self-assembly of phenylalanine-based molecules
TRANSCRIPT

Self-Assembly of Phenylalanine-Based MoleculesHelen W. German,*,† Sahin Uyaver,*,‡ and and Ulrich H. E. Hansmann*,†
†Department of Chemistry & Biochemistry, University of Oklahoma, 101 Stephenson Parkway, Norman, Oklahoma 73019-5251,United States‡Faculty of Applied Sciences, Istanbul Commerce University, 1 Inonu Str, Maltepe, 34843 Istanbul, Turkey
ABSTRACT: Using molecular dynamics, we study the self-assembly of phenylalanine with charged end-groups at varioustemperatures and concentrations. As in the case ofdiphenylalanine, we observe the formation of nanotubes;however, phenylalanine aggregates in layers of four, not six,molecules. The observed aggregates are consistent with recentexperimental measurements of fibrils obtained from mice withphenylketonuria. We investigate the stability and themechanism by which these tubular structures form and discusspotential toxicity mechanisms.
■ INTRODUCTION
Many biological processes require the self-assembly ofbiomolecules into ordered structures. For example, actinfilaments, which are essential for muscle contraction, are builtout of G-actin monomers self-assembling into long polymerscalled F-actin.1 Another example is the assembly of hemoglobinsubunits into the quaternary structure that is necessary for thetransport of oxygen. The self-assembly of amyloidogenicproteins into toxic oligomers or fibrils is associated with certainhuman illnesses, including Alzheimer’s disease. In materialscience, artificial polypeptides and synthetic compoundsnicknamed foldamers have been designed to assemble into awide variety of supramolecular materials such as nanowires.2,3
The common characteristic in all of these examples is thehierarchical assembly of monomers into new, compact, andcomplex structures that are stabilized by noncovalentinteractions. These hydrogen bonding, π-stacking, and electro-static interactions between the monomers are comparativelyweak but sum up to a strong driving force4 that is controlled bythe composition of the monomers.Important structural elements are aromatic rings that allow
for assembly of building blocks through π-stacking.5 Forinstance, diphenylalanine is known to form nanostructures suchas tapes, vesicles, tubes, and spheres.6,7 In particular,diphenylalanine nanotubes have been extensively studied by avariety of techniques under different solvent conditions8 andtemperatures.9 These nanotubes, appearing in sizes up to 2000nm diameter, are characterized by a hexagonal symmetry andT-shaped orientation of the two aromatic rings.10 Theirpiezoelectric activity,11 tunable electronic properties,12 and anextremely large Young’s modulus of 20 GPa13 make thesenanomaterials interesting candidates for a large range ofapplications including drug delivery, chromatography, tissueengineering,14 and microelectronics.9
The self-assembly of diphenylalanine is not entirelysurprising as diphenylalanine is a key motif of the fibril-forming
Aβ-peptide implicated in Alzheimer’s disease. Surprisingly,however, single phenylalanine molecules can also formnanofibrils.15 Both synthesized fibrils and those isolated frommice with phenylketonuria exhibit amyloid-like character-istics.15 In humans, phenylketonuria is a genetic disorderwhere the inability to metabolize phenylalanine leads toabnormally high concentrations of the amino acid that causemental retardation.16 The mechanism by which the phenyl-alanine accumulation causes the symptoms of phenylketonuriais not known. While an amyloid-like mechanism had notpreviously been postulated, Adler-Abramovic et al. havedemonstrated the cell toxicity of phenylalanine fibrils andtheir presence in the brains of mice with phenylketonuria.15
Unlike the crystal form of phenylalanine, the structure ofthese potentially toxic phenylalanine aggregates has not beenresolved. In the crystal structure,17,18 rows of monomersarrange as closely packed bilayers stabilized by hydrogen bondswith the termini in one row interacting with the ones in thenext. Aromatic rings are oriented perpendicular to the bilayerand interact by π-stacking with the rings assuming a T-shapedorientation; that is, the phenylalanine forms plates parallel tothe bilayers in which the aromatic rings are protected from thesurrounding. This structure differs significantly from nanofibrilsobserved by Adler-Abramovic et al.15 in molecular dynamicstrajectories of 27 phenylalanine molecules simulated in animplicit solvent with neutral N-terminal end groups, that is,conditions as expected for high pH values.15 Instead of thepreviously described crystal structure, they observe a ladder-likestructure of low stability with counterions interspersed,15 whose
Special Issue: 25th Austin Symposium on Molecular Structure andDynamics
Received: July 31, 2014Revised: October 27, 2014Published: October 27, 2014
Article
pubs.acs.org/JPCA
© 2014 American Chemical Society 1609 dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−1615

distribution of interatomic distances with peaks around 5 and11 Å agrees with experimentally observed electron diffractionpattern. The charged carboxyl terminals face each other and arebridged by counterions of opposite charge. Because of thisarrangement, the hydrophobic aromatic rings are in contactwith the surrounding water, an energetically unfavorablesituation that explains the observed short life times for thisstructure despite their transient stabilization by π−πinteractions. Geometries that optimize the energetics ofphenylalanine assemblies have also been studied by densityfunctional calculations. Three dominant structures were foundfor a system of four interacting phenylalanines.19 One interactsonly through the backbone with no indication of π stabilization.Another shows all four aromatic rings oriented and spaced in amanner typical for π stacking. The third structure, the globalminimum, contains one T-shaped interaction similar to thecrystal structure of benzene. However, the phenylalaninemolecules in these calculation had capped end-groups, andtheir energetics are therefore different from that in the bilayerfound by Adler-Abramovic et al.15
