internalization of enos via caveolae regulates paf-induced inflammatory hyperpermeability to...
TRANSCRIPT

1
INTERNALIZATION OF eNOS VIA CAVEOLAE REGULATES PAF-INDUCED
INFLAMMATORY HYPERPERMEABILITY TO MACROMOLECULES
Fabiola A. Sánchez, David D. Kim, Ricardo G. Durán, Cynthia J. Meininger1, and Walter N.
Durán
From Program in Vascular Biology, Department of Pharmacology and Physiology, UMDNJ -
New Jersey Medical School, 185 South Orange Ave, Newark, NJ 07101-1709; 1Department of
Systems Biology and Translational Medicine, Texas A & M Health Science Center, Temple, TX
76504
Running Title: eNOS endocytosis and hyperpermeability
Address correspondence to:
Fabiola A. Sánchez, PhD
185 S. Orange Ave., MSB H-638
Newark, NJ 07101-1709
Phone: 973-972-3981
Fax: 973-972-7950
Email: [email protected]
Articles in PresS. Am J Physiol Heart Circ Physiol (August 15, 2008). doi:10.1152/ajpheart.00629.2008
Copyright © 2008 by the American Physiological Society.

2
ABSTRACT
Endothelial nitric oxide synthase (eNOS) is thought to regulate microvascular permeability via
NO production. We tested the hypotheses that expression of eNOS and eNOS endocytosis by
caveolae are fundamental for appropriate signaling mechanisms in inflammatory endothelial
permeability to macromolecules. We used bovine coronary postcapillary venular endothelial
cells (CVEC) because these cells are derived from the microvascular segment responsible for
transport of macromolecules in inflammation. We stimulated CVEC with platelet-activating
factor (PAF) at 100 nM and measured eNOS phosphorylation, NO production and CVEC
monolayer permeability to FITC-dextran-70. PAF translocated eNOS from plasma membrane to
cytosol, induced changes in the phosphorylation state of the enzyme and increased NO
production from 4.3 ± 3.8 to 467 ± 22.6 nM. PAF elevated CVEC monolayer permeability to
FITC-dextran-70 from 3.4 ± 0.3 x 10-6
cm/s to 8.5 ± 0.4 x 10-6
cm/s. Depletion of endogenous
eNOS with siRNA abolished PAF-induced hyperpermeability demonstrating that expression of
eNOS is required for inflammatory hyperpermeability responses. Inhibition of the caveolar
internalization by blocking caveolar scission using transfection of dyn2K44A (dynamin
dominant negative mutant) inhibited PAF-induced hyperpermeability to FITC-dextran-70. We
interpret these data as evidence that 1) eNOS is required for hyperpermeability to
macromolecules, and 2) internalization of eNOS via caveolae is an important mechanism in the
regulation of endothelial permeability. We advance the novel concept that eNOS internalization
to cytosol is a signaling mechanism for onset of microvascular hyperpermeability in
inflammation.
Key Words: endothelial nitric oxide; endothelial cells; microvascular permeability; eNOS
translocation; acetylcholine; platelet-activating factor

3
INTRODUCTION
Increased microvascular permeability (hyperpermeability) is a hallmark of inflammation.
Hyperpermeability to macromolecules is a process that occurs in vivo mainly at postcapillary
venules. While the significance of endothelial nitric oxide synthase (eNOS)-derived nitric oxide
(NO) in the regulation of microvascular transport across postcapillary venules is an
experimentally supported emerging concept (16, 27, 34, 36), the mechanisms by which eNOS
controls microvascular permeability are poorly understood.
Pro-inflammatory agonists that increase permeability - such as vascular endothelial growth factor
(VEGF), bradykinin (BK), and platelet activating factor (PAF) - stimulate signaling cascades that
converge on eNOS (20), activate the enzyme and cause NO production (9). The activation of
eNOS proceeds mainly through phosphorylation of the enzyme at serine 1177 (Ser1177
); (7, 12),
and de-phosphorylation at threonine 495 (Thr495
); (8). Interestingly, acetylcholine (ACh), an
agent that causes vasodilation but does not alter microvascular permeability (27), induces exactly
the same changes in phosphorylation of eNOS (30). These observations suggest that
phosphorylation of eNOS per se is not a determinant of the functional consequences of eNOS-
derived NO.
We reasoned that location of eNOS may contribute to determine the functional significance of
eNOS-derived NO. eNOS is found in plasma membrane (mostly in caveolae) and Golgi in
control endothelial cells (EC). It is established that eNOS can produce NO regardless of its
location, even though its efficiency may vary (5, 37). Also, eNOS can translocate, upon

4
stimulation, from plasma membrane to intracellular compartments (10, 23, 30). This movement
has been associated with depalmitoylation of eNOS (35), and more recently, it has been
suggested that caveolae may serve as a vehicle for eNOS translocation (4).
The functional significance of eNOS internalization or traffic, if any, has not been previously
investigated. Work from Michel at al shows eNOS movement to the cytosol after VEGF
application, an agonist that induces hyperpermeability (10) and we demonstrated that PAF-
induced eNOS preferential internalization to cytosol was associated with hyperpermeability to
macromolecules (30). Based on these and the above-mentioned observations, we hypothesized
that eNOS expression is required for development of hyperpermeability and that eNOS
internalization to NO-receptors serves to determine the functional significance of eNOS-derived
NO in response to PAF, a pro-inflammatory autacoid. The experiments reported herein were
designed to test these hypotheses in bovine coronary endothelial cells derived from postcapillary
venules (CVEC; (31). We confirm in endothelial cells that eNOS expression is required for an
increase in permeability and propose, for the first time, that eNOS internalization via caveolae
plays a functional role in determining PAF-induced hyperpermeability to macromolecules.
MATERIALS AND METHODS
Antibodies. We used mouse anti-human eNOS, mouse anti-phospho-human eNOS(Ser1177
),
mouse anti-phospho-human eNOS(Thr495
) and rabbit anti-caveolin antibodies from BD
Biosciences (San Jose, CA). All the above-mentioned mouse antibodies recognize the
corresponding eNOS epitopes in the bovine CVEC.

