one hundred twenty-one dystrophin point mutations detected from stored dna samples by combinatorial...
TRANSCRIPT

One Hundred Twenty-One Dystrophin PointMutations Detected from Stored DNA Samples byCombinatorial Denaturing High-Performance LiquidChromatography
Annalaura Torella,* Amelia Trimarco,*Francesca Del Vecchio Blanco,* Anna Cuomo,*Stefania Aurino,*† Giulio Piluso,* Carlo Minetti,‡
Luisa Politano,§ and Vincenzo Nigro*†
From the Dipartimentos di Patologia Generale,* and Medicina
Sperimentale,§ Seconda Universita degli Studi di Napoli, Naples,
the Telethon Institute of Genetics and Medicine,† Naples; and the
Universita degli Studi di Genova,‡ Istituto Giannina Gaslini,
Genua, Italy
Duchenne and Becker muscular dystrophies arecaused by a large number of different mutations inthe dystrophin gene. Outside of the deletion/duplica-tion “hot spots,” small mutations occur at unpredict-able positions. These account for about 15 to 20% ofcases, with the major group being premature stopcodons. When the affected male is deceased, carriertesting for family members and prenatal diagnosisbecome difficult and expensive. We tailored a cost-effective and reliable strategy to discover point muta-tions from stored DNA samples in the absence of amuscle biopsy. Samples were amplified in combina-torial pools and tested by denaturing high-perfor-mance liquid chromatography analysis. An anoma-lous elution profile belonging to two different poolsunivocally addressed the allelic variation to an un-ambiguous sample. Mutations were then detectedby sequencing. We identified 121 mutations of 99different types. Fifty-six patients show stop codonsthat represent the 46.3% of all cases. Three non-obvious single amino acid mutations were consid-ered as causative. Our data support combinatorialdenaturing high-performance liquid chromatogra-phy analysis as a clear-cut strategy for time andcost-effective identification of small mutations whenonly DNA is available. (J Mol Diagn 2010, 12:65–73; DOI:10.2353/jmoldx.2010.090074)
Duchenne (DMD [MIM 310200]) and Becker musculardystrophies (BMD [MIM 300376]) are allelic inheriteddisorders of muscle. They affect males in �99% ofcases, being transmitted as X-linked recessive traits.1
The DMD gene spans 2.2 million bp of genomic DNA
on the X chromosome, and the 14-kb transcript en-codes a full-length protein (dystrophin) of 427 kd(Dp427m). Both DMD and BMD arise due to mutationsat the dystrophin gene locus, which comprises 79 ex-ons and eight tissue-specific promoters. The mostcommon mutations are large intragenic deletions orduplications, encompassing one or more exons, butpoint mutations are about 15 to 20% of cases, with themajor group being premature stop codons.2–9
Patients and their families confer great value to mu-tation detection for genetic counseling, but also fortherapeutic options, since there are claims of novelmutation-targeted treatments.10 –12 Unfortunately, veryoften muscle biopsies are not possible because theaffected family member is deceased. We have tailoreda cost-effective and reliable strategy to discover pointmutations from DNA samples. Based on the sensitivityof denaturing high-performance liquid chromatogra-phy (DHPLC) to detect mutations, especially in A/T-richsequences, such as the dystrophin gene,6,7 we devel-oped a combinatorial DHPLC approach to screenpooled samples.
Materials and Methods
Patients
We used archive DNA samples from six different cen-ters: Laboratory of Molecular Biology, Scientific Insti-tute E. Medea, Lecco; Department of Neurological andPsychiatric Sciences, University of Padua; Instituteof Neurology, Catholic University, Policlinico Gemelli,Rome; Muscular and Neurodegenerative Disease Unit,Giannina Gaslini Institute, University of Genova; Depart-
Supported by grants from Telethon-UILDM GUP04008 (2005–2007) andTIGEM-11B and TIGEM-C20B, Ministero dell’Istruzione dell’Universita edella Ricerca (MIUR: PRIN 2004 and 2006) (to V.N. and C.M.), Ministerodella Salute (d.lgs 502/92), Ricerca d’Ateneo (to V.N. and L.P.).
A.T. is a fellow of the Luigi Califano Foundation.
Accepted for publication July 20, 2009.
Address reprint requests to Professor Vincenzo Nigro, M.D., Laborato-rio di genetica medica, Dipartimento di Patologia Generale, SecondaUniversita degli Studi di Napoli, S. Andrea delle Dame, via L. De Crecchio7, 80138 Napoli, Italy. E-mail: [email protected] or [email protected].
Journal of Molecular Diagnostics, Vol. 12, No. 1, January 2010
Copyright © American Society for Investigative Pathology
and the Association for Molecular Pathology
DOI: 10.2353/jmoldx.2010.090074
65

ment of Experimental Medicine, Cardiomyology and Medi-cal Genetics, Second University, Naples; and Centro deEstudos do Genoma Humano, Instituto de Biociencias Uni-versidade de Sao Paulo, Brasil. Diagnosis was determinedby clinical features consistent with DMD or BMD, along withan X-linked family history. Informed consent was obtainedfrom patients, when possible, according to the guidelines ofEurobiobank or Telethon.
Archive Samples
One hundred fifty-three DNA archive samples werestored in Tris-EDTA at 4°C. Fifteen were extracted byphenol-chloroform before 1994, whereas 31 were ex-tracted from 1994 to1999, and 46 from 2000 to 2004(Figure 1). More recent samples (from 2005 to 2007)were extracted using a FlexiGene DNA kit (Qiagen,Hamburg, Germany). Old samples were often recov-ered as dry pellets. In this case, we rehydrated thepellet. We evaluated the DNA integrity by 0.6% aga-rose gel electrophoresis. We did not re-precipitate anyof the samples. When required, we performed a pre-amplification step using the GenomiPhi HY DNA am-plification kit (GE Healthcare, Chalfont St. Giles, UK),according to the manufacturer’s instruction. This kitprovides microgram quantities of DNA from nanogramamounts of starting material in only a few hours. Thelimit of polymerase chain reaction (PCR) product sizeusing this archived DNA was about 1000 bp.
Sample Optimization
Each DNA sample was diluted to a final concentrationof 30 ng/�l, and 1 �l was used in each pool. To controlfor the possibility of unequal PCR product yield, shorttandem repeat (STR) polymorphic markers DXS8015-HEX and DXS1204-FAM (Table 1) were amplified fromsingle and pooled DNA templates, in a final reactionvolume of 20 �l, by using 0.5 �mol/L each markerprimer, buffer LB 10� [200 mmol/L Tris; 100 mmol/L
Hepes; 25 mmol/L MgSO4 � 7 H2O; 100 nm KCl; 100mmol/L (NH4)2 SO4], 0.25 �mol/L each dNTP, 0.5 UAmpliTaq Gold (Applied Biosystems, Foster City, CA).
