mutational analysis of the affinity maturation of antibody 48g7 1 1 edited by d. c. rees
TRANSCRIPT

Article No. jmbi.1999.3197 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 294, 1191±1201
Mutational Analysis of the Affinity Maturation ofAntibody 48G7
Priscilla L. Yang1 and Peter G. Schultz1,2*
1Department of ChemistryUniversity of CaliforniaBerkeley, CA, 94720, USA2Department of Chemistry, TheScripps Research Institute, LaJolla, CA, 92037, USA
E-mail address of the [email protected]
Abbreviations used: Fab, antigenCDR, complementarity-determining
0022-2836/99/501191±11 $30.00/0
The af®nity maturation of antibody 48G7 from its germline predecessor48G7g has been studied at a molecular level through a combination ofstructural and biochemical means. Each of the nine somatic mutationsaccumulated during af®nity maturation has been assessed for gain orloss of function in both the germline and af®nity-matured antibodies.Individual somatic mutations were found to be either positive or neutralin their effects on af®nity for hapten JWJ1, with a marked context-depen-dence for some sites of mutation. In a number of cases signi®cant coop-erativity was found between pairs of somatically mutated residues.Interpretation of the structural changes introduced by many of the pointmutations has been possible due to the availability of high-resolutioncrystal structures of 48G7g and 48G7, and mechanisms by which thesestructural changes may result in enhanced af®nity for hapten have beenidenti®ed. Precise dissection of structure-function relationships in thissystem provides additional insights into the role of cooperativity in theevolution of antibody af®nity. Comparison of 48G7 with previouslycharacterized systems provides a varied view of the structure-functionmechanisms by which the humoral immune system produces largeincreases in af®nity.
# 1999 Academic Press
Keywords: antibody; af®nity maturation; somatic hypermutation;molecular evolution; structure-function
*Corresponding authorIntroduction
The ability of the mammalian humoral immuneresponse to produce high-af®nity receptors for vir-tually any antigen derives from its ability to gener-ate a large library of antibodies encompassing abroad range of speci®cities and then to identifylibrary members with high af®nity for a givenimmunogen. The diversity has multiple sources:®rst, in the assembly of functional antibody genesfrom V, D and J gene segments to create a germlineantibody repertoire whose diversity arises from thecombinatorial nature of its assembly as well asfrom imprecision in the joining process; second, inthe introduction of point mutations to the germlinerepertoire during af®nity maturation. Selection ofhigh-af®nity receptors is then achieved by theexpansion of clones on the basis of antibody af®-nity for the immunogen (Green et al., 1998; Wagner& Neuberger, 1996). Af®nity maturation is an inte-
ing author:
-binding fragment;region.
gral part of the humoral immune response, andcharacterization of this process is crucial to ourunderstanding of the system's remarkable ef®cacy.As a prototypical example of molecular evolution,af®nity maturation may also provide valuableinsights into other such systems, including theevolution of enzymes by natural selection as wellas by in vitro selection methods (Schultz & Lerner,1995).
We and others have begun to characterize thestructural basis of af®nity maturation. Previouslystudied systems include af®nity-matured anti-bodies elicited by p-nitrophenylacetyl (Allen et al.,1988; Alzari et al., 1990; Berek et al., 1985; Evenet al., 1985; Grif®ths et al., 1984; Lozano et al., 1993;Mariuzza et al., 1985; Nakayama et al., 1993;Neuberger & Milstein, 1995; Torigoe et al., 1995),phenyloxazalone (Alzari et al., 1990; Even et al.,1985; Grif®ths et al., 1984; Lozano et al., 1993;Mariuzza et al., 1985), and phenylarsonate haptens(Casson & Manser, 1995a,b; Parhami-Seren et al.,1990; Sharon, 1990; Strong et al., 1991; Wong et al.,1995; Wysocki et al., 1987), as well as transitionstate analogues for carbonate hydrolysis (Pattenet al., 1996; Wedemayer et al., 1997a,b), Diels-Alder
# 1999 Academic Press

1192 Mutational Analysis of the Af®nity Maturation of Antibody 48G7
(Romesberg et al., 1998; Spiller et al., 1997), andoxy-Cope rearrangement reactions (Ulrich et al.,1997; Ulrich & Schultz, 1998). Immunological evol-ution in these systems has been traced by cloningand characterization of the relevant germline ante-cedent, including measurement of hapten-af®nityand/or catalytic ef®ciency. For a number of thesesystems, high-resolution crystal structures of theaf®nity-matured and/or germline antibodies havebeen interpreted with biochemical data to assignfunctional signi®cance to speci®c changes in struc-ture. Individual somatic mutants generated by site-directed mutagenesis and intermediate speciessuch as hybrids made by chain swaps have beenexamined to characterize the evolutionary land-scape separating the germline and af®nity-maturedantibodies. Here, we describe a detailed study ofthe evolution of af®nity in one antibody system,the esterolytic antibody 48G7. Characterization ofindividual somatic mutants and their reversionmutant counterparts has allowed more precise dis-section of structure-function relationships and alsoprovided unique insights into the evolutionarypathways utilized by the humoral response,including the extent of additivity (and cooperativ-ity) that occurs in af®nity maturation.
Catalytic antibody 48G7 was originally isolatedusing a p-nitrophenyl phosphonate hapten, JWJ1(Jacobs, 1990), that mimics the transition state forthe corresponding ester (or carbonate) hydrolysisreaction (Figure 1). The immunological evolutionof this catalyst has been studied through the clon-ing and biochemical characterization (Patten et al.,1996) of its germline predecessor, 48G7g, and theelucidation of high-resolution crystal structures forgermline and af®nity-matured antibodies in com-plex with JWJ1 as well as in unliganded form(Wedemayer et al., 1997a,b). The nine somaticmutations introduced during maturation improveaf®nity for hapten by 3 � 103-fold, with 48G7 and48G7g exhibiting KD values of 50 mM and 16 nM,respectively. Mapping of the three light-chain andsix heavy-chain somatic mutations within thestructures of the antibody-hapten complexes inprevious work (Patten et al., 1996; Wedemayeret al., 1997a,b) revealed that none of the mutationsdirectly contacts the hapten. Rather, structuralchanges introduced at distant locations must some-how be propagated to the hapten-binding site. In
our subsequent studies we have begun to delineatesome of these somewhat subtle structure-functionrelationships.
Results and Discussion
Re-examination of structural data
Although structures of 48G7g and 48G7 unli-ganded antibodies and structures of 48G7g-JWJ1and 48G7-JWJ1 complexes have been described(Wedemayer et al., 1997a,b), we found it informa-tive to re-examine them during the course of ourstudy. Rather than recapitulate previous con-clusions, we describe here structural features thatmay be important to hapten-af®nity but that mayhave been overlooked in previous analyses.
The germline and af®nity-matured antibodiesbind different conformations of the hapten.Although the position of the phosphonate group isessentially the same in the two structures, thelocations of the p-nitrophenyl group and the linkermoiety differ (Figure 2). In the 48G7-JWJ1 complex,the phenyl ring lies diagonally in the binding sitewith the nitro group juxtaposed to CDR L1 andthe aromatic ring itself surrounded by residuesArg46L, Leu89L, Tyr91L, His35H, Tyr100H andGly101H. The linker, surrounded by a quartet oftyrosine residues (32L, 91L, 99H and 100H), is nearlyvertical in the path it follows to the top of the com-bining site. We have termed this arrangement thehigh-af®nity conformation of the hapten. In con-trast, 48G7g buries the phenyl group almost verti-cally in a pocket lined by Leu89L, Arg96L, Phe98L,His35H, Val37H, Trp47H and Trp104H; the linker isenclosed by a shell formed by Arg46L, Tyr91L,Tyr94L, Tyr33H and Tyr99H. We refer to this as thelow-af®nity conformation of the hapten.
