lysine methylation modulates the protein-protein interactions of yeast cytochrome c cyc1p
TRANSCRIPT

Proteomics 2015, 0, 1–11 1DOI 10.1002/pmic.201400521
RESEARCH ARTICLE
Lysine methylation modulates the protein–protein
interactions of yeast cytochrome C Cyc1p
Daniel L. Winter, Dhanushi Abeygunawardena∗, Gene Hart-Smith, Melissa A. Erceand Marc R. Wilkins
Systems Biology Initiative, School of Biotechnology and Biomolecular Sciences, University of New South Wales,Sydney, Australia
Received: November 3, 2014Revised: February 2, 2015
Accepted: March 2, 2015
In recent years, protein methylation has been established as a major intracellular PTM. It hasalso been proposed to modulate protein-protein interactions (PPIs) in the interactome. To in-vestigate the effect of PTMs on PPIs, we recently developed the conditional two-hybrid (C2H)system. With this, we demonstrated that arginine methylation can modulate PPIs in the yeastinteractome. Here, we used the C2H system to investigate the effect of lysine methylation.Specifically, we asked whether Ctm1p-mediated trimethylation of yeast cytochrome c Cyc1p,on lysine 78, modulates its interactions with Erv1p, Ccp1p, Cyc2p and Cyc3p. We show thatthe interactions between Cyc1p and Erv1p, and between Cyc1p and Cyc3p, are significantly in-creased upon trimethylation of lysine 78. This increase of interaction helps explain the reportedfacilitation of Cyc1p import into the mitochondrial intermembrane space upon methylation.This first application of the C2H system to the study of methyllysine-modulated interactionsfurther confirms its robustness and flexibility.
Keywords:
Cytochrome c / Lysine methylation / Protein–protein interactions / Systems biology /Two-hybrid
� Additional supporting information may be found in the online version of this article atthe publisher’s web-site
1 Introduction
Systems biology has shifted the focus of molecular biologyfrom the study of single genes or proteins to their interre-lations in the context of systems. One of the objectives ofsystems biology is to catalog all proteins of an organism andtheir interactions, which has become possible with the adventof large-scale technologies such as yeast two-hybrid screens [1]and affinity purification in association with MS [2]. The visu-alization of all the known protein–protein interactions (PPIs)generates networks called interactomes [3]. The interactome
Correspondence: Professor Marc R. Wilkins, The University ofNew South Wales - School of Biotechnology and BiomolecularSciences School of Biotechnology & Biomolecular Sciences, TheUniversity of New South Wales, Sydney, New South Wales, 2052AustraliaE-mail: [email protected]
Abbreviations: C2H, conditional two hybrid; BACTH, bacterialadenylate cyclase two hybrid; IMS, intermembrane space; 3met,trimethylation; Ox, oxidation; XIC, extracted ion chromatogram
of an organism is a very dynamic system [4, 5]. Among thefactors that modulate the interactome, it has been proposedthat PTMs, mediated by modifying enzymes, are major mod-ulators of PPIs [6, 7]. In fact, many examples are knownwhereby PTMs can create motifs that are recognized by pro-tein interaction domains [8].
In recent years, protein methylation has emerged as a ma-jor intracellular PTM [9–11]. Erce et al. proposed that methy-lation is of considerable impact in the interactome and plays arole in regulating PPIs that extends beyond the histone code[12]. Evidence of this is that an increasing number of non-histone methyltransferases and substrate proteins have beenidentified [11] and that several of the substrates form highlyconnected hubs in the interactome [12] (Fig. 1A).
In the past, the effect of PTMs on PPIs has been investi-gated by techniques such as co-immunoprecipitation [13,14],far western blotting [15], modifications of the yeast two-hybrid
∗Current address: Dhanushi Abeygunawardena, Developmental andStem Cell Biology Division, Victor Chang Cardiac Research Institute,Darlinghurst, NSW 2010, AustraliaColour Online: See the article online to view Figs. 1 and 5 in colour.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2 D. L. Winter et al. Proteomics 2015, 0, 1–11
Figure 1. The conditional two-hybrid system can be used toprobe PTM-mediated PPIs. (A) The yeast lysine methylproteome,embedded within a kinase-substrate network. Blue diamonds,lysine methyltransferases; orange diamonds, kinases; blue cir-cles, lysine methylated proteins; purple circles, arginine methy-lated proteins; gray nodes, interaction partners; orange edges,phosphorylation; blue edges; methylation; gray edges, PPIs. TheCyc1p interactions are shown in detail. (B) The bacterial two-hybrid (C2H) system can be used to investigate the interactions ofsuch networks. In the classical bacterial two-hybrid (BACTH) sys-tem, two partner proteins might appear not to interact and yielda false negative. (C) The additional multiple cloning sites (whitesections) allow for the cloning and co-expression of a modify-ing enzyme. Upon modification of the prey and/or bait protein,the interaction is restored. Conversely, a PTM might block aninteraction.
system [16], and tethered catalysis in yeast and mammaliansystems [17–19]. Recently, Erce et al. developed the condi-tional two-hybrid (C2H) system [20]. Based on the bacterialadenylate cyclase two-hybrid (BACTH) system [21, 22], thebait and prey proteins are co-expressed in the presence orabsence of a modifying enzyme (Fig. 1B and C). This allowsthe detection of increases or decreases in interaction due tothe bait and/or prey protein carrying a PTM. To date, it hasshown that a number of PPIs in the yeast interactome aremodulated by arginine methylation [20, 23].
In Saccharomyces cerevisiae, cytochrome c is expressed fromthe CYC1 gene and is immediately trimethylated at lysine 78
(often referred to as lysine 72 by homology to vertebrates)by the cytochrome c methyltransferase Ctm1p. Ctm1p is co-ordinately regulated with Cyc1p [24, 25]. Cyc1p is then lo-calized in the intermembrane space (IMS) of mitochondriawhere it undergoes a final maturation step, the attachmentof a heme c, which is carried out by the cytochrome c hemelyase Cyc3p and flavoprotein Cyc2p [26, 27]. In its matureform, Cyc1p can realize its function as a component of therespiratory chain, carrying electrons between complexes IIIand IV of the electron transport chain. In yeast, Cyc1p is alsoinvolved in the response to oxidative stress through its inter-action with cytochrome c peroxidase Ccp1p [28–30] and theimport of proteins to the IMS through its interaction withthiol oxidase Erv1p [31–33].
Methylation of cytochrome c does not occur in higher eu-karyotes, but plays a significant although nonessential role inyeast, facilitating its transport into the IMS [34]. Methylationis also known to change some aspects of Cyc1p biochemistry,such as its pI [35], its geometry [36], and to affect its conforma-tional changes, including the pH-induced alkaline transition[37, 38]. Furthermore, analysis of the Cyc1p structure revealsthat trimethyllysine 78 is critically positioned across the hemecrevice loop, thus possibly playing a role in the dynamics ofopening the crevice and regulating function [37]. To date,only the interaction of cytochrome c with cytochrome c per-oxidase has been studied in detail. Several structural studieswith cytochrome c from different organisms, including yeast,revealed that conformational changes, which might be mod-ulated by methylation of lysine 78 [37], play a significant roleduring this interaction [39–42].
Here, we investigated whether trimethylation of lysine 78in Cyc1p, catalysed by the methyltransferase Ctm1p, affectsits interactions with Erv1p, Ccp1p, Cyc2p, and Cyc3p. Wefound that the interaction of Cyc1p and Erv1p, and the in-teraction of Cyc1p and Cyc3p, increase upon methylation.These experiments also generated further insights into theC2H system and on the role of methylation in regulatingPPIs.
