preparation of murine b7.1-glycosylphosphatidylinositol and transmembrane-anchored staphylococcal...
TRANSCRIPT

Preparation of Murine B7.1-Glycosylphosphatidylinositol and Transmembrane-Anchored Staphylococcal EnterotoxinA Dual-Anchored Tumor Cell Vaccine and its Antitumor Effect
Pingyong Yi, M.D., Ph.D.1
Hai Yu, M.D., Ph.D.1
Wenxue Ma, M.D., Ph.D.2
Qingqing Wang, M.D., Ph.D.1
Boris R. Minev, M.D.2
1 Institute of Immunology, Zhejiang University,Hangzhou, China.
2 Cancer Center, University of California San Diego,La Jolla, California.
Supported by Grant 39770837 from the NationalNatural Science Foundation of China and by Grants399131 and 301580 from the Zhejiang ProvincialNatural Science Foundation.
The authors thank Dr. Joyce Solheim (Eppley In-stitute for Cancer Research, University of NebraskaMedical Center, Omaha, NE) for her assistance inthe preparation of the current article.
Dr. Ma and Dr. Yi contributed equally to this article.
Address for reprints: Wenxue Ma, M.D., Ph.D.,Cancer Center, University of California San Diego,9500 Gilman Drive, La Jolla, CA 92093; Fax: (858)822-1327; E-mail: [email protected]
Received September 1, 2004; revision receivedNovember 30, 2004; accepted December 3, 2004.
BACKGROUND. The authors have previously reported a tumor cell vaccine modified
with superantigen staphylococcal enterotoxin A (SEA) and its antitumor effect. The
tumor cell vaccines modified with multiple immune activators frequently elicited
stronger immune responses against established tumors than single-modified vac-
cines.
METHODS. The authors explored the effectiveness of a tumor cell vaccine trans-
duced with immune activators, dual-modified using the protein transfer tech-
nique. First, a glycosylphosphatidylinositol (GPI)-anchored murine B7.1 (mB7.1-
GPI) and a transmembrane-anchored SEA (TM-SEA) were genetically generated.
Then, the murine lymphoma EL4 cells were dual modified with the incorporation
of mB7.1-GPI and TM-SEA onto the cell surface. Flow cytometry and laser confocal
microscopy showed that the incorporation of B7.1 and SEA molecules onto EL4
cells was quite stable.
RESULTS. The dual-modified tumor cell vaccine EL4/mB7.1-GPI � TM-SEA elicited
significantly stronger antitumor immune responses both in vitro and in vivo when
compared with the single-modified tumor cell vaccines EL4/mB7.1-GPI and EL4/
TM-SEA.
CONCLUSIONS. The results of the current study validated the novel approach for
preparing tumor cell vaccines modified with dual immune active molecules using
the protein transfer technique, and supported the feasibility and effectiveness of
the dual-modified tumor cell vaccine. Cancer 2005;103:1519 –28.
© 2005 American Cancer Society.
KEYWORDS: tumor vaccine, immunotherapy, murine B7.1, staphylococcal entero-toxin A, protein transfer.
The mechanisms for tumor cells to escape immune surveillanceinclude tumor antigen-specific tolerance,1 local immune dysfunc-
tion in tumor-infiltrating mononuclear cells,2 defective antigen pre-sentation processes,3,4 down-regulated surface major histocompatib-ity complex (MHC) molecules, and lack of costimulatory molecules.5,6
Introduction of immunostimulatory molecules such as MHC Class I,MHC Class II, and B7.1 to the tumor cells and use of these surface-modified tumor cells as vaccines can induce antitumor immunity, asdemonstrated by tumor rejection.7,8 These approaches frequentlyinvolve gene transfection of target cells, which is time consuming. Inaddition, the primary tumor cells often do not grow very well in vitro,and the gene expression is unstable.9 Therefore, the practical appli-cation of this strategy to induce antitumor immunity is limited.
1519
© 2005 American Cancer SocietyDOI 10.1002/cncr.20943Published online 28 February 2005 in Wiley InterScience (www.interscience.wiley.com).

Alternatively, surface molecules can be intro-duced to tumor cells through the so-called “proteintransfer” approach.10,11 One of these approaches is theglycosylphosphatidylinositol (GPI)-anchoring tech-nique. Using this technique, the DNA sequence of thetarget protein is fused with a GPI signal sequence, andthe purified, recombinant GPI-linked fusion proteinsare incubated with tumor cells. With this approach,the target protein can be anchored to the extracellularmembranes of the tumor cells. An in vitro study hasdemonstrated that GPI-B7.1 can be anchored to thetumor cells and used to stimulate lymphocyte prolif-eration.12
Bacterial superantigens (SAgs), including staphy-lococcal enterotoxin A (SEA), staphylococcal entero-toxin B, and toxic shock syndrome toxin-1, are wellknown as very potent activators of T cells that canelicit strong immune responses both in vitro and invivo. However, SAgs, as prokaryotic proteins, are notwell suited to a GPI-signal-sequence-fusion strategyfor cell membrane anchoring, because GPI linkagerequires eukaryotic processing. We had developed astrategy for passively attaching SEA to tumor cells, inwhich the SEA gene was fused with the transmem-brane (TM) region sequence of the protooncogenec-erb-B2. The purified TM-SEA protein could be effec-tively anchored to tumor cells.13 The vaccine derivedfrom these tumor cells can stimulate proliferation oflymphocytes in vitro and induce a systemic antitumorimmunity in vivo.
The escape of tumor cells from immune surveil-lance can involve several mechanisms, which indi-cates that a single immune activator may be less ef-fective than multiple activators at inducing sufficientimmune response against tumor cell growth. Severalstudies have shown that tumor cell vaccines createdby multiple-gene transfection can induce much stron-ger antitumor immunity than those made by single-gene transfection.14 –16 However, multiple-gene trans-fection is difficult technically and less practicable.
