genetic studies on the ghrelin, growth hormone secretagogue receptor (ghsr) and ghrelin o-acyl...
TRANSCRIPT

R
Ga
BD
a
ARRAA
KGGGGGGCG
C
0d
Peptides 32 (2011) 2191–2207
Contents lists available at SciVerse ScienceDirect
Peptides
j ourna l ho me pa ge: www.elsev ier .com/ locate /pept ides
eview
enetic studies on the ghrelin, growth hormone secretagogue receptor (GHSR)nd ghrelin O-acyl transferase (GOAT) genes
oyang Liu, Edwin A. Garcia, Márta Korbonits ∗
epartment of Endocrinology, Barts and the London School of Medicine, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK
r t i c l e i n f o
rticle history:eceived 9 March 2011eceived in revised form 3 September 2011ccepted 6 September 2011vailable online 12 September 2011
eywords:hrelin
a b s t r a c t
Ghrelin is a 28 amino acid peptide hormone that is produced both centrally and peripherally. Regulatedby the ghrelin O-acyl transferase enzyme, ghrelin exerts its action through the growth hormone secreta-gogue receptor, and is implicated in a diverse range of physiological processes. These implications haveplaced the ghrelin signaling pathway at the center of a large number of candidate gene and genome-widestudies which aim to identify the genetic basis of human heterogeneity. In this review we summarizethe available data on the genetic variability of ghrelin, its receptor and its regulatory enzyme, and theirassociation with obesity, stature, type 2 diabetes, cardiovascular disease, eating disorders, and reward
HSRhrelin receptorrowth hormone secretagogue receptorhrelin O-acyl transferase (GOAT)enetic variability
seeking behavior.© 2011 Elsevier Inc. All rights reserved.
andidate gene studiesenome-wide studies
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21921.1. Ghrelin gene structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21921.2. Ghrelin receptor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21921.3. Introduction to genetic studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2193
2. Obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21932.1. Association studies on ghrelin and obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21932.2. Association studies on GHSR and obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21962.3. Genome-wide studies on ghrelin/ghrelin receptor and obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21972.4. Summary of association studies on obesity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2197
3. Stature. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21973.1. Association studies on ghrelin/ghrelin receptor and stature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21973.2. Genome-wide studies on ghrelin/ghrelin receptor and stature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21983.3. Summary of association studies on stature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2198
4. Diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21984.1. Association studies on ghrelin and type 2 diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21984.2. Association studies on ghrelin receptor and type 2 diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21994.3. Genome-wide studies on ghrelin and GHSR and type 2 diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22004.4. Summary of association studies on type 2 diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2200
5. Cardiovascular disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22005.1. Association of ghrelin polymorphisms with cardiovascular health . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2200
5.2. Association of GHSR polymorphisms with cardiovascular health .5.3. Genome-wide studies on ghrelin/ghrelin receptor and cardiovasc5.4. Summary of association studies on cardiovascular health . . . . . . . . .
∗ Corresponding author. Tel.: +44 20 7882 6238; fax: +44 20 7882 6197.E-mail address: [email protected] (M. Korbonits).
196-9781/$ – see front matter © 2011 Elsevier Inc. All rights reserved.oi:10.1016/j.peptides.2011.09.006
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2201ular health . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2201
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2201

2192 B. Liu et al. / Peptides 32 (2011) 2191–2207
6. Eating disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22016.1. Association of ghrelin polymorphisms with eating disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22016.2. Association of GHSR polymorphisms with eating disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22026.3. Association of GOAT polymorphisms with eating disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22026.4. Summary of association studies on eating disorders . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2202
7. Reward seeking behavior . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22027.1. Association studies on ghrelin and ghrelin receptor with alcohol dependence. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2202
8. Rare variants in ghrelin and GHSR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22038.1. Rare ghrelin gene variants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22038.2. Rare ghrelin receptor gene variants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2203
9. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2204 . . . . . .
1
lGfw
iGsarp
tatr
1
lit[acfiPt2st1gottatr[erirfnF
rapid homologous desensitization via clathrin-mediated endocy-tosis once stimulated by acyl ghrelin, followed by slow recyclingback to the surface [16]. GHSR-1b mRNA is widely expressed[34], but there is little data for the actual protein expression of
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
Ghrelin is a 28 amino acid peptide hormone and the naturaligand for the Growth Hormone Secretagogue Receptor (GHSR).hrelin was first identified by Kojima et al. and although initially
ound to be produced in the stomach it was not long before ghrelinas identified in a wide range of cell types and tissues [34,60,63].
Ghrelin has been associated with a vast number of effects in var-ous systems of the body. In addition to mediating GH release viaHSR, it has been further implicated in the regulation of appetite,ecretion of gastric acid, gut motility, regulation of cell prolifer-tion, apoptosis, inflammation, sleep, memory retention, anxiety,eward mechanisms, reproduction, lactation, cardiovascular out-ut and blood pressure [42,61,74,93].
In the current review we will summarize the available data onhe genetic variability of ghrelin, its receptor as well as ghrelin O-cyl transferase (GOAT) and their relationship with obesity, stature,ype 2 diabetes (T2D), cardiovascular disease, eating disorders, andeward seeking behavior.
.1. Ghrelin gene structure
One must consider the structural complexity of the ghrelin geneocus and its products to fully appreciate the effect of variationsn the ghrelin gene (Fig. 1a). Ghrelin is derived from the post-ranscriptional processing of the 117 amino acid pre-proghrelin60]. Human pre-proghrelin gene (GHRL) is located on the shortrm of chromosome 3 (3p25–26), and was initially thought toonsist of four exons, however, subsequent experiments identi-ed a number of additional upstream exons in humans [110].re-proghrelin gives rise to a number of products through alterna-ive splicing and post-transcriptional processing; the N-terminal3 amino acid signal peptide is cleaved to leave a 94 amino acidegment termed proghrelin peptide. Proghrelin is cleaved fur-her to give rise to the 28 amino acid ghrelin (encoded by exons
and 2) and a 66 amino acid C-terminal peptide termed C-hrelin [110]. C-ghrelin contains a 23 amino acid sequence termedbestatin [137]. Administration of obestatin to adult rats was ini-ially shown by Zhang et al. to reduce food intake in a dose andime dependent manner, to reduce ghrelin-induced weight gain,nd to slow down gastric emptying via inhibition of jejunal con-ractility [137]. The anorexic effect of obestatin was subsequentlyeplicated again by Zhang et al. as well as a few other groups14,112,136], but the majority of studies have found obestatin toxert no influence on food intake and body weight alone, noreduces ghrelin induced food intake [36,45,66,91,111]. Zhang et al.nitially reported obestatin to act via an orphan G-protein coupled
eceptor called GPR39; however, this group as well as others haveailed to reproduce this finding [45,136]. A number of ghrelin singleucleotide polymorphisms (SNPs) have been identified as shown inig. 2.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2205
1.2. Ghrelin receptor
The growth hormone secretagogue receptor gene (GHSR) islocated at chromosome 3q26.31 and encodes a G-protein coupled7 trans-membrane receptor [46]. The gene consists of 2 exonsseparated by 1 intron (Fig. 3a). Exon 1 codes the 5′ untranslatedregion (UTR) and transmembrane regions I–V. Exon 2 codes fortransmembrane regions VI and VII. The GHSR gene encodes 2 typesof GHSR mRNA through alternative splicing – GHSR-1a and GHSR-1b [85]. GHSR-1a contains all 7 transmembrane domains andpossesses high affinity binding to ghrelin, while GHSR-1b mRNAis encoded by exon 1 and the intronic region. GHSR-1a undergoes
Fig. 1. (a) Pre-proghrelin and its products. UTR, untranslated region numbers inboxes mark exons, while numbers underneath denote amino acids. (b) Ghrelintertiary structure model [9].

B. Liu et al. / Peptides 32 (2011) 2191–2207 2193
Prep roghreli n (chromosome 3p25.3)
5' 1 2 3 4
285bp 117b p 109b p 145b p
-501A>C rs26802
5' 3'1 2 3 4
62G>T rs35683
265A>T Gln90Le u rs468467 7
-1062G>C rs26311
1500G>C rs375577 7
-604G>A rs27647-994C>T rs2631 2 T>C rs207535 6
C>T rs42451
A>G rs35680-1500G>C rs375577 7
408C>A Leu72Me t rs696217
A>G rs35680
d com
tis
tGfsaa
tTfecdisiGnct
1
o(gt
wbbiapaFa
a
Fig. 2. Preproghrelin gene an
his receptor. The physiological role of GHSR-1b is not clear, annhibitory role of the GHSR-1a and GHSR-1b heterodimer has beenuggested in in vitro studies [76].
Although ghrelin mRNA shows a very widespread tissue dis-ribution, expression of the functional ghrelin receptor subtype,HSR-1a, seems to be more restricted with highest concentrations
ound in the CNS and the pituitary, and lower levels of expres-ion in other peripheral organs including adrenal, cardiac, thyroidnd adipose tissue. It is possible that ghrelin may also act throughdditional undiscovered receptor(s).
Ghrelin undergoes O-n-octanoylation at the serine 3 residue;his converts the inactive des-acyl ghrelin into the acyl ghrelin.his process is mediated by the GOAT enzyme and is requiredor the successful activation of GHSR-1a [41,60]. GOAT mRNA isxpressed mainly in the stomach and pancreas, with smaller con-entrations present in bone [35,41]. When fasting mice for 36 h,es-acyl ghrelin is continuously produced. However, because there
s reduced expression of gastric GOAT, acyl ghrelin levels remainstable. On feeding, the presence of dietary medium chain fatty acidsncreases ghrelin activation [57]. This suggests that the activity ofOAT may be more important in the determination of ghrelin sig-aling than ghrelin synthesis. The GOAT gene (Mboat4) located onhromosome 8p12 may therefore represent a novel candidate inhe search for genetic determinants of complex phenotypic traits.
.3. Introduction to genetic studies
Genetic association studies on complex human diseases, basedn candidate genes, whether family based or non-family basedcase control and cohort) studies, or hypothesis generatingenome-wide association studies (GWAS) are effective strategieso study the effect of genetic variation on complex traits.
Historically, case–control association studies has been the mostidely performed, with significant differences in allele frequency
etween cases and controls being used as evidence of an alleleeing involved in disease susceptibility. An observed association
n this situation will be the result of one of 3 situations [113]. Thellele itself may indeed be functional, and directly contribute to thehenotype. The allele may be in linkage disequilibrium (LD) withn allele at another locus that in turn directly affects phenotype.
inally, it is possible for the association to be due to chance, orrtifact.Family based association studies have been used because theyvoid false positive findings created by population stratification.
346G>A A 51G l 3491134 1346G>A Arg51G ln rs3491134 1
mon SNPs with ID numbers.
The most popular method is the transmission-disequilibrium test(TDT); these however possess their own set of weaknesses [17]. Foreach case–control pair, genotypes from 3 people (2 parents and oneproband) are needed. This study design therefore possesses two-thirds the genotyping efficiency of one which uses all outcomesand genetic information from each participant. Indeed, it has beencalculated that case–control association studies are more power-ful than family-based association studies for the detection of weakgenetic effects [104].
In contrast to candidate gene studies which are hypothesis-driven, genetic studies can also be carried out on the genome-widelevel in the form of hypothesis-generating genome-wide associ-ation or genome-wide linkage studies. Genome-wide studies cancomprehensively scan the genome for susceptibility factors towardobservable traits; however, higher costs and the potential for pop-ulation stratification to generate false positive results are some ofits disadvantages.
