hidroquimica del karst completo
TRANSCRIPT

HIDROQUÍMICA DEL KARST
Parte 1
QUÍMICA DEL AGUA KÁRSTICA
J.R. FAGUNDO CASTILLO Centro Nacional de Investigaciones Científicas
Ministerio de Educación Superior La Habana, Cuba
Parte 2 HIDROQUÍMICA DEL KARST EN CLIMAS EXTREMOS
J.R. FAGUNDO CASTILLO
Centro Nacional de Investigaciones Científicas Ministerio de Educación Superior
La Habana, Cuba
J.J. VALDÉS RAMOS Instituto de Geofísica y Astronomía
Ministerio de Ciencia, Tecnología y Medio Ambiente La Habana, Cuba
J.E. RODRÍGUEZ RUBIO
Instituto de Geografía Ministerio de Ciencia,Tecnología y Medio Ambiente
La Habana, Cuba
Edición: Vivian Ferrera León Ofelia Flores Valdés Imprime: Servicio de Reprografía de la Facultad de Ciencias de Granada

PRÓLOGO
Entre las más profundas satisfacciones de mi vida es ver cómo después de la fundación de la Sociedad Espeleológica de Cuba -15 de enero de 1940- se fue formando toda una legión de jóvenes exploradores y amantes de la naturaleza y como al calor de aquella institución juvenil se fueron nucleando grupos aficionados que con el decursar del tiempo se fueron convirtiendo en magníficos profesionales, de valía tanto en su Patria como en el extranjero.
Tal es el caso de los espeleólogos Juan Reynerio Fagundo Castillo, Doctor en Ciencias, especialista del Centro Nacional de Investigaciones Científicas , Julio Valdés Ramos, Ingeniero Geofísico, Doctor en Ciencias Matemáticas y especialista del Instituto de Geofísica y Astronomía del Ministerio de Ciencia, Tecnología y Medio Ambiente y Javier Rodríguez Rubio, Doctor en Ciencias Geográficas, especialista del Instituto de Geografía también perteneciente al mismo Ministerio . Graduados de la Universidad de La Habana, se destacaron en el ámbito de la espeleología y la carsología, sobre todo en las investigaciones de la acción de la corrosión de los paisajes cársicos de distintos climas, tarea que sistematizaron magistralmente en el proyecto PIGEK (Programa Internacional sobre la Génesis y Evolución del Karst) de la Comisión Físico-Química e Hidrogeología del Karst de la Unión Internacional de Espeleología.
El proyecto PIGEK hizo posible que el Pan de Guajaibón, en la Cordillera de Guaniguanico, provincia de Pinar del Río, fuera seleccionado para instalar allí el polígono experimental representativo de las condiciones tropicales, en colaboración con espeleólogos polacos de la Universidad de Silesia, dirigidos por el científico Marian Pulina.
No pocas veces tuve el privilegio de visitar el polígono del Guajaibón, unas veces descendiendo allí desde helicóptero o bien en incursiones por tierra para ver y aprender de los compañeros cubanos y polacos que al mismo tiempo que arrancaban secretos arcanos a la naturaleza, forjaban la invariable amistad internacionalista.
Para el desarrollo de dicho proyecto fueron seleccionados también como polígonos representativos de fenómenos cársicos en condiciones polares, el carso en hielo del glaciar Werenskiold y las regiones cercanas al fiordo Hornsund, en la isla de Spitsbergen en el océano Glacial Ártico, a donde Fagundo y Valdés acudieron con el mismo entusiasmo tropical que los hizo permanecer largas temporadas en el Pan de Guajaibón de su amada Cuba.
Otras regiones estudiadas fueron el carso hidrotermal de los Rodopes, Bulgaria, y el paleocarso de Silesia, Polonia, paisajes donde fueron madurando esta obra.
Durante cinco años, cubanos, polacos y búlgaros unidos en la investigación, obtuvieron resultados, los cuales sirvieron a Fagundo Castillo, Valdés Ramos y a Rodríguez Rubio para escribir la segunda parte de este libro titulada Hidroquímica del Karst en Climas Extremos, tarea difícil por su carácter pioneril, puesto que excepto los estudios iniciales del profesor alemán Herbert Lehman, con quien tuve el honor de colaborar en sus andanzas carsológicas por la Sierra de los Órganos, los problemas de la disolución cársica apenas se habían desarrollado en Cuba y, se puede decir, en el mundo. Es justo reconocer aquí el aporte a estos estudios de los carsólogos polacos, búlgaros y soviéticos, con los cuales colaboraron Fagundo, Valdés y Rodríguez.
En la primera parte del libro, Química del Agua Kárstica, Fagundo aborda los aspectos teóricos relacionados con la hidroquímica del carso y presenta ejemplos de Cuba y otras regiones del

mundo. Por la unidad que presentan ambos trabajos, la Editorial, consideró acertadamente que debían integrarse en un mismo libro que recibió el título de Hidroquímica del Karst.
Los resultados obtenidos y presentados en este libro por los investigadores cubanos servirán
de gran ayuda a los futuros estudios relacionados con la hidroquímica de las rocas cársicas, tanto en los países tropicales como templados o polares, y en el caso de Cuba esta obra será de obligada consulta a todos aquellos investigadores que se dedican al estudio de las aguas subterráneas, a la denudación química de nuestras amplísimas y bellas zonas calcáreas.
Esta obra de Juan Reynerio Fagundo, Julio Valdés Ramos y Javier Rodríguez es otra prueba más del alto desarrollo científico alcanzado por Cuba en la esfera nacional e internacional bajo la Sociedad Socialista.
La Habana, 28 de septiembre de 1994 Antonio Núñez Jiménez

AGRADECIMIENTOS
Al Dr. Antonio Pulido Bosch, Profesor Titular de Hidrogeología, por la revisión del trabajo y
su recomendación para que fuera editado en la Universidad de Granada. Al Profesor Dr. Francisco González Lodeiro, Vicerector de Investigación y Relaciones
Internacionales de la Universidad de Granada, por conceder la financiación que ha permitido la edición.
Al Dr. Carlos Gutiérrez Calzado, Director del Centro Nacional de Investigaciones Científicas por el valioso apoyo que ha brindado a la actividad.
A los colegas del GTICEK (Grupo de Trabajo Internacional sobre Cuencas Experimentales en el Karst), quienes dieron su aliento y facilidades para su elaboración.
Al Profesor Dr. Marian Pulina y el colectivo polaco con los cuales llevamos a cabo el Programa PIGEK (Programa Internacional sobre la Génesis y Evolución del Karst), parte de cuyos resultados se reflejan en el libro.
Al Dr. Antonio Núñez Jiménez y otros pioneros de los estudios karstológicos en Cuba. Al Lic. Carlos Andino, quien revisó la primera versión que no pudo ser publicada por la
Editorial Científico-Técnica de Cuba, por las dificultades impuestas por el período especial. A Vivian Ferrera, Ofelia Flores, María Victoria Sánchez y Leonardo Flores, quienes
contribuyeron de manera determinante en el trabajo de edición y diseño de la obra. A tantos compañeros de la Sociedad Espeleológica de Cuba, del Centro Nacional de
Investigaciones Científicas (Ministerio de Educación Superior), del Ministerio de Ciencia Tecnología y Medio Ambiente, así como del Instituto Nacional de Recursos Hidráulicos, con quienes hemos compartido buenos ratos de labor científica y recreativa en relación al karst a lo largo de estos años.
Por último, los autores desean expresar su gratitud a todos aquellos que de alguna manera u otra han contribuido a la publicación de esta obra.

ÍNDICE PARTE 1. QUÍMICA DEL AGUA KÁRSTICA. J.R. Fagundo. Introducción Capítulo 1. Fundamentos químico-físicos de la disolución de los minerales por las aguas naturales Propiedades disolventes del agua pura Agua natural Ley de acción de masas Algunas consideraciones termodinámicas acerca de los sistemas acuosos Relación entre la energía libre y la constante de equilibrio Constante de equilibrio y solubilidad Disolución incongruente de los minerales El pH del agua Equilibrio de los carbonatos Formación de iones complejos o pares iónicos Variación de la constante de equilibrio con la temperatura Sistemas abierto y cerrado respecto al CO2 Efecto de ion común Efecto salino o de fuerza iónica Potencial de oxidación-reducción Intercambio iónico y adsorción Capítulo 2. Proceso de disolución y precipitación de los minerales de las rocas karstificables Medición de la agresividad de las aguas kársticas Intensidad de la denudación química Factibilidad de las rocas al proceso de disolución Difusión del CO2 en el agua Generación de CO2 en el suelo Otras fuentes generadoras de CO2 Disolución y reparto de los carbonatos en las aguas naturales Disolución de los carbonatos en presencia de otros iones Cinética y velocidad de disolución de los carbonatos Corrosión mediante procesos microbiológicos y bioquímicos

Corrosión por efecto de mezcla de agua Procesos de mezcla de agua por intrusión marina en acuíferos kársticos Cambios en la concentración de la calcita disuelta por las aguas naturales Deposición de la calcita a partir de aguas sobresaturadas Deposición de calcita en manantiales y cursos superficiales Deposición de calcita y aragonito en cavidades subterráneas Proceso de karstificación en yeso Capitulo 3. Caracterización y evolución químico-física de las aguas kársticas Análisis químico y calidad de las aguas Estudio del comportamiento hidrodinámico de los acuíferos kársticos a partir de las respuestas naturales en las surgencias Empleo de relaciones iónicas Representación gráfica de la composición química de las aguas Clasificación de las aguas desde el punto de vista geológico y karstológico Clasificación de las aguas por su uso Clasificación hidroquímica Método de clasificación de Alekine Método de clasificación de Shchoukarev Método de clasificación de Kurlov Método de clasificación de Palmer Método de clasificación de Shoeller Empleo de métodos numéricos Factores que determinan la composición y evolución de las aguas kársticas Patrones hidrogeoquímicos y control de la composición química de las aguas Kársticas Variación espacial y temporal de la composición química de las aguas kársticas Variación de la composición química del agua kárstica a causa del impacto antropogénico
Zonas de protección y control sistemático de la calidad de las aguas kársticas

PARTE 2. HIDROQUÍMICA DEL KARST EN CLIMAS EXTREMOS. J.R. Fagundo, J.J. Valdés, J.E. Rodríguez. Capítulo 1. Expedición Spitsbergen'85 Breve recuento Materiales y métodos Resultados de la caracterización hidroquímica Área del Grondfjord Área del fiordo Hornsund Grado de agresividad de las aguas y denudación química Variación temporal de las propiedades químico-físicas de las aguas Patrones hidrogeoquímicos y relaciones empíricas entre las concentraciones iónicas y la conductividad eléctrica Capítulo 2. Comportamiento hidroquímico del karst tropical de Cuba Breve descripción de los casos de estudio Cuenca del río Cuyaguateje Cuenca del río San Marcos
Meseta del Guaso Llanura kárstica meridional del occidente de Cuba Región de San Antonio de los Baños Llanura meridional de Pinar del Río Karst litoral del sur de Matanzas Intensidad de la denudación química Capítulo 3. Comparación entre las regiones de clima polar y tropical Clasificación numérica Consideraciones finales Bibliografía

HIDROQUÍMICA DEL KARST
Parte 1
QUÍMICA DEL AGUA KÁRSTICA
J.R. FAGUNDO CASTILLO Centro Nacional de Investigaciones Científicas
Ministerio de Educación Superior La Habana, Cuba

Parte 1
QUÍMICA DEL AGUA KÁRSTICA
INTRODUCCIÓN
El objetivo de la primera parte de este libro, es ofrecer al lector los fundamentos químico-físicos de la disolución de los minerales constitutivos de las rocas karstificables y exponerle los efectos que determinan la composición y evolución química de las agua kársticas. El mayor énfasis se hace en las aguas que drenan los macizos kársticos carbonatados, ya que en Cuba más del 65 porciento del territorio está constituido por este medio geológico.
Los términos carso o karst provienen del nombre de una región calcárea en la meseta del Jura (en los Alpes Dináricos), donde una parte del macizo carbonatado denominado Karst o Crasu, se encuentra en territorio Yugoslavo, mientras que la otra parte, nombrada Carso, se ubica en Italia.
Esta región se caracteriza por el predominio del escurrimiento subterráneo sobre el superficial y por presentar un paisaje típico, donde son más abundantes las formas del relieve originadas por la erosión química que las debidas a la erosión mecánica. Entre las formas de erosión química se distinguen: lapiaz (o diente de perro), dolinas, poljes, uvalas, ponores (sumideros), surgencias, resurgencias, cuevas, etcétera. En las zonas montañosas los escarpes suelen ser pronunciados, especialmente en áreas tropicales donde es típico el denominado tower karst o mogote. En las zonas llanas litorales de los terrenos kársticos del trópico son comunes los tipos de dolinas conocidas como cenotes o casimbas.
La ciencia que estudia este tipo de geomorfología se denomina carsología o karstología. En Cuba se prefiere usar el primer término por ser más fonético, al igual que carso en lugar de karst, mientras que en otros países de habla hispana como México o España se emplean los términos homólogos derivados del Karst.
Las regiones kársticas suelen presentar en mayor proporción, minerales del tipo soluble como calcita, dolomita, yeso o halita. La disolución de estos minerales por las aguas naturales esta sujeta a leyes termodinámicas y controlada por la presencia de gases en las aguas, los cuales son generados fundamentalmente, por la descomposición bacteriana de las plantas y pasan al seno de la solución acuosa durante el escurrimiento de las precipitaciones por la zona del suelo. Por todo ello el proceso de adquisición de la composición química de las aguas kársticas es complejo y en él intervienen una

serie de factores de tipo quimicofísico, geológico, hidrogeológico, climático, microbiológico y antrópico. Los mismos serán objeto de análisis en este libro.
En las regiones donde predominan minerales menos solubles en que se presentan rocas de origen volcánico o metamórficos, sedimentos de tipo arenoso, arcilloso o margoso constituidos por aluminosilicatos, también pueden formarse algunas formas de disolución denominadas pseudocársicas o pseudokársticas. Además existen formas que fueron originadas en épocas pretéritas o se han formado en el presente por acción de los glaciares. Se denominan paleocársicas y glaciocársicas respectivamente o por los términos homólogos paleokársticas y glaciokársticas.
El karst fue definido originalmente sobre la base de las formas típicas presentes en la región de la meseta del Jura. A partir de la década del 50, se añadieron al concepto los fenómenos relacionados con la disolución de los minerales y el tipo de drenaje. El karst también ha sido considerado el resultado de un proceso de interacción de las aguas meteóricas con ese medio. No obstante, como ha señalado el profesor Eraso, una vez creadas estas formas, las mismas juegan un importante papel en el desarrollo ulterior de los procesos kársticos. Este especialista ha indicado que constituye un desenfoque dialéctico el desarrollo de los estudios kársticos desde la óptica de algunas de las disciplinas particulares que se relacionan con éste (Geomorfología, Geología, Química, Hidrología, Hidrogeología, Ingeniería Geológica, Ingeniería Sanitaria, etcétera), insistiendo en la necesidad de abordar los aspectos del karst de manera polidisciplinaria.
Hoy día cualquier investigación en el campo de las geociencias, requiere un enfoque multidisciplinario. Con ese estilo ha trabajado precisamente el colectivo del que forman parte los autores de este libro, que durante muchos años han llevado a cabo estudios sistemáticos en cuencas kársticas experimentales en Cuba, con la participación de diferentes especialistas de distintos países, relacionados con la dinámica y evolución del karst en diferentes condiciones climáticas, así como con los cambios en la calidad del agua debidos a la actividad del hombre en el karst.
Este libro pretende servir como herramienta de trabajo a aquellos investigadores que en forma teórica o aplicada se relacionan con la Hidroquímica de las regiones kársticas.

Capítulo 1. Fundamentos químico-físicos de la disolución de los minerales por las aguas naturales

Propiedades disolventes del agua pura
El agua es un disolvente universal de las sustancias inorgánicas. Esa propiedad está relacionada con las características de la molécula de agua, las cuales son muy diferentes a las de otra molécula con un peso atómico semejante.
El agua pura está constituida por moléculas, donde dos átomos de hidrógeno se encuentran unidos a uno de oxígeno (H2O), separados entre sí 105° (Figura 1.1a).
Figura 1.1 Representación de: a) la molécula de agua; b) proceso de disolución de un cristal de NaCl por los dipolos de agua.
Esta distribución hace que parte de la molécula posea una carga parcial positiva y la otra una
carga parcial negativa, lo que le confiere propiedades de dipolo. Cuando un mineral se introduce en

el agua, los dipolos de ésta tienden a separar los iones de carga contraria, disminuyendo las fuerzas electrostáticas que tienden a unirlas en virtud de las propiedades del enlace iónico. En el caso del cristal de cloruro de sodio (NaCl), por ejemplo, este proceso puede representarse como se indica en la figura 1.1b. Los dipolos se intercalan de tal forma, que las cargas de signo contrario son capaces de atraerse cada vez menos.
En el caso de otros iones más débilmente unidos, mediante enlaces de tipo iónico, como es el caso del cristal de calcita, este proceso es más lento, de ahí su pobre solubilidad en agua pura.
Este proceso de disolución, cuando es llevado a cabo por las aguas naturales sólo con la participación de éstas, se denomina físico (Bögli, 1980) y se diferencia del denominado químico, en que este último es más complejo, puesto que intervienen otros factores.
Las bases teóricas que fundamentan el proceso de disolución de los minerales por las aguas naturales, se han desarrollado por diferentes investigadores (Hutchinson, 1957; Roques, 1962 a,b,c; Roques, 1964; Langmuir, 1964; Garrels y Christ, 1965; Stumm y Morgan, 1970). En este texto se presentan algunos aspectos de esta teoría, por lo que para un conocimiento más amplio remitimos al lector a las obras citadas.
Agua natural
Antes de abordar las teorías que sustentan los procesos de disolución de los minerales, se debe discutir el concepto de agua natural. Según plantean algunos especialistas (Stumm y Morgan, 1970), se entiende por agua natural un sistema de cierta complejidad, no homogéneo, que puede estar constituido por una fase acuosa, una gaseosa y una o más fases sólidas.
El sistema agua natural puede ser no homogéneo, aunque puede estar constituido por subsistemas homogéneos. De esta manera, el océano posee cuencas profundas y prácticamente aisladas con composición química definidas. En algunos casos, como en los lagos y las presas, las aguas pueden estar estratificadas, en la superficie la temperatura puede ser más elevada y la densidad menor que en la profundidad. En las aguas subterráneas que se encuentran en contacto con el mar, se pueden presentar también apreciables diferencias de acuerdo con la profundidad.
En general, las aguas subterráneas poseen una composición química que es el resultado de un proceso complejo de interacciones, donde primeramente las aguas procedentes de las precipitaciones (lluvia o nieve) adquieren los gases que se originan en la zona del suelo en el proceso de descomposición y respiración de la materia orgánica y luego, reaccionan con los minerales que subyacen en el medio rocoso.
La composición química de las aguas subterráneas, al cabo de un determinado tiempo, se encuentran en equilibrio químico-físico con el contenido de gases y de fases sólidas disueltos. Estos equilibrios dependen de la temperatura y de la presión del sistema y cualquier cambio en esas condiciones produce una variación en la composición química, dando lugar a una mayor disolución de los minerales o a la precipitación de estos por recombinación iónica.
Ley de acción de masas
La disolución de un mineral o la precipitación a partir de una solución saturada está regida por la ley de acción de masas, la cual se aplica, además para otros procesos de interacción químico físicos. En general, puede formularse a partir de la expresión siguiente:
a A + b B ... = c C + d D ... (1.1)

como: K (C) (D)(A) (B)
c d
a b= (1.2)
donde: K: Constante termodinámica de equilibrio, también denominada como constante de estabilidad, así como constante del producto de solubilidad (reacciones de disolución) y constante de disociación (reacciones de disociación química). ( ): Actividad química. A y B: Compuestos reaccionantes. C y D: Productos de la reacción. a, b, c y d: Número de moles.
La actividad de una sustancia x se define como:
(x) = γx mx (1.3) donde: mx: Molalidad de la sustancia x. γx : Coeficiente de actividad de la sustancia x.
Las concentraciones, que serán representadas en este libro por corchetes [x], suelen ser iguales a las actividades cuando las soluciones son diluidas. En otros textos las actividades se expresan por ai, {i} e incluso [x], prefiriendo utilizar los paréntesis (x) para las concentraciones en lugar de las actividades.
Las molalidad se puede calcular a partir de:
m ppm 10PFx
x3
x
=−
(1.4)
donde : ppm: Concentración de x, en partes por millón (mg/L). PF: Peso fórmula de x, en g.
Para el cálculo del coeficiente de actividad de un ion, se suele utilizar la ecuación de Debye-Hückel:
− =logAZ
1 + a Bxi2
i0γ
µµ
(1.5)
donde: γx: Coeficiente de actividad. A y B: Parámetros que dependen de la temperatura (Tabla 1.1). Zi: Valencia del ion i. ai°: Diámetro eficaz del ion (Tabla 1.2). µ: Fuerza iónica.

Temperatura (00C) Parámetro A Parámetro B (10-8)
0 0,4883 0,3240 10 0,4960 0,3258 20 0,5042 0,3273 25 0,5085 0,3281 30 0,5130 0,3290 40 0,5221 0,3305 50 0,5319 0,3321 60 0,5425 0.3338
Tabla 1.1 Valores de los parámetros A y B de la ecuación de Debye-Hückel
aiº Iones y pares iónicos 2,5 NH4
+
3,0 K+, Cl-, NO3-
3,5 OH-, HS-
4,0 SO42- , PO4
3-
4,0-4,5 Na+, HCO3-, CaHCO3
+, HSO4-, NaSO4
-, KSO4-
4,5 CO32- , NaCO3
-
5,0 Sr2+ , Ba2+ , S2-
6,0 Ca2+ , Fe2+ , Mn2+ , MgHCO3 +
9,0 H+, Al3+ , Fe3+
Tabla 1.2 Valores del diámetro eficaz aiº de la ecuación de Debye-Hückel para diferentes patrones y pares iónicos
La fuerza iónica se puede determinar por la ecuación de Lewis y Randall (1923):
µ ==∑1
2 Z mi2
i 1
n
x (1.6)
La ecuación de Debye-Hückel se cumple satisfactoriamente cuando la fuerza iónica es menor
de 0,1, lo que corresponde a un agua natural con mineralización alrededor de 5 000 y 8 000 ppm. Cuando µ es mayor de 0,1 y menor de 0,5 se recomienda el uso de la variante de Davis:
Log AZ (1
0,21)i i2γ
µµ
= −+
− (1.7)
Los coeficientes de actividad de las principales especies constitutivas de las aguas naturales
tienden a disminuir, aunque no en forma lineal, al incrementarse la fuerza iónica.
Algunas consideraciones termodinámicas acerca de los sistemas acuosos
Desde el punto de vista termodinámico, existe una diferencia apreciable entre sistemas aislados, cerrados y abiertos. Se considera un sistema aislado, aquel que no puede intercambiar calor,

trabajo o materia con el medio circundante. El sistema se considera cerrado, cuando puede intercambiar energía, pero no materia con el medio, y abierto, cuando puede intercambiar tanto energía como materia. Para ejemplarizar tales sistemas termodinámicos en el ámbito de las aguas naturales, se utilizará el diagrama presentado por Stumm y Morgan (1970), el cual se reproduce en la figura 1.2.
La figura 1.2a, representa un sistema cerrado desde el punto de vista termodinámico, donde
las masas de sus constituyentes son fijas, es decir, no intercambia materia con los alrededores. Este sistema está constituido por una fase (solución acuosa), la cual no está sujeta a reacciones químico-físicas con ninguna fase gaseosa o sólida, es decir, representa un sistema idealizado; un ejemplo de este sistema puede considerarse una solución acuosa de electrólitos en un recipiente de laboratorio mantenido a temperatura y presión atmosféricas, utilizada para el estudio de los equilibrios químicos. Los datos termodinámicos (calor de reacción, entalpía, energía libre) obtenidos en estos experimentos, se pueden utilizar en la interpretación de modelos reales más complejos.
El diagrama b (Figura 1.2) representa un sistema cerrado constituido por una fase gaseosa y una líquida. Ambas fases pueden intercambiar materia entre sí. Este es el caso, por ejemplo, de un sistema formado por agua (H2O) y dióxido de carbono (CO2). Tanto las moléculas de H2O como de CO2 pueden producir reacciones de intercambio entre las dos fases:
CO2 (g) + H2O (l) = CO2 (ac) (1.8) CO2 (ac) +H2O(l) = HCO3
- (ac) + H+ (ac) (1.9)
El diagrama c (Figura 1.2) representa un sistema cerrado que comprende una fase acuosa y otra sólida, esta última parcialmente disuelta en agua. Este es el caso, por ejemplo, de la disolución de anhidrita en agua:

(1.10) CaS (s) O Ca (ac) SO (ac)4H2 2
42⎯ →⎯⎯ ++ −
Anhidrita
El diagrama ch (Figura 1.2) representa un sistema cerrado constituido por fases liquidas, gaseosas y sólidas que interactúan entre sí. Tal es el caso de las reacciones que intervienen en el sistema CO2-H2O-CaCO3 :
CO2(g) + H2O (l) + CaCO3 (S) = Ca2+ (ac) + 2 HCO3- (ac) (1.11)
Calcita Cuando las reacciones químicas en este sistema (reacción 1.11) tienden a ir hacia la
derecha se produce la disolución de la calcita, mientras que cuando tienden a ir hacia la izquierda, ésta precipita. La ocurrencia de uno u otro evento dependerá de la presión parcial del gas y de la temperatura.
El diagrama d (Figura 1.2) representa un sistema cerrado similar a c, donde interviene más de una fase sólida. Como ejemplo de este caso puede considerarse las reacciones entre los minerales moscovita y caolinita, así como entre caolinita y Gibbsita:
KAl3 SiO10 (OH) (s) + H+ (ac) = 3
2 H2O (l) + 32 Al 2 Si2 O5 (OH)4 (s) + K+ (ac) (1.12)
Moscovita Caolinita Al2 Si2 O5 (OH)4 (s) + 5 H2O (l) = 2 H4SiO4 (ac) + Al4 O3 . 3 H2 O (s) (1.13) Caolinita Gibbsita
El diagrama e (Figura 1.2) representa un sistema cerrado similar a d, donde interviene más de una fase sólida. Como ejemplo de este sistema se puede tomar la disolución simultánea de los minerales calcita (reacción 1.11) y dolomita por un agua natural que posee un cierto contenido de CO2: CO (g) + H O (l) + CaMg(CO3 )2 (s) = Ca2+ (ac) + Mg 2+ (ac) + 4 HCO3
- (ac) (1.14) 2 2 Dolomita Relación entre la energía libre y la constante de equilibrio
Desde el punto de vista termodinámico, el estado de equilibrio es un estado de máxima estabilidad, hacia el cual un sistema químico-físico cerrado se dirige mediante procesos irreversibles. En general, los cambios en este caso tienden a un decremento en la función de la energía, conocida como energía libre de Gibbs. En estas condiciones de mínima energía se obtiene una máxima entropía en el sistema.
La fuerza directriz en las reacciones químicas es comúnmente representada por la energía libre de Gibbs (Gr), para un sistema a temperatura y presión constantes, la cual representa los cambios en la energía interna por unidad de masa, y es una medida de la capacidad de una reacción para donar trabajo no mecánico.
La condición de equilibrio químico puede definirse como: Σ energía libre de - Σ ###energía libre de = 0 (1.15)

los productos los reaccionantes
Los reaccionantes y los productos en la ecuación 1.15 se consideran a temperatura y presión definidas en las condiciones de equilibrio. A partir de esta definición se pueden relacionar los cambios de energía libre con la constante de equilibrio. Para ello es necesario previamente, definir la energía libre estándar Gf° (en algunos textos aparece como Ff°). Esta magnitud se puede considerar como la energía libre de la reacción que es capaz de producir 1 mol de sustancia a partir de los elementos estables bajo las condiciones definidas para los estados estándares.
La energía libre estándar de formación de un elemento estable es tomada como cero por convención; por ejemplo la del hidrógeno, y como 1, la energía estándar del agua pura a la temperatura y presión de la reacción. En general, en el Handboock de Química Física (Robert, Melvin y Williams, 1983) y libros especializados, aparecen los valores de Gf° de las diferentes especies químicas.
Los cambios de energía libre estándar de una reacción pueden ser explicados como la suma de las energías libres de formación de los productos en sus estados estándares, menos las energías de formación de los reaccionantes también en estos estados. Gf° = Σ Gf Productos - Gfº Reaccionantes (1.16) donde: Gf°: Energía libre estándar.
Para una reacción general (1.1), los cambios de energía libre de la reacción están relacionados
con los cambios en la energía libre estándar y las actividades de cada uno de los reaccionantes, así como de los productos sometidos a las mismas condiciones de temperatura y presión, mediante la expresión:
Gr Gr RTln (C) (D)(A) (B)
Gr RTlnQoc d
a bo= + = + (1.17)
donde: R: Constante de los gases ideales. T: Temperatura, en °K. Q: Constante de reacción. Gr: Energía de reacción. Gr°: Energía estándar de reacción.
Para una reacción que procede espontáneamente Gr < 0. Si Gr > > 0 la reacción sólo puede ocurrir en dirección inversa y si Gr = 0, no procede en ninguna dirección, lo que equivale a la condición de equilibrio. Por tanto, en estas últimas condiciones, sustituyendo en 1.2:
Gr° = - RT ln K (1.18)
El cociente de reacción Q (ecuación 1.17) se relaciona con las velocidades de reacción directa e inversa (entre los reaccionantes y los productos respectivamente) mediante la expresión:
γγ
d
i
d
i
KK
1Q
KQ
= = (1.19)

donde: γd: Coeficiente de actividad de los reaccionantes. γi: Coeficiente de actividad de los productos. kd: Constante de velocidad de los reaccionantes. ki: Constante de velocidad de los productos. Si existe el equilibrio, de modo que K = Q.
El cociente de reacción también se expresa como:
{ }Q (C) (D) ........(A) (B) ........
i ii
c d
a b= =∏ (1.20)
En condiciones diferentes del equilibrio (condiciones de desequilibrio), Gr ≠ 0:
(1.21) { }Gr Gr RT lno i i
i= + ∏
Constante de equilibrio y solubilidad
En las reacciones de disolución de minerales, la constante de equilibrio se conoce comúnmente como constante del producto de solubilidad (KPS). El logaritmo inverso de esta constante de equilibrio se denomina pK y tiene una gran significación geoquímica:
pK = - log K (1.22)
La solubilidad de un mineral en agua pura se puede calcular a partir de la constante del
producto de solubilidad; por ejemplo, la disolución de la anhidrita, la cual puede representarse mediante la ecuación química de disociación (1.10). En este caso, la constante del producto de solubilidad de la anhidrita (Kanh) es: Kanh = (Ca2+) (SO4
2-) = 3,4 . 10-5 (1.23)
Si x es la solubilidad del CaSO4 en moles por litro, las concentraciones de los iones Ca2+ y SO4
2- también serán x, puesto que x moles de CaSO4 se disuelven para producir x ion gramos de Ca2+ y x ion gramos de SO4
2-. Al sustituir en 1.23, la solubilidad será: x = (3,4 . 10-5 )1/2 = 5,9 . 10-3 moles. Como 1 mol de CaSO4 = 100 g/L, x = 590 g/L. La solubilidad de un mineral en las aguas naturales es mucho más compleja de lo que se
deduce de las expresiones termodinámicas para el agua pura. La misma está determinada por la temperatura, la presión parcial de los gases disueltos, el pH, el potencial redox, el estado de división de las partículas, la concentración relativa de otros iones, sin considerar los aspectos de tipo no químico-físicos. En el medio natural estos elementos están relacionados de tal manera que no puede predecirse exactamente la forma en que ocurrirá el proceso.
En la tabla 1.3 se presenta la solubilidad en agua a 25 °C de varios minerales constitutivos de las rocas karsificables, el valor del pK, así como las reacciones químicas correspondientes. Su

análisis pone de manifiesto que el pK es un pobre indicador de la solubilidad relativa de los minerales, lo cual se debe a que en las reacciones de equilibrio las actividades de los iones o moléculas se encuentran elevados a una potencia igual al número de moles en la reacción de disolución balanceada. Por ejemplo, la solubilidad de la calcita en agua pura a pCO2 1 bar (1 atm) es de 500 mg/L, mientras que la solubilidad de la dolomita bajo las mismas condiciones es cercana a este valor (480 mg/L), a pesar de que las constantes de equilibrio difieren en un orden de 8. La similar solubilidad se debe a que, en este caso, el término (CO3
2-) se encuentra elevado a la segunda potencia en la dolomita y a la primera potencia en la calcita.
Mineral Reacción química pK Solubilidad Concentración (mg/L)
Halita NaCl = Na++ Cl- -1.6 360 000 5-10 000 Silvita KCl = K++ Cl- -0.9 244 000 0-10 000 Anhidrita CaSO4 = Ca2+ + SO4
2- 4.5 Yeso CaSO4 . 2H2O = Ca2+ +
SO42- + 2H2O
4.6 2400 0-1 500
Calcita CaCO3 = Ca2+ + CO32- 8.4 100*,480** 10-300
Dolomita CaMg(CO3)2 = Ca2++ Mg2+ + 2CO3
2-17.0 90*, 500** 10-300 CaCO3
Sílice amorfa SiO2 + 2H2O = H4SiO4 120 1-65 Cuarzo SiO2 + 2H2O = H4SiO4 37.0 12 1-12 Gibbsita Al2O3 . 2H2O + H2O = 2Al3+
+ 6 OH- 0.001 Trazas
* multiplicado por 10-3 bar ** multiplicado por 10-1 bar Tabla 1.3 Reacción química, pK, solubilidad y concentración en las aguas meteóricas de algunos minerales constitutivos de las rocas, que se disuelven congruentemente y pueden originar formas kársticas o pseudokársticas.
En relación al efecto de la solubilidad de los minerales sobre el paisaje kárstico se puede indicar que a causa de la alta solubilidad de las rocas salinas, los depósitos de estos minerales son rápidamente destruidos por la acción disolvente de las aguas meteóricas, en estos casos sólo se pueden apreciar algunos afloramientos poco voluminosos de halita o silvita. En ocasiones desempeñan un papel importante en los procesos de karstificación de tipo intersticial.
Los afloramientos de anhidrita y yeso son algo más comunes en la naturaleza, los procesos de karstificación son muy intensos en las rocas ricas en estos minerales. En ocasiones se forman cavernas de grandes magnitudes y depresiones kársticas en terrenos donde abunda el yeso. Sin embargo, algunas cuevas en yeso no alcanzan mayores dimensiones a causa de la destrucción de las galerías, como consecuencia de la intensa corrosión química.
Las calizas y dolomías constituyen los afloramientos más comunes de las rocas kársticas. La solubilidad y contenido en las aguas es muy similar en los minerales calcita y dolomita, constitutivos respectivamente de estas rocas.
Las areniscas silíceas y las cuarcitas son frecuentes en los afloramientos. El contenido de SiO2 en las aguas procedentes de estas rocas suelen ser mayores que los encontrados en las aguas que se originan en las rocas carbonatadas, aunque en ocasiones existe algún solapamiento en los rangos de concentración.
En virtud de la relativa poca solubilidad no predominan las formas kársticas en el paisaje de estos terrenos, sólo se destacan algunas formas pseudokársticas a pesar de que la distribución de este

tipo de roca es muy abundante en la naturaleza. Las formas de disolución más significativas están asociadas a las aguas termales, ya que la solubilidad de la cuarcita se incrementa rápidamente con la temperatura, siendo superior a la de la calcita a 100 ºC.
Disolución incongruente de los minerales
Cuando los productos de una reacción de disolución son todos especies iónicas, se dice que la disolución es congruente. Cuando por el contrario, la disolución de un mineral da lugar a especies iónicas y moléculas no solubles, se dice que la disolución es incongruente. Este último es el caso de muchos aluminosilicatos; por ejemplo, la ecuación química correspondiente a la disolución incongruente de la albita, un aluminosilicato de sodio:
NaAlSi3O8 (s) + CO2 (g) + 11
2 H2O (l) = Na+ (ac) + HCO3- (ac) + 2H4SiO4 (l) +
Albita 12 Al2Si2O5(OH)4 (s) (1.24)
Caolinita En presencia de CO2 el agua disuelve el mineral albita, liberando sodio, bicarbonato y ácido
silícico en el seno de la solución, mientras deposita el mineral arcilloso caolinita. Este proceso ocurre corrientemente como resultado del intemperismo de las rocas graníticas. Al aplicar la ley de acción de masas en condiciones de equilibrio químico, se obtiene:
K (Na )(HCO )(H SiO )pCOalb
3 4 42
2
=+ −
(1.25)
donde: Kalb: Constante del producto de solubilidad del mineral albita.
En el proceso de disolución de los carbonatos se suelen producir disoluciones incongruentes
cuando están presentes calcita y dolomita (Picknett, 1972; Wigley, 1973 a) o calcita y yeso (Wigley, 1973 b). Las aguas en estos casos pueden disolver secuencial o sucesivamente estos minerales. En términos de constantes de equilibrio y actividades, la disolución de la calcita y la dolomita se pueden expresar por: Kc = (Ca2+) (CO3
2-) (1.26) Kd = (Ca2+) (Mg2+) (CO3
2-)2 (1.27)
Las solubilidades de la calcita y la dolomita serán entonces, respectivamente: xc = (Kc)
1/2 y xd = (Kd)1/4 y la relación de solubilidades de ambos será:
xx
kk
d
c
d
c
12
= (1.28)
Por tanto, de acuerdo con los valores de Kd y Kc , si T < 10°C, Kd
1/2 > Kc, siendo la dolomita más soluble que la calcita. Si por el contrario T > 10 °C y Kd
1/2 < Kc, la calcita es más soluble que la dolomita.
Lo expuesto en el párrafo anterior implica que donde coexistan ambos minerales a bajas temperaturas, cuando el agua se encuentre saturada respecto a la dolomita, será sobresasaturada

respecto a la calcita, precipitando este último mineral a medida que se disuelve el primero; en este caso el agua disuelve incongruentemente a la dolomita. Si por el contrario, este proceso ocurre a una temperatura superior a 10 °C, se producirá la disolución de la dolomita en forma incongruente.
Si el proceso de disolución de ambos minerales tiene lugar secuencialmente, entonces puede ocurrir la disolución incongruente, con independencia de la temperatura del agua. El pH del agua
Un agua pura disuelve con facilidad sólo aquellos minerales como los de tipo salino o los sulfatos, donde el proceso es de tipo físico. En la mayoría de los casos, sin embargo, la solubilidad de un mineral se incrementa considerablemente en presencia de ácidos; por ejemplo, en un litro de agua a 25 °C, sólo se disuelven 12 mg de calcita, mientras que en presencia de un ácido fuerte, la solubilidad aumenta de 25 000 a 30 000 veces.
La medida del grado de acidez del agua es el pH, el cual se define como el logaritmo inverso de la actividad del ion hidrógeno o hidronio (H+ ó H3O
+ ), que resulta de la ionización de un ácido cualquiera; se expresa en moles por litro, aunque al medirse en equipos electrónicos con electrodos de referencia, la acidez se expresa en unidades de pH, por lo general entre 0 y 14, ésta se calcula de la manera siguiente:
pH = -log (H+) (1.29)
Aunque el agua pura está constituida fundamentalmente por moléculas de H2O una pequeña
porción de la misma se compone de H+ y OH-. Esta proporción está controlada por la reacción de disolución:
H2 O (l) = H+ (ac) + OH- (ac) (1.30) la cual para una temperatura dada se encuentra en equilibrio dinámico.
La constante de equilibrio correspondiente, se denomina constante de disociación del agua y
se puede expresar por:
Kw = (H+)(OH-) (1.31) donde: Kw: Constante de disociación del agua a 25 °C (Kw = 10-14)
En el agua pura (H+) = (OH-) = (Kw1/2 ) = 10-7, por lo que teóricamente su pH debe ser 7. En
la realidad, a causa de que ésta se encuentra en contacto con la atmósfera es capaz de disolver CO2, adquiriendo un pH inferior a ese valor.
El valor del pH de muchas aguas naturales que interactúan con los minerales varía en un estrecho intervalo, por lo general entre 6 y 9, lo cual entre otras causas, se debe a la gran distribución de las rocas carbonatadas y al carácter ácido-básico de las mismas, a través del sistema de equilibrios químicos que se establecen entre CO2, HCO3
- y CO3 2-.

Algunos manantiales calientes de origen volcánico poseen una elevada acidez, por la presencia de HCl y SO2 . Ácidos libres también pueden pasar al agua como resultado del vertimiento de aguas residuales.
El agua de las precipitaciones al pasar por la zona del suelo, donde es abundante el CO2, adquiere un pH relativamente bajo, del orden de 4.5. Luego por interacción con las rocas carbonatadas éste tiende a elevarse hasta cerca de 7. Cuando esta interacción se produce durante un tiempo prolongado el agua incrementa su contenido de iones HCO3
-, adquiriendo además iones CO32-
. En esas condiciones el pH puede alcanzar un valor cercano a 8.4. Por lo general, en los arroyos y ríos de las regiones húmedas no kársticas el pH varía entre 5 y 6.5, mientras que en las regiones kársticas húmedas este valor suele estar comprendido entre 7 y 8. Las aguas marinas tienden a poseer un pH cercano a 8.
Equilibrio de los carbonatos
Las leyes termodinámicas han encontrado una elegante aplicación en la disolución de las calizas y dolomías, donde tienen lugar los sistemas de equilibrios CO2-H2O-CaCO3 y CO2 -H2O-CaMg(CO3)2 respectivamente. La disolución de los minerales constitutivos de las rocas carbonatadas comprende una serie de procesos físicos y químicos donde intervienen estados gaseosos, líquidos y sólidos a través de interfases aire-agua-roca y un sistema de equilibrios químicos. Para el caso de la calcita, estos se pueden resumir de la manera siguiente:
1. Difusión del CO2 en agua durante las precipitaciones:
CO2 (g) = CO2 (ac) (1.32) aire disuelto físicamente
La concentración del CO2 en el agua es función de la presión parcial del gas (pCO2) en la atmósfera en contacto con la solución acuosa y se determina por la ley de Henry. pCO2 = D (H2 CO3) (1.33) donde: D: Coeficiente de difusión, constante para una temperatura dada.
2. Formación de ácido carbónico (durante las lluvias o en la atmósfera del suelo):
CO2 (ac) + H2 O (l) = H2CO3 (l) (1.34) disuelto CO2 químicamente físicamente disuelto La constante de equilibrio correspondiente (Kb) es:
K (H CO )pCOb
2 3
2
= (1.35)

3. Disociación del ácido carbónico:
H2CO3 (ac) = H+ (ac) + HCO3
- (ac) (1.36)
HCO3- (ac) = H+ (ac) + CO3
2- (ac) (1.37) y las constantes de equilibrio son:
K (H )(HCO )H CO1
3
2 3
=+ −
(1.38)
K (H )(CO )HCO2
32
3
=+ −
− (1.39)
4. Disolución de los cristales de CaCO3
CaCO3 (s) = Ca2+(ac) + CO3
2- (ac) (1.40) Calcita
En condiciones de equilibrio: Kc = (Ca2+)(CO3
2-) (1.41) El ion CO3
2- creado en este paso se combina con el H+, según el equilibrio 1.41 para dar HCO3-.
5. Disociación de la molécula de agua (reacción 1.30): H2O (l) = H+ (ac) + OH- (ac) (1.30) donde la constante de disociación de ésta se puede observar en la ecuación 1.31: Kw = (H+)(OH-) (1.31)
Al integrar las ecuaciones químicas 1.34, 1.36, 1.37 y 1.40, se obtiene la ecuación que representa la disolución de la calcita (ecuación 1.11): CO2 (g) + H2O (l) + CaCO3 (s) = Ca2+ (ac) + 2HCO3
- (ac) (1.11) Calcita
Para satisfacer la condición de neutralidad eléctrica de la solución, es necesario que la suma de las cargas de los cationes sea igual a la de los aniones. En términos de concentraciones esto se puede expresar por: 2 [Ca2+] + [H+] = [HCO3
-] + 2 [CO32-]+ [OH-] (1.42)

Como las concentraciones de CO32-, H+ y OH- son despreciables en condiciones habituales
(para un pH menor de 8,4), respecto a las de Ca2+ y HCO3-, la expresión 1.42 se puede reducir a:
2 [Ca2+] = [HCO3
-] (1.43)
Si se desea conocer las concentraciones de las especies químicas involucradas en el sistema de equilibrios químicos, esto es [CO2], [H2CO3], [H
+], [Ca2+], [HCO3-], [CO3
2-] y [OH-] es necesario resolver el sistema constituido por las ecuaciones de equilibrios químicos (1.31, 1.35, 1.38, 1.39, 1.41) y la de balance de carga (1.42) o su aproximada, la ecuación 1.43. Para facilitar el tratamiento se debe adoptar que la solución es diluida, de manera que las actividades son iguales a las concentraciones.
La concentración del ion HCO3 - se puede calcular al despejar ésta de la ecuación 1.39:
[ ] [ ][ ]HCO
CO H
K33
2
2
−− +
= (1.44)
y la de CO3
2- a partir de 1.41
[ ] [ ]CO KCa3
2 c2
−+
= (1.45)
Si se toma en cuenta la ecuación 1.43 y se sustituye el valor de [HCO3
-] en función de [CO3
2-], según la ecuación 1.44 se obtiene:
[ ] [ ][ ]2 Ca
K H
K Ca2 c
22
++
+= (1.46)
al despejar [H+] en la ecuación 1.46,
[ ] [ ]1
HK
2K Cac
22 2+ +
= (1.47)
si se aplica logaritmos y se toma en cuenta la ecuación 1.29, queda: pH = - 2 log [Ca2+ ] + B (1.48) donde: B: Magnitud que es función de K2 y Kc.
La expresión 1.48 corresponde a las ecuaciones teóricas que fundamentan los diagramas de Tillman-Trombe (Trombe, 1952) y Roques (1969), usados para el cálculo de la agresividad de las aguas kársticas (Figura 1.3). Como se puede apreciar, la ecuación 1.48 corresponde a una función logarítmica.

Figura 1.3 Diagrama de Tillman-Trombe.
Muxart (1972), mediante gráficos semilogarítmicos representó estos diagramas (Figura 1.4) como líneas rectas. Los valores experimentales de B (ecuación 1.48) fueron hallados en función de la temperatura, según: log B = - 0,028 6 log T + 1,065 (1.49)

Para determinar la concentración de H2CO3 en función de Ca2+, se toman en cuenta las ecuaciones 1.38 y 1.43, quedando:
[ ] [ ][ ]2 Ca
K H COH
2 1 2 3++
= (1.50)
El valor de [H+] puede expresarse en función de [Ca2+] mediante la ecuación 1.47,
quedando:
[ ] [H KK
.2 Ca2
c
2 2+ = ]+ (1.51)
Si se despeja [H2CO3] en la ecuación 1.50 y se sustituye el valor de [H+ ]en la expresión 1.51:
[ ] [H CO 4KK K
Ca2 32
1 c
2 3= + ] (1.52)
y expresando la concentración de ácido carbónico en función de pCO2 de la atmósfera en contacto con la solución, se obtiene:
[ ]pCO 4DKK K
Ca22
1 2
2 3= + (1.53)
donde: D: Coeficiente de difusión del CO2.
Si se aplica logaritmos a la ecuación 1.53 se obtiene: log pCO2 = 3 log[Ca2+]+ C (1.54) donde: C: Magnitud que es función de D, K1, K2 y Kc.

Figura 1.4 Diagrama de Muxart pH en función de CaCO3.
La expresión 1.54 corresponde a la ecuación teórica que sirvió de base para la confección de otros diagramas para la medición de la agresividad de las aguas kársticas (Figura 1.5), elaborados por Roques (1969 a) y Muxart (1972). Esta última investigadora, encontró mediante experimentos de laboratorio, que:
log C = 0,695 log T - 9,55 (1.55)

Figura 1.5 Diagrama de Muxart p CO2 en función de CaCO3.

A manera de resumen en relación a las deducciones anteriores, las ecuaciones que permiten hallar todas las incógnitas del sistema CO2-H2 O-CaCO3 son: 1.31, 1.44, 1.45, 1.47, 1.50 y 1.53.
Para resolver el sistema de ecuaciones expuesto en el párrafo anterior es necesario fijar una de las variables, por ejemplo el pH. El contenido relativo de CO2, HCO3
- y CO32-, también puede
deducirse en función del pH a partir de estas ecuaciones y representarse en forma gráfica (Figura 1.6).
En este tratamiento (Figura 1.6) sólo se tomó en cuenta la disolución de la calcita por el agua natural en ausencia de otros minerales. Sin embargo, lo más común en la naturaleza es que este proceso ocurra en presencia de otros minerales, principalmente dolomita, yeso y halita. En esas condiciones se debe tener en cuenta en los cálculos las reacciones de disociación y los equilibrios químicos correspondientes: CaMg(CO3)2 (s) = Ca2+ (ac) + Mg2+ (ac) + 2 CO3
2- (ac) (1.56) Dolomita Kd = (Ca2+)(Mg2+)(CO3
2-)2 CaSO4.2 H2O (s) = Ca2+ (ac) + SO4
2- (ac) + 2 H2O (l) (1.57) Yeso Ky = (Ca2+) (SO4
2- ) (1.58) Kh = (Na+)(Cl-) (1.59)

Figura 1.6 Concentraciones relativas de CO2 , HCO3 - y CO3
2- en función del pH..

Formación de iones complejos o pares iónicos En el proceso de disolución de los minerales por las aguas naturales, además de los iones
libres comúnmente conocidos (HCO3- , CO3
2-, Cl-, NO3-, SO4
2-, Ca2+, Mg2+, Na+ y K+), se originan en cantidades muy pequeñas los denominados iones complejos y pares iónicos: CaHCO3
+, CaCO3º, MgHCO3
+, MgCO3º, NaCO3-, NaHCO3º, CaSO4º, MgSO4º, NaSO4
-, HSO4- y otros. A pesar de su
pequeña concentración, cuando no se consideran en los cálculos cuantitativos de la agresividad de las aguas kársticas se pueden cometer errores.
Se ha demostrado que la adición de pequeñas cantidades de MgCl2, incrementa considerablemente la solubilidad de la calcita como consecuencia de la formación de MgHCO3
+ y MgCO3º, los cuales tienden a perturbar el equilibrio eléctrico entre Mg2+ y Cl-, provocando un aumento en la disolución de la calcita (Roques, 1964; Muxart (1972).
Wigley (1973 b), al estudiar las aguas procedentes de manantiales en cuencas desarrolladas en los yesos y carbonatos de la Columbia Británica con un contenido de sólidos solubles del orden de 1 700 mg/L, encontró que 70,6 % de los iones de Ca2+ aparecían como iones libres, mientras que 26,7 % se encontraban en forma de pares iónicos, como SO4
2-, 1,7 % como HCO3
- y 1 % como CO32-. En el caso del ion Mg2+, la relación entre iones libres y pares
iones era más o menos semejante. El cálculo de las concentraciones de los iones complejos y pares iónicos involucrados en los
sistemas CO2-H2O-CaCO3 y CO2-H2O-CaMg(CO3)2, se puede realizar mediante métodos numéricos (Fagundo et al, 1986). Para ello es necesario tomar en consideración un sistema de ecuaciones de equilibrio químico, que contemple tanto los iones libres como los iones complejos o pares iónicos, estos últimos se relacionan a continuación:
K (Ca )(HCO )(CaHCO )3
23
3
=+ −
+ (1.60)
K (Ca )(CO )(CaCO )4
232
3o=
+ −
(1.61)
K (Mg )(HCO )(MgHCO )5
23
3
=+ −
+ (1.62)
K (Mg )(CO )(MgCO )6
232
3o=
+ −
(1.63)
K (Na )(CO )(NaCO )7
32
3
=+ −
− (1.64)
K (Na )(HCO )(NaHCO )8
3
3o=
+ −
(1.65)

K (Ca )(SO )(CaSO )9
24
2
4o=
+ −
(1.66)
K (Mg )(SO )(MgSO )10
24
2
4o=
+ −
(1.67)
K (Na )(SO )(NaSO )11
42
4
=+ −
− (1.68)
K (K )(SO )(KSO )12
42
4
=+ −
− (1.69)
K (H )(SO )(HSO )13
42
4
=+ −
− (1.70)
Por último, habrá que considerar, además las ecuaciones de balance de masas siguientes:
m Ca2+ total = m Ca2+ + m CaHCO
3+ m CaCO3º + m CaSO4 (1.71)
m Mg2+ total = m Mg2+ + m MgHCO3+ + m MgCO3
º + m MgSO42- (1.72)
m Na+ total = m Na+ + m NaCO3- + m NaHCO3
º + m NaSO4-
(1.73) m K+ total = m K+ + m KSO4- (1.74)
m SO42- total = m SO4
2- + m CaSO4º + m MgSO4
º + m NaSO4- + m KSO4
-
+ m HSO4-
(1.75)
m HCO3- total = m HCO3
- + m CaHCO3+ + m MgHCO3
+ + m CaCO3º + m MgCO3º
+ m CO32- + m NaCO3
- + m NaHCO3º (1.76) Variación de la constante de equilibrio con la temperatura
La constante K varía con la temperatura, para conocer su valor a la temperatura del agua se puede utilizar la expresión de Vant Hoff:
dlnKdT
HRT
o
2=∆
(1.77)
donde: ∆H°: Entalpía de reacción. R : Constante de los gases ideales. T : Temperatura.

En la tabla 1.4 se presentan las constantes de equilibrio para el sistema de disolución de los minerales carbonatados, el yeso y la halita. Del análisis de la misma se infiere que las ecuaciones correspondientes a la disociación del H2 CO3 en HCO3
- (1.36), del HCO3- en CO3
2- (1.37) y de la disolución del yeso en agua (1.58) son más solubles a altas temperaturas, mientras que las de disociación del CO2 en agua (1.34) y las de disolución de los minerales de calcita (1.40), dolomita (1.56) y halita (1.59) son favorables a bajas temperaturas.
Constante de
equilibrio
Temperatura (°C)
pki 0 5 10 15 20 25 30 50 70 90 pkb 1,12 1,19 1,21 1,34 1,41 1,47 1,52 1,72 1,85 1,92 pk1 6,58 6,52 6,46 6,42 6,38 6,35 6,33 6,29 6,32 6,28 pk2 10,63 10,56 10,49 10,43 10,38 10,33 10,29 10,17 10,15 10,14 pkc 8,38 8,39 8,41 8,42 8,45 8,49 8,57 8,66 8,85 9,36 pkarg 8,22 8,24 8,26 8,28 8,31 8,34 8,37 8,54 8,73 9,02 pkd 16,56 16,71 16,75 16,89 17,00 17,90 pky 4,65 4,61 pkh -1,52 -1,58 pkCaCO3 1,40 pkMgHCO3
+ 0,95 pkCaCO3
º 3,1 pkMgCO3
º 3,4 pkCaSO4
º 2,31 pkMgSO4
º 2,5 pkNaSO4
- 0,96 pkKSO4
- 1,27 pkHSO4
- -0,25 pkNaCO3
- 2,00 pkNaHCO3 0,72
Tabla 1.4 Variación de las constantes de equilibrio de los sistemas del carbonato, sulfato y cloruro con la temperatura
En la figura 1.7 se ilustra la variación de la solubilidad del yeso y la calcita con la temperatura. Este comportamiento demuestra la complejidad de los procesos kársticos en condiciones naturales. En muchas regiones kársticas desarrolladas en yeso, las aguas adquieren una concentración de CaSO4 superior a 2 000 mg/L. En los terrenos kársticos carbonatado, por lo general, la concentración de CaCO3 mayores de 350 mg/L se deben a la influencia de sulfatos o cloruros. Sistemas abierto y cerrado respecto al CO2
Si en el proceso de disolución de los minerales carbonatados por las aguas naturales existe un suministro abundante de CO2, que puede considerarse constante durante la reacción, éste procede rápidamente hasta la saturación. En este caso la reacción tiene lugar en condiciones de sistema abierto respecto al CO2 (Figura 1.8a). Si por el contrario, la disolución de los carbonatos por las aguas naturales se produce en virtud del suministro inicial de CO2, que luego no se repone en el transcurso de la reacción, ésta se produce en condiciones de sistema cerrado respecto al CO2.