Overlays of the af®nity-matured and germlineantibody crystal structures suggest that light-chainmutations modulate af®nity via rearrangement ofside-chains while heavy-chain mutations lead tosigni®cant reorganization of both backbone andside-chains. We have sought to map the observablechanges in antibody-hapten interactions and af®-nity more precisely to individual somaticmutations. Af®nities of individual somatic andreversion mutations of 48G7g and 48G7, respect-ively, were assessed by Scatchard analysis of sur-
Figure 1. The 48G7 antibody-catalysed ester and carbonatehydrolysis reaction and presumedtransition state with phosphonatehapten JWJ1.

Figure 2. Antibodies 48G7g (blue) and 48G7 (red)complexed with hapten JWJ1. Residues are labelledaccording to Kabat et al. (1991). Arg96L and His35H
form an oxyanion hole for the phosphonate group inboth complexes. Tyr33H forms a hydrogen bond to thephosphonate group in the af®nity-matured complex butpacks against the methylene linker in the germline com-plex. The hapten occupies non-equivalent conformationsin the two complexes. The conformation observed inthe germline complex (blue) is referred to as the lower-af®nity conformation, while that observed in theaf®nity-matured complex (red) is referred to as thehigher-af®nity conformation.
Figure 3. Somatic mutations of the light chain. Thenitro group of the high-af®nity conformation of JWJ1(red) clashes sterically with the side-chain of Ser34L andArg46L of the germline antibody. Somatic mutationAsp55LHis is associated with a 1.6 AÊ displacement ofthe Arg46L guanidinium group towards His55L andaway from the nitro group of the high-af®nity confor-mation of JWJ1. Somatic mutation Ser34LGly alleviatessteric clash by removal of the hydroxyl side-chain.Residue 30L is solvent-exposed and presumably formspart of the interface with the conjugating protein orpeptide.
Mutational Analysis of the Af®nity Maturation of Antibody 48G7 1193
face plasmon resonance measurements. Equili-brium af®nity constants (KA) were used to calcu-late equilibrium dissociation constants (KD), freeenergies of binding (�G), and changes in freeenergy of binding (��G) for site-directed mutantsrelative to the germline or af®nity-matured anti-body from which they were derived. The func-tional changes produced by individual mutationswere then interpreted structurally through examin-ation of the available crystal structure.
Light-chain somatic mutations:complementarity to the nitro group
The functional importance of the Ser! Glysomatic mutation at residue 34L was directlysuggested by structural data: removal of the side-chain at this position would prevent steric clashwith the nitro group of the hapten in the high-af®-nity conformation, whereas the presence of a side-chain at position 34L would favour complexationof 48G7g with the hapten in its low-af®nity confor-mation (Figure 3). This hypothesis was con®rmedby the behaviour of the single somatic and rever-sion mutants which respectively produce ÿ0.6 and�1.0 kcal/mol changes in free energy of binding(Table 1).
The effects of somatic mutations at positions 30L
and 55L on af®nity were not easily predicted onthe basis of structure (Wedemayer et al., 1997a).The importance of the mutation at residue 55L wasdemonstrated by the 2.0 kcal/mol gain in freeenergy of binding associated with the somaticmutation Asp55LHis introduced to 48G7g and the0.9 kcal/mol loss of free energy of bindingobserved for the His55LAsp reversion mutation of48G7 (Table 1). The modulation of af®nity by thissite may be mediated by the Arg46L-Asp55L salt-bridge in the 48G7g-JWJ1 complex. Loss of thissalt-bridge due to the Asp55LHis mutation shiftsthe guanidinium group 1.6 AÊ away from the hap-ten-binding site and apparently allows binding ofthe high-af®nity conformation of the hapten.
The Ser30LAsn somatic mutation has virtuallyno effect on the af®nity of 48G7g (��Gÿ0.1 kcal/mol, Table 1) whereas the Asn30LSerreversion mutation produces a 1.5 kcal/mol loss infree energy of binding. The context-dependence ofmutations at site 30L implies that the changes infunction it causes are modulated by structuralchanges introduced at other sites of somaticmutation.
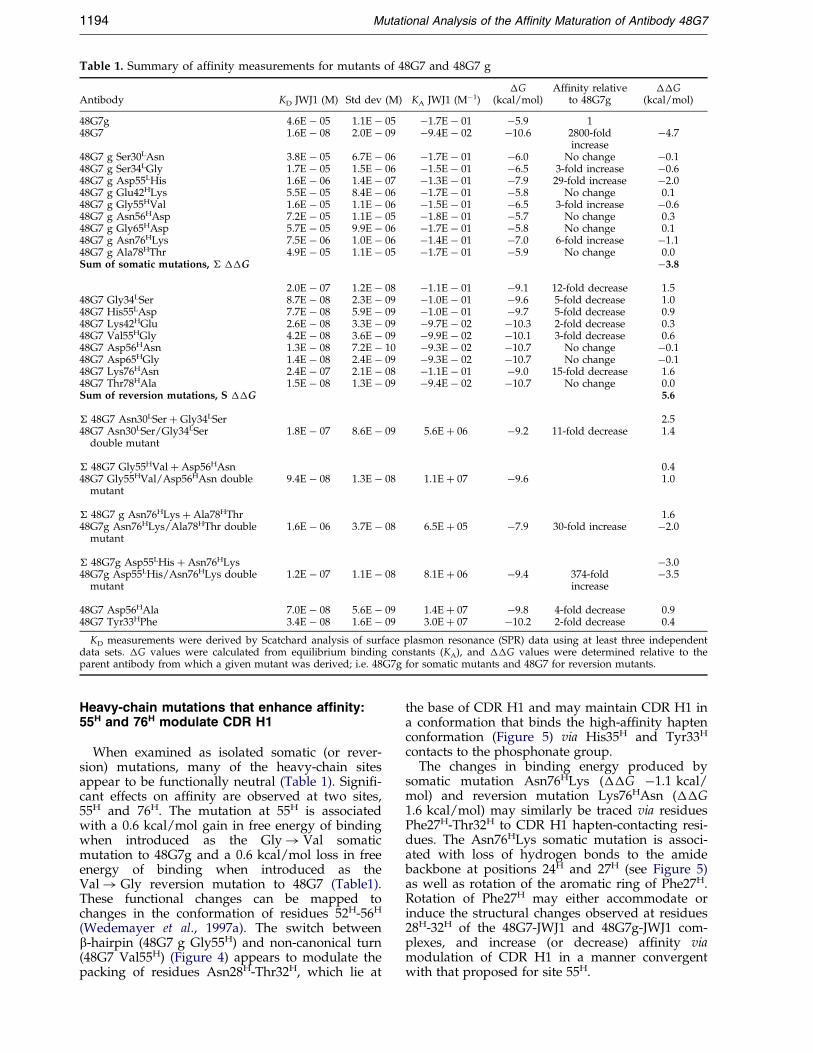
Table 1. Summary of af®nity measurements for mutants of 48G7 and 48G7 g
Antibody KD JWJ1 (M) Std dev (M) KA JWJ1 (Mÿ1)�G
(kcal/mol)Affinity relative
to 48G7g��G
(kcal/mol)
48G7g 4.6E ÿ 05 1.1E ÿ 05 ÿ1.7E ÿ 01 ÿ5.9 148G7 1.6E ÿ 08 2.0E ÿ 09 ÿ9.4E ÿ 02 ÿ10.6 2800-fold
increaseÿ4.7
48G7 g Ser30LAsn 3.8E ÿ 05 6.7E ÿ 06 ÿ1.7E ÿ 01 ÿ6.0 No change ÿ0.148G7 g Ser34LGly 1.7E ÿ 05 1.5E ÿ 06 ÿ1.5E ÿ 01 ÿ6.5 3-fold increase ÿ0.648G7 g Asp55LHis 1.6E ÿ 06 1.4E ÿ 07 ÿ1.3E ÿ 01 ÿ7.9 29-fold increase ÿ2.048G7 g Glu42HLys 5.5E ÿ 05 8.4E ÿ 06 ÿ1.7E ÿ 01 ÿ5.8 No change 0.148G7 g Gly55HVal 1.6E ÿ 05 1.1E ÿ 06 ÿ1.5E ÿ 01 ÿ6.5 3-fold increase ÿ0.648G7 g Asn56HAsp 7.2E ÿ 05 1.1E ÿ 05 ÿ1.8E ÿ 01 ÿ5.7 No change 0.348G7 g Gly65HAsp 5.7E ÿ 05 9.9E ÿ 06 ÿ1.7E ÿ 01 ÿ5.8 No change 0.148G7 g Asn76HLys 7.5E ÿ 06 1.0E ÿ 06 ÿ1.4E ÿ 01 ÿ7.0 6-fold increase ÿ1.148G7 g Ala78HThr 4.9E ÿ 05 1.1E ÿ 05 ÿ1.7E ÿ 01 ÿ5.9 No change 0.0Sum of somatic mutations, � ��G ÿ3.8
