molecular dynamics of the full-length p53 monomer
TRANSCRIPT

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
RepoRt
Cell Cycle 12:18, 1–11; September 15, 2013; © 2013 Landes Bioscience
RepoRt
www.landesbioscience.com Cell Cycle 1
Introduction
The TRp53 gene, implicated in cancer development as well as in infertility,1,2 is the most frequently mutated in all human cancers; see http://p53.fr or http://p53.iarc.fr.3-5
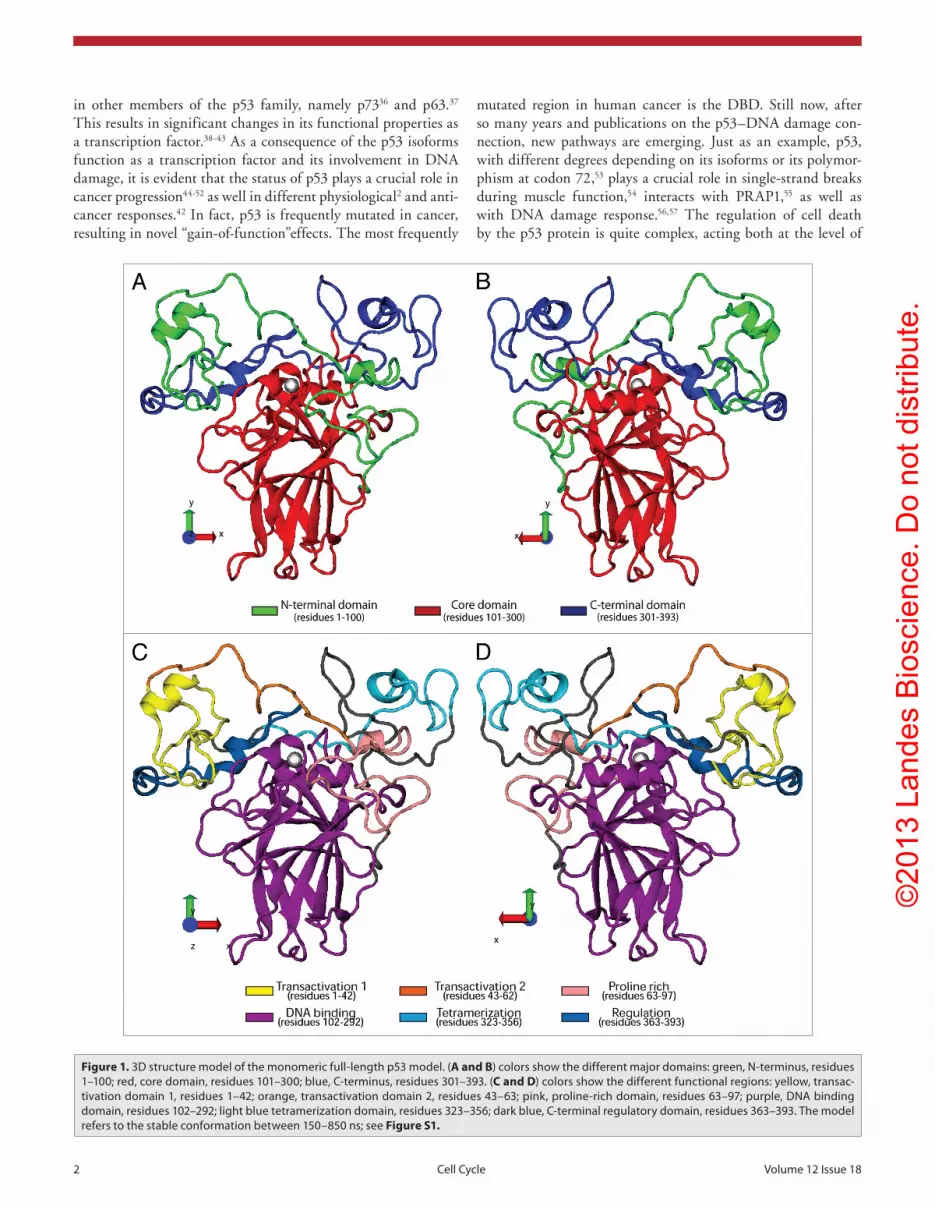
The tumor suppressor protein p53 is composed of three large domains: (1) the intrinsically flexible N-terminal region, which includes 2 transactivation domain (TAD) and a proline-rich region (PRO); (2) the central core region, which contains the DNA binding domain (DBD) region with a coordinated Zn ion between β sheet folds; (3) the C-terminal region, which includes a tetramerization domain (TET) and regulatory C terminus domain (REG; see Fig. 1). The binding to DNA is mediated by recognition of specific sequence motif interacting directly with the DBD.6 Conversely, the transcriptional function requires the physical interactions of the 2 TAD with the transcriptional machinery. p53 is in fact a powerful transcription factor, and, consequently, it drives a large number of promoters,7 depend-ing on specific activators.8-11 Possibly the most studied promoter target is p21,12-14 but new targets emerge as, for example, the
connection between IL-7Ra and telomer erosion15 or the silenc-ing of repeats and noncoding RNA.16 As a consequence of the transactivation of so many different promoters so far described, p53 is able to activate different biochemical pathways affecting the regulation of life and death of the cell. A recently discov-ered, relevant set of novel p53 targets include the metabolism of the cell,17,18 for example the pathways of mevalonate,19,20 ser-ine,21 malate,22 and glutaminolysis.23 This results in the regula-tion of cell death24 or cell cycle.25-27 A relevant metabolic role of p53 is exerted during hypoxia, as oxygen relative pressure reaches particularly lower levels at the center of the tumors, where p53 protein interplays with mTOR28 as well as with the more classic pathways of HIF-1a and VHL.29 A distinct role occurs during re-oxygenation, as during miocardial infartion or trombosis in the central nervous system.30 In both cases p53 status is a relevant factor. Although abnormal protein variants were described several decades ago,31-34 only recently has a distinct promoter been identi-fied,35 codifying of the full-length p53 or Δ0p53 (promoter 1) or for the Δ33p53 and Δ60p53 (promoter 2), similar to what occurs
*Correspondence to: Giovanni Chillemi; Email: [email protected]; Gerry Melino; Email: [email protected]: 08/06/2013; Accepted: 08/15/2013http://dx.doi.org/10.4161/cc.26162
Molecular dynamics of the full-length p53 monomer
Giovanni Chillemi1,*, pavel Davidovich2, Marco D’Abramo1, tazhir Mametnabiev2, Alexander Vasilievich Garabadgiu2, Alessandro Desideri3, and Gerry Melino4,5,*
1CINeCA; Rome, Italy; 2Laboratory of Molecular pharmacology; Saint-petersburg technological Institute; St. petersburg, Russia; 3Biology Department; University of Rome “tor Vergata”; Rome, Italy; 4Biochemistry Laboratory; IDI-IRCCS and Department of experimental Medicine and
Surgery; University of Rome “tor Vergata”; Rome, Italy; 5Medical Research Council; toxicology Unit; Leicester University; Leicester, UK
Keywords: p53, cell death, DNA damage, cancer, p73, p63
Abbreviations: DBD, DNA binding region; ED, essential dynamics; MD, molecular dynamics; TET, tet-ramerization domain; PRO, proline rich region; RE, responsive element; RMSD, root mean square deviation;
RMSF, root mean square fluctuations; SAXS, small-angle X-ray scattering; TAD, transactivation domain
the p53 protein is frequently mutated in a very large proportion of human tumors, where it seems to acquire gain-of-function activity that facilitates tumor onset and progression. A possible mechanism is the ability of mutant p53 proteins to physically interact with other proteins, including members of the same family, namely p63 and p73, inactivating their function. Assuming that this interaction might occurs at the level of the monomer, to investigate the molecular basis for this interaction, here, we sample the structural flexibility of the wild-type p53 monomeric protein. the results show a strong stability up to 850 ns in the DNA binding domain, with major flexibility in the N-terminal transactivations domains (tAD1 and tAD2) as well as in the C-terminal region (tetramerization domain). Several stable hydrogen bonds have been detected between N-terminal or C-terminal and DNA binding domain, and also between N-terminal and C-terminal. essential dynamics analysis highlights strongly correlated movements involving tAD1 and the proline-rich region in the N-terminal domain, the tetramerization region in the C-terminal domain; Lys120 in the DNA binding region. the herein presented model is a starting point for further investigation of the whole protein tetramer as well as of its mutants.
