s-phase kinase protein 2 is an attractive therapeutic target in a subset of diffuse large b-cell...
TRANSCRIPT

Journal of PathologyJ Pathol 2008; 216: 483–494Published online 26 August 2008 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/path.2433
Original Paper
S-phase kinase protein 2 is an attractive therapeutictarget in a subset of diffuse large B-cell lymphoma
S Uddin,1† A Hussain,1† M Ahmed,1 A Belgaumi,2 F Al-Dayel,3 D Ajarim,4 P Bavi1 and KS Al-Kuraya1*1Department of Human Cancer Genomic Research, Research Center, King Fahad National Center for Children’s Cancer & Research, King FaisalSpecialist Hospital and Research Centre, Riyadh, Saudi Arabia2Paediatric Haematology-Oncology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia3Department of Pathology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia4King Faisal Cancer Centre, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
*Correspondence to:KS Al-Kuraya, MD, FCAP,Department of Cancer GenomicResearch Program, King FaisalSpecialist Hospital and ResearchCancer, MBC#98-16, PO Box3354, Riyadh 11211, SaudiArabia.E-mail: [email protected]
†Joint first author.
No conflicts of interest weredeclared.
Received: 27 June 2008Revised: 27 July 2008Accepted: 15 August 2008
AbstractS-phase kinase protein 2 (SKP2), an F-box protein, targets cell-cycle regulators includingcycle-dependent kinase inhibitor p27KiP1 via ubiquitin-mediated degradation. SKP2 isfrequently overexpressed in a variety of cancer cells and has been implicated in oncogenesis;however, its role in diffuse large B-cell lymphoma (DLBCL) has not been elucidated.Therefore, we investigated the role of SKP2 and its ubiquitin-proteasome pathway in alarge series (301) of DLBCL patient samples and a panel of DLBCL cell lines. Usingimmunohistochemistry, SKP2 was detected in 41.6% of DLBCL tumours and was inverselyassociated with p27Kip1 protein level. The DLBCL subset with high SKP2 and low p27Kip1showed a strong correlation with the proliferating index marker Ki-67 (p < 0.0001) andalso with the germinal centre phenotype (p = 0.0147). Treatment of DLBCL cell lineswith bortezomib or expression of SKP2-specific siRNA causes down-regulation of SKP2and accumulation of p27Kip1, leading to suppression of growth by inducing apoptosis.Furthermore, treatment of DLBCL cells with bortezomib causes apoptosis via involvingthe mitochondrial pathway and activation of caspases. Finally, treatment of DLBCL cellswith bortezomib down-regulated the expression of XIAP, cIAP1, and survivin. Altogether,these results suggest that SKP2 and the ubiquitin-proteasome pathway may be a potentialtarget for therapeutic intervention in DLBCL.Copyright 2008 Pathological Society of Great Britain and Ireland. Published by JohnWiley & Sons, Ltd.
Keywords: DLBCL; SKP2; apoptosis; NHL therapy; tissue microarray
Introduction
B-cell lymphoma represents the malignant counterpartof a normal B cell arrested at specific maturationalstages. Diffuse large B-cell lymphoma (DLBCL) isconsidered to be the most common type of lym-phoma in adults, accounting for 30–40% of casesof non-Hodgkin lymphoma [1,2]. The incidence ofDLBCL is about 40–60% of all adult non-Hodgkinlymphomas in Saudi Arabia [3]. The reason for thehigh rate of DLBCL in the Saudi Arabian popula-tion is not known; however, recent studies suggest thatdifferences in the molecular signature, compared withwestern populations, account for this incidence [4–6].Although patients with DLBCL are potentially curablewith combination chemotherapy, the disease provesfatal in approximately 50% of patients [7]. The causeof most DLBCLs remains unknown; however, severalstudies suggest that dysregulated survival/apoptosis ordefective repair pathways are involved [8–10].
The proteasome is an intracellular multi-catalyticcomplex which together with the two regulatorsPA28 (also called proteasome 11S) and PA700 (alsocalled proteasome 19S) forms the 26S proteasome[11,12]. This constitutes the ubiquitin–proteasomesystem (UPS) and regulates a number of intracellularproteins that govern the cell cycle, tumours, growth,and survival via the degradation of a number of differ-ent polypeptides important for cell cycle progressionand apoptosis [13–16]. The cyclin-dependent kinaseinhibitor p27Kip1 is a critical cell-cycle regulatorthat is frequently altered in human cancer, includinglymphoma [17]. The main rate-limiting regulator forp27Kip1 degradation has recently been identified as anSCF (Skp1–Cullin–F-box)-type ubiquitin ligase com-plex that contains S-phase kinase protein 2 (SKP2) asthe specific substrate-recognition subunit [18–20].
Recently, it has shown that decreased p27Kip1level in cancer cells is associated with enhancedubiquitin-dependent degradation and that increasedSKP2 overexpression is linked to a poor prognosis
Copyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.www.pathsoc.org.uk
484 S Uddin et al
[21]. Therefore, restoration of p27Kip1 by inhibitingSCFSKP2 activity has been proposed as a novel ther-apeutic strategy. Recently, the proteasome inhibitorbortezomib (Velcade) has been approved by the FDAfor the treatment of multiple myeloma and mantle celllymphoma patients who have received at least oneprior therapy.
We therefore analysed the SKP2 and p27 expressionstatus in 301 patients with DLBCL and investigatedany potential correlations with clinicopathological fea-tures. Furthermore, we provide evidence demonstrat-ing that bortezomib treatment of DLBCL cell linesresults in growth arrest and elevated p27Kip1 proteinlevels through down-regulation of SCFSKP2 ubiquitinligase, SKP2, implicating a role for the SKP2/p27Kip1signalling pathway in DLBCL pathogenesis and thepotential therapeutic value of the proteasome inhibitorin the treatment of DLBCL.
Materials and methods
Construction of tissue microarrays (TMAs)
TMAs were constructed as described previously [22].Briefly, three tissue cylinders of diameter 0.6 mm werepunched from archival paraffin blocks of 301 DLBCLpatients and transferred into a recipient paraffin block
using a semi-automatic precision instrument (BeecherInstruments, Silver Spring, MD, USA).
Patient samples
Three hundred and one cases of de novo DLBCLdiagnosed between 1987 and 2006 and reclassifiedaccording to the WHO criteria [23] were collectedfrom the Department of Pathology at the King FaisalSpecialist Hospital and Research Centre. Archivalparaffin blocks and clinical data were obtained byreviewing the charts, according to the regulations ofthe hospital institutional review board. Patients werestaged by means of computerized imaging and bonemarrow biopsy.
