leptin repletion restores depressed {beta}-adrenergic contractility in ob/ob mice independently of...
TRANSCRIPT

J Physiol 565.2 (2005) pp 463–474 463
Leptin repletion restores depressed β-adrenergiccontractility in ob/ob mice independentlyof cardiac hypertrophy
Khalid M. Minhas1, Shakil A. Khan1, Shubha V. Y. Raju1, Alexander C. Phan1, Daniel R. Gonzalez1,Mike W. Skaf1, Kwangho Lee1, Ankit D. Tejani1, Anastasies P. Saliaris1, Lili A. Barouch1,Christopher P. O’Donnell2, Charles W. Emala4, Dan E. Berkowitz3 and Joshua M. Hare1
1Division of Cardiology, 2Division of Pulmonary Medicine, Department of Medicine and 3Department of Anaesthesiology and Critical Care Medicine,Johns Hopkins Medical Institutions, Baltimore, MD, USA4Department of Anaesthesiology, Columbia University College of Physicians and Surgeons, New York, NY, USA
Impaired leptin signalling in obesity is increasingly implicated in cardiovascularpathophysiology. To explore mechanisms for leptin activity in the heart, we hypothesizedthat physiological leptin signalling participates in maintaining cardiac β-adrenergic regulationof excitation–contraction coupling. We studied 10-week-old (before development of cardiachypertrophy) leptin-deficient (ob/ob, n = 12) and C57Bl/6 (wild-type (WT), n = 15) miceat baseline and after recombinant leptin infusion (0.3 mg kg−1 day−1 for 28 days, n = 6 ineach group). Ob/ob-isolated myocytes had attenuated sarcomere shortening and calciumtransients ([Ca2+]i) versus WT (P < 0.01 for both) following stimulation of the β-receptor (withisoproterenol (isoprenaline)) or at the post-receptor level (with forskolin and dibutryl-cAMP).In addition, sarcoplasmic reticulum (SR) Ca2+ stores were depressed. Leptin replenishment inob/ob mice restored each of these abnormalities towards normal without affecting gross (wallthickness) or microscopic (cell size) measures of cardiac architecture. Immunoblots revealedalterations of several proteins involved in excitation–contraction coupling in the ob/ob mice,including decreased abundance of Gsα-52 kDa, as well as alterations in the expression ofCa2+ cycling proteins (increased SR Ca2+-ATPase, and depressed phosphorylatedphospholamban). In addition, protein kinase A (PKA) activity in ob/ob mice was depressedat baseline and correctable towards the activity found in WT with leptin repletion, a findingthat could account for impaired β-adrenergic responsiveness. Taken together, these data reveala novel link between the leptin signalling pathway and normal cardiac function and suggest amechanism by which leptin deficiency or resistance may lead to cardiac depression.
(Resubmitted 16 February 2005; accepted after revision 7 March 2005; first published online 10 March 2005)Corresponding author J. M. Hare: The Johns Hopkins Medical Institutions, Cardiology Division, 720 Rutland Avenue,Ross 1059, Baltimore, MD 21205, USA. Email: [email protected]
Obesity has substantial health implications, predisposingindividuals to cardiovascular diseases such as hyper-tension, hyperlipidemia, atherosclerosis and congestiveheart failure (Eckel et al. 2002). An emerging themein obesity-related cardiovascular pathophysiology isthat neurohormonal pathways that regulate adiposehomeostasis also have cardiovascular activity, disruptionof which may contribute to cardiovascular dysfunction(Alpert, 2001; Barouch et al. 2003; Sader et al. 2003).
K. M. Minhas, S. A. Khan and S. V. Y. Raju contributed equally to thiswork.
Recent epidemiological studies support increased cardio-vascular risk in obese subjects independent of bloodpressure, left ventricular hypertrophy (LVH), diabetesmellitus or underlying organic heart disease (Kenchaiahet al. 2002). Thus, underlying signalling derangementsmay be the proximate cause of cardiac dysfunction,not obesity and its haemodynamic consequences,per se.
In this regard, there is growing interest in the cardio-vascular activity of the leptin signalling pathway (Nickolaet al. 2000; Illiano et al. 2002; Barouch et al. 2003;Rajapurohitam et al. 2003) because leptin deficiencyor resistance, both causes of obesity (Considine et al.
C© The Physiological Society 2005 DOI: 10.1113/jphysiol.2005.084566

464 K. M. Minhas and others J Physiol 565.2
1996; Bray & York, 1997; Montague et al. 1997), mayalso participate in cardiovascular disease (Pladevall et al.2003; Sader et al. 2003). Leptin, a 167 amino acidpolypeptide synthesized predominantly in adipose tissue,has widely distributed receptors (Sweeney, 2002). Indeed,cardiac myocytes also express leptin receptors, whichare coupled to signalling pathways that influence bothmyocardial contractility (Nickola et al. 2000; Woldet al. 2002) and cellular growth (Barouch et al. 2003;Rajapurohitam et al. 2003; Xu et al. 2004). We haverecently shown that mice lacking leptin (ob/ob) or itsreceptor (db/db) develop cardiac hypertrophy, reversibleby leptin repletion (in the case of ob/ob), but notweight loss alone, strongly supporting a role for leptin inmaintaining normal cardiac architecture (Barouch et al.2003).
Another key cardiovascular phenotype which leptinhas the potential to influence is β-adrenergic signal trans-duction. In adipose tissue, leptin regulates β-adrenergicsignalling which in turn mediates lipolysis and thermo-genesis (Collins et al. 1994; Collins & Surwit, 2001;Bachman et al. 2002). As depressed β-adrenergic contra-ctility is a hallmark phenotype of the failing myocardium(Bristow et al. 1990), we tested whether leptin influencescardiac β-adrenergic inotropic responses.
Methods
Animals
We studied ob/ob mice and their wild-type (C57Bl/6 J,WT) controls (Jackson Laboratory, Bar Harbour, ME,USA). The ob/ob mice were backcrossed on a C57Bl/6background greater than 30 generations, resulting inobese mice that are statistically 99.9% similar to WT atall unlinked loci. Animals were housed under diurnallighting conditions and allowed food and tap waterad libitum. Animals were killed by cervical dislocationafter deep anaesthesia with isoflurane by inhalation. TheInstitutional Animal Care and Use Committee of TheJohns Hopkins University School of Medicine approvedall protocols and experimental procedures.
Exogenous leptin infusion
Effects of leptin treatment were evaluated in both ob/oband WT mice. Six-week-old mice were anaesthetized with2% isoflurane for pump implantation. Leptin was infusedfor 28 days using 100 µl osmotic minipumps (replacedafter 14 days), implanted subcutaneously in the inter-scapular area (Alzet, Palo Alto, CA, USA), which deliveredcontinuous recombinant mouse leptin (Amgen, ThousandOaks, CA, USA; 0.3 mg kg−1 day−1) (Breslow et al. 1999;Barouch et al. 2003).
Echocardiography
Echocardiographic assessments were performed in ob/ob,WT and leptin-treated mice at 6 and 10 weeks of age. Micewere anaesthetized with 1–2% isoflurane. Studies wereperformed using a Sonos 5500 Echocardiogram (Agilent)with a 15 MHz linear transducer. Anterior wall thickness(AWT), posterior wall thickness (PWT), end-diastolic(EDD) and end-systolic (ESD) left ventricular dimensionswere recorded from M-mode images using averagedmeasurements from three to five consecutive cardiaccycles. Relative wall thickness (RWT) was calculated usingRWT = (AWT + PWT)/EDD.
