raut C, Remick SC, Robson M, Rutkowski P, Salgado R, Schapira L, Schernham-mer E, Schlumberger M, Schmoll HJ, Schnipper L, Sessa C, Shapiro CL, Steele J, Sternberg CN, Stiefel F, Strasser F, Stupp R, Sullivan R, Tabernero J, Travado L, Verheij M, Voest E, Vokes E, Von Roenn J, Weber JS, Wildiers H, Yarden Y. (2016). ESMO / ASCO Recommendations for a Global Curriculum in Medical Oncology Edition 2016. ESMO Open, 1: e000097
Dmytrus J, Matthes-Martin S, Pichler H, Worel N, Geyeregger R, Frank N, Frech C, Fritsch G. (2016). Multi-color immune-phenotyping of CD34 subsets reveals unexpected differences between various stem cell sources. Bone Marrow Transplant, 51: 1093-1100
Duregger K, Hayn D, Nitzlnader M, Kropf M, Falgen-hauer M, Ladenstein R, Schreier G. (2016). Electronic Patient Reported Outcomes in Paediatric Oncology - Applying Mobile and Near Field Communication Technology. Stud Health Technol Inform, 223: 281-288
Ebinger S, Özdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M, Dworzak M, Lutz C, Turati VA, Enver T, Horny HP, Sotlar K, Parekh S, Spiekermann K, Hiddemann W, Schepers A, Polzer B, Kirsch S, Hoffmann M, Knapp B, Hasenauer J, Pfeifer H, Panzer-Grümayer R, Enard W, Gires O, Jeremias I. (2016). Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell, 30: 849-862
Ebner H, Hayn D, Falgenhauer M, Nitzlnader M, Schleiermacher G, Haupt R, Erminio G, Defferrari R, Mazzocco K, Kohler J, Tonini GP, Ladenstein R, Schreier G. (2016). Piloting the European Unified Patient Identity Management (EUPID) Concept to Facilitate Secondary Use of Neuroblastoma Data from Clinical Trials and Biobanking. Stud Health Technol Inform, 223: 31-38
Eder J, Kammerstatter M, Erhart F, Mairhofer-Muri D, Trautinger F. (2016). Illness Perception in Primary Cutaneous T-cell Lymphomas: What Patients Believe About Their Disease. Acta Derm Venereol, 96: 381-385
Eder J, Rogojanu R, Jerney W, Erhart F, Dohnal A, Kitzwögerer M, Steiner G, Moser J, Trautinger F. (2016). Mast Cells Are Abundant in Primary Cutaneous T-Cell Lymphomas: Results from a Computer-Aided Quantitative Immunohistological Study. PLoS One, 11: e0163661
Fritsch G, Frank N, Dmytrus J, Frech C, Pichler H, Witt V, Geyeregger R, Scharner D, Trbojevic D, Zipperer E, Printz D, Worel N. (2016). Relevance of flow cytometric enumeration of post-thaw leuco-cytes: influence of temperature during cell staining on viable cell recovery. Vox Sang, 111: 187-196
Grausenburger R, Bastelberger S, Eckert C, Kauer M, Stanulla M, Frech C, Bauer E, Stoiber D, von Stackel-berg A, Attarbaschi A, Haas OA, Panzer-Grümayer R. (2016). Genetic alterations in glucocorticoid signaling pathway components are associated with adverse prognosis in children with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia. Leuk Lymphoma, 57: 1163-1173
Grelpois G, Sabbagh C, Cosse C, Robert B, Chapu-is-Roux E, Ntouba A, Lion T, Regimbeau JM. (2016). Management of Uncomplicated Acute Appendicitis as Day Case Surgery: Feasibility and a Critical Analysis of Exclusion Criteria and Treatment Failure. J Am Coll Surg, 223:694-703
Grevener K, Haveman LM, Ranft A, van den Berg H, Jung S, Ladenstein R, Klco-Brosius S, Juergens H, Merks JH, Dirksen U. (2016). Management and Outcome of Ewing Sarcoma of the Head and Neck. Pediatr Blood Cancer, 63: 604-610
