response to long-term growth hormone therapy in patients affected by rasopathies and growth hormone...
TRANSCRIPT

�
CLINICAL REPORT
Response to Long-Term Growth Hormone Therapyin Patients Affected by RASopathies and GrowthHormone Deficiency: Patterns of Growth, Pubertyand Final Height Data
Federica Tamburrino,1* Dino Gibertoni,2 Cesare Rossi,3 Emanuela Scarano,1Annamaria Perri,1 Francesca Montanari,1 Maria Pia Fantini,2 Andrea Pession,1
Marco Tartaglia,4,5 and Laura Mazzanti11Pediatric Endocrinology and Rare Diseases Unit, Department of Pediatrics, S.Orsola-Malpighi University Hospital-University of Bologna,
Bologna, Italy2Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy3Department of Medical Genetics, S.Orsola-Malpighi University Hospital, University of Bologna, Bologna, Italy4Hematology, Oncology and Molecular Medicine, Istituto Superiore di Sanit�a, Rome, Italy5Malattie Genetiche e Malattie Rare, Ospedale Pediatrico Bambino Ges�u IRCCS, Rome, Italy
Manuscript Received: 3 March 2015; Manuscript Accepted: 4 July 2015
Conflict of interest: None.
Grant sponsor: Telethon-Italy; Grant number: GGP13107; Grant sponsor:
Ricerca Finalizzata-Ministero della Salute; Grant number:
RF-2011-02349938.�Correspondence to:
Federica Tamburrino, Pediatric Endocrinology and Rare Diseases,
Department of Pediatrics, S.Orsola-Malpighi University Hospital-Uni-
versity of Bologna, Via Massarenti, 11, 40138, Bologna, Italy.
E-mail: [email protected]
Article first published online in Wiley Online Library
(wileyonlinelibrary.com): 00 Month 2015
DOI 10.1002/ajmg.a.37260
How to Cite this Article:Tamburrino F,Gibertoni D, Rossi C,
Scarano E, Perri A, Montanari F, Fantini
MP, Pession A, Tartaglia M, Mazzanti L.
2015. Response to long-term growth
hormone therapy in patients affected by
RAsopathies and growth hormone
deficiency: Patterns of growth, puberty and
final height data.
Am J Med Genet Part A 9999A:1–9.
RASopathies are developmental disorders caused by hetero-
zygous germline mutations in genes encoding proteins in the
RAS-MAPK signaling pathway. Reduced growth is a common
feature. Several studies generated data on growth, final height
(FH), and height velocity (HV) after growth hormone (GH)
treatment in patients with these disorders, particularly in
Noonan syndrome, the most common RASopathy. These
studies, however, refer to heterogeneous cohorts in terms
of molecular information, GH status, age at start and length
of therapy, and GH dosage. This work reports growth data in
88 patients affected by RASopathies with molecularly con-
firmed diagnosis, together with statistics on body propor-
tions,
pubertal pattern, and FH in 33, including 16 treated with GH
therapy for proven GH deficiency. Thirty-three patients
showed GH deficiency after pharmacological tests, and
were GH-treated for an average period of 6.8� 4.8 years.
Before starting therapy, HV was �2.6� 1.3 SDS, and mean
basal IGF1 levels were �2.0� 1.1 SDS. Long-term GH thera-
py, starting early during childhood, resulted in a positive
height response compared with untreated patients (1.3 SDS in
terms of height-gain), normalizing FH for Ranke standards
but not for general population and Target Height. Pubertal
timing negatively affected pubertal growth spurt and FH,
with IGF1 standardized score increased from �2.43 to �0.27
SDS. During GH treatment, no significant change in bone age
velocity, body proportions, or cardiovascular function was
observed. � 2015 Wiley Periodicals, Inc.
Key words: RASopathies; noonan syndrome; growth hormone;
final height; puberty
2015 Wiley Periodicals, Inc.
INTRODUCTION
Short stature is a cardinal features of RASopathies, a family of
clinically related autosomal dominant disorders caused by hetero-
1

2 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
zygous germline mutations in genes coding for transducers
participating in the RAS-MAPK signaling pathway [Tartaglia
and Gelb, 2010]. RASopathies are multisystemic disorders that
share facial dysmorphism, failure to thrive, congenital heart disease
andhypertrophic cardiomyopathy, ectodermal, and skeletal anom-
alies, variable cognitive involvement, and susceptibility to certain
malignancies asmajor characteristics, even though each feature has
a variable prevalence in individual syndromes [Tartaglia et al.,
2011; Rauen, 2013]. Some are clinically and genetically homoge-
neous, such as Costello syndrome (CS, MIM 218040) and Noonan
syndrome-like disorder with loose anagen hair (NS/LAH, MIM
607721), also known as Mazzanti syndrome, which are caused by a
narrow spectrum of mutations in HRAS [Aoki et al., 2005] and
SHOC2 [Cordeddu et al., 2009], respectively. Others exhibit a
particularly variable phenotype, which reflects a more complex
genetic basis, as documented in Noonan syndrome (NS, MIM
163950), the most common RASopathy, which is caused by
mutations in several genes (PTPN11, RIT1, SOS1, KRAS, NRAS,
RAF1, BRAF, MEK1) [Tartaglia et al., 2011; Rauen, 2013].