Figura 1.7 Solubilidad de la calcita y el yeso en agua entre 2 y 25 °C y 1 bar (1 atm) de presión.

Figura 1.8 Disolución de la calcita: a) en condiciones de sistema abierto respecto al CO2; b) en condiciones de sistemas cerrado con respecto al CO2 (Freeze y Cherry,1980)
En el laboratorio la condición de disolución en sistema abierto se consigue aplicando al
reactor una presión de CO2 constante con la ayuda de un manómetro. También, dejando el reactor abierto en contacto con la pCO2 de la atmósfera.
En los experimentos de laboratorio la condición de disolución en sistema cerrado se logra inyectando una cantidad de gas al reactor, donde se efectúa el experimento y luego se cierra el reactor, aislándolo del CO2 atmosférico. También esto se logra, al poner la atmósfera del reactor en contacto con otro gas como nitrógeno. En estas circunstancias, la cantidad de carbonatos disueltos al alcanzar el equilibrio es menor, mientras el pH se eleva más que en las condiciones de sistema abierto (Figura 1.8b).
En la naturaleza tienden a primar, por lo general, las condiciones de disolución de los carbonatos en sistema abierto o más bien de tipo intermedio entre sistema abierto y cerrado. Las características del proceso natural difieren significativamente del simulado en el laboratorio, lo cual se debe a que en el medio natural existe una gran variabilidad en el contenido de CO2, como resultado del aporte en forma irregular de diferentes fuentes y su evolución en el karst.
Como ejemplos de disolución de carbonatos en la naturaleza, los cuales tienden a semejar las condiciones de sistema abierto se pueden citar:
1. Cuando el agua procedente de las precipitaciones se infiltra lentamente en un suelo kárstico con buen drenaje y abundantes oquedades aireadas, adquiere una elevada pCO2 y la difusión del gas en el agua se produce como si el suministro del mismo fuera constante. Si en esas condiciones el agua interactúa con rocas carbonatadas subyacentes, evoluciona adquiriendo un

elevado contenido de Ca2+, Mg2+ y HCO3-. En esas condiciones el pH se va incrementando a
medida que avanza la disolución de los carbonatos hasta alcanzar un valor cercano a 8. 2. La disolución en lagos, presas y estanques, donde el CO2 se toma directamente de la
atmósfera y se utiliza por las aguas para disolver las paredes rocosas. Como ejemplos de disolución de carbonatos en la naturaleza, los cuales tienden a producirse
en condiciones de sistema cerrado respecto al CO2 se pueden considerar los siguientes: 1. La disolución de las calizas en la zona freática profunda (zona de circulación y saturación
profunda del karst). A esta región pueden llegar las aguas kársticas, conteniendo aún una elevada pCO2. En esas condiciones, las aguas continúan disolviendo los carbonatos sin el aporte de nuevos suministros de gas.
2. Cuando un agua procedente de las precipitaciones se pone en contacto con una determinada pCO2 en la zona del suelo, luego se infiltra a través de una cobertura no kárstica (por ejemplo: depósitos de tipo fluvial o fluvioglaciar) y, por último alcanza las secuencias carbonatadas. El agua en esas condiciones disuelve la roca, consumiendo el CO2 que originalmente adquirió en la zona del suelo, sin recibir apenas aportes nuevos de este gas.
3. Cuando un agua superficial procedente de una región no kárstica intercepta un macizo carbonatado. En su recorrido hipogeo disuelve los carbonatos a expensas de la pCO2 inicial de la fuente epigea. En la práctica sin embargo, esto ocurre en condiciones intermedias entre sistemas abierto y cerrado, porque en el curso transfluente la corriente también recibe aportes autóctonos no despreciables, con otro contenido de CO2.
Efecto de ion común
Cuando un agua natural pasa por un terreno donde existen minerales con iones comunes a los que ésta lleva disuelto en su seno, disminuye su capacidad para disolver esos minerales; por ejemplo: si un agua natural se encuentra saturada en calcita y en su movimiento pasa por un terreno rico en yeso, se tendrán las reacciones 1.40 y 1.57.
De acuerdo con el principio de Lechatelier, los iones Ca2+ adicionales procedentes del equilibrio de la disolución del yeso, tenderán a llevar la primera reacción química (1.40) hacia la izquierda, depositándose CaCO3. En términos de molalidades y coeficientes de actividades, la constante de equilibrio toma el valor siguiente:
Kc = γ Ca2+ γ CO3
2- [ Ca2+] [CO32- ] (1.78)
La disolución del yeso da lugar a que el producto de las actividades γCa γCO3 decrezca, pero por la contribución de Ca2+ procedente del yeso, el producto de las concentraciones [Ca2+][CO3
2-] se incrementa. Por tanto, para que la solución se mantenga en equilibrio con respecto a la calcita, debe producirse la precipitación de CaCO3.
La adición de 100 mg/L de Ca2+ procedente del yeso a 10 °C, reduce el contenido de CaCO3 del agua kárstica desde 100 mg/L a 66 mg/L. En general, cuando la fuerza iónica del agua es menor de 0,1, la solubilidad en relación a la calcita y la dolomita se reduce considerablemente si la misma se pone en contacto con un afloramiento de yeso.
En concordancia con el principio químico-físico del efecto de ion común, Gerstenhauer y Pfeiffer (1966) demostraron, mediante experimentos de laboratorio, que un contenido pequeño de

MgCO3 en las calizas reducía significativamente la cantidad de CaCO3 disuelto. Ellos encontraron que en 28 h se disolvían 17 ppm de calcita en ausencia de MgCO3, 6 ppm en presencia de 2 % de este mineral y sólo 3 ppm cuando la calcita contenía 5 % de impureza. Efecto salino o de fuerza iónica
Si se compara la solubilidad de los minerales en agua pura con la de un agua que contiene iones no comunes al proceso de disolución de ese mineral, se demuestra que la salinidad incrementa la solubilidad del mismo. Esto se debe a que, como consecuencia del incremento de la concentración, aumenta la fuerza iónica (µ), como se demuestra en la ecuación 1.6 y se produce una disminución del coeficiente de actividad, como se deduce de la ecuación 1.5 de Debye-Hückel. Este fenómeno se denomina efecto salino o de fuerza iónica.
En la figura 1.9 se ilustra este efecto para el caso de la disolución del yeso en aguas con diferente contenido de NaCl disuelto. A medida que aumenta la concentración de NaCl en la solución, aumenta la solubilidad del mineral, a causa de que disminuye el producto de los coeficientes de las actividades de los iones calcio y sulfato γCa2+ γ SO4
2- al incrementarse µ.
Figura 1.9 Incremento de la solubilidad del yeso por efecto salino o de fuerza iónica (Freeze y Cherry,1980) Potencial de oxidación-reducción

Muchas reacciones químicas que tienen lugar en el medio acuático y en especial en el sistema de las aguas subterráneas, implican trasferencia de electrones entre constituyentes disueltos, gases o sólidos. Como resultados de estas transferencias se producen cambios en los estados de oxidación-reducción de los reaccionantes y los productos.
Los elementos de valencia múltiples son susceptibles de intervenir en las reacciones de oxidación-reducción, mediante las cuales una molécula se reduce (oxidante) al tomar los electrones que le cede la otra (reductora), la cual se oxida. La reacción también es capaz de proceder en dirección contraria, de modo que este proceso se puede representar mediante la ecuación de equilibrio siguiente:
Oxidante + ne = Reductor (1.79) donde ambas moléculas (oxidante-reductor) constituyen un par redox. El proceso ocurre con la intervención de ambos tipos de moléculas, una oxidante y otra reductora, para que se pueda producir el intercambio de electrones. La reacción es más representativa mediante la interacción de dos sistemas redox: a Oxidante 1 + ne = a Reductor 1 (1.80) b Reductor 2 + ne = b Oxidante 2 (1.81) ------------------------------------------------------------- a Oxidante + b Reductor = a Reductor + b Oxidante (1.82)
La fortaleza del par redox se mide por el potencial de oxidación-reducción o potencial
redox, mediante la ecuación de Nernst:
E E RTnF
ln (Oxidante)(Reductor)h h
o= + (1.83)
donde: Eh : Potencial de oxidación-reducción o potencial redox Ehº: Constante que depende de la naturaleza del oxidante y del reductor y se refiere a las condiciones estándares F : Constante de Faraday n : Número de electrones intercambiados
Si se expresa el logaritmo en forma decimal, se obtiene:
E E 0,23log (Oxidante)(Reductor)h h
o= + (1.84)
El potencial de oxidación-reducción se mide en voltios, a través de milivoltímetros de campo es posible obtener este valor en las aguas naturales.
La proporción de iones presentes en solución en forma de uno u otro de los estados de oxidación-reducción se puede deducir a partir de la fórmula 1.82. Para el caso de los equilibrios de los sulfatos, por ejemplo, se obtienen las expresiones siguientes:

E 0,51 0,03log (S )(H )h
2
= −−
+ (1.85)
E 0,28 0,03log (HS )(H )h = − −
−
+ (1.86)
E 0,13 0,03log (H S)(H )h
2= − + (1.87)
correspondientes respectivamente a los pares redox: S2- (ac) - e = S (s) (1.88) HS- (ac) - e = S (s) + H+ (ac) (1.89) H2S (g) - 2 e = S (s) + 2 H+ (ac) (1.90)
Mediante las ecuaciones 1.88, 1.89 y 1.90 se puede calcular la concentración de los compuestos del azufre en sus diferentes formas en función del pH.
Aunque las soluciones acuosas no contienen electrones libres, también es conveniente expresar el proceso de oxidación-reducción mediante un par redox, como si estas reacciones se produjeran por separado. Además, se suele utilizar la magnitud pE para describir la actividad relativa de los electrones. Por definición:
pE = - log (e) (1.91)
El pE es una magnitud adimensional análoga a la expresión pH, utilizada para describir la actividad del protón o ion hidrógeno. El pE de una solución mide su tendencia oxidante o reductora.
Al igual que se asigna arbitrariamente el valor G° = 0 para la reacción de hidratación (H+ + H2O = H3O
+), el cambio de energía libre para la reducción de H a H2 (H+ e = 1/2 H2 (g)) es también cero; pE y pH son funciones de energía libre involucradas en la transferencia de un electrón y un protón respectivamente.
En forma general, una ecuación de tipo redox se puede formular mediante la expresión:
a A + b B + n e = c C + d D (1.92) y de acuerdo con la ley de acción de masas:
K (C) (D)e (A) (B)
c d
n a= b (1.93)
Para ilustrar la aplicación de la ley de acción de masas a las reacciones redox en condiciones
de equilibrio químico, se puede tomar, por ejemplo, la oxidación del ion ferroso a férrico por el oxígeno libre disuelto en una solución acuosa: 1/2 O2 (g) + 2 H+ (ac) + 2 e = H2O (l) (reducción) (1.94) 2 Fe2+ (ac) = 2 Fe3+ (ac) + 2 e (oxidación) (1.95)

-------------------------------------------------------------------------------------- 1/2 O2(g) + 2 Fe2+ (ac) + 2 H+ (ac) = 2 Fe3+ (ac)+ H2O (l) (1.96) Para la determinación de la constante de equilibrio en las reacciones redox, se toma como convenio expresar las ecuaciones de cada par redox en la forma reducida. Las formas oxidadas y los electrones se colocan a la izquierda y las formas reducidas a la derecha. Al aplicar la ley de acción de masas a las ecuaciones anteriores (1.94 y 1.95), para la condición de equilibrio a 25 °C, se obtienen:
K 1(pO ) (H )(e)
102
241,35
12
= + = (1.97)
K (Fe )(Fe )(e)
102
312,53= =
+
+ (1.98)
Si las reacciones redox (1.80 y 1.81) se encuentran en equilibrio y se conoce la
concentración de hierro, el pO2, así como pH, el pE obtenido experimentalmente es el mismo en ambos casos.
En los sistemas acuáticos, existe una interdependencia entre el pH y el pE, porque en muchos procesos donde intervienen especies disueltas se producen transferencias de protones o electrones. En condiciones de equilibrio, las reacciones asociadas al pH pueden ser expresadas en forma gráfica mediante los denominados diagramas Ep vs pH, desarrollados por Pourbaix (1963), tal como se ilustra en la figura 1.10.

Figura 1.10 Diagrama pE vs pH (Freeze y Cherry, 1980). Intercambio iónico y adsorción
Los materiales geológicos porosos están compuestos de un porcentaje apreciable de partículas de tamaño coloidal que tienen la capacidad de intercambiar iones adsorbidos en su superficie.
Como resultado del proceso de intemperismo de las rocas por los agentes exógenos se originan los suelos, constituidos por minerales de tipo arcilloso. Estos minerales se subdividen en cinco grupos: caolinita, montmorillonita, ilita, clorita y vermiculita; los cuales se caracterizan por

presentar tamaños de partículas del orden de los coloides, originando procesos de intercambio iónico entre las arcillas de la zona del suelo y las aguas que discurren a través de las mismas.
En la superficie de estos minerales se originan cargas eléctricas. En su interacción con el agua los sitios cargados adquieren grupos OH- que quedan adsorbidos en la superficie del mineral y otras capas de cationes, así como aniones que tienden a ser también adsorbidos en la zona adyacente a la capa hidroxilada, estableciéndose una carga neta que puede ser positiva o negativa, en dependencia de las condiciones ácido-básicas presentes en el medio (Parks, 1967). Si el pH es bajo, prevalece una carga superficial positiva, mientras que a pH elevados se desarrolla una carga superficial negativa, por tanto, los minerales arcillosos tenderán a adsorber cationes o aniones en dependencia del pH.
La capacidad de intercambio catiónico (CIC) de un material geológico coloidal, se expresa por el número de miliequivalentes de cationes que pueden ser intercambiados en una muestra con una masa seca de 100 g. El ensayo para la determinación de la CIC consiste en saturar los sitios intercambiables por tratamiento con una solución concentrada de acetato de amónio; ajuste del pH a 7; eliminación del ion NH4
+ adsorbido, lavando con una solución concentrada de NaCl (el Na+ reemplaza al NH4
+en los sitios intercambiables) y finalmente, se determina el contenido de Na+ de la solución de lavado después de alcanzado el equilibrio.
El concepto de CIC puede ilustrarse en el mineral montmorillonita, en el cual 0,67 moles de Mg2+ sustituyen isomórficamente al Al3+, esto puede expresarse como: CI (Si8)(Al3,33 Mg0,67)O20 (OH)4, donde CI representa los cationes intercambiables. Se puede asumir que los CI están constituidos sólo por Na+ y tomar el peso fórmula de la montmorillonita que es 734. Para balancear las cargas negativas causadas por la sustitución de Mg2+, se requiere en este caso de 0,67 moles de cationes de Na+ por 734 g de arcilla. En términos de las unidades empleadas para expresar el valor de la CIC se obtiene:
CIC 0,67734
x10 x100 91,5meq / 100g3= =
Para aplicar la ley de acción de masas a un proceso de intercambio iónico en condiciones de
equilibrio, se asume que el sistema de intercambio consta de dos fases discretas, la fase en solución y la fase intercambiable, constituyendo esta última todo o parte del medio poroso. El proceso de intercambio iónico se representa por: aA + bB(ad) = aA(ad) + bB (1.99) donde: A y B: Iones intercambiables. a y b: Moles de los reaccionantes. B(ad) y A(ad): Iones adsorbidos.
Al aplicar la ley de acción de masas se obtiene:

K(A ) (B)(A) (B )A B
(ad)a b
a(ad)
b− = (1.100)
En el caso del intercambio entre Na+ y Ca2+, el cual es muy importante en muchos sistemas de
aguas naturales, la ecuación de intercambio es: 2 Na+ + Ca(ad) = Ca2+ + 2 Na(ad) (1.101) y la de equilibrio químico correspondiente:
K(Ca )(Na )(Na )(Ca )Na Ca
2(ad)
2
(ad)−
+
+= (1.102)
La relación de las actividades iónicas en solución se puede expresarse en términos de
molalidades y coeficientes de actividades:
[ ][ ]
[ ][ ]
BA
B BA A
b
a
b b
a a=γγ
(1.103)
Vanselow propuso que las actividades de los iones adsorbidos se consideraran iguales a las
fracciones molales (Freeze y Cherry, 1980). En el caso de la ecuación 1.03, éstas serían:
[ ][ ] [ ]
NA
A BA =+
(1.104)
[ ][ ] [ ]
NB
A BB =+
(1.105)
donde: NA y NB : Fracción molar de los iones A y B respectivamente, los cuales se expresan en moles de ion adsorbidos.
La ecuación de equilibrio químico (1.100) se transforma entonces en:
[ ][ ]
kB B NA A N
(A B)
b bA
a(ad)
a aB
b(ad)
− =γγ
(1.106)
Vanselow y otros investigadores han encontrado, en forma experimental, que en muchos procesos de intercambio iónico con minerales arcillosos k(A-B) es constante, y se le ha denominado coeficiente de selectividad. Además, se ha extendido el uso del coeficiente de actividad de los iones adsorbidos, en forma análoga al coeficiente de actividad del soluto.
γA(A )N (ad)
(ad)(ad)
A
= (1.107)

γB(B )N (ad)
(ad)(ad)
B
= (1.108)
Al aplicar la ley de acción de masas en las ecuaciones 1.107 y 1.08; quedan relacionadas
la constante kA-B con el coeficiente de selectividad:
K A (ad)B (ad)
k(A B)
a
b (A B)− −=γγ (1.109)

Capítulo 2. Procesos de disolución y precipitación de los minerales de las rocas karstificables.

Medición de la agresividad de las aguas kársticas. Johnston y Williamson (1916) desarrollaron un trabajo teórico bastante completo sobre el
sistema CO2-H2O-CaCO3, así como crearon las bases para la interpretación de los procesos de la disolución de los carbonatos. A principios de la década del treinta, Tillman (1932) determinó la saturación límite de una solución de CaCO3, en relación con el contenido de dióxido de carbono (CO2), el pH y la temperatura. Como resultado de este trabajo se creó un diagrama de estabilidad de los compuestos del carbono posteriormente popularizado por Trombe (1952), quien lo utilizó para medir la agresividad de las aguas kársticas.
Los experimentos de Tillman se basan en la relación que existe entre dureza (CaCO3), CO2 y pH. Sin embargo, en condiciones naturales estas relaciones no resultan muy satisfactorias, como se demuestra en los resultados efectuados en el período 1969-1970 en los Tatras Occidentales por Pulina (1974). Otros investigadores como Laptiew, Miller, Sztieriny y Florove, Trombe, Wlasova y colaboradores, Swysen (entre 1939 y 1971, en Pulina, 1974); así como Roques (1962 a, b; 1964), realizaron experimentos, en laboratorio, sobre la disolución de los carbonatos, similares a los efectuados por Tillman, donde se tomó en cuenta, además de las relaciones del CaCO3 con el CO2 y la temperatura, los diferentes tipos petrográficos, así como los aspectos relacionados con la cinética del proceso de disolución.
Los estudios teóricos y experimentales mencionados en el párrafo anterior no llegaron a resolver los problemas, que ocurren en la naturaleza en relación a la disolución de los carbonatos. Los experimentos se efectuaron en condiciones de sistema cerrado, que son menos típicos en condiciones naturales. A pesar de que estos trabajos permitieron obtener el límite teórico de saturación de CO2 para diferentes temperaturas, no ofrecieron la posibilidad de medir la velocidad de estos procesos. En trabajos posteriores se han abordado estos aspectos (Roques, 1969 a, b; 1973; Roques y Ek, 1973).
Un método de medición de la agresividad del agua se basa en la denominada agresividad potencial, la cual consiste en determinar la posibilidad que tiene un agua en un momento dado, para disolver una cierta cantidad de carbonato.
Para la determinación cuantitativa de la agresividad potencial de las aguas kársticas, se ha utilizado tradicionalmente el denominado diagrama de Tillman-Trombe o la versión modificada por Roques (1972).
Para la determinación cuantitativa de la agresividad en condiciones de sistema cerrado, se ha empleado el diagrama de Tillman-Trombe modificado por Schmit. Este método gráfico se utilizó para clasificar la región de Piryny, en Bulgaria (Markowicz, Popov y Pulina, 1972). También se midió la agresividad potencial según establece el método de Hayers (Markowicz y Pulina, 1979). En el mismo, cierta cantidad de mármol o roca carbonatada de la región objeto de estudio es dejada en contacto con las aguas de los arroyos de esa zona, dentro de recipientes (en los mismos sitios de estudio) a la temperatura de esas aguas. Al cabo de cierto tiempo se mide la cantidad de CaCO3 disuelto. El proceso de disolución en sistema abierto o intermedio entre sistema abierto y cerrado respecto al CO2 , tal como ocurre en la naturaleza es menos conocido, por la variabilidad de los elementos químico-físicos que intervienen en el camino que sigue un agua kárstica durante su trayectoria.
Las observaciones realizadas por Pulina en Siberia y los experimentos llevados a cabo en la Universidad de Irkucku (Pulina, 1971), basados en mediciones de la cantidad de CaCO3 disuelto por las aguas en su infiltración vertical, pusieron de manifiesto que esta magnitud era prácticamente

perpendicular a la curva de saturación en el diagrama de Tillman-Trombe. Resultados similares fueron encontrados en los Sudetes, Tatras y Vercour (Pulina, 1971).
Sobre la base de este comportamiento, observado en un gran número de mediciones en diferentes regiones, se propuso el desarrollo de experimentos de laboratorio en condiciones de sistema abierto. Roques (1964) y Markowicz (1968) realizaron los cálculos teóricos en estas condiciones y posteriormente Muxart (1969; 1972) trabajó en las condiciones experimentales, proponiendo un nuevo método gráfico para determinar la agresividad del agua. El mismo se basa en relaciones de tipo lineal, mediante papel semilogarítmico entre el pH y el CaCO3 y mediante papel doble logarítmico entre CO2 y el CaCO3 (ver figuras 1.5 y 1.6). A pesar de las ventajas que presenta este método, los resultados obtenidos no reflejan con toda precisión el proceso de disolución de los carbonatos.
Otra vía alternativa para evaluar la capacidad de disolución de los carbonatos por las aguas naturales, ha sido el empleo de los denominados índices de saturación. Roques (1972) propuso:
∆pH = pHm - pHe (2.1) donde : ∆pH: Diferencia entre el pH medido en el agua (pHm) y el pH en el equilibrio (pHe).
Al aplicar el sistema de equilibrio de los carbonatos, Roques llegó a la expresión:
∆pH pHm logK logK log(Ca ) log(HCO ) log (1 K )(SO )c 2
23
5
42= − + + + +
−+ −− (2.2)
donde: C: Magnitud que expresa la influencia de los pares iónicos MgHCO3
+, MgCO3 0, CaHCO3
+ y CaCO3
0. Roques encontró que C = 0.051 526 (M Mg2+ ) 0,505 662
Kc, K1 y K5: Constantes de equilibrio.
Si pH > 0, pHm > pHe, la solución se encuentra saturada. Si pH < 0, pHm < pHe la solución se encuentra insaturada, tendiendo a disolver más carbonato.
La magnitud ∆pH tiene la misma significación que los índices de saturación definidos por Back y colaboradores (1966), los cuales son los siguientes:
RSC log (Ca )(CO )K
23
2
c
=+ −
(2.3)
RSD log (Ca )(Mg )(CO )K
2 23
2 2
d
=+ + −
(2.4)

RSY log (Ca )(SO )K
24
2
y
=+ −
(2.5)
donde: RSC: Relación de saturación de la calcita. RSD: Relación de saturación de la dolomita. RSY: Relación de saturación del yeso. Kd y Ky Constantes del producto de solubilidad de la dolomita y el yeso respectivamente.
En forma general, estos índices (ecuaciones 2.3, 2.4 y 2.5) se suelen expresar mediante la
expresión:
SI log KK
IAP
eq
= (2.6)
donde: SI: Índice de solubilidad. KIAP: Producto de actividad iónica. Keq : Constante termodinámica de equilibrio del mineral.
Con el objetivo de facilitar los cálculos, los índices de Back, así como las incógnitas del sistema de equilibrios químicos se han resuelto mediante la implementación de métodos de computación (Jacobson y Langmuir, 1975; Kempe, 1975 a; Fagundo y Valdés, 1975; Wigley, 1977; Fagundo et al, 1986; Alvarez et al, 1993). Los resultados de este tratamiento se han expresado gráficamente en función de la dureza y de la conductividad eléctrica (Fagundo, 1982), tal como se ilustra en la figura 2.1.
Se ha demostrado (Rodríguez, Fagundo y Spassov, 1991) que existe buena correlación matemática y no son apreciables las diferencias entre los índices RSC y ∆pH. A partir de las ecuaciones teóricas del sistema de equilibrios de los carbonatos, fueron deducidas expresiones numéricas de pH y log pCO2 en función de la conductividad eléctrica de las aguas kársticas (Fagundo, 1982), las cuales permiten controlar el contenido de CO2 disuelto y el valor del pH mediante mediciones con conductímetros de campo (Figuras 2.2 y 2.3).
La mayoría de los especialistas de los países occidentales han utilizado el método de Back para medir la capacidad de las aguas para disolver los carbonatos. Según Ford (1985), en Estados Unidos y Canadá los investigadores han hecho sus cálculos sobre la base del algoritmo elaborado por Wigley (1977).
Ford ha destacado que los métodos gráficos son desconocidos o no empleados por los científicos de los países occidentales, a pesar de las ventajas que ofrecen los mismos para evaluar en forma rápida la cantidad de CaCO3 que se disuelve o deposita a lo largo de la trayectoria de una corriente kárstica, así como para estimar la evolución del pH de las aguas. Esta ventaja sobre los métodos basados en los índices de saturación de Back, también se ha resaltado por Bakalowicz (1979).

Figura 2.1 Representación de los índices de saturación de las aguas (RSC, RSD y RSY) en función de la conductividad eléctrica: cuenca del río Cuyaguateje (Pinar del Río).

Figura 2.2 Diagrama de pH en función de la conductividad eléctrica.

Figura 2.3 Diagrama de pCO2 en función de la conductividad eléctrica.
En Europa la mayoría de los karstólogos prefieren emplear los métodos gráficos, aunque también se han empleado los métodos computarizados, los cuales calculan los índices de saturación de la calcita, la dolomita, el yeso, así como de otros minerales.
En relación a los distintos métodos propuestos para medir el grado de agresividad de un agua, así como a algunos aspectos relacionados con la misma, se puede señalar lo siguiente:
1. A pesar de las facilidades que ofrecen los métodos actuales basados en índices calculados por computación, los mismos sólo reportan cifras abstractas que tienen valor relativo, pero que no reflejan la cantidad exacta de CaCO3 disuelto o precipitado, ni el proceso a lo largo de la trayectoria del agua.
2. El cálculo de la agresividad en los restantes tres métodos se basan en el diagrama de Tillman-Trombe modificado. De los trabajos efectuados en los laboratorios para simular el proceso de disolución de los carbonatos, los llevados a cabo por Muxart se acercan más a las condiciones en que estos ocurren en la naturaleza. Estos trabajos han hecho posible una corrección más precisa de los diagramas de saturación.

3. Los resultados obtenidos empíricamente, mediante un número elevado de mediciones en diferentes regiones de clima templado, demuestran que las aguas en su camino de evolución, tienden a seguir una dirección perpendicular a las curvas de saturación.
4. La determinación cualitativa de la agresividad no ofrece dificultades apreciables, tanto mediante los métodos gráficos como los computarizados.
5. Las mayores diferencias entre los distintos métodos se obtienen en aguas muy agresivas (con poca mineralización y bajo pH) y las mínimas se obtienen en aguas altamente mineralizadas.
6. Para que la determinación de la agresividad de un agua kárstica refleje el verdadero camino seguido por ésta en su evolución, es necesario que los análisis químicos se realicen en el campo y las muestras se tomen secuencialmente, primero en el momento en que el agua hace contacto con el macizo carbonatado, si es posible en el interior de los macizos y por último en las emergencias. Si el estudio se hace a lo largo de un perfil vertical, las muestras se deberán tomar a diferentes niveles. Los estudios llevados a cabo en el laboratorio, tendientes a simular estos procesos naturales, son muy difíciles a causa de la complejidad de los mismos.
7. La evolución química que sufre un agua kárstica ha sido poco estudiada en los países tropicales mediante los métodos gráficos de Tillman-Trombe o sus análogos modificados, comúnmente empleados en Europa. La principal dificultad que presenta la medición de la agresividad de las aguas kársticas tropicales, es la rapidez con que el agua evoluciona hacia la saturación.
Cuando las aguas son tomadas en las emergencias kársticas en esta región, por lo general, se encuentran saturadas o sobresaturadas. La materia orgánica, los procesos microbiológicos, así como el efecto de mezcla de agua, desempeñan en esta región un importante papel.
Intensidad de la denudación química
El proceso de tipo exógeno o de intemperismo, que ocurre por acción de las aguas sobre los macizos carbonatadas y que da como resultado la formación del karst, se conoce como denudación química. La intensidad con que ésta ocurre ha sido objeto de estudio de muchos investigadores, como son: Corbel (1959) y Pulina (1974; 1992). El método consiste en calcular la cantidad de CaCO3 eliminado del macizo kárstico por las aguas en un determinado período de tiempo.
El método propuesto por Pulina, actualmente uno de los más utilizados por los especialistas, es el siguiente: Dm = a . dT . q (2.7) donde: Dm: Denudación química, en m3/km2 ó mm/ 1 000 años. a: Coeficiente que para rocas carbonatadas toma el valor 12,6. dT: Mineralización de las aguas producida por la interacción agua-roca, en mg/L, siendo: dT = T - Ta T : Mineralización de las aguas en las emergencias. Ta: Mineralización de las aguas de las precipitaciones. q : Módulo de escurrimiento, en L/s, siendo:

q 1000 QS
=
Q: Caudal o gasto de la emergencia, en m3/s. S: Área de la cuenca de captación, en km2.
El empleo de este método (ecuación 2.7) u otros similares, ha permitido medir en forma cuantitativa la intensidad de los procesos de denudación química en diferentes regiones kársticas (Pulina, 1974; Sauro y Meneghel, 1986; Pulina et al, 1984; Fagundo et al, 1986; Rodríguez et al, 1989; 1991; 1992; 1993).
Un sistema computarizado denominado SAPHID (Sistema Automatizado para el Procesamiento de Datos Hidrogeológicos), ha sido implementado recientemente para calcular la intensidad de la denudación química y el balance hídrico en cuencas experimentales kársticas (Vinardell, Rodríguez y Fagundo, 1991). Factibilidad de las rocas al proceso de disolución
Un factor determinante en todo proceso de disolución de minerales, es sin duda, el estado de división de la partícula. Es bien conocido que mientras más superficie de exposición presenten los cristales al ataque del agua, más rápidamente se producirá su disolución, deteniéndose este proceso al alcanzar el agua su condición de saturación.
En la naturaleza, la factibilidad de un macizo carbonatado a la disolución química o karstificación, viene dada por la composición de las calizas; su estado de agrietamiento, porosidad y textura; el estado de yacencia de las secuencias carbonatadas y no carbonatadas, así como otros aspectos relacionados con la tectónica; el tiempo de contacto de las aguas con las rocas, la presencia o no de suelos y sus tipos; el contenido de materia orgánica en el suelo y sus condiciones pedoclimáticas; la actividad del hombre y otros factores.
La textura de las calizas al igual que la de otras rocas, está determinada por el carácter espacial de los cristales o elementos componentes. lo cual se puede deber al resultado de procesos de recristalización, presentándose como una textura de tipo cristalina, granular o microcristalina.
La porosidad de la roca está relacionada con la textura y depende de la relación entre el volumen de huecos respecto al volumen total de la misma. La porosidad se mide en tanto porciento de espacios vacíos o poros en relación al volumen total de masa de roca.
El proceso de karstificación se hará más o menos factible de acuerdo con la naturaleza, dimensiones, repartición y relaciones mutuas de oquedades.
Se han definido dos tipos de huecos: primarios y secundarios, los primeros se originaron durante el proceso de formación de las rocas, mientras que los segundos, son el resultado de los cambios sufridos por las mismas con posterioridad a su formación.
Algunos especialistas han considerado la existencia en el karst de varios tipos de oquedades, por ejemplo, el karstólogo español Fernández Rubio (1975), ha postulado lo siguiente:
1. Poros intersticiales: Porosidad primaria que se reduce frecuentemente por disposición de calcita durante la diagénesis o subsecuentemente.
2. Fisuras y fracturas: Facilitan el desarrollo de la porosidad secundaria por la acción disolvente del agua circulante a través de éstas. La estratigrafía también facilita la anisotropía primaria favorable para la circulación, aunque en menor grado que las fracturas.

3. Cavidades: La circulación del agua en las calizas agranda las oquedades originales y crea cavidades que tienden a integrarse direccionalmente. Estas cavidades adoptan formas diferentes en las distintas zonas hidrogeológicas.
La geomorfóloga inglesa Sweeting (1965), ha señalado que existen más de 500 tipos de calizas del Carbonífero en el norte de Inglaterra, cuyas porosidades varían entre 2 y 25 %. De acuerdo a sus estudios, en las micritas la porosidad media es de 2 % o menos, en las espartitas ésta varía entre 5 y 8 %, así como en las biomicritas, las cuales alcanzan entre 15 y 25 %. Estas últimas calizas son alrededor de dos veces más solubles que las espartitas y las micritas para un tiempo de contacto, así como condiciones de disolución similares. Las aguas que drenan las biomicritas alcanzan una dureza del orden de 160 a 180 ppm de CaCO3, mientras las que se escurren a través de las espartitas alcanzan sólo 80 ppm, en similares condiciones de tiempo y precipitaciones. Difusión del CO2 en el agua
El contenido medio de CO2 en el aire atmosférico es, según Hutchinson (1957), de 3,3 . 10-4 atm (bar), que de acuerdo con Bakalowicz (1979) corresponde a 0,6 mg de CO2/L de aire. Este valor varía en relación a la altitud y latitud. En las zonas montañosas esta magnitud es menor, del orden de 2 . 10-4 atm (Renault, 1979), al nivel del mar adquiere 10-4, mientras que en las zonas urbanas se eleva a 7 . 10-4 (Bögli, 1969; Dupessy y Lambert, 1978). En el suelo la cantidad de CO2 es mucho mayor, pudiendo alcanzar 0,1 atm (Bögli, 1980; Bachelier, 1968; Miotke, 1971; 1974).
El contenido del CO2 del aire de las cuevas en distintos países ha sido objeto de estudio de muchos karstólogos (Renault, 1971; 1979; Ek, 1969; 1979; Ek et al, 1968; 1969; 1981), así como su variación estacional. En general, la cantidad de este gas en las cavidades kársticas depende de la distancia de la fuente generadora (el suelo), la morfología de la cueva, las características de circulación del aire, la altura, el clima de la región y otros factores.
La disolución de CO2 en el agua es un fenómeno físico, que como se indicó en el capítulo 1 (ecuación 1.33) de este libro, está controlado por la ley de Henry: pCO2 = D (H2 CO3 ) donde: D: Coeficiente de difusión que depende de la temperatura. (H2 CO3 ): Actividad del CO2 disuelto en agua.
El coeficiente D determina la transición del CO2 del aire al agua, siendo su valor: Daire = 1 cm2/s (25 °C) Dagua = 0,95 . 104,5 cm2/s (0 °C)
Esto significa que la velocidad de difusión del CO2 es 10 000 veces menor en el agua que en
el aire. A partir de la ecuación 1.33 se deduce la expresión formulada por Trombe (1952): CO2 (g/L) = M pCO2 (2.8) donde: M: Coeficiente de difusión
Si se considera que un litro de CO2 pesa 1,964 g y que 44 es el peso molecular se obtendrá:

M 1D
1,96444
0,04636D
= = (2.9)
Los valores de M reportados por Roques (1964) difieren relativamente muy poco de los calculados por Trombe (1952) para temperaturas entre 0 y 25 °C. Mediante la expresión 2.8 se han podido crear nomogramas que permiten calcular la presión de CO2 en la atmósfera, en contacto con el agua, en función de la concentración del gas disuelto (Figura 2.4).

Figura 2.4 Nomograma para el cálculo de del contenido de CO2 de la atmósfera en contacto con el agua, en función del contenido del gas disuelto en la misma (Bögli, 1980). Generación de CO2 en el suelo

La fuente esencial generadora de CO2, que el agua adquiere en su recorrido subterráneo, es sin duda alguna el suelo (Schoeller, 1941; 1950).
El CO2 de la atmósfera del suelo puede exceder teóricamente, según Roques (1962 a) hasta 300 veces y según Trombe (1952), hasta 700 veces el del aire atmosférico. Las mediciones efectuadas por Bögli (1969) arrojaron un contenido de CO2 en el suelo de 10,2 %, que comparado con el de la atmósfera del aire (0,035 %), representa unas 291 veces más.
El mecanismo mediante el cual este gas se produce en la biósfera comprende la fotosíntesis de la materia orgánica, la respiración de las plantas y la ulterior descomposición de éstas; proceso que se lleva a cabo en virtud de la acción de las bacterias y otros microorganismos, lo cual se puede representar de la forma siguiente:
fotosíntesis n CO2+ n H2O ↔ n CH2O respiración descomposición
Durante el proceso de descomposición de las plantas, la actividad enzimática de las bacterias y otros microorganismos transforma la materia orgánica producida por la fotosíntesis en sustancias húmicas: ácidos húmicos y fúlvicos, los cuales a su vez, se descomponen produciendo CO2 y agua. Este proceso se conoce como humificación.
La respiración de las raíces de la macroflora y la actividad de los microbios descomponiendo la materia orgánica son procesos productores de CO2. De ahí la importancia que tienen los factores ecológicos en la formación del karst (Bárány-Kevei, 1992).
Bachelier (1968 a, b; 1971), da una información precisa y cuantitativa sobre el mecanismo de producción de CO2 en el suelo. Los factores determinantes en este proceso son, entre otros, los relacionados con el efecto del clima (temperatura, humedad); los que dependen de la naturaleza física del suelo (aeración, granulometría) y los dependientes de la composición química (presencia de minerales solubles, de elementos como el calcio y el hierro que facilitan la estabilidad estructural del suelo). Relacionados con estos factores se encuentran además, la presencia de abundante materia orgánica y de microorganismos (Bakalowicz, 1979).
En general, el contenido de CO2 del suelo es muy variable, influyendo además el tipo de suelo, textura, horizonte, profundidad, drenaje, cobertura vegetal, flora y fauna presentes, así como la duración de los períodos alternativos de las estaciones climáticas.
El efecto del clima sobre la producción de CO2 en el suelo, se determinó cuantitativamente de manera directa, a través del estudio de la variación estacional del contenido de CO2 del suelo a lo largo de un año y en forma indirecta, a través de la determinación del contenido de carbonatos disueltos en las aguas. Los experimentos llevados a cabo por Bachelier (1971) ponen de manifiesto, que la máxima cantidad de gas se produce en verano y la mínima en invierno a temperaturas inferiores a 0 °C.
Otro elemento del clima que ejerce un papel importante en el proceso de generación de CO2
es la humedad, cuyo valor óptimo según Bachelier es de 20 a 40 %. Un exceso por encima de este valor limita la actividad biológica.
Currie (1961) ha señalado que los factores pedológicos que tienden a facilitar la difusión del CO2 y el drenaje de las aguas, favorecen la producción de este gas en el suelo.

El efecto del pH y la composición química del suelo sobre la generación de CO2 se ha evaluado por Miotke (1974), quien encontró que cuando el pH del suelo se encuentra cercano a la neutralidad o es ligeramente básico, la actividad biológica es más eficiente que cuando el pH es ácido. La presencia de calcio en cantidades apreciables tiende también a facilitar la producción de CO2 en el suelo, puesto que permite la respiración homogénea de los complejos orgánicos en los distintos perfiles del suelo (Vedy et al, 1979).
Schoeller (1941; 1950) encontró que la cantidad de CO2 de las aguas subterráneas decrece con la altura y hacia las zonas desérticas, alcanzando un máximo en las regiones mediterráneas.
Miotke (1974), comparando distintos suelos de regiones kársticas, encontró valores máximos de CO2 en los suelos tropicales de Puerto Rico y Jamaica (0,6 a 6.10-2 atm), y mínimos en la región fría de las montañas Rocosas del Canadá (de 0,04 a 0,5.10-2 atm), así como Alaska (de 0,04 a 0,05.10-2 atm). En contraste con estos resultados, en las mediciones de Ca2+ y Mg2+ efectuadas en aguas kársticas, no encontró diferencias significativas entre clima tropical y templado. La opinión de este investigador es que el comportamiento del CO2 en esas aguas está relacionado con la cobertura vegetal del suelo y las reacciones cinéticas asociadas con el tiempo de residencia del agua en el acuífero.
Smith y Atkinson (1976) han reportado rangos medios de pCO2 entre 0,2 y 11,0 % en regiones tropicales, con un valor extremo de 17,5 %; mientras que en las zonas de clima templado reporta valores entre 0,1 y 3,5 %, así como contenidos ocasionalmente extremos de 10 %. En las regiones árticas se han observado valores de pCO2 entre 0,1 y 0,2 %, mientras que en la tundra alpina éstos han alcanzado entre 0,01 y 0,5 % (Miotke, 1974).
Las mediciones del contenido de CO2 en suelos de regiones kársticas situados en climas extremos, polar y tropical, en los ejemplos de Spitsbergen y Cuba, efectuados en forma indirecta a través de las mediciones de CO2 en el agua (Fagundo, Valdés y Pulina, 1987; 1990), demuestran que el proceso de generación de este gas es mucho más intenso en condiciones tropicales que en condiciones polares, tal como se puede observar en la tabla 2.1. De la misma forma el contenido medio de CaCO3 medido en las aguas cubanas en superior al de las aguas de esa región polar.
Situación Fecha País CO2
(mg/L) CaCO3 (mg/L)
Cuenca del río Cuyaguateje 12-8-70 Cuba 17,4 174 Cuenca del río San Marcos 10-9-86 Cuba 19,8 230 Cuenca del río Guaso 10-2-88 Cuba 19,8 175 Región de Gandpassed 8-9-85 Spitsbergen 2,6 32 Región de Gandpassed 10-9-85 Spitsbergen 5,9 45 Región de Gandpassed 24-9-85 Spitsbergen 3,7 40
Tabla 2.1 Contenidos de CO2 y CaCO3 en manantiales de regiones tropicales (Cuba) y polares (Spitsbergen) Los relativamente altos contenidos de dureza observado en algunas aguas de Spitsbergen
están asociados al origen profundo de las mismas (en este caso las aguas de tipo termomineral), al estado de trituración mecánica de las rocas por la acción de los glaciares (lagos en las morrenas) o al efecto crioquímico que se produce al acercarse el invierno polar (fenómeno observado en las aguas del río Brattegg, las aguas que drenan de la cueva Kvisla y en otras aguas de las regiones estudiadas). El mayor contenido de CO2 fue observado en las aguas procedentes de la tundra (vegetación enana

del círculo polar), lo cual demuestra que a pesar de encontrarse congelados los suelos polares (permafrost), es significativa en los mismos la actividad microbiológica.
Sin embargo, en ocasiones se han reportado mayores contenidos de CaCO3 en algunas aguas kársticas de regiones templadas, cuyos suelos poseen una pCO2 inferior al de las regiones tropicales. En esos casos influyen factores tales como las condiciones de circulación de las aguas en condiciones de sistema abierto o cerrado, así como la mayor cantidad de detritos de calcita en el suelo (Drake, 1984). Un agua que disuelve los minerales en condiciones de sistema cerrado respecto al CO2, incrementa menos el contenido de CaCO3 disuelto que la que lo hace en condiciones de sistema abierto, independientemente de la latitud en que se encuentre.
Ford y Williams (1989), prefieren utilizar los términos sistemas coincidente y subsecuente en lugar de abierto y cerrado respectivamente, tomando como base los carbonatos disponibles en el cuerpo rocoso para la disolución, su porosidad, la capa de cobertura del karst y la pCO2.
Algunos investigadores han reportado relaciones matemáticas empíricas que expresan la pCO2 en función de la temperatura o de la evapotranspiración. Drake y Wigley (1975) encontraron que en las aguas kársticas procedentes de manantiales en calizas y dolomías de Estados Unidos y Canadá se cumple la relación matemática:
log pCO2 = - 2 + 0,04 T (2.11) donde: T: Temperatura anual, en °C.
Sin embargo, Bakalowicz (1976) plantea que la temperatura media anual no ofrece una estimación adecuada de la pCO2, por la amplia variación que sufre aquel parámetro durante las distintas estaciones del año.
Drake (1983) encontró que la ecuación 2.11 da buenos resultados si los valores de la pCO2
son tomados de mediciones en el suelo, en lugar del CO2 de las aguas carbonatadas. Este investigador realizó mediciones desde el ártico hasta el trópico, los resultados reales fueron más concordantes que los calculados mediante la expresión 2.12 cuando las precipitaciones medias eran pobres. log pCO2 = 2,55 + 0,004 P (2.12) donde: P: Precipitaciones, en mm.
También se obtuvo buena correlación con la evapotranspiración, la cual está relacionada con la duración de la estación de cultivo y donde es más favorable la generación del CO2 en el suelo:
log p CO2 = - 3,47 + 2,09 (1 - e - 0,001 72AET) (2.13) donde: AET: Evapotranspiración media anual.
La abundancia de materia orgánica como factor de generación de CO2 también se ha evaluado de forma cuantitativa. Según Paterson (1972) de 2 a 10 L/m2 de CO2 por día se produce en un suelo donde las plantas crecen vigorosamente.

Bray (1977 a) ha estudiado en forma indirecta este efecto, determinando la demanda química de oxígeno en las aguas kársticas (por oxidación con permanganato). Encontró que existía una correlación entre la demanda química de oxígeno y la agresividad de las aguas. Este tipo de mediciones ha sido más ampliamente utilizado para medir el efecto del impacto ambiental en el karst (Veccholli et al, 1984; Legrand, 1984; Laurent, 1985; Hemmel, 1990) y evaluar la capacidad autodepuradora de los cursos receptores de residuales urbanos e industriales.
Otras fuentes generadoras de CO2
El CO2 de las fuentes magmáticas puede penetrar en los acuíferos que se encuentran en áreas de reciente actividad volcánica (White, 1977). En el karst, en virtud de su fisuración, el CO2 disuelto en fluidos hidrotermales puede ascender desde niveles muy profundos y combinarse con las aguas meteóricas. Según Bakalowicz (1979), éste es el caso de las emergencias kársticas de Lez y Lerón en la región de Montpelier, donde la pCO2 de las aguas alcanzan de 0,5 a 0,1 atm, mientras que las aguas más superficiales de la misma región poseen una pCO2 inferior a 0,01 atm.
Según Liskowski (1975), mediante los movimientos epirogénicos que tienen lugar en la corteza terrestre, en los desplazamientos tectónicos de tipo isostáticos y en los movimientos exógenos de las masas rocosas, se puede crear CO2 capaz de disolver carbonatos a grandes profundidades. Un proceso de tipo triboquímico similar fue realizado mediante experimentos de laboratorio (Peters, 1962). La ecuación química es la siguiente:
CaCO3 (s) + Energía de = CaO (s)+ CO2 (s) + Calor (2.14) frotamiento Por esta vía se obtuvo 2 cm3 de CO2 a partir de 5 g de calcita pura a 25 °C.
Otro proceso en el cual se puede obtener CO2 de origen no pedológico, es por medio de la
acción directa de ácidos fuerte sobre los carbonatos: 2 HCl (l) + CaCO3 (s) = Ca2+ (ac) + 2 Cl- (ac) + H2 O (l) + CO2 (g) (2.15) H2SO4+CO2(l) + CaCO3 (s) = Ca2+ (ac) + SO4
2- (ac) + H 2 O (l) + CO2(g) (2.16) 2 HNO3 (l)+ CaCO3 (s) = Ca2+ (ac) + NO3- (ac) + H2 O (l) + CO2 (g) (2.17)
Este tipo de reacción ha provocado en algunas ocasiones, cambios apreciables en la calidad del agua por vertimiento de residuales industriales ácidos en el karst, dando lugar a una corrosión acelerada.
Con gran frecuencia se produce en Europa y Estados Unidos lluvias ácidas, con un pH entre 3.5 y 6.4, como consecuencia de la contaminación ambiental. Las principales fuentes de esta acidez la constituyen el H2SO4 , CO2 y el HNO3 obtenidos por la oxidación en la atmósfera del SO2 y el nitrógeno respectivamente, en los procesos de combustión industrial.
Las cantidades de ácidos fuertes que atacan las rocas carbonatadas por esta vía, no obstante, son pequeñas. Las mediciones efectuadas en las aguas que llegan a las cavidades kársticas en regiones sujetas a lluvias ácidas resultan neutralizadas por el medio carbonatado durante el recorrido

hipogeo (Kogovsek y Kempe, 1989). Las mayores afectaciones por lluvias ácidas se producen sobre los bosques, creando en ocasiones verdaderas catástrofes ecológicas.
Una vía más frecuente mediante la cual se generan ácidos fuertes en las aguas naturales es por oxidación de las piritas y otros minerales sulfurosos, proceso que es acelerado por los microorganismos. La reacción correspondiente se puede expresar esquemáticamente por:
4 FeS2 (s)+ 15
2 O2 (g) + 23 H2 O (l) + 28 Fe2+ (ac) = 2 Fe(OH)3 (s) + 30 Fe2+ (ac)+ 8 SO42- (ac)+34 H+(ac) (2.18)
Mediante este proceso (ecuación 2.18) se pueden formar aproximadamente 8 ion gramos de
H+ por cada mol de FeS2 oxidado. Muchas aguas kársticas con elevados contenidos de ion SO42- poseen además elevadas concentraciones de CaCO3, disuelto, como resultado de una producción adicional de CO2 originado por la acción de los ácidos fuertes, creados en el proceso de oxidación de las piritas sobre los carbonatos.
En la literatura karstológica se reportan, a veces, algunos procesos espeleogenéticos que se deben a la presencia de gases y fluidos ricos en CO2 y H2S. Por ejemplo, Hill (1987) explica el desarrollo de grandes y exóticas cavernas como las de Carlsbad en Nuevo México, mediante la ocurrencia de la reacción de reducción siguiente:
CnHm (l) + SO42-(ac) = H2S (g) + CO2 (g)+ H2 O (l) (2.19) Petróleo Según este investigador, en la zona profunda de la cuenca se ponen en contacto los fluidos
petrolíferos con las rocas ricas en sulfato, generando cantidades apreciables de H2S, que ascienden a la superficie y se disuelven en las aguas meteóricas. En el seno del agua, este gas se oxida parcialmente a H2SO4, de la forma siguiente: H2S (g)+ 2 O2 (g) = H2SO4 (l) (2.20) y en parte se disocia según: H2S (g) = H+ (ac) + HS- (ac) (2.21)
De esta forma se genera CO2,, H2SO4 e ion H+ libre, capaces de disolver considerable cantidad de calcita, la cual es parcialmente reemplazada por el yeso de la manera siguiente:
CaCO3 (s)+ H2SO4 (l) + 2 H2 O (l) = CaSO4 2 H2 O (s) + H+ (ac) + HCO3
- (ac) (2.22)
La presencia de una elevada densidad de cavernamiento en las montañas de Harz (Alemania), se ha relacionado con el intemperismo de la siderita contenida en el macizo carbonatado (Kempe,1975 a). En este proceso, por cada mol de siderita oxidada se debe formar un mol de CO2, según:

4 FeCO3 (s) + O2 (g)+ n H2 O (l) = 2 Fe2 O3 n H2 O (s) + CO2(g) (2.23) Siderita Disolución y reparto de los carbonatos en las aguas naturales
La disolución de los carbonatos por las aguas naturales es un proceso complejo donde intervienen muchos factores, de ahí que, a pesar de los muchos trabajos teóricos y experimentales desarrollados, los resultados no son totalmente concordantes con los que se obtienen en la naturaleza. Esto no sucede así con la disolución de los cloruros y los sulfatos, por ejemplo, donde la correspondencia es satisfactoria.
En un agua destilada desprovista de CO2 se disuelven 14,3 mg/L de CaCO3. En contraste con esto, en semejantes condiciones es posible la disolución de 320 g/L de NaCl y 2,1 g/L de CaSO4 (Sokolow, 1962). Cuando se eleva la temperatura del agua excenta de CO2, la solubilidad se incrementa.
El agua destilada en equilibrio con el CO2 atmosférico (3,2.10-4 atm) es capaz de disolver 60 mg de CaCO3 a 20 °C según la ecuación 1.11.
Como el contenido de CO2 disminuye con la temperatura, la cantidad de CaCO3 también será menor a temperaturas más elevadas, a menos que este efecto sea compensado con un contenido mayor de CO2 en el agua o una presión más elevada en la atmósfera que se encuentra en contacto con el agua.
Para alcanzar la máxima disolución de la calcita en las aguas, lo cual corresponde a un contenido de CaCO3 del orden de 350 a 400 mg/L, en virtud del sistema CO2-H2O- CaCO3, es necesaria una pCO2 de 0,02 a 0,1 atm (ver figura 2.4).
En las aguas kársticas con mineralización inferior a 0,5 g/L, la pCO2 y la temperatura son los dos factores determinantes en la disolución de los carbonatos. En aguas altamente mineralizadas hay que considerar además otros factores, tales como la actividad iónica, la presencia de iones foráneos (cuya influencia es despreciable en las aguas de baja mineralización), el suministro de ácidos orgánicos e inorgánicos ajenos al sistema de los carbonatos, otras fuentes generadoras de CO2 y ácidos fuertes, así como el efecto de mezcla de aguas.
La disolución de los carbonatos está controlada cinéticamente por el contenido de CO2. El CO2eq (en equilibrio) se ha asociado al CaCO3 disuelto, mientras que la pCO2 remanente se ha relacionado con el CO2 agresivo, capaz de seguir disolviendo los carbonatos.
El esquema siguiente resume la repartición del CO2 en el sistema CO2 -H2 O-CaCO3.