2. Obesity
2.1. Association studies on ghrelin and obesity
Obesity is a complex trait resulting from interactions betweengenetic, behavioral and environmental factors. Ghrelin, the growthhormone secretagogue, has been found to exhibit central andperipheral effects on appetite, carbohydrate metabolism, gastroin-testinal motility and secretion. Ghrelin links together regulatorymechanisms for growth hormone secretion with energy balance,and is therefore a candidate gene for obesity in humans.
Several early studies comprising of case–control associationstudies with geographically matched controls, as well as familialstudies, identified the presence of ghrelin gene sequence vari-ants through direct sequencing, and sought for the presence ofassociations between these variations and an obesity related phe-notype [25,43,64,121]. Sequence variations which were identifiedin these studies included three amino acid substitutions (Arg51Gln,Leu72Met, and Gln90Leu; Fig. 2) and a number of other SNPs.
Codon 51 codes for the last amino acid of the mature ghrelinproduct. Ukkola et al. identified 6 obese Swedish women who car-ried the 51Gln allele while none were present in the lean control
group, suggesting that this gene variation could increase obesityrisk. The same subjects self-reported to have possessed higherbody weights at the ages of 20, 30 and 40 years than those withthe wild type 51Ala allele [121]. However, this finding was not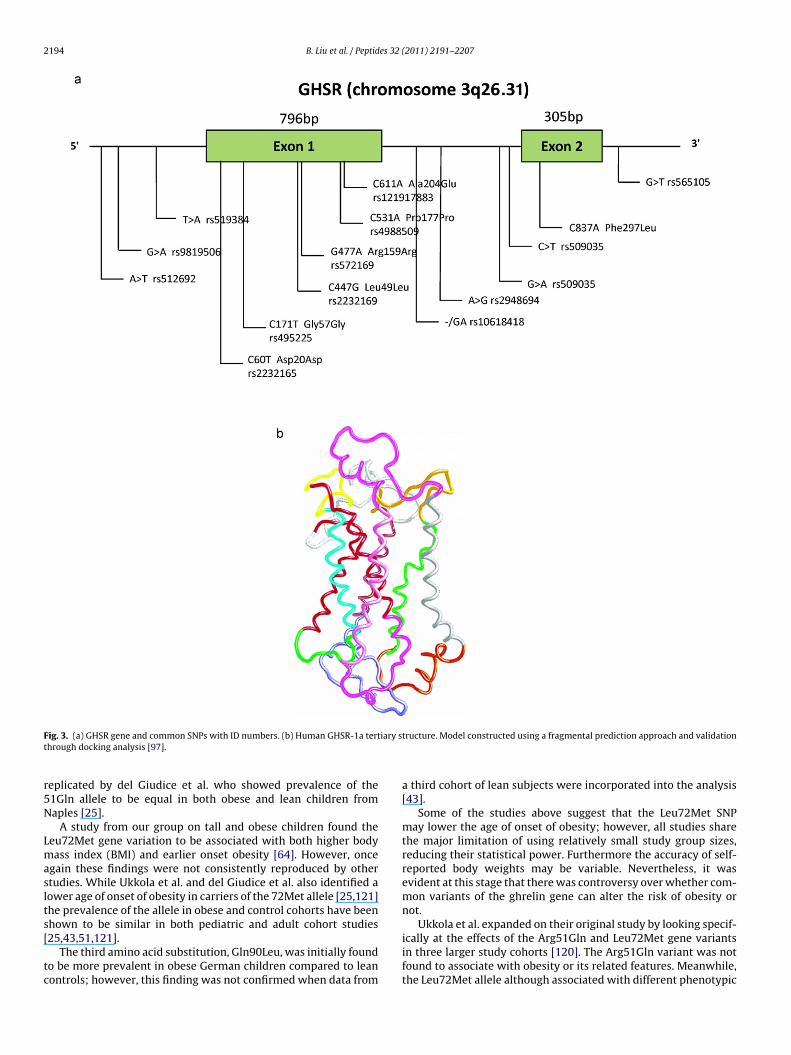
2194 B. Liu et al. / Peptides 32 (2011) 2191–2207
F iary st
r5N
Lmaslts[
tc
ig. 3. (a) GHSR gene and common SNPs with ID numbers. (b) Human GHSR-1a terthrough docking analysis [97].
eplicated by del Giudice et al. who showed prevalence of the1Gln allele to be equal in both obese and lean children fromaples [25].
A study from our group on tall and obese children found theeu72Met gene variation to be associated with both higher bodyass index (BMI) and earlier onset obesity [64]. However, once
gain these findings were not consistently reproduced by othertudies. While Ukkola et al. and del Giudice et al. also identified aower age of onset of obesity in carriers of the 72Met allele [25,121]he prevalence of the allele in obese and control cohorts have beenhown to be similar in both pediatric and adult cohort studies
25,43,51,121].The third amino acid substitution, Gln90Leu, was initially foundo be more prevalent in obese German children compared to leanontrols; however, this finding was not confirmed when data from
tructure. Model constructed using a fragmental prediction approach and validation
a third cohort of lean subjects were incorporated into the analysis[43].
Some of the studies above suggest that the Leu72Met SNPmay lower the age of onset of obesity; however, all studies sharethe major limitation of using relatively small study group sizes,reducing their statistical power. Furthermore the accuracy of self-reported body weights may be variable. Nevertheless, it wasevident at this stage that there was controversy over whether com-mon variants of the ghrelin gene can alter the risk of obesity ornot.
Ukkola et al. expanded on their original study by looking specif-
ically at the effects of the Arg51Gln and Leu72Met gene variantsin three larger study cohorts [120]. The Arg51Gln variant was notfound to associate with obesity or its related features. Meanwhile,the Leu72Met allele although associated with different phenotypic
es 32 (
ctltnp
etitt
go[sarrht
agcfgpmcfi
gsritepataatotilmieo
iivraiatai7o
B. Liu et al. / Peptid
haracteristics in two of the study cohorts, was overall protec-ive against obesity related characteristics by being associated withower BMI, visceral fat mass, and respiratory quotient. It is impor-ant to note that these two study cohorts are made up of mainlyon-obese individuals; perhaps the effects of common ghrelinolymorphisms in obese and non-obese subjects differ.
By suggesting that the 72Met allele, rather than increasing thearliness of obesity development, actually protects against obesity,he later study from Ukkola et al. has raised more questions regard-ng the effect of the Leu72Met gene variation. Several groups aimedo tackle these questions by performing gene association studies onheir own study cohorts [65,127].
Case association studies looking at one or more of the commonhrelin gene variations have been carried out on obese versus non-bese (as well as diabetic versus non-diabetic) cohorts of European39,71], Korean [55], and Canadian [84] origins with study groupizes ranging from 500 to 1500 subjects. None of these studies wereble to identify a statistically significant association with obesityisk. One study had been calculated to possess 80% power to detect aelative risk of above 1.9 for obesity or T2D, although as the authorsave recognized themselves, a negative finding does not rule outhe possibility for a lower magnitude of risk increase [71].
One Italian study on children and adolescents reported thatlthough the prevalence of all 3 previously identified commonhrelin gene variants were present at a similar rate in the obese andontrol cohorts, those with the 72Met allele did have a higher rate ofamily history of obesity. Furthermore when analysing the controlroup separately, those with the wild type 72Leu allele tended toossess lower BMI [127]. A Japanese study later reported that adulten who were carriers of the 72Met allele experienced greater
hanges in body weight and waist-hip ratio since the age of 18, anding not present in women [65].
These studies suggest that common genetic variations of thehrelin gene do not influence obesity risk directly and the fewtudies where a positive association was identified are likely toepresent a false positive finding. With regards to the widely var-ed results for the Leu72Met SNP, one possible explanation forhe observed data is to think of the 72Leu and 72Met alleles asxerting an influence over one’s susceptibility to environmentalressures on body weight: individuals possessing the 72Met allelere more receptive than those with the wild type 72Leu alleleo environmental pressures. This might explain why the 72Metllele has been associated with a lower age of obesity onset and
family history of obesity (where environmental pressures pushoward an increased in body weight), but also a reduced risk ofbesity and lower BMI in other populations (where environmen-al pressures push toward a decrease in body weight). This theorys further supported by the larger change in body weight throughife (longer time for environmental pressures to act) in Japanese
en who possesses the 72Met allele. To better substantiate thisdea, prospective interventional studies are needed, where theffects of specific alterations in environment or behavior could bebserved.
The Finnish Diabetes Prevention Study (DPS) was a random-zed controlled clinical trial in which 522 overweight patients withmpaired glucose tolerance (IGT) were assigned to either the inter-ention or control group [119]. Those in the intervention groupeceived individualized counseling aimed at increasing physicalctivity and healthy eating, while control subjects received genericnformation about diet and exercise. From the DPS, Kilpelainen etl. found that carriers of the 72Met allele who managed to increaseheir physical activity level the most, were able to lose more weight
nd waist circumference than those who increased physical activ-ty levels to a lesser degree. Meanwhile subjects homozygous for2Leu experienced weight loss which were independent of the levelf physical activity [53].2011) 2191–2207 2195
Ghrelin stimulates the release of GH, and GH in turn possesseslipolytic activity [52]. One can speculate that physical activity isable to alter ghrelin regulatory mechanisms more so in individualspossessing certain ghrelin alleles than others. Indeed, although theDPS did not measure circulating levels of ghrelin in its subjects,the phenomenon of both short term [80] and long term [30,73]physical exercise regulating circulating ghrelin levels have beendocumented elsewhere.
Another possible explanation for the observed inconsistenciesin the Leu72Met results is the presence of LD with a locus whichdoes influence obesity risk. Ando et al. demonstrated the presenceof LD between the Leu72Met locus and a 3056T>C substitution inintron 2 of the ghrelin gene. In 264 female Japanese university stu-dents, while both 72Met and 3056C minor alleles were associatedwith elevated acylated ghrelin following correction for BMI, onlythe 3056C allele was significantly associated with anthropometricmeasurements as well as self-reported past BMI. The LD with the3056 loci may help to explain why varied association results havebeen obtained toward Leu72Met; however, neither loci was foundto be significantly associated with BMI, waist circumference andpercentage body fat in a similar sized study by Takezawa et al. on233 obese Japanese subjects [116].
The significance of the 72Met and 3056C sequence variationsneeded to be verified in a larger study for increased power andreduced probability of false positive findings. Dossus et al. carriedout such a study on a selected 3748 members of the EuropeanProspective Investigation into Cancer and Nutrition (EPIC) cohort.Unfortunately, after studying 11 ghrelin SNPs, this study failedto provide the much needed clear-cut conclusion. The Leu72Metand 3056T>C gene variants were found to possess non-significantassociation for higher BMI, suggesting the need for more powerfulstudies. Meanwhile 4 other ghrelin SNPs were found to be signif-icantly associated with higher (rs3755777 and rs10490815) andlower (rs171336 and rs171407) BMI. However, a high false pos-itive report probability (>25%) prevents one from drawing firmconclusions on these associations [27].
Up until 2006, no genetic variations outside the coding regionof the ghrelin gene with a regulatory function affecting ghrelinplasma levels have been published. Vartiainen et al. hypothesizedthat variations in upstream regions of the ghrelin gene may influ-ence ghrelin expression, which in turn may be related to BMI. TheFinnish study identified 11 SNPs, of which the -501A>C sequencevariation showed borderline statistical significance for lower BMI(p = 0.055). Male but not female carriers of the -501A allele also hadhigher mean waist circumference than subjects homozygous forthe minor allele [124]. However, none of the positive phenotypicassociations were replicable in a Czech study [47]. In reply, Var-tiainen et al. were quick to point out that differences in the genepool between Finish and Czech populations may contribute to thediscrepancy in findings [123].