2.0E ÿ 07 1.2E ÿ 08 ÿ1.1E ÿ 01 ÿ9.1 12-fold decrease 1.548G7 Gly34LSer 8.7E ÿ 08 2.3E ÿ 09 ÿ1.0E ÿ 01 ÿ9.6 5-fold decrease 1.048G7 His55LAsp 7.7E ÿ 08 5.9E ÿ 09 ÿ1.0E ÿ 01 ÿ9.7 5-fold decrease 0.948G7 Lys42HGlu 2.6E ÿ 08 3.3E ÿ 09 ÿ9.7E ÿ 02 ÿ10.3 2-fold decrease 0.348G7 Val55HGly 4.2E ÿ 08 3.6E ÿ 09 ÿ9.9E ÿ 02 ÿ10.1 3-fold decrease 0.648G7 Asp56HAsn 1.3E ÿ 08 7.2E ÿ 10 ÿ9.3E ÿ 02 ÿ10.7 No change ÿ0.148G7 Asp65HGly 1.4E ÿ 08 2.4E ÿ 09 ÿ9.3E ÿ 02 ÿ10.7 No change ÿ0.148G7 Lys76HAsn 2.4E ÿ 07 2.1E ÿ 08 ÿ1.1E ÿ 01 ÿ9.0 15-fold decrease 1.648G7 Thr78HAla 1.5E ÿ 08 1.3E ÿ 09 ÿ9.4E ÿ 02 ÿ10.7 No change 0.0Sum of reversion mutations, S ��G 5.6
� 48G7 Asn30LSer � Gly34LSer 2.548G7 Asn30LSer/Gly34LSer
double mutant1.8E ÿ 07 8.6E ÿ 09 5.6E � 06 ÿ9.2 11-fold decrease 1.4
� 48G7 Gly55HVal � Asp56HAsn 0.448G7 Gly55HVal/Asp56HAsn double
mutant9.4E ÿ 08 1.3E ÿ 08 1.1E � 07 ÿ9.6 1.0
� 48G7 g Asn76HLys � Ala78HThr 1.648G7g Asn76HLys/Ala78HThr double
mutant1.6E ÿ 06 3.7E ÿ 08 6.5E � 05 ÿ7.9 30-fold increase ÿ2.0
� 48G7g Asp55LHis � Asn76HLys ÿ3.048G7g Asp55LHis/Asn76HLys double
mutant1.2E ÿ 07 1.1E ÿ 08 8.1E � 06 ÿ9.4 374-fold
increaseÿ3.5
48G7 Asp56HAla 7.0E ÿ 08 5.6E ÿ 09 1.4E � 07 ÿ9.8 4-fold decrease 0.948G7 Tyr33HPhe 3.4E ÿ 08 1.6E ÿ 09 3.0E � 07 ÿ10.2 2-fold decrease 0.4
KD measurements were derived by Scatchard analysis of surface plasmon resonance (SPR) data using at least three independentdata sets. �G values were calculated from equilibrium binding constants (KA), and ��G values were determined relative to theparent antibody from which a given mutant was derived; i.e. 48G7g for somatic mutants and 48G7 for reversion mutants.
1194 Mutational Analysis of the Af®nity Maturation of Antibody 48G7
Heavy-chain mutations that enhance affinity:55H and 76H modulate CDR H1
When examined as isolated somatic (or rever-sion) mutations, many of the heavy-chain sitesappear to be functionally neutral (Table 1). Signi®-cant effects on af®nity are observed at two sites,55H and 76H. The mutation at 55H is associatedwith a 0.6 kcal/mol gain in free energy of bindingwhen introduced as the Gly! Val somaticmutation to 48G7g and a 0.6 kcal/mol loss in freeenergy of binding when introduced as theVal! Gly reversion mutation to 48G7 (Table1).These functional changes can be mapped tochanges in the conformation of residues 52H-56H
(Wedemayer et al., 1997a). The switch betweenb-hairpin (48G7 g Gly55H) and non-canonical turn(48G7 Val55H) (Figure 4) appears to modulate thepacking of residues Asn28H-Thr32H, which lie at
the base of CDR H1 and may maintain CDR H1 ina conformation that binds the high-af®nity haptenconformation (Figure 5) via His35H and Tyr33H
contacts to the phosphonate group.The changes in binding energy produced by
somatic mutation Asn76HLys (��G ÿ1.1 kcal/mol) and reversion mutation Lys76HAsn (��G1.6 kcal/mol) may similarly be traced via residuesPhe27H-Thr32H to CDR H1 hapten-contacting resi-dues. The Asn76HLys somatic mutation is associ-ated with loss of hydrogen bonds to the amidebackbone at positions 24H and 27H (see Figure 5)as well as rotation of the aromatic ring of Phe27H.Rotation of Phe27H may either accommodate orinduce the structural changes observed at residues28H-32H of the 48G7-JWJ1 and 48G7g-JWJ1 com-plexes, and increase (or decrease) af®nity viamodulation of CDR H1 in a manner convergentwith that proposed for site 55H.

Figure 4. Structural differences between 48G7g-JWJ1(blue) and 48G7-JWJ1 (red) complexes associated withthe Gly55HVal and Asn56HAsp somatic mutations.Mutation of Gly55H to valine leads to a change in turnconformation. Residues comprising the turn undergosigni®cant displacement. Pro52aH in particular under-goes a 4 AÊ displacement of its Ce. The side-chain ofThr32H moves to accommodate the new position ofPro52aH (not shown here for purposes of clarity). For-mation of a salt-bridge between Arg50H and the somati-cally mutated Asp56H alters the Arg50H-Tyr33H
interface such that a new contact between Tyr33H andthe phosphonate group of the hapten is formed in theaf®nity-matured complex (red).
Figure 5. Changes in residues 28H-32H and CDR H1that are associated with somatic mutations in the heavychain. Germline residue Asn76H (blue) hydrogen bondsto the backbone of Ala24H and Phe27H, and loss of thesebonds in the af®nity-matured complex (red) is correlatedwith large structural changes in residues Phe27H-Thr32H
(side-chains not shown) and high-af®nity binding. Resi-due Thr78H packs against Met34H in the af®nity-matured complex (red). The inability of germline residueAla78H to form a similar contact leads to a hydrophobicinteraction between Phe27H and Met34H that mayreinforce the Asn76H-Ala24H and Asn76H-Phe27H hydro-gen bonds that are correlated with low-af®nity binding.
