octahedral cobalt complexes in dipolar aprotic solvents. viii. the proton magnetic resonance spectra...
TRANSCRIPT

OCTAHEDRAL COBALT COMPLEXES IN DIPOLAR APROTIC SOLVENTS
VIIL* THE PROTON MAGNETIC RESOKANCE SPECTRA O F SOME OCTAHEDRAL
cis- AND trans-BISETHYLE~'EDIAMINECOBALT(III) ISOMERS IN NX-DINIETHYL-
FORMAMIDE, ,IN-DIMETHYLACETaMIDE, AND DIMETHYL SULPHOXIDE
By I . R. LANTZEE? and D. W. WATTS?
[Manuscript received August 8 , 19661
The proton magnetic resonance spectra of a series of octahedral bisethylene- diaminecobalt(~~~) complexes in the dipolar aprotic solvents NN-dimethylformamide, NN-dimethylacetamide, and dimethyl sulphoxide have been examined. Resonances due to the protons attached to the nitrogen atoms of the ethylenediamine ligands are easily observed in these solvents, and their chemical shifts are reported, together with some similar data determined in deuterium oxide, and 95% sulphuric acid.
The pattern of the nitrogen proton resonances is dependent on the stereo- chemistry of the complex. Using these differences in resonance, the cis configuration of some recently synthesized solvent complexes has been contirmed.
The observed resonances are assigned to the particular protons in most cases.
We wanted to confirm the stereochemistry of some labile reaction products1 which were difficult to isolate, and it appeared likely that n.m.r. spectroscopy would distinguish cis and t rans isomers in the solvents used. Clifton and Pratt2 have shown that in determinations of the proton magnetic resonance spectra of ammine and ethylenediaminecobalt(~~~) complexes in deuterium oxide, the nitrogen protons show chemical shifts dependent on the geometry of the complex. Protons attached to a nitrogen atom t rans to an acido or chloro ligand in either a pentaammine or bisethylenediaminecobalt(n~) complex resonated a t higher field strengths than the protons bonded to a nitrogen atom cis to these ligands.
The cis-bisethylenediaminecobalt(~n) complexes reported2 had two peaks attri- butable to nitrogen protons, while the t rans isomer showed one peak due to nitrogen protons. The absorption of the t rans isomer was at a field strength in between those of the two absorptions of the isomeric cis complex. The ethylenediamine complexes also showed resonances for the carbon protons.
Similar geometry dependent shifts of the nitrogen proton resonances were observed3 with pentaammine cobalt(m) complexes using 97% sulphuric acid as the solvent. Sulphuric acid offered the advantage that it did not absorb in the region
* Part VII, Aust. J. Chem., 1966, 19, 1411. t School of Chemistry, University of Western Australia, Nedlands, W.A.
1 Lantzke, I. R., and Watts, D. W., unpublished data. a Clifton, P., and Pratt, L., Proc. chem. Soc., 1963, 339.
Jolly, W. L., Harris, A. D., and Briggs, T. S., Inorg. Chem., 1965, 4, 1064.
Aust. J . Chem., 1967, 20, 35-52

36 I. R. LANTZKE &D D. W. WATTS
of interest. In addition, exchange of the nitrogen protons with the solvent did not lead to disappearance of the NH resonances (as it does with D,O), and many com- plexes are much more soluble in sulphuric acid than in water.
The resonances of the carbon protons of a number of eth~lenediamine,~ and substituted eth~lenediamine~.~ complexes have been discussed in relation to the structure and configuration of a variety of chelate rings.
We have been studying reactions of octahedral bisethylenediamine complexes in the dipolar aprotic solvents NN-dimethylformamide (DMF),7 NN-dimethylacet- a i d e (DMA),s and dimethyl sulphoxide (DMS0).9 In particular, we have been inteerested in the solvent containing species chlorodimethylformamidebisethylene- diaminecobalt(n~) ion, [CoCl(DMF) en,I2+, chlorodimethylacetamidebisethylenedi- aminecobalt(111) ion, [CoCl(DMA) en,12+, and chloro(dimethy1 su1phoxide)bisethylene- diaminecobalt(~~~) ion [CoCl(DMSO) en,12+, and their solvolytic intercon~ersion.~ From a consideration of their infrared and visible spectra these complexes have been tentatively assigned a cis configuration although as yet they have not been resolved. Their p.m.r. spectra are of the type expected for cis isomers and so the assignment of a cis structure to these complexes is now confirmed.
We have also examined some of these complexes in deuterium oxide and 95% sulphuric acid. Because of the reduced complexity of solvent resonances, deuterium oxide offers some advantage in the determination of the structure of soluble com- plexes which do not aquate rapidly in that the better resolved carbon proton reson- ances can be used. Although solvent resonances in 05% sulphuric acid do not interfere, the complexes investigated react with the solvent and thus the results are of no use in the determination of structure.
Deutereted (d,) dimethyl sulphoxide and other deuterated dipolar aprotic solvents would have considerable advantages in that solvent resonances would not complicate the interpretation.
(a) Compounds
Most of the compounds used in this work are well known. The preparation, purification, and analysis of complexes previously used are as reported in the references recorded in the tabulated results (Tables 1-3).
The preparation and analytical results of new compounds are as follows:
cis-Chloroaquobisethylenediaminecobalt(~~~) nitrate percblorate, cis-[CoCl(OH,) en,]NO,ClO,, was prepared as the bromide monohydrate by Werner's10 method and converted into the nitrate perchlorate by dissolving 3 g of this compound in 20 ml of cold water. The solution was filtered end a large amount of solid lithium perchlorate added to the filtrate with vigorous stirring. The vessel was cooled in an ice-bath. As soon as the solution was saturated with perchlorate ion
Powell, D. B., and Sheppard, N., J. chem. Soc., 1959, 791. Day, R. J., and Reilley, C. N., Analyt. Chem., 1964, 36, 1073. Legg, J. I., and Cooke, D. W., Inorg. Chem., 1965, 4, 1576. Lantzke, I. R., and Watts, D. W., Aust. J. Chem., 1966, 19, 949. Fitzgerald, W. R., and Watts, D. W., Aust. J. Chem., 1966, 19, 935. Chin, L. F., Millen, W. A,, and Watts, D. W., Aust. J. Chem., 1965, 18, 453.
lo Werner, A., Liebigs Ann., 1912, 386, 1.