A further point to note is the potential difference in gene-association findings between males and females, a view which wasreinforced later by a Japanese study which reported a different pro-moter sequence variation (-1062G>C) to be significantly associatedwith anthropometric measurements in female but not male sub-jects [116]. Furthermore, ghrelin knock out in male but not femalemice leads to reduced body weight gain and adiposity when fed ahigh fat diet [132]. Based on these studies, we can hypothesize thatghrelin gene variations act differently in male and female subjects,possibly due to the presence of different ghrelin compensatorymechanisms.
In the study by Vartiainen et al. ghrelin gene sequence vari-
ations were not noted to significantly associate with changes infasting levels of ghrelin [124]. It is therefore difficult to envisagethe mechanism by which the ghrelin promoter variant could exertphenotypic effects. It is possible that the fasting ghrelin levels
2 es 32
miic
tlaecdtitmswscPva(-dsttahtgtti[
plTlshsmt
2
fMsil
mhsst
Grto
196 B. Liu et al. / Peptid
easured in this study may not accurately represent the 24 hntegrated area-under-the-curve (AUC) of ghrelin; furthermoret should also be remembered that only the acylated ghrelinomponent of total ghrelin levels is active at the ghrelin receptor.
Measuring 24 h ghrelin levels in humans obviously exerts logis-ical challenges especially when working with large cohort sizes,eaving many authors to use fasting ghrelin levels instead. As anlternative to this, den Hoed et al. measured postprandial lev-ls of ghrelin and other hormonal regulators of appetite. It isertainly true that humans spend a significant proportion of theay in the postprandial state. As ghrelin has also been showno increase the perception of hunger and food intake [22,133],t is therefore possible for genetic variations in ghrelin to affecthe postprandial regulation of orexigenic and anorexigenic hor-
ones, leading to a change in the duration of the perceived hungertate and thus obesity risk. In 103 Western European subjects, itas found that carriers of the -501C ghrelin allele tended to pos-
ess higher postprandial peptide tyrosine-tyrosine (PYY) responsesompared to those homozygous for the wild type -501A allele [26].YY is an anorexic hormone; higher postprandial PYY levels pro-ides a possible link for the association between the -501C allelend lower BMI observed in the study from Vartiainen et al. [124]although it should be noted that no actual association between501A>C and BMI was identified in this study). Similarly, postpran-ial response in plasma PYY was shown to be associated with aequence variation in the ghrelin receptor gene – homozygotes ofhe major G allele at the 477 position in the GHSR gene had sta-istically higher PYY responses than those carrying the minor Allele [26]. This finding concurs with a higher level of perceivedunger, disinhibition and dietary restraint noted in subjects withhe 477A GHSR allele. Furthermore in the same study, postprandialhrelin levels were associated with sequence variants in the lep-in receptor (LEPR) and PYY gene, showing inter-regulation amonghese hormones. Indeed it has been shown that PYY can directlynhibit ghrelin activity in the arcuate nucleus of the hypothalamus103].
The work from den Hoed et al. demonstrate, perhaps unsur-risingly, that genetic variations in ghrelin or its receptor affects
evels of other hormones involved in the regulation of food intake.he exact mechanism via which these effects are mediated may ateast partially be due to differences in the gene promoter activity. Atudy from our group have shown that there are 3 ghrelin promoteraplotypes present at a frequency of >5% prevalence in Caucasianubjects [32]. Out of these 3 haplotypes, only 1 contains the -501Cinor allele, and this haplotype possesses 36% higher activity than
he most common wild type haplotype.
.2. Association studies on GHSR and obesity
GHSR antagonists have been shown to reduce ghrelin inducedoot intake, adiposity and worsening of glycaemic control [5,106].
eanwhile GHSR gene knock out in mice has been shown to reduceeveral of ghrelin’s effects including increased energy intake,ncreased body weight and adiposity, and increases in anticipatoryocomotor activity to scheduled food [13,139].
GHSR-1a has high constitutive activity and GHSR receptorRNA concentrations have been found to increase up to 8 fold in the
ypothalamus following fasting [44,54]. These findings togetheruggest that mutations in the GHSR gene which alter the expres-ion, constitutive activity or ligand interaction of GHSR 1a will havehe potential to significantly affect phenotype.
The earliest study, carried out by Wang et al. identified 2
HSR SNPs (171T>C rs495225, and 477G>A rs572169). The authorseported that the 171T allele tended to be more prevalent in obesehan lean young German subjects. However, an association withbesity risk was not supported by TDT on 387 obesity trios where
(2011) 2191–2207
similar rates of transmission to obese and non-obese offspring werepresent [128].
A subsequent study by Baessler et al. detected pairwise LDbetween 5 GHSR SNPs (rs509035, rs572169, rs519384, rs512692,and rs863441) in both American and German study cohorts. Fromthese 5 SNPs, haplotype 1 with a frequency of 0.69 and haplo-type 2 with a frequency of 0.25, were transmitted more often thanexpected by chance from parents to affected offsprings. Haplotype1 was labeled as a susceptibility allele and haplotype 2 as a non-susceptibility allele for obesity. The definition of obesity in thisstudy was a BMI greater than 32 kg/m2, non-obese were definedas a BMI lower than 28 kg/m2, and subjects with a BMI between 28and 32 kg/m2 were classified as unknown. The definition of obe-sity in this study is arbitrary and these considerations should beincluded in comparison with other studies. The TDT study was per-formed in American population from Winsconsin. Baessler et al.also found association in a second cohort of German subjects in amatched case–control association study [7].
The above two studies show that, similar to ghrelin’s story, asso-ciation studies on GHSR and obesity have not been able to produceconsistent results. Although different SNPs were studied by thetwo groups above with the exception of rs572169, this should notprevent a direct comparison of results given the presence of LDdetected by Baessler et al. The study from Wang et al. used rela-tively small cohort sizes which may have limited the power of thestudy. The familial study on American subjects by Baessler et al.suggest strong and obvious relationships between numerous com-mon sequence variations and BMI; eliciting the same finding in anindependent German population, and the stepwise nature of therelationship between the minor alleles and increased obesity riskreinforces the association even further [7].
A study from our group screened for the association ofthree common haplotype-tagging SNPs (171T>C rs495225; 447C>Grs2232169; 477G>A rs572169) with obesity in children and adultsof the British population. Marginal associations were found for theT171C and BMI, and between G477A and IGF-I levels. No associationwas observed when adult populations were combined for individ-ual sequences variations, or whole haplotypes [31]. This study wasperformed in 5807 British adults and 807 British children, BMI washandled as a quantitative trait against genetic markers on the GHSRgene. Despite sufficient statistical power, a role for the GHSR genein childhood and adult obesity cannot be confirmed in these largecohorts. Because differences in allele frequencies between popu-lations can influence the outcome of genetic association studies,we also have performed genetic case–control association studiesin other European populations. A second study from our group on1275 French cases and 1059 French controls, and 453 German obesechildren and 435 German lean controls, found marginal associa-tions for the 477G allele of the GHSR gene in the French arm withan odds ratio (OR) of 1.73 for obesity that disappeared after statis-tical correction and which could not be replicated in the Germancase–control study [39].
There are mild but important differences in study design andstatistical approach between the family based study by Baessleret al. [7] and our association studies on British [31] and Europeanpopulations [39]. A call toward larger cohorts is definitely a con-cluding message from these studies, since sample size significantlyinfluences statistical power. We would like to highlight a carefulobservation about the metrics used to analyze the trait under inves-tigation. For example in the Baessler study [7], BMI was defined asa discrete variable; unaffected individuals had a BMI lower than28 kg/m2, while in our large British cohort studies BMI was treated
as a continuous quantitative variable. There is no doubt that anindividual with a BMI of 27 is already overweight. Methodologi-cal differences can influence final outcomes in genetic associationstudies. An arbitrary definition of normality against and unbiased
es 32 (
sap
rarotfs
luafnrroeresitasGia
2
wgiig
ia[rec
2
tna
crthsSs
mda
B. Liu et al. / Peptid
tatistical assessment of a variable are clear examples of geneticssociation study designs to be considered in the dissection of com-lex traits.
Most recently, Gjesing et al. carried out the largest ghrelineceptor association study to date [33]. In 15,854 unrelated middleged Danes, 7 SNPs (rs1403637, rs1916345, rs509035, rs2948694,s572169, rs495225) were genotyped for association testing withbesity. Both individual variant and whole haplotype analysis failedo reveal consistent association with measures of obesity, rein-orcing the idea that common polymorphisms of GHSR are notignificant contributors to polygenic obesity.
Finally, another approach was used by Mager et al.; instead ofooking for genetic association of the GHSR gene in the Finnish pop-lation, they addressed the possibility of using markers in this genes predictors of therapy in dietary and exercise regimens aimingor weight reduction. Mager et al. looked at 11 GHSR SNPs span-ing the 5′–3′ region of GHSR (rs6772676, rs9881097, rs474225,s490683, rs11918879, rs477441, rs9819506, rs519384, rs495225,s509035, rs565105). At baseline, no significant differences werebserved between the genotypes for obesity related traits. How-ver, during the 3 years intervention, subjects homozygous fors490683 minor C allele lost significantly more weight than oth-rs. The rs490683 SNP is putatively located within the core bindingequence of the transcription factor nuclear factor-1. In vitro bind-ng experiments of nuclear extracts to oligonucleotides containinghe different sequence variations revealed that the rs490683 Gllele possesses higher protein binding than the C allele [81]. Thisuggests individuals homozygous for the C allele will have reducedHSR expression and thus a reduction in GHSR constitutive signal-
ng. This study illustrates the possibility of using genetic markerss a guide for therapy.
.3. Genome-wide studies on ghrelin/ghrelin receptor and obesity
The ghrelin locus have not been identified in any large genome-ide studies on BMI and obesity, including a meta-analysis of
enome-wide linkage studies comprising data on over 31,000ndividuals [109], meta-analysis of genome-wide association stud-es comprising over 32,000 individuals [130], and a single largeenome-wide association study on over 34,000 individuals [118].
On the other hand, several genome-wide linkage studies havedentified the chromosome 3q24–28 region to be significantlyssociated with an increased risk for obesity or diabetes traits58,79,102,126,134]. The GHSR gene (3q26.31) is a likely candidateesponsible for this association, alternatively the ADIPOQ gene thatncodes the adiponectin protein at chromosome 3q27 is anotherandidate for this positive association [105].
.4. Summary of association studies on obesity
Great efforts have been expended in detecting genetic varia-ions contributing to susceptibility to obesity. Family based andon-family based (case control and cohort) studies have produced
mixture of inconsistently replicated results.For ghrelin, several early studies conducted with relatively small
ohort sizes suggests that ghrelin alleles do not directly increaseisk of developing obesity, but rather affects one’s susceptibilityo environmental pressures. The lack of direct effect on obesity riskas been confirmed by the majority of the larger studies; and analy-is of data from the prospective interventional Diabetes Preventiontudy suggests that ghrelin alleles control the degree of weight lossubjects experience as a result of changes in physical exercise.
Results of another series of studies suggests that ghrelin pro-oter variants alter ghrelin expression, and this is linked to
ifferent post-prandial responses of the anorexic hormone PYY,nd perceived levels of hunger, disinhibition and dietary restraint.
