protein folding and ramachandran plot
TRANSCRIPT

SEMINAR PRESENTATION
ON
PROTEIN FOLDING AND
RAMACHANDRAN PLOTSubmitted to Submitted by
Richa yadav mam Shayaba
M.Sc Biotechnology

➢ WHAT ARE PROTEINS➢ WHAT IS PROTEIN FOLDING➢ PROTEIN FODING DEPENDS UPON➢ ANFINSEN EXPERIMENT➢ FOUR STAGES OF PROTEIN FOLDING➢ MECHANISM OF PROTEIN FODING ➢ RAMACHANDRAN PLOT ➢ REFERENCE

Proteins are long chains of amino acids put together in a specific sequence to perform a particular function in the body.
Proteins are the polymer of amino acids. Proteins are the building blocks of life. They are vital for the growth of cells and
tissue repair. Proteins are involved in structure, transport,
storage, metabolism, cell signaling and many other processes.
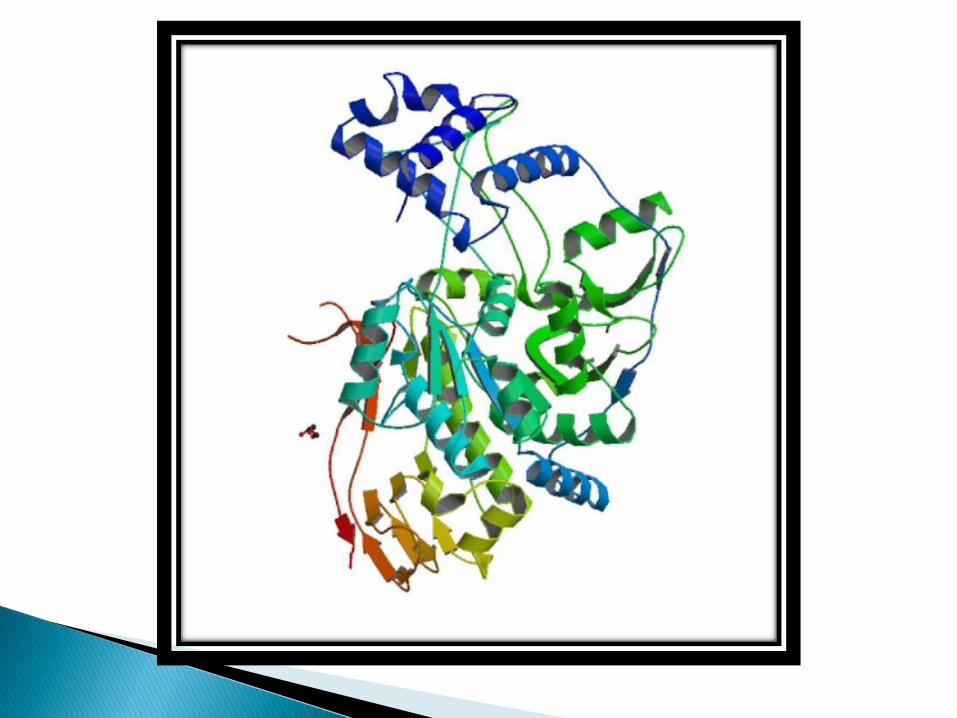

Protein foding is a process by which a polypeptide chain folds to become a biologically active protein in its native 3D structure.
Protein structure is crucial to its function.
Folded proteins are held together by various molecular interactions.

During translations, each protein is synthesized as a linear chain of amino acids or a random coil which does not have a stable 3D structure.
The amino acids in the chain eventually interact with each other to form a well defined, folded protein.
Failure to fold properly produces inactive or toxic proteins that malfunction and cause a number of diseases.

The process of protein folding depends upon:
❑THE SOLVENT
❑THE CONCENTRATION OF SALT
❑THE PH
❑THE TEMPERATURE
❑MOLECULAR CHAPERONES

