hair phenotype in non-syndromic deafness
TRANSCRIPT

International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–1285
Hair phenotype in non-syndromic deafness
T. Volo a,*, T. Sathiyaseelan b, L. Astolfi c, V. Guaran c, P. Trevisi a, E. Emanuelli a, A. Martini a
a Otolaryngology and Otosurgery Unit, University Hospital of Padova, Italyb Family Medicine and Travel Clinic, Roslyn Building Calgary, Canadac Bioacustic Laboratory, University of Padova, Italy
A R T I C L E I N F O
Article history:
Received 12 January 2013
Received in revised form 8 May 2013
Accepted 9 May 2013
Available online 14 June 2013
Keywords:
Non-syndromic hearing loss
Congenital hearing loss
GJB2 gene
Gap junction
Connexin 26
Hair phenotype
A B S T R A C T
The GJB2 gene is located on chromosome 13q12 and it encodes the connexin 26, a transmembrane
protein involved in cell–cell attachment of almost all tissues. GJB2 mutations cause autosomal recessive
(DFNB1) and sometimes dominant (DFNA3) non-syndromic sensorineural hearing loss. Moreover, it has
been demonstrated that connexins are involved in regulation of growth and differentiation of epidermal
tissues. Hence, mutations in GJB2 gene, which is responsible for non-syndromic deafness, may be
associated with an abnormal skin and hair phenotype.
We analyzed hair samples from 96 subjects: a study group of 42 patients with hearing impairments of
genetic origin (38 with a non-syndromic form, 4 with a syndromic form), and a control group including
54 people, i.e. 43 patients with other, non-genetic hearing impairments and 11 healthy volunteers aged
up to 10 years old. The surface structure of 49 hair samples was normal, whereas in 45 cases it was
altered, with a damaged appearance. Two hair samples were considered unclassifiable: one from the
patient heterozygotic for the pendrin mutation (Fig. 2C), the other from a patient from Ghana with a
R134W mutation (Fig. 2D). Among the 43 altered hair samples, 31 belonged to patients with connexin
mutations and the other 12 came from patients without connexin mutations.
� 2013 Elsevier Ireland Ltd. All rights reserved.
Contents lists available at SciVerse ScienceDirect
International Journal of Pediatric Otorhinolaryngology
jo ur n al ho m ep ag e: ww w.els evier . c om / lo cat e/ i jp o r l
1. Introduction
In 2005, the World Health Organization (WHO) estimated that12.5 million people worldwide suffered from bilateral profoundhearing loss.
Congenital hearing loss is the hereditary sensorial defect mostcommonly observed in newborns. No data are available onworldwide prevalence of congenital deafness. In Italy the overallprevalence is 0.78 per 1000 for males and 0.69 per 1000 for females[1].
Genetic factors are involved in the pathogenesis of congenitalhearing loss in at least 50% of cases.
Despite an extraordinary genetic heterogeneity, up to 50% ofpatients with autosomal recessive non-syndromic hearing lossreveal mutations in one particular gene, GJB2, which encodes forthe gap junction connexin 26, involved in inner ear homeostasis.
GJB2 mutations have been widely studied in recent years, butthere are still some doubts about the precise mechanism
* Corresponding author at: Department of Otolaryngology and Otosurgery Unit,
University Hospital of Padova, Via Giustiniani, 2, 35128 Padova, Italy.
Tel.: +39 328 891 9154.
E-mail address: [email protected] (T. Volo).
0165-5876/$ – see front matter � 2013 Elsevier Ireland Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.ijporl.2013.05.010
responsible for the hearing impairment correlating with a mutatedconnexin 26 [2–4].
Our incomplete knowledge of the gap junction in the cochleaderives mainly from the fact that what we know about thedistribution of connexin in the inner ear has been learned fromanimal models (mainly rodents). Our understanding of thephysiology of connexin relies on the assumption that it wouldhave a similar distribution in humans, but genetic studies have alsoidentified phenotypic discrepancies between mouse and humanears [5,6].
Recent studies [6–9] have shown that mutations in the GJB2gene responsible for non-syndromic deafness may be alsoassociated with an abnormal skin and hair phenotype. It has beendemonstrated that connexins are involved in regulation of growthand differentiation of epidermal tissues, and gap junctions arefound abundantly both in inner ear and epidermal tissues.