2011) 2191–2207 2197
Once again, the association between ghrelin promoter variants andobesity risk is controversial, but this model does contribute to theidea that ghrelin gene variations alter the effects of environmentalfactors; specifically on how well we behave when faced with anabundance of foods.
The ghrelin receptor possesses high constitutive activity andexperiences large changes in the level of its expression with fast-ing. The distribution of the ghrelin receptor is also much restrictedcompared to that of ghrelin, suggesting additional undiscoveredreceptors for ghrelin. These findings make the GHSR gene an inde-pendent candidate in the search for causes of obesity risk.
The results of one study strongly suggest that a common GHSRhaplotype was associated with obesity in an American family basedstudy, as well as an independent German population based study.Studies on whole haplotypes are generally better than studieson single nucleotide changes as potential interactions betweendifferent nucleotide variations are controlled for. However, thisconclusion was not confirmed by numerous other case–control andfamily based studies, including a UK based study which possessed a98% chance of replicating the positive association results, and a sig-nificantly larger study performed on over 15,000 Danish subjects.
The presence of a gene-environment interaction was detectedas part of the Finnish DPS for GHSR. One promoter sequence varia-tion which seems to exert greater binding to the TF nuclear factor1, is associated with a greater amount of weight loss with lifestyleintervention during the 3 year follow up. Thus GHSR gene varia-tions may exert similar effects to those found in ghrelin in terms ofaltering gene-environment interactions.
The lack of consensus between genome-wide association andcandidate gene association studies is a widely known phenomenon.However, in this case, the conclusions drawn from case associationand GWAS seem to be in agreement over the lack of consistent asso-ciation between ghrelin and obesity risk. The GHSR gene is in closephysical proximity to an obesity risk locus detected from genome-wide studies. However, case–control association studies have failedto support GHSR as the responsible gene, suggesting that the nearbyadiponectin gene may play a bigger role in obesity risk.
In summary, evidence does suggest that common variants ofthe ghrelin and GHSR genes may be involved in modifying sus-ceptibility to environmental pressures of weight gain or loss inhumans. The mechanism of this relationship is no doubt com-plex, and may benefit from intermediate phenotypes for complextraits, large study cohorts with attention to genetic stratification,genome-wide association studies, and the continued search for newvariants in extreme phenotypes and families with a clear Mendelianpattern of inheritance of the trait or an unusual early onset of thetrait.
3. Stature
3.1. Association studies on ghrelin/ghrelin receptor and stature
According to family and twin studies, it has been estimated thatgenetic makeup is responsible for up to 80% of the inter-individualvariation in stature [95]. Ghrelin and growth hormone secreta-gogues induce the release of GH by the anterior pituitary [60].GH stimulates the hepatic synthesis of insulin-like growth factor-I(IGF-I), insulin-like growth factor binding protein 3 (IGFBP3) andacid labile subunit (ALS). GH stimulates linear bone growth bothdirectly and indirectly via IGF-I [92]. Ghrelin is an independentregulator of GH secretion by the pituitary apart from hypothala-mic control and hence it has been postulated as a candidate gene
for stature in human.Due to its longer half life, IGF-I has commonly been used as asurrogate marker for GH. However, it is important to note that IGF-I levels are also influenced by metabolic factors independent from

2 es 32
Gt
UaVa7sab
aesvtlsc
laWhwwtblF
afimnecuvt
pmG
GfawIafiswfigFvtp
3
a
198 B. Liu et al. / Peptid
H. Several ghrelin case–control association studies have looked athe association of ghrelin sequence variants and serum IGF-I levels.
Among the numerous case association studies carried out bykkola et al. in 276 black subjects the Leu72Met sequence vari-nt was reported to associate with higher serum IGF-I levels [120].ivenza et al. showed in obese Caucasians that carriers of the 72Metllele possesses higher IGF-I than subjects homozygous for the2Leu allele [127]. Poykko et al. studied the effects of the Arg51Glnequence variation in 1045 Finnish subjects; carriers of the 51Glnllele possesses significantly lower concentrations of IGF-I, bothefore and after adjustment for age, BMI, sex, and study group [98].
The results of these studies suggest that common genetic vari-nts of ghrelin may be involved in determination of stature viaffects on the GH/IGF-I axis. However, it should be noted that theize of study cohorts, particularly in 2 of the studies, are small andulnerable to type I errors. In addition, although IGF-I is currentlyhe best indicator for serum GH levels, it is far from perfect. Veryow levels of total IGF-I can be considered as definitive evidence forevere GH deficiency, but the relationship between IGF-I and GHloser to the normal range is less clear cut [2].
Apart from the direct role in GH secretion, additional evidenceinking GHRL with human stature originates from a genetic link-ge study on 573 British families participating in the Diabetes UKarren 2 study where a region on chromosome 3p26 was found to
ave a log of odds (LOD) score for stature of 3.17 at the genome-ide level [131]. This lead our group to genotype five common SNPshich captures most of the genetic diversity of the GHRL gene. After
reating height as a quantitative trait, no evidence for associationetween the GHRL gene with stature was documented [40]. Simi-
ar findings were echoed by a subsequent case–control study in therench population [38].
Studies examining for a direct association between genetic vari-tions of ghrelin and height have failed to reveal any positivendings, suggesting that common variations in ghrelin are notajor contributors to height determination. This, however, does
ot rule out the presence of more subtle effects on stature. Forxample, recent GWAS have identified weak associations betweenommon variants in several genes and height in the general pop-lation; the effects of these common variants are weak (geneticariations in the GDF5 and HMGA2 genes explain 0.1–0.8% of theotal variation in height in the general population) [75,108].
For the GHSR gene, initial association was observed for a 2 baseair intronic deletion (rs10618418) that did not stand after adjust-ent for BMI leading the authors to conclude a minor role for theHSR gene in human stature [38].
Three of the GHSR SNPs (T171C, rs495225; C447G, rs2232169;477A, rs572169) were also studied in a separate UK based study
rom our group. In two adults and one pediatric cohort, associ-tion of these genetic variations were studied against height asell as IGF-I. In 811 adults, the 477A allele was associated with
GF-I levels; while in 4996 adults of a separate cohort, 477A wasssociated with lower height. However, there were no consistentndings between the 2 adult groups, and in a combined analysis noignificant associations were detected. No significant associationsere detected in the pediatric cohort [31]. The lack of consistentnding between GHSR gene variants and IGF-I in studies from ourroup is consistent with the study from Vartiainen et al. on 192innish adults [125]. Together these studies suggest that commonariations of the GHSR gene do not contribute in large propor-ions toward variations in IGF-I or stature in adult and pediatricopulations.
.2. Genome-wide studies on ghrelin/ghrelin receptor and stature
In a UK based GWAS comprising 1377 siblings with diabetes, locus near the ghrelin gene on chromosome 3p26 was found to
(2011) 2191–2207
influence stature (LOD score 3.17) [131]. Although this finding wasnot subsequently replicated by multiple GWAS each with data from7000 to 14,000 subjects [20,129], a recently published large GWASwith over 180,000 subjects did identify a SNP (rs572169) locatedwithin the GHSR gene as a locus with the 5th highest p-value out ofthe reported 180 loci which possessed significant association withheight. In addition, to prevent false positive associations that are theresult of population stratification, this finding was also confirmedin a family based analysis featuring 7336 individuals [70].
3.3. Summary of association studies on stature
Height is a highly heritable complex trait. It has been reportedthat transgenic mice over-expressing GHSR in hypothalamicGH-releasing hormone neurons undergo increased post-weaninggrowth rates, while rats expressing reduced level of GHSR in thehypothalamus possess shorter nose–tail length [67,83]. The ghrelinsystem stimulates the release of GH, which in turn regulates bonegrowth, and may therefore contribute to the genetic determinationof height.
Several studies have identified an association between IGF-I lev-els and genetic variations in the ghrelin gene; however, the size ofthese studies were relatively small. Furthermore, lower serum IGF-I have only been linked to short stature in patients with growthhormone deficiency, but this relationship in the normal populationis less obvious.
Numerous smaller studies looking at the association of com-mon ghrelin and GHSR genetic variations with height in both adultand pediatric populations have failed to identify a positive associa-tion. However, a large GWAS with data from over 180,000 subjectsfound GHSR as one of the 180 genes influencing height, possess-ing the fifth lowest p-value for the association [70]. This studyconfirms the notion that common variants of the GHSR gene docontribute toward the variance in adult height, but the magnitudeof its effect is likely to have been too small for earlier studies to con-sistently detect. Future research efforts in the area of ghrelin/GHSRand height should therefore aim to take this into consideration inorder to design studies of appropriate power.
4. Diabetes
4.1. Association studies on ghrelin and type 2 diabetes
T2D is a disorder of glucose metabolism, featuring a spectrumof insulin resistance, and is often associated with obesity. Bothghrelin and GHSR-1a expression have been identified in the humanpancreas, suggesting a possible relationship between ghrelin/GHSRpolymorphisms and glucose metabolism [24,34]. The exact rela-tionship between ghrelin and glucose metabolism is complex, withboth short and long term effects, and have been reviewed in depthelsewhere [107]. It may be sufficient to say that numerous groupshave found ghrelin to increase insulin resistance. Ghrelin knock-out in mice results in enhanced insulin sensitivity [115], while inhumans, fasting ghrelin correlates negatively with insulin levels,and positively with insulin resistance [99,101], and administrationof ghrelin to humans induces hyperglycaemia and hypoinsuli-naemia [15]. Although curiously, one study found higher fastingghrelin levels to be associated with a lower prevalence of T2D andlower insulin sensitivity [99].
Studies looking at the association between ghrelin sequencevariations and the risk of T2D can be divided up into those whichlooked at association with glucose and insulin levels, either in the
fasting state, or following oral glucose tolerance test (OGTT), someof which then continued to quantify insulin resistance throughthe homeostatic model assessment method (HOMA-IR); and thosewhich compared the prevalence of T2D directly.
es 32 (
ssJCscaafo
9gtiiodbasarm
iiosafit
sgpa
BtipdscPTT
s2alothHfi
fStbt
B. Liu et al. / Peptid
Three rather similar case-association studies measured fastingerum glucose, insulin levels, and HOMA-IR values in differenttudy cohorts. The cohorts used in the studies included 264apanese female university students aged 19–23 [3], 2413 adultaucasians [12], and 2228 adult Japanese subjects [65]. All threetudies failed to identify any significant association between indi-ators of glucose metabolism and the Leu72Met SNP. One studylso looked at the 3056T>C gene variant, again unable to identifyny significant association [3]. These studies all suggest that in theasting state, ghrelin gene variants do not affect glucose metabolismr insulin sensitivity.
In contrast, a fourth study carried out by Zavarella et al. on00 overweight/obese Caucasian subjects and 500 healthy geo-raphically matched controls reported the 72Leu and 72Met alleleso display a co-dominant phenotypic effect, with an increas-ng number of 72Met alleles being associated with lower fastingnsulin and HOMA-IR [135]. A second SNP located in the 5′UTRf the ghrelin gene (-604C>T) possessed a similar trend forecreasing insulin levels and HOMA-IR. A strong LD was observedetween these two SNPs, and significant associations with insulinnd HOMA-IR were present during haplotype analysis. Obe-ity is known to reduce insulin sensitivity; it is possible that
reduction in insulin sensitivity is required in order for theelatively small ghrelin polymorphism phenotypic effects to beeasured.The idea that these ghrelin minor alleles are associated with
ncreased insulin sensitivity (and therefore protective against T2D)s further supported by a study from our group where 41 tall andbese children were selected to undergo OGTT [64]. Although noignificant differences in glucose or insulin levels were observedt the beginning of the test (equating to the fasting state), 30 minollowing the start of the OGTT, the rise in insulin levels were signif-cantly lower in carriers of the 72Met allele than those possessinghe wild type genotype.
