pathophysiological mechanisms and consequences of cardiovascular calcifications: role of uremic...
TRANSCRIPT

Annales Pharmaceutiques Françaises (2009) 67, 234—240
GENERAL REVIEW
Pathophysiological mechanisms and consequences ofcardiovascular calcifications: Role ofuremic toxicity
Mécanismes physiopathologiques et conséquences des calcificationscardiovasculaires : rôle de la toxicité urémique
J.-M. Chillona,b,c, A. Mozara,b, I. Sixa,b,J. Maizela,b,c, J.-M. Bugnicourtc, S. Kamela,b,c,M. Slamaa,b,c, M. Braziera,b,c, Z.A. Massya,b,c,∗
a Inserm ERI-12, Amiens, Franceb Faculty of Medicine/Pharmacy, UPJV, Amiens, Francec Amiens University Hospital, Amiens, France
Received 19 February 2009; accepted 6 April 2009Available online 23 May 2009
KEYWORDSChronic kidneydisease;Uremic toxicity;Cardiovascularcalcification;Osteoclast-likedifferentiation;Osteoblast-likephenotype
Summary Chronic kidney disease (CKD) represents an accelerated model of the active car-diovascular calcification process. Recent data from our laboratory indicate the presence of apossible vascular remodeling leading to vascular calcification similar to that observed in bonetissue, and emphasize the role of uremic toxicity. Uremic serum not only induces differentiationof smooth muscle cells into an osteoblast-like phenotype but also inhibits the differentiationof monocyte-macrophages cells into osteoclasts. The imbalance between the two processes invascular walls in favor of osteoblast-like formation could lead to calcification. Cardiovascu-lar calcification may contribute to the high rate of cardiovascular disease in patients with CKD.However, uremic toxicity, which participates in the pathogenesis of cardiovascular calcification,seems to have independent effects on vascular walls, at least in the early stages of CKD. We
recently reported that functional (i.e. endothelial dysfunction) rather than structural changes,including vascular calcification, may contribute to the aortic hemodynamic changes observedduring early CKD. Uremic toxicity also appears to be associated with calcification of intracra-nial arteries. Knowledge concerning the pathogenesis and consequences of cardiovascular∗ Corresponding author. Division(s) of Clinical Pharmacology and Nephrology, Amiens University Hospital, avenue Réné-Laennec,80054 Amiens, France
E-mail address: [email protected] (Z.A. Massy).
0003-4509/$ — see front matter © 2009 Elsevier Masson SAS. All rights reserved.doi:10.1016/j.pharma.2009.04.001

Pathophysiological mechanisms a
MOTS CLÉSInsuffisance rénalechronique ;Calcificationscardiovasculaires ;Toxicité urémique ;Ostéoblastes ;Ostéoclastes
ouvre de nouvelles perspectives de traitements pharmacologiques susceptibles de les préveniret/ou de les traiter et donc de diminuer la mortalité et la morbidité cardiovasculaire, non
en ITou
ttaImvao
lcethiLvgtvbichc
seulement chez les patients© 2009 Elsevier Masson SAS.
Cardiovascular calcifications are frequent in the generalpopulation and are associated with an increased cardio-vascular risk [1—6]. Degenerative calcific stenosis of theaortic valve is the most frequent valvular cardiomyopathyin industrialized countries, with a prevalence of 2% in theNorth American population [7]. The process of cardiovas-cular calcification is accelerated and amplified in patientswith chronic kidney disease (CKD) [8,9]. In these patients,cardiovascular calcifications constitute an increased riskof cardiovascular mortality and morbidity [10,11]. Cardio-vascular calcifications are accompanied by deposition ofmineralized matrix proteins in vessels or aortic valves. Thesecalcium deposits are essentially localized in the media ofvessel walls, but also in subintimal atherosclerotic plaques[12,13]. These two types of deposits are often observedsimultaneously in humans, and their pathogenesis probablyshares common cellular and molecular mechanisms [12].
Pathophysiological mechanisms
Matrix deposits in the vessel walls may result from an activecellular process with transdifferentiation of smooth musclecells (SMC) into osteoblast-like cells leading to depositionof an osteogenic type of extracellular matrix, which is at
least partly mediated by activation of the apoptotic process[14]. The inducers (e.g. inorganic phosphate) of dediffer-entiation of the SMC into osteoblast-like cells appear toact via a common mechanism that involves regulation of aspecific transcriptional factor of the osteoblast-like pheno-rowt[
RC mais aussi dans la population générale.s droits réservés.
ype: Core binding factor a1 (Cbfa-1) [15,16]. This specificranscriptional factor of the osteoblasts is involved in thectivation of various genes (osteopontin, osteocalcin, typecollagen) essential for osteoblastic differentiation fromesenchymal progenitors. This active process may be aggra-
ated by reduction of calcification inhibitors such as fetuin And/or matrix GLA protein (MGP) and/or osteopontin and/orsteoprotegerin [12,17,18].
