glutamine protects mitochondrial structure and function in oxygen toxicity
TRANSCRIPT

Glutamine protects mitochondrial structureand function in oxygen toxicity
SHAMA AHMAD,1 CARL W. WHITE,1 LING-YI CHANG,2
BARBARA K. SCHNEIDER,1 AND CORRIE B. ALLEN1
Departments of 2Medicine and 1Pediatrics, National JewishMedical and Research Center, Denver, Colorado 80206Received 8 May 2000; accepted in final form 20 October 2000
Ahmad, Shama, Carl W. White, Ling-Yi Chang, Bar-bara K. Schneider, and Corrie B. Allen. Glutamine pro-tects mitochondrial structure and function in oxygen toxicity.Am J Physiol Lung Cell Mol Physiol 280: L779–L791,2001.—Glutamine is an important mitochondrial substrateimplicated in the protection of cells from oxidant injury, butthe mechanisms of its action are incompletely understood.Human pulmonary epithelial-like (A549) cells were exposedto 95% O2 for 4 days in the absence and presence of glu-tamine. Cell proliferation in normoxia was dependent onglutamine, and glutamine deprivation markedly acceleratedcell death in hyperoxia. Glutamine significantly increasedcellular ATP levels in normoxia and prevented the loss ofATP in hyperoxia seen in glutamine-deprived cells. Mito-chondrial membrane potential as assessed by flow cytometrywith chloromethyltetramethylrosamine was increased byglutamine in hyperoxia-exposed A549 cells, and a glutaminedose-dependent increase in mitochondrial membrane poten-tial was detected. Glutamine-supplemented, hyperoxia-ex-posed cells had a higher O2 consumption rate and GSHcontent. Electron and fluorescence microscopy revealed that,in hyperoxia, glutamine protected cellular structures, espe-cially mitochondria, from damage. In hyperoxia, activity ofthe tricarboxylic acid cycle enzyme a-ketoglutarate dehydro-genase was partially protected by its indirect substrate, glu-tamine, indicating a mechanism of mitochondrial protection.
human; airway; epithelium; adenosine 59-triphosphate; a-ke-toglutarate dehydrogenase; mitochondrial membrane poten-tial
HYPEROXIA CAUSES reactive oxygen species-mediated in-jury to lung cells that may contribute to the pathogen-esis of various lung diseases (18, 23, 35, 45). In addi-tion, elevated concentrations of O2 in cells in vitrocause inhibition of cellular proliferation and lunggrowth by effecting changes in mitochondrial metabo-lism and respiration and by causing DNA damage (10,38). Hyperoxia rapidly inhibits aconitase, the initialenzymatic step in the tricarboxylic acid (TCA; Krebs)cycle both in cultured cells and in the lungs of rats (19)and newborn primates (35). Cellular respiration de-clines in parallel with the loss of aconitase activity incells cultured in hyperoxia, and inhibitors of aconitasecause a similar decrease in respiration in cells cultured
under normal O2 tensions (19). One mechanism bywhich cultured cells adapt to the stress of hyperoxicexposure is through increased glycolysis (4, 24). Invivo, the lungs of rats adapted to hyperoxia have in-creased total activity of hexokinase (3), the rate-limit-ing step in glycolysis in the rat lung (42). In addition, anovel isoform, hexokinase II, is expressed in the lungsof these rats, and there also is increased expression ofhexokinase III, a nuclear isoform.
Although impairment of the aconitase step occursrapidly during hyperoxic exposure, inhibition of subse-quent steps in the TCA cycle pathway occurs later andless completely (24). The alternate substrate glu-tamine enters the TCA cycle subsequent to the aconi-tase step. Therefore, we hypothesized that increasedutilization of glutamine could contribute to hyperoxicadaptation.
Mitochondria are a potential target of injury by ox-ygen radicals, and an alteration in mitochondrial mem-brane function is an important component of oxidativestress in cells (5, 52). Because the mitochondrial mem-brane potential (MMP) in situ is a measure of theenergetic state of the cell as well as a sensitive indica-tor of mitochondrial function, we assessed the electri-cal potential across the inner mitochondrial membraneof air- and O2-exposed human pulmonary epithelial-like (A549) cells. This was done by the use of flowcytometry and the specific dye chloromethyltetrameth-ylrosamine (CMTMRos; MitoTracker Orange, Molecu-lar Probes, Eugene, OR). It was found that the innerMMP was profoundly affected by the presence orabsence of glutamine, a mitochondrial substrate, inthe growth medium of O2-exposed A549 cells. Wealso performed a series of experiments to evaluate theeffect of glutamine supplementation on survival,growth, cell and organelle morphology, ATP content,respiration, a-ketoglutarate dehydrogenase activity,glutamine consumption, and cellular glutathione(GSH) content of these cells in hyperoxia. Our findingsindicate that cells cultured in air in the absence ofglutamine can survive, but not proliferate, and pre-serve mitochondrial integrity. In hyperoxia, on the
Address for reprint requests and other correspondence: C. W.White, 1400 Jackson St., Rm. J101, Denver, CO 80206 (E-mail:[email protected]).
The costs of publication of this article were defrayed in part by thepayment of page charges. The article must therefore be herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. Section 1734solely to indicate this fact.
Am J Physiol Lung Cell Mol Physiol280: L779–L791, 2001.
1040-0605/01 $5.00 Copyright © 2001 the American Physiological Societyhttp://www.ajplung.org L779

other hand, these cells can neither proliferate nor sur-vive, and their death is preceded by degeneration oftheir mitochondria. Paradoxically, cells provided withglutamine utilized the amino acid at a considerablyincreased rate in hyperoxia compared with cells ex-posed to normal O2 tensions.
METHODS
Cells and culture. The human epithelial-like lung carci-noma cell line A549 was obtained from the American TypeCulture Collection (Manassas, VA). The cells were grown in100-mm Falcon tissue culture dishes in 10 ml of F-12Kgrowth medium (GIBCO BRL, Life Technologies, Grand Is-land, NY) containing 10% fetal calf serum, 100 U/ml ofpenicillin, 100 mg/ml of streptomycin, 20 mM glucose, and 2mM glutamine incubated at 37°C under a humidified atmo-sphere of air containing 5% CO2. A549 cultures were rou-tinely passaged by trypsinization and subcultured at aninitial plating density of 0.5 million cells/plate. Hyperoxicexposures were maintained at Denver’s atmospheric pres-sure (635 mmHg) and performed in a humidified airtightplastic incubator chamber (Billups Rothenberg, Del Mar, CA)gassed with 95% O2-5% CO2, and the cultures were incu-bated at 37°C. For experiments, custom-made F-12K medium(GIBCO BRL) without L-glutamine was used. It was supple-mented with fresh L-glutamine for the studies involving theabsence and presence of glutamine. Fresh medium was sup-plied daily during the hyperoxic exposures.
Small-airway epithelial cells (SAECs) were purchased asfrozen primary cultures from Clonetics (Walkersville, MD).They were cultured in 100-mm Falcon tissue culture dishesin 10 ml of the supplier’s SAEC basal medium supplementedwith gentamicin, amphotericin B, bovine pituitary extract,hydrocortisone, human epithelial cell growth factor, epineph-rine, transferrin, insulin, retinoic acid, triiodothyronine, andbovine serum albumin as per the supplier’s recommenda-tions. Although human SAECs can be maintained in culturefor five to six passages with the supplier’s serum-free me-dium, all experiments presented here were performed withSAECs that had been passaged a maximum of four times toensure no loss of phenotype. Cultures were split at 80–90%confluence by trypsin digestion and subcultured in the sameSAEC medium.
Measurement of MMP. MMP was estimated by the uptakeof a fixable dye, CMTMRos (MitoTracker Orange, MolecularProbes) according to the method of Macho et al. (30). Thirtyminutes before the end of hyperoxic exposure, the mediumoverlying the cells was replaced with gas-conditioned (air- orO2-containing) medium containing 150 nM CMTMRos andreturned to the incubator. At the end of the exposure, themedium over the cells was aspirated, and the cells werewashed once with phosphate-buffered saline (PBS) and thenharvested with a dye-containing trypsin-EDTA solution. Thecells were pelleted at 200 g for 10 min, and the supernatantwas removed. The cell pellet was resuspended in 1 ml of PBSand then fixed with 1 ml of 8% paraformaldehyde (ElectronMicroscopy Sciences, Fort Washington, PA) in PBS (pH 7.4).After incubation in the dark at room temperature on a shakerfor 30 min, the cells were kept on ice and analyzed with anEPICS XL flow cytometer (Coulter, Hialeah, FL) operated byCoulter’s System II software and incorporating an argonlaser (488 nm, 15 mW) for excitation. MitoTracker Orangefluorescence was assessed in FL2 (575-nm band-pass filter).Carbonyl cyanide m-chlorophenylhydrozone (CCCP; Calbio-chem, San Diego, CA) dissolved in dimethyl sulfoxide(DMSO) was added along with the dye MitoTracker Orange
for MMP inhibition studies. List mode files were collected foreach sample and transferred to a Macintosh G3 computer forsubsequent analysis. Mean fluorescence intensity (MFI) ofthe cells was used as the primary index for comparison ofMMP. Corrections for any changes in MFI due to forwardlight scatter (FS) were done by plotting MFI against FS. Theslope (MFI/FS) of these distributions for each sample, calcu-lated from the regression analysis and analysis of covariance(ANCOVA) with JMP software (SAS Institute, Cary, NC),was considered a better indicator of MMP.
Cellular O2 consumption. Cellular O2 consumption wasmeasured in a custom-built six-place respirometer. Eachchamber of this apparatus consisted of a glass water-jacketedcell (Gilson Medical Electronics) fitted with a Clark-stylepolarographic O2 electrode (model 5331, Yellow Springs In-struments, Yellow Springs, OH). The six chambers were fixedon a multiposition electromagnetic stir plate (Cole-Parmer,Vernon Hills, IL) placed within a tissue culture incubatormaintained at 37°C. The six electrodes were connected to achemical microsensor II (Diamond General Development,Ann Arbor, MI) through a 10-channel multiplexer. Channelselection and data collection were achieved by using Lab-VIEW software (National Instruments, Austin TX). Eachelectrode was preequilibrated with 1.4 ml of medium. Next, 1million cells were added in a 100-ml volume, and the stopperwas placed in the chamber. O2 concentration was measuredfor ;1 h, and the slopes representing O2 consumption werecalculated. The medium O2 saturation values were calcu-lated with the phenazine methosulfate-NADH method asdescribed by Robinson and Cooper (40).
Electron microscopy. For electron-microscopic analysis,control and hyperoxic cells with and without glutamine werefixed in 2% glutaraldehyde in 0.085 M sodium cacodylatebuffer, pH 7.4, containing 0.05% CaCl2. After being scraped,the fixed cells were suspended in fresh fixative and pelletedat 300 g for 10 min. After dehydration and embedding inresin, thin sections were cut with a Reichart Ultra Cut Emicrotome. The sections were collected on 0.4% Formvar-coated, 100-mesh circular grids (3-mm diameter) and stainedwith 2% uranyl acetate and 2% lead citrate for 15 min. Thesections were examined for mitochondria with a PhilipsCM-10 electron microscope at 8 kV, and the images werephotographed.