Isolated myocyte isolation
Cardiac myocytes were prepared from 10-week-oldWT and ob/ob mice, as previously described by Khanet al. (2003). Hearts were perfused with Ca2+-freebicarbonate buffer containing (mmol/l): 120 NaCl,5.4 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5.6 glucose, 20NaHCO3, 10 2,3-butanedione monoxime (BDM; Sigma),and 5 taurine (Sigma), gassed with 95% O2–5%CO2, and digested with collagenase type 2 (1 mg ml−1;Worthington Biochemicals, Lakewood, NJ, USA) andprotease type IV (0.1 mg ml−1; Sigma). Myocytes wereobtained by mechanically disrupting digested hearts,followed by filtration, centrifugation and suspension in0.125 Ca2+ Tyrode solution containing (mmol/l): 144NaCl, 1MgCl2, 10 Hepes, 5.6 glucose, 5 KCl adjusted toa pH of 7.4 with NaOH. Myocytes were resuspendedfirst in 0.250 mmol l−1 Ca2+ Tyrode solution, then in0.5 mmol l−1 Ca2+ Tyrode solution, and finally stored inTyrode solution containing 0.5 mmol l−1 probenecid and1.8 mmol l−1 Ca2+. Mechanical studies were completedwithin 6 h after isolation at room temperature.
Cell shortening and Ca2+ transient measurements
Mechanical properties of myocytes were assessed usinga video-based myocyte sarcomere spacing acquisitionsystem (IonOptix, Milton, MA, USA). Myocytes wereincubated with 5 µmol l−1 fura-2 AM (Molecular Probes)for 10 min then transferred to a lucite chamber on the stageof an inverted microscope (Nikon TE 200), continuouslysuperfused with Tyrode solution containing 1.8 mmol l−1
Ca2+ and 0.5 mmol l−1 probenecid, stimulated at 1 Hz.Myocyte length and width, and sarcomere length (SL)were recorded with an IonOptix iCCD camera. Changein average SL was determined by fast Fourier transform ofthe Z-line density trace to the frequency domain and SLshortening was calculated as (diastolic SL − systolic SL)/diastolic SL.
Intracellular Ca2+ concentrations [Ca2+]i weremeasured using the Ca2+-sensitive dye fura-2 and a
C© The Physiological Society 2005
J Physiol 565.2 Deranged β-adrenergic signalling in ob/ob mice 465
Table 1. Ventricular parameters and cell size in 10-week-old mice
WT − leptin WT + leptin ob/ob − leptin ob/ob + leptin
Mice (n) 6 11 6 7Body weight (g) 21.4 ± 1.5 20.3 ± 1.0 49.2 ± 1.3∗ 23.6 ± 1.3AWT (mm) 0.67 ± 0.04 0.62 ± 0.03 0.69 ± 0.05 0.66 ± 0.03RWT (mm) 0.49 ± 0.04 0.50 ± 0.03 0.55 ± 0.08 0.45 ± 0.03EDD (mm) 2.87 ± 0.12 2.69 ± 0.10 2.92 ± 0.14 3.08 ± 0.11Myocyte length (µm) 132.9 ± 2.4 129.4 ± 2.0 131.7 ± 2.8 131.3 ± 2.0Myocyte width (µm) 27.4 ± 0.7 27.6 ± 0.7 28.1 ± 0.7 27.8 ± 0.8
∗P < 0.01 versus WT − leptin, WT + leptin, and ob/ob + leptin. AWT, anterior wall thickness;PWT, posterior wall thickness; EDD, end-diastolic diameter. Relative wall thickness (RWT) wascalculated using RWT = (AWT + PWT)/EDD.
dual-excitation spectrofluorometer (IonOptix),alternately excited with a xenon lamp at wavelengthsof 365 and 380 nm. The emission fluorescence wasreflected through a barrier filter (510 ± 15 nm) to aphotomultiplier tube. The ratio of the photon live countdetected by excitation at 365 nm compared with 380 nmrepresents the fura-2 fluorescence ratio. IntracellularCa2+ concentrations were compared at baseline and peakcontraction to determine the calcium transient.
Myocyte stimulation
Myocyte contractile function was assessed with(a) isoproterenol (Sigma, 10−8–10−6 mol l−1), anon-selective β-receptor agonist; (b) forskolin (Sigma,10−8–10−6 mol l−1), a direct activator of adenylylcyclase; and (c) dibutryl cAMP (Sigma, 10−3 mol l−1), aphosphodiesterase-resistant cAMP analog. The myocyteswere continuously perfused, at approximately 2 ml min−1,with increasing doses of the activators until they reacheda steady state. Caffeine (10 mmol l−1) was administeredrapidly, after a 10 s pause, to estimate sarcoplasmicreticulum (SR) calcium stores.
Membrane preparation
Pieces of ventricular tissue (∼20 mg) from 10-week-oldWT − leptin, WT + leptin (n = 6 each), ob/ob − leptin,and ob/ob + leptin (n = 4 each) mice were thawed (from−80◦C storage) in buffer A (5 mmol l−1 Tris, pH 7.4,0.25 mol l−1 sucrose, 1 mmol l−1 MgCl2, 1 mmol l−1
EDTA, 10 µmol l−1 PMSF (phenylmethylsulphonylfluoride)) and homogenized using high speed cuttingblades (Tissuemizer, Tekmar Co., Cincinnati, OH, USA)on top speed for 45 s. Homogenates were subjected tocentrifugation at 1000 g for 10 min at 4◦C. The super-natant was recovered and subjected to centrifugation at45 000 g for 25 min at 4◦C. The pellet was resuspended in5 ml of buffer B (50 mm Tris, pH 7.4, 10 mmol l−1 MgCl2,1 mmol l−1 EDTA, 10 µmol l−1 PMSF) and centrifugedagain at 45 000 g for 25 min at 4◦C. The pellet was washedan additional time in 25 ml buffer B. The final pellet was
resuspended in 0.5 ml buffer B and used immediately inadenylyl cyclase assays.
Adenylyl cyclase assays
Triplicate samples were incubated for 10 min at 37◦Cand contained the indicated effector along with10–20 µg of membrane protein, 25 mmol l−1 Tris, pH 7.5,5 mmol l−1 MgCl2, 0.5 mmol l−1 EDTA, 1 mmol l−1
cAMP, 1 mmol l−1 ATP, 32P-α-ATP (0.5–1.5 µCi tube−1,800 Ci mmol−1), 5 µmol l−1 PMSF, 7 mmol l−1 creatinephosphate, 50 µg ml−1 creatine phosphokinase and0.25 mg ml−1 BSA in a final volume of 100 µl. Adenylylcyclase activity was measured under basal conditions(no added effectors) or in the presence of 1 µmol l−1
forskolin. Reactions were terminated by the additionof 100 µl of stop buffer (50 mmol l−1 Hepes, pH 7.5,2 mmol l−1 ATP, 0.5 mmol l−1 cAMP, 2% SDS, 1 µCi ml−1
3H-cAMP (37 Ci mmol−1)). Newly synthesized 32P-cAMPwas separated from the precursor 32P-α-ATP bysequential column chromatography over dowex (Bio-Rad