Primer Design
Genomic sequence for Dp427m, the main dystrophinisoform found in muscle, was obtained from GenBank(NM 004006.1). Its exon 1 encodes a unique N-ter-minal MLWWEEVEDCY amino acid sequence and isexpressed in the skeletal muscle and heart.
For each dystrophin exon and muscular promoter aprimers pair was designed using the Primer 3 softwarepackage with the following criteria: product size be-tween 200 and 400 bp, primer size between 24 and 28nucleotides, and melting temperature between 58°Cand 62°C (Table 2).
Primer pairs were chosen to include flanking-intronsequence. Primer sequences were checked byBLASTn to avoid matching with repeated human se-quences or covering single nucleotide polymorphismsin the vicinity of exon sequences. Only in the case ofexon 26, we designed two primer pairs that split it intotwo overlapping fragments. Following these require-ments, we created a series of amplicons, all with thesame melting characteristics. All were amplified usingthe same PCR conditions. Primers were synthesized byMWG Biotech AG, Ebersberg, Germany. All PCR sharethe same conditions (95°C 30 seconds, 60°C 90 sec-onds, 68°C 90 seconds for 33 cycles).
Amplification of Genomic DNA
PCR reactions were set up semiautomatically using anautomatic liquid handling Eppendorf epMotion and384/96-well plates. DNA was amplified in a final reac-tion volume of 18 �l by using 30 ng of genomic DNA foreach pool, buffer LB [20 mmol/L Tris; 10 mmol/LHepes; 2.5 mmol/L MgSO4 � 7 H2O; 10 nm KCl; 10mmol/L (NH4)2 SO4], 1.5 mmol/L MgCl2, 0.25 �mol/Leach dNTP, 0.5 �mol/L each primers, 0.5 U AmpliTaqGold (Applied Biosystems).
WAVE System DHPLC Analysis
The dystrophin exons and flanking intronic sequencesand the muscular promoter were analyzed using high-throughput denaturing high-performance liquid chro-matography (HT-DHPLC). PCR products were directlyanalyzed. Using pooled samples a preliminary annealingstep is not required. The system is based on DHPLC. TheWAVE DHPLC system is an ion-pair, reverse-phase HPLC
Table 1. STR Markers
DXS1204-FAM DXS8015-HEX
F Primer: 5�-ATGAACCCTTAACTCATTTAGCAGG-3� F Primer: 5�-AGTCTTCTCAGGCCAGAGC-3�R Primer: 5�-AGCNTGCACCAACATGCC-3� R Primer: 5�-AGGACCAACTTTCACATGC-3�Length: 237–251 bp Length: 174–190 bp
F, forward; R, reverse.
Figure 1. Extraction dates of DNA samples.
66 Torella et alJMD January 2010, Vol. 12, No. 1
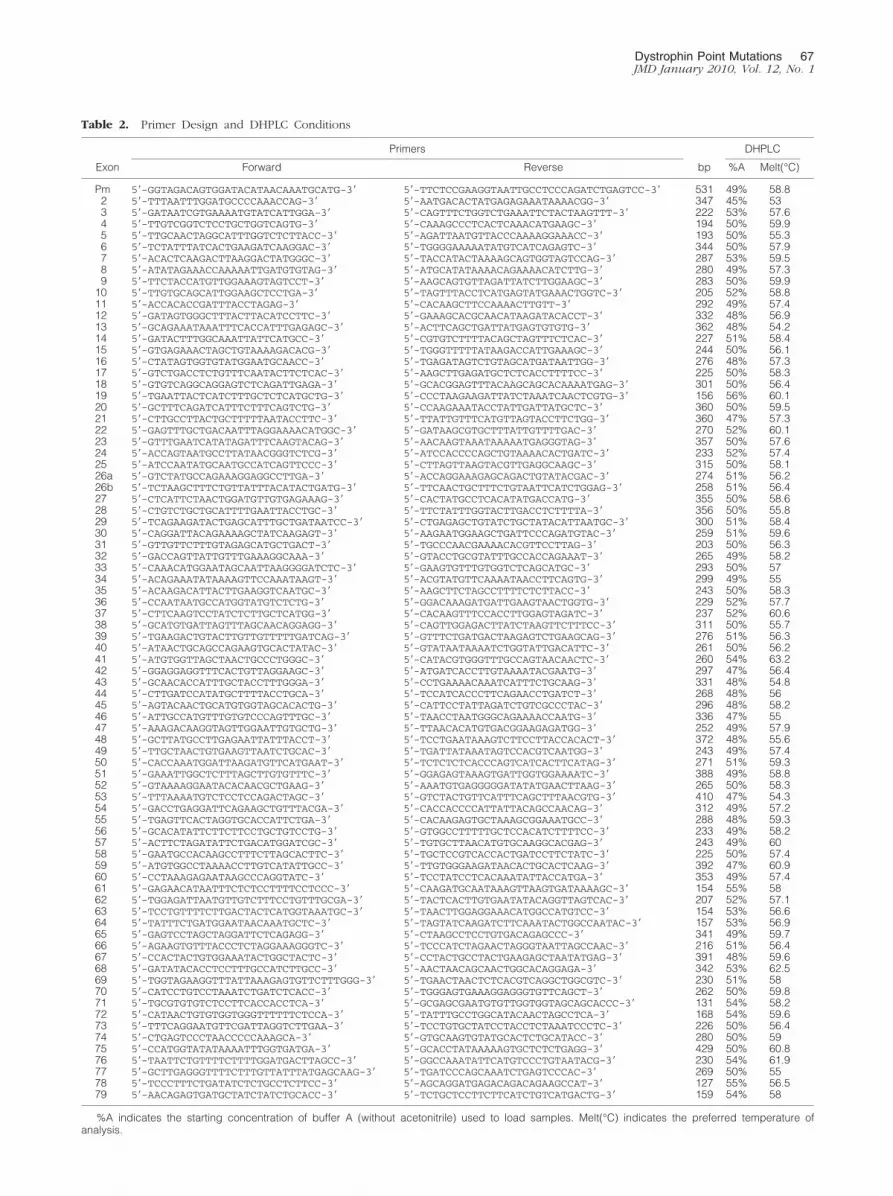
Table 2. Primer Design and DHPLC Conditions
Exon
Primers
bp
DHPLC
Forward Reverse %A Melt(°C)
Pm 5�-GGTAGACAGTGGATACATAACAAATGCATG-3� 5�-TTCTCCGAAGGTAATTGCCTCCCAGATCTGAGTCC-3� 531 49% 58.82 5�-TTTAATTTGGATGCCCCAAACCAG-3� 5�-AATGACACTATGAGAGAAATAAAACGG-3� 347 45% 533 5�-GATAATCGTGAAAATGTATCATTGGA-3� 5�-CAGTTTCTGGTCTGAAATTCTACTAAGTTT-3� 222 53% 57.64 5�-TTGTCGGTCTCCTGCTGGTCAGTG-3� 5�-CAAAGCCCTCACTCAAACATGAAGC-3� 194 50% 59.95 5�-TTGCAACTAGGCATTTGGTCTCTTACC-3� 5�-AGATTAATGTTACCCAAAAGGAAACC-3� 193 50% 55.36 5�-TCTATTTATCACTGAAGATCAAGGAC-3� 5�-TGGGGAAAAATATGTCATCAGAGTC-3� 344 50% 57.97 5�-ACACTCAAGACTTAAGGACTATGGGC-3� 5�-TACCATACTAAAAGCAGTGGTAGTCCAG-3� 287 53% 59.58 5�-ATATAGAAACCAAAAATTGATGTGTAG-3� 5�-ATGCATATAAAACAGAAAACATCTTG-3� 280 49% 57.39 5�-TTCTACCATGTTGGAAAGTAGTCCT-3� 5�-AAGCAGTGTTAGATTATCTTGGAAGC-3� 283 50% 59.9