Fluorescence microscopy. A549 cells were seeded onto22-mm glass coverslips in six-well plates at a density of 3 3105 cells/well in both the presence and absence of glutamine.Hyperoxic exposures were performed as described in Cellsand culture. At the end of exposure, dye (MitoTracker Or-ange, Molecular Probes) dissolved in DMSO and diluted withwarm medium to a concentration of 150 nM was added. Thecells were incubated with the dye for 30 min at 37°C underthe appropriate conditions (21 or 95% O2). The coverslipswith adherent cells were then rinsed with PBS and fixed in4% paraformaldehyde for 10 min. After being rinsed, eachcoverslip was mounted upside down onto a glass slide withaqueous antifade mounting medium (ProLong, MolecularProbes) and allowed to dry overnight. An Olympus Vanox-Tfluorescent microscope attached to a digital camera (Cooke,Auburn Hills, MI) was used to examine the fixed cells. Im-ages were recorded with Slide Book 2.6.5.5 software (Intelli-gent Imaging Innovations, Denver, CO) on a Macintosh G3computer.
Biochemical assays. Cellular growth was measured withthe MTT assay (34) with the water-soluble tetrazolium salt3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide(Sigma). Cells were plated in 96-well half-area tissue cultureplates (Costar 3696), and the medium was replaced by 100 ml of
L780 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
a 1:1 serum- and phenol red-free DMEM-F-12 medium mixture.Fifty microliters of MTT (4 mg/ml) in the serum- and phenolred-free DMEM-F-12 medium mixture were added, and theplate was incubated for 4 h at 37°C. The purple formazancrystals thus formed were dissolved in 50 ml of DMSO, and theoptical density of the wells on the plate was read at 540 nm witha plate reader.
Trypan blue exclusion was performed by adding 25 ml of0.1% trypan blue solution to 100 ml of cells suspended in PBS,and the cells that excluded the dye were counted with ahemacytometer.
For propidium iodide (PI) staining of nonviable cells, ;106
cells were suspended in 1 ml of PBS, and PI (2 mg/ml finalconcentration) was added. After incubation for 5 min on ice inthe dark, flow cytometric analysis was performed.
The protein content of cells was measured with a DCprotein assay kit (Bio-Rad Laboratories, Hercules, CA). ForATP analysis, the cells were harvested, an extract was pre-pared as previously described (4), and total cellular ATPcontent was estimated with a luciferase-luciferin kit (Analyt-ical Luminescence Laboratory, Sparks, MD). The glutaminecontent of the medium was estimated by cation-exchangechromatography on a Beckman 6300 amino acid analyzer ina lithium citrate buffer (33).
For the measurement of a-ketoglutarate dehydrogenaseactivity, air- and O2-exposed cells were washed with PBS andharvested in 1.0 ml of ice-cold Tris zHCl buffer (25 mM Tris,pH 7.4, supplemented with 0.25 M sucrose, 2 mM EDTA, 10mM K2HPO4, 5.0 mM MgCl2, 2.0 mM KCN, and 2 mMglutamine). After centrifugation at 12,000 g for 1 min, thepellet obtained was suspended in 200 ml of the above bufferand sonicated at 10% (setting 3) power three times in 10-sbursts with a model 50 sonic dismembranator (Fisher Scien-tific). a-Ketoglutarate dehydrogenase activity was measuredessentially as described by Bergmeyer (9), at 340 nm and inthe presence of 2 mM NAD1, 20 mM coenzyme A, 2 mM KCN,200 mM thiamine pyrophosphate, and 2 mM a-ketoglutarate.The assay buffer contained 25 mM Tris zHCl, 0.25 M sucrose,2 mM EDTA, 10 mM K2HPO4, and 5.0 mM MgCl2.
The total intracellular GSH was determined with the 5,59-dithio-bis(nitrobenzoic acid)-glutathione reductase recyclingassay (7, 13). A549 cells were harvested in PBS from differentincubation conditions and transferred to microcentrifugetubes. A volume of 200 ml of 2.5% sulfosalicylic acid with 0.2%Triton X-100 was added to the pellet and centrifuged. Thesupernatant was harvested, and 30 ml from each tube weretransferred to a 96-well plate. After this step, 140 ml of 0.3mM NADPH in stock buffer solution (125 mM sodium phos-phate and 6.3 mM EDTA, pH 7.5) and 100 ml of glutathionereductase (1 U/ml; Sigma) were added to each well. Finally,the substrate 5,59-dithio-bis(nitrobenzoic acid) (20 ml of a 6mM solution; Sigma) was added to the reaction. The absor-bance at 405 nm of each well was read with a microplatereader. GSH was quantified with a GSH standard curve.
Statistical analysis. All statistical calculations were per-formed with JMP software (SAS Institute, Cary, NC). Meanswere compared by one-way analysis of variance followed bytwo-tailed t-test for comparison between two groups and theTukey-Kramer test for multiple comparisons. A P value of,0.05 was considered significant.
RESULTS
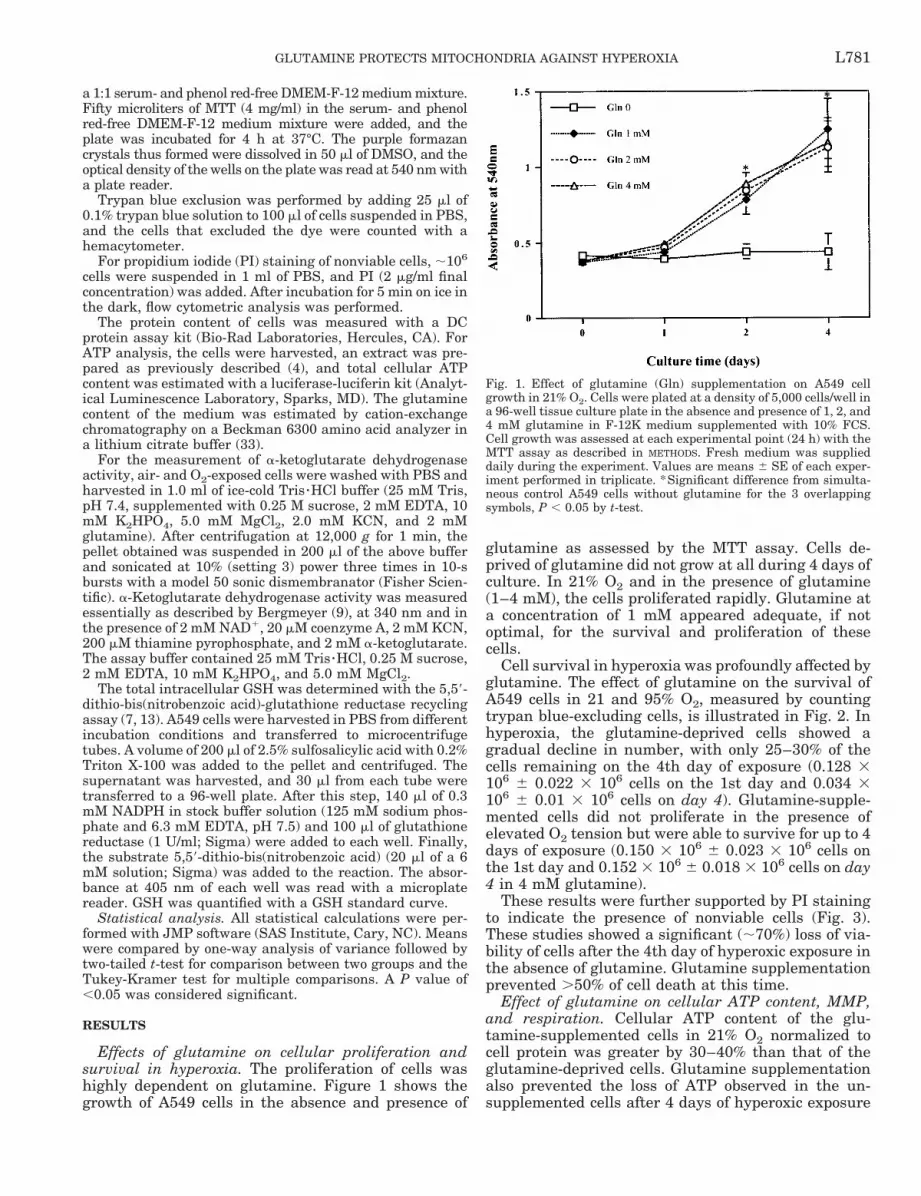
Effects of glutamine on cellular proliferation andsurvival in hyperoxia. The proliferation of cells washighly dependent on glutamine. Figure 1 shows thegrowth of A549 cells in the absence and presence of
glutamine as assessed by the MTT assay. Cells de-prived of glutamine did not grow at all during 4 days ofculture. In 21% O2 and in the presence of glutamine(1–4 mM), the cells proliferated rapidly. Glutamine ata concentration of 1 mM appeared adequate, if notoptimal, for the survival and proliferation of thesecells.
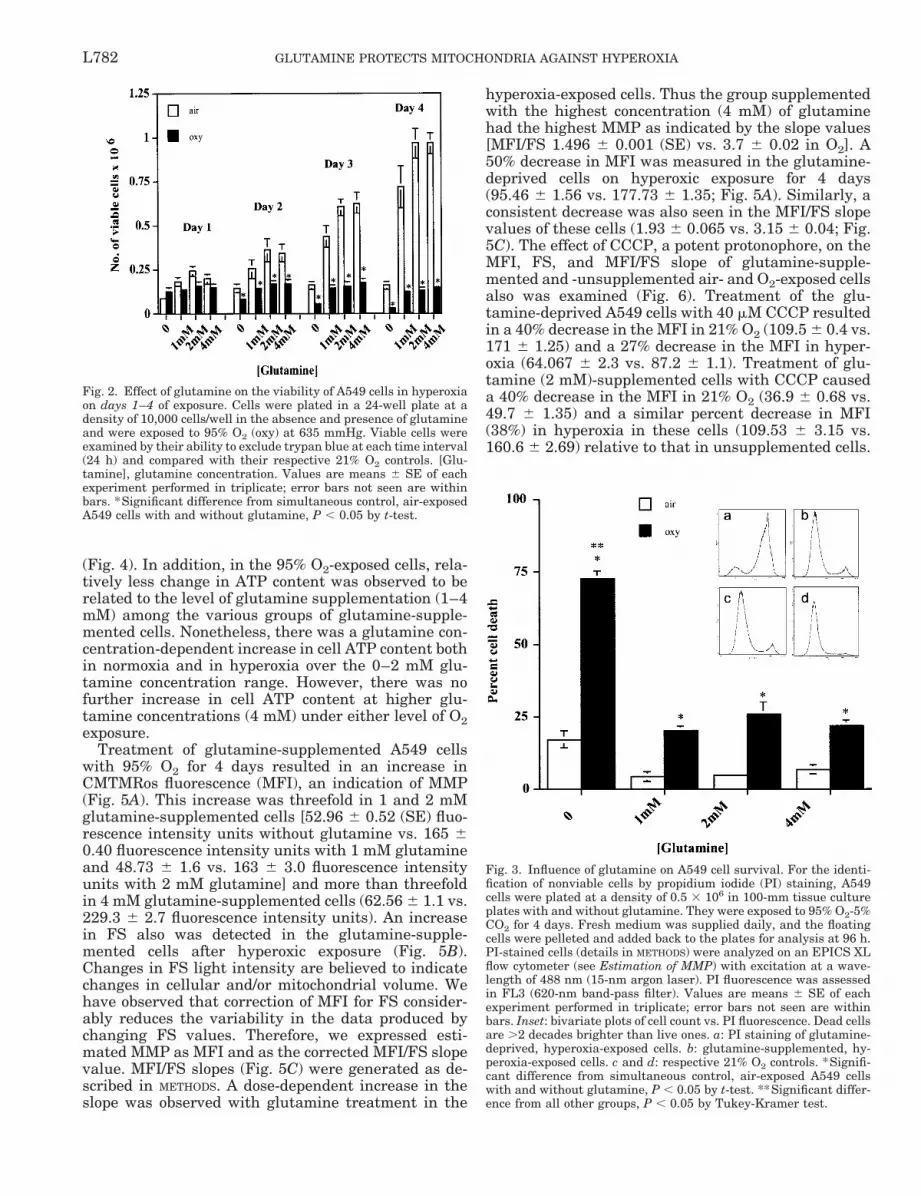
Cell survival in hyperoxia was profoundly affected byglutamine. The effect of glutamine on the survival ofA549 cells in 21 and 95% O2, measured by countingtrypan blue-excluding cells, is illustrated in Fig. 2. Inhyperoxia, the glutamine-deprived cells showed agradual decline in number, with only 25–30% of thecells remaining on the 4th day of exposure (0.128 3106 6 0.022 3 106 cells on the 1st day and 0.034 3106 6 0.01 3 106 cells on day 4). Glutamine-supple-mented cells did not proliferate in the presence ofelevated O2 tension but were able to survive for up to 4days of exposure (0.150 3 106 6 0.023 3 106 cells onthe 1st day and 0.152 3 106 6 0.018 3 106 cells on day4 in 4 mM glutamine).
