prevalence and diversity of avian influenza viruses in environmental reservoirs
TRANSCRIPT

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
Prevalence and diversity of avian influenza viruses inenvironmental reservoirs
Andrew S. Lang,1 Anke Kelly2 and Jonathan A. Runstadler2
Correspondence
Jonathan A. Runstadler
1Department of Biology, Memorial University of Newfoundland, St John’s, NL A1B 3X9, Canada
2Institute of Arctic Biology, PO Box 757000, University of Alaska Fairbanks, Fairbanks, AK 99775,USA
Received 10 August 2007
Accepted 14 September 2007
Little is known about the ecology and evolution of avian influenza in the natural environment,
despite how these affect the potential for transmission. Most work has focused on characterizing
viruses isolated from hosts such as waterfowl, and there have also been several instances of
isolation and detection from abiotic sources such as water and ice. We used RT-PCR to amplify
and characterize the influenza virus sequences present in sediments of ponds that are used
heavily by waterfowl. The detection rate of influenza virus was high (.50 %). Characterization of
the viruses present by sequencing part of the haemagglutinin (HA) gene showed that there is a
diverse collection of viruses in these sediments. We sequenced 117 partial HA gene clones
from 11 samples and detected four different HA subtypes (H3, H8, H11 and H12), with
approximately 65 % of clone sequences being unique. This culture-independent approach was
also able to detect a virus subtype that was not found by sampling of birds in the same
geographical region in the same year. Viruses were detected readily in the winter when the ponds
were frozen, indicating that these sediments could be a year-to-year reservoir of viruses to
infect birds using the ponds, although we have not shown that these viruses are viable. We
demonstrate that this approach is a feasible and valuable way to assess the prevalence and
diversity of viruses present in the environment, and can be a valuable complement to more difficult
viral culturing in attempting to understand the ecology of influenza viruses.
INTRODUCTION
There is increased recent concern about the possible arrivalin North America of highly pathogenic strains of avianinfluenza viruses (Chen et al., 2005, 2006; Ferguson et al.,2005; Liu et al., 2005; Olsen et al., 2006; Savill et al., 2006).Alaska is an intersection of migratory bird routesoriginating throughout the world, including North andSouth America, Asia, Australia and Africa. Millions ofmigratory birds arrive in Alaska to breed each spring, andmany of these breeding migrants are waterfowl. There hasbeen previous work to isolate and characterize influenzaviruses from waterfowl in Alaska (Ito et al., 1995;Runstadler et al., 2007; Spackman et al., 2005), and theseefforts continue. The information gained about influenzaviruses in these birds is important for understanding thebiology of influenza viruses and their spread amongst birdsfrom different locations. This, in turn, is important forunderstanding the public health risks associated with these
viruses and their potential transmission to agriculturalspecies. Unfortunately, the role of the physical andbiogeochemical environment as an integral part of thistransmission is poorly understood.
We recently described samples collected from an importantwaterfowl-breeding wetland area, the Minto Flats StateGame Refuge in the interior region of Alaska (Runstadleret al., 2007). Cloacal swabs were taken from 880 ducks inthe summer of 2005 and screened for the presence ofinfluenza viruses. Over 25 % of the samples were found tobe positive for influenza virus (Runstadler et al., 2007).Culturing and subsequent subtyping of viruses from asubset of these samples revealed five different haemagglu-tinin (HA)/neuraminidase (NA) subtypes (H3N6, H3N8,H4N6, H8N4 and H12N5). This work demonstrated anoverall high rate of virus infection in birds at this locationand showed that a diverse array of subtypes was present.This particular location was also the focus of a previousstudy between the years 1991 and 1994 (Ito et al., 1995),where six virus subtypes were isolated from 391 waterfowlfaecal samples from Minto Lake.
Here, we report efforts to characterize the occurrence anddiversity of influenza viruses in the sediments of pondsused by a wide variety of migratory waterfowl. Located
The GenBank/EMBL/DDBJ accession numbers for the sequencesreported in this paper are EU086918–EU087180.
A supplementary figure showing sampling locations for this study andsupplementary tables identifying viruses included in the H3 and H11phylogenetic analyses are available with the online version of this paper.
Journal of General Virology (2008), 89, 509–519 DOI 10.1099/vir.0.83369-0
0008-3369 G 2008 SGM Printed in Great Britain 509

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
within the city of Fairbanks, Alaska, is the Creamer’s FieldMigratory Waterfowl Refuge, a location that is used heavilyby migratory waterfowl during both the spring and fallmigration periods. The waterfowl, regularly numbering inthe thousands each day, are largely concentrated at threesmall ponds; samples were collected from these threeponds, beginning in the fall migration period of 2005,through the winter and into the spring migration period of2006. We used RT-PCR to target the matrix (M) and HAgenes of influenza viruses in RNA extractions from thesediment samples. The diversity of sequences in thesesamples was extremely high and included four different HAsubtypes. Most, but not all, of the HA sequences were mostsimilar to viruses isolated from ducks in Alaska in 2005.This study demonstrates that environmental sampling is avaluable technique to assess the diversity of influenzaviruses in specific geographical or environmental locationsto complement other approaches, but without the need formore difficult and time-dependent bird sampling andscreening of cloacal swabs by real-time PCR or culturing.Viruses were readily detectable in samples collected in themiddle of winter and early spring before the arrival ofmigrants. Therefore, as has been suggested for lake water(Webster et al., 1992), viruses could be persisting insediments and be a source of infection for new birds insubsequent years.
A recent summary of influenza infections by subtype indifferent wild bird species (Olsen et al., 2006) shows apredilection for some viral subtypes in specific groups ofmammals and birds. If, particularly in the case of migratorybirds, animals are dispersed in time from a potential ‘hotzone’ of virus infection, such as a migratory stopover, thenthe environmental persistence of viruses may play a strongecological role in transmission. Animals utilizing an areawhere persistence in environmental reservoirs is possiblemay experience increased viral exposure and thereforegreater potential for infection and also reassortment. Thebehaviour of the virus outside the host could therefore playa major role in interspecies infection and viral ecology andevolution (Kuiken et al., 2006).
METHODS
Sample collection and RNA isolation. Samples were collected
from three ponds within the Creamer’s Field Migratory Waterfowl
Refuge, located within the city of Fairbanks, Alaska (see
Supplementary Fig. S1, available in JGV Online). Sediment samples
were collected from the surface of the pond sediments within 1 m of
the pond edge. On one occasion, when the ponds had been drained of
all water, we were able to collect samples from further away from the
pond edges. Sediments were collected into sterile, 50 ml, plastic screw
cap tubes and stored at 4 uC briefly or at 280 uC until processed.