GJB2 mutations have also been identified in syndromicdisorders with hearing loss associated with various skin diseasephenotypes. GJB2 mutations associated with skin disease are, ingeneral, transmitted with a dominant inheritance pattern. Non-syndromic deafness is caused prevalently by a loss of function,while literature evidences suggest for syndromic deafness amechanism based on gain of function. The spectrum of skinmanifestations associated with some mutations seems to have avery high phenotypic variability. Why some mutations can lead to

T. Volo et al. / International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–1285 1281
widely varying cutaneous manifestations is poorly understood andin particular, the reason why the skin disease-deafness phenotypesdiffer from each other thus remains unclear [10].
The aim of the present work was to establish whether the GJB2gene mutations that cause a gap junction dysfunction in thecochlea may also affect gap junctions in the hair, resulting inparticular hair phenotypes.
We used scanning electron microscopy to compare images ofthe morphology and mineral surface composition of hair fromhearing-impaired patients with and without associated GJB2mutations.
2. Materials and methods
We performed a case-control study on 107 patients undergoingcochlear implantation at the Sant’Anna University Hospital in thedepartment of Audiology from March 2008 to January 2010.
For control purposes we considered hair samples obtained from11 healthy volunteers of similar age with no hearing loss or chronicdiseases, and no GJB2 mutations.
Patients’ files were analyzed in accordance with Italian privacylaw and the hospital’s regulations.
The personal and clinical details considered to describe thesample of patients were as follows: age, gender, cause(s) ofdeafness, genetic mutations, family history of hearing impair-ments, parental consanguinity, hair color, thickness and shape andnationality.
Each patient underwent audiometric assessment and geneticanalysis. Images of the patients’ hair samples underwent scanningelectron microscopy (SEM), and energy dispersive spectroscopy(EDS).
Direct DNA sequencing of the GJB2 gene (analyzing thewhole coding region, including the promoter and non-codify-ing regions) was done at the molecular genetics laboratory inFerrara on genomic DNA obtained from blood samples from allpatients and controls. PCR products were screened formutations by DHPLC (denaturing high-performance liquidchromatography) and SSCP (single strand conformationalpolymorphism). All variant profiles were characterized bysequencing the product of a second PCR amplification using anABI PRISM 3130.
Hair samples were obtained from all patients and controls, andanalyzed under the scanning electron microscope (SEM); a lengthof hair (1 cm long) was cut from near the root with stainless steelscissors. Subjects were asked to not wash their hair with anyshampoo or soap and not to use any hair products for at least 3 daysprior to the collection.
For each subject, we examined the microphotographs of twohair (considering the basal part in three cases and apical part inone).
Table 1Different mutations in GJB2 and GJB6 genes and their relative frequency in the sample
Mutations in GJB2
Homozygosis 35delG/35delG
R143W/R143W
Compound heterozygosis 35delG/M34T
35delG/R184P
35delG/V95M
35delG/L90P
35delG/DConnexin30
Simple heterozygosis M34T
IVS1+1G->A
S139C
T55N
Total
We classified the surface of the hair as normal (intact cuticle) orchanged.
Then, the elements in the hair were further analyzed usingenergy-dispersive X-ray spectroscopy (EDS), where the digitalimage of the intensity of the X-ray characteristic of the atomicstructure of a given element provides a semiquantitative map oftheir distribution on the surface of the sample.
Correlations were drawn using the STATISTICA 7.1 software(Stat Soft Italy), which enabled us to measure the distribution ofthe sample, the significance of the outcomes of ANOVA, and theparametric (Pearson’s r) and nonparametric (Spearman’s) correla-tion indexes.
3. Results
We initially analyzed hair samples from 107 patients, but had toexclude the samples from 22 because the details emerging fromtheir genetic analysis were incomplete.
The final sample consisted of 96 subjects, i.e. a study group of 42patients with hearing impairments of genetic origin (38 with anon-syndromic form, 4 with a syndromic form), and a controlgroup including 54 people, i.e. 43 patients with other, non-genetichearing impairments and 11 healthy volunteers aged up to 10years old.
The median age of the 96 participants was 9 years and 5 months(SD � 11 years 6 months); 85% of the subjects were less than 10 yearsold and 15% were over ten; the youngest was 1 year and 8 months old,and the oldest was 59.