Christian Anfinsen studied the refolding of protein ribonuclease A. Ribonuclease 124 amino acid residues and four disulphide linkages.
In the presence of urea, a denaturant, and beta-Mercaptoetanol, a reducing agent, ribonucleaseis denatured and the disulphide bonds are broken.
When the protein is allowed to renature by removing the denaturant and the reductant, the protein regain its native conformation, including four correctly paired disulphide bonds.
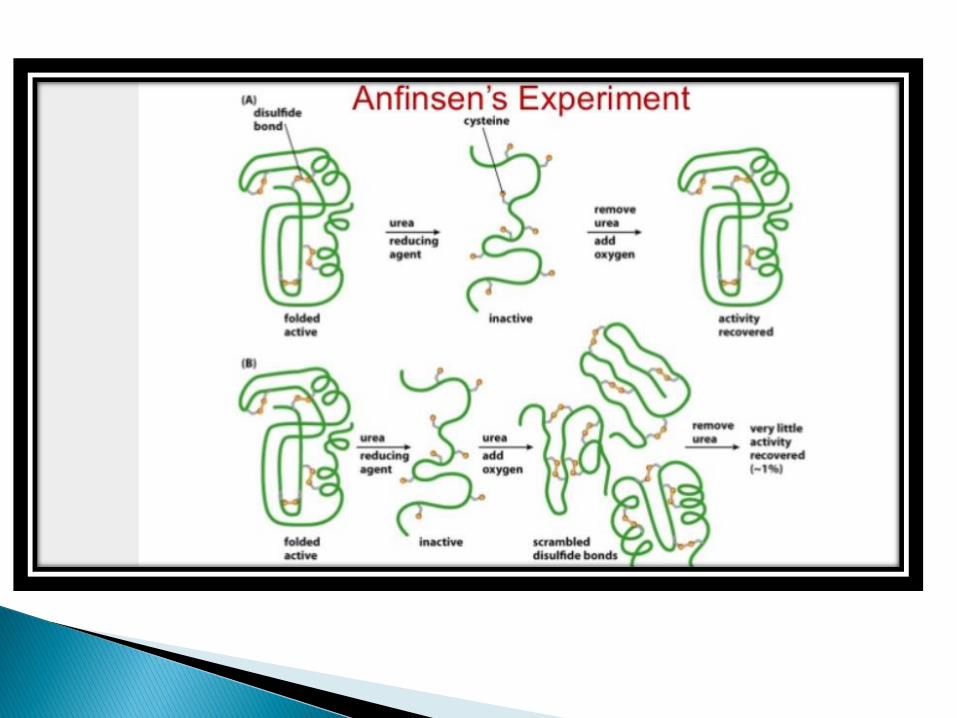

There are several models to explain folding. According to one model, folding is initiated by a spontaneous collapse of the unfolded polypeptide chain into a partly organized globular state, mediated by hydrophobic interactions among nonpolarresidues[HYDROPHOBIC COLLAPSE].
The collapsed state is referred to as a moltenglobule.
This state is clearly different from the native and the denatured state.
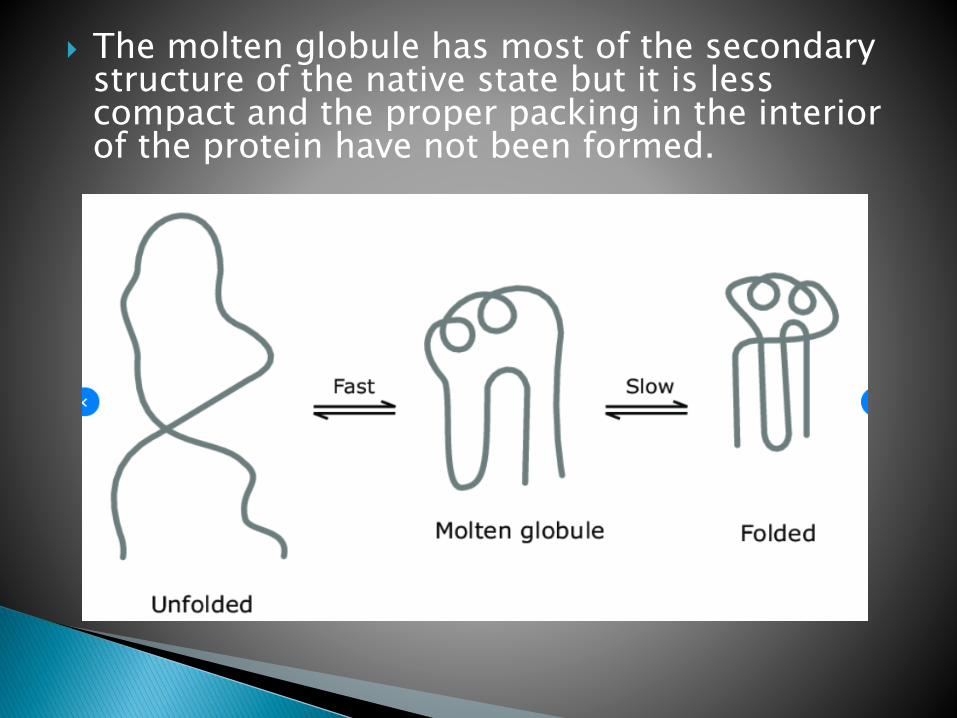
The molten globule has most of the secondary structure of the native state but it is less compact and the proper packing in the interior of the protein have not been formed.

