linkage disequilibrium maps constructed with common snps are useful for first-pass disease...
TRANSCRIPT

www.elsevier.com/locate/ygeno
Genomics 84 (20
Linkage disequilibrium maps constructed with common SNPs
are useful for first-pass disease association screens
P. Taillon-Millera,1, S.F. Sacconeb,1, N.L. Sacconec,1, S. Duana, E.F. Klossa,
E.G. Lovinsd, R. Donaldsona, A. Phongd, C. Had, L. Flagstada, S. Millera,
A. Drendela, D. Lindd, R.D. Millera, J.P. Riceb, P-Y. Kwokd,e,*
aDepartment of Dermatology, Washington University School of Medicine, St. Louis, MO 63110, USAbDepartment of Psychiatry, Washington University School of Medicine, St. Louis, MO 63110, USAcDepartment of Genetics, Washington University School of Medicine, St. Louis, MO 63110, USA
dCardiovascular Research Institute, University of California at San Francisco, Long 1332A, San Francisco, CA 94143, USAeDepartment of Dermatology, University of California at San Francisco, Long 1332A, San Francisco, CA 94143, USA
Received 10 August 2004; accepted 10 August 2004
Available online 17 September 2004
Abstract
To develop an efficient strategy for mapping genetic factors associated with common diseases, we constructed linkage disequilibrium
(LD) maps of human chromosomes 5, 7, 17, and X. These maps consist of common single nucleotide polymorphisms at an average
intermarker distance of 100 kb. The genotype data from these markers in a panel of American samples of European descent were analyzed to
produce blocks of markers in strong pair-wise LD. Power calculations were used to guide block definitions and predicted that high-level LD
maps would be useful in initial genome scans for susceptibility alleles in case-control association studies of complex diseases. As anticipated,
LD blocks on the X chromosome were larger and covered more of the chromosome than those found on the autosomes.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Polymorphism; Single nucleotide; Linkage disequilibrium; Genomics; Sample size
Introduction
With the finished reference human genome sequence
available and haplotype maps for human panels from three
continents being constructed [1], there is intense discussion
and debate on how to take advantage of the genomic
information to study common diseases with a genetic
approach. In an ideal world, the functions of all the genes
and their regulatory elements would be known and the
entire genome of an individual could be sequenced quickly
and inexpensively. Therefore, one could obtain the com-
plete genome sequences of a group of patients suffering
0888-7543/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.ygeno.2004.08.009
* Corresponding author. Fax: (415) 476 2283.
E-mail address: [email protected] (P-Y. Kwok).1 These authors contributed equally to this publication.
from a disease and compare them to those of a group of
appropriately selected control individuals and identify genes
or regulatory elements with functional differences between
the two groups. But until this level of biological under-
standing and technical capability is reached, one has to rely
on indirect tools in the search for disease-predisposing
alleles. While genetic linkage studies of families have been
very successful in identifying genes responsible for simple
traits, it has been proposed that association studies using
single nucleotide polymorphisms (SNPs) will be most
useful to identify genes involved in complex diseases [2].
Association testing relies on the principle that nearby SNPs
are often in linkage disequilibrium (LD); however, LD is
not a simple function of the distance between markers.
Instead, one observes a complicated pattern of regions of
extensive LD punctuated by regions of no LD across the
genome [3–9]. This pattern of LD is a result of many
04) 899–912

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912900
factors, including genetic drift, admixture, migration,
population structure, variable mutation rates, variable
recombination rates, gene conversion, and natural selection
[10].
Defining the patterns of LD and the underlying haplotype
structure across the human genome is expected to aid in
elucidating the causes of complex human diseases by
helping researchers design association studies that make
efficient use of SNP markers [11]. Several groups have
demonstrated that the haplotype structures are complex,
requiring many markers to define the major haplotypes
found in even small regions of the genome (10 to 20 kb)
[4,6,8,9,12,13]. What is often overlooked, however, is the
fact that many contiguous bhaplotype blocksQ are highly
correlated and that markers in neighboring haplotype blocks
are often in strong LD with each other [5,9]. These
observations prompted us to evaluate whether high-level
LD maps would capture useful long-range LD structure and
allow selection of representative btagQ SNPs in regions of
strong LD. A hierarchical strategy of performing initial
genome scans with reduced-density maps in case-control
association studies could then provide an efficient way to
identify genetic factors associated with common diseases. In
this strategy a secondary screen would likely be required to
localize a disease variant to a specific gene once a region of
association has been detected. Variants in regions with no
LD could be missed in the initial pass, as would those with
very low frequency or multiple alleles, but these cases will
be difficult with any screening strategy. For such cases a
second pass with increased density in regions of low LD
might be necessary. However, a whole genome scan
following this approach could potentially use many fewer
SNPs than has been previously proposed, thus making such
studies practical. The approach could also be used to study
genetic effects that have been localized to chromosomal
regions in prior family studies, or to survey candidate genes
of special interest, as it provides a means for prioritization of
SNPs for follow-up genotyping.
To develop this approach, we present the LD maps of
human chromosomes 5, 7, 17, and X (covering ~20% of the
human genome) consisting of common SNPs chosen
according to allele frequency data generated from three
panels of 42 individuals (European, Asian, and African
American) and genotyped in a panel of 94 American
samples of European descent. LD block definitions were
guided by power calculations, which predict that reduced-
Table 1
Characteristics of SNPs analyzed
Chromosome Chromosome
length (Mb)
SNPs
genotyped
5 179.7 1442
7 156.4 1753
17 82.3 662
X 149.5 1487
density maps will be useful in genetic analyses of common
diseases.
Results
We selected 5344 common SNPs (each with previ-
ously estimated minor allele frequencies of z10%) in
each of three panels of individuals of European, Asian,
and African descent (a list of SNPs is provided in
Supplemental Tables S1a-S1d) across human chromosomes
5, 7, 17, and X. These SNPs were genotyped in 94
genetically unrelated samples from the CEPH collection
(Supplemental Tables S2a and S2b) using a primer
extension assay with fluorescence polarization detection
[14]. On the X chromosome, only males were genotyped so
we were able to determine complete haplotypes across the
chromosome (Supplemental Table S3). The genotypes
generated from the autosomal SNPs are given in Supple-
mental Tables S4a–S4c.
For each chromosome, pair-wise linkage disequilibrium
between all markers with minor allele frequencies (MAF)
z10%was measured usingD,DV, and r2. Because the resultswere very similar between SNPs with MAFz10% and those
with MAF z20%, only the data from the SNPs with MAF
z20% will be presented. The LD results from all pair-wise
comparisons with SNPs with MAFz10% are not given with
this publication but are available at http://snp.wustl.edu/
snp_research/ld_blocks; edited results for markers withMAF
z20% are also available at this Web site.