5
Cell culture and transfection. We grew CVEC in Dulbeccco’s modified Eagle’s Medium
supplemented with 10% fetal bovine serum; 20 units/ml sodium heparin, 50 ug/ml penicillin, 50
µg/ml streptomycin and 10 µg/ml neomycin. All the experiments were performed using passage
3-7 CVEC. Using electroporation, we transfected CVEC with cDNA for the dominant negative
mutant of dynamin 2 coupled to green fluorescent protein (dyn2K44A; kindly provided by Dr.
Mark McNiven, Mayo Clinic College of Medicine, Rochester, MN). Briefly, we grew cells to
70-80% confluence, trypsinized them and re-suspended them in 100 µl of nucleofection solution.
We, then, added 1 µg of dyn2K44A cDNA to the cells. We electroporated CVEC using a basic
nucleofector kit for primary mammalian endothelial cells. We applied program T23 from Amaxa
Biosystems (Gaithersburg, MD). We electroporated CVEC with nucleofection solution and with
the empty vector as a control. The experiments were carried out 72 h after transfection. We
verified the expression of transfected dyn2K44A by the cellular fluorescence.
eNOS siRNA transfection. We used specific siRNA (Ambion, Austin, TX), as designed by Zhang
et al (37), to deplete eNOS. We transfected CVEC with siRNA using lipofectin and following
the protocol recommended by the manufacturer (Invitrogen, Carlsbad, CA). We lysed the cells at
pre-determined times and performed western blotting against eNOS to confirm depletion of the
protein.
NO measurements. We measured NO production using NO-sensitive recessed-tip
microelectrodes (1). We used 100% nitrogen and 400 and 800 parts per million NO in nitrogen to
establish a calibration range for NO of 0-600-1,200 nM (in saline). We placed coverslips
containing confluent CVEC in a perfusion chamber. We superfused the cells with media

6
supplemented with L-arginine at a rate of 1 ml/min. We administered PAF (Sigma Chemicals, St.
Louis, MO) through a side-port in the perfusion tubing to achieve a PAF concentration of 100
nM in the chamber.
Imunofluorescence microscopy. We fixed confluent monolayers of CVEC grown on glass
coverslips with 3% paraformaldehyde for 15 minutes, permeabilized them with 0.5% PBS-Triton
for 5 minutes, blocked them in 1% PBS-BSA for 30 minutes and then incubated them with anti-
eNOS antibodies and Alexa Fluor secondary antibodies. We examined the CVEC with an
inverted fluorescence microscope (Axiovert 200 M; Zeiss).
Detergent free purification of caveolae enriched membrane fractions. We grew CVEC in 100
mm tissue culture dishes (2 plates per treatment). After stimulation with 100 nM PAF, we
homogenized and sonicated the cells according to described protocols (21). We placed cell
lysates adjusted to 45% sucrose in a volume of 1.5 ml adding buffer 2 (90% sucrose/ 25 mM
MES, pH 6.5/ 150 mM NaCl, and inhibitors of proteases and phosphatases) at the bottom of a
4.5 ml centrifuge tube. We layered 1.5 ml of each buffer 3 (35% sucrose/ 250 mM sodium
carbonate, pH 11/ 25 mM MES/ 150 mM NaCl) and buffer 4 (5% sucrose/ 250 mM sodium
carbonate, pH 11/ 25 mM MES/ 150 mM NaCl) on top of it, and centrifuged the samples at
44,000 rpm for 18 h in a Beckman L8-70M Ultracentrifuge equipped with a SW60Ti rotor. We
collected 12 fractions from the top to the bottom of each tube. An equal volume of each
fraction
was used for Western blot analysis and probed for eNOS and caveolin-1.

7
Western blot analysis. We grew CVEC to confluence in 100mm plates. Immunoblot analyses of
protein expression and phosphorylation were assessed as we described previously (2, 30).
Densitometric analyses of Western blots were performed using the NIH Image J program.
Measurement of monolayer permeability. We determined control and PAF-stimulated
permeability to fluorescein isothiocyanate dextran 70 KDa (FITC-Dx-70, a macromolecule that
mimics albumin) across confluent CVEC monolayers using an established method (2, 30). We
obtained samples for baseline permeability every 15 minutes from the abluminal chamber for a
period of 60 minutes. After addition of PAF to both sides of the chamber (final concentration =
100 nM), we collected samples for additional 60 minutes.
Statistical analysis. Data are presented as mean ± S.E.M. Groups were analyzed for differences
by one-way ANOVA followed by Tukey-Kramer’s test. Significance was accepted at p < 0.05.
RESULTS
PAF stimulates eNOS phosphorylation and NO production in CVEC. Because we used
endothelial cells derived from postcapillary venules, our first step was to confirm that CVEC
express eNOS and that the enzyme can be activated to produce NO. We verified the expression
of eNOS in CVEC by immunofluorescence. Figure 1A shows immunofluorescent localization of
eNOS preferentially in the plasma membrane and Golgi, in agreement with the typical
distribution of eNOS described in endothelial cells (13, 14, 30). We corroborated eNOS
activation in response to PAF by measuring NO production and eNOS phosphorylation.