AWT RWT
WT
ob/ob
-10
-5
0
5
10
15
% C
hang
e A
fter
Lept
in In
fusi
on
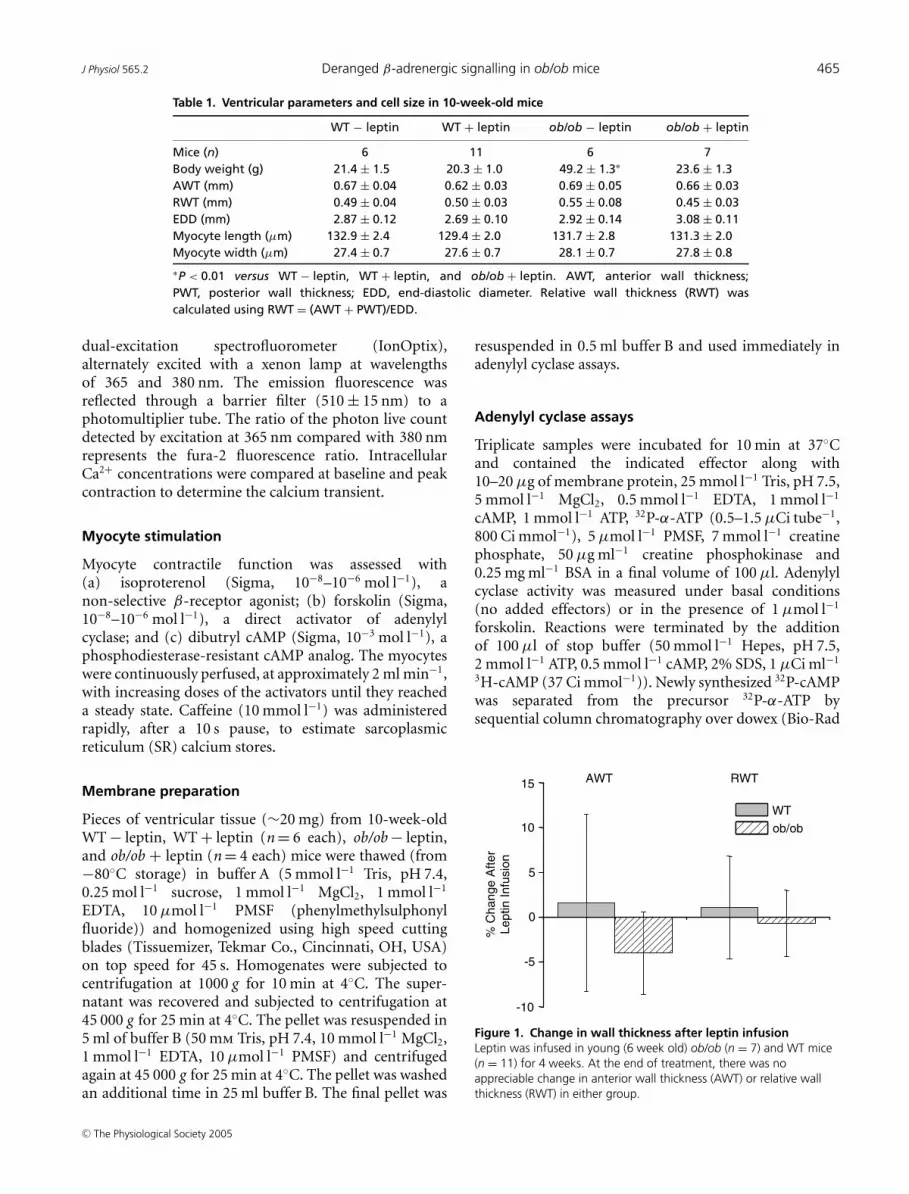
Figure 1. Change in wall thickness after leptin infusionLeptin was infused in young (6 week old) ob/ob (n = 7) and WT mice(n = 11) for 4 weeks. At the end of treatment, there was noappreciable change in anterior wall thickness (AWT) or relative wallthickness (RWT) in either group.
C© The Physiological Society 2005
466 K. M. Minhas and others J Physiol 565.2
Table 2. Baseline myocyte characteristics at 1 Hz
WT − leptin WT + leptin ob/ob − leptin ob/ob + leptin
Mice (n)∗ 14 7 10 6Diastolic SL (µm) 1.72 ± 0.01 1.74 ± 0.01 1.72 ± 0.01 1.74 ± 0.01SL shortening (%) 2.9 ± 0.2 2.6 ± 0.2 3.1 ± 0.2 2.7 ± 0.2[Ca2+]i (%) 22.0 ± 1.4 24.9 ± 1.1 21.0 ± 1.3 17.9 ± 1.4
∗5–7 cells were studied per heart. Sacromere shortening (SL shortening), (diastolic SL – systolicSL)/diastolic SL; [Ca2+]i, change in the ratio of the photon live count detected by excitation at365 nm compared with 380 nm during contraction.
Laboratories) and alumina (Sigma-Adlrich) as describedpreviously (Salomon et al. 1974), using recovery of3H-cAMP to monitor individual column efficiency(Emala, 1997). Eluted radioactivity was quantified byliquid scintillation.
Protein kinase A (PKA) activity
PKA activity was measured using a non-radioactivefluorescent detection assay (Promega Corporation). In10-week-old WT− leptin, WT+ leptin (n = 6 each), ob/ob− leptin, and ob/ob + leptin (n = 4 each) mice, heartswere removed rapidly after killing, rinsed several timesin ice-cold 1 × PBS and connective tissue, aorta and atriaremoved. The left ventricle was frozen in liquid nitrogenand stored at −80◦C. On the day of the experiment,left ventricular tissue was homogenized with a Polytron(3 × 15 s) in 1 ×cell lysis buffer (Cell Signalling Tech.). Thehomogenization buffer also contained 1 mm PMSF, andProtease Inhibitor Cocktail (Roche Diagnostics GmbH).The homogenate was centrifuged at 4◦C at 14 000 gfor 30 min. The supernatants were recovered and theirconcentrations were determined using BCA reagent(Pierce Biotechnology) and bovine serum as a standard.
A portion of the cAMP-dependent protein kinasecatalytic subunit was diluted to 2 µg ml−1 in PKA dilutionbuffer (350 mm K3PO4, pH 7.5 and 0.1 mm DTT) and wasused as positive control. For each sample, PKA reaction5 × buffer, PKA-specific peptide substrate (PepTag® A1peptide Promega or Poration), PKA activator 5 × solutionwith peptide protection solution, and water were mixedand kept on ice. The samples were incubated at roomtemperature for 30 min, loaded onto a 0.8% agarosegel (prepared in 50 mm Tris-HCl, pH 8.0), and run at135 V for 30–40 min. The bands were visualized underultraviolet light, quantified by spectrophotometry, andtheir absorbance read at 570 nm. Finally, the PKA activitywas calculated using Beer’s Law in accordance with themanufacturer’s instructions.
Western blots
As previously described (Khan et al. 2003), Westernblot analysis was performed on total protein lysateprepared from 10-week-old WT − leptin, WT + leptin,
ob/ob − leptin, and ob/ob + leptin (n = 5 in each group)mice for all proteins tested except β2-adrenergicreceptor, for which microsomal preparations were used(centrifugation of total lysate at 100 000 g for 1 h). Proteinconcentration was assayed for loading with bicinchoninicacid (Pierce Biotechnology, Rockford, IL, USA) andequal amounts were resolved with polyacrylamide gels(Invitrogen Life Technologies, Carlsbad, CA, USA).Proteins were then transferred to nitrocellulose orpolyvinylidene difluoride membrane. The followingantibodies were used in 1 : 500–1 : 1000 dilutions:sarcoplasmic reticulum (SR) Ca2+-ATPase, phospho-lamban (PLB), Giα, and β1-adrenergic receptor (AffinityBioreagents Inc.), phospho-phospholamban (P-PLB)(Upstate biotechnology), β2-adrenergic receptor (SantaCruz Biotechnology, Inc.), and Gsα (United StatesBiological). Polyclonal anti-p38 MAP kinase antibody(1 : 1000 dilutions, Santa Cruz Biotechnology, Inc.) wasused to normalize protein quantity. Bands were visualizedby chemiluminescence (SuperSignal Substrate kit, Pierce)and quantified using the NIH Imaging software.
Plasma measurements
Plasma glucose, leptin, insulin and triglycerides levelswere measured by radioimmunoassay (Linco DiagnosticServices).
Statistical analysis
Data are reported as mean ± s.e.m. Statistical significancewas determined by one-way or two-way ANOVA whereappropriate and Student-Newman-Keuls post hoc test(GraphPad Instat, STATA, and SAS statistical software).P values less than 0.05 were considered significant.
Results
Cardiac structure in young ob/ob mice
In order to separate the effects of leptin signallingon β-adrenergic inotropic responses from cardiachypertrophy, we studied young ob/ob mice at an agepreceding development of LVH (Barouch et al. 2003).Overall, cardiac structure and isolated myocyte size weresimilar at 10 weeks of age in WT − leptin (n = 6) and
C© The Physiological Society 2005
J Physiol 565.2 Deranged β-adrenergic signalling in ob/ob mice 467
ob/ob − leptin (n = 6) mice (Table 1). Importantly, leptinrepletion (WT + leptin; n = 11 and ob/ob + leptin; n = 7)did not affect either gross (wall thickness, Fig. 1) or micro-scopic (n = 50–60 cells per strain) measures of cardiacarchitecture. Taken together, these findings establish theyoung ob/ob mice as a valid model to study impaired leptinsignalling without the confounding influence of cardiachypertrophy.