OCTAHEDRAL COBALT COMPLEXES. VIII 37
a large excess of lithium nitrate was added. On stirring and scratching the walls of the beaker, the nitrate perchlorate salt was precipitated. This was recrystallized by dissolving in a minimum volume of water, filtering into a beaker containing solid lithium perchlorate, cooling the mixture in ice, and adding a few drops of concentrated nitric acid. The cis-[CoCl(H,O) en,]N03C10, crystallized out on scratching the walls of the beaker (Found: coordinated C1, 9.0. Cdc. for cis-[CoCl(H,O) en,]N03C10,: coordinated C1, 9.0%).
cis- and trans-Chloronitrobisethylenediaminecobalt(~~~) perohlorates, cis-[CoCl(NO,) en,]- C10, and trans-[CoCl(KO,) en,]ClO,, were obtained as the chloride and nitrate salts respectively, by the methods of Werner,lo and recrystallized as the perchlorates from water (Found: Co, 16.3. Calo. for cis-[CoCl(NO,) en,]ClO,: Co, 16.4%. Found: Co, 16.4. Calc, for trans-[CoCl(NO,) en,]- C10,: Co, 16.4%).
cis- and trans-Dinitrobisethylenediaminecobalt(~~~) perchlorates, (cis-[Co(NO,), en,]C10, and trans.[Co(NO,), en,]C10,) were obtained as the nitrite and nitrate salts respectively by the methods of Werner1° and recrystallized as the perchlorates (Found: C, 13.3; H, 4.7; N, 22.7. Calc. for cis-[Co(SO,),en,]ClO,: C, 13.0; H, 4.4; N, 22.7%. Found: C, 14.4; H, 4.9; Tu', 29.2. Calc. for trans-[Co(NO,), en,]N03: C, 14.4; H, 4.8; N, 29.4%).
cis- and trans-Diazidobisethylenediaminecobalt(~~~) perchlorates (cis-[Co(N,), en,]C10, and trans-[Co(S,), en,]ClO,) were prepared as the nitrate and azide saltsll and recrystallized as the perchlorates (Found: C, 15.0; H, 5.0; N, 47.4. Calo. for cis-[Co(N,), en,]NO,: C, 14.8; H, 6.0; N, 47.4%. Found: C, 13.6; H, 4.6. Calc. for trans-[Co(N,),en2]C10,: C, 13.3; H, 4.5%).
trans-Dithiocyanatobisethylenediaminecobalt(~~~) thiocyanate, trans-[Co(NCS), en,]SCN, was prepared by evaporating to a small volume a saturated solution of trans-[CoCl, en,]Cl and excess potassium thiocyanate. The red crystals obtained on cooling were washed with ethanol and ether and dried at 1 mm pressure for 24 hr (Found: C, 24.1; H, 4.8; N, 27.7; S, 27.2. Calc. for trans-[Co(NCS), en,]SCN: C, 23.8; H, 4.5; N, 27.7; S, 27.2%).
cis-Bisdimethylformamidebisethylenediaminecobalt(~~~) perchlorate (cis-[Co(DMF), en,]- (ClO,),) was prepared by adding two moles of silver perchlorate to each mole of trans-[CoCl, en,]- ClO,, dissolved in a minimum volume of DXF, warming at 60' for 20 min and filtering off the silver chloride. The complex was obtained as an oil by the addition of 4-5 volumes of ethanol and then a large excess of ether to the filtrate. The supernatant liquid was decanted and the oil crystallized from water, using lithium perchlorate to salt out the complex.
The anhydrous compound cis-[Co(DMF), en,](ClO,), was obtained on drying for 24 hr a t 110' (Found: Co, 9.4. Calc. for cis-[Co(DMF), en,](C104),: Co, 9.5%).
cis-Bis(dimethy1 sulphoxide)bisethylenediaminecobalt(~~~) perchlorate (cis-[Co(DMSO), en,]- (ClO,),) was prepared in a similar manner to that of the cis-bisdimethylformamidebisethylene- diaminecobalt(~~~) perchlorate, except that dimethyl sulphoxide was used as the solvent. This compound is explosive on heating or exposure to concentrated acid and should be handled with care andin small amounts (Found: Co, 9.2. Calc. for cis-[Co(DMSO), en,](ClO,),: Co, 9.3%).
Trisethylenediaminecobalt(~~~) chloride perohlorate monohydrate ([Co en,]C1(C1O4),,H,O) and trisethylenediaminecobalt(111) perchlorate ([Co en,](ClO,),) were prepared from trisethylene- diaminecobalt(111) chloride obtained by the method of Work.la The chloride perohlorate salt was obtained by adding lithium perchlorate to a saturated aqueous solution of the chloride, cooling, and scratching the walls of the container. The yellow crystalline solid was filtered, washed with ethanol and ether, and dried a t 0.1 mm pressure for 36 hr (Found: C1-, 7.5. Calo. for [Co en3]C1(C104),,H,0 : C1-, 7.5%).
The perchlorate salt was prepared in solution by treating a saturated solution of the chloride perchlorate, in the appropriate solvent, with an equimolar quantity of silver perchlorate. The precipitated silver chloride was removed by atration.
l1 Staples, P. J., and Tobe, M. L., J. chem. Soc., 1960, 4813. l2 Work, J. B., Inorg. Synth., 1946, 2, 221.

38 I. R. LaNTZKE AND D. W. WATTS
The purity of compounds was also confirmed by comparison of their visible and u.v. spectra with the published spectra (where the compounds had been prepared previously). This was particularly necessary for the nitro and azido complexes, with which combustion analysis was difficult because of the violence of their pyrolysis.
Perchlorate salts of the complexes were used in the dipolar aprotic solvents (except where noted in the tables), because of their greater solubility, and because it has been shown that perchlorates do not readily form ion pairs.ls
( b ) Solutions
(i) Solvents
Deuterium oxide supplied by the Australian Institute of Nuclear Science and Engineering (99.75% purity) was used diluted to 95% deuterium oxide.
Sulphuric acid (95%; Cuming Smith & Mount Lye11 Analytical Reagent) was used without further purification.
NN-Dimethylformamide, NN-dimethylacetamide, and dimethyl sulphoxide were purified by the methods described before.l8
(ii) Preparation of Solutions
All solutions were prepared immediately before measuring the spectrum. The solutions of the solid complexes were prepared by weighing a known amount of the solid into a vessel, and adding a known small amount (0. 10%) of the internal reference (see below) and then a known volume (usually 1 ml) of the appropriate solvent. As no allowance has been made for volume changes on mixing, the molar concentrations given are accurate to only c. 3%. Solutions of liquid solutes were prepared by volume to volume dilution.
To simplify comparisons most solutions were prepared at about 0. liv~ concentration (the approximate concentration of a saturated solution of several of the less soluble solutes).
(iii) Nuclear Magnetic Resonance
The nuclear magnetic resonance spectra were recorded on a Varian A60 spectrometer operating at 60 Mc/s. The temperature of the probe was c. 35'. Spectra were recorded at a 250-sec sweep rate and generally with an amplification of 16.
Chemical shifts were measured relative to sodium 3-(trimethylsily1)-1-propanesulphonate (NaTMS) as internal standard. The chemical shifts are expressed in terms of 6 (p.p.m.). 911 the shifts measured were on the low-field side of NaTMS. The areas under the curves were determined by the resonance integrator, but because of the broadness of the peaks it was frequently impossible to obtain a linear base line, so that the assessment of areas was of low accuracy. Because of the absence of an internal standard the calculation of the number of protons was relative and based on the knowledge of the total number of nitrogen protons and the total integrated area of proton resonance. I n the particular cases of complexes containing dimethylformamide the aldehyde proton of the dimethylformamide was used as an additional standard. I n some cases less than eight nitrogen proton resonances were observed in DMF and DMSO as solvent. In these cases the high-field resonances were obscured by solvent spinning sidebands; but they were always observed in DMA as solvent.
A check on the correctness of the assessments of area was provided by some experiments to determine the concentration of trans-[CoCl, en,]+ in synthetic mixtures of cis- and trans- [CoCl, en,]+, in DMSO as solvent. Provided more than 5% of trans isomer were present in the mixture, machine integrals of the area under the low-field resonance of the cis isomer and the resonance of the trans isomer could be separated. Calculations of % trans in the mixture using these areas and assuming two protons for the cis resonance and eight protons for the trans resonance gave results within &2% of the actual composition.
la Millen, W. A,, and Watts, D. W., Aust. J. Chem., 1966, 19, 43.

