liquid-crystalline main-chain polymers with a poly(p-phenylene terephthalate) backbone, 8. synthesis...
TRANSCRIPT

Macromol. Chem. Phys. 195, 1305 -1317 (1994) 1305
Liquid-crystalline main-chain polymers with a poly@-phenylene terephthalate) backbone, 8 a)
Synthesis and characterization of polyesters with 3-ethoxypropoxy and 6-(pentyloxy)hexyloxy side chains
Frans I? M. Mercx, Alois H. A. Tinnemans, Sebastiaan B. Damman*
TNO Plastics and Rubber Research Institute, P. 0. Box 6031, 2600 JA Delft, The Netherlands
(Received: June 22, 1993)
SUMMARY: The influence of replacing a methylene group in the middle of the side chain of alkoxy-
substituted poly@-phenylene terephtha1ate)s by an oxygen atom was investigated by differential scanning calorimetry (DSC), rheological measurements and X-ray diffraction. Due to the presence of the oxygen atom the side chains of the polymer with long side chains (PTAlZ(0)HQ) lose their ability to crystallize. Except for this difference and a shift of the transition temperatures to lower temperatures, this polymer shows a similar phase behaviour compared to the corresponding alkoxy-substituted polyester PTA12HQ. The polymer with short side chains, FTA6(0)HQ, shows a similar behaviour as the corresponding polymer without an extra oxygen atom, PTA6HQ. Only the transition to the nematic mesophase is shifted to lower temperatures.
Introduction
The development of melt-processable liquid-crystalline (LC) polymers has received much attention in the past years. Melt-processability of a rigid rod polymer is usually attained by disrupting the regular structure of the rigid main chain by means of random copolymerization and/or the introduction of crankshafts ‘1 or flexible spacers’). Most commercially available thermotropic polymers are based on these principles. Another possibility is the introduction of flexible side chains onto the rigid aromatic backbone.
For polyesters with a rigid poly@-phenylene terephthalate) (abbreviated as pPT) backbone, mainly a l k ~ l ~ - ~ ) and alkoxy6-s) side chains were employed. For poly@-phenylene 2,5-dialkoxyterephthalate)s (abbreviated as PTAnHQ) a layered mesophase is observed if the side chains exceed a certain length (m 2 5 ) . In this layered mesophase the side chains interdigitate and the main chains form layer^^-^). The distance between these layers is proportional to the length of the side chains. In addition to this layered mesophase, a nematic phase is found for alkoxy side chains comprising 5 or 6 carbon Shorter side chains only render a nematic phase7). Most of the above mentioned alkoxy substituted pPT’s show two stable solid-phase structures at room temperature, denoted A and B7x9). Depending on the length of the side chains a separate melting endotherm of the alkoxy side chains can be found.
a) Part 7: cf. ref.’’)
0 1994, Huthig & Wepf Verlag, Basel CCC 1022-1 352/94/$08.00

1306 F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
Scheme I:
OR
L J n
FTA6HQ; R = -C,H,, FTAIZHQ; R = -C,,HZs FTA6(0)HQ; R = -(CHz)3-O-C2HS FTAlZ(0)HQ;R = -(CH,),-O-C,H,,
The polymers PTA6HQ and PTAl2HQ (see Scheme I ) were extensively studied in the past. For PTA6HQ a phase behaviour as depicted in Fig. 1 was proposed I*). At the main chain melting point, T,,, (180"C), the room temperature structure (A or B, depending on thermal history) transforms to the layered mesophase L,. The ordering of the main chains in layers disappears at 230°C where the nematic mesophase is formed. At a temperature of 285 "C the parallel ordering of the main chains is lost and an isotropic phase is formed.
The phase behaviour of PTA12HQ is shown in Fig. 2. At room temperature three different layered phases A, B and L, can be found, which differ in the amount of ordering of main and side chains (for details, see ref. 13)). At the main chain melting point of 170°C the layered mesophase is formed and at 240°C the isotropic phase is reached.
r m 1" Ii I: 18OOC :: 23OOC :: 285°C or^, L, t- - N I
Fig. 1 . Scheme of the phase behaviour of PTA6HQ powdersT2), showing the subsequent transitions from the two room temperature modifications A and B to the layered mesophase L,, the nematic mesophase N, and the isotropic phase I
The introduction of other elements in the side chain (i. e. 0 or F) and/or more bulky substituents can have a profound influence on the transition temperatures and phase behaviour of LC polyesters'. 14) . For LC polyimides I*) the replacement of the aliphatic side chains by oligo(oxyethy1ene) side chains was shown to suppress side chain crystallization. For the polymers PTA6HQ and PTA12HQ such a replacement might have a similar effect. This paper deals with the synthesis of the polymers PTA6(0)HQ and PTA12(0)HQ. Phase behaviour, solid state structure and rheology will be compared with the corresponding polymers of about the same side chain length, PTA6HQ and FTA12HQ.

