marinobufagenin induces increases in procollagen expression in a process involving protein kinase c...
TRANSCRIPT

Health Science Campus
FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Sciences
Molecular Mechanism of Fibrosis and Central Role of Cardiotonic Steroids
in Uremic Cardiomyopathy
Submitted by:
Jihad Elkareh
In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences
Examination Committee Signature/Date
Major Advisor: Joseph Shapiro, M.D.
Jiang Liu, Ph.D.
Sonia Najjar, Ph.D.
Zi-Jian Xie, Ph.D.
Academic Advisory
Committee:
Sandrine Pierre, Ph.D.
Senior Associate Dean College of Graduate Studies
Michael S. Bisesi, Ph.D.
Date of Defense: May 15, 2008

Molecular Mechanism of Fibrosis and Central Role of Cardiotonic
Steroids in Uremic Cardiomyopathy
Jihad Victor Elkareh
University of Toledo
2008

DEDICATION
Totus tuus.
ii

ACKNOWLEDGEMENTS
I would like to acknowledge the sacrifice, support, encouragement, guidance, and love of
my Mother, Father, and Brother.
I would like to emphasize the commitment, concern, consideration, kindness, cordiality,
gentleness, wisdom, support and friendship of Dr. Joseph Shapiro as well as the care,
insight, assist, support and friendship of Drs. Sonia Najjar, Zi-Jian Xie, Jiang Liu, and
Sandrine Pierre.
I would like to recognize the support of my great friends and co-workers in the laboratory
over the years: Amjad Shidyak, David Kennedy, Nasser El-Okdi, Steven Haller, Sleiman
Smaili, Adnan Alsaka, Imad Hariri, Vanamala Raju, Liang Wu, Haiping Cai, Sandeep
Vetteth, Ajay and Veena Belvadi, Shalini Gupta, Erin Crawford, Yasser Aldhamen,
Thomas Blomquist, Carol Woods, Tomasa Guerrero.
I would like to highlight the support, training and companionship of other faculty and
mentors from whom I have learned a lot: Drs. Sankaridrug Periyasamy, Larisa Fedorova,
James Willey, Bashar Kahaleh, Maurice Manning, Deepak Malhotra, Sadik Khuder, Julie
Westerink, Alexei Bagrov, Olga Fedorova, and Sumudra Periyasamy.
iii

TABLE OF CONTENTS
Dedication ……………………………………………………………………..…………ii
Acknowledgements ……………………………………………………………..….……iii
Table of Contents………………………………………..………………….……..……...iv
Introduction …………………………………………………………….……...………….1
Literature ……………………………………………………………….…………………4
Manuscript 1: Marinobufagenin Stimulates Fibroblast Collagen Production and Causes
Fibrosis in Experimental Uremic Cardiomyopathy……………………………………...20
Manuscript 2: Partial Nephrectomy as a Model for Uremic Cardiomyopathy in the
Mouse…....……………………………………………………….……..…………….....64
Manuscript 3: Marinobufagenin Induces Increases in Procollagen Expression in a Process
Involving Protein Kinase C and Fli-1: Implications for Uremic
Cardiomyopathy……………………………………………………….……….…..……89
Summary ………………………………………………………………...…………….124
Conclusions …………………………………………………………….………………128
References ……………………………………………………………….…..…………130
Abstract ……………………………………………………………………..…….……146
iv

1
INTRODUCTION
Cardiac disease is responsible for the high mortality seen in patients suffering from
kidney diseases. (1) Impairment of cardiac function is mainly caused by myocardial
fibrosis, which is considered the end stage of heart disease. A major biochemical marker
of fibrosis would be excessive accumulation of collagen. (3-5)
Transforming growth factor beta (TGF-β) has been established as a key player in the
development of fibrosis in many diseases. However, some literature has shown that
certain forms of fibrosis, such as atherosclerotic fibrosis, are TGF-β independent. (6) The
renin-angiotensin-aldosterone system has been shown to exert profibrotic effects as well.
Aldosterone seems to induce cultured fibroblasts to produce more collagen-1 while
administration of spironolactone attenuates cardiac fibrosis in vivo.(7)
Other studies emphasize the role of the transcription factor Fli-1 in fibrosis. Fli-1
normally competes with Ets-1 in a Sp-1 dependent manner, shifting the balance between
stimulating and repressing the Col 1A2 promoter. (2) Jinnin et al showed that the protein
kinase PKCδ regulates the expression of collagen-1 through changing the equilibrium
between Ets-1 and Fli-1.(8)
Under these conditions, it has been observed that cardiotonic steroids, which bind to the
plasmalemmal Na/K-ATPase, accumulate in renal failure.These steroids have been
shown to specifically inhibit the sodium pump, as well as circulate at higher levels in
patients with kidney disease.

2
In patients suffering from uremic cardiomyopathy, marinobufagenin (MBG), a
cardiotonic steroid, has been established as a natriuretic hormone that increases Na
excretion when renal elimination of Na is impaired.
Our laboratory has observed that increases in circulating cardiotonic streoids were both
necessary and sufficient to induce systemic oxidant stress, cardiac hypertrophy, and
diastolic dysfunction seen with experimental uremia. More importantly, administration of
MBG for 4 weeks caused comparable increases in plasma MBG as did the partial
nephrectomy at 4 weeks. On the other hand, immunization against MBG attenuated the
cardiac hypertrophy, impairement of diastolic function, systemic oxidant stress, and
cardiac fibrosis seen with partial nephrectomy.
Therefore, based on data from our laboratory as well as the extensive literature
concerning fibrosis, we propose the central hypothesis: Circulating cardiotonic steroids
bind to the Na/K-ATPase and produce increased collagen production in a process
involving Fli-1, and PKCδ as part of the resultant signaling cascade. This signaling
cascade plays a crucial role in the pathogenesis of cardiac fibrosis, a major phenotype of
chronic renal failure.
In order to investigate this central hypothesis, we first examined whether cardiotonic
steroids, particularly MBG, signal through the Na/K-ATPase, directly stimulating cardiac
fibroblast collagen production. Next, the partial nephrectomy model in the rat, which
extensively simulates experimental uremia with cardiac abnormalities as seen in renal

3
failure, was applied to the mouse, which we postulated would yield similar results.
Employing this model, we show the relevance of Fli-1 in the negative regulation of
collagen, thus revealing the molecular mechanisms by which cardiotonic steroids
contribute to the cardiac fibrosis present in uremic cardiomyopathy.

4
LITERATURE
Epidemiology of Cardiovascular Disease in Chronic Kidney Disease
Chronic kidney disease has been identified as a major risk factor for cardiovascular
disease. Because it is estimated that 20 million adults suffer from chronic kidney disease
in the United States, Medicare is expected to spend 28 billion dollars by 2010 in an effort
to treat this disease.(26,27) There is a vicious cycle between the kidney and the heart
which led many scientists and doctors to term the relationship between these two vital
organs as “cardiorenal syndrome”.(26, 28, 29) Other studies showed that the mortality
rate is still high in patients with end stage renal disease (ESRD) including patients on
Hemodialysis (HD). In fact, 25-50% of patients under dialysis develop cardiac failure,
and more than 50% of the mortalities result from cardiac related causes. (30, 31, 114)
These findings suggest that HD can not replace normal renal function and this tightly
correlates renal disorders with cardiac malfunctions. Dialysis is capable of inducing
myocardial ischemia thus leading to heart failure. (32, 33) It is believed to be pro-
arrhythmogenic since it induces arrhythmias and causes increase in QT interval and QT
dispersion. (34) In addition to the arrhythmia, diastolic dysfunction and left ventricular
hypertrophy are very common in patients with ESRD, despite treatment with HD. (35-39)
In 1998, the National Kidney Foundation issued a report emphasizing the high risk of
cardiovascular disease in chronic kidney disease patients.(30) Clinically, this is called
uremic cariomyopathy and is characterized by diastolic dysfunction and left ventricular

5
hypertrophy.(1) At present, the molecular basis of uremic cardiomyopathy is poorly
understood. It is known to be characterized by systemic oxidant stress, diastolic
dysfunction, and left ventricular hypertrophy, and cardiac fibrosis. (40) Anemia,
hypertension, parathyroid hormone, the renin-angiotensin-aldosterone system (RAAS),
and lipid abnormalities have been implicated as potential pathogenetic factors in uremic
cardiomyopathy. Treatment with recombinant erythropoietin or a parathyroidectomy has
shown regression in left ventricular hypertrophy one of the major symptoms of uremic
cardiomyopathy.(41)
Components of Fibrosis
Impairment of cardiac function is mainly caused by myocardial fibrosis, and is
considered the end stage of heart disease with symptoms including cardiac dilatation,
reduction in contractility and relaxation, with progressive structural and functional
alterations.(3-5) Fibrosis is the result of an imbalance between synthesis and degradation
of the extracellular matrix (ECM). This disparity leads to abnormal remodeling of ECM.
(2, 42, 43) In fact, extensive research has shown a deposition of collagen, a key marker of
fibrosis, in the left ventricular causing hypertrophy of the heart and in some cases an
increase in blood pressure. (6)
The ECM or connective tissue, is a structural entity that supports the cell, which is
composed of three major classes of biomolecules: stuctural proteins (collagen and
elastin), specialized proteins (fibrillin, fibronectin and laminin), and proteoglycans.

6
Matrix metalloproteases (MMPs) also called matrixins, which are responsible for
degradation and remodeling of the ECM. They have been classified in six subgroups:
collagenases, gelatinases, stromelysins, matrilysins, membrane-type MMPs, and other
MMPs.(44)
Collagen, the major component of ECM, has a balanced turnover (synthesis and
degradation) that is estimated to be 80-120 days in cardiac fibroblasts. The turnover of
collagen is regulated by MMPs and their inhibitors TIMPs (tissue inhibitors of
metalloproteases). On a regular basis, fibroblasts control collagen turnover, nevertheless
during pathologic circumstances, fibroblasts are transformed into myofibroblasts. These
myofibroblasts can derive from two other sources: the bone marrow of stem cells, where
they are referred as fibrocytes and the epithelial cells in a process know as EMT
(epithelial- mesenchymal transition). (45-47) The transition from fibroblasts to
myofibroblasts is mainly regulated by TGF-β and the renin-angiotensin-aldosterone
system (RAAS)(45). When myofibroblasts are activated via its respective stimulants,
MMPs are produced as a result (46). According to Berk et al, an imbalance in the ratio of
MMPs to TIMPs might explain the left ventricular dilatation and the reduced ejection
fraction in congestive heart failure. Therefore, left ventricular hypertrophy can be seen
where there is ECM accumulation in the heart, whereas a transition to systolic failure can
be observed in hearts that have an increase in ECM degradation.(45)
Once an injury occurs, the regenerative/wound healing phase takes place, and injured
cells are replaced by cells from the same type in a progression which leaves no evidence
of scarring. On the other hand, the repair process might occur in a different way such as

7
the replacement of normal parenchymal tissue by connective tissues would potentially
leave a permanent scar from the substantial remodeling of the ECM. “Out of control
wound healing” is referred at this point to as fibrosis or fibroplasia, which can be
preceded by inflammation, although it is not driven by it at all times. (46) This in part
explains why anti-inflammatory drugs alone can not treat fibrosis efficiently. Before the
scarring stage, when fibrosis is reversible, the molecules of choice that have been used so
far were MMP inhibitors. Unfortunately, these molecules showed many side effects such
as tendonitis which limited the pharmacological development of MMP inhibitors. (45,
46)
Pathogenetic Mechanism in Fibrosis
TGF-β/BMP/Smad System:
Increases in collagen formation in the tissue could be explained as a combination of
transforming growth factor (TGF-β- dependant and TGF-β− independent) mechanisms.
Orlandi and colleagues showed that ischemia associated fibrosis appears to be TGF-β
dependent, while aging related fibrosis and atherosclerosis appears to be TGF-β
independent. (6) TGF-β stimulation of collagen-1 production involves a very well
characterized signal transduction cascade, which involves the essential participation of
Smad proteins. (48-58) Bone morphogenic proteins (BMP) which also involves Smad
proteins can stimulate fibrosis. The TGF-β/BMP/Smad system has been well studied over

8
the years and is believed to be involved in collagen regulation. TGF-β binding to TGF-β
receptors leads to full assembly of the receptors which can activate Smad2, and Smad3.
Inhibitory Smads, which are stimulated by several cytokines, antagonize the assembly of
the TGF-β receptor complex. TGF-β signals through Smad2 and 3 whereas BMPs signal
through Smad1, 5, and 8. Assembly of Smad4 (co-Smad) with active or phospho-Smad2
or 3 enters the nucleus and acts as a transcription factor. (51) Although multiple aspects
of collagen synthesis are affected by the TGF-β/BMP/Smad system, transcriptional
regulation of collagen synthesis constitutes a major component of this important pathway.
(59)
Renin-Angiotensin-Aldosterone System (RAAS):
Every component of RAAS exerts profibrotic effects on the cells. For instance, renin,
independently of its enzymatic action to enhance angiotensin II synthesis, increases the
synthesis of collagen I and fibronectin. (60) Angiotensin II enhances fibrosis directly
through the angiotensin II type 1 receptors, and indirectly through the induction of TGF-
β, which is considered the major RAAS hormone responsible for cardiac fibrosis.
Likewise, angiotensin II induces the differentiation of fibroblasts to myofibroblasts in a
pathway involving ROS and the activation of p38 MAPK, and in an additional pathway
involving tyrosine kinase mediated by c-Src. Finally, the mineralocorticoid steroid
hormone, aldosterone, has been a focus of recent investigation, especially in light of

9
spironolactone and eplerenone, two of its inhibitors. Susic et al showed that long-term
therapy with the mineralocorticoid receptor antagonist eplerenone reduced myocardial
fibrosis, and improved left ventricular function and coronary hemodynamics in
spontaneous hypertensive rats. In other literature, supraphysiological levels of
aldosterone seem to induce cultured fibroblast to produce more collagen-1 while
administration of spironolactone attenuate cardiac fibrosis in vivo.(7) Consequently,
therapies that target the TGF-β/BMP/Smad system or RAAS might be effective in the
treatment of fibrosis in congestive heart failure and other heart diseases.
Fli-1/Ets-1 System:
Fli-1 (Friend leukemia virus integration 1) is member of the Ets (E26 Transformation
Specific) transcription factor family of proteins and belongs to the ERG subfamily.(61)
All Ets transcription factors are defined by a winged helix-turn-helix DNA binding
domain (ets domain) that is specific to DNA element containing a purine-rich sequence
GGAA/T.(62) In the Fli-1 sequence, the 3’ ets domain (amino acids 277-360) is
responsible for sequence specific DNA-binding activity, whereas the 5’ ets domain
(amino acids 121-196) and the FLS (Fli-1 specific region) (amino acids 205-292) contain
the sequence responsible for transcriptional activation.(61) An analysis of the secondary
structure of Fli-1 showed the presence of helix1-loop-helix2 (H-L-H) structures in the 5’
and 3’ ets domains and revealed the presence of turn-loop-turn (T-L-T) structures in the
FLS domain as well as the CTA (caboxy-terminus) (amino acids 402-452)(61) Fli-1 was

10
commonly found as a site for retroviral integration in Friend-virus induced
erythroleukemias. (61, 63) Fli-1 was also found highly expressed in vascular endothelial
cells and hematopoietic cells. (64)
After the cloning of mouse Fli-1, the human Fli-1 gene was isolated by sequence
homology. The sequence analysis showed a high level of conservation between the
rodent and human Fli-1.(61, 65, 66) While Fli-1 is on chromosome 9 in the mouse and on
chromosome 11q23 in the human, it is located 240kb from the Ets-1 locus on both
chromosomes. Fli-1 encodes for a 452aa protein: p51 and a 419aa protein: p48. (61, 63,
66) According to Sarrazin et al, the synthesis of the 51 and 48 isoforms were initiated
from codons AUG +1 and AUG +100 of Fli-1 respectively.(67)
It is well known that in the human body there is always equilibrium between stimulating
and repressing factors. In particular, in organ fibrosis there is a competition between pro-
fibrotic and anti-fibrotic signaling pathways. Czuwara-Ladykowska and colleagues
emphasize the role of the Fli-1 in fibrosis: Fli-1 normally competes with Ets-1 in a Sp-1
dependent balance between stimulating and repressing the Col 1A2 promoter. (2) Most of
the work has been done in dermal fibroblasts showing that Ets-1 and Fli-1 have
antagonistic effects on the collagen gene promoter and they compete with each other in
the regulation of this promoter.(figure 1) (2)
In recent years, a lot of research has been conducted by Kahaleh and colleagues on the
role of Fli-1 as a suppressor of collagen transcription in scleroderma fibroblasts. In fact in
systemic sclerosis or scleroderma, which is an autoimmune disease, there is an abnormal

deposition of collagen on the skin. (68, 69) Studies have shown that in scleroderma
patients, Fli-1 gene is repressed due to epigenetic mechanisms notably a DNA
methylation and a histone deacetylation at the Fli-1 gene. (69).On the other hand,
Trojanowska et al reported a reversible acetylation of Fli-1 on the lysine residue due to
TGF-β stimulation in human dermal fibroblasts. PCAF (p300/CBP-associated factor),
which is a transcription factor that has histone acetyltransferase activity, interacts with
Fli-1 on lysine 380 leading to Fli-1 acetylation.(62) Like most of the acetylation that
occurs on the lysine residue, an impairement of Fli-1 DNA-binding to the Col 1A2
promoter resulted in a decrease in the inhibitory effect Fli-1 has on Collagen.(62, 70)
According to the authors, the TGF-β/ Smad pathway seems to be responsible for the
dissociation of Fli-1 from the collagen promoter, which explains the fibrosis seen in
systemic sclerosis.(62)
Figure 1: Schematic suggesting competition between Fli-1 and Ets-1 at the level of the Col1a2 promoter to determine whether transcription is repressed or stimulated. HDAC1– histone deacetylase 1. Adapted from reference. (2)
11

12
PKC isoforms and Role in Fibrosis:
In the late seventies, the protein kinase C (PKC ) has been identified and since then
extensive work has been done on this family of serine/threonine kinase especially its role
in exocytosis, growth, differentiation, and modulation of ion channels and receptors.(71)
The PKC family can be divided into four groups: conventional/classical PKCs
(PKCα, PKCβ1, PKCβ2, and PKCγ) , novel PKCs ( PKCδ, PKCε, PKCη, and PKCθ),
atypical PKCs (PKCζ and PKCλ/τ) , and the recently discovered PKCs (PKCμ and
PKCv).(72, 73) These twelve isozymes are implicated in many cellular responses in
normal lung function such as: permeability (PKCα), contraction (PKCβ2), migration
(PKCδ), hypertrophy (PKCη), proliferation (PKCε) apoptosis (PKCζ), and secretion
(PKCv). (73)
Recent literature suggests a mechanism of action that involves phospolipase C, which
performs a catalytic mechanism and generates inositol triphosphate (IP3) and
diacylglycerol (DAG), which then cause PKC enzymes (key targets of DAG) to
recognize and phosphorylate human EGFR at the threonine-654, (74) inducing the
internalization of EGFR to be recycled to the plasma membrane.(75)
On the other hand, six isozymes have been found to have different roles in the heart.
PKCε and PKCδ are both activated by the same receptor Gαq, however PKCδ
preferentially activates p38, MAPK, and JNK while PKCε activates p42/44 MAPK. In

13
fact, PKCε is considered being the PKC that protects the heart from ischemic damage.
Many studies showed evidence that PKCε is required to produce protection from
ischemic damage in animal models like mice and in isolated myocytes. Also, ψεRACK, a
selective agonist for PKCε, could be used in therapeutics in treating ischemic heart
disease in humans.(73) In contrast, PKCδ is associated with increased collagen
deposition and impairment of diastolic function. There is a possible correlation between
PKCε and PKCδ expression levels. When PKCε expression levels are low such as in a
knock out animal model, PKCδ expression levels appear to compensate this deficiency
leading to upregulation of PKCδ which is closely associated with heart fibrosis.(72)
The Protein Kinase C family of proteins mediates the specific activation of a variety of
transcription factors like NFkB and Ap1, but little is known whether it can be involved in
the activation of Ets Family, and specifically Fli-1. Nevertheless, Jinnin et al showed that
PKCδ regulates the expression of collagen-1 through shifting the balance of Ets-1 and
Fli-1.(8) Moreover, Jimenez et al. proposes that PKCδ inhibitors could suppress fibrosis,
indicating that PKCδ participates in the upregulation of collagen gene transcription. (25)
In addition, they suggest that the activity of several Na/K-ATPase inhibitors such as
Noradrenaline and phorbol 12-myristate 13-acetate (PMA) is stimulated by PKC
activation. (76) We noticed that the Fli-1 relationship to collagen metabolism and fibrosis
in general has been carefully examined in dermal fibroblasts, and very recently in lung
fibroblasts (42), but little is known about its role in creating fibrosis in other organs like
the heart and the kidney.