8
Application of 100 nM PAF rapidly increased NO production by CVEC from 4.3 ± 3.8 nM to
467.0 ± 22.6 nM (mean ± SEM; p < 0.05; Figure 1B). After quantification of eNOS
phosphorylation, we calculated the ratio of phosphorylated eNOS to total eNOS at control (time
= 0), 0.5, 1.0 and 3.0 minutes after PAF. Figure 1C (upper panel) shows that PAF significantly
increased phosphorylation of eNOS at Ser1177
as early as 0.5 minutes. The phosphorylation at
Ser1177
remained elevated up to minute 3.0. PAF-induced de-phosphorylation of eNOS at Thr495
was significant 1 minute after application of the autacoid and showed a trend for returning
towards baseline levels at 3 minutes. Figure 1C (lower panel) displays western blots illustrating
changes in eNOS phosphorylation at Ser1177
and Thr495
. Interestingly, NO production reached a
maximum approximately 3 minutes after the application of PAF, indicating a close temporal
correlation between eNOS phosphorylation and NO production in CVEC.
PAF-stimulated hyperpermeability is regulated by eNOS-derived NO in CVEC. To correlate
molecular signals with functional end-points, we investigated PAF-induced hyperpermeability to
FITC-Dx-70 in CVEC monolayers. PAF significantly increased permeability to FITC-Dx-70
from 3.4 ± 0.3 x 10-6
cm/s to 8.5 ± 0.4 x 10-6
cm/s (Fig. 2A). This change was rapid inasmuch as
a change in the slope of flux of FITC-Dx-70 was detected in the first sample taken at 5 minutes
after PAF application. To test that the increase in permeability is mediated by eNOS, we
depleted eNOS from CVEC using specific siRNA (37). Figure 2B shows that eNOS expression
decreases as early as 16 hours after transfection of CVEC with eNOS siRNA and illustrates that
maximal depletion of eNOS occurred at 72 hours using 30 nM eNOS siRNA. We chose this
maximal depletion time-point and siRNA concentration to perform our measurements of CVEC
monolayer permeability to FITC-Dx-70. Scrambled siRNA, used as control, does not decrease

9
the normal expression of eNOS in CVEC. Figure 2C shows that the basal permeability is normal
in eNOS-depleted CVEC. However, the hyperpermeability response to PAF is completely absent
in eNOS-depleted CVEC (Fig. 2C).
PAF induces eNOS trafficking in CVEC. Having demonstrated that eNOS is required for
development of an increase in CVEC monolayer permeability to macromolecules, we
investigated whether or not translocation of eNOS is also required for PAF-induced
hyperpermeability. As a first approach, we verified eNOS translocation in CVEC using
immunofluorescence microscopy and purification of caveolae enriched membrane fractions.
Figure 3A shows eNOS is located in the plasma membrane and Golgi in control CVEC, and
illustrates that PAF stimulates eNOS translocation from plasma membrane. Since caveolin is a
widely used marker for caveolae, we confirmed the information provided by microscopy by
isolating caveolin-containing lipid rafts fractions and probing for eNOS and caveolin by western
blotting. Figure 3B shows that, under baseline conditions, eNOS is distributed in fractions 7-12,
and is greatly enriched in fractions 7 and 8. Baseline caveolin is enriched in the same fractions.
Three minutes after PAF application, eNOS is found mainly, if not only, in heavier fractions
(particularly in fraction 12). Taken together, the microscopy and lipid rafts data indicate that
PAF induces eNOS translocation in CVEC.
eNOS internalization via caveolae and PAF-induced hyperpermeability. Because PAF-induced
eNOS preferential translocation to cytosol is associated with PAF-induced hyperpermeability
(30), we hypothesized that stimulated trafficking of eNOS-containing caveolae may serve to
deliver eNOS to subcellular effectors which determine the functional outcome of the initial

10
stimulus. To test this hypothesis, we inhibited eNOS internalization by transfecting CVEC with
dyn2K44A, a dominant negative mutant of dynamin-2 (18, 24, 25). Figure 4A shows the
expression of dyn2K44A in CVEC. The efficiency of transfection, assessed by cell counting
(fluorescent versus total cells), was 60%. Transfection of CVEC with dyn2K44A completely
prevented PAF-induced hyperpermeability, whereas CVEC transfected with the empty vector
(used to deliver dyn2K44A) developed a hyperpermeability response to PAF (Figure 4B),
showing that transfection of CVEC with the empty vector did not inhibit PAF-induced
hyperpermeability.
To further verify the efficacy of dyn2K44A in preventing scission of caveolae from plasma
membrane in CVEC, we collected, at the end of the experiment, the snapwell inserts containing
the CVEC monolayers used to measure transport of FITC-DX-70 and examined the location of
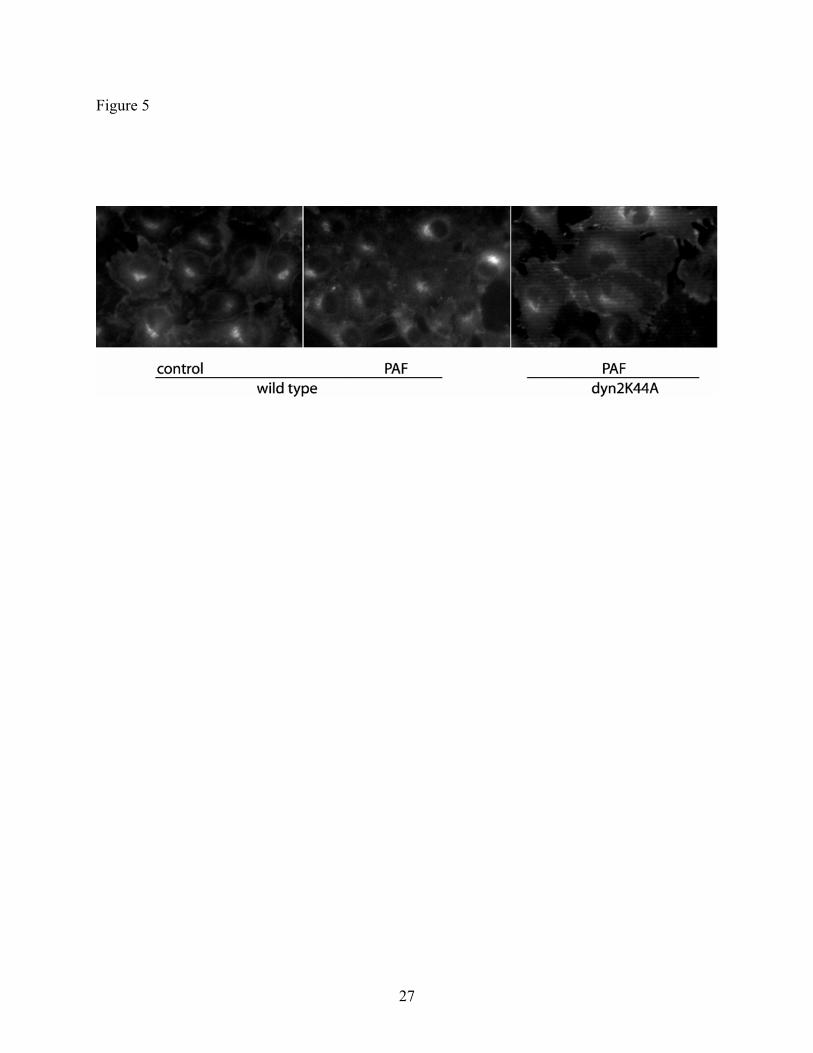
eNOS by indirect immunofluorescence microscopy. Figure 5 displays the images obtained from
the experimental snapwells. Control CVEC show eNOS located in the cell membrane and Golgi.
Non-transfected cells show that 100 nM PAF causes translocation of eNOS to subcellular
locations, as eNOS appears diffuse in the cytosol. Importantly, CVEC transfected with
dyn2K44A demonstrate that eNOS is located at the cell membrane and Golgi after application of
100 nM PAF, indicating that dyn2K44A is highly efficacious in preventing movement of eNOS
from the cell membrane (caveolae) to cytosol.