Liquid-crystalline main-chain polymers with a . . . 1307
Fig. 2. Scheme of the phase behaviour of PTA12HQ fibres19). The two room temperature structures A and B can transfer to the intermediate phase A at the respective side-chain disordering temperatures T,(A) and T,(B). At the main-chain melting temperature T, the layered mesophase L, is formed. The isotropic phase I is reached at the clearing temperature Ti. By quenching from the layered mesophase L, the ‘frozen in layered mesophase’ L, is obtained, which slowly transforms to the modification B
Results and discussion
Synthesis and thermal stability
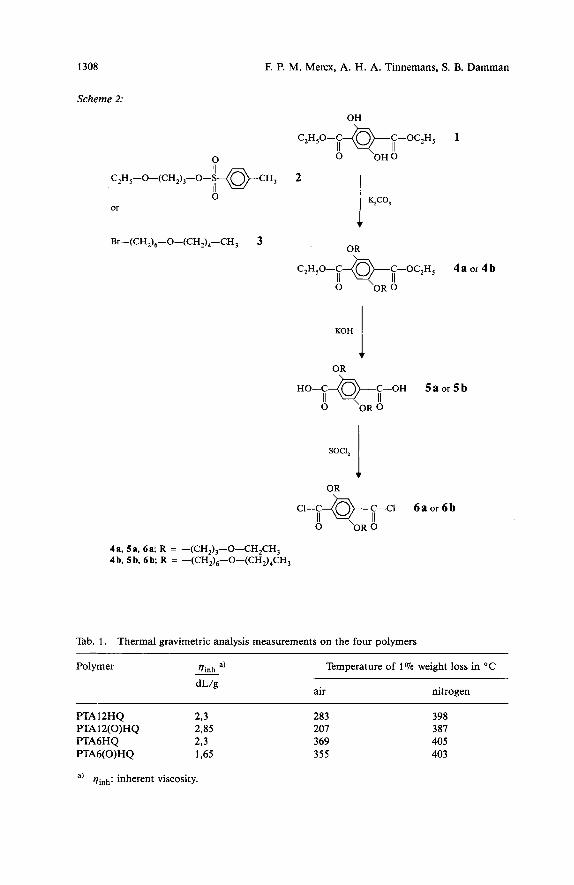
The monomers 6 a and 6b were obtained according to Scheme 2. 3-Ethoxy-1-propa- no1 was converted to the tosylate 1 6 ) 2, whereas the 6-bromohexyl pentyl ether (3) was synthesized by reacting sodium pentanolate with 1,6-dibromohexane. These com- pounds were used for the 0-alkylation of diethyl2,5-dihydroxyterephthalate (1) follow- ing the Claisen method6,”). The 0-alkylated diesters 4a and 4b were saponified using a 30 wt.-Yo aqueous potassium hydroxide solution to yield the corresponding acids 5a, 5 b, which in turn were converted to the acyl chlorides 6a, 6 b by refluxing with an excess of thionyl chloride6,12). Polymerization of 6 a and 6b with hydroquinone was carried out in solution according to known methods6p12). The molecular weights of the obtained polymers, as indicated by the inherent viscosity in chloroform, were sufficiently high in order to exclude serious effects of the molecular weight on the transition temperatures (2,15 to 2,85 dL/g for FTAl2(O)HQ and 1,48 to 1,65 dL/g for PTA6(0)HQ).
The thermal stabilities of FTA12(O)HQ and FTA6(0)HQ were evaluated by the use of thermal gravimetric analysis (TGA). The temperature at which 1070 of weight loss had occurred is shown in Tab. 1. For comparison the same measurements were performed on PTAIZHQ and PTA6HQ. The introduction of an extra oxygen atom has a severe influence on the thermal stability in air; especially the polymer with long side chains, PTA12(0)HQ, shows a strongly reduced thermal stability. The thermal stability in nitrogen, however, remains unchanged.
1308
Scheme 2:
F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
OH
0 II /I
K P , I c,H,-o-(cH,),-o-s-Q-cH, 2 0
or
OR Br-(CH,),-0-(CH,),-CH, 3
C,H~O-C*-OC,H~ II 4 a or 4 b
0 O R 0
OR
HO-C-&-OH II 5 a or 5 b
0 O R 0
I SOCI,
OR
4s. 5a, 621; R = -(CH,),-0-CH,CH, 4b, fib, 6b; R = -(CH,)6-O-(CH2),CH,
Tab. 1. Thermal gravimetric analysis measurements on the four polymers
Polymer -a' Temperature of 1 % weight loss in "C dL/g
air nitrogen
PTAl2HQ 2 3 PTA12(0)HQ 2,85 FTA6HQ 2,3 PTA6(0)HQ 1,65
a) qinh: inherent viscosity.