A
1.55
1.80
SL
(µm
)
1.2
1.7
[Ca2+
] i
(365
/380
nm
)Wild Type Obese
B
0
150
300
450
600
†
*
*
**
WT - LeptinWT + Leptinob/ob - Leptinob/ob + Leptin
bl 10-8 10-7 10-6
SL
shor
teni
ng(%
cha
nge
from
bas
elin
e)
0
50
100
150
200
bl 10-8 10-7 10-6
†****
[Ca2+
] i(%
cha
nge
from
bas
elin
e)
†
*
*
**
WT - LeptinWT + Leptinob/ob - Leptinob/ob + Leptin
†****
[Isoproterenol] (M) [Isoproterenol] (M)
250 (msec)
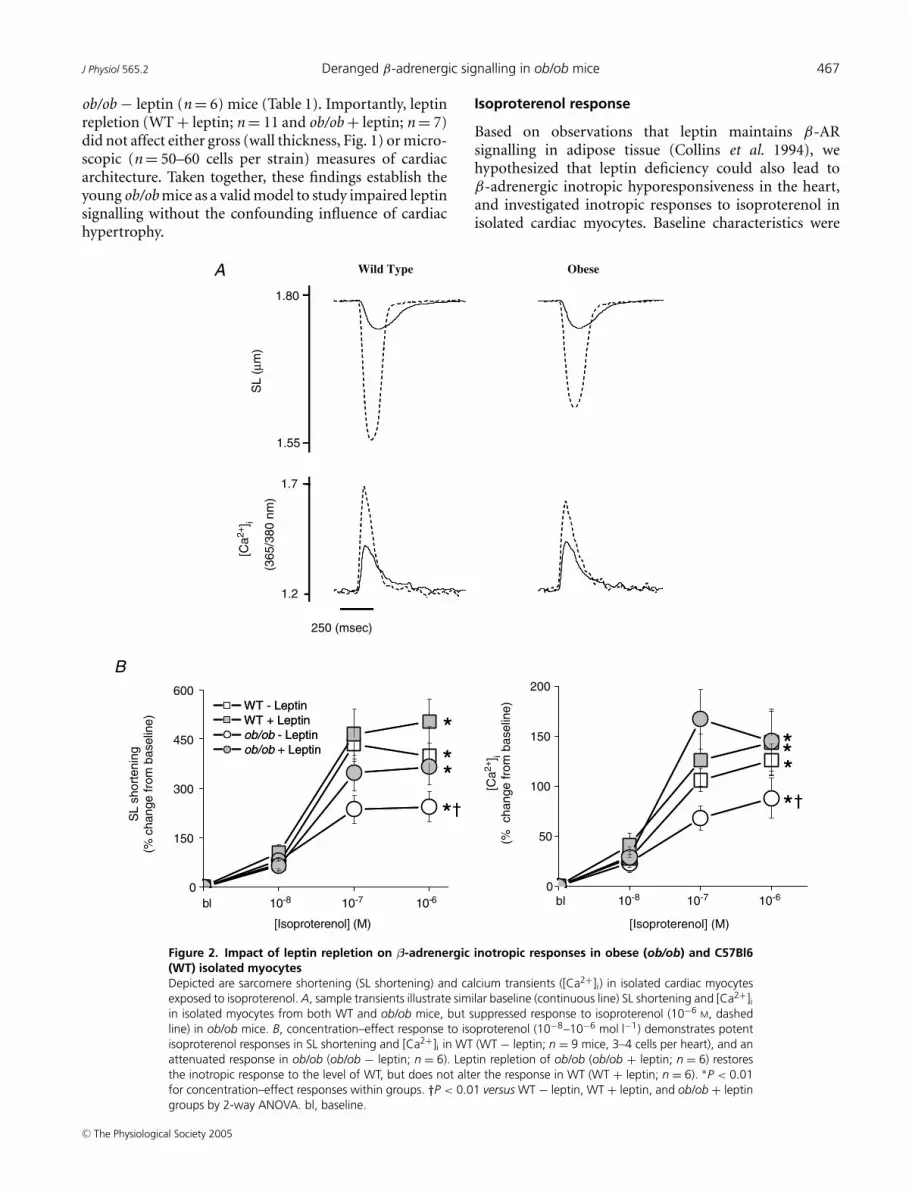
Figure 2. Impact of leptin repletion on β-adrenergic inotropic responses in obese (ob/ob) and C57Bl6(WT) isolated myocytesDepicted are sarcomere shortening (SL shortening) and calcium transients ([Ca2+]i) in isolated cardiac myocytesexposed to isoproterenol. A, sample transients illustrate similar baseline (continuous line) SL shortening and [Ca2+]iin isolated myocytes from both WT and ob/ob mice, but suppressed response to isoproterenol (10−6 M, dashedline) in ob/ob mice. B, concentration–effect response to isoproterenol (10−8–10−6 mol l−1) demonstrates potentisoproterenol responses in SL shortening and [Ca2+]i in WT (WT − leptin; n = 9 mice, 3–4 cells per heart), and anattenuated response in ob/ob (ob/ob − leptin; n = 6). Leptin repletion of ob/ob (ob/ob + leptin; n = 6) restoresthe inotropic response to the level of WT, but does not alter the response in WT (WT + leptin; n = 6). ∗P < 0.01for concentration–effect responses within groups. †P < 0.01 versus WT − leptin, WT + leptin, and ob/ob + leptingroups by 2-way ANOVA. bl, baseline.
Isoproterenol response
Based on observations that leptin maintains β-ARsignalling in adipose tissue (Collins et al. 1994), wehypothesized that leptin deficiency could also lead toβ-adrenergic inotropic hyporesponsiveness in the heart,and investigated inotropic responses to isoproterenol inisolated cardiac myocytes. Baseline characteristics were
C© The Physiological Society 2005

468 K. M. Minhas and others J Physiol 565.2
*
*
Forskolin (10-6 mol l-1)
*
**
*
*
*
††
SL
Sho
rten
ing
(% C
hang
e F
rom
BL)
SL
Sho
rten
ing
(% C
hang
e F
rom
BL)
[Ca2+
] i(%
Cha
nge
from
BL)
[Ca2+
] i(%
Cha
nge
from
BL)
0
150
300
450
600
0
50
100
150 WT - LeptinWT + Leptinob/ob - Leptinob/ob + Leptin
WT - LeptinWT + Leptinob/ob - Leptinob/ob + Leptin
*
*
0
100
200
300
400
500
0
50
100
150
200
*
**
**
*
†
†
Dibutryl cAMP (10-3 mol l-1)
A
B
BL 0.3uM 1uM 3uM 10uM0
100
200
300
400
500
600*
Forskolin
AC
act
ivity
% c
hang
e fr
om B
L
WT - LeptinWT + Leptinob/ob - Leptinob/ob + Leptin
C
Figure 3. Impact of forskolin and dibutryl cAMP on inotropic responses, and adenylyl cycalse (AC) activityA, SL shortening and [Ca2+]i were attenuated in myocytes from ob/ob (ob/ob − leptin; n = 5 mice, 3–4 cells perheart) compared with WT (WT − leptin; n = 5) controls in response to forskolin (10−6 mol l−1). Leptin treatmentof ob/ob mice (ob/ob + leptin; n = 3) reverses this abnormality (∗P < 0.001 versus baseline, and †P < 0.001 versusWT − leptin, WT + leptin and ob/ob + leptin), but does not affect the WT response (WT + leptin; n = 3).B, responses to dibutryl cAMP were suppressed in ob/ob mice and restored to WT levels by leptin. ∗P < 0.01 versusaseline, and †P < 0.05 versus WT − leptin, WT + leptin and ob/ob + leptin. C, basal and forskolin-stimulated ACactivity was unchanged in hearts from WT (n = 6) and ob/ob (n = 4) mice, and was unaffected with leptin repletion:WT + leptin (n = 6) and ob/ob + leptin (n = 4) (P = NS between groups). ∗P < 0.05 versus basal for dose–effectresponses within all groups.