Most of the patients were Italian (78.8%), 12% came from otherEuropean countries, 4% from Asia, 3% from Africa and 1% fromAmerica.
With the exception of 4 cases of progressive hearing loss, all thepatients had bilateral, symmetrical, congenital, profound hearingimpairments. Just one out 4 patients with progressive form washomozygotic for the 35delG mutation. There were no cases ofparental consanguinity, while 10 patients had a family history ofdeafness with at least one member of their family suffering fromhearing loss.
No correlations emerged between hair color or shape and thehair phenotypes except for the fact that 45% of the patients under10 years old with a connexin mutation had very thin hair.
Among the 96 subjects, 38 had a GJB2 gene mutation. All of the38 patients with non-syndromic form had a mutation on GJB2gene. Among the 4 patients with the syndromic form (one relatedto the Waardemburg syndrome, one to the Cockayne sindrome oneto the Opitz syndrome and one to the DOOR syndrome) no one hada GJB2 mutation. Among the 58 who had no GJB2 gene mutations,there was 1 deaf patient with a pendrin gene mutation (G334A) inheterozygosis, not previously described in the literature.
We found 11 different genotypes for the GJB2 allele. The mostfrequent mutation was 35delG; homozygotic patients with
.
No. of patients and relative frequency Total
26 (30.6%) 27 (28.1%)
1 (1.2%)
1 (1.2%) 6 (8.8%)
2 (2.4%)
1 (1.2%)
1 (1.2%)
1 (1.2%)
1 (1.2%) 5 (5.2%)
2 (2.4%)
1 (1.2%)
1 (1.2%)
38 (39.6%)
T. Volo et al. / International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–12851282
35delG/35delG accounted for 30% of the whole sample, whilepatients heterozygotic for the 35delG mutation represented 6% ofthe sample. The genotypes found and their relative frequency areshown in Table 1.
4. Results of scanning electron microscopy
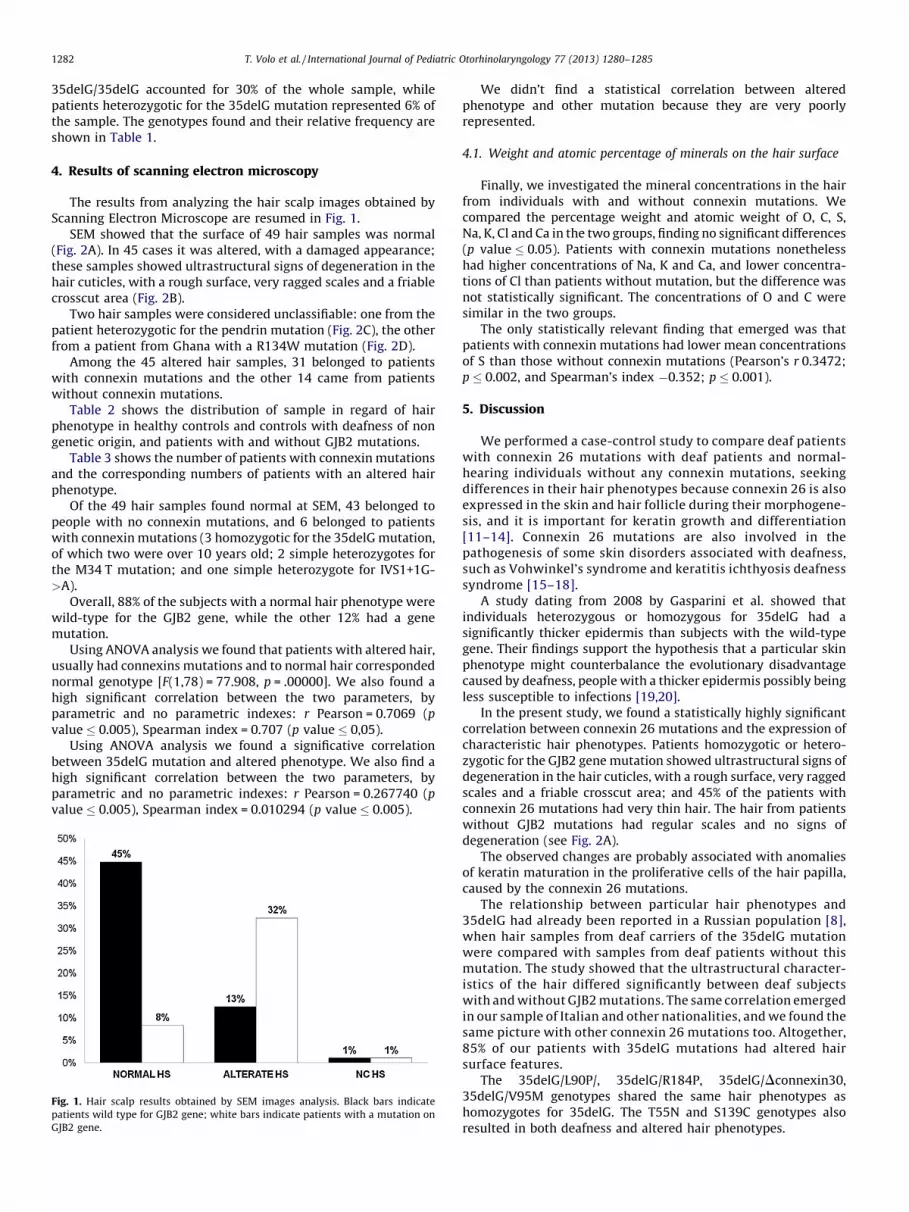
The results from analyzing the hair scalp images obtained byScanning Electron Microscope are resumed in Fig. 1.
SEM showed that the surface of 49 hair samples was normal(Fig. 2A). In 45 cases it was altered, with a damaged appearance;these samples showed ultrastructural signs of degeneration in thehair cuticles, with a rough surface, very ragged scales and a friablecrosscut area (Fig. 2B).
Two hair samples were considered unclassifiable: one from thepatient heterozygotic for the pendrin mutation (Fig. 2C), the otherfrom a patient from Ghana with a R134W mutation (Fig. 2D).