Osteoclast differentiation is closely correlated with cel-ular contacts between osteoclast and osteoblast precursorsells in bone tissue (RANK/RANK-L system). The pres-nce of both monocyte-macrophages cells that are ableo differentiate directly into osteoclasts [19] and SMC thatave an osteoblast-like phenotype and well-known factorsnvolved in the osteoclast differentiation (such as RANK-, M-CSF or pro-inflammatory cytokines) in the calcifiedascular wall strongly suggests the presence of osteoclasto-enesis in arterial walls [20,21]. The coexistence of thesewo differentiated cell types raises the hypothesis of aascular remodeling process similar to that observed inone tissue. The imbalance between the two processesn favor of the osteoblast-like phenotype could lead toalcification [22,23]. Recent preliminary data support thisypothesis by suggesting the presence, although rare, ofells with an osteoclast phenotype in the calcified arte-ial walls [18,24]. In osteoporosis, there is an imbalance
nd consequences of cardiovascular calcifications 235
calcification derived from the uremic model therefore opens up new perspectives for pharma-cologic treatments that may also help to prevent and/or treat cardiovascular calcification, andconsequently cardiovascular mortality and morbidity, not only in CKD patients but also in thegeneral population.© 2009 Elsevier Masson SAS. All rights reserved.
Résumé L’insuffisance rénale chronique (IRC) est un modèle accéléré de calcification cardio-vasculaire. Des résultats récents de notre laboratoire suggèrent l’existence d’un remodelagevasculaire, similaire à celui observé dans l’os, qui serait responsable du développement descalcifications cardiovasculaires. Ces mêmes résultats mettent en avant le rôle des toxinesurémiques. En effet, le sérum de patients urémiques induit la différenciation des cellulesmusculaires lisses en cellules avec un phénotype similaire à celui des ostéoblastes, et parallèle-ment inhibe la différenciation des monocytes-macrophages en ostéoclastes. Le déséquilibreentre ces deux processus dans la paroi vasculaire, en faveur de la formation de cellules avecun phénotype similaire à celui des ostéoblastes, pourrait être responsable de la genèse etde la pérennisation des calcifications. Les calcifications cardiovasculaires peuvent contribuerau taux élevé de maladies cardiovasculaires chez les patients atteints d’IRC. Cependant, lestoxines urémiques, qui contribuent au développement des calcifications cardiovasculaires,semblent avoir des effets indépendants sur la paroi vasculaire dès le début de l’IRC. Nousavons ainsi montré que, dans les stades précoces d’IRC, des modifications fonctionnelles(dysfonction endothéliale) plutôt que des altérations structurales contribuent aux anomalieshémodynamiques aortiques. La toxicité urémique semble également associée au développe-ment des calcifications au niveau des artères intracrâniennes. La connaissance des mécanismesresponsables du développement des calcifications cardiovasculaires et de leurs conséquences
f bone remodeling in favor of osteoclast hyperactivity,hich is responsible for accelerated bone demineraliza-
ion, not compensated by increased osteoblastic activity25]. The fact that sufficient numbers of osteoclasts have
2
ncsaebtdccoufs[pmddR
f(upipb
sood(prtTPtclociststrpt
FpsssEmup(
36
ot been identified in calcified vascular tissue may indi-ate that osteoclast-like differentiation is inhibited in thesepecific conditions or that osteoclast-like cells do not play
role in this pathological process. We therefore hypoth-size that the development of arterial calcifications maye associated not only with enhanced SMC dedifferentia-ion into osteoblast-like phenotype, but also with decreasedifferentiation of monocyte-type osteoclast precursors intoells able to limit the development of vascular calcifi-ations by resorbing mineralized matrix. Results alreadybtained by our group show that inorganic phosphate, atremic concentration, significantly inhibits osteoclast dif-erentiation and bone resorption via selective inhibition ofeveral RANKL-dependent intracellular signaling pathways26]. Osteoclastogenesis inhibition induced by inorganichosphate could therefore constitute a complementaryechanism by which inorganic phosphate predisposes to car-iovascular calcifications. However, other uremic toxins mayecrease osteoclast precursor differentiation in response toANKL.