Immunohistochemistry (IHC)
TMA slides were processed and stained manually.The IHC protocol was followed as mentioned before[24]. The streptavidin–biotin peroxidase techniquewith diaminobenzidine as chromogen was applied. Theprimary antibodies were diluted in a 1% solution ofbovine serum albumin in PBS and incubated overnightat room temperature. The primary antibodies used,their dilutions, and cut-off levels for evaluation arelisted in the Supporting information, SupplementaryTable 1. For antigen retrieval, Dako Target Retrieval
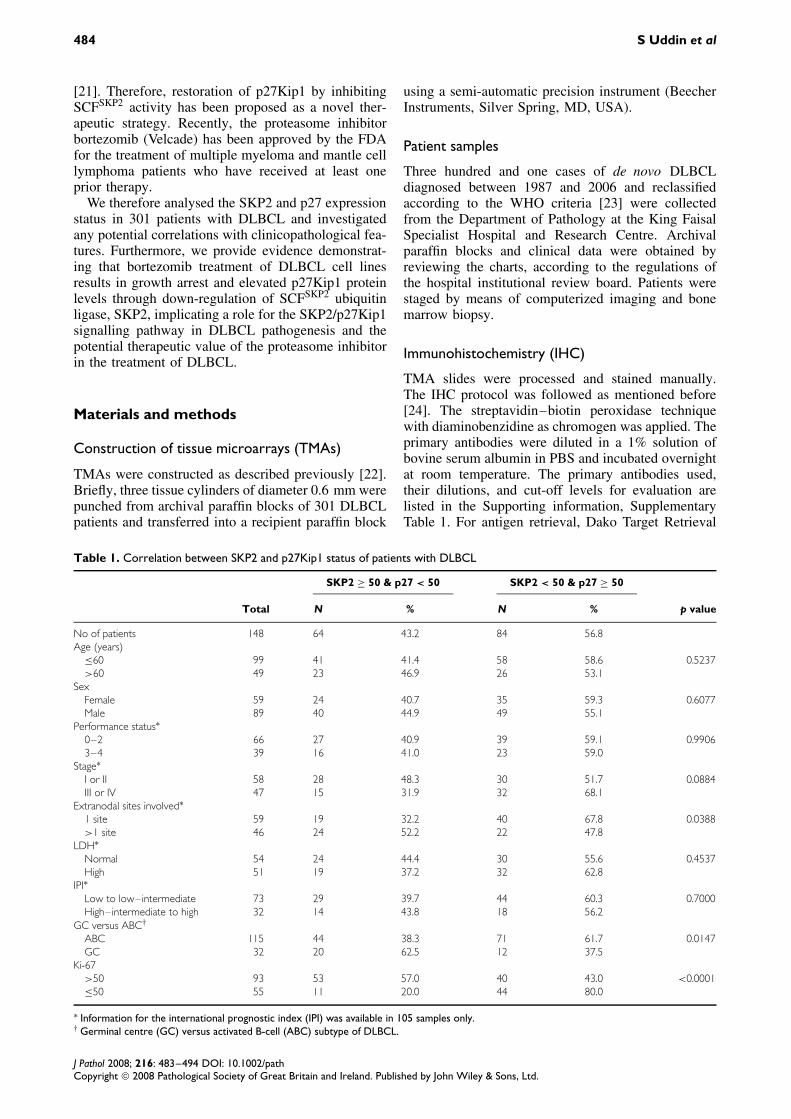
Table 1. Correlation between SKP2 and p27Kip1 status of patients with DLBCL
SKP2 ≥ 50 & p27 < 50 SKP2 < 50 & p27 ≥ 50
Total N % N % p value
No of patients 148 64 43.2 84 56.8Age (years)
≤60 99 41 41.4 58 58.6 0.5237>60 49 23 46.9 26 53.1
SexFemale 59 24 40.7 35 59.3 0.6077Male 89 40 44.9 49 55.1
Performance status∗0–2 66 27 40.9 39 59.1 0.99063–4 39 16 41.0 23 59.0
Stage∗I or II 58 28 48.3 30 51.7 0.0884III or IV 47 15 31.9 32 68.1
Extranodal sites involved∗1 site 59 19 32.2 40 67.8 0.0388>1 site 46 24 52.2 22 47.8
LDH∗Normal 54 24 44.4 30 55.6 0.4537High 51 19 37.2 32 62.8
IPI∗Low to low–intermediate 73 29 39.7 44 60.3 0.7000High–intermediate to high 32 14 43.8 18 56.2
GC versus ABC†
ABC 115 44 38.3 71 61.7 0.0147GC 32 20 62.5 12 37.5
Ki-67>50 93 53 57.0 40 43.0 <0.0001≤50 55 11 20.0 44 80.0
∗ Information for the international prognostic index (IPI) was available in 105 samples only.† Germinal centre (GC) versus activated B-cell (ABC) subtype of DLBCL.
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

Bortezomib-induced growth inhibition of DLBCL 485
Solution, pH 9.0 (catalogue number S2368) was used,and the slides were treated in a microwave oven at750 W for 5 min and then at 250 W for 20 min.The Dako Envision Plus System kit was used asthe secondary detection system, with DAB as chro-mogen. All slides were counterstained with haema-toxylin, dehydrated, cleared, and mounted. Negativecontrols included replacement of the primary antibodywith no reacting antibodies of the same species. Onlyfreshly cut slides were stained simultaneously, to min-imize the influence of slide ageing and to maximizereproducibility of the experiment.
Because intensity varies between cases due to dif-ferent tissue preservation, only the relative proportion(percentage) of positively stained tumour cells andthe findings were recorded in 10% increments. Foreach case, the core with the highest percentage oftumour cells stained was used for analysis. Cases wereconsidered positive if 50% or more of tumour cellsstained positive for p27Kip1 and SKP2. Normal SKP2expression was defined as less than 50% of tumourcells showing SKP2 expression and normal p27Kip1expression was considered as ≥50% of tumour cellsshowing p27Kip1 expression. This cut-off was cho-sen based on prior analysis on some of the markersand a similar cut-off used by others with TMA [25].The expression of CD10, BCL6, and MUM1 was con-sidered to classify tumours as germinal centre B-cell(GC)-like DLBCL and activated B cell (ABC), apply-ing the decision tree described earlier [26].
Statistical analysis
The software used for statistical analysis was Statview5.0 (SAS Institute Inc, NC, USA). Chi-square testswere used to examine relationships between nominalvariables. The limit of significance for all analyses wasdefined as a p value of 0.05; two-sided tests were usedin all calculations. Spearman’s rho correlation coef-ficient was used to describe the correlation betweenSKP2 and p27Kip1.
Cell culture
The human DLBCL cell lines SUDHL4, SUDHL5,SUDHL8, and OCI-LY19 were obtained fromDeutsche Sammlung von Mikroorganismen und Zel-lkulturen (DSMZ), Braunschweig, Germany. All thecell lines included in this study were derived fromthe germinal centre. The cell lines were cultured inRPMI 1640 medium supplemented with 20% (v/v)fetal bovine serum (FBS), 100 U/ml penicillin, and100 U/ml streptomycin at 37 ◦C in a humidified atmo-sphere containing 5% CO2. All experiments were per-formed in RPMI 1640 containing 5% serum.
Reagents and antibodies
Bortezomib (Velcade) was a gift from MilleniumPharmaceuticals Inc, Cambridge, MA, USA). MG132
was obtained from Calbiochem (San Diego, CA,USA). SKP2, p27Kip1, Bax, cytochrome c, caspase-3anti-PARP, Ub (ubiquitin) and beta-actin antibodieswere purchased from Santa Cruz Biotechnology, Inc(Santa Cruz, CA, USA). Bax 6A7 monoclonal anti-body was purchased from Sigma Chemical Co (StLouis, MO, USA) The anti-cleaved caspase-3 and anti-Bid antibodies were purchased from Cell SignalingTechnologies (Beverly, MA, USA). Caspase-8, cIAP,survivin, and XIAP antibodies were obtained fromR&D Systems (Minneapolis, MN, USA). AnnexinV was purchased from Molecular Probes (Eugene,OR, USA). JC1 was purchased from Alexis (SanDiego, CA, USA). The apoptotic DNA-ladder kit wasobtained from Roche (Penzberg, Germany).