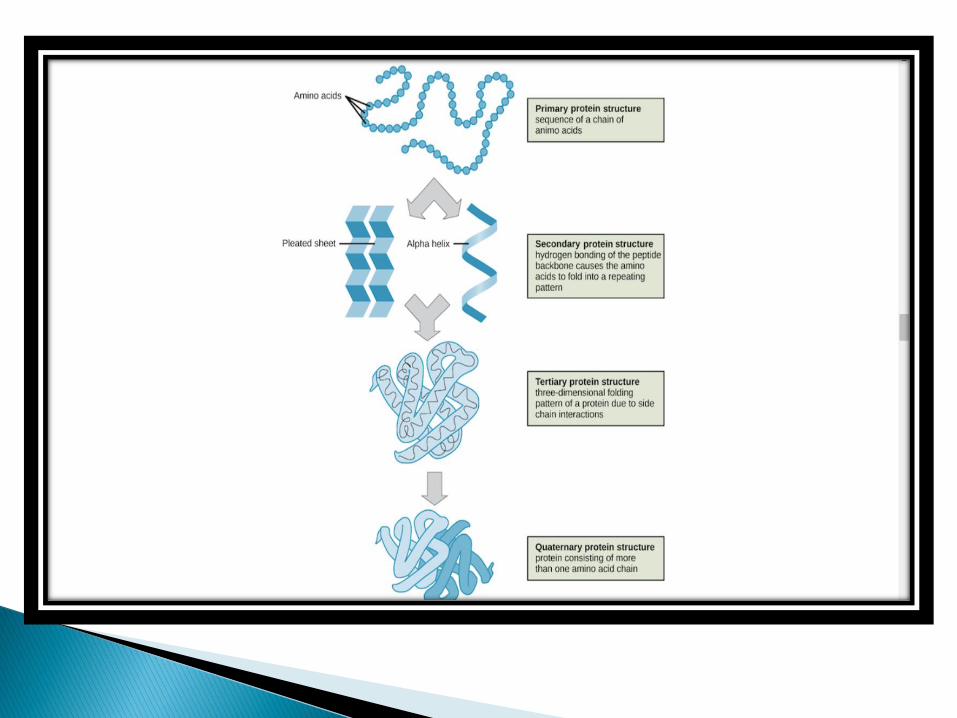
❖ PRIMARY STRUCTURE
❖ SECONDARY STRUCTURE
❖ TERTIARY STRUCTURE
❖ QUATERNARY STRUCTURE

The folding of a protein is a complex process, involving four stages, that gives rise to various 3D protein structures essential for diverse function in the human body.
The structure of a protein is hierarchically arranged, from a primary to quaternary structure.
The wide variation in amino acid sequences accounts for the different conformations in protein structure

Primary structure of a protein is linear amino acid sequence, determine its native conformation.
The specific amino acid residues and their position in the polypeptide chains are the determining factor for which portions of the protein fold closely together and form its three dimensional conformation.
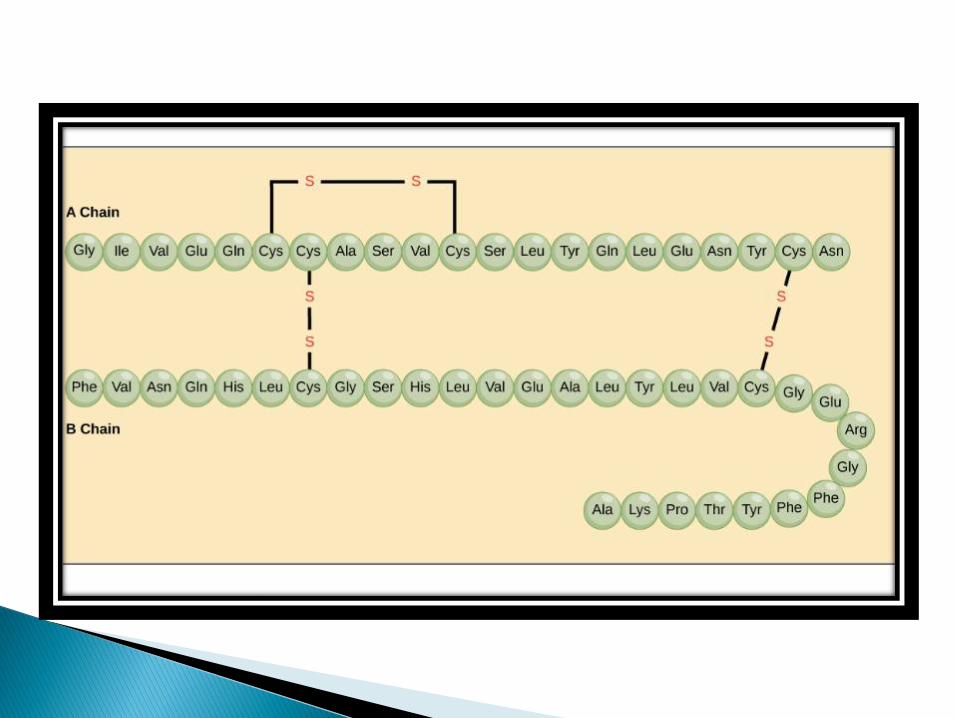