Various studies reported data on spontaneous growth in
patients with NS [Witt et al., 1986; Ranke et al., 1988; Shaw
et al., 2007], but growth standards for patients with molecularly
confirmed diagnosis have been generated only recently [Malaquias
et al., 2012]. The collected data for NS indicate that the growth
pattern is characterized by birth weight (W) and height (H) within
normal limits, followed by a rapid height loss of 1–1.5 SD in the first
year of life. After 2–4 years, mean H follows the 3rd centile until
puberty, which is generally delayed by about two years and
characterized by a low peak height velocity (HV). Similarly,
bone age tends to be delayed about 2 years starting from 4 years
[Ranke et al., 1988; Otten et al., 2007]. In subjects with cardiofa-
ciocutaneous syndrome (CFCS, MIM 115150), measurements at
birth tend to be normal, but postnatally failure to thrive (78%)
commonly occurs due to severe feeding difficulties. Two thirds of
individuals exhibit stature below the 3rd centile [Allanson et al.,
2011], and W is also generally below the normal growth curve
[Roberts et al., 2006]. In contrast, prenatal overgrowth with
relatively high birth W (>50th centile in 89%) [Hennekam,
2003] is typical of CS, likely caused by fetal hydrops [Lin et al.,
2009]. Weight loss in the first days after birth is due the resolution
of edema and the severe swallowing and sucking problems [Gripp
et al., 2012], already present in prenatal period, as evidenced by
polyhydramnios. The severe failure to thrive has a low point at
around age 12 months [Sammon et al., 2012]. Similar other
RASopathies, delayed bone age is common. Mean adult H of
138 cm was reported [Van Eeghen et al., 1999]. In Noonan syn-
drome with multiple lentigenes (NSML; previously referred to as
LEOPARD syndrome), birth W is normal or above average in 1/3
[Digilio et al., 2006]. Retardation of growth is reported in about
25% below the 3rd centile in H, and final height (FH) is 85% below
the 3rd centile [Gorlin et al., 1971; Voron et al., 1976; Sarkozy et al.,
2008]. Short stature has been described in a significant proportion
(< 23%) of patients affected by Legius syndrome [Brems et al.,
2012]. In NS/LAH, short stature is often associated with proven
growth hormone deficiency (GHD) [Mazzanti et al., 2006;
Cordeddu et al., 2009; Mazzanti et al., 2013]. Data on spontaneous
growth and growth hormone (GH) response have been reported in
a few children [Mazzanti et al., 2006; Capalbo et al., 2012;
Malaquias et al., 2012; Mazzanti et al., 2013]. Malaquias et al.
[2012] described four subjects with markedly reduced growth and
low BMI. Consistent with these data, we recently reported short
stature in these children, approximately -3 SDS in height compared
to the general Italian population standards [Mazzanti et al., 2013].
The cause of short stature in RASopathies remains poorly
understood. Different mechanisms have been reported, including
GH deficiency [Cotteril et al., 1996; Romano et al., 1996; Padidela
et al., 2008], neurosecretory dysfunction [Tanaka et al., 1992;
Noordam et al., 2001], or GH resistance [Binder et al., 2005;
Ferreira et al., 2005; Limal et al., 2006; Bertelloni et al., 2013]. The
analysis of efficacy of GH treatment has provided contradictory
results [Kirk et al., 2001; Osio et al., 2005; Otten et al., 2007;
Romano et al., 2009; Lee et al., 2012; Choi et al., 2012], and a
mean H gain ranging between 0.6 and 2 SDS has been reported
[Dahlgren et al., 2009]. GH secretion status and GH treatment in
RASopathies is still a matter of debate. Published data are
difficult to compare due to the heterogeneous protocols, as
well as different cohort selection criteria and composition. Of
note, only a few studies reported FH data on GH-treated
RASopathy subjects. In NS/LAH, proven GH deficiency
(GHD) has been described [Mazzanti et al., 2006; Cordeddu
et al., 2009], as the occurrence of very low IGF1 levels [Mazzanti
et al., 2013]. In these patients, FH was �2.34� 0.12 SDS, after
long-term of GH-therapy [Mazzanti et al., 2013]. Similarly, GH
deficit has been reported in patients with CFCS [Legault et al.,
2001; Stein RI et al., 2004; Armour et al., 2008]. In CS, some
patients were GH-treated with variable benefit [Legault et al.,
2001; Kerr et al., 2003; Stein et al., 2004], however, the risk of
hypertrophic cardiomyopathy, obstructive apneas and tumors
requires attention regarding the use of GH therapy
[Kerr et al., 2003].
In this paper, we report growth data in 88 patients affected by
RASopathies with molecularly confirmed diagnosis, followed for a
long period in our Pediatric Rare Disease Outpatient Clinic. In
particular, we report data on growth, body proportions, pubertal
pattern, and first FH results in 33 subjects, including 16 treatedwith
GH-therapy for proven GH deficiency.
PATIENTS AND METHODS
The study cohort includes patients with clinical features fulfilling
criteria for RASopathies. The clinical diagnosis was molecularly
confirmed. Patients were recruited at the Pediatric Rare Disease
Outpatient Unit of S.Orsola–Malpighi University Hospital of
Bologna, Italy, from 2001 to 2014, and followed till they reached
adulthood.
Molecular analyses were performed by Sanger sequencing the
entire coding sequences of the PTPN11, SOS1, KRAS, NRAS,
HRAS, RIT1, SHOC2, SPRED1, BRAF, RAF1, MAP2K1, and
MAP2K2, on the basis of the clinical diagnosis.
Anthropometric measurements were compared with the
standard growth curves for the general Italian population [Cacciari
et al., 2006] and for NS [Ranke et al., 1988], and were expressed as
SD scores. Growth velocity SDS were calculated on Tanner charts
[Tanner et al., 1966].