RNA was extracted from 2 g sediment sample by using an RNA
PowerSoil Total RNA isolation kit (MoBio) according to the
manufacturer’s recommendations. The resulting nucleic acid eluate
was then treated with 1 unit RNase-free DNase I (New England
Biolabs) according to the manufacturer’s instructions, followed by
extraction with buffered phenol/chloroform (1 : 1) and ethanol
precipitation (Sambrook & Russell, 2001). The precipitated RNAwas dissolved in 20 ml DEPC-treated water (Ambion) and stored at
280 uC.
Amplification of influenza virus sequences. RNA isolated fromsediments was screened for the presence of influenza virus sequences
by RT-PCR targeting part of the M gene. The primers M52C (59-CTTCTAACCGAGGTCGAAACG-39) and M253R (59-AGGGCATT-
TTGGACAAAKCGTCTA-39) (Fouchier et al., 2000) were used toamplify a 244 bp product from the M1 portion of the M segment. For
the HA gene, the primers HA-1134F (59-GGAATGATHGAYGG-NTGGTATGG-39) (Phipps et al., 2004) and Bm-NS-890R (59-ATA-
TCGTCTCGTATTAGTAGAAACAAGGGTGTTTT-39) (Hoffmann
et al., 2001) were used to amplify an approximately 640 bp regionof the HA-2 portion of the HA segment. The above primers were used
in RT-PCR with the SuperScript III One-Step RT-PCR system withPlatinum Taq DNA polymerase (Invitrogen). The reactions contained
1 ml RNA, each primer at 0.4 mM and 1 ml enzyme mix in 16 bufferin a total volume of 25 ml. For M sequence amplification, the thermal
cycler conditions were 42 uC for 30 min, then 94 uC for 4 min,followed by 35 cycles of 94 uC for 1 min, 45 uC for 1 min and 72 uCfor 1 min, and a final 7 min incubation at 72 uC. For HA sequenceamplification, the thermal cycler conditions were 42 uC for 30 min,
then 94 uC for 4 min, followed by 35 cycles of 94 uC for 1 min, 45 uCfor 1 min and 72 uC for 2 min, and a final 7 min incubation at 72 uC.
For the HA sequence amplification reactions, a second round of PCRwas used to increase the amount of material available for analyses.
These reactions were performed with either the same primers as were
used in the RT-PCR (HA-1134F and Bm-NS-890R) or with primerHARKs, a shortened version of the HA-specific reverse primer HAR K
(Bragstad et al., 2005) with non-influenza sequences removed (59-AGTAGAAACAAGGCTGTTTT-39), in place of Bm-NS-890R. Both
primer sets were used in parallel and the reactions that gave betteramplification were used for the subsequent cloning step. The entire
initial RT-PCR product was run on a 1 % agarose gel and DNA wasvisualized with ethidium bromide staining. Plugs were removed from
bands at the correct location on the gel (approx. 640 bp) with a sterilePasteur pipette and collected in a sterile microfuge tube containing
100 ml sterile distilled water. The tubes were then heated at 80 uC for20 min and 50 ml liquid was collected. This eluate was used for a
second round of PCR with the following conditions: 1 ml eluate, each
primer at 0.4 mM, 0.5 units Platinum Taq DNA polymerase, 0.4 mMeach dNTP, 2 mM MgCl2 and 16 Platinum Taq polymerase buffer in
a final volume of 25 ml. The thermal cycler conditions were 94 uC for5 min, followed by 35 cycles of 94 uC for 1 min, 45 uC for 1 min and
72 uC for 1 min, and a final 7 min incubation at 72 uC.
During the initial screening of the RNA extracts for the M gene, a
control reaction was also performed with every sample in parallel totest for potential inhibition of the PCR by factors in the purified
RNA. The control reactions contained primers targeting small subunitrRNA, uni-for (59-TGCCAGCAGCCGCGGTA-39) and uni-rev (59-
GACGGGCGGTGTGTACAA-39) (Vaisvila et al., 2001). It wasassumed that every RNA extraction from the sediment samples
would contain at least bacterial rRNA; therefore, a negative result inthis reaction was taken as evidence of failure of the RT-PCR process
itself and not necessarily a genuine influenza-negative sample.
The methods used here to amplify viral sequences with RT-PCR
products do introduce errors into the final cloned sequences (Brachoet al., 1998), and these cannot be distinguished specifically from
mutations introduced by the viral RNA polymerase, which has an
estimated error rate of 261023 per position per generation (Websteret al., 1992).
DNA cloning and sequencing. The RT-PCR and PCR productswere cleaned with a MinElute PCR purification kit (Qiagen) and
A. S. Lang, A. Kelly and J. A. Runstadler
510 Journal of General Virology 89

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
cloned with the TOPO-TA system (Invitrogen) according to the
manufacturers’ recommendations. The resulting colonies were
screened by colony PCR according to the manufacturer’s directions
(Invitrogen). Products of interest from the colony PCRs were
sequenced with a BigDye Terminator v. 3.1 cycle sequencing kit
(Applied Biosystems) according to the manufacturer’s recommenda-
tions. The sequencing reactions were analysed on an ABI 3100
sequencer (Applied Biosystems) at the University of Alaska Fairbanks
DNA Core Facility.
Sequence analyses, alignments and phylogenetics. The nuc-
leotide sequences of all M and HA clones analysed in this study have
been deposited in GenBank under accession numbers EU086918–
EU087180. Sequences were compared with those in GenBank by using
the MEGABLAST BLASTN algorithm (Altschul et al., 1990). Nucleotide
sequence alignments were done with CLUSTAL_X v. 1.81 (Chenna et al.,
2003; Thompson et al., 1997) and these alignments were used for
subsequent phylogenetic analyses. Bayesian maximum-likelihood
trees were constructed with MrBayes v. 3.1.2 (Huelsenbeck &
Ronquist, 2001) and 4 000 000 generations. Neighbour-joining trees
were constructed with PAUP v. 4.0 (Swofford, 2000) and bootstrap
values were calculated based on percentages of 10 000 replicates.
RESULTS
M sequence prevalence and diversity
In total, 91 samples were collected on 16 different daysbetween 12 August 2005 and 18 May 2006 (Table 1). Ten ofthese samples (11 %) did not produce a product with therRNA control primers and so we assume that these wereinhibited for RT-PCR (these samples are still included inTable 1). Of the 81 remaining samples, 45 (55.6 %) werepositive for the influenza virus M gene by RT-PCRscreening. We cloned the M RT-PCR products from 12
of these positive samples and sequenced at least ninedifferent clones from each cloned sample, with a total of145 clones sequenced overall. In the 145 clones, we found52 different sequences. Some sequences differ from eachother at only a single base position, and it is possible thatsome of the observed differences in sequences resultedfrom errors introduced by the polymerase enzymes(Bracho et al., 1998).