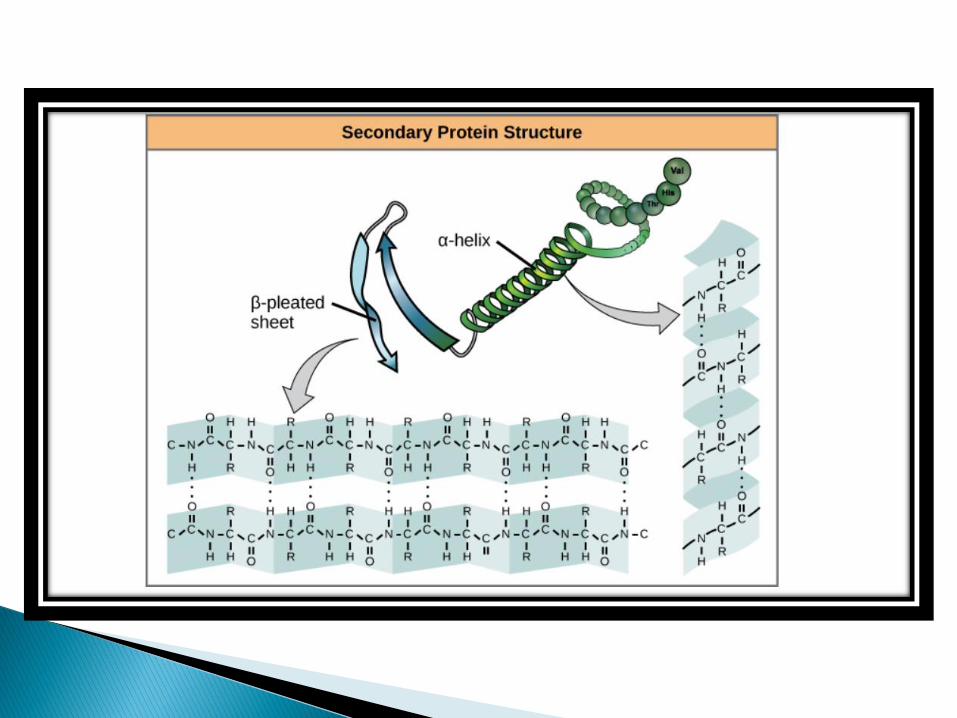
The formation of secondary structure is the first step in the folding process that a protein takes to assume its native structure.
Characteristics of secondary structure are the structures known as alpha helices and beta sheets that fold rapidly because they are stabilized by intra-molecular hydrogen bonds, as was first characterized by LINUS PAULING.

Formation of intra-molecular hydrogen bonds provides another important contribution to protein stability.
Protein secondary structure takes on the three forms
➢ALPHA HELIX
➢ BETA SHEET
➢ TURNS

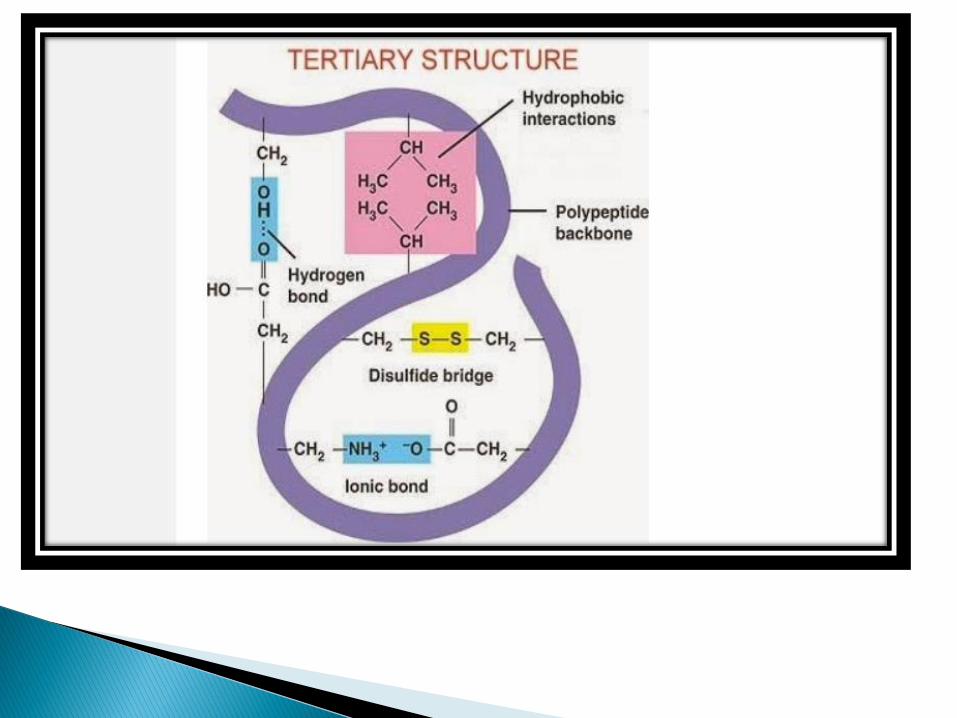
The term tertiary structure refers to the unique three dimensional conformations that globular proteins assume as a consequence of the interaction between the side chains in their primary structure.
The following types of covalent and non covalent interactions stabilize tertiary structure.
✓ hydrophobic interactions [major form of non covalent interaction]