2 Materials and methods
2.1 Bacterial strains and media
The Escherichia coli strains DH5� [F− deoR endA1 recA1relA1 gyrA96 hsdR17(rk
−, mk+) supE44 thi-1 phoA �(lacZYA-
argF)U169 �80lacZ�M15 �] (�-Select Gold Efficiency, Bi-oline), RosettaTM 2 (DE3) [F− ompT hsdSB(rB
− mB−) gal
dcm (DE3) pRARE2 (CamR)], and DHM1 (DE3) pRARE [F−
glnV44(AS) recA1 endA gyrA96 thi-1 hsdR17 spoT1 rfbD1 cya-854 pRARE (CamR)] were used in this study for cloning, pro-tein expression, and carrying out two-hybrid assays, respec-tively. The cultures were grown in LB10 media (10 g/L NaCl,10 g/L tryptone, 5 g/L yeast extract) at 37�C with shaking(180 rpm) unless otherwise stated. Chloramphenicol(17 �g/mL), ampicillin (50 �g/mL), and/or kanamycin (50�g/mL) were added to the medium when required.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 0, 1–11 3
2.2 Construction of bacterial two hybrid
co-expression vectors
pDuetMCS2T18, pRSFT25MCS1, DuetT18Gcn4, RsfT25Gcn4, and RsfT25Snp1 vectors were readily available fromprevious work [20, 23] (Supporting Information Table 1).To construct vectors for Ctm1p, Cyc1p-T18, T25-Ccp1p,T25-Cyc2p, and T25-Cyc3p expression, gene cassettes wereamplified from S. cerevisiae strain BY4741 [MAT� his3�1leu2�0 met15�0 ura3�0] genomic DNA. For T25-Erv1pexpression, the gene cassette was amplified from cDNA.PCR products were cleaned with the HiYieldTM Gel PCRDNA Fragments Extraction Kit (RBC Bioscience), digestedwith the relevant restriction enzymes (New EnglandBioLabs) (Supporting Information Table 2) in the appro-priate buffer and cleaned once more. pDuetMCS2T18and pRSFDuetT25MCS1 were digested with the relevantrestriction enzymes and treated with Antarctic phosphatase(New England BioLabs). The digested PCR products wereinserted in either MCS1 or MCS2 of pDuetMCS2T18 orpRSFDuetT25MCS1 with the Quick LigationTM Kit (NewEngland BioLabs) to generate the plasmids pETDuetCTM1,pETDuetCYC1T18, pETDuetCTM1_CYC1T18, pRSF-DuetT25ERV1, pRSFDuetT25CCP1, pRSFDuetT25CYC2,and pRSFDuetT25CYC3. Ligated products were transformedinto E. coli cloning strain DH5�. Cloning of inserts into theirrespective vectors was verified by colony PCR and capillarysequencing.
2.3 Mutagenesis
To generate the Cyc1pK78A mutant, which lacks the methy-lation site of native Cyc1p, two PCR products were ampli-fied from S. cerevisiae strain BY4741 genomic DNA usingthe pairs of primers CYC1-pETDuet Forward/CYC1K78A Re-verse and CYC1K78A Forward/CYC1-pETDuet Reverse. Theplasmids pDuetMCS2T18 and pETDuetCTM1 were ampli-fied with the pair of primers pETDuet Forward/pETDuetReverse (Supporting Information Table 2). The two CYC1products were mixed with either linearized plasmid and thethree fragments were assembled by Gibson assembly withthe Gibson Assembly R© Cloning Kit (New England BioLabs)to generate the plasmids pETDuetCYC1(K78A)T18 and pET-DuetCTM1_CYC1(K78A)T18, which contained a mutatedversion of the CYC1 gene cassette. The two adjacent pointmutations replaced a lysine codon (AAG) by an alanine codon(GCG) at position 232 (residue 78). Correct mutagenesis wasverified by capillary sequencing.
2.4 Protein expression, purification, and separation
Relevant plasmids were co-transformed into the E. coli expres-sion strain RosettaTM 2 (DE3). Production of recombinantproteins was obtained by induction with 1 mM of isopropyl
�-D-1-thiogalactopyranoside (IPTG) at 18�C overnight. Whenno pull-down was required, cells were harvested and resus-pended in 6× SDS sample buffer (350 mM Tris-HCl pH6.8, 30% (v/v) glycerol, 10% (v/v) SDS, 0.6 M DTT, 0.012%bromophenol blue) diluted at a 1:5 (buffer:water) ratio and2.5 M urea, boiled for 15 min and centrifuged at 16 000 × g for30 min at room temperature. For pull-downs, cells were re-suspended in calmodulin-binding buffer (50 mM Tris-HClpH 8.0, 1 mM imidazole, 10% (v/v) glycerol, 2 mM CaCl2,150 mM NaCl, 1 mM magnesium acetate, 0.1% (v/v) Triton-X100) along with cOmplete EDTA-free protease inhibitor(Roche). Cell suspensions were sonicated (Branson sonifier)to generate a whole cell lysate and centrifuged at 22 000 × g for30 min at 4�C. Thirty microliters of Calmodulin Affinity Resinsepharose beads (Stratagene) per microliter of lysate were in-cubated with the cleared cell lysates overnight at 4�C. Theflow-through was removed to a clean tube and retained. Beadswere washed five times with 1 mL of calmodulin-bindingbuffer before eluting with 30 �L of calmodulin elution buffer(50 mM Tris-HCl pH 8.0, 2 mM EDTA, 500 mM NaCl).Lysates and eluates were separated by 1-D SDS-PAGE on4–12% Bis-Tris gels (Invitrogen) in 1X NuPAGE R© MES SDSRunning Buffer (Invitrogen) according to manufacturer’s in-structions for further analysis.
2.5 Western blotting
Following protein separation by 1-D SDS-PAGE, sampleswere electroblotted onto a PVDF membrane (Immobilon-PSQ
Membrane, PVDF, 0.2 �m, Millipore) via western transfer intransfer buffer (25 mM bicine, 25 mM bis-tris, 1 mM EDTA,20% (v/v) methanol) at 590 mA for 70 min. For His-tag de-tection, membranes were analyzed with the Penta·His HRPConjugate Kit (Qiagen) according to manufacturer’s instruc-tions (1:10 000 in the provided Blocking Reagent Buffer).Lysine trimethylation was analyzed with an anti-N,N,N-trimethyllysine rabbit polyclonal antibody (ImmuneChem,Catalog # ICP0601) at a 1:2000 dilution in 1% BSA in PBS-T.Antibody binding was visualized via chemiluminescence us-ing Western Blotting Luminol Reagent (Santa Cruz Biotech-nology) and the Fujifilm LAS-3000 Imager.
2.6 Mass spectrometry
Gel bands to be analyzed were excised from 1D gelsand destained, reduced, and alkylated following the pro-cedure described by Shevchenko et al. [43]. Tryptic diges-tions and the preparation of proteolytic peptide samplesfor MS analysis were performed following techniques de-scribed in Hart-Smith et al. [44]. Samples were analyzedvia Liquid Chromatography-Collision Induced Dissociation-Tandem Mass Spectrometry (LC-CID-MS/MS) on an LTQOrbitrap Velos Pro (Thermo Scientific) hybrid linear iontrap mass spectrometer as described in Pang et al. [45].
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

4 D. L. Winter et al. Proteomics 2015, 0, 1–11
Protein identification was performed using the databasesearch program MASCOT (version 2.3, Matrix Science)against the Swiss-Prot database. The following search param-eters were used: instrument type; ESI-TRAP, precursor toler-ance; ± 4 ppm, MS/MS tolerance; ± 0.4 Da, variable modifi-cations; acrylamide (C), carbamidomethyl (C), oxidation (M),methylation (K), dimethylation (K), and trimethylation (K),enzyme specificity; trypsin, number of missed trypsin cleav-ages; 2. Peptide identifications were considered to be highconfidence if they were statistically significant (p<0.05) ac-cording to the MASCOT expect metric.