An association with serum insulin physiology does not neces-arily translate into an association with T2D. For this reason, manyroups also sought a direct association between T2D and ghrelinolymorphisms. The majority of these studies did so by recruiting
T2D cohort and a glucose-tolerant control cohort.A case–control association study on 850 Caucasian subjects by
erthold et al. reported that the 72Met ghrelin allele decreaseshe risk of T2D with an OR of 0.63. This finding remained signif-cant in a multivariate analyses following correction for BMI, bloodressure, and family history of T2D [10]. No clear relationship wasetected between genotype and fasting ghrelin concentration orerum glucose, again supporting the notion that post-prandial glu-ose metabolism is the factor being affected. A second study byoykko et al. studied the Arg51Gln SNP in 1045Caucasian subjects.he 51Gln allele was found to associate with an increased risk of2D [98].
However, these positive findings were not reproduced by othertudies, including a Danish case–control study with 557 type
diabetics and 233 geographically matched controls, as wells a similar study on 856 subjects from diabetic Amish fami-ies. Significant associations were not detected between severalther ghrelin SNPs (Leu72Met, Gln90Leu) and T2D [71,114]. Inhe latter study, the 72Met allele did associate significantly withigher serum glucose, but the lack of association with insulin,OMA-IR and T2D suggest that this may be a false positivending.
Two Korean studies published around the same time alsoailed to identify any association between T2D and the Leu72Met
NP [21,55]. However, one study did find diabetic carriers ofhe 72Met allele to possess lower serum creatinine, indicative ofetter renal function [55]. Mechanistically, this finding is unlikelyo be directly related to ghrelin’s effect on insulin resistance as2011) 2191–2207 2199
similar associations for macroangiopathy and retinopathy werenot present. The second Korean group looked at three ghrelinpromoter SNPs (-1500C>G, -1062G>C, -994C>T), and was againunable to identify any significant association with T2D. In vitroexperiments did; however, show that the -1062G allele possessesa 1.7 fold higher promoter activity than that of the -1062C allele. Itis interesting to find that a 1.7 fold difference in promoter activitydoes not translate into a significant phenotypic effect and supportsthe notion that ghrelin is not a major contributor to diabetesdevelopment, although one must remember that increased pro-moter activity does not necessarily equate to increased proteinexpression [21].
Takezawa et al. proposed that the causative loci for diabetic riskwas not located around Leu72Met, but rather the 3056T>C, locus towhich Leu72Met is in LD. In 115 Japanese men, the wild type 3056Tallele was reported to associate with increased T2D risk [116]. Thisassociation was not present in women, in whom an associationwith obesity and fat metabolism was identified instead [116]. If adifference between men and women does indeed exist, combined-sex data analysis may dilute any underlying associations and canpotentially explain some of the observed discrepancies betweenstudies.
A study from our group expanded upon earlier studies throughwhole haplotype analyses in order to negate the effects of pop-ulation stratification. In a study of 610 T2D patients and 820non-diabetic controls, no significant associations were observedbetween T2D risk and 5 ghrelin SNPs (-604A>G, -501C>A,Leu72Met, Gln90Leu, G62T). One ghrelin haplotype; however, wasreported to be marginally associated with protection against T2D.The promoter found within this haplotype was associated with a45% reduction in promoter activity compared to that of the wildtype haplotype [32].
4.2. Association studies on ghrelin receptor and type 2 diabetes
Studies searching for associations between diabetes and GHSRare much more limited in number when compared to its ligandcounterpart. The earliest study, by Vartiainen et al. compared theprevalence of GHSR SNPs between cohorts with low and highIGF-I levels [125]. Decreased IGF-I have been associated with T2Dand diabetes related complications [48]. Vartiainen et al. identi-fied five conservative gene variations within exon 1 of GHSR, fromwhich the wild type 477G and 171C alleles were found to asso-ciate with the highest area under the insulin curve (AUCIN) valuespost-OGTT [125]. This suggests that common GHSR SNPs may influ-ence post-prandial glucose metabolism. No association with IGF-Iwere identified – a finding in line with functional studies that showexogenous ghrelin induces hyperglycaemia independently of theGH axis [107]. Carriers of the 477G allele did possess the lowestlevels of plasma IGFBP-1; insulin has been recognized as a potentdown-regulator of IGFBP-1 expression thus reinforcing the highAUCIN finding [125].
As part of the Finnish DPS, Mager et al. looked at the associationof 7 GHSR gene variations with glucose metabolism in 507 subjectswith impaired glucose tolerance. No significant associations werepresent at the beginning of the study; during the 3 years follow upperiod, significant associations developed between measures ofglucose metabolism and GHSR SNPs (promoter region rs6772676,rs490683, and intronic rs509035). One question which remainsunanswered in this study, was why associations with glucosemetabolism were absent at the beginning of the study, but devel-oped with follow up when analyzing control and interventional
cohorts together. Rs490683 is situated at a putative nuclear factor1 binding site. Mager et al. showed that nuclear proteins bind tothe rs490683-G allele with much higher affinity compared to the Callele and therefore can potentially increase GHSR transcription as
2 es 32
aiabGs
cTetfndFddSeth
4d
ci8oito
4
paHdcostsof
opiea
Timn
mrgss
200 B. Liu et al. / Peptid
mechanism for exerting phenotypic effects. On the other hand,t is important to mention that Mager et al. failed to identify anssociation with T2D directly [81]. This latter finding is supportedy a genetic association study from our group which looked atHSR promoter variations in 610 diabetic and 820 non-diabeticubjects.
One question which presents itself is how to interpret a signifi-ant association with measures of glucose metabolism, but not with2D. It is possible that GHSR gene promoter variations do indeedxert some physiological effect on glucose metabolism; however,he size of this effect is insufficient to translate into a significant riskactor for T2D with the size of the study populations used. Alter-atively GHSR promoter variations may simply not contribute toiabetic risk, and this is supported by 2 findings from our group.irstly, different GHSR promoter haplotypes possessed significantlyifferent basal and ligand-stimulated promoter activity in vitro, andespite this difference no association with T2D were identified.econdly, even if GHSR promoter variants did significantly influ-nce diabetic risk, this influence would not be widespread due tohe over-representation (96%) of the GHSR promoter by a singleaplotype [32].
.3. Genome-wide studies on ghrelin and GHSR and type 2iabetes
GWAS have not identified the ghrelin or GHSR gene as aausative factor of T2D. However, this situation is far from unique;n an analysis study carried out by Lillioja and Wilton results from3 linkage reports on T2D risk loci failed to overlap with the resultsf GWAS [78]. This finding may reflect sample size insufficiencyn the majority of studies which have rarely used cohorts largerhan a few thousand subjects, as well as the complex etiologyf T2D.
.4. Summary of association studies on type 2 diabetes
Functional and gene knock out studies have shown ghrelin toossess a definite role in glucose metabolism, exerting both acutend chronic effects independent of the GH signaling pathway [107].owever, results of genetic association studies have been lessefinitive. Studies have been unable to identify any consistent asso-iations between ghrelin SNPs and measures of glucose metabolismr T2D. The use of obese versus non-obese study cohorts, and mea-urement of serum levels in the fasting versus fed states may helpo explain some but not all of these observed inconsistencies. Onetudy even suggests that there is a difference between the effectsf ghrelin polymorphisms in men and women – an area needingurther exploration.
One particular SNP, Leu72Met, has been the focus of numer-us studies. This amino acid is located outside the mature ghrelinroduct and thus not involved in receptor binding. In vitro stud-
es exploring how this SNP can alter ghrelin signaling will help tostablish whether inconsistent associations with the Leu72Met SNPre due to type I errors, or power insufficiency in certain studies.
Studies looking at the association of GHSR polymorphisms and2D have been very limited in number; while these studies havedentified a significant association between certain GHSR SNPs and
easures of glucose metabolism, no studies have identified a sig-ificant association with T2D directly.
From the above we can conclude that common genetic poly-
orphisms of ghrelin and GHSR are not major contributors of T2Disk; this is further supported by the lack of positive findings fromenome-wide studies, although more powerful gene associationtudies with larger study cohorts may be able to identify moreubtle effects exerted by the ghrelin/GHSR loci in the future.
(2011) 2191–2207
5. Cardiovascular disease
5.1. Association of ghrelin polymorphisms with cardiovascularhealth
GHSR is expressed on blood vessels as well as autonomic pre-ganglionic neurons of the spinal cord. Experiments in rats haveshown that ghrelin acts on sympathetic pre-ganglionic neurons toincrease blood pressure and on blood vessel receptors to decreaseblood pressure [29]. Studies which measured serum ghrelin lev-els in hypertensive and normotensive human subjects have foundserum ghrelin level to increase in hypertension, possibly as a phys-iological attempt to counteract the high blood pressure [94,99].GHRL polymorphisms which alter ghrelin activity may thereforebe associated with increased or decreased risk for essential hyper-tension.
In one study, the Arg51Gln SNP was found to associate withhypertension; the 51Gln allele was more prevalent in hyperten-sive than normotensive Finnish subjects. There were no associationbetween Arg51Gln and actual blood pressure measurements, andthis may be expected given the use of antihypertensive medicationsin the hypertensive cohort [98]. Increasing numbers of the 51Glnallele have been shown to be associated with stepwise reductionin serum ghrelin levels [99]. The 51Arg amino acid is the target sitefor endoprotease cleavage and the presence of a 51Gln amino acidcan theoretically reduce endoprotease binding, leading to reducedserum ghrelin levels.
A population based study compared the distribution ofLeu72Met alleles in Danish hypertensive and normotensive sub-jects. This study was unable to identify any significant associationbetween Leu72Met and risk of hypertension [12]. In a separatestudy on obese Swedish subjects, the 72Met allele was found toassociate with reduced prevalence of self-reported hypertension,stroke or electrocardiogram changes in women only [120]. Theresults of this latter study contrasted with that of the Finnish DPS.522 adults with impaired glucose tolerance were followed up fora period of three years, and the phenotypic associations of fourcommon GHRL SNPs (-604G>A, -501A>C, Leu72Met, and Gln90Leu)were studied. The most frequently occurring haplotype composedfrom the wild type alleles of each SNP was found to associatewith lower blood pressure measurements at baseline and through-out the 3 years follow-up. This finding remained significant afterexcluding all patients who were on antihypertensive medication.Although an association with the prevalence of hypertension wasnot present at the beginning of the study, a significant associationdid develop during years 1–3 of follow-up [82].
This discrepancy in results, whereby two different studiesmanaged to identify the same GHRL SNP possessing opposite asso-ciations with hypertension risk may be explained by the resultsof a recent study from Berthold et al. In a case–control associa-tion study of 1143 hypertensive and 1489 normotensive Caucasiansubjects, haplotype analysis showed that in the presence of the72Leu allele, presence of -501C increased hypertension risk withOR 1.303, whereas -501A decreased hypertension risk. However,when the 72Met allele was present in a haplotype, the -501C alleledecreased hypertension risk with OR 0.496 [11]. A study from ourgroup showed that different GHRL haplotypes containing differentcombinations of four GHRL SNPs (-501A>C, Leu72Met, Gln90Leu,62G>T) possessed up to 45% difference in promoter activity [32]. Itwill be interesting to study how these GHRL SNPs interact to resultin different promoter activity. An effect on post-transcriptionalprocessing and protein trafficking should also be sought. Never-
theless, it seems likely that different GHRL haplotypes leads toaltered central and peripheral ghrelin expression, thereby influ-encing hypertensive risk. Berthold et al. also confirmed the earlierobservation that the 51Gln allele is more prevalent in hypertensive
es 32 (
sp
5h
ttdcbtoalpdmmistmtiihGttwhtv
nobtctottadsdhlltisaslcf
5c
D
B. Liu et al. / Peptid
ubjects, further reinforcing the link between ghrelin polymor-hisms and blood pressure.