14
Na/K-ATPase Function
First discoverd in 1957, (77) the Na/K-ATPase has been identified as an enzyme present
in most cells. (78) From the outer medulla of pig kidneys, Hebert et al. showed the the
Na/K-ATPase structure was elucidated via electron crystallography at 9.5 A. (79) It
belongs to the P-Type ATPases which contain Na/K-ATPase, H/K-ATPase, SERCA
(sarcoplasmic endoplasmic reticulum calcium ATPase) and PMCA (plasma membrane
calcium ATPase). Among these ion pumps, Na/K-ATPase is the only one that has the
ability to bind cardiotonic steroids such as ouabain and marinobufagenin. (80) The P-
Type ATPases consists of two monoconvalently linked α and β subunits, which are
essential for the assembly of the functional enzyme. As for the α subunit (112 KDa) it is
considered the catalytic subunit because it contains the ATP and binding sites for digitalis
and other ligands.(81-83) There is 78% similarity among all α isoforms (α1, α2, α3, α4 ),
and the α2 and α3 isoforms are expressed in neuronal tissue, skeletal muscle, and cardiac
myocytes. The α1 isoform is found in all cells which makes it difficult to study the other
isoforms in a specific organ. (84, 85) The main function of the Na/K-ATPase is to
catalyse the active exchange of three cytoplasmic Na+ for two extracellular K+ across the
plasma membrane via hydrolysis of ATP. (78, 86) In our laboratory we found a rise in
cardiac tissue calcium concentrations, thus causing increases in myocardial contractility
when the Na/K-ATPase is inhibited with digitalis(22). Additional studies showed that

15
the enzyme is also a signal transducer involved in regulation of multiple secondary
messengers, gene-regulatory pathways, and pathways including activation of Src, EGFR,
Ras, PKC, MAPKs, and intracellular ROS production. (10-19, 83, 87,114) Since it
communicates with the nucleus, the pump plays a role in the regulation of gene
expression and cell proliferation. When ouabain binds to the pump, the Na/K-ATPase
interacts with neighboring membrane proteins and organizes a cytosolic cascade of
signaling complexes to send messages to other intracellular organelles. This includes
activation of Src, Ras, and MAPKs, transactivation of EGFR, increased production of
ROS, intracellular [Ca2+] and myocardial contractility. (10, 12, 14, 15, 20,114) This
model has been shown in different cell lines such as neonatal rat cardiac myocytes,
smooth muscle cells, and kidney tubular cells. (11, 13, 17, 24, 92, 93, 96)
There has been recently an interest towards endocytosis of the Na/K-ATPase and the role
it could play in signal transduction through the pump. In fact, dopamine and parathyroid
hormone stimulation induces endocytosis of the plasmalemmal Na/K-ATPase. (99, 100)
Depending on the cell type, this endocytosis is related to either PKC or PKA activation
and requires phosphorylation of specific amino acids in the α−chain of the Na/K-ATPase.
(101, 102). Liu et al have demonstrated that cardiotonic steroids could stimulate
internalization of the Na/K-ATPase in proximal tubular cells in vivo and in porcine
proximal tubular (LLC-PK1) cells in vitro (13, 21, 22, 24, 96,114) . Moreover,
internalization of the Na/K-ATPase was associated with accumulation of both the
cardiotonic steroids, and the Na/K-ATPase itself (13). This process was found to require
intact caveolar architecture, via caveolin1, a scaffold protein that belongs to this caveolar

16
compartment. Cardiotonic steroids stimulate a clathrin-dependent endocytosis pathway
which translocates the Na/K-ATPase to intracellular compartments under the activation
of Src. (24).
Circulating cardiotonic steroids and mammalian physiology:
For more than two centuries a cardiotonic steroids, have been used to treat congestive
heart failure. In 1953, Szent-Györgyi and Schatzmann revealed that cardiotonic steroid
are specific inhibitors of the sodium pump and that the digitalis receptor is the Na/K-
ATPase.(103) Before ouabain, marinobufagenin or digoxin were shown to be involved in
the sodium pump, researchers referred constantly to ‘’3rd factor’’ a substance that inhibits
the Na/K-ATPase.(104, 105, 114) In the early 80s doctors found increased levels of
digoxin and some antibodies in chronic renal failure patients not taking the digoxin
medication, which they later termed ‘’false positive’’ digoxin. (106, 107, 114) Since then
extensive work has been done to find the involvement of cardiotonic steroids in uremic
cardiomyopathy and its symptoms like hypertension and hypertrophy of the heart.
Extensive researches have been shaped through ouabain which is derived from plant
tissue and is considered the cardiotonic steroids of choice to study in most laboratories. A
compound that is immunologicaly quite similar to plant derived ouabain (ouabain like
compound or OLC) can be detected in a number of mammalian tissues, like in the
hypothalamus of cattle. OLC is an optical isomer of ouabain derived from plants. (108,

17
114) Ouabain (figure 2) exhibits high affinity for the α2/α3 Na/K-ATPase isoforms.
Since the tubular cells of the kidney in mammals express mainly the α1 isoform, ouabain
turn out to be insensitive in this case. Furthermore, endogenous ouabain levels were not
increased by chronic administration of high salt diet or by acute saline plasma volume
expansion.(109)
Not all the hypertension effects could be attributed to OLC; therefore researchers started
thinking of another sodium pump inhibitor family. Marinobufagenin (3, 5-dihydroxy-14,
15-epoxy bufodienolide) (figure 3) which is a member of the bufodienolides family of the
Na/K-ATPase inhibitors has been later identified. Bufodienolides differ from
cardenolides in having a doubly unsaturated six-membered lactone ring.
Marinobufagenin in its endogenous form is a potent vasoconstrictor and has been
extracted from some amphibians (certain species of toads). (110, 111) Marinobufagenin-
like compounds were later found in plasma and urine of humans, dogs, mice, and rats.
MBG is considered a natriuretic hormone that increases Na excretion when renal
elimination of Na is impaired by loss of renal function and this is seen in patients
suffering from uremic cardiomyopathy. In addition, patients under a high salt diet were
observed to have increased MBG in the system to eliminate the excess of sodium.
Although these cardiotonic steroids circulate with concentrations in the high pM to low
nM range, they are able to inhibit the sodium pump and have very significant
physiological effects.These hormones showed increases in the growth of rat cardiac
myocytes grown in culture. (112, 114) We have shown substantial increases in blood
pressure by only 4 weeks with MBG infusion. (9) Although ouabain and MBG have very

18
similar inhibitory effects on the pump, it is believed that their impact on fibrosis in the
heart and other tissues may vary depending on the amount of administration of the
cardiotonic steroid and its duration.
It has been established that in an acute salt loading, endogenous ouabain acts essentially
as a neurohormone in the hypothalamus to activate central and adrenal angiotensin II
which in turn stimulate the sympathetic nervous system through norepinephrine that
triggers increase plasma levels of MBG. Hence, the end result involving ouabain and
MBG will produce a 35 mmHg rise in the arterial blood pressure and a 45% inhibition of
the Na/K-ATPase. (41, 109)
Many speculated that age associated increases in systolic blood pressure in older humans
is correlated to circulating cardiotonic steroids. Recently, it has been shown that MBG
excretion is associated to increases in sodium excretion and is inversely related to age or
age-dependent increases in salt sensitivity. On the other hand, endogenous ouabain
excretion did not correlate with age, sodium excretion or salt sensitivity.(113) We can
conclude that both cardenolides and bufadienolides are involved in the regulation of
hypertension.

Figure 2: Chemical structure of Ouabain
Figure 3: Chemical structure of Marinobufagenin
19

20
Marinobufagenin Stimulates Fibroblast Collagen Production and Causes Fibrosis in
Experimental Uremic Cardiomyopathy
Jihad Elkareh PharmD1, David J. Kennedy PhD1, Belvadi Yashaswi MD1,
Sandeep Vetteth MD1, Amjad Shidyak MD1, Eric G.R. Kim1, Sleiman Smaili MD1,
Sankaridrug M. Periyasamy PhD1, Imad M.Hariri1, Larisa Fedorova PhD1,
Jiang Liu MD, PhD1, Liang Wu MD1, M. Bashar Kahaleh MD1, Zijian Xie PhD1,
Deepak Malhotra MD, PhD1, Olga V. Fedorova PhD2, Vladimir A. Kashkin MD, PhD2,
Alexei Y. Bagrov MD, PhD2, and Joseph I. Shapiro MD1
1The Departments of Medicine and Pharmacology, University of Toledo College of
Medicine and 2Laboratory of Cardiovascular Science, National Institute on Aging,
Baltimore, MD
Hypertension. 2007;49:215-224.

21
Corresponding Author:
Joseph I. Shapiro, M.D.
Chairman, Department of Medicine
University of Toledo College of Medicine
3120 Glendale Avenue, Toledo, Ohio 43614-5809
Phone: (419) 383-6030 FAX: (419) 383-6244
Email: [email protected]
Short Title: Cardiotonic steroids stimulate collagen production
Key Words: Cardiomyopathy; Renal Failure, TGF-β, Cardiotonic Steroids; Reactive
Oxygen Species; Fibrosis

22
Abstract
Introduction: We have observed recently that experimental renal failure in the rat is
accompanied by increases in circulating concentrations of the cardiotonic steroid,
marinobufagenin (MBG), and substantial cardiac fibrosis. We performed the following
studies to examine whether MBG might directly stimulate cardiac fibroblast collagen
production.
Methods: In vivo studies were performed using the 5/6th nephrectomy model of
experimental renal failure (PNx), MBG infusion (MBG), PNx after immunization against
MBG, and concomitant PNx and adrenalectomy. Physiological measurements with a
Millar catheter and immunohistochemistry were performed. In vitro studies were then
pursued with cultured isolated cardiac fibroblasts.
Results: We observed that PNx and MBG increased MBG levels, blood pressure, heart
size, impaired diastolic function, and caused cardiac fibrosis. PNx after immunization
against MBG and concomitant PNx and adrenalectomy had similar blood pressure as
PNx but less cardiac hypertrophy, diastolic dysfunction, and cardiac fibrosis. MBG
induced increases in procollagen-1 expression by cultured cardiac fibroblasts at 1 nM
concentration. These increases in procollagen expression were accompanied by increases
in collagen translation and increases in procollagen-1 mRNA without any demonstrable
increase in procollagen-1 protein stability. The stimulation of fibroblasts with MBG
could be prevented by administration of inhibitors of tyrosine phosphorylation, Src
activation, epidermal growth factor receptor transactivation, and N-acetyl cysteine.

23
Conclusion: Based on these findings, we propose that MBG directly induces increases in
collagen expression by fibroblasts, and we suggest that this may be important in the
cardiac fibrosis seen with experimental renal failure.

24
Introduction
Cardiac disease is directly responsible for the extremely high morbidity and
mortality seen in patients with end-stage renal disease.1 Clinically this cardiac disease of
renal failure, also called uremic cardiomyopathy, is characterized by diastolic dysfunction
and left ventricular hypertrophy in the setting of systemic oxidant stress.2 Although a
number of potential mechanisms, such as elevations in parathyroid hormone,
hypertension, and anemia, have been implicated as contributors to the cardiac disease
seen in this setting, its pathogenesis is still a bit unclear.
On this background, we have demonstrated previously that the cardiotonic steroid
marinobufagenin (MBG), signaling through the Na/K-ATPase, is directly responsible for
many features of experimental uremic cardiomyopathy induced by partial nephrectomy
(PNx) in the rat.3 Specifically, we noted that both rats subjected to PNx, as well as rats
given MBG supplementation by minipump, developed considerable cardiac hypertrophy
and fibrosis by four weeks, whereas rats immunized against MBG and subsequently
subjected to PNx had attenuation of these changes. From these data, we formulated the
hypothesis that MBG might directly induce cardiac fibroblasts to produce collagen, thus
producing much of the cardiac fibrosis seen with experimental renal failure. To test this
hypothesis and to determine the molecular basis by which this occurred, the following
studies were performed.

25
Methods
Animals
Male, Sprague–Dawley rats were used for all of the studies. All of the animal
experimentation described in the article was conducted in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory Animals using protocols
approved by the Medical University of Ohio Institutional Animal Use and Care
Committee.
Experimental Groups
Briefly, Sprague–Dawley rats weighing ≈ 250 g at the time of surgery were subjected to
either sham surgery with no MBG infusion (Sham), sham surgery with placement of a
minipump infusing MBG at 10 μg/kg per day (MBG), PNx, and PNx after immunization
against MBG (PNx-IM). MBG of extremely high purity (>99%) was isolated from the
venom of Bufa marinus by Kennedy et al.3 In addition to these maneuvers, a group of
PNx animals was subjected to adrenalectomy as well (PNx-ADx).
The heart weight normalized to body weight, left ventricular hemodynamics (eg, τ value,
slope of regression line fit to end diastolic pressure versus end diastolic volume generated
by inferior vena cava occlusions, all determined with a Millar catheter), plasma [MBG]
(determined after extraction on a C-18 column using DELPHIA as described
previously3), aldosterone (determined with ELISA kit 10004377, Cayman Chemical) and
cardiac immunohistochemistry (vida infra) were assessed 4 weeks after surgery.

26
Isolated Cardiac Fibroblasts
Preparation of adult rat cardiac fibroblasts was performed as described previously by
Brilla et al4 with modifications.
Western Blot Analysis
Western blot analysis was performed on protein isolated from tissue homogenates, cell
culture whole cell lysates, or nuclear extracts as described previously.2
Collagen Synthesis
Collagen synthesis rates were determined by the method of Nishida et al5 with
modifications.
Quantitative Measurement of Collagen-1 mRNA
Standardized RT-PCR was used to measure gene expression, with GAPDH transcript
used as the housekeeping gene, as reported previously.6
Statistical Analysis and Expanded Methods
Statistical analysis and details regarding experimental groups and specific measurements
are provided in an online supplement (http://hyper.ahajournals.org).

27
Results
Effect of Experimental Renal Failure and MBG on Blood Pressure, Cardiac
Hemodynamics, and Fibrosis
In the current in vivo studies (data summarized in the Table), we observed that MBG
levels were increased in PNx- and MBG-treated rats compared with sham-operated
controls. We also saw that both PNx and MBG rats had higher systolic blood pressure
than controls and that PNx-IM rats had statistically similar systolic blood pressure values
as seen with PNx. Using the Millar pressure/volume sensor catheter rather than
echocardiography in our previous report, 3 we observed that PNx induced decreases in
end systolic volume and end diastolic volume, as well as increased ejection fraction
compared with sham-operated controls. The end systolic volume and end diastolic
volume were greater, and the ejection factor values were lower in PNx-IM as compared
with PNx. Active relaxation assessed by τ was found to be impaired by both PNx and
MBG compared with shamoperated controls, with PNx-IM showing lower values than
PNx. Using pressure volume loops generated during vena cava occlusions (representative
tracings shown in Figure 1a), we noted that the end diastolic pressure volume relationship
(an inverse measurement of passive compliance) was increased in PNx- and MBG-treated
animals compared with controls, whereas PNx rats had a lower end diastolic pressure
volume relationship than PNx. Both PNx and MBG treatment increased the heart
weight/body weight ratio compared with sham-operated controls, whereas PNx-IM
animals had lower values than PNx (Table). Examining the ventricular myocyte cross-

28
sectional area determined on trichrome images, we noted that PNx and MBG infusion
both induced marked increases, whereas the myocyte cross-sectional area in PNx-IM was
considerably smaller than that seen with PNx alone (Figure 1b).
Analyzing the immunohistochemistry results, heart tissues from rats subjected to
MBG and PNx showed marked increases in collagen-1 and α smooth muscle actin
staining. Immunization against MBG attenuated these increases (Figure 1c and 1d).
Western blot analysis confirmed that PNx and MBG had 2 to 2.5 times the expression of
procollagen-1 and α smooth muscle actin seen with sham-operated controls, whereas
PNx-IM expression of both procollagen-1 and α smooth muscle actin was substantially
less than that seen with PNx (Figure 1e and 1f ).
To determine the molecular mechanism underlying this fibrosis, we examined the
expression of several proteins important in fibroblast activation. Specifically, we
examined tissue levels of transforming growth factor (TGF)-β, Smad 2/3, and Smad 4, as
well as pSmad 2/3. We did not detect significant differences among the experimental
groups in the cardiac expression of these proteins (Figure 2a–2d).
A separate group of animals (N=11) was also subjected to PNx-ADx with
physiological replacement of glucocorticoids and aldosterone.7 These animals developed
a similar degree of hypertension compared with PNx but were noted to have much lower
plasma MBG and aldosterone levels, as well as substantially lower heart/weight body
weight ratio compared with PNx alone (Table). Moreover, these animals subjected to
PNx-ADx had almost no evidence for cardiac fibrosis based on trichrome staining or

29
immunohistochemistry staining for collagen-1 or α smooth muscle actin (see the online
data supplement).
Effect of Cardiotonic Steroids on Fibroblast Collagen Expression
To further examine the molecular basis of this cardiac fibrosis, isolated cardiac
fibroblasts were subjected to increasing doses of MBG (10-10, 10-9, and 10-8 M). After 24
hours of exposure to 10-9 and 10-8 M MBG, procollagen content determined by Western
blot was increased ≈ 2 fold (both P<0.01; Figure 3a). This phenomenon was not specific
for MBG; other cardiotonic steroids also induced similar increases in procollagen content
(Figure 3b). Of interest, the threshold for effect for MBG seemed to be between 10-10 and
10-9 M, whereas for ouabain, which circulates at similar concentrations in uremic rats, the
threshold was ≈10 times higher (ie, between 10-9and 10-8 M). For both MBG and
ouabain, the threshold for inducing collagen expression was log units below the doses
necessary for detectable effects on 86Rb uptake in these cells (Figure 3c). In parallel
studies examining radiolabeled proline incorporation into collagen, we observed that 10-9
and 10-8 M MBG induced significant increases in both proline incorporation into total
protein, both matrix and supernatant. Using collagenase digestion, we observed that the
vast majority of the proline incorporation was into collagen (Figure 3d). Using
standardized RT-PCR, we observed a doubling of mRNA for collagen-1 at 24 hours in
response to 10 nM of MBG (Figure 3e). However, we did not detect any increases in

30
procollagen stability (determined by examining procollagen-1 expression after exposure
to cycloheximide) in response to this concentration of MBG (Figure 3f).
Effect of Inhibition of Na/K-ATPase Signaling on MBG-Stimulated Collagen
Expression
To examine whether cardiotonic steroids induced collagen synthesis by signaling through
the Na/K-ATPase, we performed the following studies. First, we used pharmacological
antagonism at several steps in the Na/K-ATPase cascade. Specifically, we used
pharmacological antagonism of Src activation with PP2, nonspecific tyrosine kinase
inhibition with herbimycin, inhibition of EGFR transactivation with AG1478, and
nonspecific antioxidant administration with N-acetyl cysteine. Each of these maneuvers
prevented MBG stimulation of collagen synthesis (Figure 4a). To confirm these data, we
also examined radiolabeled proline incorporation in response to MBG in the presence and
absence of either PP2 or N-acetyl cysteine. As was the case for procollagen expression,
both PP2 and N-acetyl cysteine prevented increases in proline incorporation into collagen
in the primary fibroblast cultures (Figure 4b). Next we performed studies in the SYF and
SYF+ cells (details available in the online supplement). SYF+ cells responded to MBG
and ouabain in a very similar way as the primary cardiac fibroblast cultures with respect
to upregulation of procollagen expression, whereas the SYF cells had essentially no
response to either MBG or ouabain (Figure 4c).

31
Relationship between TGF-β and MBG-Stimulated Collagen Production
To further examine the molecular mechanisms by which cardiotonic steroids induce
collagen production in fibroblasts, we examined the effects of MBG on TGF-β
expression, as well as the expression of Smad 2/3, Smad 4, and pSmad 2/3. As was the
case for the in vivo experiments described earlier, we did not observe significant changes
in TGF-β, Smad 2/3, Smad 4, or pSmad 2/3 expression in vitro (Figure 5a). Next, we
examined whether TGF-β induced collagen production and whether there was synergism
between TGF-β and MBG. In the primary cultured cells, we saw similar effects of TGF-β
(5 ng/mL) on procollagen expression as observed with cardiotonic steroids; however, we
did not note any synergism between TGF-β (5 ng/mL) and MBG (10 nM). However, it is
important to point out that we never completely serum starve the primary cultures, and
because serum is always present, some TGF-β is always present.
To address this further, we also examined the effect of the TGF-β receptor antagonist,
SB431542, on MBG stimulated collagen production. Interestingly, SB431542 at 100-
μmol/L concentration did not reduce procollagen expression below baseline on our
Western blots (Figure 5b) but did decrease radiolabeled proline incorporation below that
seen with control cells (Figure 5c). The SB431542 completely blocked both TGF-β and
MBG (10 nM) stimulation of collagen expression and radiolabeled proline incorporation
(Figure 5b and 5c).