10 5�-TTGTGCAGCATTGGAAGCTCCTGA-3� 5�-TAGTTTACCTCATGAGTATGAAACTGGTC-3� 205 52% 58.811 5�-ACCACACCGATTTACCTAGAG-3� 5�-CACAAGCTTCCAAAACTTGTT-3� 292 49% 57.412 5�-GATAGTGGGCTTTACTTACATCCTTC-3� 5�-GAAAGCACGCAACATAAGATACACCT-3� 332 48% 56.913 5�-GCAGAAATAAATTTCACCATTTGAGAGC-3� 5�-ACTTCAGCTGATTATGAGTGTGTG-3� 362 48% 54.214 5�-GATACTTTGGCAAATTATTCATGCC-3� 5�-CGTGTCTTTTACAGCTAGTTTCTCAC-3� 227 51% 58.415 5�-GTGAGAAACTAGCTGTAAAAGACACG-3� 5�-TGGGTTTTTATAAGACCATTGAAAGC-3� 244 50% 56.116 5�-CTATAGTGGTGTATGGAATGCAACC-3� 5�-TGAGATAGTCTGTAGCATGATAATTGG-3� 276 48% 57.317 5�-GTCTGACCTCTGTTTCAATACTTCTCAC-3� 5�-AAGCTTGAGATGCTCTCACCTTTTCC-3� 225 50% 58.318 5�-GTGTCAGGCAGGAGTCTCAGATTGAGA-3� 5�-GCACGGAGTTTACAAGCAGCACAAAATGAG-3� 301 50% 56.419 5�-TGAATTACTCATCTTTGCTCTCATGCTG-3� 5�-CCCTAAGAAGATTATCTAAATCAACTCGTG-3� 156 56% 60.120 5�-GCTTTCAGATCATTTCTTTCAGTCTG-3� 5�-CCAAGAAATACCTATTGATTATGCTC-3� 360 50% 59.521 5�-CTTGCCTTACTGCTTTTTAATACCTTC-3� 5�-TTATTGTTTCATGTTAGTACCTTCTGG-3� 360 47% 57.322 5�-GAGTTTGCTGACAATTTAGGAAAACATGGC-3� 5�-GATAAGCGTGCTTTATTGTTTTGAC-3� 270 52% 60.123 5�-GTTTGAATCATATAGATTTCAAGTACAG-3� 5�-AACAAGTAAATAAAAATGAGGGTAG-3� 357 50% 57.624 5�-ACCAGTAATGCCTTATAACGGGTCTCG-3� 5�-ATCCACCCCAGCTGTAAAACACTGATC-3� 233 52% 57.425 5�-ATCCAATATGCAATGCCATCAGTTCCC-3� 5�-CTTAGTTAAGTACGTTGAGGCAAGC-3� 315 50% 58.126a 5�-GTCTATGCCAGAAAGGAGGCCTTGA-3� 5�-ACCAGGAAAGAGCAGACTGTATACGAC-3� 274 51% 56.226b 5�-TCTAAGCTTTCTGTTATTTACATACTGATG-3� 5�-TTCAACTGCTTTCTGTAATTCATCTGGAG-3� 258 51% 56.427 5�-CTCATTCTAACTGGATGTTGTGAGAAAG-3� 5�-CACTATGCCTCACATATGACCATG-3� 355 50% 58.628 5�-CTGTCTGCTGCATTTTGAATTACCTGC-3� 5�-TTCTATTTGGTACTTGACCTCTTTTA-3� 356 50% 55.829 5�-TCAGAAGATACTGAGCATTTGCTGATAATCC-3� 5�-CTGAGAGCTGTATCTGCTATACATTAATGC-3� 300 51% 58.430 5�-CAGGATTACAGAAAAGCTATCAAGAGT-3� 5�-AAGAATGGAAGCTGATTCCCAGATGTAC-3� 259 51% 59.631 5�-GTTGTTCTTTGTAGAGCATGCTGACT-3� 5�-TGCCCAACGAAAACACGTTCCTTAG-3� 203 50% 56.332 5�-GACCAGTTATTGTTTGAAAGGCAAA-3� 5�-GTACCTGCGTATTTGCCACCAGAAAT-3� 265 49% 58.233 5�-CAAACATGGAATAGCAATTAAGGGGATCTC-3� 5�-GAAGTGTTTGTGGTCTCAGCATGC-3� 293 50% 5734 5�-ACAGAAATATAAAAGTTCCAAATAAGT-3� 5�-ACGTATGTTCAAAATAACCTTCAGTG-3� 299 49% 5535 5�-ACAAGACATTACTTGAAGGTCAATGC-3� 5�-AAGCTTCTAGCCTTTTCTCTTACC-3� 243 50% 58.336 5�-CCAATAATGCCATGGTATGTCTCTG-3� 5�-GGACAAAGATGATTGAAGTAACTGGTG-3� 229 52% 57.737 5�-CTTCAAGTCCTATCTCTTGCTCATGG-3� 5�-CACAAGTTTCCACCTTGGAGTAGATC-3� 237 52% 60.638 5�-GCATGTGATTAGTTTAGCAACAGGAGG-3� 5�-CAGTTGGAGACTTATCTAAGTTCTTTCC-3� 311 50% 55.739 5�-TGAAGACTGTACTTGTTGTTTTTGATCAG-3� 5�-GTTTCTGATGACTAAGAGTCTGAAGCAG-3� 276 51% 56.340 5�-ATAACTGCAGCCAGAAGTGCACTATAC-3� 5�-GTATAATAAAATCTGGTATTGACATTC-3� 261 50% 56.241 5�-ATGTGGTTAGCTAACTGCCCTGGGC-3� 5�-CATACGTGGGTTTGCCAGTAACAACTC-3� 260 54% 63.242 5�-GGAGGAGGTTTCACTGTTAGGAAGC-3� 5�-ATGATCACCTTGTAAAATACGAATG-3� 297 47% 56.443 5�-GCAACACCATTTGCTACCTTTGGGA-3� 5�-CCTGAAAACAAATCATTTCTGCAAG-3� 331 48% 54.844 5�-CTTGATCCATATGCTTTTACCTGCA-3� 5�-TCCATCACCCTTCAGAACCTGATCT-3� 