identification of an estrogen-regulated circadian mechanism necessary for breast acinar...
TRANSCRIPT

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 1
Cell Cycle 11:19, 1-10; October 1, 2012; © 2012 Landes Bioscience
RepORt RepORt
*Correspondence to: Nicoletta Sacchi; Email: [email protected]: 07/31/12; Revised: 08/22/12; Accepted: 08/23/12http://dx.doi.org/10.4161/cc.21946
Introduction
Estrogens, including 17 β-estradiol (E2), the most potent estro-gen produced in the body, are hormones that play a pivotal role in development and in sexual and reproductive functions. Moreover, estrogens are indispensable for a variety of biological processes in different systems, including the cardiovascular, mus-coskeletal, immune and central nervous system.1 Estrogens also play a critical role in mammary gland development and in the initiation and progression of breast cancer.2
Estrogen signaling is extremely complex.3 Canonical estro-gen receptors (ERs), estrogen receptor α (ERA) and estrogen receptor β (ERB), mediate estrogen action at both genomic and non-genomic levels. Specifically, ERs by involving cell-specific, co-regulatory proteins regulate the transcription of several genes in the genome of cells of different tissues, including mammary cells.
Altered estrogen receptor α (eRA) signaling and altered circadian rhythms are both features of breast cancer. By using a method to entrain circadian oscillations in human cultured cells, we recently reported that the expression of key clock genes oscillates in a circadian fashion in eRA-positive breast epithelial cells but not in breast cancer cells, regardless of their eRA status. Moreover, we reported that ERA mRNA oscillates in a circadian fashion in eRA-positive breast epithelial cells, but not in eRA-positive breast cancer cells. By using eRA-positive HMe1 breast epithelial cells, which can be both entrained in vitro and can form mammary gland-like acinar structures in three-dimensional (3D) culture, first we identified a circuit encompassing eRA and an estrogen-regulated loop consisting of two circadian clock genes, peR2 and BMAL1. Further, we demonstrated that this estrogen-regulated circuit is necessary for breast epithelial acinar morphogenesis. Disruption of this circuit due to eRA-knockdown, negatively affects the estrogen-sustained circadian peR2-BMAL1 mechanism as well as the formation of 3D HMe1 acini. Conversely, knockdown of either peR2 or BMAL1, by hampering the peR2-BMAL1 loop of the circadian clock, negatively affects eRA circadian oscillations and 3D breast acinar morphogenesis. to our knowledge, this study provides the first evidence of the implication of an eRA-circadian clock mechanism in the breast acinar morphogenetic process.
Identification of an estrogen-regulated circadian mechanism necessary for breast acinar morphogenesis
Stefano Rossetti,† Francesca Corlazzoli,† Alex Gregorski, Nurul Hidayah A. Azmi and Nicoletta Sacchi*
Department of Cancer Genetics; Roswell park Cancer Institute; Buffalo, NY USA
†these authors contributed equally to this work.
Keywords: estrogen, estrogen receptor alpha (ERA), circadian clock, PER2, BMAL1, breast epithelial cells, 3D acinar morphogenesis
Abbreviations: 3D culture, three-dimensional culture; BMAL1, brain and muscle aryl hydrocarbon receptor nuclear translocator (ARNT)-like; ctrl, control; E2, 17β-estradiol; ERA, estrogen receptor alpha; ERE, estrogen response element; IBC, invasive
breast cancer; NRE, nuclear receptor response element; MEBM, mammary epithelial basal medium; MEGM, mammary epithelial growth medium; PER2, period 2; qRT-PCR, quantitative RT-PCR; RU, relative units; RARA, retinoic acid receptor alpha;
RORA, RAR-related orphanr alpha; shRNA, short hairpin RNA
Thi
s m
anus
crip
t ha
s be
en p
ublis
hed
onlin
e, p
rior
to
prin
ting
. Onc
e th
e is
sue
is c
ompl
ete
and
page
num
bers
hav
e be
en a
ssig
ned,
the
cit
atio
n w
ill c
hang
e ac
cord
ingl
y.
Estrogen-regulated target genes display estrogen receptor consen-sus sequences (EREs) in their promoters or regulatory regions, but they can also carry non-canonical ERE sequences.4 The genomic cartography and regulation of a large number of genomically reg-ulated estrogen-responsive genes continue to advance thanks to global technologies and bioinformatics strategies, thus providing templates for rapidly identifying genomic networks under estro-genic control in different normal and pathological tissues.5-8
In addition to this classical genomic action, ERs can also act in a non-genomic fashion.9,10 Apparently, stimulation of estro-genic non-genomic signaling occurs independently of nuclear localization and transcriptional effects at canonical ER genomic targets. Rapid, membrane-initiated, estrogen-triggered responses can involve spliced variants of canonical ERs, such ERA-3611,12 or non-canonical estrogen receptors, such as the G-protein-coupled receptor 30 (GPR30).13 The interplay of genomic and
©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
2 Cell Cycle Volume 11 Issue 19
of the hypothalamus and is mainly entrained by light.20 Apparently, women exposed to artificial light, or with jobs that can alter their circadian rhythms, are predisposed to breast cancer.19 The peripheral effect of altered circadian rhythms in human breast would be due to the fact that the central clock can entrain periph-eral clocks in tissues.20 In a recent study, we were able to demonstrate that even human breast epithelial cells in culture maintain a func-tional cell clock that can be entrained by serum shock treatment.21 The ability to entrain both non-cancer and cancer breast epithelial cells in vitro enabled us to demonstrate the hypothesis that estrogen-ERA sig-naling interplays with the molecular clock, thus adding another layer of complexity to estrogen biology. This hypothesis is based not only on the observation that key clock genes are controlled by estrogen,22 but also on our observations that upon in vitro entrainment, under physiological estrogen levels, ERA-positive mam-mary epithelial cells (e.g., HME1 cells) display circadian oscillations of clock genes, while both ERA-positive and ERA-negative breast cancer cells display a disrupted circadian clock.21 Moreover, while in HME1 cells ERA mRNA oscillates in a circadian fash-ion, in ERA-positive breast cancer cell lines, with an aberrant clock, ERA mRNA does not undergo circa-dian oscillation.21
Based on the ERA expression level alone, it has always been assumed that ERA-mediated estrogen signal-ing is not impaired in ERA-positive breast cancer cells. However, lack
of accumulation of ERA mRNA in a circadian fashion in ERA-positive breast cancer cells made us reason that estrogen signal-ing might be affected, albeit differently than in ERA-negative breast cancer cells, also in ERA-positive breast cancer cells. By exploiting, as a port of entry, the genomic regulatory connection between ERA and the direct ERA-target clock gene PER222 (Fig. 1), and the regulatory connection between PER2 and BMAL123 (Fig. 1), we embarked on testing the hypothesis of a mutual regu-lation between estrogen-ERA signaling and the circadian clock.