We sought to develop a new approach, other thangene transfection, to prepare surface-modified tumorvaccines in which two different immune-active mole-cules are anchored to the tumor cell membrane. In thecurrent study, the tumor cells were incubated withfusion protein GPI-linked mouse B7.1 (mB7.1-GPI)and TM-SEA. Thus, those two molecules were pas-sively attached to tumor cells by the protein transfertechnique, and the dual-modified tumor cell vaccinewas prepared. We investigated the feasibility of theapproach for preparing a dual-anchored tumor cellvaccine, and the effectiveness of the tumor cell vac-cine made by this novel approach in an animal model.
MATERIALS AND METHODSCell LinesThe EL4 (T-cell lymphoma) cell line, derived from theC57BL/6 mouse strain, YAC-1, a natural killer cell(NK)-sensitive lymphoma cell line of A/S (H-2a) origin,and the Chinese hamster ovary (CHO) cell line were allobtained from the American Type Culture Collection(ATCC; Rockville, MD). HO-8910 (human ovarian car-cinoma) cell line was provided by the Institute forCancer Research of Zhejiang Province, China. All thesecell lines were cultured in complete RPMI-1640 me-dium supplemented with 100 U/mL penicillin, 100�g/mL streptomycin, and 10% fetal calf serum (FCS).All culture media were purchased from Gibco-BRL(Gaithersburg, MD) and FCS was provided by theShanghai Institute of Biological Products (Shanghai,China).
AnimalsFemale C57BL/6 mice, 6 – 8 weeks old, were obtainedfrom Sipper-BK Experimental Animal Company(Shanghai, China) and housed in specific pathogen-free conditions at the animal center of Zhejiang Uni-versity School of Medicine in accordance with institu-tional guidelines. Mice were housed 5 per cage in a12-hour light/dark cycle at an ambient temperature of22 � 2 °C and humidity of 50 � 10% with food andwater ad libitum. Cages were changed twice weekly toensure hygienic conditions. The animals were allowedto acclimate to the facility for 2 weeks before random-ization into different experimental groups.
Tumor Model and Experimental DesignTo establish the tumor model, EL4 cells cultured for 3days were harvested and washed twice with phos-phate-buffered saline (PBS). 1 � 106 EL4 cells in 100�L of PBS were subcutaneously injected in the rightrear flank of each C57BL/6 mouse. Tumor growth wasmonitored by measuring tumors in 2 dimensions us-ing a digital caliper on every other day, beginning 7days after inoculation. Tumor size was calculated us-ing the formula, 1/2 (length � width).17
To test the effectiveness of the dual-modified EL4/mB7.1-GPI � TM-SEA vaccine, the tumor-bearingmice were randomized on the third day of posttumorcell inoculation to the following groups with eachgroup consisting of 8 animals. On the seventh dayafter tumor cell inoculation, the tumor-bearing micein the groups were injected with 100 �L PBS, EL4tumor cell vaccine, EL4/mB7.1-GPI tumor cell vac-cine, EL4/TM-SEA tumor cell vaccine, or EL4/mB7.1-GPI � TM-SEA tumor cell vaccine, each vaccine sus-pension containing 1 � 106 cells in 100 �L PBS.
1520 CANCER April 1, 2005 / Volume 103 / Number 7

The tumor sizes were measured on Day 25 aftertumor cell inoculation (before any deaths occurred) toensure inclusion of the data from all the mice.
AntibodiesAnti-mouse B7.1 (rat immunoglobulin G 2 alpha[IgG2�], Pharmingen, San Diego, CA), rabbit anti-SEA(Toxin Technology, Sarasota, FL), goat anti-rabbitIgG/TRITC, goat anti-rat IgG/horseradish peroxidase(HRP), and goat anti-rat IgG/fluorescein isothiocya-nate (KpL, Gaithersburg, MD) were used.
Construction, Expression, and Purification of Murine B7.1Glycosylphosphatidylinositol and Transmembrane-Anchored Staphylococcal Enterotoxin AThe DNA sequence encoding the first 247 amino acidsof mB7.118 was amplified by polymerase chain reac-tion (PCR) from pLNSX-mB7.1, a gift provided by Dr.L. Chen (Bristol-Myers Squibb Pharmaceutical Re-search Institute, Seattle, WA).5 5�-GGAATTCGCTATG-GCTTGCAATTGTCAG-3� with an EcoR I restrictionsite (underlined) and 5�-CGCAGGCGGTGTATGTGT-TCTTGCTATCAGG-3� were the sense and antisenseprimers, respectively, for amplifying the mB7.1 se-quence. The DNA sequence encoding the signal forGPI anchor attachment from human placental alka-line phosphatase (hPLAP)-119 was amplified usingDNA extracted from human placental tissue. 5�-GAT-AGCAAGAACACATACACCGCCTGCGACCT-3� and 5�-ATCTCGAGTCAGGGAGCAGTGGCCGTCT-3� with anXho I restriction site (underlined) were the sense andantisense primers, respectively, for amplifying GPI.The 2 amplified gene sequences were annealed toform a chimeric GPI-anchored mB7.1 molecule by theSOE method.20 The resulting chimera was cloned inthe pGEM-T vector (Promega, Madison, WI), sub-cloned in the neomycin-resistant plasmid pcDNA3.1(�) (Invitrogen, Carlsbad, CA), and confirmed by se-quencing analysis. The recombinant plasmid wastransfected into CHO cells by using Lipofectamine2000 reagent (Invitrogen), and transfectants were se-lected with G418 (Gibco/BRL) at a concentration of600 �g/mL. Expression of the mB7.1-GPI fusion pro-tein on the surface of CHO cells was confirmed by flowcytometry, phosphatidylinositol-specific phospho-lipase C treatment, and confocal microscopy. ThemB7.1-GPI fusion protein was purified by anti-mB7.1-monoclonal antibody-CNBr-Sepharose 4B (Pharma-cia, Piscataway, NJ) affinity chromatography, then an-alyzed by sodium dodecyl sulfate-polyacrylamide gelelectrophoresis (SDS-PAGE) and Western blotting.