©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
2 Cell Cycle Volume 12 Issue 18
in other members of the p53 family, namely p7336 and p63.37 This results in significant changes in its functional properties as a transcription factor.38-43 As a consequence of the p53 isoforms function as a transcription factor and its involvement in DNA damage, it is evident that the status of p53 plays a crucial role in cancer progression44-52 as well in different physiological2 and anti-cancer responses.42 In fact, p53 is frequently mutated in cancer, resulting in novel “gain-of-function”effects. The most frequently
mutated region in human cancer is the DBD. Still now, after so many years and publications on the p53–DNA damage con-nection, new pathways are emerging. Just as an example, p53, with different degrees depending on its isoforms or its polymor-phism at codon 72,53 plays a crucial role in single-strand breaks during muscle function,54 interacts with PRAP1,55 as well as with DNA damage response.56,57 The regulation of cell death by the p53 protein is quite complex, acting both at the level of
Figure 1. 3D structure model of the monomeric full-length p53 model. (A and B) colors show the different major domains: green, N-terminus, residues 1–100; red, core domain, residues 101–300; blue, C-terminus, residues 301–393. (C and D) colors show the different functional regions: yellow, transac-tivation domain 1, residues 1–42; orange, transactivation domain 2, residues 43–63; pink, proline-rich domain, residues 63–97; purple, DNA binding domain, residues 102–292; light blue tetramerization domain, residues 323–356; dark blue, C-terminal regulatory domain, residues 363–393. the model refers to the stable conformation between 150–850 ns; see Figure S1.
©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 3
autophagy,58 lysosomes,59 or at the core machinery of pro-grammed cell death.60-65 In addition to DNA damage response and cell death, p53 plays a crucial role in regulating cellular senes-cence66-68 by interacting, for example, with MageA2,69 PATZ1,70 4E-BP1,71 mTOR,72,73 highlighting the vast complexity of this crucial regulation.
As the p53 proteins have different isoforms, and it is frequently mutated in cancer,74 in order to understand its function with the related transactivation rules and superactivating sequences,75 it is crucial to understand its structural interactions and dynamic function.76-78 p53 is active as a tetramer and its structure bound to DNA has been resolved in the truncated core domain form,79 even though only the full-length protein produces the maxi-mum bending and twisting of the consensus DNA RE.80 The full-length p53 has been resolved only in its tetrameric form by a combination of NMR, small-angle X-ray scattering, electron microscopy, and FRET,81-86 showing that in absence of DNA, an open cross-shaped structure is formed, with loosely coupled dimers interacting via the core domain, whereas the structure rigidifies upon DNA binding and becomes more compact.
Because of its structural flexibility, most experimental struc-tural studies have addressed only single domains or portions of them. In particular, the 2 TADs in the N-terminal domain were solved by NMR87,88 and X-ray diffraction;89 the core domain by X-ray diffraction90-92 and NMR93; the tetramerization region in the C-terminal domain by NMR94 and X-ray diffraction.91 The quaternary structure of the p53 tetramer in complex with DNA has been determined by a combination of SAXS, NMR, and EM.95 FRET experiments have been performed on the N-terminal plus DBD form (residues 1–292) and on the full-length in tetrameric form.82 However, there are intrinsically dis-ordered regions, functional to the biological role of p53, that were not resolved by any technique.
Full-length p53 in its monomeric form is therefore a particu-larly elusive protein to be structurally studied with experimental methods. Several in silico studies have been performed, but all focused on single domains or fragments of p53. In particular, the p53 core domain has been the most studied both for its biological role in transcription process96-100 and in cancer-related mutations.101-104 MD conformational analyses were also performed for the N-terminal recognition α helix,105-108 and for C-terminal fragments.109,110
In order to study the entire monomeric protein behavior and its inter domain interactions, we have built a model for the full-length p53 (residues 1–393). The structure of unresolved fragments (10%) and dis-ordered links between the domains have been predicted and merged with the experimental data. Here, we have accrued out a 850 ns-long molecular dynamics (MD) simulation to validate the stability of the model and predict its structural and dynamic properties.
Results
By means of the root mean square deviation (RMSD) analysis we measure how much the instant
conformation, sampled by the MD simulation, differs from the starting structure. Figure S1 shows the RMSD as a function of simulation time for the C-α atoms of the protein. As expected, the N- and C-terminal domains undergo large conformational rearrangements when compared with the core domain, resulting in a constant increase of the RMSD up to 120 ns. After this time, a plateau is reached, and the value oscillates around 10 Å until the end of the simulation. We have therefore excluded the first 150 ns of simulation (red line) and performed all the following analyses on the last 700 ns, i.e., from 150 to 850 ns.
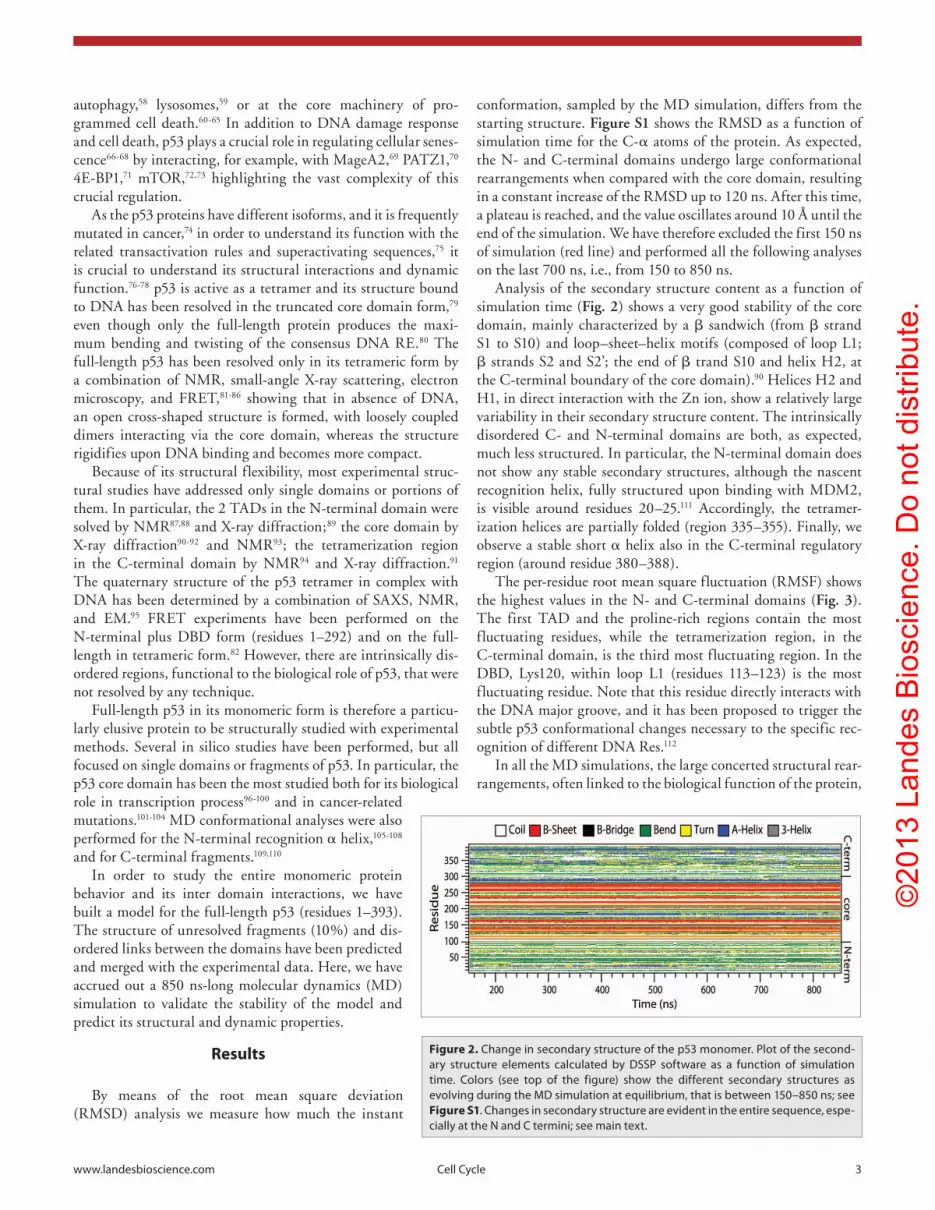
Analysis of the secondary structure content as a function of simulation time (Fig. 2) shows a very good stability of the core domain, mainly characterized by a β sandwich (from β strand S1 to S10) and loop–sheet–helix motifs (composed of loop L1; β strands S2 and S2’; the end of β trand S10 and helix H2, at the C-terminal boundary of the core domain).90 Helices H2 and H1, in direct interaction with the Zn ion, show a relatively large variability in their secondary structure content. The intrinsically disordered C- and N-terminal domains are both, as expected, much less structured. In particular, the N-terminal domain does not show any stable secondary structures, although the nascent recognition helix, fully structured upon binding with MDM2, is visible around residues 20–25.111 Accordingly, the tetramer-ization helices are partially folded (region 335–355). Finally, we observe a stable short α helix also in the C-terminal regulatory region (around residue 380–388).