Table 1 summarizes the statistics of the SNPs genotyped
in this study. As predicted, 5098 (N95%) of the SNPs
genotyped were indeed common in the CEPH panel we
studied, with MAF z10%. In fact, 4405 (83%) of the
SNPs had a MAF z20% in the CEPH panel. The mean
intermarker distance between the SNPs with MAF z20%
was found to be 151, 106, 148, and 124 kb in
chromosomes 5, 7, 17, and X, respectively. But the mean
intermarker distance can be skewed by large gaps between
sequencing contigs, the centromeric region, and regions in
which very few SNPs were available at the time of this
study. Fig. 4c shows that for the complete marker set 15%
were closer than 5 kb, about 50% were closer than 25 kb,
and more than 70% (3130 SNPs, MAF z20%,) were
within 100 kb of their nearest neighbor. When only these
3130 SNPs are considered, in fact the mean intermarker
SNPs with
MAF z10%
SNPs with
MAF z20%
Mean intermarke
distance (kb)
MAF z20%
1389 1188 151
1659 1470 106
638 550 148
1412 1197 125
r
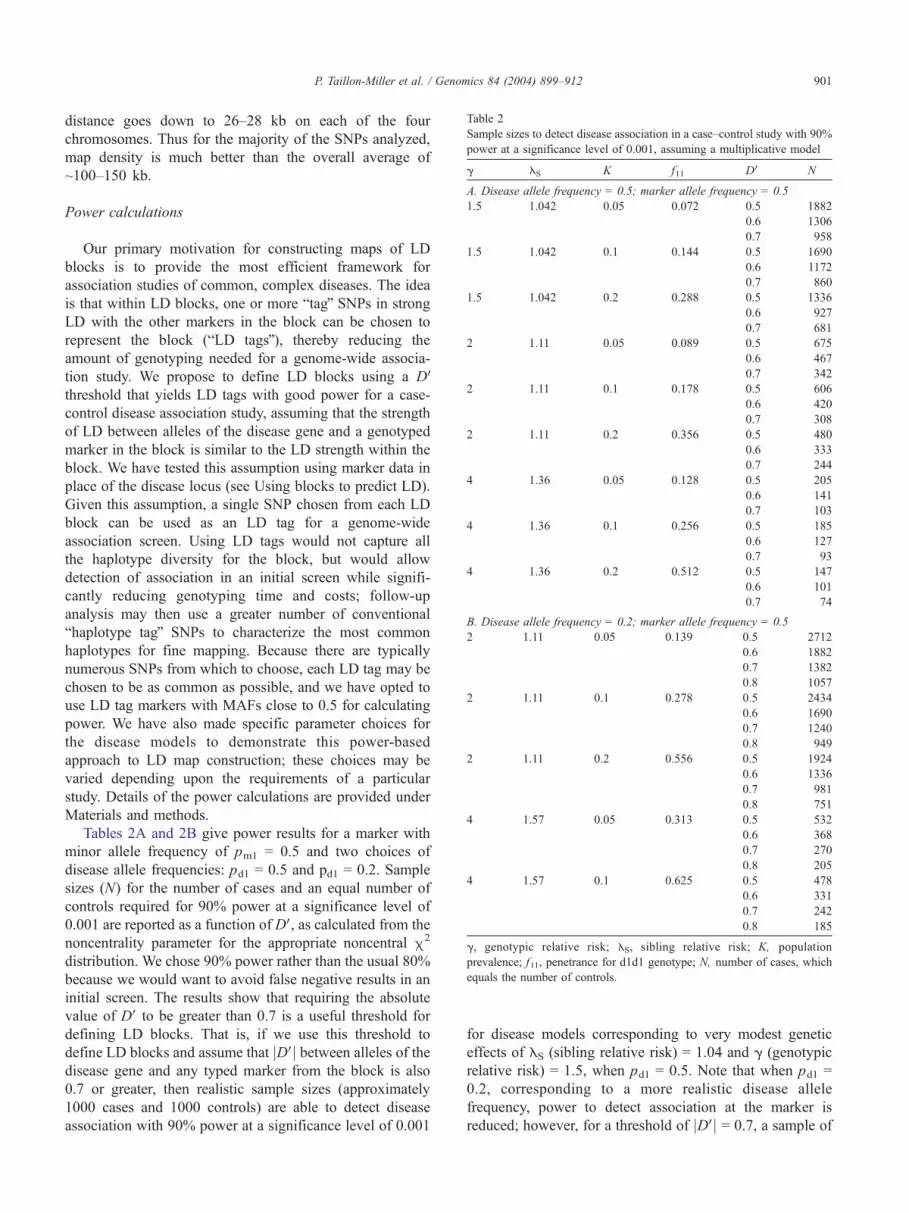
Table 2
Sample sizes to detect disease association in a case–control study with 90%
power at a significance level of 0.001, assuming a multiplicative model
g ES K f11 DV N
A. Disease allele frequency = 0.5; marker allele frequency = 0.5
1.5 1.042 0.05 0.072 0.5 1882
0.6 1306
0.7 958
1.5 1.042 0.1 0.144 0.5 1690
0.6 1172
0.7 860
1.5 1.042 0.2 0.288 0.5 1336
0.6 927
0.7 681
2 1.11 0.05 0.089 0.5 675
0.6 467
0.7 342
2 1.11 0.1 0.178 0.5 606
0.6 420
0.7 308
2 1.11 0.2 0.356 0.5 480
0.6 333
0.7 244
4 1.36 0.05 0.128 0.5 205
0.6 141
0.7 103
4 1.36 0.1 0.256 0.5 185
0.6 127
0.7 93
4 1.36 0.2 0.512 0.5 147
0.6 101
0.7 74
B. Disease allele frequency = 0.2; marker allele frequency = 0.5
2 1.11 0.05 0.139 0.5 2712
0.6 1882
0.7 1382
0.8 1057
2 1.11 0.1 0.278 0.5 2434
0.6 1690
0.7 1240
0.8 949
2 1.11 0.2 0.556 0.5 1924
0.6 1336
0.7 981
0.8 751
4 1.57 0.05 0.313 0.5 532
0.6 368
0.7 270
0.8 205
4 1.57 0.1 0.625 0.5 478
0.6 331
0.7 242
0.8 185
g, genotypic relative risk; ES, sibling relative risk; K, population
prevalence; f11, penetrance for d1d1 genotype; N, number of cases, which
equals the number of controls.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 901
distance goes down to 26–28 kb on each of the four
chromosomes. Thus for the majority of the SNPs analyzed,
map density is much better than the overall average of
~100–150 kb.
Power calculations
Our primary motivation for constructing maps of LD
blocks is to provide the most efficient framework for
association studies of common, complex diseases. The idea
is that within LD blocks, one or more btagQ SNPs in strong
LD with the other markers in the block can be chosen to
represent the block (bLD tagsQ), thereby reducing the
amount of genotyping needed for a genome-wide associa-
tion study. We propose to define LD blocks using a DVthreshold that yields LD tags with good power for a case-
control disease association study, assuming that the strength
of LD between alleles of the disease gene and a genotyped
marker in the block is similar to the LD strength within the
block. We have tested this assumption using marker data in
place of the disease locus (see Using blocks to predict LD).
Given this assumption, a single SNP chosen from each LD
block can be used as an LD tag for a genome-wide
association screen. Using LD tags would not capture all
the haplotype diversity for the block, but would allow
detection of association in an initial screen while signifi-
cantly reducing genotyping time and costs; follow-up
analysis may then use a greater number of conventional
bhaplotype tagQ SNPs to characterize the most common
haplotypes for fine mapping. Because there are typically
numerous SNPs from which to choose, each LD tag may be
chosen to be as common as possible, and we have opted to
use LD tag markers with MAFs close to 0.5 for calculating
power. We have also made specific parameter choices for
the disease models to demonstrate this power-based
approach to LD map construction; these choices may be
varied depending upon the requirements of a particular
study. Details of the power calculations are provided under
Materials and methods.
Tables 2A and 2B give power results for a marker with
minor allele frequency of pm1 = 0.5 and two choices of
disease allele frequencies: pd1 = 0.5 and pd1 = 0.2. Sample
sizes (N) for the number of cases and an equal number of
controls required for 90% power at a significance level of
0.001 are reported as a function of DV, as calculated from the
noncentrality parameter for the appropriate noncentral m2
distribution. We chose 90% power rather than the usual 80%
because we would want to avoid false negative results in an
initial screen. The results show that requiring the absolute
value of DV to be greater than 0.7 is a useful threshold for
defining LD blocks. That is, if we use this threshold to
define LD blocks and assume that jDVj between alleles of thedisease gene and any typed marker from the block is also
0.7 or greater, then realistic sample sizes (approximately
1000 cases and 1000 controls) are able to detect disease
association with 90% power at a significance level of 0.001
for disease models corresponding to very modest genetic
effects of ES (sibling relative risk) = 1.04 and g (genotypic
relative risk) = 1.5, when pd1 = 0.5. Note that when pd1 =
0.2, corresponding to a more realistic disease allele
frequency, power to detect association at the marker is
reduced; however, for a threshold of jDVj = 0.7, a sample of

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912902
approximately 1000 cases and 1000 controls still gives 90%
power at a significance level of 0.001 for disease models
corresponding to a slightly larger sibling relative risk of 1.11
and g = 2.
Needless to say, for disease genes with stronger genetic
effects, smaller sample sizes are sufficient; alternatively,
larger sample sizes will yield comparable power even if
smaller thresholds for jDVj are chosen (cf. the table entries
for g = 4). Additional power calculations indicate that with a
threshold of jDVj = 0.7, reasonable power to detect genetic
effects for a common disease is maintained for small
departures of the marker allele frequency from 0.5. For
example, consider a disease with prevalence of 0.2 and g
ranging from 2 to 3. If pm1 = 0.4 and pd1 = 0.2, and
considering both possible phases of co-occurrence of the
alleles, the range of sample sizes runs from N = 229 to N =
1451. If the prevalence is lowered to 0.1, sample sizes are
still under 2000 cases and 2000 controls. Thus as long as
LD block tags can be chosen to have allele frequency close
to 0.5, reasonable power is maintained.
