multi-spectroscopic study of fe(ii) in silicate glasses: implications for the coordination...
TRANSCRIPT

Geochimica et Cosmochimica Acta, Vol. 69, No. 17, pp. 4315–4332, 2005Copyright © 2005 Elsevier Ltd
Printed in the USA. All rights reserved
doi:10.1016/j.gca.2005.01.008
Multi-spectroscopic study of Fe(II) in silicate glasses: Implications for the coordinationenvironment of Fe(II) in silicate melts
WILLIAM E. JACKSON,1 FRANCOIS FARGES,1,2 MARK YEAGER,3 PATRICIA A. MABROUK,3 STÉPHANIE ROSSANO,2
GLENN A. WAYCHUNAS,4 EDWARD I. SOLOMON,3 and GORDON E. BROWN JR.1,5,*1Department of Geological and Environmental Sciences, Stanford University, Stanford, CA 94305-2115, USA
2Laboratoire des Géomatériaux, Université Marne-la-Vallée and CNRS FRE 2455, 77454 Marne la Vallée cedex 2, France3Department of Chemistry, Stanford University, Stanford, CA 94305-5080, USA
4Earth Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA5Stanford Synchrotron Radiation Laboratory, SLAC, MS 69, Menlo Park, CA 94025, USA
(Received July 18, 2003; accepted in revised form January 18, 2005)
Abstract—The coordination environment of Fe(II) has been examined in seven anhydrous ferrosilicate glasses at298 K and 1 bar using 57Fe Mössbauer, Fe K-edge X-ray near edge structure (XANES), and extended X-rayabsorption fine structure (EXAFS), UV-Vis-NIR, and magnetic circular dichroism (MCD) spectroscopies. Glassesof the following compositions were synthesized from oxide melts (abbreviation and nonbridging oxygen:tetrahedralcation ratio (NBO/T) in parentheses): Li2FeSi3O8 (LI2: 1.33), Rb2FeSi3O8 (RB2: 1.33), Nal.08Fel.l7Si3.l3O8 (NAl:1.09), Nal.46Ca0.24Fel.08Si2.97O8 (NC6: 1.38), Nal.09Ca0.51Fe0.72Si3.10O8 (NC2: 1.15), Na0.99Ca0.92Fe0.24 Si3.17O8
(NCl: 1.04), and Na0.29Mg0.53Ca0.52Fe0.56Al0.91Si2.44O8 (BAS: 1.05). Mössbauer, XANES, and EXAFS informa-tion suggests that iron is dominantly ferrous in all glasses (�10 atom% Fe(III)) with an average first-neighbor Fe(II)coordination varying from � 4 to 5.2 (�0.2) oxygens. The UV-Vis-NIR spectrum of each sample exhibits intenseabsorption centered near 8100–9200 cm�1 and weak absorption near 5000 cm�l, which cannot be assignedunambiguously. The MCD spectrum of NC6 glass, which is the first such measurement on a silicate glass, showsthree transitions at �8500 cm�1, �6700 cm�1, and �4500 cm�1. The behavior of these MCD bands as a functionof temperature (1.6 K to 300 K) and magnetic field strength (1 T to 7 T) indicates that they most likely arise fromthree distinct Fe(II) sites with different ground states, two of which are 5-coordinated and one of which is4-coordinated by oxygens.
The combined results suggest that Fe(II) predominantly occupies 5- and 4-coordinated sites in each glass,with the ratios differing for the different compositions. Small amounts of 6-coordinated Fe(II) are possible aswell, but primarily in the more basic glass compositions such as BAS. The substitution of Li(I) for Rb(I) inthe M2FeSi3O8 base glass composition causes a weakening of the average Fe(II)-O bond, as indicated by thelonger Fe(II)-O distance in the latter. The basalt composition glass was found to have the largest Fe(II) sitesrelative to those in the other glasses in this study. A bond valence model that helps predict the coordinationnumber of Fe(II) in silicate glasses is proposed. The structural information extrapolated to Fe(II)-bearing meltsis parameterized using bond valence theory, which helps to rationalize the melt-crystal partitioning behavior
0016-7037/05 $30.00 � .00
of ferrous iron in natural and synthetic melt-crystal systems. Copyright © 2005 Elsevier Ltd
1. INTRODUCTION
The local coordination environments of first-row transitionmetals in silicate glasses and melts are poorly constrained anddifficult to predict due to the possibility of a range of coordi-nation geometries, including some not commonly found incrystalline silicates, such as trigonal bipyramids (e.g., Fe(II):Jackson et al., 1991, 1993; Ni(II): Galoisy and Calas, 1993;Farges et al., 1994; and Ti(IV): Bugaev et al., 2004) or squarepyramids (e.g., Ti(IV): Farges et al., 1996; Farges and Brown,1997; Romano et al., 2000). Knowledge of the local coordina-tion environments of cations in silicate glasses and melts isnecessary for a fundamental understanding of their physicaland thermodynamic properties (Burns, 1993). For example, theratio of cations in tetrahedral vs. octahedral sites influences thepolymerization, viscosity, density, and heat capacity of silicatemelts (Mysen, 1988, 1991).
Among the first-row transition metals, iron is the most abundant
* Author to whom correspondence should be addressed ([email protected]).
4315
in Earth materials and, consequently, the most often studied insilicate glasses and melts. Ferrous and ferric iron can coexist insilicate glasses and melts, their ratio being a function of oxygenfugacity, temperature, and melt composition (Fudali, 1965; Sack etal., 1980; Kilinc et al., 1983; Mysen et al., 1985a; Borisov andShapkin, 1990; Kress and Carmichael, 1991; Moore et al., 1995;Baker and Rutherford, 1996; Wilke et al., 1999, 2001, 2002).Ferric iron is generally thought to populate tetrahedral sites inNa-bearing silica-rich melts and perhaps a combination of tetra-hedral and octahedral sites in Ca-bearing silica-rich melts (Levy etal., 1976; Brown et al., 1978; Mysen et al., 1984; Mysen et al.,1985a, 1985b; Mysen and Virgo, 1989; Mysen, 1991; Wang et al.,1995). However, in situ heat capacity measurements have beeninterpreted to suggest that Fe(III) may also occupy more highlycoordinated sites (Lange and Navrotsky, 1993; Tangeman andLange, 1998; Tangeman et al., 2001). Similarly, the coordinationenvironment of Fe(II) is still debated. It has been proposed fromMössbauer (Virgo and Mysen, 1985), vibrational (Mysen et al.,1982), and optical absorption studies (Boon and Fyfe, 1972; Bellet al., 1976; Goldman and Berg, 1980; Nolet, 1980; Fox et al.,
1982; Lefrère, 2002) that Fe(II) is dominantly (i.e., �95%) octa-
4316 W. E. Jackson et al.
hedrally coordinated in silica-rich glasses. However, more recentMössbauer studies suggest the presence of less highly coordinatedFe(II) species in silicate glasses (Alberto et al., 1996; Dunlap,1997; Dunlap et al., 1998; Rossano et al., 1999). In addition, FeK-edge extended x-ray absorption fine structure (EXAFS), FeK-edge X-ray absorption near edge structure (XANES), and 57FeMössbauer spectroscopic studies of Na2Fe(II)Si3O8 andK2Fe(II)Si3O8 glasses (Waychunas et al., 1988, 1989), Fe K-edgeEXAFS and XANES studies of MFe(II)0.5Si2.5O6 glasses (M �K, Na, Cs: Henderson et al., 1991, 1995; see also Iwamoto et al.,1987; Bonnin-Mosbah et al., 2001), and vibrational and Fe K-edgeEXAFS studies of M2SiO4 glasses and melts (M � Fe(II), Mn(II),Mg, Ca: Cooney and Sharma, 1990; Jackson, 1991; Jackson et al.,1993) all suggest that Fe(II) is dominantly tetrahedrally or penta-hedrally coordinated by oxygens in these glass compositions.Recent molecular dynamics calculations on Fe(II)-bearing anhy-drous silicate melts suggest the presence of a continuum of Fe(II)sites, with first-neighbor coordination ranging from 4 to 6 oxy-gens, but dominantly 4 and 5 oxygens (Rossano et al., 2000,2002). When dissolved water is present in such glasses, however,Fe(II) is thought to be dominantly 6-coordinated by oxygens, asshown in a recent study that used Mössbauer spectroscopy andhigh-resolution Fe K-edge XANES spectroscopy (Wilke et al.,1999).
To provide a more definitive description of the averagecoordination environment of Fe(II) in silica-rich glasses, wehave examined seven anhydrous ferrosilicate glasses using FeK-edge EXAFS, Fe K-edge XANES, Mössbauer, and opticalspectroscopies, including magnetic circular dichroism (MCD)methods. The compositions of these glasses are given below inthe sample preparation section. Because EXAFS analysis issensitive to nongaussian distributions of interatomic distancesaround an absorber as a result of dynamical or static positionaldisorder effects, we have fit the EXAFS data using the cumu-lant expansion method (Crozier et al., 1988) which accounts forsuch anharmonic effects. We have also carried out a shape-independent fitting (Rossano et al., 1999) of the Mössbauerdata collected in this study to obtain improved estimates of therange of sites occupied by iron in these glasses. The MCDmeasurements, which are the first on Fe(II) in a silicate glass,were carried out to help constrain interpretations of UV-vis-NIR spectra of ferrosilicate glasses. The implications of ourfindings for the coordination environments of Fe(II) and Ni(II)in silicate glasses and melts of geochemical interest and formelt-crystal partitioning of Fe(II) and NI(II) are discussed.
2. EXPERIMENTAL
2.1. Sample Preparation
The glasses were synthesized from reagent grade oxides and ironmetal. Iron was added in the form of equimolar proportions of pow-dered Fe metal and Fe2O3—a procedure that provided confirmation thatiron redox equilibrium was attained in the melt. The mixtures were thenground under ethanol, placed in iron crucibles, and heated for severalhours in a vertical, controlled-atmosphere (Ar or N2) Deltech furnace to�700 K above the estimated glass transition temperatures of theseglasses (500 –1100 K; see below). The MoSi heating elements(coated with SiO2) of the Deltech furnace and the use of a metalliciron crucible resulted in appropriate oxygen fugacities in the meltsso that the oxidation state of iron was dominantly 2�, as confirmed
by subsequent Mössbauer studies on these glasses (see below). Themelts were quenched to glasses by placing the bottom portion of thecrucible in water, with care taken not to introduce water into themelts. This procedure resulted in an estimated quench rate of �102
K/s. The samples were reground and remelted for a total of threemelting cycles to increase compositional homogeneity. The compo-sitions of the five Na-Ca-ferrosilicate glasses prepared in this man-ner were determined by electron microprobe analysis, and the re-sulting cation proportions are reported here relative to eightoxygens, with sample designations preceding each composition:NAl (Nal.08Fel.l7Si3.l3O8), NC6 (Nal.46Ca0.24Fe1.08Si2.97O8), NC2(Nal.09Ca0.51Fe0.72Si3.10O8), NCl (Na0.99Ca0.92Fe0.24Si3.17O8), andBAS (Na0.29Mg0.53Ca0.52Fe0.56A10.91Si2.44O8). The compositions ofLI2 and RB2 glasses were not determined by electron microprobeanalysis and are reported as nominal compositions: LI2(Li2FeSi3O8) and RB2 (Rb2FeSi3O8). Minor Cr2O3 (0.3 wt.%) wasincorporated into NAl, NC6, and BAS glasses, and�2.0 wt.% Cr2O3 was found in NCl and NC2 glasses as a conse-quence of the liquid reacting with the iron crucibles which containedsmall concentrations of Cr. The ratio of nonbridging oxygens pertetrahedron (NBO/T: Mysen, 1988, assuming all Fe is divalent, andbehaves as a network modifier) is as follows: LI2 and RB2 (NBO/T� 1.33), NA1 (NBO/T � 1.09), NC6 (NBO/T � 1.38), NC2(NBO/T � 1.15), NC1 (NBO/T � 1.04), and BAS (NBO/T � 1.05).Glass transition temperatures were estimated using the approach ofToplis (1998), assuming that Fe(II) connects to nonbridging oxy-gens (NBOs) in a way similar to Ca(II). Estimated Tg values for theglasses are 500 –550 K (RB2 and LI2), 1050 K (BAS), 900 K (NA1),650 K (NC6), 750 K (NC2), and 800 K (NC1), all with estimateduncertainties of �50 K. Although the fictive temperatures of theseglasses have not been determined experimentally, given the quenchrates of our glass syntheses (estimated to be �102 K/s, which istypical of standard laboratory quench rates; see Dingwell, 1995), itis unlikely that the fictive temperatures differ from the estimatedglass transition temperatures by more than 50 –100 K. X-ray dif-fraction and scanning electron microscopy showed that the glassescontained no detectable crystalline phases.