The per-residue root mean square fluctuation (RMSF) shows the highest values in the N- and C-terminal domains (Fig. 3). The first TAD and the proline-rich regions contain the most fluctuating residues, while the tetramerization region, in the C-terminal domain, is the third most fluctuating region. In the DBD, Lys120, within loop L1 (residues 113–123) is the most fluctuating residue. Note that this residue directly interacts with the DNA major groove, and it has been proposed to trigger the subtle p53 conformational changes necessary to the specific rec-ognition of different DNA Res.112
In all the MD simulations, the large concerted structural rear-rangements, often linked to the biological function of the protein,
Figure 2. Change in secondary structure of the p53 monomer. plot of the second-ary structure elements calculated by DSSp software as a function of simulation time. Colors (see top of the figure) show the different secondary structures as evolving during the MD simulation at equilibrium, that is between 150–850 ns; see Figure S1. Changes in secondary structure are evident in the entire sequence, espe-cially at the N and C termini; see main text.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
4 Cell Cycle Volume 12 Issue 18
are disguised by a great number of small irrelevant fluctuations that can be eliminated performing an essential dynamics (ED) analysis.113 ED is based on the diagonalization of the covariance matrix built from the atomic fluctuations after the removal of the translational and rotational movement, and it allows the separa-tion of the few 3N directions along which the majority of the protein motion is defined. The RMSF of the C-α atoms along the first 3 eigenvectors (Fig. S2) shows that the widest correlated movements in p53 involve the regions identified as the most fluc-tuating: i.e., TAD1 and PRO in the N-terminal domain, TET in the C-terminal domain; Lys120 in the DBD. Interestingly, the second and third eigenvectors uncouple the N-terminal domain motions, being dominated by the TAD1 or PRO fluctuations, respectively. Projections of the C-α atoms onto the eigenvectors show that nearly 20% of the whole p53 motion is described by the first eigenvector, and projections along this eigenvector show that this correlated motion is mainly contributed by the N and C terminus (Fig. 4A and B). ED results on p53 monomer allowed us to carry on further analysis to better characterize the main protein motions. The simulation time spent by the protein in a specific region along the ED eigenvectors gives us an estimation of the free energy value of the visited structural basin. The free energy maps of the projection of the trajectory on the essential subspace along eigenvectors 1–2 (Fig. S3A) shows that one major basin is present, spanning from −4 and 3 (eigevector 1) and −2 and 4 (eigenvector 2). The same analysis performed only on the core domain shows a much smaller conformational basin (Fig. S3B), demonstrating that the N- and C-terminal domains are responsible for the widest protein movements .
A number of conformations have been extracted from the minimum free energy region in the middle of the basin of Figure S3A, or along its border at the extreme of the eigenvector sub-space, and the electrostatic potential distribution has been cal-culated. Two representative conformations (identified with the 2 arrows in Fig. S3A) have been chosen and their positive and nega-tive iso-surfaces are shown in Figure 5. Figure 5A, representative
of the most stable free energy region, shows that a positive 3-fin-ger surface is formed by 3 lysine residues: 372 and 373 located in the Regulation region and 120 in the DBD, but a contribution originates also from His365, His368, and Lys370. These results indicate how the p53 different domains can cooperate in their interaction with negative partners such as the DNA. Figure 5B, chosen at the extreme along the first eigenvector subspace, shows a negative bulge formed by both TAD1 and TAD2 residues. The insert of Figure 5C highlights how the tip of the bulge is formed by the nascent helix responsible for MDM2 recognition, around residues 20–25. Therefore, wide structural fluctuations, second-ary structure variability, and electrostatic fields can provide an efficient mechanism used by p53 in looking for the right molecu-lar partner.
Global correlated motions in proteins are naturally driven by local protein–protein interactions, and so the network created by the most stable hydrogen bonds inter- and intra-domains has been analyzed. Despite the described large flexibility of the N-terminal domain, several “anchor points” between the N-terminal and the core domain are present. Three hydrogen bonds, in particular, have residence time longer than 80% of the simulation time (bold in Table S1), thus maintaining a tight interaction between residues 93–96, at the C terminus of the last proline (Pro92) of the PRO region, and 2 loop 1 residues (Thr170 and Val172), plus Arg213, located in the loop between S6 and S7, all in the DBD. Figure 4C shows a representative snapshot for these contacts with the involved lateral chains colored in orange and yellow for the N-terminal and core domain, respectively. Ser95 and Thr170 interact via a main chain hydrogen bond presents for 54% of the simulation time. Other N-terminal residues, contacting the DBD for long simulation times, are located in the TAD2. Gln52, Trp53, and Asp57 form direct hydrogen bonds for more than 50% of the simulation time with His178 (underlined in Table S1 and highlighted in Fig. 4D), close to His179 that binds the Zn ion, and therefore likely playing a role in the maintenance of the metal coordination. Moreover, Gln52 interacts with Arg181, by means of 2 hydrogen bonds, with percentage of simulation time between 35 and 40. The interface between the C-terminal and the core domain is less structured, with no contacts lasting for more than 40% with the exception of the contact between His179 and Leu383 (underlined in Table S2), present for 67% of simulation time. The REG region in the C-terminal domain forms very stable interactions with TAD1 in the N-terminal domain. In detail, Ser6 and His368 forms a direct hydrogen bond for more than 91% of simulation time (bold in Table S3), while Gln5-Thr377 and Ser6-Ser371 contacts are present for 74 and 68% of simulation time, respectively (underlined in Table S3). It is worth noting that helix 1 in the DBD is at the center of a network of interactions that involves both the TADs and the REG regions, acting as a sensor to their structural changes.
Regarding the intra domain interactions, the core domain shows a rich network of very stable hydrogen bonds (87 and 23 hydrogen bonds with residence time greater than 70 and 98%, respectively, see Table S4), in line with its structural role. The high flexibility of the N- and C-terminal domain does not allow for a large number of stable intra domain hydrogen bonds.
Figure 3. per-residue root mean square fluctuations. the values for each residue are colored in green, red, and cyan for the N-terminal, core and C-terminal domain, respectively. Larger degrees of fluctua-tions and secondary structure changes are evident in the functional domains, see Figure 1. the DBD, in red, is the most stable region with the most frequently mutated residues in human cancers located in the more stable areas.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 5
Figure 4. p53 structure along the first essential Dynamics eigenvector. (A and B) projection of the p53 structure along the first eigenvector. the three struc-tures relative to average, minimum and maximum eigenvalues are represented. (C and D) Representative MD snapshots with highlighted the lateral chain of the resi-dues involved in long residence hydrogen bonds between the N-terminal and the core domain; see also the Supplementary Tables. Colors are as in Figure 1A and B.
Figure 5. electrostatic potential surface around p53. (A) Representative struc-ture of the most stable region in the free energy plot (see Fig. S3). (B and C, insert) Representative structure of the basin boundaries in the free energy plot. the red and blue surfaces correspond to electrostatic potentials of −2.5 and +2.5 Kt/e, respectively.
However 7 and 5 hydrogen bonds are observed in the N- and C-terminal domains, respectively, with residence time greater than 70% (Tables S5 and S6). Note that while the N-terminal intra domain bonds are located all along the domain, 4 out of the 5 stable interactions of the C-terminal domain bind REG residues, the fifth bond being between Arg335 and Glu339 of the TET region.
Discussion
p53 seems evolved from a common ancestor of the p63/p73 proteins, from which a new gene, specifically dedicated to DNA damage response, with less exons, and reduced numbers of residues, has merged.114-116 Although p53 and p63/p73 diverged during evolu-tion, their relationship is still very strong and relevant. Both in cancer progression, metastasis, and in physi-ological development, all members of the family inter-act and regulate each other.117,118 This is, for example, evident in epidermis,119 as well as in DNA damage response.120 The p53 protein is highly conserved in its structure from C. elegans, D. melanogaster to H. sapi-ens.121-126 While the DBD domain is highly conserved among vertebrates and invertebrates, the C termi-nus varies, resulting in a change from dimeric struc-ture to a tetramer in the vertebrates.127-129 The more ancient members of the family include p73, involved in cancer,130 neurodevelopment,131,132 and aging,133 and p63, involved in epidermal development,119,134-136 can-cer,137-141 reproduction,142 and heart development.143 Understanding the structural restrain of its structure is pivotal to understand the function of p53144-146 as well as its potential therapeutic exploitation.147,148 The regulation of p53 protein half-life is crucial to his function149,150 and, consequently, for cancer progres-sion.151-153 This proteosomal degradation is indeed a powerful therapeutic target.154-158
The full-length p53 tetramer, bound to DNA, acquires different quaternary conformations82 where the C-terminal and DBD directly interact; the N-terminal seems to only weakly associate with DBD, and no direct interactions between N-terminal and C-terminal are observed. Additional conformations were detected at the monomeric level. In vitro, there is a realistic possibility for hetero-tetramer formation among the p53/p63/p73 family members.159 The p53 tetramer seems to bind with the DBD REs with sym-metric 10 bp sequences, in 4 classes of structures, consistent with a model where p53 slides along DNA via its C termini while the DBD hopping on/off searches for the responsive Res.81 The p73 tetramer is somehow similar, but it seems much more sensitive to spacer length.160 Limited evidence is available on the physical interaction between p53, or mutant p53, with the other mem-ber of the family, namely p63. There is, however, a robust evi-dence of function interaction and immunoprecipatation.140,161,162
To approach this question, we first created a model for the full-length p53 and performed a full structural and dynamic valida-tion by molecular dynamics.
The in silico results show a core domain with a stable struc-tural role, while intrinsically flexible N- and C-terminal domains explore a large conformational space in search of compatible part-ners, i.e., other proteins in the case of the N-terminal domain; DNA and pX3 proteins in the case of the C-terminal domain.

