plantilla tesis a4 - tdx (tesis doctorals en xarxa)
TRANSCRIPT

Methods to model and assess protein-DNA and protein-
protein interactions in the context of gene regulation
Alberto Meseguer Donlo
TESI DOCTORAL UPF / 2021
Thesis supervisor
Dr. Baldomero Oliva Miguel
Structural Bioinformatics Lab (SBI)
Research Program on Biomedical Informatics (GRIB)
Department of Experimental and Health Sciences (CEXS)

ii

iii
A Mama, a Papa y a Javier


v
In appreciation
This work was supported by the grant associated to the project “Cómo se forma el oído interno: genes, neuronas y forma 3D (MINECO - BFU2014-53203-P-)” and the grant FI-AGAUR (convocatòria d’ajuts destinats a universitats, centres de recerca i fundacions hospitalàries per contractar personal investigador novell per a l’any 2018). I would like to start by thanking my thesis supervisor, Prof. Baldo Oliva, for being a great supervisor and an example to follow, both inside and outside the lab. It is fair to say that Baldo is at least as responsible as I am for the work here presented. I also would like to thank the other people from the SBI lab: to Quim for being my desktop buddy for the whole PhD, to Oriol for starting this project and for helping me during all the PhD, and to Emre for his wise counsels in moments of need. Other people from the SBI lab I would like to thank are Patri, Narcís, Rubén, Filip, Altair, Laura, Cristiano and Jaume. Finally, I would like to thank also to Rubén Vicente for guiding my first steps in the world of science with a lot of generosity. I feel very grateful for having had the chance of teaching at the human biology and bioinformatics degrees. Again, this would not have been possible without Baldo giving me the opportunity to start teaching and being himself a reference to follow as teacher. At the end, I have learnt that I love teaching by teaching the same lessons that Baldo thought me years ago. I have lots of friends to thank at this point, and I am very happy to say that you are too many people. I think that there is not enough space to talk about all of you. Anyway, you are very important for me and I will make sure that you know that (if you don’t know it yet) in future beers, parties, gatherings, travels, summer camps, poetry slams, lindy hop jams, mountain routes, boat trips, farming volunteer experiences, concerts, yoga lessons, ping pong matches, hangovers, debate contests, dinners with peanut butter deserts, did I say beers?, dance lessons, lord of the rings marathons, music fests, jam sessions, disguise parties, mornings at IKEA, new year’s eve dance routines, werewolf games, and a long line of etceteras. Finally, I would like to thank my family, specially to my mum, my dad and my brother Javier; for taking care of me as good as somebody can take care of another person. Gabriel Garcia Marquez once wrote “Love is as important as food, but it doesn’t nourish”. Not a problem, my family gives me both things.


vii
Abstract The cells in out body are able of having completely different behaviors and shapes, although they contain the same genetic material. Among the mechanisms that allow this diversity we find transcription factors (TFs). TFs are proteins that bind specific DNA sequences in our genome and modulate the expression of nearby genes. The choice of what genes are regulated by a TF is based in the specificity and affinity of a TF for specific DNA sequences. On the other hand, the effect on gene expression, either it is an increase or a decrease, is usually mediated by interactions between the TF and other proteins. In this thesis we are presenting computational tools to study interactions between TF and DNA, and between TFs and other proteins. For TF-DNA interactions we have developed statistical potentials scoring functions and a platform to make structural models for these interactions. By combining these two tools, we have developed a method to predict the DNA binding preferences of TFs. For interactions between TFs and other proteins we have developed a method to model and estimate the affinity of the interaction.
Abstract in other lenguage Las células de nuestro cuerpo son capaces de tener comportamientos y morfologías completamente diferentes, pese a contener el mismo material genético. Entre los mecanismos que permiten esta diversidad encontramos los factores de transcripción (FTs). Los FTs son proteínas capaces de unirse a secuencias específicas de ADN en nuestro genoma y modular la expresión de los genes cercanos. La elección de que genes son regulados por cada FT viene dada por su especificidad y afinidad por ciertas secuencias de ADN. Por otro lado, el efecto sobre la expresión génica, ya sea su incremento o su disminución, suele estar mediado por interacciones entre el FT y otras proteínas. En esta tesis presentamos herramientas computacionales para estudiar interacciones entre FTs y ADN, y entre FTs y otras proteínas. Para las interacciones entre FTs y ADN hemos desarrollado un sistema de puntuación basado en potenciales estadísticos y una plataforma para hacer modelos estructurales de estas interacciones. Hemos combinado estas dos herramientas para desarrollar un método que predice las preferencias de unión a ADN de FTs. Para las interacciones entre FTs y otras proteínas hemos desarrollado un método que modela y estima la afinidad de la interacción.


ix
Preface
I am a horrible cook. This means that if I want to bake a cake, I need to follow the instructions very carefully to come up with an eatable cake. If no instructions are available, my absolute lack of cooking instincts will lead me to mess with ingredient amounts and oven time, resulting in a non-eatable cake. Think of the two cakes I have talked so far, the eatable and the non-eatable, both are made of the same ingredients and yet they are so different. This is exactly what happens with our cells. Just as it happens with cakes, all our cells contain the same ingredients. We can think of our genome as the shelve where we store all our available ingredients, the genes, and they are the same for all our cells. However, by expressing different genes at specific rates and times, our cells are able to achieve completely different behaviors and shapes. This is at the base of development and physiological processes, but also in disease. It is quite straightforward to think that if we mess with gene expression, since it is at the basis of all physiological processes of our body, diseases are going to emerge. The mechanisms that regulate gene expression are diverse and complex. One of these mechanisms are transcription factors (TFs). TFs are proteins that bind specific DNA sequences in our genome and modulate the expression of nearby genes. TFs are involved in interactions with both DNA (to find their binding sites) and other proteins (to modulate gene expression). Both of these interactions are complex, TFs can recognize several DNA sequences and interact with several proteins. Besides, these interactions involve a tradeoff between affinity and specificity. The way in which the scientific community has addressed the study of TFs has been mainly through experimental techniques. Many techniques have appeared to both characterize TF-DNA and protein-protein interactions. The experimental characterization of TF-DNA interactions usually yields a DNA binding motif represented as a position weight matrix. The experimental characterization of protein-protein interactions usually yields pairs of interacting proteins and in some cases their interacting affinity. The problem with experimental techniques is that they are expensive and time consuming. On the other hand, the number of experiments that could be conducted to characterize TF-DNA or protein-protein interactions is close to infinite. In this situation it is relevant to develop computational methods that help experimental procedures to fill the gaps in our knowledge. In this thesis we have developed computational tools to study TFs at both their TF-DNA and protein-protein interactions. Our computational tools address the problem of predicting the binding affinity of interactions as well as obtaining structural models for these interactions.


xi
Table of contents
Pag.
Abstract vii
Preface ix
List of figures xiii
List of tables xiv
1. Introduction 1
1.1 - Biological relevance of gene regulation 1
1.2 - Protein-DNA interactions in transcriptional regulation 1
1.2.1 - Transcription factors 2
1.2.1.1 - Functional classification of TFs 3
1.2.1.2 - Structural classification of TFs 4
1.2.2 - Cis-regulatory elements 10
1.3 - Methods to characterize protein-DNA interactions 11
1.3.1 - Experimental in vivo methods 11
1.3.2 - Experimental in vitro methods 14
1.3.3 - Computational methods 18
1.3.4 - Databases of protein-DNA interactions 21
1.4 - Protein-Protein interactions in transcriptional regulation 23
1.4.1 - Transcription Co-Factors 23
1.5 - Methods to characterize protein-protein interactions 24
1.5.1 - Experimental methods 24
1.5.2 - Computational methods 29
1.5.3 - Databases of protein-protein interactions 33
1.6 - Methods to characterize molecular interactions with atomic resolution
33
1.7 - Motivation of this thesis 36
2. Objectives 37

xii
3. Results 39
3.1 - Methods to model and assess protein-DNA interactions 39
3.1.1 - ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements
40
3.1.2 - On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF
118
3.1.3 - Short comment on: On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF
167
3.2 - Methods to model and assess protein-protein interactions 172
3.2.1 - Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions
173
3.2.2 - Prediction of protein–protein binding affinities from unbound protein structures
193
4. Discussion 215
5. Conclusion 223
6. Appendix 225
7. References 229

xiii
List of figures Pag.
Fig. 1. In vivo methods to characterize protein-DNA interactions.
12
Fig. 2. In vitro methods to characterize protein-DNA interactions.
14
Fig. 3. Methods to characterize protein-protein interactions.
25

xiv
List of tables
Pag.
Table 1. Computational methods to identify DNA binding proteins.
19
Table 2. Computational methods to predict protein-DNA interfaces.
20
Table 3. Computational methods to predict protein-DNA binding sites.
21
Table 4. Databases of protein-DNA interactions.
22
Table 5. Computational methods to predict protein-protein interactions.
31
Table 6. Computational methods to predict interacting interfaces of protein-protein interactions.
31
Table 7. Computational methods to predict interacting affinity of protein-protein interactions.
32
Table 8. Computational methods to predict the effect of mutations in protein-protein interactions.
32
Table 9. Databases of protein-protein interactions.
33

1
1. Introduction
1.1 - Biological relevance of gene regulation Thanks to gene regulation the different cells of our body can be extremely diverse in shape and function while containing the same genetic material. Through gene regulation, cells control where, when and in what amounts proteins are produced. In prokaryotes gene regulation allows bacteria to adapt to changes in environmental conditions. One example of this is how bacteria can switch metabolic pathways thanks to the regulation of the lac operon in response to nutrient availability (1). In unicellular eukaryotes gene regulation also allows cells to adapt to changes in environmental conditions. One example of this is how yeast under heat shock stress are able to trigger the expression of chaperones, that are proteins that will prevent or reverse the aggregation of proteins due to heat (2). In multicellular eukaryotes gene regulation is at the basis of development and physiology (3). Regarding human health, if gene expression is dysregulated it can give rise to many diseases (4–6). Gene regulation can take place at different points from transcription to the moment when the expressed protein is fully functional (7). We can classify the different stages at what gene regulation can happen in 4 phases: Transcriptional, post-transcriptional, translational and post-translational. Transcriptional regulation involves many elements whose integration leads to a fine control of what genes are transcribed. These elements are transcription factors (TFs), cis-regulatory elements, chromatin packaging, genome architecture or DNA chemical modifications, among others (8). Post-translational regulation involves the transport, maturation, splicing and decay of mRNAs (9,10). Besides, at this step mRNAs can be targeted for degradation via RNA interference (11). Translational regulation takes place mainly during the initiation of translation, when the translation machinery ensembles (12). Post-translational regulation involve the chemical modifications that proteins need to be functional (13). It also includes the degradation of proteins, which helps to control the amount of functional proteins (14). In this thesis we will focus on transcriptional regulation in eukaryotic multicellular organisms.
1.2 - Protein-DNA interactions in transcriptional regulation There are several types of proteins that interact with DNA molecules: TFs, nucleosomes, DNA replicating machinery or DNA repairing machinery, among others. In this thesis, we will focus on studying TFs, their DNA binding preferences and their role in transcriptional regulation. On the other hand, there are several types of DNA elements (also known as cis-Regulatory Elements) that can interact with TFs: promoters, enhancers, silencers or insulators, among others (15).

2
1.2.1 - Transcription Factors TFs are proteins that bind DNA in a sequence-specific way and regulate transcription (16). TFs carry out these actions by using two different domains: the DNA binding domain (DBD) and the transactivation domain (16). The DBD is responsible for the recognition and binding to specific DNA sequences. This binding happens thanks to the chemical affinity between the DBD and specific DNA sequences. The transactivation domain is responsible for the regulation of transcription (17,18). For most eukaryotic TFs it is thought that this regulation happens thanks to the recruitment of co-factors (19). This recruitment can lead to chromatin remodeling through histone modification (17,20,21) or directly to the recruitment of transcriptional machinery (22). TFs that bind a DNA sequence not necessarily have to have an effect on transcription (16). This fact is in consistence with the RNAPII-centric cooperative model. This model states that Cis-regulatory elements work together to modulate the local concentrations of TFs, RNA polymerase II (RNAPII) and other cofactors. These local concentrations of regulatory proteins will rule the rate of transcription of genes whose transcription start site is close in space (23). TFs recognize a set of similar binding sites rather than one specific sequence. These TF-binding sites are usually small, taking from 6 to 12 bases in most of the cases (16). TF binding sites can be represented as a consensus sequence, which is the most likely sequence to be bound by that TF. TF binding sites can also be represented by PWMs, which are currently the most used representation for TF binding sites. A PWM is a matrix of frequencies (or scores) where each position in the binding site has a value for each one of the four nucleotides (24). PWMs are usually obtained from experimental methods by aligning sequences bound by the same TF (25). A common use of PWMs is to scan DNA sequences and search for TF binding sites. However, this strategy is prone to produce a high number of false positives (26). This poor performance may be explained by the limitations of PWMs, such as assuming independence between the different positions in the PWM (16,24). Besides, the binding of TFs to their binding sites can be influenced by other factors rather than sequence, such as nucleosome occupancy, DNA methylation or cooperativity between TFs (16). Nucleosome occupancy impairs the binding of many TFs to the DNA (27,28). Most TFs have higher affinity for free DNA than for nucleosome-bound DNA (28). Besides, in nucleosome-bound DNA half of the DNA surface is blocked by the nucleosome and is physically inaccessible to TFs (28). DNA methylation can modify the binding affinity of most TFs for DNA sequences (29). DNA methylation usually takes place in cytosines at position 5 in CpG dinucleotides (29). It is thought that although 60%–80% of CpGs in the genome are methylated, DNA methylation is absent or reduced in regions rich in TFs binding such as promoters, enhancers or CpG islands (30). TF cooperativity is the process by which two or more TFs cooperate to identify a complex binding site (16,31–33). TF cooperativity allows the integration of inputs from different signal transduction pathways (32). TF cooperativity is a common feature in eukaryotic

3
gene regulation (33). In a recent work, Jolma et al produced abundant SELEX data on pairs of cooperative TFs (31). They showed that, in most of the cases, TF cooperativity is mediated by the DNA while in few cases it is mediated by interactions made between the TFs regardless the DNA (31). Since binding sites for cooperative TFs usually overlap, the flanks of individual binding sites overlap as well. This makes cooperative binding sites to be slightly different from individual binding sites. As a result, Jolma et al hypothesize that cooperative binding sites could be recognized by individual TFs, but with low affinity (31).
1.2.1.1 – Functional classification of TFs According to their effect in transcription, TFs can be classified into activators or repressors (34). Activators increase the transcription of some genes while repressors do the opposite. This classification has been repeatedly questioned because the same TF can act as an activator or a repressor depending on the context (16,35,36). Activators increase transcription by recruiting co-activator complexes that modify chromatin or by directly recruiting the transcriptional machinery (37). Repressors decrease transcription by recruiting co-repressors that modify chromatin via histone modification and/or nucleosome remodeling (37). According to their capacity to bind to nucleosome-bound DNA, TFs can be classified into pioneers, settlers and migrants (34,38). Pioneer TFs are able to bind to nucleosome-bound DNA (27), while settler and migrant TFs only bind free DNA (39). The difference between settler and migrant TFs is that settlers always bind to their binding sites as long they are in free DNA, while migrant TFs may not do that because they depend on co-factors to identify their binding sites (39). Pioneer TFs can induce cell fate changes by binding and opening regions of closed chromatin. One example of this are Oct4, Sox2 and Klf4; these TFs are members of the Yamanaka factors that are able to induce pluripotency in fully differentiated cells such as fibroblasts (27,40). It is thought that TFs are organized in a hierarchical way, where pioneer TFs open packed chromatin and this is populated by settler and migrant TFs (39).

4
1.2.1.2 – Structural classification of TFs We can classify TFs according to the structural similarity of their DBD (16,18,41). This structural classification is what we use to define the TF families that appear in this thesis. Here we describe in detail de DBDs of the TF families with more relevance in this thesis:
C2H2 ZF
Cis2-His2 zinc finger (C2H2-ZF) proteins are the largest family of TFs in higher metazoans and humans. The DBDs of C2H2-ZF proteins are composed by small domains called zinc fingers (42). Each zinc finger consists of a beta-beta-alpha structure stabilized by a zinc ion, where residues on the surface of the alpha-helix contact the DNA through the major groove (43). Each zinc finger is able to recognize DNA sequences of 3 nucleotides and, by combining adjacent zinc fingers, C2H2-ZF proteins are able to recognize long and complex DNA patterns (42). PDB available structures: 101 PBM available data in CisBP: 298
Homeodomain
Homeodomain proteins are one of the most studied and largest families of TFs in humans (16). The DBDs of homeodomain proteins is made of 3 helices and around 60 amino acids. Helix number 3 makes contacts with the major groove of a DNA molecule (44). PDB available structures: 112 PBM available data in CisBP: 419

5
bZIP
There are 60 basic region/leucine zipper motif proteins (bZIP) in human (16). bZIP proteins interact with the DNA by creating homo or heterodimers. The DBD of bZIP proteins consists of two long alpha helices that contact the DNA as if they were chopsticks. Each one of these alpha helices belongs to the different monomers of the bZIP dimer. One part of these helices is basic and makes contacts with the DNA. The other part contains repeats of leucines or other hydrophobic residues and it is used to dimerize the bZIP dimer (45). PDB available structures: 73 PBM available data in CisBP: 144
bHLH
Basic helix-loop-helix protein (bHLH) is the largest family of transcription factors in eukaryotes (46), with around 110 members in human (16). bHLH proteins interact with DNA by creating homo or heterodimers. The N-terminal region of the DBD is a basic helix and it is involved in interacting with the DNA. The C-terminal region of the DBD is a helix-loop-helix motif that contains hydrophobic residues to allow dimerization (46). PDB available structures: 55 PBM available data in CisBP: 113

6
Nuclear receptor
Proteins from the nuclear receptor family are both hormone receptors and transcription factors. Their activity as transcription factors is regulated by the binding of hormones or other ligands (47). There are around 50 nuclear receptors (16). Nuclear receptors have a domain for binding ligands, a DBD and a transactivation domain. The DBD consists of 2 zinc fingers and a C-terminal extension. In each of the zinc fingers there are 4 cysteines that interact with a zinc ion (48). PDB available structures: 176 PBM available data in CisBP: 82
ETS
E26 transformation-specific (ETS) family represent around 30 TFs in human (16). Their DBD is about 90 residues long and it has winged helix-loop-helix structure. Half of the members of the ETS family have an N-terminal domain whose function is to establish PPIs (49). PDB available structures: 43 PBM available data in CisBP: 24

7
Forkhead
Forkhead proteins has more than 2000 members identified in more than 108 species of animals and fungi (50), and around 50 members in human (16). Their DBD consists of, going from N-terminal to C-terminal, three alpha helices, three beta sheets and two loops. The third alpha helix interacts with the DNA through the major groove, while the junction between helix 3 and 2 and the C-terminal loops interact with the minor groove (50). PDB available structures: 31 PBM available data in CisBP: 63
Sox
Proteins from the Sox family play a key role during development (51). There are around 30 members of this family in human (16). Sox proteins interact with DNA through their HMG domain. This domain consists of three alpha helices that interact with the minor groove. This interaction causes a deformation on the DNA, making the minor groove wider and bending the major groove (51). PDB available structures: 57 PBM available data in CisBP: 42

8
Rel
TFs from the Rel family control gene expression in response to environmental agents like cytokines (52). There are around 10 members of this family in human (16). Some Rel proteins can bind the DNA as dimers and some can do it as monomers. Dimeric Rel proteins recognize DNA motifs of 10 to 11 nucleotides long (52). PDB available structures: 49 PBM available data in CisBP: 8
Myb/SANT
The Myb/SANT family is a large family of transcription factors that is largely expanded in plants (53). This family has around 15 members in humans (16). Their DBD consists of four imperfect sequence repeats of around 52 residues, where each repeat makes 3 alpha helices. The third helix of each repeat makes contacts with the major groove of a DNA molecule (53). PDB available structures: 48 PBM available data in CisBP: 140

9
GATA
Proteins from the GATA family play a crucial role during embryonic development (54). This family has around 10 members in human (16). The DBD of the GATA proteins consists of two zinc fingers. One of the two zinc fingers binds the DNA binding site while the other can either bind this same sequence or interact with other DNA sequences or protein partners (54). PDB available structures: 18 PBM available data in CisBP: 54
IRF
Interferon regulatory factors (IRF) are TFs that regulate the expression of interferon upon viral infections, among other functions (55). This family has around 10 members in human (16). Their DBD consists of alpha/beta architecture with four helices, four anti-parallel sheets and three loops. The third alpha helix makes contacts with the major groove, while the other helices and the loops contact the surrounding areas (55). PDB available structures: 13 PBM available data in CisBP: 14

10
T-box
Proteins from the T-box family play an important role in embryonic development (56). This family has around 15 members in human (16). Proteins from this family recognize palindromic DNA sequences by binding the DNA as dimers. Their DBD takes 229 residues and it establishes contacts through the DNA minor groove (56). PDB available structures: 16 PBM available data in CisBP: 21
1.2.2 - Cis-regulatory elements Cis-regulatory elements are the DNA elements located in the genome that interact with the transcriptional machinery and their regulators (36). The main cis-regulatory elements are promoters, enhancers, silencers and insulators. These elements can be separated by thousands of basepairs and yet be close in space due to chromatin conformation, allowing the interaction between them.
Promoters Promoters are the DNA region that contain the transcription start site of a gene and its surroundings (36). In promoters is where the whole regulatory input of genes is integrated into transcriptional activity (15). They contain binding sites that are required to ensemble the pre-initiation complex (36). Besides, they can contain other TF binding sites and interact with other cis-regulatory elements such as enhancers or silencers. Promoters can be classified into strong and weak. Strong promoters are those that can recruit the transcriptional machinery just by themselves, while weak promoters require the interaction with other regulatory elements to recruit the transcriptional machinery (57). Promoters can also be classified into focused and disperse. Focused promoters have one or several transcription start sites over a short region of DNA. Disperse promoters have transcription start sites across DNA regions of 50 to 100 basepairs (36). The TATA box promoter, that has been widely studied, is an example of focused promoter (36).
Enhancers Enhancers are DNA regions that increase the transcriptional activity of the genes that are close in space. They carry this function by enhancing the ensemble of the pre-initiation complex in the promoter. Enhancers require the

11
binding of TFs to be functional. Enhancers are strongly conserved across evolution and are involved in gene regulation for specific cell lineages (36).
Silencers Silencers are DNA regions that decrease the transcriptional activity of the genes that are close in space. Silencers can impair transcription actively by binding repressive TFs that will interfere with the assembly of the pre-initiation complex. Silencers can also impair transcription passively by preventing activator TFs to bind into their corresponding binding sites (36).
Insulators Insulators are DNA regions that limit the interactions that can take place between other cis-regulatory elements (36,58). Insulators are responsible to set the boundaries between chromatin domains, also called TADs (Topologically Associated Domains) (59). TADs are chromatin regions that are enriched in contacts within themselves while having scarce contacts with other chromatin regions (60). Therefore, cis-regulatory elements within the same TAD will be likely to interact, and cis-regulatory elements in different TADs will not. It has been found that genes within the same TADs tend to coordinate their transcription (61). TAD organization is a well conserved feature across evolution and is usually associated with cell-type specific expression patterns (59). In vertebrates, a common feature of insulators is to contain CTCF binding sites (36,58). CTCF is a TF from the C2H2-ZF family that plays a key role in genome organization through its role in insulators (58).
1.3 – Methods to characterize protein-DNA interactions
1.3.1 – Experimental in vivo methods ChIP related methods In ChIP (Chromatin InmunoPrecipitation) cells are treated with formaldehyde. This crosslinks proteins that interact with DNA to their DNA binding sites in vivo. Then, chromatin is broken by sonication into fragments that are around 200-600 bp long. Next, an antibody specific for the protein of interest is used to immunoprecipitate the protein of interest that will be bound to its corresponding DNA binding site. Finally, the crosslink is reversed, and the DNA sequences are isolated and amplified to create a library. In ChIP-seq this library is sequenced and each DNA read is located in a reference genome (62). ChIP-seq can be improved by using lambda endonuclease to digest the DNA of the crosslinked fragments that are not bound to a protein. This procedure is called ChIP-exo and it produces DNA fragments that are around 25-50 bp long. This substantially reduces the size of the sequenced DNA fragments allowing the

12
identification of DNA binding sites with higher precision (63). ChIP-nexus is a more robust and reproducible version of ChIP-exo, where DNA amplification is more efficient by circularizing DNA fragments (64). ChIP and ChIP related methods have been the predominant methods to profile protein-DNA interactions (PDIs) (65,66).
Fig 1. In vivo methods to characterize protein-DNA interactions.
DamID In DamID (DNA adenine methyltransferase identification) E. coli adenine methyltransferase (Dam) is fused to the TF of interest. This fusion protein is expressed at low levels. When the fusion protein comes close to DNA it methylates adenines in close GATC motifs. Methylated sequences can be identified by using a methylation-sensitive endonuclease and a ligation via PCR; and then they are sequenced. In comparison with other in vivo techniques, DamID has the quality of not using antibodies. Therefore, the quality of the experiment does not depend on having an efficient antibody for the TF of interest. Another quality of DamID is its versatility. Depending on what protein Dam is fused with, more chromatin features than just TF binding sites can be identified: if Dam is fused to RNA polymerase II, it identifies transcriptionally active sites; if it is fused to RNA-binding proteins, it identifies RNA-DNA interactions; if it is tethered to a specific locus, it can identify long range DNA interactions; if it is split and each half is fused to a TF, it can identify TF co-binding events; if it is not fused to any protein, it profiles chromatin accessibility; and if it is bound to a tracer, it can be used for live imaging. On the other hand,

13
the resolution of DamID results depends on GATC motif availability across the genome (66). CUT&RUN In CUT&RUN (Cleavage Under Targets and Release Using Nuclease) unfixed nuclei are immobilized on magnetic beads. Then, these nuclei are incubated with antibodies specific for one TF and with a chimeric fusion of protein A and MNase. Protein A binds to the antibodies, while MNase cleavages DNA sequences that are close by in presence of calcium ions. Therefore, the antibodies will bind to the TF of interest and the fusion between protein A and MNase will bind to the antibody. By doing so, this technique targets MNase activity to the binding sites of the TF of interest. When adding calcium ions to the medium, the MNase will cleavage the DNA neighboring the TF binding site. Then, the protein-DNA complex, bound to the antibody and to the protein A-MNase fusion protein, will exit the nucleus by diffusion and reach the supernatant. Then the DNA in these complexes is collected and sequenced. CUT&RUN provides a good alternative to ChIP because instead of doing a crosslinking step it is performed in situ. This allows CUT&RUN to make a chromatin mapping with high resolution and to describe local chromatin features (67). Footprinting: ATAC-seq, DNAse-seq, DMS-seq In footprinting methods, we identify what genome regions are bound by proteins and what regions are not. They are non-targeted methods, this means that don’t provide results for any protein in particular, but for all of them at the same time. Footprinting methods use tools to introduce cleavages or chemical modifications in the DNA at chromatin sites that are not occupied by proteins. Then, the resulting DNA sequences are sequenced and mapped against a reference genome. Therefore, footprinting reveals non occupied chromatin regions as those regions where cleavages or chemical modifications occur. There are different footprinting methods depending on what strategy is used to cleavage or modify the DNA, here we will review DNAse-seq, ATAC-seq and DMS-seq. In DNAse-seq DNA cleavage is done using DNase I. It requires that cell nuclei are isolated and then treated with DNAse I (68). In ATAC-seq (Assay for Transposase Accessible Chromatin using sequencing) unoccupied chromatin regions are found using the Tn5 transposase. Tn5 is a prokaryotic and hyperactive transposase that integrates adapter sequences into open chromatin regions. After the treatment with Tn5, the genome of the treated cells is sequenced and integrations made by Tn5 are identified (69). In DMS-seq (DiMethyl Sulfate sequencing) cells are treated with DMS (DiMethyl Sulfate). DMS produces N7-methylguanine and N3-methyladenine when reacting with double-stranded DNA. This reaction can only happen in DNA regions that are

14
not occupied by proteins. After the DMS treatment, DNA is cleavaged at methylated sites by β-elimination. In comparison with other footprinting methods, DMS-seq can be applied without performing nuclei isolation because DMS is able to permeate in live cells (70). MPRA In MPRA (massively parallel reporter assay) hundreds of thousands of DNA sequences containing a library of regulatory elements are synthesized. These DNA sequences are tagged with a DNA barcode and introduced into plasmids. This pool of plasmids is co-transfected into the population of cells that will be assessed in the experiment. Then, mRNA in those cells is sequenced and the levels of tagged transcripts are used as a measure of the expression of the transfected plasmids. MPRA is a non-targeted method, this means that is not providing results about any TF in particular, instead of that it measures the overall transcriptional activity in the cells under study (71).
1.3.2 – Experimental in vitro methods
Fig 2. In vitro methods to characterize protein-DNA interactions.

15
Protein binding microarrays In Protein Binding Microarrays (PBM) the DBD of a protein of interest is tested against an array of double-stranded DNA sequences. This DBD is tagged with GST (Glutatione S-Transferase N-terminal), which is recognized by a fluorescent anti-GST antibody. Fluorescence intensity is used as a measure of the affinity between the double-stranded DNA proves and the tested DBD. These arrays contain all possible double-stranded DNA sequences of 8 nucleotides (8-mers) without including palindromic sequences. Besides, each 8-mer is repeated 32 times (16 for palindromic sequences) across the array (25). This provides redundancy when assessing the binding affinity between the tested DBD and the different 8-mers. By selecting and aligning top scoring 8-mers, PBM results can be used to build PWMs (72). One advantage of PBMs is that is testing all possible 8-mers and providing either PWMs and precise data for each 8-mer. HT-SELEX In HT-SELEX (High Throughput Systematic Evolution of Ligands by EXponential enrichment) one protein is incubated with a pool of random DNA sequences. Then, the DNA sequences bound to the protein are purified and amplified using PCR and incubated again with the same protein. This process is repeated several times until sequences that bind the protein with high affinity are isolated (73–75). Several modifications of HT-SELEX have been developed to assess complex PDIs: CAP-SELEX (to assess cooperative binding of TFs into the same DNA binding site) (31), methylation-sensitive SELEX (to assess binding in methylated binding sites) (29) and Nucleosome CAP-SELEX (to assess binding in DNA sequences bound to nucleosomes) (28). One drawback of HT-SELEX and its derived methods is that it generates results biased towards high affinity binding sites (76). Bacterial and yeast one-hybrid In one-hybrid assays a fusion protein made of the DBD under study and a trans-activation domain is introduced into cells. These cells contain into their genetic material a bait sequence followed by two reporter genes. This method evaluates the capacity of the DBD and the bait DNA sequence to interact. If this interaction happens, the trans-activation domain triggers the expression of the reporter genes. One of these reporters is a positive reporter and its expression leads to cell growth. By selecting the cell colonies that grow and sequencing their bait sequences we are able to identify what DNA sequences are bound by the DBD under study (77–79). In bacterial one-hybrid this workflow is applied in E. coli cells and the trans-activation domain used is the omega subunit of the bacterial RNA polymerase. The bait sequence and the reporter genes are introduced into bacterial cells as

16
plasmids. The reporter genes used are HIS3 and URA3. HIS3 is used as positive reporter and its expression allows the cell colony to grow by overcoming the inhibition driven by 3-amino-triazole, a compound present in the culture medium. URA3 is used as negative reporter and its expression leads to cell death in presence of 5-fluoro-orotic acid. The expression of URA3 is used to discard self-activating bacterial colonies, that would lead to false positives. TF binding sites are identified by sequencing the bacterial colonies that can grow when expressing the transfected fusion protein and that die in presence of 5-fluoro-orotic acid (77,78). In yeast one-hybrid this workflow is applied in S. cerevisiae cells and the trans-activation domain used is the one of Gal4. The bait sequence and the reporter genes are integrated into the yeast genome. The reporter genes used are HIS3 and LacZ and both of them are used as positive reporters. HIS3 is used in the same way as in bacterial one-hybrid while LacZ produces a blue compound from the X-gal present in the culture medium. TF binding sites are identified by sequencing the yeast colonies that turn blue (79). SMiLE-seq In SMiLE-seq (selective microfluidics-based ligand enrichment followed by sequencing), PDIs are identified using a microfluidics-based approach. A mixture of the TF of interest, the target dsDNA, and a nonspecific competitor poly-dIdC is introduced in the microfluidics device. If TF–DNA complexes are made, they get trapped in a PDMS (polydimethylsiloxane) membrane while unbound molecules are washed away. After that, DNA sequences involved in complexes with TFs are collected, amplified by PCR and sequenced. In comparison with other methods, one of the advantages of SMiLE-seq is its ability to identify low and medium affinity PDIs. Besides, SMiLE-seq is not limited by the size of the assayed DNA sequences (76). MITOMI In MITOMI (Mechanically-Induced Trapping of Molecular Interactions) PDIs are identified using a microfluidics-based approach. The microfluidic devices used in MITOMI contain thousands of unit cells, where each unit cell can measure one PDIs. In each unit cell there are a DNA chamber and a protein chamber. Whether the chambers are opened or closed depends on micromechanical valves included into the microfluidic device. The DNA chambers contain DNA sequences that are 70 bp long and that are made by overlapping 8-mer de Brujin sequences. Each DNA sequence is repeated at least two times in the same device. These DNA sequences are labeled with Cy5 (a fluorescent dye) on one of their terminals. The protein chambers contain the TF of interest in solution. These TFs are synthesized in vitro and are labeled with BODIPY (another fluorescent dye). Then, the two chambers are opened and the DNA sequence and the proteins are incubated together. After incubation, unbound

17
proteins and DNA sequences are washed away. Protein-DNA affinity is measured as the ratio between DNA fluorescence and protein fluorescence in the protein chamber. In comparison with other methods, one of the advantages of MITOMI is its ability to identify low and medium affinity PDIs (80). DAP-seq In DAP-seq (DNA affinity purification sequencing) the TF under study is incubated with a library of genomic DNA. The TFs are synthesized in vitro and include an affinity tag. This affinity tag is used to immobilize the TFs into beads. Then, TFs bound to beads are incubated with the library of genomic DNA fragments. Finally, the unbound DNA is washed away and the bound DNA is collected, amplified by PCR and sequenced. DAP-seq can be modified to ampDAP-seq to assess the effect of cytosine methylation in protein-DNA binding. In ampDAP-seq, methylated cytosines are removed from the library of genomic DNA fragments by using PCR. Then, by comparing the results obtained by ampDAP-seq and by ordinary DAP-seq we are able to assess the effect of cytosine methylation in PDIs (81). EMSA In EMSA (Electrophoretic Mobility Shift Assay) protein-DNA complexes and their corresponding unbound DNA sequences undergo electrophoresis in a polyacrylamide or agarose gel. Since protein-DNA complexes have a higher molecular weight, they migrate more slowly across the gel. Therefore, when by seeing the bands in the get we can identify what of the assayed proteins and DNA sequences are interacting. Although EMSA is mainly a qualitative approach for measuring PDIs, it can be modified to assess binding stoichiometries, affinities and kinetics (82). ATI In ATI (Active TF Identification) a library of random DNA sequences of 40 basepairs long is incubated with the nuclear extract from a specific cell type. Then, protein-DNA complexes are identified by electrophoretic mobility shift assay (EMSA). The proteins bound to DNA are collected and their amino acid sequence is revealed by mass spectrometry. The DNA sequences bound to proteins are collected, amplified by PCR and sequenced. ATI is a method designed to assess TF activity in different cell types or in cells under different environmental conditions. It is not just finding DNA binding sites for different TFs, but also quantifying TF abundance and finding out what TFs are more relevant for gene regulation in each cell type (83).

18
1.3.3 – Computational methods Identification of DNA binding proteins Most computational methods that predict what proteins are able to bind DNA are based on analyzing protein sequences (84–103). These methods take advantage of the evolutionary relationships between protein sequences or of the physicochemical properties of amino acids. Then, they analyze data on protein sequences by different machine learning methods. Some other methods are based on analyzing structural data (104), with machine learning algorithms as well. Structure-based methods usually provide more reliable predictions than sequence-based methods, but their applicability is lower than sequence-based methods because of the limited availability of protein structures (89,102). Prediction of protein interfaces to interact with DNA molecules In this section we will focus on methods that given a protein are able to identify the region of this protein that is able to interact with a DNA molecule. These methods can be classified into those that take into account only sequence information (105–116) and those that use protein structure (117–121). In a similar way to methods to predict DNA binding proteins, sequence-based methods have higher applicability while structure-based methods yield more reliable predictions (114). Sequence-based methods make predictions by using machine learning algorithms (105–116). Structure-based methods use either machine learning (117–119) classifiers or pseudo-energy scoring functions and force fields (120,121). Characterization of TF binding sites In this section we will focus on methods to identify binding sites of TFs on DNA sequences. The computational characterization of TF binding sites depends of the available knowledge on the protein under study. If we know the genes regulated by this protein, we can search for enriched DNA sequences in the promoters and enhancers of such genes using a motif discovery algorithm (122). If we know the binding motif of this protein (represented for example as a PWM), we can scan DNA sequences and search for matches of this binding motif (123). This last method, although widely used, is prone to yield many false positives. This happens because some binding sites may not be accessible to DNA binding proteins because of chromatin features, such as nucleosome occupancy (124). Also, many TF binding motifs are highly degenerate, which also leads to a high number of false positives (125).

19
Name Data Method features
Lu et al (84) Sequence Random forest classifier.
CNN-BiLSTM (85) Sequence Convolutional neural networks.
Wang et al (86) Sequence Random forest and support vector machine classifiers.
DeepDRBP-2L (87) Sequence Neural Networks and Long Short-Term Memory.
DNABP (88) Sequence Random forest classifier.
DP-binder (89) Sequence Random forest and support vector machine classifiers.
Chauhan et al (90) Sequence Deep neural networks.
FC- SVM (91) Sequence Support vector machine.
FKRR-MVSF (92) Sequence Fuzzy Kernel Ridge Regression model based on Multi-View Sequence Features.
gDNA-Prot (93) Sequence Support vector machine.
HMMPred (94) Sequence Extreme gradient boosting and support vector machine.
Ding et al (95) Sequence Fuzzy Multiple Kernel Support Vector Machine.
MsDBP (96) Sequence Deep neural network.
PredDBP-Stack (97) Sequence Hidden markov models and ensemble learning.
You et al (98) Sequence Selective ensemble algorithm.
PredPSD (99) Sequence Gradient tree boosting.
SDBP-Pred (100) Sequence Support vector machine.
IDBP-DEP (101) Sequence Support vector machine.
StackDPPred (102) Sequence Support vector machine.
TargetDBP (103) Sequence Support Vector Machine Recursive Feature Elimination with Correlation Bias Reduction.
Wang et al (104) Structure AdaBoost classifier.
Table 1. Computational methods to identify DNA binding proteins.

20
Name Data Method features
PrPDH (105) Sequence Machine learning models, namely, support vector machine (SVM), random forest, Naïve Bayes and k-nearest neighbor.
DQPred-DBR (106) Sequence Evolution-based features and dynamic models.
CNNsite (107) Sequence Convolutional neural networks.
DRNApred (108) Sequence Two layered logistic regression.
EL_PSSM-RT (109) Sequence Ensemble learning.
ENSEMBLE-CNN (110) Sequence Convolutional Neural networks.
Zhang et al (111) Sequence Adaptive Synthetic Sampling and neural networks.
Shen et al (112) Sequence Multi-scale Local Average Blocks algorithm.
iProDNA-CapsNet (113) Sequence Capsule neural network.
TargetDNA (114) Sequence Support vector machines
funDNApred (115) Sequence Fuzzy cognitive maps, support Vector Machines, Naïve Bayes and k-Nearest Neighbor.
DNAPred (116) Sequence Ensembled hyperplane-distance-based support vector machine.
PDNAsite (117) Structure Latent Semantic Analysis and ensemble learning.
PDRLGB (118) Structure Light gradient boosting machine.
NucBind (119) Structure Support vector machines and comparison with structural templates.
PADA1 (120) Structure Atomistic force field (related to foldX).
JET2DNA (121) Structure Clustering and pseudo-energy scoring functions.
Table 2. Computational methods to predict protein interfaces to interact with DNA molecules.

21
If there is no data on what genes are regulated by one protein or what are its DNA binding preferences, we can use computational methods based on sequence (41,126–128) or structural information (125,129–131). Sequence-based methods usually encode sequence information into data that is processed by a machine learning approach to yield predictions (127,128). Another sequence-based method to search for homologous proteins with available experimental information about their DNA binding preferences. This last approach has proven to be successful at high degrees of homology and it allows to cover many proteins for which we did not have available information (41,126). Structure-based methods use knowledge-based potentials and/or atomistic force fields to predict the DNA binding preferences of proteins (125,129–131).
Name Data Method features
Cis-BP (41) Sequence Homology inference (also known as nearest neighbour)
Cis-BP (126) Sequence Similarity regression
DRAF (127) Sequence Random forest classifier.
Dang et al (128) Sequence Jensen–Shannon Divergence and random forest classifier.
Xu et al (125) Structure Knowledge-based potentials.
EMQIT (129) Structure Boltzmann formula-based scores.
Qin et al (130) Structure Knowledge-based potentials.
Farrel et al (131) Structure Knowledge based and atomistic energy terms.
Table 3. Computational methods to predict TF binding sites.
Other methods Computational tools have been designed to predict binding affinities between proteins and DNA sequences (132,133). These tools are based on protein structure and combine knowledge-based potentials and force fields with machine learning classifiers to make their predictions. Besides, tools to engineer TF are starting to appear (134). In Dutta et al, structural information of Zif268 and neural networks are used to predict amino acid sequences that recognize specific 9 basepairs DNA sequences. The predicted amino acid sequences should fit into a canonical 3 C2H2-ZF DBD, such as the one of Zif268 (134).
1.3.4 – Databases of protein-DNA interactions There are databases with available information regarding the DNA binding preferences of many proteins. They are a valuable source of data and can complement or provide an alternative for many computational methods. The two
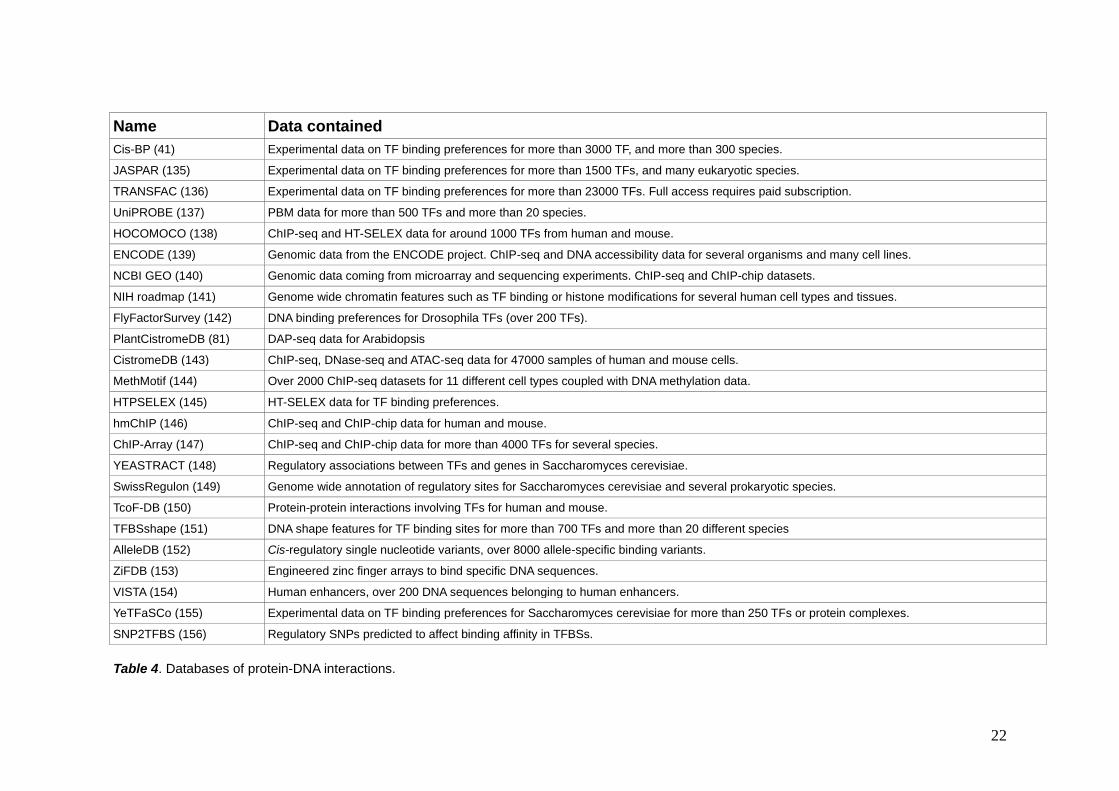
22
Name Data contained
Cis-BP (41) Experimental data on TF binding preferences for more than 3000 TF, and more than 300 species.
JASPAR (135) Experimental data on TF binding preferences for more than 1500 TFs, and many eukaryotic species.
TRANSFAC (136) Experimental data on TF binding preferences for more than 23000 TFs. Full access requires paid subscription.
UniPROBE (137) PBM data for more than 500 TFs and more than 20 species.
HOCOMOCO (138) ChIP-seq and HT-SELEX data for around 1000 TFs from human and mouse.
ENCODE (139) Genomic data from the ENCODE project. ChIP-seq and DNA accessibility data for several organisms and many cell lines.
NCBI GEO (140) Genomic data coming from microarray and sequencing experiments. ChIP-seq and ChIP-chip datasets.
NIH roadmap (141) Genome wide chromatin features such as TF binding or histone modifications for several human cell types and tissues.
FlyFactorSurvey (142) DNA binding preferences for Drosophila TFs (over 200 TFs).
PlantCistromeDB (81) DAP-seq data for Arabidopsis
CistromeDB (143) ChIP-seq, DNase-seq and ATAC-seq data for 47000 samples of human and mouse cells.
MethMotif (144) Over 2000 ChIP-seq datasets for 11 different cell types coupled with DNA methylation data.
HTPSELEX (145) HT-SELEX data for TF binding preferences.
hmChIP (146) ChIP-seq and ChIP-chip data for human and mouse.
ChIP-Array (147) ChIP-seq and ChIP-chip data for more than 4000 TFs for several species.
YEASTRACT (148) Regulatory associations between TFs and genes in Saccharomyces cerevisiae.
SwissRegulon (149) Genome wide annotation of regulatory sites for Saccharomyces cerevisiae and several prokaryotic species.
TcoF-DB (150) Protein-protein interactions involving TFs for human and mouse.
TFBSshape (151) DNA shape features for TF binding sites for more than 700 TFs and more than 20 different species
AlleleDB (152) Cis-regulatory single nucleotide variants, over 8000 allele-specific binding variants.
ZiFDB (153) Engineered zinc finger arrays to bind specific DNA sequences.
VISTA (154) Human enhancers, over 200 DNA sequences belonging to human enhancers.
YeTFaSCo (155) Experimental data on TF binding preferences for Saccharomyces cerevisiae for more than 250 TFs or protein complexes.
SNP2TFBS (156) Regulatory SNPs predicted to affect binding affinity in TFBSs.
Table 4. Databases of protein-DNA interactions.

23
databases more used in this thesis are Cis-BP (41) and JASPAR (135). Cis-BP contains binding motifs obtained by different experimental techniques such as PBM, ChIP-seq or HT-SELEX, among others. Cis-BP contains binding motifs for more than 4500 TFs from more than 700 species. Besides, Cis-BP allows to search for homology inferred motifs, increasing the applicability of these binding motifs to more than 390000 TFs (41,126). JASPAR also contains binding motifs obtained by experimental procedures such as PBM, ChIP-seq or HT-SELEX. JASPAR contains more than 1500 binding profiles organized into different vertebrate taxons. JASPAR also has a tool to infer binding motifs based on homology (135).
Other relevant databases of experimental binding motifs are TRANSFAC (136), UniPROBE (137) and HOCOMOCO (138). Some databases collect data from big consortia devoted to genome annotation such as ENCODE (139), NCBI GEO (140) and NIH roadmap (141). These databases contain large datasets on ChIP-seq as well as information on chromatin features such as histone modifications or chromatin accessibility. Some databases contain binding motifs for specific species such as FlyFactorSurvey (142) for Drosophila or PlantCistromeDB (81) for Arabidopsis. Other databases containing experimental binding motifs are CistromeDB (143), MethMotif (144), HTPSELEX (145), hmChIP (146), ChIP-Array (147). Finally, there are databases that contain information that is useful to study PDIs and yet do not contain TF binding motifs (see table) (148–156).
1.4 – Protein-Protein interactions in transcriptional regulation Protein-Protein Interactions (PPIs) play a fundamental role in the regulation of many biological processes (157), among which there is transcription. There are many types of proteins whose interactions play a role in transcriptional regulation: transcription co-factors, transcriptional machinery, histones and other chromatin structural proteins, among others. In this thesis we will focus on transcription co-factors and their role in transcriptional regulation.
1.4.1 – Transcription Co-Factors Transcription co-factors are proteins that regulate transcription by interacting with TFs or other co-factors (150). Usually, many co-factors associate into multi-subunit complexes (158–161). These complexes are able to include different combinations of subunits as well as sharing subunits with other complexes. This modularity allows their regulation to be highly precise and dynamical (159). Some co-factors are found to be redundant in terms of structure and function since they play similar roles in similar or even the same complex (159,160). It is hypothesized that many co-factors operate in a synergistic way allowing the integration of inputs from different regulatory pathways (159).

24
Depending on their effect on transcriptional activity, co-factors can be classified into co-activators or co-repressors (162). Co-activators can play a role in the different stages of transcriptional activation: removal or repressors, recruitment of activators, nucleosome removal, recruitment of transcriptional machinery, formation of the pre-initiation complex, transcription initiation and productive elongation of the RNA molecule (158). Co-repressors can regulate transcription by the following mechanisms: histone deacetylation, histone methylation, chromatin remodeling, DNA methylation and interfering with the assembly of activator machinery by competitive binding (162). Besides, some co-factors called adapters can act either as co-activators or co-repressors (160). Depending on the regulatory machinery they interact with, co-factors can be classified into general co-factors and regulator-associated co-factors. General co-factors interact with the basal transcriptional machinery and usually are present in the transcription of most genes (160). One example of general co-factors are those involved in the assembly of the pre-initiation complex at the TATA-box promoter (161). Regulator-associated co-factors are recruited by gene-specific regulators. Usually, they are able to modify histones or to remodel chromatin. Regulator-associated co-factors have been found to interact with the basal transcriptional machinery as well (160).
1.5 – Methods to characterize protein-protein interactions 1.5.1 – Experimental methods Yeast two-hybrid (Y2H) In Y2H the assayed proteins are fused to a specific DBD and to a transcriptional activation domain, respectively. Therefore, the interaction between the assayed proteins targets the transcriptional activation domain to a specific DNA binding site. This leads to the expression of downstream reporter genes. Y2H is a simple and economical method that can be easily set up in most laboratories. On the other hand, Y2H can have a high false-positive rate because the assayed proteins are overexpressed and this can lead to non-specific interactions (163,164). Membrane yeast two-hybrid (MYTH) MYTH is a method designed to identify PPIs that happen between membrane proteins. In MYTH, the assayed proteins are bound to two fragments of the ubiquitin protein, respectively. The N-terminal fragment is called Nub and the C-terminal fragment is called Cub. Cub is engineered to contain a transcription factor in its C-terminal. When the assayed proteins interact Nub and Cub come together creating a pseudo-ubiquitin molecule.

