phenyl-n-tert-butyl nitrone attenuates methamphetamine-induced depletion of striatal dopamine...
TRANSCRIPT

SYNAPSE 24~173-181 (1996)
a-Phenyl-N-tert-Butyl Nitrone Attenuates Methamphetamine-Induced Depletion
of Striatal Dopamine Without Altering Hyperthermia
G.D. CAPPON, H.W. BROENING, C. PU, L. MORFORD, AND C.V. VORHEES Division of Deuelopmental Biology, Children‘s Hospital Research Foundation, and Neuroscience Graduate
Program and Department of Pediatrics, Uniuersity of Cincinnati, Cincinnati, Ohio 45229
KEY WORDS Methamphetamine, Dopamine, Neurotoxicity, Free Radicals, a-phe nyl-N-tert-butyl nitrone (PBN), Rats
ABSTRACT Methamphetamine (MA) administration to adult rats (4 x 10 mgkg s.c.) induces neurotoxicity predominately characterized by a persistent reduction of neostriatal dopamine (DA) content. Hyperthermia following MA administration potenti- ates the resulting DA depletion. DA-derived free radicals are postulated to be a mecha- nism through which MA-induced neurotoxicity is produced. The spin trapping agent PBN reacts with free radicals to form nitroxyl adducts, thereby preventing damaging free radical reactions with cellular substrates. MA with saline pretreatment (Sal-MA) reduced neostriatal DA by 55% (P < 0.01 vs. Sal-Sal). MA with PBN pretreatment (PBN- MA) at 36 or 60 mgkg reduced neostriatal DA by 36 and 22%, respectively (P < 0.05 and P < 0.01 vs. Sal-MA) indicating partial protection. PBN pretreatment did not alter MA-induced hyperthermia. Thus, PBN does not attenuate MA-induced neurotoxicity by reducing MA-induced hyperthermia. These results support a roIe for free radicals in the generation of MA-induced dopaminergic neurotoxicity. 0 1996 Wiiey-Liss, h c .
INTRODUCTION Methamphetamine (MA) has been shown to be neuro-
toxic in a variety of species. The administration ofeither a single large dose (100 mgkg) or multiple lower doses of MA (4 x 10 mgkg) produces a characteristic pattern of monoamine neurotoxicity in mice, rats, and primates (Hotchkiss and Gibb, 1980; Ricaurte e t al., 1982; Seiden et al., 1976; Wagner et al., 1980). MA-induced neurotox- icity is typically demonstrated by reductions in neostri- atal content of dopamine (DA) and serotonin (5-HT) (Green et al., 1992) along with a decrease in tyrosine hydroxylase (TH) activity (Trulson et al., 19871, the rate limiting enzyme in the DA biosynthetic pathway. Other characteristics associated with MA-induced neurotoxic- ity include loss of DA reuptake sites (Wagner et al., 1980), decreases in TH and 5-HT immunareactivity (Hotchkiss and Gibb, 1980; Hotchkiss e t al., 1979), and increased expression of glial fibrillary acidic protein (GFAP) (Pu and Vorhees, 1993). GFAP is produced dur- ing reactive astrogliosis and is a marker of neuronal injury (O’Callaghan, 1991). Current evidence demon- strates a correlation between the magnitude of hyper- thermia attained in the course of MA administration and the severity of the resulting DA depletion (Bowyer 0 1996 WILEY-LJSS, INC.
e t al., 1994). Administering MA at an elevated ambient temperature of 26.5-30°C results in enhanced DA depletion (Bowyer et al., 1994), and treatments which prevent hyperthermia attenuate MA-induced DA deple- tion (Ali et al., 1994; Bowyer e t al., 1992).
Reactive oxygen species, which include oxygen free radicals, hydroxyl radicals, lipid hyperoxides, and hy- drogen peroxide are believed to be the etiological agents in the development of several neuropathological condi- tions. Free radicals readily oxidize endogenous mole- cules altering their biological activity and causing cellu- lar damage or death. The generation of oxygen free radicals has been proposed as a mechanism for MA- induced neurotoxicity (De Vito and Wagner, 1989a; Gio- vanni et al., 1995). DA readily oxidizes to form poten- tially toxic metabolites such as quinones, that may un- dergo redox cycling to produce hydroxyl radical, superoxide radical, and hydrogen peroxide (Graham et al., 1978; Maruyama et al., 1993; Slivka and Cohen,
Received October 3, 1995; accepted in revised form February 28, 1996.
Address reprint requests to Charles V. Vorhees, P h D , Division of Develop- mental Biology, Children’s Hospital Research Foundation, 3333 Burnet Ave., Cincinnati, OH 45229-3039.

174 G D. CAF'PON ET AL
1985). Under normal conditions, protective mecha- nisms remove these reactive molecules. However, in situations of DA overflow, oxidative metabolism of DA may generate enough free radicals to overcome cellular protective systems and damage DA neurons (Chiueh et al., 1993). MA administration to IIA neurons in vitro generates oxygen radicals and reactive metabolites along with neurite degeneration (Cubells et al., 1994). Treatment of rats with antioxidants such as ascorbic acid, ethanol, mannitol, or vitamin E, attenuate MA- induced reductions of striatal DA concentrations (De Vito and Wagner, 1989a,b; Wagner et al., 1985), support- ing a role for free radicals in MA-induced neurotoxicity. Although these drugs attenuate MA-induced DA deple- tion, some of them (e.g., ethanol) inhibit MA-induced hyperthermia, which may explain their neuroprotective effects by a non-specific mechanism (Miller and O'Cal- laghan, 1994).
a-Phenyl-N-tert-butyl nitrone (PBN) is a free radical spin trapping compound which was initially developed to detect and identify short lived Sree radicals (Janzen and Blackburn, 1969). PBN reacts with free radicals to yield a nitroxyl product that can then be detected with electron spin resonance trapping techniques (Janzen and Blackburn, 1969). PBN is a lipophilic molecule which is rapidly absorbed and readily crosses the blood- brain barrier reaching maximum tissue concentration in approximately 20 min, where it remains with a half- life of approximately 130 min (Chen et al., 1990a,b; Cheng et al., 1993). Studies have demonstrated that PBN offers protection against age-related increases in markers of oxidative damage (Carney et al., 1991) and other oxidatively damaging events such as cardiotoxic- ity induced by doxorubicin (Jotti et al., 1992) or expo- sure to phenytoin (Liu and Wells, 1994). It has been shown that PBN administration inhibits oxidative dam- age associated with ischemia-reperfusion brain injury in gerbils (Carney et al., 1991; Phillis and Clough-Helf- man, 1990). I t is hypothesized that PBN reacts with free radicals, preventing them from interacting with biological compounds thereby interrupting the chain of reactions which leads to tissue damage (Floyd and Carney, 1992).