These results were further supported by PI stainingto indicate the presence of nonviable cells (Fig. 3).These studies showed a significant (;70%) loss of via-bility of cells after the 4th day of hyperoxic exposure inthe absence of glutamine. Glutamine supplementationprevented .50% of cell death at this time.
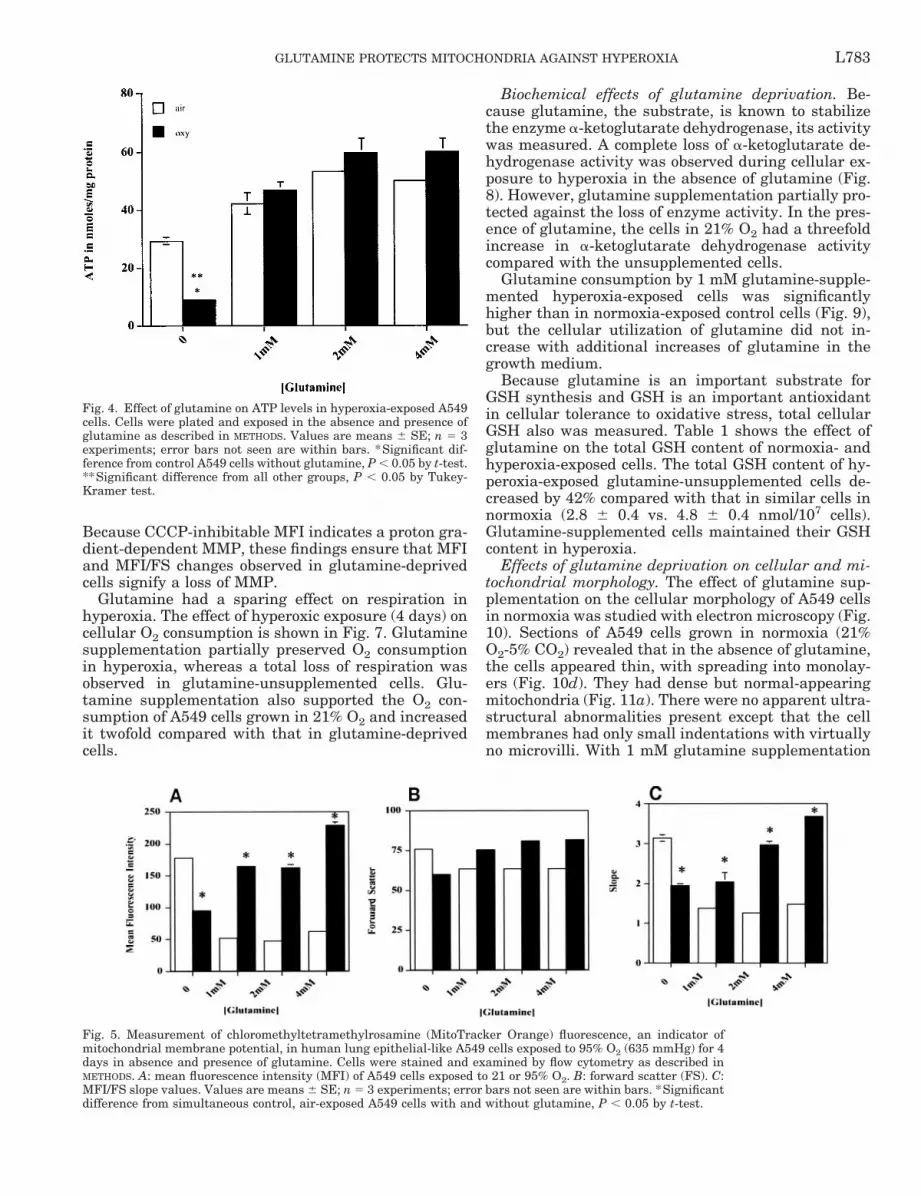
Effect of glutamine on cellular ATP content, MMP,and respiration. Cellular ATP content of the glu-tamine-supplemented cells in 21% O2 normalized tocell protein was greater by 30–40% than that of theglutamine-deprived cells. Glutamine supplementationalso prevented the loss of ATP observed in the un-supplemented cells after 4 days of hyperoxic exposure
Fig. 1. Effect of glutamine (Gln) supplementation on A549 cellgrowth in 21% O2. Cells were plated at a density of 5,000 cells/well ina 96-well tissue culture plate in the absence and presence of 1, 2, and4 mM glutamine in F-12K medium supplemented with 10% FCS.Cell growth was assessed at each experimental point (24 h) with theMTT assay as described in METHODS. Fresh medium was supplieddaily during the experiment. Values are means 6 SE of each exper-iment performed in triplicate. *Significant difference from simulta-neous control A549 cells without glutamine for the 3 overlappingsymbols, P , 0.05 by t-test.
L781GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
(Fig. 4). In addition, in the 95% O2-exposed cells, rela-tively less change in ATP content was observed to berelated to the level of glutamine supplementation (1–4mM) among the various groups of glutamine-supple-mented cells. Nonetheless, there was a glutamine con-centration-dependent increase in cell ATP content bothin normoxia and in hyperoxia over the 0–2 mM glu-tamine concentration range. However, there was nofurther increase in cell ATP content at higher glu-tamine concentrations (4 mM) under either level of O2exposure.
Treatment of glutamine-supplemented A549 cellswith 95% O2 for 4 days resulted in an increase inCMTMRos fluorescence (MFI), an indication of MMP(Fig. 5A). This increase was threefold in 1 and 2 mMglutamine-supplemented cells [52.96 6 0.52 (SE) fluo-rescence intensity units without glutamine vs. 165 60.40 fluorescence intensity units with 1 mM glutamineand 48.73 6 1.6 vs. 163 6 3.0 fluorescence intensityunits with 2 mM glutamine] and more than threefoldin 4 mM glutamine-supplemented cells (62.56 6 1.1 vs.229.3 6 2.7 fluorescence intensity units). An increasein FS also was detected in the glutamine-supple-mented cells after hyperoxic exposure (Fig. 5B).Changes in FS light intensity are believed to indicatechanges in cellular and/or mitochondrial volume. Wehave observed that correction of MFI for FS consider-ably reduces the variability in the data produced bychanging FS values. Therefore, we expressed esti-mated MMP as MFI and as the corrected MFI/FS slopevalue. MFI/FS slopes (Fig. 5C) were generated as de-scribed in METHODS. A dose-dependent increase in theslope was observed with glutamine treatment in the
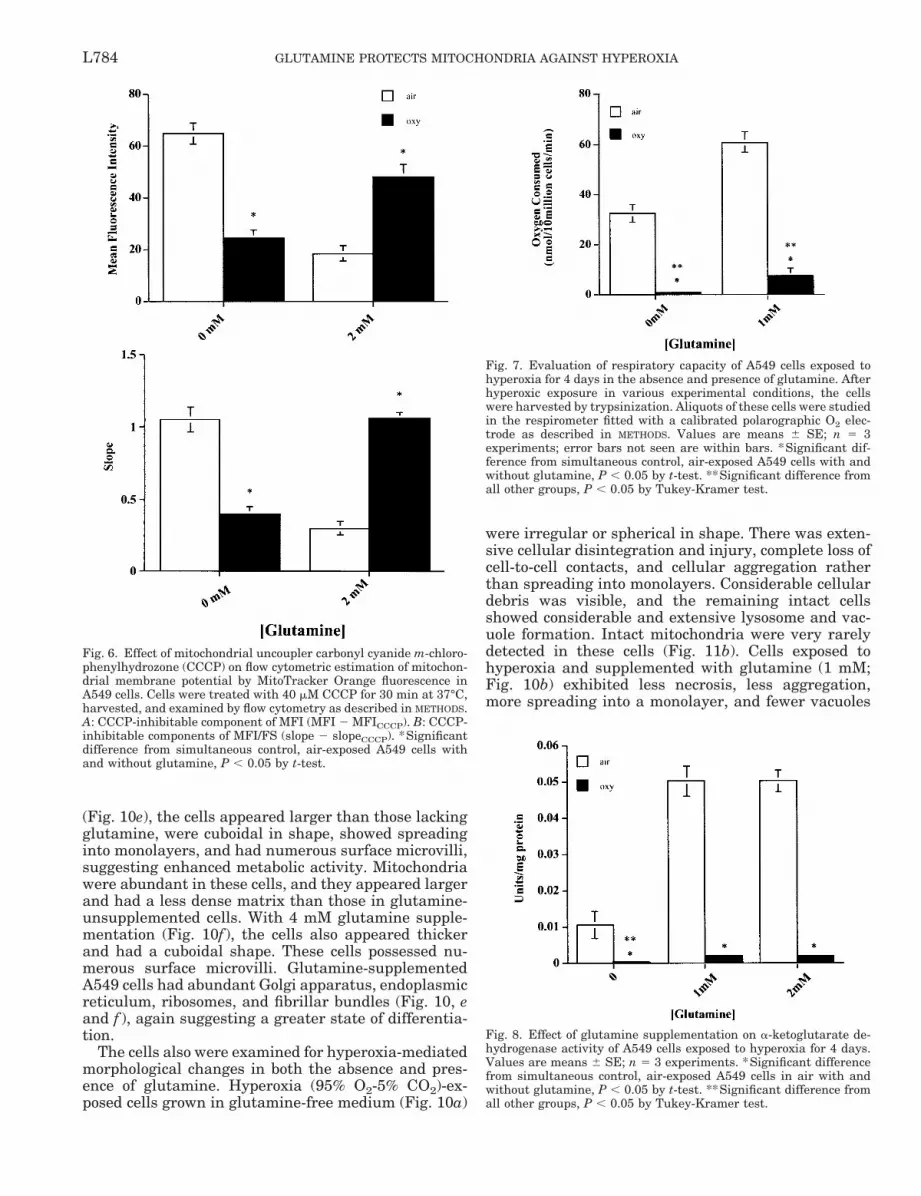
hyperoxia-exposed cells. Thus the group supplementedwith the highest concentration (4 mM) of glutaminehad the highest MMP as indicated by the slope values[MFI/FS 1.496 6 0.001 (SE) vs. 3.7 6 0.02 in O2]. A50% decrease in MFI was measured in the glutamine-deprived cells on hyperoxic exposure for 4 days(95.46 6 1.56 vs. 177.73 6 1.35; Fig. 5A). Similarly, aconsistent decrease was also seen in the MFI/FS slopevalues of these cells (1.93 6 0.065 vs. 3.15 6 0.04; Fig.5C). The effect of CCCP, a potent protonophore, on theMFI, FS, and MFI/FS slope of glutamine-supple-mented and -unsupplemented air- and O2-exposed cellsalso was examined (Fig. 6). Treatment of the glu-tamine-deprived A549 cells with 40 mM CCCP resultedin a 40% decrease in the MFI in 21% O2 (109.5 6 0.4 vs.171 6 1.25) and a 27% decrease in the MFI in hyper-oxia (64.067 6 2.3 vs. 87.2 6 1.1). Treatment of glu-tamine (2 mM)-supplemented cells with CCCP causeda 40% decrease in the MFI in 21% O2 (36.9 6 0.68 vs.49.7 6 1.35) and a similar percent decrease in MFI(38%) in hyperoxia in these cells (109.53 6 3.15 vs.160.6 6 2.69) relative to that in unsupplemented cells.