25
Fig 3. Methods to characterize protein-protein interactions.
This pseudo-ubiquitin protein is recognized by the endogenous deubiquitinating enzymes that will cleave the Cub C-terminal, releasing the transcription factor bound to its C-terminal. Then, the transcription factor goes to the nucleus and triggers the expression of reporter genes. Strengths and limitations of MYTH are similar to the ones of Y2H. One variation of MYTH is the Mammalian membrane two hybrid (MaMTH), where the same principle of ubiquitin fragments is used to assess PPIs in membranes (163,165,166). Luminescence-based mammalian interactome mapping (LUMIER) In the LUMIER assay one protein is fused to Renilla luciferase and the other protein is linked to an affinity tag. These constructs are overexpressed in cells

26
and then these cells are lysed. From this lysis the protein bound to an affinity tag is immunoprecipitated using an antibody specific for its tag. It the two proteins interact, the second protein will have been co-immunoprecipitated. Since the second protein is bound to luciferase, the interaction is measured by the luciferase activity in the immunoprecipitated fraction. This approach is easy to apply and can be set up in most laboratories. As drawback it has that immunoprecipitation is done after cell lysis, and this can disrupt low affinity PPIs (163,167). Mammalian protein–protein interaction trap (MAPPIT) In MAPPIT, PPIs are identified taking advantage on the cytokine signal transduction pathway in mammalian cells. One of the two proteins in the interaction is bound to a cytokine receptor deficient for STAT3 binding, while the other protein is bound to functional STAT3 recruitment sites. The interaction of the two assayed proteins leads to a fully functional cytokine receptor, that will trigger the expression of reporter genes via the JAK kinase pathway. This method is easy to perform and does not requires specific equipment. On the other hand, MAPPIT will only work with interactions that happen in the cytoplasmic submembrane region (163,168). Kinase substrate sensor (KISS) In KISS, we apply the two-hybrid technology in the context of a mammalian cell. One of the two proteins in the interaction is bound to the kinase domain of TYK2, while the other is bound to a cytokine receptor carrying TYK2 substrate motifs. The interaction of the two assayed proteins leads to the expression of reporter genes via the STAT3 pathway. This method works for both membrane and cytosolic membranes. Besides, it is sensitive enough to detect changes due to pharmacological or physiological changes. On the other hand this method depends on the STAT3 pathway, so the assayed proteins should not affect this pathway (163,169). Bimolecular fluorescence complementation (BiFC) In BiFC, we split a fluorescent protein into two non-fluorescent fragments. Each one of these fragments are bound to the two assayed proteins. If the interaction between these proteins happens, it should create a fluorescent complex that can be identified by microscopy or flux cytometry. This method provides spatial information about in which subcellular compartment is this interaction happening. Besides, this method is highly sensitive, allowing the detection of interactions at physiological levels as well as low affinity interactions. On the other hand, it may happen that fused proteins are not functional or that fluorescent fragments interact in a non-specific way leading to false positive fluorescent signals (163,170).

27
Fluorescence resonance energy transfer (FRET) and Bioluminescence resonance energy transfer (BRET) In FRET, we take advantage on the energy transfer from an excited fluorophore donor to a close acceptor molecule. One of the proteins in the interaction is fused to a donor, while the other is fused to an acceptor. If the two proteins interact, there will be a transfer of radiation from the donor to the acceptor, leading to a different emission pattern of radiation that can be identified with microscopy. FRET can have the drawback of giving a strong background signal. BRET is a similar method thought to overcome this last limitation. In BRET, Renilla luciferase is used as radiation donor while GFP or YFP are used as radiation receptors. FRET and BRET provide information on the subcellular location at which the interaction is happening, as well as at what moment the interactions are happening. On the other hand, FRET and BRET can have strong technical demands and only interactions that bring the two fluorophores very close will lead to a FRET signal (163,171). FRET can also be used to measure the affinity of a PPI (172). Affinity purification–mass spectrometry (AP-MS) In AP-MS, one of the two proteins in the interaction is bound to a solid support while the other is on a soluble phase. Captured proteins from the soluble phase are split into smaller peptides and analyzed by mass spectrometry. AP-MS can be also performed with native protein baits or with standardized epitope tags. This method allows high-throughput analysis of the interactions happening in a cell. On the other hand, this method cannot detect low affinity interactions. Also, it does not provide spatial neither temporal information about the interactions (163,173). Proximity-dependent biotin identification coupled to mass spectrometry (BioID-MS) In BioID-MS, we take advantage of the biotin/streptavidin system for affinity capture. Cells are modified to express one protein bound to a prokaryotic biotin ligase. This construct will tag with biotin the proteins that interact with it. Then, biotin-labeled proteins will be isolated using a biotin/streptavidin biotin affinity capture approach. Finally, the isolated proteins are analyzed by mass spectrometry. One advantage of this method in comparison with AP-MS is that the biotin labeling happens before the cell lysis. Therefore, the interactions reported by this method take place in their natural cellular context. On the other hand, the biotin ligase is a big protein, this can make that the resulting fusion protein is not completely functional (163,174).

28
Proximity ligation assay (PLA) In PLA, we take advantage of labeling the proteins of interest with conjugation proves. Conjugation proves consist of antibodies conjugated with DNA oligonucleotides. When the assayed proteins come close together, their DNA oligonucleotides serve as a template for the synthesis of a circular DNA. This circular DNA is amplified and, since it contains many repetitive elements, is identified with fluorescent complementary oligonucleotide proves. PLA can detect PPIs with single molecule resolution and locate them in cells and tissues. On the other hand, PLA is expensive and highly dependent on antibody quality and enzyme activity. This is why it cannot be applied in a high-throghput way (163,175).
Ligand–receptor capture – trifunctional chemoproteomics reagents (LRC-TriCEPS) LRC-TriCEPS is a method focused on the identification of ligand/receptor interactions. TriCEPS is a reagent consisting on three parts: one that binds ligands containing an amino group, another that binds glycosylated receptors on live cells, and another with a biotin tag. This reagent links covalently ligands and their corresponding receptors. Finally, the ligand receptor complexes are isolated using the biotin tag. This method can detect ligand/receptor interactions without using genetic manipulations. On the other hand, this method only works with N-glycosilated receptors and it fails when the receptor needs to associate to other cell structures to bind the ligand (163,176). Avidity-based extracellular interaction screen (AVEXIS) AVEXIS is a assay designed to identify novel extracellular receptor ligand interactions. This system requires that the assayed proteins are secreted. One of the assayed proteins is labeled with biotin while the other is labeled with a β-lactamase and a peptide to induce pentamerization. The resulting complexes are isolated using the biotin tag. Finally, interactions are detected thanks to the β-lactamase in an ELISA-like procedure. This assay can detect very weak PPIs with a low false-positive rate. Besides, this method can be adapted to work in a higher throughput scale. On the other hand, it does not work with membrane embedded proteins and it can be very time consuming (163,177). Isothermal titration calorimetry Isothermal titration calorimetry is a technique used to measure the affinity between proteins in a PPI. This technique uses two cells with a volume around 1 ml. One of the cells is filled with water or buffer solution and it is used as a reference. The other cell is filled with a solution of one of the interacting proteins under study. Then, the other protein involved in the interaction is injected in this

29
second cell. By measuring the changes in temperature happening in the cells, it is possible to calculate the change on free energy that takes place when a PPI is formed (178). Surface plasmon resonance Surface plasmon resonance is a technique used to measure the affinity between proteins in a PPI. In surface plasmon resonance we take advantage of the surface plasmon polariton. The surface plasmon polariton is a non-radiative electromagnetic wave that propagates in the boundary between a conductor material and an external medium such as air or water. This electromagnetic wave is very sensitive to changes in the surface of the conductor material, such as the association of molecules. In surface plasmon resonance the conducting surface is coated with one of the proteins from the interaction under study. Then, the other protein involved in the interaction is released into the media. The affinity of this interaction will be reflected in a perturbation of the surface plasmon wave, that will be identified by a detector. From this perturbation we can calculate the change on free energy that takes place when a PPI is formed (179). Stopped-flow spectroscopy Stopped-flow analysis provides kinetic data about PPIs such as the dissociation constant, from which the binding affinity between proteins can be calculated. Stopped-flow machines store solutions of the two proteins under study in separated syringes. Then, the content of both syringes is mixed and placed inside an optical cell of small volume. In this cell, changes in absorbance or fluorescence are detected in interactions that are few milliseconds old (180).
1.5.2 – Computational methods Prediction of PPIs Computational methods to predict PPIs can be classified into methods based on sequence and methods based on structure. Methods based on sequence are based on the principle that homologous proteins tend to share the same PPIs (181). This principle can be related to the idea that proteins sharing structural features tend to share similar PPIs without needing to be close homologs (182). Methods based on structure rely on homology modeling and docking to identify PPIs and provide models of them (183–188). This strategy provides more reliable results than sequence-based methods, although due to the scarce availability of structural templates they cannot be applied to many PPIs (189,190).

30
Prediction of interacting interfaces Computational methods to predict interacting PPI interfaces can be classified into those that use sequence data and those that use 3D structural data (191). Sequence-based methods use evolutionary conservation of residues as well as information from their chemical properties. Structure-based methods can be divided into those that only take into account structural features for individual residues, such as secondary structure or the degree of exposure, and those that use data from homologous structures (191). Many methods in this field combine several of these strategies (192) with machine learning classifiers such as neural networks (193) or random forests (194,195). Prediction of interaction affinity Computational methods to predict the affinity in PPIs usually rely on 3D structural data, and most of the times require the structure of the PPI complex (196). They predict affinity by applying scoring functions on the native structure of the PPI complex (197). These scoring functions can be based on statistical potentials (198), on atomic physical interactions (199) or on surface complementarity (200). These methods usually account for conformational changes using molecular dynamic simulations or simplified models, such as Poisson-Boltzmann/Surface Accessibility to Solvent (201–204). Some of these methods take into account the role of non-interacting regions (205–208). Most of the computational methods to predict the affinity in PPIs have poor accuracy when tested against large datasets (209).
Prediction of the effect of mutations Computational methods that predict the effect of mutations in PPIs affinity can be classified into methods that use sequence data or methods that use 3D structural data (157). Methods using sequence data usually are faster than methods that use structural data. Besides, they can be applied to a larger number of PPIs because they do not depend on the availability of structural data. On the other hand, sequence-based methods are not as reliable as structure-based methods and their performance depends strongly on the data used for training (157,210). Structure-based methods are more reliable but require the structure of the interacting proteins, which reduces their coverage, and they are computationally demanding (189,190). Structure-based methods that sample the conformational space of the PPI have a higher computational cost (157). Examples of this approach are thermodynamic integration and free energy perturbation methods (211). There are other molecular mechanical methods that are less computational demanding (212,213), such as Poisson-Boltzmann/Surface Accessibility to Solvent (214). Another strategy used by some structure-based methods is to use statistical potentials scoring functions (215–218). Besides, many methods use machine learning algorithms to combine some of the different strategies here explained (189,190,219–221).
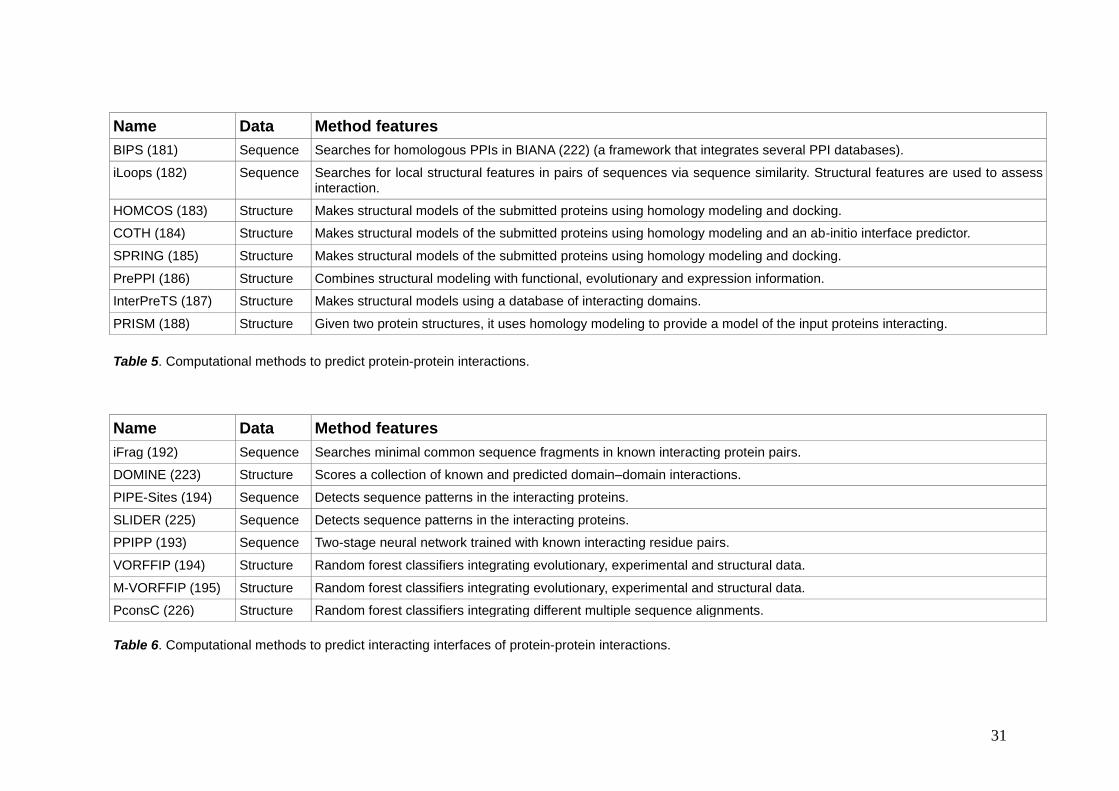
31
Name Data Method features
BIPS (181) Sequence Searches for homologous PPIs in BIANA (222) (a framework that integrates several PPI databases).
iLoops (182) Sequence Searches for local structural features in pairs of sequences via sequence similarity. Structural features are used to assess interaction.
HOMCOS (183) Structure Makes structural models of the submitted proteins using homology modeling and docking.
COTH (184) Structure Makes structural models of the submitted proteins using homology modeling and an ab-initio interface predictor.
SPRING (185) Structure Makes structural models of the submitted proteins using homology modeling and docking.
PrePPI (186) Structure Combines structural modeling with functional, evolutionary and expression information.
InterPreTS (187) Structure Makes structural models using a database of interacting domains.
PRISM (188) Structure Given two protein structures, it uses homology modeling to provide a model of the input proteins interacting.
Table 5. Computational methods to predict protein-protein interactions.
Name Data Method features
iFrag (192) Sequence Searches minimal common sequence fragments in known interacting protein pairs.
DOMINE (223) Structure Scores a collection of known and predicted domain–domain interactions.
PIPE-Sites (194) Sequence Detects sequence patterns in the interacting proteins.
SLIDER (225) Sequence Detects sequence patterns in the interacting proteins.
PPIPP (193) Sequence Two-stage neural network trained with known interacting residue pairs.
VORFFIP (194) Structure Random forest classifiers integrating evolutionary, experimental and structural data.
M-VORFFIP (195) Structure Random forest classifiers integrating evolutionary, experimental and structural data.
PconsC (226) Structure Random forest classifiers integrating different multiple sequence alignments.
Table 6. Computational methods to predict interacting interfaces of protein-protein interactions.
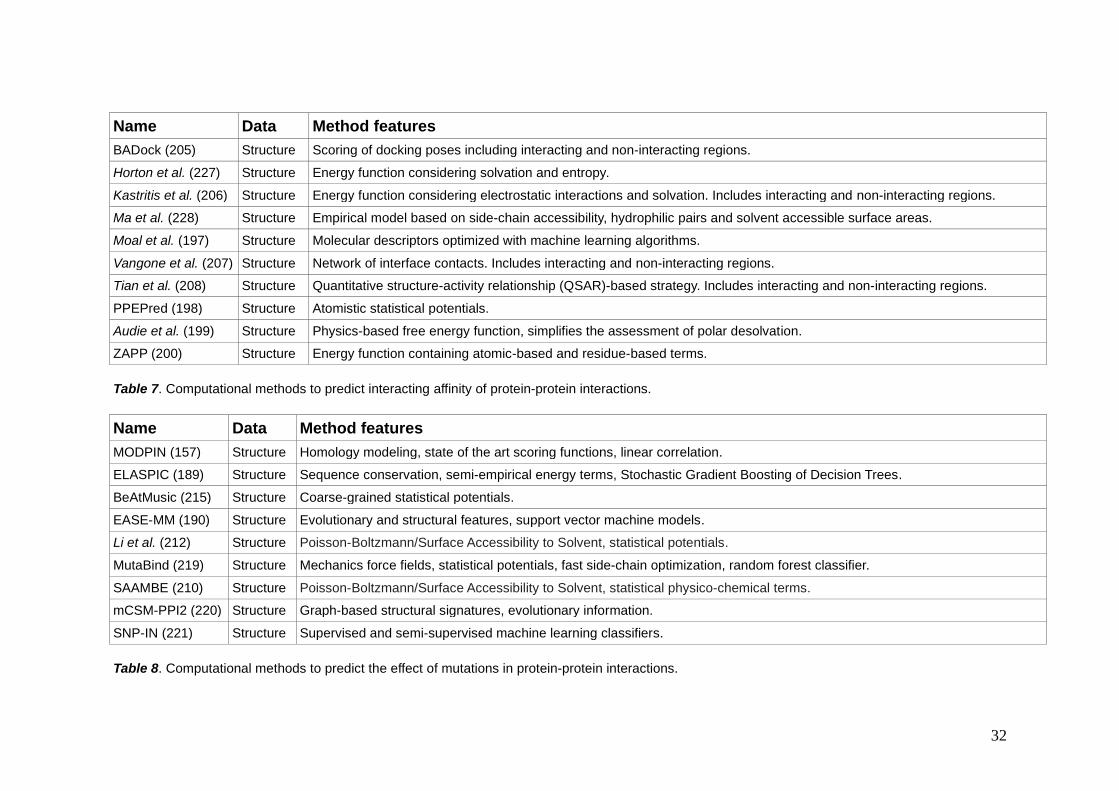
32
Name Data Method features
BADock (205) Structure Scoring of docking poses including interacting and non-interacting regions.
Horton et al. (227) Structure Energy function considering solvation and entropy.
Kastritis et al. (206) Structure Energy function considering electrostatic interactions and solvation. Includes interacting and non-interacting regions.
Ma et al. (228) Structure Empirical model based on side-chain accessibility, hydrophilic pairs and solvent accessible surface areas.
Moal et al. (197) Structure Molecular descriptors optimized with machine learning algorithms.
Vangone et al. (207) Structure Network of interface contacts. Includes interacting and non-interacting regions.
Tian et al. (208) Structure Quantitative structure-activity relationship (QSAR)-based strategy. Includes interacting and non-interacting regions.
PPEPred (198) Structure Atomistic statistical potentials.
Audie et al. (199) Structure Physics-based free energy function, simplifies the assessment of polar desolvation.
ZAPP (200) Structure Energy function containing atomic-based and residue-based terms.
Table 7. Computational methods to predict interacting affinity of protein-protein interactions.
Name Data Method features
MODPIN (157) Structure Homology modeling, state of the art scoring functions, linear correlation.
ELASPIC (189) Structure Sequence conservation, semi-empirical energy terms, Stochastic Gradient Boosting of Decision Trees.
BeAtMusic (215) Structure Coarse-grained statistical potentials.
EASE-MM (190) Structure Evolutionary and structural features, support vector machine models.
Li et al. (212) Structure Poisson-Boltzmann/Surface Accessibility to Solvent, statistical potentials.
MutaBind (219) Structure Mechanics force fields, statistical potentials, fast side-chain optimization, random forest classifier.
SAAMBE (210) Structure Poisson-Boltzmann/Surface Accessibility to Solvent, statistical physico-chemical terms.
mCSM-PPI2 (220) Structure Graph-based structural signatures, evolutionary information.
SNP-IN (221) Structure Supervised and semi-supervised machine learning classifiers.
Table 8. Computational methods to predict the effect of mutations in protein-protein interactions.

33
1.5.3 – Databases of protein-protein interactions PPIs identified experimentally are stored in databases for the use of the scientific community. Databases can be differentiated by the degree of detail at which the interactions are described. The highest degree of detail corresponds to databases such as the PDB (229), where the atomic details of the interaction are described. However, most of the identified PPIs are binary interactions without atomic details. PPI databases can also differ in whether they only contain experimentally determined PPIs or if they also contain computationally predicted interactions. Databases such as STRING (230), FPCLASS (231), IID (232) contain both experimental and computationally predicted PPIs. Other PPI databases include interactions between proteins and other molecules such as IntAct (233). Other PPI databases focus in specific types of proteins such as MatrixDB (234) (focused on proteins from the extracellular matrix) or InnateDB (235) (focused on immune related proteins).
Table 9. Databases of protein-protein interactions.
1.6 – Methods to characterize molecular interactions with atomic resolution X-ray crystallography X-ray crystallography is the most widely used method to determine molecular interactions with high resolution. Around 88% of all structures stored in the PDB (229) have been determined by X-ray crystallography (last accessed September 2020). X-ray crystallography is an experimental technique that consists in
Name Data contained
BioGRID (236) Experimental data.
DIP (237) Experimental data.
FPCLASS (231) Computational data.
HPRD (238) Experimental data.
IID (232) Computational and Experimental data.
InnateDB (235) Experimental data, focused on immune-related PPIs.
IntAct (233) Experimental data.
IrefWeb (239) Experimental data.
MatrixDB (234) Experimental data, focused on extracellular matrix PPIs.
MINT (240) Experimental data.
STRING (230) Computational and Experimental data, includes functional protein–protein associations.

34
directing an X-ray beam to a crystallized macromolecule. This will give way to an X-ray diffraction pattern that will be informative for the electronic density of the macromolecule. From this information, we can identify the position of the different atoms and bonds (241). On the other hand, X-ray crystallography has limitations such as providing structures with not biologically relevant contacts (242) or that do not represent in vivo conditions. X-ray crystallography cannot be applied to all macromolecules, since not all macromolecules can be crystallized (243). Nuclear magnetic resonance spectroscopy Nuclear Magnetic Resonance (NMR) takes advantage of the interaction between the magnetic movement of atom nuclei and a magnetic field. By applying pulses of magnetic fields, the nucleus of the macromolecule under study will get excited leading to chemical shifts. The analysis of these chemical shifts is used to calculate the distances between atoms and finally build a 3D structure of the macromolecule (244). This technique is applied to molecules in solution, that in comparison with X-ray crystallization is more similar to an in vivo situation. Around 7% of all structures stored in the PDB are obtained by NMR (PDB). Besides, NMR can be used to analyze interfacial residues and their solvent accessibility with amide hydrogen exchange experiments (245). On the other hand, NMR cannot be applied to big complexes (246). Cryo-EM tomography In cryo-electron microscopy (Cryo-EM) tomography, the macromolecule of interest is inspected by an electron microscope at cryogenic temperature. This method provides structures of low resolution in comparison with the two previous methods (243). However, cryo-EM is very useful when providing the size and shape of complex macromolecules, as it could be a protein macrocomplex or an enhanceosome. Computational modeling From all the methods in this list, computational modeling is the only one that is not experimental. However, is a method that can provide models of molecular interactions with high resolution. We can classify these computational methods in three categories: comparative modeling, docking and ab initio modeling. Comparative modeling consists in using the structure of a close homologous as a template to reconstruct the structure of the macromolecule of interest (247). The theoretical principle supporting this approach is that structure is widely conserved between homologous proteins, even far homologous (i. e. the twilight zone) (248). This principle also applies when working with interactions of macromolecules. In the case of PDIs, the interacting conformation is conserved across homologous TFs, while the DNA molecule usually adapts to the

35
conformation set by the TF. For these cases we use templates of proteins interacting with DNA molecules (the comparative modeling of PDIs is extensively revised in the section of the thesis focused on PDIs). In the case of PPIs, the interacting conformation is conserved between pairs of homologous interacting proteins. For these cases we use templates of pairs of interacting proteins (157) (the comparative modeling of PDIs is extensively revised in the section focused on PPIs). The main limitation of comparative modeling is that there are not available templates for all proteins or interactions involving proteins. Docking consists in exploring the whole conformational space of two molecules based on surface complementarity. Docking programs take as input the separated structures of the interacting macromolecules (249). These structures can be obtained experimentally or modeled. In the case of proteins, these can be modeled using comparative modeling or ab initio approaches. In the case of DNA, it can be modeled computationally using the X3DNA suite (250). Docking programs generate a huge number of conformers for an interaction between macromolecules. Most of these conformers do not represent the native conformation of the interaction under study. Therefore, docking programs require scoring functions to score the generated conformers and then being able to distinguish near-native conformers from wrong conformers. Docking has been way more used to study PPIs than PDIs. Methods distinguishing the right conformer from the whole ensemble generated by docking have low accuracy (251). That is why comparative modeling is more reliable in order to get accurate models of macromolecular interactions. A recently discovered application of docking is to use the ensemble of conformers generated by docking to predict the binding affinity between pairs of interacting proteins (182,205,252,253). Ab initio modeling consists in the modeling of a structure without using the structure of a homologous protein, but only its amino acid sequence (254,255). This is challenging because the number of conformations that have to be explored is huge and because for each conformation free energies in solvent must be computed (255). The performance of ab inito approaches has been assessed by the CASP (Critical Assessment of Techniques for Protein Structure Prediction) competition (256). In CASP12 (2016) approaches based on the threading and assembly of small protein fragments, such as I-TASSER or ROSETTA, constituted the state-of-the-art methods for ab initio modeling (255–258). However, the performance of these methods is far from perfect (258–260). In the two last CASP editions, a deep learning based method called alphafold has achieved an outstanding performance, clearly surpassing competing methods (259,260). Alphafold uses deep learning to compute potentials from multiple sequence alignments. These potentials are used to predict distances and torsions between amino acids leading to the modeling of the protein structure (259,260).

36
1.7 – Motivation of this thesis This thesis tackles two main subjects: PDIs and PPIs, and how these affect transcriptional regulation. PDIs are fundamental to understand how TF find their DNA binding sites. Yet, the binding preferences of many eukaryotic TFs remain unknown (41). Experimental techniques to identify TF binding sites are expensive and time consuming (42). Besides their role in gene regulation, some TFs are potential bioengineering tools that could be used as endonucleases. This is the case of TFs from the C2H2-ZF family that are able to recognize long and specific DNA patterns, allowing to target the endonuclease activity to specific sites in the genome (261). To tackle PDIs, we developed tools to predict the binding preferences and to make structural models of PDIs. We first developed tools for all TF families. Soon we saw that the performance of our predictive tools was very poor for the C2H2 ZF family. Since the C2H2 ZF family has high structural variability and to their relevance as gene regulators and potential bioengineering tools we created specific predictive tools for this family (42). On the other hand, we developed ModCRE, a structure homology-modeling approach to predict TF binding to cis-regulatory elements. ModCRE joins our predictive methods for the C2H2 ZF family and for the other TF families. Besides, ModCRE includes an automated pipeline to create structural models of complex TF-DNA interactions. Regarding PPIs, these are fundamental to understand the regulatory effect of most TFs since most eukaryotic TFs carry out their regulatory role through the recruitment of co-factors (19). Knowing or estimating the affinity of such PPIs would help enormously the research on gene regulation, as well as knowing the effect of mutations affecting PPIs. However, experimental techniques providing this knowledge are expensive and time consuming (262). Therefore, the development of computational tools to complement experimental techniques is foremost. To tackle PPIs we developed MODPIN, a framework that models PPIs and assesses their binding affinity. We successfully applied MODPIN to predict the effect of mutations in PPIs (157). We also reviewed the role of non-interacting regions in PPIs and the methods that use them to predict PPI binding affinities.

37
2. Objectives
This thesis aims the following objectives:
1. To develop statistical potentials to study protein-DNA binding affinity.
2. To develop a computational pipeline to make structural models of PDIs.
3. To predict the DNA binding preferences of TFs as PWMs.
4. To develop a method to predict protein-protein binding changes of affinity caused by mutations.
5. To develop a computational pipeline to make structural models of PPIs.
The achievement of these goals comprises several milestones:
• Build and update databases of structural and experimental data on PDIs.
• Develop and update statistical potentials addressed as scoring functions for PDIs.
• Generate specific statistical potentials scoring functions for PDIs for TFs of the C2H2 ZF family using bacterial one-hybrid experimental data.
• Generate a computational pipeline to generate structural models of PDIs including binary interactions, interactions of several TFs within a DNA fragment and interactions involving TFs and nucleosome bound DNA.
• Building and analyzing PWMs out of statistical potentials scoring functions and structural models.
• Validate the PWMs with experimental motifs from JASPAR and Cis-BP databases.
• Study correlations between state-of-the-art scoring functions and their experimental binding affinity for PPIs.
• Predicting the effect of mutations on the loss/gain binding of PPIs
• Generate a computational pipeline to generate structural models of PPIs.

38

39
3. Results
3.1 - Methods to model and assess protein-DNA interactions PDIs are fundamental to understand gene regulation, especially those mediated by TFs. However, the DNA binding preferences for many TFs are unknown (41) and experimental techniques are expensive and time consuming (42). In this scenario the development of computational tools to fill the gap in TF knowledge can be very useful for the scientific community. Next, we expose our works on modeling and assessing PDIs. The two first works are focused on TFs from the C2H2-ZF family. We developed tools to model protein-DNA complexes and to predict TF binding sites as PWMs. We further applied these tools to study the case of CTCF, a C2H2-ZF protein with a relevant role in genome architecture. The last work covers all the main TF families. As we did for C2H2-ZF proteins, we developed tools to model protein-DNA complexes and to predict TF binding sites as PWMs.
Manuscripts presented in this section:
Fornes O, Meseguer A, Molina-Fernández R, Bonet J, Oliva B. ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements. (To be submitted) Meseguer A, Årman F, Fornes O, Molina-Fernández R, Bonet J, Fernandez-Fuentes N, et al. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. NAR Genom Bioinform [Internet]. 2020 Sep 1 [cited 2020 Sep 2];2(3). Available from: https://academic.oup.com/nargab/article/2/3/lqaa046/5866110 Meseguer A, Molina-Fernández R, Fernandez-Fuentes N, Fornes O, Oliva B. Short comment on: On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF.

40
3.1.1 – ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements
Fornes O, Meseguer A, Molina-Fernández R, Bonet J, Oliva B. ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements. (To be submitted)

41
ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements
Fornes O1,2†, Meseguer A1†, Molina-Fernández R1, Aguirre-Plans J1, Bonet
J1,3, Oliva B1*
1Structural Bioinformatics Lab (GRIB-IMIM), Department of Experimental and
Health Science, University Pompeu Fabra, Barcelona 08005, Catalonia, Spain
2Centre for Molecular Medicine and Therapeutics, BC Children's Hospital
Research Institute, Department of Medical Genetics, University of British
Columbia, Vancouver, BC V5Z 4H4, Canada
3Laboratory of Protein Design & Immunoengineering, School of Engineering,
Ecole Polytechnique Federale de Lausanne, Lausanne 1015, Vaud,
Switzerland
†O.F. and A.M. contributed equally to this work
*To whom correspondence should be addressed: [email protected]
ABSTRACT
The knowledge on transcription factor (TF) binding sites is key to
understand gene regulation. However, the binding preferences for most of
eukaryotic TFs are unknown. In this context, the development of
computational tools as a complement to experimental procedures for
characterizing TF-binding sites is foremost. In this work, we present
ModCRE, a structure homology-modeling approach to predict TF binding
to cis-regulatory elements. ModCRE combines structural information and
protein binding microarray data to predict the binding preferences of TFs
and model TF-DNA interactions. ModCRE was applied to the following
tasks: 1) discriminate bound from unbound PBM 8-mers; 2) predict
JASPAR profiles from experimental methods excluding PBMs; 3) predict
profiles of non-redundant TFs and compare them to the profiles of the
closest non-redundant homologs; and 4) model the structure of the INFβ
human enhanceosome. Thanks to an automated homology modeling
pipeline for TF-DNA complexes, ModCRE could be applied at large-scale
in order to fill the existing gaps in gene regulatory networks.

42
KEYWORDS Transcription factors, structural bioinformatics, gene regulation. Introduction
Knowledge of transcription factor (TF) binding sites, the locations at which TFs bind to DNA in the genome, is key to understanding how genes are regulated. Characterizing TF-binding sites is foremost in order to understand how genes are regulated. TF-binding sites are represented by probabilistic models called position weight matrices (PWMs) that represent the diverse DNA sequences that can be recognized by a TF (1). In the past decade, the appearance of high-throughput techniques (2–6) has allowed the characterization of TF-binding sites at large-scale. However, experimental protocols are both laborious and difficult to apply, as it is suggested by the very small fraction of eukaryotic TFs that have been profiled (7). As an alternative, computational tools can be employed. A successful approach to predict PWMs for a TF is the structural analysis of its complex with DNA using statistical potentials (8). In this work, we present ModCRE, a structure homology-modeling approach to predict TF binding to cis-regulatory elements. ModCRE integrates structural information and protein binding microarray (PBM) data into statistical potentials. ModCRE also contains an automated homology-based pipeline to make structural models of TF-DNA interactions. By combining the modeling of TF-DNA interactions with statistical potentials, ModCRE is able to infer TF-binding sites as well as to detect TFs able to bind a particular DNA sequence. Methods Software The following software was used in this study: DSSP (version CMBI 2006) (9) to obtain protein structural features; X3DNA (version 2.0) (10) to analyze and generate DNA structures; matcher and needle, from the EMBOSS package (version 6.5.0) (11), to obtain local and global alignments, respectively; BLAST (version 2.2.22) (12) to search homologs of a given query (target) protein sequence; MODELLER (version 9.9) (13) to construct structural models; and the programs FIMO and TOMTOM from the MEME suite (14) to scan a DNA sequence with a Position-Weight Matrix and to compare two PWMs, respectively. CD-HIT has used to generate non-redundant sets of TFs (15). Databases Atomic coordinates of protein complexes are retrieved from the PDB repository (16) and protein codes and sequences are extracted from UniProt (January 2019 release) (17). We generate an internal database of structures with all the structures in the PDB containing TF-DNA interactions. Binding information of

43
TFs is retrieved from protein binding microarray (PBM) experiments from the Cis-BP database (version 2.00) (7). PBM experiments indicate the binding affinity between TFs and DNA 8-mers with the E-score value, that can go from -0.50 to 0.50. We classify PBM 8-mers into positive 8-mers (those binding to TFs with high affinity) are those with scores above 0.45, while negative 8-mers are those with scores below 0.37. Moreover, since PBMs do not specify whether an 8-mer is being recognized through the forward or reverse strand, we classified both strands of an 8-mer as either positive or negative. Unclassified 8-mers were considered dubious and discarded. Interface and triads of protein-DNA structures We define triads as a type of contacts between the protein and the double-strand DNA helix. Triads are formed by three residues: one amino acid and two contiguous nucleotides of the same strand. The distance associated with a triad is defined by the distance between the Cβ atom of the amino acid residue and the average position of the atoms of the nitrogen-base of the two nucleotides plus their complementary pairs in the opposite strand of the helix (8). The triad also has an associated amino acid residue number in the protein and a dinucleotide position in the DNA, defined by the sequence position of the first nucleotide of the dinucleotide (e.g. a triad with amino acid residue number p, dinucleotide in position q and associated distance d is represented as (triad, d, p, q)). Specific features can be added on a triad, defining an extended-triad (etriad). These features are 1) for the amino acid: hydrophobicity, surface accessibility and secondary structure (determined with DSSP); and 2) for the dinucleotide: nitrogenous bases, the closest strand, the closest groove and the closest chemical group to the amino acid. Statistical potentials We use the definition of statistical potentials described by Feliu et al (18) and Fornes et al. (8) to define several scoring functions for the interaction between a protein and a DNA binding site using contact triads. We use the distribution at distances up to 30 Å of triads to calculate the statistical potentials. We create potentials using either bins or accumulative distance intervals. The total potential of an interaction is calculated as the sum of the potentials of all triads, or triads with their environmental features (etriads). In the case of etriads, the completeness of the reference dataset is not sufficient to sample all possible combinations. We use interactions from PBM to extend the number of interacting triads (see further details and supplementary methods). Besides, we transform the statistical potentials into Z-scores (see further), to simultaneously identify the best distance associated with a triad and the best pair formed by one amino acid and one dinucleotide.

44
Z-scores The optimal condition of a statistical potential often yields a minimum. However, the minimum is not necessarily negative. The variability of signs of the potentials affects the criterion of quality of the scores. We define Z-scores in order to follow a criterion that incorporates the sign. We wish that the Z-score identifies simultaneously the best distance associated with a triad and the best pair formed by one amino acid and one dinucleotide. Consequently, we construct a Z-score function for any type of score using a standard normalization with respect the average of all amino acid types (see details in supplementary). Structural modeling of TF-DNA complexes Given a TF, different structural models of the TF in complex with DNA were obtained as follows: In step 1, the TF sequence was scanned for putative homologs in the set of known structures using BLAST. In step 2, we only used as templates those BLAST hits ensuring an alignment with enough percentage of sequence identity (above the twilight-zone curve (19)) and without gaps in the interface region (this last condition can be skipped if requested by the user). In step 3, the TF was realigned to the template sequences using MATCHER from the EMBOSS package (11). In step 4, each alignment was used to create an optimized structural model of the TF using MODELLER. This procedure can produce several models of the same TF. In addition, for TFs of the bHLH and bZIP families, since they recognize DNA as homo- or heterodimers, for each selected hit the dimer was modeled as follows: First, if the hit was already a homodimer, we used it as template. Otherwise, we searched the closest structural dimer to the hit in the set of dimers using TM-align and used the found dimer as new template. Then, the TF was realigned to both template chains (i.e. step 3) in order to generate a homodimer using MODELLER (i.e. step 4). The modeling of the DNA was done by applying the mutate_bases program of the X3DNA package (10) on the template structure. This procedure changes the nucleotides in a DNA structure without modifying the conformation of the DNA molecule. This makes that if the DNA adopts any conformation when interacting with a TF, this conformation is preserved upon modeling. This approach requires that all the templates used for TF-DNA modeling contain both a TF and a double stranded DNA molecule. The modeling of several TFs interacting with the same DNA molecule was done by superimposing models of binary interactions (either protein-DNA or protein-protein interactions) on top of a reference DNA molecule. TF-DNA models were obtained as explained previously. Protein-protein models were obtained by using MODPIN, a computational suite for the modeling and prediction of binding affinities for protein-protein interactions (20). The modeling of TFs interacting with nucleosome-bound DNA was made by superimposing models of TF-DNA binary interactions on top of the DNA molecule of the PDB structure 6FQ5. This

45
PDB structure contains a canonical nucleosome from which we extract the DNA molecule. All superimpositions were made using biopython (21). Use of experimental TF-DNA interactions to calculate statistical potentials We use a mapping function that associates the amino acids of the TF assayed in PBM experiments with the amino acids in a template structure. Similarly, we also require a mapping between nucleotides in positive DNA 8-mers from PBM experiments and the nucleotides in the template structure. The mapping of amino acids is done according with a sequence alignment between the assayed TF and the template TF. This alignment is provided by BLAST when searching for templates in our database of structures. The mapping of nucleotides is done by aligning the DNA 8-mers with the DNA nucleotides in the template structure as follows: First, we constructed a multiple sequence alignment (MSA) around the most dominant DNA 8-mer of the TF according to the PBM (i.e. the DNA 8-mer with the highest E-score). Using that DNA 8-mer as seed, the other positive DNA 8-mers of the TF were incorporated to the MSA if: 1) they were single-nucleotide variants of seed P8M; or 2) they included a continuous gap of a maximum of 2 nucleotides at 3’ or 5’ side and no mismatches with the seed P8M. Second, if the sequence of the core DNA interface of the template was found among the DNA 8-mers of the MSA, the DNA 8-mers of the MSA could be realigned to it. Otherwise, the modeling using this template was aborted. Scoring TF-DNA binding First, we calculate the interface and extract all etriads associated with distances shorter than 30Å. Then, the score of the interaction is defined as the sum of the scores (i.e. a specific potential) of all etriads with their associated distances. The same approach is applied for Z-scores. We can obtain the score of a TF without knowing the structure of the TF-DNA binary complex if it can be modelled (see details in supplementary). Construction of PWMs using TF-DNA structural models Given the modelled structure of TF-DNA complex, we obtain the PWM using statistical potentials Z-scores. Since we have developed several types of statistical potentials scoring functions, we chose for each TF family the potential that optimizes PWM prediction (see methods: Optimizing PWM prediction parameters). Once the type of potential is chosen, we use the Z-score of the corresponding ES3DCdd scoring function, as defined in supplementary. We collect the set of etriads, their associated distances, and the associated amino acid and dinucleotide positions. We obtain a test set with all possible DNA sequences of the binding site. We calculate the score of any sequence of the test using the ZES3DCdd scoring function (see details in supplementary for a heuristic approach when the binding size is longer than 9 bases). We normalize the scores from 0 to 1 as indicated in the supplementary materials, equation 51. Then, we rank the normalized scores and select only the DNA sequences producing the top scores over a cut-off threshold (i.e. 0.95). This cut-off

46
threshold is also optimized for each TF family (see methods: Optimizing PWM prediction parameters). This produces an alignment, and we use it to calculate the PWM, which we name theoretical PWM. The prediction of PWMs for the C2H2 ZF family was made with the statistical potentials from a previous work (22). Optimal conditions to predict PWMs using the structure of a TF (grid search) We optimized the parameters to predict PWMs using structural models (ModCRE) specific for each TF family and also for a general TF or unknown/undefined DNA binding protein. These parameters are: 1) the type of frequencies used to calculate the potentials (either by bins or accumulated); 2) modifying the frequencies to include the interactions of residues introduced with the probability of substitution (we apply a Taylor polynomial approach to modify the statistical potential); 3) the dataset of structures to calculate the potentials, using only structures from PDB or with models that include data on binding from PBM; 4) using all known protein structures that bind DNA or only those specific of the same family and fold; 5) the maximum contact distance to include the interacting residues of the interface; and 6) the cut-off threshold used to include DNA sequences in a multiple sequence alignment of the DNA binding site and get the PWM. The selection of best parameters is illustrated in supplementary figure S1A. The cut-off threshold varies between 0.7 and 1.00 in steps of 0.01. The selection of the cut-off threshold is illustrated in supplementary figure S1B. We calculate the accuracy of each parameter combination with the members of the same TF family by comparison of the predicted PWMs with the PWM motif of the Cis-BP database (7). Then we select the parameters that maximize the accuracy of the whole family with the following conditions: 1) maximum number of significant good predictions according to TOMTOM score; 2) best TOMTOM scores when a similar number of significant solutions are achieved; and 3) the lowest value of the threshold, when several similar solutions are obtained. Results Analysis of the statistical potentials We have constructed several statistical potentials to describe the interaction between TFs and DNA molecules. We first created a potential that could be applied to any protein interacting with DNA, called general potential. Then we created specific potentials for each TF family. To create family specific potentials, we had to split the input structural data into the different families. We first created potentials that only contained structural data from the PDB. However, these potentials could not cover the entire spectrum of amino acid – dinucleotide contacts due to data scarcity. To overcome this limitation, we included PBM data into our potentials. We modeled TF-DNA interactions described by PBM and added these models to the pool of structural data used to compute the potentials. To further enlarge the set of amino acid –

47
dinucleotide contacts used to create potentials we applied the Taylor’s polynomial approach to the classic definition of potential. On top of this, we defined distance intervals that could be accumulative or bins, giving way to two different types of potentials. Finally, we applied a Z-score modification on top of the potentials here described. To sum up, we developed general and family specific potentials that contain structural data with or without PBM data; and that can be calculated with or without Taylor’s approach and with bins or accumulative distance intervals. As an example of statistical potential, we have selected the interactions of arginine (Arg) and phenylalanine (Phe) with the dinucleotide guanine-cytosine (GC). We selected these amino acids because Arg is a classical amino acid positively charged found involved in unspecific protein-DNA contacts, while Phe is a hydrophobic residue with a high tendency to make long distance protein-DNA contacts. Figure 1A-B shows the general PAIR potentials obtained with PDB structures for the interaction of dinucleotide GC with Arg and Phe, respectively. Here we can see that scores are negative for Arg interacting at close and medium distances, while for Phe scores get negative only at long distances. Figure 1C-D shows the homeodomain family specific PAIR potentials obtained with PDB structures for the interaction of dinucleotide GC with Arg and Phe, respectively. At these figures we cannot see any scoring trend across distance, probably because the potentials miss structural data to achieve precise scoring. This is supported by the fact that when we include PBM experimental data in these same potentials, clear scoring trends appear. This can be seen in figure 1E-F, where we show the homeodomain family specific PAIR potentials obtained with PDB structures and PBM experiments for the interaction of dinucleotide GC with Arg and Phe, respectively. Here we can again see the tendency of Arg for negative scores at close and medium distances while Phe yields negative scores at long distances. Note that Phe lowest score takes place at a distance around 20Å, while for general potentials the lowest score for Phe took place at distances of 30Å. This is an example of how family specific potentials may have their own structural trends that differ from the general potentials. Finally, figure 1G-H shows the general PAIR potentials obtained with PDB structures for the interaction of Arg with dinucleotides AG and CT, respectively. This last figure shows that potential is symmetric for the reversed dinucleotide. Figure 2 shows the increase of different types of contacts produced with the help of PBM data with respect to those obtained only with PDB structures. To capture the increase in contacts as well as the increase in diversity of these contacts we designed a contact abundance score (see methods). We see that for most TF families contact diversity increases when including PBM experimental data. In figure 2 we also show contact heatmaps for the TF families AP2, Homeodomain and bHLH. These heatmaps show how we populate the different amino acid – dinucleotides contacts when using only PDB structures or when using PDB structures and PBM data. For AP2 and Homeodomain TF families, PBM data substantially increases the abundance of contacts, while for bHLH this increase is more subtle. However, in all the cases

48
PBM data provides data that enables new amino acid – dinucleotides contacts to be populated. Supplementary figure S2 shows in detail a contact heatmap for the Forkhead TF family using PDB structures and PDB structures combined with PBM data. Supplementary figure S2 also shows the granularity of features covering the interaction between valine and adenosine-cytosine. Evaluation of statistical potentials by classifying DNA 8-mers We tested different statistical potentials to discern positive from negative 8-mers in PBM data (see Methods). Since some of our potentials were obtained using this same PBM data we also used this as a self-validation to ensure that PBM data was correctly included into our potentials. PBM data was extracted from the Cis-BP database, build 2.00 (7). For each positive 8-mer from PBM data we made a homology model and analyzed its extended triads (see Methods). TFs sharing more than 70% of sequence identity were excluded to avoid redundancy in the dataset. Each of these models was scored using different statistical potentials scoring functions. Also, all possible DNA 8-mers were threaded into the model structure and scored as well. For each positive 8-mer we obtained its corresponding statistical potentials score plus the scores given to 100 negative 8-mers selected at random. We obtained precision-recall curves for the capacity of statistical potentials to differentiate positive from negative 8-mers. We represent the performance of the different potentials as area under the precision-recall curve (AUPRC) (see figure 3). We tested potentials obtained using PDB data or combined PBM and PDB data; general or specific for different TF families; and obtained with or without using Taylor’s polynomial approach. All potentials tested have been obtained with accumulative distance intervals. We evaluated all TF families for which the Cis-BP database has PBM data. However, not all TF families have the same amount of PBM data available at Cis-BP. That is why we restricted the analysis for the families that had PBM results for at least 10 different TFs. Results for the discarded families can be seen in the supplementary figure S4. Figure 3 shows the results of our performance at classifying DNA 8-mers. For all transcription factor families, there is at least one statistical potential that performs clearly better than random. Random is set at an AUPRC of 0.01, as it corresponds for an unbalanced set of 1 positive per 100 negatives. There is a trend for potentials including both PDB and PBM data to perform better than potentials that only include PDB data. This suggests that the increase of data provided by PBM experiments improves the predictive power of our potentials. Also, there is a trend for potentials specific for TF families to perform better than general potentials. This suggests that if we consider the specific features each TF family has into our potentials, the predictive power increases. We can see that AUPRC values can change considerably between the different TF families analyzed. One family displaying a singular behavior are C2H2 ZF proteins. The C2H2 ZF family is the family with the worst performance in this analysis. We hypothesize that this happens because due to the structural features of the C2H2 ZF family, the inclusion of PBM data into potentials for this

49
family is prone to error. The DNA binding domains of the C2H2 ZF proteins are made of several small domains called zinc fingers. Each zinc finger recognizes 3 nucleotides, and by placing many zinc fingers together C2H2 ZF proteins are able to recognize complex DNA sequences (22). For C2H2 ZF proteins with more than 3 zinc fingers it is not possible to know what is the combination of zinc fingers that is interacting with the DNA 8-mer in a PBM experiment. Therefore, the models we create from PBM data to increase the amount of amino acid – dinucleotide contacts are imprecise. We solved the limitations related with the C2H2 ZF family by creating new potentials using bacterial one-hybrid data as a source of experimental data, this is explained in previous work (22). Prediction of experimental PWMs We tested our statistical potentials to predict PWMs from TFs present at the JASPAR database (23). We used the latest version of JASPAR2020 (last consulted in September 2020) consisting of 1934 PWMs. From the entire dataset of JASPAR, we discarded the TFs that had more than one PWM in the database, this left us with 1364 TFs. Finally, we made predictions only for the TFs for which we could obtain homology models, 1210 TFs in total (88.7% of the analyzed dataset). For each prediction we obtained the corresponding amino acid sequence from UniProt and used it to obtain 100 homology models. These models were made using as many templates as possible. If not 100 templates were available, this number of models was obtained by generating different models with the same template (see methods: Structural modeling of TF-DNA complexes). For each model we obtained a predicted PWM (see Methods: Construction of PWMs using TF-DNA structural models). We compared the 100 predicted PWMs with the experimental PWM using TOMTOM (14). From each of these comparisons, we obtained the P-value provided by TOMTOM that we used as a measurement of similarity between predicted and experimental PWMs. At the end, for each analyzed TF we have a distribution of 100 P-values. In figure 4 we show the average -Log10(P-value) for each analyzed TF from the JASPAR database. We can see that performance can change considerably between the different TF families or even within the same family. For some TF families, almost all averaged predictions have P-values below 0.05, while for others TF families most predictions are above the 0.05 threshold. Interestingly, if we compare the results of averaging the 100 predicted PWMs with the results of the best PWM from the 100 predictions (see supplementary figure S6), the results improve considerably. This indicates that for most of the analyzed TFs (74.8%) we are able to predict at least one PMW whose comparison P-value is below the 0.05 threshold. The potentials used to predict the PWMs of the C2H2 ZF family are no longer the PBM-based potentials used in figure 3, but the bacterial one-hybrid based potentials developed in a previous work (22). The TF families shown in figure 4 have PDB data for at least 10 different TFs. Results for the discarded families can be seen in the supplementary figure S5.

50
Comparing ModCRE to the nearest neighbor approach We compared our predicted PWMs with the PWMs obtained by another bioinformatical method: the nearest neighbor approach (also called closest homolog approach). The nearest neighbor approach consists of taking the experimental PWM of the closest homolog available for a TF of interest (7). The performance of this approach depends on the degree of homology between the two TFs involved: close homologs are supposed to have more similar DNA binding domains and this should be reflected in a higher similarity of their PWMs. On the other hand, distant homologs are expected to have less similarity in their PWMs. To compare our predictions with the nearest neighbor approach we designed the next experiment: For each TF of the PBM data in the Cis-BP database used as query we compare its sequence with the rest of TFs using MMseq2. We split the matches into subsets of TFs which sequence aligned with the query with a maximum sequence identity (%ID). Then, we analyze the results in bins between 90% and 15%. Using the results of each bin, we predict several PWMs using ModCRE and the potential templates (see Methods: Construction of PWMs using TF-DNA structural models) and another using the nearest neighbor approach (this can also be a collection if we use more than one neighbor). Once a PWM, or a collection of PWMs is predicted, we rank all the PWMs in the dataset according to the score of TOMTOM (14). We identify the ranking position (or positions, in the case of a collection) of the experimental (true) PWM of the query TF. Out of this rank we obtain a score (see supplementary methods) that is directly proportional to the accuracy to find the correct PWM among the top ranking positions. Figure 5 shows the results of our comparison with the nearest neighbor approach. We see that the nearest neighbor approach performs better than ModCRE at high percentages of sequence identity. As the degree of identity decreases, the performance of the nearest neighbor decreases as well, while the performance of ModCRE remains stable. For some TF families ModCRE outperforms the nearest neighbor approach at identity percentages around the 50% (forkhead, nuclear receptor, SOX). For other TF families ModCRE only outperforms the nearest neighbor approach at low identity percentages (C2H2 ZF, ETS, Homeodomain). Finally, for some TF families ModCRE never achieves to outperform the nearest neighbor approach (bHLH, bZIP). Using the rank-enriched approach to identify TF binding motifs The rank-enriched approach takes advantage of using a collection of PWMs of a single TF. Consider the problem from the previous result section: given a database of PWMs and a TF belonging to this database, we want to identify the PWM of this TF using only information from its homologs. We can either use the nearest neighbor approach and work with one or several PWMs of the neighborhood of TFs, or to use ModCRE to predict a collection of PWMs.