MTT assays
The anti-proliferative effects of bortezomib againstdifferent DLBCL cell lines were determined by theMTT dye uptake method, as described earlier [27].
Annexin V staining
DLBCL cell lines were treated with different con-centrations of bortezomib as described in the figurelegends. Cells were harvested and the percentage ofapoptosis was measured by flow cytometry after stain-ing with fluorescein-conjugated annexin V and pro-pidium iodide (PI) (Molecular Probes), as describedpreviously [27].
Cell cycle analysis
Cell lines were either treated with or without borte-zomib for 24 h and the cells were washed once withphosphate-buffered saline (PBS) and resuspended in500 µl of hypotonic staining buffer (250 mg of sodiumcitrate, 0.75 ml of Triton X, 25 µg of propidiumiodide, 5 µg of ribonuclease A, and 250 ml of water)and analysed by flow cytometry as described previ-ously [28].
DNA laddering
DNA laddering experiments were performed asdescribed previously [27]. Briefly, 2 × 106 cells weretreated with 10 and 25 nM bortezomib for 24 h andthe cells were harvested and resuspended in 200 µlof 1× PBS. Two hundred microlitres of lysis buffercontaining 6 M guanidine–HCl, 10 mM urea, 10 mM
Tris–HCl, and 20% Triton X (v/v), pH 4.4 was addedto the cells and the mixture was incubated for 10 minat room temperature. DNA was extracted using aRoche apoptotic kit and 2 µg of DNA was elec-trophoresed on a 1.5% agarose gel containing ethidiumbromide at 75 V for 2 h and visualized using a UVlight source.
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

486 S Uddin et al
Cell lysis and immunoblotting
Cells were treated with bortezomib as described in thefigure legends and lysed as previously described [29].Briefly, cell pellets were resuspended in phosphory-lation lysis buffer (0.5–1.0% Triton X-100, 150 mM
NaCl, 1 mM EDTA, 200 µM sodium orthovanadate,10 mM sodium pyrophosphate, 100 mM sodium flu-oride, 1.5 mM magnesium chloride, 1 mM phenyl-methylsulphonyl fluoride, and 10 µg/ml aprotonin).Equal amount of proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoridemembrane (Immobilion, Millipore, etc). Immunoblot-ting was performed with different antibodies andvisualized by an enhanced chemiluminescence (ECL;Amersham, Illinois, USA) method.
Detection of Bax conformational changes
This assay was performed as described previously[27]. Briefly, following treatment with bortezomib forindicated time points, cells were harvested and washedwith PBS, after which they were lysed with Chapslysis buffer [10 mM HEPES (pH 7.4), 150 mM NaCl,1% Chaps] containing protease inhibitors. Proteinconcentrations were assessed by Bradford assay and500 µg of total protein was incubated with 2 µg ofanti-Bax 6A7 monoclonal antibody for 2 h at 4 ◦C.Following incubation, 25 µl of protein G-beads wasadded to the reaction and incubated overnight on ashaker with gentle agitation. Following five washesin Chaps lysis buffer, samples were separated bySDS-PAGE, transferred, and immunoblotted using Baxpolyclonal antibody.
Assay for cytochrome c release
Release of cytochrome c from mitochondria wasassayed as described earlier [30]. Briefly, cells weretreated with or without bortezomib as described in thefigure legends and centrifuged at 1000 g for 10 min.Cell pellets were resuspended in 5 volumes of hypo-tonic buffer and incubated for 15 min on ice. Cellswere homogenized and lysates were centrifuged topellet nuclei and unbroken cells. Supernatants werecollected by centrifugation and the resulting mitochon-drial pellets were resuspended in lysis buffer. Twentymicrograms of proteins from the cytosolic and mito-chondrial fractions of each sample was analysed byimmunoblotting using an anti-cytochrome c antibody.
Measurement of mitochondrial potential using theJC-1 (5,5′,6,6′-teterachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) assaykit
1 × 106 cells were treated with bortezomib for 24 h.Cells were washed twice with PBS and suspendedin mitochondrial incubation buffer (Alexis Corp, SanDiego, CA). JC1 was added to a final concentration of10 µM and cells were incubated at 37 ◦C in the dark for
30 min. Cells were washed twice with PBS and resus-pended in 500 µl of mitochondrial incubation buffer,and the mitochondrial membrane potential (percentageof green and red aggregates) was determined by flowcytometry as described previously [31].
Results
Expression of SKP2 and p27Kip1 in DLBCLtumours
Levels of SKP2 and p27Kip1 were examined byimmunohistochemistry in a series of 301 DLBCLs.Representative information was available in 286/301spots for SKP2 expression and 253/301 spots forp27Kip1. A typical case, shown in Figure 1A, illus-trates the decrease in nuclear staining of p27Kip1,increased levels of SKP2 and Ki-67 in a tumoursample compared with low SKP2, Ki-67, and highp27Kip1 expression in another DLBCL tumour sam-ple. SKP2 overexpression was seen in 119/286 (41.6%)and loss of p27Kip1 expression was seen in 121/253(47.8%) DLBCL samples analysed. SKP2 expressionshowed a significant inverse correlation with p27Kip1expression (ρ = −0.1409; p = 0.0259, Spearman’srho correlation, see Supporting information, Supple-mentary Figure 1), which is in concordance with previ-ous findings [32]. We further stratified all our DLBCLcases into four groups depending on the presenceof SKP2 expression and p27Kip1 expression sta-tus: SKP2 overexpression/normal p27Kip1 group (n =46); normal SKP2/normal p27Kip1 group (n = 84);SKP2 overexpression/low p27Kip1 expression group(n = 64); and DLBCLs with normal SKP2 expres-sion/low p27Kip1 expression (n = 56). Of these fourgroups, only the two subgroups showing inverse asso-ciations between SKP2 and p27Kip1 expression wereselected for further analysis. The clinicopathologicaland immunohistochemical analyses of the above sub-set of 148 DLBCL patients are summarized in Table 1.Such stratification showed a significant associationbetween the SKP2 overexpression/loss of p27Kip1DLBCL group and involvement of more than oneextranodal site (p = 0.0388) and overexpression ofthe proliferative marker Ki-67 (p < 0.0001). Thegerminal centre (GC) subtype of DLBCLs showedhigher SKP2 and low p27Kip1 levels comparedwith the activated B-cell (ABC) subtype of DLBCLs(p = 0.0147).
