human topoisomerase iiα and iiβ interact with the c-terminal region of p53
TRANSCRIPT

wItrc
s
Experimental Cell Research 255, 86–94 (2000)doi:10.1006/excr.1999.4772, available online at http://www.idealibrary.com on
Human Topoisomerase IIa and IIb Interactwith the C-Terminal Region of p53
Ian G. Cowell,*,1 Andrei L. Okorokov,† Sarah A. Cutts,† Kay Padget,* Margaret Bell,*Jo Milner,† and Caroline A. Austin*,2
*School of Biochemistry and Genetics, The Medical School, University of Newcastle, Newcastle upon Tyne NE2 4HH, United Kingdom;
and †YCR p53 Research Group, Department of Biology, University of York, York YO1 5DD, United KingdomThe p53 tumor suppressor protein is a critical regula-tor of cell cycle progression and apoptosis following ex-posure of cells to DNA damaging agents such as ionizingradiation or anticancer drugs. An important group ofanticancer drugs, including compounds such as etopo-side and doxorubicin (Adriamycin), interacts with DNAtopoisomerase II (topo II), causing the accumulation ofenzyme-DNA adducts that ultimately lead to double-strand breaks and cell death via apoptosis. Human topoIIb has previously been shown to interact with p53, and
e have extended this analysis to show that both topoIa and IIb interact with p53 in vivo and in vitro. Fur-hermore, we show that the regulatory C-terminal basicegion of p53 (residues 364–393) is necessary and suffi-ient for interaction with DNA topo II. © 2000 Academic Press
Key Words: topoisomerase; p53; DNA repair; tumoruppressor; DNA damage.
INTRODUCTION
Type II DNA topoisomerases (EC 5.99.1.3) are ATP-dependent enzymes that catalyse topological changesin DNA [1]. The enzymes are homodimers and act as aDNA gate, passing one duplex DNA segment through atransient, enzyme-bridged break in a second duplex.Human DNA topoisomerase II (topo II) is of consider-able clinical importance as it is the molecular target foranticancer drugs such as etoposide, doxorubicin(Adriamycin), and amsacrine which interrupt enzy-matic DNA breakage-reunion, leading to the formationof “cleavable complexes,” cytotoxic lesions that ulti-mately lead to cell death [1, 2]. In mammals, there aretwo separately encoded isoforms, termed topo IIa andIIb [3–10]. Both forms have been shown to mediatecytotoxicity [11]. Efficient execution of the apoptotic
1 Current address: Division of Integrative Biology, The Roslin In-stitute (Edinburgh), Roslin, Midlothian EH25 9PS.
2 To whom correspondence and reprint requests should be ad-dressed at School of Biochemistry and Genetics, The Medical School,University of Newcastle, Newcastle upon Tyne NE2 4HH. Fax: 0191
222 7424. E-mail: [email protected].860014-4827/00 $35.00Copyright © 2000 by Academic PressAll rights of reproduction in any form reserved.
pathway in response to DNA-damaging cytotoxicagents such as etoposide and doxorubicin (Adriamycin)has been shown to be dependent upon p53 [12, 13]. It isheld that p53 either directly or indirectly senses suchDNA damage, is activated, and sets into motion a se-ries of events leading either to G1 arrest or apoptosis[14–16]. The damage detector is exquisitely sensitive,as a lone double-strand break may be sufficient toactivate p53 [17]. The C-terminal 26 amino acid resi-due segment of p53 is implicated in recognition of dam-aged DNA [18] and subsequent activation of p53 inresponse to DNA damage. This domain possesses DNAreannealing activity, binds single-stranded DNA andDNA ends, and also negatively regulates the sequence-specific DNA-binding activity of the intact protein me-diated by the central domain (residues 102–292) [19,20].
Activated p53 is a sequence-specific transcriptionalactivator and a number of p53 target genes have beendiscovered that encode proteins which contribute tocell cycle arrest. These include p21WAF1 [21–23] whichinhibits several cyclin-CDK complexes and GADD45.In addition, a number of cellular proteins interact withp53 (see [15] for review). Most of these are connectedwith the activity of p53 as a transcriptional activator(TBP, TAFs,) or with DNA repair (ERCC2/XPD andERCC3/XPB components of TFIIH). Thus, p53 appearsto bring about G1 arrest by initiating a cell cycle arrestprogram. The mechanism by which p53 brings aboutapoptosis is less clear, although in some cells p53 in-duces expression of Bax, an apoptosis-promoting mem-ber of the BCL2 family [24]. A number of viral proteinsalso interact with p53 including SV40 large T antigenand BZLF1 an Epstein Barr virus (EBV) immediateearly protein [25].
Recently, it has been reported that DNA topoisom-erase I (topo I) is present in molecular complexes withp53 and that p53 stimulates the enzymatic activity oftopo I in vitro [26, 27]. This interaction involves theC-terminal region of p53 and it has been suggestedconsequently that topo I contributes to the p53-medi-
ated response to DNA damage. The involvement of
citwwwpN
DwC
c
t[
ao
s
(pTnpp
(rrm
a
p
p
p
s
87TOPOISOMERASE IIa AND b BIND p53
topoisomerases with p53 was extended by the observa-tion that expression of topo IIa is down-regulated byp53 [28, 29] and by the isolation of a cDNA cloneencoding the C-terminal domain of topo IIb in a cDNAlibrary screen to identify p53-interacting proteins [30].We report here that both isoforms of human topo IIinteract in vitro and in vivo with p53 and show that theC-terminal basic region of p53 (residues 364–393) isnecessary and sufficient for binding.
MATERIALS AND METHODS
Immunoprecipitation from human cell extracts. MCF7 cells hadpreviously been conditioned to DMEM and were therefore routinelycultured in DMEM containing 10% FBS and penicillin (50 U/ml)/streptomycin (50 mg/ml). Cells were grown to approximately 70%onfluence and doxorubicin (Adriamycin) was added to 1 mM wherendicated. Cells were returned to the incubator for 5 h before collec-ion. Cell extracts were prepared as follows. Plates were rinsed twiceith ice-cold phosphate-buffered saline (PBS) and the monolayersere scraped into a series of 1.5-ml microfuge tubes. Cells wereashed once with PBS before the cell pellet was resuspended in 3acked-cell vol (PCV) of buffer 1 (25 mM Hepes, pH 7.5, 600 mMaCl, 1 mM CaCl2, 1 mM MgCl2, 0.5% (v/v) Triton X-100, 1 mM
PMSF, 2 mM benzamidine, 50 mg/ml leupeptin, 2 mg/ml pepstatin).NAse I was added to 100 mg/ml and after 30 min on ice, the lysateas diluted with 6 PCV of buffer 2 (25 mM Hepes, pH 7.5, 1 mMaCl2, 1 mM MgCl2, 0.5% (v/v) Triton X-100, 1 mM PMSF, 2 mM
benzamidine, 50 mg/ml leupeptin, 2 mg/ml pepstatin). The lysate wasentrifuged at 13,000g and the cleared lysate (200–300 mg/reaction)
was used in the immunoprecipitation reactions indicated. Affinity-purified monoclonal antibodies DO-1 and 421 were obtained fromCalibiochem and were used at 0.2 mg per reaction. Polyclonal anti-opo II antisera 18511a and 18513b have been described elsewhere31] and were used at 3 ml per reaction. Polyclonal antisera IC03 and
IC08 employed as controls were raised against the transcriptionalrepressor E4BP4 [32] and were used at 3 ml per reaction. Afterntibody binding for 60 min on ice, immunocomplexes were collectedn protein A-agarose (20 ml per reaction of a 50%, v/v, suspension in
buffer 2). Immunocomplexes were washed in buffer 2 containing 200mM NaCl and captured proteins were eluted into 0.2% SDS, 1%b-mercaptoethanol, 25 mM Tris, pH 7.9, at 37°C for 5 min anddetected by Western blotting.