32
Discussion
Cardiac fibrosis is an important component of many cardiomyopathies, and it is a
very characteristic component of uremic cardiomyopathy.8,9 Our group and others have
observed that MBG and other cardiotonic steroids induce a signal transduction cascade
through the plasmalemmal Na/KATPase residing in caveolae, which results in activation
of Src, transactivation of the EGFR, generation of reactive oxygen species, and,
ultimately, activation of p42/44 mitogen-activated protein kinase.10–16 Interestingly, a
number of clinical situations associated with cardiac fibrosis other than renal failure are
associated with increased circulating concentrations of cardiotonic steroids (eg,
hypertension, primary hyperaldosteronism, and congestive heart failure)17,18 Although it
is preliminary to discuss the possible relevance of our findings to cardiomyopathies other
than renal failure, we should point out that Ferrandi et al19 have observed that antagonism
of endogenous cardiotonic steroids with PST 2238 ameliorates hypertension, as well as
cardiac hypertrophy in Milan hypertensive rats.
In the current study, we confirmed that PNx and MBG treatment induce similar
but not identical phenotypic changes in hemodynamics and cardiac morphology. It is
quite likely that some factors other than MBG contribute to the phenotypic changes seen
in PNx. That said, both PNx-IM and PNx-ADx, which reduce circulating MBG,
substantially attenuate the cardiac functional and morphological changes without
significantly affecting blood pressure. We should point out that experiments in the PNx-
ADx model were performed because we reasoned that as adrenal cells grown in culture

33
seem to make MBG, 20, 21 it was likely that this procedure would lower the circulating
levels of this hormone. However, whereas our data in the PNx-ADx animals support the
concept that the adrenal gland is the major (but not the only) site of MBG production in
vivo, it is also possible that other hormones made in the adrenal gland modulate MBG
production elsewhere. Further work will be necessary to clarify exactly where MBG is
produced under normal and pathological conditions.
With these findings implicating MBG in the pathogenesis of cardiac fibrosis, we
were particularly interested in the molecular mechanisms underlying the fibrosis.
Interestingly, evidence for increases in TGF-β or signaling through the Smad proteins
was not evident. We stress that these data do not exclude a role for TGF-β in this process,
because earlier increases in these proteins, translocation of the Smads, and/or a
permissive role for signaling through this pathway (vida infra) could certainly be present.
Based on these in vivo data, we pursued studies in isolated cardiac fibroblasts. We
observed that MBG in physiological concentrations directly stimulated the fibroblasts to
produce more collagen. This increase in collagen production was also observed with
other cardiotonic steroids, although the threshold concentration seemed to be ≈1 log unit
lower for MBG than for ouabain. We emphasize that the concentration of both MBG and
ouabain necessary to stimulate collagen expression was lower for both substances than
that needed to appreciably inhibit 86Rb uptake. Further evidence for this
phenomenon being dependent on signaling through the Na/ K-ATPase was that this
increase was prevented by reactive oxygen species scavenging, antagonism, or knockout

34
of Src, as well as prevention of EGFR transactivation, maneuvers that we have
demonstrated previously to block signal transduction through the Na/K-ATPase
signalsome.13,14,22–24 We also observed that the increases in collagen production were
associated with increases in proline incorporation, as well as increases in mRNA for
collagen-1. No increase in procollagen-1 stability could be demonstrated in response to
MBG.
Although increases in TGF-β or the Smad proteins were also absent in the
fibroblasts treated with MBG, it is important to note that the fibroblasts that we studied
were never truly serum starved. In fact, based on a Hyclone web page
(http://www.hyclone.com/pdf/atsv19n3.pdf), we would estimate that the fibroblasts were
exposed to ≥0.12 ng/mL of TGF-β even when cultured in the serum-depleted (1% FBS)
medium. This may, in part, explain why SB431542 was so effective in preventing MBG-
stimulated collagen production. Working with a similar preparation, Lijnen and Petrov25
noted that long incubations (48 hours) and high concentrations of TGF-β (15 ng/mL)
were necessary to induce maximal (2 times) increases in collagen production. We should
also note that TGF-β blockade with SB431542 actually decreased proline incorporation
below baseline, even in the setting of MBG synthesis, although this same
pharmacological maneuver only reduced procollagen expression to baseline when
measured with Western blot. We suspect that other mechanisms of regulation of collagen
synthesis (eg, procollagen stability) might come into play when the TGF-β pathway is
interrupted, although we did not explore this point further in the current studies. On

35
balance, our data argue, albeit preliminarily, against a major role for TGF-β or
upregulation of Smad proteins in cardiotonic steroid–induced increases in fibroblast
collagen production.
Our data suggest that, in our experimental rodent model, MBG is implicated in
the pathogenesis of the cardiac fibrosis, and the concentrations of MBG that develop in
this setting, as well as other cardiotonic steroids, have in vitro effects that are consistent
with this observation. One issue that immediately comes to mind is whether the clinical
use of digitalis might have similar effects. To this question, we would suggest the
following possibilities. First, it may be that the free concentrations of digoxin that occur
in vivo are not sufficient to induce substantial cardiac fibrosis. Total digoxin levels are
typically maintained <2 ng/mL in patients treated with digoxin, a concentration that
corresponds with ≈ 2.5 nM concentration. However, only 70% to 80% of the plasma
digoxin is free, and the free concentration might fall below the threshold level of digoxin
necessary to stimulate human cardiac (or other tissue) fibroblasts. Perhaps more relevant,
we observed a fairly flat dose–response curve to MBG and ouabain with respect to
stimulation of fibroblast collagen production once the threshold for an effect was
reached. We suggest that in the setting of heart failure, a condition known to have
associated increases in MBG and other cardiotonic steroids, 17 the addition of digoxin at
therapeutic doses might not have a detectable effect. Finally, we would point out that
a systemic examination of whether digoxin induces or influences cardiac fibrosis in
humans has not been thoroughly investigated, although the clinical efficacy of this agent
in treating congestive heart failure has been extensively examined.26–28 It is important to

36
note that the rate at which humans develop cardiac fibrosis seems to be considerably
slower than that seen with rodents, 29 which might further obfuscate whether digoxin has
profibrotic effects in clinical subjects.
In summary, we observed that concentrations of MBG similar to that which
develop in experimental renal failure produced increased synthesis of collagen in primary
cardiac fibroblasts grown in culture in a manner dependent on signaling through an
Na/K–ATPase–Src–EGFR–reactive oxygen species signaling cascade.13, 14, 30 Should
these data be confirmed in humans, this insight may provide useful therapeutic targets in
clinical uremic cardiomyopathy.
Perspectives
Cardiac fibrosis is an important component of cardiac diseases seen in a variety of
disease states. Our data in the experimental renal failure model suggest that cardiotonic
steroids, such as MBG, may contribute in a very substantial role in the cardiac fibrosis
seen in this setting. Because increases in MBG are likely to accompany a variety of
volume expansion states, the implications of our observations may extend to other
situations complicated by cardiac fibrosis.

37
Acknowledgment
We thank Carol Woods for her excellent secretarial assistance.
Sources of Funding
Portions of this study were supported by the American Heart Association (D.J.K.,
fellowship award from the Ohio Valley Affiliate) and the National Institutes of Health
(HL67963). This work has also been supported in part by the Intramural Research
Program, National Institute on Aging, National Institutes of Health.
Disclosures
None

38
References
1. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL,
McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L,
Spinosa DJ, Wilson PW. Kidney disease as a risk factor for development of
cardiovascular disease: a statement from the American Heart Association Councils on
Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology,
and Epidemiology and Prevention. Circulation. 2003;108:2154 –2169.
2. Kennedy D, Omran E, Periyasamy SM, Nadoor J, Priyadarshi A, Willey JC, Malhotra
D, Xie Z, Shapiro JI. Effect of chronic renal failure on cardiac contractile function,
calcium cycling, and gene expression of proteins important for calcium homeostasis in
the rat. J Am Soc Nephrol. 2003;14:90 –97.
3. Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB,
Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI.
Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of
experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495.
4. Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat
cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol.
1994;26:809–820.
5. Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M. Effects of CTGF/Hcs24, a
hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation
of osteoblastic cells in vitro. J Cell Physiol. 2000;184:197–206.

39
6. Willey JC, Crawford EL, Knight CR, Warner KA, Motten CA, Herness EA, Zahorchak
RJ, Graves TG. Standardized RT-PCR and the standardized expression measurement
center. Methods Mol Biol. 2004;258: 13–41.
7. Kwon TH, Nielsen J, Masilamani S, Hager H, Knepper MA, Frokiaer J, Nielsen S.
Regulation of collecting duct AQP3 expression: response to mineralocorticoid. Am J
Physiol Renal Physiol. 2002;283:F1403–F1421.
8. Harnett JD, Parfrey PS. Cardiac disease in uremia. Semin Nephrol. 1994; 14:245–252.
9. London GM, Parfrey PS. Cardiac disease in chronic uremia: pathogenesis. 1997;4:194
–211.
10. Huang L, Li H, Xie Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is
accompanied by changes in expression of several late response genes. J Mol Cell
Cardiol. 1997;29:429–437.
11. Liu L, Mohammadi K, Aynafshar B, Wang H, Li D, Liu J, Ivanov AV, Xie Z, Askari
A. Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am J
Physiol Cell Physiol. 2003;284: C1550–C1560.
12. Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. Multiple signal transduction
pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of
Ras and mitogen-activated protein kinases. J Biol Chem. 1998;273:15249 –15256.
13. Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A. Intracellular reactive oxygen
species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in
cardiac myocytes. J Biol Chem. 1999;274:19323–19328.

40
14. Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the
Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain
to mitogen-activated protein kinases. J Biol Chem. 2002;277:18694 –18702.
15. Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G, Ferrari P. Organ
hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and
antagonized by PST 2238. J Biol Chem. 2004;279: 33306–33314.
16. Ferrandi M, Manunta P, Rivera R, Bianchi G, Ferrari P. Role of the ouabain-like
factor and Na-K pump in rat and human genetic hypertension. Clin Exp Hypertens.
1998;20:629–639.
17. Gonick HC, Ding Y, Vaziri ND, Bagrov AY, Fedorova OV. Simultaneous
measurement of marinobufagenin, ouabain, and hypertensionassociated protein in various
disease states. Clin Exp Hypertens. 1998; 20:617– 627.
18. Fridman AI, Matveev SA, Agalakova NI, Fedorova OV, Lakatta EG, Bagrov AY.
Marinobufagenin, an endogenous ligand of alpha-1 sodium pump, is a marker of
congestive heart failure severity. J Hypertens. 2002;20:1189 –1194.
19. Ferrandi M, Barassi P, Minotti E, Duzzi L, Molinari I, Bianchi G, Ferrari P. PST
2238: a new antihypertensive compound that modulates renal Na-K pump function
without diuretic activity in Milan hypertensive rats. J Cardiovasc Pharmacol.
2002;40:881– 889.
20. Dmitrieva RI, Bagrov AY, Lalli E, Sassone-Corsi P, Stocco DM, Doris PA.
Mammalian bufadienolide is synthesized from cholesterol in the adrenal cortex by a

41
pathway that is independent of cholesterol side-chain cleavage. Hypertension.
2000;36:442– 448.
21. Fedorova OV, Agalakova NI, Talan MI, Lakatta EG, Bagrov AY. Brain ouabain
stimulates peripheral marinobufagenin via angiotensin II signaling in NaCl-loaded Dahl S
rats. J Hypertens. 2005;23:1515–1523.
22. Priyadarshi S, Valentine B, Han C, Fedorova OV, Bagrov AY, Liu J, Periyasamy SM,
Kennedy D, Malhotra D, Xie Z, Shapiro JI. Effect of green tea extract on cardiac
hypertrophy following 5/6 nephrectomy in the rat. Kidney Int. 2003;63:1785–1790.
23. Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac
Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+
and Ca2+ concentrations. J Biol Chem. 2000;275:27838 –27844.
24. Liu J, Periyasamy SM, Gunning W, Fedorova OV, Bagrov AY, Malhotra D, Xie Z,
Shapiro JI. Effects of cardiac glycosides on sodium pump expression and function in
LLC-PK1 and MDCK cells. Kidney Int. 2002;62:2118 –2125.
25. Lijnen P, Petrov V. Transforming growth factor-beta 1-induced collagen production
in cultures of cardiac fibroblasts is the result of the appearance of myofibroblasts.
Methods Find Exp Clin Pharmacol. 2002;24:333–344.
26. Ahmed A, Rich MW, Love TE, Lloyd-Jones DM, Aban IB, Colucci WS, Adams KF,
Gheorghiade M. Digoxin and reduction in mortality and hospitalization in heart failure: a
comprehensive post hoc analysis of the DIG trial. Eur Heart J. 2006;27:178 –186.
27. Young JB. Whither withering’s legacy? Digoxin’s role in our contemporary
pharmacopeia for heart failure. J Am Coll Cardiol. 2005;46: 505–507.

42
28. Gheorghiade M, Adams KF Jr, Colucci WS. Digoxin in the management of
cardiovascular disorders. Circulation. 2004;109:2959 –2964.
29. Lijnen PJ, Petrov VV. Role of intracardiac renin-angiotensin-aldosterone system in
extracellular matrix remodeling. Methods Find Exp Clin Pharmacol. 2003;25:541–564.
30. Liu J, Liang M, Liu L, Malhotra D, Xie Z, Shapiro JI. Ouabain-induced endocytosis
of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int.
2005;67:1844 –1854.

43
Table I. Effects of PNx, MBG, and PNx-IM on Hemodynamics and Plasma MBG.
Measurements Sham PNx MBG PNx-IM PNx-ADx
Plasma MBG, pmol/L 277±27 527±36† 484±47† 396±65‡ 325±65§
Plasma aldosterone, pg/mL 184±32 2012±320† 295±35§ 2492±493† 228±65§
Tail cuff measurements
Heart rate, bpm 367±7 388±9 367±9 380±6 365±7
Systolic blood pressure, mm Hg 102±2 197±6† 136±4† 180±9† 193±6†
Ventricular hemodynamics
End systolic volume, μL 70±4 35±4† 60±6 68±9§ 56±5‡
End diastolic volume, μL 190±11 151±10* 162±14 188±14‡ 185±14
Ejection fraction, % 73±1 79±2† 68±2* 71±1§ 72±2§
τ, ms 10.0±0.3 14.5±0.9† 11.3±0.3* 10.6±0.3§ 12.1±0.5†‡
EDPVR×1000, mm Hg/μL 24±2 52±4† 41±6† 31±3*§ 38±4†§
Heart weight/body weight ratio, g/kg 2.5±0.1 3.6±0.2† 2.8±0.1* 2.9±0.1†§ 3.3±0.1†‡

44
Analyses were performed 4 weeks after sham operation (Sham, n=20), partial nephrectomy (PNx, n=20), MBG infusion
(MBG, n=20), or immunization against MBG prior to partial nephrectomy (PNx-IM, n=20). Results reported as mean±SEM.
*P<0.05 vs Sham; †P<0.01 vs Sham, comparing PNX-IM and PNx-Adx to PNx; ‡P<0.05; and §P<0.01.

Figure Legends
Figure 1. a, Representative pressure–volume loops obtained during vena cava occlusion
from rats subjected to sham surgery (Sham), PNx, MBG infusion (MBG), and PNx after
immunization against an MBG–albumin conjugate (PNx-IM). Quantitative data reported
in the Table. Regression lines fit to the end systolic pressure volume relationship
(ESPVR, dotted line) and the end diastolic pressure volume relationship (EDPVR, solid
line). b, ventricular cross-sectional areas determined from trichrome stains of tissue
obtained from Sham, PNx, MBG, and PNx-IM animals (each group: N=8 animals, ≈100
measurements averaged to determine mean for each animal; data shown as the group
mean±SEM using N=8). Representative Immunohistochemistry images of cardiac tissues
stained for (c) collagen-1 and (d) α smooth muscle actin (αSMA). Counterstain for both
c and d was hematoxlyn. Western blot and corresponding densitometric analysis for (e)
procollagen-1 and (f) αSMA. Note that both collagen-1 and αSMA staining are much
more intense in the PNx and MBG groups compared with Sham, whereas the PNx-IM
staining is similar to Sham. Similarly, procollagen and αSMA expression are
substantially higher in PNx and MBG animals compared with Sham, whereas
immunization against MBG (PNx-IM) attenuated the changes seen with PNx. Data for e
and f are derived from N=6 experiments in each group and shown as mean±SEM. Sham
refers to hearts isolated from control animals, PNx refers to PNx, MBG refers to MBG
supplemented, and PNx-IM refers to animals immunized against MBG before PNx
surgery. *P<0.05, **P<0.01 vs Sham, #P<0.01 vs PNx.
45

Figure 2. Representative Western blot for and quantitative densitometric data shown for
(a) TGF-β1, (b) Smad 2/3, (c) Smad 4, and (d) pSmad 2/3. Data derived from N=6
experiments in each group and shown as mean±SEM. Note similar expression of these
proteins in all of the 4 experimental groups.
Figure 3. Representative Western blot for procollagen and quantitative densitometric
data obtained in response to different doses of (a) MBG (all N=10), (b) MBG 10 nM
contrasted with different doses of ouabain (0.1 to 100 nM) and digoxin (10 nM; all N=8).
c, ouabain sensitive 86Rb uptake as a function of MBG and ouabain concentration (N=4
at each concentration for both MBG and ouabain; data expressed as fraction of control).
d, relative proline incorporation in the supernatant and matrix, both with and without
collagenase digestion (total). Each group (controls and different doses of MBG) contains
N=7 replicants. The difference between the total and after collagenase digestion is
reported as collagen. The matrix is the sample that was obtained after removing
supernatant and scraping the culture dish. e, mRNA for collagen 1 in MBG-treated (10
nM; N=8) or control (N=8) fibroblasts. f, procollagen stability after cycloheximide
treatment. Time 0 is 1 hour after incubation with cycloheximide (20 μg/mL).
Densitometric data displayed on log10 scale. Leastsquare regression line fit to control
(CTL) and MBG data. Bars on quantitative graphs represent the mean±SEM. *P<0.05
and **P<0.01 vs control.
46

Figure 4. a, Effects of PP2 (1 μmol/L), herbimycin (1 μmol/L), AG1478 (250 nM), and
N-acetyl cysteine (2.5 mmol/L) on MBG (10 nM) stimulation of procollagen expression.
The PP2, herbimycin, AG1478, and N acetyl cysteine were administered from 2 hours
before the addition of MBG and continued throughout the 24 hours of MBG incubation
(total of 26 hours). Each bar represents the mean±SEM of N=8 experiments. b, effects of
PP2 (1 μmol/L) and N-acetyl cysteine (2.5 mmol/L) on MBG (10 nM)-stimulated proline
incorporation into collagen. Again, the PP2 and N-acetyl cysteine were added 2 hours
before exposure to MBG. Each bar represents the mean±SEM of N=5 experiments. c,
effects of MBG 10 nM on procollagen content in SYF and SYF+ cells. Representative
Western blots are shown above quantitative data. SYF blots loaded with 15 μg of protein
and SYF+ blots loaded with 10 μg of protein. Each bar represents the mean±SEM of N=6
determinations in each group. **P<0.01 vs control.
Figure 5. a, Effect of 24 hours of MBG (1 and 10 nM) on TGF-β, Smad 2/3, Smad 4, and
pSmad 2/3 expression determined by Western blot. b and c, effects of 24 hours of MBG
(10 nM), TGF-β (5 ng/mL), and the TGF- β receptor antagonist SB431542 (100 μmol/L)
on procollagen-1 expression (Western blot) and radiolabeled proline incorporation,
respectively. SB431542 was added 2 hours before exposure to either TGF- β or MBG
(total of 26-hour exposure). Each bar represents the mean±SEM of 6 to 8 determinations.
*P<0.05, **P<0.01 vs control.
47

Figure 1a
Figure 1b
48

Figure 1c
Figure 1d
49

Figure 1e
Figure 1f
50

Figure 2a
Figure 2b
51

Figure 2c
Figure 2d
52

Figure 3a
Figure 3b
53

Figure 3c
Figure 3d
54

Figure 3e
Figure 3f
55

Figure 4a
Figure 4b
56

Figure 4c
Figure 5a
57

Figure 5b
Figure 5c
58

Figure Legends (Supplements)
Figure 1: Representative immunofluorescence (top row) and corresponding trans-
illuminated phase contrast microscopic images (bottom row) obtained from primary
cardiac fibroblasts grown in culture. Note immunostaining to vimentin, a marker of
fibroblasts is quite bright whereas staining against von Willebrand factor (a marker of
endothelial cells), tropomyocin and myocin (cardiac myocyte markers) are absent.
Figure 2: Representative trichrome stained microscopic sections from rats subjected to
partial nephrectomy (PNx, N=8) and partial nephrectomy and coincident adrenalectomy
(PNx-ADx, N=11) are shown in panel (a). Panel (b) shows representative
immunohistochemistry staining against Collagen-1 (top figures) and αsmooth muscle
actin (α SMC) (bottom figures) in samples from the same groups (both N=6). Note that
PNx-ADx resulted in marked attenuation of trichrome or immunohistochemical staining
against collagen-1 and αSMA compared with PNx alone.
Figure 3: Panel (a) shows representative fluorescence images from fibroblasts loaded
with CM-DCF. Panel (b) shows quantitative data normalized to the basal level of
fluorescence obtained in control cells at the beginning of experiments. In all experiments,
loading of CM-DCF was performed in a standard manner, and at the beginning of each
set of experiments, camera and microscope settings were set for the remainder of the day.
Individual data points shown (control red circles, MBG 10nM green squares) with larger
59

symbols reflecting group mean + SEM. Regression lines fit to mean data demonstrated
statistically significant differences in both slope and intercept (both p< 0.01).
60