We therefore chose a jDVj threshold of 0.7 to define our
primary blocks, described below, to maximize the LD block
size while minimizing the sample size needed for a useful
range of disease models. We also report LD block results for
jDVj thresholds of 0.6, 0.8, 0.9, and 1.0.
Linkage disequilibrium blocks
We define a linkage disequilibrium block to be a
consecutive set of at least three markers along with a
threshold T and a disequilibrium coefficient L (DV or r2)
such that all pairs of markers in the block satisfy jLj z T. By
a weak linkage disequilibrium block we mean a set of at
least three consecutive markers along with three parameters
Tl, Tu, and F and a disequilibrium coefficient L such that all
pairs of markers in the block satisfy jLj z Tl (the lower
threshold) and at least F% of the pairs of markers satisfy
jLj z Tu (the upper threshold). Guided by our power
analyses, we will focus on LD blocks constructed using a
threshold T = 0.7. Although our power calculations were
based on DV, they may be reformulated to correspond to r2
levels via the formula r ¼ Dffiffiffiffiffiffiffiffiffiffiffiffiffip1p2q1q2
p .
Tables 3A and 3B contain summary statistics for the
average and maximum number of the LD blocks as well
as the average and maximum size of the blocks on each
chromosome as found by our algorithm for the coefficients
DV and r2, respectively. To indicate the extent of these
blocks, Table 3 also gives the percentage of the total
number of SNP markers that fall within blocks, and also
statistics Rmax, Rhap, and bcoverage,Q defined as follows.
From the point of view of reducing genotyping, the best
case scenario would be when a single SNP can be chosen
to represent each LD block; note that for SNPs not lying
within any block, no breductionQ takes place and each
such SNP must still be included in the genotyping. The
original number of SNPs is then reduced by a percentage
we call Rmax. For our chromosome X data, if there exists
a subset of SNPs in a block that determines all possible
haplotypes observed, we may choose to use this subset to
represent the block. We then obtain a set of SNPs
consisting of these representatives for every block together
with all SNPs not in blocks, and the original number of
SNPs is then reduced by a percentage we call Rhap. An
example of an LD block with significant haplotype
reduction is given in Fig. 1, in which all the haplotypes
found in the males we genotyped are unambiguously
defined by only 3 SNPs in an LD block consisting of 13
SNPs. We define the coverage to be the percentage of the
chromosome covered by blocks while ignoring gaps
greater than 100 kb. This definition allows us to account
for variable spacing of markers and focus on the parts of
the chromosomes that are most densely covered by our
SNP map. See Fig. 2 for a graph of the coverage by DVthreshold. Additional details on coverage are provided
under Materials and methods.
Chromosomal coverage
At the jDVj threshold of 0.7 the autosomes exhibit block
coverage of roughly 25%, while the coverage on the X
chromosome is substantially higher at 43%. Our definition
of coverage is in fact conservative compared to the
percentage of markers that occur within blocks, as the
latter measure is uniformly higher than our measure of
coverage (Table 3). While the percentage of markers in
blocks does not account for the variable spacing of our
SNP markers, it still provides a useful measure and reveals
that blocks are extensive across the chromosomes in these
data. With a jDVj threshold of 0.7, roughly 40% of
autosomal SNPs and 60% of chromosome X SNPs fall
within blocks. A graphical representation of the LD block
structure of all four chromosomes is given in Fig. 3. It is
clear from this figure that LD blocks are found across the
entire length of all four chromosomes and in some cases
can be extensive. The markers included in each LD block
are in Supplemental Table S5.
The reduction (Rmax) in the total number of SNPs,
obtained by taking one tag SNP per block while still
including every SNP that does not lie in a block, is
approximately 29% on the autosomes and 44% on X.
Hence, in theory, we can genotype just 71% of our markers
and still retain a significant amount of power in a genome-
wide association screen. On the X chromosome, only 53%
of the SNPs have to be genotyped in a genome-wide
association study instead of 73% of the SNPs needed to
define all the common haplotypes (e.g., for a threshold of
0.7, Rmax = 47% versus Rhap = 27%).
Overall, the coverage results were similar for the
autosomes while the coverage on chromosome X was
nearly twice as much. Indeed, even when we require that
jDVj = 1 the coverage on X is still 22%; roughly three times
that of the autosomes (Fig. 2). These observations confirm

Table 3
|DV| threshold Number
of blocks
Mean size of
blocks (SNPs)
Max size
(SNPs)
Mean
width (kb)
Max
width (kb)
% of markers
within blocks
Rmax
(%)
Rhap
(%)
Coverage
(%)
A. LD blocks using DV
Chromosome X
0.6 148 4.9 14 129.3 2410 60.65 48.3 21.6 47.4
0.7 145 4.7 13 104.7 987 56.47 44.4 20.5 43.2
0.8 137 4.5 13 91.2 987 51.13 39.7 19.9 36.5
0.9 124 4.3 13 76.3 689 44.11 33.8 18.2 28.4
1 107 4.1 12 67 689 36.51 27.6 16 21.7
Chromosome 5
0.6 111 4.3 15 92.6 536 40.49 31.1 - 28.4
0.7 106 4.3 13 82.3 536 38.13 29.2 - 25.4
0.8 101 4.1 11 69 536 34.51 26 - 22.1
0.9 87 3.8 11 47 323 28.11 20.8 - 15.8
1 64 3.5 7 26.8 171 18.69 13.3 - 6.5
Chromosome 7
0.6 158 4.1 10 79.3 636 43.61 32.9 - 27.1
0.7 149 4 11 74.1 471 40.48 30.3 - 23.4
0.8 138 3.9 10 63.5 446 36.67 27.3 - 19.9
0.9 115 3.8 11 51.7 446 30.07 22.2 - 14.9
1 76 3.4 10 40.3 446 17.76 12.6 - 8.3
Chromosome 17
0.6 58 3.9 6 71.8 426 40.91 30.4 - 29.9
0.7 53 3.8 6 90.8 963 36.55 26.9 - 26.9
0.8 45 3.6 6 52.6 339 29.45 21.3 - 19.1
0.9 39 3.5 6 47.7 339 24.73 17.6 - 15.8
1 25 3.2 4 18 74 14.36 9.8 - 5
B. LD blocks using r2
Chromosome X
0.6 98 4.2 13 73.3 700 34.5 26.3 16 21.4
0.7 91 4.1 13 72.4 689 31.08 23.5 14.9 19.3
0.8 71 4.2 11 76.5 689 24.64 18.7 13.2 15
0.9 61 3.9 9 56.3 527 19.8 14.7 11.8 10.6
1 39 3.5 8 36.2 169 11.53 8.3 8.3 5.4
Chromosome 5
0.6 61 3.9 10 59.9 525 19.78 14.7 - 11.7
0.7 52 3.6 10 46.4 366 15.91 11.5 - 8.6
0.8 39 3.6 8 42.8 366 11.7 8.4 - 5.7
0.9 28 3.2 4 28.8 222 7.58 5.2 - 3.2
1 7 3.1 4 23.1 67 1.85 1.3 - 0.8
Chromosome 7