Mutational Analysis of the Af®nity Maturation of Antibody 48G7 1195
Heavy-chain mutations that appear functionallyneutral; 42H, 56H, 65H and 78H
Although discrete changes in structure areassociated with mutations at sites 42H and 65H,comparison of 48G7g and 48G7 unliganded andhapten-bound structures suggests no obviousimpact of these structural changes upon af®nity.Individual somatic mutations Glu42HLys andGly65HAsp do not alter the af®nity of the 48G7g-JWJ1 complex, nor does the reversion mutationAsp65HGly alter the af®nity of the 48G7-JWJ1 com-plex. The Lys42HGlu reversion mutation causes aslight decrease in the af®nity (��G �0.3 kcal/mol;Table 1).
Somatic mutation at residue 56H allows for-mation of a salt-bridge between Asp56H andArg50H. Formation of a new hydrogen bondbetween the phosphonate group and Tyr33H
(Figure 4) was predicted to enhance af®nity forhapten (Wedemayer et al., 1997a); however, theAsn56HAsp somatic mutation results in a smalldecrease in af®nity (��G �0.3 kcal/mol) whenintroduced to 48G7g, and the Asp56HAsn reversionmutation has no effect on 48G7's af®nity forhapten (Table 1). This suggests that the Tyr33H-phosphonate contact observed in the af®nity-matured complex does little to boost af®nity of theantibody for its hapten. Similarly, the hydrophobicpacking interaction with Met34H gained uponsomatic mutation of Ala78H to Thr does notincrease the af®nity of 48G7g (��G ÿ0.1 kcal/
mol; Table 1) or decrease the af®nity of 48G7(��G �0.1 kcal/mol). It is possible, however, thatboth sites exert positive effects on af®nity whenplaced in the context of other somatic mutations.Sites 56H and 78H are proximal to mutations 55H
and 76H, respectively, and may augment the gain(or loss) of function produced by these sites viatheir effect on residues 27H-32H.
Context-dependence effects
In perhaps the simplest model of molecular evol-ution during af®nity maturation, all somaticmutations observed would enhance af®nity andtogether behave in an additive fashion. This simplemodel does not appear to ®t the 48G7 system,since over half of 48G7's nine sites of somaticmutation have no effect on af®nity when character-ized as the somatic mutation (30L, 42H, 65H and78H) or the reversion mutation (56H, 65H and 78H)in isolation. Isolated mutations can represent onlyone step along an evolutionary pathway, however,and the behaviour of the individual somatic andreversion mutants of 48G7g/48G7 may be mislead-ing. Cooperative interactions provide one mechan-ism by which a given mutation's effect on functioncan vary at different points along a single ormultiple evolutionary pathways and may explainthe apparent neutral behaviour of mutations atsites 56H and 78H as well as the non-complemen-tary behaviour observed for pairs of somatic and
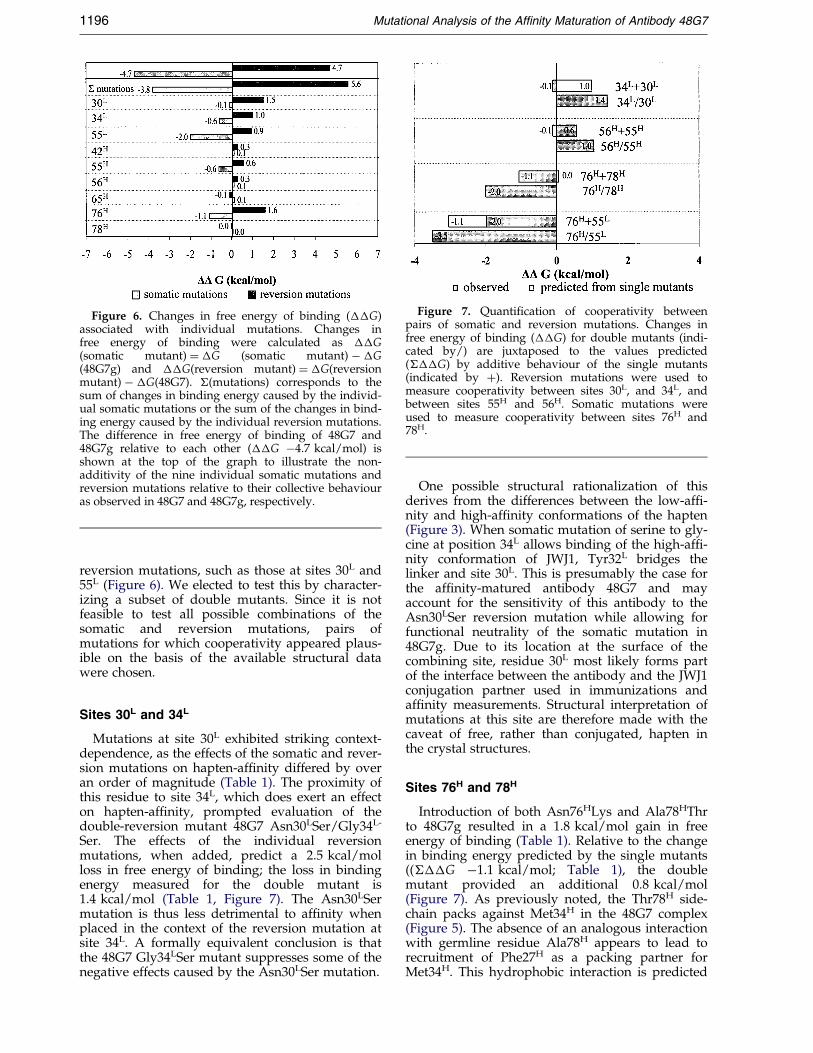
Figure 6. Changes in free energy of binding (��G)associated with individual mutations. Changes infree energy of binding were calculated as ��G(somatic mutant) � �G (somatic mutant) ÿ �G(48G7g) and ��G(reversion mutant) � �G(reversionmutant) ÿ �G(48G7). �(mutations) corresponds to thesum of changes in binding energy caused by the individ-ual somatic mutations or the sum of the changes in bind-ing energy caused by the individual reversion mutations.The difference in free energy of binding of 48G7 and48G7g relative to each other (��G ÿ4.7 kcal/mol) isshown at the top of the graph to illustrate the non-additivity of the nine individual somatic mutations andreversion mutations relative to their collective behaviouras observed in 48G7 and 48G7g, respectively.
Figure 7. Quanti®cation of cooperativity betweenpairs of somatic and reversion mutations. Changes infree energy of binding (��G) for double mutants (indi-cated by/) are juxtaposed to the values predicted(���G) by additive behaviour of the single mutants(indicated by �). Reversion mutations were used tomeasure cooperativity between sites 30L, and 34L, andbetween sites 55H and 56H. Somatic mutations wereused to measure cooperativity between sites 76H and78H.
1196 Mutational Analysis of the Af®nity Maturation of Antibody 48G7
reversion mutations, such as those at sites 30L and55L (Figure 6). We elected to test this by character-izing a subset of double mutants. Since it is notfeasible to test all possible combinations of thesomatic and reversion mutations, pairs ofmutations for which cooperativity appeared plaus-ible on the basis of the available structural datawere chosen.