Supplement Figure 1
Supplement Figure 2a
61

Supplement Figure 2b
Supplement Figure 3a
62

Supplement Figure 3b
63

Partial Nephrectomy as a Model for Uremic Cardiomyopathy in the Mouse
David J. Kennedy PhD1, Jihad Elkareh PharmD 1, Amjad Shidyak MD1,
Anna Shapiro1, Sleiman Smaili1 MD, Krishna Mutgi1, Shalini Gupta1, Jiang Tian PhD1,
Eric Morgan PhD1, Samer Khouri MD1, Christopher J. Cooper MD1,
Sankaridrug M. Periyasamy PhD1, Zijian Xie PhD1,
Deepak Malhotra MD, PhD1, Olga V. Fedorova PhD2,
Alexei Y. Bagrov MD, PhD2, and Joseph I. Shapiro MD1
1The Departments of Medicine and Pharmacology, University of Toledo College of
Medicine and 2Laboratory of Cardiovascular Science, National Institute on Aging,
Baltimore, MD
Am J Physiol Renal Physiol 294: F450–F454, 2008.
64

Corresponding Author:
Joseph I. Shapiro, M.D.
Chairman, Department of Medicine
University of Toledo College of Medicine
3120 Glendale Avenue, Toledo, Ohio 43614-5809
Phone: (419) 383-6030 FAX: (419) 383-6244
Email: [email protected]
Short Title: Mouse model for uremic cardiomyopathy
Key Words: renal failure; TGF-β; cardiotonic steroids; reactive oxygen species; fibrosis
65

ABSTRACT
Background: Because of the plethora of genetic manipulations available in the mouse,
we performed a partial nephrectomy in the mouse and examined whether the
phenotypical features of uremic cardiomyopathy described in humans and rats were also
present in the murine model.
Methods: A 5⁄6 nephrectomy was performed using a combination of electrocautory to
decrease renal mass on the left kidney and right surgical nephrectomy.
Results: This procedure produced substantial and persistent hypertension as well as
increases in circulating concentrations of marinobufagenin. Invasive physiological
measurements of cardiac function demonstrated that the 5⁄6 nephrectomy resulted in
impairment of both active and passive left ventricular relaxation at 4 wk whereas tissue
Doppler imaging detected changes in diastolic function after 6 wk. Morphologically,
hearts demonstrated enlargement and progressive fibrosis, and biochemical
measurements demonstrated downregulation of the sarcoplasmic reticulum calcium
ATPase as well as increases in collagen-1, fibronectin, and vimentin expression.
Conclusion: Our results suggest that partial nephrectomy in the mouse establishes a
model of uremic cardiomyopathy which shares phenotypical features with the rat model
as well as patients with chronic renal failure.
66

INTRODUCTION
Cardiac disease is directly responsible for the extremely high morbidity and
mortality seen in patients with end-stage renal disease (ESRD) (12). Clinically, this
cardiac disease of renal failure, also called uremic cardiomyopathy, is characterized by
left ventricular hypertrophy and diastolic dysfunction. On this background, we have
previously demonstrated that the cardiotonic steroid marinobufagenin (MBG), signaling
through the Na-K-ATPase, is responsible for many of the features of experimental uremic
cardiomyopathy induced by partial nephrectomy in the Sprague-Dawley rat (6).
Specifically, we have noted that partial nephrectomy in the rat is accompanied by
substantial elevations in blood pressure, cardiac hypertrophy, impaired left ventricular
relaxation, downregulation of the sarcoplasmic reticulum ATPase (SERCA) and cardiac
fibrosis. Except for the blood pressure elevation, immunization against MBG prevents all
of these abnormalities (2, 6).
Although the rat is an extremely useful model to study the cardiomyopathy of
renal failure (5, 6), there are many genetic manipulations which are currently available in
the mouse as well as greater ease of making additional genetic manipulations in a murine
system. Therefore, we performed the following studies to test the feasibility of studying
experimental uremic cardiomyopathy in the mouse.
67

METHODS
All animal experimentation described in the manuscript was conducted in
accordance with the National Institutes of Health Guide for the Care and Use of
Laboratory Animals using protocols approved by the University of Toledo Institutional
Animal Care and Use Committee. We performed a partial nephrectomy (PNx) in male
CD1 mice, weighing between 25 and 27 g. PNx was performed by selective cauterization
(Bovie high-temperature fine tip cautery, Aaron Medical, St. Petersburg, FL) of the entire
upper and lower poles of the left kidney, leaving a 2-mm intact segment around the hilum
(Fig. 1) as described by Gagnon and Duguid (3). This was followed by removal of the
right kidney at the same time. Following PNx or sham surgery, conscious blood pressure
was monitored weekly using the tail-cuff method (6). In a substudy to address the role of
blood pressure, n = 13 mice were given antihypertensive therapy consisting of
hydralazine (80 mg/l), reserpine (5 mg/l), and hydrochlorothiazide (30 mg/l) added to the
drinking water (15).
Some mice were subsequently anesthetized with pentobarbital sodium (50 mg/kg
ip) and studied with Doppler imaging at 4 and 6 wk following surgery or instrumented
with a Millar 1.4-Fr catheter at either 4, 6, and 8 wk for measurement of ventricular
hemodynamics [e.g., τ value, slope of regression line fit to end-diastolic pressure vs. end-
diastolic volume generated by inferior vena cava occlusions (EDPVR)] as we have
previously reported in the rat (2).
A Sonos 5500 system using a 14-MHz linear transducer (15-6L, Philips Medical
Systems, Bothell, WA) was used to perform Doppler imaging studies as described by
68

other workers (7, 13, 14). Diastolic function was assessed by examination of the early
(Ea) and late or atrial (Aa) velocity waves on the tissue Doppler imaging (TDI) studies as
well as the early (E) and atrial (A) wave on the flow Doppler studies. For the ventricular
catheter studies, diastolic function was assessed by measuring the time constant for
isovolumic relaxation (τ) to assess active relaxation and the EDPVR to assess passive
relaxation. Higher values of τ and EDPVR imply impaired active and passive relaxation,
respectively (2, 8, 10, 18).
Blood was sampled for measurement of plasma MBG concentration ([MBG]),
and the animal’s heart was removed and studied for weight, histology (trichrome staining
and morphometric analysis), and biochemical analysis as we have previously reported in
the rat (2, 6). Plasma [MBG] was determined following extraction on a C18 column using
an immunoassay employing DELPHIA as previously described (6).
Western blot analysis was performed on protein isolated from tissue homogenates
as described previously (5). Data are presented as means ± SE.
Data obtained were first tested for normality. If the data did not pass the normality
test, the Tukey test (for multiple groups) or the Mann-Whitney Rank Sum test was used
to compare the data. If the data did pass the normality test, parametric comparisons were
performed. If more than two groups were compared, one-way analysis of variance was
performed before comparison of individual groups with the unpaired Student’s t-test with
Bonferroni’s correction for multiple comparisons (16). Statistical analysis was performed
using SPSS software.
69

RESULTS
Induction of PNx resulted in rapid and sustained increases in blood pressure (Fig.
1A). Plasma [MBG] was noted to be increased about two fold 4, 6, and 8 wk following
PNx compared with sham surgery (P < 0.05 at each time; see Table 2). For examination
of left ventricular function, PNx resulted in increases in systolic function as assessed by
ejection fraction, dp/dt, and the slope of the end-systolic pressure-volume relationship
(ESPVR) following inferior vena cava constriction (Table 1). Regarding diastolic
function, both active relaxation assessed by the time constant for isovolumic relaxation
(τ) and passive relaxation assessed by the slope of the EDPVR were noted to be impaired
by PNx (representative pressure-volume loops in Fig. 1B, data in Table 1). TDI
measurements also noted trends of decreasing Ea and Aa velocities with time, but only
the Aa velocity was significantly reduced at 6 wk. Marked ventricular hypertrophy was
noted at 4, 6, and 8 wk following PNx as demonstrated by increases in the heart weight-
to-body weight ratio (see Table 3). Interestingly, end-diastolic and end-systolic volumes
were noted to be markedly reduced at 4 wk after PNx, using the Millar catheter system,
whereas these measurements appeared to increase at 6 and 8 wk. We suspect that this
“normalization” of these volumes may actually represent a conversion from concentric to
eccentric hypertrophy and a worsening of the cardiomyopathy (9), but additional studies
with longer time periods of observation will be necessary to confirm this.
Time control hearts did not demonstrate any increase in fibrosis by morphology or
any cardiac hypertrophy or increase in myocyte cross-sectional area during the course of
the study. Similarly, protein expression data were similar at the three time points studied.
70

In contrast, PNx induced activation of sarcoma viral oncogene homolog (Src) and ERK at
4, 6, and 8 wk, signal transduction steps which we have previously shown to be important
in signaling through the Na-K-ATPase (6). Decreases in both α1- and α2-Na-K-ATPase
isoform expressions were also noted as well as decreases in SERCA2a (Table 2).
Trichrome staining of histological sections demonstrated increases in myocyte cross-
sectional area as well as increased fibrosis (representative images in Fig. 2, quantitative
data in Table 3). Increases in procollagen and vimentin expression were noted as well
(Data not shown).
Interestingly, the cardiac changes induced by PNx were not measurably altered by
reductions in blood pressure achieved by the antihypertensive agents. The addition of
these agents resulted in substantial reductions in systolic BP at 4 wk (126 ± 2 mmHg, P <
0.01 vs. PNx alone) but did not substantially affect the cardiac alterations induced as
assessed by the heart weight-to-body weight ratio (4.6 ± 0.1 × 10-3, P = not significant vs.
PNx alone) or the amount of fibrosis on trichrome staining (14 ± 3%, P = not significant
vs. PNx alone) assessed after 4 wk.
71

DISCUSSION
Our laboratory has been interested in the role that cardiotonic steroids play in the
pathogenesis of uremic cardiomyopathy. Specifically, we have noted that a number of
biochemical, physiological, and morphological changes occur with PNx in the rat and
that MBG, signaling through the Na-K-ATPase, appears to be important in this process.
However, our studies are still inconclusive without the capacity to examine the actual
signaling process in vivo. We performed the studies in this report in the mouse to
establish that the same phenotypical changes followed partial nephrectomy so that we
might use a murine model for further mechanistic studies.
Our data demonstrated that the CD1 mouse responded quite similarly to PNx as
we previously reported with Sprague- Dawley rats (2, 5, 6). Specifically, we were able to
demonstrate sustained increases in conscious blood pressure and plasma [MBG], cardiac
hypertrophy, evidence for impaired active and passive relaxation and progressive cardiac
fibrosis following PNx in the male CD1 mouse. Although TDI and flow Doppler
assessments were not as sensitive as the Millar pressure catheter to changes in left
ventricular relaxation, the results of the TDI studies further demonstrated left ventricular
diastolic dysfunction consistent with uremic cardiomyopathy. This apparent insensitivity
of the echocardiographic measurements was likely a consequence of suboptimal
performance rather than an inherent limitation of this technique. In general, the
physiological, morphological, and biochemical alterations with PNx were similar to that
which we have reported in the male Sprague- Dawley rat, where suprarenal aortic
constriction was used as a control for hypertensive changes (5). Interestingly, the CD1
72

mouse also develops severe hypertension with suprarenal aortic constriction but very
little cardiac hypertrophy or fibrosis over a similar time course to what we employed in
our current study (4, 17). Moreover, in the current study, “triple” antihypertensive
therapy (15) substantially lowered blood pressure toward normal, but it did not attenuate
the cardiac hypertrophy or fibrosis induced by PNx. Thus the cardiac changes seen in this
model appear to be more dependent on the uremic milieu rather than elevations in blood
pressure alone. This report demonstrates the feasibility of pursuing detailed studies of the
molecular mechanisms utilizing knockout and “knockin” models that have already been
established in the mouse. For example, the role of caveolin-1 can be studied using the
caveolin-1 knockout mouse developed by Razani and colleagues (11). Alternatively, the
role of the Na-K-ATPase as a receptor in this process can be studied using α1-Na-
KATPase knockout (heterozygote), α2-resistant, and α1-sensitized mice created by
Lingrell and coworkers (1, 19). While these future studies are both interesting and
potentially important, we believe that this brief technical report demonstrates the
feasibility of using the mouse model for these studies as well as other investigations into
the pathogenesis of uremic cardiomyopathy.
73

ACKNOWLEDGMENTS
The authors thank Carol Woods for excellent secretarial assistance. Some of these
data were presented in abstract form at the 2006 American Society of Nephrology
Meeting, November 14–19, San Diego, CA.
GRANTS
Portions of this study were supported by the American Heart Association
(Fellowship award from the Ohio Valley Affiliate to D. J. Kennedy) and National
Institutes of Health (NIH) Grant HL-67963. This work has also been supported by the
Intramural Research Program, National Institute on Aging, NIH.
74

REFERENCES
1. Dostanic-Larson I, Lorenz JN, Van Huysse JW, Neumann JC, Moseley AE, Lingrel
JB. Physiological role of the α1- and α2-isoforms of the Na+-K+-ATPase and biological
significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp
Physiol 290: R524–R528, 2006.
2. Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S,
Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D,
Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates
fibroblast collagen production and causes fibrosis in experimental uremic
cardiomyopathy. Hypertension 49: 215–224, 2007.
3. Gagnon RF, Duguid WP. A reproducible model for chronic renal failure in the mouse.
Urol Res 11: 11–14, 1983.
4. Higashiyama H, Sugai M, Inoue H, Mizuyachi K, Kushida H, Asano S, Kinoshita M.
Histopathological study of time course changes in inter-renal aortic banding-induced left
ventricular hypertrophy of mice. Int J Exp Pathol 88: 31–38, 2007.
5. Kennedy D, Omran E, Periyasamy SM, Nadoor J, Priyadarshi A, Willey JC, Malhotra
D, Xie Z, Shapiro JI. Effect of chronic renal failure on cardiac contractile function,
calcium cycling, and gene expression of proteins important for calcium homeostasis in
the rat. J Am Soc Nephrol 14: 90–97, 2003.
6. Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB,
Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI.
75

Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of
experimental uremic cardiomyopathy. Hypertension 47: 488–495, 2006.
7. Morgan EE, Faulx MD, McElfresh TA, Kung TA, Zawaneh MS, Stanley WC,
Chandler MP, Hoit BD. Validation of echocardiographic methods for assessing left
ventricular dysfunction in rats with myocardial infarction. Am J Physiol Heart Circ
Physiol 287: H2049–H2053, 2004.
8. Nishio R, Sasayama S, Matsumori A. Left ventricular pressure-volume relationship in
a murine model of congestive heart failure due to acute viral myocarditis. J Am Coll
Cardiol 40: 1506–1514, 2002.
9. Olivetti G, Capasso JM, Meggs LG, Sonnenblick EH, Anversa P. Cellular basis of
chronic ventricular remodeling after myocardial infarction in rats. Circ Res 68: 856–869,
1991.
10. Pacher P, Mabley JG, Liaudet L, Evgenov OV, Marton A, Hasko G, Kollai M, Szabo
C. Left ventricular pressure volume relationship in a rat model of advanced aging-
associated heart failure. Am J Physiol Heart Circ Physiol 287: H2132–H2137, 2004.
11. Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F,
Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ,
Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of
hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001.
12. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL,
McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L,
Spinosa DJ, Wilson PW. Kidney disease as a risk factor for development of
76

cardiovascular disease: a statement from the American Heart Association Councils on
Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology,
and Epidemiology and Prevention. Circulation 108: 2154–2169, 2003.
13. Sebag IA, Handschumacher MD, Ichinose F, Morgan JG, Hataishi R, Rodrigues
AC, Guerrero JL, Steudel W, Raher MJ, Halpern EF, Derumeaux G, Bloch KD, Picard
MH, Scherrer-Crosbie M. Quantitative assessment of regional myocardial function in
mice by tissue Doppler imaging: comparison with hemodynamics and sonomicrometry.
Circulation 111: 2611–2616, 2005.
14. Tsujita Y, Kato T, Sussman MA. Evaluation of left ventricular function in
cardiomyopathic mice by tissue Doppler and color M-mode Doppler echocardiography.
Echocardiography 22: 245–253, 2005.
15. Vanourkova Z, Kramer HJ, Huskova Z, Vaneckova I, Opocensky M, Chabova VC,
Tesar V, Skaroupkova P, Thumova M, Dohnalova M, Mullins JJ, Cervenka L. AT1
receptor blockade is superior to conventional triple therapy in protecting against end-
organ damage in Cyp1a1- Ren-2 transgenic rats with inducible hypertension. J Hypertens
24: 2465– 2472, 2006.
16. Wallenstein S, Zucker CL, Fleiss JL. Some statistical methods useful in circulation
research. Circ Res 47: 1–9, 1980.
17. Wang QD, Bohlooly YM, Sjoquist PO. Murine models for the study of congestive
heart failure: implications for understanding molecular mechanisms and for drug
discovery. J Pharmacol Toxicol Methods 50: 163– 174, 2004.
77

18. Westermann D, Knollmann BC, Steendijk P, Rutschow S, Riad A, Pauschinger M,
Potter JD, Schultheiss HP, Tschope C. Diltiazem treatment prevents diastolic heart failure
in mice with familial hypertrophic cardiomyopathy. Eur J Heart Fail 8: 115 121, 2006.
19. Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G,
Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP.
Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J
Physiol 569: 243–256, 2005.
78

Table I. Effects of PNx on various functional parameters.
PNx Measurement Sham 4 wk 6 wk 8 wk
Doppler imaging n = 13 n = 7 n =7 ND
Ea wave (TDI endocardium), m/s 0.042±0.002 0.041±0.004 0.039±0.003
Aa wave (TDI endocardium), m/s 0.039±0.003 0.032±0.006 0.030±0.001*
S wave (TDI endocardium), m/s 0.038±0.002 0.040±0.002 0.045±0.004
E wave (Mitral inflow), m/s 1.11±0.07 1.08±0.06 1.06±0.09
A wave (Mitral inflow), m/s 0.69±0.05 0.64±0.02 0.61±0.07
Left ventricular catheter n = 28 n = 12 n = 16 n = 14
End-systolic volume, μl 6.7±0.3 3.7±0.2† 4.1±0.3† 4.2±0.3†
End-diastolic volume, μl 18.5±0.3 12.9±1.5† 16.2±1.2* 17.6±0.8
Ejection fraction, % 73±1 80±1† 82±1† 82±2†
80

τ, ms 7.5±0.3 10.5±0.4† 11.0±0.8† 10.5±0.4†
EDPVR, (mmHg/μl) × 104 310±38 563±75* 765±64† 677±76†
Values are means ± SE. TDI, tissue doppler imaging; PNx, partial nephrectomy; ND, not done; τ, time constant for isovolumic
relaxation; EDPVR, end-diastolic pressure vs. end-diastolic volume generated by inferior vena cava occlusions. Studies were
performed 4 and 6 wk, and 4, 6, and 8 wk after sham or PNx surgery for the echocardiography and ventricular catheterization,
respectively. Sham surgery mice were found to have virtually identical measurements at the different time points for both
Doppler imaging [4 (n = 6) and 6 wk (n = 7)] and ventricular catheterization studies [4 (n = 12), 6 (n = 8), and 8 wk (n = 8)].
To simplify the table, these sham surgery data have been combined. *P < 0.05 vs. sham. †P < 0.01 vs. sham.
81

Table II. Effect of PNx on various biochemical measurements.
PNx Measurement Sham (n = 28) 4 wk (n = 12) 6 wk (n = 8) 8 wk (n = 8)
Plasma [MBG], pmol/l 143±22 304±55* 331±79* 343±69*
Plasma malondialdehyde μM 2.6±0.1 2.8±0.1 2.8±0.2 3.2±0.1*
Western blot analyses n = 18 n = 6 n = 6 n = 6
pSrc/Src, fraction of control 1.0±0.04 1.35±0.08* 1.22±0.10 1.29±0.07*
pERK/ERK, fraction of control 1.0±0.02 2.18±0.04† 2.74±0.08† 1.90±0.04†
α1-Na-K-ATPase, fraction of control 1.0±0.04 0.72±0.05* 0.71±0.05* 0.60±0.06†
α2-Na-K-ATPase, fraction of control 1.0±0.07 0.73±0.04* 0.74±0.05* 0.46±0.04†
SERCA2a, fraction of control 1.0±0.06 0.74±0.04* 0.64±0.04† 0.52±0.04†
82

83
Values are means ± SE. MBG, marinobufagenin concentration; SERCA, sarcoplasmic reticulum ATPase. Studies were
performed 4 and 6 wk, and 4, 6, and 8 wk after sham or PNx surgery. Sham surgery mice were found to have virtually identical
measurements at the different time points, and to simplify the table, the sham surgery data have been combined. *P < 0.05 vs.
sham. †P < 0.01 vs. sham.