0.6 76 3.5 8 49.6 357 17.96 12.8 - 8.6
0.7 63 3.3 6 35.1 189 14.35 10.1 - 6.7
0.8 46 3.3 6 29 189 10.41 7.3 - 3.8
0.9 33 3.2 6 17 62 7.07 4.8 - 1.8
1 8 3.2 4 7.1 20 1.7 1.2 - 0.2
Chromosome 17
0.6 28 3.2 4 38.1 163 16.18 11.1 - 10.7
0.7 23 3.2 4 39.3 163 13.45 9.3 - 8.9
0.8 15 3.2 4 24.7 148 8.73 6 - 3
0.9 11 3.2 4 24.8 148 6.55 4.6 - 1.9
1 5 3.2 4 8.8 21 2.91 2 - 0.4
Rmax is the reduction, expressed as a percentage, in the total number of markers if one SNP from each block is selected and all remaining SNPs not in blocks are
also selected. Rhap is the reduction if SNPs are selected on the basis of complete haplotype determination within blocks. The coverage is the percentage of the
chromosome covered by blocks if gaps larger than 100 kb are ignored.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 903
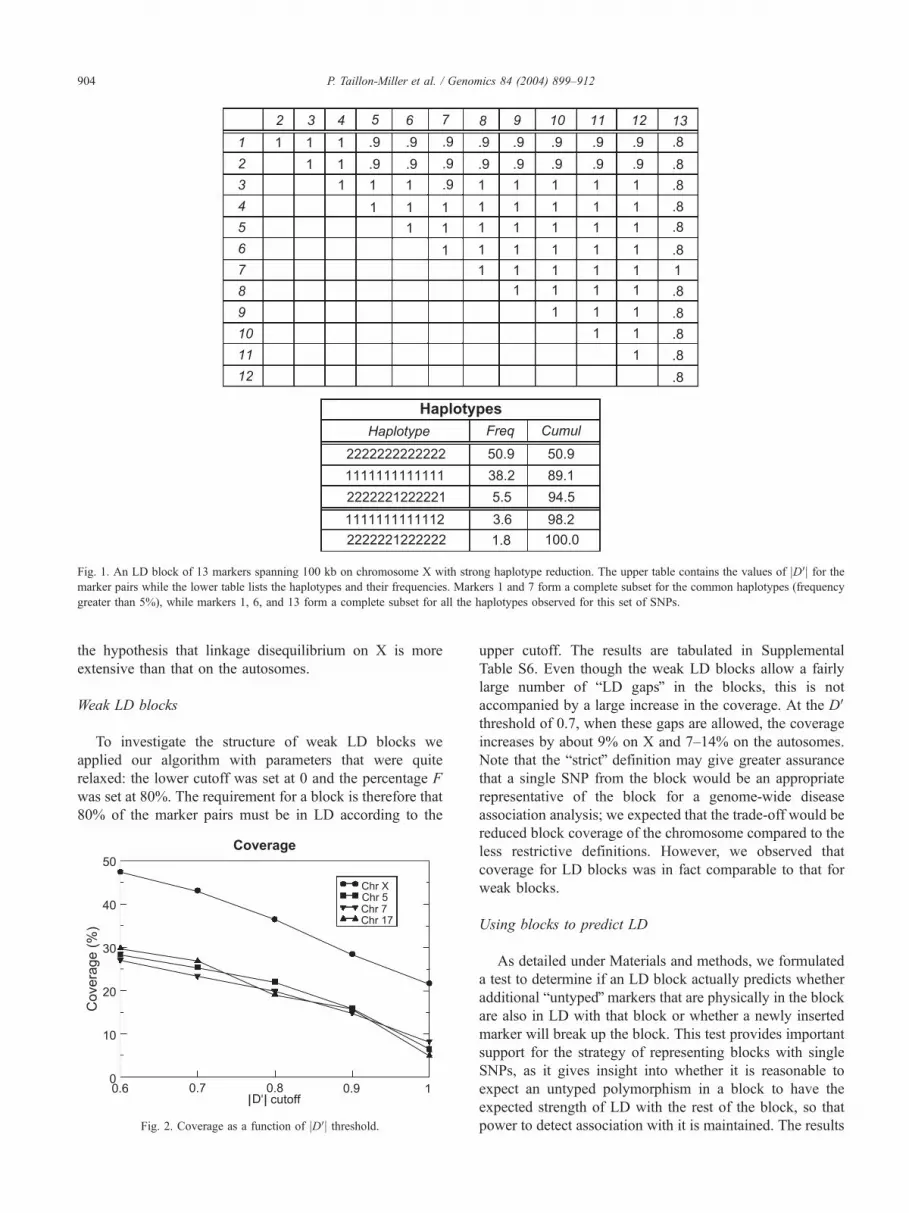
Fig. 1. An LD block of 13 markers spanning 100 kb on chromosome X with strong haplotype reduction. The upper table contains the values of jDVj for themarker pairs while the lower table lists the haplotypes and their frequencies. Markers 1 and 7 form a complete subset for the common haplotypes (frequency
greater than 5%), while markers 1, 6, and 13 form a complete subset for all the haplotypes observed for this set of SNPs.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912904
the hypothesis that linkage disequilibrium on X is more
extensive than that on the autosomes.
Weak LD blocks
To investigate the structure of weak LD blocks we
applied our algorithm with parameters that were quite
relaxed: the lower cutoff was set at 0 and the percentage F
was set at 80%. The requirement for a block is therefore that
80% of the marker pairs must be in LD according to the
Fig. 2. Coverage as a function of jDVj threshold.
upper cutoff. The results are tabulated in Supplemental
Table S6. Even though the weak LD blocks allow a fairly
large number of bLD gapsQ in the blocks, this is not
accompanied by a large increase in the coverage. At the DVthreshold of 0.7, when these gaps are allowed, the coverage
increases by about 9% on X and 7–14% on the autosomes.
Note that the bstrictQ definition may give greater assurance
that a single SNP from the block would be an appropriate
representative of the block for a genome-wide disease
association analysis; we expected that the trade-off would be
reduced block coverage of the chromosome compared to the
less restrictive definitions. However, we observed that
coverage for LD blocks was in fact comparable to that for
weak blocks.
Using blocks to predict LD
As detailed under Materials and methods, we formulated
a test to determine if an LD block actually predicts whether
additional buntypedQ markers that are physically in the block
are also in LD with that block or whether a newly inserted
marker will break up the block. This test provides important
support for the strategy of representing blocks with single
SNPs, as it gives insight into whether it is reasonable to
expect an untyped polymorphism in a block to have the
expected strength of LD with the rest of the block, so that
power to detect association with it is maintained. The results

Fig. 3. A blue diamond indicates the location and size of each LD block. The scale of the size of the LD block is located on the left y axis. The location on the
chromosome in kilobases is on the x axis. At the bottom of each graph represented by a black square is the location of each marker with a minor allele
frequency N20%. A red circle indicates the number of markers in each LD block with the scale on the right x axis. For the markers included in each LD block
and their locations on the chromosome please refer to Table S5 in the supplemental materials.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 905

Table 4
Results of test to determine if LD blocks predict LD
T1 T2 p
X 5 7 17
0.7 0.7 96.1 94.1 94.3 92.8
0.8 0.7 98.2 97.1 96.1 91.9
0.8 0.8 96.7 95.5 94.1 86.2
T1, the jDVj threshold for all pairs in the computed block of SNPs, A1, . . .,
An; T2, the jDVj threshold required for pairings with the additional SNP, B;
p, the empirical conditional probability that each pairing of B with Ai
satisfies jDVj z T2.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912906
of our test are summarized in Table 4. We see that indeed,
LD blocks are excellent predictors of the strength of LD
with other SNPs in our data. For example, on chromosome
5, if a collection of markers A1, A2, . . ., An is an LD block
with jDVj z 0.8 (the first threshold T1), then any other
marker B that is physically in this block will be, with 97.1%
certainty, in LD with A1, A2, . . ., An with jDVj z 0.7 (the
second threshold T2).
Discussion
The results of our study show that regions of strong LD
are commonly found on both autosomes and the X
chromosome with the X more extensively covered by
longer LD blocks. In fact, blocks of strong LD are easily
found with high-level SNP maps on chromosomes 5, 7,
17, and X consisting of common SNPs at ~100 kb density.
Power calculations predict that these LD blocks can be
represented by LD tag SNPs in genome-wide association
studies to identify alleles associated with common diseases
within LD blocks. As the number of common genotyped
SNPs continues to increase, we will be able to cover the
genome with LD blocks. Our results further predict that
from an overall map of 30,000 to 50,000 SNPs with minor
allele frequencies of z20% in each of the major
populations in the world, a subset of LD tag SNPs will
be useful in the initial genome-wide scanning for common
disease alleles.
The common SNPs selected for our study were identified
prior to the completion of the human genome sequence and
the launching of the International HapMap Project. Large
gaps therefore were found between contigs and the coverage
of common SNPs was incomplete. Despite these limitations,
LD blocks, defined by the DVstatistic, covered about 25% of
the autosomes and 40% of chromosome X where there was
sufficient SNP density. On average the blocks spanned
about 80 kb on the autosomes and 100 kb on X.
We designed an algorithm that reveals a block structure
whose usefulness is measured both by sharp reduction of the
number of SNPs needed in a genetic association study and
by the extent to which the LD blocks cover a chromosome.