2.7 Conditional two-hybrid assays
For drop plate assays, the relevant plasmids were co-transformed into the E. coli strain DHM1 (DE3) pRARE andcell cultures were grown overnight in LB10 media and the ap-propriate antibiotics. Two microliters of culture were droppedin duplicates on an LB10 agar plate (10 g/L NaCl, 10 g/L tryp-tone, 5 g/L yeast extract, 15 g/L) complemented with the ap-propriate antibiotics, IPTG (0.05 mM) and 5-bromo-4-chloro-3-indolyl �-D-galactoside (X-gal) (40 �g/mL), then incubatedat 30�C. The color of the colonies was compared 30 to 48 hlater. For �-galactosidase assays, relevant plasmids were co-transformed into the E. coli strain DHM1 (DE3) pRARE. Cul-tures were grown and induced with 1 mM of IPTG at 30�C for6 h. After induction, the OD600 of each culture was measured.One hundred microliters of each culture were then mixedwith 1 mL of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4,10 mM KCl, 1 mM MgSO4, 50 mM �-mercaptoethanol),20 �L of SDS 0.1% (w/v), and 40 �L of chloroform thenmixed by pipetting 15 times to lyse the cells. After allowingthe chloroform to settle, 100 �L of each lysate were mixed with20 �L of ortho-nitrophenyl-�-galactoside (ONPG) (4 mg/mL).�-galactosidase activity was assayed at 420 nm for 20 min at37�C with a SPECTRAmaxTM 340 plate reader (MolecularDevices). The whole process was conducted in 96-well mi-croplates, as described in Griffith and Wolf [46]. The activityof �-galactosidase is expressed in units of �-galactosidaseper milliliter of culture, normalized by the OD600 of thecorresponding culture, and multiplied by 1000 (arbitraryunits).
3 Results
3.1 Cyc1p-T18, Ctm1p, T25-Erv1p, T25-CCp1p,
T25-Cyc2p, and T25-Cyc3p can be expressed in
E. coli
The conditional two-hybrid system, as used here, requires theheterologous expression of yeast proteins in E. coli. To test thatall recombinant proteins used in this study could be success-fully expressed, we transformed plasmids pETDuetCYC1T18,
pETDuetCTM1, pRSFDuetT25ERV1, pRSFDuetT25CCP1,pRSFDuetT25CYC2, and pRSFDuetT25CYC3 into the E. coliexpression strain RosettaTM 2. As controls, we also ana-lyzed RosettaTM 2 and RosettaTM 2 transformed with eitherpDuetMCS2T18 or pRSFDuetT25MCS1.
In the cloning process, Ctm1p, T25-Erv1p, T25-Ccp1p, T25-Cyc2p, and T25-Cyc3p (but not Cyc1p-T18 orCyc1pK78A-T18) were fused to a 6xHis-tag at the N-terminus.The expected sizes of these recombinant proteins were, re-spectively, 68 kDa; 47 kDa; 65 kDa; 67 kDa; and 55 kDa.Thus, to verify that these recombinant proteins could be ex-pressed in E. coli, we analyzed the corresponding samplesvia western blotting with an anti-His-tag antibody. Except forT25-Cyc2p, bands close to the expected sizes were detectedby the antibody (Fig. 2A). Faint bands of smaller molecu-lar weights were also detected, indicating some C-terminaldegradation of the recombinant proteins. Moreover, a faintband was detected around 28 kDa in the cell lysate from theRosettaTM 2 transformed with pRSFDuetT25MCS1 strain;this band most likely corresponds to the T25 fragment byitself. This is the same size as the band detected in the T25-Cyc2p lane and MS confirmed these bands to correspond tothe T25 fragment (Supporting Information Fig. 1M and 1N).This result suggests that only the T25 fragment portion ofT25-Cyc2p is correctly expressed, whereas the Cyc2p portionis poorly expressed and/or quickly degraded. As Cyc2p is amitochondrial, membrane-bound protein, it could be unsta-ble in bacterial cytosol. Nevertheless, when a 1D gel slice ofappropriate mass range was analyzed by MS, several peptidesfrom Cyc2p were identified (Supporting Information Fig. 1Iand 1J).
Cyc1p-T18 and Cyc1pK78A-T18 were not fused to His-tags.As such, we used a different approach to confirm the expres-sion of these two recombinant proteins. After extracting theproteins from the corresponding cultures, we performed apull-down using calmodulin affinity beads, as the T18 frag-ment of adenylate cyclase has affinity for calmodulin [47].In the pull-down from the RosettaTM 2 lysate, SDS-PAGEshowed three proteins were purified (Fig. 2B), indicating thata few E. coli proteins show some affinity for calmodulin. How-ever, when the pull-down was performed with the lysate fromRosettaTM 2 transformed with pDuetMCS2T18, these threebands were not visible; instead, two smaller proteins werepurified. These correspond to the T18 domain of adenylatecyclase and what is presumably a smaller degradation prod-uct. Finally, the pull-downs from RosettaTM 2 transformedwith either pETDuetCYC1T18 or pETDuetCYC1(K78A)T18show the same profile as each other: proteins of approxi-mately 30; 35; and 40 kDa were purified in both lanes. The40 kDa bands correspond to Cyc1p-T18 and Cyc1pK78A-T18.The smaller bands most likely correspond to degraded ver-sions of the recombinant protein. Altogether, the above re-sults show that all recombinant proteins used in this study canbe expressed in an E. coli host. Furthermore, all recombinantproteins were confirmed by MS (Supporting InformationFig. 1).
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 0, 1–11 5
Figure 2. Expression of recombinant proteins. (A) Plasmids for the expression of Ctm1p, T25-Erv1p, T25-Ccp1p, T25-Cyc2p, and T25-Cyc3pwere transformed into the RosettaTM 2 expression strain and protein extracts were analyzed via anti-His tag blotting after induction.All proteins were correctly expressed, except for T25-Cyc2p. (B) Plasmids for the expression of Cyc1p-T18 and Cyc1pK78A-T18 weretransformed into the RosettaTM 2 expression strain and recombinant proteins were purified via calmodulin pull-down after induction.Cyc1p-T18 and Cyc1pK78A-T18 were correctly expressed. Arrows point to the recombinant proteins unless otherwise indicated.
3.2 Ctm1p can trimethylate Cyc1p-T18, but not
Cyc1pK78A-T18, in E. coli
The conditional two-hybrid (C2H) system relies on the co-expression of an enzyme, which can post-translationally mod-ify one or both of the interacting proteins. To verify thatmethyltransferase Ctm1p is able to trimethylate Cyc1p-T18at lysine 78 when both proteins are recombinantly expressedin E. coli, the pETDuetCTM1_Cyc1T18 plasmid was trans-formed into the E. coli expression strain RosettaTM 2. As con-trols, pETDuetCyc1T18 and pETDuetCTM1 were also sepa-rately transformed into E. coli strain RosettaTM 2. This wasto ensure that Cyc1p-T18 is not methylated by any endoge-nous E. coli methyltransferase and that Ctm1p is not ableto methylate any endogenous E. coli protein; these types ofundesired methylation events could interfere with the C2Hsystem.