Table III. Effect of PNx on various morphological measurements.
PNx Measurement Sham (n = 28) 4 wk (n = 12) 6 wk (n = 8) 8 wk (n = 8)
Heart weight/body weight × 103 3.9±0.1 4.7±0.2† 4.7±0.1† 4.8±0.2†
Myocyte cross-sectional area, fraction of control 1.00±0.10 1.24±0.11 1.38±0.07* 1.45±0.10†
Cardiac fibrosis, fraction of control area 1±1 12±4* 16±5† 49±10†
Values are means ± SE. Studies were performed 4 and 6 wk, and 4, 6, and 8 wk after sham or PNx surgery. Sham surgery mice
were found to have virtually identical measurements at the different time points, and to simplify the table, the sham surgery
data have been combined. *P < 0.05 vs. sham. †P < 0.01 vs. sham.
84

FIGURE LEGENDS
Fig. 1. Partial nephrectomy (PNx) produces hemodynamic changes consistent with
fibrosis. A: blood pressure responses to PNx compared with control over the period of
study. Values are means ± SE. **P < 0.01 vs. control. B: representative pressure-volume
loops in control and PNx animals. C: representative Doppler imaging tracings [tissue
Doppler imaging (TDI; top) and flow Doppler (mitral; bottom)] in control and PNx
animals. Early (Ea), atrial (Aa), systolic (S), early (E), and atrial (A) waves annotated on
control tracings.
Fig. 2. Representative Masson’s trichrome sections of left ventricular cardiac tissue in
control animals as well as 4, 6, and 8 wk after partial nephrectomy (PNx).
85

Figure 1A
Figure 1B
86
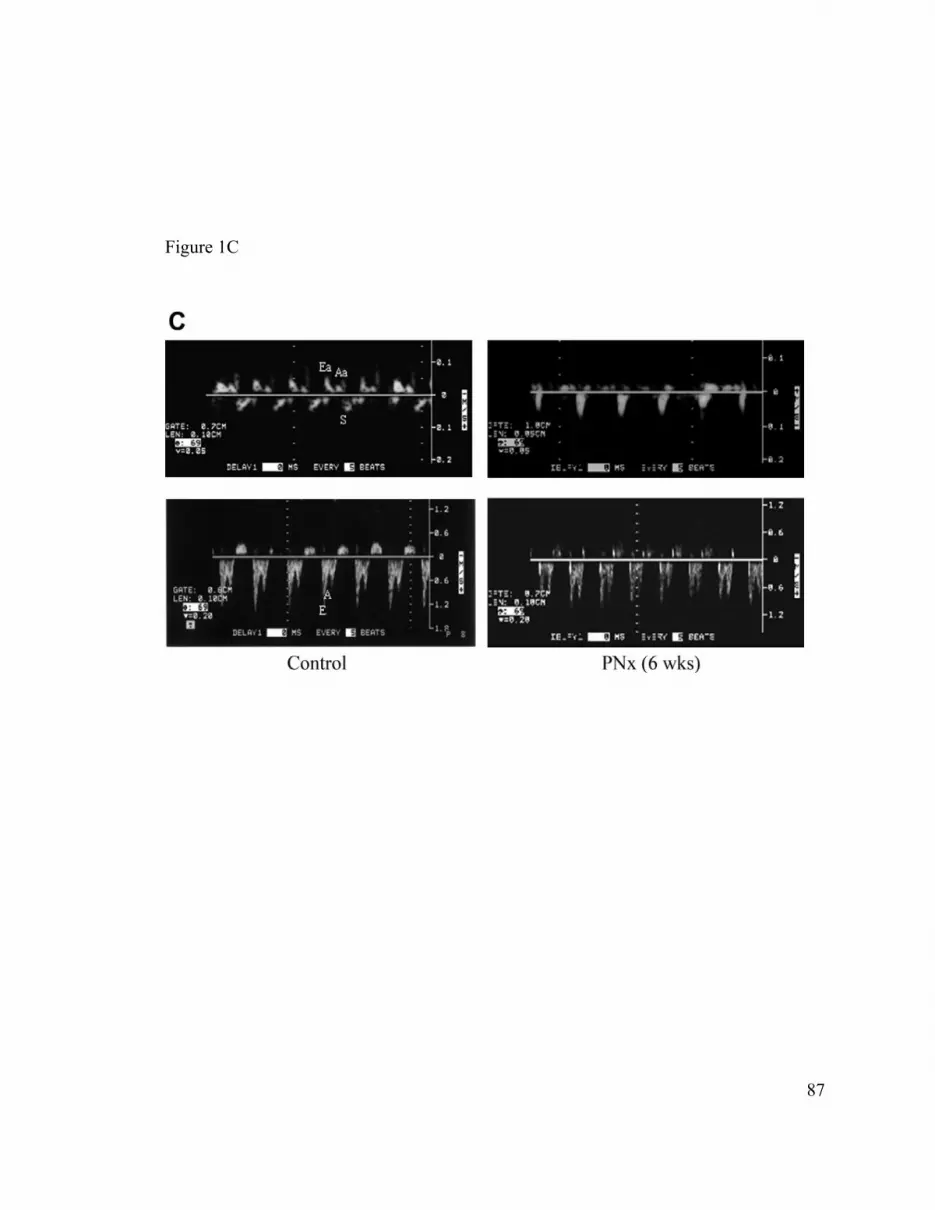
Figure 1C
87
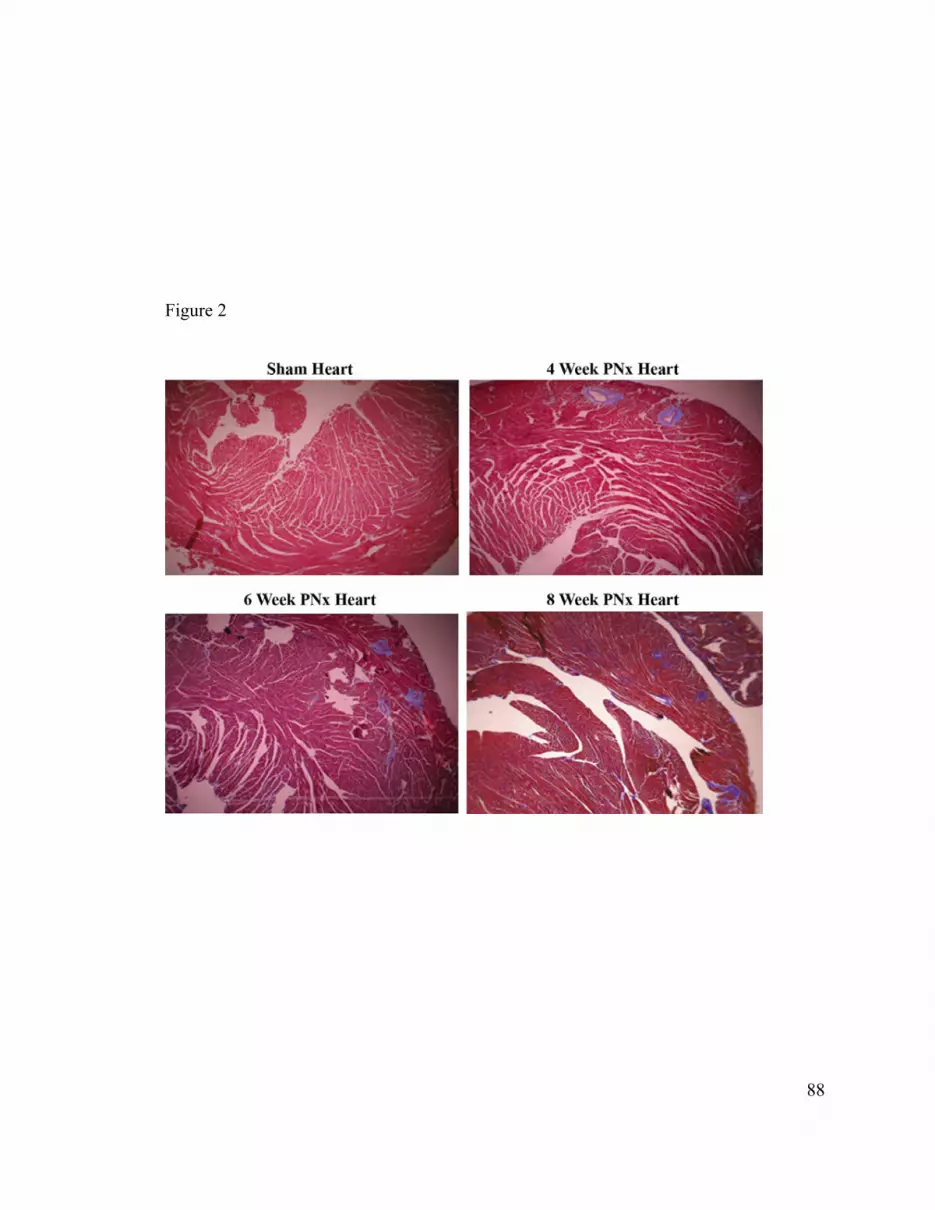
Figure 2
88

Marinobufagenin Induces Increases in Procollagen Expression in a Process
Involving Protein Kinase C and Fli-1: Implications for Uremic Cardiomyopathy
Jihad Elkareh PharmD1, Sankaridrug M. Periyasamy PhD1, Amjad Shidyak MD1,
Sandeep Vetteth MD1, Vanamala Raju MD1, Imad M.Hariri1, Nasser El-Okdi1,
Shalini Gupta1, Larisa Fedorova PhD1, Jiang Liu, MD, PhD1, Olga V. Fedorova PhD3,
M. Bashar Kahaleh MD1, Zijian Xie PhD1, Deepak Malhotra MD, PhD1,
Dennis K. Watson PhD2, Alexei Y. Bagrov MD, PhD3, and Joseph I. Shapiro MD1
1The Departments of Medicine and Pharmacology, University of Toledo College of
Medicine, 2Department of Pathology and Laboratory Medicine and Hollings Cancer
Center, Medical University of South Carolina, Charleston, SC. and 3Laboratory of
Cardiovascular Science, National Institute on Aging, Baltimore, MD
In Press
89

Corresponding Author:
Joseph I. Shapiro, M.D.
Chairman, Department of Medicine
Mail Stop #1186 Health Science Campus
University of Toledo College of Medicine
3000 Arlington Avenue
Toledo, Ohio 43614-2598
Phone: (419) 383-6030
FAX: (419) 383-6244
Email: [email protected]
Short Title: Cardiotonic steroids stimulate collagen production
Key Words: Cardiomyopathy; Renal Failure, TGF-β, Cardiotonic Steroids; Reactive
Oxygen Species; Fibrosis
90

Abstract
Background: The cardiotonic steroid, marinobufagenin (MBG) has been implicated in
the pathogenesis of cardiac fibrosis seen with experimental renal failure, and has been
shown to directly stimulate cardiac fibroblasts to produce collagen. In this manuscript, we
examined whether the transcription factor, Fli-1 might be involved in this process.
Methods: We first examined the relationship between collagen and nuclear Fli-1
expression in Fli-1 knockdown mice. Next, we used cell culture to determine whether Fli-
1 expression was altered by cardiotonic steroids and how cardiotonic steroids might alter
Fli-1 expression.
Results: The Fli-1 knockdown animals showed greater amounts of cardiac collagen
expression and fibrosis compared to wild type. This phenomenon was more pronounced
after 4 weeks of experimental renal failure induced by 5/6th nephrectomy.
Similarly, we noted that there was a strong inverse relationship between the expressions
of Fli-1 and procollagen in primary culture of rat cardiac and human dermal fibroblasts as
well as a cell line derived from renal fibroblasts. Next, we observed that in response to
MBG, decreases in nuclear Fli-1 accompanied increases in procollagen expression. After
we transfected a Fli-1 expression vector into the renal fibroblast line, basal procollagen
expression was decreased and no increases were observed in response to MBG. To
further delineate the mechanisms behind this relationship, we returned to cardiac
fibroblasts and observed that exposure to MBG was associated with a rapid translocation
of the delta isoform of protein kinase C (PKCδ) to the nucleus that appeared to peak at
91

about 15 minutes as determined with confocal immunofluorescence microscopy; this
apparent translocation was confirmed by Western blot. This PKCδ translocation could be
prevented by pharmacological inhibition of phospholipase C with U-73122, and MBG
induced increases in procollagen expression could be prevented with the PKCδ specific
inhibitor, rottlerin but not the PKCα specific inhibitor, GF109203X. Finally,
immunoprecipitation of nuclear extract following MBG treatment with an antibody to
Fli-1 resulted in increases in material immunoreactive to anti-phosphoserine (but not anti-
phosphothreonine) and 32P labeled material at the molecular weight of Fli-1.
Conclusion: We feel these data strongly support a causal relationship between decreases
in nuclear Fli-1 expression and increases in collagen production following exposure to
MBG. We further suggest that MBG stimulated activation and translocation of PKCδ
which, in turn, induced phosphorylation and downregulation of the transcription factor
Fli-1. Should these findings be confirmed, we speculate that this pathway may represent a
therapeutic target for uremic cardiomyopathy and possibly other disorders characterized
by excessive fibrosis.
92

Introduction
We have identified that the cardiotonic steroid, MBG, is responsible for many of
the clinical features of experimental uremic cardiomyopathy, and that cardiotonic
steroids, in general, directly stimulate cardiac fibroblasts to produce increased amounts of
collagen (1-3). Somewhat surprisingly, we did not observe increases in TGF-β or Smad
proteins either in vivo or in vitro with this process although we did find that a TGF-β
antagonist, SB431542, blocked stimulation of collagen production from cardiotonic
steroids (1). Because of these observations, we chose to examine other mechanisms by
which cardiotonic steroids might stimulate collagen production.
Recently, the transcription factor Friend leukemia integration-1 (Fli-1), which
belongs to the Ets family, has been identified as a negative regulator of collagen synthesis
in dermal fibroblasts (4-6). It has also been identified that stimulation of PKC,
specifically the delta isoform, can phosphorylate Fli-1 and stimulate collagen synthesis
(7). Because it has been shown that cardiotonic steroids may induce increases in protein
kinase C (PKC) activity through signaling through the Na/K-ATPase (8), we proposed to
examine whether this pathway could explain our findings.
93

Materials and Methods
Materials: MBG (>99%pure) was isolated from the venom of Bufa Marinus as
described previously (9). U-73122 (a PLC inhibitor) was purchased from Cayman
Chemical, Ann Arbor, MI. Rottlerin, (a PKCδ inhibitor), GF109203X, (a PKCα
inhibitor), and protease inhibitors were obtained from Sigma-Aldrich, St Louis, MO.
Anti-type1 collagen antibody was purchased from Southern Biotech, Birmingham, AL.
We used two sources of anti-Fli-1 antibody, both of which were polyclonal. One which
was used in the samples derived from in vitro studies was purchased from Santa Cruz,
Santa Cruz, CA whereas a rabbit anti-Fli-1 antibody which was supplied by one of the
authors (DW) was used for the Western blots derived from cardiac tissue. Anti-PKC
antibodies were purchased from BD Biosciences, San Jose, CA. Anti-phosphoserine and
anti-phosphothreonine antibodies were obtained from Calbiochem, San Diego, CA. [32P]
orthophosphoric acid was purchased from Perkin Elmer, Waltham, MA. Normal human
dermal fibroblasts were obtained from Cambrex Bioscience, Walkersville, MD, and rat
renal fibroblasts were purchased from ATCC, Manassas, VI. A pSG5 plasmid expressing
Fli-1 gene was employed as previously described by one of the authors (MBK) (5).
Animal studies: All animal experimentation described in the manuscript was conducted
in accordance with the NIH Guide for the Care and Use of Laboratory Animals using
protocols approved by the University of Toledo Institutional Animal Care and Use
(IACUC) Committee. Male mice( B6; 129SvEv-Fli-1 tm1 ) weighing between 25-30gms
were used in this study. Systolic blood pressure was measured in conscious animals using
94

tail-cuff method as previously described (10). The animals were then subjected to either
sham surgery or partial nephrectomy (PNx) as we have also previously described (10).
Following surgery, systolic blood pressure was monitored weekly until they were
sacrificed. At the end of 4 weeks, mice were anesthetized with pentobarbital sodium
(50mg/kg ip). Blood was collected for measurement of plasma MBG concentration.
Then the animal’s heart was removed, weighed and used for histological (trichrome
staining and morphometric analysis) and biochemical analysis as we have previously
reported (1-3,10). Plasma MBG was determined following extraction on a C18 column
using an immunoassay employing DELPHIA as previously described (1,3). Western blot
analysis was performed on tissue homogenates prepared from different groups as
described below.
Cell culture: Adult rat cardiac fibroblasts were isolated as previously described by
Brilla, et al.1994 (11) with modifications as previously described by us (1). Cardiac, renal
and human dermal fibroblasts were grown to confluence in DMEM with 15% FBS and
starved for 18-24 hours in a medium containing 1% FBS prior to treatment. Cardiac,
renal and human dermal fibroblasts were used up to passage 2, 10 and 7, respectively.
Protein contents of various preparations were determined using the Bio-Rad protein assay
method. U-73122, a PLC inhibitor, was used at a concentration of 10 μM in the medium.
Rottlerin, a PKC inhibitor specific for PKCδ and GF109203X, a PKC inhibitor specific
for PKCα , were used at 4 μM concentrations.
Generation of stable transfectants: Renal fibroblasts were grown to 80% confluence
and they were transfected with either pSG5 Fli-1 gene or pSG5 vector and pTargeTTM
95

vector carrying neomycin resistant gene (Promega, Madison, WI). The transfection was
carried out at a ratio of 10 parts of pSG5-Fli-1 to 1 part of pTargeTTM vector using a
transfection reagent FuGENE 6 (Roche, Nutley, NJ) according to manufacturer’s
protocol. Individual antibiotic-resistant colonies were selected and expanded in medium
containing 400ug/ml G418. The cells transfected with the pSG5-Fli-1 and a pSG5
(control) genes were analyzed for the expression of procollagen determined on whole cell
lysates and the nuclear expression of the Fli-1 protein.
Western blot analysis: Cell lysates were prepared by washing the cells twice with ice-
cold phosphate-buffered saline (PBS) and incubating them on ice for 20 minutes with
lysis buffer containing 10 mM Tris/HCl, pH 7.5, 0.5% Nonidet P40, protease and
phosphatase inhibitors. Cell lysates were collected and vortexed for 30 s and used
immediately, or stored at –80 °C. For procollagen detection, cell lysates or tissue
homogenates were dissolved in loading buffer and proteins (10ug/lane) were separated by
SDS/PAGE using 4-15% Tris-HCl precast ready gels (Bio-Rad, Hercules, CA). For Fli-1
detection, a large 10% gel was used and the protein loading was 120ug/lane for nuclear
extract or 50ug/lane for tissue homogenates. After separation, proteins were transferred
onto PVDF membranes. Membranes were blocked with 5% non-fat dry milk (Bio-Rad)
in Tris-buffered saline supplemented with 0.05% Tween-20 (TBS-T) at room temperature
for 2 hours and then incubated with primary antibody in blocking buffer at 4°C overnight.
After washing in TBS-T, membranes were incubated for 2 hours at room temperature
with horseradish peroxidase-conjugated secondary antibody in blocking buffer. After
washing in TBS, membranes were developed using ECL or ECL plus (Amersham
96

Biosciences, Piscataway, NJ). For loading controls, tubulin or actin were probed; for
both, the primary Ab was diluted 1/3000 and the secondary Ab (goat anti-mouse, Santa
Cruz) were diluted 1/2000. The images captured on x-ray film were scanned using
CanoScan Li DE 60 from Canon and quantified by using the Image J version 1.37V
software (NIH). The quantified signals were in the linear range of our detection system.
Immunostaining and confocal microscopy: Cells were fixed with cold absolute
methanol or 4% paraformaldehyde in PBS, permeabilized in permeabilization buffer
(PBS with 0.3% Triton X-100 and 0.1% BSA) for 15 minutes, and blocked with GSDB
buffer [20 mmol/L sodium phosphate, pH 7.4, with 150 mmol/L NaCl, 0.3% Triton X-
100, and 16% (v/v) filtered normal goat serum] for 30 minutes at room temperature. The
cells were then probed with primary antibody for 90 minutes at room temperature or
overnight at 4°C (rabbit polyclonal anti PKCδ antibody, Santa Cruz Biotechnology; 1:50
dilution in GSDB). After 3 washes with permeabilization buffer, the cells were incubated
with Alexa Fluor® 488-conjugated antirabbit secondary antibody for 1 hour at room
temperature. After another 3 washes, the cells were counterstained with propidium iodide
(Molecular Probes) to localize nuclei. Cells were then mounted using Prolong Anti-fade
medium (Molecular Probes) and stored at -20°C. All images were generated with a Leica
DMIRE2 confocal microscope (Wetzlar, Germany). Contrast and brightness were set to
ensure that all pixels were within the linear range. Negative controls were also performed
to verify the specificity of primary and secondary antibodies.
97

Determination of PKCδ in the nucleus: Cardiac and renal fibroblasts were grown to
confluence and starved for 18-24hrs. Cells were treated with MBG to a final
concentration of 10nM in the medium. After 15min, cells were washed with ice-cold PBS
and nuclear extracts were prepared from treated and untreated cells as described by
Wadman, et al. (12). PKCδ in the nuclear extract was determined by performing Western
blot as described above.
Immunoprecipitation and detection of phosphorylated Fli-1 by Western blot: Renal
fibroblasts were grown and starved as described above. The cells were treated for 15min
with 10nM MBG in the medium. After washing the cells in PBS, nuclear extracts were
prepared from treated and untreated cells as described by Wadman (11). Equal amounts
of proteins from nuclear extracts were used for immunoprecipitation. For
immunoprecipitation, nuclear proteins were incubated with the polyclonal anti-Fli-1
antibody conjugated to agarose beads at 4° C overnight. The following day the
immunocomplex was sedimented and washed 3-4 times with ice-cold PBS. The washed
immunocomplex was dissolved in sample buffer and the proteins were separated on a
10% gel. The separated proteins were transferred to a PVDF membrane and processed as
described under Western blot analysis. To detect whether serine or threonine molecules
are involved in phosphorylation of proteins, anti-phosphoserine or anti-phosphothreonine
antibodies were used.
98