In addition, our block definitions have the advantage of
being linked directly to the goal of using LD blocks to aid
disease association studies. As the motivation for our block
definitions differs from other important motivations such as
representation of haplotypes or study of possible evolu-
tionary models, the resulting block characteristics may differ
from those of other studies without detracting from our
conclusions. Our use of power calculations to guide block
definitions directly does rely on the assumption that the
strength of LD among the markers in the block is similar to
that of the LD that would be observed between those
markers and a disease gene if the disease gene occurred in
the block, but was untyped. We tested this assumption
directly within our data using genotyped markers in place of
the disease gene and found that for jDVj thresholds of 0.7 to
0.8, if an additional marker physically within an existing LD
block was introduced, it would satisfy the same jDVjthreshold with every block marker in the great majority of
cases. Note also that when this assumption is satisfied,
selecting a single LD tag SNP to represent the block, rather
than the more conventional approach of representing all
common haplotypes with a group of tag SNPs, may be
sufficient to detect a disease association in an initial screen,
provided appropriate parameters are chosen for the power
calculations. Further investigations that will use dense
marker data to help determine the most appropriate
strategies for SNP reduction are under way.
The above findings about the predictive ability of these
LD blocks are also consistent with our observations about
weak LD blocks. LD structure is complex and it is well
known that runs of SNPs in LD can be interrupted by SNPs
not in LD with the others [12,13]. To test this effect, we also
studied weak LD blocks, which allowed LD gaps within
blocks. Interestingly, we found that although the weak
definition would be expected to allow for larger blocks
encompassing more of the chromosome, in fact chromoso-
mal coverage increased only minimally.
The power analyses reported here considered only certain
disease models and thus should be viewed as only a guide to
selecting LD thresholds for block definition. We chose
marker allele frequencies of 0.5 for simplicity to illustrate
our approach; however, it is important to note that power is
affected by the frequency of the marker allele that tends to
co-occur with the disease allele as others have pointed out as
well [15,16]. Our overall approach provides a template
strategy to define LD blocks and prioritize SNPs for a given
disease association study; appropriate disease models and
the desired level of power may be chosen depending on the
particular study. Our report has focused on LD blocks for DVthresholds of 0.7 and above since these thresholds corre-
spond to power to detect common disease genes of modest
effect for the selected models. However, for disease genes of
stronger effects, smaller sample sizes or reduced jDVjthresholds (and therefore LD blocks having greater chro-
mosomal coverage) can yield sufficient power (Table 2).
Thus a given association genome screen can be designed
according to whether a project wishes to constrain recruiting
costs or genotyping costs.

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 907
Our hierarchical association mapping strategy is geared
toward detection of common disease alleles. The debate
about the bcommon disease, common geneQ hypothesis is
important and may be resolved by further study and
successful identification of such genes. Larger sample sizes,
stronger LD thresholds, or alternative strategies may be
necessary to allow the approach proposed here to move
beyond detecting common susceptibility alleles. Never-
theless, for detecting common alleles, a hierarchical
approach using an initial reduced-density SNP map offers
an efficient and economical alternative to large-scale
genotype studies.
In the initial phase of genetic association studies, it is
important to identify regions of the human genome
associated with a disease quickly and cost effectively.
Equally important is the ability to exclude large regions of
the genome not associated with the disease alleles. If N is
the number of individuals, M the number of markers being
genotyped, Cg the cost of genotyping a single marker for a
single individual, and CT the total cost, then CT = NMCg. N
is determined by our power calculations and M is
determined by the block structure for a given DV cutoff.Although our maps are not populated with enough SNPs to
determine M accurately, efforts such as the International
HapMap Project [1] are providing genotyping data for an
extremely dense set of SNPs across the genome. An
important next step for our approach will be to apply it to
these data to construct high-level, reduced-density associa-
tion screen maps, which can be compared to the full-density
data. Our present results strongly suggest that for the initial
genome-wide association studies of common disease,
screening maps of common SNPs at a modest density can
maintain power to detect disease association while reducing
genotyping costs compared to the approach of constructing
haplotype maps to define the common haplotypes.
Materials and methods
SNP markers
All SNPs used in this study had known frequencies,
mapped to a unique location according to the dbSNP map
(build 112), and met our criteria for primer design
(discussed below).
All SNP markers were selected from public databases or
selected and characterized for allele frequencies by our
group. They were then submitted to the SNP Consortium
(TSC) and dbSNP databases (dbSNP, http://www.ncbi.nlm.
nih.gov/SNP/; TSC, http://snp.cshl.org/).
SNPs with allele frequencies from 10 to 90% in all three
population samples of 42 individuals each (European
American, African American, and Asian American-bTheTSC setQ) were selected for genotyping. We also included a
small number of SNPs in which the allele frequencies were
between 10 and 90% in two of the three samples but the
information was not available for the third sample. In a
small number of cases from dbSNP, if the frequencies from
an equivalent set of samples met the criteria the SNP was
included.
We tracked the progress of this field by first discovering
SNPs, then characterizing SNPs, and finally taking advant-
age of the wealth of characterized SNPs available in the
public databases. We used a top-down hierarchical approach
to covering the chromosome with SNPs. SNPs were picked
across the chromosome at specific intervals without regard
for gene densities or gene locations. The first pick was
characterizing for frequency of one SNP every 25 kb across
the entire human genome with the SNPs and maps available
at that time. The SNPs with the desired frequencies were
then funneled into the genotyping project for chromosomes
X and 7. A second pick for characterization was done for
both chromosomes X and 7 at 10 kb, and chromosome X
went through a third round of characterizing SNPs at 5-kb
intervals. We also picked SNPs for chromosomes X and 7
from public databases when more characterized SNPs
became available. This study began with chromosomes X
and 7 and then later expanded to 20% of the genome with
the addition of chromosomes 5 and 17. Chromosome 5 and
17 SNPs were picked entirely from public databases that
included our own submissions. A list of the SNPs used in
this study is in supplemental Tables S1a–S1d.
SNP marker quality
All 5344 SNP assays used in this study (supplemental
Tables S1a-S1d) had a sample genotyping success rate
greater than 80%. Chromosome 7 had the best chromosome
coverage with 1753 SNPs for an average intermarker
distance of 89 kb and chromosome 5 had the least coverage
with 1442 markers and an average consecutive intermarker
distance of 125 kb. A breakdown of the number of SNPs at
10 and 20% MAF and 20% with intermarker distances b100
kb is shown in Table 1.
Fig. 4A shows the distribution of the MAF and Fig. 4B
shows the observed heterozygosity for all the SNP markers
used in this study. SNPs for this study were picked to have
a MAF of 10–50%. Although individual SNPs could have
different genotype frequencies from the predicted frequen-
cies, N95% of the SNP assays had a MAF N10% after
genotyping and N80% of the SNPs had a MAF N20% after
genotyping (Table 1). The estimate of heterozygosity
provided by dbSNP was also a good predictor of the
SNP frequency and was on average within 5% for each
bin. Between 5 and 6% of all SNPs genotyped had
heterozygosities less than 0.2 with the remaining 94–95%
being N0.2.
The distance from one SNP marker to the next is shown
in Fig. 4C. Approximately 50% of the markers are closer
than 25 kb and 70–80% of the markers are closer than 100
kb. The inset bar graph shows the 0–25,000 bp range. All
distances used were from dbSNP, build 112.