Hartz B, Marsh R, Rao K, Henter JI, Jordan M, Filipovich L, Bader P, Beier R, Burkhardt B, Meisel R, Schulz A, Winkler B, Albert MH, Greil J, Karasu G, Woessmann W, Corbacioglu S, Gruhn B, Holter W, Kühl JS, Lang P, Seidel MG, Veys P, Löfstedt A, Ammann S, Ehl S, Janka G, Müller I, Lehmberg K. (2016). The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood, 127: 3281-3290
Hayn D, Falgenhauer M, Kropf M, Nitzlnader M, Welte S, Ebner H, Ladenstein R, Schleiermacher G, Hero B, Schreier G. (2016). IT Infrastructure for Merging Data from Different Clinical Trials and Across Independent Research Networks. Stud Health Technol Inform, 228: 287-291

UNIV.-PROF. DR. OSK AR HA ASZytogenetik und molekulare Genetik

119118
Anhang
He T, Surdez D, Rantala JK, Haapa-Paananen S, Ban J, Kauer M, Tomazou E, Fey V, Alonso J, Kovar H, H, Delattre O, Iljin K. (2017, Epub 2016 Oct 17). High-throughput RNAi screen in Ewing sarcoma cells identifies leucine rich repeats and WD repeat domain containing 1 (LRWD1) as a regulator of EWS-FLI1 driven cell viability. Gene, 596: 137-146
Heller G, Topakian T, Altenberger C, Cerny-Reiterer S, Herndlhofer S, Ziegler B, Datlinger P, Byrgazov K, Bock C, Mannhalter C, Hörmann G, Sperr WR, Lion T, Zielinski CC, Valent P, Zöchbauer-Müller S. (2016). Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia, 30: 1861-1868
Hutter C, Minkov M. (2016). Insights into the pathogenesis of Langerhans cell histiocytosis: the development of targeted therapies. Immunotargets Ther, 5: 81-91
Javaheri T, Kazemi Z, Pencik J, Pham HT, Kauer M, Noorizadeh R, Sax B, Nivarthi H, Schlederer M, Maurer B, Hofbauer M, Aryee DN, Wiedner M, Toma-zou EM, Logan M, Hartmann C, Tuckermann JP, Kenner L, Mikula M, Dolznig H, Üren A, Richter GH, Grebien F, Kovar H, Moriggl R. (2016). Increased survival and cell cycle progression pathways are required for EWS/FLI1-induced malignant trans-formation. Cell Death Dis, 7: e2419
Kager L, Bruce LJ, Zeitlhofer P, Flatt JF, Maia TM, Ribeiro ML, Fahrner B, Fritsch G, Boztug K, Haas OA. (2017, Epub 2016 Oct 8). Band 3 nullVIENNA, a novel homozygous SLC4A1 p.Ser477X variant caus-ing severe hemolytic anemia, dyserythropoiesis and complete distal renal tubular acidosis. Pediatr Blood Cancer, 64: e26227
Kager L, Minkov M, Zeitlhofer P, Fahrner B, Ratzinger F, Boztug K, Dossenbach-Glaninger A, Haas OA. (2016). Two Novel Missense Mutations and a 5bp Deletion in the Erythroid-Specific Promoter of the PKLR Gene in Two Unrelated Patients With Pyruvate Kinase Deficient Transfusion- Dependent Chronic Nonspherocytic Hemolytic Anemia. Pediatr Blood Cancer, 63: 914-916
Kager L, Whelan J, Dirksen U, Hassan B, Anninga J, Bennister L, Bovee JV, Brennan B, Broto JM, Brugieres L, Cleton-Jansen AM, Copland C, Dutour A, Fagioli F, Ferrari S, Fiocco M, Fleuren E, Gaspar N, Gelderblom H, Gerrand C, Gerß J, Gonzato O, van der Graaf W, Hecker-Nolting S, Herrero-Martín D, Klco-Brosius S, Kovar H, Ladenstein R, Lancia C, LeDeley MC, McCabe MG, Metzler M, Myklebost O, Nathrath M, Picci P, Potratz J, Redini F, Richter GH, Reinke D, Rutkowski P, Scotlandi K, Strauss S, Thomas D, Tirado OM, Tirode F, Vassal G, Bielack SS. (2016). The ENCCA-WP7/EuroSarc/EEC/PROVA-BES/EURAMOS 3rd European Bone Sarcoma Networking Meeting/Joint Workshop of EU Bone Sarcoma Translational Research Networks; Vienna, Austria, September 24-25, 2015. Workshop Report. Clin Sarcoma Res, 6: 3
