identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma
TRANSCRIPT

ORIGINAL ARTICLE
Identification of candidate tumour suppressor genes frequently methylated
in renal cell carcinoma
MR Morris1,2, C Ricketts1,2, D Gentle1,2, M Abdulrahman2, N Clarke3, M Brown3, T Kishida4,M Yao4, F Latif1,2 and ER Maher1,2,5
1Cancer Research UK Renal Molecular Oncology Group, University of Birmingham, Birmingham, UK; 2Department of Medicaland Molecular Genetics, School of Clinical and Experimental Medicine, College of Medical and Dental Sciences, University ofBirmingham, Birmingham, UK; 3Genito-Urinary Cancer Research Group, Paterson Institute for Cancer Research, University ofManchester, Manchester, UK; 4Department of Urology, Yokohama City University School of Medicine, Yokohama, Japan and5West Midlands Region Genetics Service, Birmingham Women’s Hospital, Edgbaston, Birmingham, UK
Promoter region hyermethylation and transcriptionalsilencing is a frequent cause of tumour suppressor gene(TSG) inactivation in many types of human cancers.Functional epigenetic studies, in which gene expression isinduced by treatment with demethylating agents, mayidentify novel genes with tumour-specific methylation. Weused high-density gene expression microarrays in afunctional epigenetic study of 11 renal cell carcinoma(RCC) cell lines. Twenty-eight genes were then selectedfor analysis of promoter methylation status in cell linesand primary RCC. Eight genes (BNC1, PDLIM4,RPRM, CST6, SFRP1, GREM1, COL14A1 andCOL15A1) showed frequent (430% of RCC tested)tumour-specific promoter region methylation. Hyper-methylation was associated with transcriptional silencing.Re-expression of BNC1, CST6, RPRM and SFRP1suppressed the growth of RCC cell lines and RNAinterference knock-down of BNC1, SFRP1 andCOL14A1 increased the growth of RCC cell lines.Methylation of BNC1 or COL14A1 was associated witha poorer prognosis independent of tumour size, stage orgrade. The identification of these epigenetically inacti-vated candidate RCC TSGs can provide insights into renaltumourigenesis and a basis for developing novel therapiesand biomarkers for prognosis and detection.Oncogene advance online publication, 15 February 2010;doi:10.1038/onc.2009.493
Keywords: renal cell carcinoma; methylation; epige-netics
Introduction
Kidney cancers account for approximately 2% of allcancers and more than 200 000 new cases of kidney
cancer are diagnosed in the world each year (Ferlayet al., 2007). The most common form of kidney cancer inadults is renal cell carcinoma (RCC). A majority of theRCC (B75%) are classified as clear cell (conventional)and the next most frequent subtype is papillary RCC(B15% of all cases) (Mancini et al., 2008). Theprognosis of advanced RCC is poor, although newertreatments, based on knowledge of the molecularpathology of RCC may provide a basis for developingnovel approaches to therapy. Thus, the most frequentgenetic event in the evolution of sporadic clear cell RCCis inactivation of the von Hippel-Lindau (VHL) tumoursuppressor gene (TSG) (Latif et al., 1993; Foster et al.,1994; Herman et al., 1994; Clifford et al., 1998). VHLinactivation leads to stabilisation of hypoxia-induciblefactor (HIF)-1 and HIF-2 transcription factors andactivation of a wide repertoire of hypoxic response genes(Maxwell et al., 1999). HIF-mediated RCC growth maybe antagonised by multi-tyrosine kinase inhibitors suchas sunitinib and sorafenib (Chowdhury et al., 2008).Hence, identification of mechanisms of tumourigenesisin RCC can provide a basis for therapeutic intervention.Although large-scale mutation analysis studies of RCCare in progress (see http://www.sanger.ac.uk/genetics/CGP/cosmic/), with the exception of VHL, none of thethousands of genes tested to date are mutated in 415%of tumours. Epigenetic inactivation of TSGs bymethylation promoter region of CpG dinucleotides hasalso been implicated in the pathogenesis of RCC andsome important TSGs are frequently inactivated bypromoter hypermethylation but rarely mutated (forexample, RASSF1A) (Morrissey et al., 2001; Dallolet al., 2002; Morris et al., 2003). Hence, strategies toidentify epigenetically inactivated TSGs are an impor-tant approach at elucidating the molecular pathogenesisof RCC. Previously, to analyse the utility of a functionalepigenomic approach to identify candidate novelepigenetically inactivated RCC TSGs, we performed apilot study using gene expression profiling of four RCCcell lines treated with the demethylated agent 5-aza-20-deoxycytidine (Morris et al., 2005, 2008). This led us toidentify HAI-2/SPINT2 as a novel epigenetically in-activated RCC TSG (Morris et al., 2005). We nowreport the results of a large functional epigenetic screen
Received 3 June 2009; revised 6 November 2009; accepted 22 November2009
Correspondence: Professor ER Maher, Department of Medical andMolecular Genetics, University of Birmingham, Institute of Biomedi-cal Research West, Edgbaston, Birminmgham, West Midlands B152TT, UK.E-mail: [email protected]
Oncogene (2010), 1–14& 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00
www.nature.com/onc

of RCC in which 11 RCC cell lines were analysed usinga high-density gene expression microarray platform.
Results
Identification of candidate silenced genes involved in RCCEleven RCC-derived cell lines were treated with thedemethylating agent 5-aza-20-deoxycytidine (5 mM) for5 days to re-activate epigenetically silenced/downregulatedgenes and changes in gene expression were measured usingAffymetrix (Santa Clara, CA, USA) U133 plus-2 micro-arrays that contain probes for 447000 unique transcripts.To prioritise genes for methylation analysis, we initiallyfocussed on genes, which showed a 10-fold increase inexpression after de-methylation in three or more celllines. Reverse transcriptase–PCR (RT–PCR) analysesshowed a good correlation with microarray-basedestimates of changes in gene expression (see Supplemen-tary Figure 1, Figures 1a and 2a), transcripts thatrepresented hypothetical proteins were also removed. Inall, 405 unique genes were left after this filter. Next, weexcluded genes that did not have a predicted promoter-specific CpG island (as predicted by the human genomebrowser (www.genome.ucsc.edu) and Genomatix pro-
moter inspector (www.genomatix.de), 201 genes wereleft after this filter step. Genes known to be imprintedand genes that are not expressed in renal tissue (arrayexpress (www.ebi.ac.uk/arrayexpress/) were also re-moved, leaving 155 genes. Of these, 19 genes havepreviously been assessed for promoter methylation inRCC including KRT19, COL1A1 and IGFBP1 that arefrequently methylated in RCC (Ibanez de Caceres et al.,2006; Morris et al., 2008) (Supplementary Table 2 liststhe genes identified by this filtering process in full).
To evaluate the microarray fold-change filteringselection criteria, we analysed the methylation statusof three genes (LRCH1, KLF2 and FZD8) that hadexpression fold-changes of o10 (range two- to seven-fold). All three genes were unmethylated in the cell lines(Supplementary Table 1).
In total, 24 genes from our shortlist were analysed forpromoter methylation in RCC cell lines and primarytumours. In addition, we analysed promoter methyla-tion status of RPRM which, although not meeting theselection criteria had previously been shown to beinactivated by methylation in a number of other tumourtypes (Takahashi et al., 2005; Sato et al., 2006; Bernalet al., 2008). One of the 25 genes (NOG) did not showdetectable promoter region hypermethylation. Six genes(DMRTB1, SCYA20, CLDN6, TPM2L1, LOXL and
a
Figure 1 (a) RT–PCR analysis of PDLIM4, CST6 and COL14A expression. PDLIM4 expression was restored (SKRC18, SKRC54,CAKI-1)/upregulated (RCC4, KTCL140, 786-0) in 6 of 11 cell lines after treatment with the demethylating agent, 5-aza-20-deoxycytidine. CST6 expression was restored (KTCL26, SKRC18, SKRC54, 786-0, CAKI-1)/upregulated (RCC4, KTCL140,SKRC45) in 8 of 11 cell lines. Global demethylation restored expression of COL14A in 5 of 11 cell lines (KTCL140, SKRC18,SKRC45, SKRC54, 786-0). (b–d) Bisulphite sequencing analysis of PDLIM4, CST6 and COL14A1 50 CpG island regions. Primerswere designed to amplify the predicted promoter region of the 50 CpG island of PDLIM4, CST6 and COL14A. The schematicrepresents the region sequenced; vertical lines indicate individual CpGs. Sequencing indicated methylation in RCC cell lines correlatedwith gene silencing/downregulation: black circle; complete methylation; grey circle; partial methylation; empty circle; no methylation.Bisulfite sequencing of RCC tumours and adjacent normal tissue showed frequent methylation in tumours and infrequent methylationin normal tissue (see Results for details). Analysis of normal tissue derived from non-malignant kidneys indicated no CpG islandmethylation, with the exception of PDLIM4 in which three CpGs were partially and one CpG was fully methylated in one non-malignant sample (NMN2). T, tumour; N, normal tissue adjacent to tumour; NMN, non-malignant normal tissue. Representativesequencing traces are shown in Supplementary Figure 1.