The presence of influenza virus, as indicated by a positiveresult for the M gene in RT-PCR, did not show either aseasonal pattern or a pattern between the sampling sites(Table 1). In addition, multiple samples were sometimescollected on the same day from the same pond and some ofthese samples tested positive by the RT-PCR assay, whilstothers were negative for the virus.
HA sequence prevalence and diversity
We amplified and cloned the HA gene RT-PCR productsfrom 11 different samples. At least five clones weresequenced from each cloned sample, with a total of 116clones sequenced. Within these 116 clones, there were 76different nucleic acid sequences (65.5 %) and 44 differentpredicted protein sequences (37.9 %). Some sequencesdiffer from each other at only a single base position, and itis possible that some of the observed differences insequences resulted from errors introduced by the poly-merase enzymes (Bracho et al., 1998).
Four HA subtypes were identified by molecular methods inthese sequences: H3, H8, H11 and H12 (Tables 1 and 2;Fig. 1). The abundance of sequences recovered in theclones was H3.H12.H11.H8, with H3 and H12
Table 1. M gene RT-PCR results for the three ponds over the sampling period
Sample date RT-PCR results for the M gene* Occupancy of ponds HA subtypes detected
Pond 1 Pond 2 Pond 3
12 August 2005 0/1 NS NS Birds
17 August 2005 NS 1/1 0/1 Birds
24 August 2005 1/1 (0/1) 0/1 Birds H3
5 September 2005 1/1 0/1 1/1 Birds
8 September 2005 1/1 1/1 1/1 Birds H3, H11, H12
13 September 2005 1/1 0/1 1/1 Birds H3, H12
15 September 2005 1/1 0/1 0/1 Birds H3, H12
23 September 2005 1/1 0/1 1/1 Birds
26 September 2005 NS 1/4 3/4 No birds H3, H11, H12
28 September 2005 10/26 NS NS No birds
29 September 2005 NS 12/20 NS No birds
3 October 2005 NS NS 0/3 No birds
24 January 2006 NS 1/1 1/1 No birds (frozen) H3, H12
1 March 2006 (0/2) 3/3 NS No birds (frozen) H3, H8, H11, H12
23 March 2006 0/2 1/1 NS No birds (frozen)
18 May 2006 1/1 0/1 (0/1) Birds H3, H12
*No. positive samples/total no. samples taken; NS, not sampled. Parentheses indicate that all samples were also negative for the RT-PCR control
reaction.
Influenza viruses in environmental reservoirs
http://vir.sgmjournals.org 511
Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
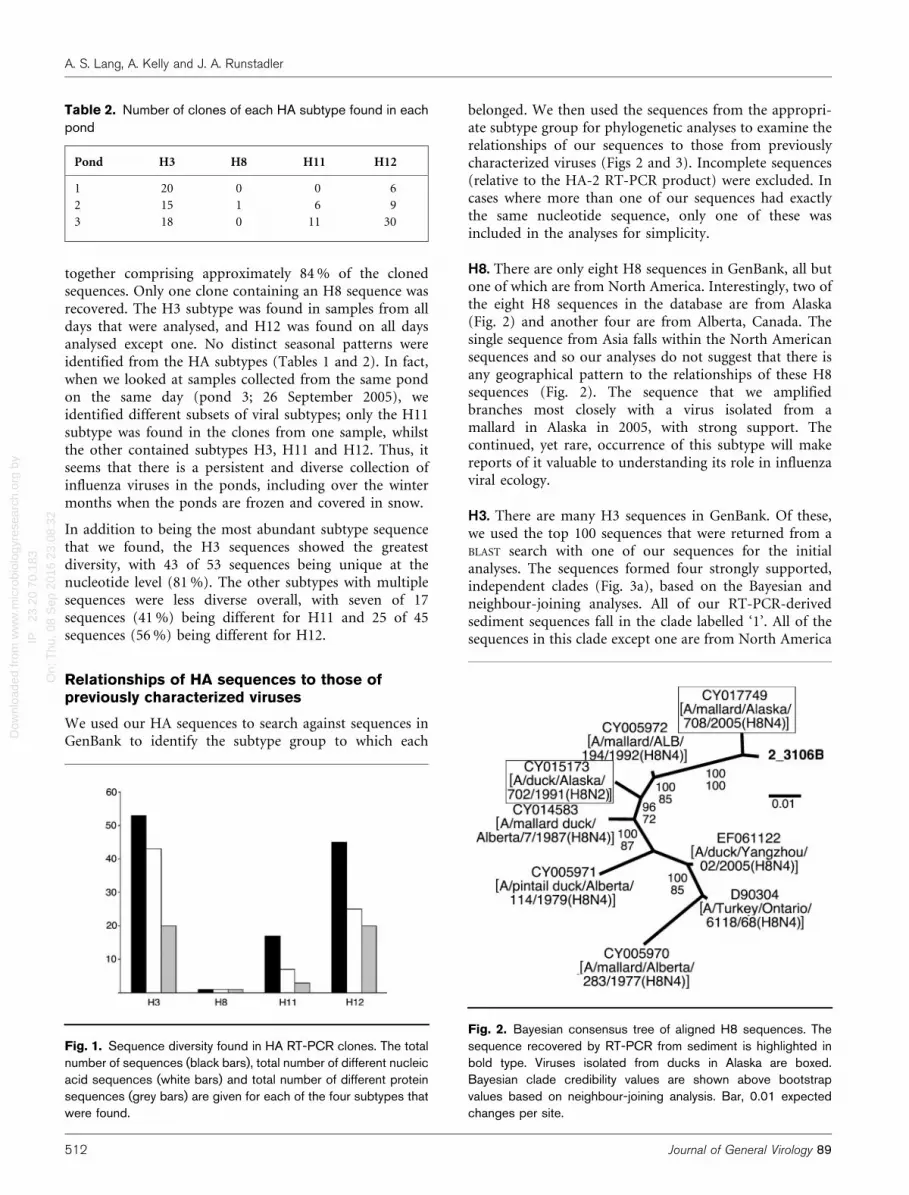
together comprising approximately 84 % of the clonedsequences. Only one clone containing an H8 sequence wasrecovered. The H3 subtype was found in samples from alldays that were analysed, and H12 was found on all daysanalysed except one. No distinct seasonal patterns wereidentified from the HA subtypes (Tables 1 and 2). In fact,when we looked at samples collected from the same pondon the same day (pond 3; 26 September 2005), weidentified different subsets of viral subtypes; only the H11subtype was found in the clones from one sample, whilstthe other contained subtypes H3, H11 and H12. Thus, itseems that there is a persistent and diverse collection ofinfluenza viruses in the ponds, including over the wintermonths when the ponds are frozen and covered in snow.