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
6 Cell Cycle Volume 12 Issue 18
The tools implemented by these p53 domains to accomplish their biological goal are great flexibility (Fig. 3), correlated movements (Fig. 4A), charged surfaces (Fig. 5), and variable secondary structure (e.g., the MDM2 recognition helix in the N-terminal domain, see Figs. 2 and 5C; the tetramerization helices in the C-terminal domain). In particular, electrostatic potential analy-sis and per-residue RMSF results (Figs. 5A and 3) are compatible with the proposed model,81 in which p53 interacts with DNA through its C-terminal domain, while Lys120 acts as a sensor, capable of triggering the conformational transition toward the specific binding of DNA Res.112
Despite the great conformational space explored by the N- and C-terminal domains as compared with the core domain (demonstrated by the free energy plot of Fig. S3A vs. S3B), they maintain stable inter domain local interactions (Fig. 4C and D; Table S1–3). It is particularly intriguing that the Zn coordinated helix H1 is at the center of these inter-domains interactions both with the N-terminal domain through His178 (Fig. 4D; Table S1) than with the REG region in the C-terminal domain through His179 (Table S2). At the same time H1 through the Zn coordi-nated cysteine residues is able to sense conformational changes in the DBD structure, both involving the β sandwich or the loop–sheet–helix regions. It is therefore tempting to hypothesize that this helix plays a relevant role in the orchestration of the complex conformational changes needed to accomplish p53 biological functions.
Materials and Methods
The model of the full-length p53 was built using the I-TASSER server service163 from the human amino acid sequence. Three tem-plate structures were used (PDB id: 3q01,91 1ycs,92 and 2fej93) for core domain prediction, and the ab initio methodology was used for the unstructured domain fragments (TAD residues 1–60 and C-terminal residues 360–391). The protein structure prediction was made with medium confidence (C-score −2.53; TM-score 0.42 −0.14) in order to consider the intrinsic flexibility of the TADs and REG. We were not interested, in fact, in obtaining the best structure for these regions, but a realistic starting one to be sampled by the MD simulation. Nevertheless, the most impor-tant 2 short α helices in the TADs, which are major involved in the “ipophilic” protein–protein interactions (e.g., p53-MDM2111 and p53-Bcl-2164), are present in the final model.
Forcefield parameters for the Zn ion 4-ligand coordination interface was obtained from Lu and co-authors,96 who performed a MD study of the core domain. All simulations were performed using Gromacs package v4.6165 with ff99SB-ILDN force field166
and periodic boundary conditions. The starting structure has been immersed in a periodic box of TIP3P water model,167 which extended from 13 Å from the solute and neutralized adding sodium ions. The PME method was used to treat the long-range electrostatics.168 Bond lengths involving bonds to hydrogens were constrained using LINCS algorithm.169 A time step of 2 fs was used. The conformational sampling was done at a temperature of 300 K using the V-Rescale algorithm. The equilibration procedure involves: (1) 2 rounds of minimizations (1500 iteration each) and dynamics (25 ps each) of the solvent and sodium ions in the bulk solvent, keeping the solute constrained to its initial position, with decreasing force constants of 500, 100, 300, 50 Kcal/(mol Å2); (2) 4 rounds of 2000 steps of minimization of whole system, where the solute restraint was kept as 100, 50, 25, and 5 Kcal/(mol Å2); (3) an unrestrained minimization of the whole system. Finally the system was heated to 300 K at constant volume and equilibrated for 300 ps at constant pressure. The production phase was started at this stage and continued up to 850 ns. The conformations were collected in the trajectories at intervals of 2 ps.
The electrostatic potential was calculated with APBS soft-ware,170 while all the other analysis were performed using the Gromacs tools. Default values of g_hbond, the Gromacs tool for hydrogen bond analysis, were used for cut-off distance and angle: i.e., 3.5 Å and 30 °. The plots were generated using VMD171 and POV-Ray (http://www.povray.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank I D’Annessa for helpful discussion and sugges-tions. This work has been supported by the Medical Research Council, UK; grant MIUR/FIRB (RBFR12BGHO) to MD; Federal Target Program grant “Research and Scientific-Pedagogical Personnel of Innovative Russia in 2009-2013”, State Contract N. 14.132.21.1334 to PD; grants from “Alleanza con-tro il Cancro” (ACC12), MIUR/PRIN (20078P7T3K_001)/FIRB (RBIP06LCA9_0023, RBIP06LCA9_0C), AIRC (2011-IG11955), AIRC 5xmille (MCO #9979), Telethon Grant GGPO9133, Min. Salute (Ric oncol 26/07), and IDI-IRCCS (RF08 c.15, RF07 c.57) to GM. Work was supported by Ministry of Education and Science of the Russian Federation (11.G34.31.0069).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26162

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 7
References1. Levine AJ, Tomasini R, McKeon FD, Mak TW,
Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12:259-65; PMID:21427767; http://dx.doi.org/10.1038/nrm3086
2. Paskulin DD, Cunha-Filho JSL, Souza CAB, Bortolini MC, Hainaut P, Ashton-Prolla P. Tp53 PIN3 and PEX4 polymorphisms and infertility associated with endometriosis or with post-in vitro fertilization implantation failure. Cell Death Dis 2012; 3:e392; PMID:23013791; http://dx.doi.org/10.1038/cddis.2012.116
3. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on Tp53 mutation patterns and tumor phenotype: lessons from recent devel-opments in the IARC Tp53 database. Hum Mutat 2007; 28:622-9; PMID:17311302; http://dx.doi.org/10.1002/humu.20495
4. Blandino G, Deppert W, Hainaut P, Levine A, Lozano G, Olivier M, Rotter V, Wiman K, Oren M. Mutant p53 protein, master regulator of human malignan-cies: a report on the Fifth Mutant p53 Workshop. Cell Death Differ 2012; 19:180-3; PMID:22095277; http://dx.doi.org/10.1038/cdd.2011.148
5. Marcel V, Olivier M, Mollereau B, Hainaut P, Bourdon JC. First International p53 Isoforms Meeting: ‘p53 isoforms through evolution: from iden-tification to biological function’. Cell Death Differ 2011; 18:563-4; PMID:21151028; http://dx.doi.org/10.1038/cdd.2010.156
6. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem 2008; 77:557-82; PMID:18410249; http://dx.doi.org/10.1146/annurev.biochem.77.060806.091238
7. Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 tran-scriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145:571-83; PMID:21565614; http://dx.doi.org/10.1016/j.cell.2011.03.035
8. Soria C, Estermann FE, Espantman KC, O’Shea CC. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 2010; 466:1076-81; PMID:20740008; http://dx.doi.org/10.1038/nature09307
9. Menendez D, Resnick MA, Haran TE. Transactivation by low and high levels of human p53 reveals new physical rules of engagement and novel super-transactivation sequences. Cell Cycle 2012; 11:4287-8; PMID:23095671; http://dx.doi.org/10.4161/cc.22467
10. Guseva NV, Rokhlin OW, Bair TB, Glover RB, Cohen MB. Inhibition of p53 expression modi-fies the specificity of chromatin binding by the androgen receptor. Oncotarget 2012; 3:183-94; PMID:22383394
11. da Costa NM, Hautefeuille A, Cros M-P, Melendez ME, Waters T, Swann P, Hainaut P, Pinto LF. Transcriptional regulation of thymine DNA glycosyl-ase (TDG) by the tumor suppressor protein p53. Cell Cycle 2012; 11:4570-8; PMID:23165212; http://dx.doi.org/10.4161/cc.22843
12. Insinga A, Cicalese A, Faretta M, Gallo B, Albano L, Ronzoni S, Furia L, Viale A, Pelicci PG. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci U S A 2013; 110:3931-6; PMID:23417300; http://dx.doi.org/10.1073/pnas.1213394110
13. Eckner R. p53-dependent growth arrest and induc-tion of p21: a critical role for PCAF-mediated histone acetylation. Cell Cycle 2012; 11:2591-2; PMID:22751433; http://dx.doi.org/10.4161/cc.21235
14. Quaas M, Müller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle 2012; 11:4661-72; PMID:23187802; http://dx.doi.org/10.4161/cc.22917