OCTAHEDRAL COBALT CONPLEXES. VIII
Dipolar aprotic solvents have proved to be suitable for the observation of the resonances of protons attached to coordinated nitrogen. The reactions of complexes in these solvents a t the temperature of the probe (35") are in general sufficiently slow for the change occurring during the preparation and recording of a spectrum (about 10 min) to be insignificant. In addition, the resonances of the nitrogen protons do not fade as happens in deuterium oxide where exchange occurs.
We have been able to distinguish cis and trans isomers of complex ions of the general formula [CoXY en21n+, where X and Y represent monodentate ligands, and en bidentate ethylenediamine. Because chemical shifts are dependent on the molecu- lar and electronic structure of the species, we have also noted some qualitative relationships between the influence of the ligands X and Y upon the nitrogen protons in cis- and trans-bisethylenediamine complexes.
In all solvents the peaks due to the nitrogen protons were broad, and no fine structure could be observed. In DMA as solvent the low-field signal of some cis isomers showed a shoulder, which may be due to unresolved fine structure. In general the peaks obtained with cis isomers were broader than those obtained with trans isomers. Thus, while the resonance of an approximately 0 . 1 ~ solution of trans-[CoCl, en,]+ ion had a width a t half-height ( W g ) of 14 c/s, W t for each of the resonances of the cis isomer under similar conditions was 19 c/s.
The broadness of these lines appears to arise from two causes: firstly, the general broadening observed with nitrogen protons, and ascribed to the quadrupole relaxation of the nitrogen-14 nucleu$* and secondly to coupling between the NH, and CH, protons. Broadening of resonances due to the nitrogen-14 quadrupole relaxation should be greater for the dipolar cis complexes than for the trans complexes because of the greater field gradient at the nitrogen atoms in the former, and the resultant increase in the nitrogen-14 quadrupole coupling energy and decrease in relaxation time. NH2,CH2 coupling was observed in the p.m.r. spectra of a number of trisethylenediamine complexes.* It must be expected to give greater line broaden- ing for the cis isomers where the CH, and NH, proton signals are already split by the asymmetric shielding.
Because of the complexity of the molecules considered here, it is not practicable at this stage to attempt a detailed analysis of the various electronic contributions to the shielding of the nitrogen protons. However, from consideration of symmetry and the nature of the Iigands it is possible to systematize much of the data obtained.
In general, it appears that the shielding of the nitrogen protons in any particular complex depends on the distance and the angle of the time-averaged position of each proton relative to the ligands X and Y and on the nature of the ligands X and Y.
That we are observing the time-averaged positions of the ethylenediamine rings in all compounds is indicated by the comparative simplicity of the spectra. This is also in agreement with other observations on chelate rings.*J6
l4 Pople, J. A,, Molec. Phys., 1958, 1, 168. l6 Gillard, R. D., and Irvine, R. ill., Chem. Rev., 1965, 65, 603.

40 I. R. LANTZKE AND D. W. WATTS
From models of octahedral trans-bisethylenediamine complexes, it can be seen that in the time-average position of the ethylenediamine molecules there are four nitrogen protons on each side of the plane of the ethylenediamine rings. In complexes of the type trans-[COX, en2ln+ all protons are equivalent, but in complexes of the type trans-[CoXY en2ln+ the protons on one side of the plane of the ethylene- diamine ring will experience a different field from those on the other side (Fig. 1).
Models of octahedral cis-bisethylenediamine complexes show that the sym- metry is greatly reduced, even if the time-averaged position of the protons is observed. I n complexes of the type cis-[COX, en2ln+, there are four pairs of equivalent nitrogen protons, but in complexes of the type cis-[CoXY en2ln+, although some protons are similarly orientated, no two protons are in identical situations.
Fig. 1.-Models of cis- and trans-bisethylenediaminecobalt(~~~) isomers.
It is possible that if the ligands X and Y differed greatly in size, inversion of the chelate ring would be hindered or even inhibited. In the case of trans complexes two separate proton resonances of equal area would be observed if inversion was inhibited, while with cis complexes a most complex pattern would result. As yet this has not been found.
I t is convenient to define two terms relating the time-averaged position of the nitrogen protons to the position of ligand X (or Y) by reference to the plane through the cobalt atom at right angles to the Co-X (or Y) bond. Nitrogen protons whose time-averaged position is on the same side of this plane as ligand X we shall describe as being "proximal" to ligand X. We will describe nitrogen protons whose time- averaged position is on the far side of the plane as being "distal" t o ligand X. The terms cis and trans will be used to describe the relationship between specific atoms or ligands, and for the overall geometry of the complex.
The results summarized in Tables 1-5 can be explained in terms of the direc- tional effect of the shielding of the ligands X and Y, the shielding of a ligand, say X, being negative (i.e. deshielding) for proximal nitrogen protons while distal nitrogen protons experience a positive shielding effect from ligand X.

OCTAHEDRAL COBALT COMPLEXES. VIII 4 1
The assignments given here have been arrived a t from a consideration of the symmetry of the various complexes, the area under each of the curves, and a com- parison with the spectra in the other solvents.
It has been impossible to use uncoordinated ethylenediamine as a standard for comparison of chemical shifts because the proton resonances in this molecule are obscured by solvent resonances. In each of Tables 2, 3, and 4 the resonances of the nitrogen protons in the complex ion [Co en3I3+ are shown for comparison.
For complexes of the type [COX, en2]+, where X is an anion, the nitrogen proton resonance of the trans form shows about the same chemical shift as the second
CHEMICAL SHIFTS OB THE NITROGEN PROTOXS OF SOME tram-[CoXY en,ln+ COMPLEXES IN S O X E DIPOLAR APROTIC SOLVENTS
The references in column 2 refer to the method of complex preparation (see Experimental)
NCS- Br- C1- NO,- NF C1- C1- C1- I -
" Very broad peak. concentrated aqueous acid.
Chloride nitrate salt.
In DMA
( P . P . ~ . )
I - b Solvent contained a small amount of Chloride perchlorate salt. Doublet.
peak of its cis isomer (when the spectrum is sufficiently resolved), so we consider that the protons, producing this resonance in the cis complexes must bear a similar relationship to both the ligands X as they do in the trans complex.
Only protons H4 and H2 (Figs. 1 and 2) satisfy this condition, there being only a 90" change between the angle of the C-N bond and the X-Co bond, the relation- ship between H 2, H 4, and the ligand X remaining otherwise the same. The increased deshielding of protons H 1 and H 3 in these cis complexes (producing the lowest field resonance) is due to the combined deshielding by both ligands X, rather than to increased shielding of protons H 4 and H 2 by the X distal to them.
The shielding effect of a ligand is greatest on protons bonded to the nitrogen atom trans to it. Thus where the resonances are sufficiently resolved to see all the proton signals (in DMA as solvent) the resonance a t highest field is due to protons HG and H 8 which are distal to both ligands, each being bonded to a nitrogen tram
Bailar, J. C., Inorg. Synth., 1946, 2, 222. l7 Lantzke, I. R., and Watts, D. W., unpublished data. 1s Lantzke, I. R., and Watts, D. W., Aust. J. Chem., 1967, 20, 173.