C© The Physiological Society 2005

J Physiol 565.2 Deranged β-adrenergic signalling in ob/ob mice 469
similar between myocytes from ob/ob and WT mice at 1 Hz(Table 2). Consistent with prior studies (Barouch et al.2002), isoproterenol (10−8–10−6 mol l−1), a non-selectiveβ-AR agonist, stimulated myocyte contraction in WTmyocytes (10−6 mol l−1; SL shortening 399 ± 89%; [Ca2+]i
126 ± 16%; Fig. 2). In contrast, this positive inotropiceffect was markedly attenuated in the ob/ob myocyteswith parallel decreases in SL shortening (10−6 mol l−1;243 ± 46%) and [Ca2+]i (88 ± 20%, both P < 0.01 versusWT).
We next assessed the impact of leptin treatment inboth ob/ob and WT mice. Leptin administration for4 weeks to WT mice did not change the response inSL shortening or [Ca2+]i (Fig. 2B). On the other hand,leptin treatment of ob/ob mice augmented β-AR-mediatedinotropic responses towards levels indistinguishable fromWT controls (10−6 mol l−1; SL shortening 365 ± 54% and[Ca2+]i 145 ± 30%).
Adenylyl cyclase (AC) activity and cAMP response
We next sought to examine downstream sites ofβ-adrenergic inotropic signalling. Direct AC activationwith forskolin (10−8 mol l−1 to 10−6 mol l−1) also led toa depressed contractile response in ob/ob compared withWT mice (Fig. 3A). Specifically, forskolin (10−6 mol l−1)led to peak SL shortening of 154 ± 34% (with [Ca2+]i of39 ± 10%) in ob/ob compared with peak SL shortening of416 ± 79% (with [Ca2+]i of 107 ± 33%) in WT (P < 0.001for both). As before, leptin treatment of the ob/ob
0
10
20
30
40
50
*
WT - Leptin WT + Leptin ob/ob - Leptin ob/ob + Leptin
prot
ein
kina
se A
act
ivity
(un
its/m
l)
Figure 4. Protein kinase A (PKA) activityAnother downstream site of β-adrenergic inotropic signalling, the PKAactivity was suppressed in ob/ob (15.07 ± 5.41 units ml−1 – thenumber of units of kinase activity per 1 ml) as compared with WT mice(41.23 ± 4.11 units ml−1). Leptin repletion restored it towards normalin ob/ob mice (31.76 ± 6.55 units ml−1) without affecting activity inWT (37.45 ± 4.49 units ml−1). ∗P < 0.05 versus WT − leptin, WT +leptin, and ob/ob + leptin.
mice fully restored these inotropic responses towardsnormal, but did not affect WT contractility. Similarly,additional post-adenylyl cyclase defects were also evidentby depressed contractile responses to dibutryl cAMP(10−3 m), a phosphodiesterase-resistant cAMP analogue(Fig. 3B). Here too, depressed SL shortening and [Ca2+]i
in ob/ob were restored to the level of WT responses byleptin. To explore underlying biochemical mechanisms forsuppressed inotropic responses, we initially tested adenylylcyclase activity in vitro. However, we found no changes inthe forskolin-stimulated (1–300 units) AC activity acrossWT and ob/ob mice with and without leptin repletion(P = NS between groups, Fig. 3C).
Protein kinase A (PKA) activity
We next examined PKA activity; it was markedlysuppressed in ob/ob (15.07 ± 5.41 units ml−1) ascompared with WT mice (41.23 ± 4.11 units ml−1,P < 0.05 versus ob/ob) (Fig. 4). Leptin repletion restoredit towards normal in ob/ob mice (31.76 ± 6.55 units ml−1,P < 0.05 versus ob/ob and P = NS versus WT) withoutaffecting activity in WT mice (37.45 ± 4.49 units ml−1).
Calcium stores
One of the key downstream organelles in β-adrenergicallystimulated cardiac excitation–contraction coupling is thesarcoplasmic reticulum (SR). SR Ca2+ stores, measuredby caffeine-induced Ca2+ release, were reduced in theob/ob (%[Ca2+]i, 64 ± 10) compared with the WT(100 ± 8%, P < 0.05) myocytes (Fig. 5). As with theinotropic responses and PKA activity, leptin treatmentof ob/ob mice restored SR Ca2+ stores towards normal
[Ca2+
] i Nor
mal
ized
(%
)
0
30
60
90
120
Caffeine Induced [Ca2+]i
*
*
**
†
*
*
**
†
WT - LeptinWT + Leptin
ob/ob + Leptinob/ob - Leptin
Figure 5. Sarcoplasmic reticulum (SR) Ca2+ storesIsolated cardiac myocytes stimulated at 1 Hz were rapidly exposed tocaffeine (10 mmol l−1) after a brief pause. SR Ca2+ reserves weremeasured as percentage change from baseline [Ca2+]i, and arepresented normalized to WT − leptin. The ob/ob (ob/ob − leptin;n = 3 mice, 3–4 cells per heart) mice had reduced SR Ca2+ storescompared with controls (WT − leptin; n = 5). Leptin treatment(ob/ob + leptin; n = 3) restored stores towards normal but did notaffect them in WT (WT + leptin). ∗P < 0.01 versus baseline, and†P < 0.05 versus WT − leptin, WT + leptin and ob/ob + leptin.
C© The Physiological Society 2005

470 K. M. Minhas and others J Physiol 565.2
(111 ± 17%, P < 0.05 versus ob/ob), but did not changestores in WT mice (112 ± 4%).
Western blot analysis
To establish additional molecular correlates of thedepressed inotropic responses, we performed immuno-blot quantification of a large panel of proteins involved inβ-adrenergic inotropic responses (see Fig. 6 and Table 3).Ob/ob mice had abnormalities at both PKA and SR levelscorrelating with the observed functional defects at theselevels. In terms of G-proteins, there were parallel decreasesin Gsα (52 kDa) and Giα, and leptin repletion restoredthe levels of Gsα (52 kDa), but not Giα, to normal inob/ob mice. At the level of SR reuptake proteins, wefound that P-PLB was decreased, consistent with depressedPKA activity. In addition, SR Ca2+-ATPase (SERCA2a)abundance was found to be increased. Here, leptin infusionrestored P-PLB towards normal and augmented levelsof SERCA2a in both ob/ob and WT hearts. Finally, the
+ +- -
64 kD
Gsα
Giα
52 kD
45 kD
6 kD
110 kD
38 kD
β1-AR
-
WT ob/ob
P-PLB
SERCA2a
Figure 6. Western blot analysis of myocardial tissue extractsfrom ob/ob and WT mice without (−) and with 4 weeks ofleptin treatment (+)Representative Western blots depict expression of key proteins inβ-adrenergic regulation of excitation–contraction coupling. Notablefindings include down-regulation of Gsα (52 kDa), Giα, and P-PLB andup-regulation of SERCA2a in ob/ob mice. Leptin repletion restoredlevels of P-PLB and Gsα (52 kDa), but not Giα, to normal in ob/obmice. In addition, leptin infusion augmented levels of SERCA2a in bothob/ob and WT hearts. See Table 3 for normalized values.
abundance of the β-adrenergic receptors was not affectedin ob/ob mice.