In the following experiments, adult rats were treated with MA or saline at an ambient temperature conducive to the production of MA-induced hyperthermia. PBN was administered 30 min prior to MA or saline, and body temperature was monitored during the acute phase of MA exposure. Rats were evaluated 3 days after treatment to determine the influence of PBN on MA- induced neurotoxicity.
MATERIALS AND METHODS Animals and drug treatment
Male Sprague-Dawley CD rats (Charles River, Ra- leigh, NC) 250-275 grams were the experimental sub- jects. The rats were housed individually in cages in conditions of controlled temperature (22 2 2°C) and
lighting (14:lO h light-dark cycle). Control and experi- mental rats were moved to a room with a temperature of 23.5"C, 30 min prior to MA administration. Food and water were given ad libitum.
Body temperatures were measured rectally every 30 min beginning one half hour prior to the first MA admin- istration and continuing until 1 h after the final MA administration (total of 7.5 h) using a digital thermome- ter (Physitemp Instruments, Clifton, NJ). Animals with a body temperature of 41.3"C or greater were immedi- ately placed on crushed ice for 30 min or until the body temperature was reduced to at least 40.0"C.
d-Methamphetamine HC1 (10 mgkg; Sigma Chemi- cal, St. Louis, MO; expressed as free base) or saline was administered S.C. four times a t 2 h intervals. PBN (36, 60, 73, or 120 mgkg; Eastman Laboratory Chemicals, Rochester, NY) or saline was administered S.C. 30 rnin prior to each MA injection.
Animals were assigned to treatment groups as fol- lows: saline-saline (n = 81, PBN 36 mgkg-saline (n = 7), PBN 60 mgkg-saline (n = 7), PBN 75 mgkg- saline (n = 3), saline-MA (n = lo), PBN 36 mgkg-MA (n = lo), PBN 60 mgkg-MA (n = lo), PBN 75 mgkg- MA (n = 8) , and PBN 120 mgkg-MA (n = 7). To deter- mine changes in striatal DA and 5-HT levels after MA exposure, animals were decapitated 3 days post-treat- ment and brains were rapidly removed and placed on ice. A brain dissection block (Zivic-Miller, Pittsburgh, PA) and razor blades were used to produce serial sec- tions (2.0 mm) from which only the caudate-putamen was dissected and retained. Tissue samples were stored a t -80°C until assayed for concentrations of DA, 3,4- dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-HT and 5-hydroxyindoleacetic acid (5- H I M ) .
Determination of caudate-putamen concentration of DA and 5HT
Monoamine content was determined using high per- formance liquid chromatography with electrochemical detection (HPLC-EC). Briefly, frozen tissue samples were weighed and sonicated in a 1 2 0 dilution (mg tis- sue/pl 0.1 N perchloric acid) containing 1 ng/b1 DHBA as an internal standard. Samples were then centrifuged at 4°C in a microcentrifuge a t 15,OOOg for 15 min. Sam- ples were filtered and 20 p1 of the supernatant was injected into the HPLC system.
The analytical column used was a Phenomenex U1- tracarb 5 pm ODS 20 column (Phenomenex Inc., Tor- rance, CAI. The HPLC system consisted of a Bioanalytic Systems PM80 solvent delivery system (BAS, West La- fayette, IN), a BAS model LC22C column heater, a BAS model LC4C ampromeric detector, and a HP 33908 integrator (Hewlett Packard, Avondale, PA). The mobile phase (pH 3.0) consisted of 70 mM KH2P04, 1 mM so- dium l-hexanesulfonic acid, 0.1 mM EDTA and 6% methanol run a t a column temperature of 40°C. All HPLC reagents and standards were purchased from
PBN AND MA-INDUCED NEUROTOXCITY
16 -
14 -
12 - - Q = u) u) = 10 -
I E OI
w s 8 -
z -
6 - 2 8 4 ;
2 -
175
0-
Sigma Chemical Co. Concentrations of DA and 5-HT were determined from standard curves generated for each analyte of interest.
Statistics Alterations in neurochemical parameters and body
temperature were determined by analysis of variance (ANOVA) followed by pairwise comparison using Dun- can's multiple range test. Mortality results were ana- lyzed using Fisher's test for uncorrelated proportions.
Immunohistochemistry Additional animals were prepared for immunohisto-
chemistry as follows: saline-saline (n = 3), PBN 60 mg/ kg-saline (n = 3), saline-MA (n = 5), and PBN 60 mg/ kg-MA (n = 5). The dosing protocol was the same as that used for the neurochemical experiment. Three days post-treatment, animals were deeply anesthetized with sodium pentobarbital and perfused transcardically with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB, pH 7.4). Following perfusion, brains were removed and postfixed in 4% PFA overnight at 4°C and then cryoprotected in 20% sucrose in PB (0.2M, pH 7.4) for 24 h a t 4°C. Frozen sections (40 pm) were coronally cut in a cryostat.
Free-floating sections were incubated with continual agitation in PBS (0.1M phosphate, pH 7.4, 0.9% NaC1) solution containing mouse anti-TH monoclonal anti- body (Incstar Corp., Stillwater, MN, at 1:10,000 dilu- tion). All antibody solutions were prepared in PBS solu- tion containing 0.2% Triton X-100 and 0.2% normal goat serum (NGS). The sections were incubated in the antibody solutions for 24 h at room temperature and then transferred to a solution (PBS with 0.2% triton and 0.2% NGS) containing biotinylated goat anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) at 1:200 dilution at room tempera- ture for 1 h. Sections were incubated in avidin-biotin peroxidase complex (ABC elite, Vector Laboratories, Inc., Burlingame, CA) solution for 1 h followed by prein- cubation with 0.05% diaminobenzidine tetrachloride (DAB) for 5 min. The peroxidase label was visualized by the addition of hydrogen peroxide (0.003% final con- centration) with a 5-7 min incubation. Between each step of the antibody reactions, the brain sections were gently rinsed with PBS solution three times, 10 min per rinse. Sections were mounted on gelatin-coated slides, dehydrated in graded alcohol solutions, and cover- slipped with DPX. Immunostaining was examined by bright-field microscopy.