Both previous numerical studies suffer from the problem thatthey do not investigate the phenylalanine assembly underphysiological conditions of pH 7, that is, with both end-groupscharged. Hence, it is not clear how relevant the previouslyobtained structures are. For this reason, we investigate in thepresent paper the self-assembly of phenylalanine with chargedend-groups at various temperatures and concentrations andcompare the obtained structures with the crystal structures ofphenylalanine18 and diphenylalanine.20 We find assemblies thatare similar to the crystal structure of phenylalanine or to thepreviously found ones by Adler-Abramovic et al.15 but onlywith low frequency. The dominant assemblies are tubularstructures with each layer consisting of four phenylalanines.Layers are separated by 5 to 6 Å, and termini of each layer areclosely packed together with the greatest distance separationoccurring across a 5 Å pore. Height and width of the assemblyare in qualitative agreement with the unit cell size as deducedfrom electron diffraction data15 We briefly discuss possiblemechanisms for the observed cell toxicity of phenylalanineaggregates.
■ METHODSOur molecular dynamics simulations rely on a semiempiricalforce field, OPLS-AA, and TIP3P water molecules asimplemented in GROMACS 4.5.5.21 This combination waschosen because it avoids biases against helical forms,22 bestgives good results for the hydration properties of aminoacids,23,24 and performs very well in free-energy calculationamino acid chain analogs.25 It was also used in recent studies onthe effect of pH on the assembly of small phenylalanine-containing peptides26 or the self-assembly of a diphenylalanine-containing peptide27 that challenges the importance of aromaticinteractions in amyloidosis. Note, however, that the previouswork by Adler-Abramovic et al.15 relied on an unspecifiedversion of CHARMM. Also, different from that study, Terminiare not capped, and each monomer is treated as a zwitterion.The molecules are simulated in a cubic box of 5 Å length ineach direction and periodic boundary conditions. We accountfor the periodicity by using the Particle Mesh Ewald (PME)algorithm for calculating electrostatic interactions.28,29 Tocompare our results with the previous work by Adler-Abramovic et al.,15 we have performed initial simulatedannealing runs for a system of same size as used by these
authors, that is, 27 monomers, leading to a concentration of360 mM. In these runs, the monomers are initially placedrandomly in the box, while in other runs (described later indetail) we start from preformed structures build out of 24monomers (corresponding to a concentration of 320 mM) putinto the center of the box. Velocities are generated by assigningvalues from a Maxwell distribution corresponding to the starttemperature. Initial configurations are first energy-minimizedusing the steepest descent minimization with a step size of 0.01ps, followed by short runs of no less than 100 ps in first NVTand then NPT ensemble controlling the temperature with aBerendsen thermostat and keeping the pressure at 1 bar usingthe Parrinello−Rahman algorithm.30 As in all our simulations,the integration step is 2 fs.Initial simulated annealing runs starting from randomized
distributions of monomers at 325 K and cooling the system to275 K over 300 ns led to ensembles of low-energy structuresthat were inspected with PyMol31 and used to build idealizedstructures with the editconf tool of GROMACS 4.5.5.21 Notethat these idealized structures are made of 24 monomers whilethe simulated annealing run used 27 monomers. This is becausewe found that the lowest energy assemblies have a four-foldsymmetry while the diphenylalanine crystal structure is knownto have a six-fold symmetry. Choosing 24 monomers allowed usto compare the stability of assemblies with both symmetries.Other idealized structures are derived directly from thediphenylalanine crystal structure (CCDC 163340).20 Forinstance, we constructed a phenylalanine structure with samesix-fold symmetry as the diphenylalanine structure by deletingevery other phenylalanine from the diphenylalanine crystalstructure and moving the remaining molecules toward eachother to give a more appropriate pore size. Once each idealizedstructure was prepared, their geometries were optimized in twoshort simulated annealing runs of one nanosecond length each.In the first run, we kept the backbone fixed while allowing theside chains to move, while in the second run we restrained theside chains but allowed the backbone atoms to move. Theresulting fully optimized assemblies, each with 24 monomers,serve as starting configurations for our main simulations, wherewe followed the systems over 200 ns and evaluated theirstability at a temperature of T = 315 K. For each system, we runthree distinct simulations with different initial velocitydistributions. This allows us to test that we reached equilibriumand guarantees three maximal independent sets of measure-ments.For our analysis, we used g_rms, g_sas, and g_hbond tools
from the GROMACS package and investigated the extent ofclustering using a script developed in-house by Sahin Uyaver.Quantities that we measured include the root-mean-squaredeviation from the start configuration (taking into account thesymmetries resulting from having 24 indistinguishable mono-mers), radius of gyration, solvent-accessible surface area,number of clusters, and size of the largest cluster. Much ofour analysis is limited to the inner two layers of our systems tominimize finite-size effects. Following previous work,12 wecalculated the size of the inner pore as the distances betweennitrogens in opposing molecules. The outer diameter iscalculated similarly based on the locations of the hydrogenson the para positions of the phenylalanine rings. Fordiphenylalanine, average coordinates of the atoms are used tocalculate distances between opposite molecules. The averagecoordinates of the nitrogens of the top (middle) layer andthose of the bottom (middle) layer are used to measure the
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151610

height between layers. Our definitions of height and the width(calculated from the outer diameter) of the assemblies arechosen to allow us to compare of our measurements with thoseof unit-cell diffraction data.