. CO2 agresivo, capaz de seguir disolviendo calizas . . . CO2 libre . . . . CO2 eq en forma de ion HCO3
- CO2 . . HCO3
- en forma de ion bicarbonato . . . CO2 combinado . . CO3
2- en muy pequeña cantidad como ion a pH cercano a la neutralidad Disolución de los carbonatos en presencia de otros iones
La presencia de iones ajenos a los sistemas CO2-H2O-CaCO3 y CO2-H2O-MgCO3, tiende a aumentar la solubilidad de los carbonatos por efecto salino o de fuerza iónica, como se pudo observar en el capítulo 1.
Mediante experimentos de laboratorio, Cigna y colaboradores (1963) demostraron que la solubilidad de la calcita puede elevarse desde 10 hasta 25 % mediante la adición de 0,1 % de NaCl. Muxart (1969; 1979) también obtuvo en el laboratorio, incrementos en la solubilidad de CaCO3 en presencia de NaCl y MgCl. En este último caso, el incremento fue aún mayor como consecuencia de la formación de los pares iónicos NaCO3
-, NaHCO30, MgCO3
0 y MgHCO3+. A similares
conclusiones había llegado Roques (1964). Este especialista resaltó el hecho de que trazas de sales extrañas, presentes en el sistema de los carbonatos, elevan apreciablemente la solubilidad del CaCO3, en la cual tienen gran significación los procesos naturales, sugiriendo una modificación de la curva de solubilidad de Tillman-Trombe utilizada para medir la agresividad de las aguas.
Si el proceso de disolución de la calcita tiene lugar en presencia de dolomita, se debe esperar por efecto de ion común, que se produzca precipitación de CaCO3 (Wigley,1973 a). También se encontró que un agua en equilibrio con la calcita pura, era capaz de precipitar CaCO3 en una zona dolomítica como resultado de la disolución incongruente de esta última.
Muxart (1975) estudió la mezcla de calcita y dolomita en proporciones similares a las que se encuentran en las dolomías, encontrando que la solubilidad de la calcita disminuye sensiblemente en presencia de CaMg(CO3)2. Sin embargo, Picknet (1972; 1977 a, b) encontró que el efecto de ion común se cumple para relaciones CaCO3:MgCO3 mayores de 0,1, no así cuando estas relaciones son del orden de 0,03 a 0,04. A esas bajas proporciones, se produce un anómalo aumento de la solubilidad de la calcita que tiene mucha importancia en los procesos kársticos.
La presencia de elementos minoritarios o trazas en las aguas naturales, reduce la solubilidad de la calcita y este efecto de inhibición se incrementa con el contenido de esas trazas. Este comportamiento se ha denominado efecto de los elementos trazas (Terjesen et al, 1961), el cual se

debe a la absorción de los iones metálicos en la superficie del cristal. En orden decreciente de su efectividad se encuentran los metales escandio, plomo, manganeso, níquel, bario y magnesio. Por ejemplo, 6 mg/L de Cu2+o 1 mg/L de Pb2+, reduce 2 veces la solubilidad de la calcita a 1 atm de pCO2. Se ha encontrado también que el ion PO4
3- ejerce un efecto inhibidor en la solubilidad de la calcita.
Cinética y velocidad de disolución de los carbonatos
En los aspectos tratados en este libro en relación al sistema de equilibrios de los carbonatos, no se ha considerado el papel del factor tiempo en el proceso de disolución de los mismos. Sin embargo, el tiempo de reacción junto a la temperatura y el contenido de CO2, son los tres principales elementos que determinan el proceso a través de las distintas fases presentes: gas, líquido y sólido. En las condiciones naturales, en un momento dado, el contenido de CO2 libre y de CO2 combinado es el resultado de la acción del tiempo, así como de las condiciones geológicas anteriores.
Los primeros trabajos realizados en los laboratorios, con el objetivo de estudiar la cinética de los procesos de disolución de los carbonatos se hicieron en condiciones "ideales", sin considerar la influencia de los elementos externos relacionados con el sistema de equilibrio de los carbonatos. Los resultados más relevantes de estos estudios han sido presentados por Bögli (1960) y Roques (1969).
El fundamento del trabajo de Bögli es de tipo teórico, en él se plantean cuatro fases de disolución. El trabajo de Roques es más complejo, éste realizó los experimentos en el laboratorio, lo fundamentó teóricamente y lo complementó mediante estudios en condiciones naturales. En el mismo se plantean tres cadenas de reacción, en la primera y tercera la cinética es de tipo heterogénea, puesto que intervienen fases gaseosa y líquida en la primera, así como líquida y sólida en la tercera. En la segunda cadena sólo interviene la fase líquida.
En la primera cadena se efectúa la difusión del CO2 gaseoso en el agua; el tiempo en que se establecen los equilibrios es un poco más lento que en la segunda, puesto que se requieren varios minutos, mientras que en la segunda los equilibrios se alcanzan prácticamente de manera instantánea. Este proceso se puede acelerar aumentando la presión del gas o la agitación del medio (Caro, 1965).
El proceso de disolución de los carbonatos por un agua que contiene un cierto contenido de CO2 ha sido estudiado además, desde el punto de vista cinético, por diferentes autores (Weyl, 1958; Curl, 1965; Roques y Ek, 1973; Berner y Morse, 1974, Plumer y Wigley, 1978; Sjoberg et al, 1984; Rauch y White, 1977; Walling y Bjerle, 1989; Compton y Unwin, 1990; Dreybrodt y Buhmann, 1991). Asimismo, Dreybrodt (1992) propuso un modelo de desarrollo de los canales kársticos a partir de las fisuras primarias de las calizas; así como un sistema computarizado para simular el proceso de ensanchamiento de una fractura por un agua agresiva a la calcita.
En determinadas condiciones experimentales (Fagundo et al, 1992), la variación en el tiempo de la concentración de los iones involucrados en el equilibrio de los carbonatos y la conductividad eléctrica puede seguir una cinética de primer orden, o calcularse mediante la expresión:
[C] = [C]eq (1 - e -ktn
) (2.24) donde: k : constante de velocidad [C] : concentraciones iónica (HCO3
-, Ca2+, Mg2+) o CE en el tiempo t [C]eq: la concentración de los mismos iones o la CE en condiciones de equilibrio

n: un exponente empírico (si n = 1, la cinética es de primer orden)
Recientemente Alvarez y Fagundo (1994) han implementado un sistema de computación (SIMUCIN), que permite simular el proceso de interacción agua-roca carbonatada, calcular las expresiones cinéticas (2.24) y comparar la velocidad de disolución de las calizas y dolomías en diferentes condiciones experimentales.
Corrosión mediante procesos microbiológicos y bioquímicos
Los microorganismos catalizan las reacciones de oxidación-reducción más importantes que tienen lugar en el medio acuático. Las bacterias constituyen los microorganismos más importantes en las aguas subterráneas. Poseen un tamaño medio entre 0,5 y 0,3 µm, no muy pequeño en comparación con las dimensiones de los poros y oquedades de muchas rocas, especialmente las karstificables, por lo cual en muchas ocasiones se pueden mover a través del karst.
Algunas rocas como las areniscas o las de tipo arcillosas, actúan como verdaderos filtros que retienen el paso de las bacterias. Las calizas por el contrario, a causa de su agrietamiento permiten la libre circulación de bacterias aún a grandes profundidades, de ahí la fragilidad del medio kástico al impacto ambiental. Además de las bacterias, pueden estar presente en este medio algas, hongos, levaduras y protozoos.
En los ríos y presas expuestos a la contaminación, así como en la zona del suelo, la actividad microbiológica es importante, siendo menor, aunque no despreciable, en la zona de las aguas subterráneas.
La capacidad catalítica de las bacterias se debe a sus enzimas, las cuales requieren de nutrientes para la síntesis y restablecimiento de las células bacterianas. En este proceso desempeña un papel importante, las sustancias capaces de almacenar y suministrar energía como la adenosina trifosfato (ATP). Estas moléculas alto-energéticas liberan la energía necesaria para la síntesis de las células bacterianas al ser hidrolizadas.
Las bacterias que necesitan oxígeno libre para su desarrollo se denominan aeróbicas; las que por el contrario, viven en ausencia de oxígeno disuelto se nombran anaeróbicas, y las que pueden vivir tanto en presencia como en ausencia de este gas se les llaman facultativas.
El contenido de oxígeno para la supervivencia de las bacterias aeróbicas es del orden de 0,05 mg/L, aunque algunas pueden vivir en concentraciones menores. En determinadas ocasiones las bacterias son capaces de adaptarse a condiciones bastante adversas y variables, como por ejemplo, elevados flujos, condiciones de pH entre 1 y 10, temperaturas entre 0 y 75 °C y salinidades del orden del agua de mar. Sin embargo, en las aguas subterráneas muchas bacterias se encuentran en cantidades despreciables, por no disponer de algunos de los elementos básicos para su desarrollo, tales como materia orgánica consumible y nutrientes adecuados, o sufren cambios de temperatura que afectan los proceso bioquímicos.
Algunas de las reacciones químicas de oxidación-reducción catalizadas por las bacterias se presentan a continuación:
1. Producción de CO2:
CH2 O (l) + O2 (g) = CO2 (g) + H2 O (l) (2.25) 2. Denitrificación:

CH2 O (l)+ 45 NO3
- (ac) = 25 N2 (g) + HCO3
- (ac) + 15 H+ (ac) + 2
5 H2 O (l) (2.26) 3. Reducción de Mn (III): CH2 O(l) + 2 MnO2 (s) + 3 H+ (ac) = 2 Mn2+ (ac) + HCO3
- (ac) + 2 H2 O (l) (2.27) 4. Reducción de Fe (III):
CH2 O(l) + 4 Fe(OH)3 (s) + 7 H+ (ac) = 4 Fe2+ (ac) + HCO3
- (ac) + 10 H2 O (l) (2.28) 5. Reducción de SO4
2-:
CH2 O (l) + 12 SO4
2-(ac) = 12 HS- (ac) + HCO3
- (ac) + 12 H+ (ac) (2.29)
6. Formación de CH4: CH2 O(l) + 1
2 H2O (l) = 12 CH4 (g) + 1
2 HCO3- (ac) + 1
2 H+ (ac) (2.30)
La ecuación 2.25 constituye la principal fuente de suministro de CO2 por vía bacteriana y requiere de la presencia de oxígeno disuelto en el agua. El CO2 se combina con el agua para formar H2CO3 que es un ácido suficientemente fuerte para disolver los carbonatos. El CH2O en estas ecuaciones representa un simple carbohidrato, aunque las reacciones también pueden ocurrir con la participación de sacáridos, polisacáridos, ácidos grasos, fenoles, aminoácidos y otros compuestos orgánicos.
En el proceso de descomposición de la materia orgánica en el suelo además de CO2 y los productos que se presentaron en las reacciones expresadas por 2.25-2.30, se forman por acción de las bacterias heterótrofas, algunos ácidos débiles como el acético, cítrico, oxálico, etcétera, que aunque son poco estables y se conservan poco tiempo en el seno del agua subterránea, son no obstante, capaces de atacar las calizas con una intensidad de hasta 10 veces mayor que la ecuación 1.11 para el CO2 (Eraso, 1981).
Por otro lado, se conoce la existencia de bacterias autótrofas que pueden fijar el nitrógeno del suelo, originando una cadena de reacciones en que se forma ácido nítrico, pasando previamente por amoniaco y nitrito. Este proceso se conoce como nitrificación de las aguas:
NH3 (g)+ 3
2 O2 (g) = HNO2 (l) + H2 (g) (2.31) HNO2 (l) + 1
2 O2 (g) = HNO3 (l) (2.32)
Los ácidos nítricos y otros compuestos nitrogenados relacionados, son capaces de disolver las rocas carbonatadas según las ecuaciones siguientes: 2 HNO2(l)+ CaCO3 (s) = Ca(NO3)2 (s) + H2 (g)+ CO2 (g) (2.33)

CH3 NH2 (l)+ 32 O2 (g) = CO2(g)+ (OH)- (ac) + NH4
+ (ac) (2.34) C5 H4 N4 O3 (s)+ 2 H2O (l) + 3
2 O2 (g) = 2 CO(NH)2 (s)+ 3 CO2 (g) (2.35) CO2 (g) + H2 O (l) + CaCO3 (s) = Ca2+ (ac) + 2 HCO3
- (ac) (2.36)
Aunque el CO2 es más soluble a bajas temperaturas, la actividad microbiológica es superior a temperaturas elevadas, lo cual trae como consecuencia que el proceso de generación de CO2 en el suelo, sea más intenso en los países tropicales (Schoeller, 1941).
En las regiones de clima tropical no se han hecho prácticamente experimentos en el campo, con la finalidad de evaluar en forma cuantitativa la acción de los procesos microbiológicos y bioquímicos en la disolución de los carbonatos, así como otras rocas karstificables.
Algunos investigadores han llevado a cabo, recientemente, un experimento de este tipo en el Pan de Guajaibón, Sierra del Rosario, (Pulina et al, 1984; 1992) donde se registró en forma continua las variaciones del pH, la conductividad eléctrica y la temperatura, así como se hicieron análisis químicos de campo de manera sistemática, donde se estudió la evolución de las aguas depositadas en las tinajitas y otras formas de corrosión kárstica, después de la lluvia. Los resultados, demuestran que la temperatura y el pH varían según ritmos diurnos, alcanzando valores máximos cuando es más intensa la actividad solar. La mineralización experimenta un incremento progresivo, aunque no en forma regular por las variaciones del pH.
En general, se puede afirmar que la formación de lapiaz, tinajitas y otras formas de corrosión de los macizos kársticos, está condicionada por la acción de las lluvias, la presencia de materia orgánica y los procesos microbiológicos.
Corrosión por efecto de mezcla de agua
El término corrosión por efecto de mezcla de agua se ha utilizado ampliamente por geomorfólogos y karstólogos (Bögli, 1971) para explicar algunos fenómenos de disolución en las rocas por aguas naturales, que desde el punto de vista de la teoría de los equilibrios de los carbonatos, se encuentran saturadas. Según este principio, un agua que ha alcanzado la saturación puede hacerse de nuevo agresiva si se mezcla con otra que se encuentra también saturada y posee un contenido de Ca2+o pCO2 distintos. Dos aguas kársticas procedentes de fuentes diferentes, se pueden encontrar saturadas en calcita, las cuales actuando por separado son incapaces de disolver más calcita. Sin embargo, al mezclarse se pueden hacer agresivas y, por tanto, el agua resultante es capaz de disolver una cantidad adicional del mineral carbonatado.
Este fenómeno en el karst, explicado en el párrafo anterior, fue descubierto por Bögli (1963; 1964; 1965) y explicado teóricamente por Ernst (1964). Según Bögli, desde el 1939 se había observado por Buneyew (en Laptiev, 1939), aunque no fue relacionado con la corrosión kárstica.
En una curva de equilibrio de CO2 vs CaCO3 (Figura 2.5), se puede observar el comportamiento de la corrosión kárstica por efecto de la mezcla de agua.

Figura 2.5 Curva de equilibrio de CO2 en función de CaCO3 que ilustra la corrosión por efecto de mezcla de agua (Bögli, 1964).
Si en la curva de Tillman-Trombe modificada por Roques (1962 a; 1964), (Figura 2.5) se toman dos aguas saturadas sobre la isoterma de 15 °C: W1 con 10 ppm de CaCO3 y 0,1 ppm de CO2; W2, con 350 ppm de CaCO3 y 174 ppm de CO2 , y se mezclan en partes iguales, se obtendrá un agua A con un contenido de 180 ppm de CaCO3 y 87 ppm de CO2. Como para esta cantidad de calcita el agua sólo requiere 27 ppm de CO2 en condiciones de equilibrio (AC), la porción AB se

requerirá para disolver el CaCO3 adicional, mientras que BC será utilizada para incrementar el valor de CO2 en equilibrio. De acuerdo con el sistema de ecuaciones de equilibrio de los carbonatos 1 mol de CaCO3 requiere 1 mol de CO2 para disolverse. Por tanto, si AB es a BD como 1:1 (44 ppm de CO2 a 100 ppm de CaCO3), BD será la cantidad de CaCO3 disuelto adicionalmente, equivalente a 65 ppm.
El gráfico (Figura 2.5) también permite calcular la cantidad de calcita disuelta si se mezclan aguas con diferente estado de agresividad, por ejemplo, una saturada y otra insaturada (W1 y W3), una saturada y otra sobresaturada (W1 y W4), así como en distintas proporciones.
Traikill (1968) encontró que, aunque mediante la mezcla de aguas de elevado contenido de CaCO3 no era posible la disolución de una cantidad adicional de calcita, se obtenía una menor sobresaturación. Picknet (1977 b) observó que cuando se mezclan aguas kársticas con diferente contenido de Mg2+ el agua resultante se hace más agresiva.
En el experimento de Muxart sobre la disolución de los carbonatos en presencia de otros iones, se obtuvo un agua agresiva por efecto de mezcla de agua, tal como se ilustra en la figura 2.6.

Figura 2.6 Experimento de Muxart (1975) donde se ilustra la variación de la calcita por efecto de mezcla con una solución que contiene Mg2+.
Como se puede apreciar (Figura 2.6), la solubilidad del CaCO3 crece con la adición de Mg2+ hasta alcanzar un valor máximo cuando la cantidad de iones es del orden de 7 %. Luego va disminuyendo a medida que sigue aumentando la cantidad de Mg2+, alcanzando la solubilidad mínima, cuando la cantidad de éste llega a 30 %, que fue la máxima cantidad utilizada en el experimento (Muxart, 1975).

Un agua en equilibrio con la calcita pura A (Figura 2.6), la cual incrementa su contenido de Mg2+ en forma progresiva, puede primero disolver (entre A y B) y luego precipitar CaCO3 (entre B y C). Si se mezclan dos aguas en equilibrio con concentraciones diferentes de Mg2+, por ejemplo Ch y D, se puede entonces obtener un agua agresiva por efecto de mezcla de agua. En ese caso el agua será capaz de disolver una cantidad suplementaria de CaCO3 equivalente a EF. Si se mezclan dos aguas en equilibrio, cuando el contenido de Mg2+ es mayor de 7 %, como por ejemplo G y H, se obtendrá un agua I que se encontrará también en equilibrio. Esta podría disolver o precipitar carbonato sólo si varían la presión o la temperatura.
Las aguas saturadas pueden también hacerse agresivas por disminución de la temperatura, porque en este caso se hace menor el CO2 disponible para la disolución de los carbonatos. Bögli (1964), denominó a este fenómeno corrosión por enfriamiento, calculó la cantidad adicional de CaCO3 que un agua puede disolver, al disminuir la temperatura de 4 a 9 °C en el intervalo de 0 a 44 °C.
La corrosión es significativa en aquellas regiones donde se producen marcadas variaciones estacionales o grandes cambios diurnos de temperatura aunque, por lo general, es menos intensa que la corrosión por mezcla de agua.
Los fenómenos de corrosión, especialmente por mezcla de aguas son, por lo general, muy comunes en el karst, tanto en la zona no saturada (conocida también como vadosa o de aeración); como en la freática o de saturación. Las características propias del karst, donde la circulación del agua ocurre a través de conductos más o menos independientes que tienden a integrarse, facilita la mezcla de diferentes aguas, las cuales aunque puedan encontrarse por separado saturadas, para determinadas proporciones de CO2-CaCO3, al mezclarse pueden hacerse de nuevo agresivas y seguir disolviendo rocas carbonatadas aún a grandes profundidades.
Julia James (1992) ha demostrado la importancia del efecto de mezcla en el desarrollo del karst, tomando como caso de estudio una cueva en Australia donde ocurren tres zonas de mezcla: una en la interfase entre el agua de escurrimiento y la zona freática salinizada; otra en la interfase vadoso-freática y una tercera en la zona más profunda inundada. La primera zona es la más activa en el proceso de disolución kárstica; en la segunda zona dominan también los procesos corrosivos, mientras que en la tercera zona, prevalecen los procesos de cristalización y precipitación de calcita.
Mediante estudios cinéticos se ha demostrado la importancia de este efecto de mezcla en los procesos de corrosión de las aguas carbonatadas (Dreybrodt, 1981). Otros investigadores también han ejemplarizado los fenómenos de mezclas de aguas, comparando su comportamiento en condiciones de sistema abierto y cerrado respecto al CO2 (Drake, 1984; Ford y Williams, 1989). Sin embargo, Plummer (1975 a) ha objetado el término corrosión por mezcla de agua, puesto que éste no incluye los incrementos en concentración, obtenidos mediante mezclas de aguas que contienen diferentes especies disueltas, donde ese incremento está asociado al efecto de iones foráneos.
Procesos de mezcla de agua por intrusión marina en acuíferos kársticos
En los acuíferos carbonatados litorales, existe un equilibrio dinámico entre el agua dulce que drena al mar a través de los conductos kársticos y el agua marina que penetra en el acuífero por los mismos conductos, con mayor extensión en los períodos secos, así como de mayor explotación del acuífero para el abasto a la población o la agricultura, en los cuales la presión hidrostática se deprime. En los períodos lluviosos y de menor explotación la presión hidrostática es mayor y se limita la entrada del agua marina.

En la zona de mezcla agua dulce-agua de mar se ponen en contacto dos fases químico-físicas muy diferentes en cuanto a su composición química, mineralización, densidad, pH, temperatura y contenido de gases disueltos. En dependencia del grado de mezcla, la litología del acuífero y otras condiciones específicas, se producirán interacciones más o menos intensas y complejas, las cuales producen cambios en la permeabilidad de las rocas, así como en la geomorfología de la región.
Como resultado de la mezcla de agua procedente del medio subterráneo y del mar, en ocasiones se origina un agua de tipo agresiva que es capaz de disolver los carbonatos, ampliando los conductos kársticos. En general, en un acuífero kástico costero se pueden distinguir tres zonas geoquímicas (Figura 2.7) con las características siguientes:
1. Zona en que prevalecen las aguas del tipo HCO3-- Ca2+ en equilibrio con la calcita.
2. Zona en que prevalecen las aguas del tipo Cl--Na+, las cuales se encuentran sobresaturadas en relación con la calcita.
3. Zona de mezcla o dispersión, donde las aguas son de los tipos HCO3- > Cl-- Ca2+ > Na+,
Cl- > HCO3- - Na+ > Ca2+ o de facies intermedias, y las cuales se suelen encontrar insaturadas
respecto a la calcita.
Figura 2.7 Zonación hidrogeológica e hidrogeoquímica de un acuífero kárstico litoral afectado por la intrusión salina.

De acuerdo con la información reportada en la literatura especializada en relación con la
geoquímica de las aguas kársticas afectadas por la intrusión marina, en muchas regiones se han obtenido aguas con un determinado grado de saturación, aguas saturadas o aguas insaturadas respecto a la calcita o la dolomita (Custodio, 1986). Por ejemplo, Back y Hanshaw (1971) encontraron altos valores de sobresaturación en aguas de pozos en las penínsulas de Yucatán y Florida que atribuyeron a este tipo de mezcla de aguas. También estos investigadores (Hanshaw y Back, 1980) observaron que la mezcla de dos aguas saturadas en calcita da lugar a un agua insaturada.

Figura 2.8 Efecto de la mezcla agua dulce-agua de mar sobre la solubilidad de la calcita a 25 °C y una pCO2
determinada (Plummer, 1975 a).

Algunos especialistas (Plummer, 1975; Plummer et al, 1975) estudiaron el comportamiento de mezclas en distintas proporciones de agua dulce con agua de mar, demostrando que algunas de estas mezclas originaban aguas agresivas (Figura 2.8). Si bien ciertas proporciones pueden originar insaturación, otras por el contrario dan lugar a aguas sobresaturadas, especialmente cuando están presentes iones Mg2+ y SO4
2+ . Estos trabajos de simulación química sirven de base para explicar el intenso desarrollo de cavidades kársticas observado por algunos investigadores en áreas kársticas costeras (Cotecchia et al, 1975; Plummer, 1976; Wigley et al, 1976; Hem, 1960).
Los estudios más completos sobre este tipo de proceso de mezcla se han realizado en las regiones kársticas costeras de Yucatán y La Florida (Back y Hanshaw,1971; Hanshaw y Back, 1980;Aguirre, 1980). En años más recientes en estos lugares se han llevado a cabo algunos estudios geoquímicos en relación con los mecanismos de interacción de las aguas subterráneas con las calizas en condiciones de mezcla (Back et al, 1986).
En las zonas costeras europeas, especialmente en la península ibérica, se reportan también algunos trabajos recientes sobre los procesos de mezcla agua dulce-agua de mar en el karst (Herman et al, 1986; Pulido Bosch, 1989), donde se analiza además el efecto que ejerce en estos procesos el problema de la sobreexplotación (Pulido Bosch, 1989; Calvache y Pulido Bosch, 1989; 1993; Custodio, 1989).
En las aguas procedentes de pozos de cuatro acuíferos kársticos desarrollados en el litoral de la región de Valencia, España, Morell y colaboradores (1986; 1988) encontraron que de un total de 38 muestras analizadas, sólo 13 estaban insaturadas respecto a la calcita. Además del mecanismo de disolución y precipitación de los carbonatos, estos investigadores estudiaron el papel que desempeñan los procesos microbiológicos de reducción de los sulfatos, el intercambio iónico y los compuestos minoritarios.
Los estudios con muestras tomadas por batometría en el acuífero kárstico litoral de la llanura meridional de Pinar del Río, demostraron que, por lo general, a medida que aumenta la profundidad del pozo, las aguas se van haciendo cada vez más saturadas hasta alcanzar un máximo, a partir del cual evolucionan hacia la insaturación o una menor saturación (Arellano y Fagundo, 1985; 1991; Arellano et al, 1989).
Analizando series cronólogicas de más de diez años, correspondientes a acuíferos kársticos litorales de Cuba sometidos a condiciones de sobreexplotación, se ha encontrado en ocasiones, tendencia a un incremento de la mineralización a pesar de mantenerse las aguas insaturadas en relación a la calcita. Algunos de estos procesos se han hecho irreversibles como consecuencia de una mayor permeabilidad en el acuífero debido a la disolución acelerada por vertimiento de residuales y efecto de fuerza iónica (Fagundo, Arellano y otros, 1991; 1993). Cambios en la concentración de la calcita disuelta por las aguas naturales
En este libro, se han discutido los diferentes efectos y mecanismos mediante los cuales las aguas adquieren un determinado contenido de CaCO3. Un resumen de los mismos se presentó por Ford y Williamsom (1989), el cual en forma esquemática se muestra en la tabla 2.2.
En general, se puede establecer que de acuerdo con los principios que establece la química-física sobre la teoría del equilibrio de los carbonatos, 14 mg/L de calcita se pueden disolver en agua pura y hasta 55 mg/L en un agua natural en equilibrio con una pCO2 atmosférica a 25 °C. Si se disminuye a 0 °C su temperatura, de acuerdo con la ley de Henry, el agua puede incrementar su solubilidad en calcita hasta 75 mg/L. Se ha reportado un incremento de 77,7 mg/L de CaCO3 al enfriarse un agua que poseía 240 mg/L de calcita disuelta, desde 20 a 10 °C (Tab. 2.2).

Dos fases Tres fases CO2 (g), H2O (l), CaCO3 (s)
1000 en agua de mar (efecto de fuerza iónica) en agua pura 14
a 250 C ³ Efecto de ³ Temperatura ³ Atmósfera estándar PCO 2 = 0.03% Sistema abierto ideal ³ CaCO3 (en mg/L) ³ ³ ³ ³ ³ ³ 75 00 C ³ 55 ³ ³ ↑
Presión de la atmósfera del suelo Sistema abierto cerrado PCO2 (6%) 450
(3%) 300 ÄÄÄÄÄÄÄÄÄ Disminución (0.3%) 150 ↓ ↑ (3%) 120 ↓ (0.03%) (0.3%) 50 (0.03%)
Corrosión por mezcla de agua Incremento de 10 mg/L por 1/2% de CO2 ↑ 66 ↑
Complejos orgánicos teórica 500 ↑ poco probable debido a la oxidación 66 ↑
Efecto de ion común Disminución ón en un 40 % ↓ 30
Tabla 2.2. Efecto de diferentes factores en la concentración de CaCO3 (en mg/L) disuelto en aguas carbonatadas (Ford y Williams,1989).
La variación en la presión hidrostática de las aguas, a causa del régimen de circulación, ejerce efectos poco apreciables en la disolución de minerales e incluso gases. Sin embargo, si se hace burbujear CO2 en un agua kárstica sometida a presión, la solubilidad de este gas se incrementa en 6 mg/L por cada 100 m de profundidad hasta alcanzar los 400 m.
A mayor profundidad, el CO2 aumenta su solubilidad en agua en 0,3 mg/L por cada 100 m de profundidad. Este efecto es responsable del elevado contenido de iones disueltos que presentan las aguas procedentes del drenaje profundo cuando emergen en los manantiales kársticos. Como aún presentan un elevado contenido de CO2 en el momento que afloran a la superficie, parte de los minerales son depositados como CaCO3 en los bordes del manantial.
El incremento de la solubilidad de la calcita por el efecto de los procesos microbiológicos y bioquímicos, fue tratado en este capítulo. Sin embargo, se debe tomar en cuenta el modo en que este proceso tiene lugar. En condiciones de sistema abierto con abundante y renovable contenido de CO2, la cantidad de calcita disuelta en el agua puede alcanzar 150; 300 ó 450 mg/L, cuando la pCO2 del suelo es de 0,3; 3 ó 46 % respectivamente, mientras que la cantidad de CaCO3 puede decrecer hasta 50 mg/L a 0,3 % de pCO2 o alcanzar sólo 120 mg/L a 3 % de pCO2, en condiciones de sistema cerrado (Bögli, 1980). (ver tabla 2.2).
Un incremento notable del contenido de CaCO3 en las aguas también puede alcanzarse en presencia de compuestos orgánicos tales como los ácidos húmicos y fúlvicos. Aunque teóricamente se ha calculado la posibilidad de un incremento de hasta 500 mg/L por esta vía, en la práctica éste es menor, a causa de los procesos competitivos de oxidación de la materia orgánica (ver tabla 2.2). La adición de iones comunes al equilibrio de los carbonatos y sulfatos, reduce hasta 40 % la solubilidad de la calcita en las aguas kársticas, tal como se ilustra en la tabla 2.2, mientras que la adición de iones foráneos a este equilibrio incrementa sensiblemente la solubilidad de este mineral. Los mayores incrementos reportados (Trailkill, 1968) se deben al resultado de la mezcla entre el

agua acuífera con el agua de mar. A 25 °C y una pCO2 elevada, propia de los suelos kársticos, la solubilidad de la calcita se puede incrementar en estas condiciones hasta 1 000 mg/L. Deposición de la calcita a partir de aguas sobresaturadas
En la superficie terrestre existe una abundante cantidad de carbonatos de origen continental, la cual se ha formado mediante un proceso que comprende tres etapas:
1. Disolución de los iones Ca2+, Mg2+ y HCO3- de las rocas por las aguas meteóricas y
juveniles. 2. Transporte de iones a una determinada distancia. 3. Precipitación de carbonato de calcio o magnesio como resultado de cambios de tipo
químico físicos, biológicos o bioquímicos ocurridos en el medio acuoso. El proceso de disolución de los carbonatos de calcio en condiciones normales ocurre
rápidamente, el CO2 disponible se consume y se eleva el pH. El proceso contrario es más lento. El agua puede permanecer sobresaturada mucho tiempo antes de que se produzca la precipitación de la calcita o el aragonito (Weyl, 1958). Si también hay magnesio presente, su precipitación como dolomita o magnesita es más lenta aún (Langmuir, 1964; Garrels, Thompson y Sievers, 1960).
El papel de los factores químico-físicos en la precipitación de los carbonatos fue estudiado por Muxart (1981). Según esta especialista, los sistemas de equilibrio CO2-H2O-CaCO3 y CO2-H2O-CaMg(CO3)2 controlan tanto la disolución de los carbonatos, como la precipitación de los mismos a partir de una solución acuosa sobresaturada. A la temperatura y presión normales de la atmósfera terrestre, el agua se puede presentar en uno de los tres estados de la materia: sólido (nieve o hielo), líquido (agua) o gaseoso (vapor). El cambio de estado en el agua por variaciones en la temperatura o presión de vapor da lugar, por lo general, a la deposición de la materia disuelta.
Al pasar un agua líquida a la fase vapor, se consume energía térmica y se produce la evaporación del solvente, así como una sobresaturación de la solución. Un cambio en las condiciones químico-físicas en este caso favorece la precipitación (temperatura, presión atmosférica, pCO2, fuerza iónica, producto de solubilidad, etcétera).
Al pasar el agua de líquido a sólido (formación de hielo o nieve), la naturaleza de los solutos presentes y las condiciones en que se produce la congelación influyen en las características químico-físicas del hielo, mediante un proceso gradual de enfriamiento, el agua va incrementando su mineralización y el hielo formado al inicio posee muy pocos minerales en su red cristalina. Con el descenso progresivo de la temperatura, las capas de hielo siguientes contendrán una cantidad mayor de soluto cristalizado. Si la solución está sobresaturada y en estado de quietud, cualquier movimiento que altere la estabilidad del proceso, provocará que la solución se congele bruscamente y los hielos formados no presentan una estructura similar a cuando el proceso ocurre en forma gradual. En sentido inverso, al descongelarse este hielo parte de los minerales atrapados no pasan a la solución acuosa en forma disuelta y esto trae como consecuencia la precipitación de minerales exógenos.
Los procesos crioquímicos, mencionados en el párrafo anterior, crean a su vez grandes cambios en las propiedades químico-físicas de las aguas originadas por descongelación de los glaciares y del permafrost (suelo permanentemente congelado) de las regiones polares. La composición química de la solución determina la estructura y las dimensiones de los cristales, también influyen las características cinéticas y termodinámicas de los equilibrios involucrados, la germinación provocada por la adición de cristales o partículas ajenas y el estado de agitación o

quietud de la solución, tal como ha quedado demostrado en los estudios cinéticos y experimentales (Giroy y Roques, 1971 a, b; 1972).
La precipitación de CaCO3 de una solución sobresaturada se puede producir de dos maneras: a temperatura constante, por disminución de la pCO2 o a pCO2 constante, por elevación de la temperatura.
Mediante experimentos de laboratorio se ha demostrado que una solución de CaCO3
expuesta a la atmósfera deposita primero calcita, luego verterita y finalmente aragonito, a partir de verterita (Roques, 1964). Los tres minerales constituyen formas alotrópicas del CaCO3, siendo la verterita la más inestable, seguida del aragonito.
Al elevarse la temperatura se incrementa la cantidad de aragonito depositado. Por esa razón, la presencia de aragonito se ha utilizado como un indicador de paleotemperatura (Moore, 1956).
La formación de aragonito también se ha asociado a la presencia de ciertos iones, tales como Mg2+, Sr2+ ó Pb2+ (Murray, 1954). En experimentos de laboratorio, se ha demostrado que la adición de Mg2+como MgCl2 incrementaba la deposición de aragonito en relación a la calcita (Roques, 1969 b).
Un agua que se encuentra saturada en relación a la calcita, al mezclarse con otra rica en iones ajenos al sistema CO2-H2O-CaCO3 puede adquirir un grado de saturación menor o incluso hacerse agresiva, en dependencia de las proporciones de la mezcla; de igual forma, la adición de iones no comunes al sistema de equilibrio de los carbonatos incrementa su solubilidad. En el caso de iones comunes a este sistema se producirá un aumento de solubilidad a bajas concentraciones y precipitación a concentraciones más elevadas. También se analizó que cuando en la disolución de los carbonatos están presentes la calcita y la dolomita, la disolución de uno de los minerales originaba la precipitación del otro.
A bajas temperaturas la dolomita es más soluble que la calcita y en el proceso de disolución del primer mineral se deposita el segundo. A temperaturas elevadas se produce el fenómeno contrario. A temperaturas del orden de 10 °C, en un sistema CO2-H2O-CaCO3-CaMg(CO3)2 la disolución incongruente de la dolomita origina una dolomita cuarcífera de fórmula: Ca1+x Mg1+x (CO3)2 que es más soluble que la dolomita ideal CaMg(CO3)2. Por el contrario, a temperatura superior ocurre la disolución incongruente de la calcita, aunque en este caso el proceso de precipitación es muy lento (Wigley, 1973 a).
Como resultado del proceso de disolución incongruente, en las regiones dolomíticas de los países fríos, las aguas naturales poseen una relación iónica Ca2+/ Mg2+ del orden de 0,59, mientras que en los países tropicales esta es del orden de 1,1 (Langmuir, 1971). Estos valores están bastante cercanos a los calculados teóricamente para la disolución incongruente a temperaturas entre 10 y 25 °C. Los experimentos llevados a cabo a 25 y 30 °C concuerdan también con estos resultados (Garrels, Thompson y Siever, 1960; Muxart, 1981).
La acumulación de formaciones de travertina fue estudiada por Nicod (1986 a) sobre la base de los factores químico-físicos a nivel local, al nivel de geosistema o hidrosistema kárstico y en relación a la variación de las condiciones durante el Holoceno como resultado de la actividad antrópica.
En las regiones donde se alternan secuencias de calizas y dolomías, las aguas en su curso pueden alcanzar el equilibrio respecto al mineral constitutivo más abundante en estas rocas en un tramo dado (calcita o dolomita). Si por ejemplo es más abundante la calcita, el agua que se mueve por esta región, al llegara la zona de las dolomías podrá disolver dolomita, porque no está saturada

con respecto a ese mineral. Como resultado del efecto de los iones comunes Ca2+ y CO3 2-
procedentes de la dolomita, el agua tenderá a sobresaturarse respecto a la calcita. Si en esas condiciones se produce la precipitación de este mineral, el agua adquirirá una mayor insaturación respecto a la dolomita y es capaz de disolver más cantidad de ésta. Este proceso de disolución incongruente de la dolomita puede originar abundantes depósitos de calcita. Se ha encontrado en el karst Dinárico, abundantes depósitos de tufa calcárea en la confluencia de dos ríos que drenan de una cuenca desarrollada en dolomías del Triásico, cuyas aguas portaban una elevada dureza de Mg2+ (Gams, 1967).
Además de los factores químico-físicos, existen otros que influyen en el proceso de deposición de minerales carbonatados. Muchos organismos vivos por ejemplo, utilizan en su metabolismo elementos de los sistemas CO2-H2O-CaCO3 y CO2-H2O-CaMg(CO3)2, produciendo la precipitación de sales de calcio y magnesio. Entre estos microorganismos, desempeñan un papel importante algunas pseudomonas, bacilos, algas azules y verdes, hongos e incluso, algunos moluscos. Sobre la base de estudios experimentales de biocristalogénesis, se observó con la ayuda de un microscopio la formación de agujas y rombohedros de calcita por la acción de microorganismos, además se encontró que en este proceso influyen condiciones ambientales similares a las que afectan las variaciones morfológicas de los depósitos, tales como grado de humedad, luminosidad y gravedad específica (Adolphe, 1981).
Muxart (1981) ha señalado que la precipitación de origen biológico depende de una serie de factores climáticos y pedoclimáticos. La presencia de clorofila en los vegetales está ligada a la fotosíntesis y la misma depende de la actividad, así como de la duración diurna de los rayos solares. Esta actividad es función a su vez de los elementos nutritivos disponibles, de la temperatura y la humedad del lugar, para esto es necesario un flujo hídrico suficiente para asegurar la supervivencia de los microorganismos. Esta investigadora destaca el papel de estos organismos en la destrucción de compuestos hidrosolubles que participan en la disolución de CaCO3 del suelo y su influencia en la generación de depósitos carbonatados.
Estudios realizados, utilizando microscopio electrónico, demuestran que en las rocas ricas en mineral dolomita, el intemperismo químico y el ataque biológico son más activos sobre CaCO3 que sobre CaMg(CO3)2 (Muxart y Girot, 1977).
Deposición de la calcita en manantiales y cursos superficiales
En muchas ocasiones las aguas kársticas adquieren un elevado contenido de CO2 en la zona del suelo y al infiltrarse a través de canales y fisuras, pueden encontrar estratos impermeables y condiciones tectónicas favorables que les permitan salir al exterior conservando aún una pCO2 elevada, muy superior a la del aire atmósférico. En esas condiciones al salir a la superficie liberan parte del CO2 disuelto hasta alcanzar la pCO2 en equilibrio a las nuevas condiciones de presión y temperatura, precipitando travertina en forma de calcita o aragonito en los bordes del manantial y especialmente en los rápidos y cascadas.
En otros casos, las aguas kársticas en su infiltración vertical hacia la zona freática, son capaces de alcanzar grandes profundidades y luego, al encontrar una falla o zona de contacto tectónico favorable, ascender hasta el exterior. En muchos casos incrementan su contenido de CO2 y el de otros gases como el H2S, por las reacciones con las piritas o en virtud del aporte de otras fuentes de tipo hidrotermal. También pueden elevar su temperatura a causa del calor geotérmico. Como consecuencia del cambio de la pCO2 y la temperatura, estas aguas son capaces de depositar

abundante cantidad de carbonato; por ejemplo, 2 t de travertina son depositadas diariamente en el manantial hidrotermal Meskountine, en Argelia (Urbain, 1953).
En un evento celebrado recientemente sobre las travertinas y la evolución del paisaje holocénico del Mediterráneo, se presentaron muchos trabajos relacionados con el origen, distribución y morfología de grandes depósitos de calcita que han tenido lugar durante el Holoceno. Adolphe (1986) relacionó el origen de los depósitos de tufa y travertina con la presencia de algas, bacteria y hongos en esos depósitos. Nicod (1986 b), atribuyó la formación de depósitos de travertina del valle de Argens a factores antrópicos y naturales, estos últimos caracterizados por la acción antagónica del proceso de creación por las algas, por un lado, y por la acción hidromecánica por el otro.
La presencia de conchas y caracoles, así como de otros organismos en los sedimentos ha permitido su datación y disposición geocronoestratigráfica por métodos palentológicos (Geovine, 1986) o por datación con C14 (Reffay et al, 1986). Esto ha permitido conocer en qué parte del Holoceno ocurrieron las máximas deposiciones. En general, estos estudios han puesto de manifiesto que durante el Holoceno, específicamente entre el Boreal y el Atlántico (9 000-5 000 a.n.e.), en Europa primó un clima seco y caliente, en esas condiciones fue más favorable el proceso de deposición de los sedimentos carbonatados (Weisrock, 1986). En los períodos Histórico y Actual se ha producido una disminución de la intensidad de este proceso, a causa de cambios climáticos y a la actividad humana que ha modificado el medio ambiente.
En la cuenca del río Cuyaguateje, en Cuba, se estudió el contenido relativo de cuarzo y caolinita de los depósitos originados durante el Cuaternario en terrazas fluviales, así como los de origen epigeo que abundan en las cuevas (Fagundo, Valdés y Pajón, 1984). También se encontró que existía una buena correlación entre los sedimentos que aparecen a los distintos niveles de terrazas y niveles de cavernamiento, lo cual fue atribuido a los cambios en el nivel de base ocurridos durante este período, como consecuencia de las fluctuaciones del nivel del mar, por los alternativos procesos de ablación y congelación ocurridos en el casquete polar.
Deposición de calcita y aragonito en cavidades subterráneas
Cuando las aguas que se infiltran a través de las grietas y fisuras de un macizo kástico interceptan una cavidad, liberan parte del CO2 disuelto hasta alcanzar la pCO2 de la atmósfera de la cueva. En muchos casos, las cuevas se encuentran aireadas y su atmósfera está en contacto con la del exterior, en esas condiciones, el agua deposita el exceso de calcita que posee en relación a la pCO2 del recinto cavernario. El CaCO3 depositado se reparte, quedando una porción en el extremo del conducto o estalactita y la otra cae al piso de la cavidad originado una estalagmita.
Además de estos tipos de espeleotemas, existen otros de las más diversas formas, cuyo origen depende de condiciones específicas de la cueva: temperatura, humedad, circulación del aire, composición litológica, cobertura pedológica y otros factores. El clima, por ejemplo, desempeña un papel determinante en el desarrollo de las denominadas anemolitas y algunos espeleotemas deformados (Choppy, 1981). En la literatura espeleológica y karstológica en general, abundan los trabajos relacionados con la clasificación y origen de los diferentes depósitos que abundan en las cuevas (Núñez Jiménez et al, 1984, Hill y Forti, 1986).
El proceso de transferencia de masa que se produce al depositarse CaCO3 por el agua kárstica que se infiltra a través de una concresión tubular, se ha estudiado por Roques (1969a).
La composición química de las aguas de degoteo y de gours, muestreadas en las cavernas presentan más bajo contenido de CO2, Ca2+ y HCO3- que las aguas de las emergencias que se encuentran en la misma región kárstica. El pH por el contrario es más elevado en las aguas de cueva

que en las de manantiales o surgencias kársticas. Por lo general, las aguas en cuevas se encuentran saturadas o sobresaturadas en relación a la calcita y sólo se hacen agresivas en los períodos lluviosos, cuando los conductos por donde se infiltra el agua son lo suficiente grandes y capaces de responder con rapidez al efecto de las precipitaciones. En esas condiciones las aguas exteriores arrastran la materia orgánica y alcanzan la cavidad con suficiente contenido de CO2 disuelto.
Aunque la calcita y el aragonito suelen ser los minerales más abundantes en las cuevas, otros en menos proporción se han encontrado tanto en depósitos hipogeos como epigeos (Hill y Forti, 1986; Shopov, 1988). En Cuba además de calcita y aragonito (Valdés y Fagundo, 1975), se ha reportado la presencia de minerales menos comunes (Pajón, Fagundo y Valdés, 1985). Proceso de karstificación en yeso
A pesar de que las rocas evaporíticas están ampliamente distribuidas en la superficie de la Tierra, muchas de éstas se encuentran en profundidad debido a que en el proceso de meteorización, la rápida disolución destruye los afloramientos superficiales. Las formas kársticas superficiales en este medio son más frecuentes en las regiones áridas (White, 1988).
Morfológicamente las regiones kársticas yesíferas presentan paisajes similares a los desarrollados en rocas carbonatadas. Las cuevas en yeso son algo diferentes a las cuevas en calizas. La química, sin embargo, es mucho más simple, no hay dependencia del pH y de la presión de CO2. El mecanismo de disolución es también diferente, la velocidad de disolución del yeso depende fuertemente de la agitación (White, 1988).
Los trabajos relacionados con la geomorfología, la hidrología y la hidroquímica de los karst en yeso son más escasos que los realizados en relación a terrenos carbonatados, incluso en España, que junto a la antigua URSS, EEUU e Italia, constituye uno de los países del mundo donde la extensión y desarrollo del karst yesífero es más importante (Calaforra y Pulido Bosch, 1989).
Como se indicó en el Capítulo 1, la disolución de los minerales evaporíticos ricos en SO42-,
yeso y anhídrita, tiene lugar mediante un proceso de disolución física donde sólo intervienen las moléculas del mineral y de H2O. De acuerdo a las leyes termodinámicas, es posible la disolución de 2,41 g/L de CaSO4 a 25 °C, siendo mayor la solubilidad a temperaturas más altas. A 58 °C y 1 atm., el yeso pierde su agua de cristalización y se transforma en anhidrita.
En muchas regiones kársticas desarrolladas en yeso, las aguas adquieren una concentración superior a 2,0 g/L, mientras que en los terrenos kársticos carbonatados, por lo general, concentraciones de CaCO3 mayores de 0,35 g/L se deben a la influencia de sulfatos o cloruros (Ford y Williams, 1989). Las aguas naturales presentan, por lo general, un contenido de SO4
2- que depende del tipo de mineral presente o de fuentes contaminantes. Perchokin (1986) reporta concentraciones de CaSO4 en aguas kársticas de fractura en macizos de yeso-anhidrita que varían entre 0.06 g/L en las depresiones rellenas con depósitos kársticos, hasta 2,4 g/L en la cima de los macizos. Forti (1988) encontró contenidos de SO4
2- entre 37 y 40 meq/L (2,58 - 2,7 g/L de CaSO4) en las aguas de la fuente de Poino, en la provincia de Reggio Emilia, las cuales variaron su concentración de Cl- de 51 a 64 meq/L (2,7 a 2,27 g/L) en el año hidrológico 1984, y según datos históricos, el contenido de NaCl de esta fuente ha sufrido una disminución desde 15 g/L en 1862 hasta 3,1 g/L en 1984.
En Cuba, donde el karst en yeso sólo ocupa una superficie ínfima del territorio, los estudios de tipo hidrológicos e hidroquímico son aún mas escasos. Sólo aparece reportado un trabajo de este

tipo, realizado en el marco de una expedición espeleológica italo-cubana al macizo yesífero de Punta Alegre en la provincia de Sancti Spíritus (Fagundo et al, 1994).
En ese sitio, la cobertura carbonatada ha sido erosionada y fragmentada, permitiendo el afloramiento de los yesos en la superficie. De ese modo la alimentación del acuífero es directa a partir de las precipitaciones, con descarga al mar a través de manantiales, algunos de los cuales afloran en el propio lecho marino. El nivel piezométrico se encuentra en contacto dinámico con las aguas marinas, las cuales invaden el acuífero yesífero a través de las grietas y conductos. Mediante un muestreo realizado en pozos y manantiales, fueron detectadas cuatro facies hidrogeoquímicas: SO4
2-- Ca2+; SO42-> Cl- - Ca2+> Na+; Cl- > SO4
2-- Na+ > Ca2+ y Cl- - Na+. La primera facies (sulfatadas cálcicas) es la más abundantes. Estas aguas se originan por
infiltración directa de las precipitaciones a través del macizo de yeso desnudo. Se caracterizan por presentar bajos contenidos de Cl- (0,06 y 0,10 g/L), CaSO4 (2,20 y 2,02 g/L) y poca mineralización (2,55 g/L de sólidos solubles totales, SST). Debido a los fenómenos de intrusión marina en el acuífero en forma difusa o a través de conductos; así como mediante interacción con halitas subyacentes, las aguas en su trayectoria vertical se mezclan en diferentes proporciones con otras aguas que poseen un elevado contenido de NaCl disuelto. Como resultado del efecto de los iones Cl- y Na+ no comunes al equilibrio involucrado en la disolución del yeso (efecto de fuerza iónica), las aguas del acuífero saturadas en yeso pueden seguir incrementando su contenido de CaSO4. En las aguas del tipo sulfatadas cálcicas, se alcanzó valores de Cl-, CaSO4 y SST de 0,49, 2,53 y 3,82 g/L, respectivamente.
Las aguas del tipo sulfato-cloruradas cálcico-sódicas presentan contenidos de Cl- entre 0,58 y 0,87 g/ L; de CaSO4 ,entre 2,40 y 2,25 g/L., y SST del orden de 3,90 g/L.
Las aguas del tipo cloruro-sulfatadas sódico-cálcicas muestreadas presentaron un contenido de Cl-, CaSO4 y SST de 4,26; 4,66 y 12,11 g/L respectivamente.
Las aguas del tipo cloruradas sódicas mostraron niveles de Cl- entre 9,05 y 13,15 g/L; de CaSO4 entre 1,50 y 2,46 g/L, y de SST entre 16,87 y 24,81 g/L, respectivamente.
Mediante experimentos de laboratorio se ha demostrado que, mientras un agua destilada en condiciones de saturación a una temperatura dada, puede disolver 2,0 g/L de CaSO4.2 H2O puro, la cantidad de yeso disuelto aumenta progresivamente hasta alcanzar 7,5 g/L con la adición de hasta 100 g/L de NaCl (Ford y Willams, 1989). Luego la solubilidad experimenta una disminución a medida que se sigue añadiendo NaCl, lo cual concuerda aproximadamente con los resultados obtenidos en el laboratorio al disolver hasta saturación un mineral puro del yacimiento de Punta Alegre con aguas con diferente concentración de NaCl disuelto (Fagundo et al, 1994).

Capítulo 3. Caracterización y evolución químico-física de las aguas kárstica

Análisis químico y calidad de las aguas
La determinación de las propiedades químico-físicas de un agua kárstica, constituye una herramienta imprescindible para los hidrólogos, hidrogeólogos, karstólogos, geomorfólogos, ingenieros sanitarios y otros especialistas relacionados con el agua.
La primera fase de todo trabajo hidrogeoquímico, comprende el estudio bibliográfico y visitas de reconocimiento en el área objeto de estudio. En esta etapa también se toman muestras, las cuales se analizan con fines de orientación.
Para la aplicación de los métodos hidrogeoquímicos en el estudio de la disolución kárstica, es necesario tomar en cuenta el balance hídrico y parámetros geoquímicos que caractericen a cada sistema, relacionando el volumen de infiltración y circulación del agua con la composición química de la misma en el área de descarga del acuífero (Garay y Morell, 1989). Con ese objetivo, en la segunda fase del trabajo se seleccionan los puntos o estaciones de observación sistemática y se instalan los equipos de registro o medición necesarios para el control de las precipitaciones, los caudales y el quimismo de las aguas.
La composición química de las aguas kársticas está controlada por los equilibrios químicos de los carbonatos y varía al cabo del tiempo. Por esta razón, los análisis químicos y las mediciones de pH, así como la conductividad eléctrica deben hacerse "in situ". En muchos países, lamentablemente se acostumbra a hacer los análisis y mediciones en el laboratorio, muchas veces hasta semanas después de tomada la muestra, cuando éstas en la práctica, carecen de valor para interpretar la naturaleza de los fenómenos kársticos.
Otra práctica inadecuada consiste en tomar la muestra dejando una cámara de aire, lo cual favorece el escape del CO2 disuelto en el agua hacia la fase gaseosa, donde este gas es más soluble. Esto da lugar a la recombinación de los iones HCO3
- y Ca2+, a la precipitación de CaCO3 (con disminución en la conductividad eléctrica) y al aumento del pH.
Los primeros métodos hechos en el campo para la caracterización de las aguas kársticas consistían en el uso de tabletas, las cuales añadidas al agua cambiaban el color (Smith, 1965). Estas fueron empleadas por los geomorfólogos ingleses en sus primeros estudios para la determinación de la denudación química en regiones kársticas. Algunas variantes de este método, se han continuado empleando por especialistas de diferentes países (Gams, 1979; Garay y Morell, 1989).
La mayoría de los karstólogos norteamericanos y algunos europeos han utilizado hasta años recientes algunas mediciones de campo, principalmente la temperatura, el pH y la conductividad eléctrica, así como mediciones de HCO3
-, Ca2+ y Mg2+ por análisis volumétrico (Corbel, 1959; Ek, 1973). Los cálculos del grado de agresividad del agua se han realizado por medio de computación (Wigley, 1977), empleando los índices de saturación de la calcita, la dolomita y la p CO2 en equilibrio.
Bray (1977 b) y Fagundo (1982) han sugerido el uso de la conductividad eléctrica para medir la agresividad de un agua. Estos últimos métodos se basan en la determinación previa de correlaciones lineales entre la conductividad eléctrica, la dureza o las concentraciones de HCO3
-, Ca2+ y Mg2+. El empleo de ecuaciones de correlación matemática entre la conductividad eléctrica y algunos parámetros químico-físicos como la mineralización (Bakalowicz, 1974), ha permitido calcular la cantidad de calcita disuelta en las aguas que emergen de un macizo carbonatado.