11
DISCUSSION
Our results demonstrate 1) eNOS expression is required for robust hyperpermeability response to
PAF, a pro-inflammatory agent, 2) internalization of eNOS is an important element of the signals
determining the permeability-enhancing function of eNOS-derived NO, and 3) traffic of eNOS
via caveolae is associated with the onset of hyperpermeability.
Translocation of eNOS from plasma membrane to subcellular compartments in response to
agonists has been reported to occur via enzyme depalmitoylation (29, 35) and/or in association
with internalization via caveolae (4) in endothelial cells derived from large vessels. A few
reports have advanced speculations concerning functional consequences of eNOS translocation
(15, 37). Our work is the first to suggest a close association between eNOS translocation via
caveolae and increase in endothelial monolayer permeability to macromolecules. In addition, our
data strongly indicate that eNOS translocation is required to stimulate the onset of PAF-induced
hyperpermeability.
While our results are strengthened by the fact that we obtained them in endothelial cells derived
from the microvascular segment normally involved in enhancing transport of macromolecules in
inflammation, preferential translocation of eNOS to the cytosol by agents that cause
hyperpermeability appears to be a property shared by other endothelial cells in culture. VEGF
(10) and BK (23), two well characterized hyperpermeability enhancing agents, cause eNOS
translocation to cytosol. In contrast, acetylcholine – an agent that causes vasodilation via eNOS-
derived NO but does not alter permeability - induces preferential translocation of eNOS to the

12
Golgi region (30). The consistency of eNOS translocation to defined subcellular locations in
response to agents that induce different functional end-points argues against the interpretation
that eNOS movement reflects regular protein traffic, and argues in favor of a response to specific
external stimuli or agonists.
Pharmacologic inhibition of eNOS blocks hyperpermeability induced by PAF, VEGF and
histamine in vivo and in cultured EC (2, 20, 27, 33, 36). In mice, deletion of the gene encoding
for eNOS leads to loss of microvascular hyperpermeability responses to PAF (16) and to VEGF
(11). We confirm in CVEC monolayers that eNOS expression is an absolute requirement for
development of increased endothelial permeability to macromolecules in response to PAF.
Interestingly, loss or depletion of eNOS does not influence baseline permeability in mice and in
CVEC. It seems that baseline permeability is maintained at a set-point by a number of redundant
mechanisms while the ability to respond to pro-inflammatory agents (PAF, VEGF) is exquisitely
sensitive to or dependent on mechanisms based on functional eNOS.
Based on our results, we propose eNOS internalization via caveolae as a novel mechanism in
endothelial regulation of microvascular permeability to macromolecules in response to PAF, a
well known pro-inflammatory agent. Our data support the concept that eNOS-derived NO serves
as an onset signal for hyperpermeability. This assessment is based on the close temporal
correlation among the changes induced by PAF on eNOS activation (Ser1177
phosphorylation at
0.5 – 1.0 minutes; Thr495
de-phosphorylation at 1.0 minute), NO production (peaking at about 1-
3 minutes) and increases in permeability to FITC-Dx-70 detected shortly after application of
PAF.

13
The concept that eNOS internalization to subcellular compartment plays a role in determining
stimulus-initiated function serves to complement the fund of knowledge demonstrating activation
of eNOS through phosphorylation or de-phosphorylation at several sites (7, 8, 12). While
phosphorylation is clearly important as a mechanism to activate eNOS, it has been difficult to
ascribe functional specificity to it inasmuch as agents that stimulate different functional
outcomes do phosphorylate eNOS at the same sites, as is the case for example for PAF and ACh
(30). Internalization of eNOS may serve as a mechanism to bring the enzyme in close contact
with soluble guanylyl cyclase (sGC) – its main effector – or an as yet unidentified subcellular
effector.
Our data support the concept that eNOS translocation and its association with elevation in
permeability involves internalization of eNOS via caveolae. This idea is based on the ability of
dyn2K44A to block the scission of caveolae from the plasma membrane, an event intimately
linked to inhibition of PAF-induced hyperpermeability to macromolecules. Even though we
cannot completely rule out the contributions of endocytosis via coated-pit vesicles, it is normally
accepted that caveolae cover about 85% of endothelial cell membranes (26). Thus, we interpret
our results with dyn2K44A as an inhibition of caveolar internalization. It is known, mainly
through in vitro experiments, that dynamin2 interacts with eNOS (3). For this reason, it could be
argued that dyn2K44A blocks eNOS directly. However, it has been pointed out that dyamin2 and
eNOS co-localize mainly in Golgi and that the eNOS binding domain with dynamin resides at a
location different from the K44 mutation (3, 4). Thus, it appears safe to conclude that relevant
action of dyn2K44A is to prevent the separation of caveolae from the plasma membrane. Our
data (Figure 5) supports this conclusion as it shows that eNOS stays at the plasma membrane
after PAF challenge in dyn2K44A-transfected CVEC. However, a recent article reports that