Karim-Kos HE, Hackl M, Mann G, Urban C, Woehrer A, Slavc I, Ladenstein R. (2016). Trends in incidence, survival and mortality of childhood and adolescent cancer in Austria, 1994-2011. Cancer Epidemiol, 42: 72-81
Kosulin K, Berkowitsch B, Lion T. (2016). Modified pan-adenovirus real-time PCR assay based on genome analysis of seventy HAdV types. J Clin Virol, 80: 60-61
Kosulin K, Dworzak S, Lawitschka A, Matthes-Le-odolter S, Lion T. (2016). Comparison of different approaches to quantitative adenovirus detection in stool specimens of hematopoietic stem cell trans-plant recipients. J Clin Virol, 85: 31-36
Kosulin K, Geiger E, Vecsei A, Huber WD, Rauch M, Brenner E, Wrba F, Hammer K, Innerhofer A, Pötschger U, Lawitschka A, Matthes-Leodolter S, Fritsch G, Lion T. (2016). Persistence and reactiva-tion of human adenoviruses in the gastrointestinal tract. Clin Microbiol Infect, 22: 381.e1-8
Kovar H, Amatruda J, Brunet E, Burdach S, Cidre-Aranaz F, de Alava E, Dirksen U, van der Ent W, Grohar P, Grünewald TG, Helman L, Houghton P, Iljin K, Korsching E, Ladanyi M, Lawlor E, Lessnick S, Ludwig J, Meltzer P, Metzler M, Mora J, Moriggl R, Nakamura T, Papamarkou T, Radic Sarikas B, Rédini F, Richter GH, Rossig C, Schadler K, Schäfer BW, Scotlandi K, Sheffield NC, Shelat A, Snaar-Jagalska E, Sorensen P, Stegmaier K, Stewart E, Sweet-Cor-dero A, Szuhai K, Tirado OM, Tirode F, Toretsky J, Tsafou K, Üren A, Zinovyev A, Delattre O. (2016). The second European interdisciplinary Ewing sarcoma research summit--A joint effort to decon-structing the multiple layers of a complex disease. Oncotarget, 7: 8613-8624
Kowalczyk JR, Samardakiewicz M, Pritchard-Jones K, Ladenstein R, Essiaf S, Fitzgerald E, Petrarulo G, Vassal G. (2016). European Survey on Standards of Care in paediatric oncology centres. Eur J Cancer, 61: 11-19
Kromp F, Ambros IM, Weiss T, Bogen D, Dodig H, Berneder M, Gerber T, Taschner-Mandl S, Ambros PF, Hanbury A. (2016). Machine Learning Frame-work incorporating Expert Knowledge in Tissue Image Annotation. Proceedings of the International Conference on Pattern Recognition, Cancun.
Kuttke M, Sahin E, Pisoni J, Percig S, Vogel A, Kraemmer D, Hanzl L, Brunner JS, Paar H, Soukup K, Halfmann A, Dohnal AM, Steiner CW, Blüml S, Basilio J, Hochreiter B, Salzmann M, Hoesel B, Lametschwandtner G, Eferl R, Schmid JA, Schab-bauer G. (2016). Myeloid PTEN deficiency impairs tumor-immune surveillance via immune-checkpoint inhibition. Oncoimmunology, 5: e1164918
Lewington V, Lambert B, Poetschger U, Sever ZB, Giammarile F, McEwan AJ, Castellani R, Lynch T, Shulkin B, Drobics M, Staudenherz A, Ladenstein R. (2017, Epub 2016 Sep 24). 123I-mIBG scintigraphy in neuroblastoma: development of a SIOPEN semi-quantitative reporting method by an inter-national panel. Eur J Nucl Med Mol Imaging, 44: 234-241
Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, Huber KV, Schmitner N, Kimmel RA, Romanov RA, Sturtzel C, Lardeau CH, Klugham-mer J, Farlik M, Sdelci S, Vieira A, Avolio F, Briand F, Baburin I, Májek P, Pauler FM, Penz T, Stukalov A, Gridling M, Parapatics K, Barbieux C, Berish-vili E, Spittler A, Colinge J, Bennett KL, Hering S, Sulpice T, Bock C, Distel M, Harkany T, Meyer D, Superti-Furga G, Collombat P, Hecksher-Sørensen J, Kubicek S. (2017, Epub 2016 Dec 1). Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell, 168: 86-100

121120
Anhang
Lion T. (2017, Epub 2016 Nov 12). Human Fungal Pathogen Identification: Methods and Protocols: Springer New York.