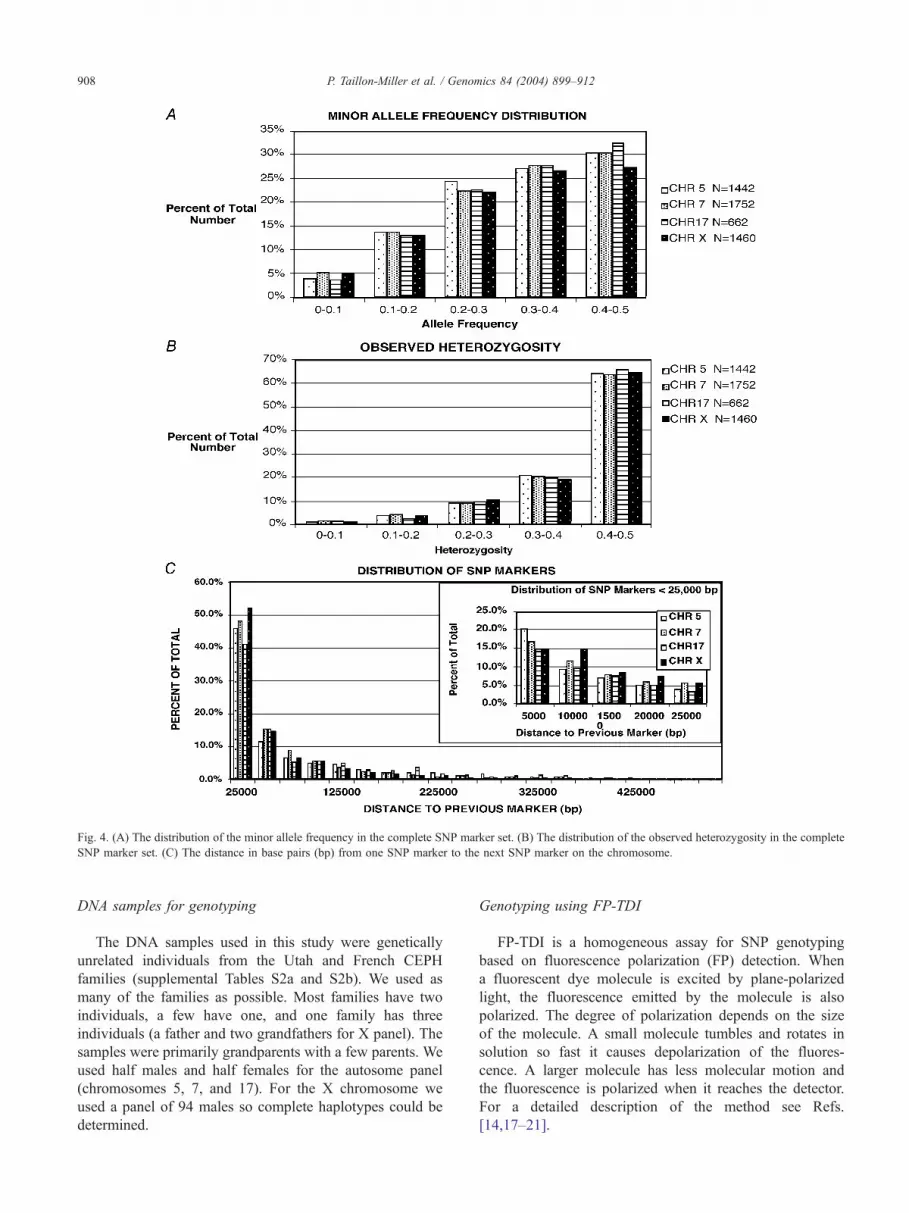
Fig. 4. (A) The distribution of the minor allele frequency in the complete SNP marker set. (B) The distribution of the observed heterozygosity in the complete
SNP marker set. (C) The distance in base pairs (bp) from one SNP marker to the next SNP marker on the chromosome.
P. Taillon-Miller et al. / Genomics 84 (2004) 899–912908
DNA samples for genotyping
The DNA samples used in this study were genetically
unrelated individuals from the Utah and French CEPH
families (supplemental Tables S2a and S2b). We used as
many of the families as possible. Most families have two
individuals, a few have one, and one family has three
individuals (a father and two grandfathers for X panel). The
samples were primarily grandparents with a few parents. We
used half males and half females for the autosome panel
(chromosomes 5, 7, and 17). For the X chromosome we
used a panel of 94 males so complete haplotypes could be
determined.
Genotyping using FP-TDI
FP-TDI is a homogeneous assay for SNP genotyping
based on fluorescence polarization (FP) detection. When
a fluorescent dye molecule is excited by plane-polarized
light, the fluorescence emitted by the molecule is also
polarized. The degree of polarization depends on the size
of the molecule. A small molecule tumbles and rotates in
solution so fast it causes depolarization of the fluores-
cence. A larger molecule has less molecular motion and
the fluorescence is polarized when it reaches the detector.
For a detailed description of the method see Refs.
[14,17–21].

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 909
The FP-TDI assay requires three unlabeled oligonu-
cleotides for each SNP. Two serve as PCR primers and
the third is a SNP probe that is complementary to the
template sequence with its 3V end annealed to the target
one base before the polymorphic site. The entire reaction
is done in one reaction tube without separation or
purification.
Because of this simplicity the FP-TDI assay is
extremely flexible and can be adapted to any number of
assays and to all levels of throughput. To meet the needs
of the project outlined here we increased our throughput to
a level that was capable of producing 4 million genotypes/
year. This was accomplished by creating a modular
approach to genotyping. Each equipment portion of the
module consisted of a Perkin-Elmer (Boston, MA, USA)
Evolution P3 liquid handling machine with a b96 well-
head,Q 24 384-well thermocycler blocks, one Perkin-Elmer
fluorescence polarization 384-well plate reader, and two
low-speed centrifuges with deep well buckets. With
efficient use of time and equipment up to two runs of
24 � 384 samples or 18,432 genotypes can be accom-
plished in a 10-h day with three people working staggered
schedules.
Genotyping procedure
All pipetting steps were done by hand with a Rainin
electronic multichannel pipettor, the Perkin-Elmer Apricot
personnel pipettor, the Robbins Hydra, or preferably with
the Perkin-Elmer Evolution P3 depending on the stage of
the project.
All PCRs and SNP detection reactions were designed
for standard melting temperature (Tm) and used standard
cycling conditions. The two PCR primers and the one SNP
detection primer were ordered arrayed in a 96-well tray at
a standard concentration for ease of handling. Primers were
ordered as 6 nmol lyophilized in a 96-well plate and then
150 Al water was added to each well to make a 40 AMstock solution.
We also prepared in advance the DNA template. To make
dried DNA plates 3 Al (0.8 ng/Al) of DNA from a 96-well
DNA plate was dispensed to all four quadrants of 384-well
black plates with the Perkin–Elmer Evolution P3 using a 96-
tip head. The plates were spun down and air-dried at room
temperature overnight. The plates were stacked and wrap-
ped with plastic film and kept desiccated at room tempe-
rature until needed. One allele combination per day was
genotyped whenever possible.
PCR
Volumes are for one reaction. Samples contained
genomic DNA dried previously, 0.5 Al 10� PCR buffer
(200 mM Tris-HCl (pH 8.4) and 500 mM KCl), 0.25 AlMgCl2 (50 mM), 0.1 Al dNTPs (2.5 mM), 0.02 Al any Hot
Start Taq (5 U/Al), 2.13 Al water, final volume 3.0 Al. Then 3Al of primers (40 AM) was added.
PCR thermocycling program
The cycling program was 1 cycle of 958C for 2 min; 35
cycles of 928C for 10 s, 588C for 20 s, 688C for 30 s; and 1
cycle of 688C for 10 min. The temperature of the
thermocycler was reduced to 48C until samples were
removed for the next step.
SNP detection reactions
For remaining the steps we used the AcycloPrime-FP
SNP Detection System Kit (Perkin–Elmer Life Sciences).
EXO-SAP reaction to remove excess primers and
unincorporated dNTPs
The 10� PCR clean-up reagent was diluted with PCR
clean-up buffer provided by the kit. Of this, 2 Al was addedto each well, and the plates was spun down.
EXO-SAP cycling program
The cycling program was 378C for 60 min, 808C for 15
min, and the temperature of the thermocycler was reduced to
48C until samples were removed for the next step.
TDI reaction mix for SNP detection
For one reaction, the volumes were 2.0 Al 10� buffer,
0.05 Al AcycloPol, 1.0 Al terminator mix, and 4.95 Al water,for a total volume of 8.00 Al.
Pipetting steps for TDI (SNP detection)
Five microliters of 1 AM SNP primers was dispensed into
the relevant quadrants of a 384-well plate, and the plate was
spun down. Eight microliters of TDI mix was dispensed into
the plate, and again the plate was spun down.
TDI cycling program
The cycling program was 1 cycle of 958C for 2 min and
10–40 cycles of 958C for 15 s, 558C for 30 s (20 cycles are
standard). The temperature of the thermocycler was reduced
to 48C until samples were removed for the next step.
Reading plates
To read the plates, they were spun down and stacked in
the Perkin-Elmer Victor or Perkin-Elmer EnVision stacker.
Plates were read for both dyes. Analysis of the data was
accomplished with the Excel Macro software provided by
the manufacturer. Data were then saved as a tab-delimited
text document that could be imported into the linkage
disequilibrium analysis software. Our lab currently uses the
SNPScorer software, which outputs results directly into a
database.
Primer design for FP-TDI
To achieve the high throughput needed for this project
all primers were designed with a strict criterion for Tm
and all reactions were run under a single set of
parameters.

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912910
PCR primer design
PCR primers were designed using Primer3, release 0.9
(with code available at http://www.genome.wi.mit.edu/
genome_software/other/primer3.html Ref. [22]) using the
parameters previously described [23]. Minor changes for
this application follow: TARGET = SNP_Position-20 bases,
20 bases; PRIMER_OPT_TM = 55, PRIMER_MAX_TM =
56, PRIMER_MIN_TM = 54; PRIMER_PRODUCT_
SIZE_RANGE = 80–400; PRIMER_PRODUCT_OPT_
SIZE = 250.