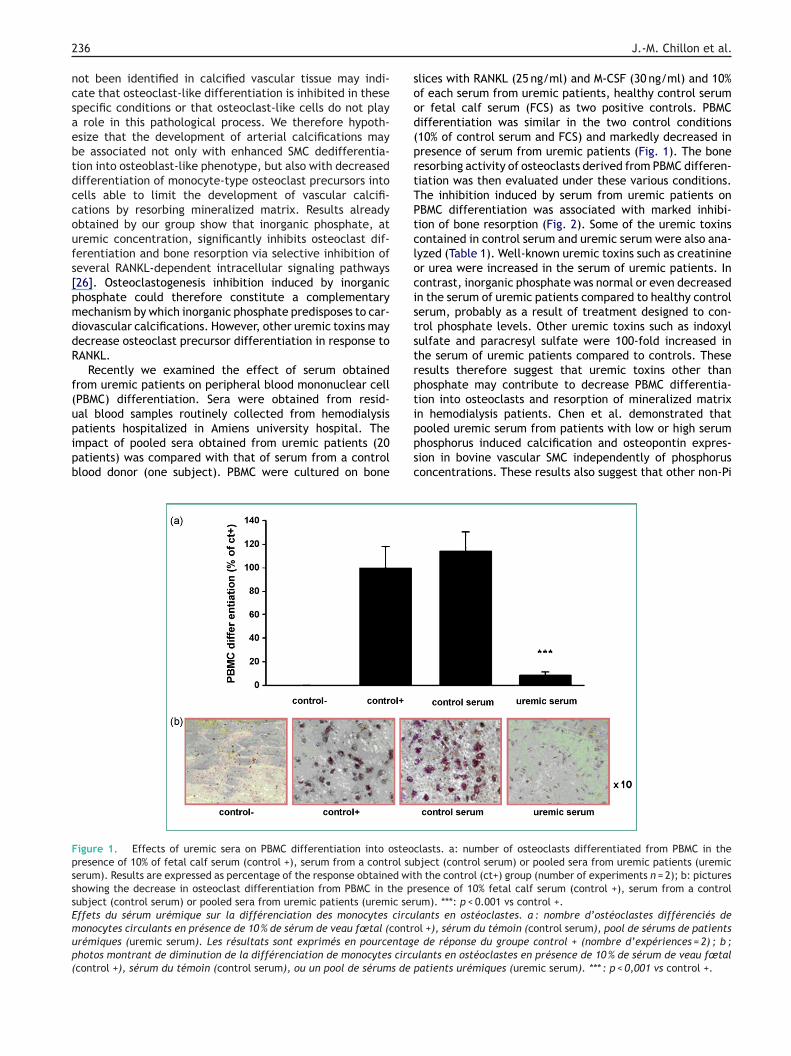
Recently we examined the effect of serum obtainedrom uremic patients on peripheral blood mononuclear cellPBMC) differentiation. Sera were obtained from resid-
al blood samples routinely collected from hemodialysisatients hospitalized in Amiens university hospital. Thempact of pooled sera obtained from uremic patients (20atients) was compared with that of serum from a controllood donor (one subject). PBMC were cultured on boneippsc
igure 1. Effects of uremic sera on PBMC differentiation into osteoresence of 10% of fetal calf serum (control +), serum from a control suerum). Results are expressed as percentage of the response obtained withowing the decrease in osteoclast differentiation from PBMC in the prubject (control serum) or pooled sera from uremic patients (uremic serffets du sérum urémique sur la différenciation des monocytes circuonocytes circulants en présence de 10 % de sérum de veau fœtal (contrrémiques (uremic serum). Les résultats sont exprimés en pourcentagehotos montrant de diminution de la différenciation de monocytes circucontrol +), sérum du témoin (control serum), ou un pool de sérums de
J.-M. Chillon et al.
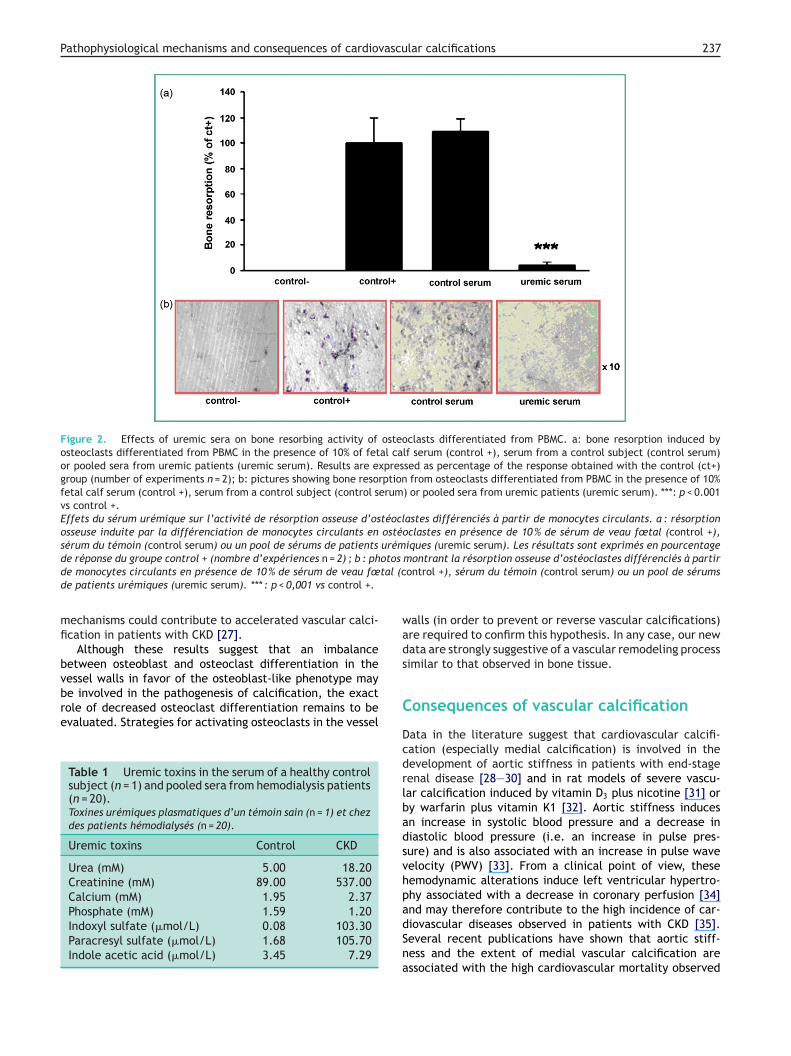
lices with RANKL (25 ng/ml) and M-CSF (30 ng/ml) and 10%f each serum from uremic patients, healthy control serumr fetal calf serum (FCS) as two positive controls. PBMCifferentiation was similar in the two control conditions10% of control serum and FCS) and markedly decreased inresence of serum from uremic patients (Fig. 1). The boneesorbing activity of osteoclasts derived from PBMC differen-iation was then evaluated under these various conditions.he inhibition induced by serum from uremic patients onBMC differentiation was associated with marked inhibi-ion of bone resorption (Fig. 2). Some of the uremic toxinsontained in control serum and uremic serum were also ana-yzed (Table 1). Well-known uremic toxins such as creatininer urea were increased in the serum of uremic patients. Inontrast, inorganic phosphate was normal or even decreasedn the serum of uremic patients compared to healthy controlerum, probably as a result of treatment designed to con-rol phosphate levels. Other uremic toxins such as indoxylulfate and paracresyl sulfate were 100-fold increased inhe serum of uremic patients compared to controls. Theseesults therefore suggest that uremic toxins other thanhosphate may contribute to decrease PBMC differentia-ion into osteoclasts and resorption of mineralized matrix