Sites 30L and 34L
Mutations at site 30L exhibited striking context-dependence, as the effects of the somatic and rever-sion mutations on hapten-af®nity differed by overan order of magnitude (Table 1). The proximity ofthis residue to site 34L, which does exert an effecton hapten-af®nity, prompted evaluation of thedouble-reversion mutant 48G7 Asn30LSer/Gly34L-
Ser. The effects of the individual reversionmutations, when added, predict a 2.5 kcal/molloss in free energy of binding; the loss in bindingenergy measured for the double mutant is1.4 kcal/mol (Table 1, Figure 7). The Asn30LSermutation is thus less detrimental to af®nity whenplaced in the context of the reversion mutation atsite 34L. A formally equivalent conclusion is thatthe 48G7 Gly34LSer mutant suppresses some of thenegative effects caused by the Asn30LSer mutation.
One possible structural rationalization of thisderives from the differences between the low-af®-nity and high-af®nity conformations of the hapten(Figure 3). When somatic mutation of serine to gly-cine at position 34L allows binding of the high-af®-nity conformation of JWJ1, Tyr32L bridges thelinker and site 30L. This is presumably the case forthe af®nity-matured antibody 48G7 and mayaccount for the sensitivity of this antibody to theAsn30LSer reversion mutation while allowing forfunctional neutrality of the somatic mutation in48G7g. Due to its location at the surface of thecombining site, residue 30L most likely forms partof the interface between the antibody and the JWJ1conjugation partner used in immunizations andaf®nity measurements. Structural interpretation ofmutations at this site are therefore made with thecaveat of free, rather than conjugated, hapten inthe crystal structures.
Sites 76H and 78H
Introduction of both Asn76HLys and Ala78HThrto 48G7g resulted in a 1.8 kcal/mol gain in freeenergy of binding (Table 1). Relative to the changein binding energy predicted by the single mutants((���G ÿ1.1 kcal/mol; Table 1), the doublemutant provided an additional 0.8 kcal/mol(Figure 7). As previously noted, the Thr78H side-chain packs against Met34H in the 48G7 complex(Figure 5). The absence of an analogous interactionwith germline residue Ala78H appears to lead torecruitment of Phe27H as a packing partner forMet34H. This hydrophobic interaction is predicted

Mutational Analysis of the Af®nity Maturation of Antibody 48G7 1197
to act cooperatively with the hydrogen bondsbetween Asn76H and the amide backbone ofAla24H and Phe27H in maintaining residues 28H-35H in an arrangement that binds the low-af®nityconformation of the hapten.
Sites 55H and 56H
The absence of a change in af®nity for the48G7 g Asn56HAsp and 48G7 Asp56HAsn mutantswas initially surprising, since the somatic mutationappeared to allow formation of a new hydrogenbond between Tyr33H and the phosphonate groupof the hapten (Figure 4). Since the structural effectsof heavy-chain mutations in general appear to con-verge upon CDR H1 with a high likelihood ofinterplay amongst them, it seemed possible thatthe essentially wild-type af®nities observed for the48G7g Asn56HAsp and 48G7 Asp56HAsn mutantsmight be explained by the absence of complemen-tary structural changes caused by other somaticmutation. Alternatively, the differences betweenthe germline and af®nity-matured complexesobserved in the static crystallographic picture andattributed to changes at residue 56H lack functionalsigni®cance. To eliminate this possibility, we ®rstsought to verify the importance of each of the mol-ecular interactions (Asp56H-Arg50H salt-bridge,Tyr33H-phosphonate group hydrogen bond)through which residue 56H is believed to affecthapten-binding. Mutation of Tyr33H to Phe in theaf®nity-matured antibody resulted in removal ofthe side-chain hydroxyl group and led to a small,but measurable, decrease in af®nity (��G �0.4kcal/mol; Table 1) for 48G7. The importance of thehydrogen bond between Tyr33H and the phospho-nate group is further substantiated by the observed®vefold decrease in catalytic ef®ciency observedfor this mutant (Patten et al., 1996). Similarly, thebehaviour of 48G7 Asp56HAla (��G �0.9 kcal/mol; Table 1) con®rmed the importance of dipoleinteractions between this site and Arg50H. We con-clude that the static quality of crystal structuresmay preclude our observation of an Asn56H-Arg50H dipole interaction that is nevertheless func-tionally signi®cant in solution and that can beablated by mutation of 56H to alanine.
Residue 55H was then examined as a possiblepartner for cooperative interactions with 56H, sincechanges in the conformation of the turn in which56H resides appear to be induced by mutations at55H. Simple additivity for the single reversionmutations at sites 55H and 56H predicts a 0.4 kcal/mol loss in free energy of binding, but reversion ofboth sites is experimentally more detrimental thanpredicted: the actual loss of binding energyobserved for 48G7 Val55HGly/Asp56HAsn is1.0 kcal/mol, with an additional 0.6 kcal/mol inbinding free energy lost due to cooperativitybetween the sites (Table 1, Figure 7).
Sites 55L and 76H
Somatic mutations at sites 55L and 76H cause thelargest changes in af®nity (��G 5 1 kcal/mol)observed for individual somatic or reversionmutations in the 48G7 system. The changes infunction associated with each site appear to bemediated by structurally distinct mechanisms:while mutations at site 55L affect complementarityto the nitro group of the hapten (Figure 3),mutations at residue 76H appear to affect contactsof the phosphonate group to Tyr33H and His35H
via changes in the packing of residues 29H-32H
(Figure 5). The 55L/76H pair was therefore pre-dicted to be a good contrast to the other pairs ofmutations characterized, the expectation being thatAsp55LHis and Asn76HLys somatic mutationswould behave independently and exhibit additivebehaviour when characterized as the doublemutant. Modest cooperativity was observed in theform of an additional 0.4 kcal/mol in free energyof binding (��G ÿ3.5 kcal/mol; Table 1) for the48G7g Asp55LHis/Asn76HLys double somaticmutant (Figure 7). Residues 55L and 76H illustratethat structural changes can amplify (or diminish)the biochemical effects originating at structurallydistant sites.
Although somatic mutations at sites 42H and 65H
appear to be functionally neutral, it is clearly poss-ible that they assume a gain of function phenotypewhen paired with other mutations of the nine-member set. Mutations that are truly neutral maybe identi®ed in future studies through recombina-tion of 48G7 and 48G7g genes by PCR shuf¯ingtechniques followed by analysis of recombinantclones using phage-display methods.
Affinity maturation as a process ofmolecular evolution
Af®nity maturation has been studied to varyinglevels of structural and biochemical detail using anumber of systems. The earliest studies of nitro-phenyl (Allen et al., 1988; Mizutani et al., 1995;Nakayama et al., 1993; Torigoe et al., 1995; Yuhaszet al., 1995), phenyloxazolone (Alzari et al., 1990;Berek et al., 1985; Even et al., 1985; Grif®ths et al.,1984; Lozano et al., 1993; Mariuzza et al., 1985;Neuberger & Milstein, 1995), and phenylarsonate-elicited systems (Casson & Manser, 1995a,b;Parhami-Seren et al., 1990; Sharon, 1990; Stronget al., 1991; Wong et al., 1995; Wysocki et al., 1987)utilized families of antibodies generated with thesame hapten. Since such families included severalsubsets of a large set of somatic mutations, func-tional and structural differences at the level ofsingle mutations could not be examined and thefunctional signi®cance of a given site of somaticmutation had to be inferred on the basis of aresidue's proximity to the hapten-binding site.Structural data were limited, precluding detectionof changes in global structure during af®nity matu-

1198 Mutational Analysis of the Af®nity Maturation of Antibody 48G7
ration and forcing the assumption that somaticmutations were not context-dependent.