Among the 45 altered hair samples, 31 belonged to patientswith connexin mutations and the other 14 came from patientswithout connexin mutations.
Table 2 shows the distribution of sample in regard of hairphenotype in healthy controls and controls with deafness of nongenetic origin, and patients with and without GJB2 mutations.
Table 3 shows the number of patients with connexin mutationsand the corresponding numbers of patients with an altered hairphenotype.
Of the 49 hair samples found normal at SEM, 43 belonged topeople with no connexin mutations, and 6 belonged to patientswith connexin mutations (3 homozygotic for the 35delG mutation,of which two were over 10 years old; 2 simple heterozygotes forthe M34 T mutation; and one simple heterozygote for IVS1+1G->A).
Overall, 88% of the subjects with a normal hair phenotype werewild-type for the GJB2 gene, while the other 12% had a genemutation.
Using ANOVA analysis we found that patients with altered hair,usually had connexins mutations and to normal hair correspondednormal genotype [F(1,78) = 77.908, p = .00000]. We also found ahigh significant correlation between the two parameters, byparametric and no parametric indexes: r Pearson = 0.7069 (p
value � 0.005), Spearman index = 0.707 (p value � 0,05).Using ANOVA analysis we found a significative correlation
between 35delG mutation and altered phenotype. We also find ahigh significant correlation between the two parameters, byparametric and no parametric indexes: r Pearson = 0.267740 (p
value � 0.005), Spearman index = 0.010294 (p value � 0.005).
Fig. 1. Hair scalp results obtained by SEM images analysis. Black bars indicate
patients wild type for GJB2 gene; white bars indicate patients with a mutation on
GJB2 gene.
We didn’t find a statistical correlation between alteredphenotype and other mutation because they are very poorlyrepresented.
4.1. Weight and atomic percentage of minerals on the hair surface
Finally, we investigated the mineral concentrations in the hairfrom individuals with and without connexin mutations. Wecompared the percentage weight and atomic weight of O, C, S,Na, K, Cl and Ca in the two groups, finding no significant differences(p value � 0.05). Patients with connexin mutations nonethelesshad higher concentrations of Na, K and Ca, and lower concentra-tions of Cl than patients without mutation, but the difference wasnot statistically significant. The concentrations of O and C weresimilar in the two groups.