Here, we show that ERA knockdown, by preventing estrogen-mediated PER2 transcriptional activation, affects the PER2-BMAL1 loop of the HME1 molecular clock. Conversely, we show
non-genomic estrogen actions ultimately impinges on transcrip-tion, which makes the interpretation of estrogen action in normal and pathological settings quite complicated.
We find it attractive to decipher how estrogen signaling, which is relevant in mammary gland development, physiology and breast cancer, is integrated with the circadian clock. Our interest stems from growing evidence that associates breast cancer with aberrant circadian rhythms.14-19 This association has led to the hypothesis that altered circadian rhythms, by altering nuclear hormone levels and signaling, can negatively affect the metabolism and physiol-ogy of hormone-dependent tissues, such as the breast tissue. The central clock is located in the suprachiasmatic nucleus (SCN)
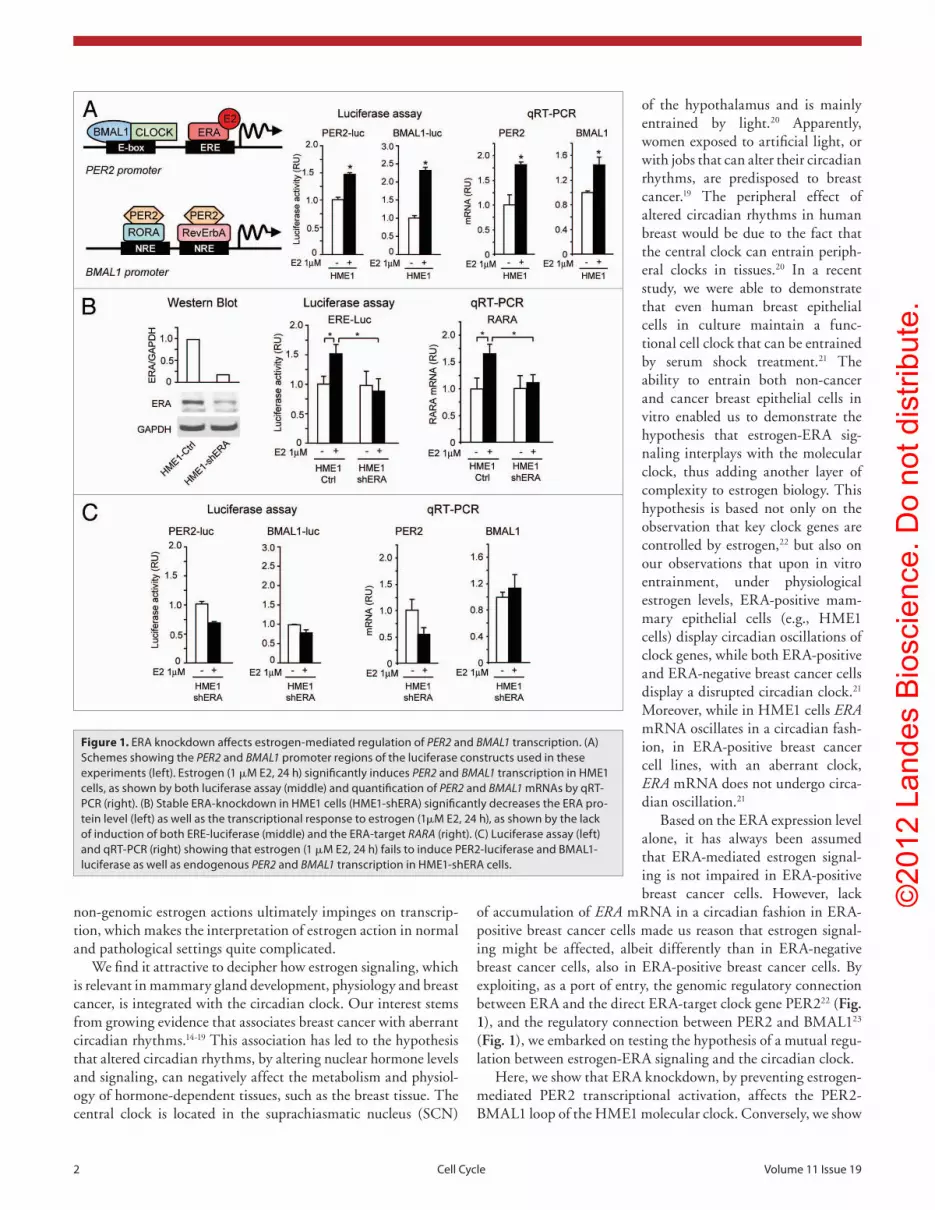
Figure 1. eRA knockdown affects estrogen-mediated regulation of PER2 and BMAL1 transcription. (A) Schemes showing the PER2 and BMAL1 promoter regions of the luciferase constructs used in these experiments (left). estrogen (1 μM e2, 24 h) significantly induces PER2 and BMAL1 transcription in HMe1 cells, as shown by both luciferase assay (middle) and quantification of PER2 and BMAL1 mRNAs by qRt-pCR (right). (B) Stable eRA-knockdown in HMe1 cells (HMe1-sheRA) significantly decreases the eRA pro-tein level (left) as well as the transcriptional response to estrogen (1μM e2, 24 h), as shown by the lack of induction of both eRe-luciferase (middle) and the eRA-target RARA (right). (C) Luciferase assay (left) and qRt-pCR (right) showing that estrogen (1 μM e2, 24 h) fails to induce peR2-luciferase and BMAL1-luciferase as well as endogenous PER2 and BMAL1 transcription in HMe1-sheRA cells.

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 3
has a functional cellular clock.21 Indeed, pharmacological admin-istration of estrogen (1 μM E2, 24 h) was sufficient to induce the expression of luciferase reporters driven by either the PER2 promoter, containing the ERE element, or the BMAL1 promoter, containing the NRE-binding sites (Fig. 1A, middle). The same treatment significantly increased also the level of endogenous PER2 and BMAL1 mRNAs in HME1 cells (Fig. 1A, right).
We next set out to knockdown ERA in HME1 cells and test whether this could affect the induction of PER2 and BMAL1 by estrogen. Stable HME1 transfection with a construct expressing a short hairpin RNA targeting ERA mRNA efficiently decreased the ERA protein level in HME1-shERA cells relative to cells stably transfected with a control scrambled shRNA sequence (HME1-Ctrl cells) (Fig. 1B, left). Importantly, the decrease in ERA was sufficient to impair ERA genomic function, as it sig-nificantly counteracted both the induction of an ERE-driven luciferase reporter gene (Fig. 1B, middle) and the transcription of the prototypic direct ERA-target gene RARA30 (Fig. 1B, right) in response to estrogen treatment (1 μM E2, 24 h). Apparently, ERA knockdown in HME1-shERA cells prevented estrogen-induced transcription of both PER2 and BMAL1, as shown by PER2-luciferase and BMAL1-luciferase assays (Fig. 1C, left) and endogenous PER2 and BMAL1 mRNAs quantification (Fig. 1C, right).