51
As we did in the previous results section, we split the results in bins for a maximum %ID between the query and the neighbours/templates. We do the ranking as above for each predicted PWM within the collection. Next, we calculate the enrichment of solutions (i.e. those PWMs thar are more frequently selected with the predicted PWMs). Finally, we obtain a new ranking based on the top solutions with higher enrichment. As above, the ranking positions were transformed into scores using the same formula that we used in the nearest neighbor analysis. For more information on the prediction by enrichment approach check the supplementary methods. We name this approach as “rank-enriched” solutions. Figure 6 shows our results for the evaluation of the rank-enriched approach. We see that both approaches, nearest neighbor and ModCRE, have a high degree of success if we apply them in combination with the rank-enriched approach. The rank-enriched approach increased the performance of our predictions as it can be seen when comparing figures 5 (without rank-enriched approach) and 6 (with rank-enriched approach). As we saw previously, the nearest neighbor tends to perform better at high %ID bins, while ModCRE has a more stable performance as identity percentage decreases. In this analysis, most TF families show a better performance for ModCRE than for the nearest neighbor approach at medium or low identity percentages. By comparing these results with the ones in figure 5 we can conclude that when applying the rank-enriched scoring approach the performance of ModCRE improves almost for all bins in comparison with the nearest neighbor approach. Scanning DNA sequences and modeling cooperative TF-DNA interactions We developed an automated pipeline that, given a DNA sequence and a specie, is able to find and model TF binding sites according with our predictive methods. This approach is based on the idea that homologous proteins tend to have similar PWMs. This pipeline starts with the search of TF binding sites using of a precomputed database of PWMs. These PWMs are derived from experimental structures from the PDB (16). For each of these PDB structures, we identify homologous sequences in the UniProt database (17). Once TF binding sites are identified, these can be associated to the corresponding homologous proteins belonging to the specie that we are interested in. For each homologous protein at a TF binding site, we obtain a statistical potentials score. This score may help us to identify what homolog protein is more likely to be bound at a particular TF binding site. This pipeline also allows to visualize protein-protein interactions involving TFs and transcriptional co-factors as a network. Interactions between TFs and transcription co-factors are retrieved from the TcoF-DB database (24). All the interactions identified by this pipeline, both protein-DNA and protein-protein interactions, can be modeled using our homology modeling pipeline. Besides, we can integrate all these interactions into a single model, allowing to show several proteins interacting with the same DNA molecule (see methods: Structural modeling of TF-DNA complexes).

52
We used this pipeline to model the structure of the INFβ enhanceosome (figure 6). The INFβ enhanceosome is the ensemble of TFs and Cis-regulatory elements that come together in the enhancer of the INFβ gene (25). The TFs that come together at the INFβ enhanceosome are ATF-2, Jun, IRF-3, IRF-7, NFKβ-1 and RelA. By having knowledge of the TFs implied in the INFβ enhanceosome and the relative order in which they bind to the DNA, we were able to recreate a structural model very similar to the experimental one provided by Panne (25) (see supplementary figure S8). The main difference between Panne’s model and ours is that we identified the binding sites belonging to IRF-7 as binding sites for IRF-3. This difference happened because IRF-7 did not pass our homology threshold when assigning what TFs were suitable homologs for each TF binding site. Discussion TF-binding specificities are foremost to understand gene regulation. Still, the binding preferences for most eukaryotic TFs are unknown (7). In this regard, the development of computational tools as a complement to experimental procedures is foremost. In this work we have presented ModCRE, a structure homology-modeling approach to predict TF binding to cis-regulatory elements. ModCRE integrates structural and PBM data into statistical potentials and combines homology modeling with statistical potentials to predict PWMs. As we have seen by our comparisons with the Cis-BP and JASPAR databases, ModCRE is a successful approach to predict PWMs for TFs. However, the application of ModCRE has limitations. Its main limitation is that it can only be applied to TFs for which we have a structure interacting with DNA. This structure can be either experimental or obtained via homology modeling. The scarcity of experimental structures, which can be applied directly or as templates for homology modeling, is limiting the amount of TF to which we can apply ModCRE. We studied the applicability of ModCRE by looking at for how many TFs from the UniProt database we could obtain homology models, the results for this analysis are shown in supplementary figure S3. This analysis shows that ModCRE can be applied to most TFs in the UniProt database. Similarly, when predicting PWMs from the JASPAR database we could apply ModCRE to 88.7% of the analyzed dataset. ModCRE’s homology modeling pipeline can be a valuable resource for the scientific community, independently from its implication in the prediction of PWMs. Structural models for TF-DNA interactions can provide fundamental information to understand TF function and behavior. Our pipeline can model complex TF-DNA interactions involving cooperative TF-DNA binding (as can be seen in figure 6), protein-protein interactions involving TFs and transcription co-factors or TFs interacting with nucleosome-bound DNA. Interestingly, a recent work linked the capacity of TFs to interact with nucleosome-bund DNA with the conformation TFs undergo when interacting with nucleosome-bound DNA (26). We hypothesize that our homology modeling pipeline could be used to

53
interrogate the structural basis of TF interaction with nucleosome-bound DNA. Finally, ModCRE’s modeling pipeline can be of great help to understand the effect of mutations that happen in TFs. Just by modeling the TF, we can see if the mutated amino acid is in close contact with DNA. Also, by predicting the PWM of the mutated TF, we can estimate the relevance of the mutation in terms of TF-DNA binding. One strength of ModCRE is that allows to obtain a collection of PWMs for a single TF. Theoretical PWMs are obtained from structural models. Therefore, ModCRE can make different structural models of the TF of interest using different templates. The structural variability in this collection of models will be reflected in the variability in the collection of PWMs. As we saw when presenting the rank-enriched approach, working with a collection of PWMs can increase substantially the performance when identifying TF binding motifs. We hypothesize that this principle can be used also to identify TF binding sites across DNA sequences. A collection of PWMs could be used to find binding sites that match not one, but most of the PWMs in the collection. This looks as a promising and fast strategy to overcome the big number of false positives that are found when scanning a DNA sequence with one PWM (27,28). Using a collection of PWMs is in consonance with the idea that TFs can interact with the DNA adopting different conformations (29). We have seen that ModCRE can have better predictive performance for some TF families than for others. Even in some cases, ModCRE can have variability within the same TF family or even when analyzing the same TF. This last case happened when predicting PWMs from the JASPAR database: for one single TF we were making both accurate and inaccurate predictions. However, if we were able to choose the best prediction, we would be able to make significantly accurate predictions for 74.8% of the JASPAR dataset. We hypothesize that by analyzing features of the models used to create the PWM and of the PWMs themselves we can differentiate reliable predictions from non-reliable predictions. Further work will be needed to confirm or reject this hypothesis.

54
References 1. Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory
elements. Nat Rev Genet. 2004 Apr;5(4):276–87.
2. Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science. 2007 Jun 8;316(5830):1497–502.
3. Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006 Nov;24(11):1429–35.
4. Hallikas O, Taipale J. High-throughput assay for determining specificity and affinity of protein-DNA binding interactions. Nat Protoc. 2006 Jun;1(1):215–22.
5. Meng X, Wolfe SA. Identifying DNA sequences recognized by a transcription factor using a bacterial one-hybrid system. Nat Protoc. 2006 Jun;1(1):30–45.
6. Deplancke B, Dupuy D, Vidal M, Walhout AJM. A Gateway-Compatible Yeast One-Hybrid System. Genome Res. 2004 Oct;14(10b):2093–101.
7. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell. 2014 Sep;158(6):1431–43.
8. Fornes O, Garcia-Garcia J, Bonet J, Oliva B. On the Use of Knowledge-Based Potentials for the Evaluation of Models of Protein–Protein, Protein–DNA, and Protein–RNA Interactions. In: Advances in Protein Chemistry and Structural Biology [Internet]. Elsevier; 2014 [cited 2019 Sep 12]. p. 77–120. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128001684000044
9. Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–637.
10. Lu X-J, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3(7):1213–27.
11. Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics. 2000 Jun 1;16(6):276–7.
12. Altschul SF, Gertz EM, Agarwala R, Schäffer AA, Yu Y-K. PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009 Feb 1;37(3):815–24.
13. Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Current Protocols in Protein Science. 2016;86(1):2.9.1-2.9.37.
14. Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015 Jul 1;43(W1):W39-49.
15. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012 Dec 1;28(23):3150–2.
16. Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017 Jan 4;45(Database issue):D271–81.

55
17. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019 Jan 8;47(D1):D506–15.
18. Feliu E, Aloy P, Oliva B. On the analysis of protein-protein interactions via knowledge-based potentials for the prediction of protein-protein docking. Protein Sci. 2011 Mar;20(3):529–41.
19. Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999 Feb;12(2):85–94.
20. Meseguer A, Dominguez L, Bota PM, Aguirre-Plans J, Bonet J, Fernandez-Fuentes N, et al. Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions. Protein Sci. 2020 Aug 14;
21. Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009 Jun 1;25(11):1422–3.
22. Meseguer A, Årman F, Fornes O, Molina-Fernández R, Bonet J, Fernandez-Fuentes N, et al. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. NAR Genom Bioinform [Internet]. 2020 Sep 1 [cited 2020 Sep 2];2(3). Available from: https://academic.oup.com/nargab/article/2/3/lqaa046/5866110
23. Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020 Jan 8;48(D1):D87–92.
24. Schmeier S, Alam T, Essack M, Bajic VB. TcoF-DB v2: update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017 04;45(D1):D145–50.
25. Panne D. The enhanceosome. Curr Opin Struct Biol. 2008 Apr;18(2):236–42.
26. Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, et al. The interaction landscape between transcription factors and the nucleosome. Nature. 2018;562(7725):76–81.
27. Tran NTL, Huang C-H. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol Direct. 2014 Feb 20;9:4.
28. Medina-Rivera A, Abreu-Goodger C, Thomas-Chollier M, Salgado H, Collado-Vides J, van Helden J. Theoretical and empirical quality assessment of transcription factor-binding motifs. Nucleic Acids Res. 2011 Feb;39(3):808–24.
29. Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009 Jun 26;324(5935):1720–3.
56
Figures
Figure 1: Statistical energy profiles PAIR obtained with general and homeodomain specific potentials. Each plot displays profiles for the default potential computed using accumulative distance intervals with and without applying Taylor’s approach (in purple and red, respectively) and bins distance intervals with and without applying Taylor’s approach (in blue and yellow, respectively). (A) Profile of Arg–GC PAIR score using general potentials obtained from PDB structures. (B) Profile of Phe–GC PAIR score using general potentials obtained from PDB structures. (C) Profile of Arg–GC PAIR score using homeodomain specific potentials obtained from PDB structures. (D) Profile of Phe–GC PAIR score using homeodomain specific potentials obtained from PDB structures. (E) Profile of Arg–GC PAIR score using homeodomain specific potentials obtained from PDB structures and PBM experiments. (F) Profile of Phe–GC PAIR score using homeodomain specific potentials obtained from PDB structures and PBM experiments. (G) Profile of Arg–AG PAIR score using general potentials obtained from PDB structures. (H) Profile of Arg–CT PAIR score using general potentials obtained from PDB structures.

57
Figure 2: Contact abundance scores for the different TF families using only PDB structures and using PDB structures plus PBM experimental data. Heatmap plots showing contact abundance are shown for families AP2, Homeodomain and bHLH. For a detailed view and explanation of these heatmap plots check supplementary figure S2.

58
Figure 3: Heatmap of the performance classifying PBM 8-mers using statistical potentials. Performance is measured with the area under the precision-recall curve (AUPRC). This validation is done using an unbalanced set with 1 positive per 100 negatives. Random performance is indicated at a AUPRC value of 0.01. The evaluated potentials are shown on the horizontal axis, while the corresponding TF families are shown across the vertical axis. TF families included in this plot have available PBM data for 10 or more non-redundant TFs. TF are considered redundant if their percentage of identical residues is higher than 70%.

59
Figure 4: Boxplots showing the average accuracy of predicted PWMs for the proteins from the JASPAR database. For each JASPAR protein we make 100 different predictions. Accuracy in the predictions is assessed by the minus logarithm of the P-value of the comparison between the predicted PWM and its corresponding experimental PWM (P-values provided by TOMTOM from the MEME suite). For each JASPAR protein we average the corresponding -log10(P-values). Averaged -log10(P-values) are shown in logarithmic scale on the X-axis, while the different TF families are shown across the Y-axis. Each individual average -log10(P-value) is indicated as red dots across the boxplots. We indicate an acceptance threshold (P-value = 0.05) with a vertical red line. TF families included in this plot have available PBM data for 10 or more non-redundant TFs. TF are considered redundant if their percentage of identical residues is higher than 70%.

60
Figure 5: Results of the comparison between ModCRE and the nearest neighbor approach. We show results for all the analyzed TFs from the Cis-BP database (global) and for some specific TF families. The normalized score for the different approaches is indicated on the Y-axis, while the % of sequence identity is shown in the X-axis. The plots display lines united by dots indicating the mean value of each distribution. In the global plot we also show the number of analyzed TFs on top of each boxplot.

61
Figure 6: Results of the rank-enriched approach applied to ModCRE and the nearest neighbor approach. We show results for all the analyzed TFs from the Cis-BP database (global) and for some specific TF families. The normalized score of the different approaches is indicated on the Y-axis, while the % of sequence identity is shown in the X-axis. The plots display lines united by dots indicating the mean value of each distribution. In the global plot we also show the number of analyzed TFs on top of each boxplot.

62
Figure 7: Outline of the modeling pipeline for protein-DNA complexes applied to the modeling of the INFβ-enhanceosome. Protein-DNA complexes are predicted by scanning a DNA sequence with a database of structure-based PWMs. Protein-DNA complexes can be selected for modeling from the data displayed as binding sites across the DNA (down left) or as a network of protein-DNA and protein-protein interactions (down right). While the first option only allows to include protein-DNA interactions in the final model, the second allows to further include transcription co-factors in the final model.

63
SUPPLEMENATRY MATERIAL FOR “ModCRE: a structure homology-modeling approach to predict TF binding to cis-regulatory elements”
Fornes O1,2†, Meseguer A1†, Molina-Fernández R1, Aguirre-Plans J1, Bonet
J1,3, Oliva B1*
1Structural Bioinformatics Lab (GRIB-IMIM), Department of Experimental and
Health Science, University Pompeu Fabra, Barcelona 08005, Catalonia, Spain
2Centre for Molecular Medicine and Therapeutics, BC Children's Hospital
Research Institute, Department of Medical Genetics, University of British
Columbia, Vancouver, BC V5Z 4H4, Canada
3Laboratory of Protein Design & Immunoengineering, School of Engineering,
Ecole Polytechnique Federale de Lausanne, Lausanne 1015, Vaud,
Switzerland
Annex of results Assessing the similarity between JASPAR and CisBP JASPAR and CisBP are the two main databases that have been used in this work as a reference of TF binding data. We assessed to what extent these two databases may have discrepancies in their results. We found that JASPAR and CisBP shared 308 TFs belonging to the major TF families studied in this work. Since more than one binding motif can be obtained for the same TF, these TFs added up to 404 binding motifs. To assess how similar were the results for these common TFs, PWMs from TFs belonging to the same TF family we compared between the two databases using TOMTOM. From each comparison we obtained a P-value, from which we obtained a -Log10(P-value) score. We also included the predictions of ModCRE in this analysis by comparing our predictions to the results in the JASPAR database. Therefore, in this analysis we have two types of comparisons: ModCRE compared with JASPAR, and CisBP compared with JASPAR. Regarding ModCRE, for each TF we obtained 100 models and for each model we predicted one PWM. Then, the 100 PWM were compared with the corresponding experimental PWM from the JASPAR database. From each comparison we obtained a P-value, from which we obtained a -Log10(P-value) score. The results of this analysis are shown in the supplementary table S1. In this table we show the average -Log10(P-value) for each TF family analyzed. Interestingly, the similarity between JASPAR and CisBP was not as high as we expected. Even, for some TF families we found that ModCRE predictions were closer to the results of JASPAR than the results of CisBP. These cases are

64
highlighted in green in the supplementary table S1. These results are consistent with the idea that different experiments may produce different results. This may happen because of the biases that each experimental technique has, as well as the differences in experimental conditions. This is addressed in supplementary figure S7, where we show cases of agreement and disagreement between JASPAR and CisBP results and our predictions from ModCRE. This variability in our sources of reference can be addressed by ModCRE thanks to its capacity to generate collections of variable PWMs. We show an example of this in supplementary figure S8. In this figure we have two binding motifs from the JASPAR database for the same TF: POU6F1 from Homo sapiens. One of the experimental motifs recognizes the DNA sequence TAATTA, while the other recognizes the sequence TAATGAG. Interestingly, ModCRE is able to predict one PWM that matches the first experimental PWM (its preferred DNA sequence is TAATTA) and another PWM that matches the second experimental PWM (its preferred DNA sequence is T-AGAA). At this point we cannot know what of the two PWMs is more correct, since both fit one experimental PWM. Here we hypothesize that probably both PWMs are correct and that they should be used together as a collection to find TF binding sites. This is consistent with our results using the rank-enriched approach, where we saw that using collections of PWMs increases substantially our predictive power.

65
Supplementary figures
Figure S1: Outline of the parameter optimization for PWM prediction (Grid Search). We test all combinations between the next parameters: data type (PDB or PBM), data specificity (general or family), Taylor’s approach, distance intervals (accumulative or bins), maximum contact distance and acceptance threshold. (A) Representation of the different optimized parameters. (B) Representation of optimization of the acceptance threshold, optimal combination of parameters is highlighted in red.

66
Figure S2: Heatmap plots of the number of amino acid—dinucleotide contacts and their environments (etriads) at distance shorter than 30A in a logarithmic scale. Detailed view of a cell of the heatmap is shown in the right side of each heatmap. Each square inside the cell shows the number of extended-triads (in logarithmic scale) for a specific amino acid—dinucleotide (the example uses valine, Val and adenosine-cytosine, AC) and their environments. Amino acid environments are: hydrophobicity (P as polar, N non polar), surface accessibility (E if exposed, B if buried) and secondary structure (E for β-strand, H for helix and C for coil). Dinucleotide environments are: type of nitrogenous bases (U for purine, I for pyrimidine), closest DNA strand (F for forward, R for reverse), closest DNA groove (A for major, I for minor) and closest chemical group (B if phospho-ribose backbone atoms, N if nucleobase). (A) Extended-triads obtained from PDB structures for the forkhead family. (B) Extended-triads obtained from PDB structures and PBM experiments for the forkhead family.

67
Figure S3: Number of proteins (A) and domains (B) to which we can apply ModCRE to make PWM predictions. The different TF families are indicated in the vertical axis while the number of proteins or domains to which ModCRE can be applied is indicated in the horizontal axis in logarithmic scale. For each family we represent in blue all available sequences in UniProt that match the PFAM hidden markov model of that TF family. From all these proteins we represent in green the proteins that can be modelled without gaps in regions with defined secondary structure at the protein-DNA interface. In red we represent all the proteins that can be modelled only with gaps in regions with defined secondary structure at protein-DNA interfaces.
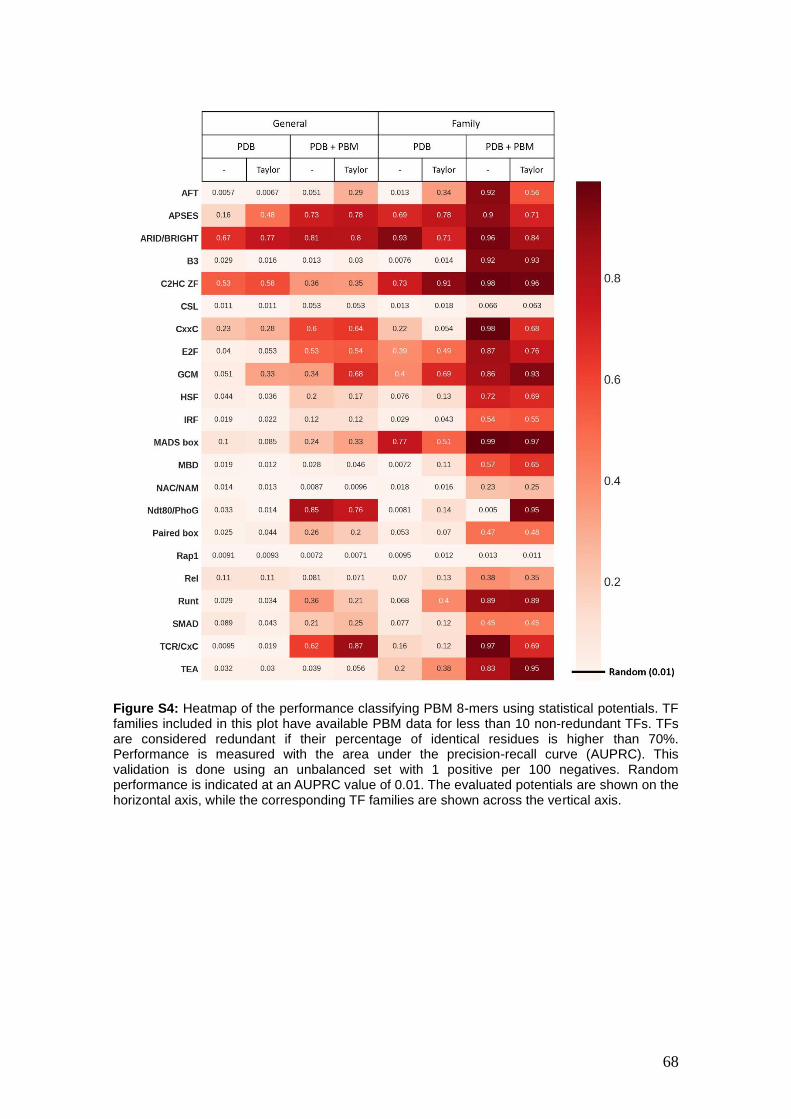
68
Figure S4: Heatmap of the performance classifying PBM 8-mers using statistical potentials. TF families included in this plot have available PBM data for less than 10 non-redundant TFs. TFs are considered redundant if their percentage of identical residues is higher than 70%. Performance is measured with the area under the precision-recall curve (AUPRC). This validation is done using an unbalanced set with 1 positive per 100 negatives. Random performance is indicated at an AUPRC value of 0.01. The evaluated potentials are shown on the horizontal axis, while the corresponding TF families are shown across the vertical axis.
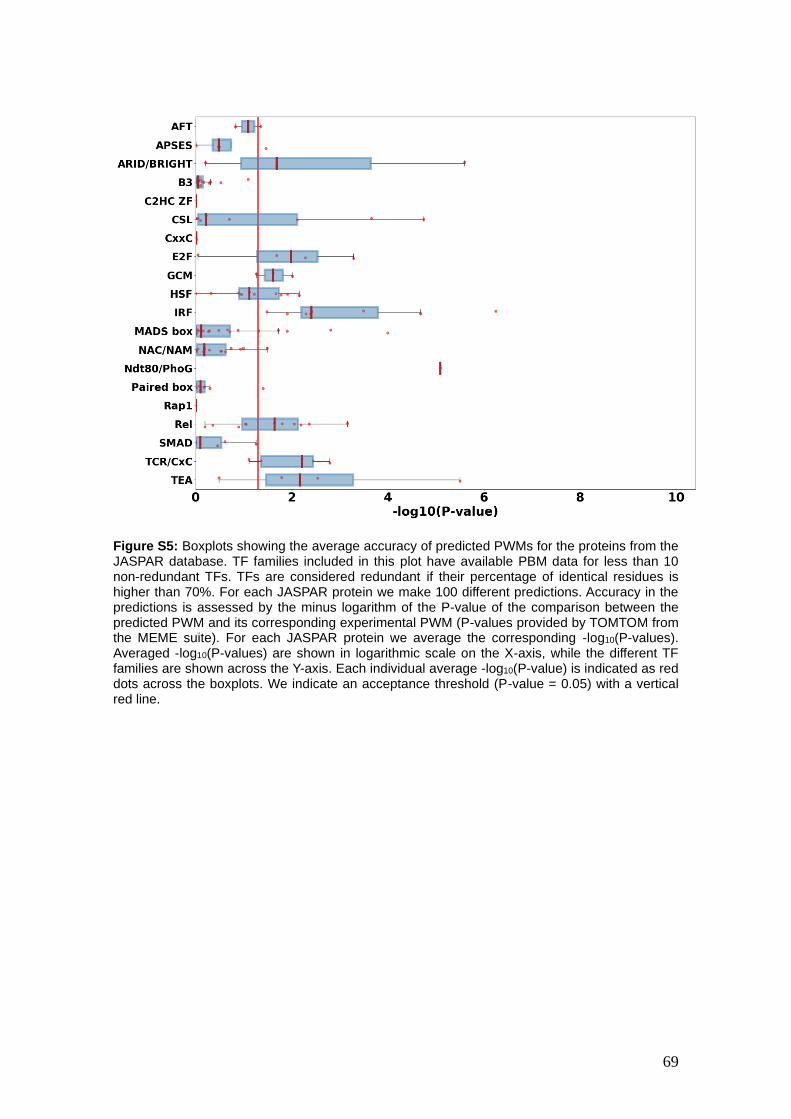
69
Figure S5: Boxplots showing the average accuracy of predicted PWMs for the proteins from the JASPAR database. TF families included in this plot have available PBM data for less than 10 non-redundant TFs. TFs are considered redundant if their percentage of identical residues is higher than 70%. For each JASPAR protein we make 100 different predictions. Accuracy in the predictions is assessed by the minus logarithm of the P-value of the comparison between the predicted PWM and its corresponding experimental PWM (P-values provided by TOMTOM from the MEME suite). For each JASPAR protein we average the corresponding -log10(P-values). Averaged -log10(P-values) are shown in logarithmic scale on the X-axis, while the different TF families are shown across the Y-axis. Each individual average -log10(P-value) is indicated as red dots across the boxplots. We indicate an acceptance threshold (P-value = 0.05) with a vertical red line.
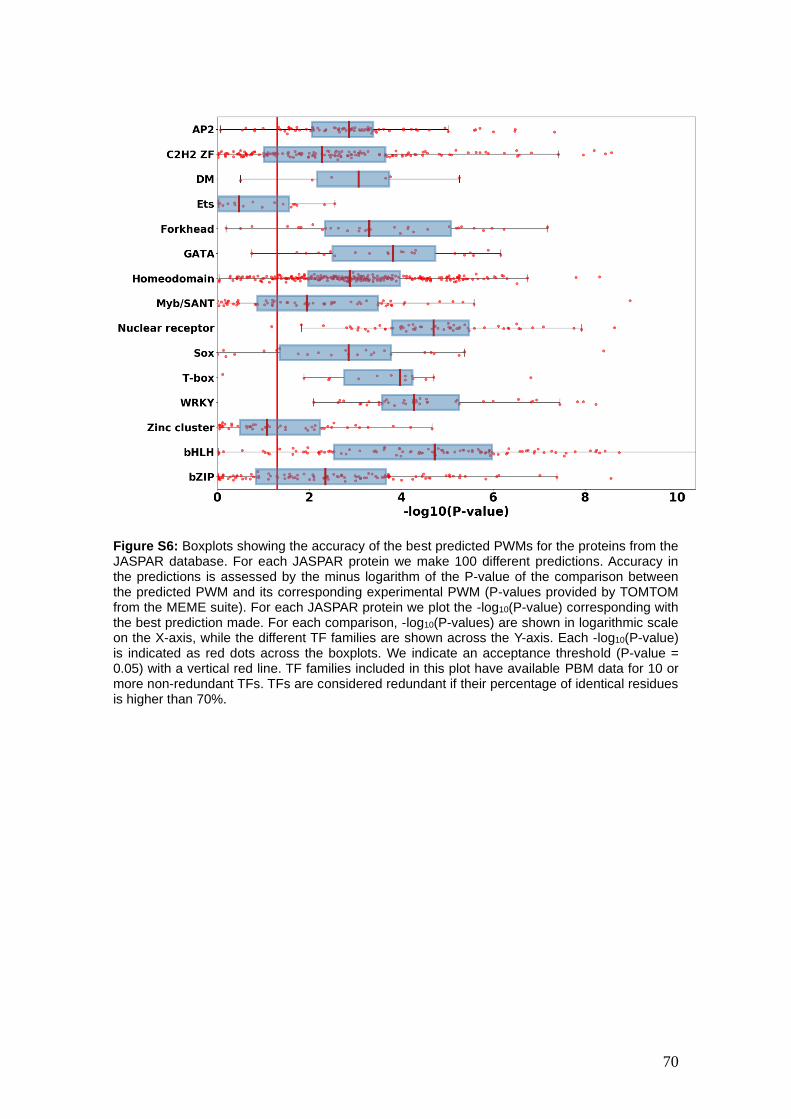
70
Figure S6: Boxplots showing the accuracy of the best predicted PWMs for the proteins from the JASPAR database. For each JASPAR protein we make 100 different predictions. Accuracy in the predictions is assessed by the minus logarithm of the P-value of the comparison between the predicted PWM and its corresponding experimental PWM (P-values provided by TOMTOM from the MEME suite). For each JASPAR protein we plot the -log10(P-value) corresponding with the best prediction made. For each comparison, -log10(P-values) are shown in logarithmic scale on the X-axis, while the different TF families are shown across the Y-axis. Each -log10(P-value) is indicated as red dots across the boxplots. We indicate an acceptance threshold (P-value = 0.05) with a vertical red line. TF families included in this plot have available PBM data for 10 or more non-redundant TFs. TFs are considered redundant if their percentage of identical residues is higher than 70%.
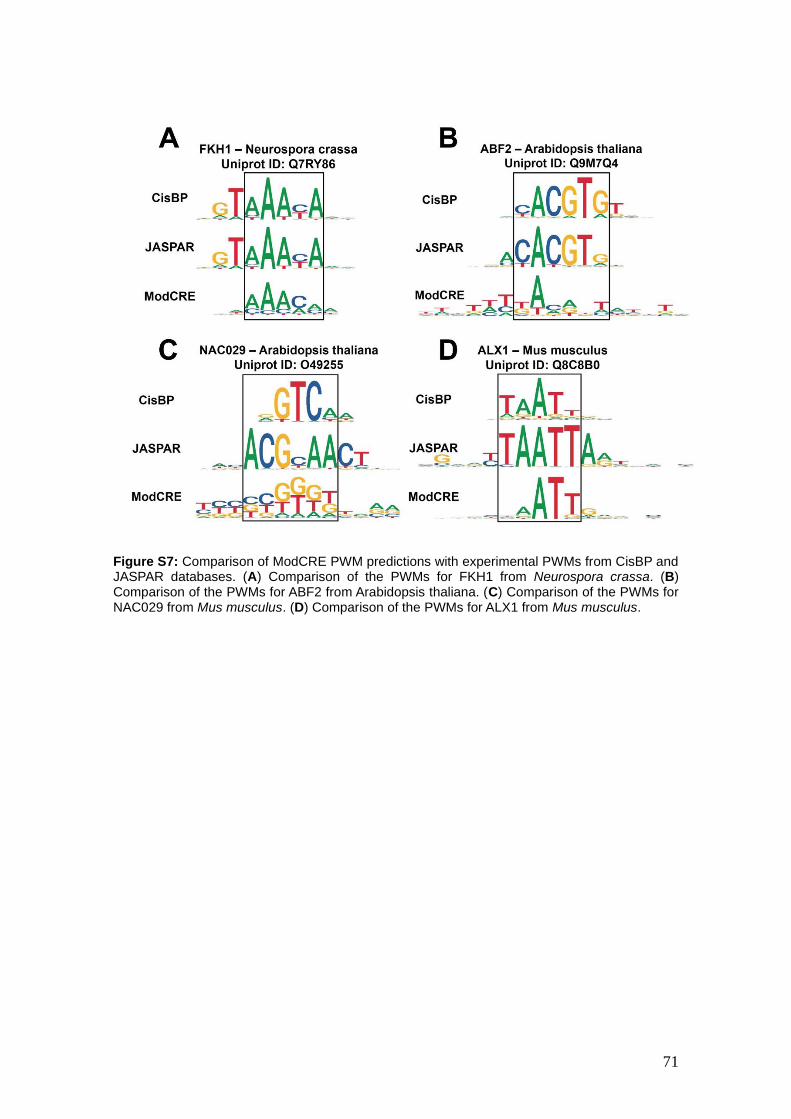
71
Figure S7: Comparison of ModCRE PWM predictions with experimental PWMs from CisBP and JASPAR databases. (A) Comparison of the PWMs for FKH1 from Neurospora crassa. (B) Comparison of the PWMs for ABF2 from Arabidopsis thaliana. (C) Comparison of the PWMs for NAC029 from Mus musculus. (D) Comparison of the PWMs for ALX1 from Mus musculus.
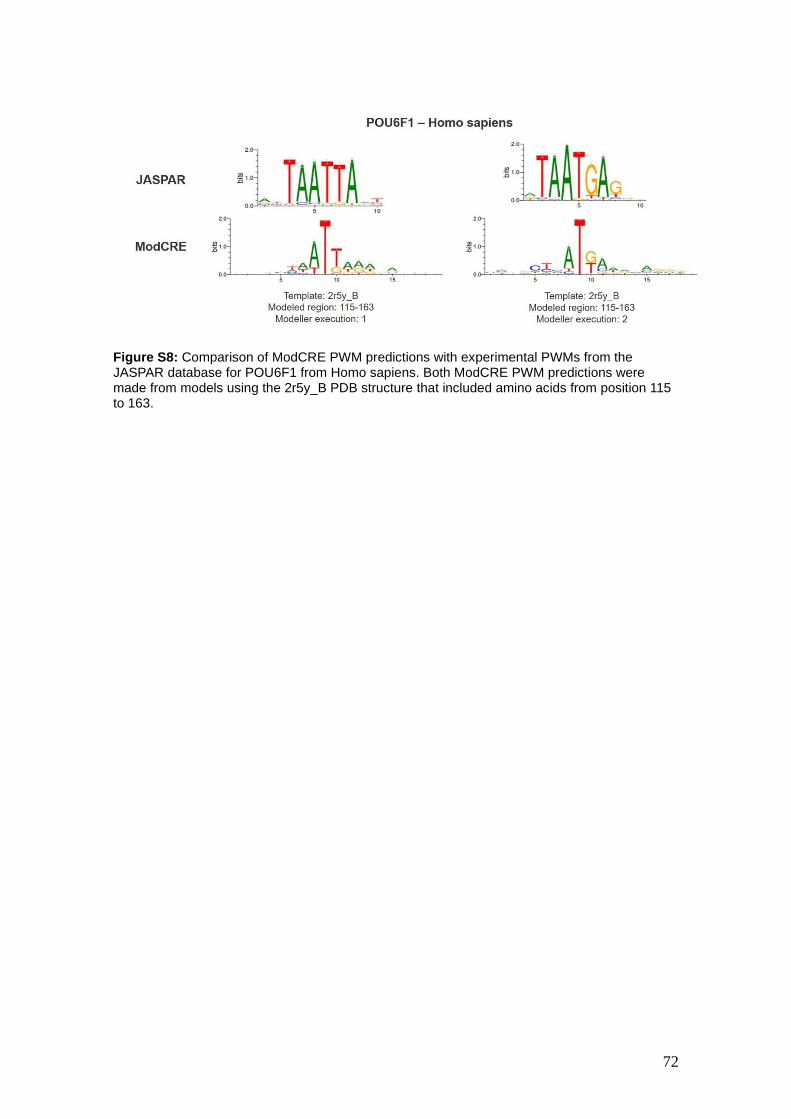
72
Figure S8: Comparison of ModCRE PWM predictions with experimental PWMs from the JASPAR database for POU6F1 from Homo sapiens. Both ModCRE PWM predictions were made from models using the 2r5y_B PDB structure that included amino acids from position 115 to 163.

73
Figure S9: Comparison of our structural model of the INF-b enhanceosome (in orange) and the model provided by Panne D (25) (in blue).

74
Table S1: Results of our comparison of PWMs between ModCRE, JASPAR and CisBP for each TF family. In this table we only analyze TFs that are present at both JASPAR and CisBP databases. It includes the names of TF families, the number of TFs in the family and the number of motifs in the family (see that for one TF we can have more than one motif). Average ModCRE shows the average -Log10(P-value) of the comparisons between ModCRE predictions and JASPAR PWMs. Average CisBP shows the average -Log10(P-value) of the comparisons between CisBP PWMs and JASPAR PWMs. We highlight in green the cases for which ModCRE predictions outperformed CisBP experimental results.
TF family #TFs #Motifs Average ModCRE Average CisBP
AP2 2 3 0,018 0,467
B3 1 1 0,350 0,111
C2H2_ZF 48 52 1,152 4,870
DM 4 5 3,497 5,208
Ets 16 17 2,772 9,826
Forkhead 14 15 3,544 6,994
GATA 5 7 1,664 1,412
GCM 2 2 2,526 5,930
HSF 3 3 1,442 1,671
Homeodomain 101 119 2,611 4,483
Homeodomain-POU 15 17 1,229 5,926
Homeodomain-Paired_box 23 32 1,036 2,919
IRF 8 10 3,274 3,651
MADS_box 1 2 0,466 0,464
Myb/SANT 4 4 0,836 0,689
Nuclear_receptor 9 19 6,524 9,211
Paired_box 1 2 2,170 5,543
Rel 1 1 9,661 20,000
Runt 1 1 5,532 17,689
Sox 6 7 2,126 4,317
T-box 3 3 4,578 13,250
TCR/CxC 2 2 0,000 2,543
Zinc_cluster 4 4 0,713 1,327
bHLH 19 26 3,062 7,485
bZIP 15 50 3,531 6,534

75
Supplementary methods Index 1. Software requirements 2. Databases 3. Interface and triads of protein-DNA 4. Statistical potentials 5. Solving scarcity of data by using Taylor’s polynomial series approach 6. Z-scores 7. General and family-specific potentials 8. Structural modeling of TFs complexed with DNA
a. Protein monomer/dimer modeling b. Protein homo/hetero dimer modeling c. DNA modeling d. Modeling protein-protein interactions e. Modeling C2H2 ZF structures for B1H
9. Use of experimental TF-DNA binding to calculate statistical potentials 10. Prediction of the PWM using the sequence of a TF
a. Straight-forward prediction b. Prediction by enrichment
11. Scoring TF-DNA binding with structure 12. Construction of PWMs using TF structures 13. Predicting the PWM with the structure of a TF
a. Straight-forward prediction b. Prediction by enrichment c. Comparison of different sets and methods to predict PWMs
14. Optimal conditions to predict PWMs using the structure of a TF (grid search) 15. Scanning of binding sites and TF clusters along a DNA sequence
a. Scanning of DNA binding domains b. Score per Nucleotide: profiles of a DNA binding site c. TF profiles along a DNA fragment d. Prediction of TFs that bind a DNA fragment e. Clusters of TFs and complexes of regulatory elements.
16. Modeling a selected TF-complex of a cluster of close TFs in a DNA fragment 17. Testing the applicability of ModCRE on the UniProt database
1. Software requirements We require the following software: DSSP (version CMBI 2006) (1) provides protein structural features; X3DNA (version 2.0) (2) is used to analyze and generate DNA structures; matcher and needle, from the EMBOSS package (version 6.5.0) (3), produces local and global alignments, respectively; BLAST (version 2.2.22) (4) and MMseqs2 (5) are employed to search homologs of a target protein; MODELLER (version 9.9) (6) is used to create structural models with all the templates similar to our target; CE-align algorithm (7), as

76
implemented in PyMOL (version 1.5) (8), is used for structural superimpositions of DNA to merge complexes and TMalign (9) to superimpose similar TF folds; and the programs FIMO and TOMTOM from the MEME suite (10) are used to scan a DNA sequence with a Position-Weight Matrix and to compare two PWMs, respectively. CD-HIT has used to generate non-redundant sets of TFs (11). 2. Databases Structural information is retrieved from the PDB repository (12) and protein codes and sequences are extracted from UniProt (January 2019 release) (13). We select all transcription factors as defined in CIS-BP database (version 2.00) (14) to generate the internal database of structures. We distinguish monomer and homodimer TF structures, accepting that some TF families often act as homodimers (“B3”, “bHLH”, “bZIP”, “Leafy”, “MADS box”, “Rel”, “Nuclear Receptor”, “STAT”, and “Zinc cluster”). We extend the set with all other structures that interact, with more than 5 contacts, with a double strand DNA molecule (see further for the definition of contacts in section 3) and name the set PDBDNA. We rearrange the set of structures by separating them in chains and constructing a set of structures formed by single protein-chains interacting with a double-strand helix (single-chain PDBDNA). We use the program TMalign to compare structures of proteins of the single-chain PDBDNA set by superimposing them. We group each structure with all others matching a good superimposition in a set named “folds”. For example, code 1PUF_A that identifies protein-chain A of 1PUF in PDB produces the group named “1PUF_A folds”. The superposition is considered good if it produces a TM-score higher than 0.5. We say that all TF-DNA structures belonging in the same group of folds have the same fold. TF-DNA binding information is retrieved from the CIS-BP database (version 2.00). For each TF in the database, we retrieve the name (Gene Name, Accession Number, Ensembl, and/or any other code to associate with the protein sequence), the specie, the family group and the Position Weight Matrix (PWM). We also retrieve for each TF the list of 8–mers evaluated in the PBM experiment with the corresponding E–score values. The name and species of the TFs are used to obtain the Entry code from UniProt and the corresponding protein sequence. We use the E–scores of the 8–mers to classify them in positive bindings (E-score > 0.45) and negative bindings (E-score < 0.37). We use the PWM of the TF assigned in CIS-BP to select the correct orientation of the 8-mer strand (i.e. we only use the 8-mer sequence of the strand corresponding to the best match with the PWM). We select the 8-mer with the best score out of all 8-mers of a TF as the best bound DNA sequence. Then, we use the best bound sequence to align all other 8-mers considered positive using the program needle of EMBOSS package (3). All 8-mers in the positive binding set aligning with one or more gaps with the best bound sequence are removed from the set. The trimming of the set of positives allows us to have a set of 8-

77
mers, all of them with the correct strand orientation and without gaps that can be used to generate a PWM based on the 8-mers of the PBM. Binding information of Zinc-finger family C2H2-ZF is retrieved from bacteria one-hybrid (B1H) experiments (15). The experiment distinguishes between Zinc-finger individual domains at the C-tail (F3 domain) and inner domain (F2 domain). The experiment performs the screening of all 64 possible 3bp targets for interactions with C2H2-ZF domains from multiple large protein libraries based on Zif268 structure with six variable amino acid positions on each individual domains F2 and F3 (16). 3. Interface and triads of protein-DNA structures
The interface between a protein (e.g. transcription factor) and DNA is defined by the residues (amino-acids and nucleotides) in contact. A general approach for protein-protein interactions is to consider that two residues are in contact if the distance between a pair of atoms from each residue is shorter than 5Å. Usually, a shell around the interface is defined by residues in contact at distance 12 Å. One interface is larger than other if the number of pairs of residues in contact is larger. For TFs in the same fold (i.e. as defined in section 2), we compare the similarity between interfaces of two protein-DNA structures by checking the number of common pairs of residues in both interfaces. The score of similar-interface between interfaces is calculated as the percentage of common pairs of residues in both interfaces with respect to the smallest interface (with independence of their residue-number and nucleotide position in their respective structures, but also independent of the distance as long as they form part of the interface). We define triads as a type of contacts between the protein and the double-strand DNA helix. Triads are formed by three residues: one amino-acid and two contiguous nucleotides of the same strand. The distance associated with a triad
is defined by the distance between the C atom of the amino acid residue and the average position of the atoms of the nitrogen-base of the two nucleotides plus their complementary pairs in the opposite strand of the helix (17). The triad also has an associated amino-acid residue number in the protein and a dinucleotide position in the DNA, defined by the sequence position of the first nucleotide of the dinucleotide. For the sake of the comparison of interfaces, we define the interface between a protein (e.g. transcription factor) and DNA as: the set of triads with associated distances shorter than 15 Å, and their associated amino-acid residue number and dinucleotide position (e.g. a with amino-acid residue number , dinucleotide in position and associated distance is represented as
). This definition is extended up to 30 Å in section 8c on the
application of structural modeling. Amino-acid residue number and dinucleotide position are specific of the structure, consequently they are irrelevant for the comparison of interfaces and are not taken into account to calculate the score

78
of similar-interface (see further). However, this definition of interface may be too rigorous when we have to compare two interfaces and the number of triads is too short. Therefore, we force requiring a minimum of 10 triads with their associated distances and positions of amino-acids and dinucleotides. If this minimum number of contacts is not achieved, we increase the cut-off distance (i.e. 15 Å) in steps of 1 Å until 10 or more triads can be assigned to the interface or we have reached a maximum of 30 Å. Specific features can be added on a triad, defining an extended-triad:
1) Hydrophobicity of the amino-acid. Amino-acid residues are split in Polar (P): {Arg, His, Lys, Asp, Glu, Ser, Thr, Asn, Gln, Cys, Gly} and Non-polar (N): {Ala, Ile, Leu, Met, Val, Phe, Trp, Tyr, Pro}.
2) Surface accessibility of the amino-acid. We use DSSP to calculate the percentage of accessibility of the residue in the unbound structure of the protein. If the percentage is smaller than 50%, the amino-acid is buried (B), otherwise it is exposed (E).
3) Secondary structure of the amino-acid. We use DSSP to calculate the secondary structure of the protein. The amino-acid of the triad is either in regular secondary structure (H if in α-helix, E if in β-strand), or in a non-regular secondary structure (C)
4) Nitrogenous bases: We classify nucleotides by their nitrogenous bases in two types, purines (U): {A, G} and pyrimidines (Y): {C, T}.
5) Closest strand. We use X3DNA to define the strands forward and reverse of the DNA. Next, we calculate the distance of all atoms of the two nucleotides to the Cβ of the amino-acid. We define the strand closest to the amino-acid (i.e. with the atom at minimum distance) as either the strand of the two nucleotides of the triad or the strand of their complementary pair in the opposite strand, which can be either forward (F) or reverse (R).
6) Closest Groove. We calculate the distances between the C of the amino-acid and the closest phosphates of the dinucleotides in both strands (i.e. the strand of the two nucleotides of the triad and its complementary). We calculate the positions of the closest phosphates in both strands (let be Pf and Pr, backbone phosphates of nucleotides f and r, respectively). We select the closest phosphate of both and its corresponding strand. Let assume that Pf is the closest phosphate and define its strand as “s”, being “S” the opposite strand. Then, we consider the set of backbone phosphates in “S” around the position complementary of nucleotide “f” (6 nucleotides up and down). Depending on their distance to “f” (towards 22Å is a major groove and towards 12 Å a minor groove), we classify them as part of the minor or major groove with respect to nucleotide “f”. This is a classification of 12 nucleotides around the complementary of “f” in two groups: 1) set at large distance (i.e. major groove); and 2) set at short distance (i.e. minor groove). Necessarily, Pr is in the list classified in major or minor groove. We use the classification of Pr to define the type of the closest groove of the amino-acid (i.e. we should say that this is the groove faced by the amino-

79
acid, defined by the pair Pf and Pr, in closest proximity to the amino-acid). The closest groove is defined as major groove (A) if Pr is in the list classified in major, otherwise it is defined as minor groove (I).
7) Chemical group of the nucleotides. We distinguish two main chemical groups of each nucleotide, the nitrogenous base (N) and the backbone (B) that includes the phosphate and sugar. We calculate the distances
between the C of the amino-acid and the atoms of the two nucleotides and their complementary. We select the atom with the shortest distance as the closest atom between the nucleotides and the amino-acid. We define the chemical group of the nucleotides of the triad as the chemical group to which belongs the closest atom (i.e. N or B).
Added features of triads can also be used on their own as feature-triads (or environment triads), and every extended-triad has an associated feature-triad, both associated with the same distance, amino-acid number and dinucleotide position. As an example, let be a lysine residue and two nucleotides, adenosine and guanosine, forming the triad [K,(AG)] at 15.6Å, with lysine in residue number 32 and adenosine in 5, described as ([K,(AG)], 15.6, 32, 5). If lysine surface is mainly exposed to solvent, in a α-helix conformation and the closest strand of DNA is the forward strand, the closest atom of the two nucleotides is a phosphate and the amino-acid faces the minor groove, the extended-triad is [{K,(p-H-E)},{(AG),(UU-F-I-B)}], where added features are (p-H-E) for the amino-acid and (UU-F-I-B) for the dinucleotide. This produces a feature-triad defined as [(p-H-E),( UU-F-I-B)] at 15.6Å. We require to define some functions on the sets of triads, extended-triads and feature-triads to extract some of the values collected from a complex structure and apply other functions:
The same functions are applied to extended-triads and feature-triads accordingly modified. For example, we first apply for all triads with d<15Å in order to calculate the score of similar-interface. Still, if any of the interfaces has less than 10 different elements (i.e. ) the score of similar-interface may not be significant (e.g. if we compare one interface with only one element and another with many, the coincidence of one of them already achieves a score of similar-interface of 100%). Therefore, we use a less rigorous approach to calculate the interface by increasing the cut-off distance until both interfaces have at least 10 elements to perform the comparison and calculate the score of similar-interface. We also define functions to substitute some of the elements of a triad, extended-triad or feature-triad (the example is given for etriads without loss of generality):

80
1) is a function that substitutes amino-acid residue “a” of the
etriad by amino-acid-residue “r”, with the corresponding change of hydrophobicity but preserving the rest of features and measures associated with the triad.
2) is a function that substitutes dinucleotide by , in , with the corresponding change of
nitrogenous bases and preserving the rest of features and measures associated with the triad.
4. Statistical potentials We use the definition of statistical potentials described by Feliu et al (18) and Fornes et al. (17) to define several scoring functions for the interaction between a protein and a DNA binding site using contact triads. We use triads, their associated measures (i.e. distance, amino-acid number and dinucleotide position) and their added features to calculate the frequencies per distance in bins of 1Å (i.e. intervals [0,1], [1,2], [2,3], [3,4], [4,5] etc.) up to 30 Å. From now, we use a distance of 1Å to define the bins without loss of generality, but we can use 2 Å and 3 Å, respectively defining sample bins in intervals [0,2], [2,4], [4,6], [6,8] etc. and [0,3], [3,6], [6,9], [9,12] etc. up to 30 Å. We also calculate the frequencies using the distance as cut-off (i.e. the frequency of triads at distance shorter than “d”, with d= 1,2,3,4, etc.). We also call these distance cut-offs accumulative distance intervals. We increase the cut-off distance in 1 Å without loss of generality, but we can increase the cut-off by 2 Å or 3 Å (i.e. defining cut-offs respectively at 2,4,6,etc. and 3,6,9,12, etc.). To obtain the frequencies, we first calculate the size (cardinality, defined by the function “Card”) of the sets of triads, associated with a distance (d), taken from the set of structures of protein-DNA interactions (PDBDNA) and grouped by their associated distance, limited to a maximum of 30Å (i.e. with ). The set of triads, associated with distances, amino-acid residue-number and dinucleotide position, is named 3Dset. Then, frequencies are defined using functions defined in section 3 as follows:
(eq. 1) (eq. 2)
Where is a triad associated with a distance d, amino-acid residue-number and dinucleotide position , taken from the set 3Dset, ND is defined using bins and Nc using cut-offs. Similarly to 3Dset we define the sets e3Dset and f3Dset for extended-triads (etriad) and feature-triads (ftriad), and calculate LD, Lc , MD and Mc with as:
(eq. 3) (eq. 4)
(eq. 5) (eq. 6)

81
Then, we define the frequencies (F for triads, G for extended-triads and H for feature-triads) as:
(eq. 7)
(eq. 8)
(eq. 9)
Where N can be ND or Nc, L can be LD or Lc , and M can be MD or Mc, depending on the approach to group the triads. This definition forces us to consider independent the groups obtained by cut-offs, instead of using the ratios with respect to the limit at 30 Å. We tested in artificial data that this approach preserves the curve of the statistical potential similar to the classical definition by bins of distances, but it’s less affected by the scarcity of data. To define a reference-state for the statistical potential, we require two more frequencies, one for ND and another for Nc , using the total number of triads in the database (triads):
(eq. 10)
Where , and it’s easy to proof that:
(eq.11) Where ftriads is the set of feature-triads and etriads the set of extended-triads, with and . Using these definitions and following previous works (17), we define the potentials E3DC, ES3DC and PAIR per triad and distance d, using the round value of d (i.e. k), as follows:
(eq.12)
(eq. 13)
(eq. 14)
Where, F, G, H and O are frequencies calculated by bins or using cut-offs. The total potential of an interaction is calculated as the sum of the corresponding potential of all triads, feature-triads and extended-triads at distances shorter than 30Å. We use each potential to score the quality (or potentiality) of the interaction. Let be I, E and D the sets defined respectively as the set of all triads, extended-triads and feature-triads with their associated distances (d), amino-acid residue number (p) and dinucleotide position (q) in the

82
binary interaction of a TF-DNA structure. Therefore, we define the energy-based scores (as they are based on total potentials) as:
(eq. 15)
(eq. 16)
(eq. 17)
Similarly, we also use the potentials defined for triads, ftriads and etriads as scores of the quality of a single interaction between one dinucleotide and one amino-acid residue. Then, the potential ES3DC of an extended-triad associated with distance d can be rewritten as:
(eq. 18) Where . We then define two scoring terms, one distance independent (ES3DCdi) and another distance dependent (ES3DCdd), as follows:
(eq.19)
(eq. 20) Using eq. 8, eq. 19 and eq. 20, we rewrite as:
(eq. 21) And the global energy-based scores:
(eq.22)
We consider ES3DCdd to evaluate the quality of an interaction because it is more specific than PAIR, as it uses extended-triads and distances. It is also highly sensible, because it is normalized over the total of triads at a given distance with independence of the features. However, we have to note that this can only be considered as a scoring of quality, because the complete statistical potential requires the other terms E3DC and ES3DCdi to complete ES3DC in equation 21. Finally, due to the scarcity of data, the curves of statistical potentials may be jagged. Therefore, we use a sliding window of approximately W samples defined by distances (by bins or cut-off) to smooth the potential curves. Let be a distance-dependent score, as defined previously, then we define the smoothed score, as:

83
(eq.23) Where and are defined as:
Where, is the total of bins between and with defined score
( ), which is around W (i.e. )
5. Solving scarcity of data by using Taylor’s polynomial series approach The main problem of using extended-triads instead of triads is the size of data required to fill the distribution by distances for all types. The current number of structures is not enough to complete such a large distribution. Consequently, for some extended-triads the distribution by their associated distances is scarce and discontinuous. In order to fill these gaps, we propose an approximation based on a Taylor’s polynomial approach of the statistical potentials. We generalize the approach over a potential function P as follows: Let P be a potential defined over extended-triads (i.e ES3DC, ES3DCdd or ES3DCdi), then it can always be expressed in terms of a function of the extended-triad with associated distance d (rounded to ), and a constant (Q) independent of the extended-triad:
(eq. 24) We approach for an extended-triad, with amino-acid residue “a” and associated distance rounded to k, as:
(eq. 25)
Where, is function calculated with raw data and without approximation,
is the function defined in section 3 to substitute amino-acid residue “a” of the etriad by amino-acid-residue “r”; is the probability of substituting amino-acid residue “r” by “a”; A is the set of 20 amino-acids and
is the set of all amino-acids except “a”. According with these definitions, if there is no data for we approach it by the weighted average of all other potential amino-acid residues (“r”) for which this data exist, located in the same featured triad and associated distance, that can substitute amino-acid “a”. We use the substitution matrix BLOSUM62 to calculate the probability of substitution of “r” by “a”.

