new insight in coordination of vic-dioximes: bis and tris( e, e-dioximato)ni(ii) complexes
TRANSCRIPT

Available online at www.sciencedirect.com
www.elsevier.com/locate/ica
Inorganica Chimica Acta 361 (2008) 2225–2235
New insight in coordination of vic-dioximes: Bis- andtris(E,E-dioximato)Ni(II) complexes
Fatma Yuksel a,b, Ays�e Gul Gurek a, Mahmut Durmus� a, _Ilke Gurol c, Vefa Ahsen a,c,*,Erwann Jeanneau b, Dominique Luneau b,*
a Gebze Institute of Technology, Department of Chemistry, P.O. Box 141, 41400 Gebze, Kocaeli, Turkeyb Universite Claude Bernard Lyon1, Laboratoire des Multimateriaux et Interfaces (UMR 5615), Campus de La Doua, 69622 Villeurbanne, France
c TUBITAK-Marmara Research Center, Materials Institute, P.O. Box 21, 41470 Gebze, Kocaeli, Turkey
Received 28 May 2007; received in revised form 9 November 2007; accepted 18 November 2007Available online 22 November 2007
Abstract
N,N0-Bis[allylamino]glyoxime, N,N0-bis[anilino]glyoxime, and N,N0-bis[1,2,3,4-tetrahydro-5-naphthalenamino]glyoxime have beenprepared from corresponding amines and (E,E)-dichloroglyoxime. These ligands gave orange-red compound with NiCl2 in less acidicmedium (pH � 5) that are bis(E,E-dioximato)nickel(II) complexes {[(E,E)-Ni(HL)2]} (1a–3a) and green compounds in acidic medium(pH � 2) that are tris(E,E-dioximato)nickel(II) dichloride complexes {[(E,E)-Ni(LH2)3]Cl2} (1b–3b). The crystal structures of all com-plexes have been determined by X-ray diffraction on a single crystal. The study of absorption spectra of these two types of complexesshows that they may be converted to each other by addition of acids (1a–3a) or bases (1b–3b) and there is no way for the amphi form.� 2007 Elsevier B.V. All rights reserved.
Keywords: Dioxime; Nickel complex; X-ray; Molecular structure; Chromism
1. Introduction
vic-Dioximes have been abundantly used as chelatingagent in coordination chemistry since the early 1900s [1–4]. Due to the two oximate groups, there are three geomet-rical isomers of the vic-dioxime that have to be consideredfor coordination. They are the anti-(E,E), amphi-(E,Z) andsyn-(Z,Z) forms as shown in Fig. 1 [5–7]. Coordination viathe N,N-chelation of the anti form (E,E) is however, themost generally encountered. This affords bis(E,E-dioxi-mato) complexes that are square planar with transitionmetal cations such as nickel(II), copper(II) palladium(II)and cobalt(III), or square pyramidal and octahedral com-
0020-1693/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.ica.2007.11.019
* Corresponding authors. Address: Gebze Institute of Technology,Department of Chemistry, P.O. Box 141, 41400 Gebze, Kocaeli, Turkey.Tel.: +90 2626053106; fax: +90 2626053101 (V. Ahsen); fax: +33 472 431160 (D. Luneau).
E-mail addresses: [email protected] (V. Ahsen), [email protected] (D. Luneau).
plexes when there are extra axial ligands [7–11]. The stabil-ity of the planar structure is due to intramolecular O–H� � �O hydrogen bonding between the two anti ligands(Scheme 1). This coordination mode is well supported bynumerous X-ray crystal structures of bis(E,E-dioximato)complexes (anti).
In the case of nickel(II), these bis(E,E-dioximato) com-plexes {[(E,E)-Ni(HL)2]} are orange-red, square planar,and thus diamagnetic [1,2,12–14]. As illustrative is the wellknown dimethylglyoximato nickel(II). There are, however,some vic-dioximato nickel(II) complexes that are green col-ored and paramagnetic. The latter are generally attributedto the bis-coordination of two vic-dioximate ligands via theNO-chelation of the amphi form (E,Z) in a tetrahedralarrangement [15–25]. In contrast with the bis(E,E-dioxi-mato) coordination mode [(E,E)-Ni(HL)2] there arealmost no structural evidences of the bis(E,Z-dioximato)coordination mode [(E,Z)-Ni(HL)2]. To the best of ourknowledge, the only reported crystal structure of a bis(E,Z-dioximato) complex is those of bis(d-camphorquinone

NH2R
Cl
Cl N
N
OH
OH
EtOH
NH
NH
R
RN
N
O
O
H
H
N
N
O
O
Ni
NH
NH
R
R
NH
NH
R
R
NH
NHR
R
N
OH
N
N
OH
OH
Ni
N
OH
N
N
OH
OHNH
NH
R
R
NHN
NH N
OH
OH
R
R
+
1-3
1a-3a
1b-3b
1, 1a,1b
2, 2a, 2b
3, 3a, 3b
Compound R
NiCl2NiCl2
2Cl-
Scheme 1. Routes for the synthesis of the ligands and their complexes.
R
R N
N
OH
OH
R
R N
N
OH
OHR
R N
NOHOH
Anti (E,E) Amphi (E,Z) Syn (Z,Z)
Fig. 1. Geometric isomers of symmetrically substituted vic-dioxime.
2226 F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235
dioximato)nickel(II) complex which is indeed green andparamagnetic [16]. While most of the reported [bis(E,Z-dioximato)nickel(II)] have been established from the factthey were synthesized from the amphi form (E,Z) of theligands, [15–21] there are, however, some reports that[(E,Z)-Ni(HL)2] complexes were obtained from [(E,E)-Ni(HL)2] complexes by treatment in acidic medium [22–26]. Pedersen and Larsen were the first to report that solu-tion of [(E,E)-Ni(HL)2] complexes changes colour, goingfrom red to green, by acid addition. As this colour changewas followed by a change in magnetic properties, from dia-magnetic to paramagnetic, [22] they suggested that squareplanar [(E,E)-Ni(HL)2] complexes isomerize to tetrahedral[(E,Z)-Ni(HL)2]. Although this conversion has not beeninvestigated thoroughly; most of the following works havethen rely on this study to explain the green nickel(II) com-plexes from (E,E) vic-dioxime. However this explanation
had many drawbacks such as (i) it is not understandablethat the oximic oxygens of the [(E,E)-Ni(HL)2] do notback-protonate in acidic medium; (ii) it is also surprisingthat dioximate ligands isomerizes from anti to amphi,whereas we would expect the anti isomer to be the morestable form in acidic medium [17,18].
We also currently admitted this attribution of the orange-red form as the [bis(E,E-dioximato)nickel(II)] complexesand the green one as indicative exclusively of [(E,Z)-bis(dioximato)nickel(II)] complexes. This confidence hasbeen greatly affected when we recently come to synthesizeseveral nickel(II) complexes with different oxamidoximeligands that were green and paramagnetic but whose crystalstructure showed no [bis(E,Z-dioximato)nickel(II)] com-plexes but instead [tris(E,E-dioximato)nickel(II)]2+ entities{[(E,E)-Ni(LH2)3]Cl2}. There have been a few previousreports of [(E,E)-Ni(LH2)3]2+ complexes in the literature[27–29]. In 1980 Endres et al. already reported [27] thatthe reaction of an (E,E)-oxamide oxime ligand withNiCl2 � 6H2O in small amount of alcohol gives green, octahe-dral (E,E)-tris(oxamide oximato)nickel(II) dichloride. Butin more diluted alcohol, the same precursors produce squareplanar, brick-red (E,E)-bis(oxamide oximato)nickel(II)[30]. A more interesting point was that tris(E,E-oxamideoximato)nickel(II) dichloride changes to bis(E, E-oxamideoximato)nickel(II) in neutral or slightly basic solution [30]This result and ours show that the [(E,Z)-Ni(HL)2] com-plexes could no more be considered as the only possibilityfor the green nickel(II) complexes of vic-dioxime.
The confusion between both [(E,Z)-Ni(HL)2] and[(E,E)-Ni(LH2)3]2+ forms comes from the fact that bothhave green color and very similar spectroscopic data. Inthis paper we try to clarify this confusion. We synthesizedthe [(E,E)-Ni(HL)2] and [(E,E)-Ni(LH2)3]Cl2 complexes ofthree different (E,E)-oxamide oxime derivatives. All com-plexes are novel and have been characterized using differentspectroscopic techniques. We established their crystalstructures from X-ray diffraction on single crystal and weinvestigated the dependence of the three forms (bis-anti,bis-amphi and tris-anti) with the pH of the reaction mediumby studying their UV spectra versus the pH.
2. Results and discussion
2.1. Synthesis
2.1.1. Ligands
For this work, we synthesized three different (E,E)-oxamide oxime ligands and their two series of nickel(II)complexes. The routes for the synthesis of all compoundsare summarized in Scheme 1.