.2. Association of GHSR polymorphisms with cardiovascularealth
Left ventricular hypertrophy (LVH) is an independent risk fac-or for cardiovascular disease. GHSR is expressed in myocardialissue and drives the production of GH mediated by ghrelin in car-iac myocytes. Baessler et al. selected 109 cases with LVH and 355ontrols without LVH from a cohort of 1230 subjects from Augs-urg, Germany and studied variation on the regulatory region ofhe GHSR gene [8]. A major caveat of this study was the inclusionf subjects on antihypertensive medication and type 2 diabetics,s these factors can compound the influence of genetic factors oneft ventricular mass (LVM). Despite this limitation and the com-lexity of the variable under study (LVM), the allele frequenciesiffered significantly between the groups when considering singlearkers and persisted in the haplotype analysis. The most com-on haplotype in this population was found to be most frequent
n the control group and inversely associated with LVH when con-idering the variable as a quantitative trait in the entire cohort. Onhe contrary the second most common haplotype was found to be
ore frequent in the case group and positively related with LVM inhe entire cohort. These results are suggestive for a role of ghrelinn the heart; however, if an effect is present, it is likely to be anndirect one. There is room to speculate that subjects carrying theaplotype linked to increased LVM should have higher levels ofHSR expression, and on the contrary subjects carrying the protec-
ive allele should have a lower expression. Further studies aimingo assess the expression levels at the cell membrane of the GHSRith radio-ligand binding assays in normal endomyocardial and ineart failure samples, and correlation with the genotype profiles onhe GHSR could give a mechanistic insight into the role of geneticariants of the GHSR in LVM.
GHSR gene variants have also been studied as markers for coro-ary artery diseases (CAD) based on evidence for association fromne meta-analysis that included four GWAS suggesting 3q26–27 toe linked with coronary ischaemic diseases [19]. Myocardial infarc-ion (MI) is a multifactorial trait and in the study of Baessler et al.onfounding factors such as obesity, hypertension, hypercholes-erolemia, T2D and smoking are well illustrated [6]. In the selectionf the cases for MI it can be argued that those subjects selected inhese studies are the ‘survivors’ of an acute coronary syndrome orhe subjects that received immediate health care at the time of thecute coronary syndrome. MI can also be manifested as suddeneath with the loss of alleles non measurable in the associationtudy. Meanwhile, for the selection of the controls, in terms of car-iovascular risk, to avoid confounding variables, this group mustave a higher risk when compared with the general population but
ower risk when compared with the case group. To overcome theseimitations of association studies in MI, Baessler et al. conductedwo case–control studies. The first study consisted of 864 subjectsn each group, using post-MI patients as the cases. Meanwhile, theecond study consisted of 826 subjects in each group, and used theffected relatives of MI patients from the first study as cases. In bothtudies, significant associations were observed between GHSR hap-otypes comprising five SNPs spanning the length of the gene, andardiac disease. p-values remained significant even after adjustingor classical cardiovascular risk factors.
.3. Genome-wide studies on ghrelin/ghrelin receptor and
ardiovascular healthHypertension was one of the first traits studied under GWAS.espite significant evidence of association from candidate gene
2011) 2191–2207 2201
studies, neither GHRL nor GHSR have been identified as a significantlocus in genome-wide studies. This includes two large scale meta-analyses with data from 34,433 and 29,136 individuals respectively[77,90].
5.4. Summary of association studies on cardiovascular health
Functional experiments have demonstrated that ghrelin clearlypossess the ability to influence blood pressure. It was thereforetempting to hypothesize that GHRL polymorphisms may influ-ence hypertensive risk. This hypothesis has been supported by theresults of several case–control association studies whereby mul-tiple GHRL SNPs have demonstrated a difference in distributionpatterns in hypertensive and normotensive study cohorts. GHSRpolymorphisms on the other hand have been found to be signif-icantly associated with LVH and MI/CAD; an effect independentof any effects on blood pressure [6,8,19]. Clarification of the cel-lular mechanism in which different gene alleles can exert thesephenotypic effects will help to confirm these findings.
6. Eating disorders
6.1. Association of ghrelin polymorphisms with eating disorders
Genetic factors have been estimated to explain over 50% of thevariance of eating behavior in both subjects with, and without eat-ing disorders [59,117].
Bulimia nervosa (BN) is an eating disorder characterized byepisodes of binge eating followed by feelings of guilt, which thepatient overcomes through certain acts such as purging, use oflaxatives, or diuretics. In patients with BN, fasting ghrelin levelshave been shown to be higher than that of controls, and the post-prandial suppression of ghrelin is attenuated [62,87]. This suggeststhat ghrelin polymorphisms may be involved in the pathogenesisof bulimia nervosa through promotion of binge eating behavior.The results of one case-association study on Japanese female sub-jects seem to support this hypothesis. Two ghrelin SNPs, Leu72Metand 3056T>C, found to be in LD, were more prevalent in 108 sub-jects with BN than 300 unrelated female controls [4]. Meanwhile,three case–control association studies and one family trios studyon Caucasian subjects failed to confirm these positive findings, withghrelin SNPs present at a similar frequency in BN and healthy con-trol subjects [18,56,88]. Overall, the sizes of all these study cohortsare comparatively small in the field of ghrelin case-associationstudies. Nevertheless it seems likely that ghrelin polymorphismsare not major contributors to BN development, at least in Caucasianpopulations. Ghrelin may contribute toward BN risk in Japanesepopulations, where different frequencies of ghrelin polymorphismsare reported (e.g., ghrelin 72Met allele present in 20% of Japaneseversus 10% of Caucasian subjects) [4,88].
Anorexia nervosa (AN) is a condition characterized by a dis-torted image of oneself leading to severe calorie restriction, plus incertain cases, the use of strenuous exercise for purposes of weightreduction. High levels of ghrelin have been found in AN patients[122]. This high ghrelin level could be the result of a compensatorymechanism, used by the subject to stimulate feeding; alternatively,a loss of function within the ghrelin system may contribute towardcalorie restriction in AN patients.
One set of case–control association, and family trios study onEuropean subjects failed to identify an association between ghrelinpolymorphisms and AN [18]. Meanwhile, a familial study on 114
French probands did report preferential transmission of Leu72Metpolymorphism to offspring with AN, specifically those diagnosedwith the binging/purging, but not restrictive, AN subtype [23].However, case-association studies on 228 Japanese patients [4] and
2202 B. Liu et al. / Peptides 32 (2011) 2191–2207
ith ID
1atsaav
6
psJsap
6
iasrr
6
elGmtwti
Fig. 4. (a) GOAT (MBOAT4) gene and location of common SNPs w
73 Caucasian patients with AN [88] failed to detect a significantssociation of ghrelin SNPs with either subtype of AN. Based onhese studies, it is unlikely for ghrelin polymorphisms to contributeignificantly toward overall AN development risk. The one positivessociation with the binging/purging subtype of AN may represent
false positive finding, although ideally this conclusion should beerified by a larger study.
.2. Association of GHSR polymorphisms with eating disorders
Only one study searching for an association between GHSRolymorphisms and eating disorders has been carried out. In thistudy, the 171CC genotype was more frequently present in 116apanese patients with BN compared to 284 unrelated controlubjects (p = 0.035, OR = 2.05, Bonferroni correction p = 0.070). Nossociation for AN, AN subtypes, or eating disorders in general wereresent for this 171T>C SNP [86].
.3. Association of GOAT polymorphisms with eating disorders
One group has recently reported that GOAT polymorphisms maynfluence AN risk. In cohorts composed of 543 German AN patientsnd 612 controls, the effect of six haplotype-tagging SNPs weretudied. In a recessive model of inheritance, genotype G/G at SNPs10096097 (Fig. 4a) was found to be associated with an increasedisk of AN [89].
.4. Summary of association studies on eating disorders
The clear relationship between the ghrelin signaling system andating behavior, as well as the presence of higher serum ghrelinevels in patients with eating disorders, makes ghrelin, GHSR, andOAT good candidates genes for eating disorder risk. However, theajority of candidate-gene association studies have not been able
o identify ghrelin polymorphisms as being significantly associatedith either BN or AN in Caucasian populations. A significant rela-
ionship may be present in Japanese populations, but further workn this area is needed to verify this.
numbers. (b) Human GOAT putative tertiary structure model [1].
For GHSR and GOAT, only one candidate-gene study is availablefor each gene. Significant associations are found by these studieswith BN and AN respectively, however, these findings need to bereplicated by others in order to rule out false positive findings.Genome-wide studies looking at eating disorders again have beenrelatively scarce compared to those which studied obesity, dia-betes, and stature. Studies that are available have not identifiedthe ghrelin system as a significant contributor to eating disorderrisk.
7. Reward seeking behavior
7.1. Association studies on ghrelin and ghrelin receptor withalcohol dependence
The mesolimbic pathway is a reward pathway which begins inthe dopaminergic neurons of the midbrain ventral tegmental area(VTA). Central administration of ghrelin, possibly acting via GHSR-1a receptors in the VTA and laterodorsal tegmental area [37,138],have been shown to increase dopamine release from this rewardpathway [49]. This suggests that the ghrelin axis may influencereward seeking behavior.
Alcohol, which has been shown to activate the same dopamin-ergic pathway [72], requires the presence of an intact ghrelin axisin order to exert its effects on reward [50]. From these studies it isreasonable to hypothesize that ghrelin may be involved in alcoholdependence.
To test this hypothesis, Landgren et al. compared the preva-lence of six GHRL and four GHSR-1a SNPs in 417 Spanish individualsdivided into cohorts of alcohol abstainers, moderate drinkers andheavy drinkers [69]. Allele A of rs2232165 in the GHSR-1a genewas associated with heavy alcohol consumption; a finding whichis independent of smoking status or gender.
However, a positive association with significant alcohol depen-
dence was not confirmed by the same group in a second study on113 Caucasian females with alcohol dependence, and 212 femalecontrols with a low level alcohol consumption. Instead, weak asso-ciations between GHSR and GHRL haplotypes and the measured
B. Liu et al. / Peptides 32 (2011) 2191–2207 2203
Fig. 5. GHSR and location of rare GHSR SNPs highlighted in blue, including: Trp2Stop [96], Ser84Ile [100], Ala169Thr [100], Val182Ala [100], Ala204Gln [96,128], Phe279Leu[ , Val1t this fi
odd
gdttm
8
8
gbsssait
8
rsf
iAaaPdb
28,96,128], Arg237Trp [96], Ala358Thr [100]. Minor alleles of Ser84Ilem Ala169Thrional delays in growth and puberty. (For interpretation of the references to color in
bjectives of protection against alcohol dependence, type 2 alcoholependence, paternal alcohol dependence and increased with-rawal symptoms were noted [68].
These early studies suggest that genetic variation within thehrelin axis may influence reward seeking behavior and drugependency, albeit on small magnitudes. Confirmation of this rela-ionship through more powerful studies, and future exploration ofhe in vivo effects of safe ghrelin antagonists may produce beneficial
ethods of tackling drug dependency.