84
If , we select the amino-acid “b” for which we get the maximum value of , this is:
; and
(eq.26) If , we cannot do the approach because at this distance we have no information of any other residue. Otherwise, if , we rewrite eq. 25 as:
(eq. 27)
Where is the set of all amino-acids except “b” and “a”. Then, we define P0 as the potential obtained without approximation and obtain
as a function of P0:
(eq. 28) We rewrite max as a function of the potential:
(eq. 29) And we also define the difference of the potential by substituting amino-acid “a” by any residue “r” with respect to the substitution with the maximum potential (i.e. amino-acid “b”), as:
(eq. 30) Then, substituting eq. 27 in eq. 24, and using eq. 28, eq.29 and eq. 30, results in:
(eq. 31)
Here we apply Taylor’s polynomial series of the logarithm function, assuming that “max” is higher than any other value for the rest of residues and we use Maclaurin series (i.e. centered in 0):
Then, we rewrite eq. 31 using only the first term of Maclaurin series as:

85
(eq.32)
Similarly, if , we can use the same definitions of max and “b” , and P0 to rewrite eq. 24 as:
(eq. 33) And using eq. 28, eq. 29 and eq.30, applying Maclaurin polynomial series:
(eq.34)
With the requirement that in order to satisfy the conditions applied for the Taylor polynomial-series approach. Finally, as in section 5, potential curves are smoothed by using a sliding window of W samples defined by distances (by bins or cut-off) to smooth the potential curves. 6. Z-scores We have to note that frequencies are always smaller than 1, consequently their logarithm is smaller than 0. However, statistical potentials are not necessarily negative, because by definition the use of a reference state is required, which is obtained by the sum of all triads at a given distance (also ftriad or etriad, depending on the potential of interest). Consequently, the comparison with a reference state can change the sign of the final sum of terms (for example, PAIR and ES3DC can be negative while E3DC may be positive). The variability of signs of the potentials affects the criterion of quality of the scores, which may become unclear. Nevertheless, indistinctly of the sign, the best interaction between an amino-acid and a dinucleotide is produced at the distance where PAIR and ES3DC are minimum, because it implies the highest frequency of a triad (or etriad) with respect to all triads (or feature-triads). For example, ES3DCdd has a minimum for the highest frequency of an extended-triad with respect to all triads. We then define z-scores in order to follow a criterion that incorporates the sign to score the quality of the interaction between an amino-

86
acid and a dinucleotide as a function of the distance. We wish that the z-score identifies simultaneously the best distance associated with a triad and the best pair formed by one amino-acid and one dinucleotide. Consequently, we construct a zscore function with any type of score, applying without loss of generality on an extended-triad (etriad) and an associated distance d, as:
(eq. 35)
Where: A is the set of all amino-acids (i.e. ), and we use the classical functions of average ( ) and standard deviation ( ), defined as:
Notice that we use the function as above and that we use the term score instead of potential (P in the previous section) to emphasize that this is used only as a criterion of the quality of the interaction. It is easy to proof that this definition satisfies our requirements for the new function zscore: 1) the minimum value of score produces a negative value of zscore (and vice-versa, the maximum yields positive); 2) as the zscore compares the score of a particular amino-acid with all other, the etriad becomes ranked from minimum to maximum allowing us to select the best residue among amino-acids in the same position, depending on the quality criterion of the score. Finally, if we choose to smooth the curves dependent on distance (see section 4), the zscore has to be calculated first with the unsmoothed scores and subsequently smoothed to avoid introducing biases. 7. General and family-specific potentials One of the requirements for the construction of the statistical potentials is to remove redundancies of complex structures. The motivation that instigates such reduction is the presence of structures of transcription factors of the same family, with very similar sequences in both protein and DNA binding sites. These structures would introduce an important bias in the statistical potentials, limiting the capacity to score and predict the bindings. Therefore, we define two different types of potentials: 1) general potentials, requiring the minimum bias that avoids redundancies of similar interactions; and 2) family-specific potentials that allow the similarity but can only be applied on TFs for which we

87
know their family (i.e. the group of folds in which they belong). For “general potentials”, in order to obtain an unbiased set of TF-DNA structures, we reduce the set of triads and their associated distances, amino-acid residue-number and dinucleotide position (3Dset) by excluding complexes of structures with more than 50% of score of similar-interface. For family-specific potentials we wish to accept an important degree of similarity that avoids too similar or redundant structures. Consequently, for “family-specific potentials” we use groups of folds and exclude complexes with more than 80% of score of similar-interface between interfaces of TFs in the same group of fold. We note that “family-specific potentials” are constructed specifically for each group of folds. Besides differences on the construction of potentials, there is also an important difference on its application. The “general potential” can be applied to all TF structures automatically without further requirements, while for the application of “family-specific potentials” on a TF-DNA interaction we first need to assign the structure to one of the groups of folds (see section 2 on “Databases”), then we apply the “family-specific potential” obtained for this fold. On the one hand, if the structure of the TF-DNA structure is known, we can assign the “fold” group straight-forward. On the other hand, If the structure of the TF-DNA structure is not known but has been modelled, then we apply the “family-specific potential” of the template with which the TF-DNA interaction was modelled. 8. Structural modeling of TFs complexed with DNA Given the sequence of a TF (named protein target) and a DNA binding site fragment (named DNA target), we have developed an automated method to obtain the structure of the complex by means of homology modelling using the program MODELLER (6). First, we search potential templates of the protein target among the set of sequences with known structure in PDBDNA using BLAST (19). We select the first match, with the highest score and the minimum E-value, to define a fragment of the sequence of the target localized by the number of one residue in the N-tail position and another in the C-tail position. Next, we select all codes of the matches of BLAST with E-value smaller than 0.1, where the sequence fragment of the TF in the match overlaps in more than 50% with the selected fragment of the target in the first match. We define the minimum position at the N-tail (MinNt) and maximum position at the C-tail (MaxCt) of the target sequence using the BLAST alignments with the sequences of the selected codes. We collect the structures and sequences of the selected codes and align the sequences with the fragment of the target between MinNt and MaxCt using the program matcher of the EMBOSS package (3). We select only those sequences where the alignment with the fragment of the target produces a sufficiently high percentage of identical residues as a function of the length of the alignment (i.e. in agreement with the curve of the Twilight-Zone described by Rost (20). The final selected sequences correspond to the templates for modelling the structure of the sequence fragment of the target between MinNt and MaxCt. We use the alignment obtained with matcher and the sequence and structure of each template to construct one or more

88
models with MODELLER (depending on the total number of models we wish to obtain). Each model is based on a different template and is stored with the name of the TF target, the code of the selected template, the start and end residues of the fragment (MinNt and MaxCt), and a number for the collection (in case we model more than one structure per template). We include the DNA structure of the template in the model of the target as a co-factor (heteroatom). After modeling the main fragment (sequence of the protein target defined between MinNt and MaxCt), we proceed similarly with the remaining fragments of the protein target (i.e. maximum two fragments: between 1 and MinNt, and between MaxCt and the last residue of the target). This implies an iterative procedure by which the TF target is split and modelled, producing models for one (if no more matches are obtained with BLAST) or several domains. Finally, we keep only those models with more than 5 contacts between the protein and the DNA structure to proceed with further modelling and analyses. In particular, we still need to model the structure of the DNA target and substitute the structure of DNA of the template (see further).
a. Protein monomer/dimer modeling
When facing the selection of templates to model the conformation of a TF, the automated approach can select a template acting in the form of a dimer. However, as we select sequences among all chains of known structures, the automated approach models the structure as a monomer, one for each chain from the same PDB file of the template. In order to use the correct form, i.e. the expected for a particular family of TF, we have predefined those families that usually act as dimers (see section 2): “B3”, “bHLH”, “bZIP”, “Leafy”, “MADS box”, “Rel”, “Nuclear Receptor”, “STAT”, and “Zinc cluster”. By default, we proceed by modelling the homodimer or monomer structure as the corresponding conformation in the template, unless the user specifically requires a specific model. In order to model the homodimer, we select only homodimer template structures and duplicate the sequence alignment of the monomer sequence between the target and each template; then, we run MODELLER (6) with each template as defined in the examples of multiple-chain modelling.
b. Protein homo/hetero dimer modeling
To model a heterodimer (or force a homodimer) we first need to select dimer templates out of all potential templates of the target. We use a file with the two sequences of the dimer in FastA format. Each sequence of the target dimer (both are the same in case we model a homodimer) is aligned and scored with the sequence of the potential templates using BLAST (see above). We select only those templates sharing the same PDB code from PDBDNA, but different chain code for each sequence of the target. This implies that we select a dimer complex structure and each sequence can be modelled with a different chain. Besides, in contrast to monomer modelling, only the first sequence of the file can be split and subsequently tested and

89
modelled (see above). This restriction is forced in order to avoid multiple solutions, because the application is specifically selected by a user who decides the sequences in the input and expects an answer in the shortest time (i.e. this is applicable only in a web-service). We realign each sequence of the target with the corresponding sequences of the template using matcher and run MODELLER as defined in multiple-chain modelling including the DNA structure as heteroatoms.
c. DNA modeling
After modeling several conformations of the protein target with each template and bound with the same DNA of the corresponding template, we still need to substitute the DNA structure in the model by the DNA of the target. With this purpose, we define some previous requirements to do the modeling of the DNA target: 1) the target DNA sequence belongs in the strand defined by the first DNA chain in the PDB format file, which is named chain of reference; 2) the target DNA sequence corresponds only to the DNA binding site, oriented from 5’ to 3’ of the chain of reference, numbering the nucleotides in increasing order; 3) we assume that the binding site sequence is limited between 5’ and 3’ positions by the region in contact with the target protein (i.e. the DNA region before 5’ and after 3’ is not in contact or not in the interface); and 4) the model of DNA preserves the same conformation as in the original template. We first assume that the sequence of the DNA target has the same length of the binding site (i.e. all other potential nucleotide residues before and after the DNA target sequence are not in contact with the TF). We model the DNA structure by substitution of the nucleotides in each model with the corresponding nucleotides of the target. We use the program X3DNA to substitute the nucleotides of one strand and automatically model its corresponding pair. Therefore, the alignment between the DNA sequence of the target and the sequence of the corresponding DNA strand in the template is required to apply the substitution. To obtain this alignment, we first trim the DNA structure of the model constructed in sections “8.a” or “8.b” (note that they still belong on the structure of the DNA original template) by removing all nucleotides that are not in contact with the TF before 5’ and after 3’ of the chain of reference. The alignment is then obtained by the exact match between the length of the DNA sequence of the template and the length of the DNA target sequence, yielding a one to one correspondence in the same order of the nucleotide sequence. Alternatively, instead of trimming the DNA of the chain of reference from the template, we can suggest the user to extend the DNA target sequence in order to preserve a one to one correspondence between the sequences of the DNA target and the DNA template (i.e. the length of the DNA sequence of the interface is shown in the web-service). If the length of the DNA sequence of the target is longer than the length of the DNA sequence in contact with the TF, we need to define the margins of

90
the binding site. We trim the DNA structure in the model by removing the nucleotides before and after the binding site defined by the contacts with the TF as before in the directions of 5’ and 3’. However, in order to consider the impact of long-range interactions, we extend the definition of the interface up to 30Å, as used in the definition of the statistical potentials, and all nucleotides in triads with associated distances under 30Å are considered in contact with the TF. If the length of the DNA target is still different than the length in the structure it implies the sequence is incorrect or incomplete and the modeling stops, otherwise we proceed as above.
d. Modeling protein-protein interactions.
To model a protein-protein interaction (target) we use a file with two protein-sequences (e.g. A and B) in FastA format in a similar way as for heterodimers. Each sequence is aligned and scored with the sequences of potential templates found by BLAST (see above) when searching on the complete set of structures (i.e. all structures from PDB). Both target sequences are split by parts according to the BLAST search as explained above. We select those templates that have the same PDB code but different chain for each fragment of the target sequences (one fragment of A and another of B). Each sequence-fragment is then modelled with a different chain, the use of several templates generates several models. Consequently, this procedure allows for modeling several interfaces of both sequences (A and B). We realign with matcher each sequence-fragment of the target with the corresponding sequences of the single-chain templates. Then, we use the multiple-chain approach of MODELLER to model several structures of the interactions of the split parts of both sequences. Structural models are validated by confirming that their interface has more than 5 residue-residue contacts and the number of clashes and perforations between surfaces is limited. e. Modeling C2H2-ZF structures for B1H The DNA binding sequence of each testing 3-domain C2H2-ZF protein is formed by 9bp nucleotides, a set of 3bp bound by each individual domain. For the selection of the binding sequences associated with each finger domain, we use the same sequences as in the B1H experiment (15): 1) for the selection of F2, the finger sequences in F1 (N-tail domain) and F3 (C-tail domain) involved in the interface are RSDNLRA(F1) and RSANLVR (F3), respectively binding AAG and GAG; and 2) for the selection of F3, the finger sequences in F1 (N-tail domain) and F2 (inner domain) involved in the interface are RSDELTR (F1) and RSDNLRA (F2), respectively binding GCG and AAG. The structure of Zif268 binding DNA is modelled with three different template structures to introduce structural variability: 1p47 (chain A), 1zaa (chain C) and 1g2f (chain C). We compare the sequence of Zif268 used in the experiment with the templates by a sequence alignment with CLUSTALW (21). We identify by WT the original sequence, and by F2 and F3 the sequences used for the selection of F2 and F3 binding sequences,

91
labeling by “X” the amino-acids that are modified. This alignment is shown here, highlighting in bold and red the attention of the specific binding sequence of each finger (blue box for F1, yellow box for F2 and green box for F3): F2 GTERPYACPVESCDRRFSRSDNLRAHIRIHTGQKPFQCRICMRNFSXXXXLXXHIRTHTG F3 GTERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDNLRAHIRTHTG WT GTERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDNLRAHIRTHTG 1g2f_C -MERPYACPVESCDRRFSQKTNLDTHIRIHTGQKPFQCRICMRNFSQQASLNAHIRTHTG 1p47_A --ERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTG 1zaa_C ---RPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTG *************** * ********************* * ******* F2 EKPFACDICGRKFARSANLVRHTKIHLRGS F3 EKPFACDICGRKFAXXXXLXXHTKIHLRGS WT EKPFACDICGRKFARSANLVRHTKIHLRGS 1g2f_C EKPFACDICGRKFATLHTRTRHTKIHLRQK 1p47_A EKPFACDICGRKFARSDERKRHTKIHLRQ- 1zaa_C EKPFACDICGRKFARSDERKRHTKIHLR-- ************** ******
After modeling the structure of Zif268, we complete the complex by modeling the structure of the DNA binding sequence. However, each template has DNA sequences of different length that do not correspond with the DNA used in the experiment. The full DNA sequence of the experiment is longer (29bp) than the binding (9bp), which is shown next, embedded in positions 11 to 19 (nucleotides highlighted), labelling by “N” those under test. Two DNA sequences are considered depending on the experiment, one for the selection of F2 and another for F3: F2: 5'- GCGGCCGCAAGAGNNNAAGTAACGAATTC - 3' F3: 5'- GCGGCCGCAANNNAAGGCGTAACGAATTC - 3'
The structure of the full DNA sequence bound by Zif268 is obtained with the program X3DNA (2) by modifying the DNA structure in the complex. First, we locate the 9bp binding region in the structure of the template and identify the positions at 5’ and 3’ (first and last). Next, we construct two frames of B-DNA structure with the lengths required to extend the template at 5’ and 3’ up to 29 nucleotides. The lengths in both sides depend on the location of the 9bp binding sequence and the DNA sequence of the experiment. Then, we use 3DNA/DSSR to perform a least-squares fitting that locates each base reference frame in the first and last positions. Finally, the structure of DNA is completed with the right sequence following the same procedure as above (in 8.c).
We also model several structures with the complex of Zif268 binding a non-specific DNA region using the same approach. These structures are be used as non-binding examples. The non-binding sequence is taken randomly by selecting a region of the sequence of the weak promoter GAL1, constructing 10 DNA fragments of 29bp for each known binding. A potential binding test in this region, which is part of the B1H experiment, may well represent the background. The forward weak promoter sequence of GAL1 is formed by 118bp that are shown here:

92
5'- GAGATTAAGGAGCAGAAGGGGTGACAGCCCTCCGAAGGAAGA GAGATTAAGCTCTCCTCCGTGCGTCCTCGTCTTCACCGGTCG CGTTCCTGAAACGCAGATGTGCCTCGCGCCGCACTGCTCCG - 3'
9. Use of experimental TF-DNA binding to calculate statistical potentials One of the main problems to obtain statistical potentials for all families and folds of TFs is the scarcity of known interactions. Even if we address this problem by using a theoretical approach such as the use of Taylor’s polynomial series, we still require enlarging the number of experimentally known structures. In fact, what we need to enlarge is the number of interacting triads. Therefore, here we propose to use the experimental knowledge of TF-DNA interactions to derive interacting triads without requiring the complete knowledge of TF-DNA complex structures. We then use the sets of derived triads associated with distances to calculate the statistical potentials. We select all TFs from the set of CIS-BP with experimentally known interactions with DNA by means of PBM. For each TF in this set, we collect all the 8-mers with accepted interactions and check if the structure is known. If the structure is known (i.e. there is a specific file in PDB with the structure of the interaction), we confirm that the DNA sequence extracted from the structure is among the 8-mers. This is done by a sequence alignment without intra-sequence gaps with all 8-mers classified as positive bindings. Then, we use the PWM obtained with all positive binding 8-mers to align the DNA sequence extracted from the structure and all positive binding 8-mers. We skip all alignments with intra-sequence gaps and trim the rest of alignments by removing the tails with gaps at the beginning and end. This results in an alignment with a length of maximum 8 nucleotides. For each alignment, we define a mapping function (mapD) between the fragment of the DNA sequence from the structure ( ), as seen in the alignment, and any of the positive binding 8-mer sequences ( ), as follows:
(eq.36) Where is a dinucleotide, with , is also dinucleotide, with
, is the position of the dinucleotide in the alignment between and , is the position of the dinucleotide in , is the position of the dinucleotide in , and the position of each dinucleotide is defined (i.e. equal to) the position of the first nucleotide in the DNA sequence. For the sake of simplicity, when the length of is the same as and the position in the alignment, , coincides with and (i.e. ), we write:

93
(eq.37) We can define a set of mapping functions with the alignments of all the DNA sequences extracted from the structures of TF-DNA complexes in PDBDNA that are aligned with positive binding 8-mer sequences in CIS-BP. Then, we use the dinucleotide substitution function (as defined in section 3), , of an extended-triad, etriad, containing a nucleotide which is substituted by n in the dinucleotide, to generate more extended-triads. There are still TFs from the set of CIS-BP with PBM experiments for which the structure is not known but can be modelled. This implies that the sequence of the TF can be aligned with sufficient percentage of identical residues to ensure its modeling (see above in section 8). Then, we define another mapping (mapP) between the protein sequence of the TF in CIS-BP and the TF sequence of a known structure (template), with:
(eq.38) Where, is an amino-acid residue in the sequence of a template and is the amino-acid in the sequence of a TF in CIS-BP, in position of the alignment of both TF sequences that correspond with positions and for and , respectively. Also, for the sake of simplicity, if the position in the alignment, , coincides with and (i.e. ) we write:
(eq.39) We define the set of all mapping functions with all the alignments between the sequences of TFs in CIS-BP (with PBM experiments) and the TFs with known structure (complexed with DNA). Also, we define as the set of extended-triads, extracted from the 3Dset, containing amino-acid residue (the set of 20 amino-acids) and dinucleotide . We remind the definition of as the function that substitutes amino-acid residue “a” of an extended-triad, etriad, by the amino-acid residue “r”. With all these definitions, we increase the set of extended-triads of the e3Dset to e3Dset’, using all the mapping functions mapD (simplified as ) and mapP
(simplified as ). We use the simplified maps without loss of generality, as all sequences and alignments can be renumbered. Both mappings, and
, are respectively defined with: 1) the alignments of the DNA sequences extracted from the structures in the PDB aligned with positive binding 8-mer sequences; and 2) the TFs that can be modelled using the alignment with their structural templates (the set is defined as ). The new set of extended-triads, e3Dset’, is defined as:

94
(eq. 40) And we recalculate LD and LC in equations eq.3 and eq.4 as:
(eq. 41) (eq. 42)
Similar approach is also taken for the sets of triads and ftriads, modifying accordingly the corresponding functions to substitute amino-acid and dinucleotide residues of a triad or a featured-triad. In order to avoid biases produced by binding similar sequences in the PBM experiment we exclude complexes of structures with more than 50% of score of similar-interface when applying the mapping and substitution of the dinucleotide sequence. To improve the speed of the calculation we also neglect DNA sequence bindings of the same TF modelled with the same template if it differs in less than 2 nucleotides of a previous selected sequence. However, we only use this reduction to calculate the general potential, while for family potentials we allow all DNA bindings to avoid a considerable computational cost (using a limit on 80% of score of similar-interface for members of the same fold has little effect but implies a long time of calculation). We proceed similarly with B1H experiments on C2H2-ZF family. For each finger (F2 and F3) and combination of 3bp nucleotides, we collect all protein sequences producing significant binding signal in the B1H experiment. We use the modelled structures of the DNA testing sequence of 29bp with different templates and introduce the mappings for the DNA sequence and the modified residues in F2 or F3 from the multiple sequence alignment. For the DNA sequence the mapping is on the 3bp modified nucleotides, affecting 4 dinucleotides, while for the protein sequence the mapping affects 6 amino-acids, both mappings being different for F2 and F3 selections. This is, for the DNA sequence the mapping is , where is a dinucleotide of the 3bp under test, is a dinucleotide of the 29bp of the modelled template, and the position, , is between 11 and 19 (11-13 for F3 and 14-16 for F2). For the substitution of residues of Zif268 in the interface, we require a mapping of the native sequences RSANLVR (F3) and RSDNLRA (F2), for all selections of the binding finger. This mapping is where is the position in the alignment (residues 47-53 for F2 and 75-81 for F3), , the position in the template, , depends on the template used to model Zif268 and
is the corresponding residue in the template in position of the alignment. From the template structure we extract the contacts (triads, etriads and ftriads) between amino-acids and dinucleotides and generate the statistical potentials. However, this introduces a bias by overestimating the constant amino-acids and nucleotides that have not been modified in the experiment. Therefore, only the

95
triads affecting the amino-acids and nucleotides under test are considered to generate the potentials. This is, we only consider the contacts between the 6 amino-acids labelled by “X” and dinucleotides containing one of the 3bp labelled by “N” to generate two potentials, one for F2 and another for F3 finger positions. We restrict each set of Zif268 sequences to those with highest signal of the B1H binding experiment in order to obtain potentials more specific or associated with the strongest binding. We define two thresholds based on the affinity percentile of a sequence: 1) higher than 80%; and 2) higher than 50%. To calculate the affinity percentile of a sequence, we follow the same definition as the authors (15). Each sequence in its corresponding domain has a logged and normalized frequency of its observation. Hence, the affinity percentile is defined as the sum of all other frequencies lower or equal to the frequency of the sequence (e.g. an affinity percentile of 80% implies that the sequence is on the tail with highest number of observations, around the top 20%). However, the number of selected sequences may be too different between experiments (i.e. the 3bp binding TGA may have 30 sequences with affinity percentile higher than 80, while AAA has only 2), which produces the opposite bias on the expected potential. To avoid a bias on the number of sequences selected, we force to have around 500 sequences for all 3bp experiments, by repeating as many times as we need each sequence (e.g. if only 2 sequences are selected for AAA and they are equally representative, we should repeat 250 times each). Each sequence is
then repeated , where is the set of sequences with affinity
percentile higher than 80 (or we use A50 for affinity percentile higher than 50). As a consequence of the approach, the contacts derived from the B1H experiment are limited to relatively short distances (the largest contacts are around 15-20Å). However, we note that we also use contacts extracted from other structures of the C2H2-ZF family in the PDB and from the use of PBM experiments, covering larger distances up to 30 Å. 10. Prediction of the PWM using the sequence of a TF
Let be a TF, named target TF in the set CIS-BP and with known PWM. We do a blind test assuming this PWM is unknown and develop two prediction approaches using the rest of TFs in the database, their sequences and their PWMs: 1) the first approach is straight-forward based on sequence similarity; 2) the second approach uses the enrichment of predictions. Both approaches are explained below.
a. Straight-forward prediction
The straight-forward prediction is based on sequence similarity. We use MMSeqs2 (5) to search sequences of TFs in CIS-BP that align with the sequence of target TF with sufficient percentage of identical residues. We classify the matches of this search by percentage of identical residues aligned (ID). Then, we analyze the relevance of sequence similarity on the prediction of the PWM. The group of TFs classified as “ ” is the set of TFs

96
from CIS-BP which sequence aligns with the sequence of target TF with a percentage of identic residues (ID) around in an interval of 10% (i.e.
). Let’s consider the group “ ”, the straight-forward prediction assumes that the PWM of target TF is the PWM of the TF with higher ID in the group. We define as solutions at all PWMs of TFs aligned with target TF with ID around and define as straight-forward prediction at the PWM of the TF with higher ID. We also group TF targets by ID, where a TF target belongs to a group if it has one or more solutions of TFs in group To evaluate the quality of the prediction, we compare the PWM of target TF with the PWM predicted. We use TOMTOM, from MEME suite (10), to compare two PWMs and define the score of similarity as:
(eq.43) Where is the significance of the TOMTOM comparison of both PWMs. We calculate the distribution of scores of similarities for all solutions of TF targets of CIS-BP as a function of the percentage of identical residues (i.e. grouped by ID). To analyze the results of several solutions within the same interval of ID, we calculate the maximum, the minimum and the average score of similarity of all solutions of TF targets as a function of ID. We also show the quality of the prediction by ranking the score of similarity between the prediction and all PWMs in CIS-BP. Then, we define the score of ranking by the position in the ranking of the score of similarity with the PWM of the target:
(eq. 44) Where “size” is the total number of TFs with known PWMs in CIS-BP and “rank” is the ranking of the score of similarity between the prediction and the PWM of target TF. If the between the PWM of the target and the predicted PWM is not significant (i.e. , then the score of ranking is null. We calculate the distribution of scores of ranking for all solutions of TFs of CIS-BP as a function of ID. As for the scores of similarities, we also calculate the maximum, minimum and average of all solutions of each target TF, grouped by ID. We use the score of ranking to calculate the accuracy of the prediction at different values of ID. We use two criteria to consider a prediction successful: 1) the score of ranking is (i.e. the predicted PWM is the most similar to the PWM of the target); and 2) the score of ranking is among a top threshold, allowing for acceptable errors or the incidence of other PWMs similar to the PWM of the target (e.g. top 1% means a ranking score higher or equal to 99). The accuracy of the prediction is calculated as the

97
ratio of successful predictions among the total number of TFs with a prediction. Hence, the accuracy is calculated as a function of ID as the ratio of successful predictions of TFs over all TFs with at least one solution at a given ID (i.e. the TF targets belonging in set ID). The goal of predicting a PWM is to find the potential binding site(s) of the target TF in a DNA sequence. Thus, the straight-forward prediction applies the predicted PWM of a target TF. This is the PWM of the TF which sequence is aligned with the sequence of the target showing the highest ID. We scan a DNA sequence with the selected PWM and collect the potential bindings detected with FIMO. Still, for the sake of analysis or comparison, we test the solutions (i.e. in general more than one PWM per target) for different intervals of ID.
b. Prediction by enrichment The prediction by enrichment uses the search of similar TFs of target TF as in the previous approach and it classifies the matches by percentages of identical residues aligned (ID). Then, for an interval of ID, i.e. IDa, we rank the matches as before, but instead of selecting the top ranked solution with higher ID we collect the PWMs of all solutions at IDa. We calculate the score of ranking as before but for all solutions at IDa and select the PWMs of the top ranked scores over a threshold (i.e. top ranked PWMs) for each solution. We have to note that we only select those for which the score of similarity is significant (i.e. ). By definition, the number of
selected PWMs is smaller than the total number of TFs with known PWMs in CIS-BP. Consequently, some PWMs are selected more often than others. We define the enrichment of a PWM as the percentage of the number of times the PWM has been selected:
(eq. 45) Where is the number of times PWM has been selected by any of the solutions of target TF and is the total number of solutions of the target TF at IDa. By its definition, . If the threshold of top ranked scores is as large as the whole dataset of PWMs (i.e. “size”), the enrichment would be 100 for all PWMs. Therefore, it’s clear that we need to limit this threshold by the probability to obtain an enrichment by random, otherwise the value of enrichment is meaningless. Let T be the threshold of top ranked scores to calculate the enrichment. Thus, we collect a total of PWMs and each can be selected a maximum of times. Let “size” be the total number of TFs with known PWMs in CIS-BP as before; then, the random probability to select one of them is while the probability to obtain a PWM in a group of T selected PWMs is . Let E be the enrichment of one PWM, meaning that we

98
select this PWM. Then, the probability ( ) to select
a PWM out of when we have selected T PWMs is obtained by the binomial distribution:
(eq. 46) We then require that the enrichment should be larger than the enrichment at which is higher than , and this is obtained by substituting by
in equation 46, otherwise we define it null (i.e. ).
We calculate as a function of ID the enrichment of the PWM of each target TF with at least one solution at such ID. This can be plotted to help us evaluating the quality of the enrichment on the prediction of PWMs. Enrichment values vary between 0 and 100, but we have to note that: 1) if none of the solutions produces a significant score, except its own PWM, the enrichment is null; 2) when analyzing the enrichment of the PWM of a target TF, if within the number of top selected PWMs the PWM of the target is never selected, the enrichment is null; and 3) by limiting the enrichment as a function of the number of top selected PWMs to those with more significant probability than random, some enrichments are nullified. In conclusion, neglecting all target TFs with null enrichment, the coverage of TFs that can be analyzed is downsized. To show the accuracy of the prediction, we rank the enrichment of solutions of each target TF when they are significant. We define the score of ranking of enrichment as in equation 44, using the rank of the enrichment instead of the score of similarity. The score of ranking is undefined if the enrichment is null, as this should affect the coverage, not the accuracy. Then, we calculate the score of ranking of the enrichment of one solution of each TF (target) as a function of ID. When plotting the “score of ranking of enrichment” it is also important to show the coverage, as for some TFs either their PWM is not selected among the top or the probability of achieving the corresponding rank of enrichment is not significant. Finally, we use the score of ranking of enrichment as above to calculate the accuracy of the prediction (i.e. we apply the same two criteria to consider a prediction successful). As before, the goal of predicting a PWM is to find the potential binding site(s) of the target TF in a DNA sequence. However, when applying the prediction based on enrichment, we use all solutions of target TF. We use the solutions to scan with FIMO a DNA sequence and collect the resulting fragments detected. DNA sequence fragments, sometimes overlapping or even the same, are collected as many times as they are detected by the different solutions. Then, we calculate the enrichment for each DNA fragment (or nucleotide) instead of the enrichment of a PWM. The enrichment is calculated with equation 45, using the number of times the fragment (or

99
nucleotide) has been collected over the total number of solutions. Due to the scanning with FIMO, the same nucleotide can be collected more than once by different PWMs overlapping around the same region. Therefore, it is more convenient to plot the enrichment by nucleotide along the DNA sequence (i.e. number of times a nucleotide is collected over ) than by fragments (this type of plot is defined as nucleotide profile). We also limit the enrichment by the significance of the matches of FIMO, controlling the probability of the match (using the P-value obtained by FIMO) to filter out unreliable results and ensure the quality of the prediction. As for straight-forward predictions, we can limit the number of solutions by the percentage of identical residues (ID), in order to have an additional control of the enrichment, or to analyze the quality of results and compare methods of prediction.
11. Scoring TF-DNA binding with structure
a. Scores of single domain structures Given the structure of a protein-DNA complex, either experimentally obtained (i.e. from crystallography and identified by a PDB code) or modelled (i.e. as described in section 8), we define several scores of the interaction based on statistical potentials. First, we calculate the interface of the interaction and extract all triads, extended-triads and feature-triads associated with distances shorter than 30Å, as defined in section 3. Then, the score of the interaction is defined as the sum of the scores (i.e. potential) of all triads with their associated distances (or extended-triads or feature-triads, depending on the type of score). The same approach is applied for z-scores. Let be a potential as defined in section 4, or a z-score as defined in section 5. Let C be the set of triads (extended-triads or feature-triads, depending on the definition of ) and their associated distances (d), amino-acid residue number (p) and dinucleotide position (q). The score of the interaction is defined as:
(eq.47)
We can obtain the score of a TF without knowing the structure of the TF-DNA binary complex if it can be modelled. We use the structure of a template to generate the set of triads and the mapping of amino-acids derived from a sequence alignment, mapP as defined in section 9 (eq.38), between the TF sequence and the sequence of the template. We also need the mapping of dinucleotides between the DNA sequence we wish to model and the DNA sequence in the template interface (see section 8.c). Instead of modeling the structure of the TF-DNA complex, we modify the scores in equation 47 by applying the substitution of the corresponding amino-acids, using the

100
functions defined as in section 3, and and the mappings mapP (in eq. 38) and mapD (in eq. 31) between the templates and the sequences of TF and DNA, respectively. Here, instead of using simplified mappings ( and from equations 37 and 39), we generalize the formula by defining special functions, and , to extract the dinucleotide or amino-acid positions in the DNA or protein sequences:
(eq. 48)
Where is either a dinucleotide or an amino-acid residue, and is a position of a dinucleotide or an amino-acid, respectively for DNA or protein sequences. Then we calculate the score of the interaction as:
(eq.49)
b. Multiple domain TFs
There are TF structures with more than one domain, where each domain uses a different potential to evaluate the interaction (i.e. when using family potentials and domains belong to different families). One example is the particular case of the C2H2-ZF family, where the TF has several domains like F2 (internal) and two extreme domains, at the N-tail (F1) and C-tail (F3). For TFs of the C2H2-ZF family, we generate two potentials for finger domains in F2 and fingers in F3, then we will use the F2 potential for F1 too. The application is straight forward as the score in equation 49 is the sum of the scores of triads; therefore, we apply equation 49 and each triad is calculated with the potential of the domain corresponding to the amino-acid position of the triad. However, we have to note that this approach is limited to the use of normalized scores, such as z-scores, to avoid the combination of different scales. To calculate the score, we require an input assigning the domain for each amino-acid position (i.e. for the triad) or split the structure of the TF in its domains, calculate the score of each domain separately and then sum. In general, for any TF, we split the sequence in domains (e.g. using PFAM domains) and model the structure of the complex for each individual domain. For the particular case of the C2H2-ZF family, we have to manually split the domains in first and inner domains (using the potential of F2) and the last domain (using the potential of F3).
12. Construction of PWMs using TF structures Given the structure of a protein-DNA complex, either experimentally obtained (i.e. from crystallography and identified by a PDB code) or modelled (see

101
section 8), we obtain the PWM of the TF by means of statistical potentials, using scores or zscores, calculated with the complex structure (we use the zscore of ES3DCdd as example). Let be the number of nucleotides of the DNA sequence in the complex. We use a sliding window of 8 nucleotides to generate fragments of 8 continuous nucleotides. Fragment “k” is defined as the interval of nucleotides [k,k+7], where k ranges from 1 to . If the length of the DNA sequence is shorter or equal to 8, we use only one fragment defined as the DNA sequence itself. For each fragment “k” of the DNA sequence in the complex structure ( ), we collect the set of triads, extended-triads and feature-triads with their associated distances between protein and DNA at less than 30Å and the associated amino-acid and dinucleotide positions (i.e.
, respectively), where the dinucleotide in the triad belongs in fragment “k”. We remind the substitution function from section 3, to substitute the dinucleotide of an by the dinucleotide . Similar functions are defined to substitute the dinucleotide in triads and feature-triads (these are only affected in the change of the nitrogenous bases). Then, for each fragment we obtain all possible DNA sequences with the length of the fragment (i.e. for a length of 8 residues this is 48) forming a set, named set . We define a mapping for the sequence of
between sequence position and dinucleotides (i.e.
with nucleotide in position of ). We also define a mapping
for any sequence in between sequence position and
dinucleotides (i.e. with nucleotide in position of ). We
calculate the score of any sequence on with the set of triads, extended-triads and feature-triads, using the associated distances, residue number and dinucleotide positions from the complex structure (i.e. triads, extended-triads and feature-triads as , respectively) and using the corresponding substitution function (e.g. as defined in section 3. Let be the score of application and assume we apply it on extended-triads without loss of generality, then the score of a sequence in is:
(eq. 50) Where, for each , and are calculated using the functions as defined in section 3 and the mappings defined above:
We normalize the scores of sequences in between 0 and 1, by transforming the score:

102
(eq.51) Then, we rank the normalized scores and select only the DNA sequences producing the top scores over a cut-off threshold. The cut-off is specific for the family of the TF (family threshold for PWM) if the scores are calculated with the family-specific potential, otherwise it is a general threshold (general threshold for PWM) as the scores are calculated with the general potential (see above in section 7). These thresholds are also dependent on the use of PBM data and Taylor’s approach. The selected sequences correspond to a fragment that may be only part of the original sequence length (as taken from the complex). Therefore, we complete all fragments with dummy nucleotides at 5’ and 3’. Naming the dummy nucleotide as “N”, the sequence with the original length ( ) derived of a sequence ( ) selected from is defined as where:
(eq. 52) We proceed similarly for all fragments and obtain a multiple sequence alignment with all selected extended sequences, all with the same length and without gaps. Finally, in order to calculate the percentage of each nucleotide in each position of the alignment, we neglect the dummy nucleotides (N) and we use the percentages to define the PWM. Following the approach in section 11, we also obtain the PWM of a TF without knowing the structure if this can be modelled. We require only the structure of a template to generate the set of triads and the mapping of amino-acids derived from a sequence alignment, mapP as defined in equation 38 (sections 9 and 11), between the TF sequence and the sequence of the template (we assume a simplified mapping, , without loss of generality). We don’t need to model the structure of the TF-DNA complex, we only need to modify the scores in equation 50 by applying the substitution of the corresponding amino-acids, using the function defined in section 3, , and the mapping, , and preserving the rest of definitions as in equation 50:
(eq. 53) The rest of the approach follows the same procedure for the construction of the PWM up to the full DNA sequence.

103
13. Predicting the PWM with the structure of a TF Let be a TF from the set CIS-BP with its PWM already assigned, named target TF. As in section 10 (see above), we assume the PWM of such TF to be unknown and develop two approaches to predict it. Conversely to the prediction by sequence, we use the structural models of the TF in complex with DNA. We use as above two different approaches: 1) the first approach is straight-forward, based on the use of one single model obtained with one template structure; 2) the second approach uses the enrichment of predictions using several conformations (either obtained with different templates or the same). Both approaches are explained below. For the sake of analysis, we classify all target TF sequences from CIS-BP in in groups of ID as in section 10 and obtain the predictions for each of them using their structural model.
a. Straight-forward prediction The straight-forward prediction is based on the selection of one single model as the structure of TF bound with DNA. The straight-forward option is to select as template the structure closest to target TF (i.e. with the highest percentage of identical residues in the alignment between both sequences). Then, we model the structure of the complex (see section 8) and calculate the PWM (see section 12). However, in order to analyze the results with several models, we obtain models (i.e. ) for each template, producing hundreds of conformations of the same TF and several PWMs that we name as in section 10, solutions. We evaluate the quality of the prediction using TOMTOM to compare the solutions with the PWM of target TF. We define the score of similarity as in section 10 (i.e.
) ). Then, we calculate the distribution of scores
for all TFs with known PWM in CIS-BP that can be modelled as a function of ID (i.e. with target TFs grouped by ID as in section 10, using the criteria explained on section 8 for modelling). To analyze the results, we calculate the maximum, the minimum and the average of the scores of similarities of all solutions of each target TF. We define the score of ranking by the position in the ranking of the score of similarity with the PWM of the target using equation 44 (in section 10). We calculate the distribution of scores of ranking for all solutions of TFs of CIS-BP as a function of the ID. We also calculate the maximum, minimum and average of ranking of all solutions of each target TF, grouped by ID. We use the score of ranking to calculate the accuracy of the prediction with the same criteria of success as in section 10. However, as we accept several solutions with the same template for the analysis, we use the score of ranking among a top threshold to calculate the ratio of success (i.e. if at least one of the solutions among the top selected is correct the prediction of target TF is successful). The accuracy of the prediction is calculated as the ratio of successful predictions among the total number of TFs with a prediction. The accuracy is calculated as a function of ID as the ratio of successful

104
predictions of TFs over all TFs that were used for the prediction by sequence at the corresponding ID (i.e. grouped as in section 10). The goal of predicting a PWM is to find the potential binding site(s) of the target TF in a DNA sequence. Consequently, the approach to apply the straight-forward prediction uses the predicted PWM of a target TF using one single model (i.e. obtained with the closest template). We use the PWM to scan a DNA sequence and collect the potential bindings detected with FIMO.
b. Prediction by enrichment As in section 10, the prediction by enrichment uses a few selected PWMs of a target TF for the prediction. We calculate the score of ranking as before, i.e. for each target we obtain several models and with each model a PWM, named solution. The solutions are compared with the PWMs of CIS-BP. Then, we select the PWMs of CIS-BP with the top ranked scores over a threshold (i.e. T top ranked PWMs as in section 10) for each solution. The enrichment is defined as in equation 45, where is the number of models generated for target TF ( times the number of templates). We calculate the probability ( ) to select a PWM out of when we have selected T PWMs using equation 46. As in section 10, we define null the enrichment if is higher than , being the total number of TFs in CIS-BP with known PWM and modelled (or known) structure (using the criteria explained on section 8 for modelling). We calculate the distribution of enrichment as a function of ID as before (i.e. with target TFs grouped by ID as defined in section 10). We calculate the accuracy of the prediction by ranking the enrichment of solutions of each target TF as in section 10. The score of ranking of enrichment is defined with equation 44, using the rank of the enrichment instead of the score of similarity. We calculate the score of ranking of the enrichment and coverage of the prediction of each target TF as a function of ID, following the same approach as in section 10. Then, we use the score of ranking of enrichment to calculate the accuracy of the prediction. As in section 10, we use all solutions of a target TF to predict the binding site in a DNA sequence. We use FIMO to scan the DNA sequence with each solution and collect the resulting fragments (it has to be noted that fragments are collected as many times as they are detected by the different solutions). Then, we calculate the enrichment for each DNA fragment (or nucleotide) using the number of times the fragment (or nucleotide) has been collected over the total number of solutions. We calculate and plot the enrichment by nucleotide along the DNA sequence (i.e. a nucleotide profile) as in section 10. We limit the enrichment by the significance of the matches of FIMO to filter out unreliable results and ensure the quality of the prediction.

105
c. Comparison of different sets and methods to predict PWMs We note that the comparison between results of predicting PWMs of TF targets by means of structures and by sequences is not straight forward. The quality of the PWMs predicted by sequence is determined by the PBM experiments, while for the prediction with structural models there is no experimental information. Therefore, the scores of similarities of different solutions have dissimilar ranges: while for the sequence approach the scores of similarities may reach values of 100, the solutions by structural models never reach values higher than 10. In order to compare the results of these different approaches, we need to normalize them. Given a score of similarity or a score of ranking, , we define the normalized score as in equation 51 but scaling between 0 and 100:
(eq. 54) Where and are respectively the minimum and maximum values of the scores obtained with all solutions of TF targets. The analysis and comparison of approaches can also be done by groups of TFs in families. TF targets are grouped according to their family, as defined in CIS-BP. A TF may have more than one DNA binding domain, consequently it may be ascribed to more than one different family. Then, instead of belonging in more than one group, we define the family of the TF as the sum of family names and form a new group. The values of and to normalize the scores in the family specific analysis are restricted to the specific set of TF targets, which implies varying the expectation of scores for each particular set. The approach doesn’t imply using solutions of a specific family but entails the results of TF targets of a specific family. The different accuracies proof that for some families it may be easier to correctly predict a PWM than others. Therefore, the analysis by families affects the scores of rankings and consequently the accuracy of the prediction per family. Furthermore, we calculate the average and statistical deviations of scores with the results of families as a function of ID (ID groups of TF targets defined as in section 10), with and without normalization:
(eq.55)

106
Where, is a family group of TFs, is any of the scores defined above for a TF target in set ID that is calculated by modelled structures or by sequence (see above and sections 10, respectively). Then, we calculate the accuracies of prediction per family using the scores of ranking (i.e.
) and calculate the average and deviation using all families:
(eq.56) Where is the set of all different families grouping the TF-targets.
14. Optimal conditions to predict PWMs using the structure of a TF (grid search)
We use the experimentally known PWMs in CIS-BP to optimize the structural-based prediction of PWMs. The definition of the statistical potentials and the conditions for modeling the structure of TF-DNA interactions and obtaining the PWM require some parameters (i.e the selection of cut-off and binned distances on potentials, using a general or a family-specific potential, etc.) that need to be optimized. Thus, we predict the PWM by means of structure for all TFs in CIS-BP that can be modelled (see section 12) and compare the predicted PWMs with the experimental PWMs by means of TOMTOM. We test the following the parameters/conditions:
1) Use potentials derived by PDB data only or adding PBM data. 2) Use general or family specific potentials. 3) Use Taylor's approach or none to infer triads when the amount of data
is limited. 4) Use potentials computed with bins or accumulative distance intervals. 5) Use maximum contact distance between amino acid and dinucleotide
of 15 Å, 22 Å or 30 Å. 6) Use several family and general thresholds for PWM as described
in section 12. We test values from 0.7 to 1.0 in steps of 0.01 and select the thresholds yielding the best results of the TOMTOM comparison. Among the best results of TOMTOM, we select the smallest threshold value.
We select the combination of conditions producing the best score and p-value with TOMTOM and obtain different sets of optimized parameters for all TFs grouped in different TF families as defined in CIS-BP.

107
The comparison between experimental and predicted PWMs is done using TOMTOM from the MEME suite (see sections 1 and 13). We select the parameter combination producing the largest number of solutions in a family set with a TOMTOM p-value under 0.05 (i.e. ). If more
than one combination can be accepted, we select the combination that produces the maximum average of in the set of the family.
15. Scanning of binding sites and TF clusters along a DNA sequence. When we introduce a DNA sequence as input (DNAseq), we test the capacity of one or several TFs, with known or modeled structure, to bind in one or more binding sites of the DNA. This analysis is performed by scanning and scoring the DNA sequence. We split this analysis in two main parts: 1) scanning and scoring the binding and calculating a binding profile pattern specific of a TF (sections a-c); and 2) selecting several TFs and grouping them in clusters of specific binding regions (sections d and e).
a. Scanning of DNA binding domains The DNA sequence is scanned with the PWMs obtained with all the structures of TFs and protein-DNA interactions collected from the PDB (defined in section 1 as PDBDNA). To obtain these PWMs, we need to consider that several proteins and TFs interact with a DNA sequence in the form of hetero- and/or homo- complex of proteins forming a quaternary structure. Therefore, for each complex structure of PDBDNA, we obtain one or several PWMs as follows: 1) we detect all protein chains interacting with any of the two DNA chains (i.e. strands) forming the double-strand helix; 2) we construct the structures of all combinations of protein-chains that bind the same helix of the original structure; and 3) we calculate the PWMs of these structures and store them associated with the corresponding structure (we name this set as 3D-PWM). This approach produces PWMs of individual chains and their combinations. For example, given a heterodimer with two protein chains, A and B, we obtain three different PWMs: one for the binding of chain A, another for chain B and another for the heterodimer formed by A and B. In addition, for the web service the user can also upload a specific PWM associated with a TF sequence, then the service checks the closest PWM stored in 3D-PWM associated with a structure and replaces it with the uploaded PWM for the scanning search, hence accommodating our approach to the specific needs of users. The scanning is performed with the program FIMO, using all PWMs stored in 3D-PWM set, limiting the search by a P-value threshold of significance, or with the specific selection of TFs (potentially allowing in the web service to use the corresponding PWMs uploaded by the user). We use a maximum P-value threshold of 0.05 by default, but this may be increased in order to enlarge the number of potential binding proteins.