In addition to being the most abundant subtype sequencethat we found, the H3 sequences showed the greatestdiversity, with 43 of 53 sequences being unique at thenucleotide level (81 %). The other subtypes with multiplesequences were less diverse overall, with seven of 17sequences (41 %) being different for H11 and 25 of 45sequences (56 %) being different for H12.
Relationships of HA sequences to those ofpreviously characterized viruses
We used our HA sequences to search against sequences inGenBank to identify the subtype group to which each
belonged. We then used the sequences from the appropri-ate subtype group for phylogenetic analyses to examine therelationships of our sequences to those from previouslycharacterized viruses (Figs 2 and 3). Incomplete sequences(relative to the HA-2 RT-PCR product) were excluded. Incases where more than one of our sequences had exactlythe same nucleotide sequence, only one of these wasincluded in the analyses for simplicity.
H8. There are only eight H8 sequences in GenBank, all butone of which are from North America. Interestingly, two ofthe eight H8 sequences in the database are from Alaska(Fig. 2) and another four are from Alberta, Canada. Thesingle sequence from Asia falls within the North Americansequences and so our analyses do not suggest that there isany geographical pattern to the relationships of these H8sequences (Fig. 2). The sequence that we amplifiedbranches most closely with a virus isolated from amallard in Alaska in 2005, with strong support. Thecontinued, yet rare, occurrence of this subtype will makereports of it valuable to understanding its role in influenzaviral ecology.
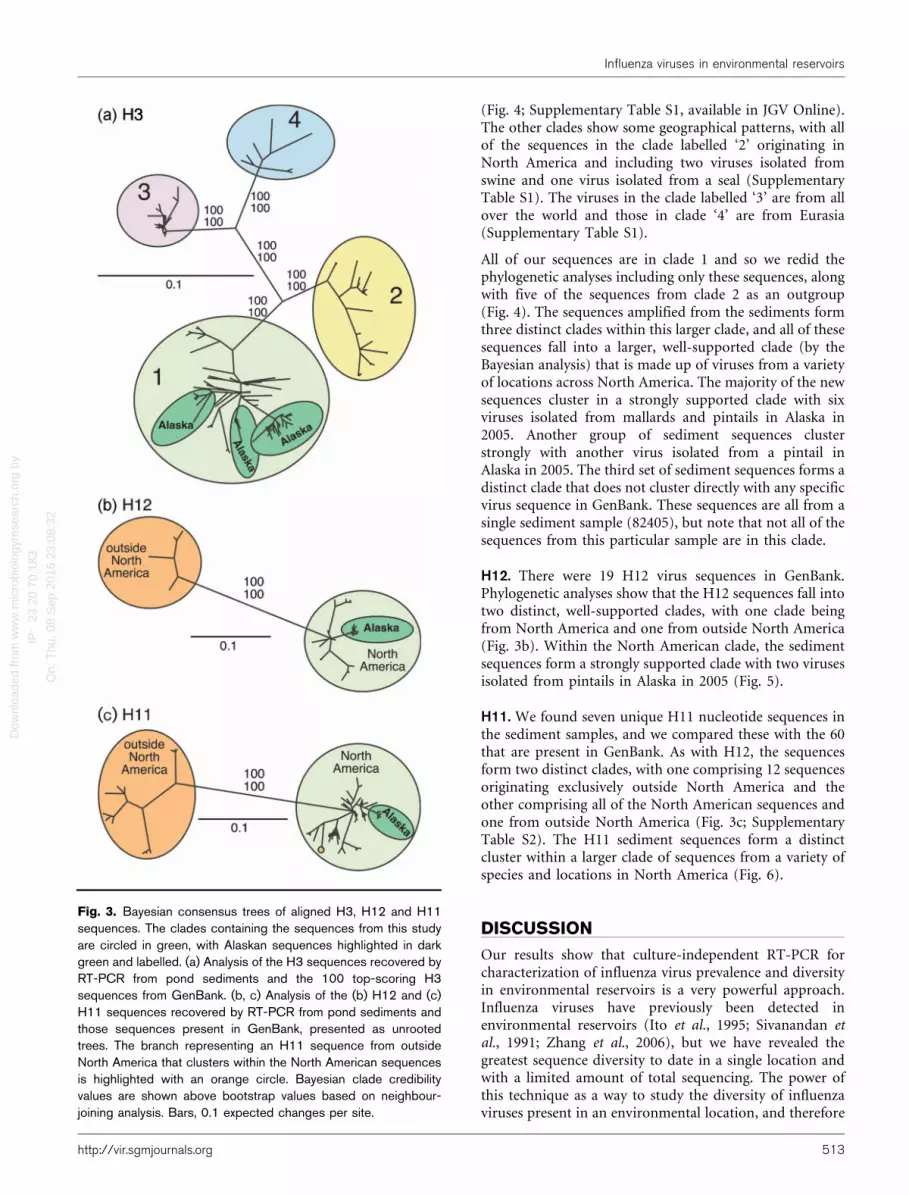
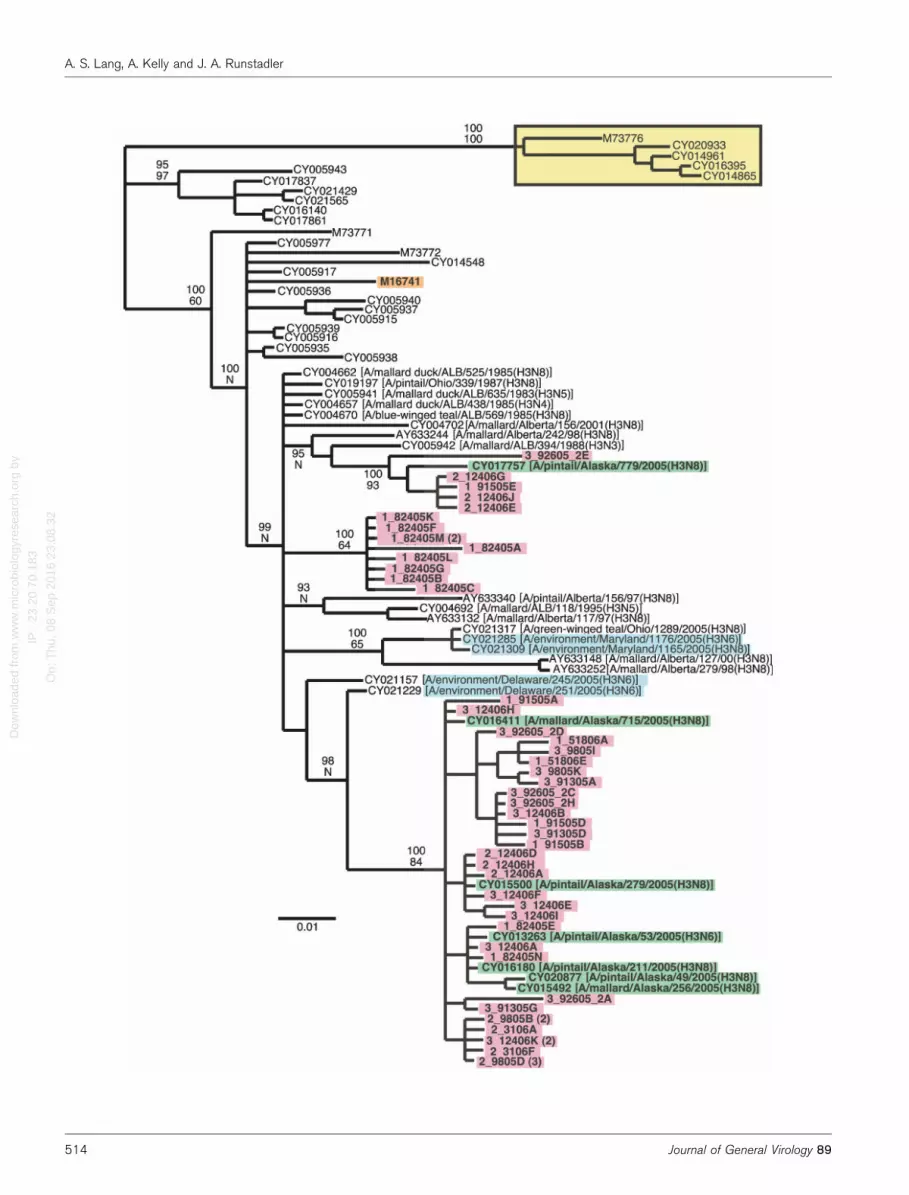
H3. There are many H3 sequences in GenBank. Of these,we used the top 100 sequences that were returned from aBLAST search with one of our sequences for the initialanalyses. The sequences formed four strongly supported,independent clades (Fig. 3a), based on the Bayesian andneighbour-joining analyses. All of our RT-PCR-derivedsediment sequences fall in the clade labelled ‘1’. All of thesequences in this clade except one are from North America
Table 2. Number of clones of each HA subtype found in eachpond
Pond H3 H8 H11 H12
1 20 0 0 6
2 15 1 6 9
3 18 0 11 30
Fig. 1. Sequence diversity found in HA RT-PCR clones. The totalnumber of sequences (black bars), total number of different nucleicacid sequences (white bars) and total number of different proteinsequences (grey bars) are given for each of the four subtypes thatwere found.
Fig. 2. Bayesian consensus tree of aligned H8 sequences. Thesequence recovered by RT-PCR from sediment is highlighted inbold type. Viruses isolated from ducks in Alaska are boxed.Bayesian clade credibility values are shown above bootstrapvalues based on neighbour-joining analysis. Bar, 0.01 expectedchanges per site.
A. S. Lang, A. Kelly and J. A. Runstadler
512 Journal of General Virology 89
Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
(Fig. 4; Supplementary Table S1, available in JGV Online).The other clades show some geographical patterns, with allof the sequences in the clade labelled ‘2’ originating inNorth America and including two viruses isolated fromswine and one virus isolated from a seal (SupplementaryTable S1). The viruses in the clade labelled ‘3’ are from allover the world and those in clade ‘4’ are from Eurasia(Supplementary Table S1).