The only statistically relevant finding that emerged was thatpatients with connexin mutations had lower mean concentrationsof S than those without connexin mutations (Pearson’s r 0.3472;p � 0.002, and Spearman’s index �0.352; p � 0.001).
5. Discussion
We performed a case-control study to compare deaf patientswith connexin 26 mutations with deaf patients and normal-hearing individuals without any connexin mutations, seekingdifferences in their hair phenotypes because connexin 26 is alsoexpressed in the skin and hair follicle during their morphogene-sis, and it is important for keratin growth and differentiation[11–14]. Connexin 26 mutations are also involved in thepathogenesis of some skin disorders associated with deafness,such as Vohwinkel’s syndrome and keratitis ichthyosis deafnesssyndrome [15–18].
A study dating from 2008 by Gasparini et al. showed thatindividuals heterozygous or homozygous for 35delG had asignificantly thicker epidermis than subjects with the wild-typegene. Their findings support the hypothesis that a particular skinphenotype might counterbalance the evolutionary disadvantagecaused by deafness, people with a thicker epidermis possibly beingless susceptible to infections [19,20].
In the present study, we found a statistically highly significantcorrelation between connexin 26 mutations and the expression ofcharacteristic hair phenotypes. Patients homozygotic or hetero-zygotic for the GJB2 gene mutation showed ultrastructural signs ofdegeneration in the hair cuticles, with a rough surface, very raggedscales and a friable crosscut area; and 45% of the patients withconnexin 26 mutations had very thin hair. The hair from patientswithout GJB2 mutations had regular scales and no signs ofdegeneration (see Fig. 2A).
The observed changes are probably associated with anomaliesof keratin maturation in the proliferative cells of the hair papilla,caused by the connexin 26 mutations.
The relationship between particular hair phenotypes and35delG had already been reported in a Russian population [8],when hair samples from deaf carriers of the 35delG mutationwere compared with samples from deaf patients without thismutation. The study showed that the ultrastructural character-istics of the hair differed significantly between deaf subjectswith and without GJB2 mutations. The same correlation emergedin our sample of Italian and other nationalities, and we found thesame picture with other connexin 26 mutations too. Altogether,85% of our patients with 35delG mutations had altered hairsurface features.
The 35delG/L90P/, 35delG/R184P, 35delG/Dconnexin30,35delG/V95M genotypes shared the same hair phenotypes ashomozygotes for 35delG. The T55N and S139C genotypes alsoresulted in both deafness and altered hair phenotypes.

Fig. 2. Hair scalp SEM images: (A) Normal hair at SEM. (B) Changed hair at SEM. (C) Hair in patient heterozigotes for a mutation of pendrin. (D) Hair in patient from Ghana with
a R134W mutation.
T. Volo et al. / International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–1285 1283
An exception was seen in two deaf patients heterozygotic forM34T, and in one deaf patient heterozygotic for IVS1+1G->Amutation, whose SEM findings showed a normal hair phenotype.
There is some controversy regarding the underlying mechanismthat induces the allelic variant M34T (the first-described autoso-mal dominant variant associated with deafness) to cause heredi-tary deafness [21,22] The debate concerns its ability to formfunctional channels, and its dominant or recessive nature. In fact,M34T has been described both as an autosomal dominant [23] andas a recessive mutation [24]. A recent study suggested that M34Tcould be regarded as a dominant allele but its activity may beaffected by modifying factors, such as an increased level of wild-type RNA expression (which may vary in different tissues) [25].This could explain the variety of clinical manifestations and whythe patients who were simple heterozygotes for M34T in thepresent study had hearing loss with a normal hair phenotype. Inaddition, the only patient heterozygotic for 35delG/M34T had adamaged hair phenotype.
Another 3 patients with DFNB1 deafness had a normal hairsurface: an explanation for this could lie in that we know that thehair contains connexin 26, but we do not know how it interactswith the other connexins expressed in human hair. Indeed,although some biophysical and regulatory characteristics are
Table 2Distribution of sample in regard of hair phenotype in healthy controls and controls
with deafness of non genetic origin, and patients with and without GJB2 mutations.