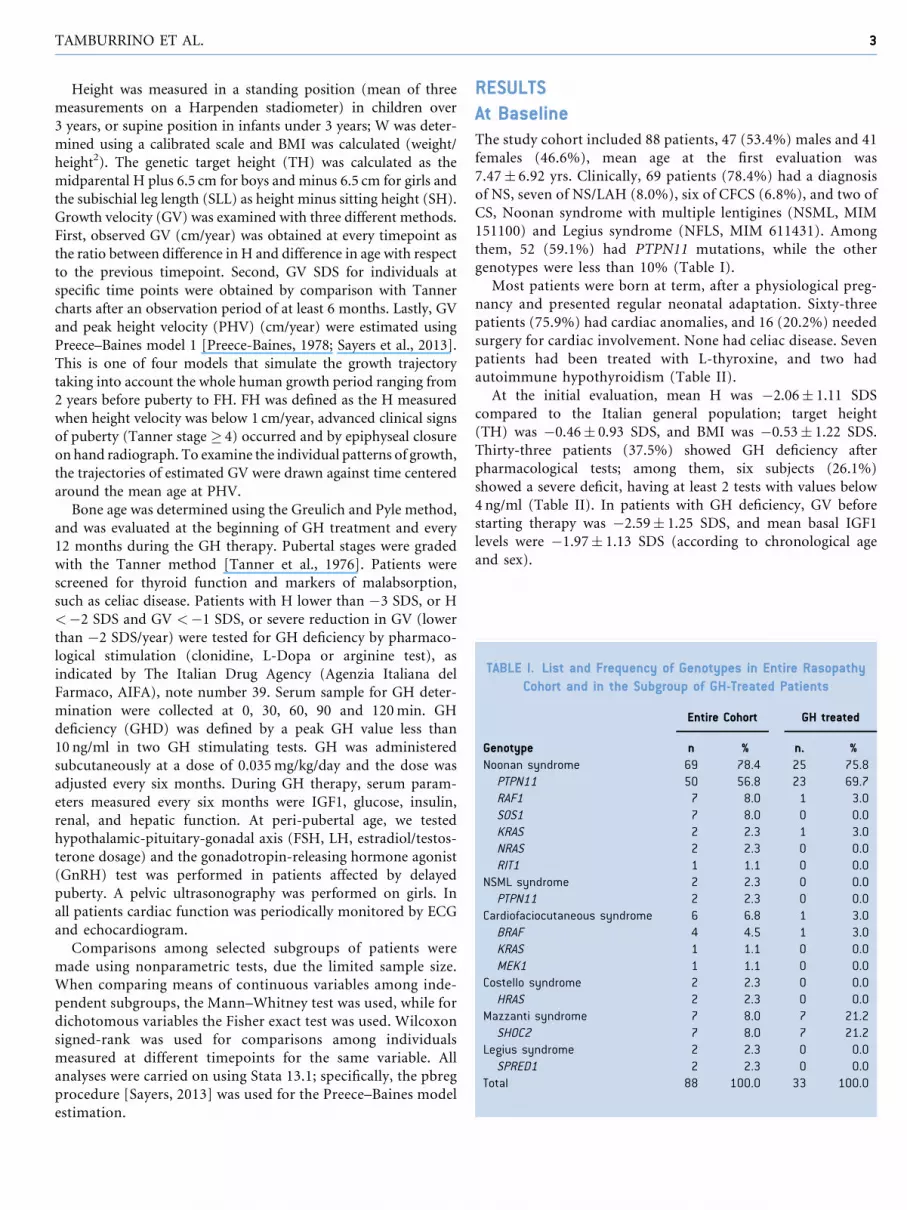
TABLE I. List and Frequency of Genotypes in Entire Rasopathy
Cohort and in the Subgroup of GH-Treated Patients
Entire Cohort GH treated
Genotype n % n. %
Noonan syndrome 69 78.4 25 75.8
PTPN11 50 56.8 23 69.7
RAF1 7 8.0 1 3.0
SOS1 7 8.0 0 0.0
KRAS 2 2.3 1 3.0
NRAS 2 2.3 0 0.0
RIT1 1 1.1 0 0.0
NSML syndrome 2 2.3 0 0.0
PTPN11 2 2.3 0 0.0
Cardiofaciocutaneous syndrome 6 6.8 1 3.0
BRAF 4 4.5 1 3.0
KRAS 1 1.1 0 0.0
MEK1 1 1.1 0 0.0
Costello syndrome 2 2.3 0 0.0
HRAS 2 2.3 0 0.0
Mazzanti syndrome 7 8.0 7 21.2
SHOC2 7 8.0 7 21.2
Legius syndrome 2 2.3 0 0.0
SPRED1 2 2.3 0 0.0
Total 88 100.0 33 100.0
TAMBURRINO ET AL. 3
Height was measured in a standing position (mean of three
measurements on a Harpenden stadiometer) in children over
3 years, or supine position in infants under 3 years; W was deter-
mined using a calibrated scale and BMI was calculated (weight/
height2). The genetic target height (TH) was calculated as the
midparental H plus 6.5 cm for boys and minus 6.5 cm for girls and
the subischial leg length (SLL) as height minus sitting height (SH).
Growth velocity (GV) was examined with three different methods.
First, observed GV (cm/year) was obtained at every timepoint as
the ratio between difference in H and difference in age with respect
to the previous timepoint. Second, GV SDS for individuals at
specific time points were obtained by comparison with Tanner
charts after an observation period of at least 6 months. Lastly, GV
and peak height velocity (PHV) (cm/year) were estimated using
Preece–Baines model 1 [Preece-Baines, 1978; Sayers et al., 2013].
This is one of four models that simulate the growth trajectory
taking into account the whole human growth period ranging from
2 years before puberty to FH. FH was defined as the H measured
when height velocity was below 1 cm/year, advanced clinical signs
of puberty (Tanner stage � 4) occurred and by epiphyseal closure
on hand radiograph. To examine the individual patterns of growth,
the trajectories of estimated GV were drawn against time centered
around the mean age at PHV.
Bone age was determined using the Greulich and Pyle method,
and was evaluated at the beginning of GH treatment and every
12 months during the GH therapy. Pubertal stages were graded
with the Tanner method [Tanner et al., 1976]. Patients were
screened for thyroid function and markers of malabsorption,
such as celiac disease. Patients with H lower than �3 SDS, or H
<�2 SDS and GV <�1 SDS, or severe reduction in GV (lower
than �2 SDS/year) were tested for GH deficiency by pharmaco-
logical stimulation (clonidine, L-Dopa or arginine test), as
indicated by The Italian Drug Agency (Agenzia Italiana del
Farmaco, AIFA), note number 39. Serum sample for GH deter-
mination were collected at 0, 30, 60, 90 and 120min. GH
deficiency (GHD) was defined by a peak GH value less than
10 ng/ml in two GH stimulating tests. GH was administered
subcutaneously at a dose of 0.035mg/kg/day and the dose was
adjusted every six months. During GH therapy, serum param-
eters measured every six months were IGF1, glucose, insulin,
renal, and hepatic function. At peri-pubertal age, we tested
hypothalamic-pituitary-gonadal axis (FSH, LH, estradiol/testos-
terone dosage) and the gonadotropin-releasing hormone agonist
(GnRH) test was performed in patients affected by delayed
puberty. A pelvic ultrasonography was performed on girls. In
all patients cardiac function was periodically monitored by ECG
and echocardiogram.
Comparisons among selected subgroups of patients were
made using nonparametric tests, due the limited sample size.
When comparing means of continuous variables among inde-
pendent subgroups, the Mann–Whitney test was used, while for
dichotomous variables the Fisher exact test was used. Wilcoxon
signed-rank was used for comparisons among individuals
measured at different timepoints for the same variable. All
analyses were carried on using Stata 13.1; specifically, the pbreg
procedure [Sayers, 2013] was used for the Preece–Baines model
estimation.