✓ Electrostatic interactions[or salt bridges]
✓ Hydrogen bonds✓ Van der waals force of interactions✓ Covalent bond[disulfide bond]

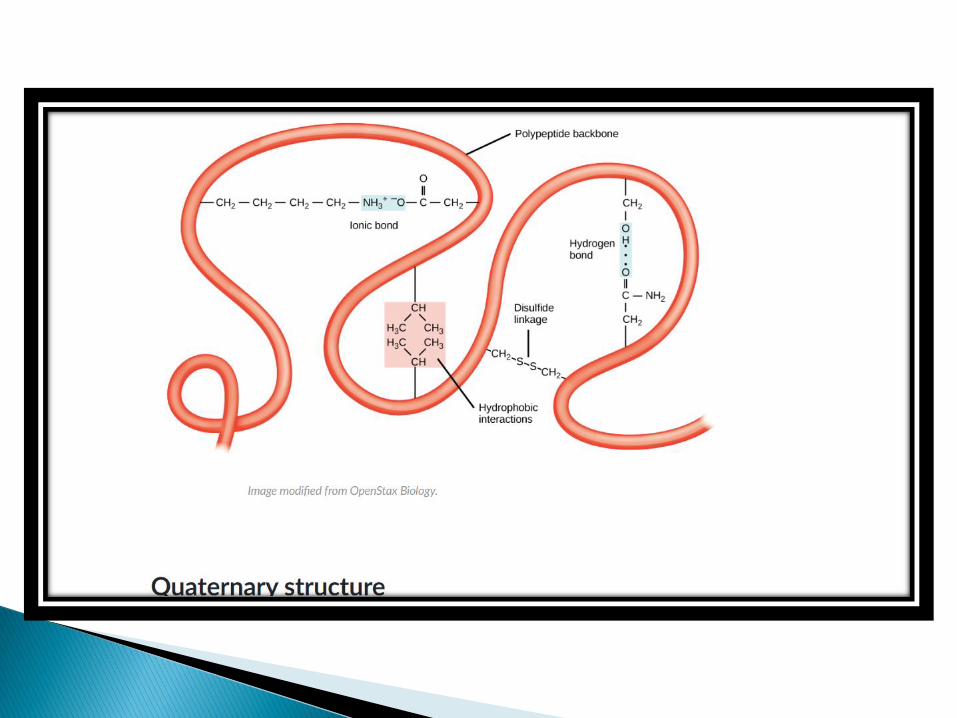
Many proteins like haemoglobin, are composed of two or more polypeptide chains.
These proteins are called multimeric proteins and each polypeptide chain is called a subunit.
Polypeptide subunits assembles to form quaternary structure and are held together by non covalent [such as hydrophobic interactions, electrostatic, hydrogen bond] as well as covalent interactions [interchaindisulfide bonds].