n hemodialysis patients. Chen et al. demonstrated thatooled uremic serum from patients with low or high serumhosphorus induced calcification and osteopontin expres-ion in bovine vascular SMC independently of phosphorusoncentrations. These results also suggest that other non-Piclasts. a: number of osteoclasts differentiated from PBMC in thebject (control serum) or pooled sera from uremic patients (uremich the control (ct+) group (number of experiments n = 2); b: picturesesence of 10% fetal calf serum (control +), serum from a controlum). ***: p < 0.001 vs control +.lants en ostéoclastes. a : nombre d’ostéoclastes différenciés deol +), sérum du témoin (control serum), pool de sérums de patients
de réponse du groupe control + (nombre d’expériences = 2) ; b ;lants en ostéoclastes en présence de 10 % de sérum de veau fœtal
patients urémiques (uremic serum). *** : p < 0,001 vs control +.
Pathophysiological mechanisms and consequences of cardiovascular calcifications 237
Figure 2. Effects of uremic sera on bone resorbing activity of osteoclasts differentiated from PBMC. a: bone resorption induced byosteoclasts differentiated from PBMC in the presence of 10% of fetal calf serum (control +), serum from a control subject (control serum)or pooled sera from uremic patients (uremic serum). Results are expressed as percentage of the response obtained with the control (ct+)group (number of experiments n = 2); b: pictures showing bone resorption from osteoclasts differentiated from PBMC in the presence of 10%fetal calf serum (control +), serum from a control subject (control serum) or pooled sera from uremic patients (uremic serum). ***: p < 0.001vs control +.Effets du sérum urémique sur l’activité de résorption osseuse d’ostéoclastes différenciés à partir de monocytes circulants. a : résorptionosseuse induite par la différenciation de monocytes circulants en ostéoclastes en présence de 10 % de sérum de veau fœtal (control +),sérum du témoin (control serum) ou un pool de sérums de patients urémiques (uremic serum). Les résultats sont exprimés en pourcentage
otostal (c
wads
de réponse du groupe control + (nombre d’expériences n = 2) ; b : phde monocytes circulants en présence de 10 % de sérum de veau fœde patients urémiques (uremic serum). *** : p < 0,001 vs control +.
mechanisms could contribute to accelerated vascular calci-fication in patients with CKD [27].
Although these results suggest that an imbalancebetween osteoblast and osteoclast differentiation in the
vessel walls in favor of the osteoblast-like phenotype maybe involved in the pathogenesis of calcification, the exactrole of decreased osteoclast differentiation remains to beevaluated. Strategies for activating osteoclasts in the vesselTable 1 Uremic toxins in the serum of a healthy controlsubject (n = 1) and pooled sera from hemodialysis patients(n = 20).Toxines urémiques plasmatiques d’un témoin sain (n = 1) et chezdes patients hémodialysés (n = 20).