RESULTS Effect of PBN and MA on caudate-putamen
DA content The effects of MA and PBN, alone and in combination,
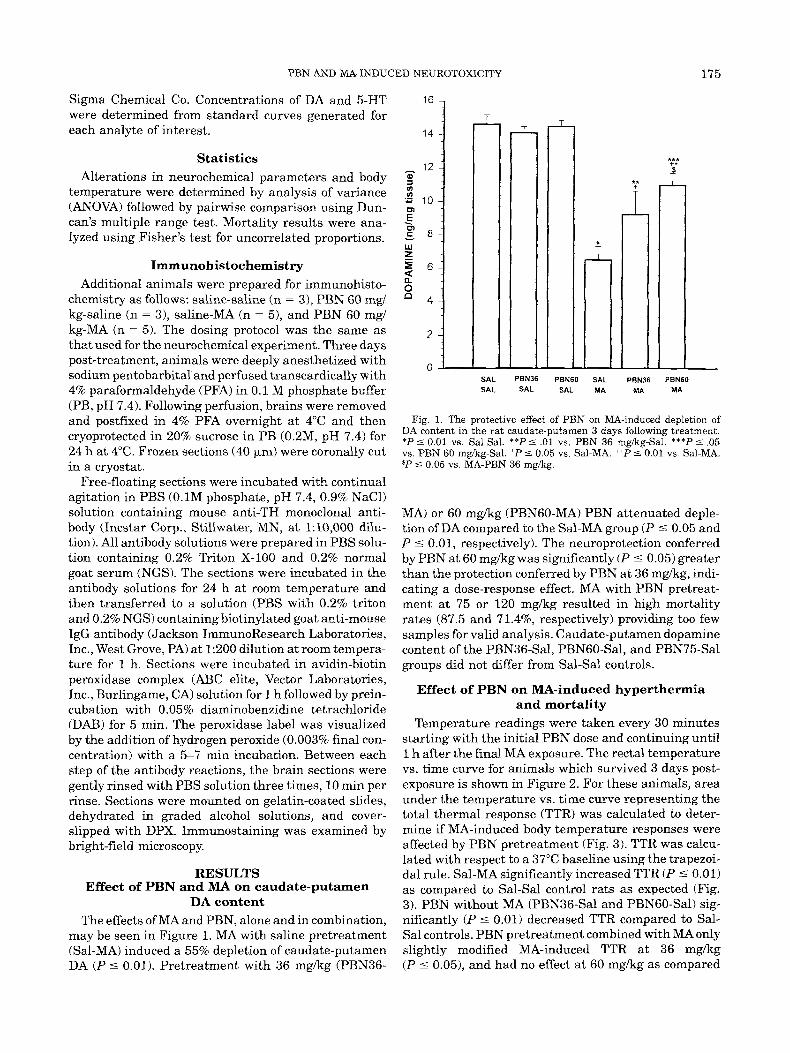
may be seen in Figure 1. NLA with saline pretreatment (Sal-MA) induced a 55% depletion of caudate-putamen DA ( P 5 0.01). Pretreatment with 36 mgkg (PBN36-
SAL PBN36 SAL SAL
T -
- PBN60 SAL
SAL MA
**
1 'BN36 MA
Fig. 1. The protective effect of PBN on MA-induced depletion of DA content in the rat caudate-putamen 3 days following treatment. *P 5 0.01 vs. Sal-Sal. **P 5 .01 vs. PBN 36 mgkg-Sal. ***P 5 .05 vs. PBN 60 mgkg-Sal. ' P 5 0.05 vs. Sal-MA. "P 5 0.01 vs. Sal-MA. $P 5 0.05 vs. MA-PBN 36 mgkg.
MA) or 60 mgkg (PBNGO-MA) PBN attenuated deple- tion of DA compared to the Sal-MA group ( P 5 0.05 and P 5 0.01, respectively). The neuroprotection conferred by PBN at 60 mgkg was significantly (P 5 0.05) greater than the protection conferred by PBN a t 36 mgkg, indi- cating a dose-response effect. M A with PBN pretreat- ment at 75 or 120 mgkg resulted in high mortality rates (87.5 and 71.4%, respectively) providing too few samples for valid analysis. Caudate-putamen dopamine content of the PBN36-Sal, PBN6O-Sal, and PBN75-Sal groups did not differ from Sal-Sal controls.
Effect of PBN on MA-induced hyperthermia and mortality
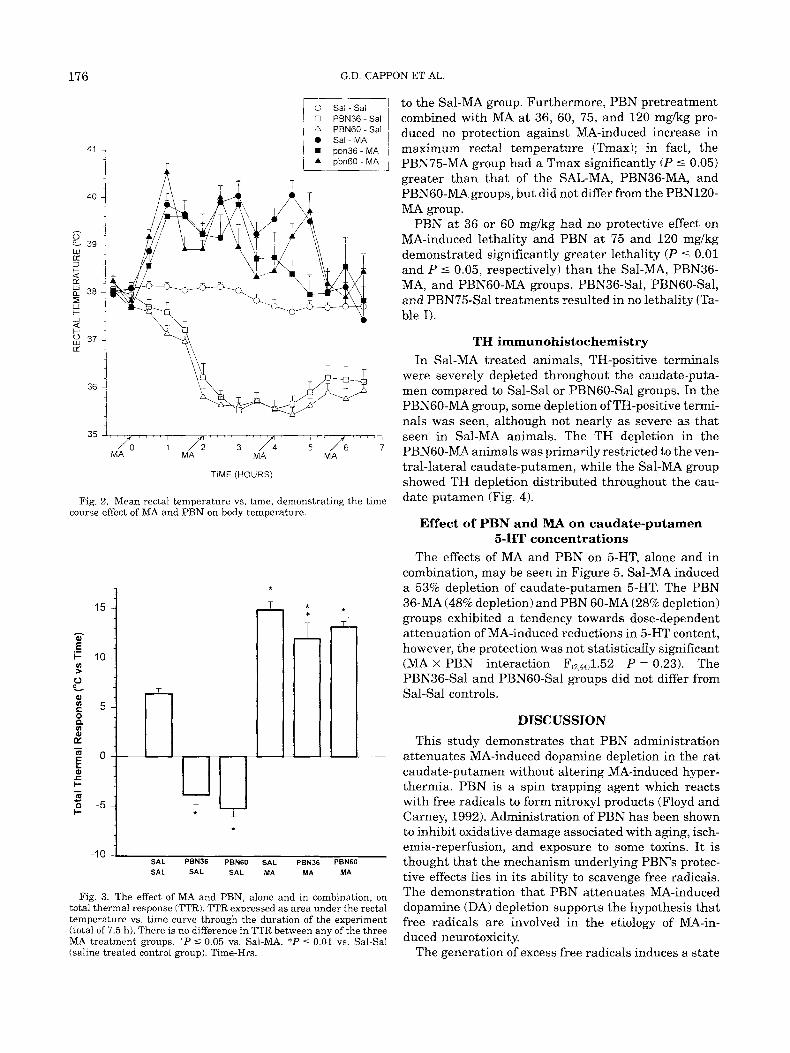
Temperature readings were taken every 30 minutes starting with the initial PBN dose and continuing until 1 h after the final MA exposure. The rectal temperature vs. time curve for animals which survived 3 days post- exposure is shown in Figure 2. For these animals, area under the temperature vs. time curve representing the total thermal response (TTR) was calculated to deter- mine if MA-induced body temperature responses were affected by PBN pretreatment (Fig. 3). TTR was calcu- lated with respect to a 37°C baseline using the trapezoi- dal rule. Sal-MA significantly increased TTR (P 5 0.01) as compared to Sal-Sal control rats as expected (Fig. 3). PBN without MA (PBN36-Sal and PBN6O-Sal) sig- nificantly (P I 0.01) decreased TTR compared to Sal- Sal controls. PBN pretreatment combined with MA only slightly modified MA-induced TTR at 36 mgkg (P 5 0.05), and had no effect at 60 mgkg as compared