. Rare variants in ghrelin and GHSR
.1. Rare ghrelin gene variants
Very few studies detected rare genetic variations in the ghrelinene. A single nucleotide substitution located in the intronic regionetween exons 1 and 2 of GHRL has been identified in two obeseubjects [121]. Another mutation was detected in a normal weightubject who possessed a 2 base pair deletion at codon 34. This framehift mutation leads to the insertion of 36 aberrant amino acids, and
stop codon at position 71, likely leading to a deficiency of ghrelinn this subject [43]. Given the rarity of these mutations, it is difficulto draw any conclusions from these reports.
.2. Rare ghrelin receptor gene variants
Several groups have reported the identification of rare GHSReceptor gene variations in certain carriers who possess either shorttature or growth hormone deficiency. These studies support a roleor GHSR in height determination.
Wang et al. screened the entire coding region of GHSR anddentified two previously uncharacterized gene variations (611C>Ala204Gln and 837C>A Phe279Leu, Fig. 5), in an obese child and
child with short normal stature respectively [128]. Both amino
cids are highly conserved among humans, swine, and rats. Thehe279Leu amino acid substitution located in transmembraneomain 6 was previously reported to decrease the specificity ofinding to a GHSR agonist in vitro, thereby providing a possible82Ala, and Ala358Thr have so far only been documented in children with constitu-gure legend, the reader is referred to the web version of this article.)
mechanism toward causation of short stature [28]. The effect ofthis gene variation may be exerted in a dominant fashion as thechild’s mother who also possessed 279Leu, was identified to be ofshort stature.
The Ala204Gln amino acid substitution located in the secondextracellular loop was studied by Pantel et al. in two unrelatedMoroccan families [96]. Out of 9 subjects heterozygous for 204Gln,6 were of short stature; this supports a dominant phenotypic effectwith incomplete penetrance. One patient homozygous for 204Glnwith short stature was also identified. In vitro studies show thatsubstitution of the apolar and neutral alanine residue by the polarand charged glutamate amino acid reduces cell surface expressionto around 20% of wild type and removes the constitutive activity ofGHSR, although ghrelin-stimulated activity remained intact.
Pantel et al. also characterized the genetics of a third family witha child with isolated growth hormone deficiency [96]. This childinherited a premature termination codon (6G>A, Trp2X) from hisasymptomatic father, and a third intracellular loop amino acid sub-stitution (709A>T, Arg237Trp) from his asymptomatic mother, andwas thus compound heterozygous for two GHSR gene variations.As a second son from the family who possessed a single Arg237Trpallele from his mother was also asymptomatic, an autosomal reces-sive pattern of inheritance is supported. In vitro studies suggest thatthe Arg237Trp gene variation allows for normal cell surface expres-sion of GHSR and its response to ghrelin, however, there is a partialloss of constitutive activity.
As growth hormone is so exquisitely linked with growth andpuberty, children with constitutional delays in growth and pubertymay therefore possess differences in the ghrelin signaling path-way. Pugliese-Pires et al. genotyped the GHSR coding region of 39such children, identifying one promoter (-6G>C) and 4 exonic sub-stitutions of highly conserved amino acids (Ser84Ile, Ala169Thr,Val182Ala, Ala358Thr) [100]. In vitro studies found the Ser84Ilesubstitution to be associated with decreased basal activity and
ligand-induced signaling which may at least be partially attributedto a marked reduction in cell surface expression. The Val182Alagene variation was similarly found to be linked with decreasedbasal and ligand-induced activity, albeit with only a tendency
2204 B. Liu et al. / Peptides 32 (2011) 2191–2207
Table 1Summary of genetic studies on ghrelin, GHSR and GOAT genes.
Candidate gene studies Genome-wide studies
GHRL GHSR GOAT GHRL GHSR
Obesity No association with obesity risk[25,39,43,47,51,55,71,84,127]
No association withobesity risk[31,33,39,128]
Not available No positive study Significant associationwith obesity atchromosome 3q24-28[58,79,102,126,134]
Association with obesity risk[3,27,64,116,120,121,124]
Association with obesityrisk [7]
Association with earlier onset of obesity[25,64,121]
Alters response toenvironmental pressures[81]
Alters response to environmentalpressures [53]
Stature Presence of association with IGF-I[98,120,127]
No association with IGF-I[31,125]
Not available Significant associationwith stature atchromosome 3p26[131], but notconfirmed for ghrelinin the same population
Significant associationbetween stature andGHSR SNP [70]
No association with stature [38,40] No association withstature [31,38]
T2D No association with glucose metabolismor insulin sensitivity [3,12,65]
Association with glucosemetabolism and insulinlevels [81,125]
Not available No positive study No positive study
Association with glucose metabolism andinsulin sensitivity in obese patients[64,135]No association with T2D[21,32,55,71,114]Association with T2D [10,98,116]
Cardiovascular health No association with hypertension risk[12]
Association with LVH [8] Not available No positive study Significant associationwith MI/CAD atchromosome 3q26–27
Association with hypertension risk[11,82,98,120]
Association with MI/CAD[19]
Eating disorder Association with BN [4] No association witheating disorders [86]
Associationwith AN [89]
No positive study No positive study
No association with BN [18,56,88]Association with AN binging/purgingsubtype [23]No association with AN [4,18,88]
Alcohol dependency Association with paternal alcohol Association with alcoholy [68,6
Not available No positive study No positive study
twwdht
wrtTpcpcto
aTiCvt
dependency [68] dependencAssociation with alcohol withdrawal [68]
oward reduced expression. Both of the latter gene variations,hich seemed to be inherited in an autosomal dominant fashionith incomplete penetrance, were identified in girls with delayedevelopment at the time of puberty (young bone age, reducedeight); however, there seemed to be sufficient catch up growtho achieve normal final heights [100].
From the above studies, we see that rare GHSR gene variationshich disturb the proper function of the receptor either through
educed cell surface expression, ligand interaction, or signalingransduction can exert marked effect on growth and development.his then leaves the important question of why common GHSRolymorphisms, some of which have been shown to possess signifi-ant differences in promoter activity, do not exert the same obvioushenotypic effects. Perhaps compensatory mechanisms are able toope with a set amount of reduced GHSR functioning, and a certainhreshold needs to be reached, before obvious detrimental effectsn height and development occur.
In terms of a relationship with obesity, Gjesing et al. identified -151T>C promoter SNP in one Danish and one Czech family.here was complete co-segregation of this variant with obesity
n the Danish family, but only incomplete co-segregation in thezech family; this may be due to incomplete penetrance of theariant. In vitro analysis shows that this variant exerts increasedranscriptional activity, likely to be due to introduction of a highly9]
specific transcription factor SP1 binding site. It is theoreticallypossible for there to be higher GHSR expression in carriers of the-151C allele, which can play a major role in appetite regulationand energy expenditure. This theory was, however, not supportedby the outcome of a Three Factor Eating Questionnaire, as wellas the lack of significant impact of the variant in a general adultDanish cohort [33]. However, this latter result may be due to thelow frequency of detected alleles, and requires confirmation in alarger study population.
9. Conclusion
Functional and gene knock out experiments have identified theinvolvement of the ghrelin signaling system in the regulation of avariety of phenotypic traits. As a result of these studies, the asso-ciation of these traits with GHRL, GHSR and more recently GOATpolymorphisms have been the subject of numerous candidate-geneassociation studies. We summarized the presented data in Table 1.
The interpretation of results from these studies has not beenstraight forward due to inconsistencies between studies - a problem
which commonly exists in the application of linkage analysis tocomplex disorders without obvious Mendelian inheritance.Despite these difficulties, we can conclude that it is likely forGHRL polymorphisms to influence hypertension risk, and may also

es 32 (
iorpttswlisofiato
pcdTs
tcsgairobsc
R
B. Liu et al. / Peptid
nfluence how body weight responds to environmental pressuresf weight gain or weight loss. From a large GWAS as well as caseeports of rare GHSR mutations, there is good evidence that GHSRolymorphisms are significant determinants of stature, however,he magnitude of this potential effect is small, as demonstrated byhe lack of consistently significant associations in candidate genetudies. GHSR polymorphisms have also been reported to associateith LVH and CAD/MI independently of conventional cardiovascu-
ar risk factors; elucidating the mechanism of these effects will aidn the confirmation of these observed associations. A single studyuggests GHSR polymorphisms may contribute toward the devel-pment of BN; however, this finding needs to be replicated beforerm conclusions can be drawn. Similarly a weak link between GHRLnd GHSR polymorphisms and reward seeking behavior (as illus-rated by alcohol dependency) may also be present, and is in needf further reinforcement.
The majority of current candidate-gene studies do not sup-ort the hypothesis of GHRL polymorphisms being significantontributors of obesity risk, T2D risk, eating disorders, or heightetermination; the same applies for GHSR and risk of obesity and2D. These conclusions are also supported by the genome-widetudies currently available.
It is still possible for GHRL and GHSR polymorphisms to con-ribute toward these phenotypic traits in magnitudes too small forurrent studies to detect. The recent GWAS on height with 180,000ubjects was able to detect many times the number of significantenes compared to smaller studies and indeed identified GHSRs one of the top candidates. This illustrates the importance ofncreasing the power of future gene association studies throughecruitment of larger study cohorts. The UK Biobank study is justne example of such a study; the recruitment process for this GWASegan in 2007 and currently over 500,000 people are involved. Thistudy can be perceived as an evolutionary step in the way we studyomplex genetic traits.
eferences
[1] Ghrelin o-acyltransferase. Model ID 331082dd3cfd07bb45fcb88e6e4cbd.http://modbase.compbio.ucsf.edu; 2010.
[2] Aimaretti G, Corneli G, Baldelli R, Di Somma C, Gasco V, Durante C, et al.Diagnostic reliability of a single IGF-I measurement in 237 adults withtotal anterior hypopituitarism and severe GH deficiency. Clin Endocrinol2003;59:56–61.
[3] Ando T, Ichimaru Y, Konjiki F, Shoji M, Komaki G. Variations in the pre-proghrelin gene correlate with higher body mass index, fat mass, and bodydissatisfaction in young Japanese women. Am J Clin Nutr 2007;86:25–32.
[4] Ando T, Komaki G, Naruo T, Okabe K, Takii M, Kawai K, et al. Possible role ofpreproghrelin gene polymorphisms in susceptibility to bulimia nervosa. AmJ Med Genet Part B-Neuropsychiatr Genet 2006;B141:929–34.
[5] Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, et al. Antago-nism of ghrelin receptor reduces food intake and body weight gain in mice.Gut 2003;52:947–52.
[6] Baessler A, Fischer M, Mayer B, Koehler M, Wiedmann S, Stark K, et al. Epistaticinteraction between haplotypes of the ghrelin ligand and receptor genes influ-ence susceptibility to myocardial infarction and coronary artery disease. HumMol Genet 2007;16:887–99.
[7] Baessler A, Hasinoff MJ, Fischer M, Reinhard W, Sonnenberg GE, Olivier M,et al. Genetic linkage and association of the growth hormone secretagoguereceptor (Ghrelin receptor) gene in human obesity. Diabetes 2005;54:259–67.
[8] Baessler A, Kwitek AE, Fischer M, Koehler M, Reinhard W, Erdmann J, et al.Association of the ghrelin receptor gene region with left ventricular hyper-trophy in the general population – results of the MONICA/KORA AugsburgEchocardiographic Substudy. Hypertension 2006;47:920–7.
[9] Beevers AJ, Kukol A. Conformational flexibility of the peptide hormone ghrelinin solution and lipid membrane bound: a molecular dynamics study. J BiomolStruct Dyn 2006;23:357–63.