El empleo de marchas analíticas de campo, se lleva a cabo desde hace muchos años por investigadores polacos y de otras nacionalidades (Markowicz y Pulina, 1979; Aminot, 1974, Krawczyk, 1992), incluyendo a los de Cuba (Pulina et al, 1984; Fagundo et al, 1981; 1986; Rodríguez et al, 1989). Las mediciones que se hacen más comúnmente en condiciones de campo son: la temperatura, el pH, la conductividad eléctrica; así como los análisis químicos de CO2 disuelto, HCO3
-, CO32- , Cl-, SO4
2- , Ca2+ y Mg2+ . Los elementos alcalinos se suelen reportar como Na+ + K+, mediante la diferencia de aniones y cationes. Además, una réplica de la misma muestra se suele preservar en el campo por adición de un ácido mineral fuerte y luego se lleva al laboratorio, donde se determina Na+ y K+ por fotometría de llama; así como los microcomponentes y elementos trazas, mediante absorción atómica, fluorescencia de rayos X u otra técnica analítica. En la actualidad se usan también los métodos de cromatografía iónica para el análisis tanto de los aniones como de los cationes en muestras de aguas y suelos.
La precisión de los análisis químicos se controla mediante diferentes métodos, entre estos la diferencia entre aniones y cationes que no debe exceder de un cierto valor. También se suele emplear la ecuación de balance:
ecat an
cat an=
−
+∑ ∑∑ ∑
200 (3.1)
donde: e: Error, en %. Σ cat: Suma de cationes, en meq/L. Σ an: Suma de aniones, en meq/L.
En aquellos casos en que el error calculado por la ecuación 3.1 sea superior a 10 %, se debe repetir el análisis o desechar la muestra.
Otro método útil para calcular el error del análisis consiste en la comparación entre la conductividad eléctrica real y la teórica (Dudley, 1972), calculada mediante la ecuación:
LogCE C CS CT i ii
n
i
n= − i
==∑∑log , log0 03
11 (3.2)
donde: CE T: Conductividad eléctrica teórica a 25 °C. CSi: Conductividad específica equivalente de cada ion i a dilución infinita y 25 °C (Tabla 3.1). Ci: Concentración de cada ion i, en meq/L.
Ion Csi (µS/cm) Ion CSi (µS/cm) HCO3- 42,4 Ca2+ 57,7
Cl- 73,5 Mg2+ 50,6 SO42- 75,5 Na+ 49,2 NO3- 69,0 K+ 72,9
Tabla 3.1. Conductividad eléctrica específica equivalente de cada ion a dilución infinita (CSi) a la concentración de 1 meq/L y a 25 °C

Para calcular el error del análisis se puede emplear la fórmula: e CE CE
CET R
R=
− 100 (3.3)
donde: CER: Conductividad eléctrica real a 25 °C.
Las determinaciones se consideran precisas cuando el porcentaje de error es menor de 5. En el caso de los análisis de campo, no se conocen los contenidos de los iones Na+ y K+, pero se puede tomar Na+ + K+ = Na+, teniendo en cuenta que en las aguas naturales Na+ > K+.
Para la determinación de la calidad de los datos hidroquímicos correspondientes a aguas termales, se han propuesto los siguientes criterios (Urbani, 1991):
- Revisión del balance de aniones y cationes. La diferencia entre ambas sumas no debería sobrepasar el 6 %. Esto no es aplicable cuando la suma de los meq/l de aniones o cationes sea menor o igual a 5, igualmente cuando el TSS sea mayor de 1 000.
- Comparación del TSS determinado y el calculado. - El TSS debería variar entre 0,55 y 0,77 x Conductividad. -(Suma de aniones/Suma de cationes en meq/L) x 100 debe ser aproximadamente igual a la
conductividad. - Sospechar del análisis si: a) Na+ = 0; b) Ca2+ = 0 y Mg2+ > 2 mg/L; c) K+ > Na+, si ambos
tienen > 5 mg/L. Hoy día, también se incluyen en las marchas analíticas aquellos indicadores de la actividad
del hombre en el karst, especialmente los componentes del ciclo de nitrógeno (NH4+, NO3
- , NO2-) y
el fósforo (ortofosfato y fosfato total), la demanda química de oxígeno (DQO) y otras técnicas empleadas en la caracterización de aguas residuales.
Otros métodos de medición empleados en los estudios del karst son los llevados a cabo con equipos electrónicos portátiles o sistemas automatizados de registro o adquisición de datos, que miden uno o varios indicadores como: temperatura, pH, conductividad eléctrica, potencial redox, oxígeno disuelto (Llorent, 1988; Matia, 1988); así como algunos iones mediante sensores y electrodos selectivos (García et al, 1977; Morell et al, 1987).
El estudio del comportamiento químico-físico de las aguas kársticas mediante análisis químico completo de campo, con registro continuo de los caudales en diferentes emergencias de una cuenca kárstica, disponiendo de la información de los elementos del clima (temperatura, humedad relativa y precipitación) se ha abordado en diferentes bases experimentales (Pulina, 1971; Bakalowicz, 1979; Paterson, 1972). En Cuba, las primeras experiencias de este tipo se llevaron a cabo en el macizo del Pan de Guajaibón, en la Sierra del Rosario (Rodríguez et al, 1989), donde se realizaron estudios relacionados con la dinámica y evolución del karst en condiciones tropicales y fue necesario hacer mediciones cuantitativas de la intensidad de la denudación química. Estudios similares, aunque con una frecuencia de observación menos sistemáticas y con menos recursos se habían hecho con anterioridad en la cuenca del río Cuyaguateje (Fagundo et al, 1981).

Estudio del comportamiento hidrodinámico de los acuíferos kársticos a partir de las respuestas naturales en las surgencias.
La investigación de la dinámica o funcionamiento del karst ha sido abordada mediante un enfoque sistémico, donde el karst es tomado como una caja negra al cual se tiene acceso a través de las funciones de entrada (las precipitaciones) y las funciones de salida (variables hidrogeológicas, hidroquímicas, hidrotérmicas e hidrobiológicas). Así es posible estudiar la estructura y grado de organización del karst a partir de las respuestas naturales de los acuíferos en las surgencias. La investigación hidrogeológica actual tiende a considerar el medio kárstico compartimentado en sistemas en el cual el flujo subterráneo se organiza para constituir una unidad de drenaje (Manguin, 1975). Según este modelo conceptual, el sistema kárstico está constituido por las siguientes partes: impluvium no kárstico, la zona de infiltración o zona no saturada y la zona saturada.
El agua en contacto con los distintos minerales constitutivos del medio rocoso por donde transita, adquiere una composición que es característica de la naturaleza del mismo, cuyo análisis puede brindar un mayor conocimiento del sistema drenado (Schoeller, 1962; Miserez, 1973; Bakalowicz, 1979; Antiguedad, 1986; Morales, 1991, Freixes, 1991). El proceso de disolución de los carbonatos por las aguas meteóricas da lugar, fundamentalmente, a iones HCO3
-, Ca2+ y Mg2+ , y sólo en ocasiones a algunas cantidades de CO3
2- . En este proceso el CO2 originado por la descomposición microbiana de la materia orgánica en el suelo, y a partir de la atmósfera directamente, en contacto con el agua y los materiales del karst, se hidrata, forma iones complejos y controla además la disociación de los carbonatos. La cinética de las reacciones H2O-CO2-MeCO3 (Me = Ca2+ o Mg2+) está determinada por la velocidad de reacción más lenta que corresponde a la interacción de la interfase líquido-sólido.
Durante las precipitaciones, el agua de infiltración conteniendo una cierta cantidad de CO2 (que depende de la materia orgánica disponible, las condiciones estructurales del suelo y del grado de desarrollo de la actividad microbiológica del mismo), disolverá más o menos calizas hasta alcanzar el equilibrio. Por tanto, el pH, el contenido de CO2, el contenido de los iones que participan en el equilibrio de los carbonatos, la dureza, la mineralización y el grado de saturación de los minerales presentes (calcita y dolomita), brindan una valiosa información sobre las características de la zona de infiltración, el modo de tránsito del agua por el macizo y el tiempo de residencia de ésta en el sistema.
Así por ejemplo, un agua insaturada con poco contenido de iones disueltos indica que el tránsito del agua a través del sistema kárstico no ha sido suficientemente largo para que se hayan establecido los equilibrios de los carbonatos. Por el contrario, si el agua está sobresaturada, posee elevados contenidos de iones disueltos, poco CO2 y altos valores de pH, indica que la misma ha permanecido suficiente tiempo en el interior del karst. No obstante, debe tenerse en cuenta que en ocasiones un alto contenido de iones disueltos pudiera estar asociado a la presencia de rocas con un alto grado de trituración o porosidad, donde es mayor la superficie de contacto agua-roca.
En ocasiones, la pérdida de CO2 en cantidades más o menos grandes provoca la recombinación iónica y precipitación de minerales, alcanzándose la saturación con menor contenido de iones disueltos, lo cual complica la interpretación del régimen de circulación hídrica y el tiempo de tránsito del agua en el sistema.
El comportamiento de los macroconstituyentes del agua no relacionados con los equilibrios de los carbonatos (Cl-, SO4
2- , Na+, K+, NO3- y SiO2) requiere un análisis particular y complementa la
información sobre el comportamiento del sistema.

El ion Cl- está asociado, fundamentalmente, a las precipitaciones y su contenido depende de la altura y la distancia del punto muestreado al mar; así como del tipo del evento lluvioso. En los procesos de evapotranspiración el contenido de este ion puede sufrir una reconcentración, especialmente en la zona epikárstica (Bakalowicz, 1979). En los pozos ubicados en las llanuras litorales, un alto contenido de Cl- puede estar asociado a la intrusión salina o los aportes de fuentes contaminantes.
Debido a que el Cl- y los iones asociados al ciclo del nitrógeno son aportados desde el exterior del sistema, su evolución temporal en las surgencias permite obtener información sobre las modalidades de infiltración del agua en el mismo y la existencia o no de elementos que favorezcan la homogeneización del quimismo del agua que entra al sistema. Además su correlación con otros elementos pueden ofrecer información sobre el origen del mismo; así como establecer o delimitar zonas de alimentación del sistema (Morales, 1991).
El contenido de NO3- en las aguas depende, fundamentalmente, de la actividad antrópica. El
SiO2, por su parte está asociado a la presencia del silicio, más abundante en las regiones no carbonatadas.
El ion Na+ puede tener un mismo origen que el Cl-, aunque debido a la capacidad de intercambio iónico de las arcillas, éstas pueden provocar diferencias significativas en las aguas respecto al contenido de aquel. El ion K+ puede sufrir grandes variaciones como resultado de las labores agrícolas a partir de los fertilizantes y de la capacidad de las raíces de las plantas en absorber este ion.
El contenido de ion SO42- en las aguas también pudiera ser aportado por la actividad
agrícola, y en ocasiones por la propia industria. En la zona del suelo, las aguas pueden adquirir considerables contenidos de este ion por disolución de sales solubles como yeso o anhidrita. La oxidación de las piritas, comúnmente asociadas a los carbonatos, también puede enriquecer el contenido de SO4
2- en el agua. Tal como ha señalado Aminot (1974), una variable hidroquímica tiene validez limitada para
fines de caracterización,y una mayor información se obtiene del estudio de la evolución temporal del quimismo de las aguas en las surgencias.
Como la composición química del agua evoluciona en su movimiento a través de los sistemas carbonatados, la información hidroquímica obtenida en puntos de control, fundamentalmente las emergencias, es función además del tiempo de residencia y las condiciones del movimiento del agua. De esta forma, la existencia de homogeneidades o heterogeneidades hidroquímicas está relacionada con la organización del drenaje del sistema. Mediante muestreos sistemáticos con frecuencia al menos quincenal o mensual, representativos de las diferentes fases hidrodinámicas (crecida, decrecida y agotamiento), algunos investigadores han podido determinar las fluctuaciones diurnas o estacionales del quimismo de las aguas kársticas (Febre, 1983; Bakalowicz, 1979; Antiguedad, 1986; 1988; Freixes et al, 1993, Morales y Antiguedad, 1993), o debido a una crecida ocasional (Bakalowicz, 1979; Fagundo, Rodríguez y Vega, 1995), estableciéndose una metodología para diferenciar el grado de organización y funcionamiento hidrodinámico de los sistemas kársticos a partir de sus respuestas naturales.
De acuerdo a la metodología propuesta por Antiguedad (1988), deben tenerse en cuenta dos aspectos: la magnitud del descenso y el tiempo de recuperación de la conductividad. El descenso de la CE es función de la dilución, una variación grande de la mineralización (conductividad) es consecuencia de la mezcla de aguas donde es importante el aporte de las aguas de infiltración rápida.

Una variación pequeña de CE indica falta de dilución, es decir, falta de mezcla importante de las aguas del manto, mas mineralizadas, con las aguas meteóricas. El tiempo de recuperación de la CE es función de la modalidad de infiltración, si éste es corto, indica el predominio de las aguas de infiltración rápida sobre las aguas de infiltración retardada. Si el tiempo de recuperación es largo, sucede lo contrario.
Empleo de relaciones iónicas
El uso de relaciones iónicas en la caracterización de las aguas naturales, también aparece reportado con frecuencia en las revistas y libros sobre el karst. Estas relaciones se expresan en miliequivalente por litro (meq/L) y van precedidas de la letra r, que significa relación, las cuales reflejan las diferentes propiedades de interacción con el medio. Entre las principales relaciones empleadas se destacan las siguientes:
r MgCa
2
2
+
+ : Refleja la relación entre la calcita y la dolomita en las aguas naturales que se
mueven por silicatos, es una expresión del grado de acidez de estas rocas.
r CaNa
2+
+ : Relación entre la calcita y la halita u otras fuentes de Na+. En las aguas kársticas
esta magnitud es muy superior a 1, excepto en aquellas que alcanzan grandes profundidades o participan en el proceso de mezcla con el agua de mar. En esos casos pueden tomar valores muy inferiores a la unidad.
r KNa
+
+ : Está relacionada con el intercambio iónico que experimentan las aguas subterráneas,
en la zona del suelo, con las arcillas y otros tipos de rocas. Se ha empleado para distinguir diferencias entre aguas superficiales (Christopher, 1975) y para interpretar el drenaje en diferentes regiones kársticas (Christopher, 1977).
r HCOCl
3−
−: Explica la relación entre los minerales calcita y halita disueltos en el agua, así
como el grado de intrusión de las aguas marinas en los karsts litorales.
r SOCl
42−
− : Relación entre el yeso y la halita u otros agentes ajenos al karst.
r Cl Na K
Cl
− − + + +
−( ) : Esta relación se utiliza para determinar el denominado índice de cambio
de base, el cual relaciona los iones cambiados en una reacción de intercambio iónico y los iones previamente existentes en el medio.
r Cl Na KSO HCO NO
− − + + +
− + − + −( )
42
3 3: Esta relación se denomina índice de desequilibrio y se ha utilizado
también por los hidrogeólogos.
Con el objetivo de caracterizar las aguas naturales con fines geológicos, también se suelen emplear otras relaciones iónicas tales como:
r LiNa
,r FeCl
, r BrCl
, r ICl
2+
+
+
−
−
−
−
−

Además se emplean en la caracterización hidrogeoquímica, los contenidos de SiO2, CO2, algunos elementos minoritarios y otros parámetros químico-físicos e hidrológicos. Algunas de estas relaciones y otras similares son utilizadas también con el objetivo de caracterizar sistemas geotérmicos (Urbani, 1991).
Algunos kárstologos españoles han empleado las relaciones iónicas señaladas anteriormente para describir las propiedades de las aguas kársticas de una cuenca determinada (Fernández Rubio y Eraso, 1975; Castillo y Eraso, 1975), así como otras adicionales:
r Cl NaCl
Ca MgNa
Ca NaNa K
− +
−
+ +
+
+ +
+ +− + +
+, ,
2 2 2
El estudio de las modificaciones producidas por las aguas marinas en los karst costeros, se ha abordado mediante la utilización de algunas relaciones iónicas (Morell et al, 1986; 1988). Todas estas expresiones de relación iónica se han implementado en el algoritmo denominado Agmar (Fagundo et al, 1986); así como en el sistema computarizado SAPHIQ (Alvarez et al, 1993). Representación gráfica de la composición química de las aguas
La representación gráfica de los datos hidroquímicos constituye una herramienta de trabajo muy eficiente en la interpretación de las propiedades de un agua, así como para hacer comparaciones. También permite ver con facilidad el comportamiento y evolución de un agua en un territorio determinado y a través del tiempo. Entre los métodos gráficos más utilizados se destacan los siguientes: diagrama de barra, diagrama circular, diagrama de Stiff, diagrama triangular, diagrama vertical.
Los diagramas de barras o de columna se han empleado ampliamente por su sencillez. La composición química se puede expresar en mg/L, meq/L o % meq/L. La forma más común consiste en presentar en la columna de la derecha los tantos porciento de los miliequivalentes de aniones en el orden rCl-, rSO4
2-, rHCO3- de arriba hacia abajo y en la columna de a izquierda, rNa+,
rMg2+, rCa2+. En la figura 3.1 se ilustra este método en la representación de algunos datos hidroquímicos.
El diagrama circular expresa la composición mediante un círculo, cuyos ángulos son proporcionales a las concentraciones y sus radios o diámetros al total de sólidos disueltos, la suma de los aniones y de los cationes es igual a 180°. Constituye uno de los métodos más útiles para expresar la composición química en el mapa de una zona.
Los mapas hidrogeoquímicos ofrecen una valiosa información sobre las relaciones existentes entre el quimismo de las aguas y las condiciones geológicas, así como físicogeográficas, cuando se superponen en el mismo, la litología presente, la red de drenaje y los diagramas circulares con la composición química de las aguas.

Figura 3.1. Representación mediante diagramas de barras de la composición química de las aguas kársticas de de la cuenca del río Cuyaguateje (Pinar del Río) a lo largo del perfil NE-SO.
El diagrama de Stiff (1951) emplea un sistema de ejes horizontales paralelos y un eje vertical.
En cada uno de estos se coloca un ion determinado. Una forma adecuada consiste en colocar en los ejes de la izquierda las concentraciones (meq/L) de los iones Na+ + K+, Ca2+ y Mg2+ de arriba hacia abajo y, en el mismo orden, en los ejes de la derecha, los iones Cl-, HCO3
- + CO32- , así como
SO42- . En España se suele tomar como segundo eje el constituido por los iones Mg2+ (izquierda) y
SO42- (derecha).
Este método permite apreciar y comparar en forma rápida los diferentes tipos de aguas (Figura 3.2), cuando éstas se encuentran en cantidades limitadas. Es especialmente útil cuando se quieren apreciar cambios en el comportamiento de un agua en determinado tiempo, por características climáticas, hidrogeológicas o efectos antrópicos (Figura 3.3).
Otro método gráfico utilizado para la representación de la composición química de un agua es el de tipo triangular. El más sencillo consiste en el empleo de dos triángulos equiláteros, uno para los aniones y otro para los cationes, donde cada vértice representa 100 % de un ion particular.
Schoeller (1962) ha propuesto el uso de un triángulo único, en el cual sobre los ejes se representa la concentración (% meq/L) de rCl- y rNa-; rHCO3
- + rCO32- y rCa2+;rSO4
2- y rMg2+. Las concentraciones aniónicas determinan un punto y las catiónicas otro. El agua queda definida por el eje que une ambos puntos.
Los diagramas triangulares de Hill y Piper tienen como ventaja en relación a los anteriores, que permiten representar un gran número de muestras en un sólo gráfico. En éstos, los triángulos de aniones y cationes ocupan los ángulos inferiores izquierdo y derecho con sus bases alineadas. La parte central del diagrama posee forma de rombo y sobre éste se proyectan los puntos de cada uno de los triángulos por medio de una recta paralela al borde superior del rombo. La intersección de

estas dos rectas representa la composición del agua con respecto a una determinada agrupación de aniones y cationes (Figura 3.4).
Por último, se debe destacar las posibilidades que ofrecen los diagramas de tipo vertical en la representación gráfica de la composición química de un grupo numeroso de muestras. En estos diagramas se coloca en el eje de ordenadas la concentración (en mg/L, meq/L o % meq/L) y en el eje de las abscisas los distintos iones presentes. El más usado de estos es la variante propuesta por Schoeller, en la cual la composición química se expresa en unidades logarítmicas (Figura 3.5).
Figura 3.2. Representación mediante diagramas de Stiff de la composición química de un agua kárstica estratificada por el efecto de mezcla: Pozo B-15, en la zona salina del plan citrícola Victoria de Girón (Bolondrón)

Figura 3.4. Representación de la composición química de las aguas de una región kárstica mediante el diagrama triangular de Hill y Piper.

Figura 3.5. Representación de la composición química de aguas de una región kárstica mediante el diagrama vertical de Shoeller.

Clasificación de las aguas desde el punto de vista geológico y karstológico
En forma general, las aguas naturales se han clasificado de formas distintas sobre la base de su génesis, tipo de roca asociada, sus características químico-físicas, agresividad, uso y otras propiedades.
Sobre la base de su génesis, se han clasificado de forma diferente por distintos investigadores, éstas pueden agruparse de la manera siguiente (White, 1963):
1. Aguas juveniles (no involucradas en la circulación atmosférica). a) Magmáticas. b) Otras aguas juveniles. 2. Aguas surgentes o reciclables (involucradas en la circulación atmosférica). a) Aguas meteóricas:
- Aguas de precipitación (lluvia o nieve). - Aguas de suelo. - Aguas subterráneas cercanas a la superficie (subsuperficiales).
b) Aguas oceánicas que penetran en los acuíferos. c) Aguas fósiles o connatas:
- De origen marino. - De origen no marino.
ch) Aguas metamórficas. - Aguas con alto contenido de CO2 y boro. - Otros tipos de aguas.
3. Aguas magmáticas No se incluyen en esta clasificación las aguas que forman parte constituyente de los
minerales. En relación a la clasificación de las aguas kársticas en particular, han primado los criterios
relacionados con las zonas hidrogeológicas del karst, así como su agresividad y propiedades químico-físicas.
Las aguas que se mueven en forma superficial o subterránea por una región kárstica, se pueden considerar de dos tipos: alóctonas y autóctonas en relación al macizo. En la tabla 3.2 se muestran las propiedades químico-físicas de algunas aguas naturales de estos dos tipos.
Se consideran aguas alóctonas al karst, aquellas que se originan en una litología tal como los granitos, los sedimentos de origen vulcanógeno, las areniscas, las arcillas y otros medios poco permeables, constituidos por minerales insolubles, principalmente aluminosilicatos. En muchos casos la composición química de estas aguas difiere poco de las precipitaciones en cuanto a su grado de mineralización y tipo de agua. En otras ocasiones, sin embargo, en virtud de los procesos de intemperismo y condiciones de flujo que tienen lugar en el medio (Figura 3.6), la composición química de estas aguas es muy diferente de la típica que debe ofrecer esa litología, aunque por lo general, presentan rasgos que las distinguen de las aguas kársticas carbonatadas.

Nº T(°C) pH CE
(µS/cm) Cl- SO42- Ca2+ Mg2+ Na++K+ CO2 HCO3
- (mg/L) (meq/L) (meq/L) (meq/L) (meq/L) (meq/L) (meq/L)
1 22,0 6,98 41 2,7 0,25 0,11 0,00 0,10 0,00 0,26 2 15,8 6,52 38 4,0 0,18 0,17 0,00 0,065 0,02 0,27 3 28,0 6,69 150 18,5 1,00 0,40 0,10 0,50 0,10 0,90 4 24,0 7,55 200 3,3 1,75 0,32 0,06 1,08 0,32 0,73 5 23,8 8,10 490 2,6 5,15 0,72 0,20 2,60 1,30 2,17 6 23,8 8,30 400 1,0 3,85 0,39 0,48 0,10 3,90 0,72 7 22,2 7,72 575 19,8 6,80 0,37 0,01 3,90 2,67 0,61 8 26,0 6,50 1290 209,0 13,60 0,45 0,14 4,78 4,62 4,79 9 27,4 6,90 1100 38,5 6,20 2,28 1,74 2,86 6,68 2,68
10 21,1 7,15 500 22,9 4,70 0,25 0,00 4,64 0,24 0,07 11 22,2 8,25 230 1,0 2,00 0,37 0,09 1,79 0,21 0,46 12 22,0 8,24 243 1,3 2,50 0,39 0,06 1,40 0,89 0,66 13 25,0 8,00 140 0,9 1,00 0,31 0,08 0,60 0,20 0,59 14 22,2 7,95 350 3,3 3,25 0,42 0,08 2,66 0,34 0,75 15 22,8 7,50 375 8,8 3,90 0,39 0,12 3,40 0,32 0,69 16 24,6 7,31 414 17,4 4,00 0,39 0,29 2,99 0,49 1,20 17 22,2 7,22 377 19,8 3,50 0,31 0,00 3,37 0,12 0,40 18 23,0 7,30 505 19,8 4,45 0,53 0,90 4,00 0,60 1,28 19 23,4 6,80 700 59,4 6,10 0,49 1,01 4,80 0,60 2,20 20 27,0 7,39 650 14,4 4,00 0,82 2,00 3,59 1,48 1,75 21 23,8 7,58 560 50,6 5,15 0,78 0,20 4,47 0,76 0,90 22 25,6 7,30 900 75,7 7,15 1,27 1,34 7,30 1,38 1,08 23 31,8 7,70 610 30,8 5,55 1,11 0,04 3,52 2,06 1,12 24 30,0 7,20 4505 49,0 3,75 1,10 35,42 28,02 6,11 6,14 25 28,0 7,83 400 1,50 3,25 0,68 0,58 2,20 0,59 1,72 26 22,6 7,90 380 1,1 4,15 0,35 0,06 3,40 0,50 0,66 27 23,0 7,35 610 18,0 4,93 0,73 0,58 4,85 0,67 0,72 28 23,0 7,40 522 13,0 4,98 0,73 0,40 3,60 1,67 0,84 29 25,0 7,60 1100 30,8 6,21 4,64 0,26 4,39 2,91 3,81 30 25,0 7,40 1000 16,5 4,25 6,30 0,36 3,88 0,88 6,15 31 25,0 7,30 9950 23,1 5,35 92,00 4,04 7,32 16,08 77,99 32 25,0 7,60 30000 30,8 5,00 271,00 20,80 13,20 52,80 230,80 Tabla 3.2. Propiedades químico-físicas de algunas aguas naturales tomadas como ejemplos, por ser representativas de diferentes condiciones hidrogeológicas. Nota: Aguas de lluvia (1-2); arroyos superficiales que discurren por: areniscas, esquistos y pizarras (3), sedimentos efusivo -sedimentarios (4-5), serpentinitas (6), sedimentos terrígeno-carbonatados (7); pozos en serpentinitas asociados al drenaje profundo (8-9); curso superficial en la zona de alimentación de un macizo kárstico (10); aguas de cueva (11-12); resurgencias y surgencias kársticas (13-15); manantiales kársticos someros (16-19); manantiales kársticos asociados al drenaje profundo (20-24); ríos superficiales kársticos (25-26): pozos en cuencas cerradas en una llanura kárstica (27-28); dolinas, casimbas y lagunas en una llanura kárstica litoral (29-32).

Figura 3.6. Representación mediante diagramas de Stiff de la composición química de aguas del drenaje alóctono al karst, que se mueven por aluminosilicatos con diferente grado de acidez: rocas ácidas (1-5), medianamente ácidas (6 y 7) y ultrabásicas(8-12).

Las aguas autóctonas de los macizos kársticos calcáreos, se caracterizan por reflejar aquellos
iones que son disueltos en el proceso de erosión química, HCO3-, Ca2+ y Mg2+ y una mineralización
no muy elevada. El contenido de SO42- y Cl- depende de la presencia de yeso o rocas salinas
respectivamente, o de procesos producidos por la actividad del hombre (Figura 3.7). En los terrenos carbonatados litorales existe una mayor o menor cantidad de iones Cl- y Na+,
en dependencia del grado de intrusión marina. Además, a partir de cierto nivel de mezcla, el contenido de Mg2+ es superior al del Ca2+, como resultado de la mayor abundancia de aquel ion en el mar, tal como se ilustra en la figura 3.8.
Las aguas kársticas también se pueden clasificar en relación con su movimiento horizontal y vertical a través del macizo. En el primer caso se distinguen las aguas: de la zona de alimentación, conducción y emisión. En el segundo caso, aguas de la zona de aeración (vadosas), de la zona de saturación (mixtas) y de la zona de saturación y circulación profunda (freáticas). Desde el punto de vista karstológico, esta zonación hidrogeológica se ha definido de forma diferente por distintos investigadores. Desde el punto de vista químico-físico el rasgo más distintivo lo ofrece el contenido de CO2 y CaCO3.


Clasificación del agua kárstica por su uso
La composición química de un agua natural, en función del uso que a la misma se le dé, se denomina calidad del agua, y existen una serie de normas que regulan las concentraciones permisibles que debe poseer cada elemento o indicador de calidad según los diferentes usos. Por ejemplo, las normas establecidas para que un agua se pueda utilizar para el abasto exigen un contenido despreciable de los componentes de los ciclos del nitrógeno y el fósforo. Sin embargo, para el riego las aguas deben poseer un alto contenido de los mismos.
Relacionadas con la calidad del agua, se encuentran las medidas a tomar en la colección de la muestra. García y Beato (1979) han propuesto un grupo de recomendaciones para el muestreo de aguas y el control de su contaminación.
Además de la calidad químico-física de las aguas es necesario controlar la calidad bacteriológica. La contaminación del agua kárstica por organismos patógenos se debe principalmente al vertimiento o percolación de residuales urbanos, o agroindustriales, puesto que este tipo de microorganismo no se origina en las condiciones naturales.
La calidad bacteriológica en ríos, lagos y aguas subterráneas, no se controla mediante la determinación de microorganismos patógenos, sino a través de la identificación de bacterias fecales, es decir, bacterias que subsisten normalmente en el tracto digestivo humano. En general, se utilizan como índice las bacterias pertenecientes al grupo coliforme. Existe una relación aproximada entre la cantidad de estas bacterias y las de tipo patógenas.
Las aguas superficiales y subterráneas de las regiones kársticas, desde el punto de vista de su uso, se clasifican de la misma forma que las restantes aguas naturales. Los usos que se hacen del agua kárstica como recurso hídrico son numerosos, si se atiende al carácter de su utilización, se pueden clasificar en diez grupos (Ferro, 1982):
1. Suministro rural y urbano. 2. Regadío para la producción agraria. 3. Abastecimiento para los procesos industriales. 4. Producción de energía mecánica y eléctrica. 5. Agua para fines terapeúticos. 6. Consumo mediante embotellamiento. 7. Carga, descarga y transportación. 8. Conservación de la flora y fauna silvestre. 9. Reproducción y consumo de plantas y animales acuáticos. 10. Agua para fines recreativos y culturales. Existe una clasificación que además de las diez consideraciones anteriores, relaciona los
principales parámetros químico-físicos y bacteriológicos utilizados en el control de la calidad de las aguas (Gutiérrez, 1982). Para la mayoría de los usos que se le da al agua, existen normas establecidas por el Instituto Nacional de los Recursos Hidráulicos (INRH), y el Ministerio de Salud Pública (MINSAP), organismos encargados de velar por la preservación de la calidad del agua.
Otro método de determinar la calidad de las aguas, consiste en evaluar un determinado índice de calidad (García, 1988), que representa la suma de diferentes indicadores del deterioro de la misma, a los cuales se les da un peso diferente en dependencia de los objetivos del control. Clasificación hidroquímica

Los criterios químico-físicos de la clasificación de las aguas kársticas no se distinguen de los utilizados para las aguas naturales en general. Se basan en el contenido de los iones más abundantes.
En la literatura aparecen numerosas clasificaciones que responden a diferentes objetivos (García, 1988; Catalán, 1988; Giménez et al, 1995)). De su análisis se puede considerar como más ventajosas las siguientes:
1. Clasificación de Alekine. 2. Clasificación de Shchoukarev. 3. Clasificación de Kurlov 4. Clasificación de Palmer. 5. Clasificación de Schoeller.
Método de clasificación de Alekine Este método toma en cuenta los tres aniones más importantes: HCO3
- + CO32- , Cl- y SO4
2- . A su vez cada clase se divide en tres grupos, según el catión que predomine (Ca2+, Mg2+ o Na+). De esta forma, las aguas se clasifican en:
Tipo: I. Aguas bicarbonatadas (predominio de HCO3
-). II. Aguas sulfatadas (predominio del SO4
2- ). III. Aguas cloruradas (predominio del Cl-). Clases: a) Aguas cálcicas (predominio del Ca2+ ). b) Aguas magnésicas (predominio del Mg2+ ). c) Aguas sódicas (predominio del Na+).
Grupos: 1. r HCO3
- > r Ca2+ + r Mg2+
2. r HCO3- < r Ca2+ + r Mg2+ < r HCO3
- + r SO42-
3. r HCO3- + r SO4
2- < r Ca2+ + r Mg2+ , es decir, r Cl- >r Na+
4. r HCO3- = 0. Este grupo no existe en el tipo I.
Gutiérrez (1982) presenta una modificación del método de Alekine, mediante el cual se
comparan los pesos equivalentes (en % meq/L) de un agua dada. Cuando un catión o anión presenta más de 50 % meq/L, el tipo de agua recibe el nombre del ion, por ejemplo bicarbonatada cálcica. Cuando un catión o anión no alcanza 50 %, pero su contenido es mayor de 33 % meq/L, el tipo de agua corresponde a un nombre combinado, por ejemplo bicarbonatada cálcico-magnesiana. Método de clasificación de Shchoukarev
Emplea como índice de clasificación los iones que se encuentran en un porcentaje superior a 25 % del total de miliequivalentes de aniones o de cationes:
1. En relación a los aniones: a) Aguas bicarbonatadas.

b) Aguas sulfatadas. c) Aguas cloruradas. ch) Aguas bicarbonatadas sulfatadas. d) Aguas bicarbonatadas cloruradas. e) Aguas sulfatocloruradas.
f) Aguas sulfatocloruradas bicarbonatadas.
2. En relación a los cationes: a) Aguas cálcicas. b) Aguas magnésicas. c) Aguas sódicas. ch) Aguas calcicomagnesianas. d) Aguas calcicosódicas. e) Aguas magnesicosódicas. f) Aguas cálcico-magnésico-sódicas.
Método de clasificación de Kurlov
Este método es similar al anterior, pero toma en cuenta para la clasificación los iones que se encuentran en un porcentaje superior al 20 % en lugar del 25 %, del total de miliequivalentes de aniones o cationes.
Una variante de los métodos de Kurlov se ha propuesto por Fagundo y Rodríguez (1992) sobre la base de las relaciones estequiométricas Na++K+:Ca2+:Mg2+:Cl-:HCO3
-:SO42-. Por ejemplo, un
agua del tipo bicarbonatada cálcica (HCO3--Ca2+) posee una relación estequeométrica 1:8:1:1:8:1.
En total, todas las aguas naturales se pueden caracterizar mediante este método, a través de 28 X 28 = 784 combinaciones numéricas o tipos de aguas, tal como se relaciona en la tabla 3.3. Estos tipos de aguas se pueden representar gráficamente mediante diagramas de Stiff para facilitar su comprensión, y corresponden a patrones hidrogeoquímicos determinados por factores geológicos, hidrogeológicos y ambientales. De ese total de tipos de aguas naturales, las que se desarrollan en los acuíferos kársticos constituyen un número más limitado.

1. Relación estequiométrica de aniones
Tipo de agua Relación Cl-:HCO3-:SO4
2-
HCO3-
Cl- SO4
2- HCO3
- > Cl- Cl- > SO4
2- HCO3
- > SO42-
Cl- > HCO3-
SO42- > Cl-
SO42- > HCO3
- HCO3
- = Cl- HCO3
- = SO42-
Cl- = SO42-
Cl- = HCO3- = SO4
2-
1:8:1 2:7:1 1:7:2 2:6:2 8:1:1 7:2:1 7:1:2 6:2:2 1:1:8 1:2:7 2:1:7 2:2:6 3:6:1 4:5:1 6:1:3 5:1:4 1:6:3 1:5:4 6:3:1 5:4:1 3:1:6 4:1:5 1:3:6 1:4:5 4:4:2 2:4:4 4:2:4 3,3:3,3:3,3
2. Relación estequiométrica de cationes Tipo de agua Relación Na+: Ca2+: Mg2+ Ca2+ Mg2+ Na++K+ Ca2+ > (Na+ + K+) (Na+ + K+) > Mg2+ Ca2+ > Mg2+ (Na+ + K+) > Ca2+ Mg2+ > (Na+ + K+) (Na+ + K+) > Ca2+ Ca2+ = Mg2+ Ca2+ = (Na+ + K+) Mg2+ = (Na++K+) Ca2+ = Mg2+ = (Na+ + K+)
1:8:1 2:7:1 1:7:2 2:6:2 1:1:8 1:2:7 2:1:7 2:2:6 8:1:1 7:2:1 7:1:2 6:2:2 3:6:1 4:5:1 6:1:3 5:1:4 1:6:3 1:5:4 6:3:1 4:5:1 3:6:1 4:1:5 1:3:6 1:4:5 2:4:4 4:4:2 4:2:4 3,3:3,3:3,3
Tabla 3.3. Relación estequiométrica entre aniones y cationes correspondientes a los 28 x 28 =784 patrones hidrogeoquímicos de las aguas naturales Método de clasificación de Palmer
Este método se fundamenta en que las características de las aguas naturales dependen de la salinidad. Considera que las sales de ácidos fuerte originan salinidad, mientras que las de los ácidos débiles dan alcalinidad, de acuerdo con este método las aguas se caracterizan por las propiedades siguientes:
S1: Salinidad primaria (por álcali). S2: Salinidad secundaria (dureza permanente).

S3: Salinidad terciaria (acidez). A1: Alcalinidad primaria (alcalinidad permanente). A2: Alcalinidad secundaria (alcalinidad temporal).
Método de clasificación de Schoeller
Este método toma en consideración el contenido (en meq/L) de los compuestos disueltos, dando el orden de importancia siguiente: cloruros > sulfatos > bicarbonatos más carbonatos > índice de intercambio de base (IIB) > relaciones entre aniones y cationes. Mediante este método se establecen los tipos de aguas siguientes:
1. En relación al cloruro (r Cl-): a) Aguas hipercloruradas (> 700 hasta saturación). b) Aguas clorohalásicas (420-700). c) Aguas cloruradas fuerte (140-420). ch) Aguas cloruradas medias (40-140). d) Aguas oligocloruradas (15-40). e) Aguas cloruradas normales (< 10). 2. En base al SO4
2- (r SO42- ):
a) Aguas hipersulfatadas (> 58). b) Aguas sulfatadas (24-58). c) Aguas oligosulfatadas (6-24). ch) Aguas sulfatadas normales (< 6). 3. En relación a la concentración de bicarbonato más carbonatos (r HCO3
- + r CO32- ):
a) Aguas hipercarbonatadas (> 7). b) Aguas carbonatadas normales (2-7). c) Aguas hipocarbonatadas (< 2).
Por último, el IIB se utiliza para indicar la relación entre los iones que se intercambian
respecto a los inicialmente presentes en el agua.
Empleo de métodos numéricos El tratamiento numérico de los datos hidroquímicos es imprescindible para poder interpretar
el objeto karstológico: el proceso de interacción de las aguas naturales con el medio geológico local, las características de un acuífero, el relieve de un paisaje kárstico (como resultado de la acción de los agentes de intemperismo y otros factores), la evolución química de un agua kárstica por la actividad del hombre, etcétera, a través de los atributos o parámetros químico y físicos: temperatura, pH, conductividad eléctrica, caudal, cantidad de CO2 disuelto, concentración de macrocomponentes, microcomponentes, indicadores de calidad, etcétera.
Los métodos matemáticos y estadístico-matemáticos, en particular, dan la posibilidad de expresar en forma compacta el objeto en relación con sus atributos. Los métodos de la Estadística Clásica o Descriptiva han sido los más empleados por los investigadores, tanto en el campo de las geociencias como en otras ciencias. Su uso ha sido especialmente adecuado en el tratamiento de las

grandes bases de datos originadas en los programas de monitoreo de la calidad del agua (Ponce, 1980; Machkova et al, 1993).
Entre las estimaciones estadísticas más empleadas para caracterizar los fenómenos naturales se encuentran: las medias aritmética y geométrica, valores máximos y mínimos, moda, varianza, desviación estándar, intervalos de confianza, modelo de distribución de datos, prueba de hipótesis, dependencia matemática entre atributos cualitativos (regresión y correlación matemática), series cronológicas y otras.
En muchos países existen redes de control de la calidad de las aguas, tanto superficiales como subterráneas, las cuales generan un gran volumen de datos hidroquímicos, hidrológicos y climáticos, cuyo procesamiento estadístico puede brindar una valiosa información acerca de las regularidades matemáticas entre las diferentes variables. Así por ejemplo, es posible encontrar algunas relaciones simples, al menos para determinados intervalos, que se aproximen a las funciones reales y permitan estimar unas variables en función de otras de más fácil medición (Guerón et al, 1993).
El desarrollo de la electrónica ha permitido la fabricación de computadoras personales y esto ha estimulado la creación y uso de software, que en forma sencilla, ofrecen a los especialistas la posibilidad de procesar grandes volúmenes de datos de medición obtenidos en las redes de observación y control sistemático.
Con el objetivo de caracterizar el comportamiento químico-físico de las aguas kársticas, se han empleado paquetes de programas comerciales con los cuales se pueden obtener matrices de correlación y estudiar el efecto recíproco de cada variable con respecto a las otras (Drake y Ford, 1974; Alvarez et al, 1993); así como correlaciones múltiples cuando una variable depende del comportamiento simultáneo de otras (Paterson, 1972).
Un sistema computarizado para la realización de mapas y diagramas hidrogeológicos ha sido reportado por Yelmos (1993), el cual permite la creación de diagramas de ensayo de bombeo, mapa de inventario de puntos de agua, diagramas de Piper-Hill, gráficos logarítmicos de Schoeller, diagramas de Stiff y mapas hidrogeoquímicos.
Además de los métodos clásicos, se emplean cada vez, con mayor frecuencia, los métodos de la Estadística Multivariada desarrollados a finales de la década del setenta. Entre estos se encuentran el denominado análisis de "cluster " (enjambres) y el análisis factorial. Muchos de estos métodos fueron aplicados en otras disciplinas de la ciencia antes que en las ciencias de la Tierra.
Los análisis de enjambres constituyen herramientas útiles para la descripción de datos cualitativos y cuantitativos. Este método consiste en seleccionar las mayores similitudes (correlaciones) entre parejas de variables (Pulido Bosch, 1989). De esta manera, se forman las primeras parejas que denotan una mayor similitud entre las mismas. A continuación se genera una nueva matriz de similitud que ya incluye a las agrupaciones previas, como si se tratase de una sola variable (Callejón et al, 1984). Una vez agrupadas todas las variables, el gráfico que resulta (dendograma) permite visualizar en forma rápida los grupos de variables afines y distinguirlos de aquellos que apenas guarden relación con el resto.
El análisis factorial es otro método de Estadística Multivariada que ha encontrado uso en las investigaciones relacionadas con el karst. Mediante este tratamiento se han estudiado los factores que controlan el comportamiento químico-físico de las aguas en las regiones carbonatadas (Jacobson et al, 1971) y aguas subterráneas en general (Dimitrov et al, 1993).
El procesamiento en el análisis factorial parte de la obtención de n autovalores y autovectores (componentes principales) de la matriz de n X n correlaciones y de la estimación de la fracción de la varianza total de los datos explicados por cada uno de tales componentes (Pulido Bosch, 1989). Estos se pueden interpretar a su vez, como nuevas "variables" no correlacionadas, las

cuales son combinaciones lineales de las variables originales, lo cual conduce a una reducción de la dimensionalidad del espacio en que se trabaja, sin pérdidas significativas de la varianza del conjunto de datos.
Para interpretar mejor los factores se ejecuta en estos una rotación varimax, mediante la cual las variables originales son proyectadas sobre los ejes. Esta operación no modifica la varianza explicada por los factores antes de la rotación.
Algunos investigadores han empleado los métodos de análisis factorial y de correlación multivariada de datos hidroquímicos, hidrológicos y climáticos con el objetivo de interpretar cómo se encuentran organizados los acuíferos kársticos (Bakalowicz, 1977; Bakalowicz y Manguin, 1980; Manguin, 1981; Manguin et Pulido Bosch, 1983). Este enfoque ha sido seguido por otros especialistas en hidrología kárstica (Callejón et al, 1984; Aguayo et al, 1988), y ecosistemas acuáticos en general (Montes et al, 1982; a, b).
Los métodos de análisis de cluster y análisis factorial fueron aplicados en Cuba por primera vez por Valdés y colaboradores para caracterizar aguas de diferente naturaleza hidrogeológica (Valdés y Fagundo,1981; Valdés y De la Cruz, 1982; De la Cruz y Valdés, 1985), y luego han sido generalizados a otros estudios relacionados con el karst (Abelló et al, 1993). En las figuras 3.9 y 3.10 se ilustra el empleo de estos métodos en la interpretación del comportamiento de las aguas de la sierra del Pan de Guajaibón y sus inmediaciones.
Figura 3.9. Dendograma que muestra los grupos numéricos determinados por el método de análisis de "cluster" (enjambres) al tratar datos hidroquímicos de la sierra del Pan de Guajaibón y sus inmediaciones.

Figura 3.10. Representación del resultado del tratamiento por análisis factorial de datos hidroquímicos de la sierra del Pan de Guajaibón y sus inmediaciones.
En las últimas décadas los análisis de correlación y espectral han constituido una de las técnicas que mejor acogida han tenido para la caracterización y cuantificación de series cronológicas de precipitaciones y caudales (Padilla y Pulido Bosch, 1993). Estos métodos han sido utilizado por muchos investigadores para describir e identificar la estructura y componentes de los acuíferos kársticos (Manguin, 1981; Manguin et Pulido Bosch, 1983; Antiguedad, 1988; Padilla, 1990; Padilla y Pulido Bosch, 1993).
Factores que determinan la composición y evolución química de las aguas kársticas
Las aguas kársticas adquieren su composición química mediante un proceso complejo, donde intervienen factores de tipo químico-físico, geológico, hidrogeológico, geomorfológico, pedológico, climático, antrópico y otros (Fagundo, 1990 a).
Los factores químico-físicos están regidos por las leyes termodinámicas que controlan la disolución de los minerales, estos factores desempeñan un papel importante en la forma en que las aguas kársticas adquieren su composición química y en el proceso de karstificación (Eraso, 1975). Entre estos se destacan: la solubilidad de los minerales, el contenido de gases disueltos, las condiciones del sistema (abierto o cerrado) en que la disolución tiene lugar, el pH, el potencial redox, el efecto salino o de fuerza iónica, el efecto de ion común y otros que se han discutido en el capítulo 1 de este libro.
Los factores geológicos se relacionan con la litología (composición de los minerales de las rocas), el estado de yacencia de las secuencias estratigráficas, la tectónica, el agrietamiento, la

textura y porosidad de las rocas, etcétera. La litología determina, por lo general, las facies hidroquímicas dominantes en una región determinada, es decir, el tipo de agua. En los terrenos kársticos carbonatados las aguas suelen ser del tipo HCO3
--Ca2+ . Los aspectos vinculados al agrietamiento y porosidad de las rocas influyen de manera determinante en el estado de división de las partículas, mientras más pequeña sea ésta, poseerá mayor superficie, facilitando la disolución del mineral. Las aguas que drenan a través de rocas calcáreas, muy trituradas por los procesos tectónicos, adquieren un contenido mayor de calcita disuelta que aquellas que drenan a través de calizas más compactas.
Los factores hidrogeológicos están relacionados con la permeabilidad del acuífero, el tipo de flujo, su velocidad, así como la zona por donde se mueve el agua (ver tabla 3.2). Todos estos aspectos inciden en el tiempo de contacto entre el agua y el mineral, por ejemplo, si el flujo tiene lugar en condiciones difusas a través de las rocas, el tiempo de interacción del agua con los minerales es más lento y, por tanto, la cantidad de minerales disueltos es mayor que si las condiciones de flujo son de tipo turbulentas a través de grietas más o menos amplias.
El contenido de CO2, la dureza y otras propiedades químico-física de las aguas kársticas difieren de acuerdo a la forma en que se mueve el agua y la zona hidrogeológica que ocupa (Figura 3.11). Estas propiedades; así como la naturaleza de los procesos de disolución de los minerales más comunes en el karst tropical de Cuba, calcita, dolomita y yeso, es discutida por Fagundo y colaboradores (1987).

En la zona de alimentación del acuífero (visto el movimiento en sentido horizontal) o en la zona de aeración o vadosa del karst (visto en sentido vertical), el contenido de CO2 de las aguas es relativamente elevado, puesto que en estos lugares ocurren intensos procesos de descomposición bacteriana de la materia orgánica. El pH en esta zona suele no ser elevado y la dureza pequeña. Los valores de la relación de saturación de la calcita y la dolomita (RSC y RSD) tienden a ser negativos, es decir, las aguas se encuentran insaturadas respecto a los minerales calcita y dolomita.
En la zona de conducción del acuífero el nivel de CO2 suele ser menor, porque una parte del gas es consumido, como consecuencia de la interacción del agua con los minerales del medio rocoso. El agua en esta zona adquiere una mayor mineralización y un pH más elevado. Esta evolución se hace más evidente si, por ejemplo, se muestrea un perfil de pozos desde la zona de alimentación hasta la de emisión del acuífero. A medida que el agua se aleja de la zona de alimentación decrece el CO2 y aumentan el pH, así como el contenido de calcita disuelta.
Las aguas en la zona de conducción se encuentran, por lo general, saturadas respecto a la calcita. Sin embargo, en las cuevas, las aguas que se infiltran (de degoteos y gours) poseen un contenido relativamente bajo, tanto del CO2 como de CaCO3, por la evaporación del gas y precipitación del CaCO3. Estas aguas se tienden a encontrar sobresaturadas respecto a la calcita.
En la zona de saturación o mixta, las aguas se encuentran casi siempre saturadas respecto a la calcita. Sin embargo, como resultado de los procesos de mezcla de aguas, éstas pueden hacerse agresivas nuevamente y continuar disolviendo minerales.
En la zona de saturación y circulación profunda, las aguas se caracterizan por presentar elevados contenidos de CaCO3 disuelto. En ocasiones aún poseen cantidades elevadas de CO2 y en los manantiales por donde emergen precipita el exceso de calcita. Además las aguas del drenaje profundo poseen, por lo general, una temperatura más elevada y estable que las de tipo meteóricas, la circulación de las primeras es más lenta y su caudal menor, el tiempo de interacción con los minerales es mayor. En estas condiciones pueden cambiar su típico patrón hidrogeoquímico o facie (agua de tipo bicarbonatada cálcica). En dependencia de los minerales presentes en las capas intersticiales o secuencias impermeables, con las cuales hacen contacto los estratos calcáreos, predominará en las aguas uno u otro ion.

Geología e
Hidrogeología T (°C) pH CO2
(mg/L) CE
(µs/cm) CaCO3 (mg/L)
Tipo de agua
Aguas no kársticas
Lluvia Efusivos Serpentinita
22,0 24,0 23,8 23,8
6,96 7,55 8,10 8,30
2,7 3,3 2,6 1,0
41 200 490 400
5 70 195 200
HCO3--Na+
HCO3--Ca2+ >Na+
HCO3--Ca2+ >Na+
HCO3--Ca2+
Aguas kársticas
Zona Alimentación Cuevas Zona aeración Zona circulación profunda
21,1 22,2 22,8
27,0
31,8 25,6 30,0
7,15 8,25 7,50
7,30
7,70 7,30 7,20
22,9 1,0 8,0
14,4
30,8 75,7 49,0
500 230 375
650
610 900
4 507
244 100 186
254
274 434
1 706
HCO3--Ca2+
HCO3--Ca2+
HCO3--Ca2+
HCO3->SO4
2- -
Ca2+ >Na+
HCO3--Ca2+>Mg2+
HCO3--Ca2+
SO42- -Ca2+
Aguas no kársticas
26,0
27.4
6,50
6.90
209,0
38.5
1 290
1 110
470
477
HCO3--Na+>Ca2+
>Mg2+
HCO3--Mg2+
Tabla 3.4. Evolución química de las aguas kársticas de montaña. En la tabla 3.4 se presentan las características químico-físicas de las aguas kársticas de
algunas regiones montañosas de Cuba, las cuales se han ordenado según su ubicación en las distintas zonas hidrogeológicas del karst. En la misma se puede apreciar la evolución que sufren estas aguas en cuanto a su composición química, a medida que en su movimiento se alejan de la zona de alimentación del acuífero.
Back y Hanshaw (1965) han sugerido que los principales procesos de interacción de las aguas naturales con el medio se pueden interpretar sobre la base de lo que se ha denominado el ciclo hidroquímico o ciclo geoquímico de las aguas. Estos procesos pueden resumirse de la manera siguiente:
a) El viento al soplar desde la tierra hacia el océano traslada Cl- , Na+ y otros componentes. b) En el proceso de condensación del agua en la atmósfera, se disuelve N2, O2 y CO2,
gases que llegan al ecosistema acuático durante las precipitaciones en forma de lluvia o nieve. c) Una cantidad adicional de CO2 se incorpora en el proceso de percolación del agua a
través del suelo rico en materia orgánica. ch) Los minerales se disuelven y liberan aniones y cationes en el seno del agua. d) Los minerales sulfurosos (piritas), se oxidan suministrando al agua SO4
2- y otros constituyentes.
e) Los cationes en solución son intercambiados con otros presentes en el suelo y las rocas. f) El SO4
2- en solución es reducido por las bacterias, generándose una cantidad adicional de CO2.

g) En la medida que se exceden sus productos de solubilidad se produce la precipitación de los minerales.
h) El agua retorna a la atmósfera por evaporación o evapotranspiración, desechando los productos químicos, o el agua retorna al océano a través de cursos superficiales o descargas subterráneas, transportando materia disuelta y en suspensión.
En un trabajo desarrollado en Australia por Chebotarev (en Frezze y Cherry, 1980), en el cual fueron muestreadas más de 10 000 aguas de pozos, se llegó a la conclusión de que en la naturaleza, las aguas subterráneas tienden a evolucionar hacia la composición del agua de mar. El observó que esta evolución química (en la dirección del flujo y el incremento de la edad geológica), en cuanto a los aniones dominantes, sigue aproximadamente las regularidades siguientes: HCO3
-
→ HCO3- > SO4
2- → SO42- > HCO3
- → SO42- > Cl- → Cl- > SO4
2-→ Cl-. Estos cambios ocurren en la medida que el agua se mueve desde zonas de grandes caudales, a
través de zonas intermedias, hasta zonas donde los flujos son escasos y el agua es vieja desde el punto de vista geológico. Esta tendencia hidrogeoquímica se ha denominado, por Schoeller, secuencia de Ignatovich y Souline en reconocimiento a estos hidrogeólogos soviéticos que llegaron a una generalización similar a la de Chebotarev.
En las regiones kársticas montañosas de Cuba, la evolución química de las aguas sigue una tendencia similar a la observada por esos investigadores, alcanzando, por lo general, las facies ricas en SO4
2- . Las aguas kársticas con elevados contenidos de Cl- se suelen encontrar en regiones llanas
que presentan gran agrietamiento o fallas, donde los horizontes acuíferos someros se ponen en contacto con otros más profundos. También en algunos pozos artesianos, que se han abierto a grandes profundidades durante los trabajos de prospección geológica o de búsqueda petrolífera; así como en las zonas litorales donde las aguas del acuífero kárstico se mezclan con las de mar y como resultado de este proceso se incrementa la solubilidad de los minerales carbonatados (efecto de mezcla de agua y efecto salino o de fuerza iónica).
En la tabla 3.5 se presentan las características químico-físicas de algunas aguas kársticas litorales. Como se puede apreciar, a medida que las mismas se acercan al mar presentan una mayor mineralización y dureza, un menor contenido de CO2 disuelto y se hacen, por lo general, más insaturadas respecto a la calcita. Geología e Hidrogeología T (°C) pH CO2
(mg/L) CE
(µs/cm) CaCO3 (mg/L)
Tipo de agua
Cuenca cerrada 23 7,35 18,0 610 276 HCO3--Ca2+
Cuenca litoral
25 25
7,60 7,30
30,8 23,1
1 100 9 950
365 1 170
HCO3->Cl--Ca2+ >Na+
Cl--Na+ Tabla 3.5. Evolución química de las aguas kársticas de llanura
Los factores de tipo geomorfológico también influyen en la composición química de las aguas, en especial, el escarpe de los macizos, el tipo de vegetación, el grado de erosión de los terrenos y la naturaleza de las propias formas del relieve kárstico. A pesar de que las formas de adsorción (dolinas, sumideros, etcétera), se pueden considerar el resultado de los procesos kársticos, una vez creadas esas formas, éstas facilitan o limitan la ulterior acción de corrosión química sobre el karst, lo cual se refleja en la composición química de las aguas.