176 G.D. CAPPON ET AL.
41 -
i
0 PBN36-Sal PBN6O - Sal ' Sal-MA 1 pbn36-MA pbn60 - MA 12-
f ' 'Y/6'' I /o ' ' . ' -I ' 72 - 3 ;' 4 35 ' ,
7 MA MA MA MA
TIME (HOURS)
Fig. 2. Mean rectal temperature vs. time, demonstrating the time course effect of MA and PBN on body temperature
-1 0 SAL PBN36 PBN60 SAL PBN36 PBNBO SAL SAL SAL MA MA MA
Fig. 3. The effect of MA and PBN, alone and in combination, on total thermal response (TTR). TTR expressed as area under the rectal temperature vs. time curve through the duration of the experiment (total of 7.5 h). There is no difference in TTR between any of the three MA treatment groups. ' P 5 0.05 vs. Sal-MA. *P 5 0.01 vs. Sal-Sal (saline treated control group). Time-Hrs.
to the Sal-MA group. Furthermore, PBN pretreatment combined with MA a t 36, 60, 75, and 120 mg/kg pro- duced no protection against MA-induced increase in maximum rectal temperature (Tmax); in fact, the PBN75-MA group had a Tmax significantly (P 5 0.05) greater than that of the SAL-MA, PBN36-MA, and PBNGO-MA groups, but did not differ from the PBN12O- MA group.
PBN a t 36 or 60 mgkg had no protective effect on MA-induced lethality and PBN at 75 and 120 mgkg demonstrated significantly greater lethality (P 5 0.01 and P 5 0.05, respectively) than the Sal-MA, PBN36- MA, and PBNGO-MA groups. PBN36-Sal, PBNGO-Sal, and PBN75-Sal treatments resulted in no lethality (Ta- ble I).
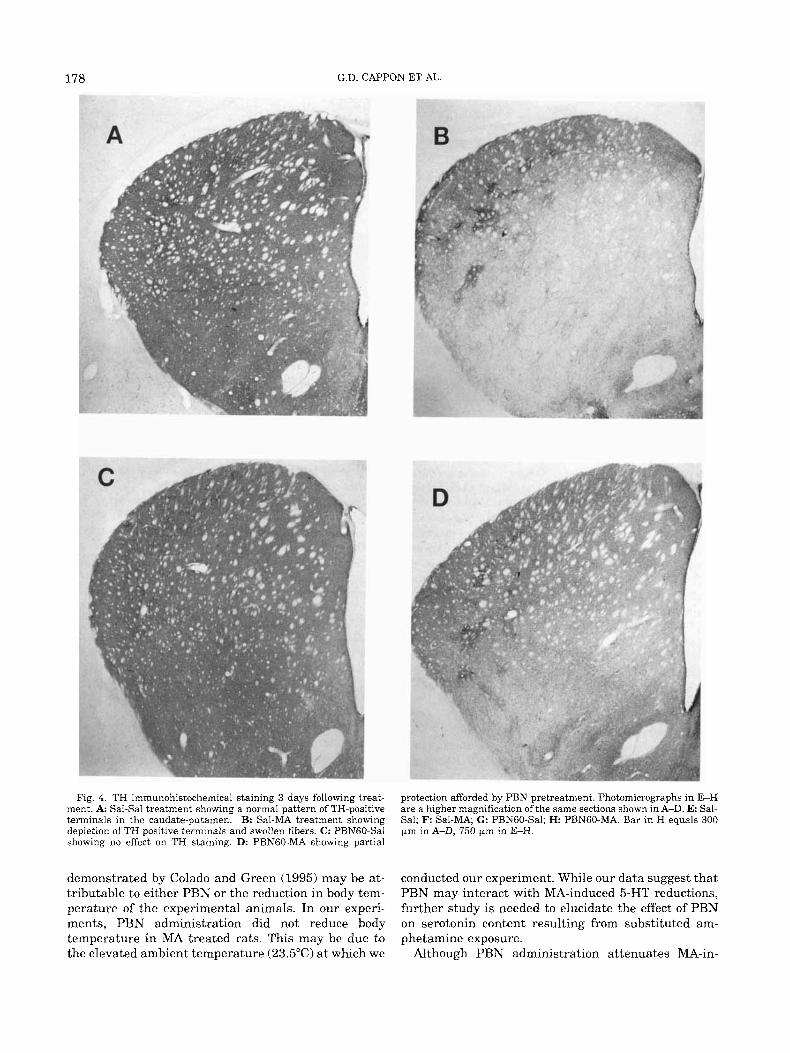
TH immunohistochemistry In Sal-MA treated animals, TH-positive terminals
were severely depleted throughout the caudate-puta- men compared to Sal-Sal or PBN6O-Sal groups. In the PBNGO-MA group, some depletion of TH-positive termi- nals was seen, although not nearly as severe as that seen in Sal-MA animals. The TH depletion in the PBNGO-MA animals was primarily restricted to the ven- tral-lateral caudate-putamen, while the Sal-MA group showed TH depletion distributed throughout the cau- date-putamen (Fig. 4).