The ligands (LH2, 1–3) were prepared as previouslyreported by the reaction of an amine derivative with(E,E)-dichloroglyoxime [31] in ethanol at room tem-perature. HCl formed during the reaction was neutral-ized by an excess of the amine for 1 and an excess ofNaHCO3 for 2 and 3. N,N0-bis[allylamino]glyoxime (1) and
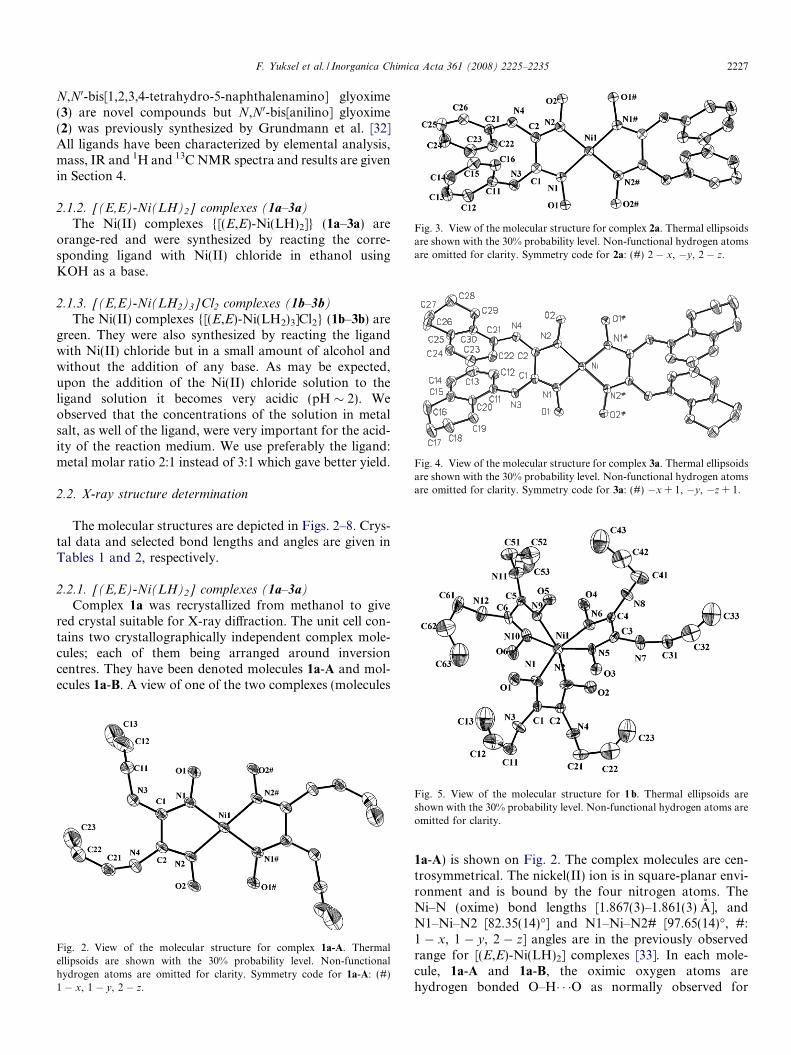
Fig. 3. View of the molecular structure for complex 2a. Thermal ellipsoidsare shown with the 30% probability level. Non-functional hydrogen atomsare omitted for clarity. Symmetry code for 2a: (#) 2 � x, �y, 2 � z.
Fig. 4. View of the molecular structure for complex 3a. Thermal ellipsoidsare shown with the 30% probability level. Non-functional hydrogen atomsare omitted for clarity. Symmetry code for 3a: (#) �x + 1, �y, �z + 1.
F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235 2227
N,N0-bis[1,2,3,4-tetrahydro-5-naphthalenamino] glyoxime(3) are novel compounds but N,N0-bis[anilino] glyoxime(2) was previously synthesized by Grundmann et al. [32]All ligands have been characterized by elemental analysis,mass, IR and 1H and 13C NMR spectra and results are givenin Section 4.
2.1.2. [(E,E)-Ni(LH)2] complexes (1a–3a)
The Ni(II) complexes {[(E,E)-Ni(LH)2]} (1a–3a) areorange-red and were synthesized by reacting the corre-sponding ligand with Ni(II) chloride in ethanol usingKOH as a base.
2.1.3. [(E,E)-Ni(LH2)3]Cl2 complexes (1b–3b)The Ni(II) complexes {[(E,E)-Ni(LH2)3]Cl2} (1b–3b) are
green. They were also synthesized by reacting the ligandwith Ni(II) chloride but in a small amount of alcohol andwithout the addition of any base. As may be expected,upon the addition of the Ni(II) chloride solution to theligand solution it becomes very acidic (pH � 2). Weobserved that the concentrations of the solution in metalsalt, as well of the ligand, were very important for the acid-ity of the reaction medium. We use preferably the ligand:metal molar ratio 2:1 instead of 3:1 which gave better yield.
2.2. X-ray structure determination
The molecular structures are depicted in Figs. 2–8. Crys-tal data and selected bond lengths and angles are given inTables 1 and 2, respectively.
2.2.1. [(E,E)-Ni(LH)2] complexes (1a–3a)
Complex 1a was recrystallized from methanol to givered crystal suitable for X-ray diffraction. The unit cell con-tains two crystallographically independent complex mole-cules; each of them being arranged around inversioncentres. They have been denoted molecules 1a-A and mol-ecules 1a-B. A view of one of the two complexes (molecules
Fig. 2. View of the molecular structure for complex 1a-A. Thermalellipsoids are shown with the 30% probability level. Non-functionalhydrogen atoms are omitted for clarity. Symmetry code for 1a-A: (#)1 � x, 1 � y, 2 � z.
Fig. 5. View of the molecular structure for 1b. Thermal ellipsoids areshown with the 30% probability level. Non-functional hydrogen atoms areomitted for clarity.
1a-A) is shown on Fig. 2. The complex molecules are cen-trosymmetrical. The nickel(II) ion is in square-planar envi-ronment and is bound by the four nitrogen atoms. TheNi–N (oxime) bond lengths [1.867(3)–1.861(3) A], andN1–Ni–N2 [82.35(14)�] and N1–Ni–N2# [97.65(14)�, #:1 � x, 1 � y, 2 � z] angles are in the previously observedrange for [(E,E)-Ni(LH)2] complexes [33]. In each mole-cule, 1a-A and 1a-B, the oximic oxygen atoms arehydrogen bonded O–H� � �O as normally observed for
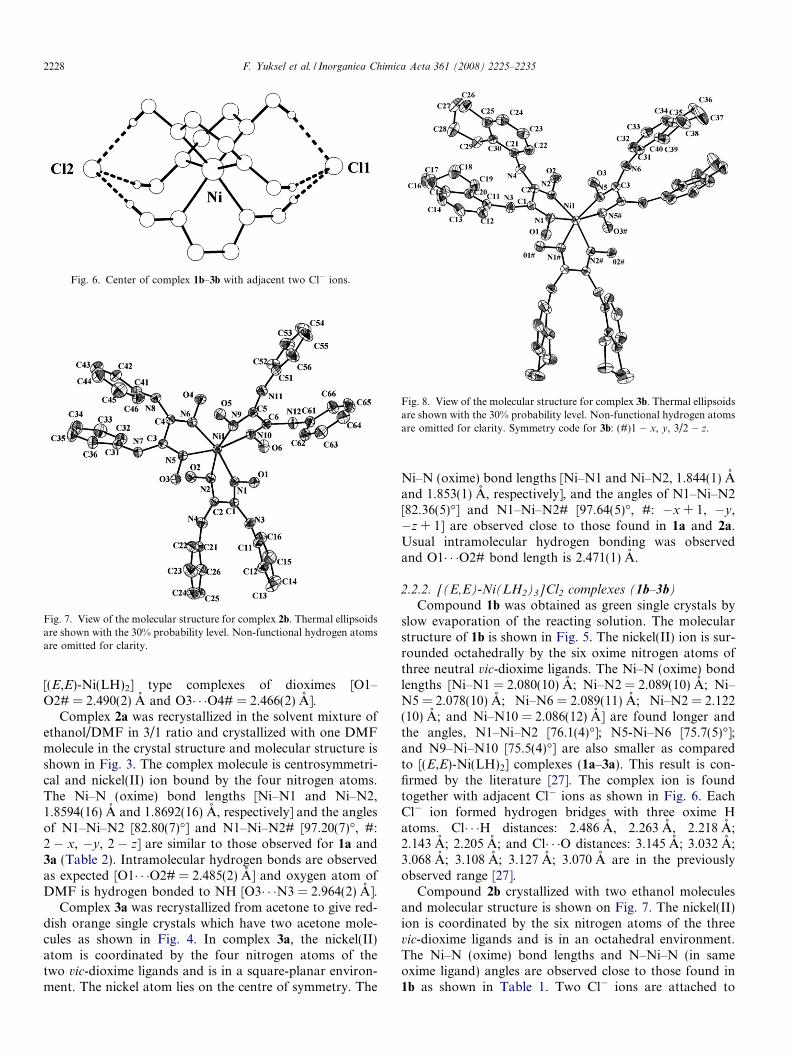
Fig. 6. Center of complex 1b–3b with adjacent two Cl� ions.
Fig. 7. View of the molecular structure for complex 2b. Thermal ellipsoidsare shown with the 30% probability level. Non-functional hydrogen atomsare omitted for clarity.
Fig. 8. View of the molecular structure for complex 3b. Thermal ellipsoidsare shown with the 30% probability level. Non-functional hydrogen atomsare omitted for clarity. Symmetry code for 3b: (#)1 � x, y, 3/2 � z.
2228 F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235
[(E,E)-Ni(LH)2] type complexes of dioximes [O1–O2# = 2.490(2) A and O3� � �O4# = 2.466(2) A].