The SEA gene21 was cloned from Staphylococcusaureus (ATCC 13565) strain. The primers 5�-GCC-GCTAGCATGAAAAAAACAGCATTTAC-3� with a Nhe I
restriction site (underlined) and 5�-GCTCTCTGCTCG-GCACTTGTATATAAA-3� were the sense and antisenseprimers for amplifying the SEA gene 13. The sequenceencoding the TM region of the c-erb-B2 gene22 wasamplified by reverse transcription-PCR from the humanovarian carcinoma cell line HO-8910.13 5�-TTTATATA-CAAGTGCCGAGCAGAGAGC-3� and 5�-AAGCTTCTT-ACATCGTGTACTTCCG-3� with a Hind III restriction site(underlined) were the sense and antisense primers, re-spectively, for amplifying the TM sequence. The twoamplified gene sequences were annealed to form a chi-meric TM-SEA molecule by the SOE method. The result-ing chimera was cloned in pGEM-T vector (Promega)and then subcloned into the pET-28a vector (EMD Bio-sciences, San Diego, CA). The recombinant pET-28a-TM-SEA13 was transfected into the host strainBL21(DE3)pLysS for expression of the TM-SEA fusionprotein. Expression of the target protein was induced byincubation at 30 °C and addition of IPTG to a finalconcentration of 1 mM. Cells were harvested after 5hours of induction, the cell pellet was resuspended in 50mL sonication buffer (300 mM NaCl/50 mM sodiumphosphate, pH 8/1%Triton X-100), and the suspensionwas frozen at �80 °C. The cell pellets were thawed andsonicated 3 times for 15 seconds on ice, centrifuged at17,800 �g for 30 minutes at 4 °C, and the supernatantwas collected for target protein purification by using theNi-NTA His.Bind resin purification system (EMD Bio-sciences).
Sodium Dodecyl Sulfate-Polyacrylamide GelElectrophoresis and Western Blot AnalysisWhole cell extracts were obtained by resuspending thePBS-washed cellular pellets in 1% SDS. The lysateswere boiled for 5 minutes, and protein amounts werequantified with protein assay reagent (Bio-Rad, Her-cules, CA) using a bovine serum albumin standardsolution as reference. Protein extracts were separatedon 10% SDS-PAGE gels and visualized by silver stain-ing, which was performed according to the manufac-turer’s instructions (Pharmacia Biotech AB). Then,they were transferred to an Immobilon P membrane(polyvinylidene difluoride; Millipore, Bedford, MA).The membranes were blocked in TBST containing 50mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.05%Tween 20 plus 5% nonfat dry milk at room tempera-ture for 30 minutes, incubated with the primary anti-bodies in a cool room (4 °C) overnight after washingthe membrane, and incubated with the second HRP-conjugated antibodies (dilution, from 1:2000 to 5000)at room temperature for 30 minutes after washing.Proteins were visualized by the ECL detection reagents(ECL kit; Amersham Pharmacia Biotech, Piscataway,NJ) after washing five times in TBST.
Dual-Modified Tumor Vaccine and Immunotherapy/Pingyong et al. 1521

The following antibodies were used for Westernblot analysis: primary antibodies of anti-mouse B7.1(rat IgG2�, Pharmingen) and rabbit anti-SEA (ToxinTechnology). Second antibodies of goat anti-mouseIgG/HRP and goat anti-rat IgG/HRP were obtainedfrom KpL.
Incorporation of Murine B7.1-Glycosylphosphatidylinositoland/or Transmembrane-Anchored StaphylococcalEnterotoxin A into Cell MembranesEL4 cells were washed 3 times with PBS/5 mM ethyl-enediaminetetraacetic acid, resuspended (5 � 106
cells/mL), and incubated with 10 �g/mL purifiedmB7.1-GPI and/or 20 �g/mL purified TM-SEA proteinfor 4 hours at 37 °C with gently shaking. The incubatedcells were washed three times, and analyzed by flowcytometric assay and confocal microscopy. EL4 cells,EL4 cells anchored with mB7.1-GPI protein, EL4 cellsanchored with TM-SEA protein, and EL4 cells an-chored with both TM-SEA and mB7.1-GPI proteinwere irradiated (50 gray) for use as tumor cell vac-cines.
Flow Cytometric AssayTumor cells (1 � 106) were washed once with 2% fetalbovine serum FBS in PBS and resuspended in 5 �Lwash buffer containing the primary antibody, incu-bated at 4 °C for 45 minutes, then washed twice withwash buffer, resuspended in wash buffer containing afluorescein-labeled secondary antibody, and incu-bated at 4 °C for 45 minutes. After 2 washes with washbuffer, the cells were resuspended in 500 �L PBS forflow cytometric analysis in a fluorescein-activated cellsorter (Beckman Coulter, Hialeah, FL).
Confocal Microscopic AnalysisCells were stained at 4 °C and fixed using 2% parafor-maldehyde in PBS for 30 minutes, then washed twicein PBS buffer. Five microliters of the cell pellet wasmixed with an equal volume of SlowFade (MolecularProbes, Eugene, OR) and placed on a glass slide. Then,the coverslip was sealed with nail polish. Fluorescencedistribution was analyzed using a laser confocal scan-ning microscope.
Measurement of Lymphocyte ProliferationThe proliferation assay used, based on the conversionof the tetrazolium salt 3, [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) to purpleformazan crystals by metabolically active cells,23 pro-vides a quantitative determination of viable cells.Splenocytes were derived from C57BL/6 mice andcocultured with irradiated tumor cells at a ratio of 5:1
for 24 hours. Cells were seeded in a 96-well plate,incubated at 37 °C in 5% CO2 for 4 hours, MTT (5mg/mL) was added to each well of cells, and the platewas incubated for 4 hours at 37 °C. The medium wasremoved, and the MTT crystals were solubilized in 0.1mL dimethylsulfoxide and subjected to centrifugationto pellet the cellular debris. Spectrophotometric ab-sorbance of each sample was measured at 570 nmusing a Spectra Microplate Reader (Bio-Tek modelEXL 800, Winooski, VT).
Cytokine Release AssayThe nonadherent splenocytes derived from C57BL/6mice, at the concentration of 1 � 107 cells/mL, werestimulated with 5 � 104 tumor vaccine cells. The su-pernatant was collected for interleukin-2 (IL-2) assayafter 24 hours, and for the interferon-gamma (IFN-�)assay after 48 hours. The concentrations of IL-2 andIFN-� were determined using a sandwich enzyme-linked immunosorbent assay kit (Eudogen, Woburn,MA).