Uremic toxins Control CKD
Urea (mM) 5.00 18.20Creatinine (mM) 89.00 537.00Calcium (mM) 1.95 2.37Phosphate (mM) 1.59 1.20Indoxyl sulfate (�mol/L) 0.08 103.30Paracresyl sulfate (�mol/L) 1.68 105.70Indole acetic acid (�mol/L) 3.45 7.29
C
DcdrlbadsvhpadSna
montrant la résorption osseuse d’ostéoclastes différenciés à partirontrol +), sérum du témoin (control serum) ou un pool de sérums
alls (in order to prevent or reverse vascular calcifications)re required to confirm this hypothesis. In any case, our newata are strongly suggestive of a vascular remodeling processimilar to that observed in bone tissue.
onsequences of vascular calcification
ata in the literature suggest that cardiovascular calcifi-ation (especially medial calcification) is involved in theevelopment of aortic stiffness in patients with end-stageenal disease [28—30] and in rat models of severe vascu-ar calcification induced by vitamin D3 plus nicotine [31] ory warfarin plus vitamin K1 [32]. Aortic stiffness inducesn increase in systolic blood pressure and a decrease iniastolic blood pressure (i.e. an increase in pulse pres-ure) and is also associated with an increase in pulse waveelocity (PWV) [33]. From a clinical point of view, theseemodynamic alterations induce left ventricular hypertro-hy associated with a decrease in coronary perfusion [34]
nd may therefore contribute to the high incidence of car-iovascular diseases observed in patients with CKD [35].everal recent publications have shown that aortic stiff-ess and the extent of medial vascular calcification aressociated with the high cardiovascular mortality observed
2
iimpoidhbraitaeccitctdntoito
onaC[qI(alahswftIl
Ic
Stcdiowalv
afu[aSc[ctsowocfiwwes
ttpelacsyTuittdabptslwtses
C
Tacas
38
n hemodialysis patients [10,36,37]. Furthermore, survivaln these patients is improved when antihypertensive treat-ents decrease PWV independently of their effects on bloodressure indicating that aortic stiffness has a major impactn survival [38]. Finally, a limited amount of data obtainedn predialysis CKD patients suggest that these structural car-iovascular alterations are not confined to patients withemodialysis, as left ventricular hypertrophy is also presentefore initiation of dialysis [29,39]. However, the exactole of vascular calcification in the observed early alter-tions has not yet been determined. In our model of CKDn apoE-/- and C57BL/6J mice, we recently demonstratedhat left ventricular hypertrophy, diastolic dysfunction andortic stiffness develop rapidly after onset of kidney dis-ase. However, these changes were not related to vascularalcification or high serum cholesterol levels or structuralhanges in the aorta. This may be related to the fact that,n contrast to other models of severe vascular calcifica-ion [32,40], we did not observed an elevation of the aorticollagen/elastin ratio in association with vascular calcifica-ion, which may be due to severe elastocalcinosis inducingestruction of elastin fibers, thereby leading to arterial stiff-ess [41]. Functional rather than structural changes mayherefore play a role in the aortic hemodynamic disordersbserved during early CKD, in contrast with the usual find-ngs in advanced CKD. One potential candidate may behe endothelial dysfunction observed in our murine modelf CKD.
Cerebral circulation is also altered during CKD. The riskf ischemic stroke is increased in CKD patients compared toon-uremic patients (4- to 10-fold increase) [42], and theylso present a poor long-term prognosis [43]. Furthermore,KD patients have a higher incidence of vascular dementia44]. Vascular calcifications in intracranial vessels are fre-uently observed on CT scan but have rarely been studied.t has been reported that intracranial artery calcificationsIAC) are frequent in a Chinese population and significantlyssociated with age, stroke history and CKD [45]. A strongink has also been demonstrated between ischemic strokend IAC [46]. In a recent clinical study, we confirmed theigh prevalence of IAC in patients with and without ischemictroke and showed, for the first time, that IAC is associatedith the presence of CKD in these patients. However, the
requency of IAC was significantly higher in stroke patientshan in non-stroke patients [47]. The physiopathology ofAC and its clinical relevance are under evaluation in ouraboratory.
mpact of the treatments on vascularalcification
everal studies in a rat model of elastocalcinosis have shownhat treatment with the ACE-inhibitor perindopril and thealcium channel blocker isradipine restored endothelium-ependent relaxation in the mesenteric beds. In contrast,sradipine (but not perindopril) prevented vascular calcium
verload [48,49]. In the same model, long-term treatmentith the peroxisome proliferators-activated receptor � lig-nd pioglitazone attenuated aortic wall elastocalcinosis andowered aortic wall stiffness, aortic pulse pressure and leftentricular hypertrophy [50].oittfi
J.-M. Chillon et al.
In order to examine the impact of various treatments ontheroma progression and/or vascular calcification, we per-ormed several studies in an apoE-/- mouse model of CKDsing the non-calcium-based phosphate binder sevelamer51], the calcium-based phosphate binder calcium carbon-te [52], the calcimimetic R-568 [53] and simvastatin [54].evelamer and R-568 delayed the progression of both aorticalcification and atherosclerosis in our uremic apoE-/- mice51,53]. In opposition to the hypothesis whereby calciumarbonate supplementation enhances vascular calcification,his salt decreased vascular calcification (despite increasingerum calcium levels) but did not prevent the progressionf atherosclerotic lesions [52]. Lastly, simvastatin treatmentas able to prevent intima calcification, despite the absencef changes in serum total cholesterol levels — suggesting aholesterol-independent action of statins on vascular calci-cation via a decrease in oxidative stress [54]. Taken as ahole, these observations are compatible with a hypothesishereby the damage caused by a combination of accel-rated calcification and uremia can be partially offset byeveral strategies.