15. Kibe R, Zhang S, Guo D, Marrero L, Tsien F, Rodriguez P, Khan S, Zieske A, Huang J, Li W, et al. IL-7Rα deficiency in p53null mice exacerbates thy-mocyte telomere erosion and lymphomagenesis. Cell Death Differ 2012; 19:1139-51; PMID:22281704; http://dx.doi.org/10.1038/cdd.2011.203
16. Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. p53 cooperates with DNA meth-ylation and a suicidal interferon response to main-tain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci U S A 2013; 110:E89-98; PMID:23236145; http://dx.doi.org/10.1073/pnas.1216922110
17. Boren J, Brindle KM. Apoptosis-induced mitochon-drial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ 2012; 19:1561-70; PMID:22460322; http://dx.doi.org/10.1038/cdd.2012.34
18. Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein A, Albanese C, et al. Dietary downregu-lation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle 2012; 11:4436-46; PMID:23151455; http://dx.doi.org/10.4161/cc.22778
19. Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 dis-rupts mammary tissue architecture via the mevalonate pathway. Cell 2012; 148:244-58; PMID:22265415; http://dx.doi.org/10.1016/j.cell.2011.12.017
20. Ginestier C, Charafe-Jauffret E, Birnbaum D. p53 and cancer stem cells: the mevalonate connexion. Cell Cycle 2012; 11:2583-4; PMID:22751434; http://dx.doi.org/10.4161/cc.21092
21. Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine star-vation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493:542-6; PMID:23242140; http://dx.doi.org/10.1038/nature11743
22. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013; 493:689-93; PMID:23334421; http://dx.doi.org/10.1038/nature11776
23. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen spe-cies. Proc Natl Acad Sci U S A 2010; 107:7461-6; PMID:20351271; http://dx.doi.org/10.1073/pnas.1002459107
24. Chang JY, Chiang MF, Lin SR, Lee MH, He H, Chou PY, Chen SJ, Chen YA, Yang LY, Lai FJ, et al. TIAF1 self-aggregation in peritumor capsule formation, spontaneous activation of SMAD-responsive pro-moter in p53-deficient environment, and cell death. Cell Death Dis 2012; 3:e302; PMID:22534828; http://dx.doi.org/10.1038/cddis.2012.36
25. Ahmed A, Yang J, Maya-Mendoza A, Jackson DA, Ashcroft M. Pharmacological activation of a novel p53-dependent S-phase checkpoint involving CHK-1. Cell Death Dis 2011; 2:e160; PMID:21593792; http://dx.doi.org/10.1038/cddis.2011.42
26. Wang Y, Zhou BP. FBW7-Aurora B-p53 feedback loop regulates mitosis and cell growth. Cell Cycle 2012; 11:4113-4; PMID:23099923; http://dx.doi.org/10.4161/cc.22607
27. Donzelli S, Fontemaggi G, Fazi F, Di Agostino S, Padula F, Biagioni F, Muti P, Strano S, Blandino G. MicroRNA-128-2 targets the transcriptional repres-sor E2F5 enhancing mutant p53 gain of function. Cell Death Differ 2012; 19:1038-48; PMID:22193543; http://dx.doi.org/10.1038/cdd.2011.190
28. Leontieva OV, Blagosklonny MV. Hypoxia and gero-suppression: the mTOR saga continues. Cell Cycle 2012; 11:3926-31; PMID:22987149; http://dx.doi.org/10.4161/cc.21908
29. Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis 2011; 2:e164; PMID:21614094; http://dx.doi.org/10.1038/cddis.2011.48
30. Gogna R, Madan E, Kuppusamy P, Pati U. Re-oxygenation causes hypoxic tumor regression through restoration of p53 wild-type conformation and post-translational modifications. Cell Death Dis 2012; 3:e286; PMID:22419115; http://dx.doi.org/10.1038/cddis.2012.15
31. Arai N, Nomura D, Yokota K, Wolf D, Brill E, Shohat O, Rotter V. Immunologically distinct p53 molecules generated by alternative splicing. Mol Cell Biol 1986; 6:3232-9; PMID:3023970
32. Conforti F, Yang AL, Agostini M, Rufini A, Tucci P, Nicklison-Chirou MV, et al. Relative expression of TAp73 and Delta Np73 isoforms. Aging-Us 2012; 4:202-5
33. Grespi F, Melino G. P73 and age-related diseases: is there any link with Parkinson Disease? Aging (Albany NY) 2012; 4:923-31; PMID:23271007
34. Tucci P. Caloric restriction: is mammalian life exten-sion linked to p53? Aging (Albany NY) 2012; 4:525-34; PMID:22983298
35. Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activ-ity. Genes Dev 2005; 19:2122-37; PMID:16131611; http://dx.doi.org/10.1101/gad.1339905
36. Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol 2008; 18:244-52; PMID:18406616; http://dx.doi.org/10.1016/j.tcb.2008.03.003
37. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ 2011; 18:1487-99; PMID:21760596; http://dx.doi.org/10.1038/cdd.2011.81
38. Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kivioja T, Ignatiev I, Kel A, Taipale J, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expres-sion analysis. Cell Death Differ 2012; 19:1992-2002; PMID:22790872; http://dx.doi.org/10.1038/cdd.2012.89
39. Lee S, Kim JY, Kim YJ, Seok KO, Kim JH, Chang YJ, Kang HY, Park JH. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ 2012; 19:1613-22; PMID:22522597; http://dx.doi.org/10.1038/cdd.2012.40
40. Blagosklonny MV. Wt p53 impairs response to che-motherapy: make lemonade to spare normal cells. Oncotarget 2012; 3:601-7; PMID:22802145
41. Pei D, Zhang Y, Zheng J. Regulation of p53: a col-laboration between Mdm2 and Mdmx. Oncotarget 2012; 3:228-35; PMID:22410433
42. Madapura HS, Salamon D, Wiman KG, Lain S, Klein G, Klein E, Nagy N. p53 contributes to T cell homeostasis through the induction of pro-apoptotic SAP. Cell Cycle 2012; 11:4563-9; PMID:23165210; http://dx.doi.org/10.4161/cc.22810
43. Rotblat B, Melino G, Knight RA. NRF2 and p53: Januses in cancer? Oncotarget 2012; 3:1272-83; PMID:23174755

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
8 Cell Cycle Volume 12 Issue 18
44. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153:449-60; PMID:23562644; http://dx.doi.org/10.1016/j.cell.2013.03.020
45. Hock AK, Vousden KH. Tumor suppression by p53: fall of the triumvirate? Cell 2012; 149:1183-5; PMID:22682240; http://dx.doi.org/10.1016/j.cell.2012.05.024
46. Berns A. Cancer: The blind spot of p53. Nature 2010; 468:519-20; PMID:21107421; http://dx.doi.org/10.1038/468519a
47. Rouaud P, Fiancette R, Vincent-Fabert C, Magnone V, Cogné M, Dubus P, Denizot Y. Mantle cell lym-phoma-like lymphomas in c-myc-3’RR/p53+/- mice and c-myc-3’RR/Cdk4R24C mice: differential oncogenic mechanisms but similar cellular origin. Oncotarget 2012; 3:586-93; PMID:22592113
48. Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010; 468:572-5; PMID:21107428; http://dx.doi.org/10.1038/nature09535
49. Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao X-H, Refetoff S, Montagna C, Di Cristofano A. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget 2011; 2:1109-26; PMID:22190384
50. Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown Swigart L, Pham DM, Seo Y, Evan GI, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010; 468:567-71; PMID:21107427; http://dx.doi.org/10.1038/nature09526
51. Krimpenfort P, Song J-Y, Proost N, Zevenhoven J, Jonkers J, Berns A. Deleted in colorectal car-cinoma suppresses metastasis in p53-defi-cient mammary tumours. Nature 2012; 482:538-41; PMID:22358843; http://dx.doi.org/10.1038/nature10790
52. Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I, Haffner-Krausz R, Jung S, et al. CKIα ablation high-lights a critical role for p53 in invasiveness control. Nature 2011; 470:409-13; PMID:21331045; http://dx.doi.org/10.1038/nature09673
53. Altilia S, Santoro A, Malagoli D, Lanzarini C, Ballesteros Álvarez JA, Galazzo G, Porter DC, Crocco P, Rose G, Passarino G, et al. Tp53 codon 72 polymorphism affects accumulation of mtDNA dam-age in human cells. Aging (Albany NY) 2012; 4:28-39; PMID:22289634
54. Fortini P, Ferretti C, Pascucci B, Narciso L, Pajalunga D, Puggioni EMR, Castino R, Isidoro C, Crescenzi M, Dogliotti E. DNA damage response by single-strand breaks in terminally differentiated muscle cells and the control of muscle integrity. Cell Death Differ 2012; 19:1741-9; PMID:22705848; http://dx.doi.org/10.1038/cdd.2012.53
55. Huang BH, Zhuo JL, Leung CHW, Lu GD, Liu JJ, Yap CT, Hooi SC. PRAP1 is a novel execu-tor of p53-dependent mechanisms in cell survival after DNA damage. Cell Death Dis 2012; 3:e442; PMID:23235459; http://dx.doi.org/10.1038/cddis.2012.180
56. Shandilya J, Wang Y, Roberts SGE. TFIIB dephos-phorylation links transcription inhibition with the p53-dependent DNA damage response. Proc Natl Acad Sci U S A 2012; 109:18797-802; PMID:23115335; http://dx.doi.org/10.1073/pnas.1207483109