Bortezomib causes cell growth inhibition andinduces apoptosis in DLBCL cells
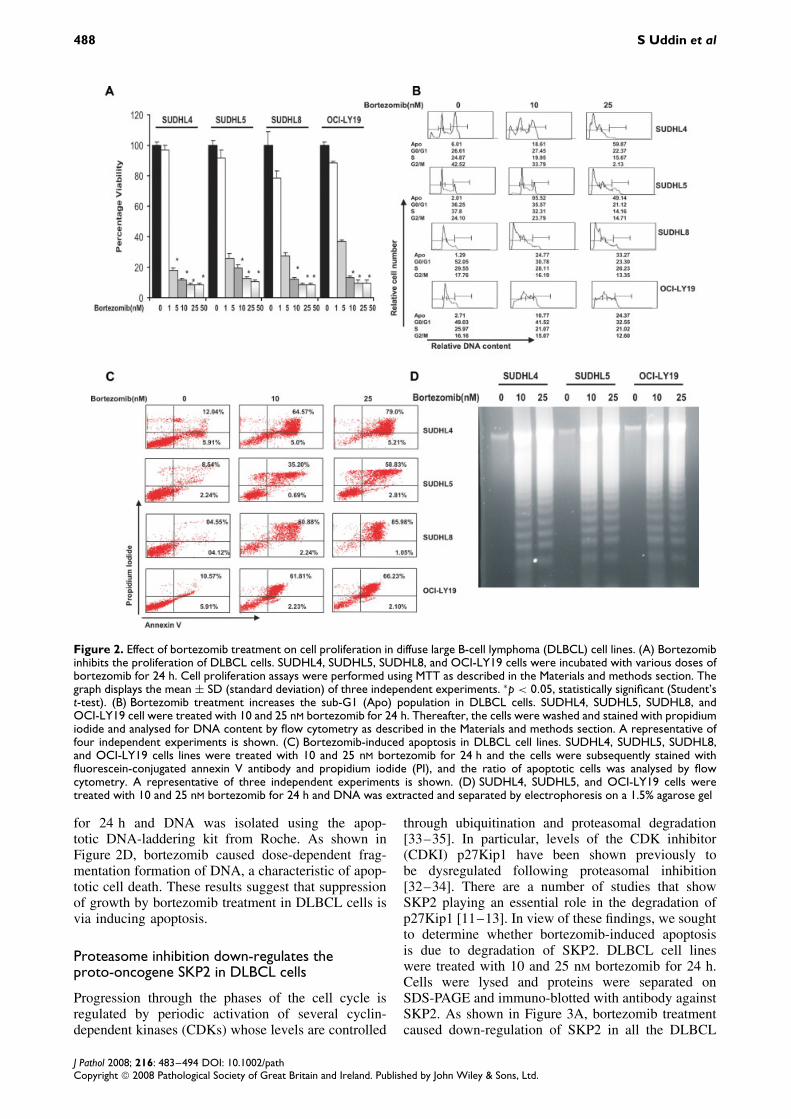
We first sought to determine whether proteasomeinhibitor bortezomib treatment leads to growth inhi-bition of the DLBCL cell lines. SUDHL4, SUDHL5,SUDHL8, and OCI-LY19 cells were treated with var-ious doses ranging from 1 to 100 nM bortezomibfor 24 h and cell proliferation was determined byMTT assays. Figure 2A shows that as the dose of
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

Bortezomib-induced growth inhibition of DLBCL 487
Figure 1. (A) Tissue microarray-based immunohistochemical analysis of SKP2, p27Kip1, and Ki-67 in DLBCL patients. DLBCLarray spot showing overexpression of SKP2 (i), low expression of p27Kip1 (ii), and high expression of proliferative marker Ki-67(iii). In contrast, another DLBCL tissue array spot shows low expression of SKP2 (iv), high expression of p27Kip1 (v), and lowexpression of Ki-67 (vi). Original magnification 20×, with the inset showing a 100 × magnified view of the same. (B) Overview ofthe tissue microarray slide containing 301 DLBCL samples
bortezomib increased from 1 to 100 nM, cell growthinhibition increased significantly in a dose-dependentmanner in all of the cell lines.
To determine whether inhibition of cell viability byproteasome inhibition was attributable to cell cyclearrest or apoptosis, DLBCL cells were treated withand without 10 and 25 nM bortezomib for 24 h. Cellcycle fractions were determined by flow cytometry.As shown in Figure 2B, the sub-G1 population ofSUDHL4 cells increased from 6.01% in the con-trol to 18.61% and 59.87% at 1 and 25 nM borte-zomib, respectively. Similar results were obtainedin SUDHL5, from 2.01% to 5.52% and 49.14%;SUDHL8, from 1.29% to 24.77% and 33.27%; andOCI-LY19, from 2.71% to 10.77% and 24.37%increase in the sub-G1 population. It has been reported
that cells with these features are those dying ofapoptosis [8]. These results indicate that DLBCL cellsunderwent apoptosis rather than cell cycle arrestfollowing bortezomib treatment.
To confirm further that this increase in the sub-G1population is indeed apoptosis, DLBCL cells weretreated with 10 and 25 nM bortezomib and apop-totic cells were assayed by annexin V/PI dual stain-ing. As shown in Figure 2C, treatment of SUDHL4,SUDHL5, SUDHL8, and OCI-LY19 cells with borte-zomib resulted in apoptosis, as demonstrated by anincrease in the population of cells in the upper rightquadrant depicting apoptotic cells. Finally, we anal-ysed DNA fragmentation, which is another hallmarkof apoptosis. SUDHL4, SUDHL5, and OCI-LY19cells were treated with 10 and 25 nM bortezomib
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
488 S Uddin et al
Figure 2. Effect of bortezomib treatment on cell proliferation in diffuse large B-cell lymphoma (DLBCL) cell lines. (A) Bortezomibinhibits the proliferation of DLBCL cells. SUDHL4, SUDHL5, SUDHL8, and OCI-LY19 cells were incubated with various doses ofbortezomib for 24 h. Cell proliferation assays were performed using MTT as described in the Materials and methods section. Thegraph displays the mean ± SD (standard deviation) of three independent experiments. ∗p < 0.05, statistically significant (Student’st-test). (B) Bortezomib treatment increases the sub-G1 (Apo) population in DLBCL cells. SUDHL4, SUDHL5, SUDHL8, andOCI-LY19 cell were treated with 10 and 25 nM bortezomib for 24 h. Thereafter, the cells were washed and stained with propidiumiodide and analysed for DNA content by flow cytometry as described in the Materials and methods section. A representative offour independent experiments is shown. (C) Bortezomib-induced apoptosis in DLBCL cell lines. SUDHL4, SUDHL5, SUDHL8,and OCI-LY19 cells lines were treated with 10 and 25 nM bortezomib for 24 h and the cells were subsequently stained withfluorescein-conjugated annexin V antibody and propidium iodide (PI), and the ratio of apoptotic cells was analysed by flowcytometry. A representative of three independent experiments is shown. (D) SUDHL4, SUDHL5, and OCI-LY19 cells weretreated with 10 and 25 nM bortezomib for 24 h and DNA was extracted and separated by electrophoresis on a 1.5% agarose gel
for 24 h and DNA was isolated using the apop-totic DNA-laddering kit from Roche. As shown inFigure 2D, bortezomib caused dose-dependent frag-mentation formation of DNA, a characteristic of apop-totic cell death. These results suggest that suppressionof growth by bortezomib treatment in DLBCL cells isvia inducing apoptosis.