Identification of candidate TSG frequently methylated in RCCMR Morris et al
2
Oncogene
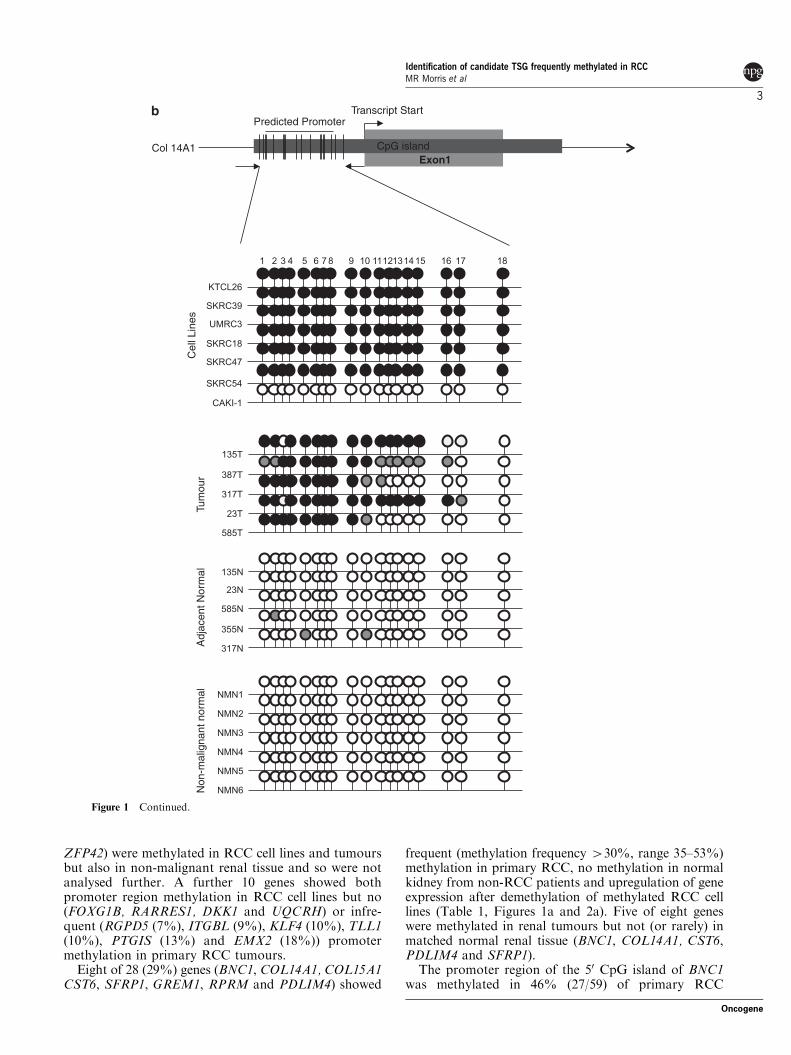
ZFP42) were methylated in RCC cell lines and tumoursbut also in non-malignant renal tissue and so were notanalysed further. A further 10 genes showed bothpromoter region methylation in RCC cell lines but no(FOXG1B, RARRES1, DKK1 and UQCRH) or infre-quent (RGPD5 (7%), ITGBL (9%), KLF4 (10%), TLL1(10%), PTGIS (13%) and EMX2 (18%)) promotermethylation in primary RCC tumours.
Eight of 28 (29%) genes (BNC1, COL14A1, COL15A1CST6, SFRP1, GREM1, RPRM and PDLIM4) showed
frequent (methylation frequency 430%, range 35–53%)methylation in primary RCC, no methylation in normalkidney from non-RCC patients and upregulation of geneexpression after demethylation of methylated RCC celllines (Table 1, Figures 1a and 2a). Five of eight geneswere methylated in renal tumours but not (or rarely) inmatched normal renal tissue (BNC1, COL14A1, CST6,PDLIM4 and SFRP1).
The promoter region of the 50 CpG island of BNC1was methylated in 46% (27/59) of primary RCC
CpG island
Transcript StartPredicted Promoter
Col 14A1Exon1
Non
-mal
igna
nt n
orm
alA
djac
ent N
orm
alTu
mou
rC
ell L
ines
Figure 1 Continued.
Identification of candidate TSG frequently methylated in RCCMR Morris et al
3
Oncogene

(Figure 2b). One of 20 (5%) adjacent normal samples(from a patient with a methylated tumour) also showedpromoter methylation. Methylation of the COL14A1promoter was detected in 44% (18/41) primary RCCtumours analysed (1/20 adjacent normal kidney samples(from a patient with a methylated tumour) and 0/6samples from patients without kidney cancer)(Figure 1b). Methylation of PDLIM4 promoter was
detected in 43% (13/30) primary RCC but not in anynormal renal tissue samples (n¼ 22) (Figure 1c). SFRP1promoter methylation was present in 34% (20/58)sporadic primary RCC but not in adjacent normaltissue samples (Figure 2b). The CST6 predicted promo-ter was methylated in 46% (28/61) primary RCC. Asmall number of normal kidney samples adjacent toRCC (11%, 4/35) showed a low level of promoter
PDLIM4
Transcript Start
Predicted promoter
CpG Island
Exon 1C
ell L
ines
Tum
ours
Adj
acen
t Nor
mal
Non
-mal
igna
nt n
orm
al
Figure 1 Continued.
Identification of candidate TSG frequently methylated in RCCMR Morris et al
4
Oncogene

methylation (all of which showed methylation in thematched tumour tissue) (Figure 1d).
Three genes, COL15A1, RPRM and GREM1 showedfrequent promoter methylation in primary RCC (53%(34/65), 44% (23/52) and 41% (11/27), respectively)(Figure 2b), but methylation was also detected relativelyfrequently in macroscopically normal renal tissue frompatients with RCC (30% (9/30), 18% (8/44), 24% (7/29), respectively (P¼ 0.034, P¼ 0.009 and P¼ 0.025,respectively compared with primary RCC)). However,there was a strong correlation between the presence of
methylation in normal renal tissue and in tumourmaterial. Thus, 8/9 normal renal tissue with COL15A1methylation in normal tissue also showed methylation inthe corresponding tumour and the corresponding figuresfor RPRM and GREM1 were 7/8 and 5/7, respectively.
Functional analysis of the tumour suppressor activityof epigenetically inactivated genesThe effect of re-expressing four of the epigeneticallysilenced RCC genes (SFRP1, RPRM, CST6 and BNC1)
2
SKRC18
KTCL26
SKRC54
RCC54
KTCL140
CAKI-1
SKRC39
581T
248T
536T
583T
151T
581N
248N
536N
Adj
acen
t Nor
mal
583T
151N
Non
-mal
igna
nt n
orm
alT
umou
rs
3 4 5 6 7 8 9 10 11 12 1314 15 161718192021 22 23 24 25 26 27 28 291
Transcript start
CpG Island
Exon 1
CST6
Predicted promoter
Cel
l Lin
es
1NMN
2NMN
3NMN
4NMN
5NMN
6NMN
Figure 1 Continued.
Identification of candidate TSG frequently methylated in RCCMR Morris et al
5
Oncogene

on in vitro growth characteristics of RCC cell lines wasassessed using colony formation and anchorage-inde-pendent growth assays.