RESULTS
At BaselineThe study cohort included 88 patients, 47 (53.4%) males and 41
females (46.6%), mean age at the first evaluation was
7.47� 6.92 yrs. Clinically, 69 patients (78.4%) had a diagnosis
of NS, seven of NS/LAH (8.0%), six of CFCS (6.8%), and two of
CS, Noonan syndrome with multiple lentigines (NSML, MIM
151100) and Legius syndrome (NFLS, MIM 611431). Among
them, 52 (59.1%) had PTPN11 mutations, while the other
genotypes were less than 10% (Table I).
Most patients were born at term, after a physiological preg-
nancy and presented regular neonatal adaptation. Sixty-three
patients (75.9%) had cardiac anomalies, and 16 (20.2%) needed
surgery for cardiac involvement. None had celiac disease. Seven
patients had been treated with L-thyroxine, and two had
autoimmune hypothyroidism (Table II).
At the initial evaluation, mean H was �2.06� 1.11 SDS
compared to the Italian general population; target height
(TH) was �0.46� 0.93 SDS, and BMI was �0.53� 1.22 SDS.
Thirty-three patients (37.5%) showed GH deficiency after
pharmacological tests; among them, six subjects (26.1%)
showed a severe deficit, having at least 2 tests with values below
4 ng/ml (Table II). In patients with GH deficiency, GV before
starting therapy was �2.59� 1.25 SDS, and mean basal IGF1
levels were �1.97� 1.13 SDS (according to chronological age
and sex).

TABLE II. Clinical Features at Birth or at Initial Evaluation of the Studied RASopathy Cohort
Number of patients n (mean) % (SD)
Males 88 47 53.4
Gestational age (wks) 76 38.57 1.81
Weight at birth (gr) 77 3241.1 541.5
Length at birth (cm) 58 49.1 2.4
Born at term 76 57 75.0
Small for gestational age 88 3 3.4
Target height (SDS Cacciari) 76 �0.46 0.93
Hypotonia 76 15 19.7
Cardiac anomalies 83 63 75.9
Pulmonary stenosis 81 29 35.8
Atrial septal defect 81 24 29.6
Ventricular septal defect 85 3 3.5
Mitral valve prolapse 81 14 17.3
Bicuspid aortic valve 81 1 1.2
Pulmonary valve dysplasia 81 14 17.3
Surgery for cardiac involvement 79 16 20.3
Peak Arginine test (ng/ml) n.v. � 10 ng/mla 31 5.12 2.60
Peak L-dopa test (ng/ml) n.v. � 10 ng/mla 19 4.38 1.90
Peak clonidine test (ng/ml) n.v. � 10 ng/mla 13 5.27 2.14
aPeaks measured on the 33 GH treated patients.
4 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
GH TherapyThirty-three patients with GH deficiency were treated for an
average of 6.76� 4.83 years. Among them, 69.7% had a
PTPN11mutation, followed by patients heterozygous for a SHOC2
mutation (21.2%). Two patients, who carried mutations docu-
mented to be associated with risk of developing hypertrophic
cardiomyopathy (p.Thr491Arg, RAF1; p.Gln257Arg, BRAF), re-
ceived GH therapy before molecular analysis was available
(Table I). At the first clinical evaluation in our Outpatient Clinic,
stature was lower for GH-treated patients compared to untreated:
�2.82� 0.78 SDS versus �1.46� 1.05 SDS for Cacciari standards
and�0.66� 0.88 SDS versus 0.56� 1.22 SDS for Ranke standards,
with both differences being significant (P< 0.001) by Mann–
Whitney test. At the beginning of treatment, mean age was
6.94� 3.58 years, and mean BMI was �0.33� 1.26 SDS. After
the first year of GH therapy, IGF1 level increased (SDS¼�0.08
� 1.26) to a significantly higher value (paired Wilcoxon test:
z¼�2.761,P¼ 0.006), andGV increased significantly (SDS¼ 1.89
� 1.52, z¼�2.66, P¼ 0.008).
Insulin resistance measured with HOMA-R was 1.17� 0.88
at the beginning of GH therapy, and 1.20� 0.61 after the first
year of GH treatment, with a not statistically significant change
(z¼ 1.363, P¼ 0.173) (Table III).
Final Height in Untreated and GH-TreatedSubjects
FH was reached by 33 patients (37.5%) at the mean age of
19.48� 5.44 years, and was �1.78� 0.90 SDS compared with
the Italian general population, and 0.35� 0.84 SDS compared
with Ranke standard. Among the patients who reached FH, 16
had been GH-treated (for 9.32� 3.97 yrs) (Fig. 1). FH in
GH-treated patients was lower (�2.21� 0.74 vs. �1.37� 0.86
SDS, using Cacciari standard; �0.03� 0.69 vs. 0.71� 0.83 SDS,
using Ranke standard), and this difference proved to be statistically
significant in Mann–Whitney test comparisons (z¼ 2.738,
P¼ 0.006, Cacciari standard; z¼ 2.576, P¼ 0.010, Ranke
standard). Sixteen patients who reached FH were female with
FH¼ 151.0� 5.7 cm (�1.89� 0.86 SDS, Cacciari standard;
0.13� 0.91 SDS, Ranke standard). The eight GH treated female
subjects reached a FH of 147.8� 2.8 cm (�2.44� 0.42 SDS,
Cacciari standard; �0.38� 0.36 SDS, Ranke standard) that was
significantly lower than the attained FH by untreated females
(154.3� 6.0cm; z¼ 2.25, P¼ 0.027 for both Cacciari and Ranke
standards). The seventeen male patients who reached FH were on
average 165.5� 5.8 cm high (�1.67� 0.86 SDS, Cacciari standard;
0.55� 0.75 SDS, Ranke standard). Eight had been treated with GH,
and reached a FH¼ 163.4� 6.6 cm that was lower than the FH
documented in untreated males (167.4� 4.4 cm), even though
such difference did not reach statistical significance (z¼ 1.347,
P¼ 0.178 for Cacciari standard; z¼ 0.915, P¼ 0.360 for Ranke
standard) (Fig. 2).