2.2. Model Compounds
Several Fe(II)- and Fe(III)-containing model compounds with Fe(II) orFe(III) in tetrahedral, trigonal bipyramidal, or octahedral coordinationgeometries were also studied by XAFS spectroscopy. The Fe(II)-bearingminerals (all unaltered by natural weathering processes and freefrom radiation damage) included a dark brown staurolite(Fe1.5Mg0.5Al9Si3.9Al0.1O22(OH)2) from North Windham, Maine, USA(13.1 wt.% [4]Fe(II)O), a sky-blue grandidierite ((Mg,Fe)Al3(BO4)(SiO4)O) from Marotrana, Madagascar (1.0 wt.% [5]Fe(II)O �0.1% Fe(III)2O3: Farges, 2001), a black fayalite ([6]Fe2SiO4) fromAllevard, Isère, France, and a light brown siderite ([6]FeCO3) fromNova Lima, Brazil. Model compounds for Fe(III) in various coordina-tion geometries with oxygen ligands included a synthetic sample of�-[4]Fe:LiAlO2: Waychunas and Rossmann, 1983), a deep purple yo-derite ((Mg,Al,[5]Fe)8Si4(O,OH)20) from Mautia Hills, Tanzania (6.1wt.% Fe2O3), a black acmite (or aegirine: Na[6]FeSi2O6) fromMont Saint-Hilaire, Québec, and a brownish andradite(Ca3
[6]Fe1.8Al0.2Si3O12) from an unknown locality in Mexico. Moredetails on these model compounds can be found in Wilke et al. (2001).
2.3. Mössbauer Spectroscopy
57Fe Mössbauer spectra were collected at 300 K from powders of theglasses with a source consisting of 57Co diffused into Pd (sourceactivity � 15 mCi). The data were collected over 512 channels inconstant acceleration symmetrical mode and averaged by folding thespectra. Fe foil served as a standard for calibration. We fit the data withtwo doublets by constraining the areas for the two components of eachdoublet to be equal while allowing the component peak half-widths andLorentzian-gaussian fractions to vary. In the final cycle, all noncon-strained parameters were allowed to vary simultaneously.
2.4. XANES Spectroscopy
Iron K-edge XANES spectra for the glass samples were collected atthe Stanford Synchrotron Radiation Laboratory (SSRL) on beamline 4-1

4317Multi-spectroscopic study of Fe(II) in silicate glasses
using a Si(220) double-crystal monochromator. All glass samples weresolid pieces of glass �25 �m thick that were cut and polished on bothsurfaces for optical absorption study. Synchrotron storage ring condi-tions were 3.00 GeV and 60–100 mA. X-ray beam harmonics wereminimized by detuning the monochromator crystals to reduce theincoming beam intensity by �50%. Iron K-edge XANES spectra werecollected in the energy-scanning mode, and all energies were calibratedwith iron metal foil, with the second inflection point on the Fe(0) foilabsorption edge set at 7111.0 eV. Vertical slits before the monochromatorwere set at 1 mm to enhance energy resolution. Energy resolution at7111.0 eV is �1.7 eV, as determined by the ability to resolve details of theXANES features for crystalline Fe(II)-containing model compounds (thecore-hole lifetime for a 1s electron of Fe is �1.2 eV: Krause and Oliver,1979). XANES spectra for each glass and crystalline model compoundwere collected from 100 eV to 25 eV below the edge using 2 s countingtimes and 5 eV steps, from 25 eV below the edge to the edge (7106–7120eV) (the pre-edge region) using 4 s counting times and 0.1 eV steps, from7120 eV to 7150 eV (the main edge region) using 2 s counting times and0.3 eV steps, and from 7150 to 7250 eV (the postedge region) using 1 scounting times and 2 eV steps.
2.5. EXAFS Spectroscopy
EXAFS data for all glass samples were collected up to 700 eV above theFe K-edge in the fluorescence-yield detection mode, using a Mn-filtered ion chamber detector (Lytle et al., 1984). To minimize self-absorption effects in the fluorescence-yield spectra, the XAFS spectrawere collected with the incident beam at 90° to the sample surface(Tröger et al., 1992) and are the averages of four scans to improve thesignal-to-noise ratio. The EXAFS oscillations are modulations of theX-ray absorption coefficient of a material on the high-energy side of anabsorption edge and arise from the interference of the ejected photo-electron wave with the backscattered photoelectron wave in the vicinityof the absorbing atom (e.g., Teo and Joy, 1981; Teo, 1986; Konings-berger and Prins, 1988). The data extend from k � 1.0 to 12.0 Å�l,where k, the photoelectron wavenumber, is calculated as k � 0.262 (E– E0), in which E is the incident photon energy and E0 is the thresholdenergy for the ejected photoelectron (chosen as the inflection point ofthe edge jump). The normalized EXAFS data were Fourier transformedover the k-range 2.5–12.0 Å�l using a Kaiser-Bessel function (usingthe XAFS 2.9 package [Winterer, 1996]). The Fourier transform datarepresent Fe(II)–X pair correlation functions, where X refers to eachsuccessive shell of backscattering neighbors. The prominent peak at�l.5–1.6 Å corresponds to the phase-shifted Fe(II)-O pair correlation.To correct for this phase-shift and extract structural information, weisolated the Fe(II)-O peak and back-Fourier transformed into k-spacefor least-squares fitting. The k3-weighted �(k) functions were modeledusing Fe-O backscattering phase-shift and amplitude functions ex-tracted from the Fe K-edge EXAFS data collected for siderite (FeCO3;Oh symmetry; [6]Fe(II)-O � 2.144 Å; Effenberger et al., 1981). Basedon fits using these backscattering phase-shift and amplitude functionsto Fe K-EXAFS data collected from additional reference compoundscontaining Fe(II) in well-characterized coordination environments (asin grandidierite, staurolite, fayalite, etc.), we can reproduce knownFe(II)-O distances to within �0.02 Å (and with a least-squares preci-sion of � �0.004 Å). In parallel, Fe(II) first-neighbor coordinationnumbers (CN) are estimated with an accuracy of �20% using anhar-monic models to derive reliable Fe-O distances and Fe CN, even inhighly disordered materials (Jackson et al., 1991; Farges et al., 1993).
2.6. Optical Spectroscopy
Optical absorption spectroscopic data were collected with a CARY17 UV/VIS/NIR spectrometer over the range 25,000–4000 cm�l fromdoubly polished slices of the seven glasses that were cut to �20–30microns thickness and mounted on infrasol glass. Data points werecollected every 2–3 cm�l with counting times of �0.2 s at each point.Baseline data were first collected from the infrasol glasses and then
subtracted from the ferrosilicate plus infrasol glass data. These datarepresent the average of 10 scans for each sample.2.7. Magnetic Circular Dichroism Spectroscopy
MCD spectroscopy is an effective method for observing ligand field(d¡d) electronic transitions in transition metal complexes. We havedeveloped a protocol using variable temperature–variable field MCDthat has proven to be quite powerful for the study of ligand-fieldtransitions in nonheme high-spin ferrous active sites in proteins (Whit-taker and Solomon, 1988; Mabrouk et al., 1991). The present studyrepresents the first application of this technique to a system of geo-chemical importance. Because MCD involves different selection rulesthan electronic absorption spectroscopy (ABS), it is possible to observetransitions in the MCD spectrum of a metal site that are difficult toobserve in ABS. A high-spin ferrous ion in an octahedral coordinationenvironment should exhibit two transitions, centered at �10,000 cm�1,and split by 1000 cm�1 to 2000 cm�1. A 5-coordinated site will alsoresult in two bands, but with a larger splitting: one near 10,000 cm�1,but possibly as low as �8000 cm�1, and the other at �5000 cm�1 orbelow. Four-coordinated high-spin ferrous sites will give rise to oneband at 4000 cm�1 to 5000 cm�1, if tetrahedral, or at higher energy ifflattened toward square planar. In addition, MCD saturation magneti-zation effects which occur at low temperature and high magnetic fieldstrength allow correlation of the excited state spectral features with theground state, even for the electron paramagnetic resonance (EPR)-inaccessible, non-Kramers S � 2 ferrous site. MCD data in the regionof 4650 cm�1 to 14,000 cm�1 were recorded using a Jasco J-200Dspectro-polarimeter equipped with an Oxford Instruments SM4-7TSpectromag superconducting magnet/cryostat with a variable temper-ature insert for operation in the 1.6 K to 300 K temperature region. Datawere recorded at up to 7 Tesla applied magnetic field. The detector wasan InSb photodiode, which was cooled to 77 K during data acquisition.
Two samples of the NC6 glass were used for MCD data collection.The first was the polished thin slice used for electronic absorption andEXAFS measurements, and the second was the powder used for Möss-bauer data collection. For the MCD experiments the powder wassuspended in mineral oil and placed between two Infrasil quartz disks,and the slice was used with no further preparation. For each spectralscan, the sample was checked for depolarization of the circularlypolarized light beam by measuring the magnetic circular dichroism ofa nickel tartrate solution placed immediately before and after thesample in the spectropolarimeter. The signal loss due to depolarizationwas �5% for each run with the powder, and �2% with the slice.
3. RESULTS
3.1. Mössbauer Spectroscopy
Figure 1 shows the experimental 57Fe Mössbauer spectra andthe best fits for six of the glasses studied: NC6, LI2, NC2, BAS,RB2, and NA1 (the Mössbauer spectrum and model obtainedfor glass NC1 are very similar to that for NA1). The results ofleast-squares fitting of the Mössbauer spectra (apparent isomershift, IS, and apparent quadrupole splitting, QS) are given inTable 1. Except for RB2 glass, which contains �20% of thetotal iron as Fe(III) (Table 1), each glass was found to be lowin ferric iron (i.e., less than 10 atom%). The fit to RB2 glassMössbauer data required constraining the areas of the ferriciron doublet peaks to be equal. This fit also required constrain-ing the Lorentzian fraction of each peak to 1.0. For each glassdata set, two Fe(II) doublets were chosen to provide a highquality fit, and we report the average (weighted by peak area)hyperfine parameters associated with each fit. There is noreason a priori to fit additional Fe(II) doublets to the spectrumcollected from each glass even though there may be more thantwo Fe(II) sites present in each sample. This conclusion followsfrom various fits of the spectra that included additional Fe(II)doublets, which did not improve the fits statistically.
Mössbauer IS values range from 0.93–1.04 mm/s (relative to
Fe metal). Average Mössbauer QS values range from 1.79–
4318 W. E. Jackson et al.
1.98 mm/s. In general, IS values are positively correlated withaverage Fe-O bond distances and, hence, iron coordinationnumbers, whereas QS values exhibit little correlation with theseparameters (Dyar, 1985; Hawthorne, 1988; Waychunas et al.,1988; Rancourt, 1994; Rossano et al., 1999; Rancourt et al.,2001). In crystalline silicates and oxides, the IS values foroctahedrally coordinated Fe(II) range from 1.10 to 1.40 mm/s,those for 5-coordinated Fe(II) range from �0.85 to 1.20 mm/s,
Fig. 1. 57Fe Mössbauer spectra for six selected ferrosilicate glasses.
Table 1. 57Fe Mössbauer spectrosc
SampleIsomer shifta
(mm/s)Quadrupole splittinga
(mm/s)
LI2 1.04 1.87RB2 0.93 1.98NA1 1.01 1.79NC6 0.98 1.81NC2 1.02 1.84NC1 0.99 1.85BAS 1.04 1.88Error (�) 0.02 0.05
a Reported as weighted values of two doublet fits relative to Fe(0).b
Values derived using shape-independent distribution (SID) model.c Parameter fixed because of high Fe(III) content.and those for tetrahedrally coordinated Fe(II) range from 0.60to �1.10 mm/s (all relative to Fe metal). The average Fe(II) ISvalues for the ferrosilicate glasses in this study lie below thosefor Fe(II) in octahedral coordination in crystalline silicates, butwell within the ranges observed for Fe(II) in both tetrahedraland 5-coordinated oxygen environments. However, these aver-age IS values do not necessarily preclude a small amount ofoctahedrally coordinated Fe(II), as we only report average ISvalues. These conclusions are supported by shape-independentdistribution (SID) models (Rossano et al., 1999), which wereused in fitting the Mössbauer spectra for three typical glasses(LI2, RB2, and NC6) to check for site distribution effects inthese glasses (Fig. 2). These 3D diagrams represent the prob-ability of the presence of Lorentzian doublets described byMössbauer parameters (IS, QS) resulting in the best agreementbetween experiment and theory. These diagrams can be repre-sented as a projection onto the IS-QS plane (see right side of Fig.2). In the diagrams calculated using this method (e.g., IS vs. QS;see left side of Fig. 2), the domain for ferric iron is located on theleft side of the plot (IS � 0.5, QS � 1), whereas that for ferrousiron is located in the center of the plot (IS � 0.5, QS �1).