■ RESULTS AND DISCUSSION
Self-assembly is a process by which the entropy of a systemdecreases. It is therefore driven by energy, and the self-assembled structure is the one with the lowest energy. Hence,self-assembly of phenylalanine can be considered a globaloptimization problem. For this reason, we have started ourstudy of the system with a series of five simulated annealingruns, starting at T = 325 K from a random structure and coolingour system of 27 phenylalanine molecules over 300 ns down toa temperature of T = 275 K. The final structures are shown inFigure 1. One run led to the formation of a structure (Figure 1e) that has some similarity to the previously observed ladder-like structure of Adler-Abramovic et al.;15 however, the terminiare closer together and the monomers are more staggered. As aconsequence, it resembles most closely the crystal structure ofphenylalanine. Three runs led to structures of similar energies(between −5640 and −5648 kJ/mol) and similar structures(shown in Figure 1a−c). These tubular assemblies are built outof layers, each made out of four phenylalanines, and differ fromboth the crystal structure and the previously observedassemblies. The fifth run led to a variant of this new tubularstructure, shown in Figure 1d, where two tubes areperpendicular to each with the aromatic rings still protectedfrom the solvent at the interface.The simulated annealing optimization indicates that the
global minimum structure, that is, the most stable form of thephenylalanine assemblies, is the newly found tubular structure.We therefore derived from the three lowest energyconfigurations of the simulated annealing runs an idealizedoptimal structure. For this purpose, we first discard threephenylalanines, reducing the number of monomers to 24,which allows us to compare more easily this structures to thesix-fold tubular form that has been previously observed fordiphenylalanine assemblies. We then maximize the overlap ofthe aromatic rings for the remaining 24 chains and increase thesymmetry of the resulting overall structure. Note that in thesimulated annealing-derived structures three of the four verticalstrands are oriented parallel with regards to the N−C termini,while the fourth strand has to be flipped to align its overall N−C direction with the others. The resulting tubular structure isshown in Figure 2 together with the crystal structures ofphenylalanine and diphenylalanine.The so-obtained idealized structure has an energy of −8434
kJ/mol and a symmetry of four when looking down theprincipal axis. Layers are 5.65 Å apart with the aromatic rings
oriented in such a way that stacking interactions are observed.Each layer has a diameter of 15.04 Å, forming a pore of size4.93 Å, which is too small for water to pass through. Height andwidth are in qualitative agreement with the unit cell size, asdeduced from electron diffraction data,15 but its height is ∼1 Ålarger and the width is 1 Å smaller. However, our width of 10.6Å is calculated from the outer diameter and does not take intoaccount the fact that the structure is not perfectly square. Notethat our structure has no similarity to the ladder-like assembliesfleetingly observed in the previous simulations.15 Its tubularstructure rather resembles the crystal structure of diphenylala-nine shown in Figure 2c, where, however, the layers are formedby six instead of four monomers. Unlike in the ladder-likeassemblies previously observed by Adler-Abramovich at al,15
the monomers are closer together in our assembly, with thelargest separation of termini occurring across the tubes pore.Besides π-stacking interactions between the aromatic rings, ourstructure is also stabilized by interactions between the termini.In the previously proposed structures of ref 15, the rings pointoutward and the electrostatic interactions, involving counter-ions, force rings and termini apart. Monomers across the longaxis are separated by 11.46 Å as opposed to <5 Å in ourstructure. Because of its larger stability, our tubular assemblywith its layers of four phenylalanine appears to be a more likelycandidate for oligomer and fibril formation. Note, however, thatwe do not observe any water molecules in our pore-likestructure. Hence, the toxicity of phenylalanine fibrils observedin Ref 15 is likely not due to tubular phenylalanine oligomersacting as pores that allow water leakage through the cellmembrane. More likely is that the np−np interaction of therings with the aliphatic chains of the lipids will separate thepolar head groups of the lipids, leading to membrane disruptionas a cause of cell toxicity.Visual analysis of the simulated annealing trajectories
indicates that the monomers start to assemble at an association
Figure 1. Final configurations as obtained in five independent simulated annealing runs. (a−c) Symmetry of the four structures. Panel D looks downthe tube axis of a symmetry of the four structures with another forming perpendicular to it. Panel e is the resulting crystal structure of phenylalanine.Green atoms are carbon, blue are nitrogen, red are oxygen, and white are hydrogen.