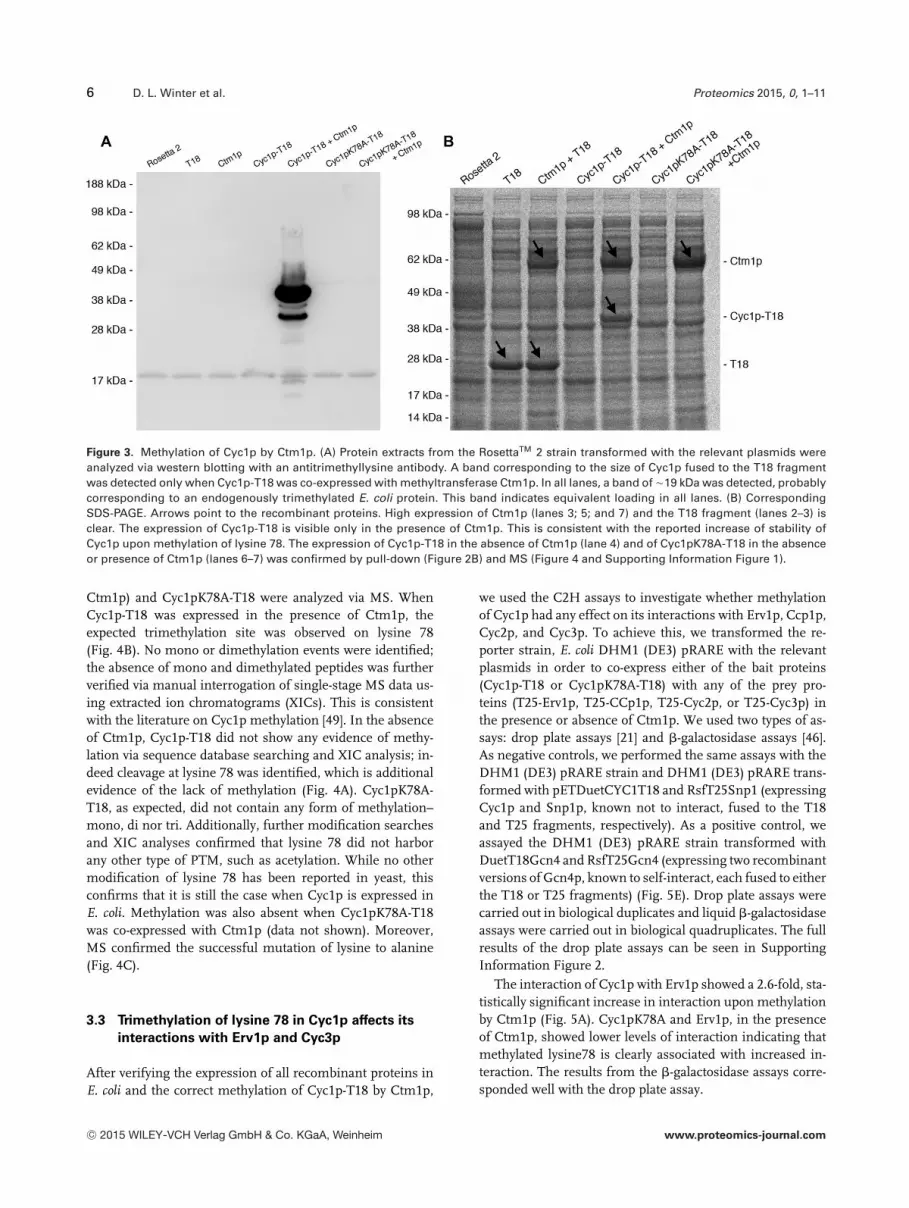
After expression of recombinant proteins in E. coli, we an-alyzed protein extracts from these cultures via western blot-ting with an anti-trimethyllysine antibody (Fig. 3A). Whencompared to RosettaTM 2 and RosettaTM 2 transformed withpDuetMCS2T18 (Fig. 3A, lanes 1–2), a band correspond-ing to the size of Cyc1-T18 appears only when Cyc1p-T18is co-expressed with Ctm1p (Fig. 3A, lane 5). This indicatesthat Ctm1p does not trimethylate any bacterial protein sub-strate when expressed in E. coli and that Cyc1p-T18 cannotbe trimethylated by any bacterial methyltransferase. The de-tected band is more intense than the corresponding band
on the 1D separation gel. This is likely due to the differ-ent methods of protein extraction: for the pull-downs, cellswere disrupted via sonication whereas for transfer to PVDFmembranes, cells were boiled in the presence of SDS, lead-ing to better protein recovery. This result also serves as fur-ther confirmation of the correct expression of both Cyc1p-T18 and Ctm1p. Finally, to ensure that the Cyc1pK78A-T18mutant cannot be trimethylated by Ctm1p, plasmids pET-DuetCYC1(K78A)T18 and pETDuetCTM1_CYC1(K78A)T18were also transformed into E. coli strain RosettaTM 2 andthe expressed recombinant proteins analyzed as above(Fig. 3A, lanes 6 and 7). In both cases, no additional bandswere visible when compared to RosettaTM 2, confirmingthat the mutation of the target lysine 78 of Cyc1p preventstrimethylation by Ctm1p. In the corresponding SDS-PAGE(Fig. 3B), which serves as a loading control for the westernblot, the expression of Ctm1p and the T18 fragment can bereadily confirmed (Fig. 3B, lanes 2; 3; 5; and 7). Strong expres-sion of Cyc1p-T18 is noticeable in the presence of Ctm1p (lane5) yet Cyc1p-T18 in the absence of Ctm1p or Cyc1pK78A-T18were of lower abundance (lanes 4; 6; and 7). Their expressionwas confirmed by pull down (Fig. 2B) and MS (Fig. 4 and Sup-porting Information Figure 1). This result is consistent withthe reported increase of stability of Cyc1p upon methylationof Cyc1p at lysine 78 [48].
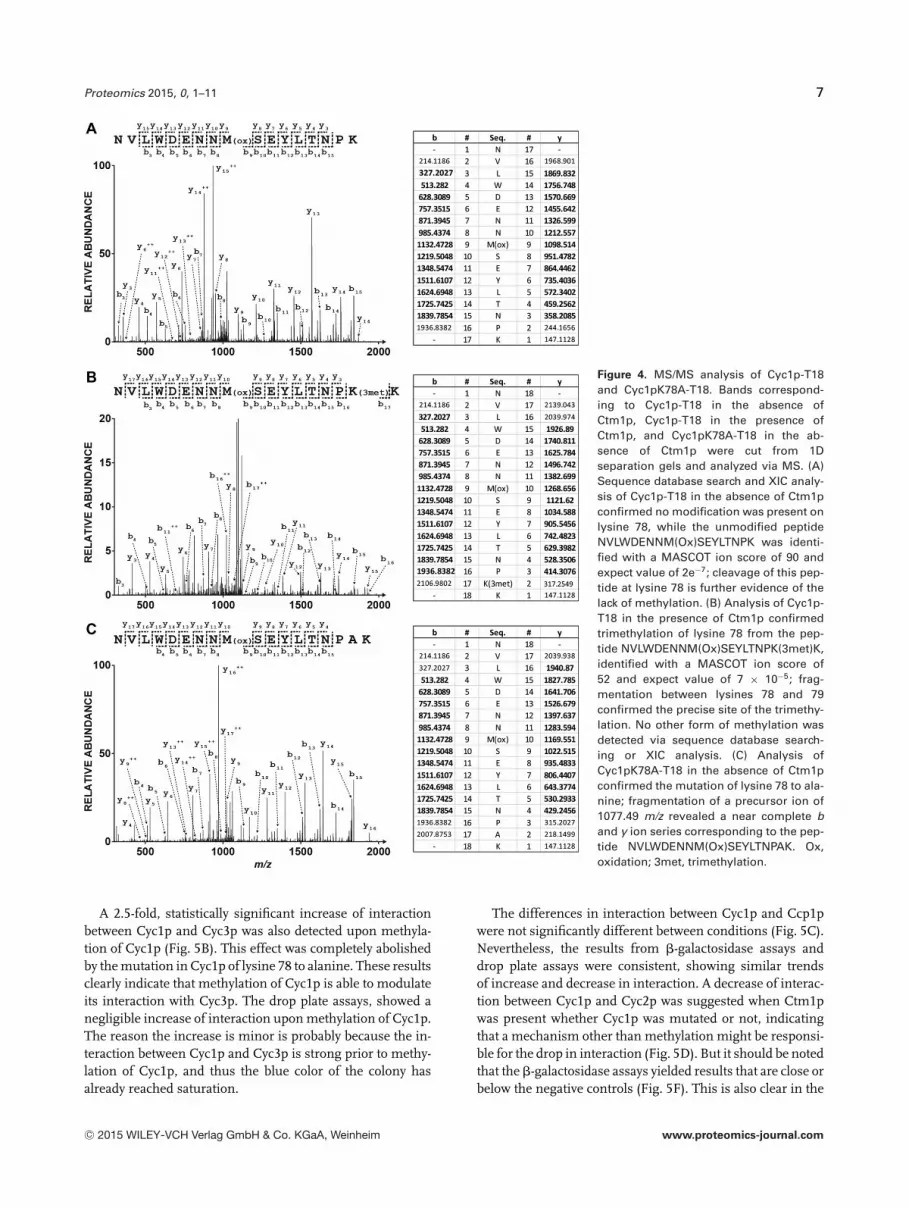
To confirm that trimethylation of Cyc1p-T18 is lo-cated on the expected residue, lysine 78, gel bands corre-sponding to Cyc1p-T18 (in the presence and absence of
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
6 D. L. Winter et al. Proteomics 2015, 0, 1–11
Figure 3. Methylation of Cyc1p by Ctm1p. (A) Protein extracts from the RosettaTM 2 strain transformed with the relevant plasmids wereanalyzed via western blotting with an antitrimethyllysine antibody. A band corresponding to the size of Cyc1p fused to the T18 fragmentwas detected only when Cyc1p-T18 was co-expressed with methyltransferase Ctm1p. In all lanes, a band of �19 kDa was detected, probablycorresponding to an endogenously trimethylated E. coli protein. This band indicates equivalent loading in all lanes. (B) CorrespondingSDS-PAGE. Arrows point to the recombinant proteins. High expression of Ctm1p (lanes 3; 5; and 7) and the T18 fragment (lanes 2–3) isclear. The expression of Cyc1p-T18 is visible only in the presence of Ctm1p. This is consistent with the reported increase of stability ofCyc1p upon methylation of lysine 78. The expression of Cyc1p-T18 in the absence of Ctm1p (lane 4) and of Cyc1pK78A-T18 in the absenceor presence of Ctm1p (lanes 6–7) was confirmed by pull-down (Figure 2B) and MS (Figure 4 and Supporting Information Figure 1).
Ctm1p) and Cyc1pK78A-T18 were analyzed via MS. WhenCyc1p-T18 was expressed in the presence of Ctm1p, theexpected trimethylation site was observed on lysine 78(Fig. 4B). No mono or dimethylation events were identified;the absence of mono and dimethylated peptides was furtherverified via manual interrogation of single-stage MS data us-ing extracted ion chromatograms (XICs). This is consistentwith the literature on Cyc1p methylation [49]. In the absenceof Ctm1p, Cyc1p-T18 did not show any evidence of methy-lation via sequence database searching and XIC analysis; in-deed cleavage at lysine 78 was identified, which is additionalevidence of the lack of methylation (Fig. 4A). Cyc1pK78A-T18, as expected, did not contain any form of methylation–mono, di nor tri. Additionally, further modification searchesand XIC analyses confirmed that lysine 78 did not harborany other type of PTM, such as acetylation. While no othermodification of lysine 78 has been reported in yeast, thisconfirms that it is still the case when Cyc1p is expressed inE. coli. Methylation was also absent when Cyc1pK78A-T18was co-expressed with Ctm1p (data not shown). Moreover,MS confirmed the successful mutation of lysine to alanine(Fig. 4C).