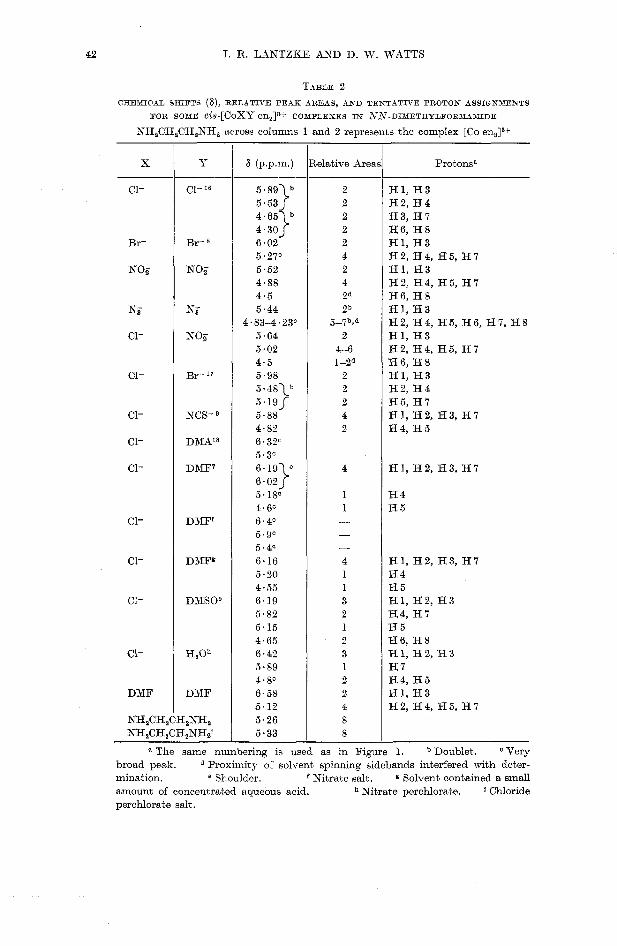
42 I. R. LANTZKE AKD D. W. WATTS
TABLE 2
CHEMICAL SHIFTS (a), RELATIVE PEAK AREAS, AND TENTATIVE PROTON ASSIGNMENTS
FOR SOME C~S-[CoXY en,]"+ CONPLEXES IN IVN-DIXETHYLFORMAXIDE
X
C1-
Br-
NO,-
N,
C1-
C1-
C1-
C1-
C1-
C1-
C1-
C1-
C1-
DMF
KH,CH,CH2NH2 NHaCH2CH,NH2'
a The same numbering is used as in Figure 1. Doublet. Very broad peak. Proximity of solvent spinning sidebands interfered with deter- mination. Shoulder. Nitrate salt. g Solvent contained a small amount of concentrated aqueous acid. Nitrate perchlorate. ' Chloride perohlorate salt.
NH,CH,CH,NH,
Y
Cl-
Br- a
NO,-
X$
NO;
Br-I?
NCS-
DMAls
DMF7
DXF'
DMFg
DAIS09
H,Oh
DMF
across columns 1
6 (p.p.m.) --
5. 8 9 I b 5 . 5 3 1
i: ::y 6.02 5 ~ 2 7 ~ 5.52 4.88 4.5 5.44
4.83-4.23' 5.64 5.02 4.5 5.98 5 . 4 8 y 5 . 1 9 1 5.88 4.82 6.32O 5.30
:::;-y 5.180 4.6' 6.4O 5.gc 5.40 6.16 5.20 4.55 6.19 5.82 5.15 4.65 6.42 5.89 4.8' 6.58 5.12 5.26 5.33
and 2 represents
Relative Areas
2 2
2 2 2 4 2 4 2d 2b
5-7b9d 2
4-6 1 -2d
2 2 2 4 2
4
1 1 -
- 4 1 1 3 2 1 2 3 1 2 2 4 8 8
the complex [Co en,l8+
Protonsa
H1, H 3 H2, H 4
H6, H 8 H3, H I
H l , H 3 H2, H4, HS, H 7 H l , H 3 H2, H4, H5, H I H6, H 8 H l , H 3 H 2, H 4, H 5, H 6, H 7, H 8 H1, H 3 H2, H4, H5, H 7 H 6 , H 8 H l , H 3 H2, H 4 H5, H 7 H1, H2, H3, H7 H4, H 5
H1, H2, H3, H 7
H 4 H 5
H1, H2, H3, H 7 H 4 8 5 H I , H2, H 3 H4, H I H 5 H6, H 8 H1, H 2 , H 3 H I H4, H 5 H l , H 3 H2, H4, H5, H I
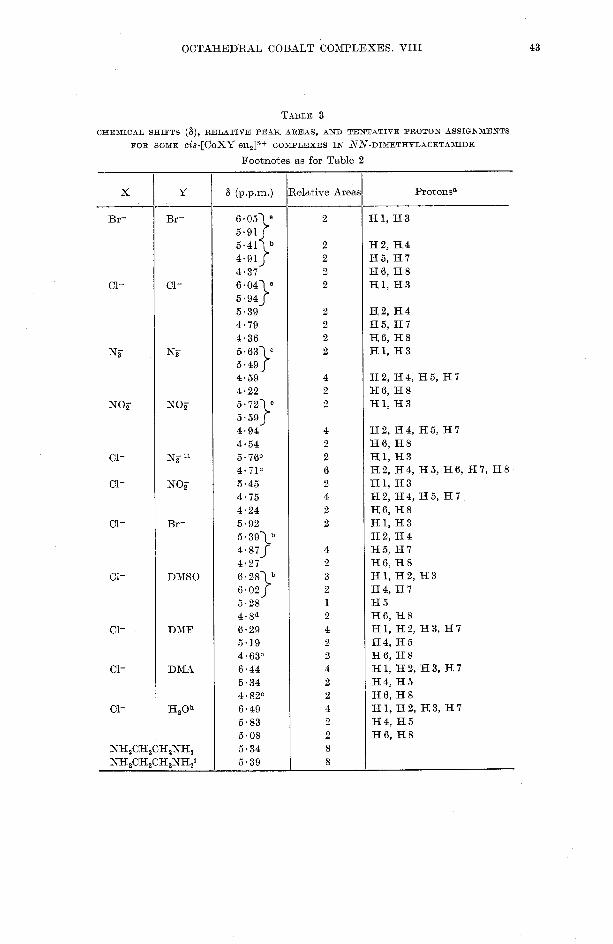
OCTAHEDRAL COBALT COXPLEXES. VI I I
Footnotes as for Table 2
elative Area, -- -
Hl, H3
H2, H4 H5, H I H6, H8 Hl , 1 - 1 8
H2, H4, H5, H7 H6, H8 H l , H3 H2, H4, H5, H6, H7, H8 H I , H3 H2, H4, H5, H7 H6, H8 H I , H3 H2, H4 H5, H7 H6, H8 H l , H2, H 3 H4, H7 H 5 H6, H8 H1, H2, H3, H7 H4, H5 H6, H8 H l , H2, H3, H7 H4, H5 H6, H8 H1, H2, H3, H I H4, H 5 H6, H8
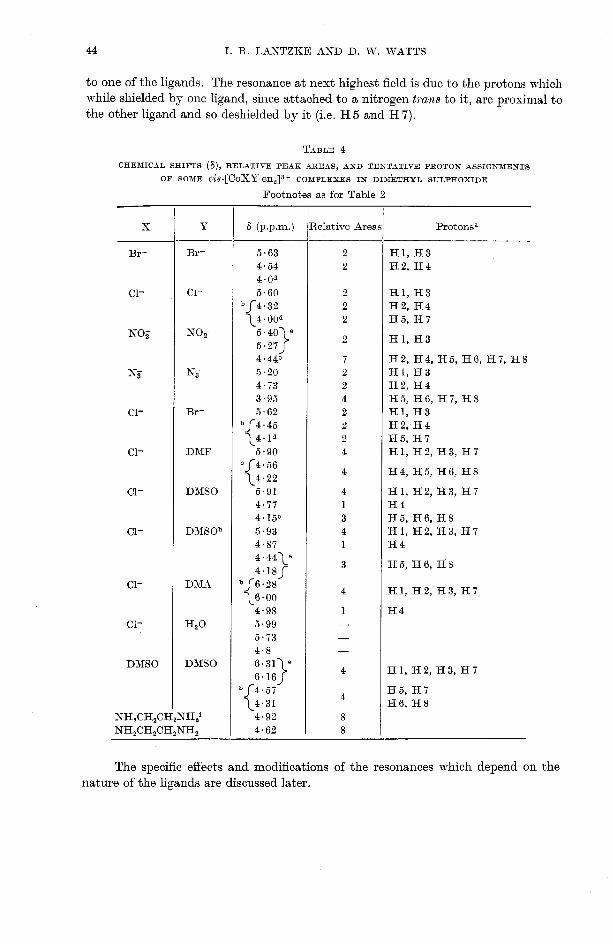
44 I. R. LAKTZKE AND D. W. WATTS
to one of the ligands. The resonance at next highest field is due to the protons which while shielded by one ligand, since attached to a nitrogen trans to it, are proximal to the other ligand and so deshielded by it (i.e. H 5 and H 7).