Figure 2. Proposed lowest energy structure of phenylalanine (a)together with the experimentally determined crystal structure ofphenylalanine (b) and the crystal structure of diphenylalanine (b).Green atoms are carbon, blue are nitrogen, red are oxygen, and whiteare hydrogen.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151611

temperature of ∼315 K. This temperature is slightly above bodytemperature and the higher temperatures (T = 310 K) used inthe previous work by Adler-Abramovthisich in their simu-lations, which in addition relied on a different force field, anunspecified version of CHARMM.15 Hence, to evaluate thestability and the mechanism of formation of our phenylalaninenanotubes, we have performed a number of molecular dynamicsimulations at this temperature, starting from the previouslygenerated energy-minimized, idealized structures. The stabilityof these assemblies with four-fold symmetry is compared withsuch assemblies with the same number of 24 phenylalanines butarranged in such a way that they resemble the six-foldsymmetry observed in the diphenylalanine crystal structure.To understand better the differences between phenylalanineand diphenylalanine assemblies, we have also probed withadditional sets of simulations the stability of diphenylalanine
assemblies with either the crystal structure (i.e., six-foldsymmetry) as start configuration or the molecules forced intothe phenylalanine nanotube structure with its four-foldsymmetry. The diphenylalanine systems are simulated at thesame temperature despite the fact that experimentally higherdissociation temperatures of ∼370 K are observed.6,9 However,similar temperature have been used by other users, even whenusing different force fields; see, for instance, ref 8(GROMOS53a6) or ref 32 (Amber ff99SB). We thereforebelieve that this temperature is also meaningful to study thestability of diphenylalanine. For our simulations, each of thefour systems is minimized with respect to their backbones andside chains and followed for 200 ns in three separate stabilitytests. We show in Figure 3 an overlay of the final configurationsof these runs with the corresponding start configurations foreach of the four cases.
Figure 3. Final configurations (tan) of three canonical molecular dynamics runs of 200 ns length with the corresponding start configurations (green)for (a) phenylalanine (F4) in the idealized four-fold structure with start and final configurations overlaid, (b) phenylalanine with an assumed six-foldstructure (F6) as experimentally observed for diphenylalanine, with the end results to the right not superimposed because symmetry was notpreserved as in the other runs, (c) diphenylalanine in the same four-fold structures (FF4) as proposed by us for F4 phenylalanine with start and finalconfigurations overlaid, and (d) diphenylalanine in its experimentally observed six-fold structure (FF6) with start and final configurations overlaid.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151612

The phenylalanine oligomer with four-fold symmetry (F4),the diphenylalanine oligomer with forced four-fold symmetry(FF4), and the diphenylalanine oligomer with six-foldsymmetry (FF6) all retained their original symmetries andgeneral structures. However, the phenylalanine oligomer withsix-fold symmetry (F6) fell apart quickly and in two of the threeruns started to reassemble into the nanotube with four-symmetry while in the third run it assumed the crystalstructure.18 The lack of stability for the F6 structure is likelydue to a pore size of 8.0[2.4] Å, which is 2 Å larger than that ofthe idealized F4 structure, which measures 6.0[0.4]Å. As aconsequence of the larger pore size, the distance between theend groups is larger for the F6 structure than for the F4structure; therefore, the stabilizing interaction between them issmaller. Hence, the difference in stability between the F4 andF6 structures demonstrates the importance of end-groupinteractions and the strong pH dependence of aggregateformation.Table 1 lists various quantities that characterize the initial
idealized and the final structures F4, F6, FF4, and FF6 asmeasured in our simulations. Note that we do not list finalvalues for the F6 structure because this oligomer is not stable,and therefore no defined final structure was observed. Note alsothat root-mean-square values are given for only the finalconfiguration. This is because their calculation is not trivialbecause one has to account for the symmetries arising from