Comparison among FH and TH was available for 28
patients, resulting in a significantly lower FH (159.2� 9.2 cm
vs. 165.1� 8.0 cm; z¼�3.746, P< 0.001 at Wilcoxon signed-
rank test using Cacciari SDS). Only six patients (21.4%) reached
a FH higher than their TH. FH was significantly lower than TH in
all subgroups of GH-treated (155.6 vs. 165.4 cm; P¼ 0.003) and
untreated patients (164.1 vs. 168.5 cm; P¼ 0.034), males (165.8
vs. 172.4 cm; P¼ 0.010) and females (151.7 vs. 161.1 cm;
P¼ 0.006).

TABLE III. Biometrical and Clinical Features at the Main Time-Points for GH-Treated Patients With RASopathies
Beginning of
treatment (n¼ 31)
After first
year (n¼ 26)
At final
height (n¼ 16)
Age (yrs) 6.94� 3.58 7.72� 3.55 18.42� 2.25
Height (SDS Cacciari) �2.82� 0.78 �2.29� 0.72 �2.21� 0.74
Height (SDS Ranke) �0.66� 0.88 �0.29� 0.84 �0.03� 0.69
BMI (SDS Cacciari) �0.33� 1.26 �0.69� 1.22 �0.48� 1.25
IGF1 (SDS) �1.97� 1.13 �0.08� 1.26 �0.25� 1.10
Velocity height (SDS) �2.59� 1.25 1.89� 1.52
HOMA-R (SDS) 1.17� 0.88 1.20� 0.61 1.59� 0.94
The number of cases in parentheses indicates the highest number of available cases at the different timepoints for the variables displayed in the table.
TAMBURRINO ET AL. 5
Height gain at FH measured in 17 subjects whose first height
measure was taken at an age lower than 13 years (females) or
15 years (males) was 0.58� 0.87 SDS and 0.55� 0.92 SDS using
Cacciari and Ranke standards, respectively. GH treated patients
had 1.28 SDS larger H gain than untreated patients (0.85 vs �0.43
Ranke SDS, respectively), which was significant byMann–Whitney
test (z¼�2.378, P¼ 0.017). The difference of H gain amongmales
and females was not significant (z¼�0.481, P¼ 0.630) (Table III)
(Fig. 3). GH-treated patients with positive H gain had a longer
duration of GH therapy (10.5 vs. 4.1 yrs). During GH-treatment,
no bone age advance was observed.
Finally, variation of IGF1 levels were examined on the six
patients who had both IGF1 measures at the beginning of the
therapy and at FH. IGF1 standardized score increased from
�2.43 to �0.27.
In our study, subjects affected by Rasopathies and GHD were
mainly represented by patients with mutation in PTPN11 and
SHOC2. In Table IV, we report clinical features and therapeutic
results of these two larger subgroups of patients: at FH three
FIG. 1. Diagram of the study cohort.
subjects affected by SHOC2 mutation showed 1 SDS larger H
gain than 10 PTPN11 mutated-patients.
Body Proportions and GH TreatmentStandardized sitting height (SH) was�3.19� 1.32 at the beginning
of treatment and �2.68� 1.01 at FH; for 7 patients with both
measures, the difference in SH was significant (z¼�2.197,
P¼ 0.028). Standardized subischial leg length (SLL) was
�2.58� 1.05 at the beginning of treatment and �1.83� 0.74 at
FH; for the 10 patients who had both measures, the difference in
SLL was significant (z¼�2.090, P¼ 0.037). Standardized stature
ratio (SH over SLL) was 1.26� 0.18 at baseline and 1.12� 0.03 at
the FH. The difference in stature ratio was not significant (z¼ 1.26,
P¼ 0.208) for eight patients who had both measures.
FIG. 2. Initial (IH) and Final (FH) height comparison by GH
treatment and sex. Heights measured by Cacciari standardized
scores. Boxplots by subgroups summarize the distributional
characteristics (boxes correspond to the inerquartile range, the
line inside the box is the median and te lines outside the box
identify adjacent values).

FIG. 3. Height Gain comparison by GH treatment; Ranke and
Cacciari standardized scores. (For description of box-plot see
fig. 2).
6 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
Pubertal DevelopmentPeak height velocity (PHV) estimated usingPreece andBainesmodel 1
wasevaluatedon11GH-treatedpatients(four femalesandsevenmales)
resulting in an averagePHVof 6.50� 1.46 cm/year. PHVwas lower for
females (6.14� 1.95) than for males (6.70� 1.24), with a non-signifi-
cant difference (z¼�0.945, P¼ 0.345). Age at PHV attainment was
significantly lower(z¼�2.457,P¼ 0.014)forfemales(11.9years) than
for males (14.6 years). Figure 4 shows the individual estimated pattern
of growth for males and females. The former reached their PHV in a
central point of their growth trajectory, while PHV was reached in the
early phase of pubertal growth in the latter.
DISCUSSION
In most patients of our RASopathy cohort, normal adaptation and
adequate anthropometric measurements at birth were observed.
TABLE IV. Comparison of Clinical Features and Therapeutic Results o
SHOC2 Pa
Clinical features of GH-treated patients by genotype
Age at initial evaluation (yrs)
Stature at initial evaluation (SDS Cacciari)
Stature at initial evaluation (SDS Ranke)
Stature at final evaluation (SDS Cacciari)
Stature at final evaluation (SDS Ranke)
Height gain (SDS Cacciari)
Height gain (SDS Ranke)
IGF at initial evaluation (SDS)
IGF at final evaluation (SDS)
Velocity height pre-therapy (SDS)
Velocity height after first year (SDS)
an¼ 23 at first evaluation; n¼ 10 at final evaluation.bn¼ 7 at first evaluation; n¼ 3 at final evaluation.
The retrospective analysis of GH response documents that at their
first evaluation, patients with GH deficiency were undersized with
respect to the general Italian population standards for their H and
TH, as well as less than 1 SDS for the NS standards.