Footnotes as for Table 2
Br-
C1-
NO,-
N,-
Ci-
C1-
C1-
C1-
C1-
C1-
DMSO
Br-
C1-
NO,
KT
Br-
DbIF
DMSO
D1\ISOh
DMA
' 3 2 0
DMSO
lelative Aree
H l , H 3 H2, H4
H l , H3
H2, H4, H5, H6, HI , H8 H l , H3 H2, H4 H5, H6, H7, H8 H l , H3 H2, H4 H5, H7 H1, H2, H3, H I
H l , H2, H3, H I H4 H5, H6, H8 H I , H2, H3, H I H4
The specific effects and modifications of the resonances which depend on the nature of the ligands are discussed later.

OCTAHEDRAL COBALT COMPLEXES. VIII 45
I n the case of complexes containing neutral or poorly shielding anionic ligands the same principles apply, although because of the absence of electric charge on these ligands their deshielding of proximal protons is greater than that observed
TABLE 5
CHENICAL SHIETS (8) OF THE ETHYLENEDIAMINE PROTONS FOR SOME cis- AND
trans-[CoXY en,ln+ COMPLEXES IN DEUTERIUM OXIDE
Footnotes a-i as in Table 2
Complex Carbon Protons Nitrogen I I
I I I Protons I
trans trans cis cis cis cis cis cis cis
C1- Cl- C1- C1- C1- DMF
C1-'C. ' DMF DMSO DMA H,Ok DMF I -
j Data reported before. Ref. 2. " Chloride salt. Overlain by CH, resonances of coordinated solvent.
with anionic ligands. As a result in the cis-[CoCl(DMF) en2I2+ ion, if the DXF is considered as ligand X (Fig. 2) the protons H 1, H 3, H 4, and H 5 are all deshielded to about the same extent. Because of the broadness of the resonances it is not
Fig. 2.-Time-averaged positions of the nitrogen protons of one of the antipodes of cis-[CoXY en,]"+.
possible to observe small differences in shielding. The two other resonances observed in this spectrum in DMF as solvent are due to protons H 2 and H 7, although without a quantitative measure of the extent of deshielding by each ligand it is not possible to decide which gives rise to which resonance.

46 I. R. LANTZKE AND D. W. WATTS
The resonance due to the remaining pair of protons (H 6 and H 8) is obscured by solvent spinning sidebands in DMF. In DMA as solvent the resonances of all eight protons are observed, but the relative shielding of the protons is modified so that protons H 2 and H 7 resonate at essentially the same field (6 5.19 p.p.m.) and the distal protons H 6 and H 8 are observed at highest field (8 4.63 p.p.m.).
Similar considerations apply to the other mono solvent complexes, with nitrogen protons proximal to the solvent ligand most deshielded, and nitrogen protons distal to both ligands X and Y a t highest field. The extent of the shielding or deshielding of the nitrogen protons proximal to one ligand and distal to the other is determined by the nature of both ligands and the solvent.
(i) NN-Dimethylformamide as Solvent
The spectrum of NN-dimethylf~rmamide~~ shows a doublet centred at 6 2.90 p.p.m. due to the non-equivalent AT-CH, protons, and a single resonance a t 6 8.02 p.p.m. due to the aldehyde proton. At the spectrum amplification necessary (16) to show the relevant resonances of the various solutes investigated, the solvent reson- ances were flanked by strong spinning sidebands. In addition the high-field resonance of the 13GH aldehyde proton doublet was observed as a small broad peak c . 98 c/s upfield from the 12C aldehyde proton signal (J(13CH) 198 c/s).~O
The solvent resonances obscured CH, and CH, proton resonances, but the region from 6 4.2 to 7.1 p.p.m. was free from interference (the 13GH resonance was not significant), and thus the resonance of the nitrogen protons of the ethylenedi- amine ligands could be readily observed. The presence of water (IM, 6 3.4 p.p.m.), dimethyl sulphoxide, and dimethylacetamide did not interfere with observation of the region between 6 4.2 and 7 .1 p.p.m.
The distinguishable nitrogen proton resonances observed with the compounds investigated are recorded in Tables 1 and 2. The simplest results are those of the trans isomers, which with the exception of the trans-[CoCl(OH,) en,],+ ion, all show a single resonance.
The trans-[CoC1(H20) en2],+ ion was prepared in situ by the addition of acid to trans-hydroxochlorobisethylenediaminecobalt(~~~) chloride monohydrate, but because of the following complications the interpretation of its spectrum was rather uncertain.
(1) The use of concentrated aqueous acids introduced both water (and hydrogen ions) to the solution and these gave rise to several variable resonances.
(2) Stoicheiometric amounts of acid did not completely protonate the trans- [Co(OH)Cl en,]+ ion; a significant proportion of the hydrogen ions pro- tonated the solvent.
(3) Excess acid collapsed the broad resonance of the trans-[CoCl(H,O) en,]2+ nitrogen protons due to their rapid exchange.
(4) Perchlorate ion caused a rapid precipitation of trans-[CoCI, en2]C10,. Forma- tion of this compound is confirmed by the appearance of a resonance a t 6 5.43 p.p.m., and by isolation and analyses.
lg "N.M.R. Spectra Catalog." Vol. 1, No. 39. (Varian Associates: Palo Alto, Cal., 1962.) ao Lauterbur, P. C., J. chem. Phys., 1957, 26, 217.

OCTAHEDRAL COBALT COMPLEXES. VIII 47
(5) Nitrate ion ion-paired with the complex and the nitrogen proton resonances were shifted to lower fields.