SNP primer design
The shortest primer with a Tm of 50–558C and a minimum
length of 16 bp and a maximum length of 40 bp was picked
on both the forward and the reverse strand. The SNP primer
is complementary to the sequence and ends with the base
adjacent to the SNP site. The Allawi and Santa-Lucia nearest
neighbor sequence-dependent thermodynamic parameters
[24] were used to determine Tm.
If both a forward and a reverse SNP primer could be
chosen in the first step, one was selected by assigning
penalties to various types of repeated sequences. This
minimized failures due to repeated sequences near the SNP.
If the penalties were equal the shortest primer was chosen
because it was the least expensive. If there was no clear
preference the default was the forward primer. Sometimes
SNP primers could not be chosen because of the melting
temperature and length criteria. This could be due to extreme
GC content, a neighboring SNP within the potential primer
site, or a reported SNP that was not a single-base biallelic
polymorphism.
In our experience, FP-TDI assays can be developed for
all SNPs found in unique sequence. Because of the stringent
design of our PCR conditions, 95% of them are successful
[25] and 80% of the FP-TDI genotyping assays work the
first time. Sixty percent of the failed assays work when
repeated with the SNP primer from the other strand, which
gives a final assay success rate of N90%. Our genotyping
error rate is b0.02%.
We periodically develop FP-TDI genotyping assays for
all uniquely mapped SNPs found in public databases. These
assays and the expanded protocols for the methods outlined
above are available at http://snp.wustl.edu.
Linkage disequilibrium analysis
Pair-wise linkage disequilibrium was computed for all
possible two-way comparisons of SNPs, with significance
levels determined by them2 statistic for the corresponding 2�2 table (1 degree of freedom). Haplotype frequencies for
autosomes were estimated using the E-M algorithm of
Excoffier and Slatkin [26] as implemented for pairs of
markers in the LDMAX program of the GOLD software
package [27]. For the X chromosome, haplotypes were
directly observable. SAS (SAS Institute, Cary, NC, USA)
together with LDMAX was used to compute m2 statistics and
linkage disequilibrium measures D, DV, and r2, defined as
follows. For two loci L1 and L2, each with two alleles 1 and 2,
let pi be the frequency of allele 1 and qi = 1 � pi be the
frequency of allele 2, at locus i (i = 1, 2). Assume pi V qi, that
is, allele 1 is the minor allele. Let pjk be the frequency of the
jk haplotype. The coefficient of disequilibrium is D = p11 �p1p2. The labeling of the alleles may affect the sign of D but
not its absolute value. The normalized disequilibrium
coefficient is obtained by dividing D by its maximum
possible (absolute) value given p1 and p2: DV= D/|D|max,
where |D|max = max ( p1p2, q1q2) if D b 0 and |D|max =
min ( q1p2, p1q2) if D N 0. The correlation coefficient
is r ¼ D=ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffip1q1p2q2
p(if we define the random variable Xi on
individuals to be 1 if the individual has allele 1 at loci i and
0 otherwise, then D = cov(X1,X2) and var(Xi) = piqi so r is
the usual correlation coefficient of these two random
variables). SNP heterozygosities were computed. Hetero-
zygosity for a biallelic marker with allele frequencies p and
q = 1 � p is given by H = 1 � p2 � q2 = 2p(1 � p).
Power calculations and linkage disequilibrium blocks
We chose to define LD blocks using threshold values for
DVthat correspond to good power for a case-control disease
association study of realistic sample size. The assumption is
that the strength of LD between alleles of the disease gene
and a genotyped marker in the block is similar to the LD
strength within the block. For such a case-control study, the
Pearson m2 test statistic for a 2 � 2 contingency table may
be used to test for allelic association. To calculate the
expected power for particular values of DV, we assumed a
range of disease models specified by disease gene frequen-
cies at a biallelic disease gene and penetrances for the three
genotype classes. To aid interpretation, we also parame-
terized each model in terms of the population prevalence (K)
of the disease, the disease gene frequency, and two of the
three penetrances. We considered both multiplicative
models and more general models, but will focus on the
multiplicative models here. Hence we assume that if d1 is
the disease allele, the penetrances for the genotype classes
are fd1d2 = g fd2d2 and fd1d1 = g2 fd2d2, where g is the
bgenotypic relative risk.Q For each multiplicative model we
computed the corresponding sibling relative risk (ES) usingcorrected formulas derived as in Risch and Merikangas [2].
We assumed marker allele frequencies to be 0.5 and
considered two cases for disease allele frequency pd1: pd1 =
0.5 (which would result in the maximum power for this
marker allele frequency) and pd1 = 0.2, corresponding to a
more realistic, common disease allele frequency. While
these are specific choices and of course do not represent the
full range of scenarios that may occur in an actual disease
association study, they provide a reasonable setting to guide
our LD block definitions. We propose this overall approach
as a general strategy for defining LD blocks for disease

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912 911
association studies; the range of models and desired power
level may then be chosen depending on the specific study.
The power calculations rely on determining the expected
marker allele frequencies in cases versus controls given a
particular disease model (penetrances), disease and marker
allele frequencies, andDVvalue (which determines haplotype
frequencies). Sham [28] derives the necessary formulas for
case and control allele frequencies in terms of haplotype
frequencies (chapter 4, section 4.6). We then used noncentral
m2 distributions to calculate sample sizes to detect associ-
ation between disease and marker in a m2 test, with specified
power and significance level. That is, for a given significance
level a, asymptotically, the m2 statistic follows a m2
distribution with 1 df under the null hypothesis and follows
a noncentral m2 with 1 df under the alternative hypothesis.
The noncentrality parameter is given by the m2 statistic
computed with parameters taking on the values correspond-
ing to the alternative hypothesis and is proportional to the
sample size2. Weir [29] discusses further details of the use of
the noncentral m2 for power in an analogous context; the
Web-interface power calculator of Purcell et al. [30] also
uses the noncentral m2 in the same way as our calculations.
After useful DV threshold values were determined from
our power calculations, LD blocks were defined using all
SNPs having a minor allele frequency of 20% or greater.
Our primary LD blocks were defined by requiring all
marker pairs in a given block to have jDVj greater than or
equal to the chosen threshold. We also studied weak LD
blocks with less restrictive definitions described below.
Algorithms for finding linkage disequilibrium blocks
Our power calculations suggest the following strategy for
reducing the number of markers genotyped for an associ-
ation study. If jDVj z T for a pair of SNPs then select only
one of them for genotyping, where T is a threshold of LD
strength to be determined from the power calculations. If the
same level of LD existed between a given SNP and several
other SNPs, then one might select that single SNP to
represent the entire group. However, by requiring the more
stringent criterion that all pairs of markers within the group
satisfy the LD threshold T, we allow ourselves more choices
for tag SNP selection and also increase our confidence that a
chosen tag SNP is likely to be a good representative, not
2 The noncentrality parameter Enc for the noncentral m2 distribution
corresponding to the alternative hypothesis, computed from the appropriate
2 � 2 table, is
knc ¼ 2N
"P M1 j caseð Þ � P M1 j controlð Þð Þ2
P M1 j caseð Þ þ P M1 j controlð Þ
þ P M2 j caseð Þ � P M2 j controlð Þð Þ2
P M2 j caseð Þ þ P M2 j controlð Þ
#
where N is the number of cases, which equals the number of controls;
P(Mi j case) is the allele frequency of Mi in cases, and P(Mi j case) is theallele frequency of Mi in controls.
only for other markers in the block but also for potentially
untyped markers in the region of the block.
We therefore define a linkage disequilibrium block to be
a consecutive (relative to the map in use) set of at least three
markers along with a cutoff threshold T and a disequilibrium
coefficient L (DVor r2) such that all pairs of markers in the
block satisfy jLj z T. Furthermore, in the interest of relaxing
the threshold requirement potentially to increase chromoso-
mal coverage and reduce the number of tag SNPs required
for a genome screen, we also examine weak LD blocks. By
a weak linkage disequilibrium block we mean a set of at
least three consecutive markers along with three parameters
Tl, Tu, and F and a disequilibrium coefficient L such that all
pairs of markers in the block satisfy jLj z Tl (the lower
threshold), and least F% of the pairs of markers satisfy jLjzTu (the upper threshold).