Lopes SS, Distel M, Linker C, Fior R, Monteiro R, Bianco IH, Portugues R, Strahle U, Saude L. (2016). Report of the 4th European Zebrafish Principal Investigator Meeting. Zebrafish, 13: 590-595
Mann G, Holter W. (2016). [Treatment of low-risk pediatric lymphocyte-predominant Hodgkin lymphoma]. Strahlenther Onkol, 192: 827-829
Minas TZ, Surdez D, Javaheri T, Tanaka M, Howarth M, Kang HJ, Han J, Han ZY, Sax B, Kream BE Hong SH, Çelik H, Tirode F, Tuckermann J, Toretsky JA, Kenner L, Kovar H, Lee S, Sweet-Cordero EA, Naka-mura T, Moriggl R, Delattre O, Üren A. (2017, Epub 2016 May 15). Combined experience of six independ-ent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget, 8: 34141-34163
Mutz CN, Schwentner R, Kauer MO, Katschnig AM, Kromp F, Aryee DN, Erhardt S, Goiny M, Alonso J, Fuchs D, Kovar H. (2016). EWS-FLI1 impairs aryl hydrocarbon receptor activation by blocking trypto-phan breakdown via the kynurenine pathway. FEBS Lett, 590: 2063-2075.
Nitzlnader M, Canete Nieto A, Ribelles AJ, Brunmair B, Ladenstein R, Schreier G. (2016). Interoperability Architecture for a Paediatric Oncology European Reference Network. Stud Health Technol Inform, 223: 39-45
Perwein T, Strehl S, König M, Lackner H, Panzer- Grümayer R, Mann G, Attarbaschi A, Urban EC, Haas OA. (2016). Imatinib-induced long-term remis-sion in a relapsed RCSD1-ABL1-positive acute lymph-oblastic leukemia. Haematologica, 101: e332-335
Pichler H, Fritsch G, König M, Daxberger H, Glogova E, Pötschger U, Breuer S, Lawitschka A, Güclü ED, Karlhuber S, Holter W, Haas OA, Lion T, Matthes-Martin S. (2016). Peripheral blood late mixed chimerism in leucocyte subpopulations following allogeneic stem cell transplantation for childhood malignancies: does it matter? Br J Haema-tol, 173: 905-917
Posch C, Moslehi H, Sanlorenzo M, Green G, Vujic I, Panzer-Grümayer R, Rappersberger K, Ortiz-Urda S. (2016). Pharmacological inhibitors of c-KIT block mutant c-KIT mediated migration of melanocytes and melanoma cells in vitro and in vivo. Oncotarget, 7: 45916-45925
Posch C, Sanlorenzo M, Vujic I, Oses-Prieto JA, Cholewa BD, Kim ST, Ma J, Lai K, Zekhtser M, Esteve-Puig R, Green G, Chand S, Burlingame AL, Panzer-Grümayer R, Rappersberger K, Ortiz-Urda S. (2016). Phosphoproteomic Analyses of NRAS(G12) and NRAS(Q61) Mutant Melanocytes Reveal Increased CK2alpha Kinase Levels in NRAS(Q61) Mutant Cells. J Invest Dermatol, 136: 2041-2048
Preuner S, Barna A, Frommlet F, Czurda S, Konstan-tin B, Alikian M, Machova Polakova K, Sacha T, Rich-ter J, Lion T, Gabriel C. (2016). Quantitative Analysis of Mutant Subclones in Chronic Myeloid Leukemia: Comparison of Different Methodological Approaches. Int J Mol Sci, 17: 642
Preuner S, Peters C, Pötschger U, Daxberger H, Fritsch G, Geyeregger R, Schrauder A, von Stack-elberg A, Schrappe M, Bader P, Ebell W, Eckert C, Lang P, Sykora KW, Schrum J, Kremens B, Ehlert K, Albert MH, Meisel R, Lawitschka A, Mann G, Panzer-Grümayer R, Güngör T, Holter W, Strahm B, Gruhn B, Schulz A, Woessmann W, Lion T. (2016). Risk assessment of relapse by lineage-specific monitoring of chimerism in children undergoing allogeneic stem cell transplantation for acute lympho blastic leukemia. Haematologica, 101: 741-746