All of our sequences are in clade 1 and so we redid thephylogenetic analyses including only these sequences, alongwith five of the sequences from clade 2 as an outgroup(Fig. 4). The sequences amplified from the sediments formthree distinct clades within this larger clade, and all of thesesequences fall into a larger, well-supported clade (by theBayesian analysis) that is made up of viruses from a varietyof locations across North America. The majority of the newsequences cluster in a strongly supported clade with sixviruses isolated from mallards and pintails in Alaska in2005. Another group of sediment sequences clusterstrongly with another virus isolated from a pintail inAlaska in 2005. The third set of sediment sequences forms adistinct clade that does not cluster directly with any specificvirus sequence in GenBank. These sequences are all from asingle sediment sample (82405), but note that not all of thesequences from this particular sample are in this clade.
H12. There were 19 H12 virus sequences in GenBank.Phylogenetic analyses show that the H12 sequences fall intotwo distinct, well-supported clades, with one clade beingfrom North America and one from outside North America(Fig. 3b). Within the North American clade, the sedimentsequences form a strongly supported clade with two virusesisolated from pintails in Alaska in 2005 (Fig. 5).
H11. We found seven unique H11 nucleotide sequences inthe sediment samples, and we compared these with the 60that are present in GenBank. As with H12, the sequencesform two distinct clades, with one comprising 12 sequencesoriginating exclusively outside North America and theother comprising all of the North American sequences andone from outside North America (Fig. 3c; SupplementaryTable S2). The H11 sediment sequences form a distinctcluster within a larger clade of sequences from a variety ofspecies and locations in North America (Fig. 6).
DISCUSSION
Our results show that culture-independent RT-PCR forcharacterization of influenza virus prevalence and diversityin environmental reservoirs is a very powerful approach.Influenza viruses have previously been detected inenvironmental reservoirs (Ito et al., 1995; Sivanandan etal., 1991; Zhang et al., 2006), but we have revealed thegreatest sequence diversity to date in a single location andwith a limited amount of total sequencing. The power ofthis technique as a way to study the diversity of influenzaviruses present in an environmental location, and therefore
Fig. 3. Bayesian consensus trees of aligned H3, H12 and H11sequences. The clades containing the sequences from this studyare circled in green, with Alaskan sequences highlighted in darkgreen and labelled. (a) Analysis of the H3 sequences recovered byRT-PCR from pond sediments and the 100 top-scoring H3sequences from GenBank. (b, c) Analysis of the (b) H12 and (c)H11 sequences recovered by RT-PCR from pond sediments andthose sequences present in GenBank, presented as unrootedtrees. The branch representing an H11 sequence from outsideNorth America that clusters within the North American sequencesis highlighted with an orange circle. Bayesian clade credibilityvalues are shown above bootstrap values based on neighbour-joining analysis. Bars, 0.1 expected changes per site.