57. Hill R, Madureira PA, Waisman DM, Lee PWK. DNA-PKCS binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget 2011; 2:1094-108; PMID:22190353
58. Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, Carrier A, Iovanna JL, Dusetti NJ. Tp53INP1, a tumor suppressor, inter-acts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ 2012; 19:1525-35; PMID:22421968; http://dx.doi.org/10.1038/cdd.2012.30
59. Malek M, Guillaumot P, Huber AL, Lebeau J, Pétrilli V, Kfoury A, Mikaelian I, Renno T, Manié SN. LAMTOR1 depletion induces p53-dependent apop-tosis via aberrant lysosomal activation. Cell Death Dis 2012; 3:e300; PMID:22513874; http://dx.doi.org/10.1038/cddis.2012.39
60. Bailey ST, Shin H, Westerling T, Liu XS, Brown M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci U S A 2012; 109:18060-5; PMID:23077249; http://dx.doi.org/10.1073/pnas.1018858109
61. Pujals A, Renouf B, Robert A, Chelouah S, Hollville E, Wiels J. Treatment with a BH3 mimetic over-comes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis 2011; 2:e184; PMID:21796156; http://dx.doi.org/10.1038/cddis.2011.67
62. Esposito F, Tornincasa M, Federico A, Chiappetta G, Pierantoni GM, Fusco A. High-mobility group A1 protein inhibits p53-mediated intrinsic apoptosis by interacting with Bcl-2 at mitochondria. Cell Death Dis 2012; 3:e383; PMID:22932725; http://dx.doi.org/10.1038/cddis.2012.126
63. Minutolo A, Grelli S, Marino-Merlo F, Cordero FM, Brandi A, Macchi B, Mastino A. D(-)lentiginosine-induced apoptosis involves the intrinsic pathway and is p53-independent. Cell Death Dis 2012; 3:e358; PMID:22833097; http://dx.doi.org/10.1038/cddis.2012.97
64. Gatta R, Dolfini D, Mantovani R. NF-Y joins E2Fs, p53 and other stress transcription factors at the apoptosis table. Cell Death Dis 2011; 2:e162; PMID:21614092; http://dx.doi.org/10.1038/cddis.2011.45
65. di Pietro A, Koster R, Boersma-van Eck W, Dam WA, Mulder NH, Gietema JA, de Vries EG, de Jong S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012; 11:4552-62; PMID:23165211; http://dx.doi.org/10.4161/cc.22803
66. Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C. MOZ increases p53 acetyla-tion and premature senescence through its complex formation with PML. Proc Natl Acad Sci U S A 2013; 110:3895-900; PMID:23431171; http://dx.doi.org/10.1073/pnas.1300490110
67. Blagosklonny MV. Tumor suppression by p53 with-out apoptosis and senescence: conundrum or rapa-log-like gerosuppression? Aging (Albany NY) 2012; 4:450-5; PMID:22869016
68. Blagosklonny MV. Cell cycle arrest is not yet senes-cence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012; 4:159-65; PMID:22394614
69. Peche LY, Scolz M, Ladelfa MF, Monte M, Schneider C. MageA2 restrains cellular senescence by targeting the function of PMLIV/p53 axis at the PML-NBs. Cell Death Differ 2012; 19:926-36; PMID:22117195; http://dx.doi.org/10.1038/cdd.2011.173
70. Cho JH, Kim MJ, Kim KJ, Kim JR. POZ/BTB and AT-hook-containing zinc finger protein 1 (PATZ1) inhibits endothelial cell senescence through a p53 dependent pathway. Cell Death Differ 2012; 19:703-12; PMID:22052190; http://dx.doi.org/10.1038/cdd.2011.142
71. Chao SK, Horwitz SB, McDaid HM. Insights into 4E-BP1 and p53 mediated regulation of acceler-ated cell senescence. Oncotarget 2011; 2:89-98; PMID:21399233
72. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY) 2012; 4:660-1; PMID:23128359
73. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY) 2012; 4:709-14; PMID:23123616
74. Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, Sundström M, Micke P, Botling J, Soussi T. Data-driven unbiased curation of the Tp53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci U S A 2012; 109:9551-6; PMID:22628563; http://dx.doi.org/10.1073/pnas.1200019109
75. Jordan JJ, Menendez D, Sharav J, Beno I, Rosenthal K, Resnick MA, Haran TE. Low-level p53 expression changes transactivation rules and reveals superac-tivating sequences. Proc Natl Acad Sci U S A 2012; 109:14387-92; PMID:22908277; http://dx.doi.org/10.1073/pnas.1205971109
76. Bista M, Freund SM, Fersht AR. Domain-domain interactions in full-length p53 and a specific DNA complex probed by methyl NMR spectroscopy. Proc Natl Acad Sci U S A 2012; 109:15752-6; PMID:22972749; http://dx.doi.org/10.1073/pnas.1214176109
77. Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggre-gation and its inhibition. Proc Natl Acad Sci U S A 2012; 109:13584-9; PMID:22869713; http://dx.doi.org/10.1073/pnas.1211550109
78. Wang G, Fersht AR. First-order rate-determining aggregation mechanism of p53 and its implica-tions. Proc Natl Acad Sci U S A 2012; 109:13590-5; PMID:22869710; http://dx.doi.org/10.1073/pnas.1211557109
79. Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol Cell 2006; 22:741-53; PMID:16793544; http://dx.doi.org/10.1016/j.molcel.2006.05.015
80. Nagaich AK, Zhurkin VB, Durell SR, Jernigan RL, Appella E, Harrington RE. p53-induced DNA bend-ing and twisting: p53 tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc Natl Acad Sci U S A 1999; 96:1875-80; PMID:10051562; http://dx.doi.org/10.1073/pnas.96.5.1875
81. Melero R, Rajagopalan S, Lázaro M, Joerger AC, Brandt T, Veprintsev DB, Lasso G, Gil D, Scheres SH, Carazo JM, et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc Natl Acad Sci U S A 2011; 108:557-62; PMID:21178074; http://dx.doi.org/10.1073/pnas.1015520107
82. Huang F, Rajagopalan S, Settanni G, Marsh RJ, Armoogum DA, Nicolaou N, Bain AJ, Lerner E, Haas E, Ying L, et al. Multiple conformations of full-length p53 detected with single-molecule f luores-cence resonance energy transfer. Proc Natl Acad Sci U S A 2009; 106:20758-63; PMID:19933326; http://dx.doi.org/10.1073/pnas.0909644106
83. Aramayo R, Sherman MB, Brownless K, Lurz R, Okorokov AL, Orlova EV. Quaternary structure of the specific p53-DNA complex reveals the mecha-nism of p53 mutant dominance. Nucleic Acids Res 2011; 39:8960-71; PMID:21764777; http://dx.doi.org/10.1093/nar/gkr386
84. Okorokov AL, Sherman MB, Plisson C, Grinkevich V, Sigmundsson K, Selivanova G, Milner J, Orlova EV. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J 2006; 25:5191-200; PMID:17053786; http://dx.doi.org/10.1038/sj.emboj.7601382

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 9
85. Cherny DI, Brázdova M, Palecek J, Palecek E, Jovin TM. Sequestering of p53 into DNA-protein filaments revealed by electron microscopy. Biophys Chem 2005; 114:261-71; PMID:15829361; http://dx.doi.org/10.1016/j.bpc.2004.12.042
86. Pham N, Lucumi A, Cheung N, Viadiu H. The tet-ramer of p53 in the absence of DNA forms a relaxed quaternary state. Biochemistry 2012; 51:8053-5; PMID:23025236; http://dx.doi.org/10.1021/bi301193k
87. Feng H, Jenkins LMM, Durell SR, Hayashi R, Mazur SJ, Cherry S, Tropea JE, Miller M, Wlodawer A, Appella E, et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009; 17:202-10; PMID:19217391; http://dx.doi.org/10.1016/j.str.2008.12.009
88. Rowell JP, Simpson KL, Stott K, Watson M, Thomas JO. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 2012; 20:2014-24; PMID:23063560; http://dx.doi.org/10.1016/j.str.2012.09.004
89. Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, et al. Single-stranded DNA mim-icry in the p53 transactivation domain interaction with replication protein A. Proc Natl Acad Sci U S A 2005; 102:15412-7; PMID:16234232; http://dx.doi.org/10.1073/pnas.0504614102
90. Cho YJ, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265:346-55; PMID:8023157; http://dx.doi.org/10.1126/science.8023157
91. Jeffery PD, Pavletich NP. Crystal structure of the tetramerization domain of the p53 tumor sup-pressor at 1.7 angstroms. Science 1995; 267:1498-502; PMID:7878469; http://dx.doi.org/10.1126/science.7878469
92. Gorina S, Pavletich NP. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 1996; 274:1001-5; PMID:8875926; http://dx.doi.org/10.1126/science.274.5289.1001
93. Cañadillas JMP, Tidow H, Freund SMV, Rutherford TJ, Ang HC, Fersht AR. Solution structure of p53 core domain: structural basis for its instabil-ity. Proc Natl Acad Sci U S A 2006; 103:2109-14; PMID:16461916; http://dx.doi.org/10.1073/pnas.0510941103
94. Clore GM, Ernst J, Omichinski JG, Kennedy WM, Sakaguchi K, Apella E, Gronenborn AM. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat Struct Biol 1995; 2:321-33; PMID:7796267
95. Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI, Valle M, Fersht AR. Quaternary structures of tumor suppres-sor p53 and a specific p53 DNA complex. Proc Natl Acad Sci USA 2007; 104:12324-9; PMID:17620598; http://dx.doi.org/10.1073/pnas.0705069104
96. Lu Q, Tan Y-H, Luo R. Molecular dynamics simu-lations of p53 DNA-binding domain. J Phys Chem B 2007; 111:11538-45; PMID:17824689; http://dx.doi.org/10.1021/jp0742261
97. Barakat K, Issack BB, Stepanova M, Tuszynski J. Effects of temperature on the p53-DNA binding interactions and their dynamical behavior: compar-ing the wild type to the R248Q mutant. PLoS One 2011; 6:e27651; PMID:22110706; http://dx.doi.org/10.1371/journal.pone.0027651
98. Santini S, Bizzarri AR, Cannistraro S. Modelling the interaction between the p53 DNA-binding domain and the p28 peptide fragment of Azurin. J Mol Recognit 2011; 24:1043-55; PMID:22038811; http://dx.doi.org/10.1002/jmr.1153
99. Madhumalar A, Smith DJ, Verma C. Stability of the core domain of p53: insights from com-puter simulations. BMC Bioinformatics 2008; 9(Suppl 1):S17; PMID:18315848; http://dx.doi.org/10.1186/1471-2105-9-S1-S17
100. John K, Alla V, Meier C, Pützer BM. GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ 2011; 18:874-86; PMID:21127500; http://dx.doi.org/10.1038/cdd.2010.153
101. Wassman CD, Baronio R, Demir O, Wallentine BD, Chen C-K, Hall LV, Salehi F, Lin DW, Chung BP, Hatfield GW, et al. Computational identifica-tion of a transiently open L1/S3 pocket for reacti-vation of mutant p53. Nat Commun 2013; 4:1407; PMID:23360998; http://dx.doi.org/10.1038/ncomms2361
102. Demir O, Baronio R, Salehi F, Wassman CD, Hall L, Hatfield GW, Chamberlin R, Kaiser P, Lathrop RH, Amaro RE. Ensemble-based computational approach discriminates functional activity of p53 cancer and rescue mutants. PLoS Comput Biol 2011; 7:e1002238; PMID:22028641; http://dx.doi.org/10.1371/journal.pcbi.1002238
103. Yang Y, Tarapore RS, Jarmel MH, Tetreault M-P, Katz JP. p53 mutation alters the effect of the esopha-geal tumor suppressor KLF5 on keratinocyte prolifer-ation. Cell Cycle 2012; 11:4033-9; PMID:22990386; http://dx.doi.org/10.4161/cc.22265
104. Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, Löschmann N, Voges Y, Breitling R, von Deimling A, Rödel F, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis 2011; 2:e243; PMID:22170099; http://dx.doi.org/10.1038/cddis.2011.129
105. Espinoza-Fonseca LM, Trujillo-Ferrara JG. Transient stability of the helical pattern of region F19-L22 of the N-terminal domain of p53: a molecular dynam-ics simulation study. Biochem Biophys Res Commun 2006; 343:110-6; PMID:16530164; http://dx.doi.org/10.1016/j.bbrc.2006.02.129
106. Huang Y, Liu Z. Anchoring Intrinsically Disordered Proteins to Multiple Targets: Lessons from N-Terminus of the p53 Protein. Int J Mol Sci 2011; 12:1410-30; PMID:21541066; http://dx.doi.org/10.3390/ijms12021410
107. Mavinahalli JN, Madhumalar A, Beuerman RW, Lane DP, Verma C. Differences in the trans-activation domains of p53 family members: a computational study. BMC Genomics 2010; 11(Suppl 1):S5; PMID:20158876; http://dx.doi.org/10.1186/1471-2164-11-S1-S5
108. Macchiarulo A, Giacchè N, Carotti A, Baroni M, Cruciani G, Pellicciari R. Targeting the con-formational transitions of MDM2 and MDMX: insights into dissimilarities and similarities of p53 recognition. J Chem Inf Model 2008; 48:1999-2009; PMID:18826207; http://dx.doi.org/10.1021/ci800146m
109. Allen WJ, Capelluto DGS, Finkielstein CV, Bevan DR. Modeling the relationship between the p53 C-terminal domain and its binding partners using molecular dynamics. J Phys Chem B 2010; 114:13201-13; PMID:20873738; http://dx.doi.org/10.1021/jp1011445
110. Gordo S, Martos V, Santos E, Menéndez M, Bo C, Giralt E, de Mendoza J. Stability and structural recovery of the tetramerization domain of p53-R337H mutant induced by a designed templating ligand. Proc Natl Acad Sci U S A 2008; 105:16426-31; PMID:18940924; http://dx.doi.org/10.1073/pnas.0805658105
111. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppres-sor transactivation domain. Science 1996; 274:948-53; PMID:8875929; http://dx.doi.org/10.1126/science.274.5289.948
112. Pan Y, Nussinov R. Lysine120 interactions with p53 response elements can allosterically direct p53 organi-zation. PLoS Comput Biol 2010; 6; PMID:20700496; http://dx.doi.org/10.1371/journal.pcbi.1000878
113. VanAalten DMF, DeGroot BL, Findlay JBC, Berendsen HJC, Amadei A. A com-parison of techniques for calculating protein essential dynamics. J Comput Chem 1997; 18:169-81; http://dx.doi.org/10.1002/(SICI)1096-987X(19970130)18:2<169::AID-JCC3>3.0.CO;2-T
114. Levine AJ. The paths to death and differentiation. Cell Death Differ 2011; 18:1391-2; PMID:21841805; http://dx.doi.org/10.1038/cdd.2011.41
115. Levine AJ. The evolution of the p53 family of genes. Cell Cycle 2012; 11:214-5; PMID:22214668; http://dx.doi.org/10.4161/cc.11.2.18899
116. Rutkowski R, Gartner A. The shark in us: learning about the evolution of the p53 regulatory circuit. Cell Cycle 2012; 11:216-7; PMID:22214666; http://dx.doi.org/10.4161/cc.11.2.18901
117. Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell Death Dis 2012; 3:e285; PMID:22419114; http://dx.doi.org/10.1038/cddis.2012.27
118. Grespi F, Amelio I, Tucci P, Annicchiarico-Petruzzelli M, Melino G. Tissue-specific expres-sion of p73 C-terminal isoforms in mice. Cell Cycle 2012; 11:4474-83; PMID:23159862; http://dx.doi.org/10.4161/cc.22787
119. Masse I, Barbollat-Boutrand L, Molina M, Berthier-Vergnes O, Joly-Tonetti N, Martin MT, de Fromentel CC, Kanitakis J, Lamartine J. Functional interplay between p63 and p53 controls RUNX1 function in the transition from proliferation to differentiation in human keratinocytes. Cell Death Dis 2012; 3:e318; PMID:22673192; http://dx.doi.org/10.1038/cddis.2012.62
120. Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, Lane DP, Callen DF. Mutant p53 uses p63 as a molecular chaperone to alter gene expres-sion and induce a pro-invasive secretome. Oncotarget 2011; 2:1203-17; PMID:22203497
121. Ou HD, Löhr F, Vogel V, Mäntele W, Dötsch V. Structural evolution of C-terminal domains in the p53 family. EMBO J 2007; 26:3463-73; PMID:17581633; http://dx.doi.org/10.1038/sj.emboj.7601764
122. Belyi VA, Levine AJ. One billion years of p53/p63/p73 evolution. Proc Natl Acad Sci U S A 2009; 106:17609-10; PMID:19826090; http://dx.doi.org/10.1073/pnas.0910634106
123. Lu F, Li YQ, Aubert I, Wong CS. Endothelial cells regulate p53-dependent apoptosis of neural progeni-tors after irradiation. Cell Death Dis 2012; 3:e324; PMID:22717579; http://dx.doi.org/10.1038/cddis.2012.59
124. Italiano D, Lena AM, Melino G, Candi E. Identification of NCF2/p67phox as a novel p53 target gene. Cell Cycle 2012; 11:4589-96; PMID:23187810; http://dx.doi.org/10.4161/cc.22853
125. Martynova E, Pozzi S, Basile V, Dolfini D, Zambelli F, Imbriano C, Pavesi G, Mantovani R. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget 2012; 3:132-43; PMID:22361592
126. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2011; 2:948-57; PMID:22248668

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
10 Cell Cycle Volume 12 Issue 18
127. Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P, et al. Biological functions of p53 iso-forms through evolution: lessons from animal and cellular models. Cell Death Differ 2011; 18:1815-24; PMID:21941372; http://dx.doi.org/10.1038/cdd.2011.120
128. Shlevkov E, Morata G. A dp53/JNK-dependant feed-back amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ 2012; 19:451-60; PMID:21886179; http://dx.doi.org/10.1038/cdd.2011.113
129. Chappell WH, Lehmann BD, Terrian DM, Abrams SL, Steelman LS, McCubrey JA. p53 expression controls prostate cancer sensitivity to chemother-apy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012; 11:4579-88; PMID:23187804; http://dx.doi.org/10.4161/cc.22852
130. Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22:2677-91; PMID:18805989; http://dx.doi.org/10.1101/gad.1695308
131. Agostini M, Tucci P, Killick R, Candi E, Sayan BS. Cervo PRdV, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proceedings of the National Academy of Sciences of the United States of America 2011; 108:21093-8.