According to these SID models, RB2 shows a wider distri-bution of iron oxidation states than NC6 or LI2, in agreementwith previous peak-fitting results. The SID models for NC6 andLI2 provide evidence for small but significant amounts of ferriciron (10 and 7 atom%, respectively) that could not be easilydetected using conventional Mössbauer peak-fitting methods.Another significant result is that for those glasses containingmore than 90% of iron as Fe(II) (all glasses except for RB2),there is a significant range of different sites around Fe(II), asshown by the shape of the Mössbauer parameters distributionfor glass LI2. This finding suggests that the structural informa-tion extracted from the other spectroscopic methods used in thisstudy represent averages of these site distributions.
3.2. X-ray Absorption Fine Structure Spectroscopy
3.2.1. Pre-edge features
The Fe K-edge XANES and pre-edge spectra collected forthe model compounds and for the seven glass samples arepresented in Figure 3A–D. The pre-edge region of the FeK-edge XANES spectrum arises mostly from bound-state andcontinuum-state electronic transitions of the core electrons
ults for seven ferrosilicate glasses.
rentzian fraction(%) RMS
MISFIT(%)
% Fe(III)b
(%)
0.43 1.28 0.14 71.0c 1.66 0.77 220.41 1.05 0.060.40 1.18 0.05 100.44 1.83 0.230.45 0.91 0.160.26 0.92 0.040.01
opy res
Lo

4319Multi-spectroscopic study of Fe(II) in silicate glasses
ejected by the incident photons (e.g., a 1s ¡ 3d transition) andcontains information on absorber atom valence, ligand type,and site geometry (Waychunas et al., 1983; Westre et al., 1997;Arrio et al., 2000; Wilke et al., 2001). In general, the greater thedeviation of the Fe(II) site from centrosymmetry, the greaterthe height of the ls ¡ 3d transition feature (Waychunas et al.,1983, 1988; Roe et al., 1984; Dräger et al., 1988; Bajt et al.,1994; Delaney et al., 1996; Heumann et al., 1997; Westre et al.,1997; Arrio et al., 2000; Galoisy et al., 2001; Wilke et al.,2001) (for Fe(II), compare siderite, fayalite, and grandidierite;for Fe(III), compare andradite with �-Fe:LiAlO2; see Fig. 3Band Table 2a). Also, the pre-edge positions for Fe(III)-bearingmodel compounds are shifted toward higher energy (by �1.4 eV,
Fig. 2. 3D Mössbauer data analysis (isomer shift and qselected glasses (left: 3D views; right: contour plots). SeLorentzian probability function.
near 7113.50 eV) as compared with Fe(II)-bearing compounds
(Fig. 3B). Because of the dependence of Fe K-XANES pre-edgecentroid position on iron oxidation state, the pre-edge position canbe used to estimate the average oxidation state of Fe as a functionof its local environment (Wilke et al., 2001; see also Berry et al.,2003).
The Fe K-XANES and pre-edge spectra for all glasses areshown in Figures 3C and 3D, respectively, and the refinedpre-edge information is listed in Table 2b. The average centroidposition of the pre-edge is close to 7112.10 � 0.05 eV (Fig. 4),suggesting the presence of dominant amounts of ferrous iron inall glasses. No clear evidence for major amounts of ferric ironwas found in any of the glasses investigated (i.e., less than 10atom% of Fe(III)), except for RB2. In this glass, a significantly
le splitting vs. Lorentzian probability function) for threeno et al. (1999), for details about the calculation of the
uadrupoe Rossa
more intense shoulder is observed at �1.0(�0.1) eV above the

4320 W. E. Jackson et al.
Fig. 3. Fe K-edge XANES spectra for the glasses and model compounds. (A) Normalized XANES spectra for selectedmodel compounds (respectively: staurolite, grandidierite, fayalite, siderite, Fe:LiAlO2, yoderite, andradite, and acmite;ferrous-bearing in black, ferric-bearing in gray; see text for details), showing their pre-edge (labeled A). (B) Normalizedpre-edge spectra for selected model compounds for Fe(II) (black) and Fe(III) (gray) in 4- (“stau” and “FeLi”), 5- (“gran”and “yode”), regular 6-coordinated (“side” and “andr”), and distorted 6-coordinated (“faya” and “acmi”) environments. A1and A2 are the pre-edge centroid positions for Fe(II) and Fe(III), respectively. (C) Normalized XANES spectra for theglasses of this study. (D) Normalized pre-edge features for the glasses of this study, showing their deconvolution intoGaussians (dashed lines) centered around A1 (indicative of ferrous iron), except for RB2 glass where a significant
contribution (�10 atom %) arising from ferric iron (centered around A2) is detected.
modeled
4321Multi-spectroscopic study of Fe(II) in silicate glasses
pre-edge maximum (Fig. 3D). Based on the calibration of ironoxidation state with XANES pre-edge position (Wilke et al.,1999, 2001), we estimate that 15(�5) atom% of the total Fe ispresent as ferric iron, which is in reasonable agreement with theresults from Mössbauer spectroscopy (22%) reported above inTable 1. The integrated areas of the normalized XANES pre-edges for the ferrosilicate glasses fall between 0.11 and 0.19(Table 2b). These values are significantly higher than those for6-coordinated oxygen environments around Fe(II), as infayalite (0.105) (Table 2a). However, they are comparable to orgreater than that of grandidierite (0.127) in which Fe(II) occu-pies a distorted 5-coordinated site (Seifert and Olesch, 1977;
Table 2a. Fe K-edge pre-edge
Sample
Pre-edge components
Normalizedheight Position (eV) Widtha
Staurolite [4]Fe(II) 0.090 7111.68 1.530.035 7113.14 1.53
Grandidierite [5]Fe(II) 0.060 7111.69 1.440.026 7113.35 1.44
Siderite [6]Fe(II) 0.018 7111.25 1.360.012 7112.09 1.360.010 7113.46 1.36
Fayalite [6]Fe(II) 0.017 7111.36 1.880.015 7111.73 1.880.014 7113.30 1.88
�-Fe:LiAlO2[4]Fe(III) 0.010 7111.73 1.63
0.164 7113.61 1.63Yoderite [5]Fe(III) 0.083 7113.39 1.77
0.009 7115.21 1.77Andradite [6]Fe(III) 0.021 7112.82 1.36
0.018 7114.28 1.36Acmite [6]Fe(III) 0.022 7112.70 1.65
0.031 7114.06 1.65Average error 0.005 0.05 0.00
a Width was allowed to vary, but with the same value for all lines.b Correlation between the experimental spectra and the sum of the
Table 2b. Fe K-edge pre-edge inf
Sample
Pre-edge components
Normalizedheight Position (eV) Widtha (eV)
LI2 0.062 7111.80 1.690.025 7113.20 1.69
RB2 0.050 7111.84 1.670.027 7113.34 1.67
NA1 0.058 7111.72 1.740.033 7113.24 1.74
NC6 0.056 7111.80 1.710.023 7113.19 1.71
NC2 0.051 7111.86 1.700.018 7113.07 1.70
NC1 0.043 7111.80 1.560.017 7112.97 1.56
BAS 0.044 7111.90 1.590.015 7113.04 1.59
Average error 0.005 0.05 0.003
a
Width was allowed to vary, but with the same value for all lines.b Correlation between the experimental spectra and the sum of the modeledMacKenzie and Meinhold, 1997; Farges, 2001). Hence, thepre-edge heights observed for the ferrosilicate glass spectra areconsistent with Fe(II) occupying a range of possible sites,which are 4- to 5-coordinated on average.
Additional constraints on these conclusions can be derivedfrom a peak-fitting analysis of the pre-edge features for theglasses. Two pseudo-Voigt functions (50% gaussian) were suffi-cient to model these pre-edges. These two components are, on theaverage, located near 7111.8(�0.1) and 7113.2(�0.2) eV, andtherefore, are separated by �1.4 eV. In the absence of signifi-cant amounts of other iron oxidation states in most of ourglasses, these two pre-edge components are related to the
ation for model compounds.
Sum ofintegrated
areas
Pre-edgecentroid
(eV)
Componentsplitting
(eV) RbIntegrated
area
0.153 0.212 7112.09 1.5 0.9990.0590.097 0.127 7112.07 1.7 0.9980.0300.029 0.063 7112.04 2.1 0.9990.0190.0150.039 0.105 7112.09 1.9 0.9990.0330.0330.020 0.350 7113.51 1.9 0.9990.3300.180 0.200 7113.58 1.8 0.9990.0200.035 0.066 7113.53 1.5 0.9990.0300.045 0.107 7113.48 1.4 0.9990.0610.005 0.005 0.05 0.2
lines.
n for seven ferrosilicate glasses.
Sum ofintegrated
areas
Pre-edgecentroid
(eV)
Componentsplitting
(eV) Rbntegrated
area
0.128 0.179 7112.20 1.4 0.9990.0510.101 0.155 7112.36 1.5 0.9980.0540.121 0.191 7112.28 1.5 0.9990.0700.116 0.164 7112.21 1.4 0.9990.0480.105 0.142 7112.18 1.2 0.9990.0370.082 0.115 7112.14 1.2 0.9980.0330.085 0.114 7112.19 1.1 0.9980.0290.005 0.005 0.05 0.2
inform
(eV)
3
ormatio
I
lines.

4322 W. E. Jackson et al.
crystal-field splitting (“10Dq”) of the 3d orbitals of Fe(II),(convoluted with, among other contributions, the probability ofthe presence of dipolar vs. quadrupolar transitions: Westre etal., 1997). The separation between the two components in-creases from 1.5 eV for Td local symmetry (tetrahedral, as instaurolite) to 2.1 eV for Oh local symmetry (octahedral, as insiderite) (see Table 2a), which is consistent with the expectedlower 10 Dq for Td vs. Oh point symmetry. The splitting for5-coordinated Fe(II) is �1.7 eV for C3v (as in Fe(II)-bearinggrandidierite: Farges, 2001). The splitting observed in the ferro-silicate glasses (� 1.1–1.5 eV) also suggests the presence of Fe(II)in sites with Td-like point symmetry. In addition, the relativeamplitudes of these two pseudo-Voigt components are too large tobe consistent with distorted octahedral geometry. This is becauseany distortion tends to increase the intensity of the second, high-energy component (compare, e.g., the pre-edge of the Fe K-XANES spectra of siderite and fayalite in Fig. 3B) so that bothpre-edge components tend to have comparable intensities, asobserved for the Fe K-XANES pre-edge spectra in numerousstructures such as olivine, fayalite, hypersthene, hedenbergite,enstatite, and hornblende (Wilke et al., 2001). Based on thispre-edge information (pre-edge centroid position, integratedarea, and components separation), only local symmetries suchas Td (as in staurolite) and C3v (trigonal bipyramid, as ingrandidierite) (or mixtures of both geometries) are consistentwith the measured pre-edge features obtained for our ferrosili-cate glasses. However, the presence of minor amounts of 6-co-ordinated Fe(II) cannot be excluded from these spectra alone.
The main-edge region of the Fe K-XANES spectra (featuresB-D in Fig. 3C) arises mostly from final state single- and multiple-scattering photoelectron processes around Fe (Paris and Tyson,
Fig. 4. Pre-edge information for various model compounds with 4-,5-, and 6-coordinated Fe(II) and Fe(III) (as well as selected binarymechanical mixtures of these compounds) vs. the glasses of this study(solid squares). This plot was modified after Wilke et al. (2001) byadding the information on the seven ferrosilicate glasses from thepresent study.
1994; Westre et al., 1997; Wu et al., 1999; Wilke et al., 2001;
Bugaev et al., 2004). In silicate glasses, where the medium-rangestructure is more disordered than in crystalline silicates, the edgeregion arises mostly from Fe-O contributions, including featuresrelated to single- and multiple-scattering (features C and D, re-spectively, in Fig. 3C). More ordered medium-range environmentsaround Fe can also contribute to feature D to make it as intense asfeature C, as observed commonly in Fe-bearing silicate glassesquenched from melts at moderate quench rates (�102 degrees/s;Wilke et al., 1999). From the relatively low intensity of feature Din the XANES spectra of our glasses, it is inferred that there is nosignificant medium-range ordering around Fe, which might havebeen the case if the quench rates had been slower or if the glasseswere poorly homogenized (Wilke et al., 1999).