Immunoprecipitation and detection of phosphorylated Fli-1 by autoradiography:
For autoradiography, renal fibroblasts were grown and starved as described above. The
starved cells were rinsed twice with phosphate free DMEM and then incubated at 37° C
in phosphate free DMEM for 30min. The cells were replaced with fresh phosphate free
DMEM containing 20uCi/ml 32P orthophosphoric acid and incubated for 2hrs. MBG was
added to the cells to a final concentration 10nM and 15min later the cells were washed
with ice-cold PBS. Nuclear extracts were prepared as described by Wadman (11). For
immunoprecipitation, nuclear extracts with equal amounts of radioactivity from treated
and untreated renal fibroblasts were incubated with the polyclonal anti-Fli-1 antibody
conjugated to agarose beads at room temperature for 2hrs. The immunocomplex was
washed with ice-cold PBS and dissolved in sample buffer and subjected to a SDS/PAGE
on a 10% gel. The gels were dried on a gel dryer and the phosphorylated proteins were
visualized by using a phosphoimager, Storm 840 from Novell and quantified by using
Image Quan TLV soft ware from Novell.
Statistical analysis: Data presented are mean± standard error of the mean. Data obtained
were first tested for normality. If the data did not pass the normality test, the Tukey test
(for multiple groups) or the Mann-Whitney Rank Sum test were used to compare the
data. If the data did pass the normality test, parametric comparisons were performed. If
more than two groups were compared, one-way analysis of variance was performed prior
to comparison of individual groups with the unpaired Student’s t-test with Bonferroni’s
correction for multiple comparisons. If only two groups of normal data were compared,
99

the Student’s t-test was used without correction (13). Statistical analysis was performed
using SPSS™ software.
100

Results
Relationship between Fli-1 and Collagen expression in cardiac tissues of wild type
and Fli-1 knockdown mice before and after nephrectomy:
We first examined whether there was a relationship between collagen production
and Fli-1 expression in cardiac tissues. We determined the expression of Fli-1 and
collagen in the hearts obtained from wild and Fli-1 knockdown (KD) mice using Western
blot. We observed that the hearts of the KD mice expressed approximately 50% of the
Fli-1 of WT hearts (figure 1a). Next, we performed either sham or partial nephrectomy
(PNx) surgery on these animals and conscious systolic blood pressure was monitored
before and after surgery as the renal failure progresses. We found that systolic blood
pressure was increased in WT as well as KD animals following PNx compared to sham
surgery. Interestingly, the increase in systolic BP was greater in the KD animals (figure
1b) compared to WT animals. When we examined the heart size in these animals, the
heart size was increased following PNx in both WT and KD animals, but the increase in
heart size was greater in the KD animals (figure 1c). Next, we examined the histology of
the hearts using trichrome staining; we noted that there was a greater degree of fibrosis
determined by the amount of blue staining in both WT and KD animals following PNx,
but the amount of blue staining was greater in the KD animals compared to the WT.
(figures 1d and 1e). Of interest, cardiac myocyte cross sectional areas were not different
between KD and WT animals either at baseline or after PNx, but the cross sectional areas
of both KD and WT increased following PNx compared to sham surgery (figure 1f). We
101

also measured collagen expression in cardiac tissues from these animals. We found that
collagen expression was high in KD compared to WT animals. This difference was more
pronounced following PNx (figure 1g). This finding was consistent with the histological
observations described above.
Relationship between procollagen and nuclear Fli-1 expression in fibroblasts from
different tissues:
We initially examined the basal expression levels of nuclear Fli-1 and procollagen
determined on whole cell lysates from fibroblasts isolated from human skin, rat heart and
kidneys and grown to confluence. We noticed indeed that the procollagen production
from cardiac fibroblasts was greater at baseline than that seen with the renal fibroblasts
which, in turn, were greater than that seen with the human dermal fibroblasts.
Interestingly, basal nuclear Fli-1 expression appeared to be strongly but inversely
correlated with the basal expression of procollagen (figure 2a). Next, we examined the
effect of MBG on procollagen and Fli-1 expression in all three fibroblasts. We noted that
MBG increased procollagen expression in the renal and dermal fibroblasts, as well as in
the cardiac fibroblasts, as we previously reported (1), (figure 2b). The threshold for
significant increases in procollagen expression was at 100 pM in the renal and dermal
fibroblasts, whereas it was 1 nM for the cardiac fibroblasts. Other cardiac glycosides
(ouabain and digoxin) also induced increases in procollagen expression in all three cell
types (data not shown) as we had previously reported in the cardiac fibroblasts (1). MBG
102

administered at a10 nM concentration resulted in marked and similar decreases in nuclear
Fli-1 expression in all fibroblasts studied (figure 2c).
Decreases in nuclear Fli-1 expression are necessary for MBG induced increases in
procollagen expression in a renal fibroblast line:
To further examine the relationship between Fli-1 expression and MBG induced
increases in procollagen production, we stably transfected the renal fibroblasts with a Fli-
1 expression vector coupled to an SV40 promoter. We found that the renal fibroblasts
transfected with Fli-1 expression vector showed marked increase in nuclear Fli-1
expression as expected. At the same time, the basal expression level of procollagen was
significantly reduced. Furthermore, when these transfected cells were exposed to MBG,
MBG did not produce an increase in procollagen expression. On the other hand, MBG
produced a significant increase in procollagen expression as well as decrease in nuclear
Fli-1 expression in control renal fibroblasts (transfected with an empty vector, figure 3).
MBG induces translocation of PKCδ into the nucleus:
In this set of experiments, we first examined the expression of various PKC
isoforms in cardiac fibroblasts. We found that the α, γ, δ, ε and λ PKC isoforms could all
be detected with Western blot. However, following MBG administration, only the PKCδ
expression was increased in the nuclear fraction. Based on the work of Jinnin and
colleagues (7), we examined the effect of rottlerin, a specific inhibitor of PKCδ on the
MBG induced expression of procollagen. We found that addition of rottlerin (4 μM),
103

resulted in marked reductions in basal procollagen expression and prevented MBG
induced increases in procollagen expression (figure 4c). In contrast, administration of
GF109203X which is specific for PKCα did not alter the change in procollagen
expression following MBG treatment for 24 hours (relative expression to control = 1.9 +
0.2, N=3).
We next used confocal immunofluorescence microscopy technique to follow the
time course of what we presumed was PKCδ translocation to the nucleus. Confocal
immunofluorescence microscopy experiments showed that that the nuclear PKCδ
expression peaked at about 15 minutes and slowly decreased over the next 24 hours of
exposure to MBG (figure 4a). An identical pattern was noted with renal fibroblasts (N=5,
data not shown). The finding of confocal experiment was supported by Western blot
experiment where we found a significant increase in cardiac nuclear PKCδ expression at
15 minutes after exposure to 10 nM MBG. This increase was blocked by co-incubation
with the PLC inhibitor, U-73122 (figure 4b). Again, a virtually identical pattern was seen
with the renal fibroblasts (N=4, data not shown).
Our next step was to examine whether the translocation of PKCδ into the nucleus
causes phosphorylation of Fli-1. To test this, cells exposed to 10nM MBG for 15min and
nuclear extracts were prepared. To enrich Fli-1, nuclear extract was incubated with
polyclonal anti-Fli-1 antibody. The resulting immunocomplex was separated on a 10%
gel and probed for phosphorylated proteins, particularly Fli-1 using an anti-
phosphoserine Ab. We found a band with greater intensity corresponding to the
molecular mass of Fli-1 (figure 5a). When the experiment was repeated in the presence
104

of rottlerin or U-73122 (both N=3), the intensity of the 50 kDa band was the same or less
than that seen with untreated cells (blot densities of 0.7 + 0.3 and 0.7 + 0.2 with rottlerin
or U-73122 alone, respectively and 0.6 + 0.3 and 0.7 + 0.2 when rottlerin or U-73122 was
combined with MBG, respectively (data expressed relative to control, both N=3), When
the immunocomplex was probed after separation of proteins with an anti-threonine Ab
we did not see any increase in intensity of the band with 50kDa mass following MBG
treatment (blot density of 0.9 + 0.2 relative to control, N=3).
To further demonstrate that MBG induced decreases in Fli-1 expression through
PKCδ mediated phosphorylation, we labeled the cells with 32P and prepared nuclear
extracts. To enrich Fli-1, nuclear extracts were immunoprecipitated with polyclonal anti-
Fli-1 antibody and the immunocomplex was subjected to gel electrophoresis. 32P labeled
proteins present on the gel were quantified using a phosphoimager. We observed an
increase in 32P labeled protein at the level corresponding to molecular mass of Fli-1
(figure 5b); this was attenuated to be comparable to that seen in control samples by co-
administration of rottlerin (blot density of 0.7 + 0.2 with rottlerin alone and 0.6 + 0.3 with
rottlerin + MBG, data expressed relative to control, N=3).
105

Discussion
Uremic cardiomyopathy is characterized by diastolic dysfunction and left
ventricular hypertrophy. Cardiac disease is responsible for the high mortality seen in
patients suffering from kidney diseases (14). Our group and others have observed that
the cardiotonic steroid marinobufagenin (MBG), signaling through the Na/K-ATPase, is
directly responsible for many features of experimental uremic cardiomyopathy. MBG
directly induces cardiac fibroblasts to produce collagen, thus producing much of the
cardiac fibrosis seen with experimental renal failure.(1,3).
Fli-1 (Friend leukemia virus integration 1) is a member of the Ets oncongene
family of proteins (15-19) and normally competes with Ets-1 in a Sp-1 dependent to
balance between stimulating and repressing the Col1a2 promoter. This transcription
factor has been clearly shown to play a role in dermal fibrosis, and a direct inhibitory
effect on collagen -1 synthesis has been demonstrated (4). In the current study, we
extended this observation to an animal model and used Fli-1 knockdown mice looking for
cardiac fibrosis. First, we demonstrated that the viable heterozygotes (Fli-1+/-) expressed
about 50% of cardiac Fli-1 compared to wild type mice. These heterozygotes also had
greater degrees of cardiac fibrosis and cardiac collagen expression compared to wild
type. This difference in the cardiac fibrosis and cardiac collagen expression between
heterozygotes and wild type was more pronounced in PNx animals’ expression both with
and without antecedent PNx surgery than wild type mice, thus supporting the concept that
106

the downregulation of nuclear Fli-1 expression induced by cardiotonic steroids plays a
significant role in the cardiac fibrosis seen in experimental renal failure (1,10).
Based on these in vivo results, we examined the relevance to different types of
fibroblasts in particular cardiac, renal, and dermal fibroblasts. First, we demonstrated an
inverse relationship between basal nuclear Fli-1 expression and collagen production in
the three types of fibroblasts. We further demonstrated that MBG induced corresponding
decreases in nuclear Fli-1 expression with increases in procollagen expression in each of
these fibroblasts, and we confirmed the importance of Fli-1 with an overexpression
transfection in the renal fibroblast cell line. These transfected cells showed higher basal
levels of nuclear Fli-1 expression and lower basal levels of procollagen expression and
did not show any significant increase in procollagen expression in response to MBG.
To further explore the signaling pathways leading to this observation, we first
looked at the protein kinase C (PKC) family of proteins that mediates the specific
activation of a variety of transcription factors. Specifically, Jinnin and colleagues have
shown that PKC-delta stimulation can phosphorylate Fli-1 and increase collagen
synthesis (7). Moreover, Watson, et al., suggested that a balance between protein kinase
and protein phosphatase regulate the phosphorylation status and effectiveness of Fli-1 in
inhibiting collagen-1 synthesis (20). We observed that MBG induced translocation of
PKCδ into the nucleus is a PLC dependent process, a finding consistent with what
Kometiani and coworkers demonstrated for ouabain induced activation of the Na/K-
ATPase signal cascade (8). U-73122 completely prevented any increase in nuclear PKCδ
whereas rottlerin prevented MBG induced increases in procollagen expression supporting
107

this concept. We further determined that it is PKCδ which phosphorylates Fli-1 as
assessed by immunoprecipitation, immunblotting and 32P labeling.
Taken together with existing literature, our findings strongly implicate the
transcription factor, Fli-1, in the progressive cardiac fibrosis seen with experimental
uremia as indicated in figure 6. If these data are confirmed by further studies, we would
suggest that the signal cascade that has been elucidated may serve as a therapeutic target
for clinical uremic cardiomyopathy.
Acknowledgements
We would like to thank Ms. Carol Woods for her excellent secretarial assistance. Portions
of this work were supported by grants from the NIH (RO1-HL67963 and P01-CA78582).
108

Literature Cited
1. Elkareh, J., Kennedy, D. J., Yashaswi, B., Vetteth, S., Shidyak, A., Kim, E. G.,
Smaili, S., Periyasamy, S. M., Hariri, I. M., Fedorova, L., Liu, J., Wu, L.,
Kahaleh, M. B., Xie, Z., Malhotra, D., Fedorova, O. V., Kashkin, V. A., Bagrov,
A. Y., and Shapiro, J. I. (2007) Hypertension 49, 215-224
2. Kennedy, D., Omran, E., Periyasamy, S. M., Nadoor, J., Priyadarshi, A., Willey,
J. C., Malhotra, D., Xie, Z., and Shapiro, J. I. (2003) J Am Soc Nephrol 14, 90-97
3. Kennedy, D. J., Vetteth, S., Periyasamy, S. M., Kanj, M., Fedorova, L., Khouri,
S., Kahaleh, M. B., Xie, Z., Malhotra, D., Kolodkin, N. I., Lakatta, E. G.,
Fedorova, O. V., Bagrov, A. Y., and Shapiro, J. I. (2006) Hypertension 47, 488-
495
4. Czuwara-Ladykowska, J., Shirasaki, F., Jackers, P., Watson, D. K., and
Trojanowska, M. (2001) J Biol Chem 276, 20839-20848
5. Wang, Y., Fan, P. S., and Kahaleh, B. (2006) Arthritis Rheum 54, 2271-2279
6. Kubo, M., Czuwara-Ladykowska, J., Moussa, O., Markiewicz, M., Smith, E.,
Silver, R. M., Jablonska, S., Blaszczyk, M., Watson, D. K., and Trojanowska, M.
(2003) Am J Pathol 163, 571-581
7. Jinnin, M., Ihn, H., Yamane, K., Mimura, Y., Asano, Y., and Tamaki, K. (2005)
Nucleic Acids Res 33, 1337-1351
8. Kometiani, P., Tian, J., Li, J., Nabih, Z., Gick, G., and Xie, Z. (2000) Mol Cell
Biochem 215, 65-72
109

9. Fedorova, O. V., Lakatta, E. G., and Bagrov, A. Y. (2000) Circulation 102, 3009-
3014
10. Kennedy, D. J., Elkareh, J., Shidyak, A., Shapiro, A. P., Smaili, S., Mutgi, K.,
Gupta, S., Tian, J., Morgan, E., Khouri, S., Cooper, C. J., Periyasamy, S. M., Xie,
Z., Malhotra, D., Fedorova, O. V., Bagrov, A. Y., and Shapiro, J. I. (2008) Am J
Physiol Renal Physiol 294, F450-454
11. Brilla, C. G., Zhou, G., Matsubara, L., and Weber, K. T. (1994) J Mol Cell
Cardiol 26, 809-820
12. Wadman, I. A., Osada, H., Grutz, G. G., Agulnick, A. D., Westphal, H., Forster,
A., and Rabbitts, T. H. (1997) EMBO J 16, 3145-3157
13. Wallenstein, S., Zucker, C. L., and Fleiss, J. L. (1980) Circ Res 47, 1-9
14. Sarnak, M. J., Levey, A. S., Schoolwerth, A. C., Coresh, J., Culleton, B., Hamm,
L. L., McCullough, P. A., Kasiske, B. L., Kelepouris, E., Klag, M. J., Parfrey, P.,
Pfeffer, M., Raij, L., Spinosa, D. J., and Wilson, P. W. (2003) Hypertension 42,
1050-1065
15. Sato, Y. (2001) Cell Struct Funct 26, 19-24
16. Truong, A. H., and Ben-David, Y. (2000) Oncogene 19, 6482-6489
17. Hromas, R., and Klemsz, M. (1994) Int J Hematol 59, 257-265
18. Dhulipal, P. D. (1997) Indian J Exp Biol 35, 315-322
19. Blair, D. G., and Athanasiou, M. (2000) Oncogene 19, 6472-6481
20. Zhang, X. K., and Watson, D. K. (2005) J Biochem 137, 297-302
21. Haas, M., Askari, A., and Xie, Z. (2000) J Biol Chem 275, 27832-27837
110

22. Kometiani, P., Li, J., Gnudi, L., Kahn, B. B., Askari, A., and Xie, Z. (1998) J Biol
Chem 273, 15249-15256
23. Mohammadi, K., Kometiani, P., Xie, Z., and Askari, A. (2001) J Biol Chem 276,
42050-42056
24. Pierre, S. V., and Xie, Z. (2006) Cell Biochem Biophys 46, 303-316
25. Yuan, Z., Cai, T., Tian, J., Ivanov, A. V., Giovannucci, D. R., and Xie, Z. (2005)
Mol Biol Cell 16, 4034-4045
111

Figure Legends
Figure 1. Characterization of cardiac fibrosis in Fli-1 knockdown animals and their
response to partial nephrectomy.
Panel a shows basal Fli-1 expression determined with Western blot
(representative blot at top, quantitative data at bottom) in cardiac tissue obtained from
N=5 wild type (WT) and Fli-1 knockdown (KD) mice. Panel b shows conscious systolic
blood pressure in WT animals exposed to sham (N=6) and partial nephrectomy (PNx,
N=8) surgery as well as KD mice exposed to sham (N=6) and PNx (N=9) surgery
determined at baseline, 1, 2, 3 and 4 weeks. Panel c shows the heart weight normalized
for body weight in these animals at 4 weeks. Panel d shows representative histology
(trichrome stain) and panel e shows the quantification of the fibrosis and panel f shows
the measurement of myocyte cross sectional area from the hearts of these mice. Panel g
shows collagen expression determined with a polyclonal antibody (representative
Western blot with dimer at about 100Kd, quantified data shown below) in these animals.
* p<0.05, ** p<0.01 vs Sham-WT, # p<0.05, ## p< 0.01 vs PNx-WT.
Figure 2. Procollagen and Fli-1 expression in different fibroblasts.
Panel a shows a representative Western blot for procollagen showing dimer
detected with polyclonal antibody at approximately 150Kd and Fli-1 detected with a
polyclonal antibody at approximately 50Kd (both shown at top) and the quantification of
these measurements in each group (N=6) obtained at baseline in rat cardiac and renal and
112

human dermal fibroblasts grown to confluence. Regression line for procollagen
expression compared to Fli-1 expression is shown with data normalized to that of cardiac
fibroblasts (r2 = 0.98, p< 0.01). Panel b shows quantitative densitometric data for
procollagen (measured by Western blot as in panel a) in response to different doses of
MBG for 24 hours (N=6). Panel c shows quantitative densitometric data for Fli-1
(measured by Western blot as in panel b) obtained in response to 10 nM MBG (N=6) for
24 hours. Bars on quantitative graphs represent the mean ± SEM. **P<0.01 vs. control.
Figure 3. Effects of MBG on procollagen and Fli-1 expression in stably transfected
rat renal fibroblasts.
MBG was administered at a 10 nM for 24 hours. Bars on quantitative graphs
represent the mean ± SEM of 6 determinations derived from Western blots as in Figure 1.
*P<0.05 and **P<0.01 vs. control.
Figure 4: Effect of MBG on nuclear PKCδ expression in cardiac fibroblasts.
Panel a shows confocal immunofluorescence micrographs of cardiac fibroblasts at
baseline and treated with 10 nM MBG for 15 min and 24 hrs. Images were also obtained
at 5 and 30 min as well as 1 and 4 hrs but not shown. Panel b shows a nuclear PKCδ
expression determined by Western blot in cells treated with 10 nM (MBG) and/or the
PLC inhibitor, U-73122 (U) at 10 μM for 15 min (N=6 in each group). Panel c shows
procollagen expression determined by Western blot (see figure 1) in cells treated with 10
113

nM MBG with or without the PKCδ inhibitor, Rottlerin 4 μM (N=6 in each group) for 24
hours. * p< 0.05, ** p< 0.01 vs control, # p<0.05, ## p<0.01 vs MBG.
Figure 5: Effect of MBG on phosphorylated Fli-1 in renal fibroblasts.
Panel a shows an immunblot against phosphoserine in nuclear extracts
immunoprecipitated with the polyclonal antibody against Fli-1; the representative blot is
shown above and quantification of N=4 determinations is shown below. Panel b shows
32P autoradiograph data obtained from cells treated with 32P labeled phosphate for 2 hours
and MBG for 15 minutes. The top shows a representative autoradiograph and the bottom
quantification of N=5 experiments. * p<0.05 vs control.
Figure 6: Schematic describing how cardiotonic induced sodium pump signaling
may result in decreases in collagen production.
In the presence of the cardiotonic steroid MBG, Na/K-ATPase is converted to a
signal transducer which complexes with Src and the EGFR. A signal cascade is initiated
which involves PLC which results in the activation of PKCδ and its translocation to the
nucleus. In the nucleus, PKCδ phosphorylates Fli-1. This, in turn, leads to more rapid
catabolism of Fli-1 and removal of Fli-1 inhibition on the Col1 promoter and, thus,
increases in collagen expression. Data supporting this schematic summarized from
references (1, 3, 21-25).
114