Plasma measurements
We confirmed adequate leptin repletion with our leptin-infusion protocol (ob/ob + leptin 12.6 ± 5.4 ng ml−1;n = 5 vs. WT − leptin 7.5 ± 1.6 ng ml−1, n = 8, P = NS)(Table 4). Additionally, other plasma indices, includingglucose (mg dl−1), insulin (ng ml−1) and triglycerides(mg dl−1) which were elevated in ob/ob − leptin wererestored towards normal with leptin repletion. Theseparameters remained unchanged in WT animals withleptin repletion (Table 4).
Discussion
The major new findings of this study are thatleptin deficiency impairs cardiac β-adrenergic inotropicresponses by a mechanism involving protein kinase Aactivity and sarcoplasmic reticulum Ca2+ reuptake.β-Adrenergic inotropic responses are depressed inob/ob mice at a young age before the developmentof LVH, in a manner completely reversible by leptinrepletion. Ob/ob mice exhibit depressed protein kinase Aactivity with a concomitant depression in phosphorylatedphospholamban and reduced SR Ca2+ stores, all ofwhich are restored towards normal with leptin repletion.Together, these results suggest novel mechanism(s) bywhich leptin deficiency or resistance may contribute toheart failure and offer insights into cardiac dysfunction inobesity.
The young ob/ob mouse model offers unique insightinto the mechanisms underlying obesity–heart failurepathophysiology. Notably, the model is normotensive andhas not developed LVH (Barouch et al. 2003). Unlikeother models of obesity (Hohl et al. 1993; Carroll et al.2002), β-adrenergic signalling and molecular findings inthe young ob/ob mice are not confounded by cardiachypertrophy, a crucial point when considering that cardiaccontractility and reserve are depressed in human obesityprior to the development of LVH (Licata et al. 1992,1995; Ferraro et al. 1996). In addition, disrupted leptinsignalling is almost universal in obesity (Considine et al.1996) and leptin signalling is linked to many effectormolecules, allowing leptin to have wide ranging effects(Cohen et al. 1996; Nickola et al. 2000; Wold et al.2002). The majority of human obesity is associated withhyperleptinaemia and leptin resistance (Considine et al.1996; Ren, 2004). However, it is important to note thatthe observed derangements are due to leptin deficiency,whether actual or perceived (leptin resistance). Thus,identifying the key mechanisms of pathophysiology inob/ob mice has the potential to offer insight into thepathophysiology of the deranged signalling pathways
C© The Physiological Society 2005

J Physiol 565.2 Deranged β-adrenergic signalling in ob/ob mice 471
Table 3. Cardiac protein abundance
WT − leptin WT + leptin ob/ob − leptin ob/ob + leptin
Mice (n) 5 5 5 5β1-AR 1.00 ± 0.10 1.03 ± 0.10 0.99 ± 0.09 1.06 ± 0.03β2-AR 1.00 ± 0.45 1.21 ± 0.61 1.11 ± 0.50 1.12 ± 0.50Gsα (52 kDa) 1.00 ± 0.14 0.85 ± 0.10 0.44 ± 0.03∗‡ 0.95 ± 0.21†Gsα (45 kDa) 1.00 ± 0.09 1.39 ± 0.47 1.40 ± 0.43 1.81 ± 0.31Giα 1.00 ± 0.11 0.66 ± 0.20 0.40 ± 0.07∗ 0.46 ± 0.20∗
SERCA2a 1.00 ± 0.27 2.94 ± 0.74∗ 4.25 ± 0.50∗ 6.30 ± 0.58∗†‡PLB 1.00 ± 0.24 1.19 ± 0.23 0.98 ± 0.17 1.00 ± 0.20P-PLB 1.00 ± 0.16 1.06 ± 0.36 0.42 ± 0.09∗‡ 0.77 ± 0.11†∗P < 0.05 versus WT − leptin; †P < 0.05 versus ob/ob − leptin; ‡ P < 0.05 versus WT + leptin. Valuesare normalized to WT − leptin.
Table 4. Plasma measurements
WT − leptin WT + leptin ob/ob − leptin ob/ob + leptin
Mice (n)∗ 8 11 5 7Glucose (mg dl−1) 203 ± 11 202 ± 9 347 ± 23∗ 192 ± 6Leptin (ng ml−1) 4.8 ± 0.7 7.5 ± 1.6 1.7 ± 0.2∗ 12.6 ± 5.4Insulin (ng ml−1) 0.47 ± 0.06 0.41 ± 0.09 4.15 ± 1.88∗ 0.35 ± 0.11Triglycerides (mg dl−1) 69 ± 7 54 ± 6 124 ± 6∗ 48 ± 5
∗P < 0.05 versus WT − leptin, WT + leptin, and ob/ob + leptin.
in human obesity. In this regard, we demonstrate thatthe ob/ob mice exhibit disrupted β-adrenergic signallingmediated by leptin deficiency and have localized the defectto the levels of PKA activity and Ca2+ cycling.
β-Adrenergic receptor and adenylyl cyclase (AC)
The manner in which obesity affects β-adrenergicsignalling remains controversial, as the few studies thathave addressed basic mechanisms have had conflictingresults. Initially, decreased β-receptor density (Bass &Ritter, 1985; Strassheim et al. 1992) was implicatedin impaired contractility; however, more recent studiesindicate that attenuated inotropy occurs independentlyof changes in β1 or β2 receptor expression (Hohl et al.1993; Ernsberger et al. 1994; Carroll et al. 2002). Ourresults are in agreement with the concept that distalsignalling transduction defects play a major role indepressed inotropic responses, as we show unchangedβ1 and β2 protein expression in ob/ob hearts. Notably,we demonstrate that leptin deficiency alters both Gs
and Gi proteins. Interestingly, leptin repletion onlyrestores depressed Gsα-52 kDa to normal without affectingeither Gi or Gsα-45 kDa, suggesting specificity of theimpact of leptin restoration. Of the G proteins thatstimulate AC, the long splice variant (Gsα-52 kDa)has been shown to have greater potency and moreconstitutive basal activity than the short splice variant(Gsα-45 kDa) (Seifert et al. 1998). These observedalterations could be an adjunct mechanism by whichleptin deficiency mediates disruption of β-adrenergic
signal transduction. Furthermore, our demonstrationof unchanged AC activity in obesity consistent withprevious reports (Carroll et al. 2002), in conjunctionwith a suppressed response to exogenous dibutryl cAMPsuggests downstream leptin-deficiency-mediated defectsin the β-adrenergic pathway.
PKA and sarcoplasmic recticulum
Cardiac β-adrenergic-mediated inotropy is greatlydependent on SR Ca2+ availability (Bers, 2002).Accordingly, we demonstrate that leptin deficiency impairsSR Ca2+ cycling as ob/ob myocytes have reduced SR Ca2+
stores and diminished levels of P-PLB, a key componentmodulating SR reuptake (Bers, 2002). Additionally, ourfindings of depressed PKA activity in ob/ob mice is pivotalas β-adrenergic activity increases contractility, at least inpart, by cAMP-mediated phosphorylation of PLB, whichremoves the tonic inhibition of PLB on SERCA2a, allowingfor increased Ca2+ reuptake (Bers, 2002). Consequently,reduced amounts of P-PLB, as seen in ob/ob, woulddisrupt β-adrenergic reserve. Reductions of P-PLB maybe secondary to diminished PKA activity, but may alsoresult from a direct action of leptin on PLB. Anotherpotential pathway of this interaction may involve leptin’saction on protein kinase C (PKC) (Ookuma et al. 1998),which has recently been shown to play a role in modulatingPLB (Watanuki et al. 2004; Braz et al. 2004). However,our findings of depressed PKA activity, lowered P-PLBexpression, and depressed Ca2+ stores associated withleptin deficiency, all of which are restored to normal
C© The Physiological Society 2005

472 K. M. Minhas and others J Physiol 565.2
with leptin repletion, strongly suggest that leptin mediatesphosphorylation of PLB probably due in large part toPKA activity. It is noteworthy that the depressed SR Ca2+
stores in ob/ob are not overcome by what appears to bea compensatory increase in SERCA2a abundance. Inter-estingly, we previously observed a similar compensatoryup-regulation of SERCA2a in NOS1 knockout mice whichalso have reduced SR Ca2+ stores (Khan et al. 2003). Leptininfusion increases SERCA2a in both WT and ob/ob miceindicating that leptin may also have non-specific effects onabundance of some (the minority) of the proteins.