Otros factores que también influyen sobre la composición química de las aguas kársticas son los de tipo pedológicos, los cuales están asociados al tipo de suelo que yace sobre las secuencias karstificables. El suelo puede ser el resultado del intemperismo de la roca o tratarse de una cobertura alóctona de origen fluvial, pluvial o glaciar; su espesor puede variar desde un grosor apreciable hasta llegar a ser muy escaso o ausente como ocurre en los karst desnudos. De sus características y condiciones pedoclimáticas depende la actividad microbiológica asociada, así como de la producción de gases y ácidos disponibles, que luego son arrastrados por las lluvias o las nieves al fundirse, haciendo posible la disolución de los minerales que forman parte del paquete de rocas kársticas subyacentes. En el caso de un karst desnudo, el agua de las precipitaciones puede adquirir el CO2 directamente de la atmósfera, pero en una proporción menor que la que se produce en el suelo.
Los factores climáticos intervienen de forma activa en la dinámica de la meteorización mecánica y química de los macizos, permitiendo en el primer caso la fragmentación, traslado y acarreo de los minerales lejos del lugar de origen, así como facilitando en el segundo caso, la disolución de los minerales de las rocas. Mientras más intensa sea la acción mecánica, más facilitará al agua su acción corrosiva.
Los elementos del clima más determinantes en el modo en que las aguas adquieren su composición química son: la temperatura, humedad relativa, intensidad y duración de las precipitaciones, intensidad y duración de las radiaciones, velocidad del aire, entre otros.
Un caso curioso de alta mineralización en las aguas, lo ofrecen los lagos morrénicos en las tierras árticas. La cobertura de nieve y hielo impide allí el desarrollo de la vegetación y la actividad microbiológica. Sin embargo, las aguas en estos lugares adquieren un alto contenido de iones disueltos por el proceso de meteorización mecánica, en virtud del movimiento de los glaciares sobre el macizo. En esas condiciones las rocas son intensamente fracturadas, facilitando la acción del agua, que a bajas temperaturas disuelven el CO2 del aire atmosférico en mayor proporción que en los países de clima más caliente. En la tabla 3.6 se presentan las principales características químico-fisicas de las aguas kársticas de las tierras polares.

Tabla 3.6. Evolucón química de las aguas kársticas polares Geología e
Hidrogeología T (°C) pH CO2
(mg/L) CE
(µs/cm) CaCO3 (mg/L)
Tipo de agua
Aguas no kársticas
Nieve Supraglaciar Subglaciar Río (cuarcita) Tundra
0 0,2
1,8
1,0 0,4
6,90 7,40
7,10
6,90 6,60
2,0 0
1,5
3,1 5,9
55 45
65
76
233
3 20
20
17 4
HCO3--Na+
HCO3->SO4
2- -Ca2+ >Ca2+
HCO3->SO4
2- -Na+ >Ca2+ HCO3
--Na+ Cl->SO4
2- -Ca2+ >Na+
Aguas kársticas
Subglaciar Río Vimsa Río Rasstupet Lago Lago en morrena Man. Rasstupet Termomineral (intrusión marina)
0 1,2 1,7 3,8 0
2,8 3,0
8,25 7,60 7,20 8,00 7,86
7,50 7,70
0 2,0 3,7 0
1,8
0,9 5,8
282 300 144 125 820
860
3 000
95 70 89 55
310
141 1 220
HCO3--Ca2+
HCO3--Ca2+ >Na+
HCO3--Ca2+
HCO3--Ca2+
SO42- >HCO3
--Ca2+
Cl--Na+ SO4
2- -Ca2+
En la actualidad, diferentes comisiones internacionales y grupos de estudio de la UNESCO
realizan investigaciones relacionadas con el impacto humano en el medio kárstico. En este sentido se destaca el papel de la Comisión de Físico-Química e Hidrología del Karst, así como otras comisiones y grupos de estudio de la Unión Internacional de Espeleología (UIS); la Comisión sobre Cambios Ambientales y Conservación de Áreas Kársticas de la Unión Internacional de Geografía (IGU), y comisiones sobre el karst de la Asociación Internacional de Hidrogeólogos (IAH) (Finlayson y Sauro, 1991). Entre los proyectos y programas que desarrollan estos organismos relacionados con el impacto del hombre sobre el karst se deben destacar: el Proyecto 299 (Geología, Clima, Hidrogeología y Formaciones Kársticas) del Programa Internacional de Correlación Geológica (IGCP); así como el Programa Internacional Geosfera-Biosfera (IGBP).

Patrones hidrogeoquímicos y control de la composición química de las aguas kársticas
A pesar de que las aguas naturales y, en particular, las aguas kársticas, adquieren su composición química mediante un proceso complejo como el que se acaba de analizar en el epígrafe anterior, donde intervienen diferentes factores, en un sitio determinado o área con cierta homogeneidad, muchos de éstos se hacen constantes y en esas condiciones la composición química absoluta del agua varía dentro de cierto rango, como consecuencia del régimen de lluvia: en los períodos lluviosos la mineralización es más pequeña que en los períodos secos. Sin embargo, la composición química relativa varía poco, las aguas en todo momento mantienen su tipo y poseen por, lo general, un mismo patrón hidrogeoquímico, tal como se ilustra en la figura 3.12, donde se relacionan en el mismo gráfico las precipitaciones, el caudal, la conductividad eléctrica y la composición química de un agua procedente de una emergencia kárstica durante un año hidrológico.
Figura 3.12. Efecto de las precipitaciones sobre el caudal, la mineralización (en términos de conductividad eléctrica) y la composición química de las aguas de la resurgencia Canilla durante un año hidrológico (1984).
El uso de relaciones matemáticas entre la concentración iónica y la conductividad eléctrica
de las aguas kársticas, se ha propuesto sobre la base de un modelo de adquisición de la composición química, similar al que ocurre en un reactor de laboratorio donde se hace pasar una corriente de

CO2 a un agua y se coloca un mineral carbonatado (Fagundo, 1985; 1990 b; Fagundo y Pajón, 1987; Pajón y Valdés, 1991; Fagundo et al, 1992).

Figura 3.13. Variación de la concentración iónica y la conductividad eléctrica durante el tiempo transcurrido en un experimento de simulación química, en el laboratorio, del proceso de disolución de una caliza pura (a); relación entre la
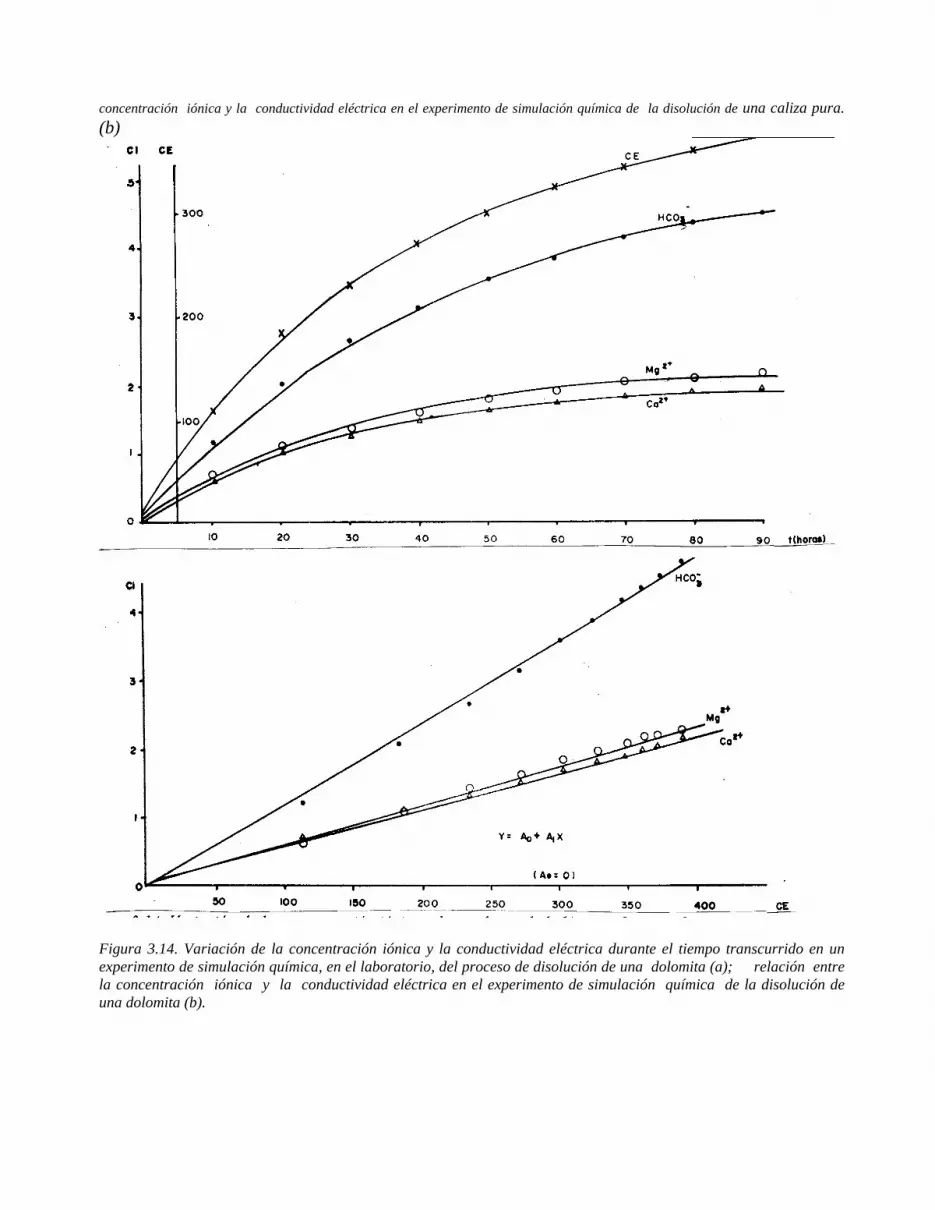
concentración iónica y la conductividad eléctrica en el experimento de simulación química de la disolución de una caliza pura. (b)
Figura 3.14. Variación de la concentración iónica y la conductividad eléctrica durante el tiempo transcurrido en un experimento de simulación química, en el laboratorio, del proceso de disolución de una dolomita (a); relación entre la concentración iónica y la conductividad eléctrica en el experimento de simulación química de la disolución de una dolomita (b).

En este proceso cinético, la velocidad de disolución del mineral depende de su composición
en la roca (litología). La concentración de los principales iones que resultan de este proceso se incrementa en el tiempo, siguiendo una función exponencial. Una función matemática similar presenta la conductividad eléctrica (Figuras 3.13a y 3.14a)
Si se grafica la relación entre la concentración de cada ion y la conductividad eléctrica se
obtiene una línea recta que pasa por el origen de coordenadas, cuya pendiente depende de la litología (Fagundo, 1990 b). Por ejemplo, en el caso de una caliza pura constituida prácticamente por mineral calcita, las pendientes correspondientes a los iones HCO3
- y Ca2+ son similares entre sí y sus magnitudes son mucho más grandes que las del ion Mg2+. En una dolomía constituida por cantidades más o menos similares de calcita y dolomita, la pendiente correspondiente al ion HCO3
- es del mismo orden que en el caso anterior, sin embargo, las pendientes correspondientes a los iones Ca2+ y Mg2+ son en este caso similares entre sí y mucho menor que la del HCO3
-, tal como se ilustra en las figuras 3.13b y 3.14b.
Si se determinan las concentraciones iónicas multiplicando las conductividades eléctricas, en cada momento en que se hizo el experimento, por las correspondientes relaciones matemáticas obtenidas, se encuentra que no existen grandes diferencias entre la composición química real y la obtenida por estos modelos matemáticos, tal como se aprecia en las figuras 3.15 y 3.16.


Figura 3.16. Variación, durante el tiempo transcurrido en el experimento de simulación química de la disolución de una dolomita, de la composición del agua del reactor, obtenida por análisis químico (izquierda) y modelación (derecha).
Basado en estos principios se elaboró un sistema automatizado (Alvarez et al, 1990) capaz de determinar en forma rápida las ecuaciones de correlación matemática entre la concentración iónica y la conductividad eléctrica, con el objetivo del ulterior control de la composición química de las aguas mediante mediciones en el campo con conductímetros portátiles. En general, cuando uno de los factores es dominante en el modo de adquisición de la composición química de las aguas, las relaciones entre las concentraciones iónicas y la conductividad eléctrica son de tipo lineal (Fagundo

et al, 1991; 1993). Si más de un factor posee un peso semejante en este proceso, las ecuaciones de segundo grado ajustan mejor. En este último caso se observa, por lo general, más de un patrón hidrogeoquímico (Fagundo y Rodríguez, 1991).
En el caso de aguas subterráneas que se encuentran estratificadas como resultado de mezcla con el mar, como sucede en los acuíferos kársticos litorales, la evolución ocurre según una serie de patrones hidrogeoquímicos en el que se va incrementando, de manera progresiva, el contenido de los iones Cl-, Na+ + K+ y en menor medida Mg2+ y SO4
2- , así como decreciendo el de los iones HCO3- y
Ca2+ (Figura 3.17). La actividad del hombre, especialmente la sobreexplotación de los acuíferos para el abasto o el regadío altera los patrones hidrogeoquímicos naturales (Fagundo et al, 1993; Ferrera, Fagundo y Benítez, 1995).
Figura 3.17 Patrones hidrogeoquímicos de las aguas del acuífero kárstico de la llanura meridional de Pinar del Río, afectado por la intrusión marina. Las concentraciones se expresan en forma relativa (suma de aniones = suma de cationes = 10 % meq/L) mediante diagramas de Stiff.
Para controlar la composición química de estas aguas mediante mediciones de conductividad
eléctrica y el empleo de ecuaciones de correlación matemática, es necesario procesar los datos por tipos de patrones hidrogeoquímicos, sobre la base de los cambios en sus relaciones iónicas:
r ClHCO
r Na KCa
−
−
+ +
++
32,

La mejor forma de hacer lo expuesto en el párrafo anterior, es mediante un sistema de reconocimiento de patrones, donde los datos originales se agrupan sobre la base de las diferentes facies hidroquímicas. Además, es necesario tener en cuenta los rangos de conductividad eléctrica en que cada pozo refleja los diferentes patrones. Esto se debe a que para un mismo rango de conductividad, un pozo situado más cerca del mar o en un área de mayor agrietamiento, reflejará facies hidroquímicas más ricas en Cl- que en HCO3
- que otro pozo más alejado del mar o situado en una zona de menor agrietamiento, el cual reflejará para el mismo rango de conductividad, facies hidroquímicas donde predomine el contenido de HCO3
- sobre el de Cl- (Fagundo y Rodríguez, 1991; 1992).
El empleo de relaciones empíricas entre la concentración iónica y la conductividad eléctrica por medio de las ecuaciones clásicas de regresión de tipo lineal o polinómica se han ensayado por algunos investigadores (Drake y Ford, 1974). Sin embargo, por esta vía se obtienen coeficientes de correlación significativos sólo para los iones mayoritarios. Además mediante estos métodos estadísticos, se obtienen expresiones que no tienen sentido físico y que dependen de los datos disponibles. Algunos especialistas han objetado el valor de este tipo de correlaciones matemáticas entre la concentración iónica y la conductividad eléctrica. No obstante, este tipo de tratamiento estadístico ofrece buenas perspectivas en el control del contenido de iones Cl- y Na+, así como la salinidad en muestras de aguas que actualmente son tomadas con hidromuestreadores verticales o batómetros (Gutíerrez et al, 1981). Variación espacial y temporal de la composición química de las aguas kársticas
Los cambios temporales en la composición química de las aguas naturales se han clasificado en función de la escala de tiempo como: cambios seculares (producidos en el orden del tiempo geológico) y cambios a corto plazo (Paces, 1980). A cada una de estas categorías le corresponden a su vez, cambios de tipo periódico, abrupto y sistemático. Los cambios seculares se refieren a la evolución del agua oceánica en el orden del tiempo geológico. Como ejemplo de este tipo de cambio se puede considerar la variación de la relación iónica Mg2+ : Ca2+, experimentada en los sedimentos carbonatados durante los últimos 2 000 millones de años.
En realidad los cambios de la composición química de las aguas a corto plazo son más estudiados, por su posible medición.
Los cambios periódicos se observan a menudo como respuesta a la radiación solar y pueden ser de tipo diurno o estacional, como ejemplo del primer caso se puede considerar las variaciones que se producen en el intervalo de 24 h en las magnitudes de la presión de CO2, las concentraciones de los iones HCO3
- y Ca2+ , así como en el grado de saturación respecto a la calcita. Estos cambios son inducidos por los organismos fotosintéticos cuya respiración durante la noche incrementan la p CO2, mientras que por el día esta actividad disminuye, causando una variación en el equilibrio de los carbonatos.
Los cambios estacionales de la composición de un agua están determinados por las variaciones en la temperatura y el régimen de precipitación en las diferentes estaciones del año. En Cuba se distinguen en la práctica dos períodos: el seco o menos húmedo, que se extiende desde noviembre a abril y el lluvioso, de mayo a octubre.
Los cambios abruptos en la composición química de las aguas naturales son causados, por lo general, por la transición de un agua de un medio ambiente a otro, cuyas propiedades químico-físicas y biológicas son muy diferentes.

Por ultimo, los cambios sistemáticos son de tipo continuo y no periódico, que se manifiestan en la variación de la masa disuelta desde un estado inicial hasta otro final.
La composición química espacial de las aguas en una región kárstica determinada, sólo da una información parcial del funcionamiento de ese karst. La variación a lo largo de uno o más años hidrológicos, así como durante los períodos de intensas lluvias, ofrece mucho más información. Las determinaciones conjuntas a lo largo del tiempo de la composición química de las aguas y el caudal en las surgencias kársticas, así como el registro simultáneo de los elementos del clima, ofrece la oportunidad de correlacionar las respuestas hídrica y química en relación a las precipitaciones, lo cual permite conocer cómo está organizado interiormente el sistema kárstico.
La variación estacional de la dureza se ha utilizado por muchos investigadores para interpretar la forma en que se mueven las aguas en el karst. Shuster y White (1971) propusieron el empleo del coeficiente de variación de la dureza en lugar de la dureza misma, como un índice para determinar si las aguas se mueven en forma difusa o a través de conductos. Las aguas que discurren por fisuras y conductos abiertos poseen un mayor coeficiente de variación, puesto que en esas condiciones se produce un mayor efecto del clima en la fluctuación de la dureza.
Otros investigadores también estudiaron la variación estacional de la dureza en diferentes regiones kársticas (Pitty, 1968; Ternan, 1972; Gams, 1976). Este método fue aplicado en Cuba, a aguas de pozos de la región de San Antonio de los Baños (Fagundo y Valdés, 1975), sin embargo, los resultados obtenidos no se corresponden con las evidencias hidrogeológicas, porque las aguas tomadas en pozos que cortan cavidades subterráneas aireadas, presentaron coeficientes de variación de la dureza inferiores a otras, tomadas en pozos donde el movimiento del agua es aparentemente más difuso.
Bakalowicz (1976) llamó la atención sobre la validez de este método en el caso que la distribución de frecuencia de la dureza sea de tipo plurimodal, lo cual es muy común en las aguas kársticas, porque éstas reciben aportes con diferentes respuestas a las precipitaciones. No obstante, este método se ha seguido utilizando, tomando en cuenta el coeficiente de variación de otras variables; por ejemplo, los coeficientes de variación de los iones mayoritarios y la conductividad eléctrica para interpretar la organización interior de tres surgencias kársticas, tomando en consideración las observaciones de Bakalowicz en cuanto a la distribución de frecuencia de esas variables (Aguayo et al, 1988).
En Cuba, además del coeficiente de variación de la dureza se han empleado los de temperatura y la conductividad eléctrica, para distinguir la forma en que tiene lugar el escurrimiento del agua a través de los macizos kársticos (Fagundo et al, 1981; 1986; Valdés y De la Cruz, 1982.
El movimiento del agua a través del medio rocoso se ha simulado por los hidrogeólogos con el objetivo de determinar las reservas de aguas subterráneas y la variación sistemática de su composición química (Paces, 1980). En acuíferos desarrollados en areniscas y otros medios isótropos, se han propuesto modelos, que se basan en ecuaciones diferenciales parciales. Estos modelos, han sido aplicados al karst, mediante una aproximación que considera lineal el movimiento de los flujos en el medio kárstico fisurado (Pérez Franco et al, 1974; Llanusa y González, 1993; Hernández, 1995). Variación de la composición química del agua kárstica a consecuencia del impacto antropogénico
Aunque algunos aspectos relacionados con el efecto negativo de la actividad del hombre en el karst se ha abordado en el capítulo 2 de este libro, al ser analizado el papel de la actividad microbiológica en la generación de CO2, así como la corrosión mediante procesos microbiológicos y

bioquímicos, por su importancia, el efecto antropogénico en el karst será objeto de un análisis complementario en este capítulo.
El agua constituye un recurso valioso que el hombre utiliza. En cierta medida el agua natural sufre un constante proceso de contaminación, en el cual parte es compensado por los procesos de autodepuración.
La contaminación es un proceso natural o artificial, mediante el cual se agrega al agua normal o beneficiosa elementos, sustancias o materia viva, que la convierte en perjudicial para todos o algunos de los diferentes usos (Ferro, 1982). Por lo general, las aguas superficiales se suelen afectar con más facilidad que las subterráneas.
La contaminación de los recursos hídricos en el karst es objeto de interés particular, a causa de la vulnerabilidad de este medio (Santo, 1991). A pesar de que en el karst el movimiento del agua es preferentemente subterráneo, este escurrimiento se realiza con preferencia a través de grietas, fisuras, conductos, así como cavidades más o menos amplias y aireadas, donde prevalecen condiciones de flujo de tipo turbulento, muy similares a las superficiales y en las mismas son factibles los procesos microbiológicos aeróbicos.
Entre los efectos negativos de la actividad del hombre, que dan lugar a la contaminación del agua kárstica se pueden considerar los siguientes:
1. Vertimiento de residuales orgánicos agroindustriales y urbanos. 2. Vertimiento de residuales inorgánicos. 3. Incremento de nitratos (nitrificación), pesticidas y otras sustancias, a causa del tratamiento químico en la agricultura. 4. Contaminación, a causa de la actividad minera. 5. Derrame de hidrocarburos. 6. Salinización de los acuíferos costeros por sobreexplotación. 7. Contaminación, a causa de la actividad del hombre, especialmente el turismo en el karst. 8. Deforestación del paisaje kárstico. Entre los efectos producidos por el vertimiento o percolación en el karst de los residuales
orgánicos y urbanos se pueden considerar los siguientes: 1. La disminución de los niveles naturales de oxígeno disuelto y aumento del contenido
de materia orgánica biodegradable en las aguas superficiales y subterráneas. 2. Eutroficación en lagos y embalses con producción de mal olor. 3. Aumento de la población microbiológica patógena, dando lugar a la aparición de
enfermedades de origen hídrico. 4. Incremento de sólidos orgánicos suspendidos y sedimentables en corrientes
superficiales. 5. Aumento del contenido de sales disueltas provocado por aportes salinos de
residuales industriales. En la literatura especializada de los años más recientes aparecen muchas citas bibliográficas,
donde se presentan los principales problemas relacionados con el efecto del impacto humano sobre el karst (Chen, 1991; James, 1991); así como con la disposición de residuales en regiones kársticas de diferentes partes del mundo (Legrant, 1985). Como colorario de estos estudios, los karstólogos coinciden en señalar que el karst es poco propicio para la disposición de este tipo de residual.
Algunos trabajos, sin embargo, ponen de manifiesto la buena capacidad autodepuradora del medio kárstico para aquellos residuales industriales ácidos procedentes de la industria del petróleo

(Lin Hua, 1991); así como para corrientes subterráneas contaminadas por la actividad del hombre (Hemmel, 1990).
En el caso de corrientes superficiales, algunos investigadores han evaluado el grado de autodepuración del karst y otros medios no kársticos en función de la remoción bacteriana (Quesada y Gutiérrez, 1982; Quesada, 1986), como vía complementaria de tratamiento de los residuales antes de su vertimiento (Gutiérrez, 1982). La disposición o infiltración de residuales urbanos y, en general, la urbanización, así como el turismo en regiones kársticas, dan lugar a un incremento de los componentes del ciclo del nitrógeno en las aguas, fundamentalmente NO3- y NO2- (Krawcyk 1990; Kranje y Kogovsek, 1990; Waterhouse, 1984).
El contenido de NO3- en las aguas potables, según establecen las normas, no debe exceder los 60 mg/L. Sin embargo, en algunas aguas kársticas se han encontrado concentraciones del orden de 500 mg/L (Smith, 1988). Mediante la actividad de fertilización agrícola, también se incorpora al karst una buena cantidad de sustancias nitrogenadas, así como fertilizantes, pesticidas y otras sustancias químicas (Pekny et al, 1987; Muler y Eingold, 1987; Pulido Bosch et al, 1993).
En Cuba, Gutiérrez y colaboradores (1982) han estudiado la vulnerabilidad de los acuíferos kársticos a los procesos de nitrificación. Según estos investigadores, los volúmenes de nitrógeno que aportan las diferentes fuentes en las áreas agrícolas son las siguientes:
Fuente Concentración (kg/ha) Fijación atmosférica 20-30 Aporte por precipitación 3-5 Aporte por riego 3-40 Aporte por residuales animales 10-30 Fertilizantes nitrogenados 60-120
El riego con las propias aguas subterráneas de la cuenca, la cual es objeto de contaminación,
constituye un elemento importante en el balance de nitrógeno. El efecto de la irrigación con efluentes sobre la calidad de las aguas kársticas subterráneas, también se ha evaluado con vistas a eliminar los efectos negativos potenciales de la contaminación bacteriológica (Kaplen, 1987). En Cuba muchas áreas agrícolas se irrigan con residuales azucareros y hasta el presente los resultados son satisfactorios.
El vertimiento de residuales inorgánicos se refleja en las aguas mediante un incremento en el contenido de compuestos metálicos (Rorsh y Llard, 1984), de éstos los más tóxicos son: plomo, mercurio, zinc y cadmio. Esto es común, por ejemplo, en muchas aguas procedentes de minas, las cuales se caracterizan por poseer valores de pH muy bajos, capaces de disolver en esas condiciones muchos microcomponentes (Reed y Singh, 1987).
La deforestación también produce efectos indeseables en el karst (Griolet, 1987). En las áreas montañosas la tala de bosques favorece el arrastre de sólidos disueltos y en suspensión, así como de la materia orgánica durante las lluvias o la fusión de las nieves, provocando cambios en la calidad del agua y sedimentación en lagos, ríos y cuevas.
La sobreexplotación de los acuíferos kársticos litorales con vistas al abasto o el regadío, trae como consecuencia que disminuya la presión hidrostática y se produzca una penetración mayor del agua de mar. Los problemas relacionados con la explotación de los acuíferos kársticos costeros son objeto de estudios en muchas áreas kársticas de Europa (Pulido Bosch, 1976; 1989; Mijatovic, 1984), Estados Unidos (Strengfield y Rapp, 1984), México (Back et al, 1986) y Cuba (Fagundo y Arellano,

1991; Fagundo et al, 1993; Barros, 1982; Laphin y Luege, 1969; Shayakubov y Morales, 1980; González y Feitó, 1988).
Relacionado con la excesiva extracción de aguas en las regiones kársticas, se encuentran los fenómenos de creación de dolinas y producción de derrumbes, al quedar evacuado un horizonte acuífero, que facilita el desprendimiento de la cobertura. Zonas de protección y control sistemático de la calidad de las aguas kársticas
La actividad del hombre causa un deterioro progresivo de la calidad de las aguas superficiales y subterráneas. A pesar de que estas últimas son menos susceptibles a la contaminación, una vez contaminadas, se hace muy difícil restablecer su calidad original (González y Jiménez, 1988 a).
Con vistas a preservar la calidad de las aguas kársticas, de forma tal que se pueda utilizar racionalmente en beneficio de la economía, se han establecido zonas de protección sanitaria, como tal se denomina el área alrededor de la toma de agua, donde se establecen diferentes tipos de regulaciones con el objetivo de evitar la contaminación y hacer mínimo el riesgo de su degradación (González y Jiménez, 1988 b).
En los últimos años, se han llevado a cabo muchas investigaciones aplicadas en el campo de la hidrogeología, con el objetivo de establecer las zonas de protección sanitarias alrededor de las fuentes de captación de interés económico social (Daoxian, 1987; Fritz y Pavicic, 1987; Kullman, 1987; Tulipano y Fidelibus, 1991).
La falta de cultura en relación con lo que debe ser la zona de protección de una fuente kárstica, hace que con frecuencia, se construyan letrinas en la misma área en que se encuentra el pozo que se utiliza para el consumo de las personas, lo cual da lugar a que por percolación pasen a la fuente gérmenes patógenos y se requiera instalar un sistema costoso de tratamiento del agua. Esta forma de contaminación era muy frecuente en muchos pueblos de Cuba hace algunos años e incidía de manera significativa en un alto índice de parasitismo en la población infantil. Con vistas al control de la calidad de las aguas kársticas, también se han elaborado políticas y sistemas de vigilancia, mediante el registro sistemático de algunos indicadores de tipo químico-físico y bacteriológico.

HIDROQUÍMICA DEL KARST
Parte 2
HIDROQUÍMICA DEL KARST EN CLIMAS EXTREMOS
J.R. FAGUNDO CASTILLO Centro Nacional de Investigaciones Científicas
Ministerio de Educación Superior La Habana, Cuba
J.J. VALDÉS RAMOS
Instituto de Geofísica y Astronomía Ministerio de Ciencia, Tecnología y Medio Ambiente
La Habana, Cuba
J.E. RODRÍGUEZ RUBIO Instituto de Geografía
Ministerio de Ciencia,Tecnología y Medio Ambiente La Habana, Cuba

Parte 2
HIDROQUÍMICA DEL KARST EN CLIMAS EXTREMOS
Capítulo 1. Expedición Spitsbergen' 85

Breve recuento
La isla de Spitsbergen, situada a 80° de latitud norte, pertenece al archipiélago de Svalbard y es una de las tierras más cercanas al Polo Norte. Limita al norte con el océano Glacial Ártico, al este con el mar de Groenlandia, al oeste con el mar de Barents y al sur con el mar de Noruega (Figura 1.1). A pesar de encontrarse en esa latitud su clima no es tan riguroso por el efecto de la Corriente del Golfo procedente del océano Atlántico. Entre los meses de junio y marzo las temperaturas medias son del orden de -11,5 °C, llegando a ser inferiores a -40 °C en el invierno. Durante el verano las temperaturas máximas medias alcanzan +4,5 °C y raras veces sobrepasan los +10 °C. En los meses de otoño, que se extienden desde agosto hasta octubre, son frecuentes los huracanes, denominados fohen, con temperaturas que suelen ser superiores a +1 °C.
Figura 1.1. Localización geográfica del archipiélago de Svalbard, donde se encuentra la isla de Spitsbergen.

Spitsbergen y todo el archipiélago constituye un territorio internacional administrado por Noruega. En la misma existen tres poblados, uno noruego y dos rusos que se dedican a la pesca y a la explotación de yacimientos de carbón. También se han instalado en la isla varias estaciones científicas polares por parte de distintas nacionalidades con el objetivo de efectuar estudios, fundamentalmente glaciológicos y meteorológicos. La expedición conformada por un grupo de científicos polacos, rusos, dos checos, un español y dos cubanos, realizaron estudios en el área denominada Grondfjord (perteneciente a la región de Nordenskiold) en la parte central de la isla y en la región aledaña al fiordo Hornsund, en la parte meridional (Figuras 1.2, 1.3 y 1.4).

Figura 1.2. Ubicación de las áreas estudiadas en la isla de Spitsbergen durante la expedición polar de 1985, auspiciada por la Universidad de Silesia: área de Grondfjord, situada a los 78° de latitud norte y el área del fiordo Hornsund, a los 77° de latitud norte, correspondientes a los polígonos experimentales ruso y polaco respectivamente.

Figura 1.3. Área de experimentación rusa en Grondfjord.

Figura 1.4. Área de experimentación polaca en el fiordo Hornsund.
Los cubanos realizaron las mediciones químico-físicas "in situ", de aguas de diferente naturaleza hidrogeológica: aguas de las zonas subglaciar, endoglaciar y supraglaciar; de arroyos y manantiales procedentes de la tundra, así como del permafrost; de nieve; de hielos y aguas subsuperficiales procedentes de los glaciares. También de lagos situados en las morrenas frontales de los glaciares.
Todo este trabajo se efectuó en condiciones muy difíciles, impuestas por el clima en esa región: bajas temperaturas, nieve, huracanes polares, desplazamiento a través de los glaciares (cuyas grietas se suelen cubrir superficialmente por finas capas de nieve), movimiento en la isla a través de helicópteros, botes y barcos, en estos últimos fue necesario abrirse paso a través de icebergs, témpanos y costras de agua marina congeladas. Las condiciones de vida fueron también muy duras, puesto que la habitación era una rústica cabaña de madera a unos 30 km de la estación científica polaca del Hornsund, sin electricidad, radio ni medios de transporte, sólo con los mínimos

recursos para la supervivencia y la seguridad contra las usuales inclemencias del tiempo y de los osos polares.
Los resultados preliminares fueron elaborados y publicados en Polonia (Valdés, Fagundo y Pulina, 1985; Fagundo, Valdés y Pulina, 1985) y Suecia (Fagundo, Valdés y Pulina, 1990). Un cuarto trabajo ya se ha publicado en una revista cubana (Fagundo, Valdés y Pulina, 1987).
El 21 de julio de 1985 viajaron hacia Varsovia los expedicionarios cubanos y de allí se trasladaron para la ciudad de Sosnowiec, donde radica la Facultad de Ciencias de la Tierra de la Universidad de Silesia. Durante un mes participaron en los preparativos de la expedición y en ese tiempo viajaron a las regiones montañosas de los Sudetes y Bielsko Biala, con el objetivo de adiestrarse en las condiciones rigurosas de trabajo de campo que debían realizar en la isla polar.
El 19 de agosto partieron hacia Moscú en compañía del profesor Marian Pulina, jefe de la expedición y el doctor Jacek Jania, glaciólogo. El primero dirige el programa polar polaco y es el coordinador principal (por Polonia) de la colaboración bilateral establecida entre la Universidad de Silesia y los organismos cubanos representados por el CNIC. El segundo es un geógrafo especializado en la geomorfología y dinámica de los glaciares de Spitsbergen.
De Moscú partió el colectivo en avión hacia Longyearbyen, capital de la isla, donde radica la administración noruega del archipiélago. Ese mismo día se trasladaron en helicóptero hacia el poblado ruso de Baretsburg, nombrado así en honor al descubridor de la isla, el danés Wilhem Barents.
En Barentsburg, los cubanos y polacos convivieron con el colectivo de geólogos rusos dirigido por el doctor Eugenio Zinger, quien desde hace muchos años se dedica al estudio de los glaciares de Svalbard. Los primeros días la representación cubana aprovechó el tiempo para hacer excursiones a las montañas aledañas al poblado, con el objetivo de lograr la necesaria aclimatación. La temperatura reinante era agradable, cercana a 0 °C.
El día 27 de agosto los cubanos, polacos y un grupo de rusos partieron en helicóptero hacia la base, al otro lado del fiordo, donde radica el polígono experimental de los científicos rusos. De ese colectivo participaron en la expedición el doctor Igor Postonov, hidrológo y jefe del grupo operativo; los licenciados Alexander Volkov y Vladimir Starizin, geólogos, el licenciado Alexander Tebenkov, glaciólogo y el licenciado Eugenio Germanov, químico. Ese mismo día el grupo hizo una excursión a la cueva de hielo de Aldegonda, situada en la lengua del glaciar del mismo nombre.
Luego se instalaron en una rústica cabaña de madera, con calefacción (de carbón de piedra y leña), al día siguiente comenzaron los trabajos de exploración y toma de muestras. A los cubanos, junto al químico ruso les correspondió realizar la cartografía de la cueva abierta en los hielos del glaciar Aldegonda (Figura 1. 5), tomar muestras de agua dentro de la misma, así como las determinaciones químico-físicas correspondientes (temperatura, pH, conductividad eléctrica y análisis químico de los macrocomponentes). Como el Sol alumbraba en el firmamento las 24 h del día, por aquella época del año, se pudieron apreciar los cambios de color en las valoraciones químicas hasta altas horas de "la noche". A los restantes miembros de la expedición correspondieron otros trabajos de tipo geomorfológico.

Figura 1.5. Cueva en hielo Aldegonda, situada en la lengua del glaciar Aldegonda, en la región de Grondfjord.
El día 29 partieron hacia el lago Congreso y tomaron muestras en éste, en arroyos y en manantiales termominerales. A diferencia de las aguas superficiales cuyas temperaturas están cercanas a 0 °C, los manantiales termales, con una composición química similar al del balneario cubano de San Diego de los Baños, poseían una temperatura de 4 °C, muy elevada para las aguas de esa latitud ártica.
Al día siguiente el colectivo de científicos emprendió la marcha hacia el Cabo Starostin, situado a unos 30 km del lago Congreso. Se continuaron los trabajos de exploración geomorfológica, cartografía, muestreo y análisis químico en la región del lago Linneo, una zona típicamente kárstica constituida fundamentalmente por calizas, dolomías y yesos.
El regreso al poblado ruso se hizo en barco el día 31 de agosto. Al día siguiente el colectivo cubano-polaco partiría en compañía de Eugenio Zinger a la base polaca en un helicóptero ruso. Durante el amanecer, Barentsburg fue azotado por el primer huracán del otoño polar, que es conocido en esas latitudes como fohen. Sus vientos alcanzan una intensidad similar a la de los ciclones tropicales y las temperaturas suelen ser relativamente elevadas, oscilando entre 0 y 10 °C sobre cero. Arrastran mucho polvo morrénico y producen gran descongelación de nieve y hielos. En

ocasiones pueden soplar hasta una semana. Aquel primero, sin embargo, fue breve, permitiendo al mediodía que pudieran volar los helicópteros, llegando a la base polaca en horas de la tarde y fueron recibidos por el jefe de la misma, ingeniero Szymanski, de la Academia de Ciencias de Polonia.
Durante el día siguiente los cubanos hicieron una excursión hasta el glaciar Hans, cuyos hielos son descargados directamente al mar por ese "río" congelado. Aquí aprovecharon la oportunidad para cartografiar otra cueva en hielo que se abría en su zona de ablación (Figura 1.6).
Al otro día, partieron los cubanos junto a Marian y Jacek hasta la playa de Hyttevika, a 5 h de camino de la base principal, donde se establecerían los cubanos en una antigua casa de noruegos cazadores de osos polares. En tanto, los polacos siguieron camino hasta otra casa utilizada como base, situada a cerca de 1 h de camino de la anterior, en la cual radicarían ellos junto a otros expedicionarios: el español Adolfo Eraso (presidente de la Unión Internacional de Espeleología), los polacos Piotor Glowacki, Stanislaw Misztal, Boleslaw Kapuscinski y Jerzy Wach, del colectivo de Pulina, así como dos checos y dos polacos más, estos últimos de la Universidad de Wroclaw.
Esta rústica vivienda, convertida en estación polar lleva el nombre de Baranowski en homenaje al glaciólogo polaco, pionero de este tipo de investigación en la isla, el cual murió en la Antártida a consecuencia de un lamentable accidente.
El día 5 de septiembre, Adolfo Eraso, Marian Pulina y los dos cubanos, viajaron hasta la zona de acumulación de los hielos del glaciar Werenskiold. Este glaciar, junto a otros sitios aledaños constituirían el objetivo del trabajo de los científicos cubanos. Durante la excursión el jefe de la expedición señaló aquellos lugares que se debían muestrear de manera sistemática con frecuencia diaria, semanal y en forma esporádica, así como también indicó los cuidados que se debían tomar durante la marcha por aquellos parajes.
Los sitios de muestreo diario serían los ríos Glaciar y Brattegg, originados, el primero en el proceso de ablación del glaciar Werenskiold y el segundo, por descongelación de las nieves y el permafrost en una cuenca aledaña. Los sitios de muestreo semanal serían varias fuentes de tipo sub y supraglaciar, previamente seleccionadas, y los restantes quedaban a la iniciativa de los investigadores cubanos.

Figura 1.6. Cueva en hielo Hans, cercana al fiordo Hornsund.

El día 12 de septiembre Jania y Pulina marcharon a trabajar a la región de Treskelen, a unos 30 km de distancia de la vivienda que ocupaban los cubanos, quienes durante ese tiempo tomaron muestras, cartografiaron otras cavernas en hielo (Figuras 1.7 y 1.8) e hicieron determinaciones químico-físicas diarias y semanales en los puntos de muestreo, así como en manantiales y arroyos de la tundra, lagos morrénicos, hielo, nieve, etcétera, en las cuencas de los ríos Gandpasset, Iskantelva, Vimsa, Nambreen y en el glaciar Torell.
Figura 1.7. Cueva en los hielos de la morrena frontal del glaciar Vimsa.

Figura 1.8. Cueva en hielo, situada en la lengua frontal del glaciar Nannbreen.
Por esa época azotó el segundo huracán polar de la temporada, con ráfagas del orden de 30 m/s (106 km/h) y temperaturas cercanas a +3 °C. Duró prácticamente dos días, destruyendo la chimenea de la casa de Hyttevika donde se guarecían los cubanos, quienes quedaron sin calefacción.
El día 20 regresaron los polacos de Treskelen con muestras de agua tomadas por ellos en esa región, éstas más las colectadas por los cubanos fueron analizadas por estos últimos.
El día 22, al cumplirse el primer mes de trabajo conjunto en la isla, se efectuó en la estación polar más distante (Baranowski), un acto solemne donde fue izada la bandera cubana, la polaca y la noruega. Durante la cena se hicieron brindis en homenaje al trabajo científico desplegado tanto en Cuba como en Spitsbergen por el colectivo cubano-polaco. Se destacó además que era la primera ocasión en que científicos cubanos trabajaban en aquella isla, aunque realmente el pionero en las excursiones al Ártico y a la Antártida había sido el profesor Antonio Núñez Jiménez, ésta era la primera expedición científica en el Ártico, así como la más larga permanencia en la región.
El día 26 se efectuó el regreso en bote, con motor fuera de borda, a la base principal, con el objetivo de viajar en helicóptero al día siguiente a Barentsburg y de allí a la capital de la isla, desde donde partirían los expedicionarios de regreso, vía Moscú.
Pero la naturaleza no quiso que esto sucediera así. Durante la madrugada azotó un nuevo huracán, prolongándose esta vez más tiempo que en las ocasiones anteriores (cuatro días). Los helicópteros no pudieron volar. Se esperó una segunda ocasión, pero tampoco fue posible por las mismas circunstancias, perdiéndose el avión ruso, que los miembros de la expedición sintieron sobrevolar por el cielo del Hornsund cubierto de nubes. No quedaba otra alternativa que esperar una embarcación, especialmente diseñada para abrirse paso entre los témpanos, que procedente de Polonia llegaría dentro de unos días para abastecer la base para el invierno.
El invierno hidrológico polar comenzó ese año el día 29 de septiembre y el acontecimiento fue celebrado en la base principal polaca. El cambio de tiempo se manifestó de inmediato. La

disminución de la intensidad de los vientos coincidió con el descenso de la temperatura por debajo de -10 °C y el aumento de las nevadas.
En esas condiciones el jefe de la expedición organizó un nuevo programa de investigación, en condiciones invernales y el colectivo se trasladó para la estación Baranowski.
La toma de muestras requería, ahora, el uso frecuente del piolet, con el objetivo de romper la gruesa capa de hielo superficial de los ríos, lagos y fuentes del glaciar. La marcha por la tundra se facilitaba, porque la misma se había prácticamente congelado. Sin embargo, la visibilidad diurna disminuía. Cada día duraba 20 min. menos que el anterior. Amanecía muy tarde y oscurecía muy temprano, se acercaba la noche polar y por esa razón, el trabajo de análisis químico había que realizarlo con mucha rapidez para poder aprovechar la luz solar. Luego dominaba la penumbra y la oscuridad, pero en compensación durante las noches la aurora boreal se exhibía en el firmamento.
El día 11 de octubre los expedicionarios se trasladaron para la estación principal y de ésta al barco que había venido a abastecer la base. A partir de entonces los polacos comenzaron a realizar estudios sobre el espesor de los hielos que se encuentran por debajo del mar en el fiordo, aprovechando la movilidad y los equipos de la nave, se desplazaron e hicieron mediciones en muchos parajes. Los cubanos en ese tiempo permanecían como espectadores y realizaban los análisis químicos, en el propio barco, de aquellas muestras que en sus excursiones a tierra hacían los polacos.
El día 16 de octubre se realizó una visita a Barentsburg y a Longyerbyen. En aquellos poblados ya la nieve cubría más de 1 m las aceras y la tundra. Las temperaturas eran muy frías.
El día 20, el último en que permanecieron los expedicionarios en la isla, se dirigieron en bote hasta la otra margen del fiordo Hornsund, abriéndose paso con los arpones entre los icebergs y costras de agua de mar congeladas. En tierra la temperatura era por debajo de -30°C, el regreso se hizo antes de que se hiciera de noche.
Al día siguiente la embarcación zarpó hacia Polonia a través del denominado Mar de Groenlandia, un mar tormentoso a causa del encuentro de las aguas polares procedentes del océano Glaciar Ártico con las cálidas del océano Atlántico.
El barco recaló en Gdynia el día 29 y el colectivo se trasladó a Sosnowiec, donde los cubanos comenzaron a elaborar los reportes de los trabajos realizados. Estos fueron interrumpidos por una invitación del profesor Pulina, viajando a los Altos Tatras donde aquel debía impartir un curso de postgrado sobre Hidrología y Geomorfología.
Al regreso terminaron de escribir los reportes del trabajo científico, los cuales contenían los resultados de los análisis químicos efectuados en la isla de Spitsbergen con una interpretación preliminar.
El día 14 de noviembre concluyó la estancia en Polonia de los dos cubanos que participaron en la expedición a la isla polar de Spitsbergen. Materiales y métodos
Las muestras fueron tomadas en frascos plásticos con cierre hermético y llenadas hasta el tope para evitar el escape de los gases disueltos. Las mismas eran representativas de aguas de distinta naturaleza hidrogeológica y glaciológica: de precipitaciones (lluvia y nieve) y hielo en la zona de alimentación, así como acumulación de los glaciares; de aguas de descongelación procedentes de la zona de ablación y de fuentes; de cuevas en hielo; del permafrost; de corrientes superficiales y subsuperficiales; de lagos existentes en las morrenas y de depresiones kársticas; de la tundra y de manantiales termominerales.

Mediante las técnicas analíticas de campo (Markowicz y Pulina, 1979), se determinó en las aguas el contenido de CO2, HCO3
-, CO3 2-, Cl-, SO4
2- , Ca 2+ y dureza, esta última expresada como la suma de Ca2+ + Mg2+ . Los iones Mg2+ y la suma de Na+ y K+ se determinaron por diferencia entre la dureza y el Ca2+, en el primer caso, y aniones y cationes en el segundo caso. Además se efectuaron mediciones “in situ” de temperatura, pH y conductividad eléctrica. La mineralización (en miligramos por litro), se expresa en función de la conductividad eléctrica mediante una ecuación matemática, la cual relaciona esta magnitud con el residuo seco obtenido, por los citados investigadores polacos al llevar a sequedad las muestras de agua natural.
Los datos se procesaron en programas de computación de tipo estadístico y mediante el paquete de programas del Sistema Automatizado para el Procesamiento de Datos Hidroquímicos (SAPHIQ) y el Sistema de Monitoreo de las Aguas (SAMA), reelaborado recientemente (Alvarez et al, 1990; 1993; Fagundo et al, 1992). Entre los programas de este sistema se encuentra: el algoritmo denominado AGMAR (Fagundo et al, 1986), basado en un modelo químico-físico de interacción agua-roca, en el cual se determinaron los valores de las relaciones de saturación de la calcita (RSC), la dolomita (RSD) y el yeso (RSY), los cuales son indicadores geoquímicos que miden la capacidad de las aguas para disolver los citados minerales; usando el Sistema Automatizado para el Monitoreo de las Aguas (SAMA), de ese paquete de programas, se determinaron además las relaciones matemáticas entre la concentración iónica y la conductividad eléctrica.
Para el caso de aguas cuya composición varía de acuerdo a la profundidad del pozo, por encontrarse en un acuífero afectado por la intrusión marina, se empleó el sistema BATOMET basado en un sistema de reconocimiento de patrones. Este sistema ha sido recientemente implementado en su versión final (Vinardell et al, 1994).
La intensidad de denudación química y el balance hídrico fueron calculados mediante el sistema computarizado SAPHID, también remozado recientemente (Vinardell et al, 1993).
Se debe señalar que la numeración de las muestras en las figuras y tabla es la misma. La mineralización en las tablas 1.3; 2.1; 2.2; 2.3; 2.4 y 2.5 se expresa en dos formas
diferentes: 1. Σm: Determinada al llevar a sequedad una muestra, la cual también puede calcularse
mediante una correlación con la conductividad eléctrica, en mg/L. 2. SST: Determinada a partir de los sólidos solubles totales, la cual se calcula mediante la
suma de todos los iones presentes en la muestra, en mg/L. El polinomio ajustado con los datos que aparecen en las tablas 1.4 y 2.6 es del tipo:
CEi = b1 . CE25 °C + b2 (CE25 °C)2 donde: CEi: Concentración iónica, en meq/L. CE25 °C: Conductividad eléctrica a 25°C, en µS/cm. b1 y b2: Coeficientes de la ecuación polinómica (el valor en las tablas 1.4 y 2.6 aparece multiplicado por el factor que se indica en ésta). Resultados de la caracterización hidroquímica
Los resultados de los análisis mencionados en el epígrafe anterior, para una mejor interpretación se representaron mediante diagramas de Stiff (1951), en el orden siguiente:

1. Área del Grondfjord. a) Glaciar Aldegonda. b) Lago Congreso. c) Lago Linneo. 2. Área del fiordo Hornsund. a) Glaciar Werenskiold. b) Región de Gandpasset-Iskantelva. c) Región de Treskelen.
Área del Grondfjord (fiordo Grande) Se encuentra situada en la región de Nordenskiold (Figura 1.3) a 70° de latitud norte. Los
estudios fueron realizados en el glaciar Aldegonda y en los alrededores de los lagos Congreso y Linneo.
En esta región afloran principalmente cuarcitas, anfíboles, mármoles, dolomías y esquistos, estos últimos, por lo general, ricos en carbón. Las secuencias se encuentran compactadas a lo largo de 25 km, con edades que van desde el Precámbrico hasta el Jurásico.
En la figura 1.9 se pueden observar los resultados, mediante diagramas de Stiff, de los análisis químicos correspondientes a esta región.
Figura 1.9. Comportamiento químico-físico de las aguas muestreadas en Grondfjord. Glaciar Aldegonda
Aldegonda es un glaciar pequeño, de unos 8 km2 de superficie, el cual termina en una lengua situada a unos 700 m detrás del fiordo. Allí se originan las aguas que drenan desde cuevas y canales superficiales abiertos en el hielo. La mayor emergencia ocurre en la denominada cueva Aldegonda. Las aguas se abren paso a través de abundantes depósitos morrénicos que indican que

unos años atrás, los hielos del glaciar descargaban directamente al mar. En su corto recorrido desde la zona de alimentación, pasa sobre estratos rocosos constituidos por mármoles y rocas silícicas del intervalo Carbonífero-Jurásico.
Las aguas del glaciar Aldegonda fueron usadas a partir de 1936 para el abasto al poblado ruso de Barentsburg, cuyos habitantes se dedican fundamentalmente a la explotación de las minas de carbón y a la pesca. Luego dejaron de usarse. En el año 1983 una expedición conjunta ruso-polaca realizó investigaciones espeleológicas (Mysztal y Pulina, 1983), hidrológicas y glaciológicas (Pulina, 1983). En las dos últimas se realizó un análisis completo en el arroyo que integra las aguas de descongelación del glaciar.
Las muestra 1-6 (ver figura 1.9) son representativas de las denominadas aguas endo y subglaciar. Ambas se diferencian entre sí desde el punto de vista químico-físico. Las primeras son del tipo bicarbonatadas sódicas con valores de conductividad eléctrica entre 40 y 45 µS/cm, los cuales reflejan el comportamiento de las precipitaciones (lluvia y nieve) con un incremento de la mineralización, por el contacto con los bloques y sedimentos que el glaciar arrastra en su movimiento. Las segundas son del tipo sulfato-bicarbonatadas cálcicas, con una conductividad que varía entre 261 y 275 µS/cm. Los valores medios de mineralización correspondientes son del orden de 26 y 166 mg/L respectivamente.
La cueva Aldegonda fue cartografiada (Figura 1.5), posee unos 500 m de galerías accesibles, desarrolladas en hielo. En comparación con el mapa previamente reportado, se evidencian cambios apreciables en la geomorfología de la cavidad desde el año 1983, lo cual demuestra la activa evolución de estos sistemas termokársticos.
Lago Congreso
La cuenca del lago Congreso se encuentra situada en un antiguo circo glaciar, en la parte superior del valle no glaciado, cuyo lecho está constituido por rocas carbonatadas del Carbonífero. Durante el verano y el otoño el lago se alimenta de aguas procedentes de la descongelación de la nieve y la fusión de parte del permafrost, así como de aguas de precipitación.
Las investigaciones en esta área se llevaron a cabo con el objetivo de complementar los estudios previamente efectuados por el colectivo polaco (Pulina, 1983). Se realizó un análisis completo a la muestra 8 (ver figura 1.9). En los restantes sitios, para su control, sólo se midió temperatura, pH, conductividad eléctrica y se determinó CO2 disuelto, así como algunas especies iónicas. Esta muestra corresponde a una emergencia que drena hacia el lago Linneo. Su composición química (sulfatada cálcica), alta mineralización (990 mg/cm) y relativamente elevada temperatura (3,8 °C), ponen de manifiesto un origen similar a las que ocurren en el lago Congreso, que son del tipo termomineral. En otros climas estas aguas tendrían una temperatura más elevada.
Las conductividades de todas las muestras fueron del mismo orden a las ya reportadas en el trabajo citado en el párrafo anterior. La del lago Congreso fue de 486 µS/cm y las restantes, tomadas en manantiales, así como en arroyos cercanos al lago, promediaron 1 400 µS/cm. Las temperaturas variaron entre 1 y 3,9 °C, relativamente elevadas respecto a las aguas muestreadas en Aldegonda, cuyo valor medio fue de 0,5 °C. Lago Linneo
Este lago se encuentra situado entre el lago Congreso y el cabo Starostin, el mismo está orientado en dirección oeste, a lo largo de un cañón de origen tectónico. Sus aguas, al igual que en el caso del lago Congreso, se originan de la descongelación de las nieves y el permafrost, así como de

las precipitaciones. En el área predominan estratos calcáreos y silícicos del Precámbrico, en su superficie abundan depósitos morrénicos con detritus de diferente origen y constitución litológica. Además del lago Linneo, existe una serie de depresiones kársticas alineadas en dirección a la costa en terrazas marinas.
En esta área se observan tres tipos diferentes de aguas: bicarbonatadas cálcicas, con conductividad eléctrica entre 125 µS/cm y 205 µS/cm, características de los lagos de las depresiones kársticas (muestras 15, 17, 18 y 19 en la figura 1.9), bicarbonatadas sódicas con conductividad de 110 µS/cm (muestra 19 de la misma figura) correspondientes a las aguas muestreadas en la zona de contacto entre las montañas y las terrazas marinas, donde la influencia de la fusión de las nieves es apreciable; y aguas del tipo sulfato-bicarbonatadas cálcicas (muestras 16 y 21), con conductividades eléctricas entre 248 y 428 µS/cm, características del lago Linneo y sus alrededores.
La composición química de las aguas de esta región está influenciada por una serie de factores: la geología local, la mezcla de aguas kársticas y termominerales que proceden de un drenaje más profundo, con nieve y hielo fundidos, así como por procesos crioquímicos, mediante los cuales la fase líquida se va enriqueciendo en componentes disueltos a medida que se congela la superficie del lago. La fase sólida, por el contrario, queda muy empobrecida en iones (Pulina, 1984).
Las evidencias de campo encontradas en las márgenes de algunos lagos de las depresiones kársticas, tales como la presencia de depósitos exógenos de anhidrita y yeso, apoyan la hipótesis del aumento de concentración por efecto crioquímico.
Area del fiordo Hornsund
La segunda área estudiada en la isla corresponde a la zona de experimentación de los investigadores polacos. Se encuentra situada en los alrededores del fiordo Hornsund, a los 77° de latitud. Pertenece a la Tierra de Wedel Jarlsberg, al suroeste de Spitsbergen (Figura 1.4). Glaciar Werenskiold
La cuenca del glaciar Werenskiold, y en particular el glaciar, se ha estudiado ampliamente a lo largo de muchos años desde el punto de vista glaciológico (Baranowski, 1977; Jania y Kolondra, 1982); climatológico (Peryema y Piasecki, 1983) e hidroquímico (Krawcyck y Pulina, 1982; 1983; Pulina et al, 1984). Sin embargo, la información sobre las regiones aledañas es escasa en la literatura.
El objetivo de este trabajo, en la zona de experimentación polaca, consistía en continuar los estudios llevados a cabo por ese colectivo en la cuenca del glaciar Werenskiold; caracterizar las aguas de ríos, arroyos, lagos y manantiales de los alrededores; estudiar el comportamiento local de las aguas de la tundra y usar esta información para comparar con otra, la cual se ha obtenido por métodos semejantes de trabajo, pero en áreas de condiciones climáticas extremas, como es el caso de la cuenca experimental del Pan de Guajaibón en Cuba, representativa de condiciones tropicales (Pulina et al, 1984; Fagundo et al, 1986; Rodríguez et al, 1989; Pulina y Fagundo, 1992).
En el área del glaciar se realizaron diferentes tipos de investigaciones como son: análisis temporal de la fluctuación de la temperatura del agua, el pH, la conductividad eléctrica, el contenido de CO2 y la dureza; el estudio, mediante análisis completo, del comportamiento químico-físico de las corrientes procedentes de la parte glaciada y no glaciada de la cuenca (ríos Glaciar y Brattegg respectivamente); así como del perfil que va desde la zona de acumulación del glaciar hasta la morrena frontal.