Immunotherapy with the Inactivated Tumor Cell VaccinesC57BL/6 mice were inoculated subcutaneously with 1� 106 EL4 cells in the right rear flank to establish themurine lymphoma model. Three days after inocula-tion, the tumor-bearing C57BL/6 mice were dividedinto 5 groups (each group containing 8 mice), andwere injected in the left rear flank using 1 of thefollowing preparations: PBS, EL4 vaccine, EL4/mB7.1-GPI vaccine, EL4/TM-SEA vaccine, or EL4/mB7.1-GPI� TM-SEA vaccine. The mice were given a total of 3injections, at 2-day intervals of tumor cell vaccine (1� 106 cells) suspended in 100 �L PBS. Tumor sizeswere expressed as the mean diameter of the longestand the shortest diameter measured with a digitalcaliper every other day. Seven days after the last vac-cine dose, three mice in each group were killed andsplenic lymphocytes were isolated for NK and CTLactivity assays. The other 5 mice in each group weremonitored for survival (for � 90 days).
Cytotoxic Assays of CTL and Natural Killer Cell ActivitySplenocytes were isolated from the mice that had beenkilled. The erythrocytes were depleted with 0.83% am-monium chloride and macrophages were removed byadherence of splenocytes on plastic plates for 2 hours.The nonadherent lymphocytes were directly used asNK effector cells. The lymphocytes were coculturedwith inactivated EL4 for 7 days in the presence ofrecombinant murine IL-2 (20 U/mL) and then col-lected as CTL effector cells. B16 cells were used as thecontrol target cells, and the NK and CTL activity wasdetermined by using the CytoTox 96 nonradioactive
1522 CANCER April 1, 2005 / Volume 103 / Number 7

cytotoxicity assay kit according to instructions fromthe manufacturer (Promega). The percentage of spe-cific lysis was determined according to the followingformula: 100 � (experimental release � spontaneousrelease)/(maximum release � spontaneous release).
StatisticsAll experiments were run in triplicate and the resultsare means � the standard deviation of triplicate de-terminations or representative data from one or twoindependent experiments. Statistical analyses wereperformed using the Student’s t test and log-rank test(for survival analysis). Differences were statisticallysignificant at P � 0.05.
RESULTSLymphocyte Proliferation and Cytokine ProductionThe authors of the current study first tested whetherEL4 cells coated with mB7.1-GPI and/or TM-SEA pro-tein could stimulate lymphocyte proliferation and re-lease of cytokines in vitro. The results showed thatEL4/mB7.1-GPI or EL4/TM-SEA cells were superior atstimulating lymphocyte proliferation, relative to EL4(Table 1). Furthermore, the production of IL-2 andIFN-� cytokines in response toEL4/mB7.1-GPI or EL4/TM-SEA was much higher than in response to EL4(Table 1, P � 0.05). The EL4/TM-SEA � mB7.1-GPIcells exhibited a greater ability than the EL4/mB7.1-GPI and EL4/TM-SEA vaccines to stimulate lympho-cyte proliferation and induce production of IL-2 andIFN-� cytokines in vitro (P � 0.05, Table 1).
Sodium Dodecyl Sulfate-Polyacrylamide GelElectrophoresis and Western Blot Analysis of PurifiedProteinFigure 1A shows the results from SDS-PAGE of thepurified recombinant mB7.1-GPI fusion protein, the
cell lysate of CHO cells transfected with pcDNA3.1(�)-mB7.1-GPI, the controls of CHO cell lysates, andthe CHO cells transfected with the pcDNA 3.1 (�)empty vector. Figure 1B shows the Western blot re-sults from the purified mB7.1-GPI protein, with theexpected size of 62 kilodalton (kD). Figure 1C showsthe Western blot results for TM-SEA fusion proteinexpressed under the induction of IPTG at differenttime points. Figure 1D shows the Western blot resultsfor the purified TM-SEA protein, with the expectedsize of 32.6 kD.
Incorporation of Purified Murine B7.1-Glycosylphosphatidylinositol and/or Transmembrane-Anchored Staphylococcal Enterotoxin A Protein into theEL4 Cell MembraneThe mouse lymphoma cell line EL4 was incubatedwith purified mB7.1-GPI and/or TM-SEA protein. Flowcytometric analysis showed that mB7.1-GPI and/orTM-SEA protein was expressed on the surface ofcoated EL4 cells. After the incubation of EL4 cells withmB7.1-GPI protein, 96.5% of the cells expressed mB7.1with a mean fluorescence intensity of 13.2. Similarly,after incubation of EL4 cells with TM-SEA protein, thepositive rate of SEA detection was 95.4%, and themean fluorescence intensity was 13.3. For EL4 cellsincubated with TM-SEA and mB7.1-GPI protein, theSEA and mB7.1 were both detected on 94% of the cells.Eight hours after incubation with mB7.1-GPI and/orTM-SEA protein, flow cytometry showed that the pro-teins were maintained on approximately 85% of theEL4 cells (data not shown). To determine the distribu-tion of mB7.1-GPI and/or TM-SEA protein on thecoated cells and the cell integrity after incubation, thecells images were visualized by laser confocal micros-copy (Fig. 2).