283 207 369 355
398 381 405 403
Liquid-crystalline main-chain polymers with a . . . 1309
.d
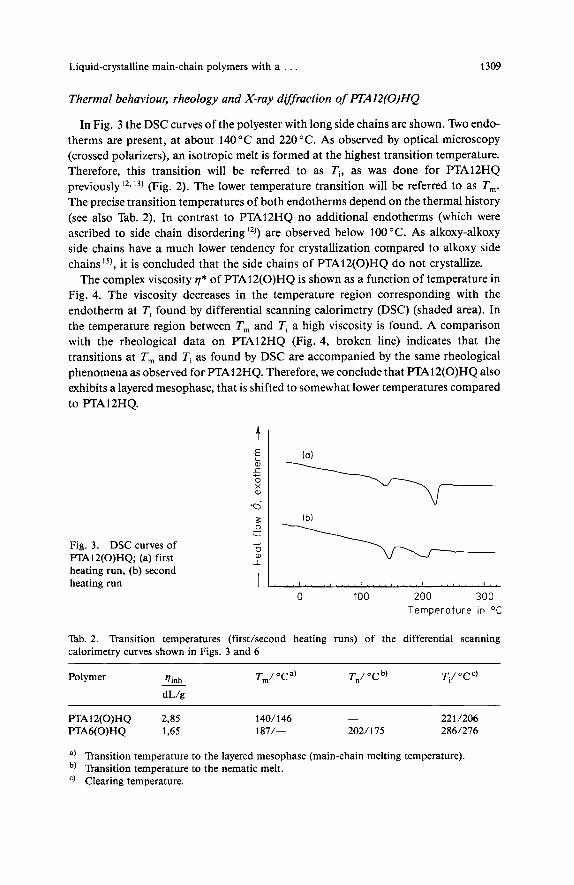
Fig. FTAlZ(0)HQ; 3. DSC curves (a) first of I
I heating run, (b) second heating run
Thermal behaviour, rheology and X-ray diffraction of pTA12(O)HQ
In Fig. 3 the DSC curves of the polyester with long side chains are shown. Two endo- therms are present, at about 140°C and 220°C. As observed by optical microscopy (crossed polarizers), an isotropic melt is formed at the highest transition temperature. Therefore, this transition will be referred to as Ti, as was done for PTA12HQ previously12.13) (Fig. 2). The lower temperature transition will be referred to as T,. The precise transition temperatures of both endotherms depend on the thermal history (see also Tab. 2). In contrast to lPTA12HQ no additional endotherms (which were ascribed to side chain disordering")) are observed below 100 "C. As alkoxy-alkoxy side chains have a much lower tendency for crystallization compared to alkoxy side chains'5), it is concluded that the side chains of PTA12(0)HQ do not crystallize.
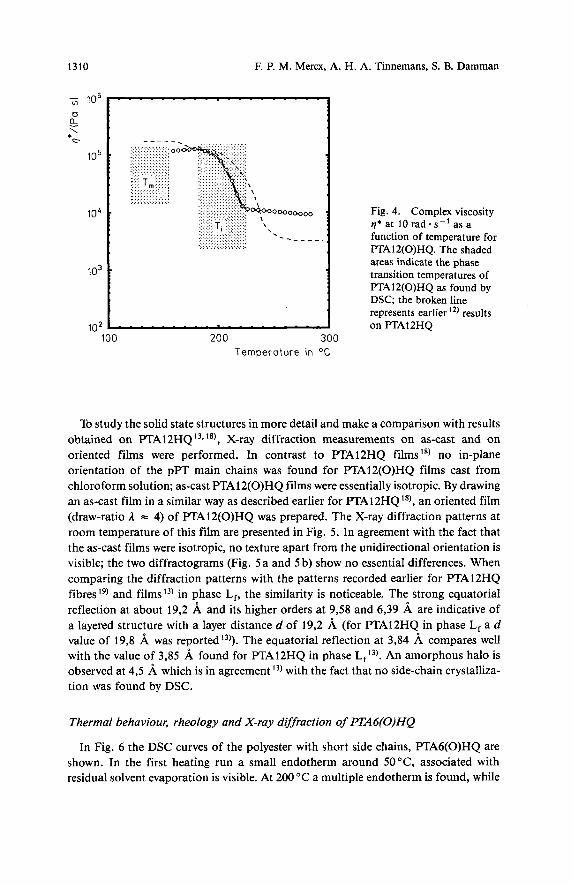
The complex viscosity q* of PTA12(O)HQ is shown as a function of temperature in Fig. 4. The viscosity decreases in the temperature region corresponding with the endotherm at Ti found by differential scanning calorimetry (DSC) (shaded area). In the temperature region between T, and Ti a high viscosity is found. A comparison with the rheological data on FTA12HQ (Fig. 4, broken line) indicates that the transitions at T, and Ti as found by DSC are accompanied by the same rheological phenomena as observed for PTA12HQ. Therefore, we conclude that PTA12(O)HQ also exhibits a layered mesophase, that is shifted to somewhat lower temperatures compared to PTAl2HQ.