The unresolved doublet observed with trans-[CoCl(H,O) en,I2+ ion probably results from the asymmetric shielding of the nitrogen protons, four of which are deshielded by the chloride ligand and four by the water ligand. The low field peak of the doublet is probably due to greater deshielding of the protons proximal to the H,O ligand, and the higher-field peak due to the nitrogen protons on the chloride side of the ethylenediamine rings.
The trans-[CoCl(NO,) en,]+ ion had a broader resonance (Wt 30 c/s) than either trans-[CoCl, en,]+ (W+ 14 01s) and trans-[Co(NO,), en2]+ (Wt 17 CIS), and could also be an unresolved doublet.
Although the resolution of spectra differed from one dipolar aprotic solvent to another, the overall form of the spectrum of each compound did not differ greatly. The spectrum of the cis-[Co(N,), en,]+ ion in DMF differed from its spectrum in DMA and DMSO. In the latter solvents there were three nitrogen proton resonances, in DMF only two; a low-field peak due to two protons and a broad peak (Wt 62 CIS), which because of solvent spinning sidebands could be integrated only as 6 A1 protons.
The solvent complexes showed greater variation. cis-[CoCl(DMF) en,]=+ ion showed three resonances due to 4, 1, and 1 protons (from low to high field), while the seventh and eighth protons were obscured by the solvent. cis-[CoCI(DMSO) en,],+ ion in DMF gave four nitrogen proton resonances; a doublet due to 3 and 2 proton resonance at lowest field, then a single proton resonance, and a 2 proton resonance at higher field. In DM80 the cis-[CoCl(DMSO) en2],+ ion showed three resonances, due to 4 protons at lowest field, then a single proton, and 3 protons (at still higher field). The cis-[CoCl(DMA) en,],+ ion gave only two resonances in DMF, and three in DMA. In this case the solvolysis of the complex by DMF was so fast that the nitrogen proton resonances were changing rapidly, and even within the 4 or 5 min required for mixing and scanning, a single proton resonance could have collapsed to such an extent that it was not detected. The greater dependence of spectra on the nature of the solvent for these dipositive ions is expected, since because of their greater charge these ions will be more strongly solvated by dipolar solvents.
Because it was necessary to add acid to the solvent to prepare trans- [CoCl(H,O) en,]2+, the same acidified solvent was used for one spectrum of the cis-[CoCl(DMF) en,],+ ion, as this material is the final product of solvolysisl of the trans-[CoCl(H,O) en,I2+ ion. In the acidified solvent the nitrogen proton resonances of the cis-[CoCl(DMF) en2I2+ ion were much sharper as shown by the following comparison of the width at half-height of the peaks, starting from the low-field side.
Anhydrous DMF (complex a t 0.27 mole 1.-l) 33, 28, 32 c/s. Acidified DMF (complex at 0.32 mole 1.-1) 25, 13, 18 c/s.
This results from the rapid chemical exchange of the nitrogen protons with those of the acid.21
Because we have been concerned with anhydrous dipolar aprotic solvents, we have not considered further the possiblity that a trace of acid would sharpen all the nitrogen proton resonances and so make the assessment of their position more precise.
Ogg, R. A., J . chem. Phys., 1954, 22, 560.

48 I. R. LANTZKE AND D. W. WATTS
As ion association has been shown to modify n.m.r. ~ p e c t r a , ~ ~ , ~ ~ it was of some interest to compare the spectra of solutions of cis-[CoCl(DMF) en2](C10,), and cis-[CoCl(DMF) en2](N03),. The perchlorate salt is unassociated in DMF while the nitrate is to some extent a~soc ia t ed .~~ The resonances of the nitrogen protons of the nitrate salt were a t lower field than those of the perchlorate, and considerably broadened. Both of these effects are to be expected for ion associated species. Because of the comparatively low solubility of the nitrate salt (saturated solution OSOVM), i t was not possible to derive any quantitative information. For the same reason i t was not possible to examine the chloride salt, the ion association constant of which is known.24
(ii) NN-Dimethylacetamide as Solvent
While the n.m.r. spectrum of NhT-dimethylacetamide consists of a number of resonances at 6<4 p.p.m., there are none at larger values of This low-field region was not obscured by small amounts of water, perckloric acid, or dimethyl sulphoxide. The presence of uncoordinated dimethylformarnide was shown by the resonance of its aldehyde proton at 6 8.16 p.p.m. The aldehyde proton of coordinated DMF resonates at higher field as is clearly shown in the spectrum of cis-[CoCl(DMF) en212+ (6 7.97 p.p.m.).
The resonances of the nitrogen protons of a number of bisethylenediamine- ooba1t(111) complexes in DNA as solvent are presented in Tables 1 and 3. In every case the general pattern was the same as that observed in NN-dimethylformamide, but in detail there were some interesting differences. In particular, all spectra were better resolved in DMA and the four anticipated proton resonances are resolved for the two complexes cis-[CoCl, en,]+ and cis-[CoBr, en2]+ (Fig. 3).
Among the trans complexes, the chloronitro complex showed a similar broad resonance to that observed in DMF, but this was a t lower field than that of either the dinitro or dichloro complexes. We cannot account for this variation.
The spectrum of the trans-chloroaquo complex proved as difficult to interpret in DMA as in DMF, and for the same reasons.
DMA proved to be the most satisfactory solvent for solvent-containing com- plexes, both because of the greater resolution of their spectra, and because solvolysis was s1ow.l As a result, in DMA the distal nitrogen protons of the cis-[CoCl(DMF) en2]2+ ion were observed, and the relative areas of the N-proton resonances of cis-[CoCl(H,O)- en2I2+ and cis-[CoCl(DMA) en2I2+ ions could be measured.
(iii) Dimethyl Sulphoxide as Solvent
Dimethyl sulphoxide shows a single resonance at 6 2.58 p.p.m. Addition of small amounts ( 0 . 5 ~ ) of water, and DMA, while producing a more complex resonance pattern at high fields, did not interfere at values of 6>4.O p.p.m. Dimethyl sulphoxide solutions containing small amounts of uncoordinated DMF (c. 5 ~ ) showed only the aldehyde proton resonance of the DMF (6 8.00 p.p.m.) a t values of 6>4.0 p.p.m. Addition of acid to DMSO resulted in a very complex resonance pattern at values
22Frenke1, G., J. chem. Phys., 1963, 39, 1614. Strengle, T. R., and Langford, C. H., J. phys. Chem., 1965, 69, 3299.
24 Lantzke, I. R., and Watts, D. W., Aust. J . Chem., 1966, 19, 969. ''N.M.R. Spectra Catalog." Vol. 2, No. 421. (Varian Associates: Palo Alto, Cal., 1963.)