Complex 2a was recrystallized in the solvent mixture ofethanol/DMF in 3/1 ratio and crystallized with one DMFmolecule in the crystal structure and molecular structure isshown in Fig. 3. The complex molecule is centrosymmetri-cal and nickel(II) ion bound by the four nitrogen atoms.The Ni–N (oxime) bond lengths [Ni–N1 and Ni–N2,1.8594(16) A and 1.8692(16) A, respectively] and the anglesof N1–Ni–N2 [82.80(7)�] and N1–Ni–N2# [97.20(7)�, #:2 � x, �y, 2 � z] are similar to those observed for 1a and3a (Table 2). Intramolecular hydrogen bonds are observedas expected [O1� � �O2# = 2.485(2) A] and oxygen atom ofDMF is hydrogen bonded to NH [O3� � �N3 = 2.964(2) A].
Complex 3a was recrystallized from acetone to give red-dish orange single crystals which have two acetone mole-cules as shown in Fig. 4. In complex 3a, the nickel(II)atom is coordinated by the four nitrogen atoms of thetwo vic-dioxime ligands and is in a square-planar environ-ment. The nickel atom lies on the centre of symmetry. The
Ni–N (oxime) bond lengths [Ni–N1 and Ni–N2, 1.844(1) Aand 1.853(1) A, respectively], and the angles of N1–Ni–N2[82.36(5)�] and N1–Ni–N2# [97.64(5)�, #: �x + 1, �y,�z + 1] are observed close to those found in 1a and 2a.Usual intramolecular hydrogen bonding was observedand O1� � �O2# bond length is 2.471(1) A.
2.2.2. [(E,E)-Ni(LH2)3]Cl2 complexes (1b–3b)
Compound 1b was obtained as green single crystals byslow evaporation of the reacting solution. The molecularstructure of 1b is shown in Fig. 5. The nickel(II) ion is sur-rounded octahedrally by the six oxime nitrogen atoms ofthree neutral vic-dioxime ligands. The Ni–N (oxime) bondlengths [Ni–N1 = 2.080(10) A; Ni–N2 = 2.089(10) A; Ni–N5 = 2.078(10) A; Ni–N6 = 2.089(11) A; Ni–N2 = 2.122(10) A; and Ni–N10 = 2.086(12) A] are found longer andthe angles, N1–Ni–N2 [76.1(4)�]; N5-Ni–N6 [75.7(5)�];and N9–Ni–N10 [75.5(4)�] are also smaller as comparedto [(E,E)-Ni(LH)2] complexes (1a–3a). This result is con-firmed by the literature [27]. The complex ion is foundtogether with adjacent Cl� ions as shown in Fig. 6. EachCl� ion formed hydrogen bridges with three oxime Hatoms. Cl� � �H distances: 2.486 A, 2.263 A, 2.218 A;2.143 A; 2.205 A; and Cl� � �O distances: 3.145 A; 3.032 A;3.068 A; 3.108 A; 3.127 A; 3.070 A are in the previouslyobserved range [27].
Compound 2b crystallized with two ethanol moleculesand molecular structure is shown on Fig. 7. The nickel(II)ion is coordinated by the six nitrogen atoms of the threevic-dioxime ligands and is in an octahedral environment.The Ni–N (oxime) bond lengths and N–Ni–N (in sameoxime ligand) angles are observed close to those found in1b as shown in Table 1. Two Cl� ions are attached to

Table 1Summary of data collection and refinement for all complexes
1a 2a 3a 1b 2b 3b
Formula C16H26N8NiO4 C28H26N8NiO4 �2(C3H7NO)
C44H50N8NiO4 �2(C3H6O)
C24H42N12NiO6 �2(Cl)
C42H42N12NiO6 �2(C2H6O) � 2(Cl)
C66H78N12NiO6 �2(Cl)
Formula weight 453.14 743.47 929.79 482.86 1030.60 1265.04T (K) 293 293 293 293 293 293Crystal system triclinic monoclinic orthorhombic triclinic monoclinic orthorhombicSpace group P�1 P121/n1 Pbca P�1 P121/c1 Pbcn
a (A) 4.847 (5) 9.2341 (4) 11.514 (8) 9.583 (7) 9.2683 (2) 16.684 (5)b (A) 13.351 (5) 12.8587 (5) 19.322 (1) 13.391 (1) 22.9694 (7) 16.121 (5)c (A) 16.733 (5) 15.0715 (6) 20.975 (1) 13.976 (1) 24.0707 (6) 24.692 (5)a (�) 106.768 (5) 89.021 (5)b (�) 90.123 (5) 102.566 (2) 82.681 (4) 91.484 (2)c (�) 95.074 (5) 80.458 (4)V (A3) 1032.3 (12) 1746.70 (12) 4666.4 (5) 1754.3 (2) 5122.6 (2) 6641 (3)Z 2 2 4 3 4 8l (mm�1) 0.98 0.62 0.47 0.76 0.55 0.43Reflections collected 7631 6997 17338 12979 20004 9104Independent reflections [R(int)] 4860 [0.023] 4079 [0.028] 5739 [0.029] 8031 [0.069] 11834 [0.056] 8335 [0.000]Data/restraints/parameters 2962/0/265 2845/0/232 5739/0/432 2359/0/373 6463/0/622 4538/0/393Goodness-of-fit on F2 1.16 1.11 0.90 1.10 1.07 1.06R(F)a 0.052 0.040 0.041 0.090 0.060 0.049Rw(F2)b 0.061 0.048 0.124 0.109 0.070 0.058
a I > 2r(I), R(F) =P
||Fo| � |Fc||/P
|Fo|.b All data Rw(F2) = [
Pw (F2
o � F2c)2/P
w(F2o)2]1/2.
F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235 2229
complex ion with hydrogen bridges as shown in Fig. 5(Cl� � �H distances: 2.428 A, 2.532 A, 2.301 A, 2.260 A;2.368 A; 2.280 A; and Cl� � �O distances: 3.016 A; 3.121 A;3.104 A; 3.075 A; 3.093 A; 3.132 A) [27]. Ethanol moleculesare found to be hydrogen bonded to the NH groups[N8� � �O7 = 3.038(2) A; N11� � �O8 = 2.900(2) A].
The molecular structure of 3b is shown in Fig. 8. Thenickel(II) ion is found octahedral coordinated with sixnitrogen atoms of three neutral vic-dioxime ligands. Bondlengths and bond angles are found similar for 1b and 2b
as shown in Table 3. Cl� ions are adhered to complexion with hydrogen bridges as shown in Fig. 5. (Cl� � �H dis-tances: 2.261 A, 2.177 A, 2.262 A, 2.260 A; 2.175 A;2.261 A; and Cl� � �O distances: 3.001 A; 3.063 A; 3.065 A;3.064 A; 3.065 A; 3.002 A) [27].
2.3. Elemental analyses, mass spectra, IR and NMR
characterizations
Many reports on nickel(II) complexes of vic-dioximerely only on elemental analysis, mass spectra, IR and 1Hand 13C NMR characterization. We present here ourresults to stress on the difficulty to ascribe the structureof the green form considering only these informations.
2.3.1. Ligands
The mass spectra (ES-MS) of 1–3 exhibit a molecularion peak at m/z 199, 271, 379 [M+H]+, respectively(Fig. S7, S10 and S13 in the ESI). In the IR spectra, NHbands were observed around 3400 cm�1. Characteristicbands of the vic-dioximes [34,35]: OH stretching vibration,C@N and N–O stretches appeared at 3310, 1650, and
925 cm�1 for ligand 1; at 3300, 1642 and 974 cm�1 forligand 2; and at 3324, 1654 and 925 cm�1 for ligand 3,respectively. The 1H and 13C NMR spectra were obtainedin CDCl3 for ligand 1, and in DMSO-d6 for ligand 2 and3. In the 1H NMR spectra, NH protons gave only one sin-glet and disappeared upon deuterium exchange. Other deu-terium exchangeable single peaks (at 9.80 ppm for ligand 1;at 10.41 ppm for ligand 2, and at 10.40 ppm for ligand 3)were ascribed to OH protons. In the 13C NMR spectra,hydroxyimino carbon atoms (C@N–O) gave one chemicalshift at 148.20 ppm for ligand 1; at 143.35 ppm for ligand2; at 143.85 ppm for ligand 3, which confirmed the anti
structure (E,E) for the vic-dioxime unit [36].