Plasmid constructs and in vitro translations. Plasmids pVA3 andTD1 encoding Gal4 DNA-binding domain-mouse p53 (residues 79–390) fusion protein and a Gal4 activation domain-SV40 large Tantigen (84–709) fusion protein, respectively, have been describedbefore [33, 34]. Plasmid pGAD-p53 encodes human p53 fused to theactivation domain of Gal4 and was constructed by ligating cDNAcontaining the entire human p53 coding sequence into the plasmidpGAD424 [35] downstream and in frame with the Gal4 activationdomain. Plasmid GBT-aCT was derived from pGBT9 [35]. A cDNAegment encoding the C-terminal domain of human topo IIa (resi-
dues 1244–1531) was ligated downstream of and in frame with theGal4 DNA-binding domain. Plasmid pGBT-bCT was constructed inthe same way for the C-terminal domain (residues 1268–1621) oftopo IIb.
Wild-type and variously mutated murine and human p53 weretranslated in vitro using a coupled transcription-translation systemPromega). In vitro translation (IVT) reactions were performed in theresence of [35S]methionine according to the suppliers instructions.he plasmids used and the proteins they encode (in brackets) areow listed: pKS 1 214 (wt human p53, hp53), pKS 1 235 (wt murine53, mp53), pKS239 (murine p53 DC, residues 1–323, mp53DC),
KS275 (murine p53 DN, mp53DN, residues 98–392), pGEM-Asp53alternatively spliced murine p53, mp53_AS), pBSSK 1 mCT9 (mu-ine p53, residues 319–392, mCT), pBSSK 1 hCT14 (human p53,esidues 319–393, hCT), pD30L2-8 (murine p53 residues 1–363,p53D30).Production of GST-topo II fusion proteins. Plasmids pGEX-aCT
nd pGEX-bCT were used to produce GST fusion proteins with theC-terminal domains of topo IIa and IIb, respectively. cDNA encodingresidues 1244–1531 (a) or 1268–1621 (b) were ligated into the mul-ticloning site of members of the pGEX family of plasmids (Pharma-cia). Escherichia coli strain BL21 was transformed with pGEX-aCT,
GEX-bCT, or pGEX-2TK and GST or fusion protein was purified asdescribed previously [32]. The resulting GSH-agarose beads werestored at 280°C until required.
In vitro binding assays. Where the products of more than one invitro translation reaction were to be compared in one experiment, anequal radioactive content of each translation product was added tothe individual binding reactions. The relative radioactive concentra-tion of the full-length protein band resulting from each IVT reactionwas assessed by SDS-PAGE and phosphorimager analysis of theresulting gels. In vitro binding reactions (GST pull-down reactions)were performed as described previously [32].
Yeast two-hybrid assays. Yeast strain HF7c [36] was transformedwith the appropriate binary combinations of pGBT9, pGBT-bCT,
GBT-aCT, pVA3 together with pTD1, pGAD-p53, and pGAD424 asdescribed previously [37]. Colonies appearing on selective mediumlacking trytophan and leucine (2TL plates) were gridded in tripli-cate onto a second 2TL plate and a plate also lacking histidine. Bothsets of plates were incubated at 30°C for 7 days.
Production of recombinant proteins. Human DNA topoisomeraseIIb was made in a yeast system and purified as in [38]. His-tagged
53 was synthesised using a bacculovirus system as in [39].Topoisomerase strand-passage assays. DNA strand-passage as-
ays were performed as described in [38].
RESULTS
Several lines of evidence suggest that the activitiesof p53 and DNA topoisomerases (type I and II) arelinked. For example, transcription from the humantopo IIa promoter is repressed by p53 in vivo and bothtopo I and the C-terminal domain of topo IIb are able tointeract with p53. We set out to investigate the biolog-ical significance of this last observation by analyzingthe interaction of wild-type and mutant forms of p53with human topo IIa and IIb.
Topo IIa and IIb Associate with p53 in Vivo inMammalian Cells
To determine whether p53 and topo II associate inmammalian cells, topo II was immunoprecipitatedfrom extracts of human MCF7 mammary carcinomacells and the resulting immunocomplexes were probedfor the presence of p53 (Fig. 1A). MCF7 cells werechosen as they contain wild-type p53 which is translo-cated to the nucleus from the cytoplasm upon DNAdamage. As expected, treatment of MCF7 cells withdoxorubicin (Adriamycin) dramatically increased thelevel of p53 found in whole cell extracts as assessed byWestern blotting with anti-p53 monoclonal antibody
DO-1 (Fig. 1A, tracks 1 and 2). Monoclonal antibodies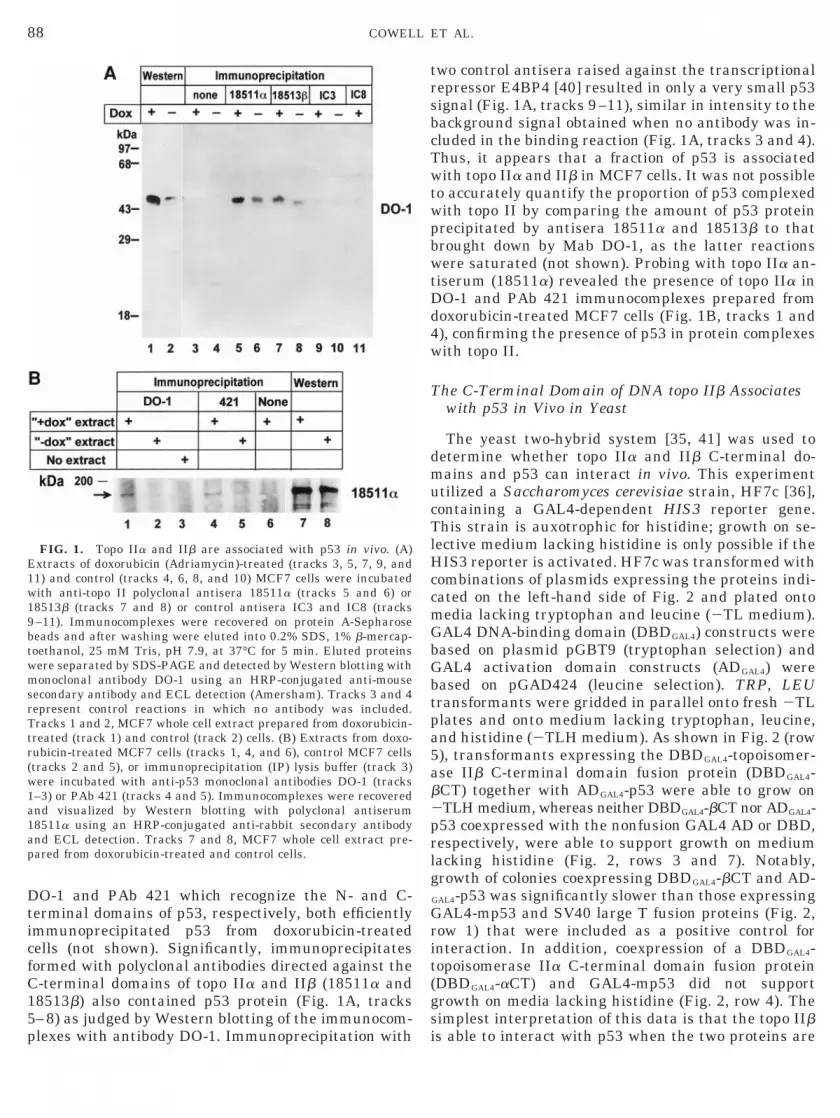