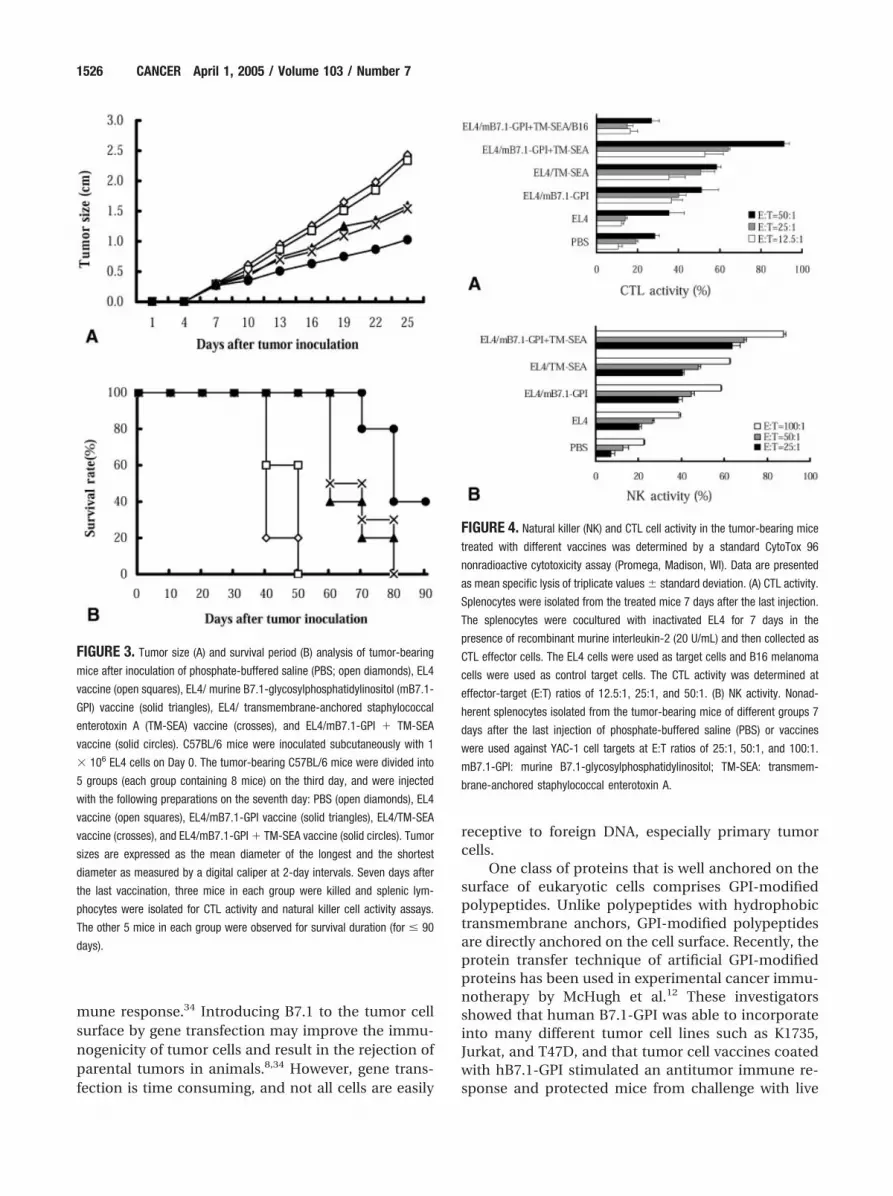
Enhanced Antitumor Effects of the Dual-Anchored EL4/Transmembrane-Anchored Staphylococcal Enterotoxin A� Murine B7.1- Glycosylphosphatidylinositol VaccineTumor growth in mice treated with the EL4/mB7.1-GPI, EL4/TM-SEA, or dual-anchored EL4/TM-SEA� mB7.1-GPI vaccines was markedly inhibited relativeto those treated with PBS or EL4 vaccine controls (Fig.3A). The antitumor effect of the dual-anchored EL4/TM-SEA � mB7.1-GPI vaccine was significantly stron-ger than that of the EL4/mB7.1-GPI or EL4/TM-SEAvaccine (P � 0.05). There were no significant differ-ences in tumor growth inhibition between the groupsreceiving the EL4/mB7.1-GPI and EL4/TM-SEA vac-cines.
Five tumor-bearing mice in each group weremonitored for their survival period. The results inFigure 3B show that lymphoma-bearing C57BL/6 mice
TABLE 1Stimulating Lymphocyte Proliferation and Cytokine ProductionIn Vitroa
Groups IL-2 IFN-� PI
A EL4 22.9 � 1.4 41 � 3.2 1.98 � 0.7B EL4/mB7.1-GPI 508.8 � 22.1b 805 � 13.5b 3.43 � 0.6b
C EL4/TM-SEA 771.3 � 11.4c 1225 � 10.9c 4.87 � 0.3c
D EL4/mB7.1-GPI � TM-SEAd 1563.6 � 49.4d 2450 � 188d 7.17 � 0.6d
IL-2: interleukin-2; IFN-�: interferon-gamma; PI: proliferation index.a IL-2 and IFN-�: mean � standard deviation pg/mL; PI: mean � standard deviation.b P � 0.05, compared with Group A.c P � 0.05, compared with Groups A and B.d P � 0.05, compared with Groups A, B, and C. Statistical analyses were performed using the Student’s
t test.
Dual-Modified Tumor Vaccine and Immunotherapy/Pingyong et al. 1523

treated with dual-anchored EL4/TM-SEA � mB7.1-GPI vaccine survived longer time than those treatedwith the EL4/mB7.1-GPI vaccine (P � 0.05), the EL4/TM-SEA vaccine (P � 0.05), and the PBS or EL4 vac-cine controls (P � 0.01), but there was no significantdifference in the survival period between the micetreated with the EL4/mB7.1-GPI and EL4/TM-SEAvaccines (P � 0.05).
Antitumor Activity Induced by Murine B7.1-Glycosylphosphatidylinositol and Transmembrane-Anchored Staphylococcal Enterotoxin A Tumor CellVaccine is Mediated by CTLs and Natural Killer CellsSplenocytes were isolated from the vaccinated mice(in triplicate) 7 days after the last therapy, coculturedwith inactivated EL4 cells (treated with mitomycin C,100 �g/mL at 37 °C for 1 hour) for 7 days in the
FIGURE 1. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis of murine B7.1-glycosylphosphatidylinositol
(mB7.1-GPI) and transmembrane-anchored staphylococcal enterotoxin A (TM-SEA) fusion proteins. (A) SDS-PAGE and silver staining of mB7.1-GPI protein
expression. (Lane 1) Purified mB7.1-GPI protein, (lane 2) cell lysate from normal Chinese hamster ovary (CHO) cells, (lane 3) cell lysate from CHO cells transfected
with pcDNA3.1 (�)-mB7.1-GPI, and (lane 4) cell lysate from CHO cells transfected with the empty vector pcDNA3.1 (�). The gel was stained by using the silver
staining system. (B) Western blot analysis of purified mB7.1-GPI protein. (Lane 1) Purified mB7.1-GPI protein and (lane 2) cell lysate from CHO cells transfected
with the empty vector pcDNA3.1 (�). Anti-mouse B7.1 (rat immunoglobulin G [IgG]2�]) and anti-rat IgG/horseradish peroxidase were used for developing the signal.