3.2.2. Extended X-ray absorption finestructure spectroscopy
Figure 5 shows the normalized EXAFS spectra and theirFourier transforms. The results of the least-squares fitting pro-cess are given in Table 3, including the average Fe(II)-O bonddistances [d(Fe-O)], a Debye-Waller factors (�2), and ananharmonic parameter C3. The average anharmonic d(Fe-O)for the ferrosilicate glasses of this study range from 1.97 Å to2.07 Å. The refined number of nearest neighbors (NN) rangesbetween 4.0 and 5.7 and are roughly consistent with the aver-age Fe-O distances refined. Based on average d(Fe(II)-O) mea-sured in various well-known model compounds, the averageNN of Fe(II) varies linearly with d(Fe(II)-O) as predicted bythe following equation : �NN� � �18.2 � 11.28 d(Fe(II)-O) (R2 � 0.93). Using this equation to estimate�NN� for our glasses, we obtain values ranging from 3.9 �0.2 [for d(Fe(II)-O) � 1.97 � 0.02 Å] to 5.1 � 0.2 [ford(Fe(II)-O) � 2.07 � 0.02 Å], which are also in reasonableagreement with the refined NN numbers except for LI2. Thus,Fe(II) is present dominantly in 4- and 5-coordinated environ-ments (on the average). Also, there is no evidence of a mixtureof 4- and 6-coordinated Fe(II), in agreement with pre-edgeinformation.
3.3. UV-Vis-NIR Spectroscopy
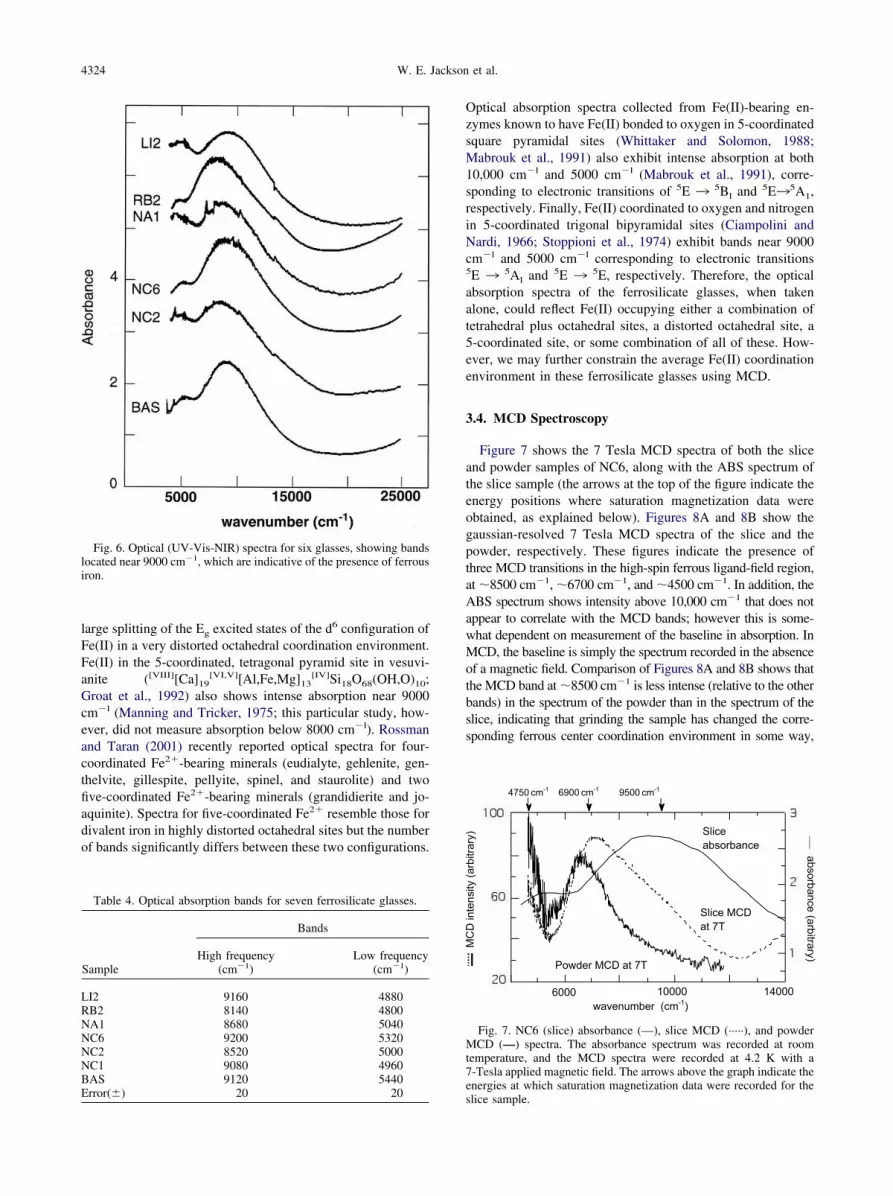
The UV-Vis-NIR data (see Fig. 6 and Table 4) exhibitintense absorption in the range 8100–9200 cm�l and weakabsorption (�10% of the area of 8100–9200 cm�l features)near 5000 cm�l. In crystalline silicates and oxides containingFe(II) in low-distortion octahedral coordination environments,absorption near 9200 cm�l has traditionally been assigned to aT2g ¡ Eg electronic transition associated with octahedrallycoordinated Fe(II). In similar phases, absorption near 5000cm�l has traditionally been assigned to an E ¡ T2 electronictransition associated with tetrahedrally coordinated Fe(II)(Burns, 1985, 1993; Rossman, 1988; Rossman and Taran,2001). The concentration of an absorbing cation on a particularsite (c) is related to absorption (A) by: c � A/�x, in which � isthe molar extinction coefficient and x is the sample thickness.From optical absorption studies of crystalline phases withknown thicknesses and concentrations of Fe(II) in specific sites,� is found to be �100 L/mol-cm for Fe(II) in tetrahedralcoordination and �1–10 L/mol-cm for Fe(II) in octahedral
coordination (Burns, 1993; Rossman and Taran, 2001). Using
that th
4323Multi-spectroscopic study of Fe(II) in silicate glasses
these values of � to interpret our data, we would conclude thatthe ratio of octahedral Fe(II) to tetrahedral Fe(II) sites in theseferrosilicate glasses is �10:1 to 100:1. However, this propor-tion of octahedrally coordinated Fe(II) is inconsistent with theobserved average Fe K-XANES pre-edge heights, d(Fe(II)-O)derived from Fe K-EXAFS, and Fe(II) Mössbauer isomer shiftvalues. To reconcile the optical absorption data with the FeK-edge XANES, K-edge EXAFS, and Mössbauer results, we
Fig. 5. (A) Normalized, k3-weighted EXAFS spectra coTheir corresponding Fourier transforms (R � indicates
Table 3. Fe K-edge EXAFS harmonic vs.
Sample
Averagenumber ofneighborsa
Average Fe-O distance
Harmonic (Å) Anharmonic (Å)
LI2 4.0 2.00 2.03RB2 4.2 1.97 2.00NA1 4.1 1.94 1.97NC6 4.7 1.96 2.02NC2 4.6 1.96 2.02NC1 5.4 2.03 2.06BAS 5.7 2.04 2.07Error
(�)0.5 0.02 0.02
a Determined in least-squares fits of EXAFS data.b
Determined by comparing EXAFS-derived d(Fe(II)-O) with correlationcompounds whose structures have been determined and refined using X-ray d
must examine alternative interpretations of the optical absorp-tion bands seen in the spectra of the ferrosilicate glasses.
It is known that coordination geometries of Fe(II) other than 6-and 4-coordinated yield similar optical absorption spectra. Forexample, the absorption spectrum of Fe(II) in the highly distortedM2 octahedral site of orthopyroxene ((Mg,Fe)SiO3: Goldman andRossman, 1977) exhibits a strong band at 10,000 cm�l and aweak band at �5000 cm�l. This absorption is related to the
at the Fe K-edge for the seven glasses of this study. (B)e distances are uncorrected for phase shifts).
onic results for seven ferrosilicate glasses.
erage coordinationnumber for Fe b
Model quality parameters
�2 (Å2) E0 (eV) �2 (anharm)
4.7 0.0144 7110.9 0.0194.3 0.0144 7111.8 0.0163.9 0.0144 7107.8 0.0154.5 0.0081 7109.5 0.0614.5 0.0121 7108.4 0.0885.0 0.0121 7113.8 0.0285.2 0.0121 7112.9 0.0030.2 0.0001 2.0
llected
anharm
Av
between d(Fe(II)-O) and coordination number of Fe(II) for modeliffraction data.
4324 W. E. Jackson et al.
large splitting of the Eg excited states of the d6 configuration ofFe(II) in a very distorted octahedral coordination environment.Fe(II) in the 5-coordinated, tetragonal pyramid site in vesuvi-anite ([VIII][Ca]19
[VI,V][Al,Fe,Mg]13[IV]Si18O68(OH,O)10;
Groat et al., 1992) also shows intense absorption near 9000cm�l (Manning and Tricker, 1975; this particular study, how-ever, did not measure absorption below 8000 cm�l). Rossmanand Taran (2001) recently reported optical spectra for four-coordinated Fe2�-bearing minerals (eudialyte, gehlenite, gen-thelvite, gillespite, pellyite, spinel, and staurolite) and twofive-coordinated Fe2�-bearing minerals (grandidierite and jo-aquinite). Spectra for five-coordinated Fe2� resemble those fordivalent iron in highly distorted octahedral sites but the numberof bands significantly differs between these two configurations.
Fig. 6. Optical (UV-Vis-NIR) spectra for six glasses, showing bandslocated near 9000 cm�1, which are indicative of the presence of ferrousiron.
Table 4. Optical absorption bands for seven ferrosilicate glasses.
Sample
Bands
High frequency(cm�1)
Low frequency(cm�1)
LI2 9160 4880RB2 8140 4800NA1 8680 5040NC6 9200 5320NC2 8520 5000NC1 9080 4960BAS 9120 5440
Error(�) 20 20Optical absorption spectra collected from Fe(II)-bearing en-zymes known to have Fe(II) bonded to oxygen in 5-coordinatedsquare pyramidal sites (Whittaker and Solomon, 1988;Mabrouk et al., 1991) also exhibit intense absorption at both10,000 cm�l and 5000 cm�l (Mabrouk et al., 1991), corre-sponding to electronic transitions of 5E ¡
5Bl and 5E¡5A1,
respectively. Finally, Fe(II) coordinated to oxygen and nitrogenin 5-coordinated trigonal bipyramidal sites (Ciampolini andNardi, 1966; Stoppioni et al., 1974) exhibit bands near 9000cm�l and 5000 cm�l corresponding to electronic transitions5E ¡
5Al and 5E ¡5E, respectively. Therefore, the optical
absorption spectra of the ferrosilicate glasses, when takenalone, could reflect Fe(II) occupying either a combination oftetrahedral plus octahedral sites, a distorted octahedral site, a5-coordinated site, or some combination of all of these. How-ever, we may further constrain the average Fe(II) coordinationenvironment in these ferrosilicate glasses using MCD.
3.4. MCD Spectroscopy
Figure 7 shows the 7 Tesla MCD spectra of both the sliceand powder samples of NC6, along with the ABS spectrum ofthe slice sample (the arrows at the top of the figure indicate theenergy positions where saturation magnetization data wereobtained, as explained below). Figures 8A and 8B show thegaussian-resolved 7 Tesla MCD spectra of the slice and thepowder, respectively. These figures indicate the presence ofthree MCD transitions in the high-spin ferrous ligand-field region,at �8500 cm�1, �6700 cm�1, and �4500 cm�1. In addition, theABS spectrum shows intensity above 10,000 cm�1 that does notappear to correlate with the MCD bands; however this is some-what dependent on measurement of the baseline in absorption. InMCD, the baseline is simply the spectrum recorded in the absenceof a magnetic field. Comparison of Figures 8A and 8B shows thatthe MCD band at �8500 cm�1 is less intense (relative to the otherbands) in the spectrum of the powder than in the spectrum of theslice, indicating that grinding the sample has changed the corre-sponding ferrous center coordination environment in some way,
Fig. 7. NC6 (slice) absorbance (—), slice MCD (·····), and powderMCD (—) spectra. The absorbance spectrum was recorded at roomtemperature, and the MCD spectra were recorded at 4.2 K with a7-Tesla applied magnetic field. The arrows above the graph indicate the
energies at which saturation magnetization data were recorded for theslice sample.
4325Multi-spectroscopic study of Fe(II) in silicate glasses
and that the band at �8500 cm�1 arises from a different site thanthe two lower energy bands.