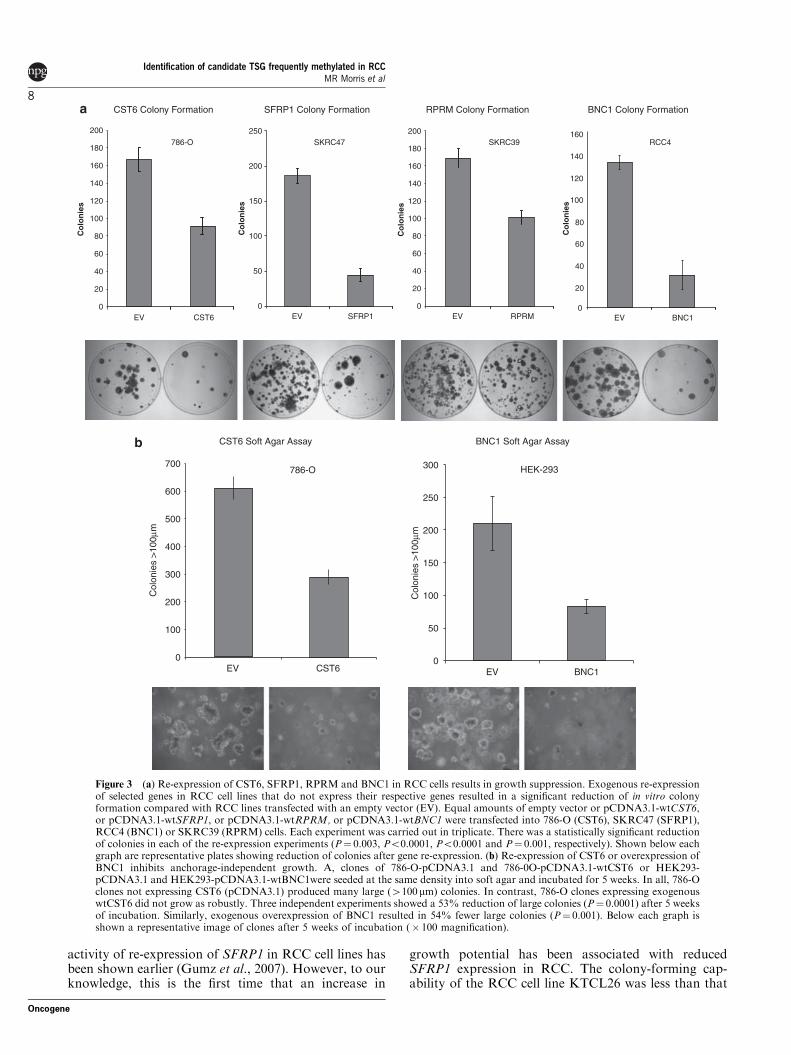
Colony formation. The tumour suppressor activity ofSFRP1, RPRM, CST6 and BNC1 was assessed byin vitro colony formation assays. After transfection ofwild-type BNC1, CST6, RPRM and SFRP1 expressionplasmids into non-expressing RCC cell lines (RCC4,786-0, SKRC39 and SKRC47, respectively) there was asignificantly reduced number of G418 resistant coloniescompared with cell lines transfected with an emptyvector control for three independent experiments(Figure 3a).
The number of BNC1 expressing RCC4 clones wasreduced by 77% compared with those transfected withan empty plasmid (s.d.¼ 10%, t¼ 12.43, Po0.0001).Re-expression of CST6 in 786-0 cells reduced theircolony-forming ability by 47% (s.d.¼ 6%, t¼ 6.629,P¼ 0.003). RPRM expressing SKRC39 clone numberswere reduced by 40% (s.d.¼ 5%, t¼ 8.328, P¼ 0.001).The number of SFRP1 expressing SKRC47 clones wasreduced by 76% compared with SKRC47 clonescontaining the empty vector (s.d.¼ 5%, t¼ 16.644,Po0.0001) (Figure 3a).
Anchorage-independent growth. The effect of re-expres-sion of, RPRM and CST6 on anchorage-independentgrowth in a soft agar colony formation assay wasassessed in stably transfected RCC cell line clones.
Colonies were counted after initial seeding and incuba-tion in soft agar for 5 weeks. Each experiment wascarried out in triplicate with three independent clones.Cells transfected with empty vector showed robustcolony growth, whereas, colony growth was greatlyreduced when CST6 was re-expressed in 786-0 cells,both the number and size of colonies was statisticallysignificantly reduced The number of colonies X100 mmwas reduced by 53% (s.d.¼ 4%, t¼ 16.15, Po0.0001) inclones expressing CST6 when compared with the controlclones (Figure 3b).
Re-expression of RPRM in SKRC39 cells did notsignificantly alter the anchorage-independent growthcapabilities of SKRC39 cells. Both clones re-expressingRPRM and those with RPRM silenced grew robustlywhen suspended in soft agar.
As the BNC1 silenced RCC4 cell line did not growwell in an anchorage-independent manner, to analysethe effect of alterations of BNC1 expression on cellgrowth, we produced BNC1 overexpressing clones ofHEK293s. Although HEK293 cells transfected with anempty vector grew robustly, BNC1 overexpressingclones produced statistically significantly fewer coloniesX100 mm (mean (±s.d.) reduction 54% (±5%), t¼ 5.92P¼ 0.001) (Figure 3b).
To analyse the possible tumourigenic advantage ofloss of expression of BNC1, SFRP1 and COL14A, weused RNA interference (RNAi) methodology to knock-down BNC1 and SFRP1 expression in HEK293s andCOL14A expression in the KTCL26 RCC cell line(insufficient knock-down of COL14A expression was
Table 1 A summary of genes identified in this study and their frequency of promoter methylation in RCC-derived cell lines, sporadic tumours,kidney tissue resected from an area adjacent to the tumour and kidney tissue from patients with no kidney cancer
Genesymbol
Accessionno.
Gene name Loci Cell linemethylationfrequency
Tumourmethylationfrequency
Adjacent kidneymethylationfrequency
Non-malignantkidney methylation
frequency
COL15A1 NM_001855 Collagen, type XV, alpha 1 9q22 7/9 35/65 (53%) 9/30 (30%) 0/6GREM1 NM_204978 Gremlin-1 15q13 7/11 11/27 (41%) 7/29 (24%) 0/6COL14A1 NM_021110 Collagen, type XIV, alpha 1 8q24 7/11 18/41 (44%) 1/20 (5%) 0/6PDLIM4 NM_003687 PDZ and LIM domain 4 5q31 5/10 13/30 (43%) 0/22 (0%) 1/6RPRM NM_019845 Reprimo, TP53-dependant G2 arrest
mediator2q23 4/9 23/52 (44%) 8/44 (18%) 0/6
SFRP1 NM_003012 Secreted frizzled-related protein 1 8p11 5/10 20/58 (34%) 0/20 (0%) 0/6CST6 NM_001323 Cystatin E/M 11q13 8/11 28/61 (46%) 4/35 (11%) 0/6BNC1 NM_001717 Basonuclin 1 15q25 5/11 27/59 (46%) 1/20 (5%) 0/6
Abbreviation: RCC, renal cell carcinoma.
Figure 2 (a) RT–PCR analysis of BNC1, SFRP1 and COL15A1, GREM1, and RPRM expression. BNC1 expression was restored(RCC4, KTCL140, SKRC18)/upregulated (SKRC45, 786-0) in 5 of 11 cell lines after treatment with the demethylating agent, 5-aza-20-deoxycytidine. SFRP1 expression was restored (RCC4, SKRC47)/upregulated (KTCL140, SKRC18, SKRC45) in 5 of 11 cell lines.COL15A1 expression was restored (RCC4)/upregulated (KTCL140, SKRC18, SKRC47, SKRC54) in 5 of 11 cell lines. GREM1expression was restored (RCC4, SKRC140, SKRC54, CAKI-1)/upregulated (SKRC39) in 5 of 11 cell lines after treatment with thedemethylating agent, 5-aza-20-deoxycytidine. Global demethylation restored expression of RPRM in 3 of 11 cell lines (KTCL26,SKRC18, SKRC39). (b) Methylation-specific PCR analysis of BNC1, SFRP1 and COL15A, GREM1, and RPRM1. MSP analysisshowed frequent (34–53%) promoter methylation in tumours, infrequent or absent methylation in adjacent tissue and no methylationin samples derived from non-malignant normal tissue (see Results for details). RCC tumour cell lines were used for each MSP assay aspositive controls for methylated and unmethylated DNA. Representative samples are shown. M, product derived from methylation-specific primers; U, products derived from unmethylated-specific primers; T, tumour sample; N, non-malignant sample; *, methylatedtumours. MSP and USP product are of a similar size. Smaller bands are primer dimmers.