having 24 indistinguishable molecules. The measured valuesquantify our results from visual inspection previously described.In particular, the values are slightly larger for FF4 than for FF6,indicating lower stability for this form. Following previousstudies,12 we measure the inner pore as the distances betweennitrogen atoms in opposing phenylalanine molecules. The outerdiameter is calculated similarly based on the locations of thehydrogens on the para positions of the phenylalanine rings. Fordiphenylalanine, averaged coordinates of the atoms are used tocalculate distances between the opposite residues. Error barsare obtained from standard deviations calculated over all pairs.For measuring the height between layers, the averagecoordinates of the nitrogens of the top (middle) layer andthose of the bottom (middle) layer are used. Shown are boththe values for the start configurations of F4, F6, FF4, and FF6and the corresponding values for the final configurations of F4,FF4 and FF6.Note that the phenylalanine with the symmetry of four has a
pore size too small (<5 Å) to fit water molecules inside.Compared with the electron diffraction data, 4.6 ± 0.06 Å tubeaxis length by 11.63 ± 0.27 Å width,15 our results show aslightly wider pore but with a more compact width. Asexpected, the diphenylalanine with a symmetry of four has alarger pore size and width (by ∼4.5 Å each), which allows forwater to exist inside the pore. Interestingly, this structure hasthe smallest average distance between layers. Increasing the
Table 1. Size, Pore Diameter, Water Content, and Root-Mean-Square Deviations (rmsd) of the Studied Assembliesa
height (Å) outer diameter (Å) pore diameter (Å) water
structure start end start end start end start end rmsd (Å)
F4 5.4 5.65[0.04] 17.61[2.09] 15.04[2.76] 5.98[0.41] 4.93[1.45] 1.5 0 2.8[1.2]F6 5.2 20.56[0.81] 7.97[2.44] 3.0FF4 5.6 5.03[0.05] 21.57[0.69] 20.23[1.22] 10.35[0.46] 9.42[1.18] 9.6 4.8 3.9[0.1]FF6 5.4 5.73[0.90] 24.00[0.27] 23.00[2.50] 12.94[0.73] 12.38[2.56] 15.9 15.3 3.0[0.6]
armsd values are for the final configurations, with the start configurations as reference.
Figure 4. Snap shots from a molecular dynamics simulation at T = 315 K that starts with a random distribution of 24 phenylalanine molecules and isfollowed over 200 ns.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151613
number of FF residues per layer to the FF6 structure widensthe pore even further, allowing for even more (three times) thenumber of water molecules to infiltrate the pore.Note also that in our molecular dynamics simulations
diphenylalanine aggregates appear to be stable at T = 315 Kin both the four-folded (FF4) and the experimentally observedsix-folded form (FF6). This is despite the slightly largerhydrophobic surface area exposed to the solvent in the FF4form (41.2 [1.3] Å2 versus 39.0 [1.5] Å2 in the FF6 form).However, the stability of the FF4 structure may just reflect tooshort simulation times. A visual inspection of the finalconfiguration shows that only two monomers are separatedfrom the FF6 structure in the three independent runs, but fourmonomers are separated in the corresponding three runs fromthe FF4 structure.The occurrence of configurations when observed over finite
times not only depends on their thermodynamic stability butalso on their kinetic accessibility. We have therefore addedanother series of 10 molecular dynamic simulations at T = 315K that start from random distributions of 24 phenylalaninemolecules and are followed over 100 ns. Snap shots from one ofthese runs are shown in Figure 4. In this run, the association isdriven by aligning of the charged termini with each other,starting after ∼20 ns. At ∼40 ns, layers of the four-foldedstructure appear. The above scenario is observed in the majorityof runs, but we found also a run where after 24 ns a ladder-likestructure forms that resembles the one found in the simulationsof Adler-Abramovich.15 However, this structure dissolves afteronly 6 ns, and the molecules reassemble later into thephenylalanine crystal structure. Hence, these runs areconsistent with our simulated annealing runs and stabilityanalysis, which indicate that the most stable form ofphenylalanine aggregates is the four-folded form of Figure 2but that aggregates can also exist in forms that resemble the oneproposed in ref 15 or the crystal structure of King et al.18