2.3.2. [(E,E)-Ni(LH)2] complexes (1a–3a)
Elemental analysis, mass, IR and NMR spectra are inagreement with the structures and are given in Section 4.The mass spectra (ES-MS) of 1a–3a (Figs. S8, S11 andS14 in the ESI) gave clearly molecular ion peak at m/z453[M]+ for 1a; at m/z 597[M]+ for 2a; and at m/z813[M]+ for 3a. In the IR spectra of 1a–3a, broad andweak OH bands and appearance of the weak band ataround 1720 cm�1 can be attributed to intramolecularhydrogen bonding [34]. The 1H and 13C NMR spectra wereobtained in CDCl3 for complexes 2a and 3a and in DMSO-d6 for complex 1a. Signals of the solvents which exist incrystal (DMF in the spectrum of 2a and acetone in thespectrum of 3a) were omitted. In the 1H NMR spectrawe did not observe the chemical shifts belonging to freeOH protons (around 10 ppm); however, the resonance ofintramolecular bonding OH–O proton signals wereobserved at 16.84 ppm for 1a; at 17.00 ppm for 2a; and
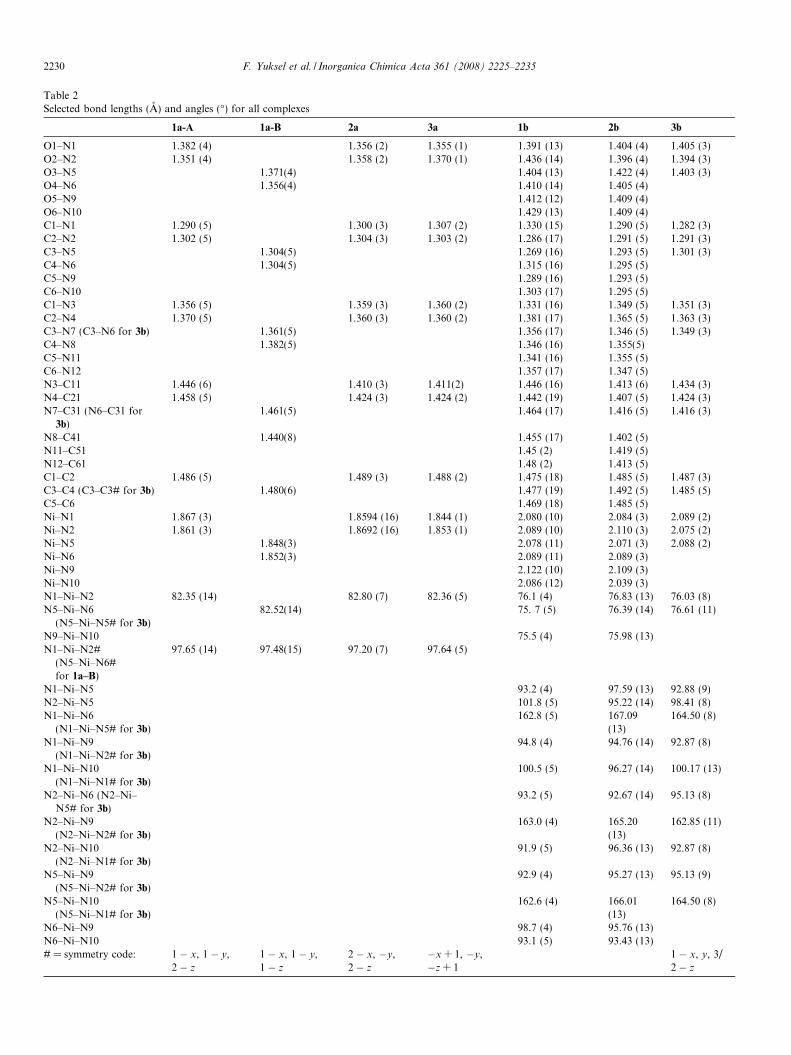
Table 2Selected bond lengths (A) and angles (�) for all complexes
1a-A 1a-B 2a 3a 1b 2b 3b
O1–N1 1.382 (4) 1.356 (2) 1.355 (1) 1.391 (13) 1.404 (4) 1.405 (3)O2–N2 1.351 (4) 1.358 (2) 1.370 (1) 1.436 (14) 1.396 (4) 1.394 (3)O3–N5 1.371(4) 1.404 (13) 1.422 (4) 1.403 (3)O4–N6 1.356(4) 1.410 (14) 1.405 (4)O5–N9 1.412 (12) 1.409 (4)O6–N10 1.429 (13) 1.409 (4)C1–N1 1.290 (5) 1.300 (3) 1.307 (2) 1.330 (15) 1.290 (5) 1.282 (3)C2–N2 1.302 (5) 1.304 (3) 1.303 (2) 1.286 (17) 1.291 (5) 1.291 (3)C3–N5 1.304(5) 1.269 (16) 1.293 (5) 1.301 (3)C4–N6 1.304(5) 1.315 (16) 1.295 (5)C5–N9 1.289 (16) 1.293 (5)C6–N10 1.303 (17) 1.295 (5)C1–N3 1.356 (5) 1.359 (3) 1.360 (2) 1.331 (16) 1.349 (5) 1.351 (3)C2–N4 1.370 (5) 1.360 (3) 1.360 (2) 1.381 (17) 1.365 (5) 1.363 (3)C3–N7 (C3–N6 for 3b) 1.361(5) 1.356 (17) 1.346 (5) 1.349 (3)C4–N8 1.382(5) 1.346 (16) 1.355(5)C5–N11 1.341 (16) 1.355 (5)C6–N12 1.357 (17) 1.347 (5)N3–C11 1.446 (6) 1.410 (3) 1.411(2) 1.446 (16) 1.413 (6) 1.434 (3)N4–C21 1.458 (5) 1.424 (3) 1.424 (2) 1.442 (19) 1.407 (5) 1.424 (3)N7–C31 (N6–C31 for
3b)1.461(5) 1.464 (17) 1.416 (5) 1.416 (3)
N8–C41 1.440(8) 1.455 (17) 1.402 (5)N11–C51 1.45 (2) 1.419 (5)N12–C61 1.48 (2) 1.413 (5)C1–C2 1.486 (5) 1.489 (3) 1.488 (2) 1.475 (18) 1.485 (5) 1.487 (3)C3–C4 (C3–C3# for 3b) 1.480(6) 1.477 (19) 1.492 (5) 1.485 (5)C5–C6 1.469 (18) 1.485 (5)Ni–N1 1.867 (3) 1.8594 (16) 1.844 (1) 2.080 (10) 2.084 (3) 2.089 (2)Ni–N2 1.861 (3) 1.8692 (16) 1.853 (1) 2.089 (10) 2.110 (3) 2.075 (2)Ni–N5 1.848(3) 2.078 (11) 2.071 (3) 2.088 (2)Ni–N6 1.852(3) 2.089 (11) 2.089 (3)Ni–N9 2.122 (10) 2.109 (3)Ni–N10 2.086 (12) 2.039 (3)N1–Ni–N2 82.35 (14) 82.80 (7) 82.36 (5) 76.1 (4) 76.83 (13) 76.03 (8)N5–Ni–N6
(N5–Ni–N5# for 3b)82.52(14) 75. 7 (5) 76.39 (14) 76.61 (11)
N9–Ni–N10 75.5 (4) 75.98 (13)N1–Ni–N2#
(N5–Ni–N6#for 1a–B)
97.65 (14) 97.48(15) 97.20 (7) 97.64 (5)
N1–Ni–N5 93.2 (4) 97.59 (13) 92.88 (9)N2–Ni–N5 101.8 (5) 95.22 (14) 98.41 (8)N1–Ni–N6
(N1–Ni–N5# for 3b)162.8 (5) 167.09
(13)164.50 (8)
N1–Ni–N9(N1–Ni–N2# for 3b)
94.8 (4) 94.76 (14) 92.87 (8)
N1–Ni–N10(N1–Ni–N1# for 3b)
100.5 (5) 96.27 (14) 100.17 (13)
N2–Ni–N6 (N2–Ni–N5# for 3b)
93.2 (5) 92.67 (14) 95.13 (8)
N2–Ni–N9(N2–Ni–N2# for 3b)
163.0 (4) 165.20(13)
162.85 (11)
N2–Ni–N10(N2–Ni–N1# for 3b)
91.9 (5) 96.36 (13) 92.87 (8)
N5–Ni–N9(N5–Ni–N2# for 3b)
92.9 (4) 95.27 (13) 95.13 (9)
N5–Ni–N10(N5–Ni–N1# for 3b)
162.6 (4) 166.01(13)
164.50 (8)
N6–Ni–N9 98.7 (4) 95.76 (13)N6–Ni–N10 93.1 (5) 93.43 (13)# = symmetry code: 1 � x, 1 � y,
2 � z
1 � x, 1 � y,1 � z
2 � x, �y,2 � z
�x + 1, �y,�z + 1
1 � x, y, 3/2 � z
2230 F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235
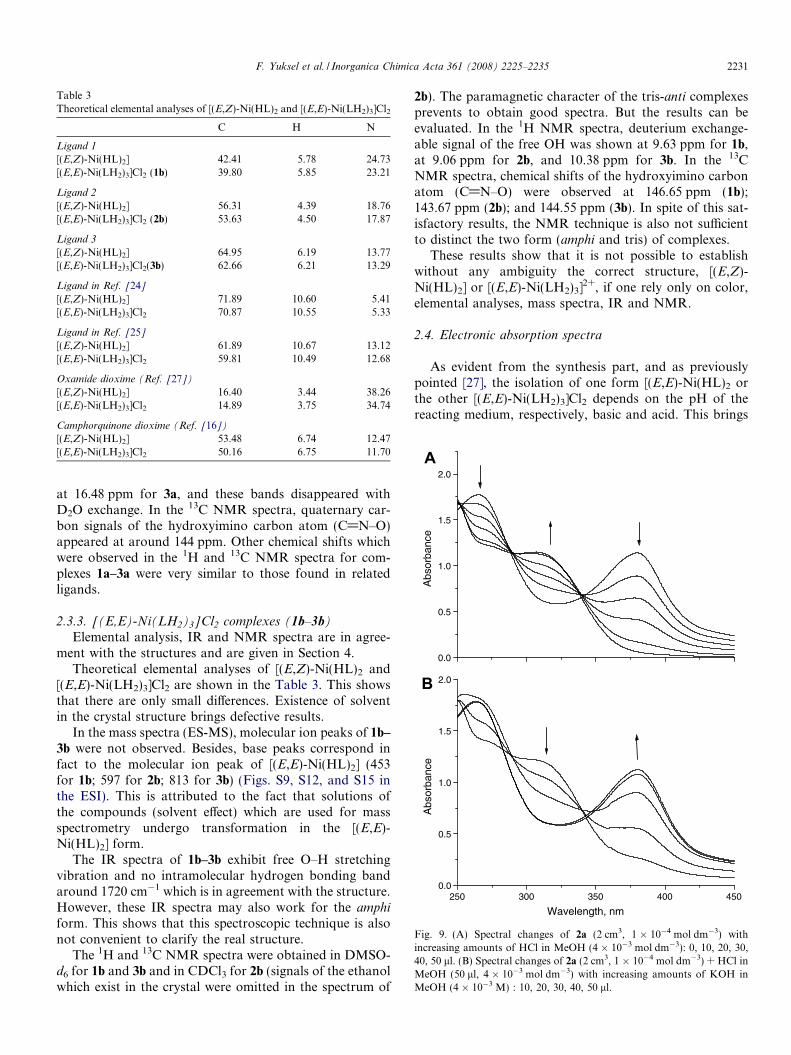
Table 3Theoretical elemental analyses of [(E,Z)-Ni(HL)2 and [(E,E)-Ni(LH2)3]Cl2
C H N
Ligand 1
[(E,Z)-Ni(HL)2] 42.41 5.78 24.73[(E,E)-Ni(LH2)3]Cl2 (1b) 39.80 5.85 23.21
Ligand 2
[(E,Z)-Ni(HL)2] 56.31 4.39 18.76[(E,E)-Ni(LH2)3]Cl2 (2b) 53.63 4.50 17.87
Ligand 3
[(E,Z)-Ni(HL)2] 64.95 6.19 13.77[(E,E)-Ni(LH2)3]Cl2(3b) 62.66 6.21 13.29
Ligand in Ref. [24]
[(E,Z)-Ni(HL)2] 71.89 10.60 5.41[(E,E)-Ni(LH2)3]Cl2 70.87 10.55 5.33
Ligand in Ref. [25]
[(E,Z)-Ni(HL)2] 61.89 10.67 13.12[(E,E)-Ni(LH2)3]Cl2 59.81 10.49 12.68
Oxamide dioxime (Ref. [27])
[(E,Z)-Ni(HL)2] 16.40 3.44 38.26[(E,E)-Ni(LH2)3]Cl2 14.89 3.75 34.74
Camphorquinone dioxime (Ref. [16])
[(E,Z)-Ni(HL)2] 53.48 6.74 12.47[(E,E)-Ni(LH2)3]Cl2 50.16 6.75 11.70
0.0
0.5
1.0
1.5
2.0
A
Abs
orba
nce
250 300 350 400 4500.0
0.5
1.0
1.5
2.0
Wavelength, nm
B
Abs
orba
nce