Approximately 37%of our entire cohort had short stature, GHD
at pharmacological tests, and very low IGF1 levels (less than �2
SDS in the general population), they were treated with GH at
standard dosage approved for children with GHD. In subjects who
started therapy before puberty, long-term GH therapy improved
their FHby 1.28 SDS over untreated patients, which is in agreement
with previously published data [Dahlgren et al., 2009]. Our data
indicated that GH therapy in these patients seemed to normalize
their stature for NS, allowing them to approximate the stature of
the untreated group of patients with normal GH secretion accord-
ing to Ranke standards, although their FH remained below the
parental H and the Italian general population standards. Such
therapeutic result were supported by IGF1 levels that increased
significantly after the first year of therapy and reached normal
values at FH, though on the limited number of patients.
During GH treatment, no significant change in bone age was
observed, and SH/SLL ratio remained stable, with no change of
body proportions. Regarding glucose metabolism, HOMA-R
was unchanged. Moreover, we did not observe a deterioration
of cardiac function and or development of hypertrophic
cardiomyopathy.
In NS patients pubertal development is typically delayed,
although not in all subjects [Romano et al., 2009]. In our subgroup
of 12 subjects who reached FH and were GH-treated, pubertal
growth showed a lowered peak, in particular in males, and a delay
in onset by about 6 months, compared to the general population
[Tanner et al., 1985]. The delayed pubertal development and the
inadequate pubertal catch-up growth could explain the impaired
FH. Our patients on GH-therapy benefitted from the pharmaco-
logical treatment if started in pre-puberty and given for a long
time. Probably, the prepubertal start of GH-treatment could
compensate the lack of a pubertal growth spurt. Pubertal behavior
in a larger cohort of patients without GH deficiency should be
clarified.
f GH-Treated Patients in Relation to Major Genotypes (PTPN11 vs
tients)
PTPN11a SHOC2b
7.45 � 3.58 5.17 � 2.62
�2.53 � 0.65 �3.45 � 0.84
�0.44 � 0.75 �1.10 � 1.14
�1.98 � 0.64 �2.34 � 0.14
0.17 � 0.67 �0.20 � 0.21
0.51 � 0.71 1.38 � 1.21
0.42 � 0.71 1.55 � 1.35
�1.76 � 1.24 �2.62 � 0.51
�0.85 � 0.91 0.09 � 1.25
�2.76 � 1.75 �2.42 � 0.56
7.88 � 1.91 8.80 � 1.39
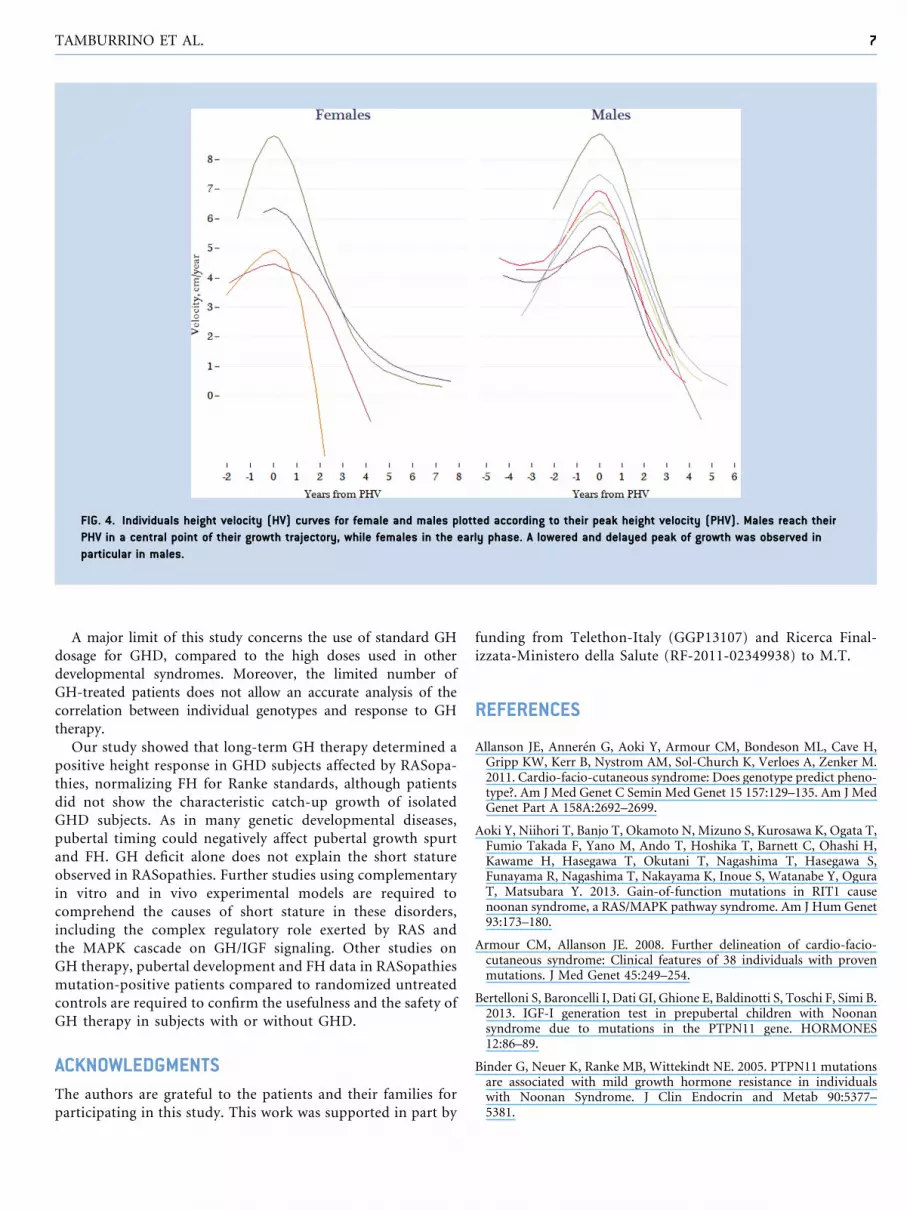
FIG. 4. Individuals height velocity (HV) curves for female and males plotted according to their peak height velocity (PHV). Males reach their
PHV in a central point of their growth trajectory, while females in the early phase. A lowered and delayed peak of growth was observed in
particular in males.
TAMBURRINO ET AL. 7
A major limit of this study concerns the use of standard GH
dosage for GHD, compared to the high doses used in other
developmental syndromes. Moreover, the limited number of
GH-treated patients does not allow an accurate analysis of the
correlation between individual genotypes and response to GH
therapy.