268 48% 5645 5�-AGTACAACTGCATGTGGTAGCACACTG-3� 5�-CATTCCTATTAGATCTGTCGCCCTAC-3� 296 48% 58.246 5�-ATTGCCATGTTTGTGTCCCAGTTTGC-3� 5�-TAACCTAATGGGCAGAAAACCAATG-3� 336 47% 5547 5�-AAAGACAAGGTAGTTGGAATTGTGCTG-3� 5�-TTAACACATGTGACGGAAGAGATGG-3� 252 49% 57.948 5�-GCTTATGCCTTGAGAATTATTTACCT-3� 5�-TCCTGAATAAAGTCTTCCTTACCACACT-3� 372 48% 55.649 5�-TTGCTAACTGTGAAGTTAATCTGCAC-3� 5�-TGATTATAAATAGTCCACGTCAATGG-3� 243 49% 57.450 5�-CACCAAATGGATTAAGATGTTCATGAAT-3� 5�-TCTCTCTCACCCAGTCATCACTTCATAG-3� 271 51% 59.351 5�-GAAATTGGCTCTTTAGCTTGTGTTTC-3� 5�-GGAGAGTAAAGTGATTGGTGGAAAATC-3� 388 49% 58.852 5�-GTAAAAGGAATACACAACGCTGAAG-3� 5�-AAATGTGAGGGGGATATATGAACTTAAG-3� 265 50% 58.353 5�-TTTAAAATGTCTCCTCCAGACTAGC-3� 5�-GTCTACTGTTCATTTCAGCTTTAACGTG-3� 410 47% 54.354 5�-GACCTGAGGATTCAGAAGCTGTTTACGA-3� 5�-CACCACCCCATTATTACAGCCAACAG-3� 312 49% 57.255 5�-TGAGTTCACTAGGTGCACCATTCTGA-3� 5�-CACAAGAGTGCTAAAGCGGAAATGCC-3� 288 48% 59.356 5�-GCACATATTCTTCTTCCTGCTGTCCTG-3� 5�-GTGGCCTTTTTGCTCCACATCTTTTCC-3� 233 49% 58.257 5�-ACTTCTAGATATTCTGACATGGATCGC-3� 5�-TGTGCTTAACATGTGCAAGGCACGAG-3� 243 49% 6058 5�-GAATGCCACAAGCCTTTCTTAGCACTTC-3� 5�-TGCTCCGTCACCACTGATCCTTCTATC-3� 225 50% 57.459 5�-ATGTGGCCTAAAACCTTGTCATATTGCC-3� 5�-TTGTGGGAAGATAACACTGCACTCAAG-3� 392 47% 60.960 5�-CCTAAAGAGAATAAGCCCAGGTATC-3� 5�-TCCTATCCTCACAAATATTACCATGA-3� 353 49% 57.461 5�-GAGAACATAATTTCTCTCCTTTTCCTCCC-3� 5�-CAAGATGCAATAAAGTTAAGTGATAAAAGC-3� 154 55% 5862 5�-TGGAGATTAATGTTGTCTTTCCTGTTTGCGA-3� 5�-TACTCACTTGTGAATATACAGGTTAGTCAC-3� 207 52% 57.163 5�-TCCTGTTTTCTTGACTACTCATGGTAAATGC-3� 5�-TAACTTGGAGGAAACATGGCCATGTCC-3� 154 53% 56.664 5�-TATTTCTGATGGAATAACAAATGCTC-3� 5�-TAGTATCAAGATCTTCAAATACTGGCCAATAC-3� 157 53% 56.965 5�-GAGTCCTAGCTAGGATTCTCAGAGG-3� 5�-CTAAGCCTCCTGTGACAGAGCCC-3� 341 49% 59.766 5�-AGAAGTGTTTACCCTCTAGGAAAGGGTC-3� 5�-TCCCATCTAGAACTAGGGTAATTAGCCAAC-3� 216 51% 56.467 5�-CCACTACTGTGGAAATACTGGCTACTC-3� 5�-CCTACTGCCTACTGAAGAGCTAATATGAG-3� 391 48% 59.668 5�-GATATACACCTCCTTTGCCATCTTGCC-3� 5�-AACTAACAGCAACTGGCACAGGAGA-3� 342 53% 62.569 5�-TGGTAGAAGGTTTATTAAAGAGTGTTCTTTGGG-3� 5�-TGAACTAACTCTCACGTCAGGCTGGCGTC-3� 230 51% 5870 5�-CATCCTGTCCTAAATCTGATCTCACC-3� 5�-TGGGAGTGAAAGGAGGGTGTTCAGCT-3� 262 50% 59.871 5�-TGCGTGTGTCTCCTTCACCACCTCA-3� 5�-GCGAGCGAATGTGTTGGTGGTAGCAGCACCC-3� 131 54% 58.272 5�-CATAACTGTGTGGTGGGTTTTTTCTCCA-3� 5�-TATTTGCCTGGCATACAACTAGCCTCA-3� 168 54% 59.673 5�-TTTCAGGAATGTTCGATTAGGTCTTGAA-3� 5�-TCCTGTGCTATCCTACCTCTAAATCCCTC-3� 226 50% 56.474 5�-CTGAGTCCCTAACCCCCAAAGCA-3� 5�-GTGCAAGTGTATGCACTCTGCATACC-3� 280 50% 5975 5�-CCATGGTATATAAAATTTGGTGATGA-3� 5�-GCACCTATAAAAAGTGCTCTCTGAGG-3� 429 50% 60.876 5�-TAATTCTGTTTTCTTTTGGATGACTTAGCC-3� 5�-GGCCAAATATTCATGTCCCTGTAATACG-3� 230 54% 61.977 5�-GCTTGAGGGTTTTCTTTGTTATTTATGAGCAAG-3� 5�-TGATCCCAGCAAATCTGAGTCCCAC-3� 269 50% 5578 5�-TCCCTTTCTGATATCTCTGCCTCTTCC-3� 5�-AGCAGGATGAGACAGACAGAAGCCAT-3� 127 55% 56.579 5�-AACAGAGTGATGCTATCTATCTGCACC-3� 5�-TCTGCTCCTTCTTCATCTGTCATGACTG-3� 159 54% 58
%A indicates the starting concentration of buffer A (without acetonitrile) used to load samples. Melt(°C) indicates the preferred temperature ofanalysis.