5p
G
pa5
prlg
19b
ap
88 COWELL ET AL.
DO-1 and PAb 421 which recognize the N- and C-terminal domains of p53, respectively, both efficientlyimmunoprecipitated p53 from doxorubicin-treatedcells (not shown). Significantly, immunoprecipitatesformed with polyclonal antibodies directed against theC-terminal domains of topo IIa and IIb (18511a and18513b) also contained p53 protein (Fig. 1A, tracks–8) as judged by Western blotting of the immunocom-
FIG. 1. Topo IIa and IIb are associated with p53 in vivo. (A)Extracts of doxorubicin (Adriamycin)-treated (tracks 3, 5, 7, 9, and11) and control (tracks 4, 6, 8, and 10) MCF7 cells were incubatedwith anti-topo II polyclonal antisera 18511a (tracks 5 and 6) or8513b (tracks 7 and 8) or control antisera IC3 and IC8 (tracks–11). Immunocomplexes were recovered on protein A-Sepharoseeads and after washing were eluted into 0.2% SDS, 1% b-mercap-
toethanol, 25 mM Tris, pH 7.9, at 37°C for 5 min. Eluted proteinswere separated by SDS-PAGE and detected by Western blotting withmonoclonal antibody DO-1 using an HRP-conjugated anti-mousesecondary antibody and ECL detection (Amersham). Tracks 3 and 4represent control reactions in which no antibody was included.Tracks 1 and 2, MCF7 whole cell extract prepared from doxorubicin-treated (track 1) and control (track 2) cells. (B) Extracts from doxo-rubicin-treated MCF7 cells (tracks 1, 4, and 6), control MCF7 cells(tracks 2 and 5), or immunoprecipitation (IP) lysis buffer (track 3)were incubated with anti-p53 monoclonal antibodies DO-1 (tracks1–3) or PAb 421 (tracks 4 and 5). Immunocomplexes were recoveredand visualized by Western blotting with polyclonal antiserum18511a using an HRP-conjugated anti-rabbit secondary antibodynd ECL detection. Tracks 7 and 8, MCF7 whole cell extract pre-ared from doxorubicin-treated and control cells.
lexes with antibody DO-1. Immunoprecipitation with
two control antisera raised against the transcriptionalrepressor E4BP4 [40] resulted in only a very small p53signal (Fig. 1A, tracks 9–11), similar in intensity to thebackground signal obtained when no antibody was in-cluded in the binding reaction (Fig. 1A, tracks 3 and 4).Thus, it appears that a fraction of p53 is associatedwith topo IIa and IIb in MCF7 cells. It was not possibleto accurately quantify the proportion of p53 complexedwith topo II by comparing the amount of p53 proteinprecipitated by antisera 18511a and 18513b to thatbrought down by Mab DO-1, as the latter reactionswere saturated (not shown). Probing with topo IIa an-tiserum (18511a) revealed the presence of topo IIa inDO-1 and PAb 421 immunocomplexes prepared fromdoxorubicin-treated MCF7 cells (Fig. 1B, tracks 1 and4), confirming the presence of p53 in protein complexeswith topo II.
The C-Terminal Domain of DNA topo IIb Associateswith p53 in Vivo in Yeast
The yeast two-hybrid system [35, 41] was used todetermine whether topo IIa and IIb C-terminal do-mains and p53 can interact in vivo. This experimentutilized a Saccharomyces cerevisiae strain, HF7c [36],containing a GAL4-dependent HIS3 reporter gene.This strain is auxotrophic for histidine; growth on se-lective medium lacking histidine is only possible if theHIS3 reporter is activated. HF7c was transformed withcombinations of plasmids expressing the proteins indi-cated on the left-hand side of Fig. 2 and plated ontomedia lacking tryptophan and leucine (2TL medium).
AL4 DNA-binding domain (DBDGAL4) constructs werebased on plasmid pGBT9 (tryptophan selection) andGAL4 activation domain constructs (ADGAL4) werebased on pGAD424 (leucine selection). TRP, LEUtransformants were gridded in parallel onto fresh 2TL
lates and onto medium lacking tryptophan, leucine,nd histidine (2TLH medium). As shown in Fig. 2 (row), transformants expressing the DBDGAL4-topoisomer-
ase IIb C-terminal domain fusion protein (DBDGAL4-bCT) together with ADGAL4-p53 were able to grow on2TLH medium, whereas neither DBDGAL4-bCT nor ADGAL4-
53 coexpressed with the nonfusion GAL4 AD or DBD,espectively, were able to support growth on mediumacking histidine (Fig. 2, rows 3 and 7). Notably,rowth of colonies coexpressing DBDGAL4-bCT and AD-
GAL4-p53 was significantly slower than those expressingGAL4-mp53 and SV40 large T fusion proteins (Fig. 2,row 1) that were included as a positive control forinteraction. In addition, coexpression of a DBDGAL4-topoisomerase IIa C-terminal domain fusion protein(DBDGAL4-aCT) and GAL4-mp53 did not supportgrowth on media lacking histidine (Fig. 2, row 4). Thesimplest interpretation of this data is that the topo IIb
is able to interact with p53 when the two proteins are
W
s(
89TOPOISOMERASE IIa AND b BIND p53
coexpressed in yeast, but that this interaction is lessefficient than that between the SV40 large T antigenand p53. It was unexpected for the DBDGAL4-aCT fusionprotein to fail to activate HIS3 expression in the pres-ence of ADGAL4-p53 since both isoforms interact withp53 in mammalian cells (see above). Possible reasonsfor this discrepancy include differential phosphoryla-tion of topoisomerase II or p53 in yeast and mamma-lian cells or sequestration or instability of the alphabut not the beta fusion protein. Alternatively a regionor regions other than or in addition to the C-terminaldomain of topoisomerase IIa may be required for inter-action with p53 in vivo (however, see below).