14
dyn2K44A can inhibit caveolar endocytosis and NO production, in response to gp60 (22). This
article did not address the mechanism by which dyn2K44A inhibits NO production nor did they
address the discrepancy that the K44A mutation site should not alter NO production. Thus, it is
plausible that dyn2K44A inhibits PAF-induced permeability simultaneously through anchoring
caveolae to plasma membrane and inhibiting caveolae-associated NO production.
Why is eNOS internalization via caveolae necessary? There are no evidence-based answers to
these questions yet. Given that NO is a highly diffusible gas, one would anticipate that location
of the source would not be essential. We and others have shown that the hyperpermeability
response is impaired in the presence of NOS inhibitors such as L-NAME and L-NMMA (2, 20,
27, 33, 36). Furthermore, in eNOS knockout mice, NO produced by other NOS-isozymes does
not restore the ability of striated muscle microvasculature to produce a robust hyperpermeability
in response to PAF (16). Thus, not only eNOS expression is needed for adequate microvascular
function but also its location is important. In regards to traffic via caveolae, we speculate that
caveolae may possess a necessary target recognizing molecule that allows eNOS to efficaciously
promote the appropriate protein-protein signaling interactions in the intracellular environment.
Another speculation is that internalization via caveolae may serve to protect eNOS from S-
nitrosylation, which inactivates the enzyme (8). In addition, we may speculate that this traffic
may serve to deliver the appropriate NO concentration to achieve the correct stimulation of sGC,
the main NO-receptor, a cytosolic protein. As it is established sGC and its product (cyclic GMP)
play a necessary role in the development of hyperpermeability (32). The mechanisms by which
sGC-cGMP activate hyperpermeability is unknown; however, cGMP may activate
phosphodiesterase 2 and induce degradation of cAMP (17). In turn, degradation of cAMP may

15
reduce the barrier properties of the microvascular EC, and thus increase permeability to
macromolecules (6, 19, 28).
In conclusion, we report and propose for the first time a novel mechanism in the regulation of
microvascular endothelial permeability. Our data strongly indicate that internalization of eNOS
via caveolae is required for PAF-induced hyperpermeability. Moderate increases in permeability
are helpful to allow the exchange of macromolecules needed for wound healing and tissue
remodeling, but a highly elevated vascular permeability may have deleterious effects. Inhibition
of caveolar endocytosis may be help to control excessive hyperpermeability in inflammation.
ACKNOWLEDGMENTS
This work was supported by NIH grant 5RO1 HL070634 and by Institutional grants from the
Department of Pharmacology and Physiology, the Dean’s Biomedical Research Support (New
Jersey Medical School) and the Foundation of the UMDNJ.

16
REFERENCES
1. Bohlen HG. Mechanism of increased vessel wall nitric oxide concentrations during
intestinal absorption. The American journal of physiology 275: H542-550, 1998.
2. Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, 2nd, and Durán WN. VEGF
increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric
oxide. Am J Physiol Heart Circ Physiol 284: H92-H100, 2003.
3. Cao S, Yao J, McCabe TJ, Yao Q, Katusic ZS, Sessa WC, and Shah V. Direct
interaction between endothelial nitric-oxide synthase and dynamin-2. Implications for nitric-
oxide synthase function. The Journal of biological chemistry 276: 14249-14256, 2001.
4. Chatterjee S, Cao S, Peterson TE, Simari RD, and Shah V. Inhibition of GTP-
dependent vesicle trafficking impairs internalization of plasmalemmal eNOS and cellular nitric
oxide production. J Cell Sci 116: 3645-3655, 2003.
5. Church JE, and Fulton D. Differences in eNOS activity because of subcellular
localization are dictated by phosphorylation state rather than the local calcium environment. The
Journal of biological chemistry 281: 1477-1488, 2006.
6. Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, and Mayadas TN.
Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor
for Rap GTPase. Blood 105: 1950-1955, 2005.
7. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, and Zeiher AM.
Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation.
Nature 399: 601-605, 1999.

17
8. Dudzinski DM, Igarashi J, Greif D, and Michel T. The regulation and pharmacology
of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46: 235-276, 2006.
9. Durán WN, Seyama A, Yoshimura K, Gonzalez DR, Jara PI, Figueroa XF, and
Boric MP. Stimulation of NO production and of eNOS phosphorylation in the microcirculation
in vivo. Microvasc Res 60: 104-111, 2000.
10. Erwin PA, Lin AJ, Golan DE, and Michel T. Receptor-regulated dynamic S-
nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. The Journal of
biological chemistry 280: 19888-19894, 2005.
11. Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang
PL, and Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial
growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A 98:
2604-2609, 2001.
12. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF,
Papapetropoulos A, and Sessa WC. Regulation of endothelium-derived nitric oxide production
by the protein kinase Akt. Nature 399: 597-601, 1999.
13. Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, and Sessa WC. Targeting of nitric
oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide
signaling. Proc Natl Acad Sci U S A 93: 6448-6453, 1996.
14. Goetz RM, Thatte HS, Prabhakar P, Cho MR, Michel T, and Golan DE. Estradiol
induces the calcium-dependent translocation of endothelial nitric oxide synthase. Proc Natl Acad
Sci U S A 96: 2788-2793, 1999.

18
15. Gonzalez E, Kou R, Lin AJ, Golan DE, and Michel T. Subcellular targeting and
agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. The Journal of
biological chemistry 277: 39554-39560, 2002.
16. Hatakeyama T, Pappas PJ, Hobson RW, 2nd, Boric MP, Sessa WC, and Durán WN.
Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol
574: 275-281, 2006.
17. Haynes J, Jr., Killilea DW, Peterson PD, and Thompson WJ. Erythro-9-(2-hydroxy-
3-nonyl)adenine inhibits cyclic-3',5'-guanosine monophosphate-stimulated phosphodiesterase to
reverse hypoxic pulmonary vasoconstriction in the perfused rat lung. J Pharmacol Exp Ther 276:
752-757, 1996.
18. Henley JR, Krueger EW, Oswald BJ, and McNiven MA. Dynamin-mediated
internalization of caveolae. J Cell Biol 141: 85-99, 1998.
19. Kooistra MR, Corada M, Dejana E, and Bos JL. Epac1 regulates integrity of
endothelial cell junctions through VE-cadherin. FEBS Lett 579: 4966-4972, 2005.
20. Lal BK, Varma S, Pappas PJ, Hobson RW, 2nd, and Durán WN. VEGF increases
permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide
synthase, and MAP kinase pathways. Microvasc Res 62: 252-262, 2001.
21. Lungu AO, Jin ZG, Yamawaki H, Tanimoto T, Wong C, and Berk BC. Cyclosporin
A inhibits flow-mediated activation of endothelial nitric-oxide synthase by altering cholesterol
content in caveolae. The Journal of biological chemistry 279: 48794-48800, 2004.
22. Maniatis NA, Brovkovych V, Allen SE, John TA, Shajahan AN, Tiruppathi C,
Vogel SM, Skidgel RA, Malik AB, and Minshall RD. Novel mechanism of endothelial nitric