Influenza viruses in environmental reservoirs
http://vir.sgmjournals.org 513
Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32A. S. Lang, A. Kelly and J. A. Runstadler
514 Journal of General Virology 89

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
present in birds that use the location, is well illustrated byone of the samples collected from pond 2 on 1 March 2006.In the RNA extracted from 2 g sediment, we were able todetect four different HA subtypes after sequencing only10 clones.
We found a remarkable amount of M gene diversity withlimited sequencing. The M primers that we used yield200 bp internal sequence in a well-conserved region of thevirus genome, and this region is a frequent target of PCR-based influenza assays (Fouchier et al., 2000; Runstadler
et al., 2007). Therefore, with this RT-PCR technique, weare detecting populations of viruses with a high degree ofdiversity in the environmental samples. The results of theM gene screening of the samples also suggest that thedistribution of viruses in the ponds is very patchy. Thereare instances where multiple samples were collected on thesame day and both positive and negative results were foundin different samples. This could result from patchydeposition of virus-containing faeces and subsequent lackof mixing, uneven degradation of virus particles, or otherbiogeochemical factors that could inhibit detection in some
Fig. 4. Bayesian consensus tree of a subset of aligned H3 sequences. The sequences in clade 1 in Fig. 3(a) are included,along with five of the sequences from clade 2 as the outgroup (highlighted in yellow). The sequences recovered by RT-PCRfrom pond sediments are highlighted in pink; if multiple clones containing the same sequences were found, the number is givenin parentheses. Viruses isolated from ducks in Alaska are highlighted in green. Viruses isolated from the environment arehighlighted in blue. All of the viruses are from North America except for the sequence highlighted in orange. Bayesian cladecredibility values are shown above bootstrap values based on neighbour-joining analysis (‘N’ indicates that an equivalent branchwas not well-supported in the neighbour-joining analysis). Identification information for viruses not labelled on the branches isprovided in Supplementary Table S1 (available in JGV Online). Bar, 0.01 expected changes per site.
Fig. 5. Bayesian consensus tree of aligned H12 sequences, with the sequences from outside North America (highlighted inorange) used as the outgroup to the North American sequences. The sequences recovered by RT-PCR from pond sedimentsare highlighted in pink; if multiple clones containing the same sequences were found, the number is given in parentheses.Viruses isolated from ducks in Alaska are highlighted in green. Bayesian clade credibility values are shown above bootstrapvalues based on neighbour-joining analysis for some of the branches. Bar, 0.1 expected changes per site.
Influenza viruses in environmental reservoirs
http://vir.sgmjournals.org 515

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
of the samples. Given this pattern, future studies shouldaim to collect a higher density of individual samples from agiven location (e.g. five samples from each pond) each timethat samples are taken.
In the RT-PCR assays that we used, the M gene targetprovided much better reliability, in terms of sensitivity andspecificity, than the HA gene. This is probably due toseveral factors, such as the smaller size of the target (200versus 600 bp) and better conservation of the primer targetsequences. This makes the M gene assay ideal for initialscreening. However, the sequence amplified in this case ismuch less informative about the viruses present. The HART-PCR was technically more problematic. Multiplerounds of amplification were usually required to obtainenough material for efficient cloning of the products.Multiple bands at sizes other than that expected, i.e.approximately 640 bp, were amplified frequently, and
non-influenza products that were the same size as theexpected influenza sequences were occasionally amplified,cloned and sequenced. These non-influenza sequences werefungal and bacterial in origin, based on BLAST searches ofGenBank. The use of more specific primer pairs that targetspecific subsets of HA subtypes may alleviate some of theseproblems.
For the HA-2 results, we found a high diversity in thesequences recovered in the clone libraries, with 76 of 116sequences (65.5 %) being unique. There were four differentsubtypes found, of which H3 and H12 were the mostabundant. The H3 sequences showed the greatest overallabundance and diversity in our study, and this is also trueof virus sequences present in GenBank (Figs 3a, 4) for thesefour subtypes. A more temporally intensive samplingscheme, combined with improving data on bird popula-tions and movements, may be able to determine whether
Fig. 6. Bayesian consensus tree of alignedH11 sequences, with the sequences fromoutside North America used as the outgroupto the North American sequences. Thesequences recovered by RT-PCR from pondsediments are highlighted in pink; if multipleclones containing the same sequences werefound, the number is given in parentheses.Viruses isolated from the environment arehighlighted in blue. Viruses isolated outsideNorth America are highlighted in orange.Identification information for viruses notlabelled on the branches is provided inSupplementary Table S2 (available in JGVOnline). Bayesian clade credibility values areshown above bootstrap values based onneighbour-joining analysis for some of thebranches (‘N’ indicates that an equivalentbranch was not well-supported in the neigh-bour-joining analysis). Bar, 0.1 expectedchanges per site.
A. S. Lang, A. Kelly and J. A. Runstadler
516 Journal of General Virology 89

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
viral diversity detected in our sequences has any relation tothe number and type of birds present at a given time or ischaracteristic of the viral populations in circulation at thetime.
Unlike the other subtypes, there have been no H11sequences reported previously from Alaska. Therefore, thisculture-independent RT-PCR method of looking for avianinfluenza in sediments was able to recover a subtype thatwas not found in a large-scale sampling of waterfowlspecies (Runstadler et al., 2007). Creamer’s Field is amigratory stopover site that attracts a great diversity ofwaterfowl, and also a great diversity of shorebirds andpasserine species in close proximity. Perhaps thesesequences originated from viruses deposited in theCreamer’s Field sediments by species that have not beensampled (heavily enough or at all) in other studies, or theculturing techniques used previously were less sensitive tothis particular virus subtype. This second explanationseems less likely, because many H11 viruses have beencultured successfully in the past. Alternatively, we could bedetecting viruses that were deposited in the sediments inprevious years and this subtype may not have beenprevalent in birds in 2005. In addition, direct samplingof birds is limited to times at which birds can be capturedefficiently and effectively; the viruses identified from suchsamples are thus temporally limited. Therefore, the H11sequences that we obtained may have been from a virusthat was prevalent in populations earlier in the season,but which had declined by the time of heavy waterfowlsampling. Moreover, they could represent viruses shedfrom birds that used locations other than the Minto Flatsarea.