Identification of candidate TSG frequently methylated in RCCMR Morris et al
6
Oncogene

obtained in HEK293 cells). Twenty-four hours afterRNAi transfection cells were seeded into 3% agar,colonies 4100 mm were counted 21 days later. In all,55% more colonies 4100 mm were produced byHEK293 cells with reduced BNC1 (s.d.¼ 11%,
t¼�6.9, Po0.0001) expression compared with cellstransfected with a control RNAi oligo (Figure 4a).Reduced expression of SFRP1 resulted in 74% morecolonies 4100 mm (s.d.¼ 6%, t¼�14.3, Po0.0001)than control cells (Figure 4a). The growth suppressing
a
b
Identification of candidate TSG frequently methylated in RCCMR Morris et al
7
Oncogene
activity of re-expression of SFRP1 in RCC cell lines hasbeen shown earlier (Gumz et al., 2007). However, to ourknowledge, this is the first time that an increase in
growth potential has been associated with reducedSFRP1 expression in RCC. The colony-forming cap-ability of the RCC cell line KTCL26 was less than that
CST6 Colony Formation
0
20
40
60
80
100
120
140
160
180
200
CST6EV
SFRP1 Colony Formation
0
50
100
150
200
250
SFRP1EV
RPRM Colony Formation
Co
lon
ies
BNC1 Colony Formation
0
20
40
60
80
100
120
140
160
EV BNC1
Co
lon
ies
0
20
40
60
80
100
120
140
160
180
200
EV RPRM
Co
lon
ies
Co
lon
ies
0
100
200
300
400
500
600
700
EV CST60
50
100
150
200
250
EV BNC1
300
Col
onie
s >
100µ
m
Col
onie
s >
100µ
m
CST6 Soft Agar Assay BNC1 Soft Agar Assay
786-O SKRC47 SKRC39 RCC4
786-O HEK-293
Figure 3 (a) Re-expression of CST6, SFRP1, RPRM and BNC1 in RCC cells results in growth suppression. Exogenous re-expressionof selected genes in RCC cell lines that do not express their respective genes resulted in a significant reduction of in vitro colonyformation compared with RCC lines transfected with an empty vector (EV). Equal amounts of empty vector or pCDNA3.1-wtCST6,or pCDNA3.1-wtSFRP1, or pCDNA3.1-wtRPRM, or pCDNA3.1-wtBNC1 were transfected into 786-O (CST6), SKRC47 (SFRP1),RCC4 (BNC1) or SKRC39 (RPRM) cells. Each experiment was carried out in triplicate. There was a statistically significant reductionof colonies in each of the re-expression experiments (P¼ 0.003, Po0.0001, Po0.0001 and P¼ 0.001, respectively). Shown below eachgraph are representative plates showing reduction of colonies after gene re-expression. (b) Re-expression of CST6 or overexpression ofBNC1 inhibits anchorage-independent growth. A, clones of 786-O-pCDNA3.1 and 786-0O-pCDNA3.1-wtCST6 or HEK293-pCDNA3.1 and HEK293-pCDNA3.1-wtBNC1were seeded at the same density into soft agar and incubated for 5 weeks. In all, 786-Oclones not expressing CST6 (pCDNA3.1) produced many large (4100mm) colonies. In contrast, 786-O clones expressing exogenouswtCST6 did not grow as robustly. Three independent experiments showed a 53% reduction of large colonies (P¼ 0.0001) after 5 weeksof incubation. Similarly, exogenous overexpression of BNC1 resulted in 54% fewer large colonies (P¼ 0.001). Below each graph isshown a representative image of clones after 5 weeks of incubation (� 100 magnification).
Identification of candidate TSG frequently methylated in RCCMR Morris et al
8
Oncogene
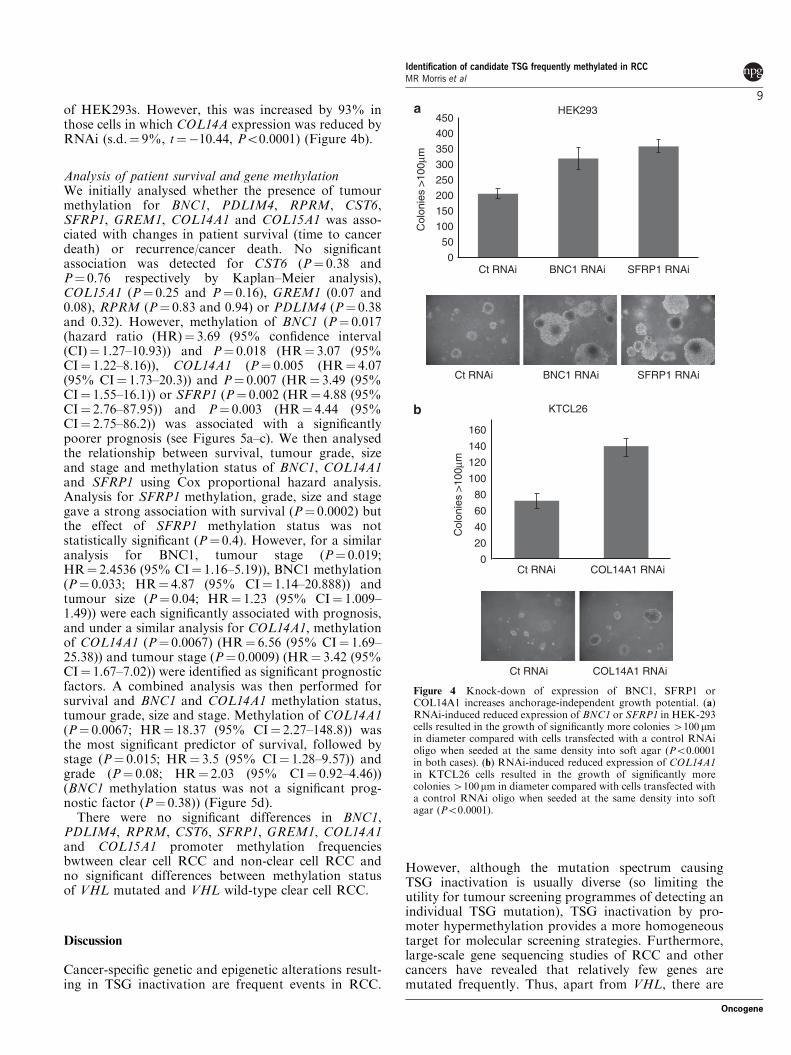
of HEK293s. However, this was increased by 93% inthose cells in which COL14A expression was reduced byRNAi (s.d.¼ 9%, t¼�10.44, Po0.0001) (Figure 4b).
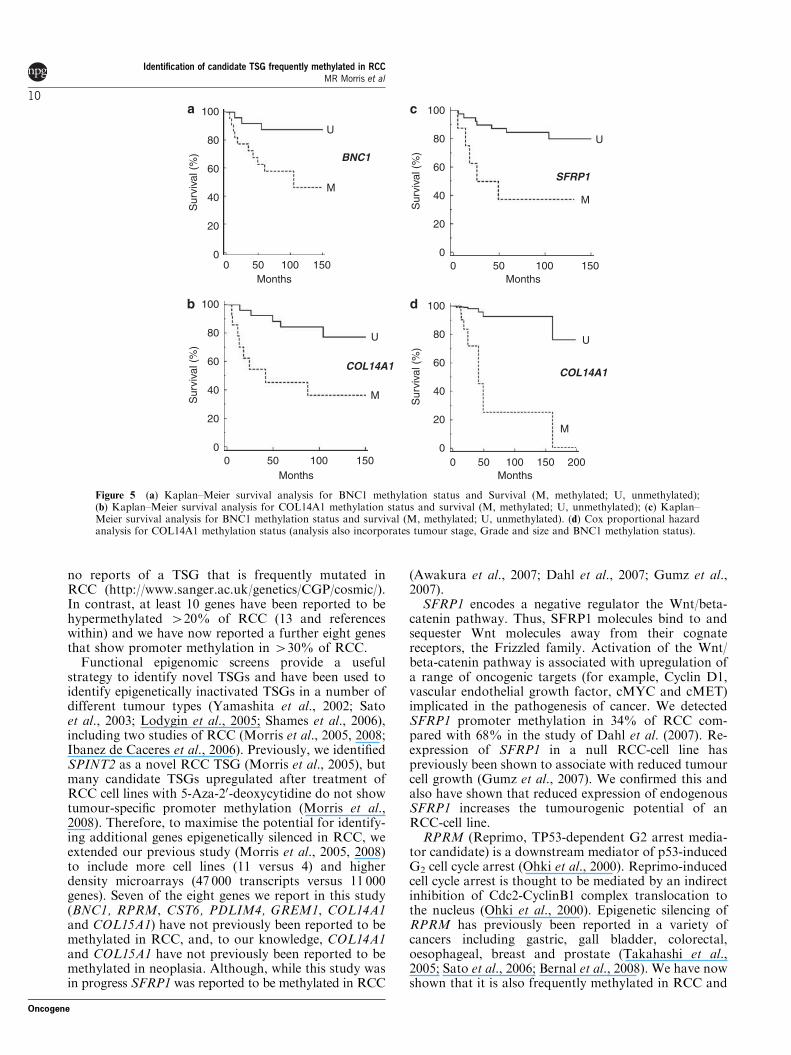
Analysis of patient survival and gene methylationWe initially analysed whether the presence of tumourmethylation for BNC1, PDLIM4, RPRM, CST6,SFRP1, GREM1, COL14A1 and COL15A1 was asso-ciated with changes in patient survival (time to cancerdeath) or recurrence/cancer death. No significantassociation was detected for CST6 (P¼ 0.38 andP¼ 0.76 respectively by Kaplan–Meier analysis),COL15A1 (P¼ 0.25 and P¼ 0.16), GREM1 (0.07 and0.08), RPRM (P¼ 0.83 and 0.94) or PDLIM4 (P¼ 0.38and 0.32). However, methylation of BNC1 (P¼ 0.017(hazard ratio (HR)¼ 3.69 (95% confidence interval(CI)¼ 1.27–10.93)) and P¼ 0.018 (HR¼ 3.07 (95%CI¼ 1.22–8.16)), COL14A1 (P¼ 0.005 (HR¼ 4.07(95% CI¼ 1.73–20.3)) and P¼ 0.007 (HR¼ 3.49 (95%CI¼ 1.55–16.1)) or SFRP1 (P¼ 0.002 (HR¼ 4.88 (95%CI¼ 2.76–87.95)) and P¼ 0.003 (HR¼ 4.44 (95%CI¼ 2.75–86.2)) was associated with a significantlypoorer prognosis (see Figures 5a–c). We then analysedthe relationship between survival, tumour grade, sizeand stage and methylation status of BNC1, COL14A1and SFRP1 using Cox proportional hazard analysis.Analysis for SFRP1 methylation, grade, size and stagegave a strong association with survival (P¼ 0.0002) butthe effect of SFRP1 methylation status was notstatistically significant (P¼ 0.4). However, for a similaranalysis for BNC1, tumour stage (P¼ 0.019;HR¼ 2.4536 (95% CI¼ 1.16–5.19)), BNC1 methylation(P¼ 0.033; HR¼ 4.87 (95% CI¼ 1.14–20.888)) andtumour size (P¼ 0.04; HR¼ 1.23 (95% CI¼ 1.009–1.49)) were each significantly associated with prognosis,and under a similar analysis for COL14A1, methylationof COL14A1 (P¼ 0.0067) (HR¼ 6.56 (95% CI¼ 1.69–25.38)) and tumour stage (P¼ 0.0009) (HR¼ 3.42 (95%CI¼ 1.67–7.02)) were identified as significant prognosticfactors. A combined analysis was then performed forsurvival and BNC1 and COL14A1 methylation status,tumour grade, size and stage. Methylation of COL14A1(P¼ 0.0067; HR¼ 18.37 (95% CI¼ 2.27–148.8)) wasthe most significant predictor of survival, followed bystage (P¼ 0.015; HR¼ 3.5 (95% CI¼ 1.28–9.57)) andgrade (P¼ 0.08; HR¼ 2.03 (95% CI¼ 0.92–4.46))(BNC1 methylation status was not a significant prog-nostic factor (P¼ 0.38)) (Figure 5d).