La variación de la conductividad eléctrica en los ríos Glaciar y Brattegg se ilustra en la figura 1.10. En ambos se observa, como tendencia, el incremento de la conductividad eléctrica a medida que se aproxima el invierno, asociado a la disminución de los caudales. Esta tendencia es ocasionalmente interrumpida por drásticas caídas, a causa de la ablación como consecuencia de los vientos fohen. El incremento al comienzo del invierno está asociado al efecto crioquímico.
Figura 1.10. Variación de la conductividad eléctrica de los ríos Brattegg y Glaciar, en el período comprendido entre septiembre y octubre de 1985.
En el período estudiado los valores más bajos, medios, y más altos de mineralización de las aguas del río Glaciar (Figura 1.11) fueron: 81, 148 y 236 mg/L (que en términos de conductividad eléctrica corresponde a 130, 239 y 380 µS/cm respectivamente), comportándose como bicarbonatadas-sulfatadas cálcicas. En el río Brattegg estos valores fueron 31, 51 y 100 mg/L (equivalentes en términos de conductividad eléctrica a 50, 82 y 162 µS/cm respectivamente) y cambiaron su tipo de bicarbonatadas sódicas, en los períodos en que predomina la ablación, a bicarbonatadas sulfatadas-cálcicas, en el período que domina la congelación.

Figura 1.11. Variación de la composición química de las aguas de los ríos Brattegg y Glaciar, en el período comprendido entre septiembre y octubre de 1985.

Figura 1.12. Ubicación de los puntos de muestreo en el glaciar Werenskiold.
En la figura 1.12 se muestra la distribución de los puntos de muestreo correspondientes a las principales descargas procedentes del glaciar, representativos de los tipos de aguas supra y subglaciar; aguas de lluvia; nieve y fïrn (hielo compacto de la zona de alimentación y acumulación del glaciar); así como de aguas tomadas en los lagos situados en las morrenas. Su composición química en forma de diagramas de Stiff se presenta en las figuras 1.13, 1.14 y 1.15.
Las aguas supra y subglaciares se diferencian entre sí, tanto en el nivel de mineralización como en el tipo químico. En las aguas de tipo supraglaciar la mineralización varió entre 16 y 167 mg/L (26 y 270 µS/cm respectivamente), donde las menos mineralizadas fueron del tipo bicarbonatadas sódicas y las más mineralizadas bicarbonatadas cálcicas.
Las aguas subglaciares presentaron menor variación de su mineralización y composición química relativa. La mineralización varió entre 112 y 171 mg/L (180 y 275 µS/cm respectivamente), en todo momento permanecieron como aguas del tipo bicarbonatadas cálcicas. Un comportamiento excepcional experimentó la fuente de la cueva Kvisla, la cual incrementó su mineralización de 62 a 1 097 mg/L (100 y 1 770 µS/cm) y cambió de bicarbonatada cálcico-sódica a bicarbonatada cálcica.
Las aguas de las precipitaciones, la nieve, el fïrn y el hielo se caracterizaron por su baja mineralización, entre 7 y 130 mg/L (12 y 210 µS/cm respectivamente), por lo general del tipo

bicarbonatadas sódicas. Las de mayor mineralización se encontraban contaminadas con polvo arrastrado desde las morrenas por los huracanes.
Las aguas de los lagos morrénicos son, por lo general, del tipo sulfato-bicarbonatadas cálcicas y poseen los mayores niveles de mineralización de las aguas estudiadas en esta área (entre 233 y 508 mg/L, equivalente a una conductividad eléctrica entre 375 y 820 µS/cm respectivamente), a excepción de la muestra tomada de agua de mar (muestra 134 en la figura 1.13).
Figura 1.13. Comportamiento químico-físico de las aguas muestreadas a lo largo del perfil que va desde la zona de acumulación del glaciar Werenskiold hasta el mar.

Figura 1.14. Variación de la composición química de las aguas del tipo: a) supraglaciar y b) subglacdiar en la cuenca Werenskiold durante el período septiembre-octubre de 1985.

Figura 1.15. Comportamiento químico-físico de las aguas muestreadas en los lagos situados en la morrena frontal del glaciar Werenskiold. Región de Gangpasset-Iskantelva

Los resultados gráficos del comportamiento químico-físico de las aguas de esta región, excluyendo las aguas de la tundra, se observan en al figura 1.16. Varios tipos de aguas, procedentes de los arroyos superficiales fueron reconocidas: bicarbonato-sulfatadas cálcico-sódicas, sódico-cálcicas y cálcico-magnesianas, así como sulfato-bicarbonatadas magnésico-cálcicas, con una mineralización media de 128 mg/L (conductividad eléctrica de 206 µS/cm). Este comportamiento refleja las diferencias litológicas de estas regiones. En unos casos predominan las cuarcitas, mientras que en otros casos, los mármoles y esquistos.
Figura 1.16. Comportamiento químico-físico de las aguas muestreadas en la región Gandpasset-Iskantelva.
Las aguas de los tipos sub y supraglaciares de la figura 1.16 (muestras 130 y 131 respectivamente), procedentes del glaciar Naambreen se comportan en forma similar a las aguas homólogas del glaciar Werenskiold, tanto en el nivel de mineralización como en el tipo químico.
Las aguas procedentes de lagos costeros son del tipo bicarbonatado-cloruradas cálcicas y tienen una mineralización entre 61 y 99 mg/L (99 y 160 µS/cm respectivamente). Estas aguas están muy influenciadas por los aerosoles marinos y la geología local.
En la figura 1.17 se presentan los resultados correspondientes a las aguas procedentes de la tundra en el área cercana a Hyttevika. Como se puede observar, no existe un tipo predominante de agua, puesto que todos los iones contribuyen de manera similar y la mineralización media es de 108 mg/L correspondiente a una conductividad eléctrica de 174 µS/cm. Estas aguas se caracterizan por poseer un bajo pH (entre 5.1 y 6.6 unidades) y relativamente alto contenido de CO2 (entre 1,3 y 5,9 mg/L), en comparación con otras aguas donde este valor es nulo (Tabla 1.1). Estos resultados demuestran que a pesar de las rigurosas condiciones del medio ambiente ártico, la influencia de los procesos biológicos es importante en la tundra.

Figura 1.17 Comportamiento químico-físico de las aguas procedentes de la tundra en la región de Hyttevika.

Muestra pH CO2 (mg/L) 135 6.50 1.5 136 6.40 2.6 137 5.40 4.6 138 5.10 5.9 139 6.20 1.3 142 6.55 4.0 143 6.35 5.0 144 6.40 4.2
Tabla 1.1. Propiedades químico- físicas de las aguas muestreadas en la tundra. Región de Treskelen
Esta región había sido estudiada previamente desde el punto de vista hidroquímico y fue muestreada por el colectivo polaco. En la figura 1.18 se muestran los resultados gráficos del comportamiento de estas aguas. Corresponden a arroyos, ríos, lagos y fuentes procedentes del área glaciada y la tundra. Presentan gran variación, tanto en tipo químico como en la
mineralización. Figura 1.18 Comportamiento químico-físico de las aguas muestreadas en la región de Treskelen.
Las de menor mineralización (con conductividades eléctricas entre 32 y 110 µS/cm) poseen, relativamente, altas concentraciones de iones sulfato, bicarbonato y sodio. Las de

mediana mineralización (con conductividades eléctricas entre 143 y 260 µS/cm), son por lo general del tipo bicarbonato-sulfatadas cálcico-magnesianas. El resto, con mineralización superior a 500 mg/L (conductividad eléctrica entre 814 y 1 373 µS/cm), varían entre bicarbonato-sulfatadas cálcico-magnésicas, sulfatadas cálcicas y sulfato-cloruradas cálcico-magnésicas. Sólo una muestra, de mediana mineralización, fue del tipo clorurada sódica. Grado de agresividad de las aguas y denudacion química
La capacidad que posee un agua natural para disolver en un momento dado un mineral carbonatado, se puede expresar en forma cuantitativa mediante la relación entre el contenido de los cationes Ca2+ y Mg2+ (expresado, por lo general, como CaCO3), el pH y el CO2 disuelto.
Sobre la base de la teoría de los equilibrios químicos en el sistema CO2-H2O-CaCO3
(Garrels y Christ,1965; Johnston y Williamson, 1916), se ha determinado teórica y experimentalmente, la saturación límite que se alcanza al ser disuelto el mineral calcita (CaCO3) por un agua natural que posee un cierto contenido de CO2 (Roques, 1964; Tillman, 1932).
Como resultado de estos trabajos (Trombe, 1952; Muxart, 1972), se han creado una serie de diagramas de estabilidad, mediante los cuales se puede expresar, en forma gráfica, el grado de agresividad de un agua. El más comúnmente aplicado, se ha popularizado por Trombe (1952) y se le conoce como diagrama de Tillman-Trombe.
Al representar gráficamente los datos de CaCO3 y pH de una muestra de agua, el punto correspondiente queda por debajo de la isoterma de saturación, el agua se encuentra insaturada respecto a la calcita. Si queda situado sobre la isoterma, se encuentra saturada y si queda por encima de ésta, sobresaturada. Este diagrama se ha utilizado ampliamente en la literatura europea. En Estados Unidos, Cuba y otros países, se han empleado más los índices de saturación definidos por Back y colaboradores (1966):
RSC Ca COKc
=+ −
log ( )(232 ) (1.1)
RSD Ca Mg COKd
=+ + −
log ( )( )( )2 232 2
(1.2)
RSY Ca SOKy
=+ −
log ( )(24
2 ) (1.3)
donde : RSC: Relación de saturación de la calcita. RSD: Relación de saturación de la dolomita. RSY: Relación de saturación del yeso. Kc: Constante del producto de solubilidad de la calcita. Kd: Constante del producto de solubilidad de la dolomita. Ky: Constante del producto de solubilidad del yeso. ( ): Actividad iónica.
El cálculo de estos índices (ecuaciones 1.1, 1.2 y 1.3) requiere de métodos numéricos complejos que actualmente, se facilitan con el uso de la computación (Jacobson y Langmuir, 1975; Fagundo y Valdés, 1975; Wigley, 1977; Fagundo, Valdés y otros, 1986; Alvarez et al, 1993). Las relaciones de saturación de la calcita, la dolomita y el yeso, se basan en comparar los productos

de actividad real y teórico de los respectivos minerales. Si el valor de estas magnitudes es menor que cero (negativo), el agua se encuentra insaturada; si es cero, se encuentra saturada, y si es mayor que cero (positivo), el agua se encuentra sobresaturada.
Otros métodos para determinar la agresividad del agua (para disolver los minerales carbonatados), se basan en la denominada capacidad potencial, la cual consiste en colocar una cierta cantidad de roca en una muestra de agua natural, a la temperatura en que se encuentra la misma en un momento dado y determinar, al cabo de varios días, la diferencia del contenido de dureza (Markowicz y Pulina, 1979).
En la figura 1.19 se muestran las propiedades de las aguas con relación al grado de saturación de la calcita, la dolomita y el yeso (Fagundo et al, 1987, los cuales se han calculado mediante el programa AGMAR (Fagundo, Valdés y otros, 1986) del sistema SAPHIQ. Estas aguas son representativas de los distintos tipos descritos en este capítulo.
Figura 1.19 Comportamiento de las aguas de la isla de Spitsbergen respecto a las relaciones de saturación de la calcita (RSC), la dolomita (RSD) y el yeso (RSY).
Como se puede apreciar en la figura 20, todas las aguas se encuentran insaturadas respecto al yeso, donde las menos insaturadas son las de los lagos morrénicos. Respecto a la calcita y a la dolomita se presentan insaturadas las aguas de hielo, nieve, arroyos, corrientes superficiales y una parte de las fuentes subglaciares. Otra parte de estas aguas, como son los lagos de las morrenas,

tienden a la saturación de estos minerales. No se observan aguas con alto grado de sobresaturación como las que se aprecian en las zonas kársticas de Cuba.
El grado de agresividad o saturación de un agua está relacionado con la cantidad de minerales que las mismas son capaces de disolver en un momento dado, es decir, mientras más agresiva se encuentre el agua, mayor es su capacidad para disolver los minerales. En general, después de la ocurrencia de precipitaciones (lluvia o nieve) en un área determinada, las aguas incrementan su agresividad y en su trayectoria, disuelven los minerales hasta alcanzar la saturación con respecto a éstos. En ese lapso, arrastran un cierto contenido de iones disueltos y sedimentos en suspensión, cuyas magnitudes se denominan denudación química y mecánica respectivamente.
La magnitud de la denudación química (Dm) en Spitsbergen ha sido determinada por varios investigadores (Corbel, 1959; Hellden, 1973; Pulina, 1971; 1974; 1984; 1992); el método propuesto por Pulina es actualmente el más utilizado por los especialistas, la cual viene definida por la expresión siguiente:
Dm a Tm q= . . (1.4) donde: Dm: Denudación química, en m3/km2 ó min./1 000 años. a: Coeficiente, que para las rocas carbonatadas toma el valor de 12.6. Tm: Mineralización de las aguas producida por la interacción agua roca ( Tm = Tm - Ta ), en la cual: Tm: Mineralización de las aguas en las emergencias, en mg/L Ta: Mineralización de las aguas de las precipitaciones, en mg/L q: Módulo de escurrimiento (q = 1 000 . Q / S ), en L/s . km2 ; en la cual: S: Area de la cuenca de captación, en km2 Q: Caudal o gasto, en m3/s.
La mineralización (Tm y Ta) se determinan a partir de su conductividad eléctrica a 25°C (Tm = 0,62 . CE 25 °C).
Los resultados más recientes de la magnitud de la denudación química determinada por los investigadores polacos en la cuenca del glaciar Werenskiold, se presentan en la tabla 1.2. Los mismos corresponden al año hidrológico 1979-1980.

Estación hidrológica Intervalo de
tiempo Número de
días Q
(m3/s) T
(mg/L) dT
(mg/L) D (m3/km2/
año) Otoño 1979 28/08-25/11 91 1,70 106,9 89,4 10,9 Invierno 79/80 26/11-20/05 176 0,05 175,5 157,5 2,0 Primavera 1980 21/05-22/07 63 5,60 61,6 44,1 12,3 Verano 1980 23/07-03/09 43 10,30 47,8 30,1 10,0 Año hidrológico polar 1989-1980
28/08-03/09
373
2,50
35,8
Tabla 1.2. Magnitud de la denudación química en el glaciar Werenskiold determinada por Pulina et. al (1984) en el año hidrológico polar comprendido entre el otoño de 1987 y el verano de 1980
La intensidad de la denudación química, durante ese año (1979-1980), alcanzó su valor máximo en la primavera, 12,3 m3/km2, en que el proceso de ablación se hizo más agudo. Durante el invierno cesó la actividad hídrica, la magnitud de la denudación química alcanzó el valor mínimo, 2,0 m3/km2. Durante el otoño y el verano polar la intensidad de la denudación química fue de alrededor de 10,9 y 12,3 m3/km2 respectivamente. El valor correspondiente al año hidrológico completo fue de 35,8 m3/km2.
Variación temporal de las propiedades químico-físicas de las aguas
Con el objetivo de apreciar mejor cómo varían las propiedades químico-físicas de las aguas al cabo del tiempo, como resultado de los cambios de las condiciones meteorológicas, se seleccionaron algunos indicadores geoquímicos (temperatura del agua, contenido de CO2, pH, dureza y las relaciones de saturación de la calcita, la dolomita y el yeso); confeccionándose diagramas de correlación gráfica de estos indicadores. En la figura 1.20 se presentan los resultados correspondientes al río Glaciar para el período comprendido entre el 6 de septiembre y el 6 de octubre de 1985.

Figura 1.20 Variación de la temperatura, el CO2 , el pH, la conductividad eléctrica, la dureza y las relaciones de saturación de la calcita (RSC), la dolomita (RSD) y el yeso (RSY) de las aguas del río Glaciar, en el período comprendido entre el 6 de septiembre y el 6 de octubre de 1985.
Como se puede apreciar en las figura 1.20, a medida que la temperatura del agua disminuye (la cual varió de 3 a 0 °C aproximadamente), se incrementó el contenido de CO2 disuelto, la mineralización (en términos de conductividad eléctrica) y la dureza, las cuales son directamente proporcionales entre sí. La variación del pH tiende, por lo general, a comportarse en forma inversa al contenido de CO2 disuelto, presentándose la forma de una de estas figuras como la imagen especular de la otra. Este comportamiento es consecuencia del proceso cinético de disolución de los minerales por las aguas naturales, las cuales poseen un cierto contenido de CO2. En virtud de este proceso, las aguas evolucionaron haciéndose menos agresivas hasta alcanzar la saturación respecto a la calcita y la dolomita. En el caso del río Brattegg, con menor contenido de sales disueltas que las del río Glaciar, las aguas evolucionaron hasta una menor insaturación.
Un comportamiento similar al explicado en el párrafo anterior se puede obtener al procesar datos hidroquímicos reportados por los investigadores polacos en otras cuencas de Spitsbergen tales como Fugleberget.
El comportamiento de los elementos hidrometeorológicos registrados por los científicos polacos en los años 1979 y 1980 en la cuenca del río Fugleberget, se puede observar, a partir de los resultados presentados en forma gráfica en una publicación de Krawcyk y Pulina (1982).

En el período del 1 de agosto al 31 de octubre, el cual abarcó las estaciones: verano, otoño, y parte del invierno hidrológico polar (este último se extendió hasta el 30 de octubre) se realizó un muestreo hidroquímico sistemático por el colectivo polaco en 1979.
En el período estudiado en 1979, las precipitaciones (en forma de lluvia o nieve) fueron más abundantes entre el 7 y el 18 de agosto y entre el 6 de septiembre y el 5 de octubre de ese año.
En ambos intervalos se produjo un incremento de los caudales de los arroyos, mientras que la mineralización no presentó una respuesta sensible tendiente a su disminución. En general, la mineralización de las aguas presentó una tendencia ascendente en la medida que se aproximaba el invierno polar.
La temperatura mantuvo una tendencia decreciente en el período comprendido entre el 1 de agosto y el 20 de octubre de 1979, sólo con algunas interrupciones de la misma, variando entre 6,2 y 0 °C. El contenido de CO2 fluctuó en ese tiempo entre 5,3 y 3,3 mg/L, sin presentar una tendencia dominante determinada. En forma semejante se comportó el pH, que varió entre 7.6 y 7.0 unidades. La conductividad eléctrica y la dureza siguieron un comportamiento similar al de la mineralización, en cuanto a sus fluctuaciones y tendencia ascendente.
En relación a los indicadores de agresividad, se pudo apreciar que en todos los casos, las aguas permanecieron insaturadas respecto a los minerales calcita, dolomita y yeso, fluctuando los valores de RSC y RSD como consecuencia de los cambios recíprocos experimentados por el CO2, el pH y la dureza. Por lo general, una menor mineralización y dureza se correspondió con una menor insaturación respecto a la calcita y la dolomita.
El período de muestreo hidroquímico sistemático llevado a cabo por los científicos polacos en 1980, se extendió desde el 4 de junio hasta el 1 de septiembre, donde tuvieron lugar las estaciones hidrológicas de la primavera, el verano y el otoño.
Las precipitaciones más abundantes se produjeron entre el 4 y el 18 de junio y entre el 20 de julio y el 5 de septiembre , lo cual provocó un aumento en los caudales de los arroyos, mientras que la mineralización de las aguas creció en el primer intervalo y luego presentó una tendencia ascendente.
Las temperaturas de las aguas en el período del 4 de junio el 1 de septiembre de 1980, tuvieron una tendencia ascendente, cercana a 0 °C en su inicio, llegaron a alcanzar hasta 8 °C, fluctuando en ese intervalo con un valor medio de 3,7 °C.
El contenido de CO2 disminuyó con el aumento de la temperatura, mientras que el pH, la dureza y las relaciones de saturación de las calcita y la dolomita crecieron, hacia el final del período, para acercarse a los valores de saturación en el caso de la calcita y a un grado de menor insaturación en el de la dolomita. El RSY presentó también una tendencia parecida a los restantes índices de agresividad, con un pico anómalo en el intermedio del intervalo.
En la cuenca de Rasstupet, el período de experimentaciones sistemáticas llevadas a cabo por el colectivo polaco, correspondió al comprendido entre el 11 de julio y el 11 de septiembre de 1978.
Los datos procesados en Rasstupet corresponden a dos tipos de aguas: ríos que se escurren por la superficie del macizo, constituido por mármoles del Precámbrico, y manantiales que emergen de ese mismo medio, cuyas aguas están en contacto con el mar a través de conductos kársticos (Leszkiewicz, 1982; Leszkiewicz et al, 1982).
En el primer caso (ríos), la temperatura del agua presentó una tendencia descendente a medida que se acercaba el período invernal. El contenido de CO2, por el contrario a lo que se observó en la región de Werenskiold, también experimentó una tendencia descendente. El pH, la mineralización y la dureza tendieron a ascender. Lo mismo sucedió con los valores de RSC y RSD,

mientras que la RSY experimentó, más bien, un comportamiento fluctuante, presentándose las aguas unas veces más insaturadas que otras respecto a este mineral en el intervalo estudiado.
En los manantiales kársticos afectados por la intrusión marina, la variación estacional de la temperatura fue despreciable, lo cual se pone de manifiesto al observar los valores relativamente bajos del coeficiente de variación de la temperatura en este sitio, en comparación con el observado en otras áreas. De igual forma se comportaron el contenido de CO2, el pH y los índices de agresividad de las aguas, sin una tendencia particular. Sólo la mineralización y la dureza tendieron a ascender a medida que se acercaba el invierno polar.
El hecho de que la alimentación de estos manantiales esté parcialmente controlada por la mezcla de éstos con el agua de mar, hace que el patrón de fluctuación de los indicadores geoquímicos seleccionados en este estudio sea más atenuado ante los cambios hidrometeorológicos externos. Patrones hidrogeoquímicos y relaciones empíricas entre las concentraciones iónicas y la conductividad eléctrica
La conductividad eléctrica de un agua natural se puede expresar, aproximadamente, como la sumatoria del producto de la concentración de cada ion por su conductividad eléctrica específica, es decir:
CE o CCi Si
i
n
25 1=
=∑ . (1.05)
donde: CE 25 °C: Conductividad eléctrica a 25 °C. Ci: Concentración iónica. Si: Conductividad eléctrica específica de cada ion a dilución infinita .
En la práctica es necesario tener en cuenta además, el efecto de concentración, cuando se quiere obtener el valor de la conductividad eléctrica teórica. De esta manera, Dudley (1972) ha propuesto para el cálculo de esta magnitud a 25 °C la relación siguiente:
log log( . ) , . logCE o CCi Si
i
nL
i
n
25 10 03
1= Ci
=∑ −
=∑ (1.6)
Muchos investigadores han encontrado relaciones matemáticas entre la conductividad
eléctrica y la mineralización (Markowicz y Pulina, 1979; Bakalowicz, 1974); la dureza (Bray, 1977) y otros parámetros químico-físicos. Las mismas se han aplicado con distintos objetivos hidrogeológicos. Sin embargo, correlaciones entre la conductividad eléctrica y la composición iónica sólo se han encontrado, en forma altamente significativa, para los iones más abundantes (Drake y Ford, 1974) y se han utilizado menos en la práctica.
Correlaciones matemáticas entre la composición química y la conductividad eléctrica de las aguas, se han utilizado por Fagundo (1985; 1990 a, b) y Fagundo y Pajón (1987); con el objetivo de caracterizar acuíferos y posteriormente, comprobar la calidad de las aguas, mediante mediciones en el campo de la conductividad eléctrica y el uso de juegos de ecuaciones matemáticas, las cuales son calculadas y almacenadas para su empleo, por el sistema de programas SAMA. Este sistema puede operar mediante dos modalidades:

1. Los datos se procesan de forma automatizada y las ecuaciones seleccionadas (por mejor
ajuste), a partir de polinomios desde uno a quinto grado, mediante un criterio estadístico (Prueba de Fisher).
2. Por tanteo, donde el usuario escoge el polinomio deseado.
Como criterio de semejanza entre los valores reales y los determinados por modelación matemática se utiliza un índice de similitud, el cual toma en cuenta el conjunto de datos hidroquímicos en lugar del coeficiente de correlación de cada ion por separado. Estas ecuaciones se han condicionado a pasar por el origen de coordenadas, teniendo en cuenta que la conductividad eléctrica de las aguas antes de tener contacto con los minerales es despreciable.
Para el procesamiento se deben utilizar datos hidroquímicos que reflejen el efecto del ciclo hidrológico, preferentemente aquellos obtenidos mediante muestreos sistemáticos a través de uno o más años hidrológicos, o en un período de tiempo en el cual se pongan de manifiesto variaciones en el quimismo como consecuencia de los cambios hidroclimáticos.
Como se ha indicado anteriormente (epígrafe variación temporal de las propiedades químico-físicas de las aguas), las aguas de la isla de Spitsbergen, sufren cambios muy drásticos en sus propiedades químico-físicas como resultado de las variaciones meteorológicas. Por esa razón, los datos hidroquímicos tomados en forma secuencial, tanto durante la expedición Spitsbergen'85, como durante otras expediciones científicas llevadas a cabo por el colectivo polaco (Tabla 1.3), ofrecen una valiosa oportunidad para estudiar la naturaleza de las correlaciones matemáticas entre la concentración iónica y la conductividad eléctrica en condiciones de cambios extremos.

Ficheros de
datos. Nº de datos
Composición química relativa HCO3
- Cl- SO42- Ca2+ Mg2+ Na+ K+
Minerali-zación
Σm TSS
Conductividad Eléctrica
Mín. Med. Máx. CV GLACIER 13 53 8 39 52 22 26 81 155 130 229 370 29,3 GLACIER1 10 53 9 38 52 20 28 123 137 130 198 250 20,3 GLACIER2 3 51 8 41 49 31 20 205 213 300 330 370 8,9 BRATTEGG 10 48 23 29 39 13 58 51 69 50 82 162 39,9 BRATEG1 8 48 24 28 26 10 64 42 60 51 67 82 17,3 BRATEG2 2 51 15 34 38 26 36 87 106 120 141 162 14,9 FUGLE1 36 74 16 10 67 9 24 109 131 110 135 192 13,8 FUGLE2 20 68 24 8 53 6 41 58 99 40 93 140 35,8 FUGLE3 7 53 38 9 36 6 58 33 60 40 54 81 13,8 FUGLE4 13 76 17 7 61 7 32 71 120 74 114 140 15,9 RASTUPZ1 10 75 11 14 60 20 20 109 196 139 176 231 17,2 RASTUPZ2 9 74 12 14 60 25 15 92 157 106 149 106 20,0 RASTUPZ3 10 14 74 12 15 21 64 428 837 179 690 1 203 33,2 RASTUPZ4 11 10 84 6 11 17 72 445 511 181 717 1 119 40,9 Tabla 1.3. Propiedades químico-físicas de las aguas muestreadas sistemáticamente durante la expedición Spitsbergen'85, así como en otras llevadas a cabo por el colectivo polaco.
En el intervalo de 130 a 370 µS/cm, en que varió la conductividad eléctrica de las aguas del río Glaciar, durante el período estudiado por el colectivo cubano-polaco, su composición relativa cambió de HCO3- > SO4
2- - Ca2+ > Na+ a HCO3- > SO4
2- - Ca2+ > Mg2+. Al ajustar los datos hidroquímicos correspondientes, por el sistema SAMA se obtuvo como resultado, que las ecuaciones de mejor ajuste fueron de segundo grado para el ion Ca2+ y de primer grado para el resto de los iones (Tabla 1.4). Ficheros de
datos. Factor Valores de los coeficientes de la ecuación polinómica
HCO3 - Cl- SO42- Ca2+ Mg2+ Na+ K+ I S
GLACIER 10-3
10-6
4,784 0,731 3,664 5,860 -4,426
2,275 2,228 0,901
GLACIER1 10-3 4,916 0,781 3,609 4,915 1,863 2,528 0,906 GLACIER2 10-3 5,531 6,784 3,412 3,520 2,086 10,218 0.871 BRATTEGG 10-3
10-5
10-7
5,535 6,784 -7,193 2,438
3,412 3,520 2,086 10,218 -5,973
0,871
BRATEG1 10-3 5,889 2,958 3,348 3,298 1,281 7,616 0,860 BRATEG2 10-3 5,204 1,557 3,528 3,927 2,820 3,543 0,847 FUGLE1 10-3 9,457 2,058 1,201 8,582 1,123 3,011 0,887 FUGLE2 10-3
10-5
9,986 6,920 1,042 8,087 0,925 12,488 0,844 -6,851
FUGLE3 10-3 7,664 6,221 1,255 5,342 1,027 8,771 0,822 FUGLE4 10-3 10,270 2,259 1,016 8,423 0,913 4,209 0,896 RASTUPZ1 10-3 10,714 1,541 2,026 6,610 3,060 2,635 0,873 RASTUPZ2 10-3 10,353 1,720 1,847 8,360 3,571 1,989 0,864 RASTUPZ3 10-3 1,243 9,322 1,378 1,535 2,095 8,313 0,869 RASTUPZ4 10-3 1,208 9,276 1,165 1,412 2,117 8,120 0,877 RAS79Z3 10-3 0,983 10,483 1,359 1,307 2,663 8,555 0,957 Tabla 1.4. Resultados del ajuste mediante polinomios de hasta quinto grado (pasan por el origen de coordenadas) de los datos hidroquímicos correspondientes a las muestras tomadas durante la expedición Spitsbergen'85 y otras efectuadas por el colectivo polaco en los alrededores del fiordo Hornsund.

El índice de similitud medio entre los datos reales y los obtenidos (Tabla 1.4), multiplicando el juego de los coeficientes correspondientes por la conductividad eléctrica es de 0,901, es decir, existe aproximadamente 90 % de semejanza entre los datos reales y los hallados por modelación matemática (Figura 1.21).
La causa de la no linealidad de todas las correlaciones se debe a la variación en el tipo de
agua, es decir, a medida que se producen los cambios hidrometeorológicos y el efecto crioquímico (Pulina, 1984). En estos casos la conductividad eléctrica no es directamente proporcional a la concentración de cada uno de los iones, porque el incremento de la conductividad puede estar asociado al aumento de algunas de las especies iónicas en particular y no a todas.
Si los datos se ajustan según su composición química relativa, se obtienen dos grupos de aguas, cuyos rangos de conductividad varían entre 130 y 250 µS/cm y entre 300 y 370 µS/cm respectivamente. Mediante la modalidad automatizada de SAMA, se obtienen ecuaciones de primer grado para todos los iones, con una similitud entre los valores reales y los hallados por modelación, algo mayor que la obtenida por las ecuaciones anteriores.
En el caso del río Brattegg, las aguas varían entre HCO3- > SO4
2- - Ca2+ a HCO3- > SO4
2- > Cl-- Na+ > Mg2+ y los rangos de conductividad entre 50 y 82 µS/cm y 120 y 162 µS/cm respectivamente. Las ecuaciones de mejor ajuste, utilizando todos los datos (N = 10) son de tercer grado para el ion Cl-, de segundo grado para el Na+ + K+ y de primer grado para el resto de los

iones, con un índice de similitud de 0,871 (Figura 1.22). Al ajustar los datos por tipos de agua, se obtienen ecuaciones de primer grado para todos los iones, con una semejanza media de 0,860 y 0,847 entre ambos, algo menor que la obtenida usando todos los datos.

Figura 1.22 Composición química de las aguas del río Brattegg, obtenida por análisis químico (izquierda) y modelos matemáticos (derecha).
Las aguas originadas por la descongelación de la nieve y el permafrost en la cuenca no glaciada de Fugleberget, en el período de estudios sistemáticos llevado a cabo por los polacos desde agosto a octubre de 1979, presentaron una composición química relativa estable, donde predominaron las del tipo HCO3
- - Ca2+ . La conductividad eléctrica varió en ese período entre 110 y 192 µS/cm.
En el período de experimentos sistemáticos correspondientes al año 1980 (cuenca de Fugleberget), variaron su tipo de HCO3
- > Cl- - Na+ > Ca2+ a HCO3- - Ca2+ > Na+ y sus rangos de
conductividad oscilaron entre 40 y 81 µS/cm y entre 74 y 140 µS/cm respectivamente. Al ajustar todos los datos hidroquímicos, se obtienen ecuaciones de segundo grado para Na+ + K+ y de primer grado para el resto de los iones, con un índice de similitud medio de 0,844 entre los datos reales y los obtenidos por modelación matemática. Si los datos se ajustan atendiendo a los tipos de aguas presentes, las ecuaciones de mejor ajuste que se obtienen son de tipo lineal, con índices de similitud medios de 0,827 y 0,896 entre ambas.
Las aguas estudiadas por los hidrólogos y glaciólogos polacos en la cuenca de Rasstupet, desde el punto de vista hidroquímico, son de dos tipos: las que se originan por escurrimiento superficial por los carbonatos metamorfizados del macizo son del tipo HCO3
- - Ca2+ , mientras que las que emergen de los manantiales, conectados al mar a través de los conductos kársticos, son del tipo Cl- - Na+. Ambos tipos de aguas, sin embargo, no sufren cambios apreciables en su composición química relativa por la acción de los agentes de intemperismo.
El ajuste matemático de los datos hidroquímicos correspondientes da por resultado, la obtención de ecuaciones de primer grado para todos los iones, cuyas pendientes son muy diferentes, lo cual se corresponde con la naturaleza hidrogeológica de los dos tipos de aguas. La semejanza media entre los datos reales y los obtenidos por modelación fue de 87 % en ambos casos. En las figuras 1.23 y 1.24 se pueden apreciar estos resultados.


Por no disponer de más series cronológicas de datos hidroquímicos de la región de
Spitsbergen, no es posible incluir en este estudio de correlaciones los datos correspondientes a otras áreas, en las cuales la población de datos resulta muy pequeña. No obstante, con el objetivo de tener una visión más completa del comportamiento químico-físico de las aguas de esta región polar, se procesaron los datos correspondientes a otros tipos de aguas de las áreas experimentales de Grondfjord y Hornsund, y de esta manera obtener, a partir de las concentraciones relativas

medias en porcentaje de miliequivalente por litro, los patrones hidrogeoquímicos correspondientes.
La información procesada, (mencionada en el párrafo anterior) sirve para inferir la evolución química de las aguas, por los complejos procesos hidrogeológicos y glaciológicos que allí tienen lugar, como resultado de las condiciones meteorológicas existentes. Los resultados correspondientes a la expedición Spitsbergen'85 se resumen en la figura 1.26, donde los patrones hidrogeoquímicos se presentan con una notación, que corresponde a la relación estequiométrica Na+ + K+: Ca2+ : Mg2+ : Cl-: HCO3
- : SO42- y donde la suma de aniones o de cationes es de 10. Esta cifra se ha
escogido para facilitar su representación gráfica y aprovechar la posibilidad que ofrece el programa de computación de dar los resultados correspondientes en porcentaje de miliequivalente por litro, para lo cual sólo debe dividir estos valores entre 10.
Figura 1.25. Patrones hidrogeoquímicos de las aguas de la región de Grondfjord muestreadas durante la expedición Spitsbergen'85.
Del análisis de la composición química relativa, representada en la figura 1.25 por diagramas de Stiff, se infiere que en la isla polar de Spisbergen existe una amplia variedad de tipos de aguas. El exámen de las tablas a las que se ha hecho referencia en este capítulo (Tablas. 1.3 y 1.4) indica además la amplitud con que varían los diferentes parámetros que caracterizan las propiedades químico-físicas de estas aguas.

Capítulo 2. Comportamiento hidroquímico
del karst tropical de Cuba

Breve descripción de los casos de estudio
El hecho de que durante la expedición Spitsbergen'85 se hayan utilizado mediciones, así como métodos de análisis químicos y físicos de las aguas, mediante técnicas semejantes a las empleadas en algunas regiones de Cuba, brinda la oportunidad de hacer estudios comparativos sobre el comportamiento químico-físico de las aguas kársticas en dos regiones con climas extremos, polar y tropical.
Un primer intento con estos objetivos fue realizado por Fagundo, Valdés y Pulina (1990), tomando como regiones de referencia la cuenca del glaciar Werenskiold, en la región cercana al fiordo Hornsund, en Spitsbergen y la cuenca del río San Marcos, en Cuba (Pan de Guajaibón), donde se encuentran ubicados los polígonos experimentales en los cuales se han llevado a cabo los estudios acerca de la dinámica y evolución del Karst en condiciones polar y tropical respectivamente. En esta ocasión, se aborda de nuevo esta temática, que desde el punto de vista científico es novedosa, abundando en aspectos no analizados con anterioridad e incluyendo otras regiones kársticas, tanto de Spitsbergen como de Cuba con el objetivo de ampliar la representatividad de las aguas kársticas correspondientes.
En este estudio además, se incluyen algunas aguas no kársticas que corresponden a los parteaguas, desde donde drenan distintas corrientes superficiales hacia áreas kársticas. Las regiones y tipos de aguas de Cuba, seleccionados, fueron las siguientes:
1. Cuenca del río Cuyaguateje. 2. Cuenca del río San Marcos. 3. Meseta del Guaso. 4. Llanura kárstica meridional del occidente de Cuba. a) Región de San Antonio de los Baños. b) Llanura meridional de Pinar del Río. c) Karst litoral del sur de Matanzas.
Cuenca del río Cuyaguateje.
Las propiedades químico-físicas y el modo en que evolucionan las aguas de la cuenca del río Cuyaguateje se han estudiado por Fagundo, Valdés, Pajón y Rodríguez (1981; 1982), las cuales se relacionan a continuación:

Características hidrogeológicas Número
de datos Nombre del fichero
1. Aguas no kársticas que discurren superficialmente por esquistos y pizarras, antes de atravesar los macizos kársticos.
15
PIZARRA
2. Aguas de infiltración tomadas en el interior de las cavidades kársticas.
33
CUEVAS
3. Aguas tomadas en las resurgencias de los macizos carbonatados, que reflejan las propiedades, tanto de las aguas alóctonas como de las autóctonas
19
SUMRES
4. Aguas tomadas en las resurgencias de los macizos carbonatados, que sólo reflejan las propiedades de las aguas autóctonas.
16
RESK
5. Manantiales y aguas de pozos en la zona de saturación de los macizos kársticos.
5 5 19
PALMERO PICAPICA MANANTIA
6. Manantiales y pozos de la zona de saturación y circulación profunda del karst.
21
BANITOS
7. Corrientes superficiales y aguas de pozos en sedimentos de la Formación Pica Pica
8
GUAYABO
8. Aguas de los ríos Cuyaguateje y Portales. 11 PORTALES
Los resultados que aparecen reportados en los trabajos publicados por ese colectivo se representan en la figura 2.1., en la cual se muestran los tipos hidroquímicos presentes en la cuenca.
En la tabla 2.1 se presentan los valores medios, mínimos, máximos, así como del coeficiente de variación de los principales indicadores geoquímicos de estas aguas.
N Nº Composición química relativa
HCO3- Cl- SO4
2- Ca2+ Mg2+ Na++K+
Minerali- zación Σm TSS
Conductividad eléctrica
Mín. Med. Máx. CV
1) 15 66 24 10 25 12 63 55 99 41 89 158 36,7 2) 19 76 20 4 51 14 25 101 137 89 163 323 36,6 3) 33 86 12 2 85 6 9 164 223 221 265 361 13,8 4) 16 87 10 3 68 13 19 208 289 185 337 534 29,2 5) 5 89 9 2 85 6 9 192 275 269 310 397 14,8 6) 5 73 7 20 75 22 3 329 418 453 531 536 8,3 7) 19 84 8 8 71 14 15 252 350 138 406 540 23,1 8) 21 68 8 24 60 21 19 318 485 473 513 575 5,0 9) 8 91 6 3 50 36 14 318 429 399 513 625 16,0 10) 11 73 9 18 66 19 15 233 210 78 375 556 31,6 Tabla 2.1. Propiedades químico-físicas de las aguas muestreadas sistemáticamente en las regiones kársticas montañosas de Cuba (cuenca del río Cuyaguateje). Ficheros de datos: 1) PIZARRA; 2) SUMRES; 3) CUEVAS; 4) RESK; 5) PALMERO; 6) PICAPICA; 7) MANANTIA; 8) BANITOS; 9) GUAYABO, 10) PORTALES
Las aguas no kársticas, que discurren en forma superficial por los sedimentos impermeables de las Alturas de Pizarras, constituidos fundamentalmente por esquistos y areniscas de la Formación San Cayetano, de edad Jurásico, se caracterizan por presentar una

baja mineralización, bajo pH, poco contenido iónico y moderado contenido de dióxido de carbono (CO2) disuelto. Son del tipo HCO3- - Na+ > Ca2+ y se encuentran insaturadas respecto a los minerales calcita, dolomita y yeso.
En la medida en que las aguas no kársticas se mueven a través de los macizos carbonatados, constituidos por calizas de edad Jurásico inferior y Cretácico (Formaciones Jagua y Guasasa), esas aguas experimentan una disminución en su contenido de CO2, incrementan su pH y dureza, se hacen más mineralizadas y menos insaturadas en unos casos, así como más saturadas en otros, respecto los minerales calcita y dolomita (con respecto al yeso se mantienen insaturadas). Pasan a ser, fundamentalmente, del tipo HCO3- - Ca2+ , aunque en el caso de las aguas tomadas en las resurgencias kársticas, cuando el grosor de los macizos no es muy grande, pueden presentar el tipo hidroquímico característico de las aguas que proceden de las Pizarras. Se comportan en realidad como aguas de mezcla.
Figura 2.1. Variación de la composición química de las aguas del río Cuyaguateje a lo largo de la cuenca,
desde las alturas de Pizarras del Sur hasta la desembocadura.
Las aguas que discurren superficial y subsuperficialmente por las secuencias terrígenas de la Formación Pica Pica del Paleógeno, adquieren una elevada mineralización por el estado de agrietamiento y condiciones de flujo en estas rocas, las cuales son del tipo HCO3
- - Ca2+ > Mg2+. Entre los diferentes tipos de aguas kársticas se observan algunas propiedades que las
distinguen entre sí; por ejemplo, las aguas de cueva se distinguen por presentar poco contenido de iones y de CO2 disueltos, así como un elevado pH. Estas aguas alcanzan la saturación respecto a la calcita en condiciones de sistema cerrado con respecto al CO2.

Las aguas procedentes de los manantiales de la zona de saturación de los macizos kársticos no difieren mucho de las aguas que emergen de las resurgencias, pero sí presentan diferencias con las que proceden de la zona de saturación y circulación profunda. Estas últimas poseen una mineralización más alta, un mayor contenido de ion SO42- y de CO2 disuelto, así como una temperatura más elevada, también se caracterizan por presentar una menor fluctuación estacional de la temperatura y la dureza.
Las aguas de los pozos ubicados en el acuífero desarrollado en las calizas del Mioceno, en la llanura que se extiende al sur de la cuenca, cerca de la desembocadura del río Cuyaguateje, se caracterizan por su alto contenido de iones Cl- y Na+, a causa de la intrusión marina en el acuífero.
Estas diferencias en las propiedades químico-físicas de las aguas de la cuenca del río Cuyaguateje, han ofrecido la oportunidad para que, de una manera no subjetiva, se hayan podido caracterizar mediante el uso de métodos de clasificación numérica (Valdés, Fagundo y Pajón, 1981).
En el caso de los ríos superficiales y aguas de cuevas, éstas se hacen insaturadas respecto a los minerales carbonatados en el período de lluvia, en la cual es arrastrada la materia orgánica de la zona del suelo. Cuenca del río San Marcos
En esta región se llevaron a cabo, desde enero de 1984 hasta diciembre de 1989, investigaciones hidrológicas e hidroquímicas por un colectivo conformado por cubanos y polacos, con el objetivo de determinar cuantitativamente la intensidad de denudación química de los macizos kársticos, en condiciones de clima tropical.
Para realizar las investigaciones expuestas en el párrafo anterior, se instalaron en los alrededores del macizo del Pan de Guajaibón, equipos para la medición y registro de las precipitaciones, así como los niveles de los ríos respectivamente. Se realizaron registros sistemáticos de la temperatura, la humedad relativa y se hicieron mediciones frecuentes de caudales, además de las determinaciones químico-físicas de las aguas. Parte de los resultados ya se han publicado (Pulina et al, 1984; Fagundo et al, 1986; 1991; 1992; 1993; Rodríguez et al, 1989; 1991; 1992; 1993; Franco et al, 1987; Pulina y Fagundo, 1992; Tys, 1992). Las aguas estudiadas se relacionan a continuación:

Características hidrogeológicas Número
de datos Nombre del fichero
1. Aguas procedentes de sedimentos efusivo-sedimentario que drenan hacia los macizos kársticos.
6 24
EFUSIVO MAMEY
2. Aguas que discurren superficialmente por rocas ultrabásicas serpentinizadas antes de integrarse al principal colector de la cuenca.
6
CAJALBAN
3. Aguas kársticas de la zona de alimentación de los macizos carbonatados
18
ALIMENTA
4. Aguas de cuevas 25 9
CUEVAGUA RIOCUEVA
5. Aguas que emergen de surgencias kársticas. 28 23 8
CANILLA ANCON ANCONII
6. Manantiales kársticos 9 24 9
FERNANDO MILCUMBRES MANGUAJA
7. Aguas del río San Marcos 8
SANMARCO
8. Aguas procedentes del drenaje profundo 12 SANDIEGO

Figura 2.2. Variación de la composición química de las aguas de la cuenca del río San Marcos y sus alrededores en la Sierra del Rosario.

En virtud de la complejidad geológica e hidrogeológica de esta región, se originan aguas de diversos tipos y propiedades químico-físicas, como se ilustra en la figura 2.2 y se presenta en la tabla 2.2. N Nº Composición química relativa
HCO3- Cl- SO42- Ca2+ Mg2+ Na++K+
Mineraliza- ción
Σm TSS
Conductividad Eléctrica
Mín. Med. Máx. CV
1) 6 85 10 5 52 16 32 195 304 123 314 540 57,7 2) 24 86 11 3 50 17 33 272 401 125 429 565 26,8 3) 6 86 9 5 3 83 14 231 324 320 374 400 7,9 4) 18 89 9 2 74 11 5 245 360 230 395 530 21,9 5) 21 82 14 4 73 10 17 171 240 185 276 375 11,2 6) 9 83 12 5 75 8 16 231 326 340 372 486 13,2 7) 28 83 12 5 70 12 18 82 265 1210 293 360 11,6 8) 23 84 10 6 74 10 16 206 300 260 232 400 12,0 9) 9 86 8 6 76 7 17 270 378 365 435 520 12,7
10) 7 76 9 15 68 12 20 258 354 370 417 480 8,3 11) 14 78 8 14 76 7 17 300 434 430 483 560 7,9 12) 9 84 7 9 71 8 21 361 530 472 582 735 14,1 13) 6 81 9 10 66 16 18 255 371 380 411 445 6,5 14) 12 9 3 88 70 13 17 2820 2772 4391 4549 4233 2,2
Tabla 2.2. Propiedades químico-físicas de las aguas muestreadas sistemáticamente en las regiones kársticas montañosas de Cuba (cuenca del río San Marcos) Ficheros de datos: 1) EFUSIVO; 2) MAMEY; 3) CAJALBAN; 4) ALIMENTA; 5) CUEVAGUA; 6) RIOCUEVA; 7) CANILLA; 8) ANCON; 9) FERNANDO; 10) ANCONII; 11) MILCUMBR; 12) MANGUAJA; 13) SANMARCO; 14) SANDIEGO.
Las aguas de la zona de alimentación de los macizos carbonatados, constituidos por calizas de edad Cretácico, poseen por lo general, un contenido alto de CO2 y bajos valores de pH, se encuentran insaturadas respecto a la calcita, la dolomita y el yeso, aunque en épocas de seca, alcanzan valores de sobresaturación respecto a los dos primeros minerales. Estas aguas son del tipo bicarbonatadas cálcicas.
Las aguas que proceden de los acuíferos no kársticos, constituidos por secuencias de origen efusivo sedimentarias, son del tipo bicarbonatadas cálcico-sódicas con baja mineralización, dureza y contenido de CO2, las mismas se encuentran insaturadas respecto a los minerales calcita, dolomita y yeso. Sin embargo, las corrientes superficiales que se mueven por estos sedimentos, incrementan sensiblemente su mineralización y dureza, como resultado de su interacción con rocas carbonatado-terrígenas que afloran a través de su curso. También son del tipo bicarbonatadas cálcico-sódicas y, por lo general, se encuentran sobresaturadas respecto a la calcita y la dolomita.
En las condiciones expuestas en el párrafo anterior, penetran en los macizos carbonatados a través de ponores; en su interior se mezclan con aguas autóctonas que se infiltran por el macizo y, en la surgencia, aparecen con menor grado de dureza y mineralización, pero con un mayor caudal y con una composición química relativa típica de un agua bicarbonatada cálcica. Al salir de los macizos, se encuentran sobresaturadas respecto a la calcita y la dolomita, excepto en los períodos de lluvias intensas. En forma similar se comportan las

aguas de las exsurgencias, cuya alimentación es sólo de tipo autóctona. Las aguas de cueva, al igual que en la cuenca del río Cuyaguateje, poseen poco contenido de CO2, elevado pH y valores relativamente moderados de mineralización y dureza. En cuanto a su tipo, son semejantes a las restantes aguas kársticas. En relación a su comportamiento frente a los minerales carbonatados y los sulfatos, estas aguas se presentan, por lo general, sobresaturadas respecto a la calcita y a la dolomita e insaturadas con respecto al yeso.
En la región de Mil Cumbres, donde las aguas de las precipitaciones se infiltran a través de secuencias carbonatado-terrígenas, las emergencias poseen un elevado contenido de CO2,
así como una mineralización y dureza relativamente altas. El contenido de SO42- es superior a las de la zona del Pan de Guajaibón. Estas aguas permanecen insaturadas respecto a la calcita, la dolomita y el yeso. En su recorrido subaéreo liberan parte del gas disuelto, precipitando CaCO3 en los rápidos y cascadas.
Las aguas superficiales asociadas a las serpentinitas de las Alturas de Cajalbana, son del tipo bicarbonatadas magnesianas y muestran elevados valores de pH. La dureza y mineralización son relativamente altas. Un pozo profundo, perforado en estas secuencias, resultó ser de tipo artesiano, sus aguas poseen una temperatura más alta, elevado contenido de CO2 disuelto, bajo pH y elevadas mineralización y durezas. Las mismas son del tipo bicarbonatadas sódico-magnesianas y se encuentran insaturadas respecto a los minerales calcita, dolomita y yeso.
El río San Marcos, principal colector de esta región, refleja en sus propiedades químico-físicas el aporte de las distintas aguas que drenan la cuenca.
Los ríos y arroyos que corren hacia el sur sobre secuencias carbonatadas y terrígeno-carbonatadas de la Sierra del Rosario, tienen un comportamiento similar a los que lo hacen por litologías semejantes hacia la vertiente norte.
Las aguas que emergen en la zona de la falla Pinar desde profundidades relativamente grandes ofrecen rasgos distintivos con elevados valores de temperatura, contenido de CO2, dureza y mineralización. Estas aguas son del tipo sulfatadas cálcicas o cálcico-sódicas y algunas poseen reconocidas propiedades mineromedicinales.
En la figura 2.3 se ilustra el comportamiento de las aguas en relación a su capacidad para disolver los minerales calcita, dolomita y yeso.

Figura 2.3 Comportamiento químico-físico de las aguas del sector de la sierra del Rosario comprendido entre las alturas de Cajalbana y la sierra de la Güira, en relación a los índices de saturación de la calcita (RSC), la dolomita (RSD) y el yeso (RSY).
Como consecuencia de las lluvias, al igual que en la Sierra de los Órganos, muchas aguas que permanecen por lo general sobresaturadas, se hacen insaturadas, principalmente las superficiales.

Figura 2.4. Efecto de las precipitaciones (P) sobre el caudal (Q), la conductividad eléctrica (CE) y la composición química de las aguas de la resurgencia Canilla, cuenca del río San Marcos, en el período comprendido entre el 18 y el 28 de septiembre de 1984.
El efecto de las precipitaciones sobre los caudales y el quimismo de las aguas, se pudo medir en forma cuantitativa, en virtud de la colaboración prestada por los guardabosques del Plan Mil Cumbres, que acopiaron en forma sistemática la información de las lluvias y los niveles de los ríos en la base experimental.