Leptin signalling
Disrupted leptin signalling can attenuate cardio-vascular function through several potential pathways.PKC (Ookuma et al. 1998) along with nitric oxide(Fruhbeck, 1999; Nickola et al. 2000), the Janus familytyrosine kinase and signal transducers and activatorsof transcription (Wold et al. 2002), mitogen-activatedprotein kinase (Wold et al. 2002; Rajapurohitam et al.2003), phosphatidylinositol 3-kinase (Cohen et al. 1996),type 3 phosphodiesterase (Zhao et al. 1998), extracellularregulated kinase, phospholipase C, insulin receptorsubstrates protein, and protein kinase B (Szanto &Kahn, 2000) (see Sweeney, 2002 for review), are justsome of the many effectors coupled to leptin signalling.These intermediate effectors influence key components ofexcitation–contraction coupling (Hare, 2003; Braz et al.2004; McDowell et al. 2004; Hare & Stanler 2005). To thebest of our knowledge, for the first time, we demonstratethe role of PKA as an intermediate effector molecule inleptin signalling, and thus offer a novel mechanism bywhich leptin attenuates β-adrenergic signal transductionin the heart.
Clinical implications
In contrast to leptin deficiency characteristic of ob/obmice, human obesity is associated with leptin resistanceand circulating leptin excess (Considine et al. 1996).Hyperleptinaemia has been described as the key driverof obesity-related cardiovascular dysfunctions, with theobserved pathology attributed to leptin resistance (seeRen, 2004 for review). The main findings of this studyare leptin-deficiency-mediated disruption of β-adrenergicsignal transduction and consequent depression ofmyocyte contractility in ob/ob mice, defects that arecorrected with leptin repletion. A key point is that thereis relative leptin deficiency downstream of the receptor inboth leptin-deficient mice and leptin-‘insensitive’ obesehumans (Sader et al. 2003). Therefore, the underlyingleptin signalling derangements are the same regardless ofthe reason behind its unavailability, whether it is leptindeficiency or resistance. Thus, to the extent that thereis ‘relative leptin deficiency’ downstream of the receptor
in the deficient state, it is attractive to speculate that thesignalling abnormalities in the ob/ob mice are clinicallyrelevant to the development of obesity-associated cardiacdysfunction. We have already extended the value of thismurine model by inducing weight loss by either re-infusingexogenous leptin or by calorific restriction (Barouch et al.2003). From a physiological standpoint these interventionsare extremely valuable in the recapitulation of the cardio-vascular benefits that could result in humans. Specifically,it should be noted that leptin infusion to ob/ob mice is thephysiological correlate of weight loss in leptin-resistantobesity, a situation in which leptin signalling is restored(Sader et al. 2003).
Limitations
Although impaired leptin signalling disrupts cardiacβ-adrenergic activity, whether this occurs entirely dueto direct effects of leptin on the heart or is partially aconsequence of sympathetic nervous system regulationby leptin is still unclear. Peripheral effects of leptin onventricular myocytes have been demonstrated (Nickolaet al. 2000) and may be responsible for cardiac changes. Atthe same time, central nervous system-mediated cardiacmodulation cannot be excluded as leptin administeredintracerebroventricularly has been shown to modify thesympathetic outflow to peripheral tissues (Dunbar et al.1997; Haynes et al. 1997). Similarly, the impact of otherhormones associated with obesity, and possibly leptin,such as cortisol, thyroxine and testosterone, cannot beignored (Cohen et al. 2001). Changes in insulin sensitivitymay also contribute to β-adrenergic hyporesponsiveness,as we (Table 4) and others (Hunt et al. 1976) have shownthat both ob/ob mice and obese humans are hyper-glycaemic and hyperinsulinaemic. In fact, the insulin andleptin hormonal signalling axes are closely interrelated(Segal et al. 1996). However, Hintz and colleagues haverecently found that insulin resistance does not alter leptinresponsiveness in cardiac myocytes (Hintz et al. 2003).Additionally, insulin treatment of diabetic animals didnot affect PKA-mediated phosphorylation and activity(Netticadan et al. 2001). Thus, it is unlikely that alteredinsulin levels account for impaired β-adrenergic signallingand our key finding of depressed PKA activity and P-PLBlevels in ob/ob mice.
Another limiting factor might be that all the functionaleffects of leptin are shown in isolated myocytes, whereasthe molecular biology data are derived from whole myo-cardium. Nonetheless, our findings are consistent betweenmolecular and functional levels.
Conclusion
Impaired leptin signalling disrupts multiple sites in theβ-adrenergic signal transduction pathway independentof hypertrophy. Our findings localize the defect to
C© The Physiological Society 2005

J Physiol 565.2 Deranged β-adrenergic signalling in ob/ob mice 473
the level of PKA activity with consequent impact onsarcoplasmic reticulum Ca2+ cycling. Although leptinabnormalities are increasingly described in obesity–heartfailure pathophysiology, the mechanisms of leptinsignalling in the heart remain poorly understood. Thecurrent findings offer novel insights into mechanisms bywhich leptin deficiency or resistance contributes to cardiacsignal transduction and contractile regulation.
Reference
Alpert MA (2001). Obesity cardiomyopathy: pathophysiologyand evolution of the clinical syndrome. Am J Med Sci 321,225–236.
Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC,Kobilka BK & Lowell BB (2002). betaAR signaling requiredfor diet-induced thermogenesis and obesity resistance.Science 297, 843–845.
Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP &Hare JM (2003). Disruption of leptin signaling contributesto cardiac hypertrophy independently of body weight inmice. Circulation 108, 754–759.
Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP,Kobeissi ZA et al. (2002). Nitric oxide regulates the heart byspatial confinement of nitric oxide synthase isoforms. Nature416, 337–339.
Bass S & Ritter S (1985). Decreased beta-adrenergic receptorbinding in obese female Zucker rats. J Auton Nerv Syst 14,81–87.
Bers DM (2002). Cardiac excitation-contraction coupling.Nature 415, 198–205.
Bray GA & York DA (1997). Clinical review 90: Leptin andclinical medicine: a new piece in the puzzle of obesity. J ClinEndocrinol Metab 82, 2771–2776.
Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky Ret al. (2004). PKC-alpha regulates cardiac contractility andpropensity toward heart failure. Nat Med 10,248–254.
Breslow MJ, Min-Lee K, Brown DR, Chacko VP, Palmer D &Berkowitz DE (1999). Effect of leptin deficiency onmetabolic rate in ob/ob mice. Am J Physiol 276,E443–449.
Bristow MR, Hershberger RE, Port JD, Gilbert EM, Sandoval A,Rasmussen R, Cates AE & Feldman AM (1990). Beta-adrenergic pathways in nonfailing and failing humanventricular myocardium. Circulation 82, I12–I25.
Carroll JF, Kyser CK & Martin MM (2002). beta-Adrenoceptordensity and adenylyl cyclase activity in obese rabbit hearts.Int J Obes Relat Metab Disord 26, 627–632.
Cohen B, Novick D & Rubinstein M (1996). Modulation ofinsulin activities by leptin. Science 274, 1185–1188.
Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P,Mombaerts P & Friedman JM (2001). Selective deletion ofleptin receptor in neurons leads to obesity. J Clin Invest 108,1113–1121.
Collins S, Daniel KW, Rohlfs EM, Ramkumar V, Taylor IL &Gettys TW (1994). Impaired expression and functionalactivity of the beta 3- and beta 1-adrenergic receptors inadipose tissue of congenitally obese (C57BL/6J ob/ob) mice.Mol Endocrinol 8, 518–527.