Dystrophin Point Mutations 67JMD January 2010, Vol. 12, No. 1

method optimized to separate heteroduplex from homodu-plex DNA fragments (Transgenomic Inc., Omaha, NE).
Sequence Analysis
PCR amplicons were purified using the EXOSAP purification kit(GE Healthcare, Chalfont St. Giles, UK): 2 �l of ExoSAP-IT wasdirectly added to 5 �l of PCR product and incubated at 37°Cfor 15 minutes. ExoSAP-IT was inactivated by heating at80°C for 15 minutes. The sequence reactions were purified
Figure 2. Quality control of PCR yield. A–C: Analysis of each individual sample using the STR DXS8015. D: Analysis of a pool containing three samples.
Figure 3. Examples of aberrant DHPLC profiles. The figure shows different DHPLC profiles with growing complexity from A to D. A: Exon 6 showed a heteroduplexin both pools 1 and 4 sharing the DNA sample TU19, in which a frameshift mutation (c.401 404 delCCAA) was detected. B: Exon 27 heteroduplexes in both pools 2and 6 sharing the DNA sample TU124, in which a splicing mutation (c.3433-1 A�G) was detected. C: Exon 29 heteroduplexes in pools 1, 4, and 5. Pools 1 and 4 sharedthe DNA sample TU181, pools 1 and 5 shared the DNA sample TU188. The same nonsense mutation (c.3940 C�T) was detected in both samples. D: Three different exon14 heteroduplexes in pools 2 and 6 and 1 and 4, corresponding to combination of a mutation (**) and a known polymorphism (*). Arrow indicates homoduplexes.
Table 3. Combinatorial Pools
Pool 1 Pool 2 Pool 3
1 4 72 5 83 6 9
Pool 4 Pool 5 Pool 6
1 2 34 5 67 8 9
Samples were divided into groups of nine. For each group wecreated six overlapping pools, each one containing three DNA samplesfrom three different patients, so that each sample was present in aunique combination of two different pools.
68 Torella et alJMD January 2010, Vol. 12, No. 1

by Applied Biosystems BigDye XTerminator purification kitto remove unincorporated dye and other contaminants.Samples were analyzed using an ABI3130xL and sequenc-ing analysis software (Applied Biosystems).
Results
We screened 153 DNA samples from unrelated DMD orBMD patients. These samples were extracted andstudied many years ago without obtaining a geneticdiagnosis (Figure 1). We preliminarily excluded dele-tions or duplications by MLPA and Log-PCR.3,4
Combinatorial Pools
To speed up the analysis and improve sensitivity, wepooled DNA samples in 17 units, each comprising samples
from nine male patients. For each unit, we assembled sixpools, each one containing DNA from three different pa-tients, so that each DNA sample was present in two differentpools and thus analyzed in duplicate (Table 3). This en-abled the parallel amplification of three DNA samples in onerun and allowed us to detect point mutations without theannealing with control DNA. To avoid pooling samples withsignificantly different PCR yield, we preliminarily geno-typed STR markers in each of the DNA samples with theABI-Prism 3130 xl using Gene Mapper software. We usedtwo different X markers (DXS8015 and DXS1204) for theamplification of separate samples to determine tandemrepeat lengths. On the basis of STR analyses, we createdthe pools by mixing three DNA samples with a differentnumber of repeats (Figure 2).
We analyzed the DMD exons, flanking intronic se-quences and the muscle-promoter using HT-DHPLC.
Table 4. Nonsense Mutations
Sample Exon DNA change Stop Protein New Disease
3761 6 c.409 G�T TAA E137X Yes DMDTU182-TU294-TU183-TU378 6 c.433 C�T TGA R145X No DMDTU139-3443 7 c.583 C�T TGA R195X No DMDTU184 10 c.1062 G�A TGA W354X No DMDTU86 10 c.1093 C�T TAA Q365X No DMDTU180 11 c.1292 G�A TGA W431X No DMDTU318 14 c.1652 G�A TGA W551X Yes DMDTU187-TU189 17 c.2125 C�T TAA Q709X No DMDTU05 19 c.2302 C�T TGA R768X No DMDG11 19 c.2380 G�T TAG E794X Yes DMDTU70 20 c.2414 C�G TGA S805X Yes DMDF1 20 c.2521 C�T TAA Q841X No DMDTU01 23 c.2956 C�T TAA Q986X No DMDTU107 23 c.3151 C�T TGA R1051X No DMDTU51-TU185 24 c.3259 C�T TAG Q1087X No DMDTU32 25 c.3409 C�T TAG Q1137X No DMDTU12 26a c.3580 C�T TAG Q1194X No DMD475 27 c.3625 C�T TAA Q1209X Yes DMDTU342 28 c.3843 G�A TGA W1281X Yes BMDTU181-TU188-TU102 29 c.3940 C�T TGA R1314X No BMDTU218 30 c.4117 C�T TAG Q1373X No DMDR46 33 c.4600 C�T TAG Q1534X No DMDTU24 34 c.4690 C�T TAA Q1564X Yes DMDTU271 35 c.4979 G�A TGA W1660X Yes BMDTU190 35 c.4996 C�T TGA R1666X No DMDTU63 37 c.5209 C�T TAA Q1737X Yes DMDTU266 39 c.5476 G�T TAA E1826X No DMD CarrierTU194 39 c.5530 C�T TGA R1844X No DMDTU60 41 c.5773 G�T TAG E1925X No DMDTU112-TU178-TU84 41 c.5899 C�T TGA R1967X No DMDTU159 42 c.6023 C�A TGA S2008X Yes DMDG2-G8-R42 46 c.6678 G�A TGA W2226X Yes DMDTU186 48 c.7006 C�T TAG Q2336X No DMDR88 57 c.8422 A�T TAG K2808X Yes DMDTU152 59 c.8713 C�T TGA R2905X No DMD3448 59 c.8880 G�A TGA W2960X Yes DMDTU02-TU157 60 c.8944 C�T TGA R2982X No DMDTU87 65 c.9461 T�A TAG L3154X No BMDTU18 68 c.9829 G�T TAA E3277X Yes BMD/DMDTU208-G13 70 c.10108 C�T TGA R3370X No DMDF4 70 c.10135 A�T TAA K3379X No DMDG3 70 c.10171 C�T TGA R3391X No DMD
Resulting TGA stop codons are indicated in bold.