Fig. 3. Influence of glutamine on A549 cell survival. For the identi-fication of nonviable cells by propidium iodide (PI) staining, A549cells were plated at a density of 0.5 3 106 in 100-mm tissue cultureplates with and without glutamine. They were exposed to 95% O2-5%CO2 for 4 days. Fresh medium was supplied daily, and the floatingcells were pelleted and added back to the plates for analysis at 96 h.PI-stained cells (details in METHODS) were analyzed on an EPICS XLflow cytometer (see Estimation of MMP) with excitation at a wave-length of 488 nm (15-nm argon laser). PI fluorescence was assessedin FL3 (620-nm band-pass filter). Values are means 6 SE of eachexperiment performed in triplicate; error bars not seen are withinbars. Inset: bivariate plots of cell count vs. PI fluorescence. Dead cellsare .2 decades brighter than live ones. a: PI staining of glutamine-deprived, hyperoxia-exposed cells. b: glutamine-supplemented, hy-peroxia-exposed cells. c and d: respective 21% O2 controls. *Signifi-cant difference from simultaneous control, air-exposed A549 cellswith and without glutamine, P , 0.05 by t-test. **Significant differ-ence from all other groups, P , 0.05 by Tukey-Kramer test.
Fig. 2. Effect of glutamine on the viability of A549 cells in hyperoxiaon days 1–4 of exposure. Cells were plated in a 24-well plate at adensity of 10,000 cells/well in the absence and presence of glutamineand were exposed to 95% O2 (oxy) at 635 mmHg. Viable cells wereexamined by their ability to exclude trypan blue at each time interval(24 h) and compared with their respective 21% O2 controls. [Glu-tamine], glutamine concentration. Values are means 6 SE of eachexperiment performed in triplicate; error bars not seen are withinbars. *Significant difference from simultaneous control, air-exposedA549 cells with and without glutamine, P , 0.05 by t-test.
L782 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
Because CCCP-inhibitable MFI indicates a proton gra-dient-dependent MMP, these findings ensure that MFIand MFI/FS changes observed in glutamine-deprivedcells signify a loss of MMP.
Glutamine had a sparing effect on respiration inhyperoxia. The effect of hyperoxic exposure (4 days) oncellular O2 consumption is shown in Fig. 7. Glutaminesupplementation partially preserved O2 consumptionin hyperoxia, whereas a total loss of respiration wasobserved in glutamine-unsupplemented cells. Glu-tamine supplementation also supported the O2 con-sumption of A549 cells grown in 21% O2 and increasedit twofold compared with that in glutamine-deprivedcells.
Biochemical effects of glutamine deprivation. Be-cause glutamine, the substrate, is known to stabilizethe enzyme a-ketoglutarate dehydrogenase, its activitywas measured. A complete loss of a-ketoglutarate de-hydrogenase activity was observed during cellular ex-posure to hyperoxia in the absence of glutamine (Fig.8). However, glutamine supplementation partially pro-tected against the loss of enzyme activity. In the pres-ence of glutamine, the cells in 21% O2 had a threefoldincrease in a-ketoglutarate dehydrogenase activitycompared with the unsupplemented cells.
Glutamine consumption by 1 mM glutamine-supple-mented hyperoxia-exposed cells was significantlyhigher than in normoxia-exposed control cells (Fig. 9),but the cellular utilization of glutamine did not in-crease with additional increases of glutamine in thegrowth medium.
Because glutamine is an important substrate forGSH synthesis and GSH is an important antioxidantin cellular tolerance to oxidative stress, total cellularGSH also was measured. Table 1 shows the effect ofglutamine on the total GSH content of normoxia- andhyperoxia-exposed cells. The total GSH content of hy-peroxia-exposed glutamine-unsupplemented cells de-creased by 42% compared with that in similar cells innormoxia (2.8 6 0.4 vs. 4.8 6 0.4 nmol/107 cells).Glutamine-supplemented cells maintained their GSHcontent in hyperoxia.
Effects of glutamine deprivation on cellular and mi-tochondrial morphology. The effect of glutamine sup-plementation on the cellular morphology of A549 cellsin normoxia was studied with electron microscopy (Fig.10). Sections of A549 cells grown in normoxia (21%O2-5% CO2) revealed that in the absence of glutamine,the cells appeared thin, with spreading into monolay-ers (Fig. 10d). They had dense but normal-appearingmitochondria (Fig. 11a). There were no apparent ultra-structural abnormalities present except that the cellmembranes had only small indentations with virtuallyno microvilli. With 1 mM glutamine supplementation
Fig. 4. Effect of glutamine on ATP levels in hyperoxia-exposed A549cells. Cells were plated and exposed in the absence and presence ofglutamine as described in METHODS. Values are means 6 SE; n 5 3experiments; error bars not seen are within bars. *Significant dif-ference from control A549 cells without glutamine, P , 0.05 by t-test.**Significant difference from all other groups, P , 0.05 by Tukey-Kramer test.
Fig. 5. Measurement of chloromethyltetramethylrosamine (MitoTracker Orange) fluorescence, an indicator ofmitochondrial membrane potential, in human lung epithelial-like A549 cells exposed to 95% O2 (635 mmHg) for 4days in absence and presence of glutamine. Cells were stained and examined by flow cytometry as described inMETHODS. A: mean fluorescence intensity (MFI) of A549 cells exposed to 21 or 95% O2. B: forward scatter (FS). C:MFI/FS slope values. Values are means 6 SE; n 5 3 experiments; error bars not seen are within bars. *Significantdifference from simultaneous control, air-exposed A549 cells with and without glutamine, P , 0.05 by t-test.
L783GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
(Fig. 10e), the cells appeared larger than those lackingglutamine, were cuboidal in shape, showed spreadinginto monolayers, and had numerous surface microvilli,suggesting enhanced metabolic activity. Mitochondriawere abundant in these cells, and they appeared largerand had a less dense matrix than those in glutamine-unsupplemented cells. With 4 mM glutamine supple-mentation (Fig. 10f ), the cells also appeared thickerand had a cuboidal shape. These cells possessed nu-merous surface microvilli. Glutamine-supplementedA549 cells had abundant Golgi apparatus, endoplasmicreticulum, ribosomes, and fibrillar bundles (Fig. 10, eand f ), again suggesting a greater state of differentia-tion.
The cells also were examined for hyperoxia-mediatedmorphological changes in both the absence and pres-ence of glutamine. Hyperoxia (95% O2-5% CO2)-ex-posed cells grown in glutamine-free medium (Fig. 10a)
were irregular or spherical in shape. There was exten-sive cellular disintegration and injury, complete loss ofcell-to-cell contacts, and cellular aggregation ratherthan spreading into monolayers. Considerable cellulardebris was visible, and the remaining intact cellsshowed considerable and extensive lysosome and vac-uole formation. Intact mitochondria were very rarelydetected in these cells (Fig. 11b). Cells exposed tohyperoxia and supplemented with glutamine (1 mM;Fig. 10b) exhibited less necrosis, less aggregation,more spreading into a monolayer, and fewer vacuoles
Fig. 6. Effect of mitochondrial uncoupler carbonyl cyanide m-chloro-phenylhydrozone (CCCP) on flow cytometric estimation of mitochon-drial membrane potential by MitoTracker Orange fluorescence inA549 cells. Cells were treated with 40 mM CCCP for 30 min at 37°C,harvested, and examined by flow cytometry as described in METHODS.A: CCCP-inhibitable component of MFI (MFI 2 MFICCCP). B: CCCP-inhibitable components of MFI/FS (slope 2 slopeCCCP). *Significantdifference from simultaneous control, air-exposed A549 cells withand without glutamine, P , 0.05 by t-test.
Fig. 7. Evaluation of respiratory capacity of A549 cells exposed tohyperoxia for 4 days in the absence and presence of glutamine. Afterhyperoxic exposure in various experimental conditions, the cellswere harvested by trypsinization. Aliquots of these cells were studiedin the respirometer fitted with a calibrated polarographic O2 elec-trode as described in METHODS. Values are means 6 SE; n 5 3experiments; error bars not seen are within bars. *Significant dif-ference from simultaneous control, air-exposed A549 cells with andwithout glutamine, P , 0.05 by t-test. **Significant difference fromall other groups, P , 0.05 by Tukey-Kramer test.
Fig. 8. Effect of glutamine supplementation on a-ketoglutarate de-hydrogenase activity of A549 cells exposed to hyperoxia for 4 days.Values are means 6 SE; n 5 3 experiments. *Significant differencefrom simultaneous control, air-exposed A549 cells in air with andwithout glutamine, P , 0.05 by t-test. **Significant difference fromall other groups, P , 0.05 by Tukey-Kramer test.