These findings demonstrate that both PER2 and BMAL1 are estrogen-regulated genes. While estrogen-induced PER2 transcription is directly regulated by ERA,22 estrogen-induced BMAL1 transcription likely is not directly mediated by ERA, due to the absence of canonical ERE-consensus sequences in the BMAL1 promoter (Fig. 1A, bottom left). Since BMAL1 is a direct PER2 target,23 its transcriptional induction by estrogen is likely due to ERA-mediated PER2 upregulation.
ERA knockdown affects the amplitude of PER2 and BMAL1 mRNA oscillation under entrainment conditions. It is known that PER2 and BMAL1, as components of the cellular clock oscil-lator, are expressed in a circadian fashion, antiphase with each other.31,32 We previously demonstrated that circadian oscillation of core clock genes, including PER2 and BMAL1, can be induced in HME1 cells upon entrainment by serum shock,21 a procedure that entails starvation in MEBM basal medium, followed by the administration of a 2 h-shock with entrainment medium (MEGM supplemented with 50% horse serum). The entrainment medium contains close to physiological concentrations of estrogen, rang-ing from 0.1 to 1 nM, as per manufacturer’s specifications. The estrogen concentration present in the entrainment medium is apparently sufficient to induce transcription of estrogen-respon-sive genes in HME1-Ctrl cells, as shown by the significantly higher activation of ERE-luciferase relative to the basal medium (Fig. 2A). This transcriptional activation must be ERA-mediated, since the level of ERE-luciferase induction was significantly less in HME1-shERA cells (Fig. 2A). Moreover, the estrogen present in the entrainment medium for just 2 h is sufficient to induce both endogenous PER2 and BMAL1 transcription in HME1-Ctrl cells, but not in HME1-shERA cells (Fig. 2B). Consistently, when we measured the circadian oscillation of PER2 and BMAL1 mRNAs in entrained cells, we observed that the oscillation amplitude of
that PER2 knockdown or BMAL1 knockdown in ERA-positive HME1 cells, significantly affect both the circadian oscillation of ERA mRNA, and the transcriptional oscillation of the ERA direct target RARA. From the first part of this study, it is plausi-ble to conclude that estrogen regulates a circuit that encompasses an ERA-controlled PER2-BMAL1 circadian loop and a PER2-BMAL1-regulated ERA circadian function. To our knowledge, this is the first evidence that mechanistically supports the exis-tence of a mutual estrogen regulation of an ERA-PER2-BMAL1 circuit, thus providing hints as to why breast cancer cells show loss of circadian rhythmicity, not only of clock genes, but also ERA and downstream ERA-targets.
As mentioned before, an altered circadian clock has been proposed as a factor that can trigger and promote breast tumor-igenesis.24 In epithelia of the normal breast tissue, there is a sub-population of ERA-positive cells that is physiologically regulated and varies with menstrual status.25 Approximately three out of four invasive breast cancers (IBCs) are ERA-positive, which is clinically important, because they exhibit estrogen-dependent growth.26,27 There is a second subpopulation of breast epithe-lial cells that lack ERA expression. It has been debated whether malignant ERA-negative primary tumors derive from a once ERA-positive cell, due to ERA loss/epigenetic silencing, or may have originated by the ERA-negative subpopulation.25,28 Loss of acinar morphogenesis is one of the first pathological signs of breast cancer initiation that can be detected by pathologists.29 A simplified model system to test for factors that can affect acinar morphogenesis is to grow cells in three-dimensional (3D) base-ment membrane culture. Here we show that differently from HME1 control cells, which can form acini with a lumen lined by polarized cells, HME1 cells with stable ERA knockdown (shERA) form acini with a filled lumen when grown under the same 3D culture conditions. Moreover, also PER2 (shPER2)- and BMAL1 (shBMAL1)-knockdown clones derived from HME1 cells can form ERA-positive 3D acini with aberrant morphology. Apparently, impairment of the PER2-BMAL1 loop, in an oth-erwise ERA-positive context, can undermine estrogen-regulated morphogenetic processes due to lack of proper circadian avail-ability of ERA, as it happens in the HME1-shERA cell context.
Results
ERA knockdown affects estrogen-mediated regulation of PER2 and BMAL1 transcription. Previously published work demon-strated that estrogen can induce PER2 transcription through direct ERA binding to an ERE-responsive element in the PER2 promoter (Fig. 1A, top left).22 PER2, in turn, was shown to pro-mote BMAL1 transcription by forming regulatory complexes with RORA and Rev-ErbA, which directly bind nuclear recep-tor response elements (NRE) in the BMAL1 promoter (Fig. 1A, bottom left).23 Based on these findings, we hypothesized that in response to estrogen, ERA regulates the cellular clock by modu-lating the transcription of both PER2 and BMAL1.
We first tested whether treatment with 17 β-estradiol (E2) can induce the transcription of PER2 and BMAL1 in the human mammary epithelial cell line HME1, which is ERA-positive and

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
4 Cell Cycle Volume 11 Issue 19
on PER2 transcription in HME1 cells. As shown for two prototypic clones, HME1-shBMAL1–5 and HME1-shBMAL1–9, stable BMAL1 knockdown efficiently decreased BMAL1 mRNA level both in non-entrained and entrained cells (Fig. 3E) and resulted in a significant attenuation of PER2 mRNA oscil-lation (Fig. 3F). Thus, PER2 and BMAL1 seem to sustain each other’s circadian oscilla-tion through a feedback loop mechanism.
In conclusion, these results demonstrate that ERA-mediated estrogen signaling con-trols, via PER2, the transcriptional regulation and circadian oscillation of the BMAL1-PER2 loop.