3.3 Trimethylation of lysine 78 in Cyc1p affects its
interactions with Erv1p and Cyc3p
After verifying the expression of all recombinant proteins inE. coli and the correct methylation of Cyc1p-T18 by Ctm1p,
we used the C2H assays to investigate whether methylationof Cyc1p had any effect on its interactions with Erv1p, Ccp1p,Cyc2p, and Cyc3p. To achieve this, we transformed the re-porter strain, E. coli DHM1 (DE3) pRARE with the relevantplasmids in order to co-express either of the bait proteins(Cyc1p-T18 or Cyc1pK78A-T18) with any of the prey pro-teins (T25-Erv1p, T25-CCp1p, T25-Cyc2p, or T25-Cyc3p) inthe presence or absence of Ctm1p. We used two types of as-says: drop plate assays [21] and �-galactosidase assays [46].As negative controls, we performed the same assays with theDHM1 (DE3) pRARE strain and DHM1 (DE3) pRARE trans-formed with pETDuetCYC1T18 and RsfT25Snp1 (expressingCyc1p and Snp1p, known not to interact, fused to the T18and T25 fragments, respectively). As a positive control, weassayed the DHM1 (DE3) pRARE strain transformed withDuetT18Gcn4 and RsfT25Gcn4 (expressing two recombinantversions of Gcn4p, known to self-interact, each fused to eitherthe T18 or T25 fragments) (Fig. 5E). Drop plate assays werecarried out in biological duplicates and liquid �-galactosidaseassays were carried out in biological quadruplicates. The fullresults of the drop plate assays can be seen in SupportingInformation Figure 2.
The interaction of Cyc1p with Erv1p showed a 2.6-fold, sta-tistically significant increase in interaction upon methylationby Ctm1p (Fig. 5A). Cyc1pK78A and Erv1p, in the presenceof Ctm1p, showed lower levels of interaction indicating thatmethylated lysine78 is clearly associated with increased in-teraction. The results from the �-galactosidase assays corre-sponded well with the drop plate assay.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Proteomics 2015, 0, 1–11 7
Figure 4. MS/MS analysis of Cyc1p-T18and Cyc1pK78A-T18. Bands correspond-ing to Cyc1p-T18 in the absence ofCtm1p, Cyc1p-T18 in the presence ofCtm1p, and Cyc1pK78A-T18 in the ab-sence of Ctm1p were cut from 1Dseparation gels and analyzed via MS. (A)Sequence database search and XIC analy-sis of Cyc1p-T18 in the absence of Ctm1pconfirmed no modification was present onlysine 78, while the unmodified peptideNVLWDENNM(Ox)SEYLTNPK was identi-fied with a MASCOT ion score of 90 andexpect value of 2e−7; cleavage of this pep-tide at lysine 78 is further evidence of thelack of methylation. (B) Analysis of Cyc1p-T18 in the presence of Ctm1p confirmedtrimethylation of lysine 78 from the pep-tide NVLWDENNM(Ox)SEYLTNPK(3met)K,identified with a MASCOT ion score of52 and expect value of 7 × 10−5; frag-mentation between lysines 78 and 79confirmed the precise site of the trimethy-lation. No other form of methylation wasdetected via sequence database search-ing or XIC analysis. (C) Analysis ofCyc1pK78A-T18 in the absence of Ctm1pconfirmed the mutation of lysine 78 to ala-nine; fragmentation of a precursor ion of1077.49 m/z revealed a near complete band y ion series corresponding to the pep-tide NVLWDENNM(Ox)SEYLTNPAK. Ox,oxidation; 3met, trimethylation.
A 2.5-fold, statistically significant increase of interactionbetween Cyc1p and Cyc3p was also detected upon methyla-tion of Cyc1p (Fig. 5B). This effect was completely abolishedby the mutation in Cyc1p of lysine 78 to alanine. These resultsclearly indicate that methylation of Cyc1p is able to modulateits interaction with Cyc3p. The drop plate assays, showed anegligible increase of interaction upon methylation of Cyc1p.The reason the increase is minor is probably because the in-teraction between Cyc1p and Cyc3p is strong prior to methy-lation of Cyc1p, and thus the blue color of the colony hasalready reached saturation.