Control 55 Patients 41
Healthy Non genetic Genetic
GJB2 Non GJB2
Altered hair surface 2 (19%) 10 (24%) 31 (82%) 2 (50%)
Normal hair surface 9 (81%) 33 (76%) 6 (16%) 1 (25%)
Unclassifiable hair surface 1 (2%) 1 (25%)
Total 11 43 38 4
specific to channels made by certain connexin types, all connexinsform channels with similar general properties, such as permeabil-ity to multiple ions, a variety of second messengers, metabolites,nutrients and other low-molecular-weight substance [26]. The hairphenotype may be normal because the keratin cells may use avariety of channels consisting of different types of connexins.
Twelve patients with a non-DFNB1 hearing impairment alsoshowed signs of hair surface degeneration. The likely reason forthis lies in that many other factors can contribute to damaging thestructure of an individual’s hair, such as nutrition or hormonestatus.
Using EDS microanalysis, we found no statistically significantdifferences in the mineral composition of the hair samples wetested (with the exception of a lower S concentration in patientswith connexin 26 mutations). This was because EDS only enablesthe mineral composition of the hair surface to be analyzed. Infuture we would suggest using other methods capable ofascertaining the mineral composition of the whole sample, e.g.X-ray powder diffraction, or the X-ray fluorescence technique [27],in which case the material being analyzed is finely ground andhomogenized before the analysis and its full thickness can bestudied [28].
Regarding the lower concentration of S, this might be the reasonwhy the hair from patients with connexin mutations appeared tobe very thin. There are reports in the literature of a lower Sconcentration being associated with trichothiodystrophy (adisease characterized by a deficient DNA damage repair systemand sulfur-deficient, brittle hair) and with a pediatric case ofkeratitis ichthyosis deafness syndrome involving hair anomaliesresembling trichothiodystrophy [29]. In these conditions, thelower S concentrations correlate with diminished cysteine levels.
As for the two hair samples that we judged as unclassifiable, onebelonged to a patient from Ghana with a R134W mutation(Fig. 2D), the other to a patient with a pendrin mutation (Fig. 2C).We hypothesized that the particular appearance of formerpatient’s hair was due to a loss of the cuticle pattern, a feature

Table 3Connexin 26 and connexin 30 mutations in patients with hair surface changes, and total numbers of patients with the same mutation.
Mutation No. of patients with connexin mutations
and altered hair surface
Total no. of patients with connexin mutations
Homozygote 35delG 23 26
Compound heterozygote 35delG/M34T 1 1
Compound heterozygote 35delG/R184P 2 2
Compound heterozygote 35delG/L90P 1 1
Compound heterozygote 35delG/Dconnexin30 1 1
Simple heterozygote S139C 1 1
Simple heterozygote M34T 0 2
Homozygote R134W N.C. (?) 1
Compound heterozygote 35delG/V95M 1 1
Simple heterozygote IVS1+1G->A 0 1
Simple heterozygote T55N 1 1
T. Volo et al. / International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–12851284
that may be seen also in Caucasians and Asians too, but that weonly found in this African patient [30]. Our sample includedanother Africa patient, from Burkina Faso, whose hair phenotypewas normal at SEM so, for the time being, we cannot say whetherour Ghanaian’s connexin mutation had a role in the patient’sparticular hair phenotype.
The hair sample from the patient heterozygotic for a pendrinmutation (G334A) was also considered unclassifiable because itshowed a specific phenotype resembling reptile skin. On EDS, thishair revealed a higher concentration of sodium than any of theother samples. Another particular feature of this patient’sphenotype was that the child had no sweat pores (in epithelialtissue organs such as the cochlea, epidermis and hair follicles). Thechild is currently being investigated for Cockayne syndrome, agenetic disease characterized by defects in DNA nucleotideexcision repair (NER). The NER pathway serves to repair DNAdamaged by the ultraviolet radiation in sunlight [31]. For thepresent, we cannot say whether the patient’s particular hairphenotype correlates with the child’s pendrin mutation or anyother variables, but we plan to conduct SEM investigations on scalphair from other patients with pendrin mutations.
6. Conclusions
Congenital deafness is an important public health issue becauseof its lifelong impact on quality of life and expensive treatment andrehabilitation programs. Non-syndromic hearing loss is the mostprevalent form worldwide, and most cases are caused by recessivemutations in the GJB2 gene [32].
This picture is reflected in our patients too, whose hearing losswas often due to GJB2 gene mutations (in 37 of 85 patients, 43.7%of the sample), followed by congenital cytomegalovirus infections(in 5 patients, 5.9%).