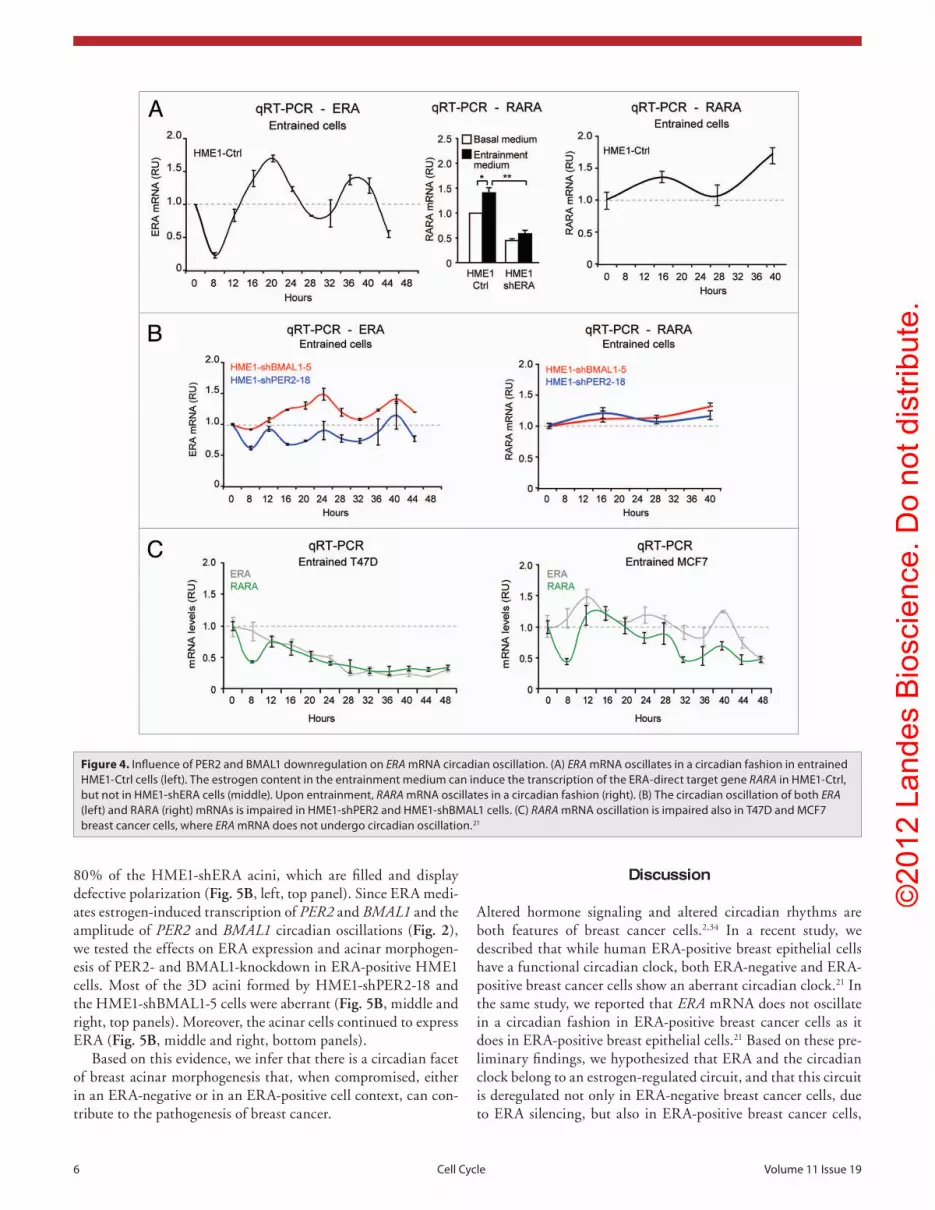
Influence of PER2 and BMAL1 downreg-ulation on ERA mRNA circadian oscillation. We have previously shown that ERA mRNA displays circadian oscillation in ERA-positive HME1 cells, but not in ERA-positive breast cancer cells, whose cellular clock is defective.21 In this study, we tested whether interference with the HME1 cellular clock, through either PER2 or BMAL1 knockdown, can influence ERA circadian oscillation.
Entrained HME1-Ctrl cells show ERA mRNA circadian oscillation with peaks at approximately 16 and 40 h (Fig. 4A, left).
The concentration of estrogen in the entrainment medium can activate the ERA function, as shown by transcriptional induc-tion of the ERA-target gene RARA (Fig. 4A, middle). The cir-cadian oscillation of ERA apparently sustains the oscillation of RARA mRNA, since both ERA and RARA mRNAs peak at the same time points (Fig. 4A, right). In contrast, HME1-shPER2 and HME1-shBMAL1 display irregular and/or damped oscil-lations of both ERA and RARA mRNAs when compared with HME1-Ctrl cells (Fig. 4B), indicating that PER2 and BMAL1-knockdown affect in a domino fashion the circadian oscillation of ERA as well as the circadian oscillation of RARA. This finding is in agreement with the observation that RARA mRNA does not oscillate in a circadian fashion in T47D and MCF7 ERA-positive breast cancer cells (Fig. 4C), which do not display ERA mRNA circadian oscillations.21
Based on these findings, ERA appears to be part of a circuit, whereby ERA controls PER2 and BMAL1 oscillation amplitude, while the PER2-BMAL1 loop keeps under control both ERA oscillation, and the oscillation of downstream ERA target genes. Interference with any of the components of this circuit affects the expression and/or the circadian oscillation pattern of the other components (Fig. 6).
Evidence that circadian components of the estrogen-reg-ulated circuit are implicated in breast acinar morphogenesis. One of the first signs of breast cancer initiation is the loss of aci-nar morphogenesis. Since both defects in estrogen-ERA signal-ing and defects in the cellular clock are common in breast cancer cells, we asked whether interference with the ERA-PER2-BMAL1
both mRNAs was significantly attenuated in HME1-shERA cells relative to HME1-Ctrl cells (Fig. 2C).
These results show that upon entrainment, ERA-mediated estrogen signal contributes to sustain the amplitude of PER2 and BMAL1 circadian oscillations.
ERA-mediated estrogen signaling controls the BMAL1-PER2 loop. The absence of EREs in the BMAL1 promoter and published evidence that PER2 controls BMAL1 transcription (Fig. 1A) made us conclude that estrogen induces BMAL1 transcrip-tion via PER2. This conclusion is further supported by the finding that transient transfection of PER2 cDNA in HME1 cells increases BMAL1-luciferase activity in a dose-dependent manner (Fig. 3A). Moreover, stable PER2-knockdown significantly reduced the lev-els of both baseline and estrogen-induced BMAL1 transcription. Prototypic HME1-shPER2–16 and HME1-shPER2–18 clones, with significantly reduced PER2 mRNA levels (Fig. 3B, left) upon entrainment maintained PER2-knockdown through the 24 and 48 h peaks (Fig. 3B, right). PER2 knockddown significantly reduced both baseline and estrogen-induced BMAL1 transcrip-tion, as shown by both BMAL1-luciferase reporter assay (Fig. 3C, left), and BMAL1 mRNA quantification in HME1-shPER2 cells (Fig. 3C, right). Remarkably, HME1-shPER2 clones displayed not only lower BMAL1 mRNA levels in non-entrained cells (Fig. 3D, left), but also a decreased amplitude of BMAL1 mRNA oscillation in entrained cells (Fig. 3D, right), thus demonstrating that PER2 is necessary to sustain BMAL1 oscillation.
Since BMAL1 is in turn necessary to maintain proper rhyth-micity of PER2,32,33 we tested the effects of BMAL1 knockdown
Figure 2. eRA-knockdown affects the amplitude of PER2 and BMAL1 mRNA oscillation under entrainment conditions. the estrogen present in the entrainment medium can induce significantly more eRe-luciferase (A) and PER2 and BMAL1 mRNA (B) in HMe1-Ctrl, relative to HMe1-sheRA cells. (C) the amplitude of PER2 and BMAL1 mRNA oscillations are significantly attenuated in HMe1-sheRA, relative to HMe1-Ctrl cells.