Our study showed that long-term GH therapy determined a
positive height response in GHD subjects affected by RASopa-
thies, normalizing FH for Ranke standards, although patients
did not show the characteristic catch-up growth of isolated
GHD subjects. As in many genetic developmental diseases,
pubertal timing could negatively affect pubertal growth spurt
and FH. GH deficit alone does not explain the short stature
observed in RASopathies. Further studies using complementary
in vitro and in vivo experimental models are required to
comprehend the causes of short stature in these disorders,
including the complex regulatory role exerted by RAS and
the MAPK cascade on GH/IGF signaling. Other studies on
GH therapy, pubertal development and FH data in RASopathies
mutation-positive patients compared to randomized untreated
controls are required to confirm the usefulness and the safety of
GH therapy in subjects with or without GHD.
ACKNOWLEDGMENTS
The authors are grateful to the patients and their families for
participating in this study. This work was supported in part by
funding from Telethon-Italy (GGP13107) and Ricerca Final-
izzata-Ministero della Salute (RF-2011-02349938) to M.T.
REFERENCES
Allanson JE, Anner�en G, Aoki Y, Armour CM, Bondeson ML, Cave H,Gripp KW, Kerr B, Nystrom AM, Sol-Church K, Verloes A, Zenker M.2011. Cardio-facio-cutaneous syndrome: Does genotype predict pheno-type?. Am J Med Genet C Semin Med Genet 15 157:129–135. Am J MedGenet Part A 158A:2692–2699.
Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Ogata T,Fumio Takada F, Yano M, Ando T, Hoshika T, Barnett C, Ohashi H,Kawame H, Hasegawa T, Okutani T, Nagashima T, Hasegawa S,Funayama R, Nagashima T, Nakayama K, Inoue S, Watanabe Y, OguraT, Matsubara Y. 2013. Gain-of-function mutations in RIT1 causenoonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet93:173–180.
Armour CM, Allanson JE. 2008. Further delineation of cardio-facio-cutaneous syndrome: Clinical features of 38 individuals with provenmutations. J Med Genet 45:249–254.
Bertelloni S, Baroncelli I, Dati GI, Ghione E, Baldinotti S, Toschi F, Simi B.2013. IGF-I generation test in prepubertal children with Noonansyndrome due to mutations in the PTPN11 gene. HORMONES12:86–89.
Binder G, Neuer K, Ranke MB, Wittekindt NE. 2005. PTPN11 mutationsare associated with mild growth hormone resistance in individualswith Noonan Syndrome. J Clin Endocrin and Metab 90:5377–5381.

8 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
Brems H, Pasmant E, Van Minkelen R, Wimmer K, Upadhyaya M, LegiusE, Messiaen L. 2012. Review and Update of SPRED1 mutations causinglegius syndrome. Human Mutation 33:1538–1546.
Cacciari E, Milani S, Balsamo A, Spada E, Bona G, Cavallo L, Cerutti F,Gargantini L, Greggio N, Tonini G, Cicognani A. 2006. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr).J Endocrinol Invest 29:581–593.
Capalbo D, Melis D, De Martino L, Palamaro L, Riccomagno S, Bona G,Cordeddu V, Pignata C, Salerno M. 2012. Noonan-like syndrome withloose anagen hair associated with growth hormone insensitivity andatypical neurological manifestation. Am J Med Genet Part A 158A:856–860.
Choi JH, Lee BH, JungCW,KimYM, JinHY, Kim JM,KimGH,Hwang JS,Yang SW, Lee J, YooHW. 2012. Response to growth hormone therapy inchildren with noonan syndrome: Correlation with or without PTPN11gene mutation. Horm Res Paediatr 77:388–393.
CordedduV,Di Schiavi E, Pennacchio LA, et al. 2009.Mutation of SHOC2promotes aberrant protein N-myristoylation and causes Noonan-likesyndrome with loose anagen hair. Nat Genet 41:1022–1026.
Cotterill AM, McKennaWJ, Brady AF, SharlandM, Elsawi M, YamadaM,Camacho-H€ubner C, Kelnar CJ, Dunger DB, Patton MA, Savage MO.1996. The short-term effects of growth hormone therapy on heightvelocity and cardiac ventricular wall thickness in children withNoonan’ssyndrome. J Clin Endocrinol Metab 81:2291–2297.
Dahlgren J. 2009. GH Therapy in noonan syndrome. Horm Res 72:46–48.
Digilio MC, Sarkozy A, de Zorzi A, Pacileo G, Limongelli G, Mingarelli R,Calabro R, Marino B, Dallapiccola B. 2006. LEOPARD syndrome:Clinical diagnosis in the first year of life. Am JMedGenet A 140:740–746.
Ferreira LV, Souza SA, Arnhold IJ, Mendonca BB. 2005. Orge AA PTPN11(protein tyrosine phosphatase, nonreceptor type 11) mutations andresponse to growth hormone therapy in children with Noonan syn-drome. J Clin Endocrinol Metab 90:5156–5160.
Gorlin RJ, Anderson RC, Moller JH. 1971. The leopard (multiple lenti-gines) syndrome revisited. Birth Defects Orig Artic Ser 7:110–115.
Gripp KW, Lin AE. 2012. Costello syndrome: A Ras/mitogen activatedprotein kinase pathway syndrome (rasopathy) resulting from HRASgermline mutations. Genet Med 14:285–292.
HennekamRC. 2003. Costello syndrome: An overview. Am JMedGenet CSemin Med Genet 117C:42–48.
Kerr B, Einaudi MA, Clayton P, Gladman G, Eden T, Saunier P, GenevieveD, Philip N. 2003. Is growth hormone treatment beneficial or harmful inCostello syndrome? [letter]. J Med Genet 40:e74.
Kirk JM, Betts PR, Butler GE, Donaldson MD, Dunger DB, Johnston DI,Kelnar CJ, Price DA,Wilton P. 2001. Short stature inNoonan syndrome:Response to growth hormone therapy. Arch Dis Child 84:440–443.
Lee PA, Ross J, Germak JA, Gut R. 2012. Effect of 4 years of growthhormone therapy in children with Noonan syndrome in the Americannorditropin studies: Web-enabled research (ANSWER) program(R)registry. Int J Pediatr Endocrinol 15.