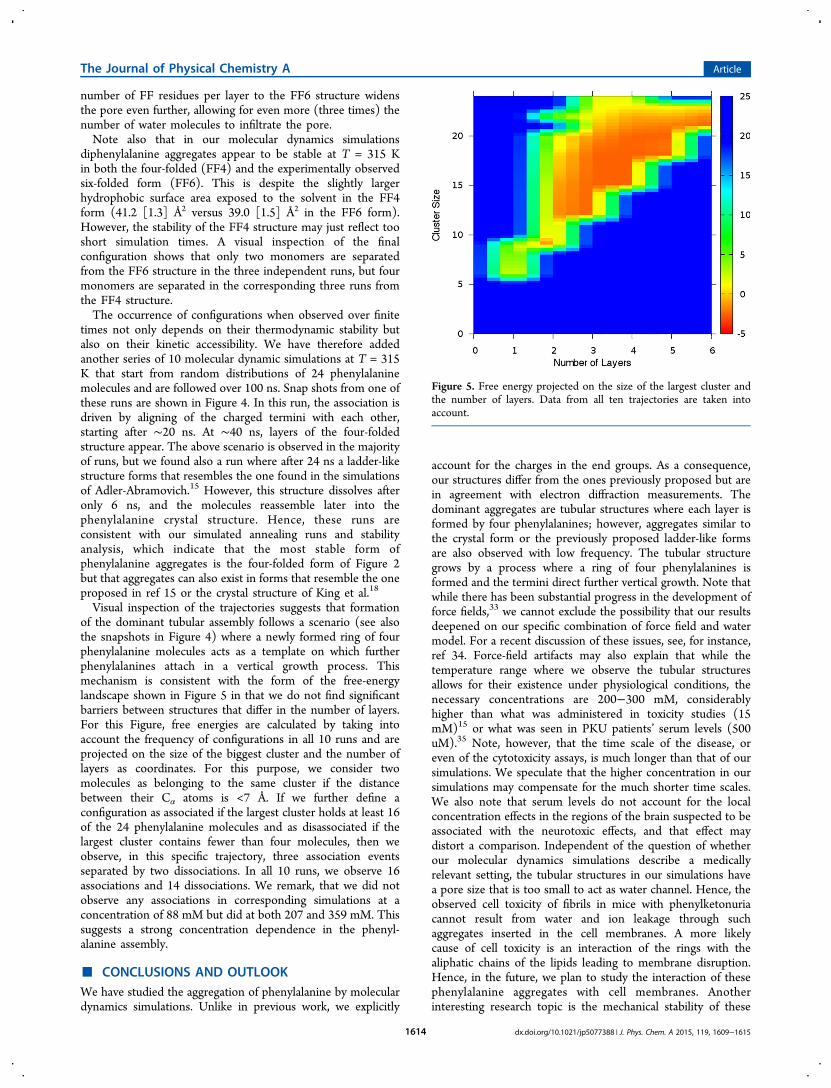
Visual inspection of the trajectories suggests that formationof the dominant tubular assembly follows a scenario (see alsothe snapshots in Figure 4) where a newly formed ring of fourphenylalanine molecules acts as a template on which furtherphenylalanines attach in a vertical growth process. Thismechanism is consistent with the form of the free-energylandscape shown in Figure 5 in that we do not find significantbarriers between structures that differ in the number of layers.For this Figure, free energies are calculated by taking intoaccount the frequency of configurations in all 10 runs and areprojected on the size of the biggest cluster and the number oflayers as coordinates. For this purpose, we consider twomolecules as belonging to the same cluster if the distancebetween their Cα atoms is <7 Å. If we further define aconfiguration as associated if the largest cluster holds at least 16of the 24 phenylalanine molecules and as disassociated if thelargest cluster contains fewer than four molecules, then weobserve, in this specific trajectory, three association eventsseparated by two dissociations. In all 10 runs, we observe 16associations and 14 dissociations. We remark, that we did notobserve any associations in corresponding simulations at aconcentration of 88 mM but did at both 207 and 359 mM. Thissuggests a strong concentration dependence in the phenyl-alanine assembly.
■ CONCLUSIONS AND OUTLOOKWe have studied the aggregation of phenylalanine by moleculardynamics simulations. Unlike in previous work, we explicitly
account for the charges in the end groups. As a consequence,our structures differ from the ones previously proposed but arein agreement with electron diffraction measurements. Thedominant aggregates are tubular structures where each layer isformed by four phenylalanines; however, aggregates similar tothe crystal form or the previously proposed ladder-like formsare also observed with low frequency. The tubular structuregrows by a process where a ring of four phenylalanines isformed and the termini direct further vertical growth. Note thatwhile there has been substantial progress in the development offorce fields,33 we cannot exclude the possibility that our resultsdeepened on our specific combination of force field and watermodel. For a recent discussion of these issues, see, for instance,ref 34. Force-field artifacts may also explain that while thetemperature range where we observe the tubular structuresallows for their existence under physiological conditions, thenecessary concentrations are 200−300 mM, considerablyhigher than what was administered in toxicity studies (15mM)15 or what was seen in PKU patients’ serum levels (500uM).35 Note, however, that the time scale of the disease, oreven of the cytotoxicity assays, is much longer than that of oursimulations. We speculate that the higher concentration in oursimulations may compensate for the much shorter time scales.We also note that serum levels do not account for the localconcentration effects in the regions of the brain suspected to beassociated with the neurotoxic effects, and that effect maydistort a comparison. Independent of the question of whetherour molecular dynamics simulations describe a medicallyrelevant setting, the tubular structures in our simulations havea pore size that is too small to act as water channel. Hence, theobserved cell toxicity of fibrils in mice with phenylketonuriacannot result from water and ion leakage through suchaggregates inserted in the cell membranes. A more likelycause of cell toxicity is an interaction of the rings with thealiphatic chains of the lipids leading to membrane disruption.Hence, in the future, we plan to study the interaction of thesephenylalanine aggregates with cell membranes. Anotherinteresting research topic is the mechanical stability of these
Figure 5. Free energy projected on the size of the largest cluster andthe number of layers. Data from all ten trajectories are taken intoaccount.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151614

aggregates and the spectrum of assemblies that could be formedby mixtures of phenylalanine and diphenylalanine aggregates.