Proff J, Walterskirchen C, Brey C, Geyeregger R, Full F, Ensser A, Lehner M, Holter W. (2016). Cyto-megalovirus-Infected Cells Resist T Cell Mediated Killing in an HLA-Recognition Independent Manner. Front Microbiol, 7: 844
Radic-Sarikas B, Tsafou KP, Emdal KB, Papamarkou T, Huber KVM, Mutz C, Toretsky JA, Bennett KL, Olsen JV, Brunak S, Kovar H, Superti-Furga G. (2017, Epub 2016 Nov 15). Combinatorial drug screening identifies Ewing sarcoma-specific sensitivities. Mol Cancer Ther, 16: 88-101
Reismüller B, Steiner M, Pichler H, Dworzak M, Urban C, Meister B, Schmitt K, Pötschger U, König M, Mann G, Haas OA, Attarbaschi A; Austrian ALL-BFM (Berlin-Frankfurt-Münster) Study Group. (2017, Epub 2016 Nov 2). High hyperdiploid acute lymphoblastic leukemia (ALL)-A 25-year population- based survey of the Austrian ALL-BFM (Berlin-Frankfurt-Munster) Study Group. Pediatr Blood Cancer, 64: e26327
Reiter M, Rota P, Kleber F, Diem M, Groeneveld- Krentz S, Dworzak M. (2016). Clustering of cell populations in flow cytometry data using a combina-tion of Gaussian mixtures. Pattern Recognition, 60: 1029-1040
Rieder FJ, Gröschel C, Kastner MT, Kosulin K, Laengle J, Zadnikar R, Marculescu R, Schneider M, Lion T, Bergmann M, Kallay E, Steininger C. (2017, Epub 2016 Aug 9). Human cytomegalovirus infection downregulates vitamin-D receptor in mammalian cells. J Steroid Biochem Mol Biol, 165: 356-362
Ruiz-Pinto S, Pita G, Patino-Garcia A, Garcia-Miguel P, Alonso J, Perez-Martinez A, Sastre A, Gomez-Mariano G, Lissat A, Scotlandi K, Serra M, Ladenstein R, Lapouble E, Pierron G, Kontny U, Picci P, Kovar H, Delattre O, González-Neira A. (2016). Identification of genetic variants in pharmacokinetic genes associated with Ewing Sarcoma treatment outcome. Ann Oncol, 27: 1788-1793
Scheer M, Dantonello T, Hallmen E, Vokuhl C, Leuschner I, Sparber-Sauer M, Kazanowska B, Niggli F, Ladenstein R, Bielack SS, Klingebiel T, Koscielniak E. (2016). Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer, 63: 1198-1206
Schüller S, Attarbaschi A, Berger A, Hutter C, Klebermass-Schrehof K, Steiner M. (2016). Hemophagocytic lymphohistiocytosis triggered by Gaucher disease in a preterm neonate. Pediatric Hematology and Oncology, 33: 462-467
Schwentner R, Herrero-Martin D, Kauer MO, Mutz CN, Katschnig AM, Sienski G, Alonso J, Aryee DN, Kovar H. (2017, Epub 2016 Dec 22). The role of miR-17-92 in the miRegulatory landscape of Ewing sarcoma. Oncotarget, 8: 10980-10993
Seidel MG, Boztug K, Haas OA. (2016). Immune Dysregulation Syndromes (IPEX, CD27 Deficiency, and Others): Always Doomed from the Start? J Clin Immunol, 36: 6-7

Einleitung
123122
Siebert N, Eger C, Seidel D, Juttner M, Zumpe M, Wegner D, Kietz S, Ehlert K, Veal GJ, Siegmund W, Weiss M, Loibner H, Ladenstein R, Lode HN. (2016). Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuro-blastoma patients treated by long-term infusion in combination with IL-2. MAbs, 8: 604-616