Dystrophin Point Mutations 69JMD January 2010, Vol. 12, No. 1

Each pool was amplified for all of the 79 dystrophingene exons and promoter. PCR products were directlyanalyzed by WAVE system using predetermined tem-perature and elution buffers concentrations (Table 2).The WAVE system provides rapid, automated scanning
for single nucleotide polymorphisms, even when thenature and location of the mutations are unknown.
DHPLC analysis of the pools allowed the unambig-uous identification of the mutant sample, avoiding thesubsequent screening of three single DNA samples.
Table 5. Frameshift Mutations
Sample Exon DNA change Protein New Disease
TU326 5 c.321 delT G109V fs X1 Yes carrierTU19 6 c.401_404 delCCAA N135V fs X5 Yes DMD3451-3453 7 c.593_594 insA H198Q fs X19 Yes DMDTU65 8 c.713_714 delTT L239A fs X7 Yes DMDTU55 11 c.1188 insT G397W fs X1 Yes DMDTU150 11 c.1181del G G394A fs X12 Yes DMDTU16 11 c.1300_1310 delCTCAGGGTAGC L434X Yes DMDTU07 12 c.1482 delG K494K fs 7 Yes DMDTU03 14 c.delGTA 1603insCT V535L fs X47 Yes DMDTU386 16 c.1859 delT L620R fs X12 Yes DMDTU23 22 c.2880 2884 delCAAAC K961L fs X5 Yes DMDTU267 22 c.2887 del T S963P fs X40 Yes DMDTU137 25 c.3285 3288 delCAGT S1096_D1097I fs X9 Yes DMDTU115 25 c.3420 del C H1140Q fs X13 Yes DMDTU27 26 c.3447 delGGlnsTT K1149N-E1150X No DMDTU177 26 c.3464 3471 del GTTGGAG G1155E fs X20 No DMDTU44 30 c.4100 delA Q1367R fs X15 Yes DMDTU103 30 c.4119 delG E1374R fs X8 Yes DMDG7 30 c.4186 insA Y1396X fs Yes DMDTU29-G10 33 c.4565delT (Stop TAA) V1522G fs X2 No DMDTU08-R49 35 c.4871_4872 delAG K1625G fs X27 Yes DMDTU13 36 c.5091 delG A1698L fs X22 Yes DMDR44 37 c.5272 _5280 del TCAGAGCTC ins CCAA S1758P fs X13 Yes DMDTU304 40 c.5606 del G R1869K fs X4 Yes DMDTU06 40 c.5697 dup A L1900I fs X5 No DMD3488 42 c.5973_5974 ins A E1992R fs X11 Yes DMDTU211 44 c.6353 delA Q2118R fs X3 Yes DMDTU04 48 c.6980del A K2329S fs X8 No DMDTU57 55 c.8081 del G F2694S fs X31 Yes DMDR37 56 c.8284 ins A I2762N fs X 10 Yes DMDTU33 58 c.8597 8598deiTT L2866R fs X28 Yes DMDTU62 59 c.8732 insA N2912Q fs X2 No DMDTU151 62 c.9204_9207 del CAAA N3068K fs X20 No DMDTU179-G1 65 c.9429_9430 del GC Q3143H fs X9 Yes DMDTU192-TU193 68 c.9926_9929 ins AAGC H3309Q fs X7 Yes BMD/DMDG12 70 c.10105 del G V3369F fs X8 Yes DMDTU214 73 c.10386 del T N3462K fs X3 Yes BMD/DMD
Table 6. Putative Splicing Defects
Sample Position DNA change Splice site New Disease
TU22-TU77 Intron 2 c.94�1 G�A Acceptor No BMDTU34 Intron 5 c.358�2 A�G Acceptor No DMDTU296 Intron 5 c.358�2 A�T Acceptor No DMDTU219 Intron 6 c.530�1 G�A Donor Yes DMD/BMDTU309 Intron 11 c.1331�2 T�C Donor Yes DMD/BMD2082 Intron 11 c.1332�9 A�G Acceptor No DMDTU124 Intron 26 c.3433�1 G�A Acceptor No DMDTU164 Exon 26 c.3603 G�A Donor Yes DMDTU105 Intron 35 c.5026�6 A�G Acceptor No DMDTU332 Intron 48 c.7098�1 G�A Donor No DMDTU379 Exon 58 c.8668 G�A Donor No DMDTU114 Intron 58 c.8668�1 G�A Donor Yes DMDTU209 Intron 58 c.8668�3 A�T Donor Yes DMD/BMDTU133 Exon 65 c.9560 A�G Donor No DMDTU54-1707 Intron 65 c.9563�1 G�A Donor No DMDTU36-TU97 Intron 70 c.10223�1 G�A Donor No DMDTU30 Intron 70 c.10223�5 G�T Donor Yes DMD
70 Torella et alJMD January 2010, Vol. 12, No. 1
The presence of a variation within a fragment appearsas altered chromatogram shapes of the two differentpools sharing the same DNA. This type of scanningunequivocally points out the patient and the fragmentfor sequence analysis (Figure 3). This approach re-duced the turnaround time and was more cost-effective.