Proteasome inhibition down-regulates theproto-oncogene SKP2 in DLBCL cells
Progression through the phases of the cell cycle isregulated by periodic activation of several cyclin-dependent kinases (CDKs) whose levels are controlled
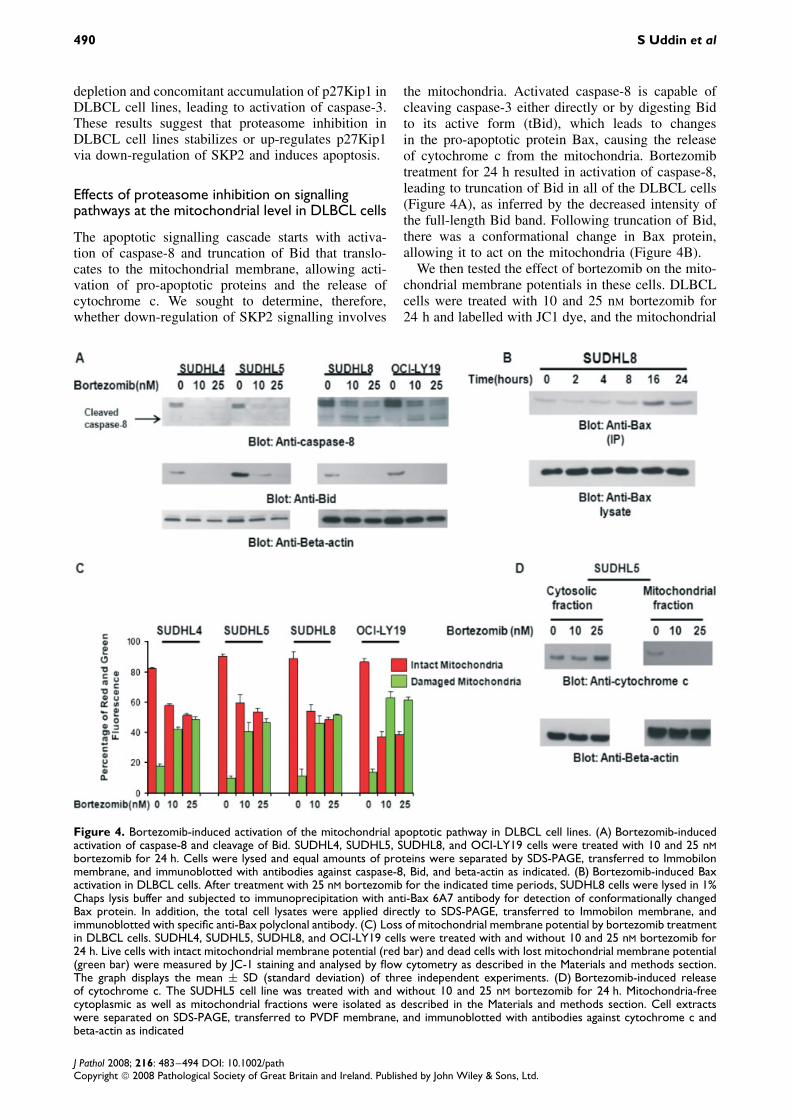
through ubiquitination and proteasomal degradation[33–35]. In particular, levels of the CDK inhibitor(CDKI) p27Kip1 have been shown previously tobe dysregulated following proteasomal inhibition[32–34]. There are a number of studies that showSKP2 playing an essential role in the degradation ofp27Kip1 [11–13]. In view of these findings, we soughtto determine whether bortezomib-induced apoptosisis due to degradation of SKP2. DLBCL cell lineswere treated with 10 and 25 nM bortezomib for 24 h.Cells were lysed and proteins were separated onSDS-PAGE and immuno-blotted with antibody againstSKP2. As shown in Figure 3A, bortezomib treatmentcaused down-regulation of SKP2 in all the DLBCL
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

Bortezomib-induced growth inhibition of DLBCL 489
Figure 3. Down-regulation of the SKP2 pathway during bortezomib treatment causes the accumulation of ubiquitinated proteinsand induced or stabilized p27Kip1. (A) Bortezomib treatment down-regulated SKP2 and increased or stabilized the level ofp27Kip1. SUDHL4, SUDHL5, SUDHL8, and OCI-LY19 cells were treated with and without 10 and 25 nM bortezomib for 24 h.After cell lysis, equal amounts of proteins were separated by SDS-PAGE, transferred to Immobilon membrane, and immunoblottedwith antibodies against SKP2, p27Kip1, and beta-actin as indicated. A representative of three different experiments is shown.(B) MG132 treatment causes down-regulation of SKP2 and up-regulation/stabilization of p27Kip1. SUDHL4 and SUDHL5 cellswere treated with and without 0.5, 1, 5, and 10 µM MG132 for 24 h. After cell lysis, equal amounts of proteins were separatedby SDS-PAGE, transferred to Immobilon membrane, and immunoblotted with antibodies against SKP2, p27Kip1, and beta-actin.(C) SKP2 SiRNA expression down-regulates SKP2 and accumulates p27Kip1. SUDHL5 and SUDHL8 cells were transfected withScrambled siRNA (100 nM) and SKP2 siRNA (50 and 100 nM) with Lipofectamine as described in the Materials and methodssection. After 48 h of transfection, cells were lysed and equal amounts of proteins were separated by SDS-PAGE, transferredto Immobilon membrane, and immunoblotted with antibodies against SKP2, P27Kip1, caspase-3, and beta-actin as indicated. Arepresentative of three separate independent experiments is depicted
cell lines, leading to up-regulation of p27Kip1. Astrong inverse relationship was found between SKP2and p27Kip1 expression after bortezomib treatment.Similar results were obtained with MG-132, anotherinhibitor of proteasome (Figure 3B). To provide directevidence that down-regulation of SKP2 up-regulates
p27Kip1 and induces apoptosis, we attempted toinhibit SKP2 expression in SUDHL5 and SUDHL8cells by the SiRNA strategy. For this purpose, SKP2activity was blocked using siRNA specifically targetedagainst SKP2. As expected, in Figure 3C, transfectionof siRNA against SKP2 resulted in SKP2 protein
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
490 S Uddin et al
depletion and concomitant accumulation of p27Kip1 inDLBCL cell lines, leading to activation of caspase-3.These results suggest that proteasome inhibition inDLBCL cell lines stabilizes or up-regulates p27Kip1via down-regulation of SKP2 and induces apoptosis.