1-1----, 1:
. . . . I . . . . I . . . . I . , . . I , . . I 1 . I ( . I I , . . 1 , .
Tab. 2. Transition temperatures (first/second heating runs) of the differential scanning calorimetry curves shown in Figs. 3 and 6
Polymer qinh T,/ 'Ca) T,/ "C b, Ti/ C c , - dL/g
~ ~~ ~
PTA12(0)HQ 2,85 140/ 146 - 22 1 /206 FTA6(0)HQ 1,65 187/- 202/175 2 8 6 / 2 7 6
a) Transition temperature to the layered mesophase (main-chain melting temperature). b, Transition temperature to the nematic melt. ') Clearing temperature.
1310
lo6 0 a \ c.
I
lo5
1oL
lo3
102 100
F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
200 300 Temperature in O C
Fig. 4. Complex viscosity q*at lOrad-s-'asa function of temperature for PTAlZ(0)HQ. The shaded areas indicate the phase transition temperatures of FTAlZ(0)HQ as found by DSC; the broken line represents earlier '*) results on PTAlZHQ
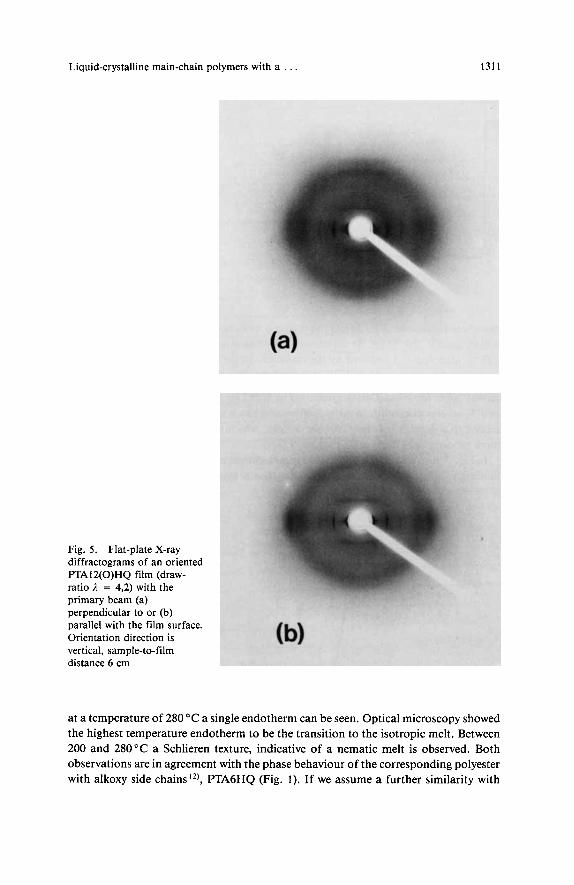
To study the solid state structures in more detail and make a comparison with results obtained on PTA12HQ"sLs), X-ray diffraction measurements on as-cast and on oriented films were performed. In contrast to PTA12HQ films'*) no in-plane orientation of the pPT main chains was found for PTAlZ(0)HQ films cast from chloroform solution; as-cast PTAlZ(0)HQ films were essentially isotropic. By drawing an as-cast film in a similar way as described earlier for PTAlZHQ I*) , an oriented film (draw-ratio ,I = 4) of PTA12(0)HQ was prepared. The X-ray diffraction patterns at room temperature of this film are presented in Fig. 5 . In agreement with the fact that the as-cast films were isotropic, no texture apart from the unidirectional orientation is visible; the two diffractograms (Fig. 5a and 5b) show no essential differences. When comparing the diffraction patterns with the patterns recorded earlier for FTA12HQ fibres 19) and films 1 3 ) in phase L,, the similarity is noticeable. The strong equatorial reflection at about 19,2 A and its higher orders at 9,58 and 6,39 A are indicative of a layered structure with a layer distance d of 19,2 A (for PTAl2HQ in phase L, a d value of 19,8 A was rep~rted'~)). The equatorial reflection at 3,84 A compares well with the value of 3,85 A found for PTA12HQ in phase L,13). An amorphous halo is observed at 4,5 A which is in agreement 1 3 ) with the fact that no side-chain crystalliza- tion was found by DSC.