132. Agostini M, Tucci P, Steinert JR, Shalom-Feuerstein R, Rouleau M, Aberdam D, Forsythe ID, Young KW, Ventura A, Concepcion CP, et al. microRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc Natl Acad Sci U S A 2011; 108:21099-104; PMID:22160706; http://dx.doi.org/10.1073/pnas.1112063108
133. Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, Federici M, Dinsdale D, Knight RA, Melino G, et al. TAp73 depletion accel-erates aging through metabolic dysregulation. Genes Dev 2012; 26:2009-14; PMID:22987635; http://dx.doi.org/10.1101/gad.197640.112
134. Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, Melino G, Aberdam D. DNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ 2011; 18:887-96; PMID:21127502; http://dx.doi.org/10.1038/cdd.2010.159
135. Marcel V, Petit I, Murray-Zmijewski F, Goullet de Rugy T, Fernandes K, Meuray V, Diot A, Lane DP, Aberdam D, Bourdon JC. Diverse p63 and p73 iso-forms regulate D133p53 expression through modu-lation of the internal Tp53 promoter activity. Cell Death Differ 2012; 19:816-26; PMID:22075982; http://dx.doi.org/10.1038/cdd.2011.152
136. Leonard MK, Kommagani R, Payal V, Mayo LD, Shamma HN, Kadakia MP. DNp63α regulates keratinocyte proliferation by controlling PTEN expression and localization. Cell Death Differ 2011; 18:1924-33; PMID:21637289; http://dx.doi.org/10.1038/cdd.2011.73
137. Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition mechanism of p63 by the E3 ligase Itch: novel strategy in the study and inhibition of this interaction. Cell Cycle 2012; 11:3638-48; PMID:22935697; http://dx.doi.org/10.4161/cc.21918
138. Salah Z, Bar-mag T, Kohn Y, Pichiorri F, Palumbo T, Melino G, Aqeilan RI. Tumor suppressor WWOX binds to DNp63α and sensitizes cancer cells to chemotherapy. Cell Death Dis 2013; 4:e480; PMID:23370280; http://dx.doi.org/10.1038/cddis.2013.6
139. Nayak G, Cooper GM. p53 is a major component of the transcriptional and apoptotic program regulated by PI 3-kinase/Akt/GSK3 signaling. Cell Death Dis 2012; 3:e400; PMID:23059819; http://dx.doi.org/10.1038/cddis.2012.138
140. Tucci P, Agostini M, Grespi F, Markert EK, Terrinoni A, Vousden KH, Muller PA, Dötsch V, Kehrloesser S, Sayan BS, et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metas-tasis in prostate cancer. Proc Natl Acad Sci U S A 2012; 109:15312-7; PMID:22949650; http://dx.doi.org/10.1073/pnas.1110977109
141. Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, Saintigny G, Dellambra E, Odorisio T, Mahé C, et al. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci U S A 2012; 109:1133-8; PMID:22228303; http://dx.doi.org/10.1073/pnas.1112257109
142. Amelio I, Grespi F, Annicchiarico-Petruzzelli M, Melino G. p63 the guardian of human reproduc-tion. Cell Cycle 2012; 11:4545-51; PMID:23165243; http://dx.doi.org/10.4161/cc.22819
143. Paris M, Rouleau M, Pucéat M, Aberdam D. Regulation of skin aging and heart development by TAp63. Cell Death Differ 2012; 19:186-93; PMID:22158419; http://dx.doi.org/10.1038/cdd.2011.181
144. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNF alpha)-induced apoptosis through SIRT1 inhibition. Cell Death Dis 2012; 3
145. Yasuda T, Oda S, Li Z, Kimori Y, Kamei Y, Ishikawa T, Todo T, Mitani H. Gamma-ray irradiation pro-motes premature meiosis of spontaneously differenti-ating testis-ova in the testis of p53-deficient medaka (Oryzias latipes). Cell Death Dis 2012; 3:e395; PMID:23034330; http://dx.doi.org/10.1038/cddis.2012.133
146. Markert EK, Levine AJ, Vazquez A. Proliferation and tissue remodeling in cancer: the hallmarks revisited. Cell Death Dis 2012; 3:e397; PMID:23034332; http://dx.doi.org/10.1038/cddis.2012.140
147. de Bie P, Ciechanover A. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ 2011; 18:1393-402; PMID:21372847; http://dx.doi.org/10.1038/cdd.2011.16
148. Michaelis M, Rothweiler F, Agha B, Barth S, Voges Y, Löschmann N, von Deimling A, Breitling R, Doerr HW, Rödel F, et al. Human neuroblastoma cells with acquired resistance to the p53 activator RITA retain functional p53 and sensitivity to other p53 activating agents. Cell Death Dis 2012; 3:e294; PMID:22476102; http://dx.doi.org/10.1038/cddis.2012.35
149. Burns DM, D’Ambrogio A, Nottrott S, Richter JD. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 2011; 473:105-8; PMID:21478871; http://dx.doi.org/10.1038/nature09908
150. Hallenborg P, Feddersen S, Francoz S, Murano I, Sundekilde U, Petersen RK, Akimov V, Olson MV, Lozano G, Cinti S, et al. Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell Death Differ 2012; 19:1381-9; PMID:22388350; http://dx.doi.org/10.1038/cdd.2012.15
151. Miliani de Marval PL, Zhang Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget 2011; 2:234-8; PMID:21406728
152. Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA, de Jong S. Disruption of the MDM2-p53 inter-action strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis 2011; 2:e148; PMID:21509038; http://dx.doi.org/10.1038/cddis.2011.33
153. Davies L, Spiller D, White MRH, Grierson I, Paraoan L. PERP expression stabilizes active p53 via modula-tion of p53-MDM2 interaction in uveal melanoma cells. Cell Death Dis 2011; 2:e136; PMID:21451571; http://dx.doi.org/10.1038/cddis.2011.19
154. Zhang L, Huang N-J, Chen C, Tang W, Kornbluth S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc Natl Acad Sci U S A 2012; 109:20931-6; PMID:23213260; http://dx.doi.org/10.1073/pnas.1212047110
155. Graves B, Thompson T, Xia M, Janson C, Lukacs C, Deo D, Di Lello P, Fry D, Garvie C, Huang KS, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimeriza-tion. Proc Natl Acad Sci U S A 2012; 109:11788-93; PMID:22745160; http://dx.doi.org/10.1073/pnas.1203789109
156. Vaseva AV, Yallowitz AR, Marchenko ND, Xu S, Moll UM. Blockade of Hsp90 by 17AAG antago-nizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis 2011; 2:e156; PMID:21562588; http://dx.doi.org/10.1038/cddis.2011.39
157. de Lange J, Verlaan-de Vries M, Teunisse AFAS, Jochemsen AG. Chk2 mediates RITA-induced apoptosis. Cell Death Differ 2012; 19:980-9; PMID:22158418; http://dx.doi.org/10.1038/cdd.2011.182
158. Bursać S, Brdovćak MC, Pfannkuchen M, Orsolić I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabušić K, Vukelić I, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essen-tial for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci U S A 2012; 109:20467-72; PMID:23169665; http://dx.doi.org/10.1073/pnas.1218535109
159. Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer forma-tion. Proc Natl Acad Sci U S A 2009; 106:17705-10; PMID:19815500; http://dx.doi.org/10.1073/pnas.0905867106
160. Ethayathulla AS, Tse P-W, Monti P, Nguyen S, Inga A, Fronza G, Viadiu H. Structure of p73 DNA-binding domain tetramer modulates p73 transactiva-tion. Proc Natl Acad Sci U S A 2012; 109:6066-71; PMID:22474346; http://dx.doi.org/10.1073/pnas.1115463109
161. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metasta-sis. Cell 2009; 137:87-98; PMID:19345189; http://dx.doi.org/10.1016/j.cell.2009.01.039
162. Muller PAJ, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009; 139:1327-41; PMID:20064378; http://dx.doi.org/10.1016/j.cell.2009.11.026
163. Roy A, Kucukural A, Zhang Y. I-TASSER: a uni-fied platform for automated protein structure and function prediction. Nat Protoc 2010; 5:725-38; PMID:20360767; http://dx.doi.org/10.1038/nprot.2010.5
164. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577-90; PMID:12667443; http://dx.doi.org/10.1016/S1097-2765(03)00050-9
165. Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29:845-54; PMID:23407358; http://dx.doi.org/10.1093/bioinformatics/btt055

©20
13 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 11
166. Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010; 78:1950-8; PMID:20408171
167. Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983:79
168. Darden T, York D, Pedersen L. Particle Mesh Ewald - an N log(N) method for Ewald sums in large sys-tems. J Chem Phys 1993; 98:10089-92; http://dx.doi.org/10.1063/1.464397
169. Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: A linear constraint solver for molec-ular simulations. J Comput Chem 1997; 18:1463-72; http://dx.doi.org/10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
170. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 2001; 98:10037-41; PMID:11517324; http://dx.doi.org/10.1073/pnas.181342398
171. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14:33-8, 27-8; PMID:8744570; http://dx.doi.org/10.1016/0263-7855(96)00018-5