(C) Western blot analysis of the TM-SEA protein from the BL21(DE3)pLysS-pET-28a-TM-SEA strain. The polyvinylidene fluoride membrane was immunoblotted with
anti-SEA raised against the following samples: 30 ng of purified SEA as positive control (lane 1), lysate from the BL21(DE3)pLysS-pET-28a vector (lane 2), lysate
from BL21(DE3)pLysS-pET-28a-TM-SEA without inducement of IPTG (lane 3), and lysate from BL21(DE3)pLysS-pET-28a-TM-SEA which was induced by IPTG with
the final concentration of 1.0 mM for 1 hours, 3 hours, 5 hours, and 7 hours, respectively (lanes 4–7). (D) Western blot analysis of the purified TM-SEA fusion protein.
The samples, in the following order, were resolved on a 12% polyacrylamide gel: purified SEA (30 ng) as positive control (lane 1), lysate supernatant of
BL21(DE3)pLysS-pET-28a-TM-SEA containing TM-SEA protein (lane 2), washed buffer from Ni-NTA resin containing unbound protein (lane 3), eluted buffer from
Ni-NTA resin containing purified TM-SEA protein (lane 4), and probed with an antiserum against SEA. kD: kilodalton.
1524 CANCER April 1, 2005 / Volume 103 / Number 7

presence of recombinant mouse IL-2 (20 U/mL,Sigma, St. Louis, MO), collected as CTL effector cells,and tested against wild-type EL4 cells as target cells,whereas B16 cells were used as control target cells.CTL activity was determined at effector:target (E:T)ratios of 12.5:1, 25:1, and 50:1 by a standard CytoTox96 nonradioactive cytotoxicity assay. Splenocytes de-rived from 3 dead mice were also used in an NK cellactivity assay (with YAC-1 cells used as the target cells)at the E:T ratios of 25:1, 50:1, and 100:1. As shown inFigure 4, lymphocytes derived from the mice treatedwith the EL4/TM-SEA � mB7.1-GPI vaccine showedthe highest CTL and NK activity compared with lym-phocytes derived from all other groups. The CTL andNK activity of the mice in the EL4/mB7.1-GPI andEL4/TM-SEA vaccine groups was also much higherthan that in the EL4 vaccine group (P � 0.05).
DISCUSSIONSAgs derived from bacterial or viral products areknown as potent activators of T lymphocytes and ef-ficient inducers of cytokine production. This propertyof SAgs has been used in cancer immunotherapy,24,25
including applications in which the SAgs were ligatedto tumor-specific monoclonal, antiidiotypic, or bi-functional antibodies for tumor-targeted T-cell activa-tion.26,27 SAgs have also been genetically transferred totumor cells, and SAgs-expressing tumor cells havebeen administered as a tumor vaccine in vivo.28,29 Inaddition, genetically engineered TM-SEA could an-chor on the surface of tumor cells and was capable of
eliciting systemic antitumor immunity without anymeasurable toxicity.17 In the current study, we showedthat the tumor vaccine EL4/TM-SEA stimulated lym-phocyte proliferation and caused the considerable re-lease of IL-2 and IFN-�. The tumor growth was signif-icantly inhibited in tumor-bearing mice treated withthe EL4/TM-SEA vaccine, and the survival period wasalso much longer than the controls. However, theantitumor effect of SAg alone is less satisfactory, es-pecially in preestablished tumors.29 A phase of im-mune exhaustion, including suboptimal production ofIFN-� and IL-2 and failure to mediate CTL activity, canbe induced by repeated administration of SAg, whichmay be due to the anergy and deletion of the respond-ing T cells.30 Our previous study showed that the an-titumor effect by fusion protein C215Fab-SEA was in-sufficient to cure tumor-bearing animals, becausecomplete tumor remission occurred in only 20% of thetreated mice.31
Other studies have shown that SAg, in combina-tion with B7 costimulation, induced a strong lympho-cyte proliferation response, accompanied by the re-lease of high concentrations of IL-2 and IFN-�.32,33
According to the “two signals” theory, T-cell activationrequires at least two distinct signals. The first is frompeptide-binding MHC molecules on antigen-present-ing cells and the second is from the interaction ofCD28 and B7.18 Absence of the second signal mayresult in T-cell clonal anergy. It was demonstrated thattumors lacking B7.1 were poorly immunogenic, andtherefore they failed to initiate an appropriate im-
FIGURE 2. Laser confocal microscopic
analysis of the EL4 cells coated with
murine B7.1-glycosylphosphatidylinositol
(mB7.1-GPI) and/or transmembrane-an-
chored staphylococcal enterotoxin A (TM-
SEA) fusion protein. (A) The mB7.1 mole-
cule was expressed on the EL4 cells
coated with the mB7.1-GPI protein (fluo-
rescein isothiocyanate [FITC] labeled). (B)
SEA antigen was expressed on the EL4
cells coated with TM-SEA protein (TRITC
labeled). (C) SEA and mB7.1 antigens were
expressed on the EL4 cells coated with
both TM-SEA and mB7.1-GPI fusion pro-
teins. (C1) mB7.1-FITC is presented in
green. (C2) SEA-TRITC is presented in red.
(C3) The merged image is presented in
yellow. Rat anti-mouse B7.1, rabbit anti-
SEA, goat anti-rabbit IgG/TRITC, and goat
anti-rat IgG/FITC were used.
Dual-Modified Tumor Vaccine and Immunotherapy/Pingyong et al. 1525
mune response.34 Introducing B7.1 to the tumor cellsurface by gene transfection may improve the immu-nogenicity of tumor cells and result in the rejection ofparental tumors in animals.8,34 However, gene trans-fection is time consuming, and not all cells are easily
receptive to foreign DNA, especially primary tumorcells.