Collins S & Surwit RS (2001). The beta-adrenergic receptorsand the control of adipose tissue metabolism andthermogenesis. Recent Prog Horm Res 56, 309–328.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A,Stephens TW, Nyce MR et al. (1996). Serumimmunoreactive-leptin concentrations in normal-weightand obese humans. N Engl J Med 334, 292–295.
Dunbar JC, Hu Y & Lu H (1997). Intracerebroventricularleptin increases lumbar and renal sympathetic nerveactivity and blood pressure in normal rats. Diabetes 46,2040–2043.
Eckel RH, Barouch WW & Ershow AG (2002). Report of theNational Heart, Lung, and Blood Institute-National Instituteof Diabetes and Digestive and Kidney Diseases WorkingGroup on the pathophysiology of obesity-associatedcardiovascular disease. Circulation 105, 2923–2928.
Emala CW (1997). Methods for the measurement of adenylylcyclase activity. In Molecular Regulation of Conscious States,ed. Lydic R, pp. 57–69. CRC Press, Boca Raton, NY.
Ernsberger P, Koletsky RJ, Baskin JS & Foley M (1994).Refeeding hypertension in obese spontaneously hypertensiverats. Hypertension 24, 699–705.
Ferraro S, Perrone-Filardi P, Desiderio A, Betocchi S, D’Alto M,Liguori L, Trimigliozzi P, Turco S & Chiariello M (1996).Left ventricular systolic and diastolic function in severeobesity: a radionuclide study. Cardiology 87, 347–353.
Fruhbeck G (1999). Pivotal role of nitric oxide in the control ofblood pressure after leptin administration. Diabetes 48,903–908.
Hare JM & Stanler JJ (2005). NO/redox disequilibrium in thefailing heart and cardiovascular system. J Clin Invest 115,509–517.
Hare JM (2003). Nitric oxide and excitation-contractioncoupling. J Mol Cell Cardiol 35, 719–729.
Haynes WG, Sivitz WI, Morgan DA, Walsh SA & Mark AL(1997). Sympathetic and cardiorenal actions of leptin.Hypertension 30, 619–623.
Hintz KK, Aberle NS & Ren J (2003). Insulin resistance induceshyperleptinemia, cardiac contractile dysfunction but notcardiac leptin resistance in ventricular myocytes. Int J ObesRelat Metab Disord 27, 1196–1203.
Hohl CM, Hu B, Fertel RH, Russell JC, McCune SA & AltschuldRA (1993). Effects of obesity and hypertension on ventricularmyocytes: comparison of cells from adult SHHF/Mcc-cp andJCR: LA-cp rats. Cardiovasc Res 27, 238–242.
Hunt CE, Lindsey JR & Walkley SU (1976). Animal models ofdiabetes and obesity, including the PBB/Ld mouse. Fed Proc35, 1206–1217.
Illiano G, Naviglio S, Pagano M, Spina A, Chiosi E, Barbieri M& Paolisso G (2002). Leptin affects adenylate cyclase activityin H9c2 cardiac cell line: effects of short- and long-termexposure. Am J Hypertens 15, 638–643.
Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ,Larson MG, Kannel WB & Vasan RS (2002). Obesity and therisk of heart failure. N Engl J Med 347, 305–313.
Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, KumarA et al. (2003). Nitric oxide regulation of myocardialcontractility and calcium cycling: independent impact ofneuronal and endothelial nitric oxide synthases. Circ Res 92,1322–1329.
C© The Physiological Society 2005

474 K. M. Minhas and others J Physiol 565.2
Licata G, Scaglione R, Avellone G, Ganguzza A, Corrao S,Arnone S & Di Chiara T (1995). Hemostatic function inyoung subjects with central obesity: relationship with leftventricular function. Metabolism 44, 1417–1421.
Licata G, Scaglione R, Paterna S, Parrinello G, Indovina A,Dichiara MA, Alaimo G & Merlino G (1992). Left ventricularfunction response to exercise in normotensive obesesubjects: influence of degree and duration of obesity.Int J Cardiol 37, 223–230.
McDowell SA, McCall E, Matter WF, Estridge TB & Vlahos CJ(2004). Phosphoinositide 3-kinase regulates excitation-contraction coupling in neonatal cardiomyocytes.Am J Physiol Heart Circ Physiol 286, H796–805.
Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H,Wareham NJ et al. (1997). Congenital leptin deficiency isassociated with severe early-onset obesity in humans. Nature387, 903–908.
Netticadan T, Temsah RM, Kent A, Elimban V & Dhalla NS(2001). Depressed levels of Ca2+-cycling proteins mayunderlie sarcoplasmic reticulum dysfunction in the diabeticheart. Diabetes 50, 2133–2138.
Nickola MW, Wold LE, Colligan PB, Wang GJ, Samson WK &Ren J (2000). Leptin attenuates cardiac contraction in ratventricular myocytes. Role of NO. Hypertension 36, 501–505.
Ookuma M, Ookuma K & York DA (1998). Effects of leptin oninsulin secretion from isolated rat pancreatic islets. Diabetes47, 219–223.
Pladevall M, Williams K, Guyer H, Sadurni J, Falces C, Ribes Aet al. (2003). The association between leptin and leftventricular hypertrophy: a population-based cross-sectionalstudy. J Hypertens 21, 1467–1473.
Rajapurohitam V, Gan XT, Kirshenbaum LA & Karmazyn M(2003). The obesity-associated peptide leptin induceshypertrophy in neonatal rat ventricular myocytes. Circ Res93, 277–279.
Ren J (2004). Leptin and hyperleptinemia – from friend to foefor cardiovascular function. J Endocrinol 181, 1–10.
Sader S, Nian M & Liu P (2003). Leptin: a novel link betweenobesity, diabetes, cardiovascular risk, and ventricularhypertrophy. Circulation 108, 644–646.
Salomon Y, Londos C & Rodbell M (1974). A highly sensitiveadenylate cyclase assay. Anal Biochem 58, 541–548.
Segal KR, Landt M & Klein S (1996). Relationship betweeninsulin sensitivity and plasma leptin concentration in leanand obese men. Diabetes 45, 988–991.
Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E& Kobilka BK (1998). Different effects of Gsalpha splicevariants on beta2-adrenoreceptor-mediated signaling. Thebeta2-adrenoreceptor coupled to the long splice variant ofGsalpha has properties of a constitutively active receptor.J Biol Chem 273, 5109–5116.
Strassheim D, Houslay MD & Milligan G (1992). Regulation ofcardiac adenylate cyclase activity in rodent models of obesity.Biochem J 283, 203–208.
Sweeney G (2002). Leptin signalling. Cell Signal 14, 655–663.Szanto I & Kahn CR (2000). Selective interaction between
leptin and insulin signaling pathways in a hepatic cell line.Proc Natl Acad Sci U S A 97, 2355–2360.
Watanuki S, Matsuda N, Sakuraya F, Jesmin S & Hattori Y(2004). Protein kinase C modulation of the regulation ofsarcoplasmic reticular function by protein kinase A-mediated phospholamban phosphorylation in diabetic rats.Br J Pharmacol 141, 347–359.
Wold LE, Relling DP, Duan J, Norby FL & Ren J (2002).Abrogated leptin-induced cardiac contractile response inventricular myocytes under spontaneous hypertension: roleof Jak/STAT pathway. Hypertension 39, 69–74.
Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF & Luo JD(2004). Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal ratcardiomyocytes. Circulation 110, 1269–1275.
Zhao AZ, Bornfeldt KE & Beavo JA (1998). Leptin inhibitsinsulin secretion by activation of phosphodiesterase 3B.J Clin Invest 102, 869–873.
Acknowledgements
This work was supported by NIH grants RO1 HL-65455 anda Paul Beeson Physician Faculty Scholars in Ageing ResearchAward, both to J.M.H.; NIH grant KO8 HL-076220 and the IrvinTalles Endourment research (J.M.H. & L.A.B.). We are indebtedto Konrad Vandegaer, Eleanor L. Pitz, and Guillermo F. Duartefor technical assistance.
C© The Physiological Society 2005