Sequence Analysis
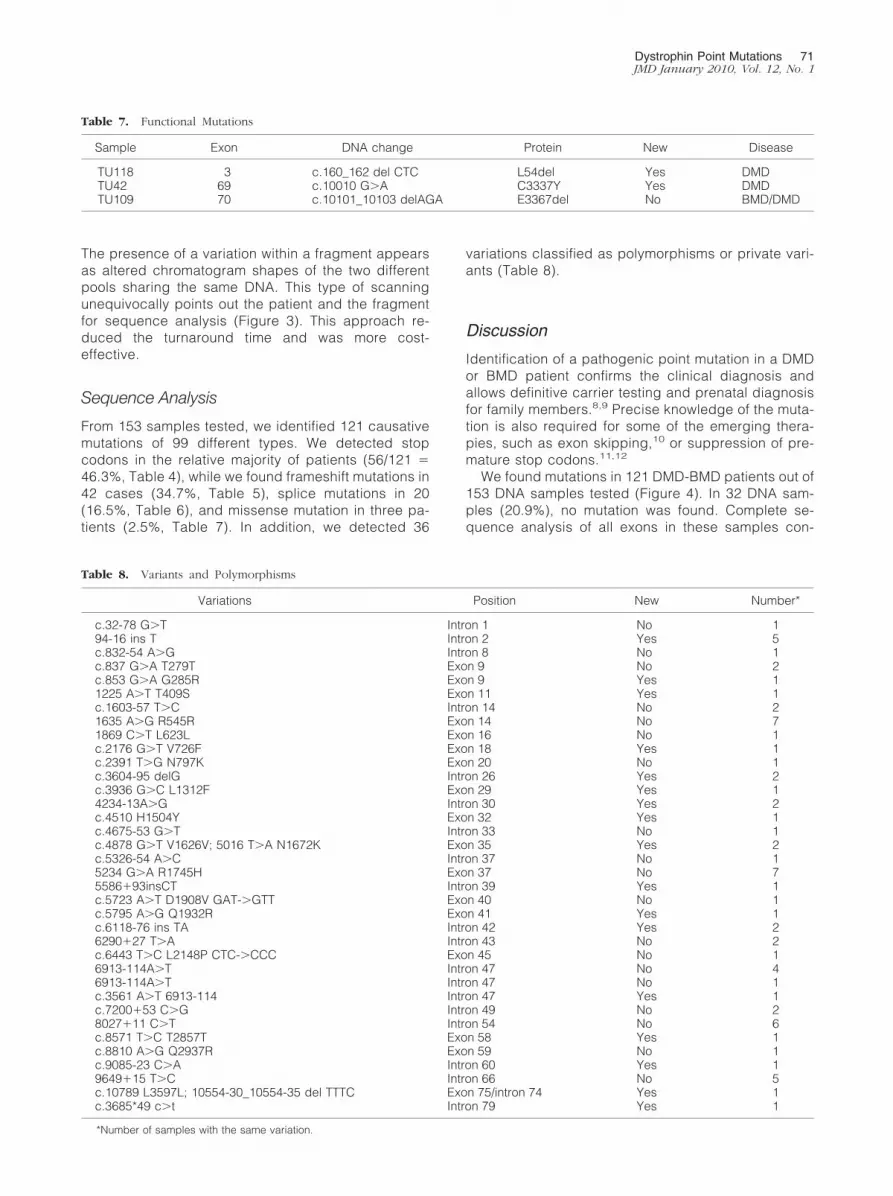
From 153 samples tested, we identified 121 causativemutations of 99 different types. We detected stopcodons in the relative majority of patients (56/121 �46.3%, Table 4), while we found frameshift mutations in42 cases (34.7%, Table 5), splice mutations in 20(16.5%, Table 6), and missense mutation in three pa-tients (2.5%, Table 7). In addition, we detected 36
variations classified as polymorphisms or private vari-ants (Table 8).
Discussion
Identification of a pathogenic point mutation in a DMDor BMD patient confirms the clinical diagnosis andallows definitive carrier testing and prenatal diagnosisfor family members.8,9 Precise knowledge of the muta-tion is also required for some of the emerging thera-pies, such as exon skipping,10 or suppression of pre-mature stop codons.11,12
We found mutations in 121 DMD-BMD patients out of153 DNA samples tested (Figure 4). In 32 DNA sam-ples (20.9%), no mutation was found. Complete se-quence analysis of all exons in these samples con-
Table 7. Functional Mutations
Sample Exon DNA change Protein New Disease
TU118 3 c.160_162 del CTC L54del Yes DMDTU42 69 c.10010 G�A C3337Y Yes DMDTU109 70 c.10101_10103 delAGA E3367del No BMD/DMD
Table 8. Variants and Polymorphisms
Variations Position New Number*
c.32-78 G�T Intron 1 No 194-16 ins T Intron 2 Yes 5c.832-54 A�G Intron 8 No 1c.837 G�A T279T Exon 9 No 2c.853 G�A G285R Exon 9 Yes 11225 A�T T409S Exon 11 Yes 1c.1603-57 T�C Intron 14 No 21635 A�G R545R Exon 14 No 71869 C�T L623L Exon 16 No 1c.2176 G�T V726F Exon 18 Yes 1c.2391 T�G N797K Exon 20 No 1c.3604-95 delG Intron 26 Yes 2c.3936 G�C L1312F Exon 29 Yes 14234-13A�G Intron 30 Yes 2c.4510 H1504Y Exon 32 Yes 1c.4675-53 G�T Intron 33 No 1c.4878 G�T V1626V; 5016 T�A N1672K Exon 35 Yes 2c.5326-54 A�C Intron 37 No 15234 G�A R1745H Exon 37 No 75586�93insCT Intron 39 Yes 1c.5723 A�T D1908V GAT-�GTT Exon 40 No 1c.5795 A�G Q1932R Exon 41 Yes 1c.6118-76 ins TA Intron 42 Yes 26290�27 T�A Intron 43 No 2c.6443 T�C L2148P CTC-�CCC Exon 45 No 16913-114A�T Intron 47 No 46913-114A�T Intron 47 No 1c.3561 A�T 6913-114 Intron 47 Yes 1c.7200�53 C�G Intron 49 No 28027�11 C�T Intron 54 No 6c.8571 T�C T2857T Exon 58 Yes 1c.8810 A�G Q2937R Exon 59 No 1c.9085-23 C�A Intron 60 Yes 19649�15 T�C Intron 66 No 5c.10789 L3597L; 10554-30_10554-35 del TTTC Exon 75/intron 74 Yes 1c.3685*49 c�t Intron 79 Yes 1
*Number of samples with the same variation.
Dystrophin Point Mutations 71JMD January 2010, Vol. 12, No. 1

firmed the DHPLC negative results. Considering that153 DNA samples correspond to 20% of all patientsthat show no deletions or duplications, about 20% of20% of patients cannot be diagnosed by DNA analysisalone. This indicates that mRNA13 or CGH array13,14
analyses can be necessary to diagnose about 4% of allDMD/BMD patients.
Notably, 22 unrelated patients shared the same mu-tations (Tables 4 – 6). Among the 56 nonsense muta-tions, 34 (60.7%) show the TGA termination codon thatis considered optimal for readthrough therapy.11 Nota-bly, among the 37 different frameshift mutations, 31(83.8%) are absent from the Leiden database.2
We identified three putative functional mutations (Table7). One is the substitution of a highly conserved cysteineat position 3337 within the second half of the dystrogly-can-binding domain. A similar mutation (C3340Y) hasbeen associated with Duchenne muscular dystrophy.15
There is the loss of aspartic acid in position 3368 withthe substitution of the glutamic acid in position 3367with aspartic acid. This produces the loss of glutamicacid in position 3367 that is known to be associatedwith a particular DMD phenotype.16
The first half of the C terminus and the cysteine-rich(D-domain; amino acid residues 3080 –3408) are highlyconserved regions of dystrophin. The region is in-volved in interactions with dystroglycan that mediatesattachment of dystrophin to the cytoplasmic surface of
the cell membrane. Deletions or chain-terminating non-sense mutations involving the D-domain usually resultin DMD.