One class of proteins that is well anchored on thesurface of eukaryotic cells comprises GPI-modifiedpolypeptides. Unlike polypeptides with hydrophobictransmembrane anchors, GPI-modified polypeptidesare directly anchored on the cell surface. Recently, theprotein transfer technique of artificial GPI-modifiedproteins has been used in experimental cancer immu-notherapy by McHugh et al.12 These investigatorsshowed that human B7.1-GPI was able to incorporateinto many different tumor cell lines such as K1735,Jurkat, and T47D, and that tumor cell vaccines coatedwith hB7.1-GPI stimulated an antitumor immune re-sponse and protected mice from challenge with live
FIGURE 3. Tumor size (A) and survival period (B) analysis of tumor-bearing
mice after inoculation of phosphate-buffered saline (PBS; open diamonds), EL4
vaccine (open squares), EL4/ murine B7.1-glycosylphosphatidylinositol (mB7.1-
GPI) vaccine (solid triangles), EL4/ transmembrane-anchored staphylococcal
enterotoxin A (TM-SEA) vaccine (crosses), and EL4/mB7.1-GPI � TM-SEA
vaccine (solid circles). C57BL/6 mice were inoculated subcutaneously with 1
� 106 EL4 cells on Day 0. The tumor-bearing C57BL/6 mice were divided into
5 groups (each group containing 8 mice) on the third day, and were injected
with the following preparations on the seventh day: PBS (open diamonds), EL4
vaccine (open squares), EL4/mB7.1-GPI vaccine (solid triangles), EL4/TM-SEA
vaccine (crosses), and EL4/mB7.1-GPI � TM-SEA vaccine (solid circles). Tumor
sizes are expressed as the mean diameter of the longest and the shortest
diameter as measured by a digital caliper at 2-day intervals. Seven days after
the last vaccination, three mice in each group were killed and splenic lym-
phocytes were isolated for CTL activity and natural killer cell activity assays.
The other 5 mice in each group were observed for survival duration (for � 90
days).
FIGURE 4. Natural killer (NK) and CTL cell activity in the tumor-bearing mice
treated with different vaccines was determined by a standard CytoTox 96
nonradioactive cytotoxicity assay (Promega, Madison, WI). Data are presented
as mean specific lysis of triplicate values � standard deviation. (A) CTL activity.
Splenocytes were isolated from the treated mice 7 days after the last injection.
The splenocytes were cocultured with inactivated EL4 for 7 days in the
presence of recombinant murine interleukin-2 (20 U/mL) and then collected as
CTL effector cells. The EL4 cells were used as target cells and B16 melanoma
cells were used as control target cells. The CTL activity was determined at
effector-target (E:T) ratios of 12.5:1, 25:1, and 50:1. (B) NK activity. Nonad-
herent splenocytes isolated from the tumor-bearing mice of different groups 7
days after the last injection of phosphate-buffered saline (PBS) or vaccines
were used against YAC-1 cell targets at E:T ratios of 25:1, 50:1, and 100:1.
mB7.1-GPI: murine B7.1-glycosylphosphatidylinositol; TM-SEA: transmem-
brane-anchored staphylococcal enterotoxin A.
1526 CANCER April 1, 2005 / Volume 103 / Number 7

wild-type tumor cells. Consistent with their findings,our study showed that the modified tumor cell vaccineEL4/mB7.1-GPI had exhibited T-cell stimulation andspecific antitumor effects in vivo.
To enhance the immunogenicity of tumor cellvaccines, we modified the whole tumor cell vaccinewith two distinct immunoactive molecules—SAgs SEAand costimulator B7.1. With the protein transfer tech-nique, the genetically engineered mB7.1-GPI and TM-SEA fusion proteins were stably anchored on the sur-face of EL4 tumor cells simultaneously. The tumorvaccine prepared using EL4 cells coated with bothTM-SEA and mB7.1-GPI fusion protein elicited a sig-nificantly stronger antitumor immune response thanthe EL4/mB7.1-GPI vaccine or EL4/TM-SEA vaccinealone, indicating that mB7.1 and SEA may be able tostimulate antitumor immune responses synergisti-cally.
Protein transfer can be applied to both solubleand membrane-associating proteins and has a num-ber of potential advantages compared with genetransfer. First, protein transfer is not dependent oncellular proliferative potential or transfectability. Sec-ond, transfection procedures are rather cumbersome,and hence coordinately expressing multiple genes inthe same cell is still, in most instances, quite challeng-ing. In contrast, protein transfer permits the simulta-neous delivery of an essentially limitless number ofproteins to cells. Third, protein transfer, unlike genetransfer, is a rapid procedure and is thus particularlywell suited for therapeutic applications.
In summary, the current study investigated a newapproach to prepare a dual immune active molecule-anchored tumor cell vaccine with the protein transfertechnique, and the results provided experimental ev-idence that supports the feasibility and effectivenessof this novel approach in cancer immunotherapy.
REFERENCES1. Qin Z, Richter G, Schuler T, et al. B cells inhibit induction of
T cell-dependent tumor immunity. Nat Med. 1998;4:627–630.
2. Miescher S, Whiteside TL, Moretta L, et al. Clonal and fre-quency analyses of tumor-infiltrating T lymphocytes fromhuman solid tumors. J Immunol. 1987;138:4004 – 4011.
3. Restifo NP, Esquivel F, Kawakami Y, et al. Identification ofhuman cancers deficient in antigen processing. J Exp Med.1993;177:265–272.
4. Seliger B, Maeurer MJ, Ferrone S. TAP off—tumors on. Im-munol Today. 1997;18:292–299.
5. Chen L, Ashe S, Brady WA, et al. Costimulation of antitumorimmunity by the B7 counterreceptor for the T lymphocytemolecules CD28 and CTLA-4. Cell. 1992;71:1093–1102.
6. Townsend SE, Allison JP. Tumor rejection after direct co-stimulation of CD8� T cells by B7-transfected melanomacells. Science. 1993;259:368 –370.
7. Ostrand-Rosenberg S, Clements VK, Thakur A, et al. Trans-
fection of major histocompatibility complex class I and classII genes causes tumour rejection. J Immunogenet. 1989;16:343–349.