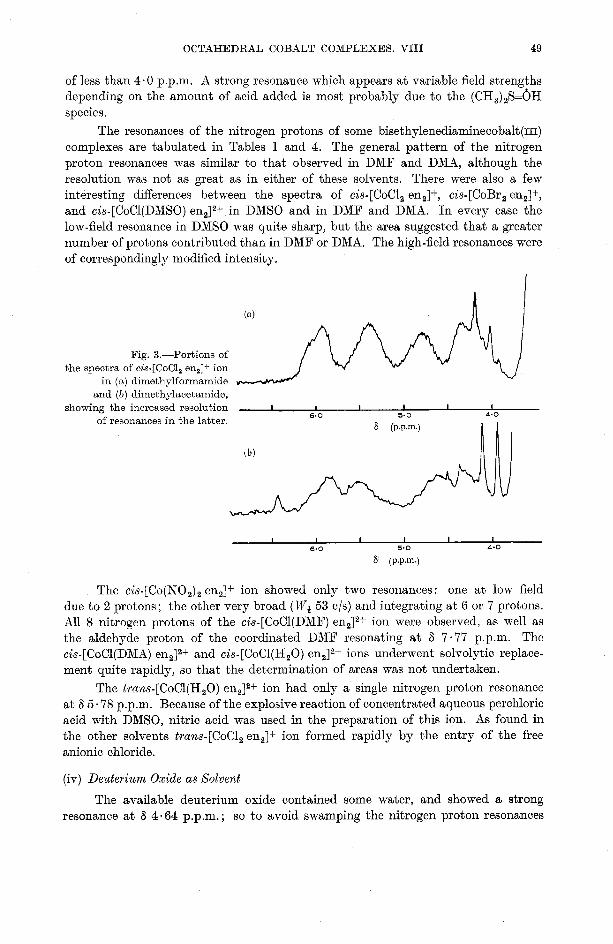
OCTAHEDRAL COBALT COMPLEXES. VIII 49
of less than 4.0 p.p.m. A strong resonance which appears a t variable field strengths depending on the amount of acid added is most probably due to the (CH,),S=OH species.
The resonances of the nitrogen protons of some bisethylenediaminecobalt(~~~) complexes are tabulated in Tables 1 and 4. The general pattern of the nitrogen proton resonances was similar to that observed in DMF and D m , although the resolution was not as great as in either of these solvents. There were also a few interesting differences between the spectra of cis-[CoCl, en,]+, cis-[CoBr, en2]+, and cis-[CoCl(DMSO) en2I2+ in DMSO and in DMF and DMA. In every case the low-field resonance in DMSO was quite sharp, but the area suggested that a greater number of protons contributed than in DMF or DMA. The high-field resonances were of correspondingly modified intensity.
Fig. 3.-Portions of the spectra of cis-[CoCl, en,]+ ion
in ( a ) dimethylformamide and (b) dimethylacetamide,
showing of the resonances increased in the resolution latter.) I I I 6 (P.P.m.) I I I
The ci~-[Co(N0,)~ en,]+ ion showed only two resonances: one a t low field due to 2 protons; the other very broad (W+ 53 01s) and integrating a t 6 or 7 protons. All 8 nitrogen protons of the cis-[CoCl(DMF) en212+ ion were observed, as well as the aldehyde proton of the coordinated DMF resonating at 6 7.77 p.p.m. The cis-[CoCl(DMA) en2],+ and cis-[CoCl(H,O) en,]2+ ions underwent solvolytic replace- ment quite rapidly, so that the determination of areas was not undertaken.
The trans-[CoCl(H,O) en,]2+ ion had only a single nitrogen proton resonance at 6 5 .78 p.p.m. Because of the explosive reaction of concentrated aqueous perchloric acid with DMSO, nitric acid was used in the preparation of this ion. As found in the other solvents trans-[CoCl, en,]+ ion formed rapidly by the entry of the free anionic chloride.
(iv) Deuterium Oxide as Xolvent
The available deuterium oxide contained some water, and showed a strong resonance a t 6 4-64 p.p.m.; so to avoid swamping the nitrogen proton resonances

50 I. R. LANTZKE AND D. W. WATTS
we did not add acid to it. As a result the nitrogen proton resonances decreased rapidly as the protons exchanged with deuterium. This was most noticeable for the high-field peaks of cis isomers.
Earlier worker^^,^^ have shown that the trans protons are more labile, and these observations confirm our assignment of the low-field resonances to the proximal protons (H 1 and H 3 of Fig. 2 when X and Y are anionic).
The proton resonances observed in deuterium oxide are recorded in Table 5. Because of the rapid disappearance of the trans nitrogen proton resonances the nitrogen proton resonances did not provide a very certain way of differentiating cis and trans isomers. The carbon proton resonances showed a difference between the cis and trans isomers examined, with the trans complexes showing a single peak and the cis complexes a complex multiplet. The trans isomer carbon proton resonances were in general a t lower field than those of the cis isomers.
(v) Sulphuric Acid as Solvent
As it seemed probable that in concentrated sulphuric acid both nitrogen and carbon protons would be unobs~ured,~ we attempted to measure the spectra of the cis- and trans-dichlorobisethylenediaminecobalt(~~~) chlorides in 95%. acid. As expected nitrogen and carbon proton resonances were observed "cleanly", but the complexes underwent a rapid solvolytic reaction, and no information could be obtained on the starting materials.
The product obtained from both the cis- and the trans-dichlorobisethylene- diaminecobalt(~~~) ions was identical within 15 min. The spectrum was characteristic of a cis complex, with resonances due to the nitrogen protons at 6 4.91 and 3-88 p.p.m., and a complex multiplet centred a t 6 2.98 p.p.m. ( W , 13 c/s) due to the carbon protons. These three resonances had areas of approximately 4 : 1 : 8, respec- tively. There were no other resonances observed to which the remaining three nitro- gen protons could be assigned. This material is possibly the complex H[Co(SO,), en,] previously suggested27 as an intermediate in the synthesis of sulphatobisethylene- diaminecobalt(111) complexes.
The assignment of 6 values was complicated by the slow decomposition of the NaTMS, but by adding a granule of solid NaTMS to the tube, recording the appro- priate region, then adding another granule and recording the same region several times, it was possible to distinguish the NaTMS resonance.
No other complexes were examined as i t seemed likely that they would all undergo a similar rapid solvolytic reaction.
(vi) General
A comparison of the chemical shifts observed with each complex (Tables 1-5) shows that the degree of resolution of the nitrogen proton resonances of cis complexes is solvent dependent, increasing in the order D20<DMSO<DMF<DMA. The increases in resolution are in part due to a wider spread of the resonances, correspond- ing to proportionately larger chemical shifts to lower fields, and in part to sharper resonances.
28 Basolo, F., and Pearson, R. G., Prog. inorg. Chem., 1962, 4, 381. 27Barraclough, C. G., and Tobe, M. L., J. ohem. Soo., 1961, 1993.
OCTAHEDRAL COBALT COMPLEXES. VIII 5 1
The resolution correlates with the decrease in dielectric constant of the solvents D,O (76.16)28>DMS0 (47.0)29>DMA (36.81)30>DMF (35.87)30, except for the reversal of DMA and DMF. The order dso correlates with the decrease of the solvation of cations3I by these solvents, which with viscosity must reflect the rate with which the ions rotate. In general double positive ions show broader resonances than the single positive ions which also corresponds to increase in solvation forces.