L784 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
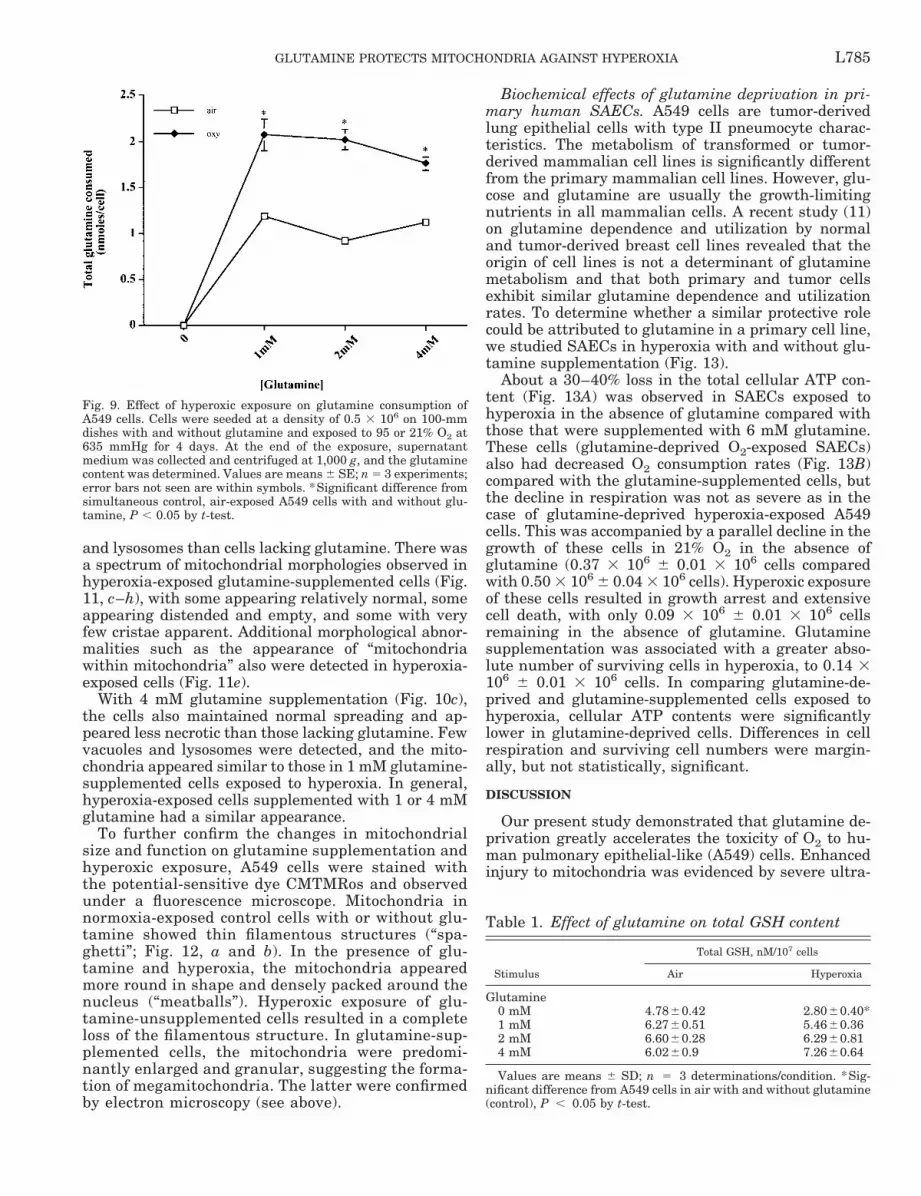
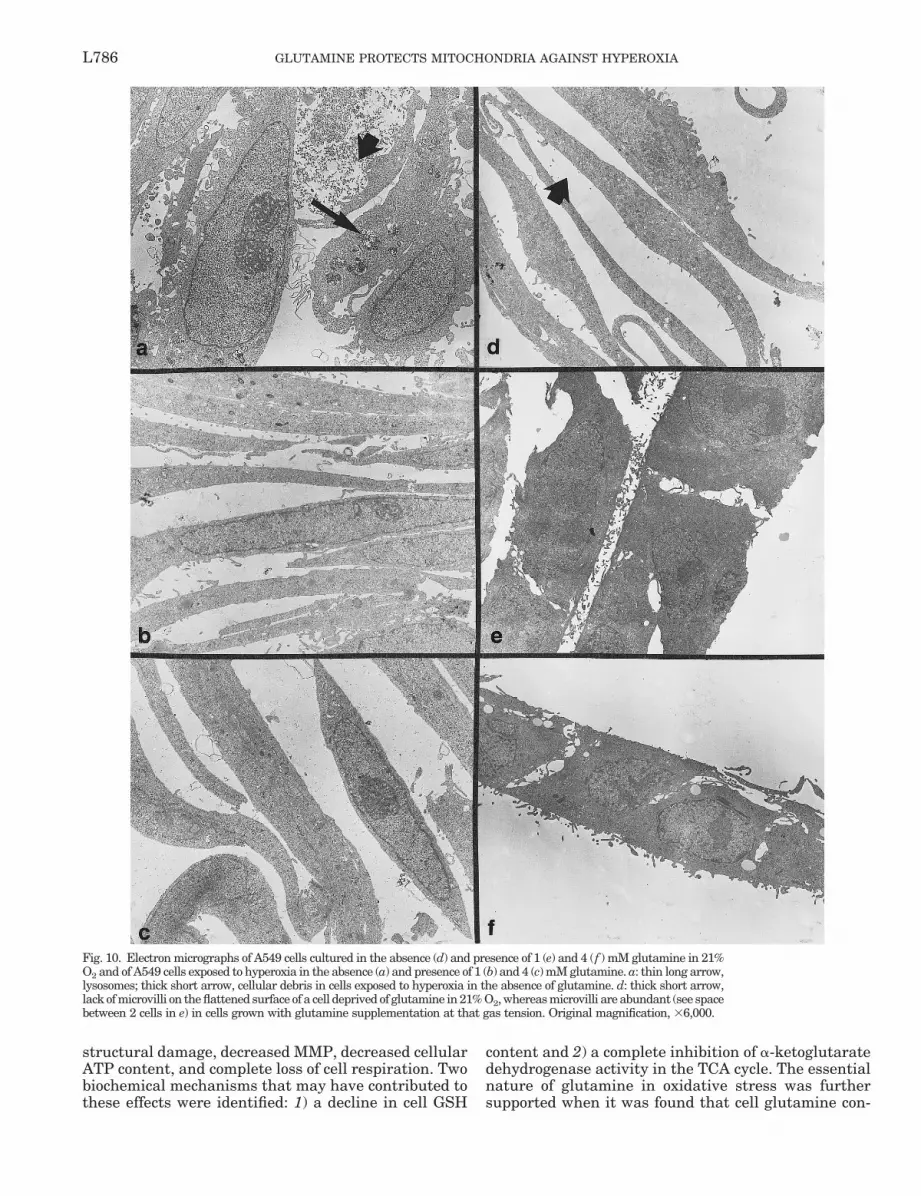
and lysosomes than cells lacking glutamine. There wasa spectrum of mitochondrial morphologies observed inhyperoxia-exposed glutamine-supplemented cells (Fig.11, c–h), with some appearing relatively normal, someappearing distended and empty, and some with veryfew cristae apparent. Additional morphological abnor-malities such as the appearance of “mitochondriawithin mitochondria” also were detected in hyperoxia-exposed cells (Fig. 11e).
With 4 mM glutamine supplementation (Fig. 10c),the cells also maintained normal spreading and ap-peared less necrotic than those lacking glutamine. Fewvacuoles and lysosomes were detected, and the mito-chondria appeared similar to those in 1 mM glutamine-supplemented cells exposed to hyperoxia. In general,hyperoxia-exposed cells supplemented with 1 or 4 mMglutamine had a similar appearance.
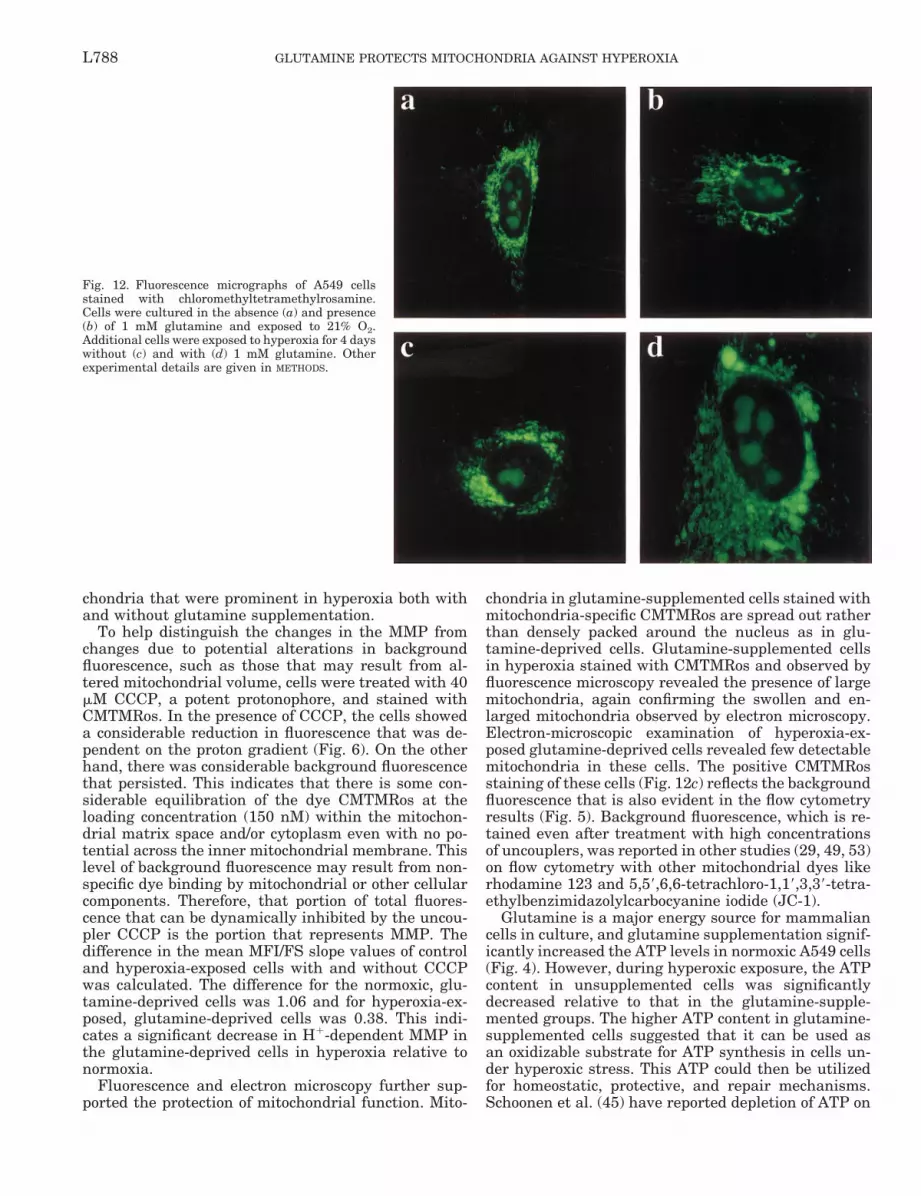
To further confirm the changes in mitochondrialsize and function on glutamine supplementation andhyperoxic exposure, A549 cells were stained withthe potential-sensitive dye CMTMRos and observedunder a fluorescence microscope. Mitochondria innormoxia-exposed control cells with or without glu-tamine showed thin filamentous structures (“spa-ghetti”; Fig. 12, a and b). In the presence of glu-tamine and hyperoxia, the mitochondria appearedmore round in shape and densely packed around thenucleus (“meatballs”). Hyperoxic exposure of glu-tamine-unsupplemented cells resulted in a completeloss of the filamentous structure. In glutamine-sup-plemented cells, the mitochondria were predomi-nantly enlarged and granular, suggesting the forma-tion of megamitochondria. The latter were confirmedby electron microscopy (see above).