ild-Type p53 and DNA-Binding Domain MutantsBind DNA topo IIa and IIb in Vitro
The sequence-specific DNA-binding activity of thep53 protein resides in the central core domain (resi-dues 100–300, see Fig. 3). Since most p53 mutationsfound in human tumors occur in this region we inves-tigated the requirement for an intact central core do-main for binding to topo II in vitro. The GST pull-downassay was used to assess binding of WT p53 and mu-tants M237R, M237I, and R273C to the C-terminaldomains of human topo IIa and IIb. These experimentsand the following ones employed two GST fusion pro-teins, GST-aCT and GST-bCT containing the C-termi-nal domains of human topo IIa (residues 1244–1531)
FIG. 2. Human p53 and topo II interact in yeast. HF7c transfor-mants harboring the indicated plasmids were gridded in an identicalpattern onto selective medium plates lacking tryptophan and leucine(2TL) or tryptophan, leucine, and histidine (2TLH). The plates wereincubated at 30°C for 8 days. GAL4 DNA-binding domain (DBD)fusion constructs were derived from the plasmid pGBT9 and GAL4activation domain (AD) plasmids were derived from plasmidpGAD424.
and IIb (residues 1258–1621), respectively. As shown
in Fig. 4, little or no p53 was retained by glutathione-agarose beads loaded with GST alone, but beads loadedwith GST-aCT or GST-bCT fusion proteins efficientlyretained wild-type and each of the mutant p53 pro-teins. Approximately 15% of the input WT p53 proteinwas retained by the fusion protein-loaded beads inthese experiments and no consistent difference be-tween the wild-type and the mutant proteins was ob-served in this respect. While mutations M237R, M237I,and R273C all result in DNA-binding deficiency, mu-tation at residue 237 (in loop 3 of the DNA-bindingdomain structure) disrupts the three-dimensional fold-ing of the central domain [42]. Residue R273 is directlyinvolved in DNA binding and contacts DNA in themajor groove and mutation at this point does not affectthe overall structure of the core domain. Thus, equallyefficient binding of WT, M237R, M237I, and R273Cimplies that a correctly folded core domain is not re-quired for binding to topo IIa and IIb.
Notably, the C-terminal domains of topo IIa and IIbappeared to bind p53 with similar affinity. Two lines ofevidence support this. First, a similar proportion of theinput p53 protein was always retained on GST-aCTand GST-bCT-loaded glutathione agarose beads (com-pare Fig. 4, tracks 9 and 13, or Fig. 5A, tracks 6 and 7,9 and 10, or 15 and 16). Secondly, the free topo IIbC-terminal domain competed equally well with immo-bilized GST-aCT and GST-bCT for binding to in vitrotranslated p53 (data not shown).
The C-Terminal Domain of p53 Is Necessary andSufficient for Binding to topo IIa or IIb
Next we tested a series of deletion mutants derivedfrom mouse p53 for binding in vitro to topo II. Ashown in Fig. 5A, both human (hp53) and mouse p53mp53) bound to immobilized human topo IIa and IIb
C-terminal domains (compare tracks 6 and 7 with 9
FIG. 3. p53 primary structure and deletion mutants. The toppart of the figure shows the organization of p53, indicating regionsresponsible for transcriptional activation (A), sequence-specific DNAbinding (D), tetramerization (T), and sequence nonspecific DNAbinding (B). The numbers above correspond to amino acid sequence.The lower part of the figure shows a series on N- and C-terminal
deletion mutants that was used in this study.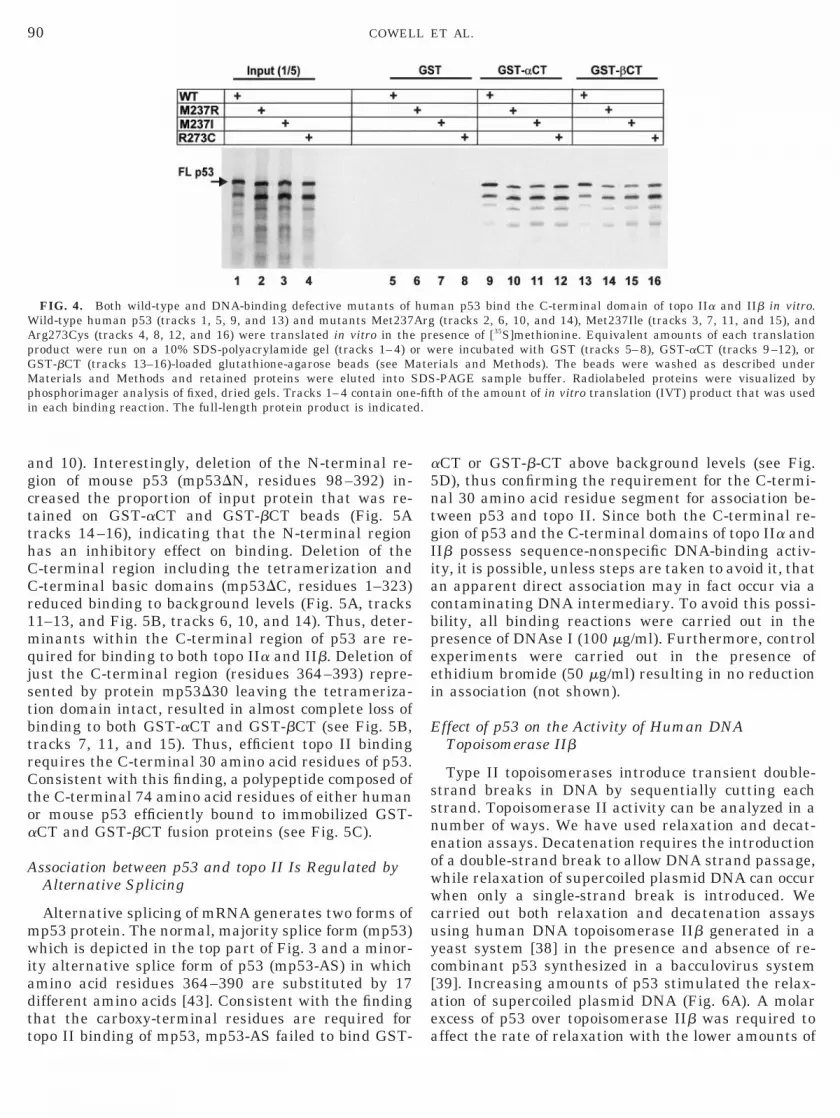
js
WApG
d.