Protein that facilitate the folding of other protein are called MOLECULAR CHAPERONES.
Chaperones act as catalysts that facilitate assembly without being part of the assembled complex.
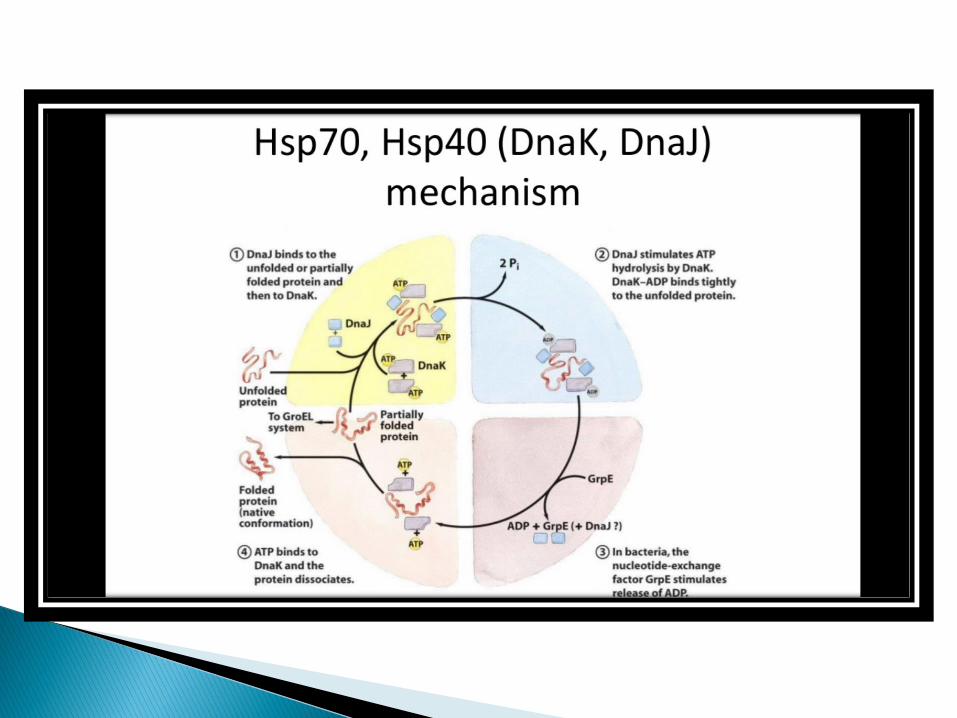
The Hsp70 and Hsp60 families of heat shock protein appear to be particularly important in the general pathways of protein folding in both prokaryotic and eukaryotic cells.
The proteins of both families function by binding to unfolded regions of polypeptides chains.

Members of the Hsp70 family stabilize unfolded polypeptide chains during translation as well as during the transport of polypeptides into a variety of subcellularcompartments, such as mitochondria and the endoplasmic reticulum.
These proteins bind to short segments of unfolded polypeptides, maintaining the polypeptide chain in an unfolded configuration and preventing aggregation.
Members of the Hsp60 family facilitate the folding of proteins into their native conformations.

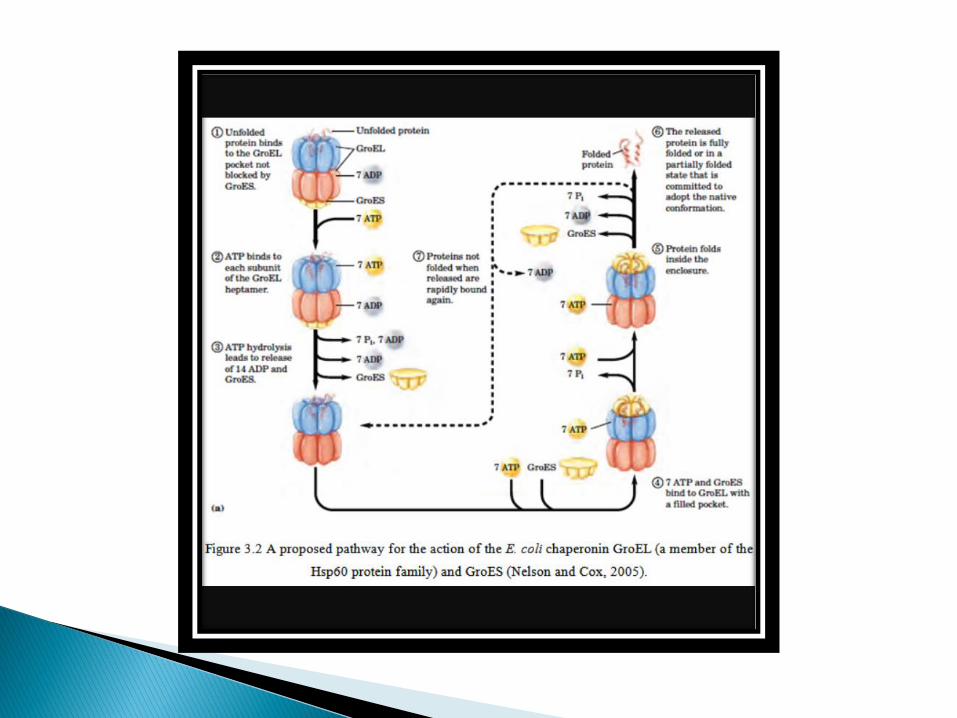
Each chaperonin consist of 14 subunits, arranged in two stacked rings to form a ‘double doughnut’ structure.
Unfolded polypeptide chains are shielded from the cytosol by being bound within the central cavity of the chaperonin cylinder.
In this isolated environment protein folding can proceed while aggregation of unfolded segments of the polypeptide chains is prevented by their binding to the chaperonin.