108
b. Score per Nucleotide: profiles of a DNA binding site. We define a nucleotide profile as a function on , , of the nucleotide position in a DNA sequence We have introduced some examples in sections 10 and 13. Here, we define score-nucleotide profiles when the function is obtained with the scores and z-scores developed in sections 4-6 with statistical potentials. Given a TF-DNA complex structure, 3D-TF, and a distance dependent score, , obtained with statistical potentials (e.g. the smoothed z-score of ES3DCdd). Let be a nucleotide position of the DNA sequence in the complex, we define a new score, in , as:
(eq.57)
Here we use extended-triads without loss of generality, although depending on the statistical potential we can use triads or feature triads instead. The set
is the set of etriads with all associated distances (i.e. any and amino-
acid residue numbers (i.e. any where dinucleotide in position implies that is the position of one of the nucleotides in the dinucleotide at (i.e.
). This new score can be normalized by considering the contribution of the nucleotide to the total score or as the percentage of the contribution of all nucleotides of the DNA sequence. Assuming that the score is the smoothed z-score of ES3DCdd and without loss of generality, the normalized nucleotide profile is:
(eq. 58) or
(eq. 59) Where for all but the extremes at 5’ and 3’, in which is 1, and
from equation 47 we write as:
(eq. 60)

109
is the set of extended-triads (with their associated distances, amino-acid numbers and dinucleotide positions) as defined in section 4 and is the length of the DNA sequence. The factor of 2 in equation 59 is produced by the fact that each nucleotide is counted twice in , with the exception of
the extreme positions in 5’ and 3’ where the nucleotides are only reckoned one time. The curve of (raw or normalized) along the positions in the DNA sequence is defined as the nucleotide profile based on 3D-TF for the (raw or normalized) potential defined in (e.g. the smoothed z-score of ES3DCdd). Hence, profiles defined upon scores derived from statistical potentials are dependent on the structure of the TF-DNA interaction complex. We have to note that, if the structure of this complex has been modelled, several models may be considered (i.e. we define the set of models of TF-DNA as ). Besides, some models may be obtained using different templates, implying that these models introduce a relevant variability on the conformational space of the TF-DNA interaction. Consequently, several nucleotide profiles of scores are accumulated for the same DNA sequence. We then calculate the average and standard deviation of for all positions along the DNA sequence with the nucleotide profiles of score based on each model structure of the set (i.e.
described as , using following equations:
(eq. 61) Equations in 61 describe two new nucleotide profile functions. The average function is defined as the nucleotide profile of (e.g. the smoothed z-score of ES3DCdd) and the RMSD defines its margins of error or variability. As seen in sections 10 and 13 and mentioned above, the nucleotide profile can also be calculated with other “scores” different than those derived by statistical potentials, for example by the enrichment or the number of times that nucleotide j is selected by several PWMs assigned to a TF (see above). This extends the definition of score nucleotide profiles to other scores different than those obtained with statistical potentials. c. TF profiles along a DNA fragment. We define TF profiles as nucleotide profiles where the value of the function is obtained with the whole interaction (or involvement) between the TF and

110
DNA that can be associated to a nucleotide position. Let be a DNA sequence (DNAseq ) and a TF with known (or predicted) PWM. We use FIMO to calculate and rank the score ( ) and significance (i.e. ) for several positions along the DNA sequence where the significance is acceptable (i.e. ). Both, score and significance, are associated with a specific interval of the DNA sequence, , that identifies the nucleotides matching the PWM in a ranking order of quality (identified by r). The position of a nucleotide, in can be matched more than once by the same PWM (depending on the limit of significance to accept the matches). Then, considering that position is matched by PWM times, and identifying the score and significance of each instance, , respectively as
and , we define the following TF profile based on the PWM:
(eq. 62)
As for nucleotide profiles, more than one PWM can be predicted for a TF, either by sequence or by structure. Consequently, if is the set of PWMs predicted for the TF, we define the TF profile of the score of FIMO and the score of significance (respectively) as:
(eq. 63) When the prediction is based on the structure of a TF-DNA complex, set is the set of PWMs obtained by the models in from the previous section. As above, we also define other two TF profiles for the variability and deviations from the average, RMSD, as:

111
(eq. 64) Another set of TF profiles is obtained by using the scores and z-scores developed in sections 4-6 with statistical potentials for the score of a TF-DNA interaction as in section 11. Let be DNAseq the DNA sequence and TF3D the known (or modelled) structure of the TF with a DNA sequence in a crystal (or the DNA in the template, if TF3D is modelled). We use TF3D to calculate the interface and determine the length, , of DNA in
contact with the TF, as in section 8. We split the sequence in fragments using a sliding window of nucleotides. Then, we define a DNA
fragment similarly as in section 12, DNAseq(k), of DNAseq in position k, as the fragment of DNAseq in the interval [k,k+ ]. We model the TF-DNA
complex structure, 3D-TFk, for any position k along the DNA sequence and calculate the binding score as in section 11 (see eq. 36) for any score or z-score. Let be the example, without loss of generality, of the smoothed z-score of ES3DCdd. This example scores the TF-DNA interaction in 3D-TFk as:
(eq. 65) Where, is the set of extended-triads (with their associated distances, amino-acid numbers and dinucleotide positions) for the complex in 3D-TFk. Let be a position in DNAseq. According to the previous definitions, we calculate a total of binding scores containing position , as if the
TF was sliding along the DNA sequence from [ , ] to [ ,
]. Then, we define the following TF profile based on TF3D,
using the smoothed z-score of ES3DCdd, as:
(eq. 66)
We normalize the score by scaling it between 0 and 1, being 1 the best score and 0 the worst. This may imply a change on the original sign of the score. For the normalization we require to calculate the scores with the best and the worst interface DNA sequences. The best score is obtained by testing the scores of the DNA sequences considering the most probable nucleotides in each position, according to the PWM obtained with the

112
structure of the TF-DNA complex (see section 12). Using in equations 47 and 49 without loss of generality, the best score is the minimum of the values tested and we write it as . Similarly, for the worst score we test the values of this function on several DNA sequences considering the less probable nucleotides in each position and we take the maximum, Then, we define the normalized TF profile based on TF3D as:
(eq.67)
As previously shown, a TF may have more than one structural model or conformation. Following the same approach as before and defining as the set of models of the TF-DNA interaction, we define without loss of generality the TF profile of the smoothed z-score of ES3DCdd as:
(eq. 68)
Where is the TF profile based on model of the
smoothed z-score of ES3DCdd. And the normalized version of eq. 68 is:
(eq.69) Following the same approach as in equation 64 we also define the TF profiles for the deviations in equations 68 and 69, respectively as:
(eq. 70)

113
d. Prediction of TFs that bind a DNA fragment
Given a DNA sequence as input, our goal is to predict the set of TFs that binds it in a cell or specific species. The first step is to collect all PWMs of TFs with known structure and scan the DNA sequence (see previous section 15.a). This identifies the potential structures and families but not yet all potential sequences. We use BLAST to collect the potential protein homologs of all structures in PDBDNA (see sections 1 and 8) and the corresponding BLAST alignments. Next, we select the proteins and their alignments and construct two mapping functions: 1) the mapping between the protein sequence and the template TF in PDBDNA (mapP); and 2) the mapping of dinucleotides in the DNA interface (mapD as defined in section 9, which is obtained with the alignment between the sequence matched by the PWM and the DNA sequence in the template). Instead of modeling the TF-DNA complex, we use the mapping functions to calculate the scores, using equation 36.b in section 11, and rank the potential binding proteins by scores. If the TF belongs to one of the families that may work as homodimer, we calculate the scores and their ranking using the structures of the monomer and the dimer.
e. Clusters of TFs and complexes of regulatory elements. Given a DNA sequence as input, we group the predicted TFs and their binding sites (see previous section 15.d) by the proximity in the DNA sequence (using as cut-off a limiting number of nucleotides, ). In case a TF acts as a dimer, we force in the same group both monomers of the dimer. The group may contain TFs with overlapping binding sites. This must be allowed because only the disposition of the structure in 3D allows us to determine if the complex can be formed. Furthermore, we add other proteins that interact with any of the TFs binding the DNA sequence. We use the database IntAct of protein-protein interactions (PPIs) to identify the potential partners that interact with the predicted Tfs and consider only those of the same species as the TFs. If two TFs belonging to different groups of TFs (and their bindings) are connected by PPIs (either interactions between TFs or with a common partner) we merge both groups in one. In order to find the connection of TFs by means of PPIs we construct a network in the following steps: 1) We define the DNA binding sites predicted as main nodes. 2) We define the TFs as nodes. They are connected to the one or more
main nodes if they are predicted to bind them. 3) We add the proteins that interact with the TFs as nodes connected to the
corresponding TF partners. The approach can be continued, adding proteins (nodes) and interactions (edges) that interact with the proteins of the network as it was left in step 3 and then iteratively continue. Two TFs are connected if they interact in the database of PPIs or there is an intermediate protein that interact with both.

114
In the web service the user can select a subnetwork of proteins in this network and model the structure of the binary interactions of TF-DNA and PPI. The ranking of the scores of TF-DNA interactions helps on this selection. Each group of the final set of groups formed by proteins and binding sites is named cluster, and the selection of elements (proteins and DNA binding sites) of a cluster in the web service is a potential TF-complex or regulatory complex.
16. Modeling a selected TF-complex of a cluster of close TFs in a DNA fragment
Following the selection of TFs of a cluster, their protein-interacting partners and DNA binding sites from sections 15.d and 15.e, we use the modelling approaches from section 8 to construct each individual binary-complex structure (i.e. PPIs and TF-DNA). To model the structure of their combination forming the TF-complex, we have first to define the expected structure of the fragment of DNA sequence that contains all the binding sites selected from the cluster. We allow for few automated options, either as B-DNA, Z-DNA or curved DNA (as in nucleosomes). The structure of the DNA fragment with the binding sites selected (i.e. a continuous interval from 5’ to 3’ taken from the original DNA sequence input, DNAseq) is modelled with X3DNA according to one of the options (this is named DNA-frame). Because the approach for modelling binary interactions (either TF-DNA or PPIs) allows for several potential conformations, it is likely that each PPI and TF-DNA complex has more than one structural model. Therefore, we produce two lists: 1) one list (TF-DNA list) with all the combinations of individual TF-DNA binary-complexes; and 2) another list (PPI list) with all combinations of conformations of the selected PPIs. The structures in each list are ranked by the maximum length of protein sequences that can be modelled, both for TF-DNA and PPI complexes. Next, for each combination of the TF-DNA list, individual binary-complexes of TF-DNA of all binding sites selected in the list are superimposed on the DNA-frame. The DNA structure of the binary-complexes of TF-DNA are removed after the superimposition, leaving each TF structure bound on the DNA-frame with the corresponding conformation as in the binary-complex (named DBD-complex and identified by a number from TF-DNA list). Often the conformation of the DNA binding domain of a TF is separated from the structure of other domains. Consequently, with the exception of few cases, it is not possible to combine the lists of PPI structures and each DBD-complex. Thus, in the web service the user can select a specific DBD-complex and one of the combinations of PPIs and handle it for his own specific modeling. Then, the selected set of PPIs and DBD-complex is understood as a model of the TF-complex. 17. Testing the applicability of ModCRE on the UniProt database We used the protein codes and sequences are extracted from UniProt (January 2019 release) (13). We also used the cross-referenced databases provided by

115
UniProt, where all the matches of hidden markov models (HMMs) from the PFAM database (22) are indicated. We searched for the matches corresponding with the HMMs of the major TF families analyzed in this work. The analyzed families and the IDs of their corresponding HMMs are shown the next table:
TF family PFAM ID TF family PFAM ID AFT PF08731 MBD PF01429
AP2 PF00847 Myb/SANT PF00249
APSES PF04383 NAC/NAM PF02365
ARID/BRIGHT PF01388 Ndt80/PhoG PF05224
B3 PF02362 Nuclear receptor PF00105
C2H2 ZF PF00096 POU PF00157
C2HC ZF PF01530 Paired box PF00292
CUT PF02376 RFX PF02257
CxxC PF02008 Rap1 PF09197
DM PF00751 Runt PF00853
E2F PF02319 SMAD PF03165
Ets PF00178 Sox PF12336
Forkhead PF00250 T-box PF00907
GATA PF00320 TBP PF00352
GCM PF03615 THAP finger PF05485
HSF PF00447 WRKY PF03106
Homeodomain PF00046 Zinc cluster PF00172
IRF PF00605 bHLH PF00010
MADS box PF00319 bZIP PF00170
Table S2. TF families for which we analyzed ModCRE applicability and the PFAM IDs of the HMMs used in this analysis.
We considered each match of a HMM with a protein sequence as one DNA binding domain (DBD) of the TF family corresponding with the HMM. Therefore, if a protein had more than one match, we considered that protein to have multiple DBDs. For all the identified domains, we searched for structural templates among the set of sequences with known structure in PDBDNA using BLAST (19). The criteria to select templates were the same than we explained previously in section 8 (Structural modeling of TFs complexed with DNA) of these supplementary methods. We considered that we can apply ModCRE to all domains with, at least, one viable template. Domains with available templates were classified into restrictive and non-restrictive. For restrictive domains we obtained templates without gaps in the template-target alignment in the regions of the protein-DNA interface with defined secondary structure. Non-restrictive domains could be modeled without applying this restriction. As expected, all restrictive domains count also as non-restrictive, because templates can be found for them if the restrictions are not applied. The analysis on DBDs is shown in supplementary figure S3B. We extended the analysis with DBDs to entire proteins (supplementary figure S3A). If one protein had at least one domain that could be modeled, we considered that we could apply ModCRE to that protein. If one protein had two DBD from different TF families with available templates, that protein counted for

116
the two TF families. If these DBD had templates under restriction, the protein would be considered to belong to the restrictive group. References 1. Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of
hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–637.
2. Lu X-J, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3(7):1213–27.
3. Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics. 2000 Jun 1;16(6):276–7.
4. Altschul SF, Gertz EM, Agarwala R, Schäffer AA, Yu Y-K. PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009 Feb 1;37(3):815–24.
5. Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017 Nov;35(11):1026–8.
6. Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Current Protocols in Protein Science. 2016;86(1):2.9.1-2.9.37.
7. Shindyalov IN, Bourne PE. A database and tools for 3-D protein structure comparison and alignment using the Combinatorial Extension (CE) algorithm. Nucleic Acids Res. 2001 Jan 1;29(1):228–9.
8. Mooers BHM. Simplifying and enhancing the use of PyMOL with horizontal scripts. Protein Sci. 2016 Oct;25(10):1873–82.
9. Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–9.
10. Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015 Jul 1;43(W1):W39-49.
11. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012 Dec 1;28(23):3150–2.
12. Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017 Jan 4;45(Database issue):D271–81.
13. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019 Jan 8;47(D1):D506–15.
14. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell. 2014 Sep;158(6):1431–43.

117
15. Persikov AV, Wetzel JL, Rowland EF, Oakes BL, Xu DJ, Singh M, et al. A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Research. 2015 Feb 18;43(3):1965–84.
16. Persikov AV, Rowland EF, Oakes BL, Singh M, Noyes MB. Deep sequencing of large library selections allows computational discovery of diverse sets of zinc fingers that bind common targets. Nucleic Acids Research. 2014 Feb 1;42(3):1497–508.
17. Fornes O, Garcia-Garcia J, Bonet J, Oliva B. On the Use of Knowledge-Based Potentials for the Evaluation of Models of Protein–Protein, Protein–DNA, and Protein–RNA Interactions. In: Advances in Protein Chemistry and Structural Biology [Internet]. Elsevier; 2014 [cited 2019 Sep 12]. p. 77–120. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128001684000044
18. Feliu E, Aloy P, Oliva B. On the analysis of protein-protein interactions via knowledge-based potentials for the prediction of protein-protein docking. Protein Sci. 2011 Mar;20(3):529–41.
19. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997 Sep 1;25(17):3389–402.
20. Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999 Feb;12(2):85–94.
21. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994 Nov 11;22(22):4673–80.
22. Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Research. 2021 Jan 8;49(D1):D412–9.

118
3.1.2 - On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF
Meseguer A, Årman F, Fornes O, Molina-Fernández R, Bonet J, Fernandez-Fuentes N, et al. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. NAR Genom Bioinform [Internet]. 2020 Sep 1 [cited 2020 Sep 2];2(3). Available from: https://academic.oup.com/nargab/article/2/3/lqaa046/5866110

119

120

121

122

123

124

125
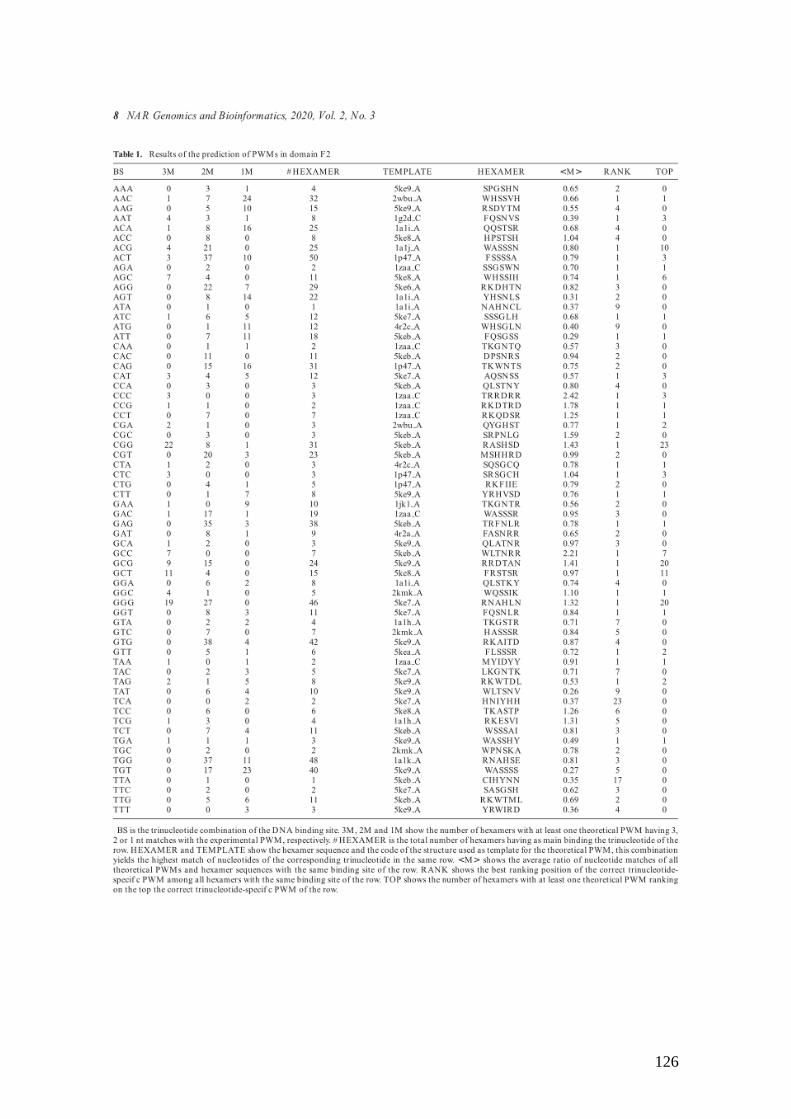
126

127

128

129

130

131

132
SUPPLEMENATRY MATERIAL FOR “On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF” Alberto Meseguer1,*, Filip Årman1,*, Oriol Fornes2, Ruben Molina-Fernandez1, Jaume Bonet3, Narcis Fernandez-Fuentes4,5, Baldo Oliva1. 1Structural Bioinformatics Lab (GRIB-IMIM), Department of Experimental and Health Science, University Pompeu Fabra, Barcelona 08005, Catalonia, Spain, 2Centre for Molecular Medicine and Therapeutics, BC Children's Hospital Research Institute, Department of Medical Genetics, University of British Columbia, Vancouver, BC V5Z 4H4, Canada, 3Laboratory of Protein Design & Immunoengineering, School of Engineering, Ecole Polytechnique Federale de Lausanne, Lausanne 1015, Vaud, Switzerland, 4Department of Biosciences, U Science Tech, Universitat de Vic-Universitat Central de Catalunya, Vic 08500, Catalonia, Spain, 5IBERS, Institute of Biological, Environmental and Rural Science, Aberystwyth University U.K
ANNEX OF RESULTS 1. Using statistical potentials to detect binding and affinities. We hypothesize that statistical potentials can differentiate binding from non-binding DNA sequences. To prove it, we calculate the difference of the scores obtained with the same hexamer sequence but different types of binding sites: DNA bindings sequences according to the B1H experiment, and potential non-binding sites (ten times larger in number to avoid any bias on the selection) retrieved from a background DNA fragment (see further in the extension of methods). We use a single template to model the structure of Zif268 bound with DNA (chain A of 1P47) for this analysis. We model the sequence of DNA according to the experiment of B1H using binding and non-binding fragments (see details in methods). We use two sets of DNA sequences, one of experimentally asserted bindings and another composed of a random selection of fragments of the sequence used as background for non-binding. We restrict the scoring to the 6 variable amino acids of the finger (hexamer fragment) in interaction with the 9-mer DNA-sequence (including the 3 variable nucleotides tested for each finger sequence) at a distance shorter than 15 Å. We calculate the difference between normalized Z-scores ( ) of two modelled structures obtained with two different DNA sequences but the same hexamer fragment. We define as score the difference between normalized Z-scores of any two models. We calculate the distribution of scores obtained between a bound and a non-bound DNA sequence (bound distribution). The distribution of the differences between unbound DNA sequences is used as background (i.e. this is a gaussian background distribution around zero). Supplementary figure S1 shows the distributions of scores for F2 and F3 domains. score distributions of

133
bound and unbound are significantly different (P-value < 0.001, using a Mann-Whitney test). 2. Selection of TFs for the comparison with JASPAR motifs.
We identify all TFs of the C2H2-ZF family analyzed in JASPAR (a total of 181). Then, we select only those formed by 3 zinc-finger domains that can be modelled with the structure of Zif268 (even if it includes other different domains). They are identified using the HMM profile of the ZF-C2H2 family (PF00096.21) from PFAM(1), selecting a list of 40 TFs (see supplementary table S3). We use all structures of the family C2H2-ZF in the PDB(2) as potential templates and select the closest homologs to model each TF. We skip models with less than 80 amino acids and only analyze proteins with 3-4 finger domains (i.e. less than 120Aa), which reduces the set to 29 TFs. No more than 40 templates are used per TF, usually between 10 and 30 and occasionally only one or two, so every TF has several models (one per template). We use the models to obtain the theoretical PWMs of each TF using all contacts under 30Å (see details in methods). For each TF we compare its motif in JASPAR with the set of theoretical PWMs. We also test the theoretical PWMs with contacts under different thresholds (ranging between 15Å to 30Å). 3. Examples of the prediction of PWMs among ortholog and paralog TFs.
The PWMs of some TFs are compared with more than one possible motif in JASPAR, often associated by some relationship in evolution (i.e. among orthologs and paralogs of different species). Examples are shown in figure S2. For example, Q43474 and Q60793 have PWMs created with templates 1p47 (chain C), 1a1i (chain A) and 1a1j (chain A) respectively similar with motifs MA0039.2, MA0039.3 and MA0039.1; or some models of P08046 and P18146 generate PWMs very similar to motifs MA0162.1, MA0162.2 and MA0162.3. Actually, P18146 corresponds to the human sequence of the protein produced by gene EGR1, P08046 corresponds to the sequence of its ortholog gene in mouse, while motif MA0162.1 corresponds to the mouse gene and MA0162.3 and MA0162.2 are the motifs in JASPAR corresponding to human. The high similarity of all of them is caused by the similarity of the DNA binding sequence, which is reflected in the structural models. Another example is shown in figure S2 to compare different motifs (MA0472.1, MA0732.1 and MA0733.1) with human and mouse sequences of non-orthologous genes EGR3 and EGR2 (Q06889 and P08152), respectively.
4. Study of biases caused by the sequence similarity between the
transcription factor and the template when comparing a theoretical PWM with its motif in JASPAR.
The statistical potentials are obtained with structures of C2H2-ZF members and experiments from B1H data of C2H2-ZF proteins. Here we test if the success of our approach depends on the similarity between the query sequence and the sequences of the structures used to create the PWMs and the statistical

134
potentials. First, we compare the sequence of the query with the sequence of Zif268, because this has been used in all the models to create the potentials using B1H experiments. Second, we compare the sequence of the query with the sequence of its template (closest homolog with available structure). These templates are used to generate the statistical potentials as well as to make the models from which we will obtain the theoretical PWMs. We calculate the similarity between the theoretical PWM and its motif in JASPAR. We measure this similarity as the logarithm of the P-value of significance. Then, we compare the sequences of the query and the template and calculate their similarity as the percentage of identical residues in the alignment. Finally, we compare the criteria of similarity to find if there is a relationship between both. Figure S3, plots A and B, show the comparison of sequence and PWMs similarities with respect to the sequence of Zif268. We observe a strong Pearson correlation statistically significant (P < 0.001) using ZES3DCF2 (0.725) and ZES3DCF3 (0.74) statistical potentials. This correlation is mainly caused by theoretical PWMs very similar to their motif in JASPAR obtained when the sequences are almost identical to Zif268, while the theoretical PWM deviates from the corresponding motif for proteins with low sequence similarity with Zif268. After removing proteins highly similar to Zif268 (> 60% sequence identity) the Pearson correlation is downgraded. Similarly, plots C and D in Figure S3, show the sequence and PWMs similarities where sequence similarities are calculated between the sequence of the TF and the template used to construct the theoretical PWM. Strong and significant Pearson correlations are also observed using ZES3DCF2 (0.70) and ZES3DCF3 (0.68) statistical potentials. Nevertheless, we also observe that TFs with sequence very different to the sequence of Zif268 yield theoretical PWMs very accurate (significantly similar to the corresponding motif in JASPAR). Similarly, theoretical PWMs of TFs, constructed with templates very different in sequence, can be very similar to their motif in JASPAR. These results demonstrate the applicability of the approach using the structures of remote homologs, or templates with low sequence similarity. Further, we have tested the bias effect on the statistical potentials for the application on human CTCF. The number of structures of CTCF used to calculate the statistical potentials is about 12% of the total of C2H2-ZF structures (13 out of 113). These structures mostly affect amino acid-dinucleotide interactions at distances between 15Å and 30Å. This may have a significant effect on the potential, so we have removed these structures and recalculated the statistical potentials, which are then free of any information on CTCFs. We compare the theoretical PWMs of the modelled structure of CTCF with motif MA0139.1. This structure contains zinc fingers 2 to 11 and its corresponding PWMs are obtained with statistical potentials ZES3DCF2 (and ZES3DCF3) at cut-off thresholds between 15 and 30 A. Besides, these PWMs are made using two types of potentials: potentials including all C2H2-ZF structures and potentials that do no include CTCF structures.

135
The best matches with the JASPAR motif of CTCF (MA0139.1) are obtained with cut-off distances between 15Å and 22Å instead of 30Å when the potential does not include structures of CTCF. Figure S5 shows the comparison of logos. We observe that the inclusion of CTCF structures is only noticed after 20Å, while the PWMs between 15Å and 20Å are mostly influenced by the B1H experiments, showing that the theoretical PWMs calculated with ZES3DCF2_all or ZES3DCF2_CTCFfree are more similar between them, but less with respect to motif MA0139.1. This test suggests that we can avoid the biases on the statistical potential, caused by structures of close homologs of a testing TF, by reducing the contacts to less than 20Å. Further, we quantify this effect by comparing the ratio on nucleotide-matches between the JASPAR motif and each theoretical PWM with respect to the cut-off distance used to calculate contacts (see table S5). Theoretical PWMs of TFs used in the comparative analysis with motifs in JASPAR (section 3 of the manuscript) are obtained with potentials ZES3DCF2 and ZES3DCF3 using cut-off distances at 15 Å, 18 Å, 22 Å, 25 Å, 28 Å and 30 Å and can be downloaded from http://sbi.upf.edu/C2H2ZF_repo. Supplementary table S4 shows the results using contacts under 18 Å. These results prove that for 8 (using ZES3DCF2) and 13 (using ZES3DCF3), out of 29 TFs, the number of theoretical PWMs significantly similar to the corresponding motif is higher than 50%. These are very significant results, because it suggests we may find the potential binding of a TF while reducing the biases on the statistical potential with a relevant degree of accuracy. However, the number of TFs is lower than when we use statistical potentials with all contacts under 30 Å. Therefore, we have tested the approach using all contacts, but with unbiased statistical potentials. To obtain unbiased statistical potentials, we first identify for all TFs under test the structures in the database of their close homologs. Then, we produce TF-specific statistical potentials without the contacts derived from their homologs in the database. For example, TFs P08046, P18146, Q06889, P08152, P11161, Q05215 are too similar (more than 50% identical residues aligned) with the sequences of the known structures of Zif268 (i.e. PDB codes 1A1J, 1AAY, 1ALI, 4X9J, 1A1K, 4R2A, 1ZAA, 1G2F, 1A1F, 1G2D, 1A1L, 1A1G, 4R2C, 1A1H). Consequently, we construct a specific statistical potential without using contacts from these structures (except those obtained by means of B1H experiments). The results are shown in tabs 5 and 6 of table S4. They proof a good consistency with the predicted PWMs when we use all known structures, with only slight decrease on the ratio of aligned nucleotides and in the number of theoretical PWMs that are significantly similar with their corresponding motif in JASPAR. 5. Analysis of proteins with C2H2-ZF domains in the PDB
We have performed a comprehensive analysis of C2H2-ZF proteins and domains in the PDB. The analysis was performed on April 24, 2020 and, for

136
reproducibility, has been deposited on Github (https://github.com/oriolfornes/C2H2-zf). We focus on proteins from UniProt (3) eukaryotic reference proteomes (one protein per gene) with C2H2-ZF domains based on PROSITE (4) predictions (profile entry PS50157). The analysis revealed that, the total number of C2H2-ZF proteins is 186,703, of which only 136 have a PDB structure; for protein-DNA complexes, the number lowers to 24. The total number of C2H2-ZF domains is 963,553, but the number of C2H2-ZF proteins with at least one domain covered by a protein-DNA complex is 19. For human, the total number of C2H2-ZF proteins and domains is 762 and 7,180, respectively, and only 11 of these proteins have domains covered by a protein-DNA complex. In summary: 1) C2H2-ZF proteins of eukaryotes average ~5 domains (~9.5 for human); 2) less than 0.1% of C2H2-ZF proteins, >80% of which are human, have a PDB structure; 3) of the previous set of proteins, 25% (~55% are human) have protein-DNA complex structure; and 4) most of the 104 C2H2-ZF/DNA complex structures identified are redundant (e.g. ~20% are from Zif268). 6. Applicability of the method on C2H2-ZF TFs from UniProt We have applied the method to each of the 186,703 C2H2-ZF proteins in UniProt reference proteomes of eukaryotes identified by PROSITE. We have been able to obtain a PIR alignment (i.e. input for MODELLER) for approximately 72% (134,399). Assuming that each PIR alignment results in at least one successful model from which to obtain a PWM, >250,000 of the 353,167 UniProt proteins with a predicted C2H2-ZF domain by PROSITE would potentially benefit from our method.
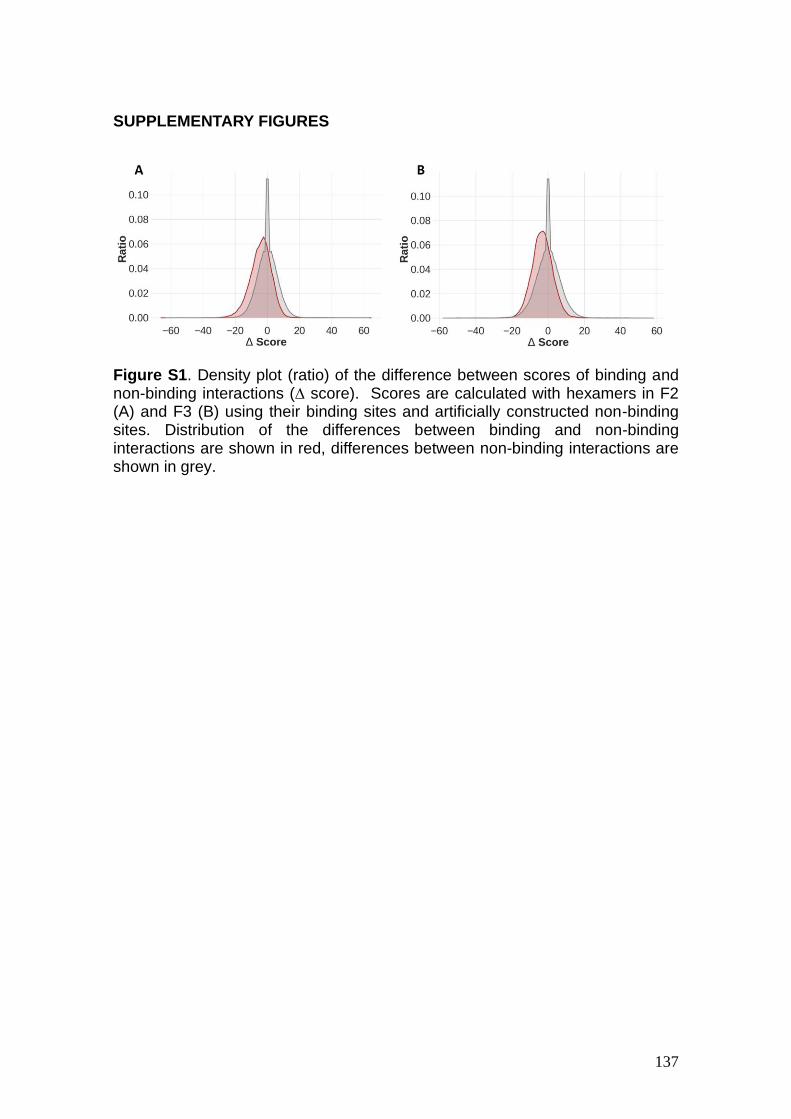
137
SUPPLEMENTARY FIGURES
Figure S1. Density plot (ratio) of the difference between scores of binding and non-binding interactions (∆ score). Scores are calculated with hexamers in F2 (A) and F3 (B) using their binding sites and artificially constructed non-binding sites. Distribution of the differences between binding and non-binding interactions are shown in red, differences between non-binding interactions are shown in grey.

138
Figure S2. Comparison between some theoretical PWMs of C2H2-ZF and their motifs in JASPAR database for ortholog and paralog examples. We use statistical potentials ZES3DCF2 to generate the theoretical PWMs. JASPAR motifs are shown at the top of each comparison. PDB codes of the templates used to construct the theoretical PWMs are also indicated. We highlight with red boxes the theoretical PWMs corresponding to the actual motif of each sequence associated with the same species (only for examples of similar motifs). (A) Examples of transcription factors with more than one motif in JASPAR for orthologous genes (P08046 corresponds with the mouse associated motif MA0162.1 and P18146 corresponds with human associated motifs MA0162.2 and MA0162.3). (B) Examples of transcription factors with different, but very similar motifs in JASPAR, using non-orthologous genes, for proteins Q06889, P11161, Q05215 and P08152 (P08152 corresponds with the same species of MA0472.1).
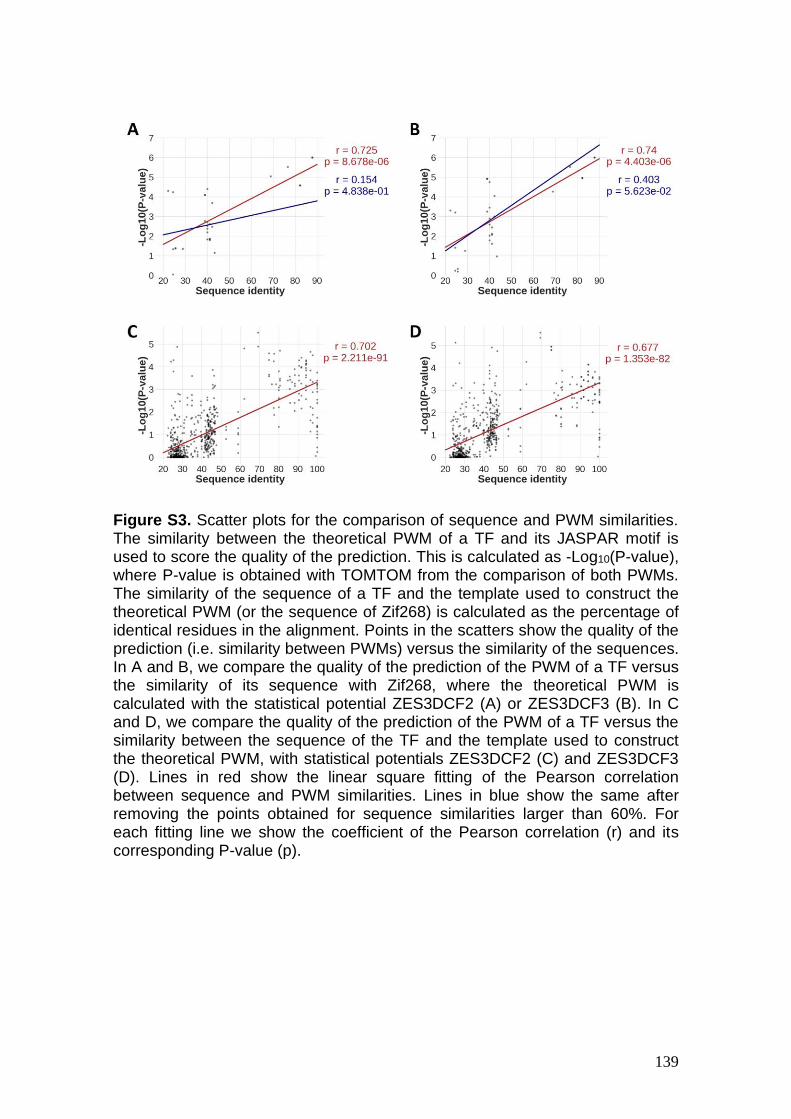
139
Figure S3. Scatter plots for the comparison of sequence and PWM similarities. The similarity between the theoretical PWM of a TF and its JASPAR motif is used to score the quality of the prediction. This is calculated as -Log10(P-value), where P-value is obtained with TOMTOM from the comparison of both PWMs. The similarity of the sequence of a TF and the template used to construct the theoretical PWM (or the sequence of Zif268) is calculated as the percentage of identical residues in the alignment. Points in the scatters show the quality of the prediction (i.e. similarity between PWMs) versus the similarity of the sequences. In A and B, we compare the quality of the prediction of the PWM of a TF versus the similarity of its sequence with Zif268, where the theoretical PWM is calculated with the statistical potential ZES3DCF2 (A) or ZES3DCF3 (B). In C and D, we compare the quality of the prediction of the PWM of a TF versus the similarity between the sequence of the TF and the template used to construct the theoretical PWM, with statistical potentials ZES3DCF2 (C) and ZES3DCF3 (D). Lines in red show the linear square fitting of the Pearson correlation between sequence and PWM similarities. Lines in blue show the same after removing the points obtained for sequence similarities larger than 60%. For each fitting line we show the coefficient of the Pearson correlation (r) and its corresponding P-value (p).
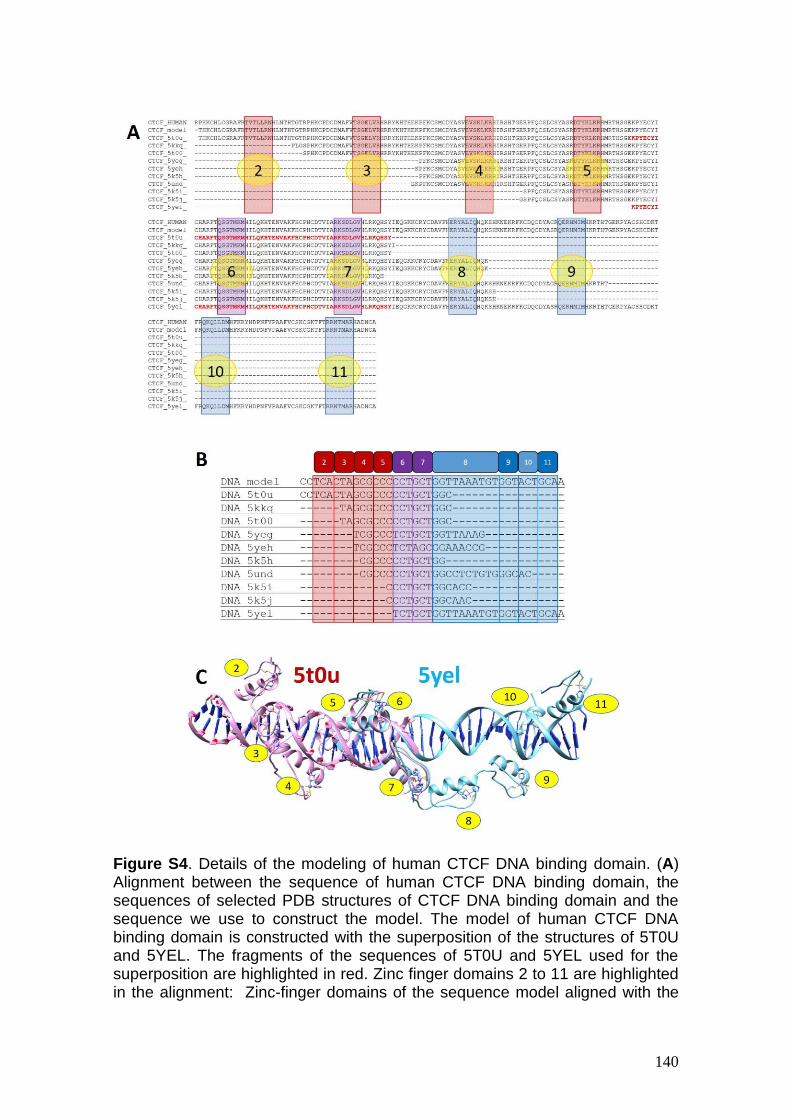
140
Figure S4. Details of the modeling of human CTCF DNA binding domain. (A) Alignment between the sequence of human CTCF DNA binding domain, the sequences of selected PDB structures of CTCF DNA binding domain and the sequence we use to construct the model. The model of human CTCF DNA binding domain is constructed with the superposition of the structures of 5T0U and 5YEL. The fragments of the sequences of 5T0U and 5YEL used for the superposition are highlighted in red. Zinc finger domains 2 to 11 are highlighted in the alignment: Zinc-finger domains of the sequence model aligned with the

141
sequence of the structure 5T0U are shown in red, zinc-finger domains aligned with the sequence of the structure 5YEL are shown in blue and zinc-finger domains aligned with both are shown in purple. (B) Alignment between the DNA sequences of the PDB structures and the sequence of DNA which structure is modelled. Nucleotides of the binding site of each finger domain are highlighted in boxes and numbered after each binding domain. (C) Ribbon plot of the modelled structure of the human CTCF-DNA complex showing zinc fingers 2 to 11.
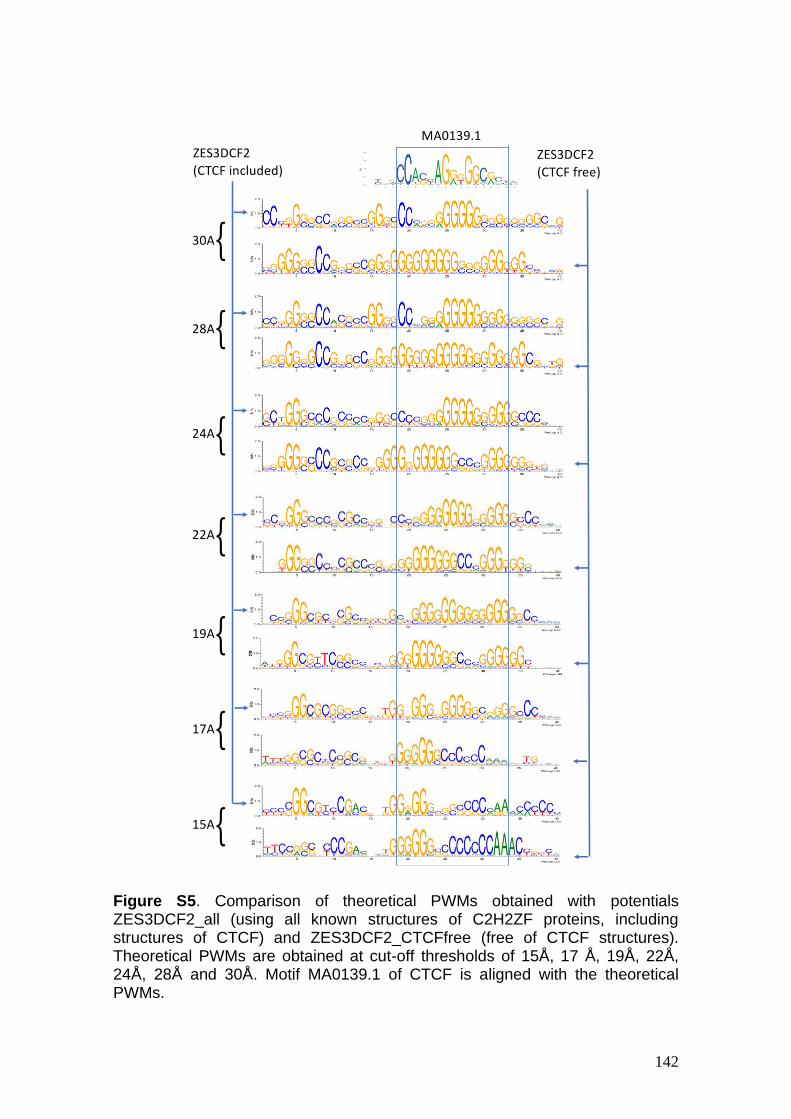
142
Figure S5. Comparison of theoretical PWMs obtained with potentials ZES3DCF2_all (using all known structures of C2H2ZF proteins, including structures of CTCF) and ZES3DCF2_CTCFfree (free of CTCF structures). Theoretical PWMs are obtained at cut-off thresholds of 15Å, 17 Å, 19Å, 22Å, 24Å, 28Å and 30Å. Motif MA0139.1 of CTCF is aligned with the theoretical PWMs.
30A{
28A{
24A{
17A{
15A{
{22A
19A{
MA0139.1ZES3DCF2
(CTCF included)ZES3DCF2
(CTCF free)

143
LEGENDS FOR SUPPLEMENTARY TABLES Table S1. Comparison of hexamer-specific and theoretical PWMs grouped by DNA binding sites. Each tab corresponds to a threshold of percentage of affinity (90%, 75% or 50%), used to obtain the results. All tabs have the data organized in the same columns. Column A shows the zinc finger used to get the potentials (either F2 or F3). Column B shows the DNA binding site. Columns R to U show the number of hexamers with a maximum number of nucleotide matches with the experimental hexamer-specific PWM (nucleotide matches can be 3, 2, 1 or 0; respectively for each column). Column X shows the total number of amino acid hexamers tested to bind the corresponding DNA binding site. Columns Y to AA show the percentage of hexamers with a maximum number of nucleotide matches with the experimental hexamer-specific PWM (nucleotide matches can be 3, 2 or 1; respectively for each column). Column AB shows the hexamer sequence for which we find a theoretical PWM with the highest number of nucleotide matches with the experimental hexamer-specific PWM. Column AC shows the structural model that produces the theoretical PWM in column AB, identified by the PDB code of the template, the hexamer sequence, and the finger domain. Column AF shows the average of nucleotide matches between all theoretical PWMs and all hexamer-specific PWMs with the same binding site of the row. Column AG shows the average score for all theoretical PWMs with the same binding site of the row. This score is obtained as the minus logarithm of the P-value obtained by TOMTOM in the comparison between theoretical and experimental hexamer-specific PWMs. Column AI shows the minimum ranking of the trinucleotide-specific PWM using all theoretical PWMs of hexamer sequences with the same binding site of the row. Column AJ shows the average ranking position of the trinucleotide-specific PWM using all theoretical PWMs of hexamer sequences with the same binding site of the row. Column AK shows the number of theoretical PWMs with the same binding site of the row that rank the trinucleotide-specific PWM in the top. Column AL shows the percentage of theoretical PWMs that are significantly similar to their corresponding experimental hexamer-specific PWM according to TOMTOM (P-value < 0.05). Column AM is one if at least one theoretical PWM among all hexamer-specific PWMs with the same binding site of the row ranks in the top the trinucleotide-specific PWM, and zero otherwise. Column AN is one if at least one theoretical PWM matches three nucleotides with the DNA binding site, and zero otherwise. Column AO is one if at least one theoretical PWM matches two nucleotides with the DNA binding site, and zero otherwise. Column AP is one if at least one theoretical PWM matches three or two nucleotides with the DNA binding site, and zero otherwise. Table S2. Comparison of hexamer-specific and theoretical PWMs. Each tab corresponds with a threshold of percentage of affinity (90%, 75% or 50%) used to obtain the results. All tabs have the data organized in the same columns. Column A shows the hexamer sequence. Column B shows the zinc finger domain where the hexamer is embedded (either F2 or F3). Column C shows the trinucleotide of the binding site. Column D shows the enlarged DNA binding site by the flanking nucleotides. Column P shows the PDB template of the

144
model that produces the PWM ranking the trinucleotide-specific PWM in the minimum position. Column Q shows the minimum position of the ranking of the trinucleotide-specific PWM. Column T shows the average ranking position of the trinucleotide-specific PWM calculated with all theoretical PWMs of the same hexamer models. Column W shows the model that produces the theoretical PWM that has the maximum number of nucleotide matches with the hexamer-specific PWM. Column X shows the maximum number of nucleotide matches between the theoretical PWM in W and the experimental hexamer-specific PWM. Column Y shows the average of nucleotide matches between the 23 theoretical PWMs constructed with the models and the experimental hexamer-specific PWM. Column AB shows the model that produces the theoretical PWM with maximum score when comparing it with the experimental hexamer-specific PWM. Column AC shows the maximum score obtained with the TOMTOM comparison between the theoretical PWMs and the experimental hexamer-specific PWM. Column AD shows the average of the scores obtained with the TOMTOM comparison of the theoretical PWMs and the experimental hexamer-specific PWM. Column AE shows the lowest P-value from the comparisons between the theoretical PWMs and the experimental hexamer-specific PWM. Table S3. Selection of C2H2-ZF proteins from the JASPAR database. Tab 1 shows the JASPAR ID and the UniProt ID for all proteins from the JASPAR database that belong to the C2H2-ZF family. Tab 2 and tab 3 show the results of the selection of proteins from the JASPAR database that according to a PFAM search have zinc finger domains. In tab 2 all proteins with at least one zinc finger domain are shown. In tab 3 results for selected proteins with only 3-4 zinc finger domains. In tab 2 and tab 3 data is organized with the same columns. Column A shows the E-value of the match between the PFAM model and the whole sequence. Column B shows the score of the match for the whole sequence. Column C shows a correction term applied to the score depending on the bias on sequence composition on the overall sequence. Column D shows the E-value of the best match between PFAM C2H2-ZF domain and the best matched region of the sequence. Column E shows the score of the best match between PFAM C2H2-ZF domain and the best matched region of the sequence. Column F shows a correction term applied to the score depending on the bias on sequence composition for the sequence region having the best match with the HMM. Column G shows the number of C2H2-ZF domains found with the PFAM model. Column H shows the natural number of column G. Column I shows the UniProt ID of the sequence studied. Table S4. Comparison of theoretical PWMs of transcription factors with their motifs in the JASPAR database. Tab 1 shows results obtained using statistical potentials based on data for domain F2 (ZES3DCF2) and cut-off distance of 30 Å. Tab 2 shows results obtained using statistical potentials based on data for domain F3 (ZES3DCF3) and cut-off distance of 30 Å. Successive odd tabs show results with statistical potential ZES3DCF2, while even tabs show results with statistical potential ZES3DCF3. Tabs 3 and 4 are obtained using the corresponding potentials and cut-off distance of 18 Å, tabs 5 and 6 are obtained with TF-specific statistical potentials where contacts from close homologs have

145
been removed. In all tabs the information is structured in the same columns. Column A shows the UniProt ID of the protein. Column B shows the JASPAR motif ID. Column D shows the model that produces the PWM with the largest number of nucleotide matches with the PWM of JASPAR. The code identifies the structural model by indicating: the UniProt ID, the first and last residue of the modelled structure and the PDB code of the template (i.e. P08046:333:420_1g2d_C_1 is the model of P08046, between amino acids 333 and 420, constructed with chain C of 1g2d as template). Column E shows the P-value of the theoretical PWM selected in D and the JASPAR PWM. Column K shows the number of models that produce a theoretical PWM significantly similar to JASPAR PWM (P-value < 0.05). Column L shows the number of complete models for the DNA binding domain of the protein under analysis. Column M shows the total number of models (complete or incomplete) for the DNA binding domain of the protein under analysis. Column N shows the average of the minus logarithm of the P-values of all theoretical compared with JASPAR motif. Column O shows the average of the number of nucleotides that match the JASPAR PWM. Column P shows the ratio of nucleotide matches over all nucleotides in the binding motif of JASPAR for all comparisons with theoretical PWMs. Column Q shows the ratio of models that produce theoretical PWMs that are significantly similar to the JASPAR PWM (P-value < 0.05). Column S is one if the ratio in Q is higher than 0.5 and a zero otherwise. Rows showing redundant motifs of sequences already analyzed on top are highlighted in yellow. Table S5. Comparison between theoretical PWMs of CTCF and MA0139.1 motif in JASPAR. Tab 1 shows results obtained using statistical potentials that include known the structures of CTCF in PDB. Tab 2 shows results obtained using statistical without contacts from structures of CTCF. In both tabs the information is structured in the same columns. Column A shows the JASPAR motif. Column B shows the model, potential and maximum distance to calculate the contacts and create the theoretical. An example of the code is 5und_F2_15, for a theoretical PWM obtained with the structure of 5UND from PDB, using potential ZES3DCF2 and cut-off distance of 15 Angstroms. Column C shows the orientation of the match between the theoretical and the experimental PWMs. If it is “+” it means that both PWMs are on the same orientation (i.e. both forward), if it is “-” it means that one of the PWMs must be reversed (i.e. one forward and the other reverse). Column D shows the TOMTOM P-value of the comparison between the theoretical PWM and the JASPAR motif. Column E shows the score of the comparison between the theoretical PWM and the JASPAR motif. Column F shows the offset between the theoretical PWM and the JASPAR motif once aligned. Column G shows the number of aligned positions between the theoretical PWM and the JASPAR motif. Column H shows the number of nucleotide matches between the theoretical PWM and the JASPAR motif. Column I shows the ratio of nucleotide matches between the theoretical PWM and the JASPAR motif with respect to the shortest binding site. Column J shows the consensus DNA sequence for the JASPAR motif. Column K shows the consensus DNA sequence for the theoretical PWM.