90 COWELL ET AL.
and 10). Interestingly, deletion of the N-terminal re-gion of mouse p53 (mp53DN, residues 98–392) in-creased the proportion of input protein that was re-tained on GST-aCT and GST-bCT beads (Fig. 5Atracks 14–16), indicating that the N-terminal regionhas an inhibitory effect on binding. Deletion of theC-terminal region including the tetramerization andC-terminal basic domains (mp53DC, residues 1–323)reduced binding to background levels (Fig. 5A, tracks11–13, and Fig. 5B, tracks 6, 10, and 14). Thus, deter-minants within the C-terminal region of p53 are re-quired for binding to both topo IIa and IIb. Deletion ofust the C-terminal region (residues 364–393) repre-ented by protein mp53D30 leaving the tetrameriza-
tion domain intact, resulted in almost complete loss ofbinding to both GST-aCT and GST-bCT (see Fig. 5B,tracks 7, 11, and 15). Thus, efficient topo II bindingrequires the C-terminal 30 amino acid residues of p53.Consistent with this finding, a polypeptide composed ofthe C-terminal 74 amino acid residues of either humanor mouse p53 efficiently bound to immobilized GST-aCT and GST-bCT fusion proteins (see Fig. 5C).
Association between p53 and topo II Is Regulated byAlternative Splicing
Alternative splicing of mRNA generates two forms ofmp53 protein. The normal, majority splice form (mp53)which is depicted in the top part of Fig. 3 and a minor-ity alternative splice form of p53 (mp53-AS) in whichamino acid residues 364–390 are substituted by 17different amino acids [43]. Consistent with the findingthat the carboxy-terminal residues are required for
FIG. 4. Both wild-type and DNA-binding defective mutants ofild-type human p53 (tracks 1, 5, 9, and 13) and mutants Met237rg273Cys (tracks 4, 8, 12, and 16) were translated in vitro in theroduct were run on a 10% SDS-polyacrylamide gel (tracks 1–4) oST-bCT (tracks 13–16)-loaded glutathione-agarose beads (see M
Materials and Methods and retained proteins were eluted into Sphosphorimager analysis of fixed, dried gels. Tracks 1–4 contain onein each binding reaction. The full-length protein product is indicate
topo II binding of mp53, mp53-AS failed to bind GST-
aCT or GST-b-CT above background levels (see Fig.5D), thus confirming the requirement for the C-termi-nal 30 amino acid residue segment for association be-tween p53 and topo II. Since both the C-terminal re-gion of p53 and the C-terminal domains of topo IIa andIIb possess sequence-nonspecific DNA-binding activ-ity, it is possible, unless steps are taken to avoid it, thatan apparent direct association may in fact occur via acontaminating DNA intermediary. To avoid this possi-bility, all binding reactions were carried out in thepresence of DNAse I (100 mg/ml). Furthermore, controlexperiments were carried out in the presence ofethidium bromide (50 mg/ml) resulting in no reductionin association (not shown).
Effect of p53 on the Activity of Human DNATopoisomerase IIb
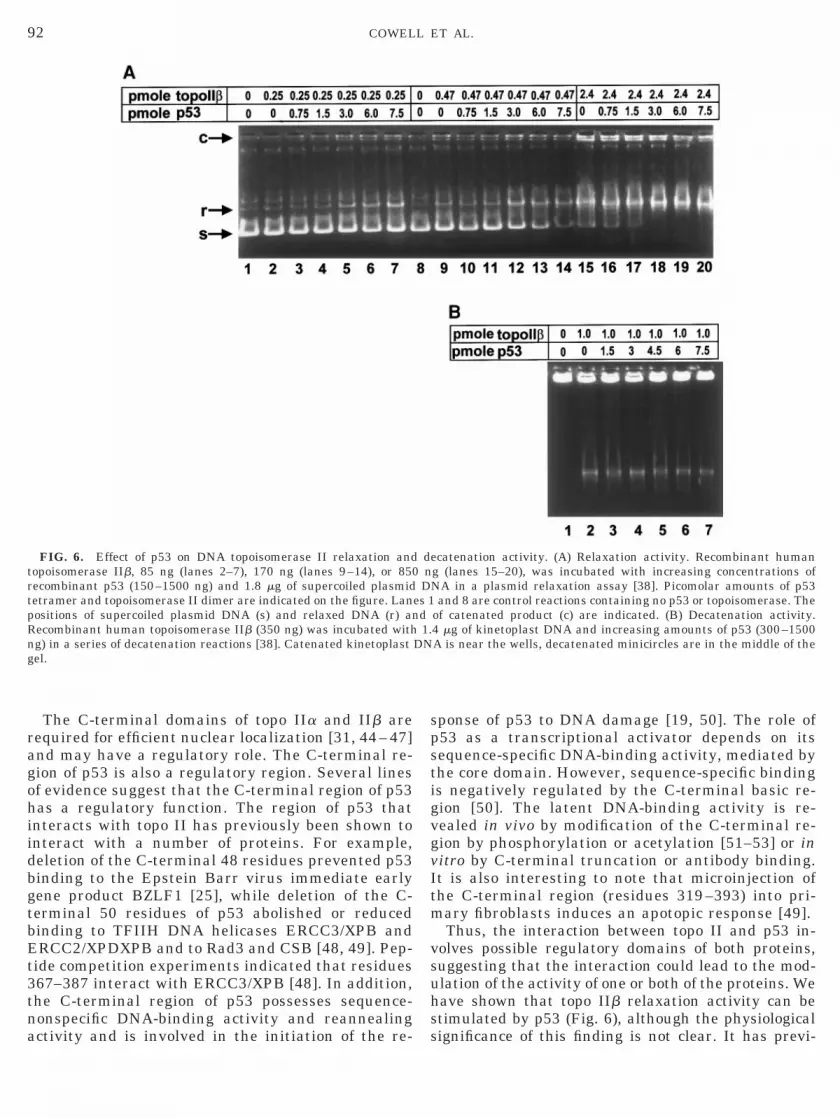
Type II topoisomerases introduce transient double-strand breaks in DNA by sequentially cutting eachstrand. Topoisomerase II activity can be analyzed in anumber of ways. We have used relaxation and decat-enation assays. Decatenation requires the introductionof a double-strand break to allow DNA strand passage,while relaxation of supercoiled plasmid DNA can occurwhen only a single-strand break is introduced. Wecarried out both relaxation and decatenation assaysusing human DNA topoisomerase IIb generated in ayeast system [38] in the presence and absence of re-combinant p53 synthesized in a bacculovirus system[39]. Increasing amounts of p53 stimulated the relax-ation of supercoiled plasmid DNA (Fig. 6A). A molarexcess of p53 over topoisomerase IIb was required to
an p53 bind the C-terminal domain of topo IIa and IIb in vitro.(tracks 2, 6, 10, and 14), Met237Ile (tracks 3, 7, 11, and 15), and
esence of [35S]methionine. Equivalent amounts of each translationere incubated with GST (tracks 5–8), GST-aCT (tracks 9–12), orrials and Methods). The beads were washed as described under-PAGE sample buffer. Radiolabeled proteins were visualized by
th of the amount of in vitro translation (IVT) product that was used
humArgpr
r wateDS-fif
affect the rate of relaxation with the lower amounts of

npawr1w
G
91TOPOISOMERASE IIa AND b BIND p53
topoisomerase IIb (Fig. 6A, lanes 1–7 and 8–14). How-ever, p53 did not stimulate the decatenation of kineto-plast DNA (kDNA) (Fig. 6B).