Effect of PBN and MA on caudate-putamen 5-HT concentrations
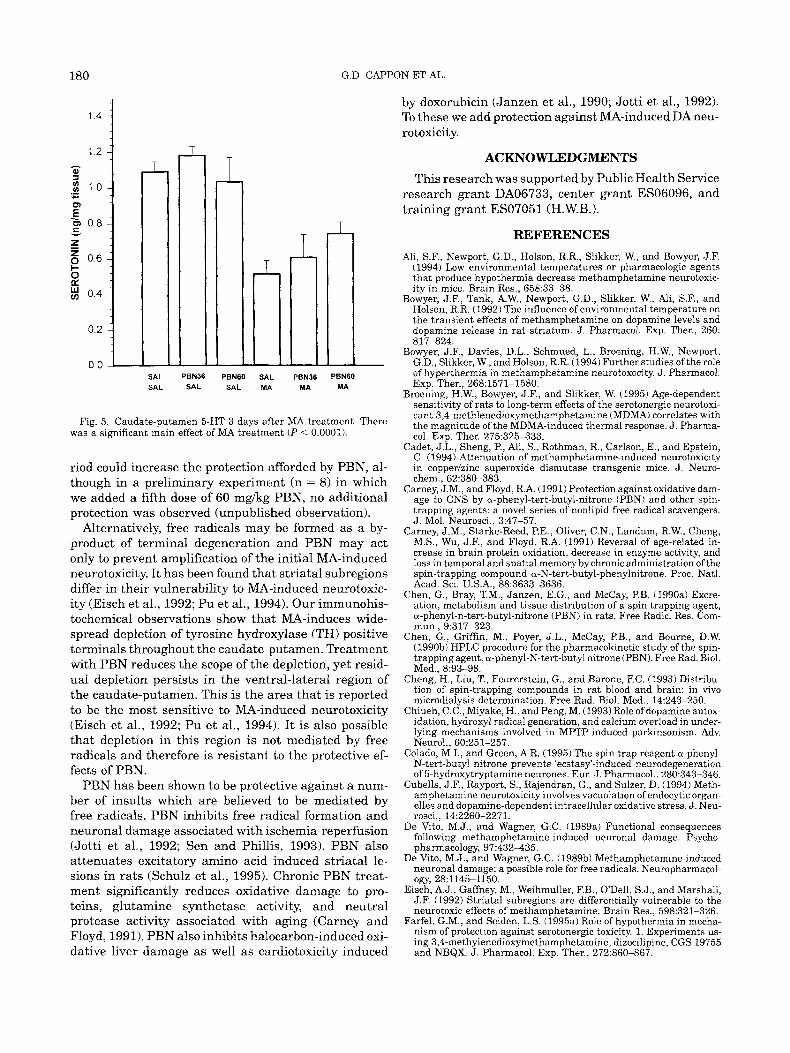
The effects of MA and PBN on 5-HT, alone and in combination, may be seen in Figure 5. Sal-MA induced a 53% depletion of caudate-putamen 5-HT. The PBN 36-MA (48% depletion) and PBN 60-MA (28% depletion) groups exhibited a tendency towards dose-dependent attenuation of MA-induced reductions in 5-HT content, however, the protection was not statistically significant (MA X PBN interaction F(2,44,1.52 P = 0.23). The PBN36-Sal and PBNGO-Sal groups did not differ from Sal-Sal controls.
DISCUSSION This study demonstrates that PBN administration
attenuates MA-induced dopamine depletion in the rat caudate-putamen without altering MA-induced hyper- thermia. PBN is a spin trapping agent which reacts with free radicals to form nitroxyl products (Floyd and Carney, 1992). Administration of PBN has been shown to inhibit oxidative damage associated with aging, isch- emia-reperfusion, and exposure to some toxins. It is thought that the mechanism underlying PBNs protec- tive effects lies in its ability to scavenge free radicals. The demonstration that PBN attenuates MA-induced dopamine (DA) depletion supports the hypothesis that free radicals are involved in the etiology of MA-in- duced neurotoxicity.
The generation of excess free radicals induces a state

PBN AND MA-INDUCED NEUROTOXICITY 177
TABLE I . Effect of PBN on MA-induced lethality and maximum rectal temperature'
Max. rectal temp. Max. rectal temp. all subjects ("C) survivors ("C) Treatment No. dosed No. survived Survivors (%)
Sali ne-saline PBN (36 mgkgl-saline PBN (60 mgkgl-saline PBN (75 mgkgl-saline Saline-MA PBN (36 mgkgl-MA PBN (60 mgkgl-MA PBN (75 mgkgl-MA PBN (120 mgkg)-MA
11 7
10 3
21 13 20 8 7
11 7
10 3
15 10 15 1 2
100 100 100 100 71.4* 76.9 75.0 12.5**** 28.6***
38.5 2 0.1 38.1 2 0.3 37.9 c 0.1 37.9 c 0.1 41.3 2 0.2** 40.9 f 0.3** 41.2 c 0.1** 41.9 2 0.2** 41.2 2 0.3**
38.5 t 0.1 38 1 ? 0.3 37.9 f 0.1 37.9 2 0.1 41.1 2 0.2** 40.6 2 0.3** 40.1 2 0.1**
-****a
40.2 2 0.9**.*"*
'There is no difference in maximum rectal temperature between any of the three MA treatment groups *P 5 0.05 vs. Sal-Sal. *"P s 0.01 vs. Sal-Sal. "7+P 5 0.05 vs. Sal-MA. ".'"P s 0.01 YS. Sal-MA.
Data not used in statistical analysis I*-**
of oxidative stress which can result in cellular damage or death. A link between MA-induced neurotoxicity and oxidative stress was first established by the demonstra- tion that the antioxidants mannitol, ethanol, vitamin E, and ascorbic acid attenuate MA-induced DA deple- tion (De Vito and Wagner, 1989a,b; Wagner et al., 1985). Additional support for the involvement of free radicals in MA-induced neurotoxicity was supplied by the dem- onstration that transgenic mice overexpressing copper1 zinc superoxide dismutase were resistant to the neuro- toxic effects of MA (Cadet et al., 1994). Methamphet- amine administration has been shown to increase neo- striatal concentrations of the salicylate reaction product dihydroxybenzoic acid, indicating increased hydroxyl radical content after MA exposure (Giovanni et al., 1995). In addition, in vivo application of MA to dopamin- ergic neurons induces vesicular vacuolization, initiating the formation of free radicals and triggering selective DA terminal degeneration (Cubells et al., 1994). These observations along with demonstrations that autoxida- tion of DA can lead to the formation of free radicals (Graham et al., 1978; Maruyama et al., 1993; Slivka and Cohen, 1985) support the involvement of free radicals in MA-induced neurotoxicity. Our data are consistent with these multiple lines of converging evidence for free radi- cal involvement in MA-induced neurotoxicity.
Methamphetamine administration induces hyper- thermia, the extent of which correlates with the severity of DA depletion (Bowyer et al., 1994). Several strategies which reduce MA-induced DA or 5-HT depletion also prevent hyperthermia (Bowyer et al., 1994; Farfel and Seiden, 1995a,b; Miller and OCallaghan, 1994). This suggests that treatments which lower body tempera- ture responses operate through a nonspecific protective mechanism by inhibiting MA-induced hyperthermia (Ali et al., 1994; Bowyer e t al., 1992). Pretreatment with PBN did not reduce the maximum core temperature or appreciably alter total thermal response induced by MA exposure. This suggests that the mechanism through which PBN is neuroprotective is not related to its ther- moregulatory effects.