■ AUTHOR INFORMATIONCorresponding Authors*E-mail: [email protected] (H.W.G.)*E-mail: [email protected] (S.U.)*E-mail: [email protected] (U.H.E.H.)NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge support from the National ScienceFoundation (research grant CHE-1266256). The simulationswere done on the BOOMER cluster of the University ofOklahoma. We thank Dr. Workalemahu Berhanu for his helpwith this work. S.U. thanks the Department of Chemistry andBiochemistry for kind hospitality during his sabbatical stay atUniversity of Oklahoma.
■ REFERENCES(1) Nelson, D. L.; Cox, M. M. Lehninger Principles of Biochemistry, 4thed.; W. H. Freeman: New York, 2005; pp183−186.(2) Miyake, H.; Tsukube, H. Coordination Chemistry Strategies forDynamic Helicates: Time-Programmable Chirality Switching withLabile and Inert Metal helicates. Chem. Soc. Rev. 2012, 41, 6977−6991.(3) Hecht, S. Construction with Macromolecules.Mater. Today 2005,8, 48−55.(4) Hill, D.; Mio, M.; Prince, R. B.; Hughes, T. S.; Moore, J. S. AField Guide to Foldamers. Chem. Rev. 2001, 101, 3393−4012.(5) Huc, I. Aromatic Oligoamide Foldamers. Eur. J. Org. Chem. 2004,17−29.(6) Adler-Abramovich, L.; Reches, M.; Sedman, V. L.; Allen, S.;Tendler, S. J. B.; Gazir, E. Thermal and Chemical Stability ofDiphenylalanine Peptide Nanotubes: Implications for Nanotechno-logical Applications. Langmuir 2006, 22, 1313−1320.(7) Guo, C.; Luo, Y.; Zhou, R.; Wei, G. Probing the Self-AssemblyMechanism of Diphenylalanine-Based Peptide Nanovesicles andNanotubes. ACS Nano 2012, 6, 3907−3918.(8) Rissanou, A. N.; Georgilis, E.; Kasotakis, E.; Mitraki, A.;Harmandaris, V. J. Effect of Solvent on the Self-Assembly of Dialanineand Diphenylalanine Peptides. J. Phys. Chem. B 2013, 117, 3962−3975.(9) Sedman, V. L.; Adler-Abramovich, L.; Allen, S.; Gazit, E.;Tendler, S. J. B. Direct Observation of the Release of Phenylalaninefrom Diphenylalanine Nanotubes. J. Am. Chem. Soc. 2013, 117, 3962−3975.(10) Gorbitz, C. H. The Structure of Nanotubes Formed byDiphenylalanine, the Core Recognition Motif of Alzheimer’s Beta-Amyloid Polypeptide. Chem. Commun. 2006, 2332−2334.(11) Kholkin, A.; Amdursky, N.; Bdikin, I.; Gazit, E.; Rosenman, G.Strong Piezoelectricity in Bioinspired Peptide Nanotubes. ACS Nano2010, 4, 610−614.(12) Andrade-Filho, T.; Ferreira, F. F.; Alves, W. A.; Rocha, A. R.The Effects of Water Molecules on the Electronic and StructuralProperties of Peptide Nanotubes. Phys. Chem. Chem. Phys. 2013, 15,7555−7559.(13) Yemini, M.; Reches, M.; Gazit, E.; Rishpon, J. PeptideNanotube-Modified Electrodes for Enzyme-Biosensor Applications.Anal. Chem. 2005, 77, 5155−5159.(14) Ellis-Behnke, R. G.; Liang, Y. X.; You, S. W.; Tay, D. K.; Zhang,S.; So, K. F.; Schneider, G. E. Nano Neuro Knitting: PeptideNanofiber Scaffold for Brain Repair and Axon Regeneration withFunctional Return of Vision. Proc. Natl. Acad. Sci. U. S. A. 2006, 103,5054−5059.(15) Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno,A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine Assembly into