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 5
apicobasal polarity, which is evidenced by the localization of the integrin at the periphery of the acini and the Golgi apparatus facing the lumen of more than 70% of the acini (Fig. 5A, top). HME1 acini are also ERA-positive, with ERA localized both in the cytoplasm and in the nucleus of most acinar cells (Fig. 5A, bottom panel). ERA-knockdown significantly diminishes ERA protein expression in the acinar cells (Fig. 5B, left, bottom panel) and impairs the process of acinar morphogenesis in more than
circuit can affect 3D HME1 acinar morphogenesis. When grown in 3D culture on reconstituted basement membrane (Matrigel), HME1 cells undergo a process of acinar morphogenesis by which single cells form spheroids that eventually develop into hollow acini lined by polarized cells. This process recapitulates many aspects of mammary gland development, and can be easily moni-tored by confocal immunofluorescence. After 10–12 d in 3D culture, HME1 acini become hollow and exhibit basolateral and
Figure 3. eRA-mediated estrogen signaling controls the BMAL1-peR2 loop. (A) transient peR2 transfection significantly promotes BMAL1-luciferase expression in HMe1 cells. (B) Stable peR2-knockdown in HMe1 cells significantly decreases PER2 mRNA level in both non-entrained and serum shock-entrained HMe1-shpeR2 cells (clones 16 and 18), relative to control HMe1 cells. (C) peR2-knockdown leads to downregulation of both BMAL1-luciferase (Left) and BMAL1 mRNA (Right) before and after estrogen treatment (1 μM e2, 24h). (D) peR2 knockdown decreases BMAL1 mRNA level both in non-entrained and entrained cells. (e) Stable BMAL1 knockdown in HMe1 cells significantly decreases BMAL1 mRNA level in both non-entrained and serum shock-entrained HMe1-shBMAL1 cells (clones 5 and 9). (F) entrained HMe1-shBMAL1 clones display reduced PER2 mRNA oscillations relative to HMe1-Ctrl cells.
©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
6 Cell Cycle Volume 11 Issue 19
Discussion
Altered hormone signaling and altered circadian rhythms are both features of breast cancer cells.2,34 In a recent study, we described that while human ERA-positive breast epithelial cells have a functional circadian clock, both ERA-negative and ERA-positive breast cancer cells show an aberrant circadian clock.21 In the same study, we reported that ERA mRNA does not oscillate in a circadian fashion in ERA-positive breast cancer cells as it does in ERA-positive breast epithelial cells.21 Based on these pre-liminary findings, we hypothesized that ERA and the circadian clock belong to an estrogen-regulated circuit, and that this circuit is deregulated not only in ERA-negative breast cancer cells, due to ERA silencing, but also in ERA-positive breast cancer cells,
80% of the HME1-shERA acini, which are filled and display defective polarization (Fig. 5B, left, top panel). Since ERA medi-ates estrogen-induced transcription of PER2 and BMAL1 and the amplitude of PER2 and BMAL1 circadian oscillations (Fig. 2), we tested the effects on ERA expression and acinar morphogen-esis of PER2- and BMAL1-knockdown in ERA-positive HME1 cells. Most of the 3D acini formed by HME1-shPER2-18 and the HME1-shBMAL1-5 cells were aberrant (Fig. 5B, middle and right, top panels). Moreover, the acinar cells continued to express ERA (Fig. 5B, middle and right, bottom panels).
Based on this evidence, we infer that there is a circadian facet of breast acinar morphogenesis that, when compromised, either in an ERA-negative or in an ERA-positive cell context, can con-tribute to the pathogenesis of breast cancer.
Figure 4. Influence of peR2 and BMAL1 downregulation on ERA mRNA circadian oscillation. (A) ERA mRNA oscillates in a circadian fashion in entrained HMe1-Ctrl cells (left). the estrogen content in the entrainment medium can induce the transcription of the eRA-direct target gene RARA in HMe1-Ctrl, but not in HMe1-sheRA cells (middle). Upon entrainment, RARA mRNA oscillates in a circadian fashion (right). (B) the circadian oscillation of both ERA (left) and RARA (right) mRNAs is impaired in HMe1-shpeR2 and HMe1-shBMAL1 cells. (C) RARA mRNA oscillation is impaired also in t47D and MCF7 breast cancer cells, where ERA mRNA does not undergo circadian oscillation.21

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 7
tumors likely evolved from ERA-positive precursors.25 Several factors can cause ERA silencing during breast cancer progres-sion. One factor is the activation of the mitogen-activated protein kinase pathway, through ligand-activated tyrosine kinase recep-tors at the cell surface.35 Epigenetic chromatin repression of ERA regulatory regions by DNA hypermethylation is another mecha-nism that can cause ERA transcriptional silencing.36 Thus, it is plausible that ERA silencing affects, in addition to many other ERA-regulated processes, the circadian clock in ERA-negative IBCs as well. Indeed, entrained ERA-negative MDA-MB-231 breast cancer cells with an epigenetically silent and hypermethyl-ated ERA37 display an aberrant circadian clock.21
Interestingly, ERA-positive breast cancer cells, such as the widely studied T47D and MCF7 cells, upon entrainment, also display an aberrant circadian clock,21 even if estrogen (E2) treat-ment induces transcriptional activation of several ERA targets, including the clock gene PER2.22 Based on our previous obser-vation that entrained T47D and MCF7 cells display aberrant circadian oscillation of ERA mRNA,21 we tested whether aber-rant circadian oscillation of ERA mRNA can be the consequence of the PER2-BMAL1 loop disruption. Upon entrainment of HME1-shPER2 and HME1-shBMAL1 clones, we detected an aberrant circadian oscillation pattern of ERA mRNA as well as aberrant circadian oscillation of the mRNA of the direct ERA-target gene RARA. Thus, it is plausible to conclude that
due to ERA aberrant circadian oscillation. In the first part of this study, we gathered evidence that supports this hypothesis.
By inducing ERA stable knockdown in ERA-positive HME1 cells (shERA), we detected the attenuation of the circadian oscillation of two key molecular clock components: PER2 and BMAL1. Next, by stable knockdown of an ERA-regulated clock gene, PER2, in HME1 cells (shPER2), we found that upon entrainment the amplitude of the circadian oscillations of BMAL1, which is under the regulation of PER2, is also sig-nificantly attenuated. Thus, estrogen-sustained PER2 expres-sion seems to be required to sustain BMAL1 oscillations. Further, by generating stable HME1-knockdown BMAL1 clones (shBMAL1), we established that BMAL1 is instead required to sustain PER2 mRNA circadian oscillation.
These findings, based on the panel of HME1-derived shERA, shPER2 and shBMAL1 clones, show that estrogen signaling sus-tains, through ERA, the amplitude of the circadian oscillations of both PER2, which is a direct ERA transcriptional target, and BMAL1, which is indirectly regulated by estrogen through PER2-RORA and PER2-RevErbA complexes.23 Further, these findings establish that PER2 and BMAL1 constitute a loop which is part of the estrogen-ERA regulated circuit (Fig. 6).