Although PBN did not interfere with MA-induced hy- perthermia, the PBN36-Sal and PBN6O-Sal groups ex- hibited a hypothermic response in comparison to the Sal-Sal group. PBN-induced hypothermia has been ob- served by other investigators (Colado and Green, 1995; Schulz et al., 1995; Yue et al., 1992). At present it is unknown why PBN produces hypothermia. The hypo- thermic effect of PBN may be related to its ability to react with free radicals or may be due to some as yet uncharacterized property.
Our results suggest that PBN administration may afford protection against 5-HT depletion in the caudate- putamen in a pattern similar to the protection afforded to DA neurons. However, the protection against MA- induced 5-HT depletion did not reach statistical signifi- cance. This is in agreement with findings showing that overexpression of copperlzinc superoxide dismutase also inhibits 5-HT depletion (Hirata et al., 1995) sug- gesting free radical involvement in MA-induced 5-HT depletion.
A recent study demonstrated that PBN attenuates the depletion of 5-HT induced by the substituted amphetamine 3,4-methylenedioxymethamphetamine (MDMA) (Colado and Green, 1995). This suggests that MDMA also produces its effects by the formation of free radicals (Colado and Green, 1995). Administration of PBN (150 mglkg i.p.) 10 min prior to and 2 h after a single dose of MDMA (10 mglkg i.p.1 prevented the loss of 5-HT reuptake sites, and partially inhibited 5-HT depletion. However, these authors also found that MDMA-induced hyperthermia was prevented by the first administration of PBN. The second dose of PBN induced a decrease in body temperature of both saline controls and MDMA-treated animals. Other studies have demonstrated that reducing core temperature pre- vents DA and 5-HT depletion induced by MDMA, MA, methylenedioxyamphetamine (MDA), fenfluramine and parachloroamphetamine (Bowyer et al., 1992; Broening et al., 1995; Farfel and Seiden, 1995a,b; Miller and O'Callaghan, 1994; Schmidt e t al., 1990). Therefore, the protection against MDMA-induced 5-HT reductions
178 G.D. CAPPON ET AL.
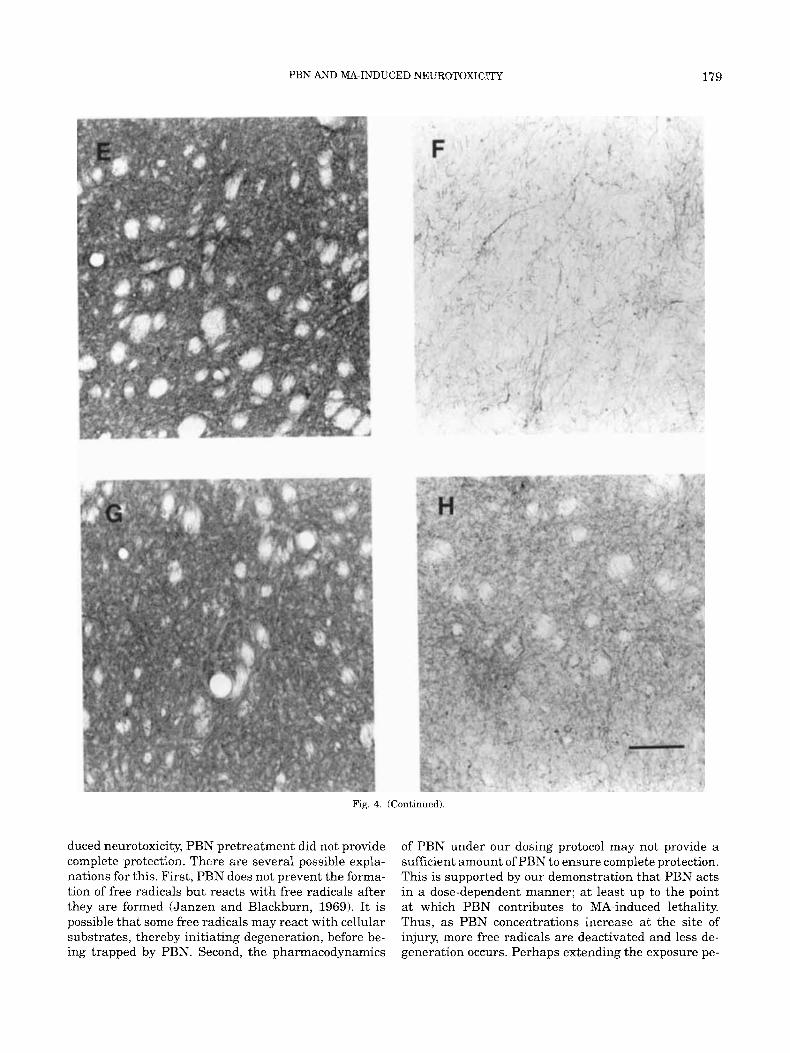
Fig. 4. TH immunohistochemical staining 3 days following treat- ment. A: Sal-Sal treatment showing a normal pattern of TH-positive terminals in the caudate-putamen. B: Sal-MA treatment showing depletion of TH positrve terminals and swollen fibers. C: PBNGO-Sal showing no effect on TH staining. D: PBNGO-MA showing partial
protection afforded by PBN pretreatment. Photomicrographs in E-H are a higher magnification of the same sections shown in A-D. E: Sal- Sal; F Sal-MA; G: PBNGO-Sal; H PBNGO-MA. Bar in H equals 300 Fm in A-D, 750 pm in E-H.
demonstrated by Colado and Green (1995) may be at- conducted our experiment. While our data suggest that tributable to either PBN or the reduction in body tem- PBN may interact with MA-induced 5-HT reductions, perature of the experimental animals. In our experi- further study is needed to elucidate the effect of PBN ments, PBN administration did not reduce body on serotonin content resulting from substituted am- temperature in MA treated rats. This may be due to phetamine exposure. the elevated ambient temperature (23.5”C) a t which we Although PBN administration attenuates MA-in-
PBN AND MA-INDUCED NEUROTOXICITY 179
Fig. 4. (Continued).
duced neurotoxicity, PBN pretreatment did not provide complete protection. There are several possible expla- nations for this. First, PBN does not prevent the forma- tion of free radicals but reacts with free radicals after they are formed (Janzen and Blackburn, 1969). It is possible that some free radicals may react with cellular substrates, thereby initiating degeneration, before be- ing trapped by PBN. Second, the pharmacodynamics
of PBN under our dosing protocol may not provide a sufficient amount of PBN to ensure complete protection. This is supported by our demonstration that PBN acts in a dose-dependent manner; at least up to the point a t which PBN contributes to MA-induced lethality. Thus, as PBN concentrations increase at the site of injury, more free radicals are deactivated and less de- generation occurs. Perhaps extending the exposure pe-
180
J
1 4 -
1 2 - - m
3 1 0 - .- c
F 2 0 8 -
z - $ 0 6 {
2 0 4 -
0 2 -
0 0
G.D. CAF'PON ET AL
- - SAL SAL
PEN36 PEN60 SAL SAL
- SAL MA
PEN36 MA
PEN60 MA
Fig. 5. Caudate-putamen 5-HT 3 days after MA treatment. There was a significant main effect of MA treatment ( P < 0.0001).
riod could increase the protection afforded by PBN, al- though in a preliminary experiment (n = 8) in which we added a fifth dose of 60 mgkg PBN, no additional protection was observed (unpublished observation).