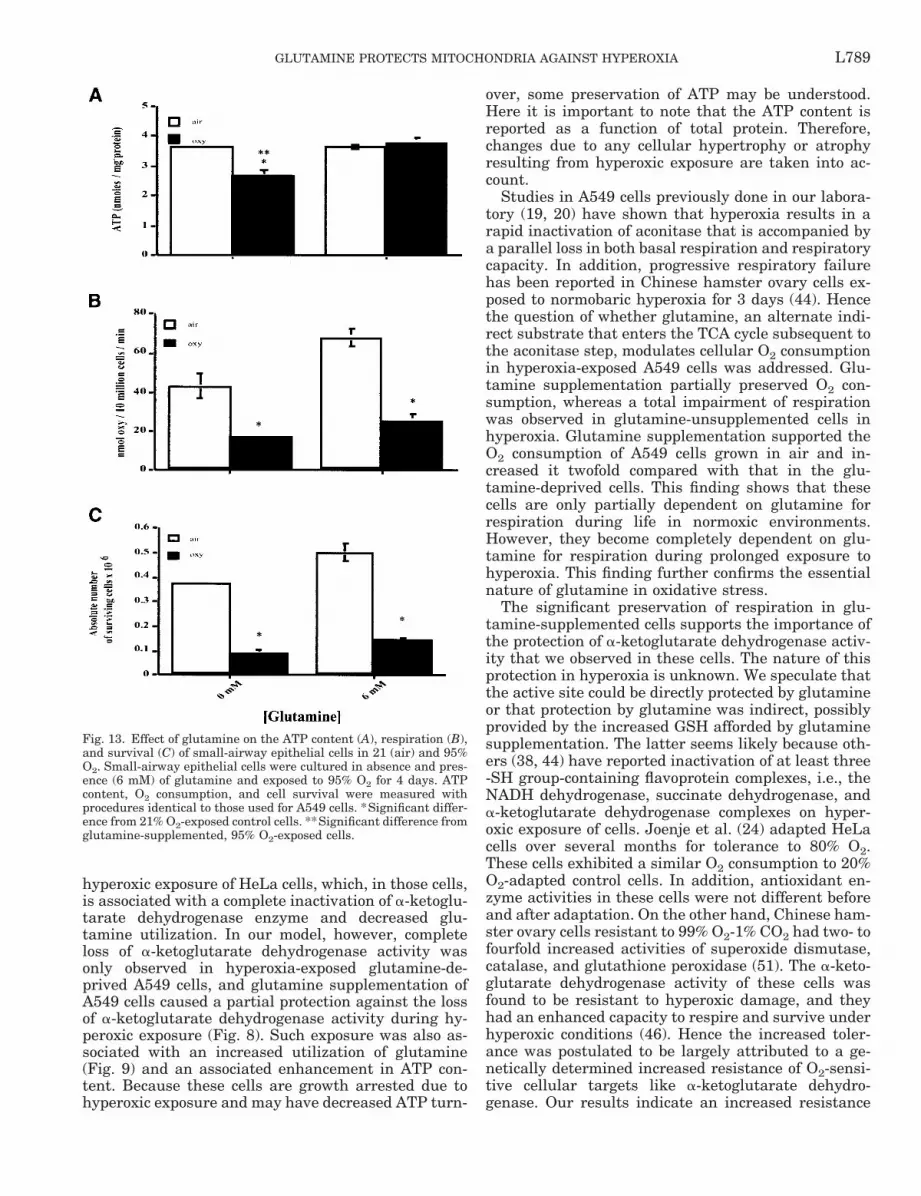
Biochemical effects of glutamine deprivation in pri-mary human SAECs. A549 cells are tumor-derivedlung epithelial cells with type II pneumocyte charac-teristics. The metabolism of transformed or tumor-derived mammalian cell lines is significantly differentfrom the primary mammalian cell lines. However, glu-cose and glutamine are usually the growth-limitingnutrients in all mammalian cells. A recent study (11)on glutamine dependence and utilization by normaland tumor-derived breast cell lines revealed that theorigin of cell lines is not a determinant of glutaminemetabolism and that both primary and tumor cellsexhibit similar glutamine dependence and utilizationrates. To determine whether a similar protective rolecould be attributed to glutamine in a primary cell line,we studied SAECs in hyperoxia with and without glu-tamine supplementation (Fig. 13).
About a 30–40% loss in the total cellular ATP con-tent (Fig. 13A) was observed in SAECs exposed tohyperoxia in the absence of glutamine compared withthose that were supplemented with 6 mM glutamine.These cells (glutamine-deprived O2-exposed SAECs)also had decreased O2 consumption rates (Fig. 13B)compared with the glutamine-supplemented cells, butthe decline in respiration was not as severe as in thecase of glutamine-deprived hyperoxia-exposed A549cells. This was accompanied by a parallel decline in thegrowth of these cells in 21% O2 in the absence ofglutamine (0.37 3 106 6 0.01 3 106 cells comparedwith 0.50 3 106 6 0.04 3 106 cells). Hyperoxic exposureof these cells resulted in growth arrest and extensivecell death, with only 0.09 3 106 6 0.01 3 106 cellsremaining in the absence of glutamine. Glutaminesupplementation was associated with a greater abso-lute number of surviving cells in hyperoxia, to 0.14 3106 6 0.01 3 106 cells. In comparing glutamine-de-prived and glutamine-supplemented cells exposed tohyperoxia, cellular ATP contents were significantlylower in glutamine-deprived cells. Differences in cellrespiration and surviving cell numbers were margin-ally, but not statistically, significant.
DISCUSSION
Our present study demonstrated that glutamine de-privation greatly accelerates the toxicity of O2 to hu-man pulmonary epithelial-like (A549) cells. Enhancedinjury to mitochondria was evidenced by severe ultra-
Fig. 9. Effect of hyperoxic exposure on glutamine consumption ofA549 cells. Cells were seeded at a density of 0.5 3 106 on 100-mmdishes with and without glutamine and exposed to 95 or 21% O2 at635 mmHg for 4 days. At the end of the exposure, supernatantmedium was collected and centrifuged at 1,000 g, and the glutaminecontent was determined. Values are means 6 SE; n 5 3 experiments;error bars not seen are within symbols. *Significant difference fromsimultaneous control, air-exposed A549 cells with and without glu-tamine, P , 0.05 by t-test.
Table 1. Effect of glutamine on total GSH content
Stimulus
Total GSH, nM/107 cells
Air Hyperoxia
Glutamine0 mM 4.7860.42 2.8060.40*1 mM 6.2760.51 5.4660.362 mM 6.6060.28 6.2960.814 mM 6.0260.9 7.2660.64
Values are means 6 SD; n 5 3 determinations/condition. *Sig-nificant difference from A549 cells in air with and without glutamine(control), P , 0.05 by t-test.
L785GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
structural damage, decreased MMP, decreased cellularATP content, and complete loss of cell respiration. Twobiochemical mechanisms that may have contributed tothese effects were identified: 1) a decline in cell GSH
content and 2) a complete inhibition of a-ketoglutaratedehydrogenase activity in the TCA cycle. The essentialnature of glutamine in oxidative stress was furthersupported when it was found that cell glutamine con-
Fig. 10. Electron micrographs of A549 cells cultured in the absence (d) and presence of 1 (e) and 4 (f ) mM glutamine in 21%O2 and of A549 cells exposed to hyperoxia in the absence (a) and presence of 1 (b) and 4 (c) mM glutamine. a: thin long arrow,lysosomes; thick short arrow, cellular debris in cells exposed to hyperoxia in the absence of glutamine. d: thick short arrow,lack of microvilli on the flattened surface of a cell deprived of glutamine in 21% O2, whereas microvilli are abundant (see spacebetween 2 cells in e) in cells grown with glutamine supplementation at that gas tension. Original magnification, 36,000.
L786 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA

sumption was doubled during exposure to hyperoxiaeven in the presence of a marked decline in a-ketoglu-tarate dehydrogenase activity. In addition, we demon-strate for the first time that glutamine can protecta-ketoglutarate dehydrogenase from inactivation un-der oxidative stress (hyperoxia).
Glutamine, an important precursor of peptides, pro-teins, and nucleotides, also serves a critical role incellular metabolic pathways (16, 36, 54). Acute glu-tamine deprivation is associated with DNA damage aswell as elevated expression of growth-arrest genes (1,21). Such deprivation often leads to a severe decline incell viability (43). Glutamine is a highly labile constit-uent of cell culture medium, and yet most media areprepackaged to contain glutamine, thus severely lim-iting shelf life. Various investigators may or may notuse fresh medium and/or add supplemental glutamine.The findings of the present study indicate that theeffects of oxidative stress on cells may be profoundlyaffected depending on the actual concentrations of glu-tamine present during such exposures.
Glutamine deprivation significantly affected thegrowth of A549 cells, and hyperoxic exposure of thesecells caused a markedly exaggerated injury. Anotherstudy (25) has also revealed the glutamine require-
ment of pulmonary endothelial cells and A549 cells forgrowth. In this study, glutamine supplementation in-creased cell proliferation in normoxia (21% O2), and 1mM glutamine was adequate to support growth. Addi-tionally, glutamine supplementation not only sup-ported the growth of A549 cells but protected themfrom hyperoxia-mediated injury and death. In the ex-periments reported here, more than two-thirds of thecell death observed could have been prevented by glu-tamine supplementation.
Lung tissue and cellular injury, growth arrest, andaccumulation of cells in the G1/G2 and S phases of thecell cycle commonly occur on hyperoxic exposures (12,22, 39, 47). In this study, several important differenceswere found in the structure of A549 cells on hyperoxicexposure in the absence and presence of glutamine.Glutamine supplementation improved the mitochon-drial viability such that, in hyperoxia, enlarged mito-chondria, a spectrum of abnormal mitochondria, andeven a few normal-looking mitochondria were ob-served. In the absence of glutamine, however, very fewmitochondrial structures could be identified after hy-peroxic exposure. Enlarged mitochondria with rup-tured membranes have been found to be a feature ofcells from lungs, hearts, and brains from hyperoxia-exposed animals (8, 37, 41). Enlarged mitochondriaalso frequently occur in hyperoxia-adapted HeLa cells,Chinese hamster ovary cells (24, 50), and other cellsstressed by oxidants. Although the enlargement of mi-tochondria likely represents injury, it also may indi-cate adaptation to compensate during oxidative stressby increasing the ratio of surface area to volume withinmitochondria.