19
oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ Res 99:
870-877, 2006.
23. Michel T, Li GK, and Busconi L. Phosphorylation and subcellular translocation of
endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 90: 6252-6256, 1993.
24. Oh P, McIntosh DP, and Schnitzer JE. Dynamin at the neck of caveolae mediates their
budding to form transport vesicles by GTP-driven fission from the plasma membrane of
endothelium. J Cell Biol 141: 101-114, 1998.
25. Parton RG, Joggerst B, and Simons K. Regulated internalization of caveolae. J Cell
Biol 127: 1199-1215, 1994.
26. Predescu SA, Predescu DN, Timblin BK, Stan RV, and Malik AB. Intersectin
regulates fission and internalization of caveolae in endothelial cells. Molecular biology of the cell
14: 4997-5010, 2003.
27. Ramírez MM, Quardt SM, Kim D, Oshiro H, Minnicozzi M, and Durán WN.
Platelet activating factor modulates microvascular permeability through nitric oxide synthesis.
Microvasc Res 50: 223-234, 1995.
28. Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, and Bos
JL. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon
stimulation of the beta 2-adrenergic receptor. J Cell Biol 160: 487-493, 2003.
29. Robinson LJ, Busconi L, and Michel T. Agonist-modulated palmitoylation of
endothelial nitric oxide synthase. The Journal of biological chemistry 270: 995-998, 1995.
30. Sánchez FA, Savalia NB, Durán RG, Lal BK, Boric MP, and Durán WN. Functional
significance of differential eNOS translocation. Am J Physiol Heart Circ Physiol 291: H1058-
1064, 2006.

20
31. Schelling ME, Meininger CJ, Hawker JR, Jr., and Granger HJ. Venular endothelial
cells from bovine heart. The American journal of physiology 254: H1211-1217, 1988.
32. Varma S, Breslin JW, Lal BK, Pappas PJ, Hobson RW, 2nd, and Durán WN.
p42/44MAPK regulates baseline permeability and cGMP-induced hyperpermeability in
endothelial cells. Microvasc Res 63: 172-178, 2002.
33. Wu HM, Huang Q, Yuan Y, and Granger HJ. VEGF induces NO-dependent
hyperpermeability in coronary venules. The American journal of physiology 271: H2735-2739,
1996.
34. Wu HM, Yuan Y, Zawieja DC, Tinsley J, and Granger HJ. Role of phospholipase C,
protein kinase C, and calcium in VEGF-induced venular hyperpermeability. The American
journal of physiology 276: H535-542, 1999.
35. Yeh DC, Duncan JA, Yamashita S, and Michel T. Depalmitoylation of endothelial
nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. The
Journal of biological chemistry 274: 33148-33154, 1999.
36. Yuan SY. Signal transduction pathways in enhanced microvascular permeability.
Microcirculation 7: 395-403, 2000.
37. Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, and Fulton D.
Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in
reconstituted endothelial cells. Arterioscler Thromb Vasc Biol 26: 1015-1021, 2006.

21
LEGENDS TO FIGURES
Fig. 1: PAF (at 100 nM) induces eNOS phosphorylation and NO production in CVEC. A)
Immunofluorescence showing eNOS expression in cell membrane and Golgi in control CVEC.
B) PAF significantly stimulated a robust increase in pericellular NO concentration. The induced
response was rapid, peaked at about 3 minutes and returned to basal levels approximately 10
minutes after PAF application (mean ± SEM, n = 6, p < 0.05). C) PAF significantly increased
phosphorylation of eNOS at Ser1177
and decreased eNOS phosphorylation at Thr495
as a function
ot time, as indicated by the ratio of phosphorylated (p-eNOS) to total eNOS (*: p<0.05, n=3).
The western blots illustrate typical examples of changes in eNOS phosphorylation at Ser1177
and
Thr495
.
Fig. 2: Depletion of eNOS inhibits PAF-induced hyperpermeability. A) 100 nM PAF
increases permeability to FITC-Dx-70 in CVEC. Data are expressed as permeability coefficients
(mean S.E.M). *: p<0.05, n = 5. B) Western blots showing depletion of eNOS as a function of
siRNA concentration and time. C) Depletion of endogenous eNOS in CVEC abrogates the
development of PAF-induced hyperpermeability. CVEC transfected with scrambled siRNA
served as a control. The increase in permeability elicited by 100 nM PAF is significant compared
to all other interventions (mean S.E.M; *: p<0.05, n = 5).
Fig. 3: PAF induces eNOS translocation in CVEC. A) Immunofluorescence images of CVEC
were obtained in control cells and after 100 nM PAF treatment. The image shows that PAF
induces the disappearance of eNOS from plasma membrane and its appearance in a diffuse