A few clinical trials have confirmed that administra-ion of phosphate chelators (such as sevelamer) can slowhe progression of vascular calcification in hemodialyzedatients [55,56]. The question of whether the reported ben-ficial effects of sevelamer on vascular calcification willead to improved clinical outcomes remains unanswered,s reported by a systematic review of the clinical effi-acy and safety of sevelamer based on 32 trials comparingevelamer to any other therapy or placebo in adult dial-sis patients including more than 4000 participants [57].he DCOR study, which evaluated a prevalent dialysis pop-lation, could not show a difference between treatmentsn mortality (primary endpoint), except for patients olderhan 65 years (secondary endpoint) [58]. Nevertheless, inhe secondary analysis of the DCOR study, there was evi-ence that sevelamer use resulted in a 10% decrease inll-cause hospitalization rate and a 12% decrease in num-er of all-cause hospital days compared with calcium-basedhosphate-binder [59]. Moreover, a recent follow-up studyo the RIND trial, by Block et al. [60], demonstrated aurvival benefit in incident dialysis subjects receiving seve-amer, although the number of patients under considerationas relatively small. Hence, suggestions for future clinical
rials in large dialysis populations, of long duration, and pos-ibly enrolling patients with higher event rates, should bencouraged to confirm the postulated beneficial effect ofevelamer treatment on patient outcomes.
onclusion
he process of cardiovascular calcification is acceleratednd amplified in CKD patients. Cardiovascular calcificationontributes to the increased risk of cardiovascular mortalitynd morbidity in these patients. Our new data are stronglyuggestive of a vascular remodeling process similar to that
bserved in bone tissue. Uremic toxicity, which participatesn the pathogenesis of cardiovascular calcification, seemso have independent effects on vascular walls, at least inhe early stages of CKD. The fact that cardiovascular calci-cation is a regulated process rather than a passive process
ascu
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
Pathophysiological mechanisms and consequences of cardiov
opens up new perspectives for pharmacological treatments.Pharmacological treatments identified in CKD patients mayalso contribute to decrease cardiovascular calcification, andconsequently cardiovascular mortality and morbidity, in thegeneral population, which shows the same trend to cardio-vascular calcification although at an older age.
References
[1] Iribarren C, Sidney S, Sternfeld B, Browner WS. Calcificationof the aortic arch: risk factors and association with coronaryheart disease, stroke, and peripheral vascular disease. JAMA2000;283:2810—5.
[2] Kondos GT, Hoff JA, Sevrukov A, Daviglus ML, Garside DB,Devries SS, et al. Electron-beam tomography coronary arterycalcium and cardiac events: a 37-month follow-up of 5635initially asymptomatic low- to intermediate-risk adults. Circu-lation 2003;107:2571—6.
[3] Li J, Galvin HK, Johnson SC, Langston CS, Sclamberg J, PrestonCA. Aortic calcification on plain chest radiography increasesrisk for coronary artery disease. Chest 2002;121:1468—71.
[4] Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Asso-ciation of aortic-valve sclerosis with cardiovascular mortalityand morbidity in the elderly. N Engl J Med 1999;341:142—7.
[5] Rosenhek R, Binder T, Porenta G, Lang I, Christ G, SchemperM, et al. Predictors of outcome in severe, asymptomatic aorticstenosis. N Engl J Med 2000;343:611—7.
[6] Wilson PW, Kauppila LI, O’Donnell CJ, Kiel DP, Hannan M, PolakJM, et al. Abdominal aortic calcific deposits are an impor-tant predictor of vascular morbidity and mortality. Circulation2001;103:1529—34.
[7] Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS,Smith VE, et al. Clinical factors associated with calcific aorticvalve disease. Cardiovascular Health Study. J Am Coll Cardiol1997;29:630—4.
[8] Braun J, Oldendorf M, Moshage W, Heidler R, Zeitler E, LuftFC. Electron beam computed tomography in the evaluation ofcardiac calcification in chronic dialysis patients. Am J KidneyDis 1996;27:394—401.
[9] Perkovic V, Hunt D, Griffin SV, du Plessis M, Becker GJ. Acceler-ated progression of calcific aortic stenosis in dialysis patients.Nephron Clin Pract 2003;94:c40—45.
[10] London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B,Adda H. Arterial media calcification in end-stage renal disease:impact on all-cause and cardiovascular mortality. Nephrol DialTransplant 2003;18:1731—40.
[11] Wang AY, Woo J, Wang M, Sea MM, Ip R, Li PK, et al. Associationof inflammation and malnutrition with cardiac valve calcifica-tion in continuous ambulatory peritoneal dialysis patients. JAm Soc Nephrol 2001;12:1927—36.
[12] Shinke T, Karsenty G. Vascular calcification — a passive processin need of inhibitors. Nephrol Dial Transplant 2000;15:1272—4.
[13] Tomson C. Vascular calcification in chronic renal failure.Nephron Clin Pract 2003;93:c124—130.
[14] Shanahan CM. Vascular calcification: a matter of damage limi-tation? Nephrol Dial Transplant 2006;21:1166—9.