Fig. 9. (A) Spectral changes of 2a (2 cm3, 1 � 10�4 mol dm�3) withincreasing amounts of HCl in MeOH (4 � 10�3 mol dm�3): 0, 10, 20, 30,40, 50 ll. (B) Spectral changes of 2a (2 cm3, 1 � 10�4 mol dm�3) + HCl inMeOH (50 ll, 4 � 10�3 mol dm�3) with increasing amounts of KOH inMeOH (4 � 10�3 M) : 10, 20, 30, 40, 50 ll.
F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235 2231
at 16.48 ppm for 3a, and these bands disappeared withD2O exchange. In the 13C NMR spectra, quaternary car-bon signals of the hydroxyimino carbon atom (C@N–O)appeared at around 144 ppm. Other chemical shifts whichwere observed in the 1H and 13C NMR spectra for com-plexes 1a–3a were very similar to those found in relatedligands.
2.3.3. [(E,E)-Ni(LH2)3]Cl2 complexes (1b–3b)
Elemental analysis, IR and NMR spectra are in agree-ment with the structures and are given in Section 4.
Theoretical elemental analyses of [(E,Z)-Ni(HL)2 and[(E,E)-Ni(LH2)3]Cl2 are shown in the Table 3. This showsthat there are only small differences. Existence of solventin the crystal structure brings defective results.
In the mass spectra (ES-MS), molecular ion peaks of 1b–
3b were not observed. Besides, base peaks correspond infact to the molecular ion peak of [(E,E)-Ni(HL)2] (453for 1b; 597 for 2b; 813 for 3b) (Figs. S9, S12, and S15 inthe ESI). This is attributed to the fact that solutions ofthe compounds (solvent effect) which are used for massspectrometry undergo transformation in the [(E,E)-Ni(HL)2] form.
The IR spectra of 1b–3b exhibit free O–H stretchingvibration and no intramolecular hydrogen bonding bandaround 1720 cm�1 which is in agreement with the structure.However, these IR spectra may also work for the amphi
form. This shows that this spectroscopic technique is alsonot convenient to clarify the real structure.
The 1H and 13C NMR spectra were obtained in DMSO-d6 for 1b and 3b and in CDCl3 for 2b (signals of the ethanolwhich exist in the crystal were omitted in the spectrum of
2b). The paramagnetic character of the tris-anti complexesprevents to obtain good spectra. But the results can beevaluated. In the 1H NMR spectra, deuterium exchange-able signal of the free OH was shown at 9.63 ppm for 1b,at 9.06 ppm for 2b, and 10.38 ppm for 3b. In the 13CNMR spectra, chemical shifts of the hydroxyimino carbonatom (C@N–O) were observed at 146.65 ppm (1b);143.67 ppm (2b); and 144.55 ppm (3b). In spite of this sat-isfactory results, the NMR technique is also not sufficientto distinct the two form (amphi and tris) of complexes.
These results show that it is not possible to establishwithout any ambiguity the correct structure, [(E,Z)-Ni(HL)2] or [(E,E)-Ni(LH2)3]2+, if one rely only on color,elemental analyses, mass spectra, IR and NMR.
2.4. Electronic absorption spectra
As evident from the synthesis part, and as previouslypointed [27], the isolation of one form [(E,E)-Ni(HL)2 orthe other [(E,E)-Ni(LH2)3]Cl2 depends on the pH of thereacting medium, respectively, basic and acid. This brings
2232 F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235
the questions that the two forms may be inter-convertiblewhen changing the pH and there may be a pH range inwhich the [(E,Z)-bis(dioximato)nickel(II)] exist. To clarifythese points we investigated the absorption spectra of bothforms in solution when changing the pH.
Thus, we investigated the behavior of the [(E,E)-Ni(HL)2] complexes (1a–3a) by first adding acid to solu-tions of the complexes to convert them in the tris-anti formthen at this point we added a base to come back at the ini-tial point. For the [(E,E)-Ni(LH2)3]Cl2 complexes (1b–3b),we first added base to convert them in the bis-anti formthen acid to come back at the initial point. We usedKOH as base and HCl or CF3COOH as acid. Figs. 9 and10 show KOH/HCl titrations of 2a and 2b, respectively.KOH/CF3COOH titrations of 2a and 2b and KOH/HCland KOH/CF3COOH titrations of 1a and 1b are given assupporting information (Figs. S1–S5 in the ESI). There isno difference between using HCl solution in methanoland CF3COOH solution in chloroform. Titrations of 3a
and 3b were not possible due to solubility problems inCHCl3/MeOH mixtures but their electronic spectra in
0.0
0.5
1.0
1.5
2.0
2.5
3.0A
Abs
orba
nce
250 300 350 400 4500.0
0.5
1.0
1.5
2.0
2.5
3.0B
Abs
orba
nce
Wavelength, nm
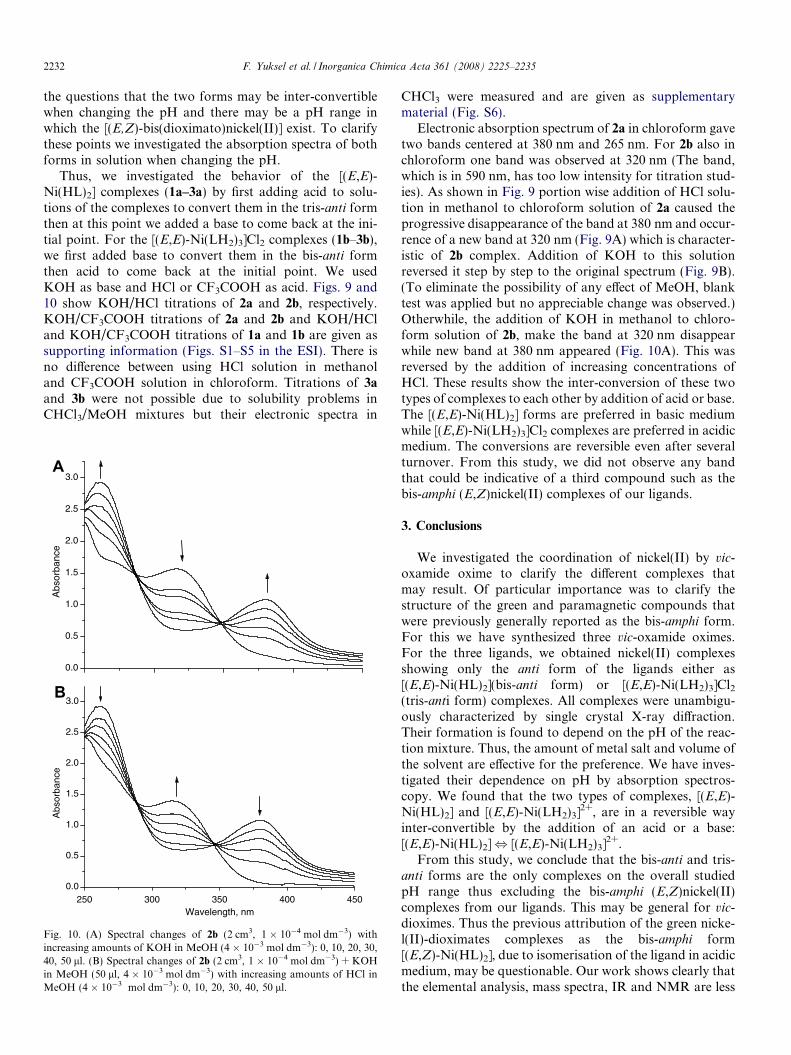
Fig. 10. (A) Spectral changes of 2b (2 cm3, 1 � 10�4 mol dm�3) withincreasing amounts of KOH in MeOH (4 � 10�3 mol dm�3): 0, 10, 20, 30,40, 50 ll. (B) Spectral changes of 2b (2 cm3, 1 � 10�4 mol dm�3) + KOHin MeOH (50 ll, 4 � 10�3 mol dm�3) with increasing amounts of HCl inMeOH (4 � 10�3 mol dm�3): 0, 10, 20, 30, 40, 50 ll.