We have recently focused our efforts oncomparison of af®nity-matured antibodies withtheir germline predecessors as well as with``sibling'' antibodies that arose from the sameV-(D)-J rearrangement but diverged duringsomatic hypermutation (Patten et al., 1996;Romesberg et al., 1998; Spiller et al., 1997; Ulrichet al., 1997; Ulrich & Schultz, 1998; Wedemayeret al., 1997a). High-resolution structural characteriz-ation of both germline and af®nity-maturedcomplexes and examination of individualmutations have enabled changes in global structureand function to be dissected into discrete changesproduced by individual somatic mutations. Ourstudies thus far present a somewhat varied pictureof the evolution of structure and function duringaf®nity maturation.
The Diels-Alder antibody 39A11 presents thesimplest case, thus far, since it acquired only twosomatic mutations during its af®nity maturationand the majority of the gain in af®nity can beattributed to only one of the mutations (Romesberget al., 1998). The oxy-Cope antibody AZ28 accumu-lated six somatic mutations over the course of itsimmunological evolution (Ulrich et al., 1997), fourof which were found to be functionally signi®cantwhen characterized in isolation. The global gainobserved when comparing af®nity-matured andgermline antibodies in this system could beaccounted for by additive behaviour of the fourindividual gain of function mutations.
The apparent absence of cooperativity in anti-bodies such as AZ28 and 39A11 may underscoreinherent biases during molecular evolution duringaf®nity maturation of antibodies. First, somaticmutation of antibody genes is not a random pro-cess, as known ``hot spots'' of mutation have beenidenti®ed (Green et al., 1998). If such ``hot spots''represent sites at which point mutations can pro-duce maximal changes in function, the process ofaf®nity maturation may be more ef®cient thanevolution by random mutation. This is supportedby studies of anti-phosphocholine antibodies, T15and D16, which share the same germline VH gene.Random introduction of somatic mutations to boththe primary (T15) and secondary (D16) responseantibodies in vitro led to >50 % of mutants withdecreased af®nity (Chen et al., 1995), indicatingthat a random somatic hypermutation mechanismwould be inef®cient. Interestingly, gain of functionmutations was observed only in the secondaryresponse antibody (D16), and never in the primaryresponse antibody (T15), implying that some lightand heavy-chain combinations are structurallyincompetent as substrates for improving haptenaf®nity via somatic hypermutation (Chen et al.,1995).
Second, fundamental differences in structuremay signi®cantly affect the evolution of a givenprotein fold. On the basis of the apparent plasticityof the antibody combining site in achieving high
af®nity for a diverse range of antigens, Schultz andcolleagues entertained the idea that the immuno-globulin fold has been evolved to adapt rapidly (inevolutionary terms) to selective pressure(Romesberg et al., 1998; Wedemayer et al., 1997a).The adaptation of the immunoglobulin superfamilyto the binding of diverse ligands (Huang et al.,1997; Yoshihara et al., 1991) illustrates an inherentplasticity of this receptor protein fold that seems tosupport this idea. One consequence of this may bethat many different subsets, each comprised byrelatively few mutations, can satisfy the pressureto evolve nanomolar af®nity during af®nity matu-ration. Thus, while cooperativity could be ben-e®cial, it is not necessary in order to survive theselection process. If the nanomolar af®nities typi-cally selected during af®nity maturation can beachieved relatively ef®ciently via additivemutations, the evolutionary time-scale may be tooshort to allow signi®cant accumulation of function-ally neutral mutations (Kimura, 1970; Zhang & Gu,1998).
The context-dependence effects and cooperativ-ity amongst somatic mutations in the 48G7 systemdemonstrate that af®nity maturation can recruitstructural changes that appear functionally neutralin isolation but that contribute to af®nity in a non-additive fashion. Cooperativity between proximalmutations (30L/34L, 55H/56H and 76H/78H), aswell as between non-proximal mutations whoseindividual members act upon different moieties ofthe hapten (55L/76H), illustrates that the sources ofa global gain in function may not always be deli-neated through local changes in structure pro-duced by individual mutations. The question thenis, what determines the extent of additivity andcooperativity during af®nity maturation?
48G7 may be considered more highly evolvedthan antibodies 39A11 and AZ28 with respect tothe number of somatic mutations it acquired aswell as with respect to the gain in af®nity achievedduring the course of its af®nity maturation. In con-trast, most products of af®nity maturation charac-terized to date represent an approximately 102-foldimprovement relative to their germline startingpoints (Allen et al., 1988; Berek et al., 1985; Maizels,1995; Pewzner-Jung et al., 1996; Romesberg et al.,1998; Sharon, 1990; Torigoe et al., 1995; Ulrich et al.,1997). Due to the unusually low af®nity (KD > 10ÿ5)of its evolutionary starting point, 48G7g, the press-ure to evolve nanomolar af®nity in the 48G7system may have necessitated exploration of a lar-ger sequence space and retention of neutralmutations in addition to gain of function somaticmutations during the course of af®nity maturation.The short time-scale over which af®nity maturationoccurs does not appear to prohibit the evolution ofcooperative interactions; rather, simple additivestructure-function relationships have evolved inthe 39A11 and AZ28 systems because they satisfythe selective pressure applied during af®nity matu-ration. The greater challenge of evolving nanomo-lar af®nity from a micromolar receptor such as

Mutational Analysis of the Af®nity Maturation of Antibody 48G7 1199
48G7g may provide the selective pressure necess-ary for evolution of cooperativity. Directed evol-ution of 48G7g and other germline antibodies byin vitro mutagenesis techniques (e.g. error-pronePCR and recombination by DNA shuf¯ing)coupled with phage display selections may allowthis to be tested. By precise characterization of thisand other antibody systems, the diversity ofresponses observed during af®nity maturation canbe assessed. By examining the co-evolution ofstructure and function during af®nity maturation,we gain insights (and inspiration) that can beapplied towards molecular evolution in other pro-tein systems.
Materials and Methods
Mutagenesis
Somatic mutations of 48G7g were generated inpUC119-48G7g, a phagemid vector created by subclon-ing of the EcoRI/SalI fragment of vector pDEI554 (Pattenet al., 1996) into pUC119. Reversion mutants of 48G7were generated and expressed in phagemid vectorpDEI440 (Patten et al., 1996; Ulrich et al., 1995). Doublereversion mutants (30L/34L, 55H/56H) and doublesomatic mutants (55L/76H, 76H/78H) were generated bysite-directed mutagenesis of templates already contain-ing one of the two mutations. Site-directed mutagenesiswas accomplished using a modi®ed version of the Kun-kel protocol (Kunkel et al., 1987). Successful mutationwas screened when possible by restriction mapping andveri®ed in all cases by dideoxy sequencing of phagemidDNA.
Expression and purification of recombinantFab fragments
Antibodies were produced as mouse-human chimericFab fragments under the control of the phoA promoterin Escherichia coli strain 25F2, largely as described byUlrich et al. (1995). 48G7 and 48G7 reversion mutantswere expressed as shake ¯ask cultures; 48G7g and48G7g somatic mutants were fermented. Fab fragmentswere extracted from harvested cells and puri®ed byUltralink2 Immobilized Protein G chromatography asdescribed (Ulrich, 1996; Ulrich et al., 1995). Alternatively,crude periplasmic extracts were dialyzed into phosphatebuffer (pH 7.0) and puri®ed by FPLC using Hi-Trap Pro-tein G columns (Pharmacia Amersham) following themanufacturer's instructions. Purity was assessed by SDS-PAGE. Puri®ed Fab was passed through an FPLC Super-ose12 column into BIAcore buffer (0.02 M Tris-HCl(pH 8.2), 0.05 M NaCl, 0.005 % (v/v) Surfactant P20)immediately prior to measurement of equilibriumbinding constants by surface plasmon resonance. Fabconcentrations were determined by measurement of UVabsorption at 280 nm (e280 64,570 Mÿ1cmÿ1). Fabs wereconcentrated with the use of Centricon 10 concentratorsas necessary.