22
fashion in cytosol. The images are representative of 3 independent experiments. B) Western blots
of isolated lipid rafts in control and PAF treated cells (Fraction 1 = lightest; fraction 12 =
heaviest). Fractions were probed against eNOS and caveolin.
Fig. 4: Inhibition of caveolar internalization decreases PAF-induced hyperpermeability in CVEC.
A) Image showing expression of dyn2K44A in transfected CVEC. B) The panel shows the
impact of CVEC transfection with dyn2K44A on permeability to FITC-DX-70. CVEC
transfected with the corresponding empty vector served as control. PAF induced a robust
hyperpermeability in the control CVEC. Transfection of CVEC monolayers with dyn2K44A
significantly inhibited the PAF-induced hyperpermeability to FITC-DX-70. (*: p<0.05 compared
to control and interventions, n=5).
Fig. 5: Dyn2K44A inhibits eNOS traffic in CVEC. The figure displays eNOS
inmunofluorescence images taken from CVEC monolayers at the end of the measurement of
permeability. In the control (baseline) CVEC, eNOS is distributed in the cell membrane and in
the Golgi area. Non-transfected CVEC show eNOS translocation to cytosol (diffuse material)
after challenge with PAF (center panel). In contrast, eNOS is efficaciously retained in the
plasma membrane in the dyn2K44A transfected CVEC after administration of 100 nM PAF.
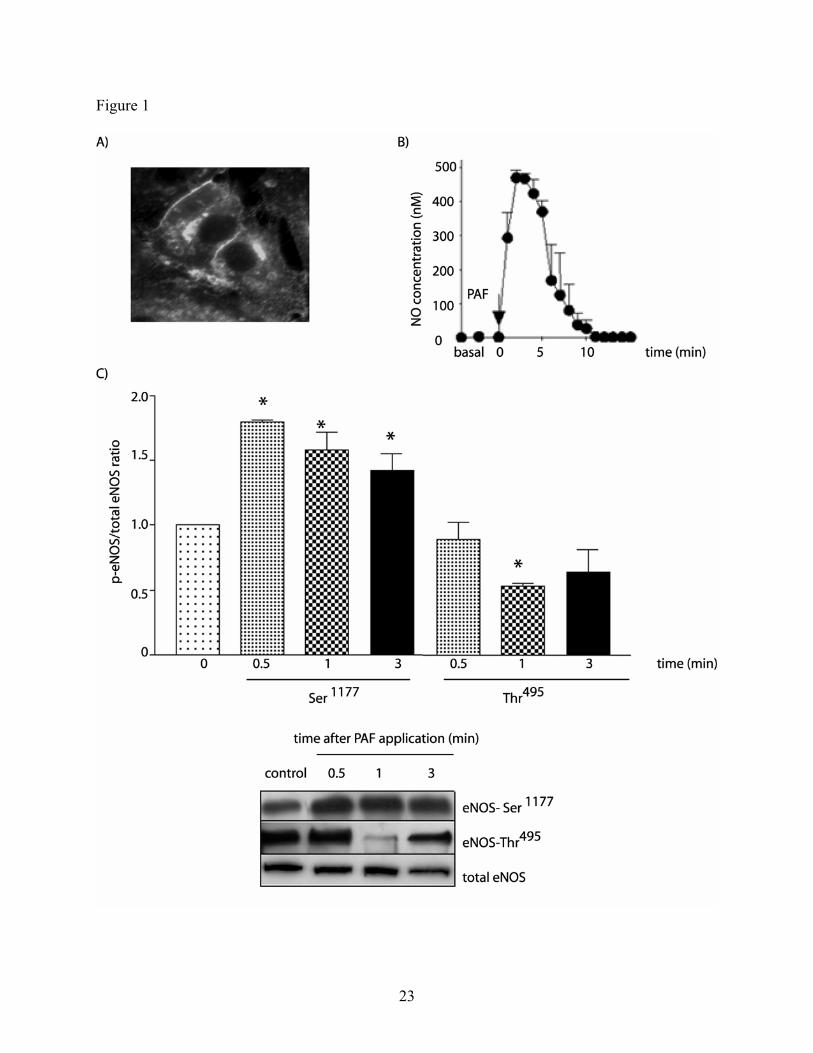
Figure 1
23
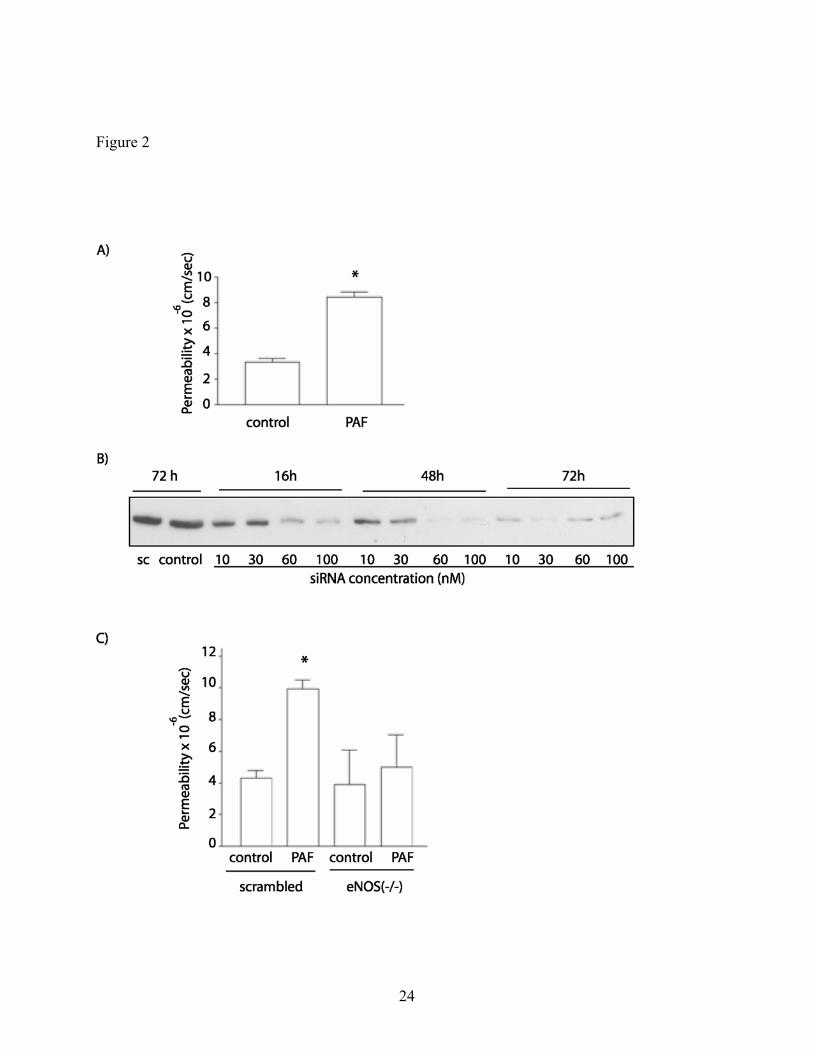
Figure 2
24
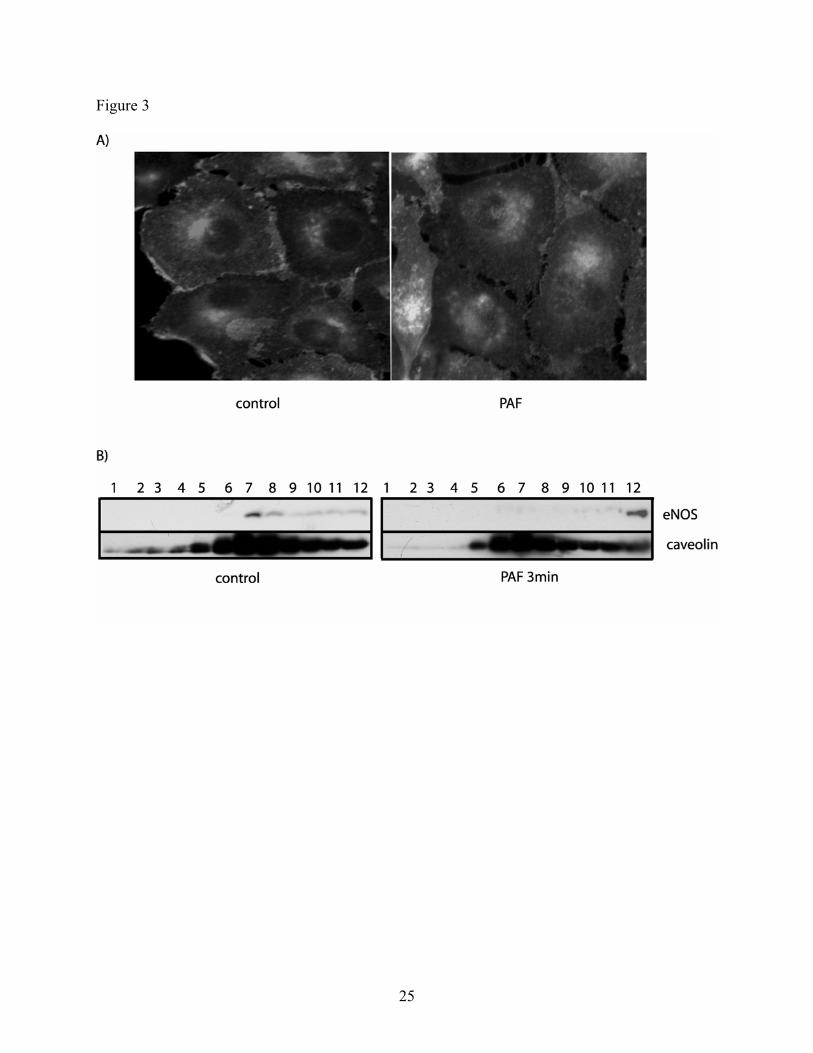
Figure 3
25
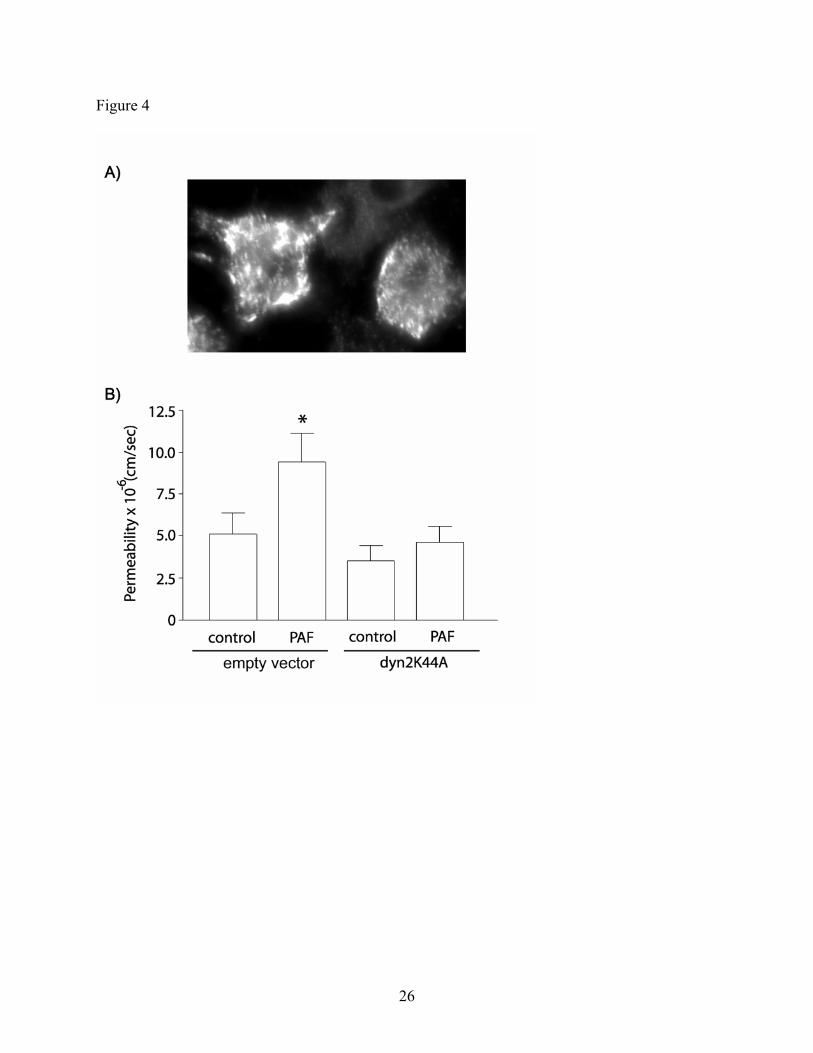
Figure 4
26
27
Figure 5