DISCUSSION
We have confirmed the observations of Yuwen etal. [30], that topo IIb interacts with p53 and we havesignificantly extended these observations. We haveshown that both the alpha and beta isoforms of hu-
FIG. 5. Both human and mouse p53 bind to the C-terminal doecessary and sufficient for binding in vitro. (A) Full-length mouse p553 (mp53DC, residues 1–323), and N-terminally truncated p53 (mp5mounts of each protein were incubated with GST, GST-aCT, or GSashed as described under Materials and Methods. Bound materia
adiolabeled proteins were visualized and quantified by phosphorima–4) allowed the determination of proportion of each input protein reith GST, GST-aCT, or GST-bCT-loaded glutathione-agarose beads
were loaded directly to tracks 1–4. (C) The C-terminal segments of huwere translated in vitro. Equivalent amounts of each protein were
ST-bCT-loaded glutathione-agarose beads (tracks 5 and 6). Boundanalysis. (D) Equivalent amounts of in vitro translated mouse p53SDS-polyacrylamide gel (tracks 1 and 2) or were incubated with GSTAfter washing, bound material was eluted into SDS-PAGE sample bby phosphorimager analysis.
man topo II associate with both murine and human
p53 in vitro using GST pull downs, demonstratingthat the interaction is evolutionarily conserved. Invivo interactions of both the alpha and beta isoformsof human topo II have been demonstrated using im-munoprecipitations from nontransfected cell lines.The interaction with human DNA topo IIb has alsobeen confirmed using the yeast two-hybrid assaysystem. The interactions involve the topo II C-termi-nal domains and the carboxyl-terminal basic regionof p53, we have mapped the p53 interaction domain
in of topo IIa and topo IIb. The C-terminal basic region of p53 ismp53), full-length human p53 (hp53), C-terminally truncated mouse
, residues 98–393) were translated in vitro (tracks 1–4). EquivalentCT-loaded glutathione agarose beads (tracks 5–16). The beads wereas eluted into SDS-PAGE sample buffer and after electrophoresisanalysis. Comparison with directly loaded translated protein (lanesed. (B) Wild-type and truncated mouse p53 proteins were incubated
acks 5–16). Bound proteins were analyzed as in (A). Input proteinsn (hCT, residues 319–393) and mouse (mCT, residues 319–390) p53ubated with GST (tracks 1 and 2), GST-aCT (tracks 3 and 4), andoteins were visualized by SDS-PAGE followed by phosphorimageralternatively spliced p53 (mp53-AS) were loaded directly onto anT-aCT, or GST-bCT-loaded glutathione-agarose beads (tracks 3–8).
er. Following electrophoresis, radiolabeled proteins were visualized
ma3 (3DNT-bl wgertain(trmainc
pror
, GSuff
to residues 364 –393.
vItm
vsuh
92 COWELL ET AL.
The C-terminal domains of topo IIa and IIb arerequired for efficient nuclear localization [31, 44 – 47]and may have a regulatory role. The C-terminal re-gion of p53 is also a regulatory region. Several linesof evidence suggest that the C-terminal region of p53has a regulatory function. The region of p53 thatinteracts with topo II has previously been shown tointeract with a number of proteins. For example,deletion of the C-terminal 48 residues prevented p53binding to the Epstein Barr virus immediate earlygene product BZLF1 [25], while deletion of the C-terminal 50 residues of p53 abolished or reducedbinding to TFIIH DNA helicases ERCC3/XPB andERCC2/XPDXPB and to Rad3 and CSB [48, 49]. Pep-tide competition experiments indicated that residues367–387 interact with ERCC3/XPB [48]. In addition,the C-terminal region of p53 possesses sequence-nonspecific DNA-binding activity and reannealing
FIG. 6. Effect of p53 on DNA topoisomerase II relaxation andtopoisomerase IIb, 85 ng (lanes 2–7), 170 ng (lanes 9–14), or 850recombinant p53 (150–1500 ng) and 1.8 mg of supercoiled plasmidtetramer and topoisomerase II dimer are indicated on the figure. Lanpositions of supercoiled plasmid DNA (s) and relaxed DNA (r) anRecombinant human topoisomerase IIb (350 ng) was incubated withng) in a series of decatenation reactions [38]. Catenated kinetoplastgel.
activity and is involved in the initiation of the re-
sponse of p53 to DNA damage [19, 50]. The role ofp53 as a transcriptional activator depends on itssequence-specific DNA-binding activity, mediated bythe core domain. However, sequence-specific bindingis negatively regulated by the C-terminal basic re-gion [50]. The latent DNA-binding activity is re-vealed in vivo by modification of the C-terminal re-gion by phosphorylation or acetylation [51–53] or initro by C-terminal truncation or antibody binding.t is also interesting to note that microinjection ofhe C-terminal region (residues 319 –393) into pri-ary fibroblasts induces an apotopic response [49].Thus, the interaction between topo II and p53 in-
olves possible regulatory domains of both proteins,uggesting that the interaction could lead to the mod-lation of the activity of one or both of the proteins. Weave shown that topo IIb relaxation activity can be
stimulated by p53 (Fig. 6), although the physiological
ecatenation activity. (A) Relaxation activity. Recombinant humang (lanes 15–20), was incubated with increasing concentrations ofNA in a plasmid relaxation assay [38]. Picomolar amounts of p53
and 8 are control reactions containing no p53 or topoisomerase. Theof catenated product (c) are indicated. (B) Decatenation activity.4 mg of kinetoplast DNA and increasing amounts of p53 (300–1500A is near the wells, decatenated minicircles are in the middle of the
dnD
es 1d1.
DN
significance of this finding is not clear. It has previ-

93TOPOISOMERASE IIa AND b BIND p53
ously been shown that p53 could stimulate the relax-ation activity of DNA topo I [26, 27, 54]. However, inboth cases a molar excess of p53 was required. DNAtopoisomerase I also interacts with the C-terminal re-gion of p53; deletion of the C-terminal 92 amino acids(299–390) abolished this interaction [27]. More re-cently the region of interaction has been narroweddown to p53 residues 302–321 [54], which is not thesame region of p53 that interacts with topo II, p53residues 364–393. Efforts to determine whether topo IIcould modulate the sequence-specific DNA-binding ac-tivity of p53 were inconclusive due the difficulty inidentifying gel retardation conditions appropriate forboth p53 and topo II.
It is plausible that an interaction between topo IIand p53 is necessary for efficient detection of topoII-mediated DNA strand breaks and their subsequentrepair or activation of the apoptotic response of the cell.Even in the absence of overt antitumor drugs, topo IImust generate a background level of strand breaks.Furthermore, xenobiotics that are topo poisons arepart of the natural environment [55]. Thus, the inter-action of p53 with topo II may represent a surveillancemechanism for topo II-mediated genomic damage.
We thank the NECRC, YCRC, and CRC for financial support.Thanks to Lorna J. Warnock for providing bacculovirus-infected cellsfrom which to purify p53.