Our findings enable us to confirm a direct correlation betweenGJB2 mutations and hair phenotypes, and suggest that furtherresearch is warranted on gap junction function in hair.
Previous works have demonstrated that a particular skinphenotype correlates with an altered connexin expression (GJB2mutation carriers have a thicker epidermis than people with the wild-type GJB2 gene), so it would seem worthwhile to conduct furtherfunctional studies on connexin gene expression in these tissues.
A better understanding of the new phenotype with the pendrinmutation, of the Ghanaian child, and of our patients with lower Sconcentrations on the surface of their hair may help to shed lighton the genotype–phenotype correlations in patients with hearingloss, and may elucidate the function of different regions ofconnexin 26.
The evidence emerging from the present study could contributein the future to a better knowledge of what happens in the earwhen connexin 26 is mutated.
Acknowledgements
The authors would like to thank Zhuravskiy S. G. for his valuablecomments during the study and Frances Coburn for providinglanguage help.
References
[1] L. Bubbico, A. Rosano, A. Spagnolo, Prevalence of prelingual deafness in Italy, ActaOtorhinol. Italy 27 (2007) 17–21.
[2] F. Gualandi, A. Martini, E. Calzolari, Progress in understanding GJB2-linkeddeafness, Commun. Genet. 6 (3) (2003) 125–132, Review.
[3] F. Gualandi, A. Ravani, A. Berto, A. Sensi, C. Trabanelli, F. Falciano, et al., Exploringthe clinical and epidemiological complexity of GJB2-linked deafness, Am. J. Med.Genet. 15 (2002) 38–45.
[4] Snoeckx Rl, Huygen Pl, D. Feldmann, S. Marlin, F. Denoyelle, J. Waligora, et al., GJB2mutations and degree of hearing loss: a multicenter study, Am. J. Hum. Genet. 77(6) (2005) 945–957.
[5] E. Gualandi, A. Ravani, A. Berto, S. Burdo, P. Trevisi, A. Ferlini, et al., Occurrence ofdel(GIB6-D13S1830) mutation in Italian non-syndromic hearing loss patientscarrying a single GJB2 mutated allele, Acta Otolaryngol. Suppl. 552 (2004) 29–34.
[6] N. Hilgert, Huentelman Mj, Thorburn Aq, E. Fransen, N. Dieltjens, M. Mueller-Malesinska, et al., Phenotypic variability of patients homozygous for the GJB2mutation 35delG cannot be explained by the influence of one major modifiergene, Eur. J. Hum. Genet. 17 (4) (2009) 517–524.
[7] P. Gasparini, P.P. Guastalla, V.I. Guerci, et al., Detection of epidermal thickening inGJB2 carriers with epidermal US, Radiology 251 (2009) 280–286.
[8] K. Heathcote, P. Syrris, N.D. Carter, M.A. Patton, A connexin 26 mutation causes asyndrome of sensorineural hearing loss and palmoplantar hyperkeratosis, J. Med.Genet. 37 (2000) 50–51.
[9] S.G. Zhuravskiy, A.A. Kurus, A.E. Taraskina, S.A. Ivannov, Ultrastructure of the hairin genetic prelingual deafness associated with 35delG mutation in the Con-nexin26 gene (GJB2), Bull. Exp. Biol. Med. 148 (2009) 79–81.
[10] S. Iossa, E. Marciano, A. Franze, GJB2 gene mutations in syndromic skin diseaseswith sensorineural hearing loss, Curr. Genom. 12 (7) (2011) 475–785.
[11] P. Lefebvre, T. Van De Water, Connexins, hearing and deafness: clinical aspects ofmutations in the connexin26 gene, Brain Res. Rev. 32 (2000) 159–162.
[12] M.B. Peterson, P.J. Willems, Non syndromic autosomal recessive deafness, Clin.Genet. 69 (2006) 371–392.
[13] G. Van Camp, R.J.H. Smith, The Hereditary Hearing Loss Homepage, 2003.[14] A. Martini, Genetica della funzione uditiva normale e patologica, OMEGA
edizioni, 2006.[15] H. Kokotas, L. Van Laer, G. Van Camp, B. Petersen, Strong linkage disequilibrium
for the frequent GJB2 35delG mutation in the greek population, Am. J. Med. Genet.14 (2008) 2879–2884.