Thermal behaviour, rheology and X-ray diffraction of PTA6(O)HQ
In Fig. 6 the DSC curves of the polyester with short side chains, PTA6(0)HQ are shown. In the first heating run a small endotherm around 50°C, associated with residual solvent evaporation is visible. At 200 "C a multiple endotherm is found, while
Liquid-crystalline main-chain polymers with a . . . 1311
Fig. 5 . Flat-plate X-ray diffractograms of an oriented F'TA12(0)HQ film (draw- ratio 1 = 4,2) with the primary beam (a) perpendicular to or (b) parallel with the film surface. Orientation direction is vertical, sample-to-film distance 6 cm
at a temperature of 280 "C a single endotherm can be seen. Optical microscopy showed the highest temperature endotherm to be the transition to the isotropic melt. Between 200 and 280°C a Schlieren texture, indicative of a nematic melt is observed. Both observations are in agreement with the phase behaviour of the corresponding polyester with alkoxy side chains "), FTA6HQ (Fig. 1). If we assume a further similarity with

1312
t E
X aJ
0 aJ
a, f
d - 2 L
c
I
I
F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
(a)
Fig. 6. DSC curves of PTA6(0)HQ; (a) first heating run, (b) second heating run ' " ' I , , , , ' I , I , ' . . , . ( . . , , I . . . . ' . . . . ' , . , .
the phase behaviour of F'TA6HQ the multiple melting endotherm in the temperature region around 200 "C can be explained as the transition to the layered mesophase at 185 "C (denoted T,) followed by the transition to the nematic mesophase at 205 "C (denoted T,). Compared to F'TA6HQ the temperature region in which the layered mesophase exists (between T, and T,) is smaller at a comparable molecular weight (about 20°C for PTA6(0)HQ, compared to 50°C for PTA6HQ"). The fact that T, and T, are less discernible in the second heating run is not yet understood. It might be related to a decrease of the molecular weight, which was shown earliert2) to have a more pronounced effect on T, than on T,.
In Fig. 7 the results of the rheological measurements on F'TA6(0)HQ are shown. The minimum in the curve in the temperature region between T, and Ti confirms the
0 a \ c
I
lo5
1oL
lo3
102 100 200 300
Temperature in "C
Fig. 7. Complex viscosity )I* at 10 rad . s - ' as a function of temperature for PTA6(0)HQ. The shaded areas indicate the phase transition temperatures of ITA6(0)HQ as found by DSC; the broken line represents earlier ") results on PTA6HQ

Liquid-crystalline main-chain polymers with a . . . 1313
existence of a nematic mesophase in this temperature region I*)). The transition to the nematic mesophase as found by DSC (shaded area) agrees well with the decrease of the melt viscosity.
As was done j9) for PTA6HQ the solid-state structure of PTA6(O)HQ is investigated by analyzing the X-ray diffractograms of oriented fibres. By spinning from the nematic mesophase at 240"C, followed immediately by draw-down (draw ratio I = 17) an oriented fibre of PTA6(0)HQ was obtained. The X-ray diffractogram of this fibre is shown in Fig. 8a. By annealing for one hour at 160°C the number of reflections
Fig. 8. Flat-plate X-ray diffractograms of an oriented PTA6(0)HQ fibre spun from the nematic mesophase; (a) as-spun fibre, (b) annealed at 160°C for one hour. Orientation direction is vertical, sample-to-film distance 6 cm

1314 F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
increases (Fig. 8b) pointing to a more ordered 3-dimensional structure. The reflections of the annealed fibre will be compared with those found earlierI9) for an annealed PTA6HQ fibre. The strong equatorial reflection at 13,3 A and its higher order at 6,63 A can be explained by a layer distance d of 13,3 A (or, in terms of the PTA6HQ indexing used before 19), the (100) reflection). The other prominent equatorial reflection at 3,67 A may very well be the (010) reflection. The strong reflection at 11,3 A on the first layer line could be the (021) reflection. All of the above mentioned reflections have very similar values compared to the annealed PTA6HQ fibre. However, as the amount of reflections is very low and the crystal structure of PTA6HQ may be complicatedz0), the assignment of a unit cell is not possible.
In contrast to what might be expected, annealing does not significantly alter the transition enthalpies of the PTA6(0)HQ fibre. This fact was, however, also observed for PTA6HQ. Apparently, the changes observed in the X-ray diagrams are minor rearrangements of the crystal packing (small longitudal shifts of the main chains relative to each other), insufficient to lead to a higher heat of melting or a higher melting temperature.
Conclusions
Replacing a methylene group in the centre of the side chains by an oxygen atom leads to small differences in phase behaviour and solid-state structures of pPTs with alkoxy side chains.
For the polymer with long side chains PTA12(0)HQ, the introduction of the ether link prevents side-chain crystallization. Compared with PTA12HQ, the LC phase is shifted to lower temperatures. The solid-state structure is very similar to the quenched mesophase of PTA12HQ, denoted phase L,.