146
EXTENSION OF METHODS 1. Software requirements We require the following software: DSSP (version CMBI 2006) (5) provides protein structural features; X3DNA (version 2.0) (6) is used to analyze and generate DNA structures; matcher and needle, from the EMBOSS package (version 6.5.0) (7), produces local and global alignments, respectively; BLAST (version 2.2.22) (8) is employed to search homologs of a target protein; MODELLER (version 9.9) (9) is used to create structural models with all the templates similar to our target; and the programs FIMO and TOMTOM from the MEME suite(10) are used to scan a DNA sequence with a Position-Weight Matrix and to compare two PWMs, respectively. 2. Databases and bacteria one-hybrid experiment Structural information is retrieved from the PDB repository (2) and protein codes and sequences are extracted from UniProt (January 2019 release) (3). We select all transcription factors of the C2H2-ZF family as defined in CIS-BP database (version 1.62) (11) with known structures in PDB to generate the internal database of structures (set PDBDNA). We rearrange the set of structures by separating them in chains and constructing a set of structures formed by single protein-chains interacting with a double-strand helix (single-chain PDBDNA). After removing structures with methylated forms of DNA in the binding site, the set is obtained with the PDB codes: '5t00', '5kkq', '1a1g', '6jnm', '4r2d', '1tf6', '4m9v', '4r2a', '1tf3', '4r2c', '4x9j', '2i13', '5und', '5ke7', '5ke8', '2drp', '5ke6', '1llm', '4m9e', '5vmv', '1p47', '5vmw', '5ei9', '5eh2', '2wbu', '6e93', '6e94', '1jk1', '1jk2', '3uk3', '5wjq', '2kmk', '6jnn', '5v3m', '6jnl', '4gzn', '5t0u', '5kea', '5keb', '1ubd', '5k5h', '5k5l', '5k5j', '5k5i', '1a1f', '1a1i', '1a1h', '1a1k', '1a1j', '1a1l', '5ke9', '4f6m', '4f6n', '2gli', '5yeg', '1un6', '5yef', '1g2f', '5yeh', '1g2d', '1zaa', '5yel', '5egb', '1aay', '4is1', '5vmu', '1f2i', '5v3j', '5yj3', '2hgh', '5vmz', '2lt7', '5vmx', '5vmy' Binding information of Zinc-finger family C2H2-ZF is retrieved from bacteria one-hybrid (B1H) experiments (12). The experiment distinguishes between Zinc-finger individual domains at the C-tail (F3 domain) and inner domain (F2 domain). The experiment performs the screening of all 64 possible 3bp targets for interactions with C2H2-ZF domains from multiple large protein libraries based on Zif268 structure with six variable amino acid positions on each individual domains F2 and F3 (13). Here, we summarize the key points of the B1H experiment of Persikov et al. (12) that we use in our work. The experiment surveys systematically the DNA-binding landscape of C2H2-ZF domains. It relies on a repertoire of protein libraries of engineered modifications of the sequence of Zif268 allowing for each of the 20 amino acids at positions -1,1,2,3,5 and 6 of the alpha-helix (i.e. the hexamer core sequence) of domains F2 and F3. In the 3D space, these

147
residues interact specifically with three nucleotides. Attached is an example for the F2 domain, with the amino acid positions indicated by numbers and the three nucleotides represented with Ns and highlighted in red:
The modified Zif268 proteins are expressed as fusions to the omega subunit of RNA polymerase, which act as activation domain of the hybrid assay. Then, each of the 64 possible 3bp DNA target sequences, located 10bp upstream of a weak promoter driving the reporter genes HIS3 and URA3, is tested; when cells are grown on minimal media requiring the activation of HIS3, only a functional protein-DNA interaction leads survival of the bacteria. The authors considered that the affinity of protein-DNA interaction was related to the growth rate in the B1H system. They suggested a computational approach to infer the affinity of a specific hexamer core sequence based on the frequency with which it was found in each of the 64 possible 3bp targets. The method for predicting the DNA- binding specificity for a core sequence was defined by the authors as the “lookup” procedure, which is based on finding the core sequence across all the protein selections. They normalized the frequencies so that they summed up to 1 across the 64 possible 3bp targets, and obtained a binding profile with the probability distribution of the preference of a core sequence for each 3bp target. We retrieved from the repository provided by the authors (http://zf.princeton.edu/b1h) the files with the logged and normalized frequencies of hexamer core sequences for each of the 64 possible 3bp targets. Finally, the affinity of the protein-DNA interaction is calculated as the affinity percentile:
Where, f(seq) is the frequency of the particular hexamer core sequence and f is any other frequency on the set of hexamer core sequences binding the same 3bp.

148
3. Interface and triads of protein-DNA structures
The interface between a transcription factor and DNA is defined by the residues (amino-acids and nucleotides) in contact. A general approach for protein-protein interactions is to consider that two residues are in contact if the distance between a pair of atoms from each residue is shorter than 5Å. We define triads as a type of contacts between the protein and the double-strand DNA helix. Triads are formed by three residues: one amino-acid and two contiguous nucleotides of the same strand. The distance associated with a triad is defined by the distance between the Cβ atom of the amino acid residue and the average position of the atoms of the nitrogen-base of the two nucleotides plus their complementary pairs in the opposite strand of the helix (14). The triad also has an associated amino-acid residue number in the protein and a dinucleotide position in the DNA, defined by the sequence position of the first nucleotide of the dinucleotide. We define the interface between a transcription factor and DNA as: the set of triads with associated distances shorter than 15 Å, and their associated amino-acid residue number and dinucleotide position (e.g. a with amino-acid residue number , dinucleotide in position and associated distance is represented as ). Specific features can be added on a triad, defining an extended-triad:
1) Hydrophobicity of the amino-acid. Amino-acid residues are split in Polar (P): {Arg, His, Lys, Asp, Glu, Ser, Thr, Asn, Gln, Cys, Gly} and Non-polar (N): {Ala, Ile, Leu, Met, Val, Phe, Trp, Tyr, Pro}.
2) Surface accessibility of the amino-acid. We use DSSP to calculate the percentage of accessibility of the residue in the unbound structure of the protein. If the percentage is smaller than 50% the amino-acid is buried (B), otherwise it is exposed (E).
3) Secondary structure of the amino-acid. We use DSSP to calculate the secondary structure of the protein. The amino-acid of the triad is either in regular secondary structure (H if in α-helix, E if in β-strand), or in a non-regular secondary structure (C)
4) Nitrogenous bases: We classify nucleotides by their nitrogenous bases in two types, purines (U): {A, G} and pyrimidines (Y): {C, T}.
5) Closest strand. We use X3DNA to define the strands forward and reverse of the DNA. Next, we calculate the distance of all atoms of the two nucleotides to the Cβ of the amino-acid. We define the strand closest to the amino-acid (i.e. with the atom at minimum distance) as either the strand of the two nucleotides of the triad or the strand of their complementary pair in the opposite strand, which can be either forward (F) or reverse (R).
6) Closest Groove. We calculate the distances between the Cβ of the amino-acid and the closest phosphates of the dinucleotides in both strands (i.e. the strand of the two nucleotides of the triad and its complementary). We calculate the positions of the closest phosphates in

149
both strands (let be Pf and Pr, backbone phosphates of nucleotides f and r, respectively). We select the closest phosphate of both and its corresponding strand. Let assume that Pf is the closest phosphate and define its strand as “s”, being “S” the opposite strand. Then, we consider the set of backbone phosphates in “S” around the position complementary of nucleotide “f” (6 nucleotides up and down). Depending on their distance to “f” (towards 22Å is a major groove and towards 12 Å a minor groove), we classify them as part of the minor or major groove with respect to nucleotide “f”. This is a classification of 12 nucleotides around the complementary of “f” in two groups: 1) set at large distance (i.e. major groove); and 2) set at short distance (i.e. minor groove). Necessarily, Pr is in the list classified in major or minor groove. We use the classification of Pr to define the type of the closest groove of the amino-acid (i.e. we should say that this is the groove faced by the amino-acid, defined by the pair Pf and Pr, in closest proximity to the amino-acid). The closest groove is defined as major groove (A) if Pr is in the list classified in major, otherwise it is defined as minor groove (I).
7) Chemical group of the nucleotides. We distinguish two main chemical groups of each nucleotide, the nitrogenous base (N) and the backbone (B) that includes the phosphate and sugar. We calculate the distances between the Cβ of the amino-acid and the atoms of the two nucleotides and their complementary. We select the atom with the shortest distance as the closest atom between the nucleotides and the amino-acid. We define the chemical group of the nucleotides of the triad as the chemical group to which belongs the closest atom (i.e. N or B)
Added features of triads can also be used on their own as feature-triads (or environment triads), and every extended-triad has an associated feature-triad, both associated with the same distance, amino-acid number and dinucleotide position. As an example, let be a lysine residue and two nucleotides, adenosine and guanosine, forming the triad [K,(AG)] at 15.6Å, with lysine in residue number 32 and adenosine in 5, described as ([K,(AG)], 15.6, 32, 5). If lysine surface is mainly exposed to solvent, in a α-helix conformation and the closest strand of DNA is the forward strand, the closest atom of the two nucleotides is a phosphate and the amino-acid faces the minor groove, the extended-triad is [{K,(p-H-E)},{(AG),(UU-F-I-B)}], where added features are (p-H-E) for the amino-acid and (UU-F-I-B) for the dinucleotide. This produces a feature-triad defined as [(p-H-E),( UU-F-I-B)] at 15.6Å. We require to define some functions on the sets of triads, extended-triads and feature-triads to extract some of the values collected from a complex structure and apply other functions:

150
The same functions are applied to extended-triads and feature-triads accordingly modified. We also define functions to substitute some of the elements of a triad, extended-triad or feature-triad (the example is given for etriads without loss of generality):
1) is a function that substitutes amino-acid residue “a” of the etriad by amino-acid-residue “r”, with the corresponding change of hydrophobicity but preserving the rest of features and measures associated with the triad.
2) is a function that substitutes dinucleotide by , in , with the corresponding change of
nitrogenous bases and preserving the rest of features and measures associated with the triad.
4. Statistical potentials We use the definition of statistical potentials described by Feliu et al (15) and Fornes et al. (14) to define several scoring functions for the interaction between a protein and a DNA binding site using contact triads. We use triads, their associated measures (i.e. distance, amino-acid number and dinucleotide position) and their added features to calculate the frequencies per distance in bins of 1Å (i.e. intervals [0,1], [1,2], [2,3], [3,4], [4,5] etc.) up to 30 Å. We also calculate the frequencies using the distance as cut-off (i.e. the frequency of triads at distance shorter than “d”, with d= 1,2,3,4, etc.). To obtain the frequencies, we first calculate the size (cardinality, defined by the function “Card”) of the sets of triads, associated with a distance (d), taken from the set of structures of protein-DNA interactions (PDBDNA) and grouped by their associated distance, limited to a maximum of 30Å (i.e. with ). The set of triads, associated with distances, amino-acid residue-number and dinucleotide position, is named 3Dset. Then, frequencies are defined using functions defined in section 3 as follows:
(eq. 1) (eq. 2)
Where is a triad associated with a distance d, amino-acid residue-number and dinucleotide position , taken from the set 3Dset, ND is defined using bins and Nc using cut-offs. Similarly to 3Dset we define the sets e3Dset and f3Dset for extended-triads (etriad) and feature-triads (ftriad), and calculate LD, Lc , MD and Mc with as:
(eq. 3) (eq. 4)
(eq. 5) (eq. 6)

151
Then, we define the frequencies (F for triads, G for extended-triads and H for feature-triads) as:
(eq. 7)
(eq. 8)
(eq. 9)
Where N can be ND or Nc, L can be LD or Lc , and M can be MD or Mc, depending on the approach to group the triads. This definition forces us to consider independent the groups obtained by cut-offs, instead of using the ratios with respect to the limit at 30 Å. We tested in artificial data that this approach preserves the curve of the statistical potential similar to the classical definition by bins of distances, but it’s less affected by the scarcity of data. To define a reference-state for the statistical potential, we require two more frequencies, one for ND and another for Nc , using the total number of triads in the database (triads):
(eq. 10)
Where , and it’s easy to proof that:
(eq.11) Where ftriads is the set of feature-triads and etriads the set of extended-triads, with and . Using these definitions and following previous works (14), we define the potentials E3DC, ES3DC and PAIR per triad and distance d, using the round value of d (i.e. k), as follows:
(eq.12)
(eq. 13)
(eq. 14)
Where, F, G, H and O are frequencies calculated by bins or using cut-offs. The total potential of an interaction is calculated as the sum of the corresponding potential of all triads, feature-triads and extended-triads at distances shorter than 30Å. We use each potential to score the quality (or potentiality) of the interaction. Let be I, E and D the sets defined respectively as the set of all triads, extended-triads and feature-triads with their associated distances (d), amino-acid residue number (p) and dinucleotide position (q) in the binary interaction of a TF-DNA structure. Therefore, we define the energy-based scores (as they are based on total potentials) as:

152
(eq. 15)
(eq. 16)
(eq. 17)
Similarly, we also use the potentials defined for triads, ftriads and etriads as scores of the quality of a single interaction between one dinucleotide and one amino-acid residue. Then, the potential ES3DC of an extended-triad associated with distance d can be rewritten as:
(eq. 18) Where . We then define two scoring terms, one distance independent (ES3DCdi) and another distance dependent (ES3DCdd), as follows:
(eq.19)
(eq. 20) Using eq. 8, eq. 19 and eq. 20, we rewrite as:
(eq. 21) And the global energy-based scores:
(eq.22)
We consider ES3DCdd to evaluate the quality of an interaction because it is more specific than PAIR, as it uses extended-triads and distances, and it is also highly sensible, because it is normalized over the total of triads at a given distance with independence of the features. However, we have to note that this can only be considered as a scoring of quality, because the complete statistical potential requires the other terms E3DC and ES3DCdi to complete ES3DC in equation 21. Finally, due to the scarcity of data the curves of statistical potentials may be jagged. Therefore, we use a sliding window of approximately W samples defined by distances (by bins or cut-off) to smooth the potential curves. Let be a distance-dependent score, as defined previously, then we define the smoothed score, as:
(eq.23)

153
Where and are defined as:
Where, is the total of bins between and with defined score
( ), which is around W (i.e. )
5. Z-scores We have to note that frequencies are always smaller than 1, consequently their logarithm is smaller than 0. However, statistical potentials are not necessarily negative, because by definition the use of a reference state is required, which is obtained by the sum of all triads at a given distance (also ftriad or etriad, depending on the potential of interest). Consequently, the comparison with a reference state can change the sign of the final sum of terms (for example, PAIR and ES3DC can be negative while E3DC may be positive). The variability of signs of the potentials affects the criterion of quality of the scores and they may become unclear. Nevertheless, indistinctly of the sign, the best interaction between an amino-acid and a dinucleotide is produced at the distance where PAIR and ES3DC are minimum, because it implies the highest frequency of a triad (or etriad) with respect to all triads (or feature-triads). For example, ES3DCdd has a minimum for the highest frequency of an extended-triad with respect to all triads. We then define z-scores in order to follow a criterion that incorporates the sign to score the quality of the interaction between an amino-acid and a dinucleotide as a function of the distance. We wish that the z-score identifies simultaneously the best distance associated with a triad and the best pair formed by one amino-acid and one dinucleotide. Consequently, we construct a zscore function with any type of score, applying without loss of generality on an extended-triad (etriad) and an associated distance d, as:
(eq. 24)
Where: A is the set of all amino-acid types (i.e. ), and we use the classical functions of average ( ) and standard deviation ( ), defined as:

154
Notice that we use the function as above and that we use the term score instead of potential (P in previous section) to emphasize that this is used only as a criterion of the quality of the interaction. It’s easy to proof that this definition satisfies our requirements for the new function zscore: 1) the minimum value of score produces a negative value of zscore (and vice-versa, the maximum yields positive); 2) as the zscore compares the score of a particular amino-acid with all other, the etriad becomes ranked from minimum to maximum allowing us to select the best residue among amino-acids in the same position, depending on the quality criterion of the score. Finally, if we choose to smooth the curves dependent on distance, the zscore has to be calculated first with the unsmoothed scores and subsequently smoothed to avoid introducing biases. 6. Structural modeling of C2H2-ZF complexes Given the sequence of a TF (named protein target) and a DNA binding site fragment (named DNA target), we obtain the structure of the complex by means of homology modelling using the program MODELLER (9). First, we search potential templates of the protein target among the set of sequences with known structure in PDBDNA using BLAST (16). We use the alignment obtained with matcher and the sequence and structure of each template to construct the models with MODELLER. The DNA binding sequence of each testing 3-domain C2H2-ZF protein is formed by 9bp nucleotides, a set of 3bp bound by each individual domain. For the selection of the binding sequences associated with each finger domain we use the same sequences as in the B1H experiment (12): 1) for the selection of F2, the finger sequences in F1 (N-tail domain) and F3 (C-tail domain) involved in the interface are RSDNLRA(F1) and RSANLVR (F3), respectively binding AAG and GAG; and 2) for the selection of F3, the finger sequences in F1 (N-tail domain) and F2 (inner domain) involved in the interface are RSDELTR (F1) and RSDNLRA (F2), respectively binding GCG and AAG.
The structure of Zif268 binding DNA is modelled with 23 different template structures to introduce structural variability, retrieved by codes: 1p47 (chain A), 1zaa (chain C), 1g2d(chain C), 1a1h(chain A), 1a1g(chain A), 1a1i(chain A), 1a1j(chain A), 1a1k(chain A), 1a1l(chain A), 1aay(chain A), 1jk1(chain A), 1jk2(chain A), 2kmk(chain A), 2wbu(chain A), 4r2a(chain A), 4r2c(chain A), 4r2d(chain A), 5ke6(chain A), 5ke7(chain A), 5ke8(chain A), 5ke9(chain A), 5kea(chain A) and 5keb(chain A) . We compare the sequence of Zif268 used in the experiment with the templates by a sequence alignment with CLUSTALW (17). We identify as WT the original sequence, and as F2 and F3 the sequences used for the selection of F2 and F3 binding sequences, labeling by “X” the amino-acids that are modified. This alignment is shown here for 1p47 used as template, highlighting in bold and red the attention of the specific binding sequence of each finger (blue box for F1, yellow box for F2 and green box for F3):

155
F2 GTERPYACPVESCDRRFSRSDNLRAHIRIHTGQKPFQCRICMRNFSXXXXLXXHIRTHTG F3 GTERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDNLRAHIRTHTG WT GTERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDNLRAHIRTHTG 1p47_A --ERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTG *************** * ********************* * ******* F2 EKPFACDICGRKFARSANLVRHTKIHLRGS F3 EKPFACDICGRKFAXXXXLXXHTKIHLRGS WT EKPFACDICGRKFARSANLVRHTKIHLRGS 1p47_A EKPFACDICGRKFARSDERKRHTKIHLRQ- ************** ******
After modeling the structure of Zif268, we complete the complex by modeling the structure of the DNA binding sequence. However, each template has DNA sequences of different length that do not correspond with the DNA used in the experiment. The full DNA sequence of the experiment is longer (29bp) than the binding (9bp), which is shown next, embedded in positions 11 to 19 (nucleotides highlighted), labelling by “N” those under test. Two DNA sequences are considered depending on the experiment, one for the selection of F2 and another for F3:
F2: 5'- GCGGCCGCAAGAGNNNAAGTAACGAATTC - 3' F3: 5'- GCGGCCGCAANNNAAGGCGTAACGAATTC - 3'
The structure of the full DNA sequence bound by Zif268 is obtained with the program X3DNA(6) by modifying the DNA structure in the complex. First, we locate the 9bp binding region in the structure of the template and identify the positions at 5’ and 3’ (first and last). Next, we construct two frames of B-DNA structure with the lengths required to extend the template at 5’ and 3’ up to 29 nucleotides. The lengths in both sides depend on the location of the 9bp binding sequence and the DNA sequence of the experiment. Then, we use 3DNA/DSSR to perform a least-squares fitting that locates each base reference frame in the first and last positions. Finally, the structure of DNA is completed with the right sequence. We model the DNA structure by substitution of the nucleotides in each model with the corresponding nucleotides of the target. We use the program X3DNA to substitute the nucleotides of one strand and automatically model its corresponding pair. In order to understand the approach, we select one of the hexamer sequences in F2, “SAGSYN” (highlighted in bold), with highest affinity for “AAA” and we use the structure of 1p47 as template. We use the program MODELLER with the alignment:
MODEL GTERPYACPVESCDRRFSRSDNLRAHIRIHTGQKPFQCRICMRNFSSAGSLYNHIRTHTGEKPFACDICGR 1p47_A --ERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGR MODEL KFARSANLVRHTKIHLRGS/....................../......................* 1p47_A KFARSDERKRHTKIHLRQ-/....................../......................*
The dots in the alignment are used to indicate the ligand, which in this case corresponds to the DNA sequence, using the slash to separate the chains. MODELLER produces a model with the anticipated sequence on the protein,

156
but the DNA sequence is still the same as in the original template (i.e. 5’-GTGGCGTGGGCGGCGTGGGCGT-3’ in chain “C” and the paired sequence in chain “D”). Then, we use the package X3DNA to modify the sequence in the structure of chains “C” and “D”, substituting the nucleotides by the expected sequence with the program “mutate_bases” from X3DNA package. The modified sequence becomes then 5’-GCGGCCGCAAGAGAAAAAGTAA-3’, where the anticipated sequence “AAA” is highlighted in bold. Finally, we use the program “fiber” from X3DNA package to complete the missing nucleotides (i.e. “CGAATTC”) with a B-DNA conformation. The modeling of any other transcription factor formed by C2H2-ZF domains is similarly done, aligning the protein sequence of the query with the best template protein sequences of transcription factors formed by consecutive C2H2-ZF domains. We filter out the alignments with gaps in the DNA binding helix of the templates. The schema for modeling is here summarized:
C2H2-Zf
Modeling of C2H2-ZF binding domains Step1: Search of similar sequences on the database of proteins with known structure of the C2H2-ZF family Step 2: Use the criteria of the twilight zone threshold to accept potential chains of templates aligned with the query. Align the sequence of the query protein and the sequence of the template with “matcher” (7). Neglect templates with gaps between query and template sequence in the fragment of the template that corresponds to the binding site Step 3: Add the sequence of DNA in the alignment formatted to run MODELLER (9) Step 4: Built the complex with MODELLER (9)
We use a database of 86 chain structures, with DNA complexed with the DNA binding domain, formed by continuous zinc-fingers, as the set of templates for the server in http://sbi.upf.edu/C2H2ZF_repo (codes of chains are also indicated):

157
'1zaa_C', '4x9j_A', '1p47_A', '1a1i_A', '1a1j_A', '1a1l_A', '1jk1_A', '4r2c_A', '5ke6_A', '5ke7_A', '1llm_C', '1llm_D', '1ubd_C', '2drp_A', '2lt7_A', '3uk3_C', '4f6m_A', '4f6n_A', '4is1_C', '5k5i_A', '5k5l_G', '5und_A', '5v3j_E', '5vmw_A', '5vmz_A', '6e93_A', '1un6_D', '5yef_A', '5yef_J', '2kmk_A', '1g2f_C', '1jk2_A', '1a1g_A', '1a1k_A', '1g2d_C', '4m9e_A', '4r2a_A', '4r2d_A', '5ke8_A', '5kea_A', '1f2i_G', '1p47_B', '1tf6_A', '2hgh_A', '4gzn_C', '5k5l_E', '5k5l_F', '5kkq_A', '5t0u_A', '5v3m_C', '5vmy_A', '5yeg_A', '5yel_B', '6jnl_A', '1un6_B', '5yef_B', '2wbu_A', '1a1f_A', '1a1h_A', '1aay_A', '5ke9_A', '5keb_A', '1f2i_H', '1tf3_A', '2gli_A', '2i13_A', '3uk3_D', '4is1_D', '4m9v_C', '5egb_A', '5eh2_F', '5ei9_E', '5k5h_A', '5k5j_A', '5t00_A', '5vmu_A', '5vmv_A', '5vmx_A', '5wjq_D', '5yeh_A', '5yj3_C', '5yj3_D', '6e94_A', '6jnm_A', '6jnn_A', '5yef_G' We also model several structures with the complex of Zif268 binding a non-specific DNA region using the same approach. These structures are used as non-binding examples. The non-binding sequence is taken randomly by selecting a region of the sequence of the weak promoter GAL1, constructing 10 DNA fragments of 29bp for each known binding. A potential binding test in this region, which is part of the B1H experiment, may well represent the background. The forward weak promoter sequence of GAL1 is formed by 118bp that are shown here:
5'- GAGATTAAGGAGCAGAAGGGGTGACAGCCCTCCGAAGGAAGA GAGATTAAGCTCTCCTCCGTGCGTCCTCGTCTTCACCGGTCG CGTTCCTGAAACGCAGATGTGCCTCGCGCCGCACTGCTCCG - 3'
7. Use of experimental TF-DNA binding to calculate statistical potentials One of the main problems to obtain statistical potentials for all families and folds of TFs is the scarcity of known interactions. We need to enlarge the number of interacting triads. Therefore, here we propose to use the experimental knowledge of TF-DNA interactions to derive interacting triads without requiring the complete knowledge of TF-DNA complex structures. We then use the sets of derived triads associated with distances to calculate the statistical potentials. Let be a TF with experimentally known interactions with several DNA sequences. We define a mapping function (mapD) between the fragment of the DNA sequence from the structure ( ), and each positive binding sequence (
), as follows:
(eq.25) Where is a dinucleotide, with , is also dinucleotide, with
, is the position of the dinucleotide in the alignment between
and , is the position of the dinucleotide in , is the position of the dinucleotide in , and the position of each dinucleotide is defined (i.e.
equal to) the position of the first nucleotide in the DNA sequence. For the sake

158
of simplicity, when the length of is the same as and the position in the
alignment, , coincides with and (i.e. ), we write:
(eq.26) We can define a set of mapping functions with the alignments of all the DNA sequences extracted from the structures of TF-DNA complexes in PDBDNA that are aligned with positive binding sequences. Then, we use the dinucleotide substitution function (as defined in section 3), , of an extended-triad, etriad, containing a nucleotide which is substituted by n in the dinucleotide, to generate more extended-triads. There are other TFs for which the structure is not known but can be modelled. This implies that the sequence of the TF can be aligned with sufficient percentage of identical residues to ensure its modeling. Then, we define another mapping (mapP) between the protein sequence of the TF with known experimental data on DNA binding and the TF sequence of a known structure (template), with:
(eq.27) Where, is an amino-acid residue in the sequence of a template and is the amino-acid in the sequence of a TF with experimental data, in position of the alignment of both TF sequences that correspond with positions and for
and , respectively. Also, for the sake of simplicity, if the position in the alignment, , coincides with and (i.e. ) we write:
(eq.28) We define the set of all mapping functions with all the alignments between the sequences of TFs with experimental annotation and the TFs with known structure (complexed with DNA). Also, we define as the set of extended-triads, extracted from the 3Dset, containing amino-acid residue (the set of 20 amino-acids) and dinucleotide . We remind the definition of as the function that substitutes amino-acid residue “a” of an extended-triad, etriad, by the amino-acid residue “r”. With all these definitions, we increase the set of extended-triads of the e3Dset to e3Dset’, using all the mapping functions mapD (simplified as ) and mapP
(simplified as ). We use the simplified maps without loss of generality, as all sequences and alignments can be renumbered. Both mappings, and
, are respectively defined with: 1) the alignments of the DNA sequences extracted from the structures in the PDB aligned with positive binding sequences; and 2) the TFs that can be modelled using the alignment with their

159
structural templates (the set is defined as ). The new set of extended-triads, e3Dset’, is defined as:
(eq. 29) And we recalculate LD and LC in equations eq.3 and eq.4 as:
(eq. 30) (eq. 31)
Similar approach is also taken for the sets of triads and ftriads, modifying accordingly the corresponding functions to substitute amino-acid and dinucleotide residues of a triad or a featured-triad. We proceed similarly with B1H experiments on C2H2-ZF family. For each finger (F2 and F3) and combination of 3bp nucleotides, we collect all protein sequences producing significant binding signal in the B1H experiment. We use the modelled structures of the DNA testing sequence of 29bp with different templates and introduce the mappings for the DNA sequence and the modified residues in F2 or F3 from the multiple sequence alignment. For the DNA sequence the mapping is on the 3bp modified nucleotides, affecting 4 dinucleotides, while for the protein sequence the mapping affects 6 amino-acids, both mappings being different for F2 and F3 selections. This is, for the DNA sequence the mapping is , where is a dinucleotide of the 3bp under test, is a dinucleotide of the 29bp of the modelled template, and the position, , is between 11 and 19 (11-13 for F3 and 14-16 for F2). For the substitution of residues of Zif268 in the interface we require a mapping of the native sequences RSANLVR (F3) and RSDNLRA (F2), for all selections of the binding finger. This mapping is where is the position in the alignment (residues 47-53 for F2 and 75-81 for F3), , the position in the template, , depends on the template used to model Zif268 and
is the corresponding residue in the template in position of the alignment. From the template structure we extract the contacts (triads, etriads and ftriads) between amino-acids and dinucleotides and generate the statistical potentials. However, this introduces a bias by overestimating the constant amino-acids and nucleotides that have not been modified in the experiment. Therefore, only the triads affecting the amino-acids and nucleotides under test are considered to generate the potentials. This is, we only consider the contacts between the 6 amino-acids labelled by “X” and dinucleotides containing one of the 3bp labelled by “N” to generate two potentials, one for F2 and another for F3 finger positions. We restrict each set of Zif268 sequences to those with highest signal of the B1H binding experiment in order to obtain potentials more specific or associated with the strongest binding. We define three thresholds based on the affinity

160
percentile of a sequence: 1) higher than 90%; 2) higher than 75%; and 3) higher than 50%. To calculate the affinity percentile of a sequence we follow the same definition as the authors(12). Each sequence, in its corresponding domain, has a logged and normalized frequency of its observation ( ). Hence, the
affinity percentile is defined as the sum of all other frequencies lower or equal to the frequency of the sequence (e.g. an affinity percentile of 90% implies that the sequence is on the tail with highest number of observations, in the top 10%). However, the number of selected sequences may be too different between experiments (i.e. the 3 nucleotides of the binding “TGA” may have 30 sequences with affinity percentile higher than 90, while AAA has only 2), which produces the opposite bias on the expected potential. To avoid a bias on the number of sequences selected, we force to have around 500 sequences for all trinucleotide-binding experiments, by repeating as many times as we need each sequence (e.g. if only 2 sequences are selected for AAA and they are equally representative, we should repeat 250 times each). Each sequence is repeated in proportion to the number of observations. This is, for a sequence with
observation , it is repeated , where is the set of
sequences with affinity percentile higher than 90 (or we use A50 for affinity percentile higher than 50, etcetera). As a consequence of the approach, the contacts derived from the B1H experiment are limited to relatively short distances (the largest contacts are around 15-20Å). However, we note that we also use contacts extracted from other structures of the C2H2-ZF family in the PDB and from the use of PBM experiments, covering larger distances up to 30 Å. 8. Scoring TF-DNA binding with structure
a. Scores of single domain structures Given the structure of a protein-DNA complex, either experimentally obtained (i.e. from crystallography and identified by a PDB code) or modelled, we define several scores of the interaction based on statistical potentials. First, we calculate the interface of the interaction and extract all triads, extended-triads and feature-triads associated with distances shorter than 30Å. Then, the score of the interaction is defined as the sum of the scores (i.e. potential) of all triads with their associated distances (or extended-triads or feature-triads, depending on the type of score). The same approach is applied for z-scores. Let be a potential or a z-score and let C be the set of triads (extended-triads or feature-triads, depending on the definition of ) and their associated distances (d), amino-acid residue number (p) and dinucleotide position (q). The score of the interaction is defined as:
(eq.32)

161
We can obtain the score of a TF without knowing the structure of the TF-DNA binary complex if it can be modelled. We use the structure of a template to generate the set of triads and the mapping of amino-acids derived from a sequence alignment, mapP as defined in section 7 (eq.28), between the TF sequence and the sequence of the template. We also need the mapping of dinucleotides between the DNA sequence we wish to model and the DNA sequence in the template interface. Instead of modeling the structure of the TF-DNA complex, we modify the scores by applying the substitution of the corresponding amino-acids, using the functions defined as in section 3,
and and the mappings mapP (in eq. 28) and mapD (in eq. 27) between the templates and the sequences of TF and DNA, respectively. Here, instead of using simplified mappings ( and ), we generalize the formula by defining special functions, and , to extract the dinucleotide or amino-acid positions in the DNA or protein sequences:
(eq. 33)
Where is either a dinucleotide or an amino-acid residue, and is a position of a dinucleotide or an amino-acid, respectively for DNA or protein sequences. Then we calculate the score of the interaction as:
(eq.34) b. Multiple domain TFs
There are TF structures with more than one domain, such as the particular case of the C2H2-ZF family, where the TF has several domains like F2 (internal) and two more domains, one at the N-tail (F1) and another C-tail (F3). However, we apply two potentials for all domains, one obtained with B1H data on finger domains in F2 and another for F3. For example, we use the statistical potential ZES3DC calculated with variant sequences in F2 domain (ZES3DCF2) or in F3 (ZES3DCF3).
9. Construction of PWMs using Zif268 structure models. Given the modelled structure of Zif268-DNA complex, we obtain the PWM by means of statistical potentials using scores or zscores (we use the zscore of ES3DCdd by default as example). We focus on the specific nucleotides for three continuous fingers (9 bases) covering the whole binding site in sliding windows. We collect the set of triads, extended-triads and feature-triads with their associated distances between protein and DNA and the associated amino-acid and dinucleotide positions (i.e. , respectively), where the dinucleotide in the triad belongs in 9 nucleotide overlapping fragments (we name them , etc.). We remind the substitution function

162
, to substitute the dinucleotide of an by the dinucleotide . Similar functions are defined to substitute the dinucleotide in triads and feature-triads (these are only affected in the change of the nitrogenous bases). Then, for each fragment , with k=1,N-8 and the binding site of length N,
we obtain a test set with all possible DNA sequences (i.e. a total of 49). We define two mappings, and , respectively for the native
fragment sequence of the model and any sequence in the test set, both between sequence position and dinucleotides (i.e. with
nucleotide in position of ). We calculate the score of any sequence of the test set with the triads, extended-triads and feature-triads, using the associated distances, residue number and dinucleotide positions from the complex structure (i.e. triads, extended-triads and feature-triads as , respectively) and using the corresponding substitution function (e.g.
. Let be the z-score of and assume we apply it on extended-triads without loss of generality, then the score of a sequence
is:
(eq. 35) Where, for each , and are calculated using the functions as defined in section 3 and the mappings defined above:
We normalize the scores between 0 and 1, using the total set of scores
obtained with all generated sequences (i.e. set ) and modifying the
sign accordingly:
(eq.36) Where we have assumed that the best score is already the maximum. For example, for we need to multiply the score by -1. Then, we rank the normalized scores and select only the DNA sequences producing the top scores over a cut-off threshold (often 0.9). This produces an alignment from which we calculate the PWM (i.e. frequencies of nucleotides in each position)

163
10. Score per Nucleotide: profiles of a DNA binding site.
We define a nucleotide profile as a function on , , of the nucleotide position in a DNA sequence Here, we define score-nucleotide profiles when the function is obtained with the scores and z-scores. Given a TF-DNA complex structure, 3D-TF, and a distance dependent score, , obtained with statistical potentials (e.g. the smoothed z-score of ES3DCdd). Let be a nucleotide position of the DNA sequence in the complex, we define a new score, in , as:
(eq.37) Here we use extended-triads without loss of generality, although depending on the statistical potential we can use triads or feature triads instead. The set is
the set of etriads with all associated distances (i.e. any and amino-acid residue numbers (i.e. any where dinucleotide in position implies that is the position of one of the nucleotides in the dinucleotide at (i.e.
). This new score can be normalized by considering the contribution of the nucleotide to the total score or as the percentage of the contribution of all nucleotides of the DNA sequence. Assuming that the score is the smoothed z-score of ES3DCdd and without loss of generality, the normalized nucleotide profile is:
(eq. 38) or
(eq. 39)
Where for all but the extremes at 5’ and 3’, in which is 1, and from
equation 37 we write as:
(eq. 40)
is the set of extended-triads (with their associated distances, amino-acid numbers and dinucleotide positions) and is the length of the DNA sequence. The factor of 2 in equation 39 is produced by the fact that each

164
nucleotide is counted twice in , with the exception of the extreme positions in
5’ and 3’ where the nucleotides are only reckoned one time.
The curve of (raw or normalized) along the positions in the DNA sequence is defined as the nucleotide profile based on 3D-TF for the (raw or normalized) potential defined in (e.g. the smoothed z-score of ES3DCdd). Hence, profiles defined upon scores derived from statistical potentials are dependent on the structure of the TF-DNA interaction complex. We have to note that, if the structure of this complex has been modelled, several models may be considered (i.e. we define the set of models of TF-DNA as ). Besides, some models may be obtained using different templates, implying that these models introduce a relevant variability on the conformational space of the TF-DNA interaction. Consequently, several nucleotide profiles of scores are accumulated for the same DNA sequence. We then calculate the average and standard deviation of for all positions along the DNA sequence with the nucleotide profiles of score based on each model structure of the
set (i.e. described as , using following equations:
(eq. 41)
Equations in 41 describe two new nucleotide profile functions. The average function is defined as the nucleotide profile of (e.g. the smoothed z-score of ES3DCdd) and the RMSD defines its margins of error or variability.
The nucleotide profile can also be calculated with other “scores” different than those derived by statistical potentials, for example by the enrichment or the number of times that nucleotide j is selected by several PWMs assigned to a TF. This extends the definition of score nucleotide profiles to other scores different than those obtained with statistical potentials. 11. Construction of the experimental PWM The experimental PWM of a Zif268 sequence obtained from B1H, with a specific hexamer fragment on F2 or F3, is calculated based on its affinities for different binding sites. The binding site is formed by thee nucleotides flanked by two fixed nucleotides (G and A for F2, and two A for F3). All binding sites targeted by a specific hexamer-fragment with affinity higher than a threshold are stored and gapless aligned to construct the PWM (e.g. the top 10% threshold uses all DNA-bound sequences with affinity percentile higher than 90%, while for a threshold of 100% we use all detected sites with any not null affinity

165
percentile). We construct experimental PWMs for top 10%, top 25%, top 50% and for all targeted sites. These experimental PWMs are also named hexamer-specific PWMs, to distinguish from PWMs obtained with other experiments or with a different approach. References of supplementary 1. El-Gebali, S., Mistry, J., Bateman, A., Eddy, S.R., Luciani, A., Potter, S.C., Qureshi, M.,
Richardson, L.J., Salazar, G.A., Smart, A. et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res, 47, D427-D432.
2. Rose, P.W., Prlic, A., Altunkaya, A., Bi, C., Bradley, A.R., Christie, C.H., Costanzo, L.D., Duarte, J.M., Dutta, S., Feng, Z. et al. (2017) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res, 45, D271-D281.
3. UniProt, C. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res, 47, D506-D515.
4. Sigrist, C.J., Cerutti, L., de Castro, E., Langendijk-Genevaux, P.S., Bulliard, V., Bairoch, A. and Hulo, N. (2010) PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res, 38, D161-166.
5. Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577-2637.
6. Lu, X.J. and Olson, W.K. (2008) 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc, 3, 1213-1227.
7. Rice, P., Longden, I. and Bleasby, A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet, 16, 276-277.
8. Altschul, S.F., Gertz, E.M., Agarwala, R., Schaffer, A.A. and Yu, Y.K. (2009) PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res, 37, 815-824.
9. Webb, B. and Sali, A. (2016) Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics, 54, 5 6 1-5 6 37.
10. Bailey, T.L., Johnson, J., Grant, C.E. and Noble, W.S. (2015) The MEME Suite. Nucleic Acids Res, 43, W39-49.
11. Weirauch, M.T., Yang, A., Albu, M., Cote, A.G., Montenegro-Montero, A., Drewe, P., Najafabadi, H.S., Lambert, S.A., Mann, I., Cook, K. et al. (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158, 1431-1443.
12. Persikov, A.V., Wetzel, J.L., Rowland, E.F., Oakes, B.L., Xu, D.J., Singh, M. and Noyes, M.B. (2015) A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Res, 43, 1965-1984.
13. Persikov, A.V., Rowland, E.F., Oakes, B.L., Singh, M. and Noyes, M.B. (2014) Deep sequencing of large library selections allows computational discovery of diverse sets of zinc fingers that bind common targets. Nucleic Acids Res, 42, 1497-1508.

166
14. Fornes, O., Garcia-Garcia, J., Bonet, J. and Oliva, B. (2014) On the use of knowledge-based potentials for the evaluation of models of protein-protein, protein-DNA, and protein-RNA interactions. Adv Protein Chem Struct Biol, 94, 77-120.
15. Feliu, E., Aloy, P. and Oliva, B. (2011) On the analysis of protein-protein interactions via knowledge-based potentials for the prediction of protein-protein docking. Protein Sci.
16. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 25, 3389-3402.
17. Thompson, J.D.H., D.G.; Gibson, T.J. & others. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res, 22, 4673--4680.

167
3.1.3 - Short comment on: On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF
Meseguer A, Molina-Fernández R, Fernandez-Fuentes N, Fornes O, Oliva B. Short comment on: On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF.

168
Short comment on: On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF Meseguer A1, Molina-Fernández R1, Fernandez-Fuentes N2, Fornes O3, Oliva B1. 1Structural Bioinformatics Group, Research Programme on Biomedical Informatics, Department of Experimental and Health Science, Universitat Pompeu Fabra, Barcelona 08003, Catalonia, Spain, 2Department of Biosciences, U Science Tech, Universitat de Vic-Universitat Central de Catalunya, Vic, Catalonia 08500, Spain, 3Centre for Molecular Medicine and Therapeutics, BC Children’s Hospital Research Institute, Department of Medical Genetics, University of British Columbia, Vancouver, BC V5Z 4H4, Canada
Corresponding authors: Baldo Oliva ([email protected]), Abstract: In the work entitled “On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF” we present a new computational method to predict the DNA-binding preferences for Cis2-His2 zinc finger (C2H2-ZF) protein-domains from their amino acid sequence or structure. The method uses the structures of protein-DNA complexes to calculate a set of knowledge-based statistic potentials. Given the low numbers of protein-DNA complexes with known structure, we supplement the set of structures with experimental yeast one-hybrid interactions of more than 170000 sequence-designed C2H2-ZF domains. We have implemented a server to model the structure of any protein-DNA complex of this family and derive its theoretical Position Weight Matrix based on the best scores of interactions calculated with the statistic potentials. The approach is validated and applied to the human sequence of CTCF. SHORT COMMENT Knowing the DNA-binding preferences of transcription factors (TFs) is foremost to understand gene regulation. Among TFs, Cis2-His2 zinc finger (C2H2-ZF) proteins are the largest family in higher metazoans (1) and in humans (2). However, the DNA-binding preferences for many C2H2-ZF proteins are still unknown (2). Since determining the DNA-binding preferences of TFs experimentally is both expensive and time-consuming (2,3), computational methods that complement experimental approaches can be very useful. We use scoring functions based on structural and experimental data, called statistic potentials (4), to predict the DNA-binding preferences of C2H2-ZF proteins. Statistic potentials are scoring functions obtained from the analysis of

169
a set of reference structures (5). From this set of reference structures, we obtain frequencies of contacts between amino acids and dinucleotides. These frequencies are then used to calculate the statistic potentials by applying the inverse of the Boltzmann equation (6). When analyzing the quality of protein structures, statistic potentials provide scores depending on how similar their contact frequencies are to the ones in the reference set (5). In the case of C2H2-ZF, family we derive specific statistic potentials as follows: The reference set is constructed by collecting the structures of the members of the C2H2-ZF family complexed with DNA. Given the fact that the number of such structures is scarce and do not cover the entire spectrum of pairs of “amino acid” with “nucleotide” contacts, we complement the reference set with structural models of protein-DNA interactions from bacterial one-hybrid experiments (4,7). The resulting statistic potentials are used to predict DNA-binding preferences of C2H2-ZF proteins from their amino acid sequence as described by means of a Position-Weight-Matrix (PWM) (4). We evaluate the performance of our approach by comparing the theoretical and experimental PWMs from bacterial one-hybrid experiments (7) and from the JASPAR database (8). Finally, we apply our approach to predict the binding preferences of CTCF, a transcriptional repressor with a key role in genome compartmentalization (9). Since no available CTCF-DNA complex structure covers the entire DNA binding domain of CTCF, this results in incomplete theoretical PWM predictions. To overcome this limitation, we model almost the entire DNA binding domain of CTCF: 10 out of 11 C2H2-ZF domains. The theoretical predictions are significantly similar to the canonical DNA-binding motif of CTCF between domains 4 to 8. Remaining domains correspond to other binding motifs upstream and downstream of the canonical binding domain (4), which has been described experimentally (10). One strength of our method is that it provides more than one PWM per TF. Over 50% of predicted PWMs are significantly similar to their corresponding experimental PWM (4). This means that, by scanning a DNA sequence with a set of PWMs of a TF, we can detect with higher reliability the binding site of a TF among the regions enriched by several matches of PWMs. Having several PWMs for one TF also implies that TFs might bind different DNA sequences using different interacting conformations. Zinc finger domains can interact with DNA in up to six different ways (or binding modes), each of which changes the orientation of the specificity residues of the finger with respect to the DNA and resulting motif (11). An example illustrating this observation is CTCF, which displays different combinations of binding motifs spaced by a variable number of nucleotides (10). Our method is fast enough to be used in large collection of proteins containing C2H2-ZF domains. Out of all the eukaryotic C2H2-ZF proteins in the UniProt database (12), more than 70% of them can be analyzed by our method. The results of our study are available at: http://sbi.upf.edu/C2H2ZF_repo. Besides the examples presented in the server, protein sequences containing the C2H2-ZF domains can be submitted to derived structural models and theoretical PWMs.

170
Figures
Figure 1: Scheme of the method. We display a partial model of the structure of human CTCF (C2H2-ZFs domains 6 to 11) binding a DNA molecule and its predicted PWM. The forward strand, oriented to calculate the PWM, is colored in the ribbon-plate. At the bottom of the figure, we show the steps to extract contact frequencies between amino acids and nucleotides to construct the statistic potentials and its application to predict a PWM by ranking the best scored DNA sequences (see further details in the original publication (4)).

171
References 1. Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10(4):252–63.
2. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell. 2018 08;172(4):650–65.
3. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014 Sep 11;158(6):1431–43.
4. Meseguer A, Årman F, Fornes O, Molina-Fernández R, Bonet J, Fernandez-Fuentes N, et al. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. NAR Genom Bioinform [Internet]. 2020 Sep 1 [cited 2020 Sep 2];2(3). Available from: https://academic.oup.com/nargab/article/2/3/lqaa046/5866110
5. Fornes O, Garcia-Garcia J, Bonet J, Oliva B. On the use of knowledge-based potentials for the evaluation of models of protein-protein, protein-DNA, and protein-RNA interactions. Adv Protein Chem Struct Biol. 2014;94:77–120.
6. Finkelstein AV, Badretdinov AYa null, Gutin AM. Why do protein architectures have Boltzmann-like statistics? Proteins. 1995 Oct;23(2):142–50.
7. Persikov AV, Wetzel JL, Rowland EF, Oakes BL, Xu DJ, Singh M, et al. A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Res. 2015 Feb 18;43(3):1965–84.
8. Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020 Jan 8;48(D1):D87–92.
9. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009 Jun 26;137(7):1194–211.
10. Nakahashi H, Kieffer Kwon K-R, Resch W, Vian L, Dose M, Stavreva D, et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013 May 30;3(5):1678–89.
11. Garton M, Najafabadi HS, Schmitges FW, Radovani E, Hughes TR, Kim PM. A structural approach reveals how neighbouring C2H2 zinc fingers influence DNA binding specificity. Nucleic Acids Res. 2015 Oct 30;43(19):9147–57.
12. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019 Jan 8;47(D1):D506–15.

172
3.2 Methods to model and assess protein-protein interactions Protein-protein interactions are fundamental for the regulatory effect of most TFs. This is because most eukaryotic TFs carry out their regulatory effect through the recruitment of co-factors (19). Knowledge on protein interacting affinities, as well as how they change upon mutations, can help to complete our knowledge on gene regulatory networks. However, providing this data experimentally is expensive and time consuming (262). Given this situation, the development of computational tools to complement experimental techniques can be very helpful for the scientific community. Next, we expose our works on modeling and assessing protein-protein interactions. The first work consists on a pipeline for modeling and predicting the affinity of PPIs called MODPIN. We apply MODPIN to predict the changes in affinity that take place upon mutations in PPIs. The second work is a review on computational methods to predict protein binding affinities from unbound protein structures. In this review we summarize several computational approaches, and we focus on approaches that use docking results to predict the binding affinities between proteins.
Manuscripts presented in this section:
Meseguer A, Dominguez L, Bota PM, Aguirre-Plans J, Bonet J, Fernandez-Fuentes N, et al. Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions. Protein Sci. 2020 Aug 14. Meseguer A, Bota PM, Fernández-Fuentes N, Oliva B. Prediction of protein–protein binding affinities from unbound protein structures. (Submitted to Methods in Molecular Biology)

173
3.2.1 - Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions
Meseguer A, Dominguez L, Bota PM, Aguirre-Plans J, Bonet J, Fernandez-Fuentes N, et al. Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions. Protein Sci. 2020 Aug 14.