The data in Figure 9 show the MCD saturation magnetizationbehavior of the slice sample at 9500 cm�1, 6900 cm�1, and4750 cm�1. These data are plotted as a saturation magnetiza-tion curve showing intensity as a function of �H/2kT, where �is the Bohr magneton (9.27 10�24 J.T�1), H is the magneticfield (in Tesla) at the temperature T, and k is Boltzmann’sconstant. Examination of Figure 9 indicates signal saturationfor the lowest energy band at 5 Tesla (�H/2kT � 1; Fig. 9A),while the two higher energy peaks do not saturate, even at 7Tesla (�H/2kT � 1.3 in Figs. 9B and 9C). The 4750 cm�1 datashow saturation at low temperature and high field, and the datataken at increasing temperatures give rise to a nested set ofcurves. This behavior is characteristic of a ground state with anon-Kramers doublet lowest in energy. The magnetization datataken at the other two energies also give rise to nested sets ofcurves, but they do not show saturation, even at the lowesttemperature (1.8 K) and highest magnetic field (7 Tesla; �H/2kT � 1.3).
As a single ferrous center can have at most two ligand-fieldtransitions in the �4500 cm�1 region, the fact that at least threebands are observed in the combination of MCD and ABS
Fig. 8. MCD spectra at 4.2 K and 7 Tesla for glass sample NC6. (A)Slice sample. (B) Powder sample. Gaussian fits for each spectrum weregenerated independently using Levenberg-Marquardt analysis.
shown in Figure 6 requires that there are at least two distinct
ferrous sites in NC6. In order for these three bands to arise fromless than three distinct ferrous sites, the bands at �6700 cm�1
and �4500 cm�1 would have to arise from the same site. Inthis case, the �6700 cm�1 band would arise from the samenon-Kramers ground state as does the �4500 cm�1 band, andthe site would be 5-coordinated, based on the energies of thebands. The fact that these transitions have different variabletemperature-variable magnetic field MCD behavior for theMCD spectra shown in Figure 7 would suggest that they havedifferent ground states and therefore correspond to differentferrous sites. However, this can be affected by transition po-larization effects. The lack of signal saturation in the data takenat 6900 cm�1 might be explained by this band having predom-inantly z-polarization. However, all possible 5-coordinated sitegeometries, from trigonal bipyramidal to square pyramidal,
Fig. 9. Saturation magnetization behavior of glass sample NC6 at (A)4750 cm�1, (B) 6900 cm�1, and (C) 9500 cm�1. MCD amplitude fora range of applied magnetic field strengths at (●) 1.8 K, (O) 2.2 K, (�)3.0 K, (□) 4.2 K, (‘) 10 K, () 25 K, and () 50 K.
would give rise to two transitions, the higher energy component

4326 W. E. Jackson et al.
of which would be dominantly xy-polarized. In the trigonalbipyramidal (D3h) case, the electronic ground state is 5E� withthe only spin-allowed excited states being 5A1= and 5E=. Thetransition to the higher energy 5A1= state is symmetry forbid-den, whereas the transition to the 5E= state is xy-polarized. Ifthe symmetry is lowered, the forbidden transition would dom-inantly mix with the allowed band and gain xy-polarization.The C4v square-pyramidal site would have a 5E ground state,with the highest energy spin-allowed transition being to the 5B1
state, which is an xy-polarized transition. Thus, the possibilityof polarization effects leading to the different saturation behav-ior in the MCD spectra shown in Figure 9 is unlikely, and thethree bands must arise from three distinct ferrous sites withdifferent ground states.
The coordination environment of the ferrous centers in thesesites may be deduced, at least in part, from the energies of thebands. The band at �4500 cm�1 is in the region where eitherthe lower energy component of a 5-coordinated site or the onlycomponent of a 4-coordinated site would appear. Because thereis no transition observed to higher energy which could beassigned as arising from the same site, this lowest energy bandrepresents a 4-coordinate site which has a geometry close totetrahedral. The band at �6700 cm�1 may be either a singletransition of a 4-coordinated site, which is flattened towardsquare planar, or the higher energy transition of a 5-coordinatedsite. In the latter case the lower energy transition would beexpected to appear at an energy below the spectropolarimetercutoff point.
The band at �8500 cm�1 most likely represents a 4- or5-coordinated site as well. Again, for the 5-coordinated possi-bility the second transition would be at lower energy. This banddoes appear in the region where the lower energy transitionassociated with a 6-coordinated distorted octahedral ferrous sitewould appear. If this site were 6-coordinated, with the higherenergy transition unobserved in the MCD, it would explain thefact that there appears to be some difference between the MCDand ABS spectra of the slice in the higher energy ligand-fieldregion in Figure 7. However, in order for this to be the case, thehigher energy transition would have to be predominantly z-polarized, which is not likely for a close to octahedral ferroussite as this should have mixed polarization. Thus, the MCDdata indicate at least three different ferrous sites with coordi-nation numbers below six.
4. DISCUSSION
4.1. Average Coordination Environment of Fe(II) inFerrosilicate Glasses
The Fe K-edge XANES spectra for the ferrosilicate glassesstudied suggest that Fe(II) occupies an average site that devi-ates markedly from centrosymmetry. The EXAFS-derivedFe(II)-O distances for each glass are consistent with an averageFe(II) site that has a coordination number smaller than 6, andthe anharmonic fit to the EXAFS spectra of these glassesconfirms this, while indicating a slightly longer averaged(Fe(II)-O) than is obtained with harmonic fits (Table 3). TheMössbauer isomer shifts for all the glasses studied are consis-tent with d(Fe(II)-O) of 2.00 to 2.02 (�0.02) Å, values typical
of a mixture of 4- or 5-coordinated Fe(II) sites. Although theUV-Vis-NIR spectra of the glasses appear to be consistent withmainly octahedral or distorted octahedral coordination of Fe(II)and minor 4-coordinated Fe(II) by conventional assessment, itis also possible to reinterpret these data as indicative of 4- and5-coordinated Fe(II), perhaps with some octahedral Fe(II). Fi-nally, the MCD results for NC6 glass suggest several differentFe(II) sites, all with coordination numbers of 4 or 5.
All spectroscopic data for the samples examined are consis-tent with Fe(II) predominantly occupying sites ranging from5-coordinated, perhaps trigonal bipyramidal, to tetrahedral inthese ferrosilicate glasses. This does not preclude the possibil-ity that Fe(II) occupies a distorted octahedral site; however, iftrue, the average d(Fe(II)-O) and Mössbauer IS values aresmaller than those observed for octahedral Fe(II) in any crys-talline phase. When Rb(I) is replaced by Li(I), we note anincrease in average Mössbauer IS, average d(Fe(II)-O), andenergy of the high-frequency optical absorption band. Wecorrelate these changes with the strong network-modifyingbehavior of Li(I) relative to Rb(I) in drawing electron densityaway from the bridging oxygen bonds (i.e., Si-O-Si) and weak-ening local Fe(II)-O bonds (e.g., de Jong and Brown, 1980;Roy and Navrotsky, 1984).
Examination of the average Fe(II) coordination numbersderived from the Fe(II)-O distances in these glasses suggestssome rough trends as a function of the relative amounts ofnetwork modifiers (Rb, Na, Li, Ca, and Mg). The Ca/Na ratiosfor NA1, NC2, and NC1 glasses are 0.0, 0.47, and 0.93,respectively, with similar NBO/T values of 1.09, 1.15, and1.04, respectively. In parallel, we observe average d(Fe-O)values of 1.97 � 0.02, 2.02 � 0.02, and 2.06 � 0.02 Å,respectively, for NA1, NC2, and NC1 glasses, which yields�NN� values of 3.9 � 0.2, 4.5 � 0.2, and 5.0 � 0.2,respectively. In BAS (with NBO/T � 1.05), which is evenricher in Ca, the average d(Fe-O) is 2.07 � 0.02 Å, yielding�NN� of 5.1 � 0.2. In contrast, the replacement of Ca � Naby Rb (RB2 glass) results in a lower d(Fe-O) (2.00 � 0.02) Åand lower �NN� (4.3 � 0.2 oxygens). Replacing Rb (RB2glass with NBO/T � 1.33) by Li (LI2 glass with NBO/T �1.33) results in a slight increase in the average d(Fe-O) from2.00 � 0.02 to 2.03 � 0.02 Å (�NN� increases from 4.3 �0.2 to 4.7 � 0.2 oxygens). From these observations, we suggestthat decreasing the effective charge of network modifiers (i.e.,reducing the ratio of divalent to monovalent cations) in theseglasses shifts the average Fe coordination number to lowervalues and that this effect is more significant than that due todifferences in NBO/T.
An optical absorption study of a labradorite feldspar((Ca,Na)Al2Si2O8) containing �0.4 wt.% FeO found an intenseabsorption band near 8500 cm�l and a weaker band near 4800cm�l in each crystallographic direction (Hofmeister and Ross-man, 1983). This study concluded that Fe(II) occupies thelarge, 8-9 coordinated A site interstitial to the silicate tetrahe-dral framework. Although this composition is relatively Al(III)-rich and Fe(II)-poor when compared with the ferrosilicateglasses examined in the present study, the optical absorptionspectra are similar. Our results, however, do not support anaverage d(Fe(II)-O) that would correspond to Fe(II) with acoordination number of 8-9 in the ferrosilicate glasses exam-ined. It is expected that if Fe(II) occupied such a large site in
the glasses, average Mössbauer IS values and EXAFS-derived
4327Multi-spectroscopic study of Fe(II) in silicate glasses
d(Fe(II)-O) would be much larger; i.e., Fe(II) in the 8-coordi-nated site of almandine garnet, Fe3Al2Si3Ol2, has d(Fe(II)-O)of 2.30 Å (Novak and Gibbs, 1971), and an IS relative to Fe(0)of 1.3 mm/s (Hawthorne, 1988; Rancourt, 1994).
Waychunas et al. (1988, 1989) studied the coordination ofFe(II) in Na2FeSi3O8 (NA2) and K2FeSi3O8 (K2) glasses by FeK-XANES, Fe K-EXAFS, and Mössbauer spectroscopies.They found the average d(Fe(II)-O) to be 2.02 � 0.02 Å (NA2)and 2.00 � 0.02 Å (K2) and average Mössbauer IS values to be1.01 � 0.02 mm/s (NA2) and 0.95 � 0.02 mm/s (K2) (relativeto Fe(0) � 0.0), and concluded that Fe(II) was dominantlytetrahedrally coordinated in these glasses. In light of resultsfrom the current study, the assignment of Fe(II) exclusively totetrahedral coordination (Waychunas et al., 1988, 1989) re-quires reinterpretation. Anharmonic corrections to the EXAFS-derived Fe(II)-O distances of the glasses examined in thepresent study (see Table 3) resulted in distances that are 0.03 to0.06 Å longer than the ones derived using the harmonic ap-proximation. Assuming similar anharmonic corrections to theFe(II)-O distances in the NA2 and K1 glasses studied byWaychunas et al. (1988), the average Fe(II)-O distances wouldhave values (2.06 and 2.04 Å, respectively) that are consistentwith average Fe(II) coordination numbers between 4 and 5. TheMössbauer IS values reported in Waychunas et al. (1988, 1989)are consistent with the values reported for the RB2 and LI2glasses in this study, where Fe(II) is coordinated by 4 to 5oxygens. These results support the prediction that cations withhigher field strength perturb bridging Si-O bonds and weaken(lengthen) Fe-O bonds, although within error a significantchange in Fe-O distance was not observed.
Anharmonic EXAFS analysis of Fe2SiO4 glass (Jackson etal., 1993) showed that the average d(Fe(II)-O) (2.02 � 0.02 Å)is slightly longer than in the melt (1.98 � 0.02 Å), indicatinga larger amount of [5]Fe(II) in the fayalite-composition glass.This result suggests that caution must be applied in inferringcoordination numbers for Fe and other first-row transitionmetals in silicate melts from spectroscopic data on these metalsin silicate glasses (cf. Keppler and Rubie, 1993). For example,Cooney and Sharma (1990) interpreted Raman spectra ofFe2SiO4 glass as indicating a coordination number for Fe(II)lower than six. However, as indicated earlier, the local coordi-nation environment of an element in a silicate glass reflects thelocal structure at a given fictive temperature.