The most important difference between PTA6(0)HQ and the reference polymer PTA6HQ is a shift of T, to lower temperatures. Consequently, the temperature region in which the nematic mesophase is found increases, while the layered mesophase is found only in a small temperature region.
Experimental part
Characterization methods
Differential scanning calorimetry (DSC) measurements were performed on a DuPont 9900 DSC, with a heating rate of 20"C/min in a nitrogen atmosphere. The peak of the melting endotherm was taken as the melting point.
Thermal gravimetric analysis (TGA) measurements were performed on a DuPont 951 thermogravimetric analyzer with a heating rate of 20 "C/min.
NMR spectra were recorded at 30°C on a Varian VXR400 spectrometer ( 'H resonance frequency 400 MHz, 13C resonance frequency 100 MHz) using a 5-mm switchable probe and a VXR data system. Tetramethylsilane (TMS) was used as internal reference.
Inherent viscosities were determined at 25 "C, with solutions of the polymer in CHCl, (2 g/L), using an Ubbelohde capillary viscosimeter.
Flat-plate X-ray diffractograms were recorded in transmission using Ni-filtered CuK, radia- tion.

Liquid-crystalline main-chain polymers with a . . . 1315
Rheological measurements were performed on a Rheometrics RDS-2 mechanical spectrometer in the parallel-plate arrangement using 25 mm plates. The parallel-plate gap spacing was 1 - 1,5 mm and the dynamic strain amplitude was 1 %. Samples were prepared by compressing dried powders to 25 mm plaques at 260°C (PTA6(0)HQ or 240°C (FTA12(0)HQ).
Materials
Diethyl 2,5-dihydroxyterephthalate (Riedel-de Haen, 98%), 3-ethoxy-1-propanol (Aldrich, 97%), p-toluenesulfonyl chloride, 1-pentanol, 1,6-dibromohexane, sodium dithionite (Aldrich, 85%) as well as acetone (Merck, p. a. quality) were used without further purification. Hydro- quinone (Merck, zur synthese) was purified by sublimation in vacuo. Thionyl chloride (Fluka, >99%), pyridine (Merck, p. a.), as well as 1,1,2,2-tetrachIoroethane (Merck, zur synthese) were distilled in vacuo prior to use.
Synthesis of monomers
3-Ethoxypropy1-p-toluenesulfDnate (2): In a 1 liter three-necked flask fitted with a magnetic stirrer and thermometer were placed, under nitrogen, 130,2 g (1,25 mol) of 3-ethoxy-1-propanol and 401 g (5.07 mol) of pyridine. After cooling this mixture to 1O"C, 230,5 g (1,21 mol) of p- toluenesulfonyl chloride were added in small portions over a 30-minute period (at such a rate that the temperature did not exceed 20°C at any time). The mixture was then stirred for 3 h at a temperature below 20 "C, after which it was diluted with 750 mL of 12 N hydrochloric acid in 2,5 L of ice water. Next, the mixture was transferred to a separatory funnel and extracted 4 times with 500 mL of diethyl ether. The combined organic layers were washed 5 times with 250 mL of water. It was then dried over anhydrous magnesium sulfate. The crude product, obtained as a colourless oil after removal of the solvent at reduced pressure, amounted to 273,7 g (91 Vo), and was used without further purification.
' H NMR (CDCI,): 6 = 1,lO (t; OCH2C_H3), 2,43 ( s ; CH,), 3,36 (4; OC_H2CH,, J = 7,O Hz), 1,89 (m; CH2C_H2CH2), 3.42 (t; C_H,0C2H,), 4,14 (t; SO2OC_H2), 7,34 and 7,79 (AB, Ar, JAB = 8.4 Hz).
Diethyl2,5-bis(3-ethoxypropoxy)terephthalate (4a): A solution of 107,8 g (0,424 mol) of 1, 263 g (1,02 mol) of 2 and 117,3 g (0,848 mol) of anhydrous potassium carbonate in 2,24 L of acetone was refluxed, under nitrogen atmosphere, for two weeks. After nearly complete reaction, as indicated by 'H NMR, the solids were filtered off and washed 3 times with small portions of acetone. From the collected pale-yellow filtrates the solvent was removed on a rotavapor, and the residual oil was dissolved in 200 mL of ethanol. Cooling to - 20°C afforded crystals which were collected in a chilled room, washed with cold ethanol, and dried in vacuo.
Yield: 66%; m.p.: 38-40°C. 'H NMR (CDCI,): 6 = 1,39 (t; COOCH2C_H,), 4,37 (4; COOC_H2CH,, J = 7,2 Hz), 1,19 (t;
OCH2C_H3), 3,49 (9; C_H,CH,, J = 7,2 Hz), 2,06 (m; CH2C_H2CH2), 3,62 (t; C_H20C2H,), 4,ll (t; ArOC_H,), 7,36 (s; H(Ar)).