174

175

176

177
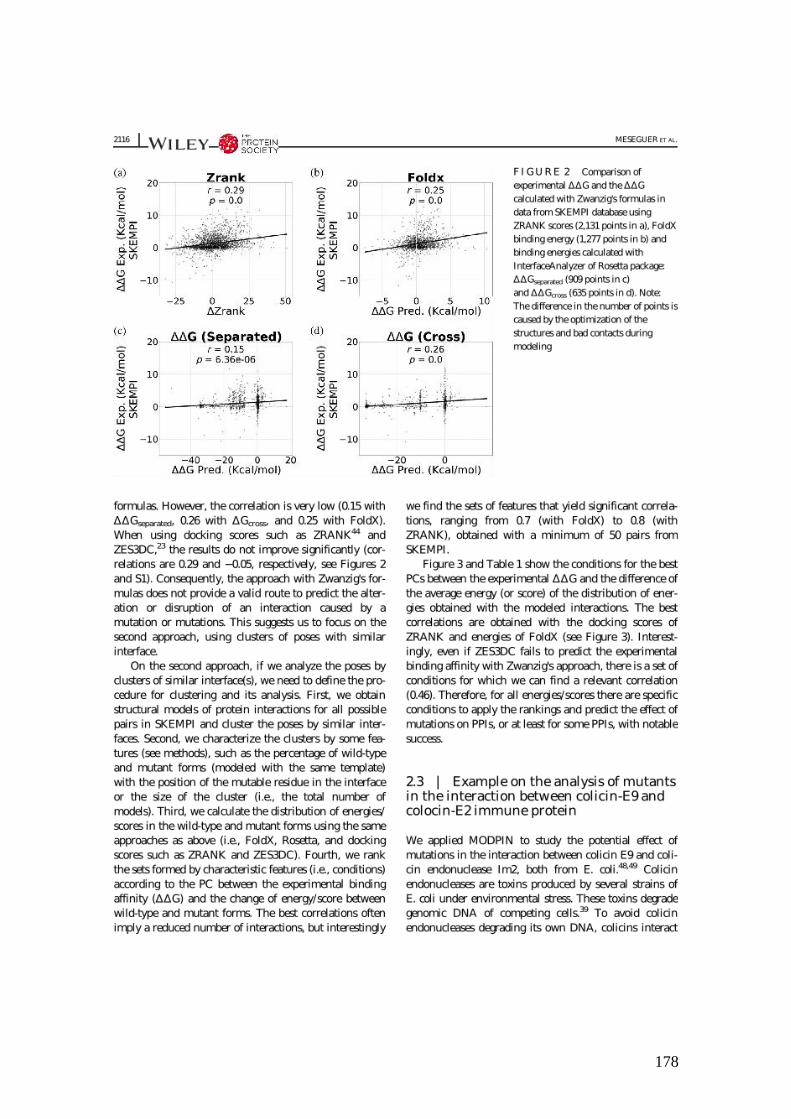
178
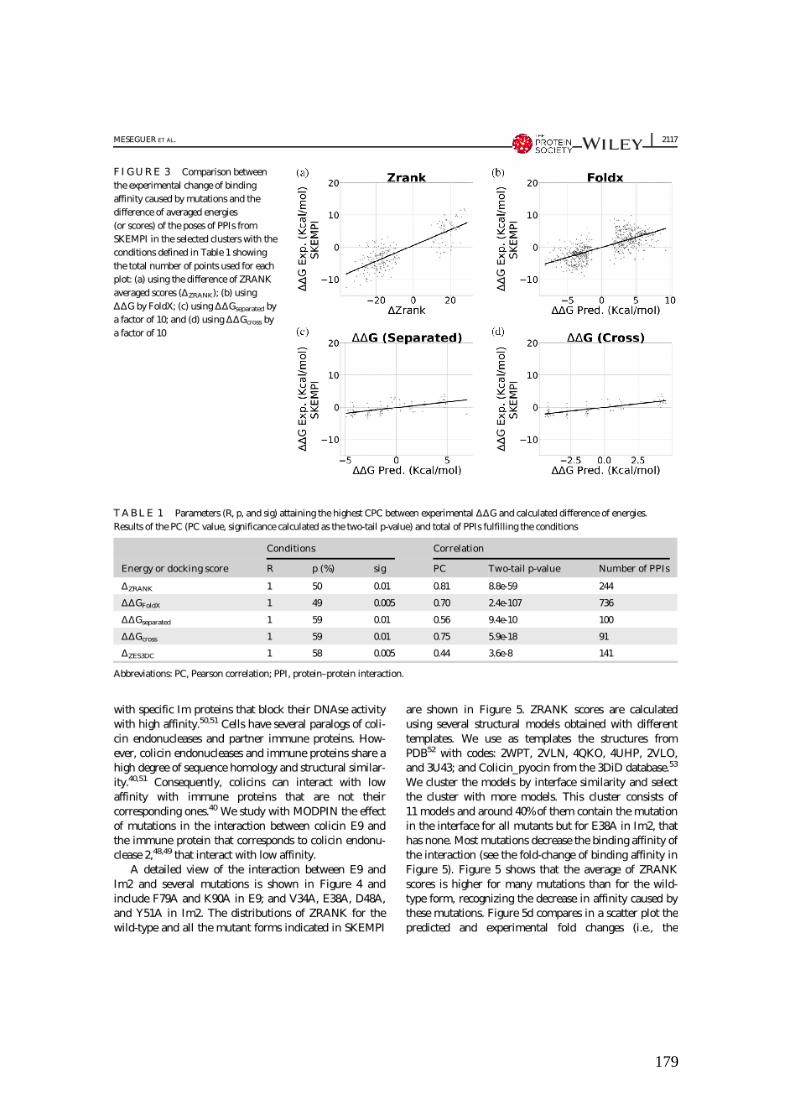
179

180
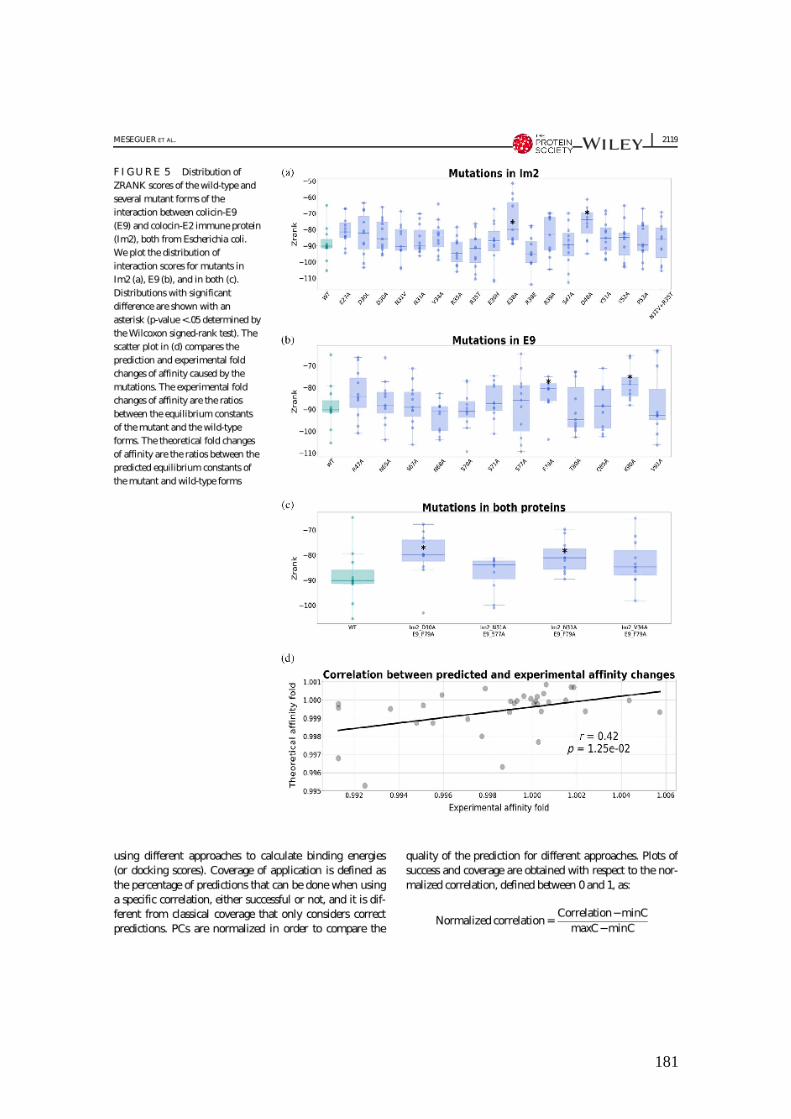
181

182

183

184

185

186

187

188

189

190

191

192

193
3.2.2 - Prediction of protein–protein binding affinities from unbound protein structures
Meseguer A, Bota PM, Fernández-Fuentes N, Oliva B. Prediction of protein–protein binding affinities from unbound protein structures. (Submitted to Methods in Molecular Biology)

194
Prediction of protein–protein binding affinities from unbound protein structures. Alberto Meseguer1, Patricia Bota1,3, Narcis Fernández-Fuentes2,3 and Baldo Oliva1
1Structural Bioinformatics Lab (GRIB-IMIM), Department of Experimental and Health Science, University
Pompeu Fabra, Barcelona 08005, Catalonia, Spain, 2Institute of Biological, Environmental and Rural
Sciences, Aberystwyth University, SY233EB Aberystwyth, United Kingdom 3Department of Biosciences, U
Science Tech, Universitat de Vic-Universitat Central de Catalunya, Vic 08500, Catalonia, Spain.
ABSTRACT Proteins are the workhorses of cells to carry out sophisticated and complex
cellular processes. Such processes require a coordinated and regulated
interactions between proteins that are both time and location specific. The
strength, or binding affinity, of protein-protein interactions ranges between the
micro- to the nanomolar association constant, often dictating the molecular
mechanisms underlying the interaction and the longevity of the complex, i.e.
transient or permanent. In consequence, there is a need to quantify the strength
of protein-protein interactions for biological, biomedical and biotechnological
applications. While experimental methods are labour intensive and costly,
computational ones are useful tools to predict the affinity of protein-protein
interactions. In this chapter we review the methods developed by us to address
this question. We will briefly present two methods to comprehend the structure
of the protein complex either derived by comparative modelling or docking.
Then we introduce BADOCK, a method to predict the binding energy without
requiring the structure of the protein complex, thus overcoming one of the major
limitations of structure-based methods for the prediction of binding affinity.
BADOCK utilizes the structure of unbound proteins and the protein docking
sampling space to predict protein-protein binding affinities. We will present step-
by-step protocols to utilize these methods, describing the inputs and potential
pitfalls as well as their respective strengths and limitations.
KEYWORKS: Protein-protein interactions; protein docking; binding affinity; protin structures; protein interfaces

195
INTRODUCTION Protein-protein interactions as basis of cellular functions Almost, if not all, cellular processes, and thus all the mechanisms that makes life possible, are underpinned by highly coordinated and regulated communications between proteins. Protein-protein interactions (PPIs) are both space and time dependent. Two proteins will not be able to interact unless they find each other and the time span of the interaction will be dictated, principally, by the strength of such association otherwise known as its binding affinity. The determination of the binding affinity of protein associations is therefore a relevant challenge that impacts in different aspects of Life Science, Biomedical and Biotechnology research where PPIs is the subject of study. Determining the binding affinity of PPIs by experimental means can be achieved with biophysical methods such as isothermal titration calorimetry, surface plasmon resonance, fluorescence, spectroscopy or stopped-flow analyses. These approaches are usually time consuming and costly. Moreover, the variety of experimental methods can produce several affinity measures, being the estimation of binding affinity dependent on the method and the context (1). Therefore, computational approaches can help, either providing a useful guidance on the context of the interaction or giving an approximation of the affinity. Among the computational methods, there are two large groups classified according to the type of input-information. On the one hand, there are the sequence-based methods. These methods rely on mathematical and machine-learning models to infer binding affinity from the protein primary structure (2,3). On the other hand, there are structure-based methods, namely they required the 3D coordinates of the protein complex to estimate the binding affinity (1,4,5). Structure-based methods are more accurate, but its main limitation is that only for few interactions are known the structures of the complexes. To overcome this limitation we developed a program, BADOCK (6), that took advantage of the accumulated knowledge on the role played by non-interface regions in the formation of the protein complexes (see next). Relevance on non-native binding interface regions in the formation of protein complexes It has been estimated that the concentration of proteins in a cell goes from 2 to 4 million proteins per cubic micron (7). Proteins contact each other continually in such a highly crowded environment, leading to a high likelihood for non-specific interactions to happen (8-11). A paradox in protein–protein interactions is how the unbound proteins of a binary complex recognize each other in such a crowded environment and how they find their best interacting interface in a short period of time (12). If finding the best interacting interface were the result of protein collisions with only one successful outcome (i.e., the native conformation of the binary complex), the formation of PPIs would require a large amount of time (12). Nevertheless, interacting proteins find each other and form a complex in a relatively short time and this is another paradox to solve after

196
Levinthal’s paradox in protein folding (13). Recent works have proposed a plausible solution to this paradox by considering that all faces of the protein surfaces, and not only their binding sites, contain information to distinguish binding from non-binding partners (6,12,14,15). First, it has been shown that distributions of docking poses can be used to distinguish interacting proteins from non-interacting proteins (6,14). Second, it has been shown that protein surfaces are under evolutionary pressure to recognize efficiently their binding partners (also known as positive design) and to avoid non-binding partners (also known as negative design) (16-20). This is in line with the work of Schweke et al. (15), proving that surface propensity to interact with ligands is shared by homologous proteins. Finally, it has been shown that protein-binding sites have similar features (21-24): they tend to be flat, hydrophobic surfaces, and provide a non-specific binding patch that is suitable for many ligands. This suggests that binding sites may not be enough to define the specificity ruling protein-protein recognition. All these findings are yet in agreement with the funnel-like intermolecular energy landscape used to describe PPIs (25,26). According to this model, PPIs reach their native-state conformation after going through several intermediate conformations (27). These conformations have lower values of free energy as long as they get close to the native-state, resembling the energy funnel model for protein folding (28). Therefore, albeit intermediate conformations involve non-native assembly regions, they are still relevant for achieving the native conformation. BADOCK: estimating binding affinity from unbound proteins With the aim to shed light into the role of non-interacting sites, we studied the formation and binding affinity of binary complexes of globular soluble proteins from its unbound proteins. We used the poses resulting from the protein-protein docking search to scout the conformational space of potential encounter complexes. The analysis of this study boosted the prediction of binding affinity using the unbound protein structures, proving its feasibility in a method named BADOCK (6). BADOCK uses docking poses generated with PatchDock (using default parameters) (29), refined and rescored with FiberDock (30) and it includes additional scores from statistical potentials (31). We classified the poses of the docking and affinity benchmark datasets (32) into four classes, depending on the orientation of the binding sites of the protein partners: two productive (near-native and face-face) and two non-productive (face-back and back-back) (Fig. 1.) Interestingly, the scores in the four classes of poses were correlated: low binding affinities in the native pose corresponds to the low energy-binding scores in the other classes, while high binding affinities in native conformations also correspond with large energy-binding scores in the other classes. Because of this feature, we could compare binding affinities of different PPIs using the distribution of scores from their docking-space search, without requiring the

197
native or near-native conformations. Using this principle, BADOCK applies a linear regression model to predict ΔG values using any of the docking scores. By default, the method uses the ES3DC statistical potential because this had the best correlation with the affinities in the benchmark (32). The method was tested using a 10-fold validation protocol in the affinity benchmark (32), achieving an average Pearson’s correlation of 0.36 (P < 0.05) with an error around 2.84 Kcal/mol (6). METHODS Predicting the structure of protein complexes by comparative modelling (MODPIN). Background The binding energy of PPIs can be predicted from the structures of their complexes and several methods are available to carry out such calculations (see introduction). If the structure of the protein complex is not available, several methods (33,34) can be used to obtain a structural model by comparative modelling (35). We have also developed a method, MODPIN, to model several conformations for each protein-protein interaction (59) (Fig. 2). These can be used to explore the conformational space of an interaction and study the association of proteins (36,37). Since PPIs can have different conformations, a set of models in the form of an ensemble of conformations is more representative than a single structure (38,39). MODPIN provides a range of metrics to account for the strength or quality of the interaction in the form of energies or docking scores (6,32,40,41). We have used MODPIN to assess the effect of mutations on the binding energy of complexes (42,43). Structural models of macro-complexes have been used by other authors to predict the binding affinity by a large variety of methods (44,45), but here we used it to understand and complement the prediction by our approach, BADOCK, without using the complex structure (see in section 2.3). Protocol Using the server:
• Go to http://galaxy.interactomix.com
• In the section INTERACTOMIX TOOLS under ‘Structural-level analyses you can access MODPIN.
• Upload a text file with the amino acid sequences of the proteins you want to model in FASTA format. An example is shown in the web page.
• Upload a text file with the protein pairs to model. In case of testing mutations this is to be indicated it in the file. An example of an input is shown in the web page.
• Choose the option to include hydrogens.

198
• Choose the option to check the quality of the structure of the interaction (i.e. the structure is relaxed with Rosetta). If this option is not selected the modelling is fast, but the quality of the model may be affected.
• Choose the option to force modelling if you wish to overwrite the previous models.
• Choose the option to include templates from the 3DiD database.
• Choose the number of models to be obtained per template.
• Choose the option to renumber the structure according to the amino acid sequence; it starts with 1 otherwise.
• Submit your job.
• The execution returns a set of structural models of PPIs.
• To further analyse the models the user is invited to the “Analysis” tool window. In this window the user is requested to upload the input files as before. The program assigns the structural models and scores the interactions by docking potentials or physic-chemical energies (the program uses the docking scores of ZRANK by default (46)). Other available scorings are: InterfaceAnalyzer of Rosetta (52), FoldX (40) and Statistical Potentials such as ES3DC (31,59).
• Structural models with similar interface are clustered and the server returns the files with the structures and scores of each cluster.
Local installation: MODPIN can also be installed locally. The scripts are available in https://github.com/structuralbioinformatics/MODPIN Predicting the structure of protein complexes by docking-based approaches (V-D2OCK). Background When neither the structure of the complex nor an acceptable template is available, the structure of the protein complex can be inferred by docking-based approaches (47). Docking consists on modelling the structure of the complex starting from the structure of each separated molecule (29). Docking algorithms produce many conformations (i.e. docking poses) that are subsequently scored to find the most preferable conformation(s) (48). Wass et al. showed that sets of docking poses could be used to discern between interacting and non-interacting proteins using the distribution of docking scores (14). Therefore, docking scores provide indications of the binding affinity between two proteins. In a previous work we developed V-D2OCK (VORFFIP Driven DOCKing), a data-driven docking algorithm (49) that uses VORFFIP (50) to identify and delineate the likely interfaces of unbound proteins and direct the docking. V-D2OCK returns a list of possible conformations of protein complexes ranked by three different scoring functions (see below). Although these scores cannot be used straight forward as binding energies or affinities, they can be used nonetheless as

199
qualitative measures to select the most likely conformation(s) and subsequently used to understand and complement the prediction of the binding energy with BADOCK. V-D2OCK’s workflow is shown in Fig. 2. Protocol
• Go to http://galaxy.interactomix.com/
• In the section INTERACTOMIX TOOLS under Structural-level analyses you can access V-D2OCK.
• V-D2OCK requires two PDB coordinate files to perform the docking. Protein A will remain static during the docking. Submit both proteins.
• Select the Scoring functions. Three choices are available to users: Patchdock score that takes into account geometrical features (29); ZRANK scoring function that combines both physics-based and knowledge-based terms (46); and (iii) ES3DC scoring function based on coarse-grained knowledge based potentials (31).
• Select the tolerance of the clustering algorithm.
• Submit the job
• The server will return a compressed file (tar) with the information on the rotation and translation matrices and a file with the scores for each of the docking poses.
Estimation of binding affinities from unbound proteins: BADOCK. Background As described in detail in the introduction, BADOCK is a method that predicts the binding affinity of binary protein-protein interactions by means of the scores after sampling the docking space with the structures of the unbound proteins. Decoys for the docking simulation are binned in four classes depending on the orientation of their respective binding sites. The correlation between energetic terms and docking scores is used to derive a linear regression model to predict the binding affinity. BADOCK’s workflow is shown in Fig. 1. Protocol Using the server:
• Go to http://sbi.upf.edu/BADOCK
• Upload two PDB files of the unbound protein-partners (Fig. 3A). These structures may correspond to a multidomain structure or the user can split the structure by domains and test separated domains at a time.
• Bookmark the page for later accession or wait until the program finishes (Fig. 3B).
• Ultimately the server will return the predicted binding affinity (Fig. 3C).
• By searching in the results page with the job ID you can access to

200
BADOCK results of prior executions (Fig. 3D).
• BADOCK his also accessible through the Galaxy InteractoMIX platform at http://galaxy.interactomix.com/tool_runner?tool_id=interactomix_BADOCK.
Local installation: The scripts and data parameters of BADOCK are available in: https://github.com/BADOCKsbi/BADOCK (6). NOTES
1. If attempting to model protein complexes with multiple domains and no suitable templates can be found for all the domains involved, then it would be advisable to divide the different monomers in domains (e.g. using PFAM (51)) and attempt the prediction with them.
2. V-D2OCK server can be accessed in two URLs, the one shown in the
protocol above or in http://www.bioinsilico.org/VD2OCK . The latter provides richer 3D visualization capabilities and docking poses are visualized with a Jmol applet.
3. Unless there is an error on the input files of the coordinates of the
proteins (i.e. usually this occurs if the input files are not in any of the standard PDB formats), V-D2OCK and BADOCK will always produce docking poses of the complex and consequently a prediction of the binding energy. If they fail, test first that the coordinates of the proteins adhere to PDB standards.
4. Optimize the structure prior to submission; there are several methods
available in Rosetta (52) (e.g. commands fixbb and relax) or in FoldX (40) (e.g. command repair_pdb).
5. Users can add hydrogens before optimization, using for example the
program Reduce (53).
6. When docking multidomain proteins, the analysis of docking solutions will show the preferences for specific domains of one or both proteins. In this case, the protein could be decomposed into domains prior to the prediction of binding affinity with BADOCK. We have to note that most protein-protein interactions are produced by the interaction between two single domains, as seen in Interactome3D (33). Therefore, attempting to calculate the interaction with single domains may be more suitable.
7. BADOCK server does not allow the user to tune parameters or choose
execution options. However, users have the option to modify the input

201
PDB files. Some suggested modifications are:
• BADOCK was implemented based on the interaction of soluble globular proteins and except for antigen-antibody interactions. For membrane proteins, the method can only be applied on the soluble globular fragment. Thus, for membrane-proteins we recommend splitting the structures by domains and test single domains with BADOCK or restrict to the soluble globular fragments. It is important to note that the method will not work for antibodies.
In the case of multidomain proteins:
• We can use MODPIN to determine whether the most probable interaction of the partners is produced by single domains or imply multidomain conformations. If the interaction is done between two single-domains, we can split the structure in domains and submit the structures of the interacting ones to BADOCK, otherwise use the largest structure available.
• If MODPIN cannot be applied, but the structures of the unbound partners are available, we can use V-D2OCK to check the type of interaction (single or multidomain) and proceed accordingly as before.
8. We can use iFrag to predict the regions of the proteins that are more likely to interact and proceed similarly as in Notes 6 and 7. IFrag is a web service that predicts regions of interaction in PPIs based on sequence information, it is available in http://sbi.imim.es/web/index.php/research/servers/iFrag (54).
9. BADOCK is also accessible through the Galaxy InteractoMIX platform: https://galaxy.interactomix.com in ‘Structure-based’ menu.
EXAMPLES Example to complement BADOCK with MODPIN MODPIN can be used to observe and analyse the conformational space of the interaction of two proteins through known interactions of homologs. It also complements BADOCK when one or both proteins of the interaction are composed by several domains. We use MODPIN to sample the conformational space of the interaction based on the homologs, recognize the implication of domains and select those relevant on performing the interaction. Here we show as example the interaction between mTOR and the target of rapamycin complex LST8 subunit (also known as the mammalian lethal with SEC13 protein 8). LST8 is a subunit of both mTORC1 and mTORC2 complexes formed with Raptor (mTOR). While the LST8 subunit is a small protein consisting of a single domain, mTOR is a large multi-domain protein. We retrieve the amino acid sequences of human mTOR and LST8 subunit from the UniProt database (55) (codes P42345 for mTOR and Q9BVC4 for LST8

202
subunit). We construct a single file with both sequences in FASTA format and input them to run MODPIN in the galaxy interactoMIX server (Fig. 4A). MODPIN produces a collection of 6 models of the PPI. We assess the structural similarity of each partner of the interaction on the set of models using the superposition with MATCHMAKER from the Chimera package(56). We observe that the structural models of the proteins involved in the interaction are highly similar between them (mean RMSD smaller than 1.5 Å and 1 Å for mTOR and LST8 subunit models, respectively). In addition, we observe that 5 out of 6 models share the same interface, where the region of mTOR in the interface involves amino acids 2262 to 2294 (Fig. 4B). This region belongs to the PI3K/PI4K domain (amino acids 2182–2516) according to the ProSite annotation provided by the UniProt database (21). We isolated the domain containing this region in a new PDB file. Then, we submitted to BADOCK the structure of the PI3K/PI4K domain and the LST8 subunit in BADOCK (Fig. 4C) and compared with the affinity predicted by using the whole structure of mTOR. The prediction of DG using the specific domain of mTOR is DG = -11.429 kcal/mol , while the prediction using the whole structure is DG = -10.779 kcal/mol. Given an acceptable error of about 3kcal/mol with BADOCK, both values are within the same range and we may use the average of both. Example to complement BADOCK with V-D2OCK We cannot apply MODPIN when the structure of an interaction is not known and there are no homologs to use as template for modelling. Then, if the structure of the unbound proteins is available, we can use a docking approach, such as V-D2OCK, to sample the conformational space of the interaction. V-D2OCK combines a prediction of interacting interfaces with rigid-body docking to generate an ensemble of conformers of a PPI. Therefore, we apply V-D2OCK to recognize the implication of domains and select those relevant on performing the interaction as above, albeit this approach is based on the sample of poses derived from docking, which may be less rigorous. We use as example the interaction between the Cyclin-dependent kinase 9 (CDK9) and the La-related protein 7 (LARP7). The unbound structures are retrieved from the PDB for CDK9(entry 3blh chain A) and two structures that correspond to two different domains for LARP7, one at the N-tail (Nt domain, formed by PFAM domains La and RRM-1, entry 4wkr chain A) and the other at the C-tail (Ct domain, formed by PFAM domain RRM-2, entry 6d12 chain A). We use V-D2OCK with default parameters to sample the docking space and obtain several potential interacting interfaces between CDK9 and the Nt domain of LARP7 (Fig. 5A) and between CDK9 and the Ct domain of LARP7 (Fig. 5C). Then, we use BADOCK to predict the binding energy using the structures of both potential interactions (Fig 5B, 5D). The predicted affinities are respectively ΔG = -8,83 kcal/mol and ΔG = -9.74 kcal/mol. Given that the error of BADOCK is about 3kcal/mol, we may use any of both predicted affinities or its average, but on the contrary to the previous example, the complete structure of LARP7 is unknown and we cannot ensure that the independent role of all domains.

203
Example to complement BADOCK with iFrag To complement the previous use of V-D2OCK, we can also use a sequence-based approach to predict the regions involved in an interaction. Here, we propose the use of iFrag, a method that predicts the regions of the interface between two proteins based on the recursion of several interacting homologs. iFrag is available in the sequence based methods in https://galaxy.interactomix.com and in http://sbi.imim.es/iFrag (54). As shown with the use of V-D2OCK and BADOCK, the fragments Nt and Ct of the structure produce comparable binding energies, but we are still unable to decide whether the interaction of the Nt fragment is produced by an specific domain or if the whole fragment is involved in the interaction. With iFrag predictions we observe that from the Nt fragment, composed by La and RRM-1 domains, the most likely responsible for the interaction with CDK9 is RRM-1. Therefore, the structure of the Nt fragment is split in domains La and RRM-1. We analyse separately the docking poses with V-D2OCK and used BADOCK to predict the binding affinity (see figure 7). The results show a similar binding energy, ΔG = -9,64 kcal/mol with the RRM-1 domain and ΔG =-9,75 kcal/mol with the La domain. In conclusion, among the different affinities we assume that the binding between LARP7 and CDK9 is about -9,64 kcal/mol.

204
FIGURES
Figure 1: BADOCK workflow. BADOCK takes as input two unbound structures. The method uses PatchDock to sample by rigid-body docking the interaction space. Docking poses are optimized using FiberDock. All docking poses are scored and used to predict ΔG. The orientations of the poses are classified as: 1) Near-Native, docking poses with a ligand-RMSD < 10Å (6); 2) Face-Face, poses where binding sites of both protein-partners face each other (i.e. they are inaccessible to other proteins); Face-Back, poses where only one binding site interacts with the protein partner (i.e. the binding site of only one of the proteins is accessible; and Back-Back, when both binding sites are accessible to interact with other protein units (see details in (6)).

205
Figure 2: MODPIN and V-D2OCK workflows. Both workflows produce conformations of PPIs. MODPIN uses the amino acid sequence of both partners as input and it uses BLAST to search homologs of both sequences with a known structure of the interaction (PPI templates). One or more structural models are obtained with MODELLER (57) for each PPI template. V-D2OCK uses two unbound structures as input, predicts the interface of their interaction and it applies a rigid-body docking with PatchDock restricted to the predicted interface.

206
Figure 3: BADOCK web service. (A) Submission page of the BADOCK web server. (B) Message sent after submission, it includes the job identification (JOB ID) and a link to access the results. (C) Results page of BADOCK web server shows the predicted ΔG for the interaction of the submitted proteins. (D) Menu of the results page: by introducing a JOB ID results can be retrieved from a previous execution.

207
Figure 4: Example of the use of BADOCK complemented with MODPIN. (A) Submission page of MODPIN in the galaxy InteractoMIX server. (B) Structure of a model of mTOR (orange and grey) interacting with the LST8 subunit of the target of rapamycin complex (blue). The region of mTOR involved in the interaction with the LST8 subunit in the majority of models obtained with MODPIN is highlighted in orange. (C) Result of BADOCK obtained with the structures of LST8 subunit and the highlighted region of mTOR.

208
Figure 5: Example of the use of BADOCK complemented with V-D2OCK. (A) Example of one of the predicted poses of the interaction between the Cyclin-dependent kinase 9 (CDK9, in red) and the Nt fragment of La-related protein 7 (LARP7, in blue). The prediction of affinity with BADOCK is shown at the bottom. (B) Example of one of the predicted poses of the interaction between the Cyclin-dependent kinase 9 (CDK9, in red) and the Ct fragment, composed by the RRM-2 domain, of LARP7 (in blue). The prediction of affinity with BADOCK is shown at the bottom.

209
Figure 6: Results page of iFrag using the sequences of LARP7 and CDK9 as example. Input sequences are shown at the top. The heatmap highlights that the interaction between LARP7 and CDK9 is likely produced by the region of domain RRM-1 of LARP7.

210
Figure 7: Example of the use of BADOCK complemented with V-D2OCK after iFrag selection of domains. (A) Predicted pose involving the interface between the Cyclin-dependent kinase 9 or CDK9 (CDK9, in red) and the RRM-1 motif of La-related protein 7 (in blue). The prediction of affinity with BADOCK is shown at the bottom. Predicted pose involving the interface between the Cyclin-dependent kinase 9 or CDK9 (CDK9, in red) and the La domain of La-related protein 7 (in blue). The prediction of affinity with BADOCK is shown at the bottom.

211
REFERENCES 1. Vangone A, Bonvin AM. Contacts-based prediction of binding affinity in protein-protein
complexes. Elife. 2015 Jul 20;4:e07454.
2. Abbasi WA, Asif A, Ben-Hur A, Minhas FUAA. Learning protein binding affinity using
privileged information. BMC Bioinformatics. 2018 Nov 15;19(1):425.
3. Yugandhar K, Gromiha MM. Feature selection and classification of protein-protein
complexes based on their binding affinities using machine learning approaches. Proteins.
2014 Sep;82(9):2088–96.
4. Horton N, Lewis M. Calculation of the free energy of association for protein complexes.
Protein Sci. 1992 Jan;1(1):169–81.
5. Moal IH, Agius R, Bates PA. Protein-protein binding affinity prediction on a diverse set of
structures. Bioinformatics. 2011 Nov 1;27(21):3002–9.
6. Marín-López MA, Planas-Iglesias J, Aguirre-Plans J, Bonet J, Garcia-Garcia J,
Fernandez-Fuentes N, et al. On the mechanisms of protein interactions: predicting their
affinity from unbound tertiary structures. Bioinformatics. 2018 15;34(4):592–8.
7. Milo R. What is the total number of protein molecules per cell volume? A call to rethink
some published values. Bioessays. 2013 Dec;35(12):1050–5.
8. McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular
model of the bacterial cytoplasm. PLoS Comput Biol. 2010 Mar 5;6(3):e1000694.
9. Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, et al. Biomolecular interactions modulate
macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife.
2016 01;5.
10. Mika JT, Poolman B. Macromolecule diffusion and confinement in prokaryotic cells. Curr
Opin Biotechnol. 2011 Feb;22(1):117–26.
11. Ellis RJ. Macromolecular crowding: an important but neglected aspect of the intracellular
environment. Curr Opin Struct Biol. 2001 Feb;11(1):114–9.
12. Planas-Iglesias J, Marin-Lopez MA, Bonet J, Garcia-Garcia J, Oliva B. iLoops: a protein-
protein interaction prediction server based on structural features. Bioinformatics. 2013 Sep
15;29(18):2360–2.
13. Levinthal C. Are there pathways for protein folding? J Chim Phys. 1968;65:44–5.
14. Wass MN, Fuentes G, Pons C, Pazos F, Valencia A. Towards the prediction of protein
interaction partners using physical docking. Mol Syst Biol. 2011 Feb 15;7:469.
15. Schweke H, Mucchielli M-H, Sacquin-Mora S, Bei W, Lopes A. Protein Interaction Energy
Landscapes are Shaped by Functional and also Non-functional Partners. J Mol Biol. 2020
Feb 14;432(4):1183–98.
16. Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid
edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002 Mar 5;99(5):2754–9.

212
17. Pechmann S, Levy ED, Tartaglia GG, Vendruscolo M. Physicochemical principles that
regulate the competition between functional and dysfunctional association of proteins.
Proc Natl Acad Sci USA. 2009 Jun 23;106(25):10159–64.
18. Deeds EJ, Ashenberg O, Gerardin J, Shakhnovich EI. Robust protein protein interactions
in crowded cellular environments. Proc Natl Acad Sci USA. 2007 Sep 18;104(38):14952–
7.
19. Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, et al. A de novo protein
binding pair by computational design and directed evolution. Mol Cell. 2011 Apr
22;42(2):250–60.
20. Garcia-Seisdedos H, Empereur-Mot C, Elad N, Levy ED. Proteins evolve on the edge of
supramolecular self-assembly. Nature. 2017 10;548(7666):244–7.
21. Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J
Mol Biol. 1999 Feb 5;285(5):2177–98.
22. Chakrabarti P, Janin J. Dissecting protein-protein recognition sites. Proteins. 2002 May
15;47(3):334–43.
23. Li X, Keskin O, Ma B, Nussinov R, Liang J. Protein-protein interactions: hot spots and
structurally conserved residues often locate in complemented pockets that pre-organized
in the unbound states: implications for docking. J Mol Biol. 2004 Nov 26;344(3):781–95.
24. Keskin O, Ma B, Nussinov R. Hot regions in protein--protein interactions: the organization
and contribution of structurally conserved hot spot residues. J Mol Biol. 2005 Feb
4;345(5):1281–94.
25. McCammon JA. Theory of biomolecular recognition. Curr Opin Struct Biol. 1998
Apr;8(2):245–9.
26. Tsai H-HG, Reches M, Tsai C-J, Gunasekaran K, Gazit E, Nussinov R. Energy landscape
of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics
simulation: significant role of Asn ladder. Proc Natl Acad Sci USA. 2005 Jun
7;102(23):8174–9.
27. Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein-protein association
kinetics. Chem Rev. 2009 Mar 11;109(3):839–60.
28. Wolynes PG. Evolution, energy landscapes and the paradoxes of protein folding.
Biochimie. 2015 Dec;119:218–30.
29. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock:
servers for rigid and symmetric docking. Nucleic Acids Res. 2005 Jul 1;33(Web Server
issue):W363-367.
30. Mashiach E, Nussinov R, Wolfson HJ. FiberDock: Flexible induced-fit backbone
refinement in molecular docking. Proteins. 2010 May 1;78(6):1503–19.
31. Feliu E, Aloy P, Oliva B. On the analysis of protein-protein interactions via knowledge-
based potentials for the prediction of protein-protein docking. Protein Sci. 2011
Mar;20(3):529–41.

213
32. Vreven T, Hwang H, Pierce BG, Weng Z. Prediction of protein-protein binding free
energies. Protein Sci. 2012 Mar;21(3):396–404.
33. Mosca R, Céol A, Aloy P. Interactome3D: adding structural details to protein networks. Nat
Methods. 2013 Jan;10(1):47–53.
34. Zhang QC, Petrey D, Garzón JI, Deng L, Honig B. PrePPI: a structure-informed database
of protein-protein interactions. Nucleic Acids Res. 2013 Jan;41(Database issue):D828-
833.
35. Poglayen D, Marín-López MA, Bonet J, Fornes O, Garcia-Garcia J, Planas-Iglesias J, et
al. InteractoMIX: a suite of computational tools to exploit interactomes in biological and
clinical research. Biochem Soc Trans. 2016 15;44(3):917–24.
36. Levy Y, Cho SS, Onuchic JN, Wolynes PG. A survey of flexible protein binding
mechanisms and their transition states using native topology based energy landscapes. J
Mol Biol. 2005 Mar 4;346(4):1121–45.
37. Andrusier N, Mashiach E, Nussinov R, Wolfson HJ. Principles of flexible protein-protein
docking. Proteins. 2008 Nov 1;73(2):271–89.
38. Stein A, Rueda M, Panjkovich A, Orozco M, Aloy P. A systematic study of the energetics
involved in structural changes upon association and connectivity in protein interaction
networks. Structure. 2011 Jun 8;19(6):881–9.
39. Goh C-S, Milburn D, Gerstein M. Conformational changes associated with protein-protein
interactions. Curr Opin Struct Biol. 2004 Feb;14(1):104–9.
40. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server:
an online force field. Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W382-388.
41. Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs
highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013
Jan;22(1):74–82.
42. Jankauskaite J, Jiménez-García B, Dapkunas J, Fernández-Recio J, Moal IH. SKEMPI
2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and
thermodynamics upon mutation. Bioinformatics. 2019 01;35(3):462–9.
43. Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, et al. The
MIntAct project--IntAct as a common curation platform for 11 molecular interaction
databases. Nucleic Acids Res. 2014 Jan;42(Database issue):D358-363.
44. Siebenmorgen T, Zacharias M. Computational prediction of protein–protein binding
affinities. WIREs Computational Molecular Science. 2020;10(3):e1448.
45. Siebenmorgen T, Zacharias M. Evaluation of Predicted Protein-Protein Complexes by
Binding Free Energy Simulations. J Chem Theory Comput. 2019 Mar 12;15(3):2071–86.
46. Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized
energy function. Proteins. 2007 Jun 1;67(4):1078–86.
47. Takemura K, Matubayasi N, Kitao A. Binding free energy analysis of protein-protein
docking model structures by evERdock. J Chem Phys. 2018 Mar 14;148(10):105101.

214
48. Barradas-Bautista D, Moal IH, Fernández-Recio J. A systematic analysis of scoring
functions in rigid-body protein docking: The delicate balance between the predictive rate
improvement and the risk of overtraining. Proteins. 2017;85(7):1287–97.
49. Segura J, Marín-López MA, Jones PF, Oliva B, Fernandez-Fuentes N. VORFFIP-driven
dock: V-D2OCK, a fast and accurate protein docking strategy. PLoS ONE.
2015;10(3):e0118107.
50. Segura J, Jones PF, Fernandez-Fuentes N. Improving the prediction of protein binding
sites by combining heterogeneous data and Voronoi diagrams. BMC Bioinformatics. 2011
Aug 23;12:352.
51. El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein
families database in 2019. Nucleic Acids Res. 2019 08;47(D1):D427–32.
52. Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, et al. The Rosetta
All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory
Comput. 2017 Jun 13;13(6):3031–48.
53. Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using
hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999
Jan 29;285(4):1735–47.
54. Garcia-Garcia J, Valls-Comamala V, Guney E, Andreu D, Muñoz FJ, Fernandez-Fuentes
N, et al. iFrag: A Protein–Protein Interface Prediction Server Based on Sequence
Fragments. Journal of Molecular Biology. 2017 Feb 3;429(3):382–9.
55. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res.
2019 Jan 8;47(D1):D506–15.
56. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF
Chimera--a visualization system for exploratory research and analysis. J Comput Chem.
2004 Oct;25(13):1605–12.
57 Webb B, Sali A Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc.
Protein Sci. 2016 86:2.9.1-2.9.37.
58 Meseguer A, Dominguez L, Bota PM, Aguirre-Plans J, Bonet J, Fernandez-Fuentes N,
Oliva B Using collections of structural models to predict changes of binding affinity in
protein-protein interactions. (In revision in Protein Science).
59 Aguirre-Plans J, Meseguer A, Molina-Fernandez R, Marín-López MA, Jumde G, Casanova
K, et al. SPServer: split-statistical potentials for the analysis of protein structures and
protein-protein interactions. BMC Bioinformatics. 2021 Jan 6;22(1):4.

215
4. Discussion Eukaryotic cells have the ability to regulate their gene expression, leading to a huge variety of cell morphologies, functions and behaviors. Part of this regulation takes place at transcription, which is in turn regulated by PDIs, protein-protein interactions, DNA chemical modifications or chromatin condensation among other mechanisms. This thesis has focused on PDIs involving transcription factors and protein-protein interactions involving transcriptional co-factors, in the context of transcriptional regulation. We developed modeling computational pipelines and scoring functions for both types of interactions. For PDIs, we developed statistical potentials scoring functions. We further applied these scoring functions to predict PWMs and find TF binding sites as well as assess TF-DNA interactions. We also developed a comparative modeling pipeline capable of modeling protein complexes bound to free DNA or nucleosome-bound DNA. For protein-protein interactions, we developed a scoring system based on linear regression and state-of-the-art scoring functions. We also developed a comparative modeling pipeline capable of modeling collections of models for the same protein-protein interaction. From this point forward, we will carry out the discussion of this thesis by assessing the contribution of our work to the scientific community and the future perspectives for the work here exposed.
4.1 - Statistical potentials: how good they are? Knowing the binding preferences of TFs is fundamental to understand gene regulation. However, the binding preferences of many TFs remain unknown (41) and their experimental determination is expensive and time consuming (42). In this situation, computational tools can be used to complement experimental techniques and fill the gap in the knowledge about TF binding preferences. We contributed to this area of knowledge by creating statistical potentials scoring functions. These functions score contacts between amino acids and dinucleotides found in structures of PDIs. By coupling these scoring functions with an automated modeling pipeline, we can predict PWMs for any TF that we can model. This is done by threading different DNA sequences into a protein-DNA structure and selecting and aligning the top scoring sequences. This strategy has been successful as we have shown in comparisons with experimental PWMs from the JASPAR and Cis-BP databases. However, the application of our methods has some limitations. First, we can only predict PWMs for TFs for which we are able to obtain a structure interacting with DNA (either experimental or obtained by comparative modeling). The scarcity of experimental structures for TF-DNA interactions makes that we cannot obtain structures for some TFs (either experimental or by modelling). However, as we studied in chapter 3.1.1 of this thesis, for most TFs

216
(from both UniProt and JASPAR databases) we were able to obtain structural models, and therefore to apply our predictive methods. Our predictive methods can be very versatile and adapt to different biological situations. Examples of this could be the prediction of binding preferences of TFs in diverse conditions such as: having mutations in the DBD, interacting with the DNA in the presence of other TFs, establishing dimers with other TFs, binding specific DNA sequences or binding nucleosome-bound DNA. Imagine that we want to assess the binding preferences of a TF in any of the situations that we just exposed. If we were to assess this experimentally, it would require conducting complex experiments in most of the cases. If we use our predictive methods, results can be obtained in a cheaper and faster way. First, we would obtain structural models for the TF-DNA interaction under study, if structural data is available. From these structural models we can predict PWMs for the TF and infer binding affinities for specific DNA sequences. Also, having structural models of TF-DNA interactions can provide information on how the interaction of a TF can change depending on mutations, interaction with other TFs or nucleosome occupancy. The predictive performance of our statistical potentials is better for some TF families than for others. Even in some cases, we can see a strong variability in performance within the same TF family. This makes us wonder if we can estimate the confidence of our predictions. First, if we know what TF families perform the best, we can already expect a level of confidence depending on the TF family analyzed. Another way to estimate the confidence of our predictions could be to compare our predicted PWMs with experimental PWMs for the same TF, if available. If our prediction is accurate, this can be an indicator of our predictive power for that TF. Then, we can trust our methods when making predictions in more complex situations such as mutations, cooperative binding or interactions with nucleosome-bound DNA. We hypothesize that the confidence of our predictions can be estimated from data related to the predicted structural models and PWMs. By using a machine learning approach, we could use this data to choose, from a collection of PWMs, the ones that resemble more to the experimental ones. This is relevant since we were able to make statistically significant predictions for at least one PWM for 74.8% of the analyzed TFs from the JASPAR database (see section 3.1.1).
4.2 - Using statistical potentials to find TF binding sites genome wide Finding TF binding sites in silico genome wide is a complex task. The most straightforward way of finding TF binding sites is to scan DNA sequences with PWMs. However, this is likely to produce lots of errors as low values for both precision and recall (263). Regarding precision, the scanning of a DNA sequence with a PWM is prone to produce lots of false positives (264,265). This happens because when scanning a DNA sequence with a PWM the topology of the DNA is not taken into account. Parts of the DNA sequence may be bound

217
by nucleosomes or be located in closed chromatin regions. If that is the case, most of the binding sites located at these regions will not be functional. That is why filtering the regions that are open chromatin and nucleosome free may help to increase the precision when finding TF binding sites. Selecting DNA regions that are conserved is also an efficient strategy to reduce false positives (263). The basis for this strategy is that TF binding sites are functional elements in the genome, therefore they should be conserved. Our methods can predict a collection of PWMs for a single TF. This provides a new strategy to search TF binding sites genome wide: find binding sites that match not one, but most of the PWMs in the collection. Theoretical PWMs are obtained from structural models. Therefore, ModCRE can make different structural models of the TF of interest using different templates. The structural variability in this collection of models will be reflected in the variability in the collection of PWMs. This looks like a promising and fast strategy to overcome the big number of false positives that are found when scanning a DNA sequence with one PWM (264,265). Using collections of PWMs is also consistent with the fact that TFs may have different experimental PWMs. This does not mean that some experimental results are wrong, but that each technique and set of experimental conditions emphasizes some aspects of the TF-DNA binding. Differences in experimental PWMs can also happen because TFs can interact with the DNA adopting different conformations (266). Besides, some TFs may have multiple or very complex DBDs. The binding preferences of such complex TFs usually are represented in more than one PWM for the same TF, leading to more discrepancy between experimental sources. On the one hand, this means that using all available experimental PWMs for a TF as a collection would be a useful way to integrate all the experimental data available into one approach. On the other hand, it means that there is not one clear reference to validate the predictions of ModCRE. Interestingly, when comparing ModCRE predictions with experimental PWMs for the same TF from diverse sources, we found predictions matching the different experimental PWMs. This suggests that ModCRE can represent the variability that we see in experimental PWMs.
4.3 - Studying protein-protein interactions using collections of structural models
The use of collections of structural models is a helpful approach to the study of binding affinities in PPIs. This is consistent with the hypothesis of “conformational selection”, where proteins can have diverse conformations and only some of them are suitable for interaction (267,268). Besides, recent works pointed out that non-interacting conformations also contain information to distinguish binding from non-binding partners (182,205,252,253). Also, working with a collection of structures enables us to use population-based statistics when analyzing the docking scores or binding energies (157). Interestingly, the

218
use of collections of models has also proven to be helpful when studying binding affinities in PDIs. Besides the estimation of binding affinities, the use of collections of structural models is also helpful to increase the number of PPIs that can be analyzed. Structure-based methods for the estimation of binding affinities usually require the structure of the PPI under study. Since the availability of experimental structures of PPIs is scarce, many structure-based methods can be applied only to a small number of PPIs. The applicability of these methods would increase if it could be applied to structures of unbound proteins, for which the availability of structural data is higher. This is the case of BADOCK (205), the main method reviewed in section 3.2.2 of this thesis. BADOCK takes advantage on the idea that non-interacting conformations also contain information about binding affinities. Therefore, it generates a collection of structural models via docking and analyzes the entire collection, without needing to know what conformations the interacting ones are. In this thesis we have used two methods to obtain collections of structural models for PPIs: homology modeling (used by MODPIN) and docking (used by BADOCK). Since homology modeling requires structures of known PPIs, its applicability is lower than the one of docking. Also, since homology modeling mimics the conformations of experimentally determined PPIs, it is restricted to generate models that are similar to interacting conformations. On the other hand, docking has more freedom to explore the conformational space, leading to collections of models where interacting conformations are scarce. Interacting conformations usually have lower energy (269) and we would expect them to have stronger changes in energy upon mutations, in comparison with non-interacting conformations. Therefore, it makes sense to use homology modeling to assess the effect of mutations, because we restrict the analysis to conformations where energy changes upon mutations will be easily detected by scoring functions. If we used docking to assess the effect of mutations, the change in energy would be diluted among non-interacting conformations, where we expect the energy change to be lower.
4.4 - Using statistical potentials and protein-protein interactions to rebuild gene regulatory networks The ultimate goal of this project is to reconstruct gene regulatory networks using in silico methods. This is a very ambitious goal, and it is hard to achieve using only the tools that we present in this thesis. Our computational tools predict TF-binding sites as well as what of these TFs interact with transcriptional co-factors. This interaction with co-factors can be helpful to determine the role of the TF as a transcriptional activator or repressor. Combining our tools with available experimental data may be a good strategy to reconstruct gene regulatory networks.

219
We propose that our tools can be used in the context of specific cell types for which gene expression and nucleosome occupancy data are available. Gene expression data (coming from RNA-seq or similar experiments) could be used to filter out TFs and co-factors that are not expressed in the cell type of interest. Nucleosome occupancy data could be used to filter out those TF-binding sites that fit into nucleosome rich regions. HiC data could also be used for this purpose by filtering out those TF-binding sites that fit into closed chromatin regions. This last filtering would not be necessary for pioneer TFs. Therefore, this strategy would also require developing criteria to distinguish pioneer from non-pioneer TFs. This is one of the future challenges of this project for which we next propose a line of work.
4.5 - Future challenges: using structural modeling to assess the binding of TF to nucleosome-bound DNA
Most TFs cannot bind, or bind with less affinity, to nucleosome-bound DNA (27,28). One of the reasons why this happens is because the nucleosome is blocking half of the DNA surface, which is physically inaccessible to TFs (28). A dataset of SELEX results assessing TF binding preferences for nucleosome-bound DNA has been recently published (28). In this work, Zhu and colleagues discuss that the binding preferences of some TFs to nucleosome-bound DNA may be explained by the interacting modes between the TF and the nucleosome-bound DNA. In particular, they describe 5 major interaction modes between TFs and nucleosome-bound DNA, being each one of these modes specific for a set of TF families (28). We hypothesize that we could use the results from Zhu and colleagues to create a computational tool to predict what TFs are able to interact with nucleosome-bound DNA, and if that is the case, predict the corresponding interaction mode. To do so, we would apply our structural modeling pipeline that is capable of modeling TFs interacting with nucleosome-bound DNA. Since our structural modeling pipeline is based on homology modeling, our predictive method would be strongly based on the idea that homologous proteins interact with nucleosome-bound DNA in the same way. This is consistent with the fact that the major interaction modes described by Zhu and colleagues are specific for certain TF families (28).
4.6 - Future challenges: statistical potentials to assess the effect of mutations in TF-DNA interactions
Mutations can affect TF-DNA interactions by modifying their chemical affinity. According to the change in affinity, TF-DNA interactions can either be created or disrupted. Besides, these mutations can happen in genes encoding TFs or in non-coding regulatory elements such as enhancers or promoters. The lost or creation of new TF-DNA interactions can lead to changes in GRNs and, in some cases, to phenotypical changes or diseases (6).

220
A future task is to use statistical potentials to predict the effect of mutations in TF-DNA interactions. This can be already done using ModCRE and comparing the binding profiles of two DNA sequences with the same TF. However, we have not validated this strategy to assess the effect of mutations in TF-DNA interactions. We hypothesize that statistical potentials may not be accurate enough to predict precise changes in affinity. This is why we plan to combine several statistical potentials scoring functions with machine learning classifiers to achieve an optimized prediction. To achieve this objective, we want to use as benchmarks the datasets developed by Fuxman-Bass et al (6), Barrera et al (270) and Shi et al (271). The dataset of Fuxman Bass et al has been obtained using yeast one-hybrid to assess the effect of mutations in DNA non-coding regions that have been associated with diseases (6). The dataset of Barrera et al has been obtained using protein binding microarrays for TFs with mutations associated with mendelian diseases (270). The dataset of Shi et al is a collection of allele-specific binding events obtained by ChIP. Allele-specific binding events happen when TFs bind preferentially to one allele in heterozygous binding sites (271). Besides, PBM raw data can also be used as benchmark to assess the effect of mutations happening in DNA binding sites, by comparing DNA 8-mers differing in only one nucleotide.
4.7 - Future challenges: statistical potentials to assess the effect of DNA methylation in TF-DNA interactions DNA can be methylated at cytosines and this modification can affect the binding of transcription factors (29). DNA methylation has been found to have a relevant role in regulating genome structure and activity (29). A dataset of SELEX results assessing TF binding preferences in the presence of DNA methylation has been recently published. In this dataset the binding preferences for several TFs are assessed as well as how these change with cytosine methylation (29). We hypothesize that we could use this dataset to create statistical potentials that evaluate the effect of cytosine methylation on TF binding. To do so, we would obtain structural models of the PDIs identified by SELEX. From these models we could extract amino acid – dinucleotide contacts and use them to create a new set of potentials. Doing this would require to slightly modify the definition of our current potentials, since these are not designed to consider cytosine methylation. One way in which these potentials could be redesigned is by including the presence of a methyl group in the environment of the dinucleotide, as well as its proximity to the amino acid involved in the contact.