The differences in interaction between Cyc1p and Ccp1pwere not significantly different between conditions (Fig. 5C).Nevertheless, the results from �-galactosidase assays anddrop plate assays were consistent, showing similar trendsof increase and decrease in interaction. A decrease of interac-tion between Cyc1p and Cyc2p was suggested when Ctm1pwas present whether Cyc1p was mutated or not, indicatingthat a mechanism other than methylation might be responsi-ble for the drop in interaction (Fig. 5D). But it should be notedthat the �-galactosidase assays yielded results that are close orbelow the negative controls (Fig. 5F). This is also clear in the
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

8 D. L. Winter et al. Proteomics 2015, 0, 1–11
Figure 5. Conditional two-hybrid analysis ofthe interactions of Cyc1p. The interactions ofCyc1p with Erv1p, Ccp1p, Cyc2p, and Cyc3pwere analyzed via conditional two-hybrid as-says. Cyc1pK78A, in the presence of Ctm1p,served as a control. (A) The interaction ofCyc1p with Erv1p is increased upon methy-lation of Cyc1p. (B) The interaction of Cyc1pwith Cyc3p is increased upon methylation ofCyc1p. (C) Methylation of Cyc1p has no ef-fect on its interaction with Ccp1p. (D) The in-teraction between Cyc1p and Cyc2p yieldedresults close to the negative controls, andwere thus inconclusive. (E) Controls. (F) All�-galactosidase assays put to scale. *p < 0.05,**p < 0.005.
drop plate assays (Fig. 5D). This result also seems to suggestthat Cyc1p and Cyc2p are not interaction partners. However,it was reported elsewhere that an interaction between Cyc1pand Cyc2p was detected via a split-ubiquitin, membrane yeasttwo-hybrid (MYTH) assay [27]. Here, the interaction mightnot have been detected due to the poor expression of Cyc2pin a bacterial host (Fig. 2A). Thus, the effect of methylationon this interaction remains unknown.
In summary, these experiments showed that methylationof Cyc1p increased its interactions with Erv1p and Cyc3pbut did not affect its interaction with Ccp1p. Nevertheless, itshould be noted that there is a reported increase of stabilityof Cyc1p upon methylation of lysine 78 [48], which we alsoobserved in the exogenous co-expression of Cyc1p-T18 andCtm1p in E. coli (Fig. 3B). It may thus be that the increase ininteraction of Cyc1p on methylation may involve an increasein affinity and/or an increase in abundance of Cyc1p; bothcan be biologically important.
4 Discussion
In this study, we investigated the effect of lysine methylationof Cyc1p on four of its PPIs. The results allow us to drawseveral conclusions on the C2H system and on the functionof methylation of Cyc1p.
This work differs from previous work with the C2H system[20, 23] in three ways. First, it focuses on a different typeof methylation, lysine methylation; this extends the list ofPTMs studied with the C2H system. Second, Cyc1p harborsonly a single PTM site, whereas proteins previously studied
harbor many more; this allows us to investigate whether theC2H system is capable of detecting differences in interactiondue to subtle changes in the chemistry of the bait or preyprotein. Finally, we used a different type of control for theexperiments. Previously, a mutated, inactivated version ofthe modifying enzyme was used as a control [20,23] whereashere, this was achieved by mutating the substrate protein.
4.1 The increased interaction of Erv1p and Cyc3p
with methylated Cyc1p is of likely functional
significance
In yeast, methylation of Cyc1p by Ctm1p facilitates its importinto the mitochondria IMS [34]. Cyc3p and Cyc2p also playa central role in the import of Cyc1p into the IMS. This hasbeen shown through the deletion of either CYC3 [26, 50] orCYC2 [51], which leads to accumulation of cytochrome c inthe cytoplasm. While the mechanism whereby methylationof Cyc1p facilitates import of Cyc1p in the IMS is largelyunknown, the role of Cyc3p and Cyc2p in the localization ofCyc1p is better understood. The addition of a heme c to Cyc1pby Cyc3p is required to trap Cyc1p in the mitochondria IMS[52]. This requires reduction of the heme prior to its additionto Cyc1p [53], which is carried out by Cyc2p [27].
Here, we show that methylation of Cyc1p increases its in-teraction with Cyc3p. This could explain the enhanced trans-port of Cyc1p into mitochondria IMS upon methylation oflysine 78. The increase in interaction might facilitate the ad-dition of the heme, and thus the trapping of Cyc1p in theIMS. Our experiments could not detect any effect of Cyc1p
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 0, 1–11 9
methylation on its interaction with Cyc2p. Thus, it remainsunknown if Ctm1p is able to affect the interaction betweenCyc1p and Cyc2p and whether this could further explain theenhanced localization of Cyc1p upon methylation.
We also show an increase in interaction between Cyc1pand Erv1p in the presence of Ctm1p. In fact, in the absenceof Ctm1p, the detected interaction shows similar levels tothe negative controls (Fig. 5F) and methylation of lysine 78appears to be necessary for the interaction. To date, no rolein the import of Cyc1p has been suggested for Erv1p. Thisincrease in interaction is thus unlikely to explain the effect ofCyc1p methylation on its localization and must play a differ-ent role. Erv1p is involved in the import of proteins into theIMS by forming a ternary complex with Mia40p and a sub-strate protein, promoting the formation of disulphide bondsin the substrate [54]. In the process, Erv1p is reduced. To re-cycle Erv1p to its oxidized form, electrons are transferred toCyc1p [33]. Thus, methylation of lysine 78, which promotesthe interaction of Cyc1p and Erv1p, might enhance the recy-cling of the Erv1p-Mia40p disulfide relay system.
Finally, we have shown that methylation of Cyc1p does notimpact its interaction with Ccp1p. This is consistent withprevious studies of the interaction of Cyc1p with Ccp1p.Structural analysis of the interaction showed that lysine 79of Cyc1p, but not lysine 78, is capable of forming a hydrogenbond with glutamate 290 of Ccp1p [41]. As such, methylationof lysine 78 is unable to affect any of the bonds between Cyc1pand Ccp1p. It is thus more likely to play a role in modulatingthe transfer of electrons between the two proteins by affect-ing the dynamics of the heme crevice loop, rather than in theinteraction itself [37].
4.2 Mutations of substrate proteins can be used as
controls for C2H experiments
Because there is no reported way to inactivate methyltrans-ferase Ctm1p, we decided to use a mutated version of thesubstrate Cyc1p, which lacked the methylation site, as a con-trol. This was easily achievable because Cyc1p contains onlyone PTM site. This strategy proved successful: we were able toobserve whether the mutation abolished the effect of Ctm1pon native Cyc1p or not. However, the mutation itself mayaffect the interaction. As such, we advise to choose mutationsthat abolish the PTM site(s) while minimizing changes in thephysicochemistry of the protein substrate. Here, we opted forthe lysine 78 to alanine mutation that has been used in pre-vious studies to investigate the effect of lysine trimethylationon the structure of Cyc1p [37].
More generally, the successful use of a mutated substrateas a control indicates that poorly characterized enzymes canbe studied with the C2H system, as long as its substrate andPTM target sites are known. It also indicates that for sub-strates with many PTM sites mediated by the same enzyme,although rarer, the individual effect of any particular site canbe studied by replacing the residue at that position, instead of
mutating the enzyme–which abolishes all PTMs. The same istrue in cases where both bait and prey proteins are substratesof the same modifying enzyme, but the investigator wishesto preserve the PTMs of either the prey or the bait.