CHCl3 were measured and are given as supplementarymaterial (Fig. S6).
Electronic absorption spectrum of 2a in chloroform gavetwo bands centered at 380 nm and 265 nm. For 2b also inchloroform one band was observed at 320 nm (The band,which is in 590 nm, has too low intensity for titration stud-ies). As shown in Fig. 9 portion wise addition of HCl solu-tion in methanol to chloroform solution of 2a caused theprogressive disappearance of the band at 380 nm and occur-rence of a new band at 320 nm (Fig. 9A) which is character-istic of 2b complex. Addition of KOH to this solutionreversed it step by step to the original spectrum (Fig. 9B).(To eliminate the possibility of any effect of MeOH, blanktest was applied but no appreciable change was observed.)Otherwhile, the addition of KOH in methanol to chloro-form solution of 2b, make the band at 320 nm disappearwhile new band at 380 nm appeared (Fig. 10A). This wasreversed by the addition of increasing concentrations ofHCl. These results show the inter-conversion of these twotypes of complexes to each other by addition of acid or base.The [(E,E)-Ni(HL)2] forms are preferred in basic mediumwhile [(E,E)-Ni(LH2)3]Cl2 complexes are preferred in acidicmedium. The conversions are reversible even after severalturnover. From this study, we did not observe any bandthat could be indicative of a third compound such as thebis-amphi (E,Z)nickel(II) complexes of our ligands.
3. Conclusions
We investigated the coordination of nickel(II) by vic-oxamide oxime to clarify the different complexes thatmay result. Of particular importance was to clarify thestructure of the green and paramagnetic compounds thatwere previously generally reported as the bis-amphi form.For this we have synthesized three vic-oxamide oximes.For the three ligands, we obtained nickel(II) complexesshowing only the anti form of the ligands either as[(E,E)-Ni(HL)2](bis-anti form) or [(E,E)-Ni(LH2)3]Cl2(tris-anti form) complexes. All complexes were unambigu-ously characterized by single crystal X-ray diffraction.Their formation is found to depend on the pH of the reac-tion mixture. Thus, the amount of metal salt and volume ofthe solvent are effective for the preference. We have inves-tigated their dependence on pH by absorption spectros-copy. We found that the two types of complexes, [(E,E)-Ni(HL)2] and [(E,E)-Ni(LH2)3]2+, are in a reversible wayinter-convertible by the addition of an acid or a base:[(E,E)-Ni(HL)2], [(E,E)-Ni(LH2)3]2+.
From this study, we conclude that the bis-anti and tris-anti forms are the only complexes on the overall studiedpH range thus excluding the bis-amphi (E,Z)nickel(II)complexes from our ligands. This may be general for vic-dioximes. Thus the previous attribution of the green nicke-l(II)-dioximates complexes as the bis-amphi form[(E,Z)-Ni(HL)2], due to isomerisation of the ligand in acidicmedium, may be questionable. Our work shows clearly thatthe elemental analysis, mass spectra, IR and NMR are less

F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235 2233
helpful and that only the crystal structure can identify thestructure.
4. Experimental
4.1. Measurements
Elemental analyses were obtained from Thermo Finni-gan Flash 1112 Instrument and FT-IR spectra wererecorded on a Bio-Rad FTS 175C FTIR spectrophotome-ter as KBr pellets. 1H and 13C NMR spectra were recordedin CDCl3 or in DMSO-d6 solutions on a Varian 500 MHzspectrometer using TMS as an internal reference. The massspectra were acquired on an LCQ-ion trap (Thermofinni-gan, San Jose, CA, USA), equipped with an electrospraysource and were recorded on a Thermo LCQ DECA XP-Max spectrometer. Electrospray full scan spectra, in therange of m/z 100–4000 amu, were obtained infusionthrough fused silica tubing at 10 ml min�1. The solutionswere analyzed in a positive mode.
Optical spectra in UV–Visible region were recorded witha Schimadzu 2001 UV PC spectrophotometer using 1 cmpathlength cuvettes at room temperature. The absorptionspectra of [(E,E)-Ni(HL)2] (1a–3a) and [(E,E)-Ni(LH2)3]Cl2(1b–3b) complexes are taken in 1 � 10�4 mol dm�3 concen-tration in chloroform. Acid-base titrations were performedwith fixed concentrations of corresponding complexes(2 cm3, 1 � 10�4 mol dm�3) and increasing amount of base(KOH/MeOH) and acid (HCl/MeOH or CF3COOH/CHCl3) solutions. (4 � 10�3 mol dm�3; 0, 10, 20, 30, 40,50 ll. It means each addition has 0.2 mol acid or base to1.0 mol complex).
4.2. Crystallography
Suitable crystals were mounted on a Nonius KappaCCDdiffractometer using Mo Ka radiation (k = 0.71073 A) andequipped with a CCD area detector. Intensities were col-lected at 150 K by means of the program COLLECT [37] witha step rotation angle of 1� for the x angle. Reflection index-ing, Lorentz-polarisation correction, peak integration andbackground determination were carried out with the DEN-ZO [38] Frame scaling and unit-cell parameters refinementswere made through the program SCALEPACK [38]. The struc-tures were solved and refined on F2 using the SHELXTL [39]for compound 3a and on F using the CRYSTALS [40] for othercompounds. All non-hydrogen atoms were refined withanisotropic thermal parameters. The hydrogen atoms wereincluded in the final refinement model in calculated posi-tions with isotropic thermal parameters. Crystal structureand refinement data for all novel compounds are summa-rized in Table 2.
4.3. Synthesis
(E,E)-dichloroglyoxime [31] was prepared according to adescribed procedure. All reagents and solvents were
reagent-grade quality and they were obtained from com-mercial suppliers and were purified as described in Perrinand Armarego [41] before use.
4.3.1. Synthesis of the ligands (H2L)
N,N0-bis[allylamino] glyoxime (1): Allylamine (6 ml,4.57 g, 80.00 mmol) was dissolved in absolute ethanol(15 ml) under argon. To this solution (E,E)-dic-hloroglyoxime (3.20 g, 20.00 mmol) in absolute ethanol(20 ml) was added drop-wise. The mixture was stirred atroom temperature overnight. Water was added drop-wiseuntil a white precipitate formed. It was filtered, washedwith water and then dried. The ligand is soluble in dichlo-romethane, n-hexane, ethanol and DMF. Yield: 1.150 g (%29) ; m.p.: 162 �C (dec.). Anal. Calc. for C8H14N4O2: C,48.47; H, 7.12; N, 28.26. Found: C, 48.20; H, 7.04; N,28.45%. IR (KBr) (cm�1) 3400 m(NH), 3310 m(OH), 3040m(Ar–CH), 2920 m(CH), 1650 m(C@N), 1470 and 1365m(CH), 925 m(N–O). MS(ES-MS), m/z (%): 199 (100)[M+H]+. 1H NMR (500 MHz, CDCl3): d = 9.80 (b, 2H,O–H), 5.91–5.98 (m, 2H, CH), 5.33 (d, 4H, @CH2), 5.25(s, 2H, NH), 3.86 (d, 4H, CH2) ppm. 13C NMR(500 MHz, CDCl3): d = 148.20 (C@N–O), 135.46 (CH),116.42 (@CH2), 45.87 (CH2) ppm.
N,N0-bis[anilino] glyoxime (2): To a solution of aniline(0.83 g, 8.92 mmol) in 30 ml of absolute ethanol was addedan excess (1.90 g) of solid NaHCO3 and mixture was stirredat room temperature for 1 h. To this mixture was addeddrop-wise the solution of (E,E)-dichloroglyoxime (0.70 g,4.46 mmol) in 20 ml of ethanol. The mixture was stirredat room temperature. After 24 h, solid NaHCO3 wasremoved by filtration and the filtrate was cooled to 4 �C.The white product was precipitated at this temperatureand filtered off and then recrystallized from ethanol(40 ml). The crystals were filtered, washed with cold etha-nol and cold diethyl ether then dried. Yield: 0.75 g (63%).m.p.: 229 �C (dec.). Anal. Calc. for C14H14N4O2: C,62.21; H, 5.22; N, 20.73. Found: C, 62.13; H, 5.32; N,20.65%, ; IR (KBr) [cm�1] 3375 m(NH), 3275 m(OH), 3025m(Ar–CH), 2890–2791 m(CH), 1642 m(C@N), 1495 and1356 m(CH), 974 m(N–O). MS(ES-MS), m/z (%): 271 (100)[M+H]+. 1H NMR (500 MHz, DMSO-d6): d = 10.41 (s,2H, O–H), 8.01 (s, 2H, NH), 6.97 (t, 4H, Ar–CH), 6.71(m, 6H, Ar–CH) ppm. 13 C NMR ( 500 MHz, DMSO-d6): d = 143.35 (C@N–O), 140.40 (ArC–NH), 128.86 (Ar–CH), 121.93 (Ar–CH), 119.60 (Ar–CH) ppm.