Legault L, Gagnon C, Lapointe N. 2001. Growth hormone deficiency incostello syndrome: A possible explanation for the short stature. J Pediatr138:151–152.
Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, Vidaud M,Le Bouc Y. 2006. Noonan syndrome: Relationships between genotype,growth, and growth factors. J Clin Endocrinol Metab 91:300–306.
Lin AE, O’Brien B, Demmer LA, Almeda KK, Blanco CL, Glasow PF, BerulCI, Hamilton R, Innes AM, Lauzon JL, Sol-Church K, Gripp KW. 2009.Prenatal features of Costello syndrome: Ultrasonographic findings andatrial tachycardia. Prenat Diagn 29:682–690.
Malaquias AC, Brasil AS, Pereira AC, Arnhold IJP, Mendonca BB, BertolaDR, Jorge AAL. 2012. Growth standards of patients with Noonan andNoonan-like syndromeswithmutations in theRAS/MAPKpathway. AmJ Med Genet Part A 158A:2700–2706.
Mazzanti L, Cacciari E, Cicognani A, Bergamascgi R, Scarano E, ForaboscoA. 2006. Noonan-like syndrome with loose anagen hair: A new syn-drome?. Am J Med Genet A 118A:279–286.
Mazzanti L, Tamburrino F, Scarano E, Perri A, Vestrucci B, Guidetti M,Rossi C, Tartaglia M. 2013. GH Therapy and first final height data inNoonan-like syndrome with loose Anagen hair (Mazzanti syndrome).Am J Med Genet Part A 161A:2756–2761.
Noordam C, van der Burgt I, Sweep CG, Delemarre-van de Waal HA,Sengers RC,Otten BJ. 2001.Growth hormone (GH) secretion in childrenwith Noonan syndrome: Frequently abnormal without consequencesfor growth or response to GH treatment. Clin Endocrinol (Oxf)54:53–59.
Osio D, Dahlgren J, Albertsson Wikland K, Westphal O. 2005. Improvedfinal height with long-term growth hormone treatment in Noonansyndrome. Acta Paediatr 94:1232–1237.
Otten J, Noordam K. 2007. Short stature in Noonan syndrome: Results ofgrowth hormone treatment. Growth Hormo Pediatr-20 years of KIGS347–355.
Padidela R, Camacho-H€ubner C, Attie KM, Savage MO. 2008. Abnormalgrowth in Noonan syndrome: Genetic and endocrine features andoptimal treatment. Horm Res 70:129–136.
Preece MA, Baines MJ. 1978. A new family of mathematical modelsdescribing the human growth curve. Ann Hum Biol 5:1–24.
Ranke MB, Heidemann P, Knupfer C, Enders H, Schmaltz AA, Bierich JR.1988. Noonan syndrome: Growth and clinical manifestations in 144cases. Eur J Pediatr 148:220–227.
Roberts A, Allanson J, Jadiko SK, Kavamura MI, Opitz JM, Youg T,Neri G. 2006. The cardiofaciocutaneous syndrome. J Med Genet43:833–842.
Romano AA, Blethen SL, Dana K, Noto RA. 1996. Growth hormonetreatment in Noonan syndrome: The national cooperative growth studyexperience. J Pediatr 128:S18–S21.
Romano AA, Dana K, Bakker B, Davis DA, Hunold JJ, Jacobs J, Lippe B.2009. Growth response, near-adult height, and patterns of growth andpuberty in patients with noonan syndrome treated with growth hor-mone. J Clin Endocrinol Metab 94:2338–2344.
SammonMR,DoyleD,Hopkins E, Sol-ChurchK, StableyDL,McGready J,Schulze K, Alade Y, Hoover-Fong J, Gripp KW. 2012. Normative growthcharts for individuals with Costello syndrome. Am J Med Genet Part A158A:2692–2699.
Sarkozy A, DigilioMC, Dallapiccola B. 2008. Leopard syndrome. Orphan JRare Dis 3:13.
Sayers A, Baines M, Tilling K. 2013. A new family of mathematical modelsdescribing the human growth curve—Erratum: Direct calculation ofpeak height velocity, age at take-off and associated quantities. Ann HumBiol 40:298–299.
Sayers A. 2013. PBREG: Stata module to fit the Preece and Baines (1978)family of growth curves and age, height, and velocity at peak heightvelocity http://ideas.repec.org/c/boc/bocode/s457612.html.
Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. 2007. The naturalhistory of Noonan syndrome: A long-term follow-up study. Arch DisChild 92:128–132.
Stein RI, Legault L, Daneman D, Weksberg R, Hamilton J. 2004. Growthhormone deficiency in costello syndrome. Am J Med Genet A129A:166–170.

TAMBURRINO ET AL. 9
Tanaka K, Sato A, Naito T, Kuramochi K, Itabashi H, Takemura Y. 1992.Noonan syndrome presenting growth hormone neurosecretory dysfunc-tion. Int Med 31:908–911.
Tanner JM, Whitehouse RH, Takaishi M. 1966. Standards from birth tomaturity for height, weight, height velocity, and weight velocity: Britishchildren 1965 Parts I and II. Arch Dis Child 41:454–471.
Tanner JM, Whitehouse RH. 1976. Clinical longitudinal standards forheight, weight, height velocity, weight velocity, and the stages of puberty.Arch Dis Child 51:170–179.
Tanner JM, Davies PS. 1985. Clinical longitudinal standards for heightand height velocity for North American children. J Pediatr 107:317–329.
Tartaglia M, Gelb BD, Zenker M. 2011. Noonan syndrome and clinicallyrelated disorders. Best Pract Res Clin Endocrinol Metab 25:161–179.
Tartaglia M, Gelb BD. 2010. Disorders of disregulated signal trafficthrough the RAS-MAPK pathway: Phenotypic spectrum and molecularmechanisms. Ann N Y Acad Sci 1214:99–121.
Van Eeghen AM, Van Gelderen I, Hennekam RCM. 1999. Costellosyndrome: Report and review. Am J Med Genet 82:187–193.
Voron DA, Hatfield HH, Kalkhoff MD. 1976. Multiple lentigines syn-drome: Case report and review of the literature. Am J Med 60:447–456.
Witt DR, Keena BA, Hall JG, Allanson AJ. 1986. Growth curves for heightin Noonan syndrome. Clin Genet 30:150–153.