There were no significant differences in BNC1,PDLIM4, RPRM, CST6, SFRP1, GREM1, COL14A1and COL15A1 promoter methylation frequenciesbwtween clear cell RCC and non-clear cell RCC andno significant differences between methylation statusof VHL mutated and VHL wild-type clear cell RCC.
Discussion
Cancer-specific genetic and epigenetic alterations result-ing in TSG inactivation are frequent events in RCC.
However, although the mutation spectrum causingTSG inactivation is usually diverse (so limiting theutility for tumour screening programmes of detecting anindividual TSG mutation), TSG inactivation by pro-moter hypermethylation provides a more homogeneoustarget for molecular screening strategies. Furthermore,large-scale gene sequencing studies of RCC and othercancers have revealed that relatively few genes aremutated frequently. Thus, apart from VHL, there are
0
50
100
150
200
250
300
350
400
450
Ct RNAi BNC1 RNAi SFRP1 RNAi
0
20
40
60
80
100
120
140
160
Ct RNAi COL14A1 RNAi
HEK293
KTCL26C
olon
ies
>10
0µm
Col
onie
s >
100µ
m
Ct RNAi
Ct RNAi
BNC1 RNAi
COL14A1 RNAi
SFRP1 RNAi
Figure 4 Knock-down of expression of BNC1, SFRP1 orCOL14A1 increases anchorage-independent growth potential. (a)RNAi-induced reduced expression of BNC1 or SFRP1 in HEK-293cells resulted in the growth of significantly more colonies 4100mmin diameter compared with cells transfected with a control RNAioligo when seeded at the same density into soft agar (Po0.0001in both cases). (b) RNAi-induced reduced expression of COL14A1in KTCL26 cells resulted in the growth of significantly morecolonies4100mm in diameter compared with cells transfected witha control RNAi oligo when seeded at the same density into softagar (Po0.0001).
Identification of candidate TSG frequently methylated in RCCMR Morris et al
9
Oncogene
no reports of a TSG that is frequently mutated inRCC (http://www.sanger.ac.uk/genetics/CGP/cosmic/).In contrast, at least 10 genes have been reported to behypermethylated 420% of RCC (13 and referenceswithin) and we have now reported a further eight genesthat show promoter methylation in 430% of RCC.
Functional epigenomic screens provide a usefulstrategy to identify novel TSGs and have been used toidentify epigenetically inactivated TSGs in a number ofdifferent tumour types (Yamashita et al., 2002; Satoet al., 2003; Lodygin et al., 2005; Shames et al., 2006),including two studies of RCC (Morris et al., 2005, 2008;Ibanez de Caceres et al., 2006). Previously, we identifiedSPINT2 as a novel RCC TSG (Morris et al., 2005), butmany candidate TSGs upregulated after treatment ofRCC cell lines with 5-Aza-20-deoxycytidine do not showtumour-specific promoter methylation (Morris et al.,2008). Therefore, to maximise the potential for identify-ing additional genes epigenetically silenced in RCC, weextended our previous study (Morris et al., 2005, 2008)to include more cell lines (11 versus 4) and higherdensity microarrays (47 000 transcripts versus 11 000genes). Seven of the eight genes we report in this study(BNC1, RPRM, CST6, PDLIM4, GREM1, COL14A1and COL15A1) have not previously been reported to bemethylated in RCC, and, to our knowledge, COL14A1and COL15A1 have not previously been reported to bemethylated in neoplasia. Although, while this study wasin progress SFRP1 was reported to be methylated in RCC
(Awakura et al., 2007; Dahl et al., 2007; Gumz et al.,2007).
SFRP1 encodes a negative regulator the Wnt/beta-catenin pathway. Thus, SFRP1 molecules bind to andsequester Wnt molecules away from their cognatereceptors, the Frizzled family. Activation of the Wnt/beta-catenin pathway is associated with upregulation ofa range of oncogenic targets (for example, Cyclin D1,vascular endothelial growth factor, cMYC and cMET)implicated in the pathogenesis of cancer. We detectedSFRP1 promoter methylation in 34% of RCC com-pared with 68% in the study of Dahl et al. (2007). Re-expression of SFRP1 in a null RCC-cell line haspreviously been shown to associate with reduced tumourcell growth (Gumz et al., 2007). We confirmed this andalso have shown that reduced expression of endogenousSFRP1 increases the tumourogenic potential of anRCC-cell line.
RPRM (Reprimo, TP53-dependent G2 arrest media-tor candidate) is a downstream mediator of p53-inducedG2 cell cycle arrest (Ohki et al., 2000). Reprimo-inducedcell cycle arrest is thought to be mediated by an indirectinhibition of Cdc2-CyclinB1 complex translocation tothe nucleus (Ohki et al., 2000). Epigenetic silencing ofRPRM has previously been reported in a variety ofcancers including gastric, gall bladder, colorectal,oesophageal, breast and prostate (Takahashi et al.,2005; Sato et al., 2006; Bernal et al., 2008). We have nowshown that it is also frequently methylated in RCC and
U
M
U
M
BNC1
COL14A1
U
M
U
M
SFRP1
COL14A1
Sur
viva
l (%
)
100
80
60
40
20
0
Sur
viva
l (%
)
100
80
60
40
20
0
Sur
viva
l (%
)
100
80
60
40
20
0
Sur
viva
l (%
)
100
80
60
40
20
00 50 100 150
Months
0 50 100 150Months
0 50 100 150Months
200
0 50 100 150Months
Figure 5 (a) Kaplan–Meier survival analysis for BNC1 methylation status and Survival (M, methylated; U, unmethylated);(b) Kaplan–Meier survival analysis for COL14A1 methylation status and survival (M, methylated; U, unmethylated); (c) Kaplan–Meier survival analysis for BNC1 methylation status and survival (M, methylated; U, unmethylated). (d) Cox proportional hazardanalysis for COL14A1 methylation status (analysis also incorporates tumour stage, Grade and size and BNC1 methylation status).
Identification of candidate TSG frequently methylated in RCCMR Morris et al
10
Oncogene

that re-expression RPRM reduces the tumour-formingproperties of RCC-derived cell lines.