Fig. 10. Bond valence models for three of the general
4.2. Pauling Bond Valence Analysis of Fe(II)Coordination Environments in Ferrosilicate Glasses
Given the EXAFS-derived average Fe-O bond distances andcoordination numbers for Fe(II) in the ferrosilicate glassesconsidered in this study, one can use the bond-valence principleof Pauling (1929) to help understand the effect of glass/meltcomposition on the coordination environment of iron as well ason the physical and thermodynamic properties of the glasses.Using an approach similar to that used in our earlier XAFSstudies of high-valent cations in silicate glasses (Farges, 1991;Farges et al., 1991, 1992; Brown et al., 1995), we can calculatebond valences of average d(Fe-O) for the different glassesusing the bond valence–bond distance correlations of Brownand Altermatt (1985). These results are summarized in Figure10 for three glass compositions. The average coordinationnumber of Fe(II) in the Rb-bearing glass is 4, which is consis-tent with the low bond valence of Rb-O bonds (s �0.1 v.u.) andthe expected high coordination number of Rb in this glass (�8).In contrast, in the Ca-bearing glass, the average coordinationnumber of Fe(II) is between 5 and 6, which is consistent withthe significantly higher bond valence of the Ca-O bond, asshown in the right-most panel of Figure 10. An intermediatesituation is seen for the Na-rich ferrosilicate glass, where thebond valence of Na-O bonds results in a preferred averagecoordination number of �5 oxygens for Fe(II).
By applying Pauling’s second rule to the connecting oxygenin the three examples shown in Figure 10, one can define aunified Pauling relationship:
�cations
S � SFe �NF
SNF �NM
SNM (1)
in which sNi, sNF, and sNM are the average bond valences forthe Fe-O, NF (network-former)-O and NM (network-modifi-er)-O bonds, respectively. Because of Pauling’s second rule,one can also write (where NFe is the coordination number of Feand ZO is the charge on oxygen):
SFe �2
NFe
and �cations
S � Z0 (2,3)
from which one can extract NFe, the predicted iron coordinationnumber
glass compositions examined in this study.

4328 W. E. Jackson et al.
NFe �2
2 � �NF
SNF � �NM
SNM
(4)
This “simple” view of the local bonding requirements aroundthe various cations in silicate glasses and melts helps explaindifferences in their physical properties, including color, den-sity, viscosity, and conductivity. For example, the optical ex-tinction coefficient for the Ca-rich ferrosilicate glass is signif-icantly smaller than for the Rb-rich glass because of thedifference in average coordination number of Fe(II), which is,in turn, dictated by the local bonding requirement around theaverage oxygen in the glass. In addition, the density of theRb-rich ferrosilicate glass should be lower than that of theCa-rich glass because of the higher packing efficiency of Fe(II)in the latter.
4.3. Implications for Fe(II) Partitioning between SilicateMelts and Crystals
There has been a great deal of interest in the partitioningbehavior of first-row transition metal cations between silicatemelts and coexisting igneous minerals since the introduction ofcrystal field theory to geochemistry (Williams, 1959; Burns andFyfe, 1964). This well-known theory provides a reasonableframework for rationalizing the order of uptake of these cationsinto crystals from silicate melts. Burns (1993) summarizedmany of the ideas and observations that have been used to builda structural view of first-row transition metal ion behavior insilicate melts, but he cautioned that much of the reasoning todate is based on observations of these ions in silicate glassesunder ambient conditions, not melts under high-temperatureconditions. The implicit assumption made in most earlier struc-tural studies of silicate glasses is that the coordination environ-ments of these ions are the same in glasses and melts of thesame composition. Williams (1959) first suggested that thepartitioning of a cation from melt to crystal usually involves anincrease in average coordination number and a decrease inaverage interatomic distance. He further speculated that first-row transition metal ions that are particularly stabilized inoctahedral coordination (e.g., Ni(II) and Cr(III)) will gain sta-bility in leaving sites of irregular coordination in the melt andentering more regular octahedral sites in crystals. This reason-ing is generally correct (except for the suggestion that the M-Odistance is generally smaller in the crystal), but its validitydepends on knowledge of average first-row transition metal ioncoordination sites in silicate melts.
Predictions of melt-crystal partitioning behavior of first-rowtransition metal ions, based on crystal field stabilization ener-gies (CFSE) derived from UV/visible spectroscopic studies ofsilicate glasses, appear to be valid only for Ni(II) and Cr(III),which have significantly higher values of CFSE in crystals thanin glasses relative to the other ions (Calas and Petiau, 1983).Both Calas and Petiau (1983) and Keppler (1992) presentindirect evidence that melt-crystal partition coefficients forfirst-row transition metal ions depend on melt polymerization,with these ions partitioning preferentially into crystals as themelts become more polymerized. An important question re-mains, however. Are the coordination environments of first-row transition metal ions in silicate glasses the same as those in
silicate melts?The coordination geometries of Fe(II) (and other relateddivalent first-row transition elements such as Ni) in anhydroussilicate glasses synthesized at 1 atm are controversial, withsome workers favoring distorted octahedral coordination fordivalent Fe and Ni (e.g., Keppler, 1992) and others favoring 4-and 5-coordinated sites (Jackson et al., 1991, 1993; Galoisy andCalas, 1991; Galoisy and Calas, 1993; Wang et al., 1995).High-temperature XAFS studies of Fe(II) and Ni(II) in anhy-drous silicate melts of several compositions (Fe2SiO4,NaSi2O5, NaSi3O7, and M2(Fe,Ni)Si3O8, where M � Li, Na,K, Rb) have shown that they are dominantly 4- or 5-coordi-nated (Waychunas et al., 1988, 1989; Jackson et al., 1991,1993; Farges et al., 1994) in agreement with Fe(II) speciationinformation for tonalitic and rhyolitic melts. These results alsocorrelate positively with the speciation of Ni(II) in albitic meltsas inferred from optical spectroscopy (Keppler and Bagdassa-rov, 1999) as well as in supercritical aqueous fluids (i.e., up to400°C@5 kbars; Hoffman et al., 1999, 2000). In contrast, inhydrated melts exposed to hydrothermal conditions (5 kbars),six-coordinated Fe(II) and Ni(II) are observed (Wilke et al.,1999; Farges et al., 2001a,b). These results indicate that Fe(II)and Ni(II) generally have lower average coordination numbersin anhydrous silicate melts than in anhydrous glasses of thesame composition. This difference is not surprising given thefact that quench rates of silicate melts in many laboratory glasssyntheses are on the order of 102–104 K/s (Dingwell, 1995),which are slow enough such that structural relaxations occurduring the quench. Direct evidence of structural relaxations inthe form of local coordination changes around Ni(II) was foundin a high-temperature XAFS study of Ni(II) in a sodium dis-ilicate glass/melt system (Farges and Brown, 1996), whichshowed that Ni(II) is 4-coordinated on average in the melt (at1250 K) and 5-coordinated on average in the glass (at 300 K).These glasses were quenched by turning off the power to theXAFS furnace (estimated quench rate of �102 K/s).
There is a clear rationale for the observed differences inFe(II) (Jackson et al., 1993) and Ni(II) (Farges and Brown,1996) coordination numbers in silicate glasses and their corre-sponding melts based on the bond valence model discussedabove. From Eqn. 4, one can predict that lower coordinationshould predominate around first-row transition elements inmelts relative to glasses or crystals at lower temperature. Thereason for this is that high temperature promotes thermal ex-pansion of bonds and, therefore, tends to lower bond valencesaround ions, especially around network modifiers whose bondsto oxygen expand more with temperature due to their weaknessrelative to network former-oxygen bonds, which expand verylittle (see Table 1 in Brown et al., 1995). As a consequence,Pauling’s second rule will be less and less satisfied with in-creasing temperature unless reorganization of the local coordi-nation environment occurs. For example, the coordinationnumber of cations such as Fe(II) and Ni(II) could decrease withincreasing temperature, resulting in shorter bonds with oxygensand higher bond valences. In contrast, no decrease in coordi-nation number of Si or Na would be expected in the glassesstudied as these ions should already have their lowest expectedcoordination numbers (4 for Si, and 5-6 for Na: Brown et al.,1995). Furthermore, Na-O bonds would be expected to expandwith increasing temperature because of their relatively low
Pauling bond strength. As a result, a decrease in coordination
4329Multi-spectroscopic study of Fe(II) in silicate glasses
number of Fe(II) is the most likely way to maintain satisfactionof Pauling’s valence rule with increasing temperature for oxy-gens bonded to Fe(II) (see Fig. 10).
The above reasoning provides some important constraints onmelt structure and on the melt/crystal partitioning behavior ofions such as Fe(II) and Ni(II). Assumptions made in the pastabout melt structure for olivine-composition melts, for exam-ple, are that (1) these melts have both octahedral (Oct) andtetrahedral (Tet) sites, by analogy with the olivine structure; (2)olivine-composition melts have a larger Oct/Tet ratio than morecompositionally complex silicate melts; and (3) Fe(II) in suchmelts occupies Oct sites (Roeder, 1974; Irvine and Kushiro,1976; Takahashi, 1978; Mysen et al., 1982). Assumption (3)has also been made for Ni(II) in olivine-composition melts(Takahashi, 1978). The data of Takahashi (1978) show theorder of melt:crystal partitioning to be Ni(II) � Co(II) � Fe(II)� Mn(II) for olivine crystals relative to coexisting(Mg0.5Fe0.5)2SiO4–K2O · 4SiO2 melts, which is the order ofthe octahedral site preference energies (OSPE) for these ions.These observations are similar to the predictions of Roeder(1974) and suggest that the cations occupy fewer octahedralsites in the melt than in the coexisting olivine crystal. Compar-ison of OSPE for Fe(II) (�0.17 eV; Burns, 1993) with kT(�0.14 eV at 1575 K, where k is the Boltzmann constant)indicates that Fe(II) should not show a strong preference foroctahedral sites in the crystal or melt at 1575 K. In contradic-tion to the above suggestions that Fe(II) occupies octahedralsites in olivine-composition melts, the high-temperatureEXAFS studies by Jackson et al. (1991, 1993) show that Fe(II)occupies mainly 4-coordinated sites in Fe2SiO4 melt. Ni(II) hasa much larger OSPE (�0.9 eV: Burns, 1993), and thus it shouldbe stabilized in octahedral sites in the melt (at 1575 K), if theyexist. More recent studies have shown that Ni(II) is dominantly4-coordinated in a variety of anhydrous sodium silicate melts(Farges et al., 1994; Farges and Brown, 1996; Farges et al.,2001a), and it is dominantly tetrahedrally and pentahedrallycoordinated in more SiO2-rich glasses (CaNiSi2O6 and(Na,K)2NiSi3O8: Galoisy and Calas, 1991, 1993).
Thus, the existing high-temperature structural data for Fe(II)and Ni(II) in a variety of silicate melts can be satisfactorilymodeled using bond valence theory which does not support thegeneral assumption that these cations are 6-coordinated insilicate melts of a wide compositional range (cf. Keppler,1992). This general assumption may only be verified by studiesof the local cation environments in silicate melts at appropriatetemperatures and pressures.
Acknowledgments—We wish to thank Carl Ponader (Corning Inc.,Corning, NY) and Jean-Marie Combes (St. Gobain SARL, Aubervil-liers, France) for their help with EXAFS data collection, and JonathanStebbins (Stanford University) and Mike Hochella (Virginia Polytech-nic Institute and State University) for helpful reviews of earlier ver-sions of this manuscript. Jonathan Stebbins is also acknowledged foruseful discussions about fictive temperatures of silicate glasses. GCAAssociate Editor Claudia Romano (Universita’ degli Studi Roma Tre)and three anonymous referees are thanked for careful reviews of ourmanuscript, which improved its clarity. We also acknowledge supportof the Stanford Synchrotron Radiation Laboratory (SSRL) staff. This
work was supported by NSF grants EAR-9305028, CHE-0089215,CHE-0431425, and INT-9726528 to G.E.B., and NIH grant GM40392to E.I.S. SSRL is supported by the U.S. Department of Energy (BESand BER) and by the National Institutes of Health.
Associate editor: C. Romano
REFERENCES
Alberto H. V., Pinto da Cunha J. L., Mysen B. O., Gil J. M., and Ayresde Campos N. (1996) Analysis of Mössbauer spectra of silicateglasses using a two-dimensional Gaussian distribution of hyperfineparameters. J. Non-Crystal. Solids 194, 48–57.
Arrio M. A., Rossano S., Brouder C., Galoisy L., and Calas G. (2000)Calculation of multipole transitions at the Fe K pre-edge throughp-d hybridization in the Ligand Field Multiplet model. Europhys.Lett. 51, 454–460.
Bajt S., Sutton S. R., and Delaney J. S. (1994) X-ray microprobeanalysis of iron redox states in silicates and oxides using X-rayabsorption near edge structure (XANES). Geochim. Cosmochim.Acta 58, 5209–5214.
Baker L. L. and Rutherford M. J. (1996) The effect of dissolved wateron the oxidation state of silicic melts. Geochim. Cosmochim. Acta60, 2179–2187.