Alternatively, free radicals may be formed as a by- product of terminal degeneration and PBN may act only to prevent amplification of the initial MA-induced neurotoxicity. It has been found that striatal subregions differ in their vulnerability to MA-induced neurotoxic- ity (Eisch et al., 1992; Pu et al., 1994). Our immunohis- tochemical observations show that MA-induces wide- spread depletion of tyrosine hydroxylase (TH) positive terminals throughout the caudate- putamen. Treatment with PBN reduces the scope of the depletion, yet resid- ual depletion persists in the ventral-lateral region of the caudate-putamen. This is the area that is reported to be the most sensitive to MA-induced neurotoxicity (Eisch et al., 1992; Pu et al., 1994). It is also possible that depletion in this region is not mediated by free radicals and therefore is resistant to the protective ef- fects of PBN.
PBN has been shown to be protective against a num- ber of insults which are believed to be mediated by free radicals. PBN inhibits free radical formation and neuronal damage associated with ischemia-reperfusion (Jotti et al., 1992; Sen and Phillis, 1993). PBN also attenuates excitatory amino acid induced striatal le- sions in rats (Schulz e t al., 1995). Chronic PBN treat- ment significantly reduces oxidative damage to pro- teins, glutamine synthetase activity, and neutral protease activity associated with aging (Carney and Floyd, 1991). PBN also inhibits halocarbon-induced oxi- dative liver damage as well a s cardiotoxicity induced
by doxorubicin (Janzen et al., 1990; Jotti et al., 1992). To these we add protection against MA-induced DA neu- rotoxicity.
ACKNOWLEDGMENTS This research was supported by Public Health Service
research grant DA06733, center grant ES06096, and training grant ES07051 (H.W.B.).
REFERENCES Ali, S.F., Newport, G.D., Holson, R.R., Slikker, W., and Bowyer, J.F.
(1994) Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxic- ity in mice. Brain Res., 658:33-38.
Bowyer, J.F., Tank, A.W., Newport, G.D., Slikker, W., Ali, S.F., and Holson, R.R. (1992) The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J. Pharmacol. Exp. Ther., 260: 817-824.
Bowyer, J.F., Davies, D.L., Schmued, L., Broening, H.W., Newport, G.D., Slikker, W., and Holson, R.R. (1994) Further studies of the role of hyperthermia in methamphetamine neurotoxoci ty. J. Pharmacol. Exp. Ther., 268:1571-1580.
Broening, H.W., Bowyer, J.F., and Slikker, W. (1995) Age-dependent sensitivity of rats to long-term effects of the serotonergic neurotoxi- cant 3,4-methlenedioxymethamphetamine (MDMA) correlates with the magnitude of the MDMA-induced thermal response. J. Pharma- col. Exp. Ther. 275:325-333.
Cadet, J.L., Sheng, P., Ali, S., Rothman, R., Carlson, E., and Epstein, C. (1994) Attenuation of methamphetamine-induced neurotoxicity in copperizinc superoxide dismutase transgenic mice. J. Neuro- chem., 62:380-383.
Carney, J.M., and Floyd, R.A. (1991) Protection against oxidative dam- age to CNS by a-phenyl-tert-butyl-nitrone (PBN) and other spin- trapping agents: a novel series of nonlipid free radical scavengers. J. Mol. Neurosci., 3:47-57.
Carney, J.M., Starke-Reed, P.E., Oliver, C.N., Landum, R.W., Cheng, M.S., Wu, J.F., and Floyd, R.A. (1991) Reversal of age-related in- crease in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound a-N-tert-butyl-phenylnitrone. Proc. Natl. Acad. Sci. U.S.A., 88:36333636.
Chen, G., Bray, T.M., Janzen, E.G., and McCay, P.B. (1990a) Excre- ation, metabolism and tissue distribution of a spin trapping agent, a-phenyl-n-tert-butyl-nitrone (PBN) in rats. Free Radic. Res. Com- mun., 9:317-323.
Chen, G., Griffin, M., Poyer, J.L., McCay, PB., and Bourne, D.W. (1990b) HPLC Drocedure for the Dharmacokinetic studv of the soin- trapping agent;a-phenyl-N-tert-hyl nitrone (PBN). F;ee Rad. Biol. Med., 8:93-98.
Cheng, H., Liu, T., Feurerstein, G., and Barone, F.C. (1993) Distribu- tion of spin-trapping compounds in rat blood and brain: in vivo microdialysis determination. Free Rad. Biol. Med., 14:243-250.
Chiueh, C.C., Miyake, H., and Peng, M. (1993) Role of dopamine autox- idation, hydroxyl radical generation, and calcium overload in under- lying mechanisms involved in MPTP-induced parkinsonism. Adv. Neurol., 60:25 1-257.
Colado, M.I., and Green, A.R. (1995) The spin trap reagent a-phenyl- N-tert-butyl nitrone prevents 'ecstasy'-induced neurodegeneration of 5-hydroxytryptamine neurones. Eur. J. Pharmacol.. 280:343-346.
Cubells, J.F., Rayport, S., Rajendran, G., and Sulzer, D. (1994) Meth- amphetamine neurotoxicity involves vacuolation of endocytic organ- elles and dopamine-dependent intracellular oxidative stress. J. Neu- rosci., 14:2260-2271.
De Vito, M.J., and Wagner, G.C. (1989a) Functional consequences following methamphetamine-induced neuronal damage. Psycho- pharmacology, 97:432435.
De Vito, M.J., and Wagner, G.C. (1989b) Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacol- ogy, 28:1145-1150.
Eisch. A.J.. Gaffnev, M.. Weihmuller. F.B.. O'Dell. S.J.. and Marshall. J.F.. (1992) Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res., 598:321-326.
Farfel, G.M., and Seiden, L.S. (1995a) Role of hypothermia in mecha- nism of protection against serotonergic toxicity. 1. Experiments us- ing 3,4-methylenedioxymethamphetamine, dizocilipine, CGS 19755 and NBQX. J. Pharmacol. Exp. Ther., 272:860-867.