Effects of proteasome inhibition on signallingpathways at the mitochondrial level in DLBCL cells
The apoptotic signalling cascade starts with activa-tion of caspase-8 and truncation of Bid that translo-cates to the mitochondrial membrane, allowing acti-vation of pro-apoptotic proteins and the release ofcytochrome c. We sought to determine, therefore,whether down-regulation of SKP2 signalling involves
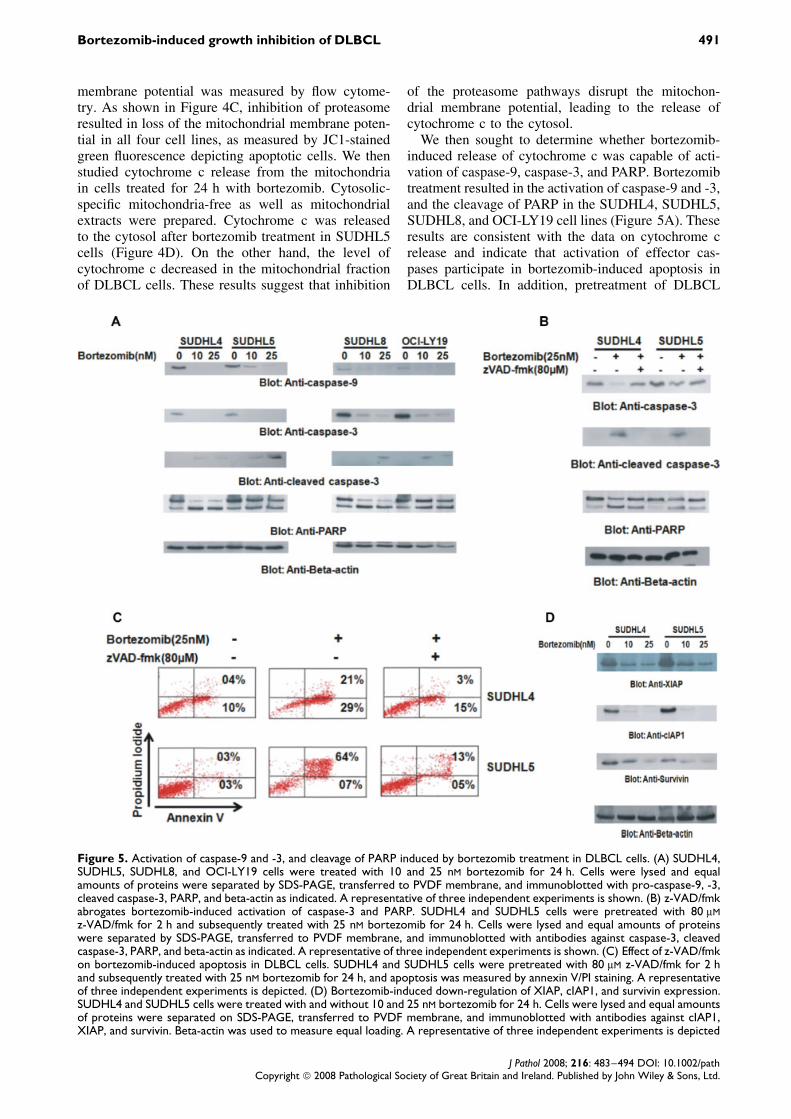
the mitochondria. Activated caspase-8 is capable ofcleaving caspase-3 either directly or by digesting Bidto its active form (tBid), which leads to changesin the pro-apoptotic protein Bax, causing the releaseof cytochrome c from the mitochondria. Bortezomibtreatment for 24 h resulted in activation of caspase-8,leading to truncation of Bid in all of the DLBCL cells(Figure 4A), as inferred by the decreased intensity ofthe full-length Bid band. Following truncation of Bid,there was a conformational change in Bax protein,allowing it to act on the mitochondria (Figure 4B).
We then tested the effect of bortezomib on the mito-chondrial membrane potentials in these cells. DLBCLcells were treated with 10 and 25 nM bortezomib for24 h and labelled with JC1 dye, and the mitochondrial
Figure 4. Bortezomib-induced activation of the mitochondrial apoptotic pathway in DLBCL cell lines. (A) Bortezomib-inducedactivation of caspase-8 and cleavage of Bid. SUDHL4, SUDHL5, SUDHL8, and OCI-LY19 cells were treated with 10 and 25 nMbortezomib for 24 h. Cells were lysed and equal amounts of proteins were separated by SDS-PAGE, transferred to Immobilonmembrane, and immunoblotted with antibodies against caspase-8, Bid, and beta-actin as indicated. (B) Bortezomib-induced Baxactivation in DLBCL cells. After treatment with 25 nM bortezomib for the indicated time periods, SUDHL8 cells were lysed in 1%Chaps lysis buffer and subjected to immunoprecipitation with anti-Bax 6A7 antibody for detection of conformationally changedBax protein. In addition, the total cell lysates were applied directly to SDS-PAGE, transferred to Immobilon membrane, andimmunoblotted with specific anti-Bax polyclonal antibody. (C) Loss of mitochondrial membrane potential by bortezomib treatmentin DLBCL cells. SUDHL4, SUDHL5, SUDHL8, and OCI-LY19 cells were treated with and without 10 and 25 nM bortezomib for24 h. Live cells with intact mitochondrial membrane potential (red bar) and dead cells with lost mitochondrial membrane potential(green bar) were measured by JC-1 staining and analysed by flow cytometry as described in the Materials and methods section.The graph displays the mean ± SD (standard deviation) of three independent experiments. (D) Bortezomib-induced releaseof cytochrome c. The SUDHL5 cell line was treated with and without 10 and 25 nM bortezomib for 24 h. Mitochondria-freecytoplasmic as well as mitochondrial fractions were isolated as described in the Materials and methods section. Cell extractswere separated on SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibodies against cytochrome c andbeta-actin as indicated
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Bortezomib-induced growth inhibition of DLBCL 491
membrane potential was measured by flow cytome-try. As shown in Figure 4C, inhibition of proteasomeresulted in loss of the mitochondrial membrane poten-tial in all four cell lines, as measured by JC1-stainedgreen fluorescence depicting apoptotic cells. We thenstudied cytochrome c release from the mitochondriain cells treated for 24 h with bortezomib. Cytosolic-specific mitochondria-free as well as mitochondrialextracts were prepared. Cytochrome c was releasedto the cytosol after bortezomib treatment in SUDHL5cells (Figure 4D). On the other hand, the level ofcytochrome c decreased in the mitochondrial fractionof DLBCL cells. These results suggest that inhibition
of the proteasome pathways disrupt the mitochon-drial membrane potential, leading to the release ofcytochrome c to the cytosol.
We then sought to determine whether bortezomib-induced release of cytochrome c was capable of acti-vation of caspase-9, caspase-3, and PARP. Bortezomibtreatment resulted in the activation of caspase-9 and -3,and the cleavage of PARP in the SUDHL4, SUDHL5,SUDHL8, and OCI-LY19 cell lines (Figure 5A). Theseresults are consistent with the data on cytochrome crelease and indicate that activation of effector cas-pases participate in bortezomib-induced apoptosis inDLBCL cells. In addition, pretreatment of DLBCL
Figure 5. Activation of caspase-9 and -3, and cleavage of PARP induced by bortezomib treatment in DLBCL cells. (A) SUDHL4,SUDHL5, SUDHL8, and OCI-LY19 cells were treated with 10 and 25 nM bortezomib for 24 h. Cells were lysed and equalamounts of proteins were separated by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with pro-caspase-9, -3,cleaved caspase-3, PARP, and beta-actin as indicated. A representative of three independent experiments is shown. (B) z-VAD/fmkabrogates bortezomib-induced activation of caspase-3 and PARP. SUDHL4 and SUDHL5 cells were pretreated with 80 µMz-VAD/fmk for 2 h and subsequently treated with 25 nM bortezomib for 24 h. Cells were lysed and equal amounts of proteinswere separated by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibodies against caspase-3, cleavedcaspase-3, PARP, and beta-actin as indicated. A representative of three independent experiments is shown. (C) Effect of z-VAD/fmkon bortezomib-induced apoptosis in DLBCL cells. SUDHL4 and SUDHL5 cells were pretreated with 80 µM z-VAD/fmk for 2 hand subsequently treated with 25 nM bortezomib for 24 h, and apoptosis was measured by annexin V/PI staining. A representativeof three independent experiments is depicted. (D) Bortezomib-induced down-regulation of XIAP, cIAP1, and survivin expression.SUDHL4 and SUDHL5 cells were treated with and without 10 and 25 nM bortezomib for 24 h. Cells were lysed and equal amountsof proteins were separated on SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibodies against cIAP1,XIAP, and survivin. Beta-actin was used to measure equal loading. A representative of three independent experiments is depicted
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

492 S Uddin et al
cells with 80 µM z-VAD/fmk, a universal inhibitor ofcaspases, abrogated apoptosis and prevented apopto-sis and caspase-3 and PARP activation induced bybortezomib (Figures 5B and 5C), clearly indicatingthat caspases play a critical role in bortezomib-inducedapoptosis in DLBCL cells.