The role of abiotic reservoirs in perpetuation andtransmission of influenza viruses is unclear (Websteret al., 1992), but viruses have previously been isolatedfrom and detected in water samples. Viruses were culturedsuccessfully from 12 of 102 water samples collectedbetween 1992 and 1994 from lakes in Alaska with breedingwaterfowl (Ito et al., 1995). Three different subtypes wereidentified in these water samples. This shows that abioticsources such as water (and sediment) could be acting asreservoirs of active viruses that can infect further birds. Ithas also been reported recently that influenza viruses havebeen detected in lake ice and water in Siberia (Zhang et al.,2006). This study only reported viruses with the H1subtype, and perhaps the more general primers used herewould allow the detection of other subtypes, if present.There was a high degree of diversity in the H1 sequencesfound, with 83 unique sequences recovered. These H1sequences showed a monophyletic pattern, similar toresults with our H12 and H11 sequences (Figs 5 and 6),but very distinct from the polyphyletic pattern that wefound for the H3 sequences (Fig. 4). There are four H3 andthree H11 virus sequences in GenBank included in ouranalyses that were isolated from environmental samples.The H11 environmental isolates all cluster strongly withtwo viruses isolated from shorebirds (Fig. 6), but the H3
viruses cluster most closely with viruses isolated fromducks and the sediment sequences reported here (Fig. 4). Astrong set of environmental data could contribute atemporal and spatial scale for virus distributions, so thatthe identification of a pathogenic virus in a bird could beused to map likely contact with other species in otherlocations.
A long-term study of the different HA subtypes that arefound in these ponds would be ideal for understanding theinflux of novel virus subtypes and possible passage ofothers out of the system. It may be possible to use thesesediments as an archive of historical influenza virusdiversity in birds using these ponds by doing linear depthprofiles with cores. Viable viruses can be found in oldsediments (up to 40 cm depth) in marine systems(Lawrence et al., 2002), and they may be equally wellpreserved here. Even if not viable, characterizing thediversity present over past time would be extremelyvaluable for understanding the ecology of influenza virusesin Alaskan waterfowl. Some sediments can harbourextremely high amounts of free DNA (Dell’Anno &Danovaro, 2005) but, to our knowledge, similarly abund-ant free RNA has not been found in natural systems. Wewould not expect extracellular RNA to be stable in thesesediments, where there is an abundant microbial com-munity that would rapidly degrade any free RNA.Therefore, we interpret the amplification of influenza virussequences to result from RNA extracted from intact virusparticles, although we have not demonstrated the presenceof infectious particles.
A limitation of this approach arises from the segmentednature of the influenza virus genome. RT-PCR can onlyprovide information about a single gene sequence at a time,and therefore it is not possible to know what combinationsof gene segments are in the actual viruses withoutculturing. It is clear that the combinations of genesegments in viruses are both important and dynamic(Brown et al., 1998; Ghedin et al., 2005; Hatchette et al.,2004; Holmes et al., 2005; Spackman et al., 2006; Websteret al., 1992). It is also unclear whether there might be a biaswith the HA-2 primers for which subtypes are amplifiedwell, but they have been used to amplify sequences from allof the subtypes H1–H15 (Phipps et al., 2004). In caseswhere there are mixtures of viruses, it is possible that somesequences would amplify better than others. This mayexplain the lack of any H4 subtype sequences in our clonecollections, despite the fact that H4 viruses were prevalentin the species of waterfowl using Creamer’s Field in 2005(Runstadler et al., 2007). Alternatively, it could be that H4subtypes are less stable in the sediments and do not persistas well, or were shed less prolifically than the othersubtypes that we did detect.
Application of this culture-independent RT-PCR approachto more samples and locations will greatly increase ourunderstanding of the prevalence and diversity of theinfluenza viruses circulating in host populations and the
Influenza viruses in environmental reservoirs
http://vir.sgmjournals.org 517

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
environments that they use. It could readily be expanded tocover analysis of more viral gene segments. In conjunctionwith the culturing of viruses, both from birds and fromenvironmental samples, this approach will offer significantcontributions to understanding influenza virus ecology andevolution.
ACKNOWLEDGEMENTS
We thank Nancy Gundlach, Lauralea Colamussi and Franziska Kohl
for help with collecting sediment samples, Danielle Mondloch for
help with the RNA extractions, screening clone libraries and
sequencing, Tom Chapman for suggestions concerning the phylo-
genetic analyses and Alex Culley for comments on the manuscript.
We also thank the anonymous reviewers for helpful comments. We
are grateful to Jason Caikowski, the Creamer’s Field Sanctuary
manager at the Alaska Department of Fish and Game, for access to the
ponds and information on waterfowl abundance. The project
described was supported by grant no. RR016466 from the National
Center for Research Resources (NCRR), a component of the National
Institutes of Health (NIH), and its contents are solely the
responsibility of the authors and do not necessarily represent the
official view of NCRR or NIH.
REFERENCES
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman,D. J. (1990). Basic local alignment search tool. J Mol Biol 215,
403–410.
Bracho, M. A., Moya, A. & Barrio, E. (1998). Contribution of Taq
polymerase-induced errors to the estimation of RNA virus diversity.
J Gen Virol 79, 2921–2928.
Bragstad, K., Jorgensen, P. H., Handberg, K. J., Mellergaard, S.,Corbet, S. & Fomsgaard, A. (2005). New avian influenza A virus
subtype combination H5N7 identified in Danish mallard ducks. Virus
Res 109, 181–190.
Brown, I. H., Harris, P., McCauley, J. & Alexander, D. (1998). Multiple
genetic reassortment of avian and human influenza A viruses in
European pigs, resulting in the emergence of an H1N2 virus of novel
genotype. J Gen Virol 79, 2947–2955.
Chen, H., Smith, G. J. D., Zhang, S. Y., Qin, K., Wang, J., Li, K. S.,Webster, R. G., Peiris, J. S. M. & Guan, Y. (2005). Avian flu H5N1
virus outbreak in migratory waterfowl. Nature 436, 191–192.
Chen, H., Smith, G. J. D., Li, K. S., Wang, J., Fan, X. H., Rayner, J. M.,Vijaykrishna, D., Zhang, J. X., Zhang, L. J. & other authors (2006).Establishment of multiple sublineages of H5N1 influenza virus in
Asia: implications for pandemic control. Proc Natl Acad Sci U S A
103, 2845–2850.
Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins,D. G. & Thompson, J. D. (2003). Multiple sequence alignment with the
CLUSTAL series of programs. Nucleic Acids Res 31, 3497–3500.