CST6 is a type 2 secreted cystatin. It inhibits cathepsinB, L, H and V, members of a family of lysosomalproteases. Cathepsins are involved in multiple biologicalprocesses such as apoptosis, intracellular protein cata-bolism and pericellular matrix re-modelling (Brommeand Kaleta, 2002). It has been suggested that animbalance of cathepsins and their respective inhibitorsmay promote tumour cell invasion (Bellail et al., 2004)and so loss of CST6 expression by epigenetic silencingmight be predicted to contribute to the malignantphenotype. Consistent with this we found that re-expression of CST6 reduced the ability of RCC cells toform colonies in an anchorage-independent manner.CST6 has also been shown to be epigenetically silencedand reduced in vitro tumourigenicity in breast cancer(Shridhar et al., 2004; Ai et al., 2006) and gliomas (Qiuet al., 2008).
PDLIM4 encodes a LIM domain candidate tumourgene mapping to 5q31.1. Although methylation ofPDLIM4 has not been reported previously in RCCtumours, Boumber et al. (2007) reported frequentPDLIM4 promoter methylation in acute myelogenousleukaemia and colon cancer. Gremlin (GREM1) is aninhibitor of transforming growth factor-b signalling thathas previously been reported to be frequently methy-lated in lung, breast and bladder cancers (Suzuki et al.,2005). We found frequent methylation of GREM1suggesting that this may contribute to the perturbedtransforming growth factor-b signalling that is detectedin many human cancers, including RCC. GREM1methylation was detected in some samples of adjacentnormal renal tissue from RCC patients with GREM1tumour methylation. Similar findings were obtained forRPRM and COL15A1, these results would be consistentwith the hypothesis that GREM1, RPRM andCOL15A1 methylation might occur as part of apremalignant field effect, as has been described inbronchial epithelium (Wistuba, 2007).
Basonuclin (BNC1) is a zinc-finger transcriptionfactor that interacts with the promoters of both RNApolymerases I and II. In silico analysis suggests thatbasonuclin target genes may be implicated in chromatinstructure, transcription/DNA-binding, adhesion/cell–cell junction, signal transduction and intracellulartransport (Wang et al., 2006). Shames et al. (2006)reported that BNC1 was methylated in breast, lung,prostate and colon cancers, suggesting that BNC1methylation might be of relevance to the diagnosis andmanagement of a range of human cancers.
Ibanez de Caceres et al. (2006) have reportedepigenetic silencing of COL1A1 in RCC. In this study,we identified frequent methylation of COL15A1 andCOL14A1 in RCC. COL15A1 encodes a nonfibrillaryproteoglycan, which is found in many tissue typesforming an integral unit of the collagenous networksubjacent to the basement membrane (Amenta et al.,2005). The COOH-terminal end of COL15A1, whichhas homology to endostatin has been shown to haveanti-angiogenic properties and is capable of inhibiting
tumour growth in a xenograph renal model (Ramchan-dran et al., 1999). Moreover, recently COL15A1 wasidentified in a fibroblast-tumour cell hybrid screen as apotent suppressor of tumour growth, and re-expressionof COL15A1 in a HeLa derivative cell line completelysuppressed tumour formation in nude mice (Harriset al., 2007). We have identified frequent COL15A1methylation in RCC (53%). Collagen XIVA1(COL14A1) is a large extracellular matrix glycoproteinthat associates with mature collagen fibrils (Schuppanet al., 1990). Although methylation of COL14A1 has notpreviously been reported in human cancer, Schuppanet al. (1990) noted frequent absence of COL14A1 in thevicinity of invading tumours such as Kaposi sarcomaand oral squamous cell carcinoma. In addition, we notedata from the Sanger cancer genome re-sequencingproject (http://www.sanger.ac.uk/genetics/CGP/Studies/)that identifies COL14A1 mutations in B1% (n¼ 4) ofRCC sequenced (n¼ 412). COL14A1 interacts withDecorin (Ehnis et al., 1997), which regulates fibrillogen-esis and downregulates activity of a number of receptors(EGFR, IGF-IR, LRP) implicated in cell growth andsurvival (Santra et al., 2002; Brandan et al., 2006;Schaefer et al., 2007).
Loss of expression of epigenetically silenced TSGs canaffect a wide variety of cellular processes, includingthose which directly affect cell growth and proliferation.We analysed whether re-expressing SFRP1, RPRM,CST6 and BNC1 in RCC cell lines would influencein vitro analyses of tumourigenesis. BNC1 has notpreviously been shown to affect tumour cell growth, butwe found that re-expression of BNC1 suppressedtumour cell growth. Similar results were found forRPRM, CST6 and SFRP1. Although RPRM and CST6have not previously been studied in RCC, these findingsare consistent with those reported in cervical and breastcancer cell lines (Ohki et al., 2000; Shridhar et al., 2004).We have also shown that reducing the expression ofBNC1, SFRP1 and COL14A1 in an embryonic kidneycell line or an RCC-cell line increased tumourogenicpotential as assessed by soft agar colony formationassays.
The methylated genes identified in this and previousstudies of RCC (Morris et al., 2003, 2005, 2008; Ibanezde Caceres et al., 2006), represent a variety of functions.However, we note that many of these genes can belinked to pathways associated with the VHL TSG thathas a gatekeeper function for RCC (analogous to that ofAPC in colorectal cancer). The VHL gene product has akey role in regulating the HIF-1 and HIF-2 and, in turn,these influence a wide repertoire of target genes.Although dysregulation of hypoxia-inducible genes(particularly those regulated by HIF-2) is stronglylinked to risk of RCC with VHL inactivation, HIF-dysregulation appears to be necessary but insufficientfor VHL-related RCC tumourigenesis (Clifford et al.,2001; Raval et al., 2005). A number of HIF-independentfunctions have been ascribed to pVHL. In the context ofCOL14A1 and COL15A1 methylation, it is interestingto note that analysis of Caenorhabditis elegans wormsmutant for VHL-1 showed a clear HIF-independent link
Identification of candidate TSG frequently methylated in RCCMR Morris et al
11
Oncogene

between pVHL and extracellular matrix function(Bishop et al., 2004). Recently, pVHL has been reportedto regulate the p53 pathway (relevant to RPRM) andthe Wnt/beta-catenin pathway (downstream of SFRP1)by targeting beta-catenin for proteasomal degradation(Chitalia et al., 2008).
The identification of frequently methylated RCCTSGs can highlight critical pathways that might betargeted for novel therapeutic interventions. In addition,the detection of methylated DNA in urine or serummight be used as biomarkers for the diagnosis, stagingor risk stratification of RCC (Battagli et al., 2003;Hoque et al., 2004; Urakami et al., 2006). VHLinactivation occurs early in tumourigenesis but, to date,has not been shown to be a significant prognosticindicator (Smits et al., 2008). Thus, given the infre-quency of mutations in other TSGs in RCC, detection ofTSG methylation would appear to presents a promisingstrategy for prognostic biomarkers in RCC. However,only a few potential RCC methylation prognosticbiomarkers have been reported (Gonzalgo et al., 2004;Breault et al., 2005; Christoph et al., 2006; Yamadaet al., 2006; Costa et al., 2007; McRonald et al., 2009).We found that methylation of BNC1, COL14A1 andSFRP1 was each associated with significantly poorerpatient survival. However, for SFRP1 this associationwas not independent of conventional prognostic factors(stage, size and grade). In multivariate analysis withconventional prognostic factors, methylation ofCOL14A1 and BNC1 were each significant predictorsof poorer survival. Combining the COL14A1 and BNC1methylation data identified COL14A1 methylation as anindependent prognostic factor with higher statisticalsignificance than stage, size and grade.
Materials and methods
Patients and samplesDNA from up to 61 primary RCCs (B80% clear cell and 20%non-clear cell) and matched adjacent macroscopically normalrenal tissue and normal renal tissue (not required for surgicalpathology) from six patients undergoing non-renal cancersurgery (mean age 57 years, range 23–79 years) were analysed.Local research ethics committees approved the collection ofsamples and informed consent was obtained from each patient.This study was conducted according to the principles expressedin the Declaration of Helsinki.
Cell lines, 5-aza-20-deoxycytidine treatment and microarrayanalysisRCC cell lines KTCL 26, RCC4, UMRC2, UMRC3,SKRC18, SKRC39, SKRC45, SKRC47, SKRC54, 786-0 andCaki-1 were routinely maintained in Dulbecco’s modifiedEagle’s medium (DMEM) (Invitrogen, San Diego, CA, USA)supplemented with 10% fetal calf serum (FCS) at 37 1C, 5%CO2. The demethylating agent 5-aza-20-deoxycytidine (Sigma,Poole, UK) was freshly prepared in ddH2O and the filtersterilised. Cell lines were plated in 75-cm2 flasks in DMEMsupplemented with 10% FCS at differing densities, dependingon their replication factor, to ensure that both control and5-aza-20-deoxycytidine treated lines reached approximately 75%
confluency at the point of RNA extraction. Twenty-four hourslater, cells were treated with 5mM 5-aza-20-deoxycytidine. Themedium was changed 24 h after treatment and then changedagain after 72 h. RNA was prepared 5 days after treatmentusing RNABee (AMS Biotechnology, Abingdon, UK). TotalRNA from all 11 cell lines ±5-aza-20-deoxycytidine was isolatedusing RNA-Bee reagent following manufacturer’s instructions(AMS Biotechnology) followed by purification using RNeasyMini-columns (Qiagen, Crawley, UK). Complementary RNAprobes were prepared using the Affymetrix protocol andhybridised to HG-U133 plus2 GeneChip oligonucleotidearrays (Affymetrix). Array hybridisation and data productionwas carried out by the CRUK Paterson Institute MicroarrayService (http://bioinformatics.picr.man.ac.uk/mbcf/).