Lack of ERA is a feature of approximately 25% of the invasive breast cancers (IBCs).26 Epidemiological, histological/pathologi-cal and molecular evidence shows that many ERA-negative breast
Figure 5. evidence that circadian components of the estrogen-regulated circuit are implicated in breast acinar morphogenesis. (A) Immunocytochem-istry followed by confocal microscopy showing control HMe1 acini after 12 d of three-dimensional (3D) culture on reconstituted basement membrane (Matrigel). In the top panel, the organization of the DApI-stained nuclei (blue) indicates whether the acini are hollow, the presence of an outer layer of integrin (green) indicates baso-lateral polarity, while the presence of the Golgi apparatus (red) in the inner portion of the acinus is a sign of apico-basal polarity. the percentage of acini with the displayed morphology is indicated in upper left corner. In the bottom panel, immunostaining with anti-eRA (green) shows eRA expression both in the cytoplasm and, to a lesser extent, in the DApI-stained nuclei (blue). the lower inset shows an enlargement of the framed area. (B) the top panels show the aberrant morphology of the HMe1-sheRA (left), HMe1-shpeR2 (middle) and HMe1-shBMAL1 (right) acini, with the relative percentage. the bottom panels show that HMe1-sheRA, HMe1-shpeR2 and HMe1-shBMAL1 aberrant acini differ in eRA expression.

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
8 Cell Cycle Volume 11 Issue 19
can have a domino circadian effect also on ERA genomic function, and likely on ERA non-genomic function.
The second hypothesis that emerged from this study is that an aberrant clock mechanism either in an ERA-negative or in an ERA-positive breast epithelial cell context can trigger breast cancer initiation. Aberrant morphological features are the first histological signs indicative of breast cancer initiation that can be detected by patholo-gists.29 We wondered whether factors affecting the PER2-BMAL1 loop in either an ERA-negative or an ERA-positive context can have an effect on acinar morphogenesis. The results shown in this study suggest that an aberrant clock, either due to ERA silencing and consequent deregula-tion of the estrogen-ERA-regulated clock mecha-nisms (e.g., the PER2-BMAL1 loop mechanism), or due to direct genetic/epigenetic mutations of clock components (e.g., PER2 or BMAL1) in ERA-positive cells, can affect the architecture of the breast acinus.
To our knowledge, this is the first study showing that in addition to metabolic and phys-iological processes, breast acinar morphogenetic processes are also under circadian regulation.
Materials and Methods
Cell culture. The human telomerase-immortal-ized breast epithelial cell strain h-TERT-HME1 (hereafter called HME1) (Clontech) and the derived clones were grown in mammary epi-thelial growth medium (MEGM) (Lonza). The human breast cancer cell lines MCF7 (ATCC, HTB-22) and T47D (ATCC, HTB-133) were grown in DMEM supplemented with 5% fetal bovine serum (FBS) (Life Technologies).
Development of stable knockdown clones by shRNA. To develop stable knockdown clones, cells were transfected with retrovi-ral constructs expressing short hairpin RNA (shRNA) sequences targeting the mRNAs of interest by using Lipofectamine 2000 (Life Technologies). For ERA knockdown, cells were transfected with pRS(HuSH)-shERA (Origene Technologies). For PER2 and BMAL1 knock-down, shRNA sequences targeting the respec-
tive mRNA (PER2: 5'-GAG TAT TAC CAG CTG CTG-3'; BMAL1: 5'-CCC TGA TGC CTC TTC TCC A-3'), or a control scrambled sequence were first cloned into the pSuper-retro vec-tor (Oligoengine) according to the manufacturer’s instructions, then transfected into HME1 cells. Transfected cells were seeded at low density and selected with 1 μg/ml puromycin, and single clones were isolated and expanded. Clones were first screened for the presence of the correct construct by PCR and sequencing,
in ERA-positive cells, the disruption of estrogen-regulated clock genes, such as PER2 and BMAL1, has a profound rebound on the circadian oscillation of ERA and, consequently, also on the circadian oscillation of downstream ERA-regulated targets, such as RARA. Even if we do not know yet the exact regulatory mech-anisms to fully explain how the PER2-BMAL1 loop is impli-cated in the circadian expression of ERA, it is conceivable that circadian ERA deregulation, due to defective clock components,
Figure 6. the eRA-peR2-BMAL1 circuit. In the presence of an intact eRA-peR2-BMAL1 cir-cuit, whereby eRA-mediated estrogen signaling affects the oscillations of PER2 and BMAL1, and the peR2-BMAL1 regulatory loop in turn affects ERA oscillations, HMe1 cells undergo proper acinar morphogenesis. Interference with any of the components of the eRA-peR2-BMAL1 circuit has a rebound on the circadian oscillation of the other components and results in impaired HMe1 acinar morphogenesis.

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
www.landesbioscience.com Cell Cycle 9
TCC ATC TGC TGC CCT G-3'). Transcript levels were quan-tified with the Delta-delta Ct method, using the housekeeping gene GAPDH (fwd: 5'-GAA GGT GAA GGT CGG AGT C-3', rev: 5'-GAA GAT GGT GAT GGG ATT TC-3') for normaliza-tion. Experiments were performed in triplicate, and significance was determined by the Student’s t-test.
Western blotting. Cells were lysed in lysis buffer (50 mM TRIS-HCl pH 7.4, 250 mM NaCl, 5 mM EDTA, 1% Triton X100) supplemented with Complete proteinase inhibitor cocktail (Roche). Proteins from total cell lysates were resolved on a SDS-PAGE gel and blotted by using standard procedures. Endogenous ERA and GAPDH were detected with anti-ERA (MC-20) and anti-GAPDH antibodies (S. Cruz Biotechnologies). After incu-bation with the primary antibody, membranes were incubated with appropriate HRP-conjugated secondary antibodies (GE Healthcare) followed by ECL detection (GE Healthcare).