In our study, Cyc1p was not modified by any E. coli en-dogenous enzyme. This ensured that no PTMs other thantrimethylation of lysine 78 interfered with our assays. How-ever, it is possible that in some cases expression of exogenousproteins in E. coli will lead to unexpected modifications. Thisis more likely to be the case with phosphorylation, as sev-eral promiscuous kinases and phosphatases exist in bacteria[55] that could potentially modify or demodify exogenous pro-teins. In the case of interfering kinases, the investigator canavoid any interference with the C2H assay in two ways: ei-ther by mutating the unexpected modification sites on thesubstrate or by identifying and knocking out the bacterialenzyme responsible for the modification. Since the latter re-quires substantial work, it should be reserved for the rarecases where a bacterial enzyme is able to modify the samesite as the exogenous enzyme being studied. In the case ofan interfering phosphatase, a phosphomimetic protein canbe expressed instead of the exogenous kinase if knocking outthe bacterial phosphatase is not possible.
4.3 �-galactosidase assays are an important
complement to drop plate assays
We employed both �-galactosidase and drop plate assays tostudy interactions. The drop plate assays are the most con-venient to prepare, requiring only overnight cultures to begrown and then plated. This simple assay is thus well suitedfor large-scale experiments. However, for strong interactionsthat are further increased upon modification of the bait orprey protein, the drop plate assays might not be sufficient todetect the increase. This is because the colonies have reachedsaturation of the blue color even prior to the addition of a mod-ifying enzyme to the system. Here, this is the case for the in-teraction between Cyc1p and Cyc3p (Fig. 5B) and in previousstudies, for Npl3p self-interaction [20]. The �-galactosidaseassay is indispensable in these cases. Moreover, it is the onlyquantitative assay out of the two, allowing for the statisticalsignificance of the change in interaction to be calculated.
4.4 Conclusion
We have extended the work with the conditional two-hybrid(C2H) system and shown that lysine methylation modulatesthe interactions of Cy1p with Erv1p and Cyc3p. These resultshelp explain the mechanism whereby methylation increasesimport of Cyc1p into the mitochondria IMS: the increasedinteraction of Cyc3p with methylated Cyc1p could facilitatethe addition of the heme to Cyc1p and thereby trap it inthe IMS. The C2H system will be useful to uncover furthermechanisms that involve the modulation of PPIs by PTMs.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

10 D. L. Winter et al. Proteomics 2015, 0, 1–11
DLW and MAE acknowledge the assistance of Dr. Cristo-pher Marquis in the preparation of the conditional two-hybridβ-galactosidase assays. DLW acknowledges support from the Uni-versity of New South Wales. MAE acknowledges support from theUniversity of New South Wales. GHS acknowledges support fromthe University of New South Wales. MRW acknowledges supportfrom the Australian Research Council, the Australian FederalGovernment EIF Super Science scheme and the New South WalesState Government Science Leveraging Fund. The authors thankDr. Ling Zhong, Ms. Sydney Liu Lau, and A/Professor MarkRaftery for their maintenance of the Orbitrap mass spectrome-ters housed at the UNSW Mark Wainwright Analytical CentreBioanalytical Mass Spectrometry Facility (BMSF).
The authors have declared no conflict of interest.
5 References
[1] Fields, S., Song, O., A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246.
[2] Rigaut, G., Shevchenko, A., Rutz, B., Wilm, M. et al., A genericprotein purification method for protein complex characteri-zation and proteome exploration. Nat. Biotechnol. 1999, 17,1030–1032.
[3] Cusick, M. E., Klitgord, N., Vidal, M., Hill, D. E., Interactome:gateway into systems biology. Hum. Mol. Genet. 2005, 14(Spec No. 2), R171–R181.
[4] deLichtenberg, U., Jensen, L. J., Brunak, S., Bork, P., Dynamiccomplex formation during the yeast cell cycle. Science 2005,307, 724–727.
[5] Komurov, K., White, M., Revealing static and dynamic modu-lar architecture of the eukaryotic protein interaction network.Mol. Syst. Biol. 2007, 3, 110.
[6] Wilkins, M. R., Kummerfeld, S. K., Sticking together? Fallingapart? Exploring the dynamics of the interactome. TrendsBiochem. Sci. 2008, 33, 195–200.
[7] Winter, D. L., Erce, M. A., Wilkins, M. R., A web of possibil-ities: network-based discovery of protein interaction codes.J. Proteome Res. 2014, 13, 5333–5338.
[8] Seet, B. T., Dikic, I., Zhou, M. M., Pawson, T., Reading proteinmodifications with interaction domains. Nat. Rev. Mol. CellBiol. 2006, 7, 473–483.
[9] Pang, C. N., Gasteiger, E., Wilkins, M. R., Identification ofarginine- and lysine-methylation in the proteome of Sac-charomyces cerevisiae and its functional implications. BMCGenom. 2010, 11, 92.
[10] Bremang, M., Cuomo, A., Agresta, A. M., Stugiewicz, M.et al., Mass spectrometry-based identification and charac-terisation of lysine and arginine methylation in the humanproteome. Mol. BioSyst. 2013, 9, 2231–2247.
[11] Guo, A., Gu, H., Zhou, J., Mulhern, D. et al., Im-munoaffinity enrichment and mass spectrometry analysisof protein methylation. Mol. Cell. Proteomics 2014, 13,372–387.
[12] Erce, M. A., Pang, C. N., Hart-Smith, G., Wilkins, M. R., Themethylproteome and the intracellular methylation network.Proteomics 2012, 12, 564–586.
[13] Petropoulos, L., Hiscott, J., Association between HTLV-1 Taxand I kappa B alpha is dependent on the I kappa B alphaphosphorylation state. Virology 1998, 252, 189–199.
[14] James, M., Nuttall, A., Ilsley, J. L., Ottersbach, K. et al.,Adhesion-dependent tyrosine phosphorylation of (beta)-dystroglycan regulates its interaction with utrophin. J. CellSci. 2000, 113, 1717–1726.
[15] Edmondson, D. G., Dent, S. Y., Identification of protein inter-actions by western analysis. Curr. Protoc. Protein Sci. 2001,19, 7.1–7.10.
[16] Osborne, M. A., Dalton, S., Kochan, J. P., The yeasttribrid system–genetic detection of trans-phosphorylatedITAM-SH2-interactions. Bio/technology 1995, 13, 1474–1478.
[17] Guo, D., Hazbun, T. R., Xu, X. J., Ng, S. L. et al., A teth-ered catalysis, two-hybrid system to identify protein-proteininteractions requiring post-translational modifications. Nat.Biotechnol. 2004, 22, 888–892.
[18] Acharya, A., Xu, X. J., Husain-Ponnampalam, R. D.,Hoffmann-Benning, S., Kuo, M. H., Production of consti-tutively acetylated recombinant p53 from yeast and Es-cherichia coli by tethered catalysis. Protein Expr. Purif. 2005,41, 417–425.
[19] Spektor, T. M., Rice, J. C., Identification and characterizationof posttranslational modification-specific binding proteins invivo by mammalian tethered catalysis. Proc. Natl. Acad. Sci.USA 2009, 106, 14808–14813.
[20] Erce, M. A., Low, J. K., Hart-Smith, G., Wilkins, M. R., A condi-tional two-hybrid (C2H) system for the detection of protein-protein interactions that are mediated by post-translationalmodification. Proteomics 2013, 13, 1059–1064.
[21] Karimova, G., Pidoux, J., Ullmann, A., Ladant, D., A bacterialtwo-hybrid system based on a reconstituted signal trans-duction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756.
[22] Battesti, A., Bouveret, E., The bacterial two-hybrid systembased on adenylate cyclase reconstitution in Escherichia coli.Methods 2012, 58, 325–334.
[23] Erce, M. A., Abeygunawardena, D., Low, J. K., Hart-Smith, G.,Wilkins, M. R., Interactions affected by arginine methylationin the yeast protein-protein interaction network. Mol. Cell.Proteomics 2013, 12, 3184–3198.
[24] Liao, H. N., Sherman, F., Yeast cytochrome c-specific protein-lysine methyltransferase: coordinate regulation with cy-tochrome c and activities in cyc mutants. J. Bacteriol. 1979,138, 853–860.