[15] Moe SM, Duan D, Doehle BP, O’Neill KD, Chen NX. Uremiainduces the osteoblast differentiation factor Cbfa1 in humanblood vessels. Kidney Int 2003;63:1003—11.
[16] Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, AebersoldR, et al. Smooth muscle cell phenotypic transition associated
with calcification: upregulation of Cbfa1 and downregula-tion of smooth muscle lineage markers. Circ Res 2001;89:1147—54.[17] Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, MahnkenAH, Bohm R, et al. Association of low fetuin-A (AHSG) concen-
[
[
lar calcifications 239
trations in serum with cardiovascular mortality in patients ondialysis: a cross-sectional study. Lancet 2003;361:827—33.
18] Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, ScullyS, et al. Osteoprotegerin reverses osteoporosis by inhibitingendosteal osteoclasts and prevents vascular calcification byblocking a process resembling osteoclastogenesis. J Exp Med2000;192:463—74.
19] Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiationand activation. Nature 2003;423:337—42.
20] Abedin M, Tintut Y, Demer LL. Vascular calcification: mecha-nisms and clinical ramifications. Arterioscler Thromb Vasc Biol2004;24:1161—70.
21] Collin-Osdoby P. Regulation of vascular calcification by osteo-clast regulatory factors RANKL and osteoprotegerin. Circ Res2004;95:1046—57.
22] Harada S, Rodan GA. Control of osteoblast function and regu-lation of bone mass. Nature 2003;423:349—55.
23] Karsenty G. The complexities of skeletal biology. Nature2003;423:316—8.
24] Doherty TM, Uzui H, Fitzpatrick LA, Tripathi PV, Dunstan CR,Asotra K, et al. Rationale for the role of osteoclast-like cells inarterial calcification. FASEB J 2002;16:577—82.
25] Rosen CJ. Clinical practice. Postmenopausal osteoporosis. NEngl J Med 2005;353:595—603.
26] Mozar A, Haren N, Chasseraud M, Louvet L, Maziere C, WattelA, et al. High extracellular inorganic phosphate concentrationinhibits RANK-RANKL signaling in osteoclast-like cells. J CellPhysiol 2008;215:47—54.
27] Chen NX, O’Neill KD, Duan D, Moe SM. Phosphorus and uremicserum up-regulate osteopontin expression in vascular smoothmuscle cells. Kidney Int 2002;62:1724—31.
28] Blacher J, Demuth K, Guerin AP, Safar ME, Moatti N, LondonGM. Influence of biochemical alterations on arterial stiffnessin patients with end-stage renal disease. Arterioscler ThrombVasc Biol 1998;18:535—41.
29] Raggi P, Bellasi A, Ferramosca E, Islam T, Muntner P, BlockGA. Association of pulse wave velocity with vascular andvalvular calcification in hemodialysis patients. Kidney Int2007;71:802—7.
30] Sigrist M, Bungay P, Taal MW, McIntyre CW. Vascular calcificationand cardiovascular function in chronic kidney disease. NephrolDial Transplant 2006;21:707—14.
31] Marque V, Van Essen H, Struijker-Boudier HA, Atkinson J,Lartaud-Idjouadiene I. Determination of aortic elastic modu-lus by pulse wave velocity and wall tracking in a rat model ofaortic stiffness. J Vasc Res 2001;38:546—50.
32] Bouvet C, Moreau S, Blanchette J, de Blois D, Moreau P. Sequen-tial activation of matrix metalloproteinase 9 and transforminggrowth factor beta in arterial elastocalcinosis. ArteriosclerThromb Vasc Biol 2008;28:856—62.
33] Safar ME, Levy BI, Struijker-Boudier H. Current perspectiveson arterial stiffness and pulse pressure in hypertension andcardiovascular diseases. Circulation 2003;107:2864—9.
34] London GM, Guerin AP. Influence of arterial pulse and reflectedwaves on blood pressure and cardiac function. Am Heart J1999;138:220—4.
35] Vanholder R, Massy Z, Argiles A, Spasovski G, Verbeke F,Lameire N. Chronic kidney disease as cause of cardio-vascular morbidity and mortality. Nephrol Dial Transplant2005;20:1048—56.
36] Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, LondonGM. Impact of aortic stiffness on survival in end-stage renaldisease. Circulation 1999;99:2434—9.
37] Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arte-rial calcifications, arterial stiffness, and cardiovascular risk inend-stage renal disease. Hypertension 2001;38:938—42.
38] Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, Lon-don GM. Impact of aortic stiffness attenuation on survival

2
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
40
of patients in end-stage renal failure. Circulation 2001;103:987—92.
39] Levin A, Singer J, Thompson CR, Ross H, Lewis M. Preva-lent left ventricular hypertrophy in the predialysis population:identifying opportunities for intervention. Am J Kidney Dis1996;27:347—54.
40] Niederhoffer N, Lartaud-Idjouadiene I, Giummelly P, DuvivierC, Peslin R, Atkinson J. Calcification of medial elastic fibersand aortic elasticity. Hypertension 1997;29:999—1006.