Figure 1a
0.0
0.2
0.4
0.6
0.8
1.0
1.2
**
WT KD
Fli-1
Figure 1b
80
100
120
140
160
180
200 Sham -W TSham -KD
PNx-W TPNx-KD
** **
****
** **
##
##
# #
Baseline 1 2 3 4
Time (weeks)
115

Figure 1c
Figure 1d
0
1
2
3
4
5
6
****
*
#
Sham-WT PNx-WT Sham-KD PNx-KD
Fli-1 KD
Sham
WT
PNx
116

Figure 1e
Figure 1f
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
Sham-WT PNx-WT Sham-KD PNx-KD
**
**
**#
0
500
1000
1500
2000
2500
3000 ** **
Sham-WT PNx-WT Sham-KD PNx-KD
117

Figure 1g
0
1
2
3
4
Sham-WT PNx-WT Sham-KD PNx-KD
****
**##
Procollagen
Figure 2a
F l i - 1 E x p r e s s io n
( R e la t i v e t o C a r d ia c )
0 1 2 3 4
Proc
olla
gen
Exp
ress
ion
(Rel
ativ
e to
Car
diac
)
Procollagen
Fli-1
Cardiac Renal Dermal
0 . 0
0 . 2
0 . 4
0 . 6
0 . 8
1 . 0
1 . 2
118
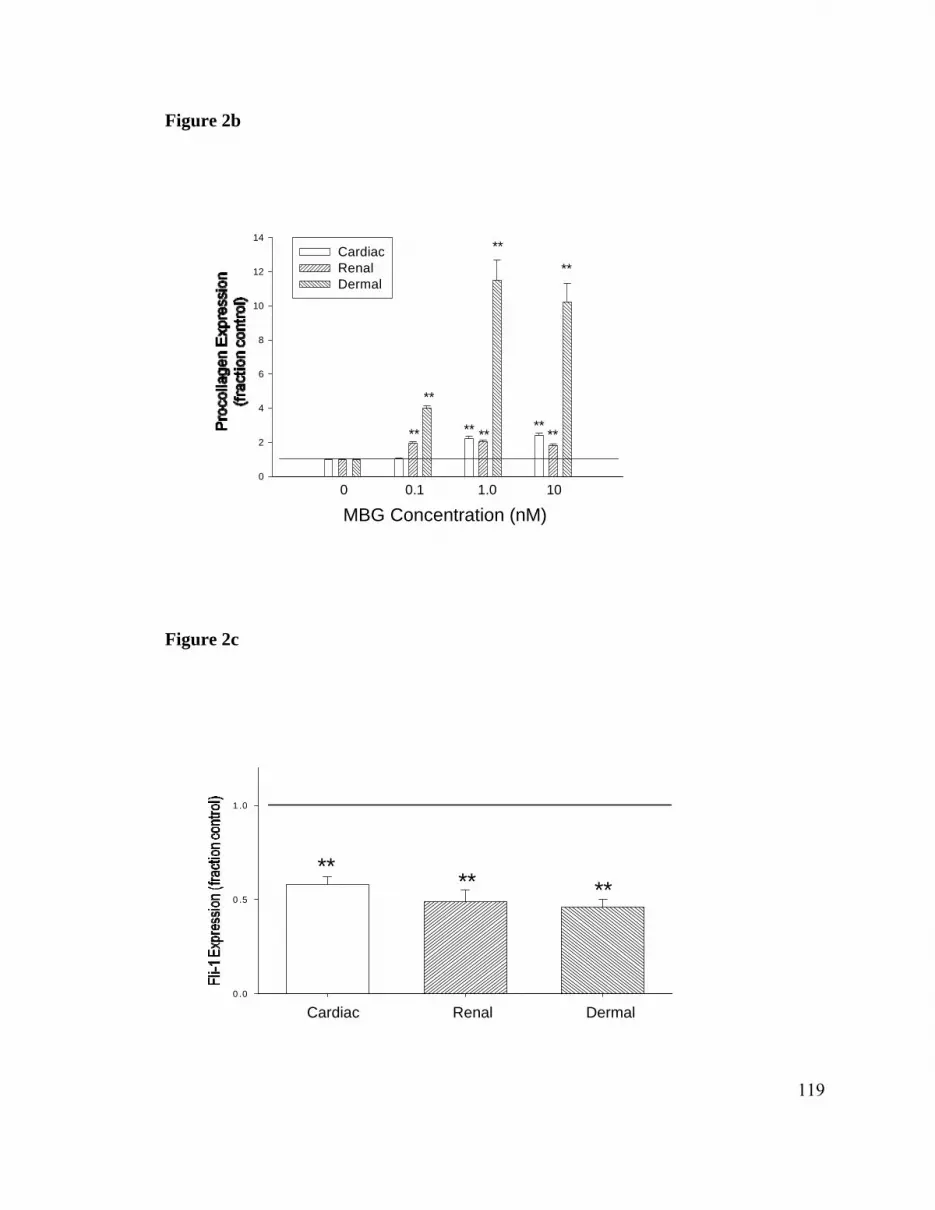
Figure 2b
0
2
4
6
8
10
12
14CardiacRenalDermal
MBG Concentration (nM)0 0.1 1.0 10
**
**
** **
**
****
**
Figure 2c
0 .0
0 .5
1 .0
** ** **
Cardiac Renal Dermal
119

Figure 3
0
1
2
3
4
P ro c o lla g e nF li-1
Control MBG Control MBG Normal Cells Transfected Cells
** ** **
* ** **
Figure 4a
Baseline MBG 15 min MBG 24 hr
120

Figure 4b
0
1
2
Control MBG U MBG+U
**
**##
**
PKCδ
Figure 4c
0
1
2
3
Control MBG R MBG+R
**
**##
**
121

Figure 5a
0
1
2
Control MBG
*
50 KD
Figure 5b
0
1
2
3
4
Control MBG
*
50 KD
122

Figure 6
Src
FibrosisEGFR
PP
P PP
PCaveolin
P PLC
MBG
Procollagen 1
PKCδ
PKCδ
Proteolysis
Fli-1
Col1 promoter
Fli-1 Procollagen 1 mRNA
123

SUMMARY
Usually the term uremic cardiomyopathy refers to patients with renal failure who develop
diastolic dysfunction , oxidant stress, left ventricular hypertrophy and cardiac fibrosis
which are most probably related to their renal disease. Increased circulation of
cardiotonic steroids such as ouabain and marinobufagenin are in part responsible for the
cardiac fibrosis seen in uremic cardiomyopathy which has been demonstrated both in
clinical and experimental renal failure.(9) It is interesting to note that collagen
production, a marker of fibrosis, is regulated by the transcription factor Fli-1 which is
regulated by the protein kinase PKCδ in human dermal fibroblasts.(8, 25) Cardiotonic
steroids might play a major role in the cardiac fibrosis seen in uremic cardiomyopathy
and the molecular mechanism of fibrosis might involve PKCδ and Fli-1.
We have recently seen that experimental renal failure in the rat is accompanied by
increases in circulating concentrations of the cardiotonic steroid, marinobufagenin
(MBG), and substantial cardiac fibrosis. In the current study, we investigated the
implication of MBG in the pathogenesis of the cardiac fibrosis. First we noticed that the
5/6th nephrectomy model of experimental renal failure PNx and MBG infusion induced
similar but not identical phenotypic changes in hemodynamics and cardiac morphology.
On the other hand, both immunization against MBG; PNx-IM and the concomitant PNx
and adrenalectomy; PNx-ADx, reduced circulating MBG and substantially attenuate the
124

cardiac functional and morphological changes without significantly affecting blood
pressure. Second, we found that MBG like his close relative ouabain induced a signal
transduction cascade through the plasmalemmal Na/KATPase residing in caveolae, which
results in activation of Src, transactivation of the EGFR, generation of reactive oxygen
species, and, ultimately, activation of p42/44 mitogen-activated protein kinase. Ergo, we
used specific antagonists for Src, EGFR and ROS and showed that these compounds
blocked signal transduction through the Na/K-ATPase and therefore inhibited the fibrosis
induced by MBG assessed by increases in procollagen-1 expression by cultured cardiac
fibroblasts. Finally, in an attempt to determine the molecular mechanism underlying this
fibrosis, we examined the expression of several proteins important in fibroblast activation
such as TGF-β and the Smad proteins. Interestingly, evidence for increases in these
proteins was not evident. We should point out that these results do not exclude a role for
TGF-β in this process, which could have occurred at an earlier or later time of the fibrosis
progression. There is a probability that other mechanisms of regulation of collagen
synthesis could take place when the TGF-β pathway is interrupted.
Next, we pursued further studies of the molecular mechanisms utilizing knockout and
“knockin” models that have already been established in the mouse but not yet in the rat
model. Because of this we performed a partial nephrectomy in the mouse and checked
whether the phenotypical features of uremic cardiomyopathy seen in humans and rats
were also present in the murine model. Interestingly, CD1 mouse responded quite
similarly to PNx as we previously reported with Sprague- Dawley rats. Particularly, we
125

demonstrated higher MBG levels in plasma and increases in blood pressure, left
ventricular hypertrophy, and cardiac fibrosis following PNx in male CD1 mice. Although
Doppler imaging tracing and flow Doppler assessments were not as sensitive as the
Millar pressure catheter to changes in left ventricular relaxation, we were able to note
evidence for impaired in active and passive relaxation evaluated by ventricular
hemodynamics measurements. We should point out that the cardiac changes seen in the
murine model seemed to be more dependent on the uremic milieu rather than elevations
in blood pressure alone. In fact, we showed that a “triple” antihypertensive therapy
considerably lowered blood pressure toward normal, but it did not attenuate the cardiac
hypertrophy or fibrosis induced by PNx in the male CD1 mice. Therefore, we
demonstrated that the CD1 mouse responded quite similarly to PNx as we previously
reported with Sprague- Dawley rats. In our next studies we decided to use the mouse
model into the pathogenesis of uremic cardiomyopathy. Specifically, we will use a knock
down murine model.
Fli-1 is a transcription factor that has clearly been shown to play a role in dermal fibrosis,
because of its inhibitory effect on collagen-1 synthesis. In the current study, we
demonstrated an inverse relationship between basal nuclear Fli-1 expression and collagen
production in cardiac and renal fibroblasts to go along with what was seen in dermal
fibroblasts. Moreover, MBG induced corresponding decreases in nuclear Fli-1 expression
with increases in procollagen expression in each of these fibroblasts. Next, we transfected
renal fibroblasts with the Fli-1 gene; these cells showed higher basal levels of nuclear Fli-
126

1 expression and lower basal levels of procollagen expression and did not show any
significant increase in procollagen expression in response to MBG. In support to the in
vitro results, we look at the significance to cardiac fibrosis using a Fli-1 knockdown
model. As expected, the heterozygous mice had greater degrees of cardiac fibrosis and
cardiac collagen expression compared to wild type mice. This observable fact was more
pronounced after 4 weeks of experimental renal failure induced by 5/6th nephrectomy in
both heterozygotes and wild type. We turned back to our in vitro experiments, to look at
the molecular mechanism behind this relationship. We noticed that exposure of cardiac
fibroblasts to MBG, for about 15 minutes, was associated with a rapid translocation to the
nucleus of a protein kinase; PKCδ. MBG induced increases in procollagen expression
could be prevented by rottlerin, a PKCδ specific inhibitor, or by U-73122 a
pharmacological inhibition of phospholipase C. We finally showed that
PKCδ stimulation by MBG phosphorylated Fli-1 on its serine residue and thus increased
collagen synthesis.
Taken together our data suggest the following pathway for cardiotonic steroids in the
regulation of cardiac fibrosis seen in patients with uremic cardiomyopathy: MBG
stimulates activation and translocation of the protein kinase PKCδ which phosphorylates
and downregulates the transcription factor Fli-1 which will lead to increase collagen
synthesis. Should these findings be well established, we speculate that this pathway may
represent a therapeutic target for uremic cardiomyopathy and other fibrotic disorders.
127

CONCLUSIONS
1. Experimental renal failure in rats and mice is accompanied by increases in
circulating concentrations of cardiotonic steroids such as marinobufagenin
(MBG). This phenomenon is also observed in patients with renal diseases.
2. MBG like other cardiotonic steroids induces a signal transduction cascade through
the plasmalemmal Na/KATPase residing in caveolae, which results in activation
of Src, transactivation of the epidermal growth factor receptor, generation of
reactive oxygen species, and activation of protein kinases.
3. In rat undergoing 5/6th nephrectomy (PNx) for 4 weeks, both immunization
against MBG, and the concomitant PNx and adrenalectomy reduced circulating
MBG and substantially attenuate the cardiac functional and morphological
changes without significantly affecting blood pressure.
4. The same phenotypical features of uremic cardiomyopathy seen in humans and
rats were also present in the murine model undertaking partial nephrectomy. The
cardiac changes seen in the mice appear to depend on the uremic milieu rather
than elevations in blood pressure alone.
128

5. Fli-1 knockdown mice had greater degrees of cardiac fibrosis and cardiac
collagen expression compared to wild type mice before and after partial
nephrectomy.
6. Renal fibroblasts transfected with the Fli-1 gene showed higher basal levels of
nuclear Fli-1 expression and lower basal levels of procollagen expression with no
significant increase in procollagen expression in response to MBG.
7. MBG stimulates activation and translocation of the protein kinase PKCδ which
phosphorylates and downregulates the transcription factor Fli-1 which will lead to
increase collagen synthesis.
129

REFERENCES
1. Elkareh, J., Kennedy, D.J., Yashaswi, B., Vetteth, S., Shidyak, A., Kim, E.G.,
Smaili, S., Periyasamy, S.M., Hariri, I.M., Fedorova, L., et al. 2007.
Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in
experimental uremic cardiomyopathy. Hypertension 49:215-224.
2. Czuwara-Ladykowska, J., Shirasaki, F., Jackers, P., Watson, D.K., and
Trojanowska, M. 2001. Fli-1 inhibits collagen type I production in dermal
fibroblasts via an Sp1-dependent pathway. J Biol Chem 276:20839-20848.
3. Wakatsuki, T., Schlessinger, J., and Elson, E.L. 2004. The biochemical response
of the heart to hypertension and exercise. Trends Biochem Sci 29:609-617.
4. Remme, W.J. 2003. Pharmacological modulation of cardiovascular remodeling: a
guide to heart failure therapy. Cardiovasc Drugs Ther 17:349-360.
5. McGuane, J.T., and Parry, L.J. 2005. Relaxin and the extracellular matrix:
molecular mechanisms of action and implications for cardiovascular disease.
Expert Rev Mol Med 7:1-18.
6. Orlandi, A., Francesconi, A., Marcellini, M., Ferlosio, A., and Spagnoli, L.G.
2004. Role of ageing and coronary atherosclerosis in the development of cardiac
fibrosis in the rabbit. Cardiovasc Res 64:544-552.
7. Funder, J. 2001. Mineralocorticoids and cardiac fibrosis: the decade in review.
Clin Exp Pharmacol Physiol 28:1002-1006.
130

8. Jinnin, M., Ihn, H., Yamane, K., Mimura, Y., Asano, Y., and Tamaki, K. 2005.
Alpha2(I) collagen gene regulation by protein kinase C signaling in human
dermal fibroblasts. Nucleic Acids Res 33:1337-1351.
9. Kennedy, D.J., Vetteth, S., Periyasamy, S.M., Kanj, M., Fedorova, L., Khouri, S.,
Kahaleh, M.B., Xie, Z., Malhotra, D., Kolodkin, N.I., et al. 2006. Central role for
the cardiotonic steroid marinobufagenin in the pathogenesis of experimental
uremic cardiomyopathy. Hypertension 47:488-495.
10. Haas, M., Askari, A., and Xie, Z. 2000. Involvement of Src and epidermal growth
factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol
Chem 275:27832-27837.
11. Haas, M., Wang, H., Tian, J., and Xie, Z. 2002. Src-mediated inter-receptor cross-
talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays
the signal from ouabain to mitogen-activated protein kinases. J Biol Chem
277:18694-18702.
12. Huang, L., Li, H., and Xie, Z. 1997. Ouabain-induced hypertrophy in cultured
cardiac myocytes is accompanied by changes in expression of several late
response genes. J Mol Cell Cardiol 29:429-437.
13. Liu, J., Periyasamy, S.M., Gunning, W., Fedorova, O.V., Bagrov, A.Y., Malhotra,
D., Xie, Z., and Shapiro, J.I. 2002. Effects of cardiac glycosides on sodium pump
expression and function in LLC-PK1 and MDCK cells. Kidney Int 62:2118-2125.
131

14. Tian, J., Gong, X., and Xie, Z. 2001. Signal-transducing function of Na+-K+-
ATPase is essential for ouabain's effect on [Ca2+]i in rat cardiac myocytes. Am J
Physiol Heart Circ Physiol 281:H1899-1907.
15. Xie, Z., Kometiani, P., Liu, J., Li, J., Shapiro, J.I., and Askari, A. 1999.
Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to
hypertrophy and its marker genes in cardiac myocytes. J Biol Chem 274:19323-
19328.
16. Xie, Z., and Askari, A. 2002. Na(+)/K(+)-ATPase as a signal transducer. Eur J
Biochem 269:2434-2439.
17. Liu, L., Mohammadi, K., Aynafshar, B., Wang, H., Li, D., Liu, J., Ivanov, A.V.,
Xie, Z., and Askari, A. 2003. Role of caveolae in signal-transducing function of
cardiac Na+/K+-ATPase. Am J Physiol Cell Physiol 284:C1550-1560.
18. Tian, J., Liu, J., Garlid, K.D., Shapiro, J.I., and Xie, Z. 2003. Involvement of
mitogen-activated protein kinases and reactive oxygen species in the inotropic
action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP)
channels. Mol Cell Biochem 242:181-187.
19. Tian, J., Cai, T., Yuan, Z., Wang, H., Liu, L., Haas, M., Maksimova, E., Huang,
X.Y., and Xie, Z.J. 2006. Binding of Src to Na+/K+-ATPase forms a functional
signaling complex. Mol Biol Cell 17:317-326.
20. Kometiani, P., Li, J., Gnudi, L., Kahn, B.B., Askari, A., and Xie, Z. 1998.
Multiple signal transduction pathways link Na+/K+-ATPase to growth-related
132

genes in cardiac myocytes. The roles of Ras and mitogen-activated protein
kinases. J Biol Chem 273:15249-15256.
21. Oweis, S., Wu, L., Kiela, P.R., Zhao, H., Malhotra, D., Ghishan, F.K., Xie, Z.,
Shapiro, J.I., and Liu, J. 2006. Cardiac glycoside downregulates NHE3 activity
and expression in LLC-PK1 cells. Am J Physiol Renal Physiol 290:F997-1008.
22. Periyasamy, S.M., Liu, J., Tanta, F., Kabak, B., Wakefield, B., Malhotra, D.,
Kennedy, D.J., Nadoor, A., Fedorova, O.V., Gunning, W., et al. 2005. Salt
loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal
tubule cells. Kidney Int 67:1868-1877.
23. Xie, Z., and Cai, T. 2003. Na+-K+--ATPase-mediated signal transduction: from
protein interaction to cellular function. Mol Interv 3:157-168.
24. Liu, J., Kesiry, R., Periyasamy, S.M., Malhotra, D., Xie, Z., and Shapiro, J.I.
2004. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1
cells by a clathrin-dependent mechanism. Kidney Int 66:227-241.
25. Jimenez, S.A., Gaidarova, S., Saitta, B., Sandorfi, N., Herrich, D.J., Rosenbloom,
J.C., Kucich, U., Abrams, W.R., and Rosenbloom, J. 2001. Role of protein kinase
C-delta in the regulation of collagen gene expression in scleroderma fibroblasts. J
Clin Invest 108:1395-1403.
26. Weiner, D.E., Tabatabai, S., Tighiouart, H., Elsayed, E., Bansal, N., Griffith, J.,
Salem, D.N., Levey, A.S., and Sarnak, M.J. 2006. Cardiovascular outcomes and
all-cause mortality: exploring the interaction between CKD and cardiovascular
disease. Am J Kidney Dis 48:392-401.
133