Sipurzynski J, Fahrner B, Kerbl R, Crazzolara R, Jones N, Ebetsberger G, Jauk B, Strenger V, Wohl-muther B, Schwinger W, Lackner H, Urban C, Holter W, Minkov M, Kager L, Benesch M, Seidel MG. (2016). Management of chronic immune thrombocy-topenia in children and adolescents: lessons from an Austrian national cross-sectional study of 81 patients. Semin Hematol, 53: Suppl 1, S43-47
Smidak R, Aradska J, Kirchberger S, Distel M, Sialana FJ, Wackerlig J, Mechtcheriakova D, Lubec G. (2016). A detailed proteomic profiling of plasma membrane from zebrafish brain. Proteomics Clin Appl, 10: 1264-1268
Stegmaier S, Leuschner I, Poremba C, Ladenstein R, Kazanowska B, Ljungman G, Scheer M, Blank B, Bielack S, Klingebiel T, Koscielniak E. (2017, Epub 2016 Sep 13). The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer, 64: 89-95
Taschner-Mandl S, Schwarz M, Blaha J, Kauer M, Kromp F, Frank N, Rifatbegovic F, Weiss T, Laden-stein R, Hohenegger M, Ambros IM, Ambros PF. (2016). Metronomic topotecan impedes tumor growth of MYCN-amplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget, 7: 3571-3586
Thompson D, Vo KT, London WB, Fischer M, Ambros PF, Nakagawara A, Brodeur GM, Matthay KK, DuBois SG. (2016). Identification of patient subgroups with markedly disparate rates of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group project. Cancer, 122: 935-945
Tischer S, Geyeregger R, Kwoczek J, Heim A, Figue-iredo C, Blasczyk R, Maecker-Kolhoff B, Eiz-Vesper B. (2016). Discovery of immunodominant T-cell epitopes reveals penton protein as a second immunodominant target in human adenovirus infection. J Transl Med, 14: 286
Valent P, Groner B, Schumacher U, Superti-Furga G, Busslinger M, Kralovics R, Zielinski C, Penninger JM, Kerjaschki D, Stingl G, Smolen JS, Valenta R, Lassmann H, Kovar H, Jäger U, Kornek G, Müller M, Sörgel F. (2016). Paul Ehrlich (1854-1915) and His Contributions to the Foundation and Birth of Trans-lational Medicine. J Innate Immun, 8: 111-120
Ventura S, Aryee DN, Felicetti F, De Feo A, Mancar-ella C, Manara MC, Picci P, Colombo MP, Kovar H, Care A, Scotlandi K. (2016). CD99 regulates neural differentiation of Ewing sarcoma cells through miR-34a-Notch-mediated control of NF-kappaB signaling. Oncogene, 35: 3944-3954
Vesely C, Frech C, Eckert C, Cario G, Mecklen-brauker A, zur Stadt U, Nebral K, Kraler F, Fischer S, Attarbaschi A, Schuster M, Bock C, Cavé H, von Stackelberg A, Schrappe M, Horstmann MA, Mann G, Haas OA, Panzer-Grümayer R. (2017). Genomic and transcriptional landscape of P2RY8-CRLF2- positive childhood acute lymphoblastic leukemia. Leukemia, Epub 2017 Jan 6
Weilner S, Schraml E, Wieser M, Messner P, Schnei-der K, Wassermann K, Micutkova L, Fortschegger K, Maier AB, Westendorp R, Resch H, Wolbank S, Redl H, Jansen-Dürr P, Pietschmann P, Grillari-Voglauer R, Grillari J. (2016). Secreted microvesicular miR-31 inhibits osteogenic differentiation of mesenchymal stem cells. Aging Cell, 15: 744-754
Weiss T, Taschner-Mandl S, Bileck A, Slany A, Kromp F, Rifatbegovic F, Frech C, Windhager R, Kitzinger H, Tzou CH, Ambros PF, Gerner C, Ambros IM. (2016). Proteomics and transcriptomics of periph-eral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia, 64: 2133-2153