REFERENCES
1. Wang, J. C. (1996). DNA topoisomerases. Annu. Rev. Biochem.65, 635–692.
2. Caron, P. R., and Wang, J. C. (1993). In “Molecular Biology ofDNA Topoisomerases” (T. Andoh, H. Ikeda, and M. Oguro,Eds.), pp. 243–263, CRC Press, Boca Raton, FL.
3. Drake, F. H., Hofmann, G. A., Bartus, H. F., Mattern, M. R.,Crooke, S. T., and Mirabelli, C. K. (1989). Biochemical andpharmacological properties of p170 and p180 forms of topoisom-erase II. Biochemistry (USA) 28, 8154–8160.
4. Drake, F. H., Zimmerman, J. P., McCabe, F. L., Bartus, H. F.,Per, S. R., Sullivan, D. M., Ross, W. E., Mattern, M. R., Johnson,R. K., and Crooke, S. T. (1987). Purification of topoisomerase IIfrom amsacrine-resistant P388 leukemia cells. Evidence for twoforms of the enzyme. J. Biol. Chem. 262, 16739–16747.
5. Chung, T. D. Y., Drake, F. H., Tan, K. B., Per, S. R., Crooke,S. T., and Mirabelli, C. K. (1989). Characterization and immu-nological identification of cDNA clones encoding two humanDNA topoisomerase II isoenzymes. Proc. Natl. Acad. Sci. USA86, 9431–9435.
6. Tsai Pflugfelder, M., Liu, L. F., Liu, A. A., Tewey, K. M., WhangPeng, J., Knutsen, T., Huebner, K., Croce, C. M., and Wang,J. C. (1988). Cloning and sequencing of cDNA encoding humanDNA topoisomerase II and localization of the gene to chromo-some region 17q21–22. Proc. Natl. Acad. Sci. USA 85, 7177–7181.
7. Austin, C. A., and Fisher, L. M. (1990). Isolation and charac-terization of a human cDNA clone encoding a novel DNA topo-isomerase II homologue from HeLa cells. FEBS Lett. 266, 115–
117.8. Austin, C. A., Sng, J.-H., Patel, S., and Fisher, L. M. (1993).Novel HeLa topoisomerase II is the IIb isoform: Complete cod-ing sequence and homology with other type II topoisomerases.Biochim. Biophys. Acta 1172, 283–291.
9. Davies, S. L., Jenkins, J. R., and Hickson, I. D. (1993). Humancells express two differentially spliced forms of topoisomeraseIIb mRNA. Nucleic Acids Res. 21, 3719–3723.
10. Jenkins, J. R., Ayton, P., Jones, T., Davies, S. L., Simmons,D. L., Harris, A. L., Sheer, D., and Hickson, I. D. (1992). Isola-tion of cDNA clones encoding the beta isozyme of human DNAtopoisomerase II and localisation of the gene to chromosome3p24. Nucleic Acids Res. 20, 5587–5592.
11. Willmore, E., Frank, A. J., Padget, K., Tilby, M. J., and Austin,C. A. (1998). Etoposide targets topoisomerase IIa and IIb inleukemic cells: Isoform-specific cleavable complexes visualizedand quantified in situ by a novel immunofluorescence tech-nique. Mol. Pharmacol. 54, 78–85.
12. Lowe, S. W., Ruley, H. E., Jacks, T., and Housman, D. E. (1993).p53-dependent apoptosis modulates the cytotoxicity of antican-cer agents. Cell 74, 957–967.
13. Lowe, S. W., Bodis, S., McClatchey, A., Remington, L., Ruley,H. E., Fisher, D. E., Housman, D. E., and Jacks, T. (1994). p53status and the efficacy of cancer therapy in vivo. Science 266,807–810.
14. Ko, L. J., and Prives, C. (1996). p53: Puzzle and paradigm.Genes Dev. 10, 1054–1072.
15. Levine, A. J. (1997). p53, the cellular gatekeeper for growth anddivision. Cell 88, 323–331.
16. Okorokov, A. L., and Milner, J. (1997). Proteolytic cleavage ofp53: A model for the activation of p53 in response to DNAdamage. Oncol. Res. 9, 267–273.
17. Huang, L. C., Clarkin, K. C., and Wahl, G. M. (1996). Sensitiv-ity and selectivity of the DNA damage sensor responsible foractivating p53-dependent G(1) arrest. Proc. Natl. Acad. Sci.USA 93, 4827–4832.
18. Lee, S., Elenbaas, B., Levine, A., and Griffith, J. (1995). p53 andits 14 kDa C-terminal domain recognise primary DNA-damagein the form of insertion-deletion mismatches. Cell 81, 1013–1020.
19. Wu, L., Bayle, J. H., Elenbaas, B., Pavletich, N. P., and Levine,A. J. (1995). Alternatively spliced forms in the carboxy-terminaldomain of p53 protein regulate its ability to promote annealingof complementary single strands of nucleic-acids. Mol. Cell.Biol. 15, 497–504.
20. Hupp, T. R., Sparks, A., and Lane, D. P. (1995). Small peptidesactivate the latent sequence-specific DNA binding function ofp53. Cell 83, 237–245.
21. Li, Y., Jenkins, C. W., Nichols, M. A., and Xiong, Y. (1994). Cellcycle expression and p53 regulation of the cyclin-dependentkinase inhibitor p21. Oncogene 9, 2261–2268.
22. El-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B.,Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W.,and Vogelstein, B. (1993). WAF1, a potential mediator of p53tumor suppression. Cell 75, 817–825.
23. Di Leonardo, A., Linke, S. P., Clarkin, K., and Wahl, G. M.(1994). DNA damage triggers a prolonged p53-dependent G1arrest and long-term induction of Cip1 in normal human fibro-blasts. Genes Dev. 8, 2540–2551.
24. Miyashita, T., and Reed, J. C. (1995). Tumor suppressor p53 isa direct transcriptional activator of the human bax gene. Cell80, 293–299.
25. Zhang, Q., Gutsch, D., and Kenney, S. (1994). Functional andphysical interaction between p53 and BZLF1: Implications for
Epstein-Barr virus latency. Mol. Cell. Biol. 14, 1929–1938.
2
2
2
3
3
3
3
3
3
3
3
4
4
94 COWELL ET AL.
26. Gobert, C., Bracco, L., Rossi, F., Olivier, M., Tazi, J., Lavelle, F.,Larsen, A. K., and Riou, J. F. (1996). Modulation of DNA topo-isomerase I activity by p53. Biochemistry 35, 5778–5786.
7. Albor, A., Kaku, S., and KuleszMartin, M. (1998). Wild-typeand mutant forms of p53 activate human topoisomerase. I. Apossible mechanism for gain of function in mutants. CancerRes. 58, 2091–2094.
8. Wang, Q., Zambetti, G. P., and Suttle, D. P. (1997). Inhibition ofDNA topoisomerase IIa gene expression by the p53 tumor sup-pressor. Mol. Cell. Biol. 17, 389–397.
9. Sandri, M. I., Isaacs, R. J., Ongkeko, W. M., Harris, A. L.,Hickson, I. D., Broggini, M., and Vikhanskaya, F. (1996). p53regulates the minimal promoter of the human topoisomeraseIIa gene. Nucleic Acids Res. 24, 4464–4470.