Lung cells exposed to hyperoxia can generate freeradicals like superoxide anion, hydroxyl, and alkylradicals via mitochondrial electron transport (18). Mi-tochondrial DNA, metabolism, and function are highlysusceptible to injury by these species. Such mitochon-drial injury can contribute to the pathogenesis of ne-crotic and apoptotic cell death (17, 28, 38). Whetherenhancement of cell survival in this study is relatedto energy homeostasis and protection of mitochon-drial function was investigated by measuring the MMPand the total ATP content of hyperoxia-exposed glu-tamine-supplemented and -unsupplemented cells. Inour studies of MMP, glutamine-deprived 21% O2-ex-posed control cells actually had higher MFI valuesthan glutamine-supplemented normoxia-exposed cells.This could be due to hyperpolarization of the mitochon-drial membrane caused by mitochondrial ATP accumu-lation in these metabolically inactive, growth-retardedcells, which may also have diminished ATP turnover.In addition, there was a remarkable increase in MFIobserved in hyperoxia-exposed glutamine-supple-mented cells (1–4 mM; Fig. 5). There were two possiblecauses for this increase in fluorescence. This could bedue to either an increase in the MMP or an increase inthe apparent MMP due to an increase in the totalnumber of mitochondria per cell (49). Indeed, electronmicroscopy revealed morphological changes in mito-
Fig. 11. Electron micrograph of mitochondria in A549 cells exposedto hyperoxia in absence (b) and presence (c–h) of glutamine. a: denseround mitochondria of 21% O2-exposed A549 cells without glutaminesupplementation. Original magnification, 347,500.
L787GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
chondria that were prominent in hyperoxia both withand without glutamine supplementation.
To help distinguish the changes in the MMP fromchanges due to potential alterations in backgroundfluorescence, such as those that may result from al-tered mitochondrial volume, cells were treated with 40mM CCCP, a potent protonophore, and stained withCMTMRos. In the presence of CCCP, the cells showeda considerable reduction in fluorescence that was de-pendent on the proton gradient (Fig. 6). On the otherhand, there was considerable background fluorescencethat persisted. This indicates that there is some con-siderable equilibration of the dye CMTMRos at theloading concentration (150 nM) within the mitochon-drial matrix space and/or cytoplasm even with no po-tential across the inner mitochondrial membrane. Thislevel of background fluorescence may result from non-specific dye binding by mitochondrial or other cellularcomponents. Therefore, that portion of total fluores-cence that can be dynamically inhibited by the uncou-pler CCCP is the portion that represents MMP. Thedifference in the mean MFI/FS slope values of controland hyperoxia-exposed cells with and without CCCPwas calculated. The difference for the normoxic, glu-tamine-deprived cells was 1.06 and for hyperoxia-ex-posed, glutamine-deprived cells was 0.38. This indi-cates a significant decrease in H1-dependent MMP inthe glutamine-deprived cells in hyperoxia relative tonormoxia.
Fluorescence and electron microscopy further sup-ported the protection of mitochondrial function. Mito-
chondria in glutamine-supplemented cells stained withmitochondria-specific CMTMRos are spread out ratherthan densely packed around the nucleus as in glu-tamine-deprived cells. Glutamine-supplemented cellsin hyperoxia stained with CMTMRos and observed byfluorescence microscopy revealed the presence of largemitochondria, again confirming the swollen and en-larged mitochondria observed by electron microscopy.Electron-microscopic examination of hyperoxia-ex-posed glutamine-deprived cells revealed few detectablemitochondria in these cells. The positive CMTMRosstaining of these cells (Fig. 12c) reflects the backgroundfluorescence that is also evident in the flow cytometryresults (Fig. 5). Background fluorescence, which is re-tained even after treatment with high concentrationsof uncouplers, was reported in other studies (29, 49, 53)on flow cytometry with other mitochondrial dyes likerhodamine 123 and 5,59,6,6-tetrachloro-1,19,3,39-tetra-ethylbenzimidazolylcarbocyanine iodide (JC-1).
Glutamine is a major energy source for mammaliancells in culture, and glutamine supplementation signif-icantly increased the ATP levels in normoxic A549 cells(Fig. 4). However, during hyperoxic exposure, the ATPcontent in unsupplemented cells was significantlydecreased relative to that in the glutamine-supple-mented groups. The higher ATP content in glutamine-supplemented cells suggested that it can be used asan oxidizable substrate for ATP synthesis in cells un-der hyperoxic stress. This ATP could then be utilizedfor homeostatic, protective, and repair mechanisms.Schoonen et al. (45) have reported depletion of ATP on
Fig. 12. Fluorescence micrographs of A549 cellsstained with chloromethyltetramethylrosamine.Cells were cultured in the absence (a) and presence(b) of 1 mM glutamine and exposed to 21% O2.Additional cells were exposed to hyperoxia for 4 dayswithout (c) and with (d) 1 mM glutamine. Otherexperimental details are given in METHODS.
L788 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA
hyperoxic exposure of HeLa cells, which, in those cells,is associated with a complete inactivation of a-ketoglu-tarate dehydrogenase enzyme and decreased glu-tamine utilization. In our model, however, completeloss of a-ketoglutarate dehydrogenase activity wasonly observed in hyperoxia-exposed glutamine-de-prived A549 cells, and glutamine supplementation ofA549 cells caused a partial protection against the lossof a-ketoglutarate dehydrogenase activity during hy-peroxic exposure (Fig. 8). Such exposure was also as-sociated with an increased utilization of glutamine(Fig. 9) and an associated enhancement in ATP con-tent. Because these cells are growth arrested due tohyperoxic exposure and may have decreased ATP turn-
over, some preservation of ATP may be understood.Here it is important to note that the ATP content isreported as a function of total protein. Therefore,changes due to any cellular hypertrophy or atrophyresulting from hyperoxic exposure are taken into ac-count.
Studies in A549 cells previously done in our labora-tory (19, 20) have shown that hyperoxia results in arapid inactivation of aconitase that is accompanied bya parallel loss in both basal respiration and respiratorycapacity. In addition, progressive respiratory failurehas been reported in Chinese hamster ovary cells ex-posed to normobaric hyperoxia for 3 days (44). Hencethe question of whether glutamine, an alternate indi-rect substrate that enters the TCA cycle subsequent tothe aconitase step, modulates cellular O2 consumptionin hyperoxia-exposed A549 cells was addressed. Glu-tamine supplementation partially preserved O2 con-sumption, whereas a total impairment of respirationwas observed in glutamine-unsupplemented cells inhyperoxia. Glutamine supplementation supported theO2 consumption of A549 cells grown in air and in-creased it twofold compared with that in the glu-tamine-deprived cells. This finding shows that thesecells are only partially dependent on glutamine forrespiration during life in normoxic environments.However, they become completely dependent on glu-tamine for respiration during prolonged exposure tohyperoxia. This finding further confirms the essentialnature of glutamine in oxidative stress.
The significant preservation of respiration in glu-tamine-supplemented cells supports the importance ofthe protection of a-ketoglutarate dehydrogenase activ-ity that we observed in these cells. The nature of thisprotection in hyperoxia is unknown. We speculate thatthe active site could be directly protected by glutamineor that protection by glutamine was indirect, possiblyprovided by the increased GSH afforded by glutaminesupplementation. The latter seems likely because oth-ers (38, 44) have reported inactivation of at least three-SH group-containing flavoprotein complexes, i.e., theNADH dehydrogenase, succinate dehydrogenase, anda-ketoglutarate dehydrogenase complexes on hyper-oxic exposure of cells. Joenje et al. (24) adapted HeLacells over several months for tolerance to 80% O2.These cells exhibited a similar O2 consumption to 20%O2-adapted control cells. In addition, antioxidant en-zyme activities in these cells were not different beforeand after adaptation. On the other hand, Chinese ham-ster ovary cells resistant to 99% O2-1% CO2 had two- tofourfold increased activities of superoxide dismutase,catalase, and glutathione peroxidase (51). The a-keto-glutarate dehydrogenase activity of these cells wasfound to be resistant to hyperoxic damage, and theyhad an enhanced capacity to respire and survive underhyperoxic conditions (46). Hence the increased toler-ance was postulated to be largely attributed to a ge-netically determined increased resistance of O2-sensi-tive cellular targets like a-ketoglutarate dehydro-genase. Our results indicate an increased resistance
Fig. 13. Effect of glutamine on the ATP content (A), respiration (B),and survival (C) of small-airway epithelial cells in 21 (air) and 95%O2. Small-airway epithelial cells were cultured in absence and pres-ence (6 mM) of glutamine and exposed to 95% O2 for 4 days. ATPcontent, O2 consumption, and cell survival were measured withprocedures identical to those used for A549 cells. *Significant differ-ence from 21% O2-exposed control cells. **Significant difference fromglutamine-supplemented, 95% O2-exposed cells.
L789GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA

conferred by glutamine to the O2-sensitive a-ketoglu-tarate dehydrogenase.
Hyperoxia-exposed A549 cells had a twofold higherrate of glutamine consumption compared with nor-moxic cells (Fig. 9). Increased glutamic acid uptake inparallel with increased GSH levels has been reportedin hyperoxia-exposed bovine pulmonary endothelialcells (14, 15). GSH and glutamine, as a precursor ofGSH, are known to play important roles in the protec-tion against oxidative stress caused by hyperoxia (6,26). In this previous study (15), a 50% decrease incellular GSH level was found on exposure of glu-tamine-deprived cells to hyperoxia. In our study,glutamine supplementation prevented hyperoxic de-pletion of GSH levels in A549 cells. Therefore, pro-tection of mitochondrial function and increased cellviability in A549 cells could be attributed, at least inpart, to the restored GSH levels. Decreased cell GSHcontents have been associated with disruption ofMMP and hence mitochondrial function during oxi-dative stress (17, 31, 53).
In this study, enhanced cell survival in hyperoxiawith glutamine supplementation could be attributed tothe protection of mitochondrial structure and function,increased ATP content, O2 consumption, and protec-tion of a-ketoglutarate dehydrogenase and cellularGSH. Studies with human SAECs indicated that glu-tamine deprivation can cause similar abnormalities inbioenergetics in primary and tumor-derived epithelialcells. Survival of A549 cells in air and the absence ofglutamine could also be due to endogenous glutaminesynthetase activity (48). Glutamine synthetase ishighly susceptible to oxidant stress (2), and throughthis mechanism, oxidant-stressed cells grown in theabsence of glutamine could be further deprived of glu-tamine. A recent report (27) also suggests that inflam-matory necrotic cell death occurring as a result ofoxidant stress in pulmonary endothelial cells could beconverted to noninflammatory apoptosis by glutaminesupplementation through increased ATP levels. More-over, glutamine supplementation in patients duringadvanced disease states is a subject of recent medicalinterest (32), and further studies in this area may beuseful.
We thank Stephanie Park for preparing the manuscript, TimPattison for help in performing the electron microscopy, and Dr.Steve Goodman for glutamine analysis.
This work was supported by National Heart, Lung, and BloodInstitute Grants HL-57144, HL-52732, and HL-56263.
S. Ahmad was a recipient of the Robert J. Suslow Fellowship inEnvironmental Lung Disease funded by National Jewish Medicaland Research Center (Denver, CO).
REFERENCES
1. Abcouwer SF, Schwarz C, and Meguid RA. Glutamine de-privation induces the expression of GADD45 and GADD153primarily by mRNA stabilization. J Biol Chem 274: 28645–28651, 1999.
2. Aksenov MY, Aksenova MV, Carney JM, and ButterfieldDA. Oxidative modification of glutamine synthetase by amyloidbeta peptide. Free Radic Res 27: 267–281, 1997.
3. Allen CB, Guo XL, and White CW. Changes in pulmonaryexpression of hexokinase and glucose transporter mRNAs in ratsadapted to hyperoxia. Am J Physiol Lung Cell Mol Physiol 274:L320–L329, 1998.