[25] Polevoda, B., Martzen, M. R., Das, B., Phizicky, E. M., Sher-man, F., Cytochrome c methyltransferase, Ctm1p, of yeast. J.Biol. Chem. 2000, 275, 20508–20513.
[26] Dumont, M. E., Cardillo, T. S., Hayes, M. K., Sherman, F., Roleof cytochrome c heme lyase in mitochondrial import andaccumulation of cytochrome c in Saccharomyces cerevisiae.Mol. Cell. Biol. 1991, 11, 5487–5496.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2015, 0, 1–11 11
[27] Corvest, V., Murrey, D. A., Hirasawa, M., Knaff, D. B. et al., Theflavoprotein Cyc2p, a mitochondrial cytochrome c assemblyfactor, is a NAD(P)H-dependent haem reductase. Mol. Micro-biol. 2012, 83, 968–980.
[28] Volkov, A. N., Nicholls, P., Worrall, J. A., The complex ofcytochrome c and cytochrome c peroxidase: the end of theroad? Biochim. Biophys. Acta 2011, 1807, 1482–1503.
[29] Charizanis, C., Juhnke, H., Krems, B., Entian, K. D., The mito-chondrial cytochrome c peroxidase Ccp1 of Saccharomycescerevisiae is involved in conveying an oxidative stress sig-nal to the transcription factor Pos9 (Skn7). Mol. Gen. Genet.1999, 262, 437–447.
[30] Kwon, M., Chong, S., Han, S., Kim, K., Oxidative stresseselevate the expression of cytochrome c peroxidase in Sac-charomyces cerevisiae. Biochim. Biophys. Acta 2003, 1623,1–5.
[31] Dabir, D. V., Leverich, E. P., Kim, S. K., Tsai, F. D. et al., A rolefor cytochrome c and cytochrome c peroxidase in electronshuttling from Erv1. EMBO J. 2007, 26, 4801–4811.
[32] Herrmann, J. M., Riemer, J., Mitochondrial disulfide re-lay: redox-regulated protein import into the intermembranespace. J. Biol. Chem. 2012, 287, 4426–4433.
[33] Hell, K., The Erv1-Mia40 disulfide relay system in the inter-membrane space of mitochondria. Biochim. Biophys. Acta2008, 1783, 601–609.
[34] Park, K. S., Frost, B., Tuck, M., Ho, L. L. et al., Enzymaticmethylation of in vitro synthesized apocytochrome c en-hances its transport into mitochondria. J. Biol. Chem. 1987,262, 14702–14708.
[35] Kim, C. S., Kueppers, F., Dimaria, P., Farooqui, J. et al., En-zymatic trimethylation of residue-72 lysine in cytochrome c.Effect on the total structure. Biochim. Biophys. Acta 1980,622, 144–150.
[36] Brems, D. N., Stellwagen, E., The effect of methylation on cy-tochrome c fragment complementation. J. Biol. Chem. 1981,256, 11688–11690.
[37] Cherney, M. M., Junior, C. C., Bowler, B. E., Mutationof trimethyllysine 72 to alanine enhances His79-heme-mediated dynamics of iso-1-cytochrome c. Biochemistry2013, 52, 837–846.
[38] Pollock, W. B., Rosell, F. I., Twitchett, M. B., Dumont,M. E., Mauk, A. G., Bacterial expression of a mitochon-drial cytochrome c. Trimethylation of lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition.Biochemistry 1998, 37, 6124–6131.
[39] Kang, C. H., Brautigan, D. L., Osheroff, N., Margoliash,E., Definitaion of cytochrome c binding domains bychemical modification. Reaction of carboxydinitrophenyl-and trinitrophenyl-cytochromes c with baker’s yeast cy-tochrome c peroxidase. J. Biol. Chem. 1978, 253, 6502–6510.
[40] Hildebrandt, P., Heimburg, T., Marsh, D., Powell, G. L., Con-formational changes in cytochrome c and cytochrome oxi-dase upon complex formation: a resonance Raman study.Biochemistry 1990, 29, 1661–1668.
[41] Pelletier, H., Kraut, J., Crystal structure of a complex be-tween electron transfer partners, cytochrome c peroxidaseand cytochrome c. Science 1992, 258, 1748–1755.
[42] Guo, M., Bhaskar, B., Li, H., Barrows, T. P., Poulos, T. L., Crystalstructure and characterization of a cytochrome c peroxidase-cytochrome c site-specific cross-link. Proc. Natl. Acad. Sci.USA 2004, 101, 5940–5945.
[43] Shevchenko, A., Wilm, M., Vorm, O., Mann, M., Mass spec-trometric sequencing of proteins silver-stained polyacry-lamide gels. Anal. Chem. 1996, 68, 850–858.
[44] Hart-Smith, G., Chia, S. Z., Low, J. K., McKay, M. J. et al., Sto-ichiometry of Saccharomyces cerevisiae lysine methylation:insights into non-histone protein lysine methyltransferaseactivity. J. Proteome Res. 2014, 13, 1744–1756.
[45] Pang, C. N., Tay, A. P., Aya, C., Twine, N. A. et al., Tools tocovisualize and coanalyze proteomic data with genomes andtranscriptomes: validation of genes and alternative mRNAsplicing. J. Proteome Res. 2014, 13, 84–98.
[46] Griffith, K. L., Wolf, R. E., Jr., Measuring beta-galactosidaseactivity in bacteria: cell growth, permeabilization, and en-zyme assays in 96-well arrays. Biochem. Biophys. Res. Com-mun. 2002, 290, 397–402.
[47] Greenlee, D. V., Andreasen, T. J., Storm, D. R., Calcium-independent stimulation of Bordetella pertussis adenylatecyclase by calmodulin. Biochemistry 1982, 21, 2759–2764.
[48] Farooqui, J., DiMaria, P., Kim, S., Paik, W. K., Effect of methy-lation on the stability of cytochrome c of Saccharomycescerevisiae in vivo. J. Biol. Chem. 1981, 256, 5041–5045.
[49] DeLange, R. J., Glazer, A. N., Smith, E. L., Identification andlocation of episilon-N-trimethyllysine in yeast cytochromesc. J. Biol. Chem. 1970, 245, 3325–3327.
[50] Dumont, M. E., Ernst, J. F., Sherman, F., Coupling of heme at-tachment to import of cytochrome c into yeast mitochondria.Studies with heme lyase-deficient mitochondria and alteredapocytochromes c. J. Biol. Chem. 1988, 263, 15928–15937.
[51] Dumont, M. E., Schlichter, J. B., Cardillo, T. S., Hayes, M.K. et al., CYC2 encodes a factor involved in mitochondrialimport of yeast cytochrome c. Mol. Cell. Biol. 1993, 13, 6442–6451.
[52] Kranz, R., Lill, R., Goldman, B., Bonnard, G., Merchant, S.,Molecular mechanisms of cytochrome c biogenesis: threedistinct systems. Mol. Microbiol. 1998, 29, 383–396.
[53] Nicholson, D. W., Neupert, W., Import of cytochrome c intomitochondria: reduction of heme, mediated by NADH andflavin nucleotides, is obligatory for its covalent linkage toapocytochrome c. Proc. Natl. Acad. Sci. USA 1989, 86, 4340–4344.
[54] Bottinger, L., Gornicka, A., Czerwik, T., Bragoszewski, P. et al.,In vivo evidence for cooperation of Mia40 and Erv1 in theoxidation of mitochondrial proteins. Mol. Biol. Cell 2012, 23,3957–3969.
[55] Jers, C., Soufi, B., Grangeasse, C., Deutscher, J., Mijakovic,I., Phosphoproteomics in bacteria: towards a systemic un-derstanding of bacterial phosphorylation networks. ExpertRev. Proteomics 2008, 5, 619–627.
C© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com