27. Elsayed, E.F., Tighiouart, H., Griffith, J., Kurth, T., Levey, A.S., Salem, D.,
Sarnak, M.J., and Weiner, D.E. 2007. Cardiovascular disease and subsequent
kidney disease. Arch Intern Med 167:1130-1136.
28. Anderson, R.N. 2002. Deaths: leading causes for 2000. Natl Vital Stat Rep 50:1-
85.
29. Schrier, R.W. 2006. Role of diminished renal function in cardiovascular
mortality: marker or pathogenetic factor? J Am Coll Cardiol 47:1-8.
30. Sarnak, M.J., Levey, A.S., Schoolwerth, A.C., Coresh, J., Culleton, B., Hamm,
L.L., McCullough, P.A., Kasiske, B.L., Kelepouris, E., Klag, M.J., et al. 2003.
Kidney disease as a risk factor for development of cardiovascular disease: a
statement from the American Heart Association Councils on Kidney in
Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and
Epidemiology and Prevention. Hypertension 42:1050-1065.
31. Weiner, D.E., Tighiouart, H., Stark, P.C., Amin, M.G., MacLeod, B., Griffith,
J.L., Salem, D.N., Levey, A.S., and Sarnak, M.J. 2004. Kidney disease as a risk
factor for recurrent cardiovascular disease and mortality. Am J Kidney Dis
44:198-206.
32. Selby, N.M., and McIntyre, C.W. 2007. The acute cardiac effects of dialysis.
Semin Dial 20:220-228.
33. Ragosta, M., Samady, H., Isaacs, R.B., Gimple, L.W., Sarembock, I.J., and
Powers, E.R. 2004. Coronary flow reserve abnormalities in patients with diabetes
134

mellitus who have end-stage renal disease and normal epicardial coronary
arteries. Am Heart J 147:1017-1023.
34. McIntyre, C.W., Burton, J.O., Selby, N.M., Leccisotti, L., Korsheed, S., Baker,
C.S., and Camici, P.G. 2008. Hemodialysis-induced cardiac dysfunction is
associated with an acute reduction in global and segmental myocardial blood
flow. Clin J Am Soc Nephrol 3:19-26.
35. Foley, R.N., Parfrey, P.S., Kent, G.M., Harnett, J.D., Murray, D.C., and Barre,
P.E. 2000. Serial change in echocardiographic parameters and cardiac failure in
end-stage renal disease. J Am Soc Nephrol 11:912-916.
36. Foley, R.N., Parfrey, P.S., Harnett, J.D., Kent, G.M., Martin, C.J., Murray, D.C.,
and Barre, P.E. 1995. Clinical and echocardiographic disease in patients starting
end-stage renal disease therapy. Kidney Int 47:186-192.
37. Stack, A.G., and Bloembergen, W.E. 2001. A cross-sectional study of the
prevalence and clinical correlates of congestive heart failure among incident US
dialysis patients. Am J Kidney Dis 38:992-1000.
38. Mitsnefes, M.M., Daniels, S.R., Schwartz, S.M., Khoury, P., and Strife, C.F.
2001. Changes in left ventricular mass in children and adolescents during chronic
dialysis. Pediatr Nephrol 16:318-323.
39. Besarab, A., and Aslam, M. 2001. Should the hematocrit (hemoglobin) be
normalized in Pre-ESRD or dialysis patients? Yes! Blood Purif 19:168-174.
40. Amann, K., and Ritz, E. 1997. Cardiac disease in chronic uremia:
pathophysiology. Adv Ren Replace Ther 4:212-224.
135

41. Mohmand, B., Malhotra, D.K., and Shapiro, J.I. 2005. Uremic cardiomyopathy:
role of circulating digitalis like substances. Front Biosci 10:2036-2044.
42. Goulet, S., Bihl, M.P., Gambazzi, F., Tamm, M., and Roth, M. 2007. Opposite
effect of corticosteroids and long-acting beta(2)-agonists on serum- and TGF-
beta(1)-induced extracellular matrix deposition by primary human lung
fibroblasts. J Cell Physiol 210:167-176.
43. Horstmeyer, A., Licht, C., Scherr, G., Eckes, B., and Krieg, T. 2005. Signalling
and regulation of collagen I synthesis by ET-1 and TGF-beta1. Febs J 272:6297-
6309.
44. Pardo, A., and Selman, M. 2006. Matrix metalloproteases in aberrant fibrotic
tissue remodeling. Proc Am Thorac Soc 3:383-388.
45. Berk, B.C., Fujiwara, K., and Lehoux, S. 2007. ECM remodeling in hypertensive
heart disease. J Clin Invest 117:568-575.
46. Wynn, T.A. 2007. Common and unique mechanisms regulate fibrosis in various
fibroproliferative diseases. J Clin Invest 117:524-529.
47. Quan, T.E., Cowper, S.E., and Bucala, R. 2006. The role of circulating fibrocytes
in fibrosis. Curr Rheumatol Rep 8:145-150.
48. Verrecchia, F., and Mauviel, A. 2002. Transforming growth factor-beta signaling
through the Smad pathway: role in extracellular matrix gene expression and
regulation. J Invest Dermatol 118:211-215.
136

49. Schiller, M., Javelaud, D., and Mauviel, A. 2004. TGF-beta-induced SMAD
signaling and gene regulation: consequences for extracellular matrix remodeling
and wound healing. J Dermatol Sci 35:83-92.
50. Bartram, U., and Speer, C.P. 2004. The role of transforming growth factor beta in
lung development and disease. Chest 125:754-765.
51. Javelaud, D., and Mauviel, A. 2004. [Transforming growth factor-betas: smad
signaling and roles in physiopathology]. Pathol Biol (Paris) 52:50-54.
52. Roberts, A.B., Russo, A., Felici, A., and Flanders, K.C. 2003. Smad3: a key
player in pathogenetic mechanisms dependent on TGF-beta. Ann N Y Acad Sci
995:1-10.
53. Schnaper, H.W., Hayashida, T., Hubchak, S.C., and Poncelet, A.C. 2003. TGF-
beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal
Physiol 284:F243-252.
54. Tabibzadeh, S. 2002. Homeostasis of extracellular matrix by TGF-beta and lefty.
Front Biosci 7:d1231-1246.
55. Ihn, H. 2002. The role of TGF-beta signaling in the pathogenesis of fibrosis in
scleroderma. Arch Immunol Ther Exp (Warsz) 50:325-331.
56. Verrecchia, F., and Mauviel, A. 2002. Control of connective tissue gene
expression by TGF beta: role of Smad proteins in fibrosis. Curr Rheumatol Rep
4:143-149.
57. Hill, C.S. 1999. The Smads. Int J Biochem Cell Biol 31:1249-1254.
137

58. Gressner, A.M., Weiskirchen, R., Breitkopf, K., and Dooley, S. 2002. Roles of
TGF-beta in hepatic fibrosis. Front Biosci 7:d793-807.
59. Zhang, Y., and Derynck, R. 2000. Transcriptional regulation of the transforming
growth factor-beta -inducible mouse germ line Ig alpha constant region gene by
functional cooperation of Smad, CREB, and AML family members. J Biol Chem
275:16979-16985.
60. Huang, Y., Wongamorntham, S., Kasting, J., McQuillan, D., Owens, R.T., Yu, L.,
Noble, N.A., and Border, W. 2006. Renin increases mesangial cell transforming
growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin
II-independent mechanisms. Kidney Int 69:105-113.
61. Truong, A.H., and Ben-David, Y. 2000. The role of Fli-1 in normal cell function
and malignant transformation. Oncogene 19:6482-6489.
62. Asano, Y., Czuwara, J., and Trojanowska, M. 2007. Transforming growth factor-
beta regulates DNA binding activity of transcription factor Fli1 by p300/CREB-
binding protein-associated factor-dependent acetylation. J Biol Chem 282:34672-
34683.
63. Ben-David, Y., and Bernstein, A. 1991. Friend virus-induced erythroleukemia and
the multistage nature of cancer. Cell 66:831-834.
64. Nakerakanti, S.S., Kapanadze, B., Yamasaki, M., Markiewicz, M., and
Trojanowska, M. 2006. Fli1 and Ets1 have distinct roles in connective tissue
growth factor/CCN2 gene regulation and induction of the profibrotic gene
program. J Biol Chem 281:25259-25269.
138

65. Prasad, D.D., Rao, V.N., and Reddy, E.S. 1992. Structure and expression of
human Fli-1 gene. Cancer Res 52:5833-5837.
66. Watson, D.K., Smyth, F.E., Thompson, D.M., Cheng, J.Q., Testa, J.R., Papas,
T.S., and Seth, A. 1992. The ERGB/Fli-1 gene: isolation and characterization of a
new member of the family of human ETS transcription factors. Cell Growth
Differ 3:705-713.
67. Sarrazin, S., Starck, J., Gonnet, C., Doubeikovski, A., Melet, F., and Morle, F.
2000. Negative and translation termination-dependent positive control of FLI-1
protein synthesis by conserved overlapping 5' upstream open reading frames in
Fli-1 mRNA. Mol Cell Biol 20:2959-2969.
68. Kubo, M., Czuwara-Ladykowska, J., Moussa, O., Markiewicz, M., Smith, E.,
Silver, R.M., Jablonska, S., Blaszczyk, M., Watson, D.K., and Trojanowska, M.
2003. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in
fibrotic scleroderma skin. Am J Pathol 163:571-581.
69. Wang, Y., Fan, P.S., and Kahaleh, B. 2006. Association between enhanced type I
collagen expression and epigenetic repression of the FLI1 gene in scleroderma
fibroblasts. Arthritis Rheum 54:2271-2279.
70. Kiernan, R., Bres, V., Ng, R.W., Coudart, M.P., El Messaoudi, S., Sardet, C., Jin,
D.Y., Emiliani, S., and Benkirane, M. 2003. Post-activation turn-off of NF-kappa
B-dependent transcription is regulated by acetylation of p65. J Biol Chem
278:2758-2766.
139

71. Dekker, L.V., McIntyre, P., and Parker, P.J. 1993. Altered substrate selectivity of
PKC-eta pseudosubstrate site mutants. FEBS Lett 329:129-133.
72. Klein, G., Schaefer, A., Hilfiker-Kleiner, D., Oppermann, D., Shukla, P., Quint,
A., Podewski, E., Hilfiker, A., Schroder, F., Leitges, M., et al. 2005. Increased
collagen deposition and diastolic dysfunction but preserved myocardial
hypertrophy after pressure overload in mice lacking PKCepsilon. Circ Res
96:748-755.
73. Dempsey, E.C., Newton, A.C., Mochly-Rosen, D., Fields, A.P., Reyland, M.E.,
Insel, P.A., and Messing, R.O. 2000. Protein kinase C isozymes and the regulation
of diverse cell responses. Am J Physiol Lung Cell Mol Physiol 279:L429-438.
74. Davis, R.J., Ganong, B.R., Bell, R.M., and Czech, M.P. 1985. sn-1,2-
Dioctanoylglycerol. A cell-permeable diacylglycerol that mimics phorbol diester
action on the epidermal growth factor receptor and mitogenesis. J Biol Chem
260:1562-1566.
75. Crotty, T., Cai, J., Sakane, F., Taketomi, A., Prescott, S.M., and Topham, M.K.
2006. Diacylglycerol kinase delta regulates protein kinase C and epidermal
growth factor receptor signaling. Proc Natl Acad Sci U S A 103:15485-15490.
76. Jimenez, R., Zarzuelo, A., Galisteo, M., and Duarte, J. 2002. Involvement of
protein kinase C and Na+/K+-ATPase in the contractile response induced by
myricetin in rat isolated aorta. Planta Med 68:133-137.
77. Skou, J.C. 1957. The influence of some cations on an adenosine triphosphatase
from peripheral nerves. Biochim Biophys Acta 23:394-401.
140

78. Blanco, G. 2005. The NA/K-ATPase and its isozymes: what we have learned
using the baculovirus expression system. Front Biosci 10:2397-2411.
79. Hebert, H., Purhonen, P., Vorum, H., Thomsen, K., and Maunsbach, A.B. 2001.
Three-dimensional structure of renal Na,K-ATPase from cryo-electron
microscopy of two-dimensional crystals. J Mol Biol 314:479-494.
80. Pierre, S.V., and Xie, Z. 2006. The Na,K-ATPase receptor complex: its
organization and membership. Cell Biochem Biophys 46:303-316.
81. Lingrel, J.B., Van Huysse, J., O'Brien, W., Jewell-Motz, E., and Schultheis, P.
1994. Na,K-ATPase: structure-function studies. Ren Physiol Biochem 17:198-
200.
82. Lingrel, J.B., Van Huysse, J., O'Brien, W., Jewell-Motz, E., Askew, R., and
Schultheis, P. 1994. Structure-function studies of the Na,K-ATPase. Kidney Int
Suppl 44:S32-39.
83. Sweadner, K.J. 1989. Isozymes of the Na+/K+-ATPase. Biochim Biophys Acta
988:185-220.
84. Lingrel, J.B., Young, R.M., and Shull, M.M. 1988. Multiple forms of the Na,K-
ATPase: their genes and tissue specific expression. Prog Clin Biol Res 268B:105-
112.
85. Lingrel, J.B., Orlowski, J., Shull, M.M., and Price, E.M. 1990. Molecular genetics
of Na,K-ATPase. Prog Nucleic Acid Res Mol Biol 38:37-89.
86. Liang, M., Tian, J., Liu, L., Pierre, S., Liu, J., Shapiro, J., and Xie, Z.J. 2007.
Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem.
141

87. Schwartz, A., Grupp, G., Wallick, E., Grupp, I.L., and Ball, W.J., Jr. 1988. Role
of the Na+K+-ATPase in the cardiotonic action of cardiac glycosides. Prog Clin
Biol Res 268B:321-338.
88. Blaustein, M.P. 1984. Sodium transport and hypertension. Where are we going?
Hypertension 6:445-453.
89. Blaustein, M.P. 1993. Physiological effects of endogenous ouabain: control of
intracellular Ca2+ stores and cell responsiveness. Am J Physiol 264:C1367-1387.
90. Allen, J., Forman, A., Maigaard, S., Jespersen, L.T., and Andersson, K.E. 1989.
Effect of endogenous vasoconstrictors on maternal intramyometrial and fetal stem
villous arteries in pre-eclampsia. J Hypertens 7:529-536.
91. Allen, J.C. 1982. Regulation of cytoplasmic Ca++ by smooth muscle membranes.
Proc West Pharmacol Soc 25:335-337.
92. Aizman, O., Uhlen, P., Lal, M., Brismar, H., and Aperia, A. 2001. Ouabain, a
steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U
S A 98:13420-13424.
93. Aydemir-Koksoy, A., and Allen, J.C. 2001. Low concentrations of ouabain
induce vascular smooth muscle cell proliferation. Cell Mol Biol (Noisy-le-grand)
47:341-345.
94. Belusa, R., Aizman, O., Andersson, R.M., and Aperia, A. 2002. Changes in
Na(+)-K(+)-ATPase activity influence cell attachment to fibronectin. Am J
Physiol Cell Physiol 282:C302-309.
142

95. Bereta, J., Cohen, M.C., and Bereta, M. 1995. Stimulatory effect of ouabain on
VCAM-1 and iNOS expression in murine endothelial cells: involvement of NF-
kappa B. FEBS Lett 377:21-25.
96. Liu, J., Liang, M., Liu, L., Malhotra, D., Xie, Z., and Shapiro, J.I. 2005. Ouabain-
induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells
requires caveolin-1. Kidney Int 67:1844-1854.
97. Mohammadi, K., Kometiani, P., Xie, Z., and Askari, A. 2001. Role of protein
kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J Biol
Chem 276:42050-42056.
98. Wang, H., Haas, M., Liang, M., Cai, T., Tian, J., Li, S., and Xie, Z. 2004.
Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J
Biol Chem 279:17250-17259.
99. Bertorello, A., and Aperia, A. 1989. Regulation of Na+-K+-ATPase activity in
kidney proximal tubules: involvement of GTP binding proteins. Am J Physiol
256:F57-62.
100. Khundmiri, S.J., Bertorello, A.M., Delamere, N.A., and Lederer, E.D. 2004.
Clathrin-mediated endocytosis of Na+,K+-ATPase in response to parathyroid
hormone requires ERK-dependent phosphorylation of Ser-11 within the alpha1-
subunit. J Biol Chem 279:17418-17427.
101. Chibalin, A.V., Ogimoto, G., Pedemonte, C.H., Pressley, T.A., Katz, A.I.,
Feraille, E., Berggren, P.O., and Bertorello, A.M. 1999. Dopamine-induced
endocytosis of Na+,K+-ATPase is initiated by phosphorylation of Ser-18 in the
143

rat alpha subunit and Is responsible for the decreased activity in epithelial cells. J
Biol Chem 274:1920-1927.
102. Done, S.C., Leibiger, I.B., Efendiev, R., Katz, A.I., Leibiger, B., Berggren, P.O.,
Pedemonte, C.H., and Bertorello, A.M. 2002. Tyrosine 537 within the Na+,K+-
ATPase alpha-subunit is essential for AP-2 binding and clathrin-dependent
endocytosis. J Biol Chem 277:17108-17111.
103. Schoner, W. 2002. Endogenous cardiac glycosides, a new class of steroid
hormones. Eur J Biochem 269:2440-2448.
104. de Wardener, H.E. 1996. Franz Volhard Lecture 1996. Sodium transport
inhibitors and hypertension. J Hypertens Suppl 14:S9-18.
105. Bricker, N.S. 1967. The control of sodium excretion with normal and reduced
nephron populations. The pre-eminence of third factor. Am J Med 43:313-321.
106. Graves, S.W. 1986. Endogenous digitalis-like factors. Crit Rev Clin Lab Sci
23:177-200.
107. Graves, S.W., and Williams, G.H. 1987. Endogenous digitalis-like natriuretic
factors. Annu Rev Med 38:433-444.
108. Kawamura, A., Guo, J., Itagaki, Y., Bell, C., Wang, Y., Haupert, G.T., Jr., Magil,
S., Gallagher, R.T., Berova, N., and Nakanishi, K. 1999. On the structure of
endogenous ouabain. Proc Natl Acad Sci U S A 96:6654-6659.
109. Bagrov, A.Y., and Fedorova, O.V. 2005. Cardenolide and bufadienolide ligands
of the sodium pump. How they work together in NaCl sensitive hypertension.
Front Biosci 10:2250-2256.
144

110. Flier, J., Edwards, M.W., Daly, J.W., and Myers, C.W. 1980. Widespread
occurrence in frogs and toads of skin compounds interacting with the ouabain site
of Na+, K+-ATPase. Science 208:503-505.
111. Bagrov, A.Y., Fedorova, O.V., Dmitrieva, R.I., Howald, W.N., Hunter, A.P.,
Kuznetsova, E.A., and Shpen, V.M. 1998. Characterization of a urinary
bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial
infarction. Hypertension 31:1097-1103.
112. Priyadarshi, S., Valentine, B., Han, C., Fedorova, O.V., Bagrov, A.Y., Liu, J.,
Periyasamy, S.M., Kennedy, D., Malhotra, D., Xie, Z., et al. 2003. Effect of green
tea extract on cardiac hypertrophy following 5/6 nephrectomy in the rat. Kidney
Int 63:1785-1790.
113. Anderson, D.E., Fedorova, O.V., Morrell, C.H., Longo, D.L., Kashkin, V.A.,
Metzler, J.D., Bagrov, A.Y., and Lakatta, E.G. 2008. Endogenous sodium pump
inhibitors and age-associated increases in salt sensitivity of blood pressure in
normotensives. Am J Physiol Regul Integr Comp Physiol 294:R1248-1254.
114. Kennedy, DJ. 2005. Cardiovascular Complications of Ischemic Renal Disease:
The Effect of Renal Dysfunction on Cardiac Disease and the Central Role of
Cardiotonic Steroids in the Pathogenesis of Uremic Cardiomyopathy. PhD
Dissertation, The University of Toledo.
145

ABSTRACT
It has been recognized that patients with chronic renal failure eventually develop diastolic
dysfunction, cardiac hypertrophy and systemic oxidant stress along with increases in
circulating concentrations of the cardiotonic steroid, marinobufagenin (MBG) in uremic
cardiomyopathy.
Because of this, we performed 5/6th partial nephrectomy in rats to study experimental
renal failure (PNx), MBG infusion, PNx after immunization against MBG, and
concomitant PNx and adrenalectomy. We also studied 5 6th partial nephrectomy as a
potential experimental renal failure model in mice. Next, we speculated a relationship
between decreases Fli-1 expression and increases in collagen production following
exposure to MBG. Therefore, we examined Fli-1 knockdown mice and compared it to
wild type mice. Physiological measurements with a Millar catheter and
immunohistochemistry were performed. In vitro studies were then pursued with cultured
isolated cardiac fibroblasts, human dermal fibroblasts, as well as a cell line derived from
renal fibroblasts.
First, in rats, we observed that PNx after immunization against MBG as well as
concomitant PNx and adrenalectomy had similar blood pressure as PNx but less cardiac
hypertrophy, diastolic dysfunction, and cardiac fibrosis. Second, in mice, the 5 6
nephrectomy resulted in impairment of both active and passive left ventricular relaxation
at four weeks as well as progressive fibrosis in the heart. Third, the Fli-1 knockdown
146

mice showed greater amounts of cardiac collagen expression and fibrosis compared to
wild type before and after 4 weeks of experimental renal failure induced by 5/6th
nephrectomy.
We realized that stimulation of cultured cardiac fibroblasts with MBG could be prevented
by administration of inhibitors of tyrosine phosphorylation, Src activation, EGFR
transactivation, and N-acetyl cysteine. Furthermore, in response to MBG, decreases in
nuclear Fli-1 accompanied increases in procollagen expression. Finally, we observed that
exposure of cardiac fibroblasts to MBG was associated with a rapid translocation of
PKCδ to the nucleus that appeared to peak at about 15 minutes as determined with
confocal immunofluorescence and Western blot.
Taken together, these data suggest that MBG directly induces increases in collagen
expression by fibroblasts in a process involving Fli-1 and PKCδ, and is in part
responsible for the cardiac fibrosis seen with experimental renal failure.
147