Modulation of the IAP family of proteins inbortezomib-induced apoptosis in DLBCL cell lines
We also examined whether bortezomib induced apop-tosis by modulating the expression of inhibitors ofapoptosis protein (IAP) family members, which ulti-mately determine the cell’s response to apoptotic stim-uli. SUDHL4 and SUDHL5 cells were treated with 10and 25 nM bortezomib for 24 h and the expressionof cIAP, XIAP, and survivin was determined usingwestern blotting. As shown in Figure 5D, bortezomibtreatment caused a dose-dependent down-regulation ofcIAP1, XIAP, and survivin. These results indicate thatIAP proteins may also be involved in bortezomib-induced apoptosis.
Discussion
Recent studies have clearly shown that SKP2 plays anessential role in the degradation of p27Kip1 [21,36].SKP2 protein enables the efficient transfer of ubiqui-tin of p27Kip1, resulting in rapid proteasome-mediateddegradation [18]. The interaction between these pro-teins was evident in a significant proportion of thetumour samples studied, whereby the expression ofSKP2 was inversely associated with p27Kip1 proteinlevels [21,37]. We therefore determined the role ofboth SKP2 and p27Kip1 in the aetiology of DLBCLand found that those DLBCL samples that exhibitedderegulation of SKP2 and p27Kip1, ie a high level ofSKP2 and a low level of p27Kip1, showed a strongcorrelation with Ki-67 and the germinal centre phe-notype, suggesting a possible role for SKP2 in thepathogenesis of this subset of DLBCLs. These clini-cal data as well as recent findings in cell lines suggesta potential role of SKP2-mediated p27Kip1 in lym-phoma [38–40]. All together, these findings promptedus to analyse p27Kip1 regulation in several DLBCLcell lines. Interestingly, we observed that DLBCLcell lines displayed lower levels of p27Kip1 and anincreased level of SKP2.
Proteasome inhibitors have been shown to stabilizea large number of cellular proteins, initially inducingcell cycle arrest and ultimately leading to programmedcell death. Our data showed that bortezomib treat-ment of DLBCL cells down-regulated the expressionof SKP2 with reciprocal up-regulation of p27Kip1, awell-known target of proteasome [21]. Inhibition ofSKP2 expression by specific SKP2 siRNA increasedp27Kip1 levels, strongly suggesting that bortezomib-mediated down-regulation of SKP2 and up-regulationofp27Kip1 likely play a significant role in the induc-tion of apoptosis in DLBCL cell lines. The precise
mechanism of bortezomib-mediated down-regulationof SKP2 is not known. Recently, SKP2 has beenshown to be down-regulated by cell adhesion to stromacells, leading to up-regulation of p27Kip1 [41]. Thesestudies further suggest that regulation of SKP2 occursat both translational and post-translational levels. Wespeculate that bortezomib-induced down-regulation ofSKP2 follows a similar mechanism; however, it needsfurther investigations. Apoptosis is a multi-step pro-cess and an increasing number of genes have beenidentified that are involved in the control or execu-tion of apoptosis [42]. Our study shows that proteaso-mal inhibition by bortezomib in DLBCL cells causedapoptosis via activation of caspase-8 and truncation ofBid that translocates to the mitochondrial membrane,allowing activation of pro-apoptotic proteins and therelease of cytochrome c into the cytosol. Releasedcytochrome c results in the formation of apoptosomeby interaction with apaf1 and caspase-9, leading tothe activation of caspase-3, eventually resulting in thecleavage of PARP in apoptotic cells — a hallmarkof apoptosis by various anti-tumour agents [43]. Fur-thermore, bortezomib treatment of DLBCL cell linesresults in down-regulation of the expression of cIAP,XIAP, and survivin, implicating a role for IAPs in theactivation of caspase-9 and -3 in bortezomib-inducedapoptosis. These data suggest that inhibition of ubiq-uitin–proteasome in DLBCL induces apoptosis viacaspase-cascade activation.
Together, our results establish that the SKP2 ubiqui-tin–proteasome plays a role in the growth and survivalof DLBCL cells. Down-regulation of SKP2 expressionleads to the accumulation and stabilization of p27Kip1,leading to the induction of apoptosis in DLBCL cellsthrough the release of cytochrome c from the mito-chondria and activation of downstream caspases. Inaddition, the DLBCL subgroup of patients with highSKP2 expression and a low level of p27Kip1 showeda strong correlation with Ki-67 and the germinal cen-tre subtype. These studies may have important impli-cations for future pre-clinical and clinical studies inDLBCL. Indeed, they may pave the way for investi-gations aimed at determining the usefulness of a novelstrategy for targeted therapy of the DLBCL subsetshowing alteration in the ubiquitin–proteasome sys-tem with inhibitors of proteasome pathways.
Acknowledgements
We would like to thank Sriraman Devarajan for clinical dataanalysis, and Saeeda Ahmed, Azadali Moorji, Valerie Atizado,Hasan Al-Dossari, and Valorie Balde for their technical assis-tance.
Supporting information
Supporting information may be found in the onlineversion of this article.
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

Bortezomib-induced growth inhibition of DLBCL 493
References
1. The Non-Hodgkin’s Lymphoma Classification Project. A clinicalevaluation of the International Lymphoma Study Group classifica-tion of non-Hodgkin’s lymphoma. Blood 1997;89:3909–3916.
2. Muris JJ, Cillessen SA, Vos W, van Houdt IS, Kummer JA, vanKrieken JH, et al. Immunohistochemical profiling of caspasesignaling pathways predicts clinical response to chemotherapyin primary nodal diffuse large B-cell lymphomas. Blood2005;105:2916–2923.
3. Bazarbashi S, De Vol E, Young S, Al-Eid H, Arteh S. CancerIncidence Report Saudi Arabia. National Cancer Registry2004;1999–2000.
4. Al-Kuraya KS, Siraj AK, Al-Dayel FA, Ezzat AA, Al-JommahNA, Atizado VL, et al. Epigenetic changes and their clinicalrelevance in Saudi diffuse large B-cell lymphoma. A molecu-lar and tissue microarray analysis of 100 cases. Saudi Med J2005;26:1099–1103.
5. Al-Kuraya K, Narayanappa R, Siraj AK, Al-Dayel F, Ezzat A, ElSolh H, et al. High frequency and strong prognostic relevanceof O6-methylguanine DNA methyltransferase silencing in diffuselarge B-cell lymphomas from the Middle East. Hum Pathol2006;37:742–748.
6. Al-Kuraya K, Siraj AK, Bavi P, Ezzat A, Al-Dayel F, Bel-gaumi A, et al. High throughput tissue microarray analysis ofFHIT expression in diffuse large B-cell lymphoma from SaudiArabia. Mod Pathol 2006;19:1124–1129.
7. Hartge P, Wang SS. Overview of the etiology and epidemiologyof lymphoma. In Non-Hodgkin’s Lymphomas, Mauch PM,Armitage JO, Coiffier B, Dalla-Favera R, Harris NL (eds).Lippincott, Williams and Wilkins: New York, 2004; 711–727.
8. Uddin S, Hussain AR, Siraj AK, Manogaran PS, Al-Jomah NA,Moorji A, et al. Role of phosphatidylinositol 3′-kinase/AKTpathway in diffuse large B-cell lymphoma survival. Blood2006;108:4178–4186.
9. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cat-toretti G, et al. NFkappaB activity, function, and target-gene sig-natures in primary mediastinal large B-cell lymphoma and diffuselarge B-cell lymphoma subtypes. Blood 2005;106:1392–1399.
10. Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma:mechanisms, consequences, and therapeutic implications. Blood2007;109:2700–2707.
11. Ciechanover A. The ubiquitin–proteasome proteolytic pathway.Cell 1994;79:13–21.
12. Ovaa H, Kessler BM, Rolen U, Galardy PJ, Ploegh HL, MasucciMG. Activity-based ubiquitin-specific protease (USP) profiling ofvirus-infected and malignant human cells. Proc Natl Acad SciU S A 2004;101:2253–2258.
13. Milano A, Iaffaioli RV, Caponigro F. The proteasome: a worth-while target for the treatment of solid tumours? Eur J Cancer2007;43:1125–1133.
14. Goldberg AL. Functions of the proteasome: the lysis at the end ofthe tunnel. Science 1995;268:522–523.
15. Cusack JC, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J,et al. Enhanced chemosensitivity to CPT-11 with proteasomeinhibitors PS-341: implications for systemic nuclear factor-kappaBinhibition. Cancer Res 2001;61:3535–3540.
16. McDade TP, Perugini RA, Vittimberga FJ Jr, Callery MP. Ubiqui-tin–proteasome inhibition enhances apoptosis of human pancreaticcancer cells. Surgery 1999;126:371–377.
17. Bai M, Tsanou E, Skyrlas A, Sainis I, Agnantis N, Kanavaros P.Alterations of the p53, Rb and p27 tumor suppressorpathways in diffuse large B-cell lymphomas. Anticancer Res2007;27:2345–2352.
18. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is requiredfor ubiquitin-mediated degradation of the CDK inhibitors p27.Nature Cell Biol 1999;1:193–199.
19. Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M,Muller U, et al. p45SKP2 promotes p27Kip1 degradationand induces S phase in quiescent cells. Nature Cell Biol1999;1:207–214.
20. Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1)ubiquitination and degradation is regulated by the SCF(Skp2)complex through phosphorylated Thr187 in p27. Curr Biol1999;9:661–664.
21. Uddin S, Ahmed M, Bavi P, El-Sayed R, Al-Sanea N, AbdulJabbar A, et al. Bortezomib (Velcade) induces p27Kip1 expressionthrough S-phase kinase protein 2 degradation in colorectal cancer.Cancer Res 2008;68:3379–3388.
22. Abubaker J, Jehan Z, Bavi P, Sultana M, Al-Harbi S, Ibrahim M,et al. Clinicopathological analysis of papillary thyroid cancerwith PIK3CA alterations in a Middle Eastern population. J ClinEndocrinol Metab 2008;93:611–618.
23. Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology andGenetics of Tumors of Hematopoetic and Lymphoid Tissues: WorldHealth Organization Classification of Tumors. IARC Press: Lyon,2001.
24. Bavi P, Jehan Z, Atizado V, Al-Dossari H, Al-Dayel F, Tulbah A,et al. Prevalence of fragile histidine triad expression in tumorsfrom Saudi Arabia: a tissue microarray analysis. Cancer EpidemiolBiomarkers Prev 2006;15:1708–1718.
25. Shapira M, Ben-Izhak O, Bishara B, Futerman B, Minkov I,Krausz MM, et al. Alterations in the expression of the cell cycleregulatory protein cyclin kinase subunit 1 in colorectal carcinoma.Cancer 2004;8:1615–1621.
26. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Dela-bie J, Ott G, et al. Confirmation of the molecular classificationof diffuse large B-cell lymphoma by immunohistochemistry usinga tissue microarray, Blood 2004;103:275–282.
27. Hussain AR, Al-Jomah NA, Siraj AK, Manogaran P, Al-Hussein K, Abubaker J, et al. Sanguinarine-dependent inductionof apoptosis in primary effusion lymphoma cells. Cancer Res2007;67:3888–3897.
28. Uddin S, Hussain AR, Manogaran PS, Al-Hussein K, Plata-nias LC, Gutierrez MI, et al. Curcumin suppresses growth andinduces apoptosis in primary effusion lymphoma. Oncogene2005;24:7022–7030.
29. Uddin S, Ah-Kang J, Ulaszek J, Mehmud D, Wickrema A.Differentiation stage-specific activation of p38 mitogen-activatedprotein kinase isoforms in primary human erythroid cells. ProcNatl Acad Sci U S A 2004;101:147–152.
30. Uddin S, Hussain AR, Al-Hussein KA, Manogaran PS, Wick-rema A, Gutierrez MI, et al. Inhibition of phosphatidylinositol3′-kinase/AKT signaling promotes apoptosis of primary effusionlymphoma cells. Clin Cancer Res 2005;11:3102–3108.
31. Uddin S, Hussain AR, Al-Hussein K, Platanias LC, Bhatia KG.Inhibition of phosphatidylinositol 3′-kinase induces preferentialkilling of PTEN-null T leukemias through AKT pathway. BiochemBiophys Res Commun 2004;320:932–938.
32. Chiarle R, Fan Y, Piva R, Boggino H, Skolnik J, Novero D, et al.S-phase kinase-associated protein 2 expression in non-Hodgkin’slymphoma inversely correlates with p27 expression and definescells in S phase. Am J Pathol 2000;160:1457–1466.
33. Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A,Lazarus DD, et al. Proteasome inhibitors: a novel class of potentand effective antitumor agents. Cancer Res 1999;59:2615–2622.
34. Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation ofp21Cip1 at the onset of mitosis: a role at the G2–M-phasetransition. Mol Cell Biol 1998;18:546–557.
35. Hideshima T, Richardson P, Chauhan D, Palombella VJ,Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drugresistance in human multiple myeloma cells. Cancer Res2001;61:3071–3076.
36. Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M,et al. The cell-cycle regulatory protein Cks1 is required forSCF (Skp2)-mediated ubiquitinylation of p27. Nature Cell Biol2001;3:321–324.
37. Chiappetta G, De Marco C, Quintiero A, Califano D, Gherardi S,Malanga D, et al. Overexpression of the S-phase kinase-associated protein 2 in thyroid cancer. Endocr Relat Cancer2007;14:405–420.
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.

494 S Uddin et al
38. Lim MS, Adamson A, Lin Z, Perez-Ordonez B, Jordan RC,Tripp S, et al. Expression of Skp2, a p27Kip1 ubiquitin ligase, inmalignant lymphoma: correlation with p27Kip1 and proliferationindex. Blood 2002;100:2950–2956.
39. Seki R, Okamura T, Koga H, Yakushiji K, Hashiguchi M, Yoshi-moto K, et al. Prognostic significance of the F-box protein Skp2expression in diffuse large B-cell lymphoma. Am J Hematol2003;73:230–235.
40. Mani A, Gelmann EP. The ubiquitin–proteasome pathway and itsrole in cancer. J Clin Oncol 2005;23:4776–4789.
41. Lwin T, Hazlehurst LA, Dessureault S, et al. Cell adhesioninduces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway inmantle cell and other non-Hodgkin’s B-cell lymphomas. Blood2007;110:1631–1638.
42. Gastman BR. Apoptosis and its clinical impact. Head Neck2001;23:409–425.
43. Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteasesof the apoptotic pathway. Oncogene 1998;24:3237–3245.
J Pathol 2008; 216: 483–494 DOI: 10.1002/pathCopyright 2008 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.