The binding of unfolded polypeptides to the chaperonin is a reversible reaction that is couple to the hydrolysis of ATP as a source of energy.
ATP hydrolysis thus drives multiple rounds of release and rebinding of unfolded regions of the polypeptide chain to the chaperonin, allowing the polypeptide regions of the polypeptide to fold gradually into the correct conformation.

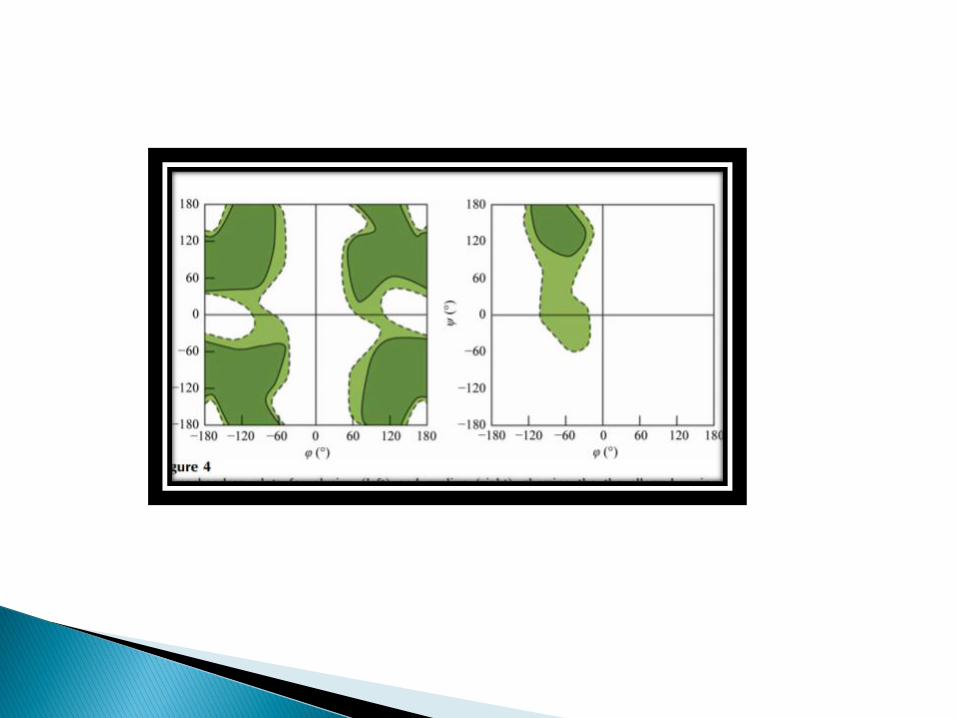
In a polypeptide the main chain N-Calpha and Calpha-C bonds relatively are free to rotate.
These rotations are represented by the torsion angles phi and psi, respectively.
G.N Ramachandran used computer models of small polypeptides to systematically vary phi and psi with the objective of finding stable conformations.