Determination of equilibrium binding constants
Measurement of equilibrium binding constants wasaccomplished by surface plasmon resonance using theBIAcore1 2000 Biosensor (BIAcore Inc., Piscataway, NJ)and CM5 chips (research grade). All coupling reactions
and experiments were performed at 25 �C. Running buf-fer for chip derivatizations was HBS (10 mM Hepes(pH 7.4), 0.15 M NaCl, 3 mM EDTA, 0.005 % SurfactantP20) Running buffer for all equilibrium measurementswas 0.02 M Tris-HCl (pH 8.2), 0.05 M NaCl, 0.005 % Sur-factant P20. Chip surfaces were stripped at the end ofeach cycle by pulses of either 0.05 M NaOH, 0.005 %Surfactant P20 or 1 M NaCl, 0.005 % Surfactant P20 for®ve minutes followed by re-equilibration with runningbuffer.
Hapten JWJ1 was coupled to the amino terminus ofresin-bound peptide (ASQYFPSQAHGSK) with the useof HOBT, DIEA and DIC using standard procedures.Following acylation, the peptide conjugate was depro-tected and cleaved, then puri®ed by reversed phaseHPLC (Vydac C18, VYDAC/The Separations Group,Hesperia, CA), freeze-dried, and stored either as a drysolid or in 1 ml aliquots of a 1 mg/ml solution in water.Resin-bound peptide was also acylated with acetic anhy-dride to generate a peptide conjugate for use in control¯ow-cells. The hapten-peptide conjugate and acetylatedpeptide were con®rmed by electrospray mass spec-trometry.
Peptide conjugates were coupled to the surface ofresearch grade CM5 sensor chips via the carboxy-term-inal lysine residue of the peptide. The sensor chip surfacewas activated by injection of 15-20 ml of a freshly pre-pared solution of 0.2 M EDAC (1-ethyl-3-(3-dimethyla-minopropyl)carbodiimide), 0.05 M NHS (N-hydroxysuccinimide ) at 2 ml/minute. Peptide-JWJ1 con-jugate and acetylated peptide were immobilized by ¯owof 15-20 ml of a 0.15 mg/ml solution of peptide-JWJ1 oracetylated peptide in coupling buffer (10 mM Hepes(pH 7.4), 0.4 M NaCl, 2.4 mM EDTA, 0.005 % SurfactantP20) over the activated chip surface at 2 ml/minute.Injections of 20 ml of peptide-JWJ1 or 15 ml of acetylatedpeptide reproducibly provided coupling densities ofapproximately 240 RU per ¯ow-cell. Surfaces werecapped by injection of 70 ml of 1 M ethanolamine (pH 9)at a rate of 5 ml/minute. Chip surfaces were conditionedby several (®ve to ten) injections of 1 mM Fab.
For af®nity measurements under equilibrium con-ditions, a solution of the appropriate Fab in BIAcore buf-fer was injected using the Quickinject mode at a ¯ow-rate of 2 ml/minute. Contact time of 12 minutes wasallowed prior to SPR measurement in order to achievecomplete equilibration of association and dissociation.Chip surfaces were regenerated at the end of each cyclewith a pulse of either 50 ml of 0.05 M NaOH in 0.005 %Surfactant P20 or 100 ml of 1 M NaCl in 0.005 % Surfac-tant P20. The signal (relative to the baseline measuredprior to injection) obtained at the end of the injection(RU) was corrected for non-speci®c binding by subtrac-tion of the signal measured in the ¯ow-cell derivatizedwith acetylated peptide (RuAc). This value(RUJWJ1 � RU ÿ RUAc) was then used in Scatchard plotsof RUJWJ1/C versus RU, where C was the concentrationof Fab; the negative slope of the plot corresponded toKA. A broad range of Fab concentrations was initiallyinjected to allow estimation af®nity. Accurate measure-ments were then performed (5three times) at ®ve to tenconcentrations bracketing the estimated KD value. Dataanalysis was performed using the KaleidaGraph1 soft-ware package (KaleidaGraph1 version 3.0.1, SynergySoftware, Reading, PA). Free energy of binding (�G)were calculated using the formula �G � ÿ RT ln(KA).Changes in af®nity produced by a given mutation(��G) were calculated by subtracting the free energy ofbinding of the mutant (�Gmutant) from the free energy

1200 Mutational Analysis of the Af®nity Maturation of Antibody 48G7
of binding of the antibody from which the mutantwas derived; e.g. G (48G7g S30LN) � �G (48G7gS30LN) ÿ �G (48G7g).
Structural analysis
PDB coordinates and electron density maps for thestructures of 48G7 and 48G7g unliganded and com-plexed with JWJ1 were obtained from the laboratory ofRaymond C. Stevens. Structures were analysed using thesoftware programs InsightII (Molecular Simulations Inc.,San Diego, CA) and O (Jones et al., 1991).
Acknowledgements
David King and Nathanael S. Gray are gratefullyacknowledged for the syntheses of peptide and haptenJWJ1, respectively. Helle Ulrich, Charles Cho, Phil Pat-ten, Ulrich Wendt and Nathanael S. Gray generouslyprovided technical assistance and extremely useful dis-cussions. This work was supported by the National Insti-tutes of Health. P.L.Y. was supported by a HowardHughes Medical Institute Pre-Doctoral Fellowship.
References
Allen, D., Cumano, A., Simon, T., Sablitzky, F. &Rajewsky, K. (1988). Modulation of antibody bind-ing af®nity by somatic mutation. Int. J. Cancer. Sup-plement, 3, 1-8.
Alzari, P. M., Spinelli, S., Mariuzza, R. A., Boulot, G.,Poljak, R. J., Jarvis, J. M. & Milstein, C. (1990).Three-dimensional structure determination of ananti-2-phenyloxazolone antibody: the role ofsomatic mutation and heavy/light chain pairing inthe maturation of an immune response. EMBO J. 9,3807-3814.
Berek, C., Grif®ths, G. M. & Milstein, C. (1985). Molecu-lar events during maturation of the immuneresponse to oxazolone. Nature, 316, 412-418.
Casson, L. P. & Manser, T. (1995a). Evaluation of lossand change of speci®city resulting from randommutagenesis of an antibody V-H region. J. Immunol.155, 5647-5654.
Casson, L. P. & Manser, T. (1995b). Random mutagen-esis of two complementarity determining regionamino acids yields an unexpectedly high frequencyof antibodies with increased af®nity for both cog-nate antigen and autoantigen. J. Expt. Med. 182,743-750.
Chen, C., Roberts, V. A., Stevens, S., Brown, M., Stenzel-Poore, M. P. & Rittenberg, M. B. (1995). Enhance-ment and destruction of antibody function bysomatic mutation: unequal occurrence is controlledby V gene combinatorial associations. EMBO J. 14,2784-2794.
Even, J., Grif®ths, G. M., Berek, C. & Milstein, C. (1985).Light chain germ-line genes and the immuneresponse to 2-phenyloxazolone. EMBO J. 4, 3439-3445.
Green, N. S., Lin, M. M. & Scharff, M. D. (1998). Somatichypermutation of antibody genes: a hot spot warmsup. Bioessays, 20, 227-234.
Grif®ths, G. M., Berek, C., Kaartinen, M. & Milstein, C.(1984). Somatic mutation and the maturation of
immune response to 2-phenyl oxazolone. Nature,312, 271-275.