N,N0-bis[1,2,3,4-tetrahydro-5-naphthalenamino]glyoxime
(3): White product was prepared according to the procedureas described for preparation of compound 2 by 1,2,3,4-tet-rahydro-5-naphthalenamine (1.0 g, 6.8 mmol), NaHCO3
(1.5 g) and (E,E)-dichloroglyoxime (0.53 g, 3.4 mmol) andthen recrystalized from methanol. Yield: 0.82 g (64%).m.p.: 248 �C (dec.). Anal. Calc. for C22H26N4O2: C, 69.82;H, 6.92; N, 14.80%. Found: C, 69.63; H, 7.04; N, 14.45%;IR (KBr) [cm�1] 3409 m(NH), 3325 m(OH), 3025 m(Ar–CH), 2927-2837 m(CH), 1654 m(C@N), 1468 and 1352m(CH), 925 m(N–O). MS(ES-MS), m/z (%): 379 (100)

2234 F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235
[M+H]+. 1H NMR (500 MHz, DMSO-d6): d = : 10.40 (s,2H, O–H), 6.96 (t, 2H, Ar–CH), 6.83 (d, 2H, Ar–CH),6.74 (s, 2H, C–NH), 6.70 (d, 2H, Ar–CH), 2.50 (t, 4H,CH2), 1.61 (m, 4H, CH2), 1.50 (m, 4H, CH2), 1.36 (t, 4H,CH2) ppm. 13 C NMR (500 MHz, DMSO-d6) d: 143.85(C@N–O), 136.95 (Ar–C), 136.61 (Ar–C), 128.74 (Ar–C),124.92 (Ar–CH), 123.64 (Ar–CH), 118.44 (Ar–CH), 29.32(CH2), 22.67 (CH2), 22.28 (CH2), 22.12 (CH2) ppm.
4.3.2. Synthesis of [(E,E)-Ni(HL)2] complexes
Bis[N,N0-bis(allylamino)glyoximato]nickel(II) (1a): To anethanol solution (10 ml) of 1 (0.200 g, 1.00 mmol) wasadded an ethanol solution (5 ml) of the NiCl2 � 6H2O(0.119 g, 0.50 mmol) with stirring at 50 �C for 15 min. Adistinct change in color and a decrease in the pH value(�2) of the solution were observed. Potassium hydroxide(0.057 g, 1 mmol) in ethanol (20 ml) was added (pH � 6).The reaction mixture was cooled to room temperature.The red product was precipitated at room temperatureand filtered off and it was dried in vacuum. Recrystalliza-tion from methanol gave red crystal suitable for X-raydiffraction. It is soluble in common organic solventssuch as ethanol, dichloromethane and diethyl ether. Yield:0.099 g (44%). m.p.: 144 �C. Anal. Calc. forC16H26N8NiO4; C, 42.41; H, 5.78; N, 24.73. Found: C,42.52; H, 5.96; N, 24.52%. IR (KBr) [cm�1] 3360 m(NH),3123 m(OH), 3020 m(Ar–CH), 2900 m(CH), 1725 m(O–H–O), 1620 m(C@N), 1410 and 1350 m(CH), 1050 m(N–O).MS(ES-MS), m/z (%): 453 (100) [M]+. 1H NMR(500 MHz, DMSO-d6): d = 16.84 (b, 2H, O–H), 6.22 (s,4H, NH), 5.81–5.89 (m, 4H, CH), 5.08 (d, 8H, @CH2),4.07 (d, 8H, CH2) ppm. 13C NMR (500 MHz, DMSO-d6): d = 144.22 (C@N–O), 137.22 (CH), 115.85 (@CH2),46.70 (CH2) ppm. UV/Vis (chloroform): kmax, nm (e, lmol�1 cm�1) = 260 (18000), 365 (12000).
Bis[N,N0-bis(anilino)glyoximato]nickel(II) (2a): The redproduct was prepared according to the procedure asdescribed for the preparation of 1a by ligand 2 (0.07 g,0.26 mmol), NiCl2 � 6H2O (0.031 g, 0.13 mmol) and KOH(0.011 g, 0.26 mmol) and then recrystallized in the solventmixture of ethanol/DMF in 3/1 ratio. Yield: 0.065 g(84%). m.p.: 259 �C (dec.). Anal. Calc. forC28H26N8NiO4.2(C3H7NO); C, 54.93; H, 5.42; N,18.84%. Found: C, 54.58; H 5.63; N, 18.54%,C28H26N8NiO4.2(C3H7NO); requires C, 54.93; H, 5.42;N, 18.84%. IR (KBr) [cm�1] 3350 m(NH), 3300 m(OH),3025 m(Ar–CH), 2890–2791 m(CH), 1710 m(O–H–O), 1577m(C@N), 1490 and 1352 m(CH), 916 m(N–O). MS(ES-MS),m/z (%): 597 (100) [M]+; 1H NMR (500 MHz, CDCl3):d = 17.00 (b, 2H, O–H), 7.04 (t, 8H, Ar–CH), 6.96 (d,4H, Ar–CH), 6.80 (s, 4H, NH), 6.58 (d, 8H, Ar–CH)ppm. 13C NMR (500 MHz, CDCl3): d = 142.95 (C@N–O), 137.72 (ArC–NH), 128.75 (Ar–CH), 124.55 (Ar–CH),122.21 (Ar–CH) ppm. UV/Vis (chloroform): kmax, nm(e, l mol�1cm�1) = 265 (17500), 380 (11000).
Bis[N,N0-bis(1,2,3,4-tetrahydro-5-naphthalenamino) gly-
oximato]nickel(II)(3a): The red product was prepared
according to the procedure as described for the preparationof 1a by ligand 3 (0.250 g, 0.66 mmol), NiCl2 � 6H2O(0.031 g, 0.33 mmol) and KOH (0.037 g, 0.66 mmol).Yield:0.199 g (74%). m.p.: 286�C (dec.). Anal. Calc. forC44H50N8NiO4 � 2(C3H6O): C, 64.59; H, 6.72; N, 12.05.Found: C, 64.32; H, 6.24; N, 12.47%. IR [cm�1] 3342m(NH), 3325 m(OH), 3027 m(Ar–CH), 2931–2861 m(CH),1709 m(O–H–O), 1566 m(C@N), 1462 and 1336 m(CH),1039 m(N–O). MS (ES-MS), m/z (%): 813 (100) [M]+. 1HNMR (500 MHz, CDCl3): d = 16.48 (b, 2H, O–H–O),6.89 (t, 4H, Ar–CH), 6.73 (d, 4H, Ar–CH), 6.62 (d, 4H,C–NH), 6.42 (s, 4H, Ar–CH), 2.55 (t, 8H, CH2), 2.17 (m,8H, CH2), 2.05 (m, 8H, CH2), 1.56 (t, 8H, CH2) ppm.13C NMR (500 MHz, CDCl3): d = 143.73 (C@N–O),137.69 (Ar–C), 135.84 (Ar–C), 129.52 (Ar–C), 125.51(Ar–CH), 125.27 (Ar–CH), 119.98 (Ar–CH), 29.64 (CH2),24.31 (CH2), 22.55 (CH2), 22.47 (CH2) ppm. UV/Vis (chlo-roform): kmax, nm (e, l mol�1cm�1) = 270 (23000), 383(16500).
4.3.3. Synthesis of [(E,E)-Ni(H2L)3]Cl2 complexes
Tris [N,N0-bis(allylamino)glyoximato]nickel(II)dichloride(1b): To an ethanol solution (5 ml) of 1 (0.200 g, 1 mmol)was added an ethanol solution (5 ml) of NiCl2 � 6H2O(0.119 g, 0.5 mmol) with stirring at 50 �C for 15 min. A dis-tinct change in color and a decrease in the pH value (�2) ofthe solution were observed. The reaction mixture wascooled to room temperature and abandoned to green crys-tal suitable for X-ray diffraction. Yield: 0.067 g (27%).m.p.: 183 �C (dec.). Anal. Calc. for C24H42N12NiO6.2(Cl);C, 39.80; H, 5.85; N, 23.21. Found: C, 39.56; H, 5.98; N,23.37 %. IR (KBr) [cm�1] 3360 m(NH), 3200 m(OH), 3020m(Ar–CH), 2930 m(CH), 1660 m(C@N), 1450 and 1320m(CH), 1010 m(N–O). 1H NMR (500 MHz, DMSO-d6):d = 9.63 (b, 6H, OH), 5.76 (b, 12H, CH and NH), 5.08(d, 12H, @CH2), 3.57 (s, 12H, CH2) ppm. 13C NMR(500 MHz, DMSO-d6): d = 146.65 (C@N–O), 137.80(CH), 115.53 (@CH2), 45.80 (CH2) ppm. UV/Vis (chloro-form): kmax, nm (e, l mol�1 cm�1) = 270 (24000), 596 (32).