[16] R. Choudry, et al., Changing patterns of gap junction intercellular communicationand connexin distribution in mouse epidermis and hair follicles during embry-ionic development, Dev. Dyn. 210 (1997) 417–430.
[17] E. Maestrini, B.P. Korge, J. Ocana-Sierra, A missense mutation in connexin26,D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’ssyndrome) in three unrelated families, Hum. Mol. Genet. 8 (1999) 1237–1243.
[18] G. Richard, F. Rouan, C.E. Willoughby, et al., Misses mutations in GJB2 encodingconnexin26 cause the ectodermal dysplasia keratits-ichtyosis-deafness syn-drome, Am. J. Human Genet. 70 (2002) 1341–1348.
[19] Y.K. Man, C. Trolove, D. Tattersall, et al., A deafness-associate mutant humanconnexin 26 improves the epithelial barrier in vitro, J. Membr. Biol. 218 (2007)29–34.
[20] C. Meyer, et al., Selection for deafness? Nat. Med. 8 (2002) 1332–1333.[21] M. Bicego, M. Beltramello, S. Melchionda, M. Carella, V. Piazza, L. Zelante, et al.,
Pathogenic role of deafness related to M34 T mutation of Connexin 26, Hum. Mol.Genet. 17 (2006) 2569–2587.
[22] I.M. Skerrett, E.M. Kasperek, M.E. Kelsell, D.P.B.J. Nicholson, Aberrant gating, butnormal expression pattern underlies the recessive phenotype of the deafnessmutant Connexin 26 M34 T, FASEB J. 18 (2004) 860–862.

T. Volo et al. / International Journal of Pediatric Otorhinolaryngology 77 (2013) 1280–1285 1285
[23] D.P. Kelsell, J. Dunlop, H.P. Stevens, N.J. Lench, N.J. Liang, J. Parry, et al., Connexin26 mutations in hereditary non-syndromic sensorineural deafness, Nature 387(1997) 80–83.
[24] M.J. Houseman, L.A. Ellis, A. Pagnamenta, D.I.W.L. Rickard, S. Osborn, H.A. Dahl,et al., Genetic analysis of the Connexin 26 M34T variant: identificatoin ofgenotype M34T/M34T segregating with mild-moderate non-syndromic sensori-neural hearing loss, J. Med. Genet. 38 (2001) 20–25.
[25] E. Wilch, M. Zhu, K.B. Burkhart, M. Regier, J.L. Elfenbein, L.A. Fisher, et al.,Expression of GJB2 and GJB6 is reduced in a novel DFNB1 allele, Am. J. Hum.Genet. 79 (2006) 174–179.
[26] L. Wiszniewski, A. Limat, J.H. Saurat, P. Meda, D. Salomon, Differental expressionof human connexins during stratification of keratynocites, J. Invest. Dermol. 115(2000) 278–285.
[27] Y. Dede, H.N. Erten, A. Zararsiz, N. Efe, Determination of trace element levels inhuman scalp hair in occupationally exposed subjects by XRF, J. Radio NuclearChem. 247 (2001) 393–397.
[28] C. Whiston, in: P. Elizabeth (Ed.), X-ray methods, Anal. Chem. (1987) (openlearning).
[29] L. Raeve, M. Bonduelle, D. Roseeuw, J. Stene, Trichothiodystrofy-like hair abnor-malities in a child with Keratitis Hychtyosis Deafness Syndrome, Pediatr. Der-matol. 25 (2008) 466–469.
[30] N.P. Khumalo, P.T. Doe, P.R. Dawber, D.J.P. Ferguson, What is normal black Africanhair? J. Am. Dermatol. 4 (2000) 815–820.
[31] K. Kraemer, N. Patronas, R. Shiffmann, B. Brooks, D. Tamura, D.I.J. Giovanna,Xeroderrma pigmentosum, trichothiodystrophy and Cockayne syndrome: acomplex genotype–phenotype relationship, Neuroscience 145 (2007)1388–1396.
[32] A. Davis, S. Hind, The impact of hearing impairment: a global health problem, Int.J. Pediatr. Otorhinolaryngol. 49 (1999) S51–S54.