4. Allen CB and White CW. Glucose modulates cell death due tonormobaric hyperoxia by maintaining cellular ATP. Am JPhysiol Lung Cell Mol Physiol 274: L159–L164, 1998.
5. Ames BN, Shigenaga MK, and Hagen TM. Mitochondrialdecay in aging. Biochim Biophys Acta 1271: 165–170, 1995.
6. Amores-Sanchez MI and Medina MA. Glutamine, as a pre-cursor of glutathione, and oxidative stress. Mol Genet Metab 67:100–105, 1999.
7. Anderson ME. Determination of glutathione and glutathionedisulfide in biological samples. Methods Enzymol 113: 548–555,1985.
8. Balentine JD. Ultrastructural pathology of hyperbaric oxygen-ation in the central nervous system. Observations in anteriorhorn gray matter. Lab Invest 31: 580–592, 1974.
9. Bergmeyer HU. Methods of Enzymatic Analysis. New York:Academic, 1974, p. 1981–1987.
10. Buckley S, Driscoll B, Barsky L, Weinberg K, Anderson K,and Warburton D. ERK activation protects against DNA dam-age and apoptosis in hyperoxic rat AEC2. Am J Physiol LungCell Mol Physiol 277: L159–L166, 1999.
11. Collins CL, Wasa M, Souba WW, and Abcouwer SF. Deter-minants of glutamine dependence and utilization by normal andtumor-derived breast cell lines. J Cell Physiol 176: 166–178,1998.
12. Crapo JD, Barry BE, Foscue HA, and Shelburne J. Struc-tural and biochemical changes in rat lungs occurring duringexposures to lethal and adaptive doses of oxygen. Am Rev RespirDis 122: 123–143, 1980.
13. Das KC, Lewis-Molock Y, and White CW. Activation ofNF-kB and elevation of MnSOD gene expression by thiol reduc-ing agents in lung adenocarcinoma (A549) cells. Am J PhysiolLung Cell Mol Physiol 269: L588–L602, 1995.
14. Deneke SM and Fanburg BL. Regulation of cellular glutathi-one. Am J Physiol Lung Cell Mol Physiol 257: L163–L173, 1989.
15. Deneke SM, Steiger V, and Fanburg BL. Effect of hyperoxiaon glutathione levels and glutamic acid uptake in endothelialcells. J Appl Physiol 63: 1966–1971, 1987.
16. Doverskog M, Ljunggren J, Ohman L, and Haggstrom L.Physiology of cultured animal cells. J Biotechnol 59: 103–115,1997.
17. Esteve JM, Mompo J, Garcia de la Asuncion J, Sastre J,Asensi M, Boix J, Vina JR, Vina J, and Pallardo FV.Oxidative damage to mitochondrial DNA and glutathione oxida-tion in apoptosis: studies in vivo and in vitro. FASEB J 13:1055–1064, 1999.
18. Freeman BA and Crapo JD. Hyperoxia increases oxygen rad-ical production in rat lungs and lung mitochondria. J Biol Chem256: 10986–10992, 1981.
19. Gardner PR, Nguyen DD, and White CW. Aconitase is asensitive and critical target of oxygen poisoning in culturedmammalian cells and in rat lungs. Proc Natl Acad Sci USA 91:12248–12252, 1994.
20. Gardner PR, Raineri I, Epstein LB, and White CW. Super-oxide radical and iron modulate aconitase activity in mamma-lian cells. J Biol Chem 270: 13399–13405, 1995.
21. Gorman L, Mercer LP, and Hennig B. Growth requirementsof endothelial cells in culture: variations in serum and aminoacid concentrations. Nutrition 12: 266–270, 1996.
22. Harris JB, Chang LY, and Crapo JD. Rat lung alveolar typeI epithelial cell injury and response to hyperoxia. Am J RespirCell Mol Biol 4: 115–125, 1991.
23. Jobe AJ. The new BPD: an arrest of lung development. PediatrRes 46: 641–643, 1999.
24. Joenje H, Gille JJ, Oostra AB, and Van der Valk P. Somecharacteristics of hyperoxia-adapted HeLa cells. A tissue culturemodel for cellular oxygen tolerance. Lab Invest 52: 420–428,1985.
25. Kang YJ, Feng Y, and Hatcher EL. Glutathione stimulatesA549 cell proliferation in glutamine-deficient culture: the effect
L790 GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA

of glutamate supplementation. J Cell Physiol 161: 589–596,1994.
26. Komadina KH, Duncan CA, Bryan CL, and Jenkinson SG.Protection from hyperbaric oxidant stress by administration ofbuthionine sulfoximine. J Appl Physiol 71: 352–358, 1991.
27. Lelli JL Jr, Becks LL, Dabrowska MI, and Hinshaw DB.ATP converts necrosis to apoptosis in oxidant-injured endothe-lial cells. Free Radic Biol Med 25: 694–702, 1998.
28. Lemasters JJ, Qian T, Bradham CA, Brenner DA, CascioWE, Trost LC, Nishimura Y, Nieminen AL, and Herman B.Mitochondrial dysfunction in the pathogenesis of necrotic andapoptotic cell death. J Bioenerg Biomembr 31: 305–319, 1999.
29. Linsinger G, Wilhelm S, Wagner H, and Hacker G. Uncou-plers of oxidative phosphorylation can enhance a Fas deathsignal. Mol Cell Biol 19: 3299–3311, 1999.
30. Macho A, Decaudin D, Castedo M, Hirsch T, Susin SA,Zamzami N, and Kroemer G. Chloromethyl-X-rosamine is analdehyde-fixable potential-sensitive fluorochrome for the detec-tion of early apoptosis. Cytometry 25: 333–340, 1996.
31. Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B,Susin SA, Zamzami N, and Kroemer G. Glutathione deple-tion is an early and calcium elevation is a late event of thymocyteapoptosis. J Immunol 158: 4612–4619, 1997.
32. Miller AL. Therapeutic considerations of L-glutamine: a reviewof the literature. Altern Med Rev 4: 239–248, 1999.
33. Moore S and Stein WH. Chromatographic determination ofamino acids by the use of automatic recording equipment. Meth-ods Enzymol 6: 819–831, 1963.
34. Morgan DM. Tetrazolium (MTT) assay for cellular viability andactivity. Methods Mol Biol 79: 179–183, 1998.
35. Morton RL, Ikle D, and White CW. Loss of lung mitochondrialaconitase activity due to hyperoxia in bronchopulmonary dyspla-sia in primates. Am J Physiol Lung Cell Mol Physiol 274: L127–L133, 1998.
36. Neermann J and Wagner R. Comparative analysis of glucoseand glutamine metabolism in transformed mammalian celllines, insect and primary liver cells. J Cell Physiol 166: 152–169,1996.
37. Noronha-Dutra AA and Steen EM. Lipid peroxidation as amechanism of injury in cardiac myocytes. Lab Invest 47: 346–353, 1982.
38. Pruijn FB, Schoonen WG, and Joenje H. Inactivation ofmitochondrial metabolism by hyperoxia-induced oxidativestress. Ann NY Acad Sci 663: 453–455, 1992.
39. Rancourt RC, Staversky RJ, Keng PC, and O’Reilly MA.Hyperoxia inhibits proliferation of Mv1Lu epithelial cells inde-pendent of TGF-b signaling. Am J Physiol Lung Cell Mol Physiol277: L1172–L1178, 1999.
40. Robinson J and Cooper JM. Method of determining oxygenconcentrations in biological media, suitable for calibration of theoxygen electrode. Anal Biochem 33: 390–399, 1970.
41. Rosenbaum RM, Wittner M, and Lenger M. Mitochondrialand other ultrastructural changes in great alveolar cells ofoxygen-adapted and poisoned rats. Lab Invest 20: 516–528,1969.
42. Salotra PT and Singh VN. Regulation of glucose metabolismin the lung: hexokinase-catalyzed phosphorylation, a rate-limit-ing step. Life Sci 31: 791–794, 1982.
43. Sanfeliu A and Stephanopoulos G. Effect of glutamine limi-tation on the death of attached Chinese hamster ovary cells.Biotechnol Bioeng 64: 46–53, 1999.
44. Schoonen WG, Wanamarta AH, van der Klei-van MoorselJM, Jakobs C, and Joenje H. Respiratory failure and stimu-lation of glycolysis in Chinese hamster ovary cells exposed tonormobaric hyperoxia. J Biol Chem 265: 1118–1124, 1990.
45. Schoonen WG, Wanamarta AH, van der Klei-van MoorselJM, Jakobs C, and Joenje H. Hyperoxia-induced clonogenickilling of HeLa cells associated with respiratory failure andselective inactivation of Krebs cycle enzymes. Mutat Res 237:173–181, 1990.
46. Schoonen WG, Wanamarta AH, van der Klei-van MoorselJM, Jakobs C, and Joenje H. Characterization of oxygen-resistant Chinese hamster ovary cells. III. Relative resistance ofsuccinate and alpha-ketoglutarate dehydrogenases to hyperoxicinactivation. Free Radic Biol Med 10: 111–118, 1991.
47. Shenberger JS and Dixon PS. Oxygen induces S-phasegrowth arrest and increases p53 and p21(WAF1/CIP1) expres-sion in human bronchial smooth-muscle cells. Am J Respir CellMol Biol 21: 395–402, 1999.
48. Souba WW, Herskowitz K, and Plumley DA. Lung glutaminemetabolism. JPEN J Parenter Enteral Nutr 14, Suppl: 68S–70S,1990.
49. Vander Heiden MG, Chandel NS, Williamson EK, Schu-macker PT, and Thompson CB. Bcl-xL regulates the mem-brane potential and volume homeostasis of mitochondria. Cell91: 627–637, 1997.
50. Van der Valk P, Gille JJ, Oostra AB, Roubos EW, Sminia T,and Joenje H. Characterization of an oxygen-tolerant cell linederived from Chinese hamster ovary. Antioxygenic enzyme lev-els and ultrastructural morphometry of peroxisomes and mito-chondria. Cell Tissue Res 239: 61–68, 1985.
51. Van der Valk P, Gille JJ, van der Plas LH, Jongkind JF,Verkerk A, Konings AW, and Joenje H. Characterization ofoxygen-tolerant Chinese hamster ovary cells. II. Energy metab-olism and antioxidant status. Free Radic Biol Med 4: 345–356,1988.
52. Wallace DC, Shoffner JM, Trounce I, Brown MD, BallingerSW, Corral-Debrinski M, Horton T, Jun AS, and Lott MT.Mitochondrial DNA mutations in human degenerative diseasesand aging. Biochim Biophys Acta 1271: 141–151, 1995.
53. Wullner U, Seyfried J, Groscurth P, Beinroth S, Winter S,Gleichmann M, Heneka M, Loschmann P, Schulz JB,Weller M, and Klockgether T. Glutathione depletion andneuronal cell death: the role of reactive oxygen intermediatesand mitochondrial function. Brain Res 826: 53–62, 1999.
54. Zielke HR, Zielke CL, and Ozand PT. Glutamine: a majorenergy source for cultured mammalian cells. Fed Proc 43: 121–125, 1984.
L791GLUTAMINE PROTECTS MITOCHONDRIA AGAINST HYPEROXIA