Tris[N,N0-bis(anilino)glyoximato]nickel(II)dichloride (2b):
Green crystals suitable for X-ray diffraction were preparedaccording to the procedure as described for the preparationof 1b by ligand 2 (0.094 g, 0.35 mmol), NiCl2 � 6H2O(0.042 g, 0.174 mmol) in 10 ml ethanol. Yield: 0.085 g(78%). m.p.: 241 �C (dec.). Anal. Calc. forC42H42N12NiO6 � 2(C2H6O) � 2(Cl): C, 53.51; H, 5.27; N,16.28. Found: C, 53.56; H, 5.06; N, 16.50%. IR (KBr)[cm�1] 3355 m(NH), 3187 m(OH), 3020 m(Ar–CH), 2930m(CH), 1657 m(C@N), 1597, 1497, 1437 and 1320 m(CH),1012 m(N–O). 1H NMR (500 MHz, CDCl3): d = 10.32 (b,6H, OH), 7.83 (br, 6H, NH); 7.11(m, 12H, ArCH), 6.75(m, 18H, ArCH) ppm. 13C NMR (500 MHz, CDCl3):d = 143.67 (C@N–O), 136.20 (ArC–NH), 130.15 (Ar–CH), 124.80 (Ar–CH), 116.65 (Ar–CH) ppm. UV/Vis (chlo-roform): kmax, nm (e, l mol�1cm�1) = 320 (15500), 590 (57).
Tris[N,N0-bis(1,2,3,4-tetrahydro-5-naphthalenamino)gly-
oximato] nickel(II) dichloride (3b): Green crystals suitable

F. Yuksel et al. / Inorganica Chimica Acta 361 (2008) 2225–2235 2235
for X-ray diffraction were prepared according to the proce-dure as described for the preparation of 1b by ligand 3
(0.250 g, 0.66 mmol), NiCl2 � 6H2O (0.078 g, 0.33 mmol)in 10 ml ethanol. Yield: 0.067 g (48%). m.p.: 260 �C(dec.). Anal. Calc. for C66H78N12NiO6.2(Cl): C, 62.66; H,6.21; N, 13.29. Found: C, 62.27; H, 6.36; N, 13.42%. IR(KBr) [cm�1] 3356 m(NH), 3123 m(OH), 3026 m(Ar–CH),2931–2859 m(CH), 1653 m(C@N), 1468 and 1335 m(CH),1017 m(N–O). 1H NMR (500 MHz, DMSO-d6): d = 10.38(s, 6H, O–H), 6.89 (t, 6H, Ar–CH), 6.76 (d, 6H, Ar–CH),6.68 (d, 6H, C–NH), 6.49 (s, 6H, Ar–CH), 2.48 (t, 12H,CH2), 1.56 (m, 12H, CH2), 1.45 (m, 12H, CH2), 1.30 (m,12H, CH2) ppm. 13C NMR (500 MHz, DMSO-d6):d = 144.55 (C@N–O), 137.64 (Ar–C), 137.26 (Ar–C),129.49 (Ar–C), 125.55 (Ar–CH), 124.42 (Ar–CH), 119.09(Ar–CH), 30.04 (CH2), 26.16 (CH2), 23.31 (CH2),22.77 (CH2) ppm. UV/Vis (chloroform): kmax, nm (e, lmol�1cm�1) = 320 (15200), 601 (58).
Acknowledgements
Travel grants between France and Turkey were spon-sored through the CNRS-TUBITAK (TBAG-U/146105T274) collaboration program. Fatma Yuksel is gratefulto the supporters of her studies in France; TUBITAK-(BI-DEB)-2219 and the Region Rhone-Alpes.
Appendix A. Supplementary material
CCDC 640846, 640847, 640848, 640849, 257321, 640850contain the supplementary crystallographic data for com-pounds 1a, 1b, 2a, 2b, 3a, 3b. These data can be obtainedfree of charge from The Cambridge Crystallographic DataCenter via www.ccdc.cam.ac.uk/data_request/cif. Supple-mentary data associated with this article can be found, inthe online version, at doi:10.1016/j.ica.2007.11.019.
References
[1] L. Tschugaeff, Chem. Ber. 40 (1907) 186.[2] C.P. Ulpiani, Gazz. Chim. Ital. 42 (1) (1912) 503.[3] G. Ponzio, Gazz. Chim. Ital. 51 (2) (1921) 213.[4] M.F. Barker, Chem. News J. Ind. Sci. 130 (1925) 99.[5] M.O. Forster, J. Chem. Soc., Trans. 83 (1903) 514.[6] G. Ponzio, Gazz. Chim. Ital. 60 (1930) 49.[7] A. Chakravorty, Coord. Chem. Rev. 13 (1974) 1.[8] O. Bekaroglu, S. Sarısaban, A.R. Koray, K. Weidenhammer, B.
Nuber, J. Weiss, M.L. Ziegler, Acta Crystallogr. B 34 (1978) 3591.[9] V. Ahsen, F. Gokc�eli, O. Bekaroglu, J. Chem. Soc., Dalton Trans. 8
(1987) 1827.
[10] Y. Gok, H. Kantekin, Polyhedron 16 (14) (1997) 2412.[11] S. Gursoy, M.B. Koc�ak, A. Cihan, A. Gul, O. Bekaroglu, Trans. Met.
Chem. 25 (2000) 474.[12] M. Durmus�, V. Ahsen, D. Luneau, J. Pecaut, Inorg. Chim. Acta 357
(2004) 588.[13] G. Gumus�, V. Ahsen, C. Lebrun, D. Luneau, J. Pecaut, New. J.
Chem. 28 (2004) 177.[14] I. Gurol, G. Gumus�, F. Yuksel, E. Jeanneau, V. Ahsen, Acta
Crystallogr. E62 (2006) m3303.[15] J.H. Boyer, G. Mamikunian, J. Org. Chem. 23 (1958) 1807.[16] (a) M.M. Sheung, R.J. Angelici, D. Powell, R.A. Jacobson, J. Am.
Chem. Soc. 100 (1978) 7068;(b) M.M. Sheung, R.J. Angelici, Inorg. Chem. 19 (1980) 363.
[17] B. Mohr, V. Enkelmann, G. Wegner, Mol. Cryst. Liq. Cryst. 281(1996) 215.
[18] Y. Gok, O. Bekaroglu, Synth. React. Inorg. Metal-Org. Chem. 11 (7)(1981) 621.
[19] M. Ertas�, A.R. Koray, V. Ahsen, O. Bekaroglu, J. Organomet. Chem.319 (1987) 197.
[20] E. Ozcan, E. Karapınar, B. Demirtas�, Trans. Met. Chem. 27 (2002)557.
[21] S. Serin, O. Bekaroglu, Z. Anorg. Allg. Chem. 496 (1983) 197.[22] B. Pedersen, E. Larsen, Acta Chem. Scand. (9) (1973) 3291.[23] R. Hirota, Y. Yoshida, Y. Iida, Polyhedron 12 (14) (1993) 1817.[24] G. Gumus�, V. Ahsen, Mol. Cryst. Liq. Cryst. 348 (2000) 167.[25] I. Gurol, V. Ahsen, Synth. React. Inorg. Metal – Org. Chem. 31 (1)
(2001) 127.[26] L.R.M. Paping, T.P.M. Beelen, C.P.J. Rummens, R. Prins, Polyhe-
dron 1 (6) (1982) 503.[27] H. Endres, T. Jannack, Acta Crystallogr. B 36 (1980) 2136.[28] D.G. Batir, L.D. Ozols, I.I. Bulgak, SU 806686, 1981, (CAN
95:34656).[29] T. Itoh, J. Toyoda, M. Tadokoro, H. Kitagawa, T. Mitani, K.
Nakasuji, Chem. Lett. (1995) 41.[30] H. Endres, T. Jannack, B. Prickner, Acta Crystallogr. B 36 (1980)
2230.[31] K.A. Lance, K.A. Goldsby, D.H. Busch, Inorg. Chem. 29 (1990)
4537.[32] C.J. Grundman, V. Mini, J.M. Dean, D. Hans, Liebigs Ann. Chem.
687 (1965) 191.[33] (a) M. Calleri, G. Ferraris, D. Viterbo, Acta Crystallogr. 22 (1967)
468;(b) L.E. Godycki, R.E. Rundle, Acta Crystallogr. 6 (1953) 487.
[34] R.C. Voter, C.V. Banks, V.A. Fassel, P.W. Kehres, Anal. Chem. 23(1951) 1730.
[35] A. Gul, O. Bekaroglu, J. Chem Soc. Dalton Trans. (1983) 2537.[36] A. Nakamura, A. Konishi, S. Otsuka, J. Chem Soc. Dalton Trans.
(1979) 488.[37] Nonius (1997–2001). COLLECT. Nonius BV, Delft, The Netherlands.[38] Z. Otwinowski, W. Minor, Methods in Enzymology, Academic Press,
NewYork, 276 (1997) 307.[39] SHELXTL; 5.030 ed.; Brucker Analytical X-ray Instruments, Inc.:
Madisson, WI, 1998.[40] P.W. Betteridge, J.R. Carruthers, R.I. Cooper, K. Prout, D.J.
Watkin, J. Appl. Crystallogr. 36 (2003) 1487.[41] D.D. Perrin, W.L. F Armarego, Purification of Laboratory Chem-
icals, second ed., Pergamon Press, Oxford UK, 1980.