221
4.8 - Future challenges: in silico design of TFs
TFs are potential bioengineering tools due to their capacity to target specific sites in the genome. This can be used for genome editing by fusing nucleases to DBDs of TFs. This fusion protein will be able to target a protein with endonuclease activity to a specific site in the genome. Many works have applied this strategy (272–281). So far the two more popular approaches to genome editing using TFs are zinc fingers nucleases and transcription activator-like effector nucleases (TALENs) (282). Zinc finger nucleases are fusion proteins that have the DBD from the C2H2 ZF family. C2H2 ZF proteins can recognize long and complex DNA binding sites by using zinc finger domains, where each of these domains recognizes 3 basepairs (42). C2H2 ZF proteins can recognize long and complex DNA sequences, which enables to target nuclease activity precisely into the genome and avoid unwanted indels. By knowing the binding preferences of individual zinc fingers and placing more or less zinc finger domains, we could hypothetically engineer C2H2 ZF proteins to recognize any DNA binding site (42). Recently, C2H2 ZF proteins have been used to develop precise endonucleases to introduce indels in the CCR5 gene which can lead to HIV cellular resistance (261). TALENs consist of a non-specific FokI nuclease domain fused to a DBD that can be customizable to recognize diverse DNA sequences. This DBD is made of highly conserved repeats derived from transcription activator-like effectors (TALEs), which are proteins secreted by Xanthomonas bacteria to alter transcription of genes in host plant cells (277). TALEs are not included in any of the major TF families that we worked with during this thesis. However, we can make models for these proteins and we can score their interactions with DNA using general potentials. We hypothesize that we could use statistical potentials to identify the optimal protein sequence for a TF to recognize a target DNA sequence. Given the structure of a TF, we can model this TF binding our target DNA sequence. From this model we obtain amino acid – dinucleotide contacts, each of which have an associated statistical potentials score. We can find what amino acid substitutions lead to an optimal score. Since amino acids make contacts with overlapping DNA regions, we cannot assume that amino acids are independent when searching for the optimal score. That is why we consider to do the scoring by patches of several amino acids that are close in space. Using this strategy with C2H2 ZF proteins seems promising since we have strong predictive tools for this TF family, and they are able to recognize long and complex DNA sequences precisely.

222

223
5. Conclusions This section describes in short the achievements of the work presented in this thesis:
i. Statistical potentials scoring functions are an excellent approach to predict TF-DNA PWMs, hinting towards its potential use on binding affinity.
ii. Both protein binding microarray and bacterial one-hybrid experimental
data can be efficiently used to extend data on statistical potentials scoring functions via homology modeling.
iii. Family-specific statistical potentials are able to capture the structural features of their corresponding TF families. This results in the fact that family-specific potentials have a higher predictive power than general potentials for most TF families.
iv. Statistical potentials scoring functions in combination with the homology
modeling pipeline can be effectively used to predict TF binding motifs using collection of models.
v. Using a collection of PWMs increases the accuracy to predict TF binding motifs. This is true for both statistical potentials predictions and for the nearest neighbor approach.
vi. We have created an homology modeling pipeline to integrate structural models of PDIs and PPIs.
vii. We have integrated PDI and PPI information into networks of TFs and transcription co-factors.
viii. Using homology modeling, state-of-the-art scoring functions for protein-
protein interactions, data clustering and correlation we were able to predict changes in binding affinity caused by mutations in protein-protein interactions.
ix. We reviewed computational methods to predict binding affinities in protein – protein interactions by scouting the conformational space of the interaction poses.

224

225
6. Appendix 6.1 - SPServer: split-statistical potentials for the analysis of protein structures and protein-protein interactions My contribution to this work was to write the tutorials of the presented application, to test extensively the application, to design the manuscript and to collaborate to write the manuscript. Abstract Background: Statistical potentials, also named knowledge-based potentials, are scoring functions derived from empirical data that can be used to evaluate the quality of protein folds and protein-protein interaction (PPI) structures. In previous works we decomposed the statistical potentials in different terms, named Split-Statistical Potentials, accounting for the type of amino acid pairs, their hydrophobicity, solvent accessibility and type of secondary structure. These potentials have been successfully used to identify near-native structures in protein structure prediction, rank protein docking poses, and predict PPI binding affinities. Results: Here, we present the SPServer, a web server that applies the Split-Statistical Potentials to analyze protein folds and protein interfaces. SPServer provides global scores as well as residue/residue-pair profiles presented as score plots and maps. This level of detail allows users to: (1) identify potentially problematic regions on protein structures; (2) identify disrupting amino acid pairs in protein interfaces; and (3) compare and analyze the quality of tertiary and quaternary structural models. Conclusions: While there are many web servers that provide scoring functions to assess the quality of either protein folds or PPI structures, SPServer integrates both aspects in a unique easy-to-use web server. Moreover, the server permits to locally assess the quality of the structures and interfaces at a residue level and provides tools to compare the local assessment between structures. SERVER ADDRESS: https://sbi.upf.edu/spserver/ . Keywords: Knowledge-based potential; Protein structure evaluation; Protein structure prediction; Protein structure quality assessment; Protein–protein evaluation; Protein–protein interaction.
Aguirre-Plans J, Meseguer A, Molina-Fernandez R, Marín-López MA, Jumde G, Casanova K, et al. SPServer: split-statistical potentials for the analysis of protein structures and protein-protein interactions. BMC Bioinformatics. 2021 Jan 6;22(1):4.

226
6.2 - Galaxy InteractoMIX: An Integrated Computational Platform for the Study of Protein-Protein Interaction Data My contribution to this work was to explain and write the contents about MODPIN (157). MODPIN is an application included inside Galaxy InteractoMIX.
Abstract Protein interactions play a crucial role among the different functions of a cell and are central to our understanding of cellular processes both in health and disease. Here we present Galaxy InteractoMIX (http://galaxy.interactomix.com), a platform composed of 13 different computational tools each addressing specific aspects of the study of protein-protein interactions, ranging from large-scale cross-species protein-wide interactomes to atomic resolution level of protein complexes. Galaxy InteractoMIX provides an intuitive interface where users can retrieve consolidated interactomics data distributed across several databases or uncover links between diseases and genes by analyzing the interactomes underlying these diseases. The platform makes possible large-scale prediction and curation protein interactions using the conservation of motifs, interology, or presence or absence of key sequence signatures. The range of structure-based tools includes modeling and analysis of protein complexes, delineation of interfaces and the modeling of peptides acting as inhibitors of protein-protein interactions. Galaxy InteractoMIX includes a range of ready-to-use workflows to run complex analyses requiring minimal intervention by users. The potential range of applications of the platform covers different aspects of life science, biomedicine, biotechnology and drug discovery where protein associations are studied.
Mirela-Bota P, Aguirre-Plans J, Meseguer A, Galletti C, Segura J, Planas-Iglesias J, et al. Galaxy InteractoMIX: An Integrated Computational Platform for the Study of Protein-Protein Interaction Data. J Mol Biol. 2020 Sep 23;166656.

227
6.3 – List of posters Meseguer A, Årman F, Fornés O, Molina R, Bonet J, Fernandez-Fuentes N, Oliva B. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. European Conference on Computational Biology. September 2020; Sitges, Spain. Meseguer A, Årman F, Fornés O, Molina R, Bonet J, Fernandez-Fuentes N, Oliva B. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. Conference on Inteligent Systems for Molecular Biology. July 2020. Meseguer A, Årman F, Molina R, Fornés O, Bonet J, Oliva B. Predicting the DNA-binding preferences of C2H2-ZF proteins combining structural and experimental data. Meeting on genomics and bioinformatics from the Catalan Society of Bioinformatics. December 2019; Barcelona, Spain. Meseguer A, Fornes O, Årman F, Bonet J, Oliva B. Predcition of transcription factor binding by structural modeling. International Society for Computational Biology meeting and European Conference on Computational Biology. July 2019; Basel, Switzerland. Fornes O, Meseguer A, Årman F, Bonet J, Oliva B. Prediction of transcription factor binding by structural modeling. Meeting on genomics and bioinformatics from the Catalan Society of Bioinformatics. December 2018; Barcelona, Spain. Fornes O, Meseguer A, Bonet J, Oliva B. Prediction of transcription factor binding by structural modeling. European Conference on Computational Biology. September 2018; Athens, Greece. Fornes O, Meseguer A, Bonet J, Oliva B. Prediction of transcription factor binding by structural modeling. Cold Spring Harbor Laboratory meeting on gene regulation. March 2018; Cold Spring Harbor Laboratory, USA. Meseguer A, Fornes O, Fuxman Bass JI, Oliva B. Predicting the impact of mutations on transcription factor binding. Meeting on genomics and bioinformatics from the Catalan Society of Bioinformatics. December 2017; Barcelona, Spain. Meseguer A, Fornes O, Fuxman Bass JI, Oliva B. Predicting the impact of mutations on transcription factor binding. RecombCG. October 2017; Barcelona, Spain. Meseguer A, Fornes O, Fuxman Bass JI, Oliva B. Predicting the impact of mutations on transcription factor binding. FEBS3+. October 2017; Barcelona, Spain.

228

229
7. References
1. Lewis M. Allostery and the lac Operon. Journal of Molecular Biology. 2013 Jul 10;425(13):2309–16.
2. Mühlhofer M, Berchtold E, Stratil CG, Csaba G, Kunold E, Bach NC, et al. The Heat Shock Response in Yeast Maintains Protein Homeostasis by Chaperoning and Replenishing Proteins. Cell Reports. 2019 Dec 24;29(13):4593-4607.e8.
3. Arda HE, Walhout AJM. Gene-centered regulatory networks. Brief Funct Genomics. 2010 Jan;9(1):4–12.
4. Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013 Oct 14;24(4):542–56.
5. Mathelier A, Shi W, Wasserman WW. Identification of altered cis-regulatory elements in human disease. Trends Genet. 2015 Feb;31(2):67–76.
6. Fuxman Bass JI, Sahni N, Shrestha S, Garcia-Gonzalez A, Mori A, Bhat N, et al. Human Gene-Centered Transcription Factor Networks for Enhancers and Disease Variants. Cell. 2015 Apr;161(3):661–73.
7. Fornés Crespo O. On the characterization of protein-DNA interactions using statistical potentials and protein-protein interactions [Internet] [Ph.D. Thesis]. TDX (Tesis Doctorals en Xarxa). Universitat Pompeu Fabra; 2015 [cited 2020 Nov 4]. Available from: http://www.tdx.cat/handle/10803/320192
8. Lelli KM, Slattery M, Mann RS. Disentangling the Many Layers of Eukaryotic Transcriptional Regulation. Annu Rev Genet. 2012;46:43–68.
9. Xiao L, Wang J-Y. Posttranscriptional Regulation of Gene Expression in Epithelial Cells by Polyamines. In: Pegg AE, Casero Jr Robert A, editors. Polyamines: Methods and Protocols [Internet]. Totowa, NJ: Humana Press; 2011 [cited 2020 Nov 4]. p. 67–79. (Methods in Molecular Biology). Available from: https://doi.org/10.1007/978-1-61779-034-8_4
10. Gonçalves V, Jordan P. Posttranscriptional Regulation of Splicing Factor SRSF1 and Its Role in Cancer Cell Biology. Biomed Res Int [Internet]. 2015 [cited 2020 Nov 4];2015. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4529898/
11. Han H. RNA Interference to Knock Down Gene Expression. Methods Mol Biol. 2018;1706:293–302.
12. Vesely PW, Staber PB, Hoefler G, Kenner L. Translational regulation mechanisms of AP-1 proteins. Mutation Research/Reviews in Mutation Research. 2009 Jul 1;682(1):7–12.
13. Flick F, Lüscher B. Regulation of Sirtuin Function by Posttranslational Modifications. Front Pharmacol [Internet]. 2012 Feb 28 [cited 2020 Nov 4];3. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3289391/
14. Hanna J, Guerra-Moreno A, Ang J, Micoogullari Y. Protein Degradation and the Pathologic Basis of Disease. Am J Pathol. 2019 Jan;189(1):94–103.
15. Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet. 2012 Mar 6;13(4):233–45.

230
16. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell. 2018 Feb;172(4):650–65.
17. Brivanlou AH, Darnell JE. Signal transduction and the control of gene expression. Science. 2002 Feb 1;295(5556):813–8.
18. Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nature Reviews Genetics. 2009 Apr;10(4):252–63.
19. Reiter F, Wienerroither S, Stark A. Combinatorial function of transcription factors and cofactors. Current Opinion in Genetics & Development. 2017 Apr 1;43:73–81.
20. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007 Feb 23;128(4):707–19.
21. Venters BJ, Pugh BF. How eukaryotic genes are transcribed. Crit Rev Biochem Mol Biol. 2009 Jun;44(2–3):117–41.
22. Sikorski TW, Buratowski S. The basal initiation machinery: beyond the general transcription factors. Curr Opin Cell Biol. 2009 Jun;21(3):344–51.
23. Andersson R, Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020 Feb;21(2):71–87.
24. Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000 Jan;16(1):16–23.
25. Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006 Nov;24(11):1429–35.
26. Jayaram N, Usvyat D, R. Martin AC. Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics. 2016 Nov 2;17(1):547.
27. Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015 Apr 23;161(3):555–68.
28. Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, et al. The interaction landscape between transcription factors and the nucleosome. Nature. 2018;562(7725):76–81.
29. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017 05;356(6337).
30. Cusack M, King HW, Spingardi P, Kessler BM, Klose RJ, Kriaucionis S. Distinct contributions of DNA methylation and histone acetylation to the genomic occupancy of transcription factors. Genome Res. 2020 Oct;30(10):1393–406.
31. Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015 Nov 19;527(7578):384–8.
32. Panne D, Maniatis T, Harrison SC. An atomic model of enhanceosome structure in the vicinity of DNA. Cell. 2007 Jun 15;129(6):1111–23.

231
33. Morgunova E, Taipale J. Structural perspective of cooperative transcription factor binding. Curr Opin Struct Biol. 2017;47:1–8.
34. Gheorghe M. Integrative approaches to study TF-DNA interactions. 2020 [cited 2020 Nov 5]; Available from: https://www.duo.uio.no/handle/10852/75931
35. Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordân R, Rohs R. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014 Sep;39(9):381–99.
36. Riethoven J-JM. Regulatory regions in DNA: promoters, enhancers, silencers, and insulators. Methods Mol Biol. 2010;674:33–42.
37. Payankaulam S, Li LM, Arnosti DN. Transcriptional repression: conserved and evolved features. Curr Biol. 2010 Sep 14;20(17):R764-771.
38. Francois M, Donovan P, Fontaine F. Modulating transcription factor activity: Interfering with protein-protein interaction networks. Seminars in Cell & Developmental Biology. 2020 Mar 1;99:12–9.
39. Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol. 2014 Feb;32(2):171–8.
40. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006 Aug 25;126(4):663–76.
41. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell. 2014 Sep;158(6):1431–43.
42. Meseguer A, Årman F, Fornes O, Molina-Fernández R, Bonet J, Fernandez-Fuentes N, et al. On the prediction of DNA-binding preferences of C2H2-ZF domains using structural models: application on human CTCF. NAR Genom Bioinform [Internet]. 2020 Sep 1 [cited 2020 Sep 2];2(3). Available from: https://academic.oup.com/nargab/article/2/3/lqaa046/5866110
43. Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212.
44. Billeter M. Homeodomain-type DNA recognition. Progress in Biophysics and Molecular Biology. 1996 Jan 1;66(3):211–25.
45. Jakoby M, Weisshaar B, Dröge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj T, et al. bZIP transcription factors in Arabidopsis. Trends in Plant Science. 2002 Mar 1;7(3):106–11.
46. Sun X, Wang Y, Sui N. Transcriptional regulation of bHLH during plant response to stress. Biochemical and Biophysical Research Communications. 2018 Sep 5;503(2):397–401.
47. Lazar MA. Maturing of the nuclear receptor family. J Clin Invest. 2017 Apr 3;127(4):1123–5.
48. Nuclear Hormone Receptors and Gene Expression | Physiological Reviews [Internet]. [cited 2020 Nov 2]. Available from: https://journals.physiology.org/doi/full/10.1152/physrev.2001.81.3.1269

232
49. Hsing M, Wang Y, Rennie PS, Cox ME, Cherkasov A. ETS transcription factors as emerging drug targets in cancer. Medicinal Research Reviews. 2020;40(1):413–30.
50. Benayoun BA, Caburet S, Veitia RA. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011 Jun;27(6):224–32.
51. Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013 Oct;140(20):4129–44.
52. Chytil M, Verdine GL. The Rel family of eukaryotic transcription factors. Curr Opin Struct Biol. 1996 Feb;6(1):91–100.
53. Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010 Oct;15(10):573–81.
54. Tremblay M, Sanchez-Ferras O, Bouchard M. GATA transcription factors in development and disease. Development. 2018 22;145(20).
55. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–55.
56. Smith J. T-box genes: what they do and how they do it. Trends Genet. 1999 Apr;15(4):154–8.
57. Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren B-Z, et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One. 2010 May 12;5(5):e10611.
58. Phillips JE, Corces VG. CTCF: Master Weaver of the Genome. Cell. 2009 Jun 26;137(7):1194–211.
59. Van Bortle K, Nichols MH, Li L, Ong C-T, Takenaka N, Qin ZS, et al. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 2014;15(5):R82.
60. Lajoie BR, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods. 2015 Jan 15;72:65–75.
61. Le Dily F, Beato M. TADs as modular and dynamic units for gene regulation by hormones. FEBS Lett. 2015 Oct 7;589(20 Pt A):2885–92.
62. Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009 Oct;10(10):669–80.
63. Rhee HS, Pugh BF. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol. 2012 Oct;Chapter 21:Unit 21.24.
64. He Q, Johnston J, Zeitlinger J. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat Biotechnol. 2015 Apr;33(4):395–401.
65. Visa N, Jordán-Pla A. ChIP and ChIP-Related Techniques: Expanding the Fields of Application and Improving ChIP Performance. In: Visa N, Jordán-Pla A, editors. Chromatin Immunoprecipitation: Methods and Protocols [Internet]. New York, NY: Springer; 2018 [cited 2020 May 2]. p. 1–7. (Methods in Molecular Biology). Available from: https://doi.org/10.1007/978-1-4939-7380-4_1

233
66. Aughey GN, Cheetham SW, Southall TD. DamID as a versatile tool for understanding gene regulation. Development. 2019 15;146(6).
67. Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017 16;6.
68. Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009 Apr;6(4):283–9.
69. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat Methods. 2013 Dec;10(12):1213–8.
70. Umeyama T, Ito T. DMS-Seq for In Vivo Genome-wide Mapping of Protein-DNA Interactions and Nucleosome Centers. Cell Rep. 2017 Oct 3;21(1):289–300.
71. Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012 Feb 26;30(3):271–7.
72. Berger MF, Bulyk ML. Universal protein binding microarrays for the comprehensive characterization of the DNA binding specificities of transcription factors. Nat Protoc. 2009;4(3):393–411.
73. Oliphant AR, Struhl K. Defining the consensus sequences of E.coli promoter elements by random selection. Nucleic Acids Res. 1988 Aug 11;16(15):7673–83.
74. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990 Aug 3;249(4968):505–10.
75. Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010 Jun;20(6):861–73.
76. Isakova A, Groux R, Imbeault M, Rainer P, Alpern D, Dainese R, et al. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat Methods. 2017;14(3):316–22.
77. Meng X, Brodsky MH, Wolfe SA. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol. 2005 Aug;23(8):988–94.
78. Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008 May;36(8):2547–60.
79. Reece-Hoyes JS, Diallo A, Lajoie B, Kent A, Shrestha S, Kadreppa S, et al. Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat Methods. 2011 Oct 30;8(12):1059–64.
80. Fordyce PM, Gerber D, Tran D, Zheng J, Li H, DeRisi JL, et al. De Novo Identification and Biophysical Characterization of Transcription Factor Binding Sites with Microfluidic Affinity Analysis. Nat Biotechnol. 2010 Sep;28(9):970–5.
81. O’Malley RC, Huang S-SC, Song L, Lewsey MG, Bartlett A, Nery JR, et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell. 2016 May 19;165(5):1280–92.

234
82. Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat Protoc. 2007 Aug;2(8):1849–61.
83. Wei B, Jolma A, Sahu B, Orre LM, Zhong F, Zhu F, et al. A protein activity assay to measure global transcription factor activity reveals determinants of chromatin accessibility. Nat Biotechnol. 2018;36(6):521–9.
84. Lu W, Song Z, Ding Y, Wu H, Huang H. A Prediction Method of DNA-Binding Proteins Based on Evolutionary Information. In: Huang D-S, Jo K-H, Huang Z-K, editors. Intelligent Computing Theories and Application. Cham: Springer International Publishing; 2019. p. 418–29. (Lecture Notes in Computer Science).
85. Hu S, Ma R, Wang H. An improved deep learning method for predicting DNA-binding proteins based on contextual features in amino acid sequences. PLoS One [Internet]. 2019 Nov 14 [cited 2020 May 5];14(11). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6855455/
86. Wang W, Sun L, Zhang S, Zhang H, Shi J, Xu T, et al. Analysis and prediction of single-stranded and double-stranded DNA binding proteins based on protein sequences. BMC Bioinformatics. 2017 Jun 12;18(1):300.
87. Zhang J, Chen Q, Liu B. DeepDRBP-2L: a new genome annotation predictor for identifying DNA binding proteins and RNA binding proteins using Convolutional Neural Network and Long Short-Term Memory. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2019;1–1.
88. Ma X, Guo J, Sun X. DNABP: Identification of DNA-Binding Proteins Based on Feature Selection Using a Random Forest and Predicting Binding Residues. PLoS One [Internet]. 2016 Dec 1 [cited 2020 May 6];11(12). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5132331/
89. Ali F, Ahmed S, Swati ZNK, Akbar S. DP-BINDER: machine learning model for prediction of DNA-binding proteins by fusing evolutionary and physicochemical information. J Comput Aided Mol Des. 2019 Jul 1;33(7):645–58.
90. Chauhan S, Ahmad S. Enabling full-length evolutionary profiles based deep convolutional neural network for predicting DNA-binding proteins from sequence. Proteins: Structure, Function, and Bioinformatics. 2020;88(1):15–30.
91. Ridok A, Widodo N, Mahmudy WF, Rifai M. FC- SVM: DNA binding Proteins prediction with Average Blocks (AB) descriptors using SVM with FC feature Selection. In: 2019 International Conference on Sustainable Information Engineering and Technology (SIET). 2019. p. 22–7.
92. Zou Y, Ding Y, Tang J, Guo F, Peng L. FKRR-MVSF: A Fuzzy Kernel Ridge Regression Model for Identifying DNA-Binding Proteins by Multi-View Sequence Features via Chou’s Five-Step Rule. International Journal of Molecular Sciences. 2019 Jan;20(17):4175.
93. Zhang Y, Wuyunqiqige, Zheng W, Liu S, Zhao C. gDNA-Prot: Predict DNA-binding proteins by employing support vector machine and a novel numerical characterization of protein sequence. Journal of Theoretical Biology. 2016 Oct 7;406:8–16.
94. Sang X, Xiao W, Zheng H, Yang Y, Liu T. HMMPred: Accurate Prediction of DNA-Binding Proteins Based on HMM Profiles and XGBoost Feature Selection [Internet]. Vol. 2020, Computational and Mathematical Methods in Medicine. Hindawi; 2020 [cited 2020 May 5]. p. e1384749. Available from: https://www.hindawi.com/journals/cmmm/2020/1384749/

235
95. Ding Y, Tang J, Guo F. Identification of DNA-Binding Proteins via Fuzzy Multiple Kernel Model and Sequence Information. In: Huang D-S, Jo K-H, Huang Z-K, editors. Intelligent Computing Theories and Application. Cham: Springer International Publishing; 2019. p. 468–79. (Lecture Notes in Computer Science).
96. Du X, Diao Y, Liu H, Li S. MsDBP: Exploring DNA-Binding Proteins by Integrating Multiscale Sequence Information via Chou’s Five-Step Rule. J Proteome Res. 2019 Aug 2;18(8):3119–32.
97. Wang J, Zheng H, Yang Y, Xiao W, Liu T. PredDBP-Stack: Prediction of DNA-Binding Proteins from HMM Profiles using a Stacked Ensemble Method [Internet]. Vol. 2020, BioMed Research International. Hindawi; 2020 [cited 2020 May 5]. p. e7297631. Available from: https://www.hindawi.com/journals/bmri/2020/7297631/
98. You W, Yang Z, Guo G, Wan X-F, Ji G. Prediction of DNA-binding proteins by interaction fusion feature representation and selective ensemble. Knowledge-Based Systems. 2019 Jan 1;163:598–610.
99. Tan C, Wang T, Yang W, Deng L. PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction. Molecules. 2020 Jan;25(1):98.
100. Ali F, Arif M, Khan ZU, Kabir M, Ahmed S, Yu D-J. SDBP-Pred: Prediction of single-stranded and double-stranded DNA-binding proteins by extending consensus sequence and K-segmentation strategies into PSSM. Analytical Biochemistry. 2020 Jan 15;589:113494.
101. Zhou L, Song X, Yu D-J, Sun J. Sequence-based Detection of DNA-binding Proteins using Multiple-View Features Allied with Feature Selection. Molecular Informatics [Internet]. [cited 2020 May 5];n/a(n/a). Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/minf.202000006
102. Mishra A, Pokhrel P, Hoque MT. StackDPPred: a stacking based prediction of DNA-binding protein from sequence. Bioinformatics. 2019 Feb 1;35(3):433–41.
103. Hu J, Zhou X, Zhu Y-H, Yu D-J, Zhang G. TargetDBP: Accurate DNA-Binding Protein Prediction via Sequence-based Multi-View Feature Learning. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2019;1–1.
104. Wang W, Langlois R, Langlois M, Genchev GZ, Wang X, Lu H. Functional Site Discovery From Incomplete Training Data: A Case Study With Nucleic Acid–Binding Proteins. Front Genet [Internet]. 2019 [cited 2020 May 6];10. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2019.00729/full
105. Zhang S, Zhao L, Zheng C-H, Xia J. A feature-based approach to predict hot spots in protein–DNA binding interfaces. Brief Bioinform [Internet]. [cited 2020 May 5]; Available from: https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbz037/5424984
106. Chai H, Zhang J, Yang G, Ma Z. An evolution-based DNA-binding residue predictor using a dynamic query-driven learning scheme. Mol BioSyst. 2016 Nov 15;12(12):3643–50.
107. Jiyun Zhou, Qin Lu, Ruifeng Xu, Lin Gui, Hongpeng Wang. CNNsite: Prediction of DNA-binding residues in proteins using Convolutional Neural Network with sequence features. In: 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2016. p. 78–85.

236
108. Yan J, Kurgan L. DRNApred, fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res. 2017 Jun 2;45(10):e84–e84.
109. Zhou J, Lu Q, Xu R, He Y, Wang H. EL_PSSM-RT: DNA-binding residue prediction by integrating ensemble learning with PSSM Relation Transformation. BMC Bioinformatics. 2017 Aug 29;18(1):379.
110. Zhang Y, Qiao S, Ji S, Zhou J. ENSEMBLE-CNN: Predicting DNA Binding Sites in Protein Sequences by an Ensemble Deep Learning Method. In: Huang D-S, Jo K-H, Zhang X-L, editors. Intelligent Computing Theories and Application. Cham: Springer International Publishing; 2018. p. 301–6. (Lecture Notes in Computer Science).
111. Zhang Y, Qiao S, Ji S, Han N, Liu D, Zhou J. Identification of DNA–protein binding sites by bootstrap multiple convolutional neural networks on sequence information. Engineering Applications of Artificial Intelligence. 2019 Mar 1;79:58–66.
112. Shen C, Ding Y, Tang J, Song J, Guo F. Identification of DNA–protein Binding Sites through Multi-Scale Local Average Blocks on Sequence Information. Molecules. 2017 Dec;22(12):2079.
113. Nguyen BP, Nguyen QH, Doan-Ngoc G-N, Nguyen-Vo T-H, Rahardja S. iProDNA-CapsNet: identifying protein-DNA binding residues using capsule neural networks. BMC Bioinformatics. 2019 Dec 27;20(23):634.
114. Hu J, Li Y, Zhang M, Yang X, Shen H-B, Yu D-J. Predicting Protein-DNA Binding Residues by Weightedly Combining Sequence-Based Features and Boosting Multiple SVMs. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2017 Nov;14(6):1389–98.
115. Amirkhani A, Kolahdoozi M, Wang C, Kurgan L. Prediction of DNA-binding residues in local segments of protein sequences with Fuzzy Cognitive Maps. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2018;1–1.
116. Zhu Y-H, Hu J, Song X-N, Yu D-J. DNAPred: Accurate Identification of DNA-Binding Sites from Protein Sequence by Ensembled Hyperplane-Distance-Based Support Vector Machines. J Chem Inf Model. 2019 Jun 24;59(6):3057–71.
117. Zhou J, Xu R, He Y, Lu Q, Wang H, Kong B. PDNAsite: Identification of DNA-binding Site from Protein Sequence by Incorporating Spatial and Sequence Context. Sci Rep. 2016 Jun 10;6(1):1–15.
118. Deng L, Pan J, Xu X, Yang W, Liu C, Liu H. PDRLGB: precise DNA-binding residue prediction using a light gradient boosting machine. BMC Bioinformatics. 2018 Dec 31;19(19):522.
119. Su H, Liu M, Sun S, Peng Z, Yang J. Improving the prediction of protein–nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics. 2019 Mar 15;35(6):930–6.
120. Blanco JD, Radusky L, Climente-González H, Serrano L. FoldX accurate structural protein–DNA binding prediction using PADA1 (Protein Assisted DNA Assembly 1). Nucleic Acids Res. 2018 May 4;46(8):3852–63.
121. Corsi F, Lavery R, Laine E, Carbone A. Multiple protein-DNA interfaces unravelled by evolutionary information, physico-chemical and geometrical properties. PLOS Computational Biology. 2020 Feb 3;16(2):e1007624.

237
122. Hashim FA, Mabrouk MS, Al-Atabany W. Review of Different Sequence Motif Finding Algorithms. Avicenna J Med Biotechnol. 2019;11(2):130–48.
123. Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015 Jul 1;43(W1):W39-49.
124. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012 Sep 6;489(7414):75–82.
125. Xu B, Schones DE, Wang Y, Liang H, Li G. A structural-based strategy for recognition of transcription factor binding sites. PLoS ONE. 2013;8(1):e52460.
126. Lambert SA, Yang AWH, Sasse A, Cowley G, Albu M, Caddick MX, et al. Similarity regression predicts evolution of transcription factor sequence specificity. Nat Genet. 2019;51(6):981–9.
127. Khamis AM, Motwalli O, Oliva R, Jankovic BR, Medvedeva YA, Ashoor H, et al. A novel method for improved accuracy of transcription factor binding site prediction. Nucleic Acids Res. 2018 Jul 6;46(12):e72–e72.
128. Dang TKL, Meckbach C, Tacke R, Waack S, Gültas M. A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence. Entropy. 2016 Oct;18(10):379.
129. Smolinska K, Pacholczyk M. EMQIT: a machine learning approach for energy based PWM matrix quality improvement. Biology Direct. 2017 Aug 1;12(1):17.
130. Qin W, Zhao G, Carson M, Jia C, Lu H. Knowledge-based three-body potential for transcription factor binding site prediction. IET Systems Biology. 2016;10(1):23–9.
131. Farrel A, Murphy J, Guo J-T. Structure-based prediction of transcription factor binding specificity using an integrative energy function. Bioinformatics. 2016 15;32(12):i306–13.
132. Corona RI, Sudarshan S, Aluru S, Guo J. An SVM-based method for assessment of transcription factor-DNA complex models. BMC Bioinformatics. 2018 Dec 21;19(20):506.
133. Yang W, Deng L. PreDBA: A heterogeneous ensemble approach for predicting protein-DNA binding affinity. Sci Rep. 2020 Jan 28;10(1):1–11.
134. Dutta S, Madan S, Parikh H, Sundar D. An ensemble micro neural network approach for elucidating interactions between zinc finger proteins and their target DNA. BMC Genomics. 2016 Dec 22;17(13):1033.
135. Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020 Jan 8;48(D1):D87–92.
136. Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006 Jan 1;34(Database issue):D108-110.
137. Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2015 Jan;43(Database issue):D117-122.

238
138. Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018 Jan 4;46(D1):D252–9.
139. Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018 04;46(D1):D794–801.
140. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013 Jan;41(Database issue):D991-995.
141. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010 Oct;28(10):1045–8.
142. Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, et al. FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res. 2011 Jan;39(Database issue):D111-117.
143. Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019 Jan 8;47(D1):D729–35.
144. Xuan Lin QX, Sian S, An O, Thieffry D, Jha S, Benoukraf T. MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 2019 08;47(D1):D145–54.
145. Jagannathan V, Roulet E, Delorenzi M, Bucher P. HTPSELEX--a database of high-throughput SELEX libraries for transcription factor binding sites. Nucleic Acids Res. 2006 Jan 1;34(Database issue):D90-94.
146. Chen L, Wu G, Ji H. hmChIP: a database and web server for exploring publicly available human and mouse ChIP-seq and ChIP-chip data. Bioinformatics. 2011 May 15;27(10):1447–8.
147. Wang P, Qin J, Qin Y, Zhu Y, Wang LY, Li MJ, et al. ChIP-Array 2: integrating multiple omics data to construct gene regulatory networks. Nucleic Acids Res. 2015 Jul 1;43(W1):W264-269.
148. Abdulrehman D, Monteiro PT, Teixeira MC, Mira NP, Lourenço AB, dos Santos SC, et al. YEASTRACT: providing a programmatic access to curated transcriptional regulatory associations in Saccharomyces cerevisiae through a web services interface. Nucleic Acids Res. 2011 Jan;39(Database issue):D136-140.
149. Pachkov M, Erb I, Molina N, van Nimwegen E. SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Res. 2007 Jan;35(Database issue):D127-131.
150. Schmeier S, Alam T, Essack M, Bajic VB. TcoF-DB v2: update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017 04;45(D1):D145–50.

239
151. Chiu T-P, Xin B, Markarian N, Wang Y, Rohs R. TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2020 08;48(D1):D246–55.
152. Chen J, Rozowsky J, Galeev TR, Harmanci A, Kitchen R, Bedford J, et al. A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat Commun. 2016 Apr 18;7:11101.
153. Fu F, Voytas DF. Zinc Finger Database (ZiFDB) v2.0: a comprehensive database of C₂H₂ zinc fingers and engineered zinc finger arrays. Nucleic Acids Res. 2013 Jan;41(Database issue):D452-455.
154. Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007 Jan;35(Database issue):D88-92.
155. de Boer CG, Hughes TR. YeTFaSCo: a database of evaluated yeast transcription factor sequence specificities. Nucleic Acids Res. 2012 Jan;40(Database issue):D169-179.
156. Kumar S, Ambrosini G, Bucher P. SNP2TFBS - a database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017 04;45(D1):D139–44.
157. Meseguer A, Dominguez L, Bota PM, Aguirre-Plans J, Bonet J, Fernandez-Fuentes N, et al. Using collections of structural models to predict changes of binding affinity caused by mutations in protein-protein interactions. Protein Sci. 2020 Aug 14;
158. Krasnov AN, Mazina MYu, Nikolenko JV, Vorobyeva NE. On the way of revealing coactivator complexes cross-talk during transcriptional activation. Cell Biosci. 2016 Feb 24;6(1):15.
159. Näär AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu Rev Biochem. 2001;70:475–501.
160. Martinez E. Multi-protein complexes in eukaryotic gene transcription. Plant Mol Biol. 2002 Dec;50(6):925–47.
161. Thomas MC, Chiang C-M. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006 Jun;41(3):105–78.
162. Stewart MD, Wong J. Nuclear receptor repression: regulatory mechanisms and physiological implications. Prog Mol Biol Transl Sci. 2009;87:235–59.
163. Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, Stagljar I. Fundamentals of protein interaction network mapping. Mol Syst Biol. 2015 Dec 17;11(12):848.
164. Hamdi A, Colas P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol Sci. 2012 Feb;33(2):109–18.
165. Snider J, Kittanakom S, Damjanovic D, Curak J, Wong V, Stagljar I. Detecting interactions with membrane proteins using a membrane two-hybrid assay in yeast. Nat Protoc. 2010 Jul;5(7):1281–93.
166. Petschnigg J, Groisman B, Kotlyar M, Taipale M, Zheng Y, Kurat CF, et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat Methods. 2014 May;11(5):585–92.

240
167. Blasche S, Koegl M. Analysis of protein-protein interactions using LUMIER assays. Methods Mol Biol. 2013;1064:17–27.
168. Lievens S, Peelman F, De Bosscher K, Lemmens I, Tavernier J. MAPPIT: a protein interaction toolbox built on insights in cytokine receptor signaling. Cytokine Growth Factor Rev. 2011 Dec;22(5–6):321–9.
169. Lievens S, Gerlo S, Lemmens I, De Clercq DJH, Risseeuw MDP, Vanderroost N, et al. Kinase Substrate Sensor (KISS), a mammalian in situ protein interaction sensor. Mol Cell Proteomics. 2014 Dec;13(12):3332–42.
170. Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–87.
171. Ciruela F. Fluorescence-based methods in the study of protein-protein interactions in living cells. Curr Opin Biotechnol. 2008 Aug;19(4):338–43.
172. Lin T, Scott BL, Hoppe AD, Chakravarty S. FRETting about the affinity of bimolecular protein-protein interactions. Protein Sci. 2018;27(10):1850–6.
173. Dunham WH, Mullin M, Gingras A-C. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 2012 May;12(10):1576–90.
174. Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012 Mar 19;196(6):801–10.
175. Koos B, Andersson L, Clausson C-M, Grannas K, Klaesson A, Cane G, et al. Analysis of protein interactions in situ by proximity ligation assays. Curr Top Microbiol Immunol. 2014;377:111–26.
176. Frei AP, Moest H, Novy K, Wollscheid B. Ligand-based receptor identification on living cells and tissues using TRICEPS. Nat Protoc. 2013;8(7):1321–36.
177. Sanderson CM. A new way to explore the world of extracellular protein interactions. Genome Res. 2008 Apr;18(4):517–20.
178. Ladbury JE, Chowdhry BZ. Sensing the heat: the application of isothermal titration calorimetry to thermodynamic studies of biomolecular interactions. Chem Biol. 1996 Oct;3(10):791–801.
179. Willander M, Al-Hilli S. Analysis of biomolecules using surface plasmons. Methods Mol Biol. 2009;544:201–29.
180. Crabtree MD, Shammas SL. Stopped-Flow Kinetic Techniques for Studying Binding Reactions of Intrinsically Disordered Proteins. Meth Enzymol. 2018;611:423–57.
181. Garcia-Garcia J, Schleker S, Klein-Seetharaman J, Oliva B. BIPS: BIANA Interolog Prediction Server. A tool for protein–protein interaction inference. Nucleic Acids Res. 2012 Jul;40(Web Server issue):W147–51.
182. Planas-Iglesias J, Marin-Lopez MA, Bonet J, Garcia-Garcia J, Oliva B. iLoops: a protein-protein interaction prediction server based on structural features. Bioinformatics. 2013 Sep 15;29(18):2360–2.
183. Kawabata T. HOMCOS: an updated server to search and model complex 3D structures. J Struct Funct Genomics. 2016 Dec;17(4):83–99.

241
184. Mukherjee S, Zhang Y. Protein-protein complex structure predictions by multimeric threading and template recombination. Structure. 2011 Jul 13;19(7):955–66.
185. Guerler A, Govindarajoo B, Zhang Y. Mapping monomeric threading to protein-protein structure prediction. J Chem Inf Model. 2013 Mar 25;53(3):717–25.
186. Zhang QC, Petrey D, Garzón JI, Deng L, Honig B. PrePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res. 2013 Jan;41(Database issue):D828-833.
187. Aloy P, Russell RB. InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics. 2003 Jan;19(1):161–2.
188. Baspinar A, Cukuroglu E, Nussinov R, Keskin O, Gursoy A. PRISM: a web server and repository for prediction of protein-protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014 Jul;42(Web Server issue):W285-289.
189. Berliner N, Teyra J, Çolak R, Lopez SG, Kim PM. Combining Structural Modeling with Ensemble Machine Learning to Accurately Predict Protein Fold Stability and Binding Affinity Effects upon Mutation. PLOS ONE. 2014 Sep 22;9(9):e107353.
190. Folkman L, Stantic B, Sattar A, Zhou Y. EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. Journal of Molecular Biology. 2016 Mar 27;428(6):1394–405.
191. Esmaielbeiki R, Krawczyk K, Knapp B, Nebel J-C, Deane CM. Progress and challenges in predicting protein interfaces. Brief Bioinform. 2016 Jan;17(1):117–31.
192. Garcia-Garcia J, Valls-Comamala V, Guney E, Andreu D, Muñoz FJ, Fernandez-Fuentes N, et al. iFrag: A Protein-Protein Interface Prediction Server Based on Sequence Fragments. J Mol Biol. 2017 03;429(3):382–9.
193. Ahmad S, Mizuguchi K. Partner-Aware Prediction of Interacting Residues in Protein-Protein Complexes from Sequence Data. PLOS ONE. 2011 Dec 14;6(12):e29104.
194. Segura J, Jones PF, Fernandez-Fuentes N. Improving the prediction of protein binding sites by combining heterogeneous data and Voronoi diagrams. BMC Bioinformatics. 2011 Aug 23;12:352.
195. Segura J, Jones PF, Fernandez-Fuentes N. A holistic in silico approach to predict functional sites in protein structures. Bioinformatics. 2012 Jul 15;28(14):1845–50.
196. Erijman A, Rosenthal E, Shifman JM. How structure defines affinity in protein-protein interactions. PLoS One. 2014;9(10):e110085.
197. Moal IH, Agius R, Bates PA. Protein-protein binding affinity prediction on a diverse set of structures. Bioinformatics. 2011 Nov 1;27(21):3002–9.
198. Su Y, Zhou A, Xia X, Li W, Sun Z. Quantitative prediction of protein-protein binding affinity with a potential of mean force considering volume correction. Protein Sci. 2009 Dec;18(12):2550–8.
199. Audie J, Scarlata S. A novel empirical free energy function that explains and predicts protein-protein binding affinities. Biophys Chem. 2007 Sep;129(2–3):198–211.
200. Vreven T, Hwang H, Pierce BG, Weng Z. Prediction of protein-protein binding free energies. Protein Sci. 2012 Mar;21(3):396–404.

242
201. Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003 Jul 18;330(4):891–913.
202. Gumbart JC, Roux B, Chipot C. Efficient determination of protein-protein standard binding free energies from first principles. J Chem Theory Comput. 2013 Aug 13;9(8).
203. Moritsugu K, Terada T, Kidera A. Energy landscape of all-atom protein-protein interactions revealed by multiscale enhanced sampling. PLoS Comput Biol. 2014 Oct;10(10):e1003901.
204. Rodriguez RA, Yu L, Chen LY. Computing Protein-Protein Association Affinity with Hybrid Steered Molecular Dynamics. J Chem Theory Comput. 2015 Sep 8;11(9):4427–38.
205. Marín-López MA, Planas-Iglesias J, Aguirre-Plans J, Bonet J, Garcia-Garcia J, Fernandez-Fuentes N, et al. On the mechanisms of protein interactions: predicting their affinity from unbound tertiary structures. Bioinformatics. 2018 15;34(4):592–8.
206. Kastritis PL, Rodrigues JPGLM, Folkers GE, Boelens R, Bonvin AMJJ. Proteins feel more than they see: fine-tuning of binding affinity by properties of the non-interacting surface. J Mol Biol. 2014 Jul 15;426(14):2632–52.
207. Vangone A, Bonvin AM. Contacts-based prediction of binding affinity in protein-protein complexes. Elife. 2015 Jul 20;4:e07454.
208. Tian F, Lv Y, Yang L. Structure-based prediction of protein-protein binding affinity with consideration of allosteric effect. Amino Acids. 2012 Aug;43(2):531–43.
209. Kastritis PL, Bonvin AMJJ. Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J Proteome Res. 2010 May 7;9(5):2216–25.
210. Petukh M, Dai L, Alexov E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. International Journal of Molecular Sciences. 2016 Apr;17(4):547.
211. Bash PA, Singh UC, Brown FK, Langridge R, Kollman PA. Calculation of the relative change in binding free energy of a protein-inhibitor complex. Science. 1987 Jan 30;235(4788):574–6.
212. Li M, Petukh M, Alexov E, Panchenko AR. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J Chem Theory Comput. 2014 Apr 8;10(4):1770–80.
213. Petukh M, Li M, Alexov E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLOS Computational Biology. 2015 Jul 6;11(7):e1004276.
214. Homeyer N, Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol Inform. 2012 Feb;31(2):114–22.
215. Dehouck Y, Kwasigroch JM, Rooman M, Gilis D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013 Jul;41(Web Server issue):W333-339.
216. Aloy P, Oliva B. Splitting statistical potentials into meaningful scoring functions: Testing the prediction of near-native structures from decoy conformations. BMC Struct Biol. 2009;9(1):71.

243
217. Feliu E, Aloy P, Oliva B. On the analysis of protein-protein interactions via knowledge-based potentials for the prediction of protein-protein docking. Protein Sci. 2011 Mar;20(3):529–41.
218. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W382-388.
219. Li M, Simonetti FL, Goncearenco A, Panchenko AR. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016 08;44(W1):W494-501.
220. Rodrigues CHM, Myung Y, Pires DEV, Ascher DB. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 2019 Jul 2;47(W1):W338–44.
221. Zhao N, Han JG, Shyu C-R, Korkin D. Determining Effects of Non-synonymous SNPs on Protein-Protein Interactions using Supervised and Semi-supervised Learning. PLOS Computational Biology. 2014 May 1;10(5):e1003592.
222. Garcia-Garcia J, Guney E, Aragues R, Planas-Iglesias J, Oliva B. Biana: a software framework for compiling biological interactions and analyzing networks. BMC Bioinformatics. 2010 Jan 27;11:56.
223. Yellaboina S, Tasneem A, Zaykin DV, Raghavachari B, Jothi R. DOMINE: a comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res. 2011 Jan 1;39(suppl_1):D730–5.
224. Amos-Binks A, Patulea C, Pitre S, Schoenrock A, Gui Y, Green JR, et al. Binding Site Prediction for Protein-Protein Interactions and Novel Motif Discovery using Re-occurring Polypeptide Sequences. BMC Bioinformatics. 2011 Jun 2;12(1):225.
225. Boyen P, Dyck DV, Neven F, Ham RCHJ van, Dijk ADJ van. SLIDER: A Generic Metaheuristic for the Discovery of Correlated Motifs in Protein-Protein Interaction Networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2011 Sep;8(5):1344–57.
226. Skwark MJ, Abdel-Rehim A, Elofsson A. PconsC: combination of direct information methods and alignments improves contact prediction. Bioinformatics. 2013 Jul 15;29(14):1815–6.
227. Horton N, Lewis M. Calculation of the free energy of association for protein complexes. Protein Sci. 1992 Jan;1(1):169–81.
228. Ma XH, Wang CX, Li CH, Chen WZ. A fast empirical approach to binding free energy calculations based on protein interface information. Protein Eng. 2002 Aug;15(8):677–81.
229. Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017 Jan 4;45(Database issue):D271–81.
230. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015 Jan;43(Database issue):D447-452.
231. Kotlyar M, Pastrello C, Pivetta F, Lo Sardo A, Cumbaa C, Li H, et al. In silico prediction of physical protein interactions and characterization of interactome orphans. Nat Methods. 2015 Jan;12(1):79–84.

244
232. Kotlyar M, Pastrello C, Sheahan N, Jurisica I. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res. 2016 Jan 4;44(D1):D536-541.
233. Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014 Jan;42(Database issue):D358-363.
234. Launay G, Salza R, Multedo D, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database: updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015 Jan;43(Database issue):D321-327.
235. Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, et al. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013 Jan;41(Database issue):D1228-1233.
236. Oughtred R, Stark C, Breitkreutz B-J, Rust J, Boucher L, Chang C, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019 08;47(D1):D529–41.
237. Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004 Jan 1;32(Database issue):D449-451.
238. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009 Jan;37(Database issue):D767-772.
239. Turinsky AL, Razick S, Turner B, Donaldson IM, Wodak SJ. Navigating the global protein-protein interaction landscape using iRefWeb. Methods Mol Biol. 2014;1091:315–31.
240. Calderone A, Iannuccelli M, Peluso D, Licata L. Using the MINT Database to Search Protein Interactions. Curr Protoc Bioinformatics. 2020 Mar;69(1):e93.
241. Kemp TJ, Alcock NW. 100 years of X-ray crystallography. Sci Prog. 2017 Mar 1;100(1):25–44.
242. Bahadur RP, Chakrabarti P, Rodier F, Janin J. A dissection of specific and non-specific protein-protein interfaces. J Mol Biol. 2004 Feb 27;336(4):943–55.
243. Cheng Y. Single-particle cryo-EM-How did it get here and where will it go. Science. 2018 31;361(6405):876–80.
244. Mlynárik V. Introduction to nuclear magnetic resonance. Anal Biochem. 2017 15;529:4–9.
245. Kalodimos CG, Biris N, Bonvin AMJJ, Levandoski MM, Guennuegues M, Boelens R, et al. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004 Jul 16;305(5682):386–9.
246. Yu H. Extending the size limit of protein nuclear magnetic resonance. Proc Natl Acad Sci USA. 1999 Jan 19;96(2):332–4.
247. Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Current Protocols in Protein Science. 2016;86(1):2.9.1-2.9.37.
248. Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999 Feb;12(2):85–94.

245
249. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W363-367.
250. Lu X-J, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3(7):1213–27.
251. Barradas-Bautista D, Moal IH, Fernández-Recio J. A systematic analysis of scoring functions in rigid-body protein docking: The delicate balance between the predictive rate improvement and the risk of overtraining. Proteins. 2017;85(7):1287–97.
252. Wass MN, Fuentes G, Pons C, Pazos F, Valencia A. Towards the prediction of protein interaction partners using physical docking. Mol Syst Biol. 2011 Feb 15;7:469.
253. Schweke H, Mucchielli M-H, Sacquin-Mora S, Bei W, Lopes A. Protein Interaction Energy Landscapes are Shaped by Functional and also Non-functional Partners. J Mol Biol. 2020 Feb 14;432(4):1183–98.
254. Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science. 2012 Nov 23;338(6110):1042–6.
255. Bradley P, Misura KMS, Baker D. Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science. 2005 Sep 16;309(5742):1868–71.
256. Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(0 9):89–99.
257. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015 Jan;12(1):7–8.
258. AlQuraishi M. AlphaFold at CASP13. Bioinformatics. 2019 Nov 1;35(22):4862–5.
259. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Tunyasuvunakool K, et al. High Accuracy Protein Structure Prediction Using Deep Learning. Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book). 2020 Dec;
260. Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020 Jan;577(7792):706–10.
261. Oakes BL, Xia DF, Rowland EF, Xu DJ, Ankoudinova I, Borchardt JS, et al. Multi-reporter selection for the design of active and more specific zinc-finger nucleases for genome editing. Nat Commun. 2016 Apr;7(1):10194.
262. Garcia-Garcia J, Bonet J, Guney E, Fornes O, Planas J, Oliva B. Networks of Protein Protein Interactions: From Uncertainty to Molecular Details. Mol Inform. 2012 May;31(5):342–62.
263. Hannenhalli S. Eukaryotic transcription factor binding sites—modeling and integrative search methods. Bioinformatics. 2008 Jun 1;24(11):1325–31.
264. Tran NTL, Huang C-H. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol Direct. 2014 Feb 20;9:4.

246
265. Medina-Rivera A, Abreu-Goodger C, Thomas-Chollier M, Salgado H, Collado-Vides J, van Helden J. Theoretical and empirical quality assessment of transcription factor-binding motifs. Nucleic Acids Res. 2011 Feb;39(3):808–24.
266. Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009 Jun 26;324(5935):1720–3.
267. Stein A, Rueda M, Panjkovich A, Orozco M, Aloy P. A systematic study of the energetics involved in structural changes upon association and connectivity in protein interaction networks. Structure. 2011 Jun 8;19(6):881–9.
268. Goh C-S, Milburn D, Gerstein M. Conformational changes associated with protein-protein interactions. Curr Opin Struct Biol. 2004 Feb;14(1):104–9.
269. Wolynes PG. Evolution, energy landscapes and the paradoxes of protein folding. Biochimie. 2015 Dec;119:218–30.
270. Barrera LA, Vedenko A, Kurland JV, Rogers JM, Gisselbrecht SS, Rossin EJ, et al. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science. 2016 Mar 25;351(6280):1450–4.
271. Shi W, Fornes O, Mathelier A, Wasserman WW. Evaluating the impact of single nucleotide variants on transcription factor binding. Nucleic Acids Res. 2016 Dec 1;44(21):10106–16.
272. Kim H, Kim J-S. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014 May;15(5):321–34.
273. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010 Sep;11(9):636–46.
274. Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annu Rev Genomics Hum Genet. 2013;14:135–58.
275. Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013 Jul;31(7):397–405.
276. Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng. 2013 Jul;110(7):1811–21.
277. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013 Jan;14(1):49–55.
278. Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014 Feb 13;156(4):836–43.
279. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013 Feb 15;339(6121):823–6.
280. Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013 Aug;194(4):1029–35.

247
281. Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013 Aug;10(8):741–3.
282. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017 Jun;16(6):387–99.