8. Hodge JW, Abrams S, Schlom J, et al. Induction of antitumorimmunity by recombinant vaccinia viruses expressing B7-1or B7-2 costimulatory molecules. Cancer Res. 1994;54:5552–5555.
9. Tykocinski ML, Kaplan DR, Medof ME. Antigen-presentingcell engineering. The molecular toolbox. Am J Pathol. 1996;148:1–16.
10. Medof ME, Nagarajan S, Tykocinski ML. Cell-surface engi-neering with GPI-anchored proteins. FASEB J. 1996;10:574 –586.
11. Medof ME, Kinoshita T, Nussenzweig V. Inhibition of com-plement activation on the surface of cells after incorpora-tion of decay-accelerating factor (DAF) into their mem-branes. J Exp Med. 1984;160:1558 –1578.
12. McHugh RS, Ahmed SN, Wang YC, et al. Construction, pu-rification, and functional incorporation on tumor cells ofglycolipid-anchored human B7-1 (CD80). Proc Natl Acad SciUSA. 1995;92:8059 – 8063.
13. Ma W, Yu H, Wang Q, et al. In vitro biological activities oftransmembrane superantigen staphylococcal enterotoxin Afusion protein. Cancer Immunol Immunother. 2004;53:118 –124.
14. Pulaski BA, Terman DS, Khan S, et al. Cooperativity ofStaphylococcal aureus enterotoxin B superantigen, majorhistocompatibility complex class II, and CD80 for immuno-therapy of advanced spontaneous metastases in a clinicallyrelevant postoperative mouse breast cancer model. CancerRes. 2000;60:2710 –2715.
15. Coughlin CM, Wysocka M, Kurzawa HL, et al. B7-1 andinterleukin 12 synergistically induce effective antitumor im-munity. Cancer Res. 1995;55:4980 – 4987.
16. Mazzocchi A, Melani C, Rivoltini L, et al. Simultaneoustransduction of B7-1 and IL-2 genes into human melanomacells to be used as vaccine: enhancement of stimulatoryactivity for autologous and allogeneic lymphocytes. CancerImmunol Immunother. 2001;50:199 –211.
17. Ma W, Yu H, Wang Q, et al. A novel approach for cancerimmunotherapy: tumor cells with anchored superantigenSEA generate effective antitumor immunity. J Clin Immunol.2004;24:294 –301.
18. Freeman GJ, Gray GS, Gimmi CD, et al. Structure, expres-sion, and T cell costimulatory activity of the murine homo-logue of the human B lymphocyte activation antigen B7. JExp Med. 1991;174:625– 631.
19. Millan JL. Molecular cloning and sequence analysis of hu-man placental alkaline phosphatase. J Biol Chem. 1986;261:3112–3115.
20. Horton RM, Hunt HD, Ho SN, et al. Engineering hybridgenes without the use of restriction enzymes: gene splicingby overlap extension. Gene. 1989;77:61– 68.
21. Betley MJ, Mekalanos JJ. Nucleotide sequence of the type Astaphylococcal enterotoxin gene. J Bacteriol. 1988;170:34 –41.
22. Coussens L, Yang-Feng TL, Liao YC, et al. Tyrosine kinasereceptor with extensive homology to EGF receptor shareschromosomal location with neu oncogene. Science. 1985;230:1132–1139.
23. Vistica DT, Skehan P, Scudiero D, et al. Tetrazolium-basedassays for cellular viability: a critical examination of selectedparameters affecting formazan production. Cancer Res.1991;51:2515–2520.
Dual-Modified Tumor Vaccine and Immunotherapy/Pingyong et al. 1527

24. Papageorgiou AC, Acharya KR. Superantigens as immuno-modulators: recent structural insights. Structure. 1997;5:991–996.
25. Kotb M. Superantigens of gram-positive bacteria: structure-function analyses and their implications for biological ac-tivity. Curr Opin Microbiol. 1998;1:56 – 65.
26. Dohlsten M, Abrahmsen L, Bjork P, et al. Monoclonal anti-body-superantigen fusion proteins: tumor-specific agentsfor T-cell-based tumor therapy. Proc Natl Acad Sci USA.1994;91:8945– 8949.
27. Sakurai N, Kudo T, Suzuki M, et al. SEA-scFv as a bifunc-tional antibody: construction of a bacterial expression sys-tem and its functional analysis. Biochem Biophys Res Com-mun. 1999;256:223–230.
28. Shrayer DP, Kouttab N, Hearing VJ, et al. Immunization ofmice with melanoma cells transfected to secrete the supe-rantigen, staphylococcal enterotoxin A. Cancer ImmunolImmunother. 1998;46:7–13.
29. Rosendahl A, Kristensson K, Carlsson M, et al. Long-termsurvival and complete cures of B16 melanoma-carrying an-
imals after therapy with tumor-targeted IL-2 and SEA. Int JCancer. 1999;81:156 –163.
30. Renno T, Hahne M, Tschopp J, et al. Peripheral T cellsundergoing superantigen-induced apoptosis in vivo expressB220 and upregulate Fas and Fas ligand. J Exp Med. 1996;183:431– 437.
31. Wang Q, Yu H, Zhang L, et al. Adenovirus-mediated intra-tumoral lymphotactin gene transfer potentiates the anti-body-targeted superantigen therapy of cancer. J Mol Med.2002;80:585–594.
32. Boise LH, Minn AJ, Noel PJ, et al. CD28 costimulation canpromote T cell survival by enhancing the expression ofBcl-XL. Immunity. 1995;3:87–98.
33. Tatsumi T, Takehara T, Katayama K, et al. Expression ofcostimulatory molecules B7-1 (CD80) and B7-2 (CD86) onhuman hepatocellular carcinoma. Hepatology. 1997;25:1108 –1114.
34. Chen L, McGowan P, Ashe S, et al. Tumor immunogenicitydetermines the effect of B7 costimulation on T cell-medi-ated tumor immunity. J Exp Med. 1994;179:523–532.
1528 CANCER April 1, 2005 / Volume 103 / Number 7