Bell P. M., Mao H. K., and Weeks R. A. (1976) Optical spectra andelectron paramagnetic resonance of lunar and synthetic glasses: Astudy of the effects of controlled atmosphere, composition andtemperature. Proc. 7th Lunar Sci. Conf., pp. 2543–2559.
Berry A. J., O’Neill H., Jayasuriya K., Campbell S. J., and Foran G. J.(2003) XANES calibrations for the oxidation state of iron in asilicate glass. Amer. Mineral. 88, 967–977.
Bonnin-Mosbah M., Simionovici A. S., Metrich N., Duraud J. P.,Massare D., and Dillmann P. (2001) Iron oxidation states in silicateglass fragments and glass inclusions with a XANES micro-probe. J.Non-Crystal. Solids 288, 103–113.
Boon J. A. and Fyfe W. S. (1972) The coordination number of ferrousions in silicate glasses. Chem. Geol. 10, 287–298.
Borisov A. A. and Shapkin A. I. (1990) A new empirical equationrating Fe3�/Fe2� in magmas to their composition, oxygen fugacityand temperature. Geochem. Int. 27, 111–116.
Brown G. E. Jr., Farges F., and Calas G. (1995) X-ray scattering andx-ray spectroscopy studies of silicate melts. In Structure, Dynamicsand Properties of Silicate Melts (eds. J. F. Stebbins, P. F. McMil-lan, and D. B. Dingwell), Reviews in Mineralogy, Vol. 32, 317–410, Mineralogical Society of America, Washington, DC.
Brown G. E. Jr., Keefer K. D., and Fenn P. M. (1978) Extended X-rayAbsorption Fine Structure (EXAFS) study of iron-bearing silicateglasses: Ion coordination environment and oxidation state. Prog.Geol. Soc. Amer. Ann. Mtg. 10, 37319.
Brown I. D. and Altermatt D. (1985) Bond valence parameters obtainedfrom a systematic analysis of the inorganic crystal structure data-base. Acta Crystallogr. B41, 244–247.
Bugaev L. A., Farges F., Rusakova E. B., Sokolenko A. P., Latokha Y.,and Avakyan L. (2004) The Fourier-transform analysis of Ti K-XANES in metamicts and glasses. Physica Scripta T115, 215–217.
Burns R. G. (1985) Thermodynamic data from crystal field spectra.Rev. Mineral. 14, 277–314.
Burns R. G. (1993) Mineralogical Applications of Crystal Field The-ory, 2nd ed Cambridge University Press, Cambridge, UK, 312 pp.
Burns R. G. and Fyfe W. S. (1964) Site of preference energy �selective uptake of transition-metal ions from magma. Science 144,1001.
Calas G. and Petiau J. (1983) Structure of oxide glasses. Spectroscopicstudies of local order and crystallochemistry. Geochemical impli-cations. Bull. Minéralogie 106, 33–55.
Ciampolini M. and Nardi N. (1966) Trigonal bibyramidal complexes ofbivalent manganese, iron and zinc with Tris(2-dimethylamino-ethyl)amine. Inorg. Chem. 5, 1150–1154.
Cooney T. F. and Sharma S. K. (1990) Structure of glasses in thesystems Mg2SiO4 - Fe2SiO4, Mn2SiO4 - Fe2SiO4, Mg2SiO4 -
CaMgSiO4 and Mn2SiO4 - CaMnSiO4. J. Non-Crystal. Solids 122,10–32.
4330 W. E. Jackson et al.
Crozier E. D., Rehr J. J., and Ingalls R. (1988) Amorphous and liquidsystems. In X-ray Absorption: Principles, Applications, Techniquesof EXAFS, SEXAFS and XANES, Chemical Analysis, Vol. 92 (eds.D. C. Koningsberger and R. Prins), pp. 373–442. John Wiley &Sons, New York.
de Jong B. H. W. S. and Brown G. E. Jr. (1980) Polymerization ofsilicate and aluminate tetrahedra in glasses, melts and aqueoussolutions: II. The network modifying effects of Mg2�, K�, Na�,Li�, H�, OH�, F�, Cl�, H2O and CO2 in silicate melts. Geochim.Cosmochim. Acta 44, 1627–1642.
Delaney J. S., Bajt S., Sutton S. R., and Dyar M. D. (1996) In situmicroanalysis of Fe3�/�Fe ratios in amphibole by X-ray absorptionnear edge structure (XANES) spectroscopy. In Mineral Spectros-copy: A Tribute to Roger Burns (eds. M. D. Dyar, C. A. McCam-mon, and M. W. Schaefer). Geochemical Society Special Publica-tion 5, pp. 289–304.
Dingwell D. B. (1995) Relaxation in silicate melts: Some applications.Rev. Mineral. 32, 21–66.
Dräger G., Frahm R., Materlik G., and Brummer O. (1988) On themultipole character of the x-ray transitions in the pre-edge structureof Fe K absorption spectra. Phys. Stat. Solidi. B 146, 287–293.
Dunlap R. A., Edelman D. A., and Mackay G. R. (1998) A Mössbauereffect investigation of correlated hyperfine parameters in naturalglasses (tektites). J. Non-Crystal. Solids 223, 141–146.
Dunlap R. A. (1997) An investigation of Fe oxidation states and sitedistributions in a Tibetan tektite. Hyperf. Interact. 110, 217–225.
Dyar M. D. (1985) A review of Mössbauer data on inorganic glasses:The effects of composition on iron valency and coordination. Am.Mineral. 70, 304–316.
Effenberger H., Mereiter K., and Zemann J. (1981) Crystal structurerefinements of magnesite, calcite, rhodochrosite, siderite, smithso-nite and dolomite with discussion of some aspects of the stereo-chemistry of calcite-type carbonates. Z. Kristallogr. 156, 233–243.
Farges F. (1991) Structural environment around Th4� in silicateglasses. Implications for the geochemistry of incompatible Me4�
elements. Geochim. Cosmochim. Acta 55, 3303–3319.Farges F. (2001) Crystal chemistry of iron in natural grandidierites: An
x-ray absorption fine structure spectroscopy study. Phys. Chem.Minerals 28, 619–629.
Farges F. and Brown G. E. Jr. (1996) An empirical model for theanharmonic analysis of high-temperature XAFS spectra of oxidecompounds with applications to the coordination environment of Niin Ni-olivine and Ni-Na-disilicate glass and melt. Chem. Geol. 127,253–268.
Farges F. and Brown G. E. Jr. (1997) Coordination chemistry of Ti(IV)in silicate glasses and melts. IV. Natural and synthetic volcanicglasses. Geochim. Cosmochim. Acta 61, 1863–1870.
Farges F., Brown G. E. Jr., Calas G., Galoisy L., and Waychunas G. A.(1994) Structural transformation in Ni-bearing Na2Si2O5 glassesand melt. Geophys. Res. Lett. 21, 1931–1934.
Farges F., Brown G. E. Jr., Navrotsky A., Gan H., and Rehr J. J. (1996)Coordination chemistry of Ti(IV) in silicate glasses and melts. II.Glasses under ambient conditions. Geochim. Cosmochim. Acta 60,3029–3054.
Farges F., Brown G. E. Jr., Petit P.-E., and Munoz M. (2001a) Tran-sition elements in water-bearing silicate glasses/melts. Part I. Ahigh resolution and anharmonic analysis of Ni coordination envi-ronments in crystals, glasses and melts.Geochim. Cosmochim. Acta86, 1665–1678.
Farges F., Guyot F., Andrault D., and Wang Y. (1993) Local environ-ment around Fe in Mg0.9Fe0.1SiO3 perovskite. An Fe K-edge XASstudy. Eur. J. Mineral. 6, 303–312.
Farges F., Munoz M., Siewert R., Malavergne V., Brown G. E. Jr.,Behrens H., Nowak M., and Petit P.-E. (2001b) Transition elementsin water-bearing silicate glasses/melts. Part II. Ni in water-bearingglasses. Geochim. Cosmochim. Acta 86, 1679–1693.
Farges F., Ponader C. W., and Brown G. E. Jr. (1991) Structural environ-ments of incompatible elements in silicate glass/melt systems: I. Zr attrace levels. Geochim. Cosmochim. Acta 55, 1563–1574.
Farges F., Ponader C. W., Calas G., and Brown G. E. Jr. (1992) Localenvironment around incompatible elements in silicate glass/melt
systems. II: U(VI), U(V) and U(IV). Geochim. Cosmochim. Acta56, 4205–4220.Fox K. E., Furukawa T., and White W. B. (1982) Transition metal ionsin silicate melts. Part 2. Iron in sodium silicate glasses. Phys. Chem.Glasses 23, 169–178.
Fudali R. F. (1965) Oxygen fugacities of basaltic and andesitic mag-mas. Geochim. Cosmochim. Acta 29, 1063–1075.
Galoisy L. and Calas G. (1991) Spectroscopic evidence for five-coordinated nickel in CaNiSi2O6 glass. Am. Mineral. 76, 1777–1780.
Galoisy L. and Calas G. (1993) Structural environment of nickel insilicate glass/melt systems. I. Spectroscopic determination of coor-dination states. Geochim. Cosmochim. Acta 57, 3613–3626.
Galoisy L., Calas G., and Arrio M.-A. (2001) High-resolution XANESspectra of iron in minerals and glasses: Structural information fromthe pre-edge. Chem. Geol. 174, 307–319.
Goldman D. S. and Berg J. I. (1980) Spectral study of ferrous iron inCa-Al borosilicate glass at room and melt temperatures. J. Non-Crystal. Solids 38/39, 183–188.
Goldman D. S. and Rossman G. R. (1977) The spectra of iron inorthopyroxene revisited: The splitting of the ground state. Am.Mineral. 62, 151–157.
Groat L. A., Hawthorne F. C., and Ercit T. S. (1992) The chemistry ofvesuvianite. Canadian Mineral. 30, 19–48.
Hawthorne F. C. (1988) Mössbauer spectroscopy. Rev. Mineral. 18,255–340.
Henderson C. M. B., Charnock J. M., Helz G. R., Kohn S. C., PattrickR. A. D., and Vaughan D. J. (1991) EXAFS in earth sciencesresearch. In XAFS Vl: X-ray Absorption Fine Structure VI. EllisHorwood Ltd. pp. 573–578.
Henderson C. M. B., Cressey G., and Redfern S. A. T. (1995) Geo-logical applications of synchrotron-radiation. Rad. Phys. Chem. 45,459–481.
Heumann D., Dräger G., and Bocharov S. (1997) Angular-dependencein the K pre-edge XANES of cubic crystals: The separation of theempty metal eg and t2g states of NiO and FeO. J. de Phys. IV France7 C2, 481–483.
Hoffmann M. M., Darab J. G., Heald S. M., Yonker C. R., and FultonJ. L. (2000) New experimental developments for in situ EXAFSstudies of chemical reactions under hydrothermal conditions.Chem. Geol. 167, 89–103.
Hoffmann M. M., Darab J. G., Palmer B. J., and Fulton J. L. (1999) Atransition in the Ni2� complex structure from six-to four-coordinateupon formation of ion pair species in supercritical water: An XAFS,NIR and MD study. J. Phys. Chem. A 103, 8471–8482.
Hofmeister A. M. and Rossman G. R. (1983) Color in feldspars. InFeldspar Mineralogy (ed. P. H. Ribbe), Reviews in Mineralogy,Vol. 2, pp. 271–280. Mineralogical Society of America, Washing-ton, DC.
Irvine T. N. and Kushiro I. (1976) Partitioning of Ni and Mg betweenolivine and silicate liquids. Carnegie Inst. Washington Yearbook75, 668–675.
Iwamoto N., Umesaki N., and Atsumi T. (1987) EXAFS and x-ray-diffraction studies of iron ions in a 0.2(Fe2O3)·0.8(Na2O·2SiO2)glass. J. Mater. Sci. Lett. 6, 271–273.
Jackson W. E. (1991) Spectroscopic studies of ferrous iron in silicateliquids, glasses and crystals. Ph.D. dissertation, Department ofGeology, Stanford University. 162 pp.
Jackson W. E., Mustre De Leon J., Brown G. E. Jr., Waychunas G. A.,Conradson S. D., and Combes J.-M. (1993) High-temperature XASstudy of Fe2SiO4 liquid: Reduced coordination of ferrous iron.Science 262, 229–232.
Jackson W. E., Waychunas G. A., Brown G. E.Jr., Mustre de Leon J.,Conradson S. D. and Combes J.-M. (1991) In-situ high temperaturex-ray absorption study of ferrous iron in orthosilicates crystals andliquids. In X-ray Absorption Fine Structure (ed. S. S. Hasnain), pp.298–301. Ellis Horwood, Chichester, UK.