41] Maizel J, Six I, Slama M, Tribouilloy C, Sevestre H, Poirot S,et al. Mechanisms of aortic and cardiac dysfunction in uremicmice with aortic calcification. Circulation 2009;119:306—13.
42] Seliger SL, Gillen DL, Longstreth Jr WT, Kestenbaum B,Stehman-Breen CO. Elevated risk of stroke among patients withend-stage renal disease. Kidney Int 2003;64:603—9.
43] Iseki K, Fukiyama K. Clinical demographics and long-term prog-nosis after stroke in patients on chronic haemodialysis. TheOkinawa Dialysis Study (OKIDS) Group. Nephrol Dial Transplant2000;15:1808—13.
44] Lass P, Buscombe JR, Harber M, Davenport A, Hilson AJ. Cog-nitive impairment in patients with renal failure is associatedwith multiple-infarct dementia. Clin Nucl Med 1999;24:561—5.
45] Chen XY, Lam WW, Ng HK, Fan YH, Wong KS. The frequency anddeterminants of calcification in intracranial arteries in Chinesepatients who underwent computed tomography examinations.Cerebrovasc Dis 2006;21:91—7.
46] Chen XY, Lam WW, Ng HK, Fan YH, Wong KS. Intracranial arterycalcification: a newly identified risk factor of ischemic stroke.J Neuroimaging 2007;17:300—3.
47] Bugnicourt JM, Chillon JM, Massy ZA, Canaple S, Lamy C,Deramond H, et al. High prevalence of intracranial artery cal-cification in stroke patients with CKD: a retrospective study.Clin J Am Soc Nephrol 2009;4:284—90.
48] Henrion D, Chillon JM, Capdeville-Atkinson C, Atkinson J.Effect of chronic treatment with the calcium entry blocker,isradipine, on vascular calcium overload produced by vitaminD3 and nicotine in rats. J Pharmacol Exp Ther 1992;260:1—8.
49] Henrion D, Chillon JM, Capdeville-Atkinson C, Vinceneux-Feugier M, Atkinson J. Chronic treatment with the angiotensin
I converting enzyme inhibitor, perindopril, protects in vitrocarbachol-induced vasorelaxation in a rat model of vascularcalcium overload. Br J Pharmacol 1991;104:966—72.50] Gaillard V, Casellas D, Seguin-Devaux C, Schohn H, Dauca M,Atkinson J, et al. Pioglitazone improves aortic wall elasticity
[
J.-M. Chillon et al.
in a rat model of elastocalcinotic arteriosclerosis. Hypertension2005;46:372—9.
51] Phan O, Ivanovski O, Nguyen-Khoa T, Mothu N, Angulo J,Westenfeld R, et al. Sevelamer prevents uremia-enhancedatherosclerosis progression in apolipoprotein E-deficient mice.Circulation 2005;112:2875—82.
52] Phan O, Ivanovski O, Nikolov IG, Joki N, Maizel J, Louvet L,et al. Effect of oral calcium carbonate on aortic calcificationin apolipoprotein E-deficient (apoE-/-) mice with chronic renalfailure. Nephrol Dial Transplant 2008;23:82—90.
53] Ivanovski O, Nikolov IG, Joki N, Caudrillier A, PhanO, Mentaverri R, et al. The calcimimetic R-568 retardsuremia-enhanced vascular calcification and atherosclerosisin apolipoprotein E-deficient (apoE-/-) mice. Atherosclerosis2008. Nov 18 Epub ahead of print.
54] Ivanovski O, Szumilak D, Nguyen-Khoa T, Nikolov IG, Joki N,Mothu N, et al. Effect of simvastatin in apolipoprotein E defi-cient mice with surgically induced chronic renal failure. J Urol2008;179:1631—6.
55] Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the pro-gression of coronary and aortic calcification in hemodialysispatients. Kidney Int 2002;62:245—52.
56] Block GA, Spiegel DM, Ehrlich J, Mehta R, Lindbergh J, Dreis-bach A, et al. Effects of sevelamer and calcium on coronaryartery calcification in patients new to hemodialysis. Kidney Int2005;68:1815—24.
57] Tonelli M, Wiebe N, Culleton B, Lee H, Klarenbach S, ShriveF, et al. Systematic review of the clinical efficacy and safetyof sevelamer in dialysis patients. Nephrol Dial Transplant2007;22:2856—66.
58] Suki WN, Zabaneh R, Cangiano JL, Reed J, Fischer D, GarrettL, et al. Effects of sevelamer and calcium-based phosphatebinders on mortality in hemodialysis patients. Kidney Int2007;72:1130—7.
59] St Peter WL, Liu J, Weinhandl E, Fan Q. A compari-son of sevelamer and calcium-based phosphate binders onmortality, hospitalization, and morbidity in hemodialysis: asecondary analysis of the Dialysis Clinical Outcomes Revisited(DCOR) randomized trial using claims data. Am J Kidney Dis
2008;51:445—54.60] Block GA, Raggi P, Bellasi A, Kooienga L, Spiegel DM. Mor-tality effect of coronary calcification and phosphate binderchoice in incident hemodialysis patients. Kidney Int 2007;71:438—41.