C&3@8 (42651) Calc. C 61,96 H 8,04 Found C 61,86 H 8,21
2,5-Bis(3-ethoxypropoxy)?erephthalic cid(5 a): A solution of 1 19,6 g (0,28 mol) of 4 a in 295 mL of ethanol was treated, under reflux conditions, with a solution of 157,4 g (2,80 mol) of potassium hydroxide in 365 mL of water for 4 h. After cooling to 0 "C, the solution was neutralized with a small excess of a 6 N solution of concentrated HCI (pH,,,, 1,3). The crude product was diluted with water, collected by filtration and, after drying, recrystallized from an ethanol/water mixture (vol. ratio 4/1).
Yield: 87%; m. p.: 114,5- 1 16 "C.

1316 F. P. M. Mercx, A. H. A. Tinnemans, S. B. Damman
'H NMR (CDCI,): 6 = 1,21 (t; OCH,C_H,), 3,52 (9; OC_H,CH,, J = 7,2 Hz), 2,17 (m, CH,C_H,CH,), 3,63 (t; C_H,OC,H,), 4,39 (t; ArOC_H,), 7,86 (s; H(Ar)).
2,5-Bis{3-ethoxypropoxy)terephthaloyl chloride (6 a): The acid 5 a could be converted to the corresponding acyl chloride 6 a by stirring with a 1 0-fold excess of thionyl chloride for 19 h at room temperature. To avoid contact with air, the excess of thionyl chloride was removed by distillation in vacuo, in such a way that the temperature did not exceed 25 "C at any time. The crude product was purified by extraction with dry hexane for 4,5 h in a previously dried soxhlet apparatus. After filtration under nitrogen atmosphere, the solvent was removed from the clear, yellow solution by distillation in vacuo, as described above, yielding yellow crystals.
Yield: 83% m.p.: 70-71 "C.
Cl&,@&I, (407,29) Calc. C 53,08 H 5,94 C1 17,41 Found C 53,ll H 5,72 C1 17,08
6-Bromohexylpentyl ether (3): In a 1 L round-bottom flask, fitted with a magnetic stirrer and reflux condenser, were placed, under nitrogen atmosphere, 1630 mL (15 mol) of I-pentanol and 573 g (2,5 mol) of sodium (cut in small pieces). The reaction mixture was held at 80 "C until all the sodium has disappeared. After cooling, this solution of sodium pentanolate was added to a solution of 610 g (2,5 mol) of 1,6-dibromohexane in 1,5 L of dry toluene. The resulting precipitate was filtered off after stirring at reflux temperature for 19 h. Distillation at reduced pressure (76-81 "C, 0,04 mmHg) of the dried filtrate yielded 195,3 g of the crude product which was contaminated with about 1'70 of 1,6-dibromohexane and 2% of the bisether (CH,(CH,),O- (CH,)60(CH2)4(CH,), as seen by gas chromatography (GC). Redistillation via a Widmer column afforded the 6-bromohexyl pentyl ether (3) which contained less than 0,02% of the starting dibromohexane. This product was used without further purification.
2,5-Bis[6-{pentyloxy)hexyloxyJterephthalic acid (5 b): A solution of 65,4 g (0,257 mol) of 1, 215,4 g (0,858 mol) of the bromide, 71,15 g (0,514 mol) of anhydrous potassium carbonate, 2,32 g of NaI (1,8 mol-% with respect to the bromide) in 1 L of methyl ethyl ketone (MEK) was refluxed, under nitrogen atmosphere, for 10 d. After nearly complete reaction, as indicated by 'H NMR, the solids were filtered off, and washed with MEK. The residual oil obtained after removal of the solvent, was treated, under reflux conditions, with a solution of 144,2 g of potassium hydroxide in 337 mL of water and 700 mL of ethanol for 2 h. After cooling to 0 "C, the solution was neutralized with a small excess of a 6 N solution of HCI. The crude product was treated twice with about 500 mL of water, and once with 500 mL of ethanol. Crystallization from a mixture of methanoVethano1 (vol. ratio 2/1) afforded 128,6 g of white crystals.
Yield: 93%; m.p.: 102-103 "C. ' H NMR (CDCI,): 6 = 0,89 (t; CH,), 1.30- 1.33 (m; CH3, 1.43- 1,67 (m; CH,), 1,93 (m,
CH ) 3,40 (t; OC_H,CH,-), 3,42 (t; OC_H,CH,-), 4,30 (t; ArOCH,), 7,86 (s, H (Ar)).