RT–PCR conditionsPCR cycling conditions consisted of 5min at 95 1C followed by30 cycles of 45 s of denaturation at 95 1C, 45 s of annealing at55–60 1C and 45 s of extension at 72 1C. Semi-quantitativeanalysis of expression was carried out using LabWorkssoftware (Ultraviolet Products, Upland, CA, USA). (RT–PCRprimers and conditions on request).
Bisulfite modification and methylation analysisBisulfite DNA sequencing was performed as described previously(Morris et al., 2005, 2008). Briefly, 0.5–1.0mg of genomic DNAwas denatured in 0.3M NaOH for 15min at 37 1C, and thenunmethylated cytosine residues were sulfonated by incubationin 3.12M sodium bisulfite (pH 5.0; Sigma)/5mM hydroquinone(Sigma) in a thermocycler (Hybaid, Franklin, MA, USA) for20 cycles of 30 s at 99 1C and 15min at 50 1C. The sulfonatedDNA was recovered using the Wizard DNA cleanup system(Promega, Southampton, UK) in accordance with the manufac-turer’s instructions. The conversion reaction was completed bydesulfonating in 0.3M NaOH for 10min at room temperature.The DNA was ethanol-precipitated and resuspended in water.
Promoter methylation analysisCpG islands were identified on the human genome browserand putative promoter regions were predicted by PromoterInspector software (Genomatix (www.genomatix.de)). Detailsof bisulphite sequencing primers and COL15A1 MSP primersare provided in Supplementary Table 1. Examples of directtumour-DNA sequencing traces are shown in SupplementaryFigure 2. Methylation-specific PCR analysis for the followinggenes was carried out using previously described MSP primers:BNC1 (Shames et al., 2006), SFRP1 (Nomoto et al., 2007) andREPRIMO (Sato et al., 2006).
Plasmid constructs and colony formation assayThe CST6, SFRP1 and REPRIMO expression constructs weremade by cloning the full-length human coding regions amplifiedfrom kidney cell lines into the EcoR1–BamHII sites ofpCDNA3.1. (Invitrogen) or pFLAG–CMV4 (Sigma) vectors.BNC1 was amplified from the IMAGE clone 40080551.Plasmid constructs were verified by sequencing. Six micro-grams of empty vector and an equal Molar amount ofexpression vector were transfected, using Fugene (Roche,Burgess Hill, UK), following the manufacturer’s instructions,into 5� 105 target cells (SKRC39, RCC4, 786-0, SKRC47 orHEK-293). Forty-eight hours after transfection, cells were seededin a serial dilution and maintained in DMEM and 10% fetalbovine serum supplemented with 1mg/ml G418 (Life Techno-logies, Paisley, UK). Surviving colonies were stained with 0.4%crystal violet (Sigma) in 50% methanol, 21 days after initial
Identification of candidate TSG frequently methylated in RCCMR Morris et al
12
Oncogene

seeding, and counted. Each transfection was carried out intriplicate. In addition, replicate experiments were carried outto obtain further clones for expression analysis. Expressionwas confirmed by RT–PCR and western blot analysis.
Anchorage-independent growth assayRCC clones stably expressing BNC1, CST6, REPRIMO or theempty vector control were suspended in 2-ml DMEM 10%FCS, 3% agar. Cells were maintained by addition of 200 ml ofDMEM 10% FCS, supplemented with 1mg/ml G418, weekly.After 5 weeks of growth, a final count of colonies wasperformed. RNAi ‘silencer select’ oligos against BNC1 (s2012),COL14A1 (s14677) and SFRP1 (s12713) or ‘silencer select’control oligo no.1 (Ambion, Warrington, UK) were trans-fected into HEK-293 or KTCL26 cells using Interferin reagent(Polyplus, Illkirch, France) following the manufacture’s instruc-tions. After 24-h incubation, cells were seeded into 2-ml DMEM
10% FCS, 3% agar. Cells were maintained by addition of200 ml of DMEM 10% FCS weekly. After 3 weeks of growth, afinal count of colonies was performed. Cells not seeded intoagar were incubated for a further 24 h before efficiency ofknock-down was assessed by RT–PCR.Statistical analysis was performed as indicated with a
significance level of 5%.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank Cancer Research UK for financial support.
References
Ai L, Kim WJ, Kim TY, Fields CR, Massoll NA, Robertson KD et al.(2006). Epigenetic silencing of the tumor suppressor cystatin Moccurs during breast cancer progression. Cancer Res 66: 7899–7909.
Amenta PS, Scivoletti NA, Newman MD, Sciancalepore JP, Li D,Myers JC. (2005). Proteoglycan-collagen XV in human tissues isseen linking banded collagen fibers subjacent to the basementmembrane. J Histochem Cytochem 53: 165–176.
Awakura Y, Nakamura E, Ito N, Kamoto T, Ogawa O. (2007).Methylation-associated silencing of SFRP1 in renal cell carcinoma.Oncol Rep 20: 1257–1263.
Battagli C, Uzzo RG, Dulaimi E, Ibanez de Caceres I, Krassenstein R,Al-Saleem T et al. (2003). Promoter hypermethylation of tumorsuppressor genes in urine from kidney cancer patients. Cancer Res
63: 8695–8699.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. (2004).
Microregional extracellular matrix heterogeneity in brain modulatesglioma cell invasion. Int J Biochem Cell Biol 36: 1046–1069.
Bernal C, Aguayo F, Villarroel C, Vargas M, Diaz I, Ossandon FJet al. (2008). Reprimo as a potential biomarker for early detection ingastric cancer. Clin Cancer Res 14: 6264–6269.
Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O’Rourke D et al.(2004). Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol 2: e289.
Boumber YA, Kondo Y, Chen X, Shen L, Gharibyan V, Konishi Ket al. (2007). RIL, a LIM gene on 5q31, is silenced by methylationin cancer and sensitizes cancer cells to apoptosis. Cancer Res 67:1997–2005.
Brandan E, Retamal C, Cabello-Verrugio C, Marzolo MP. (2006). Thelow density lipoprotein receptor-related protein functions as anendocytic receptor for decorin. J Biol Chem 281: 31562–31571.
Breault JE, Shiina H, Igawa M, Ribeiro-Filho LA, Deguchi M,Enokida H et al. (2005). Methylation of the gamma-catenin gene isassociated with poor prognosis of renal cell carcinoma. Clin Cancer
Res 11: 557–564.Bromme D, Kaleta J. (2002). Thiol-dependent cathepsins: pathophy-
siological implications and recent advances in inhibitor design. Curr
Pharm Des 8: 1639–1658.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV,
Zhou MI et al. (2008). Jade-1 inhibits Wnt signalling byubiquitylating beta-catenin and mediates Wnt pathway inhibitionby pVHL. Nat Cell Biol 10: 1208–1216.
Chowdhury S, Larkin JM, Gore ME. (2008). Recent advances in thetreatment of renal cell carcinoma and the role of targeted therapies.Eur J Cancer 44: 2152–2161.
Christoph F, Weikert S, Kempkensteffen C, Krause H, Schostak M,Kollermann J et al. (2006). Promoter hypermethylation profile of
kidney cancer with new proapoptotic p53 target genes and clinicalimplications. Clin Cancer Res 12: 5040–5046.
Clifford SC, Cockman ME, Smallwood AC, Mole DR, WoodwardER, Maxwell PH et al. (2001). Contrasting effects on HIF-1alpharegulation by disease-causing pVHL mutations correlate withpatterns of tumourigenesis in von Hippel-Lindau disease. Hum
Mol Genet 10: 1029–1038.Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER. (1998).
Inactivation of the von Hippel-Lindau (VHL) tumour suppressorgene and allelic losses at chromosome arm 3p in primary renal cellcarcinoma: evidence for a VHL-independent pathway in clear cellrenal tumourigenesis. Genes Chromosomes Cancer 22: 200–209.
Costa VL, Henrique R, Ribeiro FR, Pinto M, Oliveira J, Lobo F et al.(2007). Quantitative promoter methylation analysis of multiplecancer-related genes in renal cell tumors. BMC Cancer 7: 133.
Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck Jet al. (2007). Frequent loss of SFRP1 expression in multiple humansolid tumours: association with aberrant promoter methylation inrenal cell carcinoma. Oncogene 26: 5680–5691.
Dallol A, Forgacs E, Martinez A, Sekido Y, Walker R, Kishida T et al.(2002). Tumour specific promoter region methylation of the humanhomologue of the Drosophila Roundabout gene DUTT1 (ROBO1)in human cancers. Oncogene 21: 3020–3028.
Ehnis T, Dieterich W, Bauer M, Kresse H, Schuppan D. (1997).Localization of a binding site for the proteoglycan decorin oncollagen XIV (undulin). J Biol Chem 272: 20414–20419.
Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boye P. (2007).Estimates of the cancer incidence and mortality in Europe in 2006.Ann Oncol 18: 581–592.
Foster K, Prowse A, van den Berg A, Fleming S, Hulsbeek MM,Crossey PA. (1994). Somatic mutations of the von Hippel-Lindaudisease tumour suppressor gene in non-familial clear cell renalcarcinoma. Hum Mol Genet 3: 2169–2173.
Gonzalgo ML, Yegnasubramanian S, Yan G, Rogers CG, Nicol TL,Nelson WG et al. (2004). Molecular profiling and classification ofsporadic renal cell carcinoma by quantitative methylation analysis.Clin Cancer Res 10: 7276–7283.
Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrandSN et al. (2007). Secreted frizzled-related protein 1 loss contributesto tumor phenotype of clear cell renal cell carcinoma. Clin Cancer
Res 13: 4740–4749.Harris A, Harris H, Hollingsworth MA. (2007). Complete suppression
of tumor formation by high levels of basement membrane collagen.Mol Cancer Res 5: 1241–1245.
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S.(1994). Silencing of the VHL tumor-suppressor gene by DNA
Identification of candidate TSG frequently methylated in RCCMR Morris et al
13
Oncogene

methylation in renal carcinoma. Proc Natl Acad Sci USA 91:9700–9704.
Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, WestraWH et al. (2004). Quantitative detection of promoter hypermethyla-tion of multiple genes in the tumor, urine, and serum DNA ofpatients with renal cancer. Cancer Res 64: 5511–5517.
Ibanez de Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG,Cairns P. (2006). Identification of novel target genes by an epigeneticreactivation screen of renal cancer. Cancer Res 66: 5021–5028.
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML. (1993).Identification of the von Hippel-Lindau disease tumor suppressorgene. Science 260: 1317–1320.
Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H.(2005). Functional epigenomics identifies genes frequently silencedin prostate cancer. Cancer Res 65: 4218–4227.
Mancini V, Battaglia M, Ditonno P, Palazzo S, Lastilla G, MontironiR et al. (2008). Current insights in renal cell cancer pathology. Urol
Oncol 26: 225–238.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC,
Cockman ME et al. (1999). The tumour suppressor protein VHLtargets hypoxia-inducible factors for oxygen-dependent proteolysis.Nature 399: 271–275.
McRonald FE, Morris MR, Gentle D, Winchester L, Baban D,Ragoussis J et al. (2009). CpG methylation profiling in VHL relatedand VHL unrelated renal cell carcinoma. Mol Cancer 8: 31.
Morris MR, Gentle D, Abdulrahman M, Clarke N, BrownM, KishidaT et al. (2008). Functional epigenomics approach to identifymethylated candidate tumour suppressor genes in renal cellcarcinoma. Br J Cancer 98: 496–501.
Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, BanksRE et al. (2005). Tumor suppressor activity and epigeneticinactivation of hepatocyte growth factor activator inhibitor type2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer
Res 65: 4598–4606.Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD
et al. (2003). Multigene methylation analysis of Wilms’ tumour andadult renal cell carcinoma. Oncogene 22: 6794–6801.
Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S,Astuti D et al. (2001). Epigenetic inactivation of the RASSF1A3p21.3 tumor suppressor gene in both clear cell and papillary renalcell carcinoma. Cancer Res 61: 7277–7281.
Nomoto S, Kinoshita T, Kato K, Otani S, Kasuya H, Takeda S et al.(2007). Hypermethylation of multiple genes as clonal markers inmulticentric hepatocellular carcinoma. Br J Cancer 97: 1260–1265.
Ohki R, Nemoto J, Murasawa H, Oda E, Inazawa J, Tanaka N et al.(2000). Reprimo, a new candidate mediator of the p53-mediated cellcycle arrest at the G2 phase. J Biol Chem 275: 22627–22630.
Qiu J, Ai L, Ramachandran C, Yao B, Gopalakrishnan S, Fields CRet al. (2008). Invasion suppressor cystatin E/M (CST6): high-levelcell type-specific expression in normal brain and epigenetic silencingin gliomas. Lab Invest 88: 910–925.
Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, LuH et al. (1999). Antiangiogenic activity of restin, NC10 domain ofhuman collagen XV: comparison to endostatin. Biochem Biophys
Res Commun 255: 735–739.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL
et al. (2005). Contrasting properties of hypoxia-inducible factor 1
(HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cellcarcinoma. Mol Cell Biol 25: 5675–5686.
Santra M, Reed CC, Iozzo RV. (2002). Decorin binds to a narrowregion of the epidermal growth factor (EGF) receptor, partiallyoverlapping but distinct from the EGF-binding epitope. J Biol Chem
277: 35671–35681.Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron
JL et al. (2003). Discovery of novel targets for aberrant methylationin pancreatic carcinoma using high-throughput microarrays. CancerRes 63: 3735–3742.
Sato N, Fukushima N, Matsubayashi H, Iacobuzio-Donahue CA, YeoCJ, Goggins M. (2006). Aberrant methylation of Reprimo correlateswith genetic instability and predicts poor prognosis in pancreaticductal adenocarcinoma. Cancer 107: 251–257.
Schaefer L, Tsalastra W, Babelova A, Baliova M, Minnerup J,Sorokin L et al. (2007). Decorin-mediated regulation of fibrillin-1in the kidney involves the insulin-like growth factor-I receptorand Mammalian target of rapamycin. Am J Pathol 170:301–315.
Schuppan D, Cantaluppi MC, Becker J, Veit A, Bunte T, Troyer Det al. (1990). Undulin, an extracellular matrix glycoproteinassociated with collagen fibrils. J Biol Chem 265: 8823–8832.
Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar Net al. (2006). A genome-wide screen for promoter methylation inlung cancer identifies novel methylation markers for multiplemalignancies. PLoS Med 3: e486.
Shridhar R, Zhang J, Song J, Booth BA, Kevil CG, Sotiropoulou Get al. (2004). Cystatin M suppresses the malignant phenotype ofhuman MDA-MB-435S cells. Oncogene 23: 2206–2215.
Smits KM, Schouten LJ, van Dijk BA, Hulsbergen-van de Kaa CA,Wouters KA, Oosterwijk E et al. (2008). Genetic and epigeneticalterations in the von hippel-lindau gene: the influence on renalcancer prognosis. Clin Cancer Res 14: 782–787.
Suzuki M, Shigematsu H, Shames DS, Sunaga N, Takahashi T,Shivapurkar N et al. (2005). DNA methylation-associated inactiva-tion of TGFbeta-related genes DRM/Gremlin, RUNX3, and HPP1in human cancers. Br J Cancer 93: 1029–1037.
Takahashi T, Suzuki M, Shigematsu H, Shivapurkar N, Echebiri C,Nomura M et al. (2005). Aberrant methylation of Reprimo inhuman malignancies. Int J Cancer 115: 503–510.
Urakami S, Shiina H, Enokida H, Hirata H, Kawamoto K,Kawakami T et al. (2006). Wnt antagonist family genes asbiomarkers for diagnosis, staging, and prognosis of renalcell carcinoma using tumor and serum DNA. Clin Cancer Res 12:6989–6997.
Wang J, Zhang S, Schultz RM, Tseng H. (2006). Search for basonuclintarget genes. Biochem Biophys Res Commun 348: 1261–1271.
Wistuba II. (2007). Genetics of preneoplasia: lessons from lung cancer.Curr Mol Med 7: 3–14.
Yamada D, Kikuchi S, Williams YN, Sakurai-Yageta M, Masuda M,Maruyama T et al. (2006). Promoter hypermethylation of thepotential tumor suppressor DAL-1/4.1B gene in renal clear cellcarcinoma. Int J Cancer 118: 916–923.
Yamashita K, Upadhyay S, Osada M, Hoque MO, Xiao Y, Mori Met al. (2002). Pharmacologic unmasking of epigenetically silencedtumor suppressor genes in esophageal squamous cell carcinoma.Cancer Cell 2: 485–495.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Identification of candidate TSG frequently methylated in RCCMR Morris et al
14
Oncogene