PBN AND MA-INDUCED NEUROTOXICI'IY 181
Farfel, G.M., and Seiden, L.S. (1995b) Role of hypothermia in the mechanism of protection against serotonergic neurotoxicity. 11. Ex- periments with methamphetamine, p-chloroamphetamine, fenflura- mine, dizocilpine and dextrometorphan. J. Pharmacol. Exp. Ther., 272:868-875.
Floyd, R.A., and Carney, J.M. (1992) Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol., 32:S22-S27.
Giovanni, A., Liang, L.P., Hastings, T.G., and Zigmond, M.J. (1995) Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J. Neuro- chem., 64:1819-1825.
Graham, D.G., Tiffany, S.M., Bell, W.R., and Gutknecht, W.F. (1978) Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol., 14:
Green, A.R., De Souza, R.J., Williams, J.L., Murray, T.K., and Cross, A.J. (1992) The neurotoxic effects of methamphetamine on 5-hy- droxytryptamine and dopamine in brain: evidence for the protective effect of chlormethiazole. Neuropharmacology, 31:315321.
Hirata, H., Ladenheim, B., Rothman, R.B., Epstein, C., and Cadet, J.L. (1995) Methamphetamine-induced serotonin neurotoxicity is mediated by superoxide radicals. Brain Res., 677:345-347.
Hotchkiss, A.J., and Gibb, J.W. (1980) Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J . Pharmacol. Exp. Ther., 214: 257-262.
Hotchkiss, A.J., Morgan, M.E., and Gibb, J.W. (1979) The long-term effects of multiple doses of methamphetamine on neostriatal trypto- phan hydroxylase, tyrosine hydroxylase, choline acetyltransferase and glutamate decarboxylase activities. Life Sci., 25:1373-1378.
Janzen, E.G., and Blackburn, B.J. (1969) Detection and identification of short-lived free radicals by electron spin resonance trapping tech- niques (spin trapping). Photolysis of organolead, -tin, and -mercury compounds. J. Am. Chem. SOC., 91:4481-4490.
Janzen, E.G., Towner, R.A., and Yamashiro, S. (1990) The effect of phenyl-tert-butyl nitrone (PBN) on CCL,-induced rat liver injury detected by proton magnetic resonance imaging (MRI) in vivo and electron micropsy (EM). Free Radic. Res. Commun., 9:325-335.
Jotti, A., Paracchini, L., Perletti G., and Piccinini, F. (1992) Cardiotox- icity induced by doxorubicin in vivo: protective activity of the spin trap alpha-phenyl-tert-butyl nitrone. Pharmacol. Res., 26: 143-149.
Liu, L., and Wells, P.G. (1994) In vivo phenytoin-initiated oxidative damage to proteins and lipids in murine maternal hepatic and em- bryonic tissue organelles: potential molecular targets of chemical teratogenesis. Toxicol. Appl. Pharmacol., 125:247-255.
Maruyama, W., Takahashi, T., Minami, M., Takahashi, A., Dostert, P., Nagatsu, T., and Naoi, M. (1993) Cytotoxicity ofdopamine-derived 6,7-dihydroxy-l,2,3,4-tetrahydroisoquinolines. Adv. Neurol., 60: 224-230.
Miller, D.B., and O'Callaghan, J.P. (1994) Environment-, drug- and
644-653.
stress-induced alterations in body temperature affect the neurotox- icity of substituted amphetamines in the C57BW6J mouse. J. Phar- macol. Exp. Ther., 270:752-760.
O'Callaghan, J.P. (1991)Assessment ofneurotoxicity: use of glial fibril- lary acidic protein as a biomarker. Biomed. Environ. Sci., 4:197-206.
Phillis, J.W., and Clough-Helfman, C. (1990) Protection from cerebral ischemic injury in gerbils with the spin trap agent N-tert-butyl-a- phenylnitrone (PBN). Neurosci. Lett., 116:315-319.
Pu, C., and Vorhees, C.V. (1993) Developmental dissociation of meth- amphetamine-induced depletion of dopaminergic terminals and astrocyte reaction in rat striatum. Dev. Brain Res., 72:325-328.
Pu, C., Fisher, J.E., Cappon, G.D., andvorhees, C.V. (1994jThe effects of amfonelic acid, a dopamine uptake inhibitor, on methamphet- amine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum. Brain Res., 649:217-224.
Ricaurte, G.A., Guillery, R.W., Seiden, L.S., Schuster, C.R., and Moore, R.Y. (1982) Dopamine nerve terminal degeneration produced by high doses of methylamphetarnine in the rat brain. Brain Res., 235: 93-103.
Schmidt, C.J., Black, C.K.,Abbate, G.M., andTaylor,V.L. (1990)Meth- ylenedioxymethamphetamine-induced hyperthermia and neurotox- icity are independently mediated by 5-HT2 receptors. Brain Res.,
Schulz, J.B., Henshaw, D.R., Siwek, D., Jenkins, B.G., Ferrante, R.J., Cipolloni, P.B., Kowall, N.W., Rosen, B.R., and Beal, M.F. (1995) Involvement of free radicals in excitotoxicity in vivo. J. Neuro- chem., 64:2239-2247.
Seiden, L.S., Fischman, M.W., and Schuster, C.R. (1976) Long term methamphetamine induces changes in brain catecholamines in tol- erant rhesus monkeys. Drug Alcohol Depend., 1:215-219.
Sen, S., and Phillis, J.W. (1993) Alpha-phenyl-tert-butyl-nitrone (PBN) attenuates hydroxyl radical production during ischemia-reperfusion injury of the rat brain: an EPR study. Free Radic. Res. Commun., 19:255-265.
Slivka, A., and Cohen, G. (1985) Hydroxyl radical attack on dopamine. J . Biol. Chern., 260:15466-15472.
Trulson, M.E., Cannon, M.S., Faegg, T.S., and Raese, J.D. (1987) Tyro- sine hydroxylase immunochemistry and quantitative light micro- scopic studies of the mesolimbic dopamine system in rat brain: ef- fects of chronic methamphetamine administration. Brain Res.
529:85-90.
Bull., 18~269-277. Wagner, G.C., Ricaurte, G.A., Seiden, L.S., Schuster, C.R., Miller, R.J.,
and Westlev. J. (1980) Lone-lasting deuletions of striatal douamine and loss of iopamine uptak; sites 6llowing repeated administration of methamphetamine. Brain Res., 181:151-160.
Wagner, G.C., Carelli, R.M., and Jarvis, M.F. (1985) Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphet- amine in rats. Res. Comm. Chem. Pathol. Pharm., 47:221-228.
Yue, T., Gu, J., Lysko, P.G., Chen, H., Barone, F.C., and Feuerstein, G. (1992) Neuroprotective effect of phenyl-t-butyl-nitrone in gerbil global brain ischemia and in cultured rat cerebellar neurons. Brain Res., 574:193-197.