Witt V, Pichler H, Fritsch G, Peters C. (2016). Multiple small versus few large amount aspirations for bone marrow harvesting in autologous and allogeneic bone marrow transplantation. Transfus Apher Sci, 55: 221-224
Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, Dworzak M, Schmugge M, van den Heuvel-Eibrink M, Ussowicz M, De Moerloose B, Catala A, Smith OP, Sedlacek P, Lankester AC, Zecca M, Bordon V, Matthes-Martin S, Abrahamsson J, Kühl JS, Sykora KW, Albert MH, Przychodzien B, Maciejewski JP, Schwarz S, Göhring G, Schlegel-berger B, Cseh A, Noellke P, Yoshimi A, Locatelli F, Baumann I, Strahm B, Niemeyer CM; EWOG-MDS. (2016). Prevalence, clinical characteristics, and prog-nosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood, 127: 1387-1397
Worel N, Fritsch G, Agis H, Bohm A, Engelich G, Leitner GC, Geissler K, Gleixner K, Kalhs P, Buxhofer-Ausch V, Keil F, Kopetzky G, Mayr V, Rabitsch W, Reisner R, Rosskopf K, Ruckser R, Zoghlami C, Zojer N, Greinix HT. (2016). Plerixafor as preemptive strategy results in high success rates in autologous stem cell mobilization failure. J Clin Apher, Epub 2016 Aug 31
Zaliova M, Moorman AV, Cazzaniga G, Stanulla M, Harvey RC, Roberts KG, Heatley SL, Loh ML, Konopleva M, Chen IM, Zimmermannova O, Schwab C, Smith O, Mozziconacci MJ, Chabannon C, Kim M, Frederik Falkenburg JH, Norton A, Marshall K, Haas OA, Starkova J, Stuchly J, Hunger SP, White D, Mullighan CG, Willman CL, Stary J, Trka J, Zuna J. (2016). Characterization of leukemias with ETV6-ABL1 fusion. Haematologica, 101: 1082-1093
Zuna J, Moericke A, Arens M, Koehler R, Panzer-Grümayer R, Bartram CR, Fischer S, Fronk-ova E, Zaliova M, Schrauder A, Stanulla M, Zimmer-mann M, Trka J, Stary J, Attarbaschi A, Mann G, Schrappe M, Cario G. Implications of delayed bone marrow aspirations at the end of treatment induc-tion for risk stratification and outcome in children with acute lymphoblastic leukaemia. Br J Haematol, 173: 742-748

IMPRESSUM UNTERSTÜTZUNG
HERAUSGEBER St. Anna Kinderkrebsforschung e.V. Zimmermannplatz 10, A-1090 Wien www.ccri.at, www.kinderkrebsforschung.at
KONTAKTMag. Marion Zavadil, CCRI Sekretariat [email protected]
VERANTWORTLICH FÜR DEN WISSENSCHAFTLICHEN INHALT Univ.-Prof. Dr. Heinrich Kovar Wissenschaftlicher Leiter [email protected]
VERANTWORTLICH FÜR DEN K AUFMÄNNISCHEN INHALT Mag. Jörg Bürger, MBA Kaufmännischer Leiter ab 08/2016 [email protected]
WISSENSCHAFTSKOMMUNIK ATION & PROJEKTMANAGEMENTLisa Huto Leitung Spenden, PR & Kommunikation [email protected]
KONZEPTION & GESTALTUNG Büro X Wien www.buerox.at
FOTOGRAFIE Harald Eisenberger www.eisenberger.co.at
DRUCK Estermann GmbH www.estermann-druck.at
DOWNLOAD des Forschungsberichtes als PDF-Datei: http://science.ccri.at
SPENDENKONTOSt. Anna Kinderkrebsforschung e.V.
Bank Austria IBAN: AT79 1200 0006 5616 6600 BIC: BKAUATWW

ST. ANNA KINDERKREBSFORSCHUNGFORSCHUNGSBERICHT 2016
ST. A
NN
A K
IND
ERK
REB
SFO
RSC
HU
NG
FO
RSC
HU
NG
SBER
ICH
T 20
16kinderkrebsforschung.atscience.ccri.at