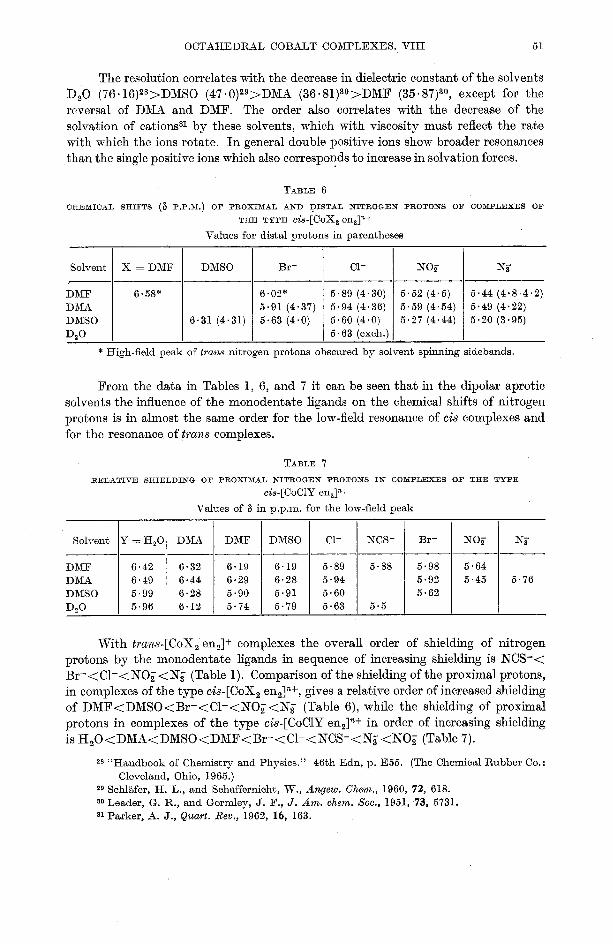
ORED.1ICAL SHIFTS (8 P.P.x.) OR PROXIMAL AND DISTAL NITROGEN PROTONS O F COMPLEXES OF
THE TYPE cis-[Coxa enaln+
From the data in Tables 1, 6, and 7 it can be seen that in the dipolar aprotic solvents the influence of the monodentate ligands on the chemical shifts of nitrogen protons is in almost the same order for the low-field resonance of cis complexes and for the resonance of trans complexes.
Values for distal protons in parentheses
With trans-[COX, en,]+ complexes the overall order of shielding of nitrogen protons by the monodentate ligands in sequence of increasing shielding is NCS-< Br-<cl-<No;<N~_ (Table 1). Comparison of the shielding of the proximal protons, in complexes of the type cis-[COX, en21n+, gives a relative order of increased shielding of DMF< DMSO <Br-< C1- <NO; <Ng (Table 6), while the shielding of proximal protons in complexes of the type cis-[CoClY en,ln+ in order of increasing shielding is H,O<DMA<DMSO<DMF<Br-<C1-<NCS-<N;<NO; (Table 7).
Solvent I
DMF DNA DNSO D2O
RELATIVE SHIELDING OF PROXIMAL NITROGEN PROTONS IN COMPLEXES O F THE TYPE
cis-[CoClY en,Y+
Values of 6 in p.p.m. for the low-field peak
28 "Handbook of Chemistry and Physics." 46th Edn, p. E55. (The Chemical Rubber Co.: Cleveland, Ohio, 1965.)
29 Schljifer, H. L., and Schaffernicht, W., Angew. Chem., 1960, 72, 618. aOLeader, G. R., and Gormley, J. F., J. Am. chern. Soc., 1951, 73, 5731. 81Parker, A. J., Quart. Rev., 1962, 16, 163.
* High-field peak of trans nitrogen protons obscured by solvent spinning sidebands.
NO:
5 .52 (4 .5 ) 5 .59 (4.54) 5 .27 (4.44)
Nb
5 .44 (4.8-4.2) 5 .49 (4.22) 5.20 (3.95)
C1-
5.89 (4.30) 5 .94 (4.36) 5 .60 (4 .0) 5 .63 (exch.)
X = DMF
6.58*
NOT
5 .64 5 .45
Solvent
DMF DMA DMSO
D2O
h ' j
5.76
C1- ~ - ~ ~ ~ - ~ - ~
5.89 5 . 9 4 5 .60 5 .63
DMSO
6 .31 (4.31)
Y = H,O
6 .42 6 .49 5 .99 5 .96
Br- -
6 ~ 0 2 ~ 5 .91 (4 .37) 5 .63 (4 .0 )
KCS-
5.88
5 . 5
Br-
5.98 5 .92 5 .62
DMA
6 .32 6 . 4 4 6 .28 6 .12
DXF
6.19 6.29 5 .90 5 .74
DMSO
6 .19 6 .28 5 .91 5 .79

52 I. R. LAPU'TZKE AND D. W. WATTS
Although the chemical shift of the low-field resonance is determined by different proportions of shielding and or deshielding by two ligands, the relative overall effectiveness of the ligands for shielding of the proximal nitrogen protons in dipolar aprotic solvents appears to be H,O<DMA<DMF<DMSO <NCS-<Br-< Cl-<NO;<N,.
This is the same order as the strength of these species as ligands in aqueous solution (i.e. DMA<DMF<DMSO<Br-<C1-<H20<SCN-<NO;<N;) as asses- sed from their stabilities, with the exception of H,O and SCN-. Since proton magnetic resonance measurements are a sensitive probe of the electronic field experienced by the protons, a correlation between bond strength and chemical shifts is to be expected.
It has been emphasized p r e v i o u ~ l y ~ ~ that estimates of the strength of water as a ligand based upon both stability measurements and nucleophilicity must lead to overestimation because of the contribution of anion s ~ l v a t i o n ~ ~ ~ ~ ~ to the stability of aquo complexes. The order given for ligand strength based on these p.m.r. results supports our own previous conclusion and correlates with the dipole moments of the ligands H,O (1.87 D) <DMA (3 79 D)<DMF (3 82 D) <DMSO (4.3 D).13 The differ- ence in order with respect to thiocyanate is not understood.
From the few high-field resonances resolved with complexes of the type cis-[COX, en,]"+ (Table 6), it appears as though the ligands capable of i~ bonding (SCN-, NO;, and N;) shield distal protons less than those which only form u bonds. There are not enough results to derive any conclusive deduction, but such a result is in keeping with the known trans effects of such ligands.
Finally, i t would be of considerable value in interpretation of substitution reactions of octahedral cobalt compounds if the permanent electronic polarization of the atoms bonded directly to the cobalt could be determined. While the protons we have been considering are only one bond distant from an atom coordinated to the cobalt, proton chemical shifts do not always correlate directly with the electro- negativity of attached groups and hence with bond polarization. In general, it is necessary to consider the magnetic effects of currents flowing in other parts of the molecule, as well as the effects of the bonding electrons.
In a t least one case we know that this more complex approach is needed. The p.m.r. spectrum of the cis-[CoCl(DMF) en,I2+ ion in DMF shows that the most deshielded protons are those proximal to the DMF ligand, and the least deshielded are the protons distal to both the Cl- and DAfF ligands. Yet from kinetic evidence7 we consider that ion pairs of chloride with cis-[CoCl(DMF) en,],+ have the C1- trans to the two monodentate ligands; that is adjacent to the two distal nitrogen protons. These kinetic results indicate a low electron density in the region of the distal protons, so the relative shielding of these distal protons must arise from contributions due to the electron charge distribution in the neighbouring atoms, not from a high electron density in the region of these protons.
The authors are indebted to Professor D. R. Stranks whose discussion initiated the work. They wish to acknowledge helpful discussion with Dr B. Johnson, Dr B. N. Figgis, Mr M. W. Jarvis, and particularly the work of Mr J. Matisons in recording the spectra.