Toxic Fibrils Suggests Amyloid Etiology in Phenylketonuria. Nat.Chem. Biol. 2012, 8, 701−706.(16) Flydal, M. I.; Martinez, A. Phenylalanine Hydroxylase: Function,Structure, and Regulation. IUBMB Life 2013, 65, 341−349.(17) Weissbuch, I.; Frolow, F.; Addadi, L.; Lahav, M.; Leiserowitz, L.Oriented Crystallization as a Tool for Detecting Ordered Aggregatesof Water-Soluble Hydrophobic Alpha-Amino Acids at the Air-SolutionInterface. J. Am. Chem. Soc. 1990, 112, 7718−7724.(18) King, M. D.; Blanton, T. N.; Korter, T. M. Revealing the TrueCrystal Structure of L- Phenylalanine Using Solid-State DensityFunctional Theory. Phys. Chem. Chem. Phys. 2012, 14, 1113−1116.(19) Pohl, G.; Plumley, J. A.; Dannenberg, J. J. The Interactions ofPhenylalanines in Beta-Sheet-Like Structures From Molecular OrbitalCalculations Using Density Functional Theory (DFT), MP2, andCCSD(T) Methods. J. Chem. Phys. 2013, 138, 245102/1−245102/5.(20) Gorbitz, C. H. Nanotube Formation by HydrophobicDipeptides. Chem.Eur. J. 2001, 7, 5153−5159.(21) Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.;Apostolov, R.; Shirts, M. R.; Smith, J. C.; Kasson, P. M.; van der Spoel,D.; et al. GROMACS 4.5: A High-Throughput and Highly ParallelOpen Source Molecular Simulation Toolkit. Bioinformatics 2013, 29,845−854.(22) Best, R. B.; Buchete, N.; Hummer, G. Are Current MolecularDynamics Force Fields Too Helical? Biophys. J. 2008, 95, L07−L09.(23) Jorgensen, W. L.; Tirado-Rives, J. The OPLS PotentialFunctions for Proteins. Energy Minimizations for Crystals of CyclicPeptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1656−1671.(24) Hess, B.; van der Vegt, N. F. A. Hydration ThermodynamicProperties of Amino Acid Analogues: A Systematic Comparison ofBiomolecular Force Fields and Water Models. J. Phys. Chem. B 2006,110, 17616−17626.(25) Shirts, M. R.; Pitera, J. W.; Swope, W. C.; Pande, V. S. ExtremelyPrecise Free Energy Calculations of Amino Acid Side Chain Analogs:Comparison of Common Molecular Mechanics Force Fields forProteins. J. Chem. Phys. 2003, 119, 5740−5761.(26) Do, T. D.; LaPointe, N. E.; Economou, N. J.; Buratto, S. K.;Feinstein, S. C.; Shea, J.; Bowers, M. T. Effects of pH and Charge Stateon Peptide Assembly: The YVIFL Model System. J. Phys. Chem. B2013, 117, 10759−10768.(27) Lakshmanan, A.; Cheong, D. W.; Accardo, A.; Di, F. E.; Riekel,C.; Hauser, C. A. E. Aliphatic Peptides Show Similar Self-Assembly toAmyloid Core Sequences, Challenging the Importance of AromaticInteractions in Amyloidosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110,519−524.(28) Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: AnN*log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys.1993, 98, 10089−10092.(29) Essmann, U.; Perera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.;Pedersen, L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys.1995, 103, 8577−8593.(30) Parrinello, M.; Rahman, A. Polymorphic Transitions in SingleCrystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52,7182−7190.(31) DeLano, W. L. PyMOL Molecular Graphics System, version1.3.0.4; Schrodinger, LLC, 2002.(32) Jeon, J.; Mills, C. E.; Shell, M. S. Molecular Insights intoDiphenylalanine Nanotube Assembly: All-Atom Simulations ofOligomerization. J. Phys. Chem. B 2013, 117, 3935−3943.(33) Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M. P.;Drar, R. O.; Shaw, D. E. Systematic Validation of Protein Force FieldsAgainst Experimental Data. PLoS One 2012, 7, e32131.(34) Piana, S.; Lindorff-Larsen, K.; Shaw, D. E. How Robust AreProtein Folding Simulations with Respect to Force Field Parameter-ization? Biophys. J. 2011, 100, L47−L49.(35) Kono, K.; Okano, Y.; Nakayama, K.; Hase, Y.; Minamikawa, S.;Ozawa, N.; Yokote, H.; Inoue, Y. Diffusion-weighted MR imaging inpatients with Phenylketonuria: Relationship Between Serum Phenyl-alanine Levels and ADC Values in Cerebral White Matter. Radiology2005, 236, 630−636.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5077388 | J. Phys. Chem. A 2015, 119, 1609−16151615