(C,(Ar)), 122,8 (Ci(Ar)), 151,7 (C2(Ar)), 163,9 (COOH). I 2 C NMR (CDCI,): 6 = 14.0, 22,5, 25,6, 25,8, 28,3, 28,8, 29,4, 29,5, 70.4 71,0, 71,2, 117,5
C&@, (538,72) Calc. C 66,89 H 9,36 Found C 66,89 H 9,22
2,5-Bis[6-{pentyloxy)hexyloxyJterephthaloyl chloride (6 b): The acid 5 b could be converted in almost quantitative yield to the corresponding acyl chloride 6b by treatment, at room temperature, with a 12-fold excess of thionyl chloride for 1 3 h. After reflux of the solution for 3 h, the excess of thionyl chloride was removed by distillation in vacuo in such a way that the temperature did

Liquid-crystalline main-chain polymers with a . . . 1317
not exceed 60 "Cat any time. The acyl chloride was purified by dissolution in 250mL of dry hexane and filtration of the turbid solution under nitrogen. After removal of the solvent at ambient temperature, the residual crystalline product was dried in vacuo (20 "C, 0,l mmHg).
Yield: 98%; m. p.: 34-35 "C.
C~OH@&~, (57522) Calc. C 62,60 H 8,41 CI 12,32 Found C 62,87 H 8,46 CI 12,39
Polymerization
Polymers were synthesized by polycondensation in solution. In a typical example 43,00 g (74,7 mmol) of 6 b, an equivalent amount of freshly sublimed hydroquinone (8,23 g) and 224 mL of 1,1,2,2-tetrachloroethane were added to a three-necked flask fitted with a stirring unit, a dropping funnel and a nitrogen inlet. The resulting inhomogeneous solution was stirred for about 40 min at room temperature. Then, 24,2 mL (0,3 mol) of pyridine was slowly added over a 10-minute period. The mixture was subsequently stirred for 3 h at elevated temperatures (100 "C). After cooling, the mixture was poured into 3 L of methanol under vigorous stirring with an Ultra Turax stirring unit, and the precipitated polymer was collected by filtration. The polymer was purified by several washing steps: suspending in 1 L of methanol followed by filtration and washing with methanol, water and methanol, respectively. The final product was obtained after drying to constant weight in vacuo at 60°C. Yield was nearly quantitative.
We are indebted to Dr. Ir. J. A. H. M. Buijs for carefully reading the manuscript. Financial support from the Dutch Ministry of Economic Affairs (IOP-PCBP 302) and DSM is gratefully acknowledged.
I ) H. N. Yoon, L. F. Charbonneau, G. W. Calundann, Adv. Mater. 4, 206 (1992) 2, W. J. Jackson Jr., H. F. Kuhfuss, J. Polym. Sci., Part A: Polym. Chem. 14, 2043 (1976) 3, J. Majnusz, J. M. Catala, R. W. Lenz, Eur. Polym. J. 19, 1043 (1983) 4, T. Heitz, P. Rohrbach, H. Hdcker, Makromol. Chem. 190, 3295 (1989) ') H.-R. Dicke, R. W. Lenz, J. Polym. Sci., Part. A: Polym. Chem. 21, 2581 (1983) 6 , M. Ballauff, Makromol. Chem., Rapid Commun. 7, 407 (1986) ') M. Ballauff, Angew. Chem. 101, 261 (1989) 8, J. M. Rodriguez-Parada, R. Duran, G. Wegner, Macromolecules 22, 2507 (1989) ') A. Adams, H. W. Spiess, Makromol. Chem., Rapid Commun. 11, 249 (1990)
lo) M. Ballauff, G. F. Schmidt, Makromol. Chem., Rapid Commun 8, 93 (1987) U. Falk, H. W. Spiess, Makromol. Chem., Rapid Commun. 10, 149 (1989)
") S. B. Damman, F. P. M. Mercx, C. M. Kootwijk-Damman, Polymer 34, 1891 (1993) 13) S. B. Damman, G. J. Vroege, Polymer 34, 2732 (1993) 14) R. Sinta, R. A. Gaudiana, R. A. Minns, H. G. Rogers, Macromolecules 20, 2374 (1987) 15) F. Helmer-Metzmann, M. Ballauff, R. C. Schulz, G. Wegner, Makromol. Chem. 190, 985
i6) R. S . Tipson, J. Org. Chem. 9, 235 (1944) 1 7 ) L. Claisen, 0. Eislieb, Justus Liebigs Ann. Chem. 401, 30 (1913) '*) S. B. Damman, F. P. M. Mercx, P. J. Lemstra, Polymer 34, 2726 (1993) ") S. B. Damman, F. P. M. Mercx, J. Polym. Sci., Part B: Polym. Phys.31, 1759 (1993) 20) P. Galda, D. Kistner, A. Martin, M. Ballauff, Macromolecules 26, 1595 (1993) * I ) J. A. H. M. Buijs, S. B. Damman, J. Polym. Sci., Part B: Polym. Phys., in press
(1 989)