En la figura 2.4, se muestra una correlación gráfica entre los parámetros hidroclimáticos y químico-físicos de las aguas de la resurgencia Canilla, una de las emergencias en las que se instaló uno de los limnígrafos, durante el período comprendido entre el 18 y el 28 de septiembre de 1984, en que tuvieron lugar fuertes aguaceros.
Como se puede apreciar en la figura 2.4, unas horas después de producirse las lluvias, se incrementó el caudal de las aguas y disminuyó la conductividad eléctrica. La concentración de todos sus iones, también disminuyó en valor absoluto, sin embargo, la composición relativa no experimentó cambios apreciables, las aguas se mantuvieron en todo momento, como bicarbonatas cálcicas.
El efecto de las lluvias es más sensible sobre el pH, el contenido de CO2 y los valores de RSC, RSD y RSY. Las aguas superficiales experimentan variaciones más apreciables en el tipo de agua, por efecto de las precipitaciones, aunque este efecto sólo se observa en los primeros momentos transcurridos después de caídas las lluvias. En ese instante las aguas tienen una gran capacidad para disolver los minerales, pero este proceso es rápido y al cabo de un breve tiempo, adquieren un alto contenido de iones disueltos alcanzando la saturación. Esto se puso de manifiesto en los experimentos efectuados con lisímetros. Meseta del Guaso
Durante el período comprendido entre el 12 de enero y el 12 de febrero de 1988, se llevó a cabo una expedición científica cubano-búlgara a la meseta del Guaso, Guantánamo, con el objetivo de realizar estudios polidisciplinarios relacionados con los recursos hídricos de esa región. Los resultados de la caracterización hidrológica e hidroquímica han sido publicados en una revista especializada (Rodríguez, Fagundo y Spassov, 1991). Estas aguas se relacionan a continuación: Características hidrogeológicas Número
de datos Nombre del fichero
1. Aguas de lluvia y de lisímetros instalados con fines experimentales.
5 5
LLUVIA LISÍMETRO
2. Ríos superficiales kársticos 13 GUASO1 3. Resurgencias y aguas de cueva 27 GUASO2 4. Manantiales de la zona de alimentación del macizo kárstico 5 GUASO3 5. Manantiales de la zona de emisión del macizo kárstico. 17 GUASO4
En la meseta del Guaso, las aguas se discurren en forma superficial o subterránea, a través de grietas y conductos kársticos, fundamentalmente en la zona de contacto entre las formaciones carbonatadas de Charco Redondo y Yateras, ambas del Paleógeno, además, intercalados con los carbonatos, aparecen sedimentos de tipo impermeables. Al pie de las elevaciones kársticas, tiene lugar uno de esos contactos tectónicos, originando una serie de manantiales, de cuyas aguas nacen los ríos Bano y Guaso.
En el macizo del Guaso se determinaron cuatro tipos de aguas, atendiendo a la clasificación hidroquímica de Schoukarev: del tipo HCO3
- > Cl- - Na+, correspondientes a las aguas de lluvia; del tipo HCO3
- > Cl- - Na+ > Ca2+ , correspondientes a las aguas de escurrimiento superficial (tomadas en los lisímetros); aguas HCO3
- - Ca2+ , correspondientes a las aguas kársticas en su conjunto; y del tipo HCO3
- > SO42- - Ca2+ > Na+, propias de los
manantiales no kársticos (ver tabla 2.3).

N Nº Composición química relativa
HCO3
- Cl- SO42- Ca2+ Mg2+ Na++K+
Mineraliza- ción
Σm TSS
Conductividad eléctrica
Mín. Med. Máx. CV 1) 5 51 49 0 17 13 70 20 25 23 32 44 21,32) 5 63 37 0 40 9 51 31 35 34 49 65 21,33) 13 89 11 0 76 17 7 171 249 255 276 356 11,44) 27 90 10 0 84 12 4 206 270 245 333 485 16,25) 5 91 9 0 81 8 11 256 358 357 413 511 8,9 6) 17 92 9 0 82 8 10 234 316 255 378 419 9,1
Tabla 2.3. Propiedades químico-físicas de las aguas muestreadas sistemáticamente en las regiones kársticas montañosas de Cuba (meseta del Guaso). Ficheros de datos: 1) LLUVIA; 2) LISIMETR; 3) GUASO1; 4) GUASO2; 5) GUASO3; 6) GUASO4
Durante la expedición se pudo determinar, en forma cuantitativa, el efecto de las lluvias sobre el quimismo de las aguas y la evolución de éstas a partir de las lluvias, hasta alcanzar la saturación respecto a la calcita (Figura 2.5).
En general, durante los aguaceros se incrementan los caudales y disminuye el contenido de
iones disueltos (Figura 2.6). Las aguas arrastran la materia orgánica y enriquecen el contenido de CO2 original extraído de la atmósfera. Estas aguas agresivas disuelven rápidamente los minerales carbonatados (calcita y dolomita), elevando su dureza y mineralización, los cuales son

directamente proporcionales entre sí. No obstante, su composición relativa experimenta poca variación, es decir, se mantiene en todo momento su mismo tipo hidroquímico.
Los cambios explicados en el párrafo anterior producen una disminución del valor del pH (Figura 2.7). Entre la ocurrencia de las lluvias y el incremento de la dureza de las aguas, la mineralización, así como en la conductividad eléctrica se experimenta un ligero desplazamiento de tiempo, que es el que necesita el agua para disolver los minerales con el contenido adicional de CO2.

Llanura kárstica meridional del occidente de Cuba
En la literatura geomorfológica, comúnmente se hace referencia al karst tropical, tomando como tipo aquellos paisajes kársticos desarrollados en las regiones montañosas, conocidos como karst de mogotes o tower karst y karst cónico o cockpit karst. Al primer tipo geomorfológico, están asociados los macizos karstificados de la cuenca del río Cuyaguateje y el Pan de Guajaibón y al segundo, la zona de Mil Cumbres, en la cuenca del río San Marcos y la meseta del Guaso. Sin embargo, los acuíferos kársticos más ricos en nuestro país, en cuanto a su potencial hídrico, no son precisamente los de montaña.
La mayor parte de los recursos hídricos subterráneos y más valiosos, se encuentran en los acuíferos desarrollados en las calizas miocénicas de las llanuras, principalmente las que se encuentran en la parte meridional del occidente de Cuba, que abarca las provincias de Pinar del Río, La Habana y Matanzas; así como en otras áreas kársticas del centro del país.
Con el objetivo de ilustrar el comportamiento hidroquímico de estas regiones, se han seleccionado algunas zonas representativas de este territorio; por ejemplo, la región de San Antonio de los Baños, en la provincia La Habana; un sector de la llanura meridional de Pinar del Río y otro, situado al sur de la provincia de Matanzas. El primer sitio tipifica el comportamiento de los karst llanos de cuencas cerradas, limitadas por secuencias más o menos impermeables. Los otros sitios seleccionados, son representativos del karst litoral con descarga libre al mar, los cuales se encuentran afectados por la intrusión marina.
Región de San Antonio de los Baños
El comportamiento químico-físico de las aguas de esta región fue estudiado por Fagundo y Valdés (1975), Valdés y Fagundo (1980), quienes realizaron muestreos mensuales durante el año hidrológico 1974, las cuales se relacionan a continuación: Características hidrogeológicas Número de
datos Nombre del fichero
Río San Antonio de los Baños, cerca del Sumidero 10 SANANTON Pozos ubicados en la zona limítrofe entre las cuencas Ariguanabo y Sur
10 10 10 10
PADIRAC TALLER CASCARRA LACEIBA
El área donde se efectuaron los estudios, es la inmediata al poblado de San Antonio de los
Baños en la provincia La Habana. La zona constituye uno de los sectores septentrionales limítrofes de la región hidrogeológica Llanura Meridional Habana-Matanzas (Molerio, 1974), la cual se encuentra situada entre las cuencas de Ariguanabo y Sur. El macizo está integrado por series carbonatadas y carbonatado-terrígenas que yacen concordantes entre sí, ocupando la porción carbonatada la parte superior del corte.
En la tabla 2.4, se muestran las propiedades químico-físicas de estas aguas. Las mismas se caracterizan por presentar más alto contenido de CO2 y de minerales disueltos que las aguas kársticas de las regiones montañosas. Esto se debe, posiblemente, a que la generación de CO2 en los suelos de las llanuras es más abundante y la disolución de los minerales tiene lugar allí en condiciones de sistema abierto respecto al CO2, puesto que las calizas del Mioceno, en la zona

del suelo poseen muchas oquedades que son ocupadas por el aire y el CO2. Las lluvias al infiltrarse en estas circunstancias se ponen en contacto con una elevada presión de CO2.
N Nº Composición química relativa
HCO3
- Cl- SO42- Ca2+ Mg2+ Na++K+
Mineralización
Σm TSS
Conductividad eléctrica
Mín. Med. Máx. CV 1) 10 74 19 7 76 10 14 368 477 509 593 703 9,7 2) 19 76 20 4 51 14 25 101 137 89 163 323 36,6 3) 33 86 12 2 85 6 9 162 223 221 265 361 13,8 4) 16 87 10 3 68 13 19 208 289 185 337 534 29,2 5) 5 89 9 2 85 6 9 192 275 269 310 397 14,8
Tabla 2.4. Propiedades químico-físicas de las aguas muestreadas sistemáticamente en las llanuras kársticas de Cuba (región de San Antonio de los Baños) Fichero de datos: 1) SANANTON; 2) PADIRAC; 3) TALLER; 4) CASCARRA;5) LACEIBA
Si se comparan las aguas, correspondientes a un perfil de pozos ubicados de norte a sur, se observa que la relación iónica Ca2+/ Mg2+ va decreciendo con la distancia, lo cual está acorde con las condiciones geológicas del corte; por ejemplo, el pozo La Ceiba, situado en el extremo meridional, donde el grosor de la serie carbonatado-terrígena (algo dolomitizada) es mayor, presenta el más bajo valor medio de esta relación, mientras que el pozo El Padirac y el propio río San Antonio, situados cerca del extremo norte del perfil, presentan los valores medios más elevados, donde precisamente las calizas se encuentran menos dolomitizadas. Llanura meridional de Pinar del Río
El comportamiento químico-físico de las aguas de este territorio, que comprende unos 4 600 km2 se ha discutido ampliamente en la literatura (Arellano, 1986; Arellano y Fagundo, 1991; Fagundo et al, 1991; 1992; 1993), con el objetivo de obtener criterios acerca de la zonación geoquímica de estos acuíferos. Estas aguas se relacionan a continuación: Características hidrogeológicas Número
de datos Nombre del fichero
Pozos de observación, utilizados para el control de la salinidad, ubicados en un acuífero kárstico afectado por la intrusión salina (muestras tomadas por batometría)
75 27
BATPRI BATPRII
En esos trabajos se emplearon diferentes parámetros hidroquímicos, relaciones iónicas y
los índices de saturación de las aguas respecto a los minerales calcita, dolomita y yeso, tomando como base los sistemas de equilibrios químicos que se establecen en el proceso de disolución de los minerales, como resultado de la interacción de las aguas naturales con las rocas kársticas.
Desde el punto de vista hidrológico, se reconocen tres cuencas, ubicadas en las porciones occidental, central y oriental de ese territorio. El horizonte acuífero principal en la llanura es del Mioceno, compuesto por una secuencia carbonatada y terrígeno-carbonatada, en la que predomina la karstificación, sobre todo en la porción oriental. El karst se encuentra confinado por una cobertura de sedimentos del Cuaternario, de baja permeabilidad y de constitución areno-arcillosa, cuyo espesor medio es de 40 m, pudiendo alcanzar hasta 80 m. La zona de alimentación del acuífero se encuentra ubicada en las regiones montañosas, así como premontañosas y la descarga de éstas es libre al mar. Las

propiedades químico-físicas de las aguas de estos acuíferos dependen de las características del drenaje en la región. De esta manera, en la red de pozos establecida para la observación sistemática de la calidad y el régimen de las aguas subterráneas, se aprecia que las mismas se encuentran estratificadas y reflejan la composición química de los diferentes horizontes del acuífero. Los horizontes más someros presentan aguas del tipo bicarbonatadas cálcicas, mientras que los más profundos, reflejan la composición del agua de mar (del tipo cloruradas sódicas). En las zonas intermedias las aguas suelen ser de los tipos HCO3
- > Cl--Ca2+ > Na+ o Cl- > HCO3
- - Na+ > Ca2+, en dependencia del grado de mezcla (Figura 2.8).
Figura 2.8 Composición química de las aguas de pozos en el acuífero kárstico de la llanura meridional de Pinar del Río.
Durante el año la composición química relativa varía en dependencia de la presión hidrostática en los acuíferos: en los meses más lluviosos se recargan, mientras que en los períodos más secos y cuando es más intensa la explotación éstos se abaten, aumentando el contenido de Cl
- y Na
+ en las aguas (Fagundo y Arellano, 1991; Fagundo et al, 1993). Estas características
obligan a mantener una política de vigilancia en cuanto a la explotación de dichos acuíferos, con el objetivo de preservar la calidad de sus aguas.
En los últimos 10 años, se ha detectado en algunos pozos del territorio, una tendencia a la salinización de las aguas, variando su composición química de HCO3
- > Cl--Ca2+ > Na+ a Cl- > HCO3
- - Na+ > Ca2+ . De seguir esta tendencia, la calidad de las aguas empeorará, tranformándose su tipo en clorurada sódica.

La disolución de los minerales en todo estos acuíferos confinados, tiene lugar en condiciones de sistema cerrado respecto al CO2, puesto que en esas condiciones las aguas subterráneas en su movimiento no reciben nuevos aportes de CO2. Un aumento en la disolución de los carbonatos, con respecto a lo previsto en el proceso de interacción agua-roca, tiene lugar en esta región, como consecuencia del denominado efecto salino. En virtud del mismo, se produce un aumento en la capacidad de disolución de los minerales, por la presencia en las aguas, de iones no comunes al equilibrio de los carbonatos, tales como el Cl- y el Na+ aportados por el mar.
Las aguas de los pozos en esta región se encuentran, por lo general, saturadas respecto a los minerales calcita y dolomita e insaturadas respecto al yeso (Figura 2.9). Un hecho curioso observado en algunos pozos es que a medida que aumenta su profundidad y, por ende, su grado de mezcla con el agua de mar, los valores de RSC y RSD indican que las aguas se van haciendo saturadas hasta alcanzar un límite de sobresaturación, después del cual se van haciendo de nuevo menos saturadas o insaturadas (Figura 2.10 ).
Este comportamiento (Figura 2.10) puede estar relacionado con los resultados obtenidos por Wigley y Plummer (1976) en experimentos a nivel de laboratorio con mezclas de aguas kársticas con agua de mar. Estos investigadores encontraron, que cuando disoluciones de minerales de diferente composición se mezclan con agua marina, las molalidades y actividades de los iones individuales en la mezcla, a menudo, son funciones no lineales de sus valores. Esta no linealidad es significativa en la determinación de los niveles de saturación.
Figura 2.9 Comportamiento químico-físico de las aguas de la llanura meridional de Pinar del Río en relación a las relaciones de saturación de la calcita (RSC), la dolomita (RSD) y el yeso (RSY).
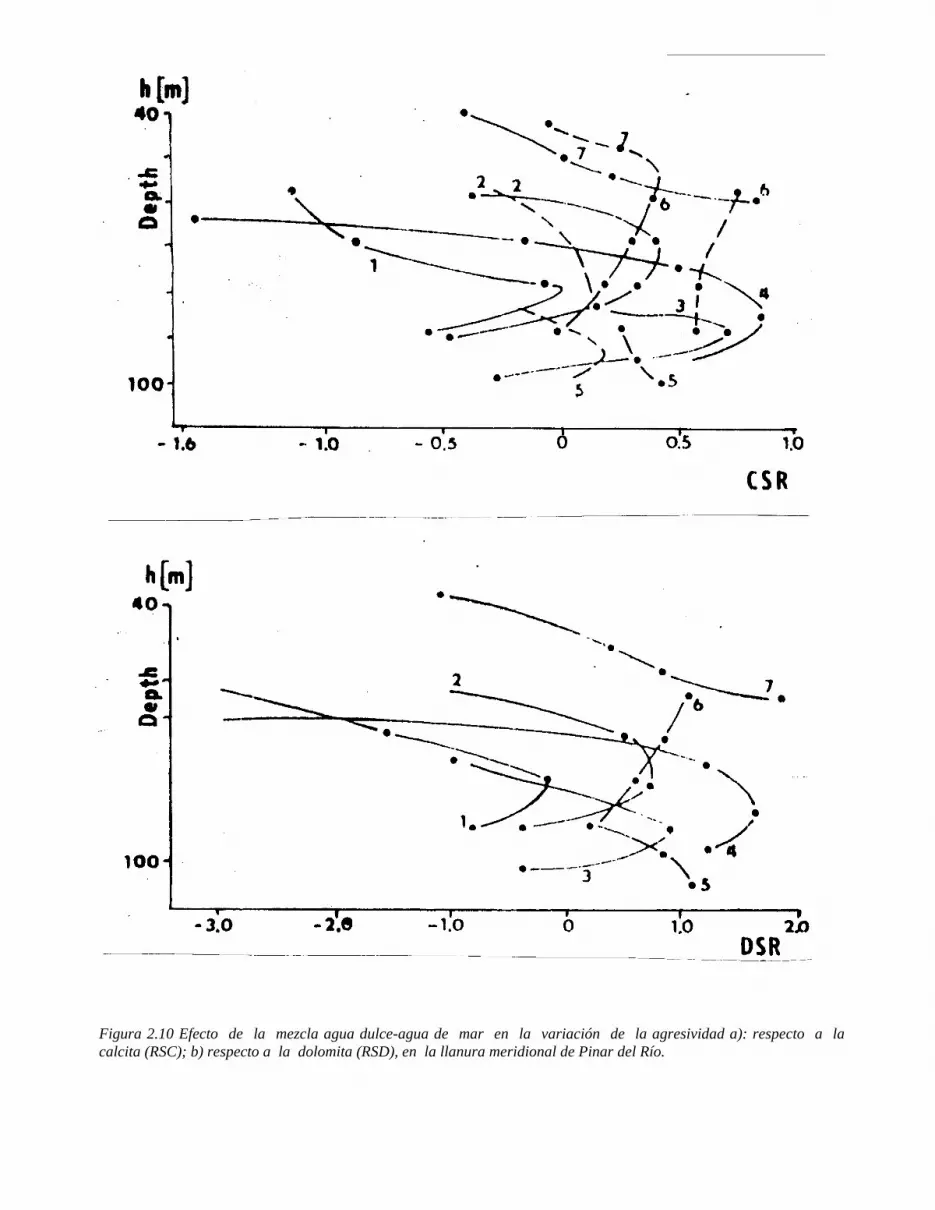
Figura 2.10 Efecto de la mezcla agua dulce-agua de mar en la variación de la agresividad a): respecto a la calcita (RSC); b) respecto a la dolomita (RSD), en la llanura meridional de Pinar del Río.

Karst litoral del sur de Matanzas
Esta región es representativa también del karst litoral, pero se diferencia de la llanura meridional de Pinar del Río, en que en este último los acuíferos se encuentran confinados, mientras que en el sur de Matanzas, las secuencias carbonatadas afloran en la superficie. El suelo es relativamente escaso y el drenaje, al igual que en el resto de la llanura litoral del occidente de Cuba, es libre, con descarga al mar. Las aguas de esta región se relacionan a continuación: Características hidrogeológicas Número
de datos Nombre del fichero
Aguas utilizadas para el regadío en las plantaciones citrícolas. 9 9 9 8 7
TRINCHER COCODRIL INFIERNO CUELLES AUSTRALI
Aguas tomadas por batometría en pozos de observación para el control de la salinidad.
47 14
BATOMMI BATOMMII
En la tabla 2.5 se presentan los valores medios, mínimos, máximos, así como del coeficiente de variación de los principales indicadores geoquímicos de estas aguas.
N Nº Composición química relativa
HCO3- Cl- SO4
2- Ca2+ Mg2+ Na++K+
Mineralización
Σm TSS
Conductividad eléctrica
Mín. Med. Máx. CV
1) 5 73 7 20 75 22 3 329 418 453 531 536 8,3 2) 19 84 8 8 71 14 15 252 350 138 406 450 23,1 3) 21 68 8 24 60 21 19 318 485 473 513 575 5,0 4) 8 91 6 3 50 36 14 318 429 399 513 625 16,0 5) 11 73 9 18 66 19 15 233 210 78 375 556 31,6 6) 6 85 10 5 52 16 32 195 304 123 314 540 57,7
Tabla 2.5. Propiedades químico-físicas de las aguas muestreadas sistemáticamente en las llanuras kársticas de Cuba (sur de Matanzas) Fichero de datos: 1) TRINCHER; 2) COCODRIL; 3)INFIERNO; 4) CUELLES; 5) AUSTRALI; 6) BOLONDRO
Durante el período comprendido entre enero de 1986 y enero de 1987, en el plan citrícola " Victoria de Girón", en Bolondrón, Matanzas, se llevaron a cabo estudios sistemáticos, con frecuencia de muestreo mensual, con el objetivo de estudiar el comportamiento químico-físico de las aguas y encontrar modelos de correlación matemática entre la concentración iónica y la conductividad eléctrica de las mismas, con vistas al posterior monitoreo y control de su calidad, mediante simples mediciones de conductividad eléctrica y el uso de las correlaciones. En general se encontró que las ecuaciones de mejor ajuste eran de segundo grado para el HCO3
- y el Ca2+ y de primer grado para el resto de los iones, cuando se procesaban todos los datos (Fagundo et al, 1993). Sin embargo, si los datos son separados por tipos hidroquímicos y procesados por separado, las ecuaciones de mejor ajuste correspondientes para cada ion son de primer grado.

En el marco de estos estudios, se puso de manifiesto que la disolución de los minerales carbonatados tiene lugar aquí, en condiciones de sistema abierto respecto al CO2 , se observó un elevado contenido de este gas disuelto en las aguas. La mineralización media y la dureza son elevadas (ver tabla 2.5) y su composición relativa, al igual que en la región litoral homóloga en Pinar del Río, refleja la mezcla del acuífero y del mar.
La estratificación de las aguas se hace más evidente en las muestras tomadas en condiciones estáticas por batometría, pero el efecto de la intrusión marina también se acusa en régimen dinámico, al someterse a explotación continua un pozo en la región. Observe en la figura 2.11, como al comienzo del bombeo en las aguas se cumple Ca2+ > Mg2+, mientras que a partir de las 24 h se invierte esta relación (Mg2+ > Ca2+ ), lo cual refleja el mayor aporte del agua marina, que se caracteriza por poseer un contenido mayor de Mg2+ que de Ca2+.

Figura 2.11 Variación de la composición química de las aguas de un pozo sometido a explotación continua en el plan citrícola "Victoria de Girón", en Bolondrón (Matanzas).

Se ha observado en nuestro país que al procesar los datos hidroquímicos (Fagundo, 1990 a, b), muestreados de manera sistemática en un mismo sitio, mediante el sistema SAMA, se obtienen ecuaciones de mejor ajuste de tipo lineal para todos los iones, cuando la litología es el factor dominante en el modo de adquisición de la composición química de las aguas (ver tabla 2.6). En estos casos la composición iónica absoluta suele variar, pero no la composición química relativa, por lo que las aguas en todo momento, tienden a mantener un mismo patrón hidrogeoquímico.
Los datos correspondientes al fichero denominado Trinchera y a las dolinas Cocodrilo, Infierno y Cuelles, así como de otros pozos (2.6), ajustaron mejor a ecuaciones de segundo grado para los iones bicarbonato y calcio y de primero para el resto de los iones. Los coeficientes de las ecuaciones correspondientes eran muy similares entre sí, lo cual permitió agrupar todos los datos (en un nuevo fichero que se denominó BOLONDRON), al ajustarse dieron ecuaciones similares a las obtenidas al ajustar los datos por separado. Sólo los datos hidroquímicos correspondientes al canal Australia, donde la composición química está más controlada por la intrusión salina, ajustaron mejor a ecuaciones de primer grado para todos los iones. Estos resultados permiten establecer un régimen de control de la calidad de las aguas en esta región, mediante mediciones de conductividad eléctrica y el uso de dos juegos de ecuaciones de correlación matemáticas (Tabla 2.6).

Tabla 2.6. Resultados del ajuste mediante polinomios de hasta quinto grado (pasan por el origen de coordenadas) de los datos hidroquímicos correspondientes a las aguas kársticas de Cuba tomadas como ejemplo en este trabajo
Fichero de datos
Factor
Valores de los coeficientes de la ecuación polinómica HCO3
- Cl- SO42- Ca2+ Mg2+ Na++K+ IS
PIZARRAS 10-3 9,414 3,430 1,365 3,596 1,637 8,976 0,776 SUMRES 10-3
10-6 8,548 3,319 0,493 5,670 1,522 3,679 0,821 -7,034
CUEVAS 10-3 9,529 1,304 0,285 5,876 2,552 2,690 0,715 RESK 10-3 9,636 1,085 0,361 7,508 1,451 2,124 0,865 PALMERO 10-3 9,862 0,990 0,251 9,456 0,625 1,021 0,826 PICAPICA 10-3 7,654 0,774 2,036 7,901 2,289 0,274 0,896 MANANTIA 10-3 8,980 0,868 0,835 7,610 1,514 1,559 0,857 BANITOS 10-3 8,570 1,081 3,039 7,620 2,739 2,331 0,839 GUAYABO 10-3 9,937 0,716 0,320 5,499 3,926 1,547 0,877 CUYAPOR 10-3 8,039 0,957 1,935 7,249 2,002 1,680 0,838 EFUSIVO 10-3 10,676 1,132 0,662 6,341 2,091 4,039 0,838 MAMEY 10-3 10,133 1,316 0,350 5,936 1,951 3,911 0,901 CAJALBAN 10-3 11,009 1,171 0,574 0,326 10,585 1,884 0,949 ALIMENTA 10-3 10,401 1,079 0,202 8,639 1,296 1,747 0,907 CUEVAGUA 10-3 9,721 1,718 0,447 8,694 1,154 2,038 0,857 RIOCUEVA 10-3 9,849 1,479 0,634 8,996 0,974 1,992 0,887 CANILLA 10-3 9,797 1,370 0,573 8,212 1,396 2,132 0,917 ANCON 10-3 9,782 1,197 0,716 8,651 1,214 1,831 0,919 FERNANDO 10-3 9,856 0,986 0,619 8,721 1,408 1,284 0,902 ANCONII 10-3 9,585 1,108 1,863 8,485 1,489 2,582 0,862 MILCUMBR 10-3 9,058 0,946 1,615 8,821 0,867 1,932 0,905 MANGUAJ 10-3 10,084 0,804 1,118 8,564 0,910 2,532 0,897 SANMARCO 10-3 10,322 1,109 1,322 8,204 2,166 2,383 0,880 SANDIEGO 10-3 0,781 2,276 7,867 6,250 1,156 1,518 0,978 LISIMETR 10-3 6,138 3,491 0,000 4,379 0,746 4,504 0,796 GUASO1 10-3 10,412 1,200 0,000 8,927 1,952 0,733 0,890 GUASO2 10-3 9,367 1,027 0,000 8,819 1,140 0,435 0,914 GUASO3 10-3 9,649 0,902 0,000 8,550 0,884 1,667 0,894 GUASO4 10-3 9,788 0,796 0,000 8,683 0,889 1,018 0,897 SANANTON 10-3 7,909 2,092 0,724 7,958 1,083 1,684 0,869 PADIRAC 10-3
10-5 7,214 2,204 0,756 14,827 0,992 1,734 0,884 -1,104
TALLER 10-3 10-5
7,375 2,204 0,755 7,673 0,963 1,643 0,889 -1,371
CASCARRA 10-3
10-611,527 2,531 1,559 7,826 1,241 0,997 0,862 -5,082
LACEIBA 10-3 9,573 1,185 0,686 7,266 3,404 0,774 0,913 TRINCHER 10-3
10-610,886 4,167 0,574 7,828 2,450 3,523 0,929 -5,122 -3,949
COCODRIL 10-3
10-69,696 4,484 0,477 5,966 2,769 3,812 0,943 -3,660 -1,854
CUELLES 10-3
10-69,961 4,071 0,608 9,122 2,589 3,529 0,939 -4,245 -4,844
AUSTRALI 10-3 4,092 4,663 1,264 3,906 2,585 3,529 0,805 BOLONDRO 10-3
10-69,885 4,180 0,538 8,371 2,585 3,639 0,906 -3,965 -3,949
El mismo procesamiento utilizado en el párrafo anterior se puede realizar con los datos hidroquímicos de las muestras tomadas usando batómetros (hidromuestreadores verticales), tanto en las llanuras kársticas litorales de Pinar del Río, La Habana, Matanzas y otras regiones de Cuba. En estos casos, sin embargo, es necesario tener en cuenta el modo en que evolucionan la aguas de acuerdo al grado de dolomitización inicial del acuífero y cómo cada pozo reconoce los

diferentes patrones hidrogeológicos, en un rango de conductividad eléctrica determinado (Fagundo y Rodríguez, 1991).
Cuando existe mezcla de aguas o cambio del quimismo, tanto en valor absoluto como relativo, las ecuaciones de mejor ajuste pueden ser de tipo no lineal (por lo general de segundo grado), para al menos algunos de los iones. Esto sucede, por ejemplo, en las aguas utilizadas para el abasto y el regadío (Fagundo et al, 1993), en la zona kárstica litoral afectada por la intrusión marina. Intensidad de la denudación química
Los estudios relacionados con la medición cuantitativa del proceso de karstificación son escasos en la literatura cubana relacionada con los recursos hídricos.
Entre los primeros intentos, en este sentido, se destacan los resultados obtenidos en la región de San Antonio de los Baños por Valdés y Fagundo (1980). Estos investigadores, utilizando las relaciones empíricas de dependencia matemática, encontradas por Barner y Morse (1974), entre la velocidad de disolución del carbonato de calcio, el pH del agua y el correspondiente a la saturación, para distintos valores de presión de CO2, calcularon la intensidad de disolución de los carbonatos para las aguas de esta región en el período estudiado. Según estos cálculos, la velocidad de disolución varió entre 0 mg/cm2 por año, en los meses de menos lluvia y 220 mg/cm2 por año, en los meses más lluviosos, con un valor medio de 31,6, mg/cm2 por año.
Durante las investigaciones sistemáticas llevadas a cabo en la cuenca del río Cuyaguateje en el período 1978-1980 (Fagundo et al, 1981), también se pudo calcular cuantitativamente la intensidad conque se disuelven los macizos kársticos. Los cálculos arrojaron, que tomando como cierre de la cuenca la estación de aforo Portales II y el río Portales, la cantidad de CaCO3 eliminado de la cuenca, es del orden de 16,3 kg/s y teniendo en cuenta la densidad de las calizas, así como suponiendo un régimen de lluvias estable en el año, similar al existente durante los dos días del mes de agosto de 1979, en los cuales se realizaron los experimentos; además de una similar composición química de las aguas, la cifra anterior correspondería a un equivalente en disolución anual de 10 525 m3 de roca, lo que representa un cubo de un volumen aproximado de 60 m3 por cada lado.
Otras determinaciones similares, se realizaron en el marco de una expedición científica cubano-búlgara a la meseta del Guaso. En esa ocasión se determinó que la intensidad de denudación química de las aguas en esta región kárstica, es del orden de 62 y 28 m3/km2 por año para los ríos Bano y Guaso respectivamente.
Estudios cuantitativos sobre la denudación química, se han efectuado con mayor sistematicidad y precisión a partir de 1984, en el polígono experimental del Pan de Guajaibón (Pulina et al, 1984). Se instalaron equipos que permitieron cuantificar las lluvias y el nivel de los ríos, además de realizar de manera sistemática, mediciones de caudales y del quimismo de las aguas.
Durante la expedición científica cubano-polaca de 1984, se calculó una intensidad de denudación química anual correspondiente a 41,5, 24,7 y 48,0 m3/km2 por año para los sistemas Ancón, Canilla y el Pan de Guajaibón en su conjunto.
Los resultados más confiables, acerca de la magnitud de la denudación química en el karst tropical de Cuba, corresponden a las mediciones realizadas en el Pan de Guajaibón en el

período 1984-1989 (Rodríguez y Fagundo, 1991; 1992; Pulina y Fagundo, 1992; Rodríguez et al, 1993), los cuales se presentan en la tabla 2.7.
Período de
observaciones Sistema S (km2) Período Q
(m3/s) T
(mg/L) dT
(mg/L) D
m3/km2.año Año hidrológico noviembre 1984-octubre 1985 (1)
Canilla
Ancón
Macizo Pan de
Guajaibón
5,1
4,35
9,45
Seco Lluvioso Año Seco Lluvioso Año Seco Lluvioso Año
0,079 0,121 0,100
0,181 0,232 0,207
0,260 0,353 0,307
186 174 181
206 206 206
187 191 194
166 154 161
186 186 186
177 117 174
16 24 48
49 63
112
31 41 72
Enero 1984-abril 1989 (2)
Canilla
Ancón
Macizo Pan de
Guajaibón
5,1
4,35
9,45
Seco Lluvioso Anual Seco Lluvioso Anual Seco Lluvioso Anual
0,071 0,175 0,123
0,173 0,322 0,248
0,244 0,497 0,371
186 174 181
2,06 2,06 2,06
197 191 194
166 154 161
186 186 186
177 171 174
15 34 49
46 87
133
28 57 85
Tabla 2.7. Magnitud de la denudación química en el macizo Pan de Guajaibón. Ref.: (1)- Rodríguez et al, 1989; (2)- Rodríguez, Fagundo y Pulina (1993).

Capítulo 3. Comparación entre las regiones de clima polar y tropical

Clasificación numérica
Uno de los aspectos más importantes en este trabajo es el análisis comparativo entre las aguas polares y tropicales desde el punto de vista de su comportamiento químico-físico. Con el objetivo de destacar los grupos naturales de aguas y su interrelación, se emplearon métodos de análisis exploratorio de datos, específicamente de clasificación numérica no supervizada.
Con esta finalidad, las aguas polares de Spitsbergen fueron comparadas con las aguas tropicales de la región del Pan de Guajaibón y sus inmediaciones en la Sierra del Rosario, Cuba, desde el punto de vista de las variables siguientes: conductividad eléctrica, CO2, HCO3
-, Cl-, SO4-2, Ca+2, Mg+2 y Na+ + K+, las cuales fueron estandarizadas a fin de que tuviesen
todas el mismo peso. Se empleó el método de Ward (1963), tomando como medida de disimilaridad el cuadrado de la distancia euclideana. El resultado de la aplicación de estos métodos consiste en un dendrograma que muestra los sucesivos pasos de función del algoritmo y los niveles del mínimo, el incremento de la función de suma de cuadrados de error dentro del grupo.
En una primera aproximación, 468 muestras de agua de Spitsbergen y Cuba, fueron procesadas respecto a las variables mencionadas en el párrafo anterior, reflejando la presencia de 3 grupos generales bien diferenciados, pero desproporcionados respecto al número de sus componentes y a su homogeneidad interna (Figura 3.1). El primero minoritario (sólo 10 elementos) y muy heterogéneo, está compuesto por 6 aguas cubanas: pozo El Sitio (3 muestras), manantial de Bermejales, manantial de San Diego y manantial de la Cueva de los Portales, y por 4 aguas polares: Treskelen (2 muestras, emergencia Kvisla y manantial Linneo). La característica hidrogeológica de las aguas de este grupo es la de tratarse de manantiales termales (es de destacar que la temperatura no fue incluida en el análisis), mayormente sulfurosas, en algunos casos de aguas provenientes de zonas afectadas por la intrusión salina; así como las muestras polares de la zona de Treskelen. Desde el punto de vista químico-físico poseen una elevada mineralización total (reflejada por la conductividad eléctrica) con un contenido predominante de sulfatos y cloruros, tratándose de aguas de los tipos sulfatada-cálcica y clorurada-sódica.

Figura 3.1. Dendograma que muestra los grupos numéricos determinados por análisis de "cluster" (enjambres) al tratar 468 datos hidroquímicos de Spitsbergen y Cuba.
Un caso interesante es el de la emergencia Kvisla (muestra 104, ver figura 1.14), la cual no responde a ninguno de los tipos anteriores. El agua más similar a ella es la muestra cubana del manantial de la Cueva de los Portales, lo cual se puede explicar por el efecto crioquímico causante de la elevada concentración iónica en la muestra, tomada en los días cercanos a la congelación total de la emergencia. En cualquier caso, el común denominador de este grupo es la muy alta mineralización a causa de diversos factores hidrogeológicos y químico-físicos, reflejados por la heterogeneidad interna del mismo.
El segundo grupo, compuesto por 35 elementos corresponde sólo a aguas de la zona polar de Rasstupet, provenientes de manantiales afectados por la intrusión salina en zonas de descarga próximas al fiordo Hornsund. Estas aguas son de una mineralización que varía en margen amplio, cuya composición es eminentemente clorurada-sódica, reflejando el patrón del agua de mar.
El tercer grupo, integrado por 423 muestras, contiene a nivel menor dos subgrupos, en cada uno de los cuales predominan las aguas de Cuba y Spitsbergen respectivamente, con algunas interdigitaciones.
La desproporcional disimilaridad entre las aguas del primer, segundo y tercer grupo, enmascaran la estructura interna de este último. Por ello se realizó un segundo paso de análisis, donde se analizaron 410 muestras eliminando del conjunto aquellas muestras del primer grupo causantes de la deformación del dendrograma, a fin de estudiar el componente mayoritario de la información.
En el dendrograma obtenido (Figura 3.2) se evidencia la presencia de dos grupos: uno compuesto por aguas cubanas en mayor proporción que las polares y otro con una composición inversa, es decir, mayor cantidad de aguas polares que cubanas.

El primer grupo corresponde básicamente a las aguas de Cuba, con su alta mineralización,
su tipo predominante bicarbonatada-cálcica, así como con el claro reflejo de las particularidades geológicas e hidrogeológicas de las distintas regiones. Las aguas polares que aparecen aquí se diferencian netamente de las aguas cubanas y no se "interdigitan" con éstas, expresándose como una clara unidad. Las muestras que presentan este comportamiento son: las de la zona del glaciar Aldegonda, el lago Linneo y las provenientes de los lagos morrénicos, las cuales presentan una mineralización relativamente mayor que el resto de las aguas de Spitsbergen.
El dendrograma refleja la diferenciación de las aguas cubanas (mayormente bicarbonatadas-cálcicas) con respecto a las polares (sulfatada-cálcica), en esta comparación no se consideran la zona de intrusión marina de Rasstupet, ni las de los manantiales termales, los cuales se encuentran muy mineralizados.
En el segundo grupo resulta interesante no sólo el que varias aguas cubanas manifiesten comportamiento "polar", sino también el que lo hagan con un muy elevado nivel de similitud respecto a aguas del medio ambiente ártico. A pesar de que éstas presentan ese comportamiento, poseen diferente naturaleza hidrogeológica (sumideros provenientes de rocas no kársticas, depósitos subterráneos y gours en cavernas, a partir de flujos rápidos en la zona de aeración del macizo kárstico, así como arroyos superficiales en zonas de rocas efusivas). Todas tienen como común denominador una relativamente baja mineralización y carga iónica.
En este segundo grupo, al igual que en primero, las aguas polares reflejan las particularidades de los diferentes medios ambientes glaciológicos e hidrogeológicos estudiados en Spitsbergen; por ejemplo, las cuencas de Rasstupet (sus aguas superficiales) y Flugleberget, ambas aparecen diferenciadas respecto a las de Brattegg, el glaciar Werenskiold y el conjunto de muestras de hielo, nieve y firn. Ello indica que el ártico es un medio lejos de ser homogéneo respecto a la dinámica del proceso de interacción agua-roca, a pesar de la existencia de un común denominador climático.

Consideraciones finales
Del análisis del comportamiento químico-físico de las aguas kársticas en las regiones de climas extremos, polar y tropical (Spitsbergen y Cuba) se pueden derivar algunas consideraciones de tipo general, relacionadas con el proceso de adquisición de la composición química, su evolución y la intensidad de la denudación química, a causa de las diferencias del clima en ambas latitudes, así como de las litologías locales presentes.
Desde el punto de vista químico-físico, las aguas en una y otra latitud actúan sobre los minerales solubles (carbonatos y sulfatos principalmente). La disolución de los minerales presentes en las rocas carbonatadas (calizas y dolomías), se facilita con la presencia en el seno del agua de cierto contenido de dióxido de carbono (CO2) adquirido del aire atmosférico y de la atmósfera del suelo, mientras que en el caso de los sulfatos y rocas salinas, el proceso de disolución de los minerales es de tipo físico (sólo interviene el agua).
En el caso del hielo, su fusión por efecto del calor da lugar en los países de clima polar y templado, a formas del relieve denominadas termokársticas por su semejanza a las del karst desarrollado en calizas. La disolución de los minerales constitutivos de las rocas solubles, se puede presentar esquemáticamente mediante las ecuaciones siguientes:
CO2 (g)+ H2O (ac)+ CaCO3 (s) = Ca2+ (ac) + 2 HCO3- (ac) (3.1) calcita 2 CO2 (g)+ 2 H2O (ac)+ CaMg(CO3)2 (s) = Ca2+ (ac)+ Mg2+ (ac) + 4 HCO3- (ac) (3.2) dolomita H2O
CaSO4 . 2 H2O (ac) = Ca2+ (ac) + SO42- (ac) (3.3) yeso H2O
NaCl (ac) = Na+ (ac) + Cl- (ac) (3.4) halita Calor H2O (s) = H2O (l) (3.5) hielo
De acuerdo a la ley de Henry, los gases son más solubles en agua a temperaturas más bajas. Por esta razón se debe esperar que por el proceso de adquisición directa del CO2 de la atmósfera, su contenido en las aguas sea mayor en las regiones polares que en las tropicales. Este hecho llevó a algunos geomorfólogos, por ejemplo, Corbel (1959) a considerar que en los países de clima frío sería más intensa la denudación química. Sin embargo, esta hipótesis se fue desechando con el tiempo, porque el proceso microbiológico de generación de CO2 en el suelo es más activo, en

condiciones pedoclimáticas favorables para este proceso, esto es, donde existan relativamente altas temperatura y humedad relativa, tal como ocurre en los suelos tropicales.
Además, en las regiones polares los suelos se encuentran prácticamente congelados (permafrost), abunda poco la materia orgánica disponible y la actividad de los microorganismos es pobre. No obstante, como se ha visto en este trabajo, las aguas procedentes de la tundra, la vegetación enana de las tierras polares, se caracterizan por presentar un contenido de CO2, que es más elevado que el de las restantes aguas de esas regiones, lo que indica, que la actividad biológica está presente aún en esas condiciones climáticas tan rigurosas.
En virtud del poco contenido de este gas en las aguas polares, la cantidad de minerales disueltos que, por lo general, resulta también pequeña, se encuentran comúnmente insaturadas en relación a los minerales calcita, dolomita y yeso. El proceso de disolución de los minerales carbonatados tiene lugar, en la mayoría de los casos, en condiciones de sistema abierto respecto al CO2.
Las aguas que presentan mayor grado de mineralización son las de los lagos de las morrenas, las termominerales de origen profundo y las que se mezclan con el agua de mar como consecuencia de la intrusión marina de los acuíferos kársticos. Este mecanismo de disolución, también se favorece por el grado de trituración mecánica, por acción de los glaciares, a que ha sido sometido el macizo.
Un hecho interesante, lo ofrece la variación temporal de la composición química de las aguas correspondientes a las corrientes superficiales originadas por la fusión de los hielos del glaciar y el permafrost. Durante la primavera y el verano, estas aguas poseen una mineralización muy baja y reflejan, en su composición química, las propiedades de la nieve y el hielo. Sin embargo, a medida que transcurre el otoño y se acerca el invierno hidrológico polar, su mineralización se incrementa y su composición relativa varía, reflejando las propiedades del medio rocoso por donde se mueven estas aguas. La causa de este comportamiento está relacionada con el efecto crioquímico, en virtud del cual una parte de la masa acuosa se congela en forma de cristales de hielo relativamente puros, y el resto se enriquece en minerales disueltos.
Las aguas en las regiones tropicales, adquieren el mayor contenido de CO2 disuelto, durante las precipitaciones, en que la materia orgánica es extraída del suelo. Luego, en un período relativamente corto este gas actúa sobre los minerales que se encuentran en contacto con el agua, los cuales se disuelven hasta su consumo. Este proceso puede ocurrir en condiciones de sistema abierto o cerrado respecto al CO2, en dependencia de que el sistema kárstico posea suelos aireados con suficientes oquedades, o por el contrario, se encuentre cubierto por depósitos poco permeables; situaciones éstas, que imponen, en el primer caso, un régimen de generación prácticamente continua del CO2 y en el segundo caso, un consumo también continuo de CO2, sin el aporte de nuevos suministros. En uno u otro caso, los minerales son disueltos en un tiempo breve hasta alcanzar la saturación. En el primer proceso, el pH adquiere al final, un valor más elevado que en el segundo.
Las aguas superficiales en Cuba, por lo general, se mantienen saturadas o sobresaturadas una gran parte del año, las cuales se hacen de nuevo agresivas en los períodos lluviosos. En esas condiciones reinician el proceso de disolución de los minerales hasta que de nuevo se saturan.
Las aguas kársticas subterráneas pueden mantener un cierto contenido de CO2 en equilibrio con los minerales disueltos a una temperatura determinada, pudiendo alcanzar un elevado grado de mineralización. Cuando emergen por un manantial o exsurgencia, parte del

gas se escapa y para alcanzar de nuevo el equilibrio de los carbonatos, el agua debe adquirir la presión de CO2 de la atmósfera, precipitando en el cauce el exceso de CaCO3 en relación con las nuevas condiciones de los equilibrios. En forma similar se producen las precipitaciones de travertina en las cavernas y conductos kársticos.
La mineralización de las aguas kársticas en Cuba, es más elevada en las regiones llanas que en las montañas, en especial, en las aguas que proceden de la zona de saturación y circulación profunda (drenaje profundo), así como la de las aguas subterráneas que se encuentran en los acuíferos litorales conectados al mar a través de conductos kársticos.
La disolución de los minerales en las rocas no solubles es por lo general de tipo incongruente, es decir, que en ese proceso una parte pasa al agua en forma de iones mientras otra, se transforma en otro mineral no soluble que queda como residuo. También en estos casos la producción de CO2 por los microorganismos facilita la disolución de los minerales, así como el cambio de pH y del potencial redox del medio, lo cual se favorece con las bacterias del suelo, transformando la valencia de los iones. Muchos elementos son movilizados por las aguas porque en el proceso de oxidación-reducción los minerales que se originan son más solubles o fáciles de arrastrar por las aguas. El proceso de disolución de algunos minerales constitutivos de las rocas no solubles se representa a continuación: 4 FeS2 (s)+ 15 O2 (g)+ 8 H2O (l) = 2 FeO3 (s) + 16 H+ (ac) + 4 SO42- (ac) (3.6) pirita hematita 2 NaAlSi3O8 (s)+ 2CO2 (g) + 11 H2O (l) = Al2Si2O5(OH)4 + 2 Na+ + 2 HCO3- albita caolinita + 4 H4SiO4 (ac) (3.7) (Mg, Fe)2SiO4 (s)+ 2 CO2 (g)+ H2 (g) + 1/2 O2 (g)+ n H2O (l) = olivino Fe2O3. n H2O (s) + 2 H2SiO3 (ac)+ 2 Mg2+ (ac) + 2 HCO3- (ac) (3.8) limonita 2 (Mg, Fe)Si2O6 (s) + CO2 (g) + n H2O (ac) = Fe2Si4O10(OH)2. n H2O (s) + piroxeno ferronontronita 2 Mg2+ (ac) + 2 HCO3- (ac) (3.9)
La intensidad de la denudación química en ambas regiones de climas extremos, depende fundamentalmente, del régimen de precipitaciones, de la temperatura y de la factibilidad de las aguas para disolver los minerales de las rocas. Este último factor esta relacionado con el estado de agrietamiento de los macizos y depende del tectonismo local o de procesos de intemperismo mecánico, como es el caso de la fricción a que se someten las rocas por los glaciares.

Las precipitaciones en las regiones polares ocurren en forma de lluvia o nieve, de acuerdo a la temperatura del aire. En los períodos menos fríos, como en el verano y, especialmente el otoño, donde además tienen lugar los huracanes polares relativamente calientes, parte de las nieves y los hielos se funden y aumenta el caudal de las corrientes superficiales y subsuperficiales. En tales circunstancias es más intensa la denudación química de esos parajes.
En las regiones tropicales, también es más intensa la denudación durante los períodos en que tienen lugar las mayores precipitaciones: durante la primavera y el verano en que ocurren con frecuencia grandes aguaceros; desde el verano hasta el otoño, al paso de los huracanes que suelen venir acompañados de intensas lluvias y en ocasiones durante los frentes fríos, que ocurren en el invierno, los cuales vienen precedidos también de lluvia.
Como ha señalado Tys (1992), lo más importante en el proceso de karstificación en las condiciones tropicales de Cuba es la cantidad y distribución de las precipitaciones anuales. Las intensas lluvias tanto en los períodos seco como lluvioso juegan un factor importante en el desarrollo de los procesos de colapso en las depresiones kársticas poligonales.
Aunque no existen muchas áreas experimentales, en las que se hayan hecho estudios cuantitativos sobre la magnitud de la denudación química, las experiencias llevadas a cabo en la cuenca del glaciar Werenskiold y en el Pan de Guajaibón, durante los años hidrológicos 1979-1980 y 1984-1985 respectivamente, indican que en esta última región es más intenso este proceso.

BIBLIOGRAFÍA
Abelló, I., Luaces, P., Fagundo, J.R. y Guerón, J. (1993). Estudio estadístico multivariado
sobre datos de las diferentes litologías de la cuenca del Río San Marcos, Sierra del Rosario, Pinar del Río, Cuba. Libro de Comunicaciones, I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 205-214.
Adolphe, J.P. (1981). Exemples de contribution microorganiques dans les contructions carbonate continentales. Actes du Coll. de l' AGF Formation Carboniques Externes, Tufs et Travertins, Paris, pp. 15-30.
Adolphe, J.P. (1986). Biocristallogeneses et ecomorphologie des tufs, travertins. Enseignements tires d' exemples naturales et experimentaux. Table Ronde sur Travertins l. s. et Evolution des Paysages Holocenes dans le domaine Mediterraneen, Provence 1985, Mediterranee, 1/2: 11-17.
Aguayo, J., Antiguedad, I., Eraso, A. y García de Cortazar, A. (1988). Respuesta hidroquímica en el tránsito de aguas bajas-aguas altas en las principales surgencias de la unidad hidrogeológica Gatzume Zestoa (Gepasova). Implicación sobre la organización del drenaje kárstico. Hidrogeología (Granada), 3: 38-49.
Aguirre, L.V. (1980). Aplicación de principios geoquímicos en la hidrología kárstica de la península de Yucatán. Ingeniería Hidráulica en México, 1(3): 21-29.
Arellano, D.M. y Fagundo, J.R. (1985). Criterio acerca de la zonación geoquímica de un acuífero cársico mediante índices químico-físicos, parámetros hidroquímicos y relaciones iónicas. Voluntad Hidráulica, 66: 2-13.
Álvarez, E. y Fagundo, J.R. (1995). SIMUCIN: Sistema para el estudio cinético y la modelación de las reacciones de disolución de minerales. En: El Karst y los Acuíferos Kársticos. Ejemplos y Métodos de Estudio. Ed. Univ. Granada: 209-213..
Álvarez, E., Vinardell, I., Fagundo, J.R., Reguera, E. y Cardoso, M. E. (1990). Evolución química y relaciones empíricas en aguas naturales. II- Sistema automatizado para el monitoreo de la aguas. Voluntad Hidráulica, 83: 15-25.
Álvarez, E., Vinardell, I., Fagundo, J.R. y Rodríguez, J.E. (1993). Sistemas automatizados para el procesamiento de datos hidroquímicos: SAPHIQ, GEOQUIM, SAMA, BATOMETER.Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 189-194.
Aminot, A. (1974). Geochimie des eaux d' aquiferes karstiques. 2- Les analyses chimiques en Hidrologie Karstique. Ann. Speleol., 29 (4): 462-486.
Antiguedad, I. (1986). Estudio hidrogeológico de la cuenca del Nervión-Ibaizabal. Contribución a la investigación de los sistemas acuíferos kársticos. Tesis Doctoral. Univ. del País Vasco, 338 pp.
Antiguedad, I. (1988). Estudio de acuíferos kársticos a partir de sus respuestas naturales. Aplicación a los sistemas del País Vasco. Rev. de la Soc. Geológica de España. Vol. 1 (1-2): 211-227.
Arellano, D.M. (1986). A study of karstic groundwater circulation using natural isotopes. Case history: Pinar del Rio Province, Cuba. Tesis de Dr. en Ciencias Geológicas, Univ. Carolina, Praga, pp. 178.

Arellano, D.M., Fagundo, J.R., Jilek, P. and Silar, J. (1989). Radiocarbon dating in a karstified coastal aquifer in Cuba. Acta Universitatis Carolinae-Geologica. Ed. Univ. Karlova (Praga) 3: 367-387.
Back, W., Cherry, R.N and Hanshaw, B.B. (1966). Chemical equilibrium between the water and minerals of a carbonate aquifer. Nat. Speleol. Soc. Bull., 28 (3): 119-126.
Back, W. and Hanshaw, B.B. (1965). Chemical Hydrology. Advances in Hydroscience, 2: 49-109.
Back, W. and Hanshaw, B.B. (1971). Comparison of Chemical Hydrology of the Carbonate Peninsula of Florida and Yucatan. J. Hydrol., 10: 330.
Bachelier, G. (1968 b). Contribution a l' etude de la mineralisation du carbone des sols, Mem. ORSTOM, 30:145.
Bachelier, G. (1971). Dynamique saisonniere de deux sols en climat tempere, RCP du C.N.R.S 40, Ecologie du sol, pp.187-253.
Bakalowicz, M. (1974). Geochimie des eaux d' aquiferes karstiques. 1. Relation entre mineralisation et conductivite. Ann. Speleol., 29 (2): 167-173.
Bakalowicz, M. (1976). Geochimie des eaux karstiques. Une methode d' etude de l' organisation des ecoulements souterrains, 2 College Hydrologie in Pays Calcaire. Fasc. 2, 3 ieme Serie. Universite de Besucon, pp. 49-58.
Bakalowicz, M. (1979). Contribution de la Geochimie des eaux a la connaissance de l'aquifere karstique et de la karstification. These D. Univ. Pierre et Marie Curie, Paris, pp. 269.
Bakalowicz, M. (1992). Geochimie des eaux et flux de matieres dissoutes. L'approche objective du role du climat dans la karstogenese. In: Karst et evolutions climatiques. Ed. J.N. Salomon; R. Maire, Presses Universitaires de Bordeaux, 61-74.
Bakalowicz, M. et A. Manguin (1980). L' aquiferes karstiques. Sa definition, ses caracteristiques et son identification. Coll. A.T.P. Hydrogeol. C.N.R.S. Montpellier 1978.. In: Bull. Soc. Geol. France, 11: 71-79.
Baranowski, B. (1977). The subpolar glaciers of Spitsbergen seen against the climate of this regions. Ed. Universitatis Wratislaviensis, Katowice, pp. 111.
Bárány-Kevei, I. (1992). Les facteurs écologiques dans la formation du karst. In: Karst et évolutions climatiques. Ed. J.N. Salomon; R. Maire, Presses Universitaires de Bordeaux, 51-59.
Bögli, A. (1960). Kalklossung und karrenbildung, Zft. fur Geomorph. Suppl., 2: 4-21. Bögli, A. (1963). Hohlenkarren. 3 th Int. Kongr. Spelol, Wien, 1961: 25-27. Bögli, A (1964). La corrosion par melange des eaux. Int. J. Speleol., 1: 61-70.
Back, W., Hanshaw, B.B., Herman, J.S. and Van Driel, J.N. (1986). Differential dissolution of a Pleistocene reef in the ground-water mixing zone of coastal Yucatan, Mexico. Geology, 14: 137-140.
Bachelier, G. (1968 a). Problemes relatifs a l'atmosphere du sol et utilisation posible d' un detector de gaz pour la mesure de su teneur en gaz carbonique, Cah. ORSTOM. Pedologie, 6 (1): 95-104.
Bakalowicz, M. (1977). Etude du degre d' organisation des ecoulements souterrains dans les aquiferes carbonates pour une methode hydrogeochimique nouvelle, C.R.Ac. Sc., pp. 2 463-2 466.
Barros, O. (1982). La intrusión salina y sus efectos en la explotación de acuíferos cársicos. Casos históricos. Coloquio Int. sobre Hidrología de la región del Caribe, La Habana, pp. 440-473.
Berner, R.A. and Morse, J.W. (1974). Dissolution kinetics of calcium carbonate. Am. J. Sci. 274: 108-131.