The third is the c.160_162 CTC deletion in exon 3 inDMD, which resulted in the loss of an evolutionaryconserved leucine in position 54 in the actin-bindingdomain. This is the same amino acid position replacedby an arginine described in a boy with Duchennemuscular dystrophy.17 Interestingly, this missense mu-tation in position 54 was questioned because it was notcompletely studied by DNA sequencing. After 16years, our findings support the causative role of thechange.
Two unusual mutations were also identified. Prema-ture stop codons in positions 1281 and 1314 associ-ated with BMD and not DMD. This could be explainedby the skipping of exon 28 or 29, respectively.18
Our DNA-based mutation screening strategy is suitablefor high-throughput applications in patients for which mRNAis unavailable. Sample pooling, together with identical PCRconditions for all fragments, were set up for the simulta-neous detection of any mutations type within exons andexon flanking regions. The preliminary analysis of singleand pooled samples by STR markers permitted us to con-firm the same PCR efficiency in all combinatorial pools. Inthe protocol, the pooling was conservative, since we onlymixed three samples. With the availability of more sensitivemethods of DNA detection (ie, multicolor fluorescence) we
Figure 4. Distribution of all causative point mutations along the dystrophin cDNA. Segments corresponding to groups of 10 exons are indicated in dark and lightgray.
72 Torella et alJMD January 2010, Vol. 12, No. 1

can foresee possibilities to pool dozens of samples withfurther impressive reduction of costs.
Acknowledgments
We thank Marina Fanin, Giuliana Galluzzi, Enzo Ricci,Federico Zara, Claudio Bruno, Sara Scapolan, MariaTeresa Bassi, and Mayana Zatz for DNA samples andAlessandra Ferlini for helpful discussion. We acknowl-edge the SUN-Naples Human Mutation Gene Bank(Cardiomyology and Medical Genetics), which is part-ner of the Eurobiobank Network.
References
1. Emery AE: The muscular dystrophies. Lancet 2002, 359:687–6952. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den
Dunnen JT: Entries in the Leiden Duchenne muscular dystrophymutation database: an overview of mutation types and paradoxicalcases that confirm the reading-frame rule. Muscle Nerve 2006,34:135–144
3. Schwartz M, Duno M: Improved molecular diagnosis of dystrophingene mutations using the multiplex ligation-dependent probe ampli-fication method. Genet Test 2004, 8:361–367
4. Trimarco A, Torella A, Piluso G, Maria Ventriglia V, Politano L, Nigro V:Log-PCR: a new tool for immediate and cost-effective diagnosis of upto 85% of dystrophin gene mutations. Clin Chem 2008, 54:973–981
5. Deburgrave N, Daoud F, Llense S, Barbot JC, Recan D, Peccate C,Burghes AH, Beroud C, Garcia L, Kaplan JC, Chelly J, Leturcq F:Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with non-sense and frameshift mutations in the DMD gene. Hum Mutat 2007,28:183–195
6. Belsito A, Politano L, Piluso G, Comi LI, Nigro V: Dystrophin genescanning by DHPLC of DMD carriers without deletions or duplica-tions. Acta Myol 1999, 3:221–223
7. Bennett RR, den Dunnen J, O’Brien KF, Darras BT, Kunkel LM:Detection of mutations in the dystrophin gene via automated DHPLCscreening and direct sequencing. BMC Genet 2001, 2:17
8. Nigro V, Politano L, Nigro G, Romano SC, Molinari AM, Puca GA:
Detection of a nonsense mutation in the dystrophin gene by multipleSSCP. Hum Mol Genet 1992, 1:517–520
9. Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR,Weiss RB: Rapid direct sequence analysis of the dystrophin gene.Am J Hum Genet 2003, 72:931–939
10. van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ,Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ,Verschuuren JJ, van Ommen GJ: Local dystrophin restoration with an-tisense oligonucleotide PRO051. N Engl J Med 2007, 357:2677–2686
11. Aurino S, Nigro V: Readthrough strategies for stop codons in Duch-enne muscular dystrophy. Acta Myol 2006, 25:5–12
12. Wilton S: PTC124, nonsense mutations and Duchenne muscular dys-trophy. Neuromuscul Disord 2007, 17:719–720
13. Bovolenta M, Neri M, Fini S, Fabris M, Trabanelli C, Venturoli A,Martoni E, Bassi E, Spitali P, Brioschi S, Falzarano MS, Rimessi P,Ciccone R, Ashton E, McCauley J, Yau S, Abbs S, Muntoni F, MerliniL, Gualandi F, Ferlini A: A novel custom high density-comparativegenomic hybridization array detects common rearrangements as wellas deep intronic mutations in dystrophinopathies. BMC Genomics2008, 9:572
14. del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, PhamHT, Wiszniewska J, Chinault AC, Beaudet AL, Eng CM: A novelcustom high density-comparative genomic hybridization array de-tects common rearrangements as well as deep intronic mutations indystrophinopathies. Hum Mutat 2008, 9:1100–1107
15. Lenk U, Oexle K, Voit T, Ancker U, Hellner KA, Speer A, Hubner C: Acysteine 3340 substitution in the dystroglycan-binding domain ofdystrophin associated with Duchenne muscular dystrophy, mentalretardation and absence of the ERG b-wave. Hum Mol Gen 1996,5:973–975
16. Becker K, Robb SA, Hatton Z, Yau SC, Abbs S, Roberts RG: Loss ofa single amino acid from dystrophin resulting in Duchenne musculardystrophy with retention of dystrophin protein. Hum Mutat 2003,21:651
17. Prior TW, Papp AC, Snyder PJ, Burghes AH, Bartolo C, Sedra MS,Western LM, Mendell JR: A missense mutation in the dystrophingene in a Duchenne muscular dystrophy patient. Nat Genet 1993,4:357–360
18. Ginjaar IB, Kneppers AL, v d Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC, Weegenaar J, den Dunnen JT, Bakker E:Dystrophin nonsense mutation induces different levels of exon 29skipping and leads to variable phenotypes within one BMD family.Eur J Hum Genet 2000, 8:793–796
Dystrophin Point Mutations 73JMD January 2010, Vol. 12, No. 1