Once a set of nonoverlapping LD blocks has been
determined we would like to know what proportion of the
chromosome is covered by LD blocks. We could divide the
sum of the widths of the blocks by the width of the
chromosome but this would be in some sense misleading
since the denominator includes blarge gapsQ such as the
centromere and gaps between contigs. Therefore, we omit
these regions and consider only that portion of the chromo-
some where the SNPs are suitably dense. More precisely,
define the densely covered region of the chromosome to be the
union of all the intervals between consecutive SNPs that are
within some distance W of each other. In other words, every
part of the densely covered region must lie between two SNPs
that are withinW.We then define the coverageC by taking the
intersection of blocks with the densely covered region and
divide the total size of this region by the size of the densely
covered region. In our analysis, we chose W to be 100 kb.
We have designed an algorithm that takes ordered
markers and produces a set of nonoverlapping blocks and
attempts to maximize the block coverage. This is consistent
with our goal of using blocks as a tool for reducing SNP
density while retaining power in an initial association
screen. We therefore wish block coverage to be as extensive
as possible to allow for maximum genotyping reduction
according to our strategy, but are not necessarily interested
in block size or blocks as indicators of haplotype diversity.
Beginning with the first marker and then proceeding, in
order, to the following markers, we find the largest block
containing each marker (these blocks may include markers
that either precede or follow the current marker). A block is
ignored if it is contained in the block previously discovered
by the algorithm. If a block overlaps with the previous block
but is not contained in it we consider the following four
options: (1) truncate the previous block by removing a
minimal number of markers so that it no longer overlaps
with the current block, (2) truncate the current block in the
same way, (3) discard the previous block, or (4) discard the
current block. The option that produces the greatest amount
of coverage is chosen, with ties yielding to the order of the
options.

P. Taillon-Miller et al. / Genomics 84 (2004) 899–912912
When haplotypes are known we are interested in finding
subsets of SNPs that determine all observed haplotypes
within blocks. We call these complete subsets, and our
algorithm for finding them consists of testing all subsets of
size 1, 2, 3, and so on, and reporting the first complete
subset found. Since for the autosomes only the genotypes
are known, this algorithm is applied only to chromosome X.
Using blocks to predict LD
One of the applications of determining LD blocks is to
reduce the number of markers required for an association
study by selecting LD tag SNPs from the blocks. An
underlying assumption is that if the disease gene is physically
in the block then it can be expected to be in LDwith a tag SNP
from the block. We have designed a test to determine if LD
blocks work as predictors this way.
Suppose A1, A2, . . ., An are SNPs that form an LD block.
If B is an additional SNP that lies physically between A1 and
An we expect that B would be part of the block; that is, B is in
linkage disequilibrium with A1, A2, . . .., An. More precisely,
we assume that jDVj z T1 for each of pairs in the set A1, A2,
. . ., An. We then determine the empirical conditional
probability P that jDVj z T2, the lower cutoff, for each of
the pairings of B with A1, A2, . . ., An. If this probability is
high then we conclude that LD blocks are good predictors of
LD with additional markers in the region. In other words, if
we already have a block, then the block structure will most
likely be preserved after new markers are added to it. We use
two cutoffs to allow for some flexibility in our test.
Acknowledgment
Some of this work was supported by grants from the
National Institutes of Health: HG01720 (P.Y.K.), DA15129
(N.L.S.), and AA07580 (S.F.S.).
Appendix A. Supplementary data
Supplementary data for this article may be found on
ScienceDirect or at http://snp.wustl.edu/snp_research/
ld_blocks.
References
[1] The International HapMap Consortium, The International HapMap
project, Nature 426 (2003) 789.
[2] N. Risch, K. Merikangas, The future of genetic studies of complex
human diseases, Science 273 (1996) 1516–1517.
[3] P. Taillon-Miller, et al., Juxtaposed regions of extensive and minimal
linkage disequilibrium in human Xq25 and Xq28, Nat. Genet. 25
(2000) 324.
[4] M.J. Daly, J.D. Rioux, S.F. Schaffner, T.J. Hudson, E.S. Lander, High-
resolution haplotype structure in the human genome, Nat. Genet. 29
(2001) 229–232.
[5] D.E. Reich, et al., Linkage disequilibrium in the human genome,
Nature 411 (2001) 199–204.
[6] S.B. Gabriel, et al., The structure of haplotype blocks in the human
genome, Science 296 (2002) 2225–2229.
[7] N. Patil, et al., Blocks of limited haplotype diversity revealed by high-
resolution scanning of human chromosome 21, Science 294 (2001)
1719.
[8] E. Dawson, et al., A first-generation linkage disequilibrium map of
human chromosome 22, Nature 418 (2002) 544.
[9] M.S. Phillips, et al., Chromosome-wide distribution of haplotype
blocks and the role of recombination hot spots, Nat. Genet. 33 (2003)
382.
[10] K.G. Ardlie, L. Kruglyak, M. Seielstad, Patterns of linkage
disequilibrium in the human genome, Nat. Rev. Genet. 3 (2002)
299–309.
[11] A.G. Clark, et al., Linkage disequilibrium and inference of ancestral
recombination in 538 single-nucleotide polymorphism clusters across
the human genome, Am. J. Hum. Genet. 73 (2003) 285–300.
[12] C.S. Carlson, et al., Additional SNPs and linkage-disequilibrium
analyses are necessary for whole-genome association studies in
humans, Nat. Genet. 33 (2003) 518–521.
[13] D.C. Crawford, et al., Evidence for substantial fine-scale variation in
recombination rates across the human genome, Nat. Genet. 36 (2004)
700–706.
[14] T.M. Hsu, P.-Y. Kwok, Homogeneous Primer Extension Assay with
Fluorescence Polarization Detection, in Single Nucleotide Poly-
morphisms: Method And Protocols, Humana Press, Totowa, NJ,
2002, pp. 177–188.
[15] K.T. Zondervan, L.R. Cardon, The complex interplay among
factors that influence allelic association, Nat. Rev. Genet. 5 (2004)
89–100.
[16] R.M. Pfeiffer, M.H. Gail, Sample size calculations for population and
family-based case-control association studies on marker genotypes,
Genet. Epidemiol. 25 (2003) 136–148.
[17] X. Chen, L. Levine, P.-Y. Kwok, Fluorescence polarization in
homogeneous nucleic acid analysis, Genome Res. 9 (1999) 492.
[18] T.M. Hsu, P.-Y. Kwok, Homogeneous primer extension assay with
fluorescence polarization detection, Methods Mol. Biol. 212 (2003)
177.
[19] T.M. Hsu, X. Chen, S. Duan, R.D. Miller, P.-Y. Kwok, Universal SNP
genotyping assay with fluorescence polarization detection, Biotechni-
ques 31 (2001) 560.
[20] P.-Y. Kwok, SNP genotyping with fluorescence polarization detection,
Hum. Mutat. 19 (2002) 315.
[21] R.A. Greene, et al., A Novel Method for SNP Analysis Using
Fluorescence Polarization. P10131, Perkin-Elmer Life Sciences,
Boston, 2001.
[22] S. Rozen, H. Skaletsky, Primer3 on the WWW for general users
and for biologist programmers, Methods Mol. Biol. 132 (2000)
365.
[23] E.F. Vieux, P.-Y. Kwok, R.D. Miller, Primer design for PCR and
sequencing in high-throughput analysis of SNPs, Biotechniques 32
(2002) S28.
[24] R. Owczarzy, et al., Predicting sequence-dependent melting stability of
short duplex DNA oligomers, Biopolymers 44 (1997) 217.
[25] R.D. Miller, S. Duan, E.G. Lovins, E.F. Kloss, P.-Y. Kwok, Efficient
high-throughput resequencing of genomic DNA, Genome Res. 13
(2003) 717.
[26] L. Excoffier, M. Slatkin, Maximum-likelihood estimation of molec-
ular haplotype frequencies in a diploid population, Mol. Biol. Evol. 12
(1995) 921.
[27] G.R. Abecasis, W.O.C. Cookson, GOLD-graphical overview of
linkage disequilibrium, Bioinformatics 16 (2000) 182.
[28] P. Sham, Statistics in Human Genetics, Arnold, London, 1998.
[29] B.S. Weir, Genetic Data Analysis II, Sinauer, Sunderland, MA,
1996.
[30] S. Purcell, S.S. Cherny, P.C. Sham, Genetic power calculator: design
of linkage and association genetic mapping studies of complex traits,
Bioinformatics 19 (2003) 149.