G.N Ramachamdran plots the phi value on the X-axis and the psi value on the Y-axis.
Plotting the torsional angles in this way graphically shows which combination of angles are possible.
The torsional angles of each residue in a peptide define the geometry of its attachment to its two adjacent residues by positioning its planar peptide bond relatives to the two adjacent planar peptide bonds, thereby the torsional angles determine the conformation of the residues and the peptide.
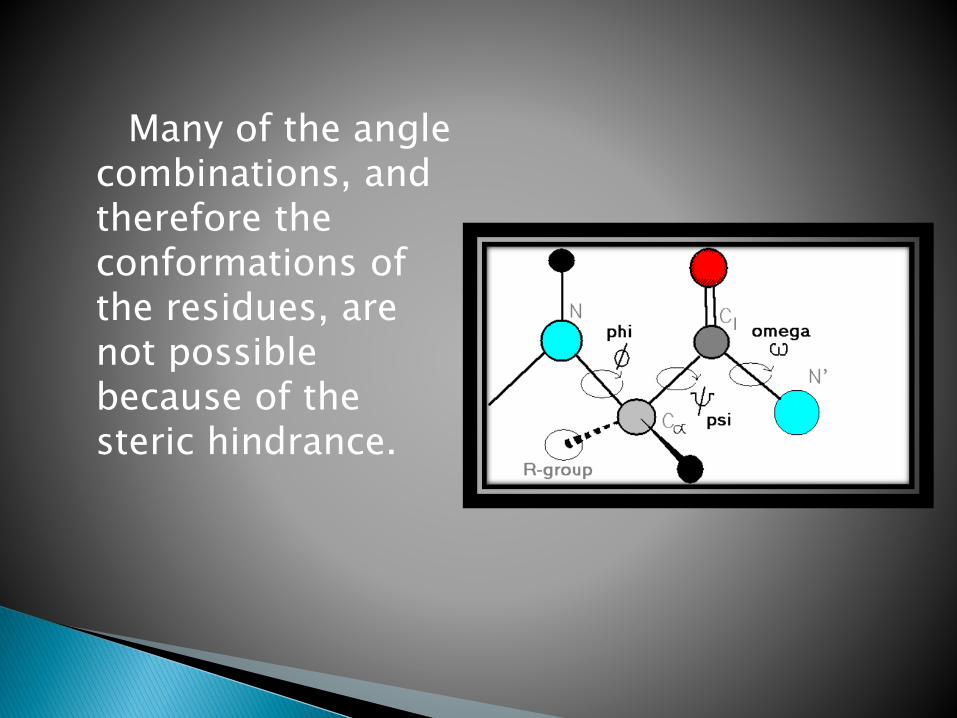
Many of the angle combinations, and therefore the conformations of the residues, are not possible because of the steric hindrance.
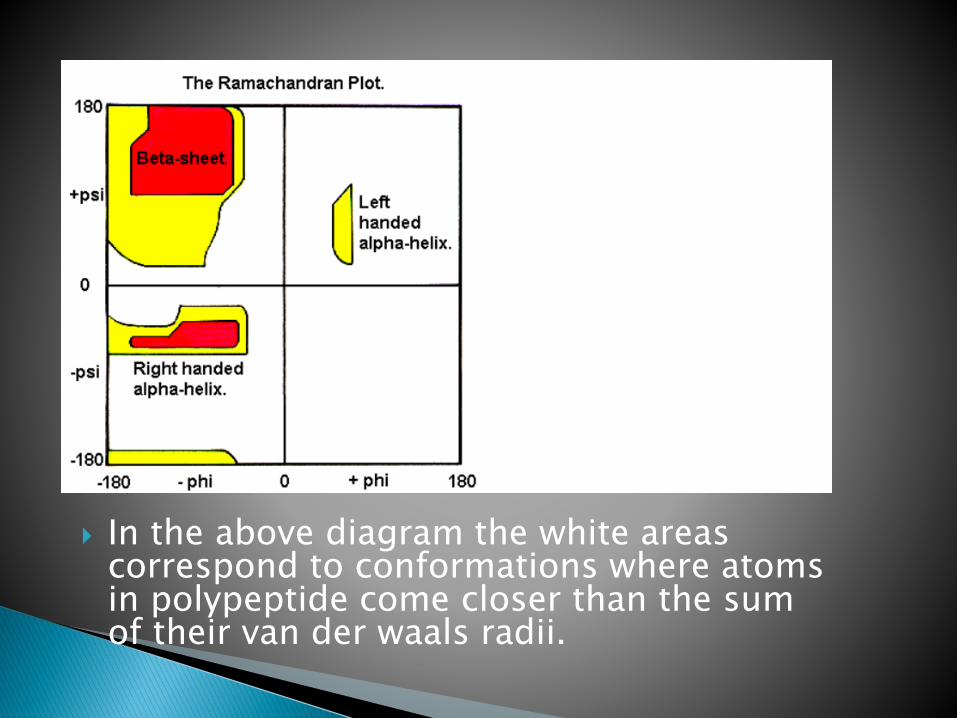
In the above diagram the white areas correspond to conformations where atoms in polypeptide come closer than the sum of their van der waals radii.

These regions are strically disallowed for all amino acids except glycine which is unique in that it lacks a side chain.
The red regions correspond to conformations where there are no steric clashes i.e these are the allowed regions namely the alpha- helical and beta-sheet conformations.
The yellow area shows the allowed regions if slightly shorter van der waals radi are used in the calculation, i.e the atoms are allowed to come a little closer together.
This bring out an additional region which corresponds to the left handed alpha helix.

L-amino acids cannot form extended regions of left-handed helix, but occasionally individual residues adopt this conformation.
These residues are usually glycine but can also be aspargine or aspartate where the side chain forms a hydrogen bond with the main chain and therefore stabilizes this otherwise unfavorable conformation.
Disallowed regions generally involve sterichindrance between the side chain C-beta methylene group and main chain atoms.

Glycine has no side chain and therefore can adapt phi and psi angels in all four quadrants of the Ramachandran plot.
Hence it frequently occurs in turn regions of proteins where any other residues would be sterically hindered.

Kumar.pranav and Mina.usha[2017], Life sciences,sitxth edition, Pathfinder publication.
Nelson.L.D and Cox.M.M[20], Principles of biochemistry,seventh edition, New York, W.H.Freemaid and company.
Cheriyedath.S, Protein folding[online] available at http;/www.news.medical.net.
Teacher krishna, Protein folding and Ramachandran plot[online] available at http;/www.slideshare.net.