Huang, Z., Li, S. & Korngold, R. (1997). Immunoglobu-lin superfamily proteins: structure, mechanisms,and drug discovery. Biopolymers, 43, 367-382.
Jacobs, J. W. (1990). Catalytic antibodies. Doctoral disser-tation, University of California at Berkely.
Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallog. sect. A, 47,110-119.
Kabat, E. A. (1991). Sequences of Proteins of ImmunologicalInterest. 5th edit. NIH publication no. 91-3242, USDept. of Health and Humans Services Public HealthService National Institutes of Health, Bethesda, MD.
Kimura, M. (1970). The length of time required for aselectively neutral mutant to reach ®xation throughrandom frequency drift in a ®nite population.Genet. Res. 15, 131-133.
Kunkel, T. A., Roberts, J. D. & Zakour, R. A. (1987).Rapid and ef®cient site-speci®c mutagenesis with-out phenotypic selection. Methods Enzymol. 154, 367-382.
Lozano, F., Rada, C., Jarvis, J. M. & Milstein, C. (1993).Af®nity maturation leads to differential expressionof multiple copies of a kappa light-chain transgene.Nature, 363, 271-273.
Maizels, N. (1995). Somatic hypermutation: how manymechanisms diversify V region sequences?. Cell, 83,9-12.
Mariuzza, R. A., Boulot, G., Guillon, V., Poljak, R. J.,Berek, C., Jarvis, J. M. & Milstein, C. (1985). Prelimi-nary crystallographic study of the Fab fragments oftwo monoclonal anti-2-phenyloxazolone antibodies.J. Biol. Chem. 260, 10268-10270.
Mizutani, R., Miura, K., Nakayama, T., Shimada, I.,Arata, Y. & Satow, Y. (1995). Three-dimensionalstructures of the Fab fragment of murine N1G9antibody from the primary immune response andof its complex with (4-hydroxy-3-nitrophenyl)ace-tate. J. Mol. Biol. 254, 208-222.
Nakayama, T., Arata, Y. & Shimada, I. (1993). A multi-nuclear NMR study of the af®nity maturation ofanti-NP mouse monoclonal antibodies: comparisonof antibody combining sites of primary responseantibody N1G9 and secondary response antibody3B62. Biochemistry, 32, 13961-13968.
Neuberger, M. S. & Milstein, C. (1995). Somatic hyper-mutation. Curr. Opin. Immunol. 7, 248-254.
Parhami-Seren, B., Wysocki, L. J., Margolies, M. N. &Sharon, J. (1990). Clustered H chain somaticmutations shared by anti-p-azophenylarsonate anti-bodies confer enhanced af®nity and ablate thecross-reactive idiotype. J. Immunol. 145, 2340-2346.
Patten, P. A., Gray, N. S., Yang, P. L., Marks, C. B.,Wedemayer, G. J., Boniface, J. J., Stevens, R. C. &Schultz, P. G. (1996). The immunological evolutionof catalysis. Science, 271, 1086-1091.
Pewzner-Jung, Y., Simon, T. & Eilat, D. (1996). Struc-tural elements controlling anti-DNA antibody af®-nity and their relationship to anti-phosphorylcholine activity. J. Immunol. 156, 3065-3073.
Romesberg, F. E., Spiller, B., Schultz, P. G. & Stevens,R. C. (1998). Immunological origins of binding andcatalysis in a Diels-Alderase antibody. Science, 279,1929-1933.

Mutational Analysis of the Af®nity Maturation of Antibody 48G7 1201
Schultz, P. G. & Lerner, R. A. (1995). From moleculardiversity to catalysis: lessons from the immune sys-tem. Science, 269, 1835-1842.
Sharon, J. (1990). Structural correlates of high antibodyaf®nity: three engineered amino acid substitutionscan increase the af®nity of an anti-p-azophenylarso-nate antibody 200-fold. Proc. Natl Acad. Sci. USA,87, 4814-4817.
Spiller, B., Romesber, F., Schultz, P. & Stevens, R. (1997).The role of af®nity maturation in a Diels Alderasecatalytic antibody. FASEB J. 11, A1326.
Strong, R. K., Campbell, R., Rose, D. R., Petsko, G. A.,Sharon, J. & Margolies, M. N. (1991). Three-dimen-sional structure of murine anti-p-azophenylarsonateFab 36-71. 1. X-ray crystallography, site-directedmutagenesis, and modeling of the complex withhapten. Biochemistry, 30, 3739-3748.
Torigoe, H., Nakayama, T., Imazato, M., Shimada, I.,Arata, Y. & Sarai, A. (1995). The af®nity maturationof anti-4-hydroxy-3-nitrophenylacetyl mouse mono-clonal antibody: a calorimetric study of the antigen-antibody interaction. J. Biol. Chem. 270, 22218-22222.
Ulrich, H. D. (1996). Recombinant catalytic antibodies,thesis, University of California at Berkeley.
Ulrich, H. D. & Schultz, P. G. (1998). Analysis of haptenbinding and catalytic determinants in a family ofcatalytic antibodies. J. Mol. Biol. 275, 95-111.
Ulrich, H. D., Patten, P. A., Yang, P. L., Romesberg, F. E.& Schultz, P. G. (1995). Expression studies of cataly-tic antibodies. Proc. Natl Acad. Sci. USA, 92, 11907-11911.
Ulrich, H. D., Mundorff, E., Santarsiero, B. D., Driggers,E. M., Stevens, R. C. & Schultz, P. G. (1997). Theinterplay between binding energy and catalysis inthe evolution of a catalytic antibody. Nature, 389,271-275.
Wagner, S. D. & Neuberger, M. S. (1996). Somatichypermutation of immunoglobulin genes. In AnnualReview of Immunology (Paul, W. E., ed.), vol. 14, pp.441-457, Annual Reviews Inc., Palo Alto, California,USA.
Wedemayer, G. J., Patten, P. A., Wang, L. H., Schultz,P. G. & Stevens, R. C. (1997a). Structural insightsinto the evolution of an antibody combining site(see comments) (published erratum appears inScience, 1997, Sep 5;277(5331):1423). Science, 276,1665-1669.
Wedemayer, G. J., Wang, L. H., Patten, P. A., Schultz,P. G. & Stevens, R. C. (1997b). Crystal structures ofthe free and liganded form of an esterolytic catalyticantibody. J. Mol. Biol. 268, 390-400.
Wong, Y. W., Kussie, P. H., Parhami-Seren, B. &Margolies, M. N. (1995). Modulation of antibodyaf®nity by an engineered amino acid substitution.J. Immunol. 154, 3351-3358.
Wysocki, L. J., Gridley, T., Huang, S., Grandea d., A. G.& Gefter, M. L. (1987). Single germline VH and Vkappa genes encode predominating antibody vari-able regions elicited in strain A mice by immuniz-ation with p-azophenylarsonate. J. Expt. Med. 166,1-11.
Yoshihara, Y., Oka, S., Ikeda, J. & Mori, K. (1991).Immunoglobulin superfamily molecules in the ner-vous system. Neurosci. Res. 10, 83-105.
Yuhasz, S. C., Parry, C., Strand, M. & Amzel, L. M.(1995). Structural analysis of af®nity maturation: thethree-dimensional structures of complexes of ananti-nitrophenol antibodies. Mol. Immunol. 32, 1143-1155.
Zhang, J. & Gu, X. (1998). Correlation between the sub-stitution rate and rate variation among sites in pro-tein evolution. Genetics, 149, 1615-1625.
Edited by D. C. Rees
(Received 3 June 1999; received in revised form 14 September 1999; accepted 17 September 1999)