Dell’Anno, A. & Danovaro, R. (2005). Extracellular DNA plays a key
role in deep-sea ecosystem functioning. Science 309, 2179.
Ferguson, N. M., Cummings, D. A. T., Cauchemez, S., Fraser, C.,Riley, S., Meeyai, A., Iamsirithaworn, S. & Burke, D. S. (2005).Strategies for containing an emerging influenza pandemic in
Southeast Asia. Nature 437, 209–214.
Fouchier, R. A. M., Bestebroer, T. M., Herfst, S., Van der Kemp, L.,Rimmelzwaan, G. F. & Osterhaus, A. D. M. E. (2000). Detection of
influenza A viruses from different species by PCR amplification
of conserved sequences in the matrix gene. J Clin Microbiol 38,
4096–4101.
Ghedin, E., Sengamalay, N. A., Shumway, M., Zaborsky, J.,Feldblyum, T., Subbu, V., Spiro, D. J., Sitz, J., Koo, H. & otherauthors (2005). Large-scale sequencing of human influenza reveals
the dynamic nature of viral genome evolution. Nature 437,
1162–1166.
Hatchette, T. F., Walker, D., Johnson, C., Baker, A., Pryor, S. P. &Webster, R. G. (2004). Influenza A viruses in feral Canadian ducks:
extensive reassortment in nature. J Gen Virol 85, 2327–2337.
Hoffmann, E., Stech, J., Guan, Y., Webster, R. G. & Perez, D. R.(2001). Universal primer set for the full-length amplification of allinfluenza A viruses. Arch Virol 146, 2275–2289.
Holmes, E. C., Ghedin, E., Miller, N., Taylor, J., Bao, Y., George, K. S.,Grenfell, B. T., Salzberg, S. L., Fraser, C. M. & other authors (2005).Whole-genome analysis of human influenza A virus reveals multiple
persistent lineages and reassortment among recent H3N2 viruses.
PLoS Biol 3, e300.
Huelsenbeck, J. P. & Ronquist, F. (2001). MRBAYES: Bayesian inference
of phylogenetic trees. Bioinformatics 17, 754–755.
Ito, T., Okazaki, K., Kawaoka, Y., Takada, A., Webster, R. G. & Kida, H.(1995). Perpetuation of influenza A viruses in Alaskan waterfowl
reservoirs. Arch Virol 140, 1163–1172.
Kuiken, T., Holmes, E. C., McCauley, J., Rimmelzwaan, G. F.,Williams, C. S. & Grenfell, B. T. (2006). Host species barriers to
influenza virus infections. Science 312, 394–397.
Lawrence, J. E., Chan, A. M. & Suttle, C. A. (2002). Viruses causing
lysis of the toxic bloom-forming alga Heterosigma akashiwo(Raphidophyceae) are widespread in coastal sediments of British
Columbia, Canada. Limnol Oceanogr 47, 545–550.
Liu, J., Xiao, H., Lei, F., Zhu, Q., Qin, K., Zhang, X.-w., Zhang, X.-l.,Zhao, D., Wang, G. & other authors (2005). Highly pathogenic H5N1
influenza virus infection in migratory birds. Science 309, 1206.
Olsen, B., Munster, V. J., Wallensten, A., Waldenstrom, J., Osterhaus,A. D. M. E. & Fouchier, R. A. M. (2006). Global patterns of influenza A
virus in wild birds. Science 312, 384–388.
Phipps, L. P., Essen, S. C. & Brown, I. H. (2004). Genetic subtyping of
influenza A viruses using RT-PCR with a single set of primers based
on conserved sequences within the HA2 coding region. J VirolMethods 122, 119–122.
Runstadler, J. A., Happ, G. M., Slemons, R. D., Sheng, Z. M.,Gundlach, N., Petrula, M., Senne, D., Nolting, J., Evers, D. L. & otherauthors (2007). Using RRT-PCR analysis and virus isolation to
determine the prevalence of avian influenza virus infections in ducksat Minto Flats State Game Refuge, Alaska, during August 2005. Arch
Virol 152, 1901–1910.
Sambrook, J. & Russell, D. W. (2001). Molecular Cloning: aLaboratory Manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring
Harbor Laboratory.
Savill, N. J., St Rose, S. G., Keeling, M. J. & Woolhouse, M. E. J.(2006). Silent spread of H5N1 in vaccinated poultry. Nature 442,
757.
Sivanandan, V., Halvorson, D. A., Laudert, E., Senne, D. A. & Kumar,M. C. (1991). Isolation of H13N2 influenza A virus from turkeys and
surface water. Avian Dis 35, 974–977.
Spackman, E., Stallknecht, D. E., Slemons, R. D., Winker, K., Suarez,D. L., Scott, M. & Swayne, D. E. (2005). Phylogenetic analyses of typeA influenza genes in natural reservoir species in North America
reveals genetic variation. Virus Res 114, 89–100.
Spackman, E., McCracken, K. G., Winker, K. & Swayne, D. E. (2006).H7N3 avian influenza virus found in a South American wild duck is
A. S. Lang, A. Kelly and J. A. Runstadler
518 Journal of General Virology 89

Dow
nloa
ded
from
ww
w.m
icro
biol
ogyr
esea
rch.
org
by
IP:
23.2
0.70
.183
On:
Thu
, 08
Sep
201
6 23
:08:
32
related to the Chilean 2002 poultry outbreak, contains genes fromequine and North American wild bird lineages, and is adapted todomestic turkeys. J Virol 80, 7760–7764.
Swofford, D. (2000). PAUP*: Phylogenetic analysis using parsimony(*and other methods), version 4.0. Sunderland, MA: Sinauer Associates.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. &Higgins, D. G. (1997). The CLUSTAL_X Windows interface: flexiblestrategies for multiple sequence alignment aided by quality analysistools. Nucleic Acids Res 25, 4876–4882.
Vaisvila, R., Morgan, R. D., Posfai, J. & Raleigh, E. A. (2001).Discovery and distribution of super-integrons among pseudomonads.Mol Microbiol 42, 587–601.
Webster, R. G., Bean, W., Gorman, O., Chambers, T. & Kawaoka, Y.(1992). Evolution and ecology of influenza A viruses. Microbiol Rev56, 152–179.
Zhang, G., Shoham, D., Gilichinsky, D., Davydov, S., Castello, J. D. &Rogers, S. O. (2006). Evidence of influenza A virus RNA in Siberianlake ice. J Virol 80, 12229–12235.
Influenza viruses in environmental reservoirs
http://vir.sgmjournals.org 519