0. Yuwen, H., Hsia, C. C., Nakashima, Y., Evangelista, A., andTabor, E. (1997). Binding of wild-type p53 by topoisomerase IIand overexpression of topoisomerase II in human hepatocellu-lar carcinoma. Biochem. Biophys. Res. Commun. 234, 194–197.
31. Cowell, I. G., Willmore, E., Chalton, D., Marsh, K. L., Jazrawi,E., Fisher, L. M., and Austin, C. A. (1998). Nuclear distributionof DNA topoisomerase IIb: A potent nuclear targeting signalresides in the C-terminal 116 amino acids. Exp. Cell Res. 243,232–240.
2. Cowell, I. G., and Hurst, H. C. (1996). Protein-protein interac-tion between the transcriptional repressor E4BP4 and the TBP-binding protein Dr1. Nucleic Acids Res. 24, 3607–3613.
3. Iwabuchi, K., Li, B., Bartel, P., and Fields, S. (1993). Use of thetwo-hybrid system to identify the domain of p53 involved inoligomerization. Oncogene 8, 1693–1696.
4. Li, B., and Fields, S. (1993). Identification of mutations in p53that affect its binding to SV40 large T antigen by using theyeast two-hybrid system. FASEB J. 7, 957–963.
5. Bartel, P. L., Chien, C. T., Sternglanz, R., and Fields, S. (1993)In “Cellular Interactions in Development: A Practical Ap-proach” (D. A. Hartley, Ed.), pp. 153–179, Oxford Univ. Press,Oxford, UK.
6. Feilotter, H. E., Hannon, G. J., Ruddel, C. J., and Beach, D.(1994). Construction of an improved host strain for two hybrid-screening. Nucleic Acids Res. 22, 1502–1503.
7. Cowell, I. G. (1996) In “Methods in Molecular Biology: cDNALibrary Protocols” (I. G. Cowell, and C. A. Austin, Eds.), Hu-mana Press, Totowa.
8. Austin, C. A., Marsh, K. L., Wasserman, R. A., Willmore, E.,Sayer, P. J., Wang, J. C., and Fisher, L. M. (1995). Expression,domain structure, and enzymatic properties of an active recom-binant human DNA topoisomerase IIb. J. Biol. Chem. 270,15739–15746.
39. Molinari, M., Okorokov, A. L., and Milner, J. (1996). Interactionwith damaged DNA induces selective proteolytic cleavage ofp53 to yield 40 kDa and 35 kDa fragments competent for se-quence-specific DNA-binding. Oncogene 13, 2077–2086.
40. Cowell, I. G., Skinner, A., and Hurst, H. C. (1992). Transcrip-tional repression by a novel member of the bzip family of tran-scription factors. Mol. Cell. Biol. 12, 3070–3077.
41. Fields, S., and Song, O. (1989). A novel genetic system to detectprotein-protein interactions. Nature 340, 245–246.
42. Cho, Y., Gorina, S., Jeffrey, P. D., and Pavletich, N. P. (1994).
Crystal structure of a p53 tumor suppressor-DNA complex:Understanding tumorigenic mutations. Science 265, 346 –355.
43. Arai, N., Nomura, D., Yokota, K., Wolf, D., Brill, E., Shohat,O., and Rotter, V. (1986). Immunologically distinct p53 mol-ecules generated by alternative splicing. Mol. Cell. Biol. 6,3232–3239.
44. Adachi, N., Miyaike, M., Kato, S., Kanamaru, R., Koyama, H.,and Kikuchi, A. (1997). Cellular distribution of mammalianDNA topoisomerase II is determined by its catalytically dis-pensable C-terminal domain. Nucleic Acids Res. 25, 3135–3142.
45. Mirski, S. E., Gerlach, J. H., Cummings, H. J., Zirngibl, R.,Greer, P. A., and Cole, S. P. (1997). Bipartite nuclear localiza-tion signals in the C terminus of human topoisomerase IIa.Exp. Cell Res. 237, 452–455.
6. Shiozaki, K., and Yanagida, M. (1992). Functional dissection ofthe phosphorylated termini of fission yeast DNA topoisomeraseII. J. Cell Biol. 119, 1023–1036.
47. Wessel, I., Jensen, P. B., Falck, J., Mirski, S. E. L., Cole,S. P. C., and Sehested, M. (1997). Loss of amino acids(1490)Lys-Ser-Lys(1492) in the COOH-terminal region of topo-isomerase IIa in human small cell lung cancer cells selected forresistance to etoposide results in an extranuclear enzyme local-ization. Cancer Res. 57, 4451–4454.
8. Wang, X. W., Yeh, H., Schaeffer, L., Roy, R., Moncollin, V., Egly,J. M., Wang, Z., Friedberg, E. C., Evans, M. K., Taffe, B. G.,Bohr, V. A., Weeda, G., Hoeijmakers, J. H. J., Forrester, K., andHarris, C. C. (1995). p53 modulation of TFIIH-associated nu-cleotide excision repair activity. Nature Genet. 10, 188–195.
49. Wang, X. W., Vermeulen, W., Coursen, J. D., Gibson, M.,Lupold, S. E., Forrester, K., Xu, G., Elmore, L., Yeh, H., Hoeij-makers, J. H., and Harris, C. C. (1996). The XPB and XPD DNAhelicases are components of the p53-mediated apoptosis path-way. Genes Dev. 10, 1219–1232.
50. Bayle, J. H., Elenbaas, B., and Levine, A. J. (1995). The car-boxy-terminal domain of p53 regulates sequence-specific DNA-binding through its nonspecific nucleic acid binding activity.Proc. Natl. Acad. Sci. USA 92, 5729–5733.
51. Hoffmann, R., Craik, D. J., Pierens, G., Bolger, R. E., and Otvos,L. (1998). Phosphorylation of the C-terminal sites of human p53reduces non-sequence-specific DNA binding as modeled withsynthetic peptides. Biochemistry 37, 13755–13764.
52. Chiarugi, V., Cinelli, M., and Magnelli, L. (1998). Acetylationand phosphorylation of the carboxy-terminal domain of p53:Regulative significance. Oncol. Res. 10, 55–57.
53. Sakaguchi, K., Herrera, J. E., Saito, S., Miki, T., Bustin, M.,Vassilev, A., Anderson, C. W., and Appella, E. (1998). DNAdamage activates p53 through a phosphorylation-acetylationcascade. Genes Dev. 12, 2831–2841.
54. Gobert, C., Skladanowski, A., and Larsen, A. K. (1999). Theinteraction between p53 and DNA topoisomerase I is regulateddifferently in cells with wild-type and mutant p53. Proc. Natl.Acad. Sci. USA 96, 10355–10360.
55. Austin, C. A., Patel, S., Ono, K., Nakane, N., and Fisher, L. M.(1992). Baicalein and related cytotoxic flavonoids unwind DNAand are novel inducers of topoisomerase II mediated DNA
cleavage. Biochem. J. 282, 883–889.Received August 3, 1999Revised version received November 8, 1999