Bögli, A. (1965). The role of corrosion by mixed water in cave forming. In: Problems of Speleological Research. Proceeding Int. Speleol. Conf. Brno. CSSR, 1964, pp. 125-131.
Bögli, A. (1971). Corrosion by mixing karst waters. Trans. Cave Res. Group G. Brit., 13 (2):109-114.
Bögli, A. (1980). Karst Hydrology and Physical Speleology. Ed. Springer-Verlag, Berlin, Heidelberg, New York, pp.254.
Bray, L.G. (1977a). The role of organic matter in limestone solution in the Ogob Feynnon DDQ Streamway. Proceeding of the 7 th Int. Speleological Congress Sheffield, England, pp. 65-68.
Bray, L.G. (1977 b). Rapid aggressiveness assessment using conductimetry. Proceeding of the 7 th International Speleological Congress, Scheffield, pp. 61-71.
Calaforra, J.M. y Pulido Bosch, A. (1989). Principales sistemas kársticos en yeso en España. En: El karst en España. Ed. J.J. Duran , J. López Martínez. Monografía de la Soc. de Geomorfología, 9: 277 - 294.
Callejón, S.M., Pulido Bosch, A. y Valenzuela, P. (1984). Aplicación de los análisis cluster al estudio de características físico-químicas de aguas subterráneas. Estudios Geol., 40: 193-200.
Caro, P. (1965). La chimie du gaz carbonique et des carbonates et les phenomenes hydrogeologiques karstiques. Chron. Hydrogeol. BRGM,7: 51-77.
Corbel, J. (1959). Erosion en terrain calcaire, Vitesse d'erosion et morphologiea. Ann. Geogr., 68: 97-120.
Cotecchia, V., Tazioli, G.S. et Tittozzi, P. (1975). Geochimica delle acques della Peninsola Salentins in relatione al rapparti tra le acque di falda, le acque marine sotterranee e il mare. Geol. Appl. e Idrogeol., 10: 1-9.
Curl, R. (1965). Solution Kinetics of Calcite. Proc. 4th Intern. Congress Speleolol. Ljubijana 1965, 3: 61-66.
Currie, J.A. (1961). Gaseous diffusion in the aeration of aggregated soils. Soil’s Science, 92: 40-45.
Custodio, E. (1989). Consideraciones sobre la sobreexplotación de acuíferos en España. En: La Sobreexplotación de Acuíferos. Ed. Inst. Tecnológico Geo Minero de España, Almería: 43-64.
Bögli,A. (1969). CO2 - Gehalte der Lift in Alpine Karstboden und Hohlen. 5 th. Int. Congr. Stuttgart, 28: 1-9.
Calvache, M.L. y Pulido Bosch, A. (1989). Estado de la sobreexplotación estacional mediante un modelo de flujo en el acuífero del río Verde (Almuñecar, Granada). En: La Sobreexplotación de Acuíferos. Ed. Inst. Tecnológico Geo Minero de España, Almeria: 21-34.
Calvache, M.L. and Pulido Bosch, A. (1993). The influence on sea-water intrusion process of a karstic massif in a detrital system.In: Some Spanish Karstic Aquifers. Ed. Univ. de Granada, 127-142.
Castillo, R. y Eraso, A. (1975). Estudio geoquímico del karst del río Duraton, relación entre litofacies y el contenido salino del agua. Ann. Speleol., 30 (4): 591-608.
Catalán, J. (1988). Química del Agua. Ed. CNIC., La Habana, pp. 422. Cigna ,A.A., Cigna, L. et Vido, L. (1963). Quelques considerations sur l' effect sel dans
la solubilite des calcaires. Ann. Speleol., (18): 185-191. Comton, R.G. and Unwin, P.R. (1990). The dissolution of calcite in aqueous solution at pH
< 4; kinetics and mechanism. Phil Trans. R. Soc. London A, 330: 1-45.
Custodio, E. (1986). Hidroquímica del karst. En: Jornadas sobre el karst en Euskadi Karstari Buruszko Ihardunaldiak Euskadin Journées sur le karst en Euskadi. Donostia-San Sebastián (España), Tomo 2, 131-179.

Chen, J. (1991). A proposal: study of sinkholes as a point-source contamination in karst terrain. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 173-175.
Choppy, J. (1981). Role du climat dans le developpment des concretions. La Grotte d' Italia, 4 (10). 45-51.
Christopher, N.S.J. (1975).The use of saturation index and potassium to sodium ratio as indicators of speleological potential, with special reference to Derbyshire. Trans. Brit. Cave Res. Assoc., 2 (1): 29-34.
Dimitrov, D., Velikov, B. and Machkova, M. (1993). Processing of ground water hydrochemical data by means of cluster and discriminant analysis. Hidrogeología (Granada) 8: 25-39.
Daoxian, Y. (1987). Some characteristic of groundwater protection in karst area. 19 th Congress of Int. Association of Hydrologists, 19 (2) 135-142.
Drake, J.J. (1983). The effect of geomorphology and seasonality on the chemistry of carbonate groundwaters. J. Hydrol., 61 (1/3): 223-226.
Drake, J.J. (1984). Theory and model for global carbonate solution by groundwater. In: Groundwater as a Geomorphic Agent. Ed. R.G. La Fleur. Univ. Hyman, London, 210-226.
Drake J.J. and D.C. Ford (1974). Hydrochemistry of the Athabasca and North Saskatchewan River in the Rocky Mountains of Canada. Water Resources Research, 10 (6): 1 192-1 198.
Dreybrodt, W. (1981). Kinetics of the dissolution of calcite and its applications to karstification, Chem. Geol., 31: 245- 261.
Dreybrodt, W. (1992). Dynamics of karstification: A model applied to hydraulic structures in karst terraines. Applied Hydrogeology, 3(92): 20-32.
Dupessy, J.C. and G. Lambert (1878). Le gaz carbonique: polluant majeur de l'atmosphere. La Recherche, 9 (91): 699-698.
Ek, C. (1973). Analysis d' eaux des calcaires Paleozoiques de la Belgique. Metodes. Techniques et Resultates. Proffessional Paper, 18: 33.
Ek, C. (1979). Variations saisonnieres des teneur in CO2 d' une Grotte Belge: Le Trou Joney a Comblain-Au-Pont. Ann. Societ. Geologique de Belgique, 102: 71-75.
Christopher, N.S.J. (1977). The relative concentration of sodium to potassium in karst and allogenic waters. Proceeding of the 7 th International Speleological Congress, Sheffield, pp. 110-113.
De la Cruz, A . y Valdés, J. J. (1985). Estudio de las aguas kársticas de la Sierra del Pan de Guajaibón y sus inmediaciones mediante métodos matemáticos exploratorios de datos. Voluntad Hidráulica, 68: 25-34.
Drake, J.J. and Wigley, T.M.L. (1975). The effect of climate on chemistry of carbonate groundwater flow in jointed limestone. Nat. Speleol. Soc. Bull., 28: 111.
Dreybrodt, W. and D. Buhmann (1991). A mass transfer model for dissolution and precipitation of calcite from solutions in turbulent motion. Chemical Geology, 90: 107-122.
Dudley, J.R. (1972). Reability of Water Analysis. General Technical Department Report. TDR No 09271, pp. 27.
Ek, C. (1969). Abondance du gaz carbonique dans les fissures de grottes. Actes 5 th. Cog. Int. Speleol., Stuttgart, 14: 1-3.
Ek, C. (1981), D.Caron et J. Roberge: La forte teneur en gaz carbonique de l' air d' une cavite du Quebec: La Grotte de Saint Leonard, Montreal. Naturaliste Can.,108:57-63.

Ek, C., Delecour F. et F. Weissen (1968). Teneur en CO2 d l'air de quelques Grottes Belges. Techique eployei et premiers resultats. Ann. Speleol., 23 (1): 243-257.
Ek, C., Gilewska S., Kaszowski L., Kobylecki, A., Oleksynowa, K. and Oleksynowa, B. (1969). Some analyses of the CO2 content of the air in five caves. Z. Geomorphol, 13 (3): 226-286.
Eraso, A. (1981). Introducción al estudio del karst. En: Lapiaces, pp. 63-91. Ernest, L. (1964). Zur Frage der Mischungskorrosion. Die Hohle 15: 71-75. Fagundo, J.R. (1982). Determinación de índices para la caracterización de aguas cársicas
mediante fórmulas semi empíricas. Coloquio Int. sobre Hidrología Cársica de la Región del Caribe, La Habana, pp. 496-509.
Fagundo, J.R. (1990 a). Evolución química y relaciones empíricas en aguas naturales. Efecto de los factores geológicos, hidrogeológicos y ambientales. Hidrogeología (Granada), 5: 33-46.
Fagundo, J.R., Arellano, D.M., Benítez, G., Surí, A., Avilé, C., Ferrera, V. y M. Torres (1993). Cambios hidrogeoquímicos por sobreexplotación de acuíferos cársicos. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 141-148.
Eraso, A. (1975). Le role des factores physico-chemiques dans le processes de karstification. Ann. Speleol., 30 (4): 567-580.
Fagundo, J.R. (1985). Caracterización de acuíferos mediante relaciones entre contenidos iónicos y parámetros quimico físicos. Revista CENIC Ciencias Químicas, 16 (2): 321-236.
Fagundo, J.R. (1990 b). Evolución química y relaciones empíricas en aguas naturales. 1- Estudio mediante simulación química del efecto de la litología. Voluntad Hidráulica, 82: 28-37.
Fagundo, J.R., Alvarez, E., Benítez, G., Ferrera , V. y Vega, J. (1992). Simulación química y matemática de la disolución de rocas carbonatadas por las aguas naturales. Proc. del XXIII Congreso Interamericano de Ingeniería Sanitaria y Ambiental, La Habana 1992, 152-157.
Fagundo, J.R., Alvarez, E., Vinardell, I y Vega, J. (1992). Control automatizado de la calidad de las aguas. Proc. del XXIII Congreso Interamericano de Ingeniería Sanitaria y Ambiental, La Habana 1992, 98-103.
Fagundo, J.R. and Arellano, D.M. (1991). Hydrogeochemical impacts of coastal karst aquifer overdrafts. Studia Carsologica (Brno), 5: 37-47.
Fagundo, J.R. y Pajón. J.J. (1987). Contribución al estudio de las relaciones lineales entre contenidos iónicos y parámetros químico-físicos. Efecto de la litología. Ingeniería Hidráulica, 6 (1): 12-29.
Fagundo, J.R., Pajón, J.M., Valdés, J.J., Rodríguez, J.E. y Arellano, D.M. (1987). Naturaleza de los procesos de disolución de los minerales calcita, dolomita y yeso en aguas naturales del occidente de Cuba. Voluntad Hidráulica, 76: 30-42.
Fagundo, J.R. and J.E. Rodríguez (1991). Hydrogeochemical and mathematical correlations in karst at the examples of the Pinar del Río Province, Cuba. Conference on Anthropogenic Impact and Environmental Changes in Karst, Brno 1990. Studia Carsologica, 4: 29-34.
Fagundo, J.R. and Rodríguez, J.E. (1992). Hydrogeochemical pattern and mathematical correlations in karst at the examples of Cuba. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 361-369; (1992): Newsletters 1992, IGCP-299, Guillin (China): 41-45.
Fagundo, J.R., Rodríguez, J.E., Benítez, G., Morera, W., Fernández, C. y Vega, J. (1993). Caracterización hidroquímica y control de la calidad de las aguas del karst de la cuenca Zapata.

Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 73-82.
Fagundo, J.R., Rodríguez, J.E., Pajón, J.M., Franco, E., Alvarez, E., Vinardell, I., Vega, J. y Benítez, G. (1991). Evolución química y relaciones empíricas en aguas naturales. 3- Cuenca del río San Marcos, Sierra del Rosario, Pinar del Río, Cuba. Voluntad Hidráulica, 85: 8-18.
Fagundo, J.R., Rodríguez, J.E., Pajón, J.M., Franco, E., Benítez, G., Rodríguez, A.C., Guerón, J. e Abelló, I. (1993). Caracterización hidroquímica de las aguas del Pan de Guajaibón y otras áreas cársicas cercanas en la Sierra del Rosario. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 43-54.
Fagundo, J.R., Rodríguez, J.E., de la Torre, J., Arencibia, J.A. and Forti, P. (1994). Hydrologic and hydrochemical characterization of Punta Alegre gypsum karst (Cuba). Congress of International Union of Hydrologist, Irán 1993. Newsletter Geology, Climate, Hydrology and Karst Formation, Guiling (China): 95-105
Fagundo, J.R., Rodríguez,J.E. y Vega, J. (1995). Contribución al conocimiento hidrodinámico de los sistemas cársicos del Pan de Guajaibón y la Meseta del Guaso a partir de datos hidroquímicos durante las crecidas. En : El Karst y los Acuíferos Kársticos. Ejemplos y Métodos de Estudio. Ed. Univ. Granada: 119-135.
Fagundo, J.R. y Valdés, J.J. (1975). Estudio químico físico de las aguas kársticas de la región de San Antonio de los Baños (La Habana) mediante el uso de modelos matemáticos. Ann. Speleol., 30 (4). 643-653.
Fagundo, J.R., Valdés, J.J, Cardoso, M.E. y de La Cruz, A. (1986). Algoritmo para el cálculo de parámetros e índices químico-físicos y geoquímicos en aguas naturales altamente mineralizadas. Revista CENIC Ciencias Químicas, 12 (1/2): : 72-76.
Fagundo, J.R. Valdés, J.J y Pajón, J.M. (1982). Estudio hidroquímico de las aguas cársicas de la cuenca del río Cuyaguateje. Coloquio Internacional sobre Hidrología de la Región del Caribe, La Habana, pp. 375-401.
Fagundo, J.R., Valdés, J.J and Pulina, M. (1985). Hydrochemical investigations in the Werenskiold Glacier basin and its surroundings. Research report, Univ. Silaski, Katowice, pp. 1-17.
Fagundo, J.R.., Valdés, J.J y Pulina, M. (1987). Resultados de las investigaciones en Spitsbergen. Ingeniería Hidráulica, 8 (3): 217-237.
Fagundo, J.R.., Valdés, J.J and Pulina, M. (1990). Hydrochemical investigations in extreme climatic areas, Cuba and Spitsbergen. International Symposium, Ciudad de la Habana 1988. In: Water Resources Management and Protection in Tropical Climates, Havana, Stockholm, pp. 45-54.
Fagundo, J.R.., Valdés, J.J., Rodríguez, J.E., Pajón, J.M., de la Cruz, A., García, A. y Pulina, M. (1986). Estudio preliminar sobre el proceso de denudación cársica en el polígono cubano-polaco del Pan de Guajaibón. Voluntad Hidráulica, 70/71: 11-15.
Febre, J.P (1983). Etude hydrogeologique de la partle sud-ouest du Causse de Martel (Quercy). Thése 3 Cycle Laboratoire de Geologie-Petrologie. Univ. Paul Sabatler Toulouse, 342 pp.
Fagundo, J.R., Valdés, J.J. y Pajón, J.M. (1984). Estudio de los sedimentos Cuaternarios de la cuenca del río Cuyaguateje mediante Espectroscopía IR y Difracción de rayos X. Voluntad Hidráulica, 63: 53-61.
Fagundo, J.R., Valdés, J.J , Pajón, J.M. y Rodríguez, J.E (1981). Comportamiento químico-físico de las aguas de la cuenca del río Cuyaguateje. Ingeniería Hidráulica, 2(3): 251-274.

Ferrera, V., Fagundo, J.R. y Benítez, G. (1995). Caracterización hidrogeoquímica de las aguas del la cuenca cársica costera Sur, Tramo Guira-Quivicán (Provincia Habana). En: El Karst y los Acuíferos Kársticos. Ejemplos y Métodos de Estudio. Ed. Univ. Granada: 227-238.
Ferro, F. (1982). Hidrología General. Ed. Científico-Técnica, Ciudad de la Habana, pp. 406. Finlayson, B. and Sauro, U. (1991). International cooperation in research on karst
environments. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 231-239.
Ford, D.C. (1985). Dynamics of the karst system: a review of some recent work in North America. Annals de la Societe Geologique de Belgique, 108: 283-291.
Gams, I. (1976). Variation of total hardness of karst waters in relation to discharge (case studied in Slovenia, Yugoslavia). Proc. Int. Symp. Karst Denudation, Ljubljana 1975, pp. 41-58.
García, J.M. (1988). El control de la contaminación de las aguas: monitoreo y estudios intensivos. Tesis Dr. Ciencias Técnicas, Ciudad de La Habana, pp. 116.
Garrels, R.M., Thomposon, M.E. and Siever, R. (1960). Stability of some carbonates at 25 °C and one atmosphere total pressure. Am. Jour. Sci., 258: 402-418.
Fernández-Rubio, R. y Eraso, A. (1975). Nuevas formas kársticas de erosión-precipitación en la cueva del agua (Granada, España). Ann. Speleol., 70 (4): 655-663.
Ford, D.C. and P. Williams (1989). Karst Geomorphology and Hydrology. Ed. Univ. Hyman, London, pp. 601.
Forti, P. (1988). La Fonti di Poino. En: Guida Alla Speleologia nel Reggiano. Ed. M. Chesi, Gruppo Speleologico Palitnologico Gaetano Chiereci: 41 - 49.
Franco, E., Fagundo, J.R. y Pajón, J. M. (1987). Resultados de los estudios hidroquímicos realizados en el Pan de Guajaibón en el período Enero 28 a Febrero 17 de 1986. Revista Reporte de Investigación ACC, Ciudad de La Habana, 11: 1-17.
Freeze, R.A. and Cherry, J.A. (1980). Groundwater. Ed. Prentice-Hall, Englewood, Cliffs, New York, pp. 604.
Freixes, A. (1991). El medio kárstico: de la investigación observacional y experimental a la modelación. Hidrogeología Subterránea y Migración de Contaminantes. Ed. A. Correig, 97-141.
Freixes, A., Monterde, M. y Ramoneda, J. (1993). Hidrología de los sistemas kársticos del Valle de Arán (Pirineos, Catalunya) Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 131-140.
Fritz, F. and Pavicic, A. (1987). Hydrogeological aspect of protection of the karst spring Jadro in Croatia. 19 th Congress of Int. Association of Hydrologists, 19 (2):161- 167.
Gams, I. (1967). On the types of tufa depositing waters and on the corrosion intensity in north western dinaric karst. Actes Coll. Hydrol. des Roches Fissurees, Dubrovnik, 2: 684- 689.
Gams, I. (1979). International comparative study of limestone solution by means of standard tablets (fist preliminary report). Actes du Symposium International sur l' erosion karstique U.I.S., Provence, Marcelle, Nimes, pp. 71-73. Garay P.e I. Morell (1989). Tasas de disolución en regiones kársticas españolas. En: El Karst en España, Monografía editada por J.J. Duran y L. López. Soc. Española de Geomorfología, pp. 257-264.
García, J.M. y Beato, O. (1979). Muestreo de las Aguas. Recomendaciones Técnicas Generales. Ed. Inst. Hidroeconomía, La Habana, pp. 25.
García, J.M., Gutiérrez, J. y Fernández, C. (1977). Determinación de sodio en aguas naturales mediante electrodos selectivos. Voluntad Hidráulica, 43: 20-25.
Garrels, R.M. and Christ, Ch.L. (1965). Solutions, Minerals and Equilibria. Ed. Harper and Row, New York, pp. 450.

Garrels, R.M., Thomson, M.E. and Siever, R. (1961). Control of carbonate solubility by carbonate complexes. Am. J. Sci., 259: 24.
Gerstenhauer, A. und Pfeiffer, R.H. (1966). Beitrage zur Frage der Losungsfreudigkeit von kalkgesteinen. Abh. Karst-Hohlenked, A. 2.
Geovine, A. (1986). Les travertine holocene de Vauvenargues (Bouches-du-Rhone). Table Ronde sur Travertins l.s. et Evolution des Paysages Holocene dans le domine Mediterraneen, Provence 1985, Mediterrane, 1/2: 81-91.
Girou, A. et Roques, H. (1971 a). Etude des cinetiques des precipitation des carbonates de calcium. Bull. de l'Association de Geograph. Francais, N 389-390: 227-233.
González, A. y Jiménez, S. (1988 a). La protección sanitaria a los acuíferos cársicos cubanos: un problema actual. I Parte. Voluntad Hidráulica, 77: 3-18.
Griolet, C.P. (1982). Effect of a deforestation on karstic groundwater quality. 19 th Congress of the Int. Association of the Hydrologist, 19 (1): 109-117.
Gutiérrez, J. (1982). Clasificación y representación gráfica de las aguas naturales. Ed. Instituto de Hidroeconomía, La Habana, pp. 27.
Gutiérrez,J, García, J. y Beato, O. (1981).Algunas experiencias obtenidas en el estudio de la calidad de las aguas subterráneas empleando hidromuestreadores verticales. Voluntad Hidráulica, 57: 43-55.
Gutiérrez, J., González, A. y García, J.M. (1982). Protección de aguas subterráneas en acuíferos cársicos contra la contaminación producida por efluentes de lagunas de estabilización. Coloquio Int. sobre Hidrología Cársica de la región del Caribe, La Habana, pp. 588-610.
Gutiérrez, J., Molerio, L. y García, J.M. (1982). Vulnerabilidad de los acuíferos kárticos a los procesos de nitrificación. Coloquio Int. sobre Hidrología Cársica de la región del Caribe, La Habana, pp. 524-536.
Girou, A. et Roques, H. (1971 b). Etude theorique de la cinetique de precipitation de carbonates de calcium. Ann. Speleol. 26 (2): 332-366.
Girou, A. et Roques, H. (1972). Etude experimentale de la cinetique de precipitation des carbonates de calcium. Ann. Speleol., 27 (2): 273-316.
González, A. y Feitó, R. (1988). Estudio mediante isótopos ambientales y datos hidroquímicos de los cambios cualitativos y el origen de las aguas subterráneas de la parte central de la cuenca Sur de Matanzas. Voluntad Hidráulica, 80: 57-66.
González, A. y Jiménez, S. (1988 b).La protección sanitaria a los acuíferos cársicos cubanos: un problema actual. II Parte. Voluntad Hidráulica, 78: 3-25.
Guerón, J., Fagundo, J.R., Abelló, I. y Ontivero, E. (1993). Utilización de las técnicas de regresión en el procesamiento de diferente naturaleza hidrogeoquímica. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 195-204.
Hanshaw, B.B. and Back, W. (1980). Chemical mass-wasting of the northern Yucatan Peninsula by groundwater dissolution. Geology, 8: 222-224.
Hellden U. (1973). Some calculations of the denudation rate in a dolomitic limestone area at Isfjord-radio, West Spitsbergen. Trans. Cave Research Group of G. Brit., 15 (2).
Hem, J.D. (1960). Chemical equilibrium diagrams for ground water system, Internat. Scientific. Hydrology Bull., 19: 45-53.
Hemmel, J (1990).: Spontaneous clarification of anthropogenically polluted subterranean streams in the moravian karst. Proceedings of the Int. Conference on Anthropogenic Impact and Environmental Change in Karst. Brno 1990. Studia Carsologica, 2 (3): 33-42.

Herman,J.S., Back, W. and Pomar, L. (1986). Speleogenesis in groundwater mixing zone. The coastal carbonate aquifers of Mallorca and Menorca, Spain. 9 th Congreso Internacional de Espeleología, Barcelona, 1: 13-15.
Hernández, A.O. (1995). Posibilidad y necesidad de la modelación regional de acuíferos cársicos como un medio poroso equivalente. En: El Karst y los Acuíferos Kársticos. Ejemplos y Métodos de Estudio. Ed. Univ. Granada: 1967-1973.
Hill, C.A. (1987). Geology of Carlsbad Caverns and other caves on the Guadalupe Mountains, New Mexico and Texas. WM Bureau Mines and Mines Resources Bull., 117: 150-187.
Hill, C. and Forti, P. (1986). Cave Minerals of the World. Ed. National Speleological Society, Huntsville, Alabama, pp. 238.
Hutchinson, G.E. (1957). A treatise on Limnology. Geography, Physics and Chemistry. Ed. Willey and Sons, New York, pp. 1 015.
Jacobson, R.L., Langmuir, D. and O'Brien, P.J. (1971). Factor analysis of carbonate groundwater chemistry, Cave and Karst Research. In: Speleology, 13 (5): 42.
Jacobson, R.L. and Langmuir, D. (1975). An accurate method for calculating saturation level of ground waters with respect to calcite and dolomite. Trans. Cave Res. Group. G. Brit., 14: 104-108.
James, J.M. (1992). Corrosion par melange des eaux dans les grottes de la plaine de Nullarbor, Australie. In: Karst et évolutions climatiques. Ed. J.N. Salomon; R. Maire, Presses Universitaires de Bordeaux, 333-348.
James, J.M. (1991). The Nullarbor carbonate aquifer, Australia: the impact of man and suggestions for future management. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 205-214.
Jania, J. and Kolondra, L. (1982). Field Investigations performed during the Glaciological Spitsbergen Expedition in Summer of 1982. Interim Report. Ed. Univ. Slaski, Katowice,pp.32.
Johnston, J. and Williamson, E.D (1916).The complete solubility curve of calcium carbonate. J. Am. Chem. Soc., 38: 5.
Kaplen, G.I. and Shandybin, V.E. (1987). The effect of irrigation with effluents on quality of groundwater. 19 th Congress of the Int. Association of Hydrologist, 19 (1): 119-124.
Kempe, S. (1975 a). A computer program for hydrochemical problems in karst water. Ann. Speleol., 30 (4): 699-702.
Kempe, S. (1975 b). Siderite-weathering, a non biogenic source of CO2. Ann. Speleol., 30 (4): 703-704.
Kogovsek, J. and Kempe, A. (1989). The influence of acid precipitations to processes in Postojnska Vama. Acta Carsologica, 18: 221-232.
Kranje, A. and Kogovsek, J. (1990). Karst water pollution through vertical percolation in Slovene Dinaric Karst. Proceeding of the Conference on Anthropogenic Impact and Environmental Changes in Karst. Brno 1990. Studia Carsologica, 2:65-73.
Krawczyk, W. (1992). Methods of field analytics of karst water. In: Hydrochemical methods in dynamic geomorphology. Scientific Works of Silesian University in Katowice, Katowice, (1254), 65-83.
Krawczyk, W. and Pulina, M. (1982). Preliminary results of hydrogeological and hydrogeochemical investigation in Fugleberget basin (SW Spitsbergen). In: Wyprawy Polarine, Ed. Universitety Slaskiewo 1977-1980. t. I, pp. 167-180,

Krawczyk, W. and Pulina, M. (1983). Hydrochemical investigations in the Werensheld Glacier basin. In: Field Investigations Performed During the Glaciological Spitsbergen Expedition in 1983. Interim Report. Ed. Univ. Slaski, Katowice, pp. 15-18.
Krawczyk, W., Pulina, M. and Tys, A. (1990). Quantitative and qualitative degradation of upper Jurassic aquifer in Olkuss-Zawiercie karst region (South Poland). Proceeding of the Conference on Anthropogenic Impact and Environmental Changes in Karst. Brno 1990. Studia Carsologica, 2: 75-86.
Kullman, E. (1987). Protection of groundwaters in fissure karst rock environments. 19th Congress of Int. Association of Hydrologists, 19 (2) 234-239.
Langmuir, D. (1964). Stability of carbonates in the system CaO-MgO-CO2-H2O. These pH. D. Geologie, Harvard Univ., pp. 143.
Langmuir, D. (1971). The Geochemistry of Some Carbonate Ground Water in Central Pennsylvania. Geochim. Cosmochim. Acta, 35: 1023.
Laphin, N.N. y Luege, J.R. (1969). Salinización de las llanuras costeras. Voluntad Hidráulica, 17: 21-31.
Laptiev, F.F. (1939). Aggressive wirkungen der wasser auf karbonat-gesteine. Gips und Beton. Tr. Spetsgeo, 1.
Laurent, E. (1985). Reflexions sur la protection des aquiferes karstiques et sur des activites humaines generatrices de karsts acceleres. Les examples du Tournaises et de Geleppe. Ann. Soc. Geol., 108: 125-135.
Legrand, H.E. (1984). Enviromental Problems in karst terrains. Hydrology of Karstic Terrains Case History, 1: 189-194.
Leszkiewicz J., J. Wach and J. Waga (1982). Karst spring at the foot of Rasstupet wall. Specification of 1978-1979. In: Wyprawy Polarine. Ed. Universytety Slaskiewo 1977-1980, t. I, pp. 181-189.
Leszkiewicz, J. (1982). Karst spring of the foot of Rasstupet wall in the South Spitsbergen in the investigations from 1978. In: Wyprawy Polarine, Ed. Universytety Slaskiewo 1977 -1980, t. I, pp.77-86.
Lewis, G.N. and Randall, M. (1923). Thermodinamics. Ed. Mc Graw Hill, New York, pp. 381.
Lin Hua, S. (1991). The function of karst aquifer to clean polluted waters in South suburb of Beijing. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 185-193.
Liszkowski J. (1975). Ist die Mischungskorrosion die einzige im pheatischen Bereich der Karstgrundwaserleiter wirksame korrosionform. Proc. 6 th Int. Congr. Speleol. III, Olomouc 1973, pp. 193-198.
Llanusa, H. y González, J. (1993). Aplicación del modelo hidrogeológico determinístico a la cuenca M-I de Matanzas. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 63-71.
Lloret, A. (1988). Sistemas automáticos de medida. En: Aguas Subterráneas. Instrumentación, Medida y Toma de Muestras. Ed. Prensa XXI S.A., 73-92.
Machkova, M., Tzankov, K., Mandadjiev,D., Velikovand, B. and Dimitrov,D. (1993). Solving an ecological problem related to the surface and groundwater quality in the Upperthacian Lowland, Bulgaria. Hidrogeología, 8: 1-11.

Manguin, A. (1975). Contribution á l'étude hydrodinamique des aquiféres karstiques. Thése Doctorat és Sciences Naturalles. Dijon. Annales Spéléologie, 29 (3): 283-332; (4): 495-601; 30 (1): 21-124.
Manguin, A. (1981). Utilisation des analyses correlatoire et spectrale dans l' approche des systemes hidrologiques. C.R. Ac. Sc., 293: 401-409.
Manguin, A. et A. Pulido Bosch: (1983). Aplication de los análisis de correlación y espectral en el estudio de acuíferos kársticos. Tecniterral, 51: 53-55.
Markowicz, M. (1968). Procesy wspolczesnej korozji krasowej masywu wapiennego Jury Czestochowskiej. Speleologia, 3: 2.
Markowicz, M., Popov, W. and Pulina, M. (1972). Comments on karst denudation in Bulgaria. Geogr. Polonica, pp. 23.
Markowicz, M. and Pulina, M. (1979). Ilosciowa pomikroanaliza chemiczna wod w obszarach krasu welanowego. Ed. Silesian Universitet, Katowice, pp. 67.
Matia, Ll. (1988). Técnicas instrumentales para el análisis de conductividad, oxígeno disuelto, pH y potencial redox en el campo. En: Aguas Subterráneas. Instrumentación, Medida y Toma de Muestras. Ed. Prensa XXI S.A., 145-204.
Mijatovic, B.F. (1984). Problems of sea water intrusion into aquifers of coastal Dinaric Karst. Hydrology of Dinaric Karst, 4: 115-140.
Miotke, F.D. (1971). Measurement of CO2 in soil atmospheres: Procedure Results and Significance in Karst Studies. Cave Research Meeting, Hamilton, Ontario, 1975, Abstract in: Cave and Research in Speleology, 13 (5): 42-43.
Miotke, F.D. (1974). Carbon dioxide and the soil atmosphere. The importance of CO2 for karst processes. Abh. Karst Hohlenkunde, A. 9: 1-49,
Miserez, J.J (1973). Géochimie des eaux du karst Jurassien. Contribution phisico-chimique a l'etude des altérations. Thése Doc. Sci. Nat 313, Neuchatel.
Molerio, L. (1974). Esquema geoespelelógico de Cuba. Ed. Dpto.de Hidrogeología, Grupo Hidráulico Nacional de Cuba, La Habana, pp. 55.
Montes, C., Amat, J.A. y Ramírez-Díaz, L. (1982a). Ecosistemas acuáticos del Bajo Guadalquivir (SW España) I. Características generales, físico-químicas y biológicas de las aguas. Studia Oecológica, 3 (1): 129-158.
Montes, C., Amat, J.A. y Ramírez-Díaz, L. (1982 b). Ecosistemas acuáticos del Bajo Guadalquivir (SW España). Variación estacional de los componentes físico-químicos y biológicos de la aguas. Studia Oecológica, 2 (1): 159-180.
Moore, G.W. (1956). Aragonite speleothems as indicators of paleotemperatures. Am. J. Sci., 254: 746-753.
Morales, I. (1991). Estudio hidrogeológico de la zona norte de Vizcaya. Tesis Doc. Sci. Geológicas. Univ. País Vasco, 503 pp.
Morales ,I y Antiguedad, I (1993). Diferenciación de tres sistemas kársticos de Bizkaia (País Vasco) a partir del análisis de sus respuestas naturales. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaumne I, Castellón (España), 215-231.
Morell, I., Giménez, E. y Esteller, M.V. (1987). Ejemplo de utilización de registros verticales de conductividad, temperatura, pH y oxígeno disuelto. Aplicación a los acuíferos de Oropesa-Torreblanca y Moncofar (Castellón). Hidrogeología, 11: 171-182.

Morell, I., Giménez,E. y Esteller, M. V. (1988). Comportamiento iónico y proceso físico-químico en acuíferos detríticos costeros de las Planas de Oropesa, Castellón y Granada (Com. Valenciana). Hidrogeología, 3: 21-33.
Morell, I., Medina, J., Pulido Bosch, A. y Fernández-Rubio, R. (1986). Caracterización de la intrusión marina en el acuífero costero de Oropesa-Torreblanca (Prov. de Castellón) en base al estudio de relaciones iónicas. Hidrogeología, 1:15-31.
Muler, J.C. and Eingold, J.C. (1987). Vulnerability of a karst terrain to groundwater contamination by the agricultural chemical ethylene dibromide Northwest Florida (USA). 19 th Congress of the Int. Association of Hydrologist, 19 (2): 240-253.
Murray, J.W. (1954). The deposition of calcite and aragonite in cave. J. Geol., 62: 481-492. Muxart, R. et Muxart, T. (1969). Contribution a l'etude de la dissolution des calcaires
dans les eaux naturalles. Ann. Speleol., 24 (4): 639-650. Muxart, T. (1972). Contribution a l' etude des courbes de solubilite de la calcite dans l' eau
en presence d' anhydride carbonique. Ann. Speleol., 27 (3): 465-478. Muxart, T. (1975). Note sur la solubilite de la dolomite et des melanges calcite + dolomite
dans l' eau. Actes Veme Cog., Suisse de Speleol, Interlaken, 1974, pp. 145-154. Muxart, T. (1979). Dissolution experimentale de la calcite et d' une dolomite dans les
solutions aqueuses d' acides organiques simples, Trav. Table franco-suisse sur le karst 1978. Bull. Ass. Fr. de Karstologie, 4: 31-37.
Muxart, T. (1981). Rappel des principaux facteurs conditionnant la precipitation des carbonates in milieu continental. Acte du Colloque de l' AGF Formation Carbonates Externes, Tufs et Travertins, Paris, pp. 119-129.
Muxart, T. et P. Girot (1977). L' alteration meteorique des roches. Pub. Dep. Geogr., Univ. Paris-Sorbonne, 4: 279.
Mysztal, S. and Pulina, M. (1983). Investigations of Glacier Caves. In: Field Investigations Performed During the Glaciological Spitsbergen Expedition in 1983. Interim Report. Ed. Univ. Slaski, Katowice, pp. 26-33.
Nicod, J. (1986). Les cascades des barrages de travertin de l' Argens superieur (Var). Mediterranee, 1/2: 71-80.
Nicod, J. (1986). Facteurs physico-chemiques de l' accumulation des formations travertineuses. Mediterranee, 1/2: 161-165.
Núñez Jiménez, A., Viña, N., Acevedo, M., Mateo, M., Iturralde, M. y Graña, A. (1984). Cuevas y Carsos. Ed. Militar, La Habana, pp. 431.
Paces, T. (1980). Kinetics of water systems. Geologica Survey of Praga, pp. 85-108. Padilla, A. (1990). Los modelos matemáticos aplicados al análisis de los acuíferos kársticos.
Tesis Doc. Iniv. Granada, pp. 267. Padilla, A. y Pulido Bosch, A. (1993). Consideraciones sobre la aplicación de los análisis de
correlación y espectral al estudio de los acuíferos kársticos. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 149-160.
Pajón, J.M., Fagundo, J.R. y Valdés, J.J. (1985). Ocurrencia de Goethita y limonita en la cueva superior del arroyo Majaguas. Sistema Cavernario Majaguas-Canteras. Pinar del Río. Voluntad Hidráulica, 68: 40-45.
Pajón, J.M. y Valdés, J.J. (1991). Simulación química de la disolución de rocas carbonatadas del macizo kárstico de Pan de Guajaibón, Sierra del Rosario, Cuba. Lapiaz Monografía III (Valencia): 25-37.

Parks, G.A. (1967). Aqueous Surface Chemistry of Oxides and Complex Oxide Minerals. In: Equilibrium Concepts in Natural Waters Systems, Advances in Chemistry, Series No 67. Ed. Am. Chem. Soc., Washington, D.C., pp. 121.
Paterson, K. (1972). Responses in the chemistry of springwater in the Oxford regions to some climatic variables. Trans. Cave Res. Group G. Brit., 14 (2): 132-140.
Pekny, J., Skoreoa, J. and Urba, J. (1987). Groundwater Protection zones and agriculture activity. 19 th Congress of the Int. Association of Hydrologist, 19 (2): 291-301.
Perchorkin, A.I and Perchorkin, J.A. (1986). Chemical composition of fracture-karst water of sulphate karsted massifs. IV Congreso Int. de Espeleología, Barcelona 1986: 79 - 82.
Pérez Franco, D. (1974). Flujo no lineal a través de rocas fisuradas y a través de rocas con canales de disolución de gran diámetro. Voluntad Hidráulica, 30: 1-3.
Peryema, J. and Piasecki, J. (1983). Meteorological Conditions in Werenskiold Glacier basin, In: Field Investigations Performed During the Glaciological Spitsbergen Expedition in 1983. Interim Report. Ed. Univ. Slaski, Katowice, pp. 12 -14.
Peters, K. (1962). Mechanochemische Reaktionen. I Symp. Zerkleinern 1962, Frankfurt A/M Weinheim: Chemie, pp. 78-98.
Picknett, R.G. (1972). The pH of calcite solutions with and without magnesium present, and the implications with regards to rejuvenated aggressiveness. Trans. Cave. Res.Group G. Brit. 14: 141-149.
Picknett, R.G. (1977 a). Rejuvenation of aggressiveness in calcium carbonate solutions by means of magnesium carbonate. Proceeding of the 7 th Int. Speleological Congr. Shiffield, England, pp.346-348.
Picknett, R.G. (1977 b). Foreign substances and calcite solubility in carbonate waters. Proceeding of the 7 th Int. Speleological Congr. Shiffield, England, pp. 349-361.
Plummer, L.N. (1975). Mixing of seawater with CaCO3 groundwaters. Geol. Soc. Am. Mem., 142: 219-236.
Plummer, L.N., Parkhurst, D.L. and Kosiur, D.R. (1975 b) MIX2: A computer program for modeling chemical reactions in natural waters. Ed. U.S. Geol. Survey W.R.I. Rep. 75-61, pp.68.
Plummer, L.N. and Wigley, T.M.L. (1978). The kinetics of calcite dissolution in CO2-water system at 5 °C to 60 °C and 0,0 to 1,0 atm. CO2. Am. J. Sci., 278: 179-216.
Pitty, F.A. (1968). Some features of calcium hardness fluctuation in two karst stream and their possible value in geohydrological studies. J. Hydrology, 6: 202-208.
Ponce, S.L. (1980). Statistical methods commonly used in water quality data analysis, WSDG Technical Paper WSDG-TP-00001. Ed. Watershed System Development Group. USDA Forest Service, Colorado, pp. 136.
Pourbaix, M. (1963). Atlas d' Equilibres Electrochemiques. Ed. Gauthiers-Villars, Paris. Pulido Bosch, A. (1976). Salinización y recarga artificial en el acuífero de Javer (Alicante).
Simposio Nacional de Hidrología, Valencia, 2: 772-787. Pulido Bosch, A. (1989). Caracterización Hidroquímica del campo de Dalías (Almería). Ed.
Inst. Andaluz de Reforma Agraria, España. pp. 265. Pulido Bosch, A (1991). Algunas reflexiones relativas a la sobreexplotación de acuíferos
kársticos. Hidrogeología (Granada), 6: 47-58. Pulina, M. (1968). Zjawiska krasowe w Wschedniej Syberiri. Prace Geogra. IG Pan 70. Pulina, M. (1971). Observations of the chemical denudation of some karst areas of Europe
and Asia. Studia Geomorphol. Carpatho-Balcanica, 5: 79-92.

Pulina, M. (1974). Denudacja Chemiczna Obszarach Krasu Weglanowego. Ed. Prace Geogr. IG PAN, Warszawa, pp. 159.
Pulina, M. (1977). Remarques sur les phenomenes karstiques dans le partie meridionale du Spitsbergen. Kras i Speleologia, 10 (1).
Pulina, M. (1983). Hydrological and Glaciological Investigations on Bertil Glacier and the Grondfjord Region. In: Field Investigations Performed During the Glaciological Spitsbergen Expedition in 1983. Interim Report. Ed. Univ. Slaski, Katowice, pp. 39-50.
Pulina, M. (1984). The effect of cryochemical processes in the glaciers and the permafrost in Spitsbergen. Polar Reseach, 5 (3/3): 137-163.
Pulina, M., Fagundo, J.R., Valdés, J.J., Rodríguez, J.E., Kozik, A., Leszkiewicz, J., Glowacki, P., Pajon, J.M., Cruz, A. and García, A. (1984). The Dynamics of Contemporary Karst Processes in the Tropical Area of Cuba. Preliminary report of the field investigations performed by the Expedition GUAJAIBON'84 in the winter season 1984. Ed. Univ. Slaski. Sosnowice, pp. 42.
Rauch, H.W. and White, W.B. (1977). Dissolution kinetics of carbonate rocks. 1. Effects of lithology on dissolution rate. Water Research, 3 (2): 381-394.
Reffay, A., Campy, M., Richard, H. et Rousseau, D.D. (1986). Les formations carbonatees du Dort N (Ain). premieres observations. Table Ronde sur Travertins l.s. et evolutions des Paysages Holocenes dans le domaine Mediterraneen, Provence 1985, Mediterranee, 1/2: 105-112.
Pulina, M. (1992). Karst denudation. In: Hydrochemical methods in dynamic geomorphology. Scientific Works of Silesian University in Katowice, Katowice, (1254), 16-39.
Pulina, M. and Fagundo, J.R. (1992). Tropical karst and chemical denudation of Western Cuba. Geographia Polonica, Warsawa, (60): 195-216.
Pulina, M., Peryema, J., Kida, J. and Krawcyk, W. (1984). Characteristic of the polar hydrological year 1979/1980 in the basin of the Werenskiold Glacier SW Spitsbergen. Polar Research, 5(3/4): 165-1982.
Quesada, V. (1986). Tasa de remoción bacteriana en el río Almendares. Voluntad Hidráulica, 70/71: 4-7.
Quesada, V. y Gutiérrez, J. (1982). Tasa de remoción bacteriana en arroyos cársicos. Caso de estudio: arroyo de los Pioneros. Coloquio Int. sobre Hidrología Cársica de la región del Caribe, La Habana, pp. 550-563.
Reed, S.M. and Singh, R.N. (1987). A geochemical survey of opencast mine in the United Kingston. Proceeding of the Hydrology of Coal Basin Int. Symp., Katowice, pp. 147-153.
Renault, P. (1971). La teneur en anhydride carboniques des atmospheres des grottes. Bull. Assoc. Geogr. Fr, 389/390: 241-245.
Renault, P. (1979). Mesures periodiques de la pCO2 dans les Grottes Francaises du Cours de les DIX Dernieres Anneis. Actes du Symposium International sur l' Erosion Karstique, UIS, pp. 13-33.
Robert, C.W., Melvin, J. A. and Williams, H.B. (1983). CRC Handbook of Chemistry and Physics. 64 th Edition. Ed. CRC Press Inc., EE.UU., pp. D43-D92.
Rodríguez, J.E., Cuellar, A., Llanes, J., Chong Li, A. y Fagundo, J.R. (1993). Hidrología de la Ciénaga de Zapata. En: Estudio Geográfico Integral Ciénaga de Zapata, 59-72.
Rodríguez, J.E. y Fagundo, J.R. (1991). Hydrology and dynamics of tropical karst processes in Cuba. Studia Carsologica (Brno) 4: 59-55; (1992). Newsletters 1992, IGCP-299, Guillin (China): 46-51.

Rodríguez, J.E., Fagundo, J.R., Cutié, F., Cruz, C. y Franco, E. (1989). Hidrología cársica del macizo del Pan de Guajaibón, Sierra del Rosario, Pinar del Río. Año hidrológico noviembre de 1984-octubre de 1985. Ed. Academia, La Habana, pp. 160.
Rodríguez, J.E., Fagundo, J.R. y Pulina, M. (1993). Hidrología y dinámica de los procesos cársicos tropicales contemporáneos de Cuba. Libro de Comunicaciones. I Taller sobre Cuencas Experimentales en el Karst, Matanzas (Cuba) 1992. Ed. Univ. Jaume I, Castellón (España), 105-114.
Rodríguez, J.E., Fagundo, J.R. y Spassov, K. (1991). Caracterización hidrológica e hidroquímica del carso de la meseta del Guaso (Guantánamo, Cuba). Lapiaz Monografía III (Valencia): 3-21.
Roques, H. (1962 a). Consideration theoriques sur la chimie des carbonates (1° memoire). Ann. Speleol., 17(1):11-41.
Roques, H. (1962 b). Consideration theorique sur la chimie des carbonates (2° memoire). Ann. Speleol., 17 (2): 241-284.
Roques, H. (1962 c). Consideration theorique sur la chimie des carbonates (3° memoire). Ann. Speleol., 17 (3):463-467.
Roques, H. (1964). Contribution a l' etude statique et cinetique des systemes gaz carbonique-eau-carbonate. Ann. Speleol., 19 (2): 255-484.
Roques, H. (1969 a). Problemes de transfert de masse poses par l' evolution des eaux souterraines. Ann. Speleol., 24 (3): 455-494.
Roques, H. (1969 b). A review of present day problems in physical chemistry of carbonates in solutions. Trans. Cave. Res. Group G. Brit., pp. 139-163.
Roques, H. (1972). Sur une nouvelle methode graphique de etude des eaux naturelles. Ann. Speleol., 27 (1): 79-92.
Roques, H. et C. Ek. (1973). Etude experimentale de la dissolution de calcaires par une eau chargee de CO2. Ann. Speleol., 24 (4): 549-563.
Rorsh, J.M. and Llard, J.W. (1984). Trace elements behaviour in a limestone aquifers. In: Hydrogeology of karstic terrain Case History, 1: 206-210.
Santo, A. (1991). Karst processes and potential vulnerability of the Campanian carbonatic aquifers: the state of knowledge. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 95-107.
Sauro, U. and Meneghel, M. (1986). Aspects of Karst erosion in the Venetian Pearl. 9th Congress Int. de Espeleología, Barcelona 1986, 1: 35-35.
Schoeller, H. (1941). L' influence du climat sur la composition chimique des eaux souterraines vadoses. Bull. Soc. Geol. Fr, 11: 267-289.
Schoeller, H. (1950). Les variations de teneurs en gaz carbonique des eaux subterraines en function de l' altitude. C.R.As. Sc., 230: 560-561.
Schoeller, H. (1962). Les eaux souterraines. Ed. Masson, Paris. Shayakubov, B. y Morales, C. (1980). Consideraciones para la explotación de acuíferos
costeros en presencia de intrusión marina. Voluntad Hidráulica, 54: 10-15. Shopov, Y.Y. (1988). Bulgarian Cave minerals. The NSS Bulletin, 50: 21-24. Shuster, E.T. and White, W.B. (1971). Seasonal fluctuation in chemistry of limestone
spring: a possible mean for characterizing carbonate aquifers. J. Hydrology, 14: 93 -128. Sjoberg, E.L. and Rickard, D.T. (1984). Temperature dependence of calcite dissolution
kinetics between 1 and 62 °C at pH 2,7 to 8,4 in aqueous solution. Geochim. Cosmochim. Acta, 48: 485-493.

Smith, D.I. (1965). Some aspects of limestone solution in Bristol Region. In: Denudation in Limestone Regions: a Symposium, The Geographical J., 131 (1): 44-49.
Smith, D.I. (1988). Carbonate aquifers in Australia. A review. In: Resource Management in limestone landscapes: international perspectives. Ed. D.S. Gillieson and D.I. Smith, Sydney, pp. 15-41.
Smith, D.I. and T.C. Atkinson (1976). Process, landforms and climate in limestone regions. In: Geomorphic and Climate. Ed. E. Derbyshire, pp. 369-409.
Sokolow, D.S. (1962).Osnownyje uslowija razwitija karsta. Geotieoltechizdat. Stiff, H.A. (1951). The interpretation of chemical water analysis by means of pattern, Jour.
Petroleum Technology, 3(10): 15-17. Strengfield,V.T. and Rapp, J.R. (1984). Salt-water encroachment in the Miami area, Dade
Country, Florida. Hydrology of Karstic Terrains, 1: 160-163. Stumm, W.S. and Morgan, J.J. (1970). Aquatic Chemistry. An Introduction Emphasizing
Chemical Equilibrium in natural water. Ed. Wiley-Interscience, New York, London, Sydney, Toronto, pp. 583.
Sweeting, M.M., Grown, G.E. et al. (1965). Denudation in Limestone Region: A Symposium. The Geographical J., 1 (3):34-36.
Terjesen, S.C., Erga, O. and Ve, A. (1961). Phase boundary processes as rate determining steps in reaction between solids and liquids. Chem. Eng. Sci., 74: 277-288.
Ternan, J.L. (1972). Comments on the use of calcium hardness variability index in the study of carbonate aquifers with reference to the Central Pennines. England, J. Hydrology 16: 317-321.
Tillman, J. (1932). Die Chemische Untersuchung von Wasser und Abwasser. Ed. Halle, W. Knapp.
Trailkill, J. (1968). Chemical and Hydrologic factors in excavations of Limestone Caves. Bull. Geol. Soc. Am., 79: 19.
Trombe, F. (1952). Traite de Speleologie. Ed. Payot, Paris, pp. 376. Tulipano, L. and Fidelibus, M.D. (1991). Modern orientation on the karstic hydrogeology:
impact on problems of groundwater protection into carbonate aquifers experience from the Apulia region. Proc. of the Int. Conference on Environmental Changes in Karst Areas. Univ. Padova (Italy) 1991; Quaderni del Dipartimento di Geografia (13): 383-398.
Tys, A. (1992). Current karst processes in the polygonal karst in the Pan de Guajaibón Massif (Sierra del Rosario, NW Cuba). In: Karst et évolutions climatiques. Ed. J.N. Salomon; R. Maire, Presses Universitaires de Bordeaux, 135-144.
Urbain, P. (1953). Contribution de l' hydrogeologie thermale a' la tectonique: l' aire d' emergence d' Hammam Meskoutine (Departament de Constantine). Soc. Geol. France. Bull. Soc. G, 3: 247-251.
Urbani, F. (1991). Geotermia en Venezuela. Ed. Fundación GEOS, Caracas (Venezuela), pp.347.
Valdés, J.J. y Fagundo, J.R. (1975). Ocurrencia de estalactitas excéntricas de aragonito en la cueva de Perfecto (Sierra de los Órganos, Pinar del Río, Cuba). Ann. Speleol., 3 (4): 761-766.
Valdés, J.J., Fagundo, J.R. y Pajón, J. M. (1981). Aplicación de métodos numéricos al estudio hidrogeoquímico de las aguas de la cuenca del río Cuyaguateje, Ingeniería Hidráulica, 2 (3): 215-286.
Valdés J.J. y Fagundo, J.R.(1980). Determinación de la capacidad de disolución de las aguas kársticas durante un año hidrológico. Ingeniería Hidráulica, 1(2): 61-77.

Valdés J.J., Fagundo, J.R. y Pulina, M. (1985). Hydrochemical investigations in the Grondfjord region. Research report, Univ. Slaski, Katowice, pp. 1-4.
Valdés, J.J. y De la Cruz, A. (1982). Caracterización geomatemática de las aguas de la cuenca del río Cuyaguateje. Coloquio Int. sobre Hidrología de la región del Caribe, La Habana, pp. 277-287.
Wigley, T.M.L. (1977). WATSPEC. A computer program for determining the equilibrium of aqueous solutions. British Geomorphological Research Group Technical Bulletin, 20: pp. 46.
Veccholli, J., Ehrlich, G.G, Godsy, E.M. and Pascale, C.A. (1984). Alterations in the chemistry of an industrial waste liquid injected into limestone near Pensacola, Florida. Hydrology of Karstic Terrains Case History, 1: 217-221.
Vedy, J.C., Duchaufour, P. et Jacquin, F. (1979). Relation entre le cycle du calcium el l' humification. C.R. As. Sc., 276: 1 665-1 668.
Vinardell, I., Alvarez, E. y Fagundo, J.R. (1995). Sistema automatizado para el control de las aguas cársicas afectadas por la intrusión marina mediante reconocimiento de patrones, BATOMET. En: El Karst y los Acuíferos Kársticos. Ejemplos y Métodos de Estudio. Ed. Univ. Granada: 251-256.
Vinardell, I., Rodríguez, J. E. y Fagundo, J.R. (1991). Sistema automatizado para el procesamiento de datos hidrológicos, SAPHID. Revista CNIC Ciencias Biológicas, 22 (3): 7-8.
Wallin, M. and Bjerle, J. (1989). Rate models of limestone dissolution: A comparison. Geochim. Cosmochim. Acta, 53: 1 171- 1 176.
Ward, J.H (1963). Hierarchical grouping to optimize an objetive function. J. Amer. Statist. Assoc., 58(301):236-244.
Waterhouse, J.D. (1984). Investigation of Pollution of the karstic aquifer of the Mount Gambeer area in South Australia. Hydrology of Karstic Terrains Case History, 1:202-205.
Weisrock, A. (1986). Variations climatiques et periodes de sedimentation carbonatees l' holocene-l' age des depots, Table Ronde sur Travertins l.s. et evolution des Paysages Holocene dans le domine Mediterraneen. Provence 1985. Mediterranee, 1/2: 165-167.
Weyl, P.K. (1958). The solution kinetics of calcite. J. Geol., 66: 163-176. White, W. (1977). Role of solution kinetics in the development of karst aquifers. In: Karst
Hydrology. Ed. S. Tolsen and F.L. Doyle, Memory 12, EE.UU., pp. 503-517. White, D.E., Hemm, J.H. and Waring, G.A. (1963). Chemical composition of subsurface
waters. In: Data Geochemistry, 6 th Ed. Geological Survey Professional Paper 400-F, pp. 67. Wigley, T.M.L.(1973 a). The incongruent solution of dolomite. Geochim. Cosmochim. Acta,
73: 1 397-1 402. Wigley, T.M.L. (1973 b). Chemical evolution of the system calcite-gypsum-water. Can. J.
Earth Sci., 10 (2): 306-315.
Wigley, T.M.L. and L.N. Plummer (1976). Mixing of carbonate waters. Geochim. Cosmochim. Acta, 40: 489-995.
Yelamos, J.G. (1994). Realización de mapas hidrogeológicos con un programa general para gráficos en dos dimensiones. Hidrogeología (Granada) 9: 1-18.

Edita: Grupo de Investigación Recursos Hídricos y Geología Ambiental ISBN: 84-921345-8-x