Keppler H. (1992) Crystal field spectra and geochemistry of transitionmetal ions in silicate melts and glasses. Am. Mineral. 77, 62–75.
Keppler H. and Bagdassarov N. (1999) The speciation of Ni and Co insilicate melts from optical absorption spectra to 1500°C. Chem.Geol. 158, 105–115.
Keppler H. and Rubie D. C. (1993) Pressure-induced coordination
changes of transition-metal ions in silicate melts. Nature 364,54–56.
4331Multi-spectroscopic study of Fe(II) in silicate glasses
Kilinc A., Carmichael I. S. E., Rivers M. L., and Sack R. O. (1983) Theferric-ferrous ratio of natural silicate liquids equilibrated in air.Contrib. Mineral. Petrol. 83, 136–140.
Koningsberger D. C. and Prins R. (1988) X-ray Absorption: Principles,Applications, Techniques of EXAFS, SEXAFS and XANES. WileyInc., 673 pp.
Krause M. O. and Oliver J. H. (1979) Natural widths of atomic K andL levels, K alpha X-ray lines and several KLL auger lines. J. Phys.Chem. Ref. Data 8, 329–338.
Kress V. C. and Carmichael I. S. E. (1991) The compressibility ofsilicate liquids containing Fe2O3 and the effect of composition,temperature, oxygen fugacity and pressure on their redox states.Contrib. Mineral. Petrol. 108, 82–92.
Lange A. R. and Navrotsky A. (1993) Heat-capacities of Fe2O3-bearingsilicate liquids. Geochim. Cosmochim. Acta 110, 311–320.
Lefrère Y. (2002) Propriétés d’ absorption optique du Fe2� and Fe3� dansdes verres d’intérêt industriel: Mesure modélisation et implicationsstructurales. Ph.D. thesis, Université de Paris 7. 243 pp. (in French).
Levy R. A., Lupis C. H. P., and Flinn P. A. (1976) Mössbauer analysis ofthe valence and coordination of iron cations in SiO2-Na2O-CaOglasses. Phys. Chem. Glasses 17, 94–103.
Lytle F. W., Greegor R. B., Sandstrom D. R., Marques E. C., Wong J.,Spiro C. L., Huffman G. P., and Huggins F. E. (1984) Measurementof soft X-ray absorption spectra with a fluorescent ion chamberdetector. Nucl. Instrum. Methods 226, 542–548.
Mabrouk P. A., Orville A. M., Lipscomb J. D., and Solomon E. I.(1991) Variable temperature variable-field magnetic circular di-chroism studies of the Fe(II) active site in metapyrocatechase:Implications for the molecular mechanism of extradiol dioxygen-ases. J. Am. Chem. Soc. 113, 4053–4061.
MacKenzie K. J. D. and Meinhold R. H. (1997) MAS NMR study ofpentacoordinated magnesium in grandidierite. Am. Mineral. 82,479–482.
Manning P. G. and Tricker M. J. (1975) Optical-absorption and Möss-bauer spectral studies of iron and titanium site-populations invesuvianites. Can. Mineral. 13, 259–265.
Moore G., Righter K., and Carmichael I. S. E. (1995) The effect ofdissolved water on the oxidation state of iron in natural silicateliquids. Contrib. Mineral. Petrol. 120, 170–179.
Mysen B. O. (1988) Structure and Properties of Silicate Melts.Elsevier, Amsterdam, 354 pp.
Mysen B. O. (1991) Relations between structure, redox equilibria ofiron and properties of magmatic liquids. In Advances in PhysicalChemistry, Vol. 9 (eds. L. L. Perchuk and I. Kushiro), pp. 41–98.Cambridge University Press, Cambridge, UK.
Mysen B. O. and Virgo D. (1989) Redox equilibria, structure, andproperties of Fe-bearing aluminosilicate melts: Relationshipsamong temperature, composition and oxygen fugacity in the systemNa2O-Al2O3-SiO2-Fe-O. Am. Mineral. 74, 58–76.
Mysen B. O., Virgo D., and Seifert F. A. (1982) The structure ofsilicate melts: Implications for chemical and physical properties ofnatural magma. Rev. Geophys. Space Phys. 20, 353–383.
Mysen B. O., Virgo D., and Seifert F. A. (1984) Redox equilibria of Fein alkaline earth silicate melts: Relationships between melt struc-ture, oxygen fugacity, T and properties of Fe-bearing silicate liq-uids. Am. Mineral. 69, 834–847.
Mysen B. O., Virgo D., Neumann E. R., and Seifert F. A. (1985a)Redox equilibria and the structural states of ferric and ferrous ironin melts in the system CaO-MgO-Al2O3-SiO2-Fe-O: Relationshipsbetween redox equilibria, melt structure and liquidus phase equi-libria. Am. Mineral. 70, 317–331.
Mysen B. O., Virgo D., Scarfe C. M., and Cronin D. J. (1985b)Viscosity and structure of iron- and aluminum-bearing calciumsilicate melts at 1 atm. Am. Mineral. 70, 487–498.
Nolet D. A. (1980) Optical absorption and Mössbauer spectra of Fe, Tisilicate glasses. J. Non-Crystal. Solids 37, 99–110.
Novak G. A. and Gibbs G. V. (1971) The crystal chemistry of thesilicate garnets. Am. Mineral. 56, 791–825.
Paris E. and Tyson T. A. (1994) Iron site geometry in orthopyroxene:Multiple scattering calculations and XANES study. Phys. Chem.
Mineral. 21, 299–304.Pauling L. (1929) The principles determining the structure of complexionic crystals. J. Am. Chem. Soc. 51, 1010–1026.
Rancourt D. G. (1994) Mössbauer spectroscopy of minerals I. Inadequacyof Lorentzian-line doublets in fitting spectra arising from quadrupolesplitting distributions. Phys. Chem. Minerals 21, 244–249.
Rancourt D. G., Fortin D., Pichler T., Thibault P. J., Lamarche G.,Morris R. V., and Mercier P. H. J. (2001) Mineralogical character-ization of a natural As-rich hydrous ferric oxide coprecipitateformed by mixing hydrothermal fluids and seawater. Am. Mineral.86, 834–851.
Roe A. L., Schneider D. J., Mayer R. J., Pyrz J. W., Widom J., and OueL. Jr. (1984) X-ray absorption-spectroscopy of iron-tyrosinate pro-teins. J. Am. Chem. Soc. 106, 1676–1681.
Roeder P. L. (1974) Activity of iron and olivine solubility in basalticliquids. Earth Planet. Sci. Lett. 23, 397–410.
Romano C., Paris E., Poe B. T., Giuli G., Dingwell D. B., and MottanaA. (2000) Effect of aluminum on Ti-coordination in silicate glasses:A XANES study. Am. Mineral. 85, 108–117.
Rossano S., Balan E., Morin G., Bauer J.-P., Brouder C., and Calas G.(1999) 57Fe Mössbauer spectroscopy of tektites. Phys. Chem. Min-eral. 26, 530–538.
Rossano S., Ramos A., and Delaye J. M. (2000) Environment of ferrousiron in CaFeSi2O6 glass: Contributions from EXAFS and moleculardynamics. J. Non-Crystal. Solids 273, 48–52.
Rossano S., Farges F., Ramos A., Delaye J. M., and Brown G. E. Jr.(2002) Bond valence in silicate glasses. J. Non-Crystal. Solids 304,167–173.
Rossman G. R. (1988) Optical spectroscopy. Rev. Mineral. 18, 207–254.
Rossman G. R. and Taran M. N. (2001) Spectroscopic standards forfour- and fivefold-coordinated Fe2� in oxygen-based minerals. Am.Mineral. 86, 896–903.
Roy B. N. and Navrotsky A. (1984) Thermochemistry of charge-coupled substitutions in silicate glasses: The systems Ml/n
n�A1O2-SiO2 (M � Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba, Pb). J. Am. Ceram.Soc. 67, 606–610.
Sack R. O., Carmichael I. S. E., Rivers M., and Ghiorso M. S. (1980)Ferric-ferrous equilibria in natural silicate liquids at 1 bar. Contrib.Mineral. Petrol. 75, 369–376.
Seifert F. and Olesch M. (1977) Mössbauer spectroscopy of grandi-dierite, (Mg,Fe)Al3SiO9. Am. Mineral. 62, 547–553.
Stoppioni P., Mani F., and Sacconi L. (1974) Halo-, hydrido- anddinitrogencomplexes of iron (II) with tritertiary phosphines. Inorg.Chim. Acta 11, 227–230.
Takahashi E. (1978) Partitioning of Ni2�, Co2�, Fe2�, Mn2� andMg2� between olivine and silicate melts: Compositional depen-dence of partition coefficient. Geochim. Cosmochim. Acta 42,1829–1844.
Tangeman J. A. and Lange R. A. (1998) The effect of Al3�, Fe3� andTi4� on the configurational heat capacities of sodium silicate liq-uids. Phys. Chem. Mineral. 26, 83–99.
Tangeman J. A., Lange R., and Forman L. (2001) Ferric-ferrous equi-libria in K2O-FeO-Fe2O3-SiO2 melts. Geochim. Cosmochim. Acta65, 1809–1819.
Teo B. K. and Joy D. C., eds. (1981) EXAFS Spectroscopy: Techniquesand Applications. Plenum Press, 275 pp.
Teo B. K. (1986) EXAFS: Basic Principles and Data Analysis. SpringerVerlag, 285 pp.
Toplis M. J. (1998) Energy barriers to viscous flow and the predictionof glass transition temperatures of molten silicates. Amer. Mineral.83, 480–490.
Tröger L., Arvanitis D., Baberschke K., Michaelis H., Grimm U., andZschech E. (1992) Full correction of the self-absorption in soft-fluorescence extended X-ray-absorption fine structure. Phys. Rev. B46, 3283–3289.
Virgo D. and Mysen B. O. (1985) The structural state of iron inoxidized vs. reduced glasses at 1 atm: A 57Fe study. Phys. Chem.Minerals 12, 65–76.
Wang Z. F., Cooney T. F., and Sharma S. K. (1995) In situ structuralinvestigation of iron-containing silicate liquids and glasses.
Geochim. Cosmochim. Acta 59, 1571–1577.
4332 W. E. Jackson et al.
Waychunas G. A., Apted M. J., and Brown G. E. Jr. (1983) X-rayK-edge absorption spectra of Fe minerals and model compounds:Near-edge structure. Phys. Chem. Minerals 10, 1–9.
Waychunas G. A., Brown G. E. Jr., Ponader C. W., and Jackson W. E.(1988) Evidence from X-ray absorption for network-forming Fe2�
in molten alkali silicates. Nature 332, 251–253.Waychunas G. A., Brown G. E. Jr., Jackson W. E., and Ponader C. W.
(1989) In-situ high temperature x-ray absorption study of iron inalkali silicate melts and glasses. Physica B 21, 144–146.
Waychunas G. A. and Rossman G. R. (1983) Spectroscopic standardfor tetrahedrally coordinated ferric iron: �-LiAlO2:Fe3�. Phys.Chem. Mineral. 9, 212–215.
Westre T. E., Kennepohl P., de Witt J., Hedman B., Hodgson K. O.,and Solomon E. I. (1997) A multiplet analysis of Fe K-edge 1s ¡
3d pre-edge features of iron complexes. J. Am. Chem. Soc. 1196297–6314.
Whittaker J. W. and Solomon E. I. (1988) Spectroscopic studies on
ferrous non-heme iron active sites: Magnetic circular dichroism ofmononuclear Fe sites in superoxide dismutase and lipoxygenase.J. Am. Chem. Soc. 110, 5329–5339.
Wilke M., Farges F., Behrens H., and Burkhard D. (1999) The effect ofwater on the local environment of Fe in silicate glasses. Eur. J.Mineral. 11 (Suppl. 1), 244.
Wilke M., Farges F., Petit P. E., Brown G. E. Jr., and Martin F. (2001)Oxidation state and coordination of Fe in minerals: An Fe KXANES spectroscopic study. Am. Mineral. 65, 713–730.
Wilke M., Behrens H., Burkhard D. J. M., and Rossano S. (2002) Theoxidation state of iron in polymerized silicate melt at 500 MPawater pressure. Chem. Geol. 189, 55–67.
Williams R. J. P. (1959) Deposition of trace elements in basic magma.Nature 184, 44.
Winterer M. (1996) The XAFS package. Proc. 9th Int. Conf. on X-rayAbsorption Fine Structure. XAFS VI, Grenoble, France; 144.
Wu Z., Bonnin-Mosbah M., Duraud J. P., Metrich N., and Delaney J. S.(1999) XANES studies of Fe-bearing glasses. J. Synchrotron Rad.
6, 344–346.