Assessment of acinar morphogenesis in three-dimensional (3D) cultures. HME1 cells and derived clones were grown in 3D cultures on reconstituted basement membrane (Matrigel) as we previously described41 and based on.42 Briefly, single cell suspen-sions were seeded on 8-well chamber slides coated with growth factor-reduced Matrigel (BD Biosciences) in MEGM plus 2% Matrigel for up to 12 d. After fixation with 4% paraformalde-hyde for 20 min., permeabilization with 0.2% Triton X100 for 15 min. and blocking with PBS plus 1% BSA, 1% FBS and 0.05% Tween 20 for 2 h, cells were incubated with the primary antibody overnight at 4°C, washed, incubated with the appropriate fluo-rescent secondary antibody for 2 h at room temperature, washed, counterstained with 300 nM DAPI (Sigma) and mounted with Vectashield (Vector Laboratories). The Golgi apparatus was iden-tified with anti-GM130 antibody (BD Biosciences), followed by goat anti-mouse Alexa Fluor 546 antibody (Life Technologies). Integrin was identified with anti-CD49f antibody (Millipore) fol-lowed by anti-rat Alexa Fluor 488 antibody (Life Technologies). ERA was detected with anti-ERA (HC-20, S. Cruz) followed by anti-rabbit Alexa Fluor 488 antibody (Life Technologies). Immunostained acini were analyzed by confocal microscopy (SP2 Spectral Confocal Microscope, Leica). A minimum of 70 acini from two independent experiments were analyzed and scored either as normal (hollow acini with polarized cells) or aberrant (filled acini and/or non-polarized cells). The morphol-ogy observed in more than 70% of the acini was considered as the prevalent morphology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank past graduate students Joseph Esposito, Silvia Pozzi and Vincenzo Gagliostro, for developing and characterizing some of the stable HME1 clones used in this study. This work was sup-ported by funding of the Department of Defense (DOD) Concept Award W81XWH0610657 (N.S.), the Breast Cancer Coalition of Rochester (N.S.), the Friends for an Earlier Test for Breast Cancer Foundation (N.S.) and the National Cancer Institute (NCI) insti-tutional grant P30 CA016056.
and then tested for knockdown efficiency by real-time PCR or western blotting (see Results).
Drugs and treatments. 17-β estradiol (E2) (Sigma) was dis-solved in ethanol and stored in aliquots at -80°C. Sub-confluent cells were treated with E2 in MEGM for 24 h.
Cell entrainment by serum shock. Cells were entrained by serum shock procedure, as we previously described,21 and based on the method originally proposed by Balsalobre et al.38 Briefly, cells were seeded onto 12-well plates at a density of 1 × 105 cells per well, allowed to grow to confluence, washed with PBS and starved over-night before the serum shock. HME1 cells were starved in mam-mary epithelial basal medium (MEBM) (Lonza), while T47D and MCF7 were starved in serum-free DMEM. Starved cells were then serum-shocked for 2 h with entrainment medium, which consisted of MEGM + 50% horse serum (Life Technologies) for HME1 cells, and DMEM + 50% horse serum, for T47D and MCF7. After the serum shock, cells were washed with PBS and returned to star-vation conditions. Time points were taken prior to the serum shock (t = 0), and every 4 or 12 h thereafter up to 48 h.
Luciferase assay. Sub-confluent cells grown in a 24-well plate were transfected with 300 ng/well of the indicated luciferase reporter vectors and 10 ng/well of pRL-TK, which constitutively expresses renilla luciferase (Promega), by using Lipofectamine LTX (Life Technologies). PGL3-PER2-luciferase, carrying the mouse Per2 promoter from -782 to +76,39 was kindly provided by Dr. H. Ueda (RIKEN Institute); pGL2-BMAL1-luciferase, car-rying the human BMAL1 promoter from -3465 to +57bp,40 was kindly provided by Dr. T. Takumi (Osaka Bioscience Institute); XETL-ERE-luciferase, carrying the estrogen response element AGGTCACAGTGACCT, was kindly provided by Dr. D. Picard (University of Geneva); the human PER2 cDNA was amplified from HME1 cells by PCR with specific primers (fwd: 5'-TCA CAG GAT CGA AGA GCA GAC G-3'; rev: 5'-CGT CTG CTC TTC GAT CCT GTG A-3') and cloned into pcDNA3.1/V5-His-TOPO (Life Technologies), as per manufacturer’s instructions. The total amount of DNA used for each transfection was kept constant by adding empty pcDNA3.1. Unless otherwise speci-fied, cells were transfected overnight and treated for 24 h before measuring luciferase activity. Luciferase activity was measured by using Dual Glow Luciferase Assay System (Promega) and normalized to Renilla Luciferase activity as per manufacturer’s instructions. Experiments were performed in duplicate, and sig-nificance was determined by the Student’s t-test.
Quantitative real-time RT-PCR (qRT-PCR). Total RNA was extracted by using Trizol (Life Technologies) and retrotran-scribed into cDNA with high capacity reverse transcription kit (Life Technologies) according to the manufacturer’s instructions. qRT-PCR was performed on an iCycler (Bio-Rad) using the cDNA as a template, iQ SYBR Green Supermix (Bio-Rad) and primers specific for ERA (fwd: 5'-AAG AGC TGC CAG GCC TGC C-3'; rev: 5'-TTG GCA GCT CTC ATG TCT CC-3'), RARA (fwd: 5'-GCC AGG CGC TCT GAC CAC TC-3'; rev: 5'-CAG GCG CTG ACC CCA TAG TGG T-3'), PER2 (fwd: 5'-AAG CAG GTG AAA GCC AAT GAA GA-3'; rev: 5'-CCA CCG CAA ACA TAT CGG CAT T-3') and BMAL1 (fwd: 5'-AGG ATG GCT GTT CAG CAC ATG-3'; rev: 5'-CAA AAA

©20
12 L
ande
s B
iosc
ienc
e. D
o no
t dis
tribu
te.
10 Cell Cycle Volume 11 Issue 19
32. Reppert SM, Weaver DR. Coordination of circa-dian timing in mammals. Nature 2002; 418:935-41; PMID:12198538; http://dx.doi.org/10.1038/nature00965.
33. Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer 2003; 3:350-61; PMID:12724733; http://dx.doi.org/10.1038/nrc1072.
34. Canaple L, Kakizawa T, Laudet V. The days and nights of cancer cells. Cancer Res 2003; 63:7545-52; PMID:14633665.
35. Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mito-gen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen recep-tor alpha-negative human breast tumors. Cancer Res 2006; 66:3903-11; PMID:16585219; http://dx.doi.org/10.1158/0008-5472.CAN-05-4363.
36. Yan L, Yang X, Davidson NE. Role of DNA meth-ylation and histone acetylation in steroid receptor expression in breast cancer. J Mammary Gland Biol Neoplasia 2001; 6:183-92; PMID:11501578; http://dx.doi.org/10.1023/A:1011308707512.
37. Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expres-sion in human breast cancer cells. Cancer Res 1994; 54:2552-5; PMID:8168078.
38. Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998; 93:929-37; PMID:9635423; http://dx.doi.org/10.1016/S0092-8674(00)81199-X.
39. Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, et al. A transcription factor response element for gene expression during circadian night. Nature 2002; 418:534-9; PMID:12152080; http://dx.doi.org/10.1038/nature00906.
40. Akashi M, Takumi T. The orphan nuclear recep-tor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat Struct Mol Biol 2005; 12:441-8; PMID:15821743; http://dx.doi.org/10.1038/nsmb925.
41. Rossetti S, Hoogeveen AT, Esposito J, Sacchi N. Loss of MTG16a (CBFA2T3), a novel rDNA repres-sor, leads to increased ribogenesis and disruption of breast acinar morphogenesis. J Cell Mol Med 2010; 14(6A):1358-70; PMID:19961547; http://dx.doi.org/10.1111/j.1582-4934.2009.00982.x.
42. Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30:256-68; PMID:12798140; http://dx.doi.org/10.1016/S1046-2023(03)00032-X.
References1. Gustafsson JA. What pharmacologists can learn
from recent advances in estrogen signalling. Trends Pharmacol Sci 2003; 24:479-85; PMID:12967773; http://dx.doi.org/10.1016/S0165-6147(03)00229-3.
2. Anderson E. The role of oestrogen and progester-one receptors in human mammary development and tumorigenesis. Breast Cancer Res 2002; 4:197-201; PMID:12223124; http://dx.doi.org/10.1186/bcr452.
3. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 2007; 87:905-31; PMID:17615392; http://dx.doi.org/10.1152/physrev.00026.2006.
4. Welboren WJ, Stunnenberg HG, Sweep FC, Span PN. Identifying estrogen receptor target genes. Mol Oncol 2007; 1:138-43; PMID:19383291; http://dx.doi.org/10.1016/j.molonc.2007.04.001.
5. Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet 2007; 3:e87; PMID:17542648; http://dx.doi.org/10.1371/journal.pgen.0030087.
6. Joseph R, Orlov YL, Huss M, Sun W, Kong SL, Ukil L, et al. Integrative model of genomic factors for deter-mining binding site selection by estrogen receptor-α. Mol Syst Biol 2010; 6:456; PMID:21179027; http://dx.doi.org/10.1038/msb.2010.109.
7. Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009; 462:58-64; PMID:19890323; http://dx.doi.org/10.1038/nature08497.
8. Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, et al. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J 2009; 28:1418-28; PMID:19339991; http://dx.doi.org/10.1038/emboj.2009.88.
9. Moriarty K, Kim KH, Bender JR. Minireview: estro-gen receptor-mediated rapid signaling. Endocrinology 2006; 147:5557-63; PMID:16946015; http://dx.doi.org/10.1210/en.2006-0729.
10. Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and non-genomic actions on target genes. Mol Endocrinol 2005; 19:833-42; PMID:15695368; http://dx.doi.org/10.1210/me.2004-0486.
11. Ohshiro K, Schwartz AM, Levine PH, Kumar R. Alternate estrogen receptors promote invasion of inflammatory breast cancer cells via non-genomic sig-naling. PLoS One 2012; 7:e30725; PMID:22295107; http://dx.doi.org/10.1371/journal.pone.0030725.
12. Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, et al. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol 2010; 24:709-21; PMID:20197310; http://dx.doi.org/10.1210/me.2009-0317.
13. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estro-gen receptor mediates rapid cell signaling. Science 2005; 307:1625-30; PMID:15705806; http://dx.doi.org/10.1126/science.1106943.
14. Li Q, Zheng T, Holford TR, Boyle P, Zhang Y, Dai M. Light at night and breast cancer risk: results from a population-based case-control study in Connecticut, USA. Cancer Causes Control 2010; 21:2281-5; PMID:20927578; http://dx.doi.org/10.1007/s10552-010-9653-z.
15. Leonardi GC, Rapisarda V, Marconi A, Scalisi A, Catalano F, Proietti L, et al. Correlation of the risk of breast cancer and disruption of the circadian rhythm (Review). [Review]. Oncol Rep 2012; 28:418-28; PMID:22664950.
16. Menegaux F, Truong T, Anger A, Cordina-Duverger E, Lamkarkach F, Arveux P, et al. Night work and breast cancer: A population-based case-control study in France (the CECILE study). Int J Cancer 2012; PMID:22689255; http://dx.doi.org/10.1002/ijc.27669.
17. Hansen J. Risk of breast cancer after night- and shift work: current evidence and ongoing studies in Denmark. Cancer Causes Control 2006; 17:531-7; PMID:16596307; http://dx.doi.org/10.1007/s10552-005-9006-5.
18. Davis S, Mirick DK. Circadian disruption, shift work and the risk of cancer: a summary of the evi-dence and studies in Seattle. Cancer Causes Control 2006; 17:539-45; PMID:16596308; http://dx.doi.org/10.1007/s10552-005-9010-9.
19. Stevens RG. Light-at-night, circadian disruption and breast cancer: assessment of existing evidence. Int J Epidemiol 2009; 38:963-70; PMID:19380369; http://dx.doi.org/10.1093/ije/dyp178.
20. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 2010; 72:517-49; PMID:20148687; http://dx.doi.org/10.1146/annurev-physiol-021909-135821.
21. Rossetti S, Esposito J, Corlazzoli F, Gregorski A, Sacchi N. Entrainment of breast (cancer) epithelial cells detects distinct circadian oscillation patterns for clock and hormone receptor genes. Cell Cycle 2012; 11:350-60; PMID:22193044; http://dx.doi.org/10.4161/cc.11.2.18792.
22. Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene 2007; 26:7916-20; PMID:17599055; http://dx.doi.org/10.1038/sj.onc.1210585.
23. Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev 2010; 24:345-57; PMID:20159955; http://dx.doi.org/10.1101/gad.564110.
24. Stevens RG. Circadian disruption and breast can-cer: from melatonin to clock genes. Epidemiology 2005; 16:254-8; PMID:15703542; http://dx.doi.org/10.1097/01.ede.0000152525.21924.54.
25. Allred DC. The origins of oestrogen receptor negative breast cancer. Breast Cancer Res 2007; 9:S20; http://dx.doi.org/10.1186/bcr1818.
26. Elledge R, Allred DC. Clinical aspects of estrogen and progesterone receptors. In: Harris J, Lippman ME, Morrow M, Osborne CK, eds. Diseases of the Breast. Philadelphia, PA: Lippincott Williams & Wilkins, 2004:601-17.
27. Masood S. Estrogen and progesterone receptors in cytology: a comprehensive review. Diagn Cytopathol 1992; 8:475-91; PMID:1396026; http://dx.doi.org/10.1002/dc.2840080508.
28. Sims AH, Howell A, Howell SJ, Clarke RB. Origins of breast cancer subtypes and therapeutic implications. Nat Clin Pract Oncol 2007; 4:516-25; PMID:17728710; http://dx.doi.org/10.1038/ncponc0908.
29. Allred DC, Mohsin SK, Fuqua SA. Histological and bio-logical evolution of human premalignant breast disease. Endocr Relat Cancer 2001; 8:47-61; PMID:11350726; http://dx.doi.org/10.1677/erc.0.0080047.
30. Laganière J, Deblois G, Giguère V. Functional genom-ics identifies a mechanism for estrogen activation of the retinoic acid receptor alpha1 gene in breast cancer cells. Mol Endocrinol 2005; 19:1584-92; PMID:15831516; http://dx.doi.org/10.1210/me.2005-0040.
31. Balsalobre A. Clock genes in mammalian periph-eral tissues. Cell Tissue Res 2002; 309:193-9; PMID:12111549; http://dx.doi.org/10.1007/s00441-002-0585-0.
All in-text references underlined in blue are linked to publications on ResearchGate, letting you access and read them immediately.