[10] Berthold HK, Giannakidou E, Krone W, Mantzoros CS, Gouni-Berthold I. TheLeu72Met polymorphism of the ghrelin gene is associated with a decreasedrisk for type 2 diabetes. Clin Chim Acta 2009;399:112–6.
[11] Berthold HK, Giannakidou E, Krone W, Tregouet DA, Gouni-Berthold I. Influ-ence of ghrelin gene polymorphisms on hypertension and atheroscleroticdisease. Hypertens Res 2010;33:155–60.
[12] Bing C, Ambye L, Fenger M, Jorgensen T, Borch-Johnsen K, Madsbad S, et al.Large-scale studies of the Leu72Met polymorphism of the ghrelin gene in
2011) 2191–2207 2205
relation to the metabolic syndrome and associated quantitative traits. DiabetMed 2005;22:1157–60.
[13] Blum ID, Patterson Z, Khazall R, Lamont EW, Sleeman MW, Horvath TL, et al.Reduced anticipatory locomotor responses to scheduled meals in ghrelinreceptor deficient mice. Neuroscience 2009;164:351–9.
[14] Bresciani E, Rapetti D, Dona F, Bulgarelli I, Tamiazzo L, Locatelli V, et al.Obestatin inhibits feeding but does not modulate GH and corticosteronesecretion in the rat. J Endocrinol Invest 2006;29:RC16–8.
[15] Broglio F, Arvat E, Benso A, Gottero C, Muccioli G, Papotti M, et al.Ghrelin, a natural GH secretagogue produced by the stomach, induces hyper-glycemia and reduces insulin secretion in humans. J Clin Endocrinol Metab2001;86:5083–6.
[16] Camina JP, Carreira MC, El Messari S, Llorens-Cortes C, Smith RG, CasanuevaFF. Desensitization and endocytosis mechanisms of ghrelin-activated growthhormone secretagogue receptor 1a. Endocrinology 2004;145:930–40.
[17] Cardon LR, Palmer LJ. Population stratification and spurious allelic association.Lancet 2003;361:598–604.
[18] Cellini E, Nacmias B, Brecelj-Anderluh D, Badia-Casanovas A, Bellodi L, BoniC, et al. Case–control and combined family trios analysis of three polymor-phisms in the ghrelin gene in European patients with anorexia and bulimianervosa. Psychiatr Genet 2006;16:51–2.
[19] Chiodini BD, Lewis CM. Meta-analysis of 4 coronary heart disease genome-wide linkage studies confirms a susceptibility locus on chromosome 3q.Arterioscler Thromb Vasc 2003;23:1863–8.
[20] Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, et al. A large-scale genome-wideassociation study of Asian populations uncovers genetic factors influencingeight quantitative traits. Nat Genet 2009;41:527–34.
[21] Choi HJ, Cho YM, Moon MK, Choi HH, Shin HD, Jang HC, et al. Polymorphisms inthe ghrelin gene are associated with serum high-density lipoprotein choles-terol level and not with type 2 diabetes mellitus in Koreans. J Clin EndocrinolMetab 2006;91:4657–63.
[22] Cummings DE, Frayo RS, Marmonier C, Aubert R, Chapelot D. Plasma ghrelinlevels and hunger scores in humans initiating meals voluntarily with-out time- and food-related cues. Am J Physiol-Endocr M 2004;287:E297–304.
[23] Dardennes RM, Zizzari P, Tolle V, Foulon C, Kipman A, Romo L, et al. Fam-ily trios analysis of common polymorphisms in the obestatin/ghrelin, BDNFand AGRP genes in patients with Anorexia nervosa: Association with sub-type, body-mass index, severity and age of onset. Psychoneuroendocrinology2007;32:106–13.
[24] Date Y, Nakazato M, Hashiguchi S, Dezaki K, Mondal MS, Hosoda H, et al.Ghrelin is present in pancreatic alpha-cells of humans and rats and stimulatesinsulin secretion. Diabetes 2002;51:124–9.
[25] del Giudice EM, Santoro N, Cirillo G, Raimondo P, Grandone A, D’Aniello A,et al. Molecular screening of the ghrelin gene in Italian obese children: theLeu72Met variant is associated with an earlier onset of obesity. Int J Obes2004;28:447–50.
[26] den Hoed M, Smeets A, Veldhorst MAB, Nieuwenhuizen AG, Bouwman FG,Heidema AG, et al. SNP analyses of postprandial responses in (an) orexigenichormones and feelings of hunger reveal long-term physiological adaptationsto facilitate homeostasis. Int J Obes 2008;32:1790–8.
[27] Dossus L, McKay JD, Canzian F, Wilkening S, Rinaldi S, Biessy C, et al.Polymorphisms of genes coding for ghrelin and its receptor in relationto anthropometry, circulating levels of IGF-I and IGFBP-3, and breast can-cer risk: a case–control study nested within the European ProspectiveInvestigation into Cancer and Nutrition (EPIC). Carcinogenesis 2008;29:1360–6.
[28] Feighner SD, Howard AD, Prendergast K, Palyha OC, Hreniuk DL, Nargund R,et al. Structural requirements for the activation of the human growth hor-mone secretagogue receptor by peptide and nonpeptide secretagogues. MolEndocrinol 1998;12:137–45.
[29] Ferens DM, Yin L, Bron R, Hunne B, Ohashi-Doi K, Kitchener PD, et al.Functional and in situ hybridization evidence that preganglionic sym-pathetic vasoconstrictor neurons express ghrelin receptors. Neuroscience2010;166:671–9.
[30] Foster-Schubert KE, McTiernan A, Frayo RS, Schwartz RS, Rajan KB, YasuiY, et al. Human plasma ghrelin levels increase during a one-year exerciseprogram. J Clin Endocrinol Metab 2005;90:820–5.
[31] Garcia EA, Heude B, Petry CJ, Gueorguiev M, Hassan-Smith ZK, Spanou A, et al.Ghrelin receptor gene polymorphisms and body size in children and adults. JClin Endocrinol Metab 2008;93:4158–61.
[32] Garcia EA, King P, Sidhu K, Ohgusu H, Walley A, Lecoeur C, et al. The role ofghrelin and ghrelin-receptor gene variants and promoter activity in type 2diabetes. Eur J Endocrinol 2009;161:307–15.
[33] Gjesing AP, Larsen LH, Torekov SS, Hainerova IA, Kapur R, Johansen A, et al.Family and population-based studies of variation within the ghrelin receptorlocus in relation to measures of obesity. PLoS One 2010;5:8.
[34] Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, et al. Thetissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R,in humans. J Clin Endocrinol Metab 2002;87:2988–91.
[35] Gomez R, Lago F, Gomez-Reino JJ, Dieguez C, Gualillo O. Expression and modu-
lation of ghrelin O-acyltransferase in cultured chondrocytes. Arthritis Rheum2009;60:1704–9.[36] Gourcerol G, Coskun T, Craft LS, Mayer JP, Heiman ML, Wang L, et al.Preproghrelin-derived peptide, obestatin, fails to influence food intake in leanor obese rodents. Obesity 2007;15:2643–52.

2 es 32
206 B. Liu et al. / Peptid[37] Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJS, et al.Distribution of mRNA encoding the growth hormone secretagogue receptorin brain and peripheral tissues. Mol Brain Res 1997;48:23–9.
[38] Gueorguiev M, Lecoeur C, Benzinou M, Mein CA, Meyre D, Vatin V, et al.A genetic study of the ghrelin and growth hormone secretagogue receptor(GHSR) genes and stature. Ann Hum Genet 2009;73:1–9.
[39] Gueorguiev M, Lecoeur C, Meyre D, Benzinou M, Mein CA, Hinney A, et al.Association studies on ghrelin and ghrelin receptor gene polymorphisms withobesity. Obesity 2009;17:745–54.
[40] Gueorguiev M, Wiltshire S, Garcia EA, Mein C, Lecoeur C, Kristen B, et al. Exam-ining the candidacy of ghrelin as a gene responsible for variation in adultstature in a United Kingdom population with type 2 diabetes. J Clin EndocrinolMetab 2007;92:2201–4.
[41] Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, et al.Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl AcadSci USA 2008;105:6320–5.
[42] Higgins SC, Gueorguiev M, Korbonits M. Ghrelin, the peripheral hunger hor-mone. Ann Med 2007;39:116–36.
[43] Hinney A, Hoch A, Geller F, Schafer H, Siegfried W, Goldschmidt H, et al.Ghrelin gene: Identification of missense variants and a frameshift mutationin extremely obese children and adolescents and healthy normal weight stu-dents. J Clin Endocrinol Metab 2002;87:2716–9.
[44] Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High consti-tutive signaling of the ghrelin receptor – identification of a potent inverseagonist. Mol Endocrinol 2003;17:2201–10.
[45] Holst B, Egerod KL, Schild E, Vickers SP, Cheetham S, Gerlach LO, et al. GPR39signaling is stimulated by zinc ions but not by obestatin. Endocrinology2007;148:13–20.
[46] Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, et al.A receptor in pituitary and hypothalamus that functions in growth hormonerelease. Science 1996;273:974–7.
[47] Hubacek JA, Adamkova V, Bohuslavova R, Suchanek P, Poledne R, Lanska V. Nosignificant association between A-501C single nucleotide polymorphism inpreproghrelin and body mass index or waist-to-hip ratio in central Europeanpopulation. Metabolism 2008;57:1016–7.
[48] Janssen J, Lamberts SWJ. The role of IGF-I in the development of cardiovasculardisease in type 2 diabetes mellitus: is prevention possible? Eur J Endocrinol2002;146:467–77.
[49] Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelinadministration into tegmental areas stimulates locomotor activity andincreases extracellular concentration of dopamine in the nucleus accumbens.Addict Biol 2007;12:6–16.
[50] Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, et al.Requirement of central ghrelin signaling for alcohol reward. Proc Natl AcadSci USA 2009;106:11318–23.
[51] Jo DS, Kim SL, Kim SY, Hwang PH, Lee KH, Lee DY. Preproghrelin Leu72Metpolymorphism in obese Korean children. J Pediatr Endocrinol Metab2005;18:1083–6.
[52] Jorgensen JOL, Vahl N, Hansen TB, Fisker S, Hagen C, Christiansen JS. Influenceof growth hormone and androgens on body composition in adults. Horm Res1996;45:94–8.
[53] Kilpelainen TO, Lakka TA, Laaksonen DE, Mager U, Salopuro T, Kubaszek A,et al. Interaction of single nucleotide polymorphisms in ADRB2, ADRB3, TNF,IL6, IGF1R, LIPC, LEPR, and GHRL with physical activity on the risk of type 2diabetes mellitus and changes in characteristics of the metabolic syndrome:The Finnish Diabetes Prevention Study. Metabolism 2008;57:428–36.
[54] Kim MS, Yoon CY, Park KH, Shin CS, Park KS, Kim SY, et al. Changes in ghrelinand ghrelin receptor expression according to feeding status. Neuroreport2003;14:1317–20.
[55] Kim SY, Jo DS, Hwang PH, Park JH, Park SK, Yi HK, et al. Preproghrelin Leu72Metpolymorphism is not associated with type 2 diabetes mellitus. Metabolism2006;55:366–70.
[56] Kindler J, Bailer U, De Zwaan M, Fuchs K, Leisch F, Strnad A, et al. Ghrelin genepolymorphisms Arg51Gln and Leu72Met in eating disorders: an associationstudy. Am J Med Genet 2006;141B:771.
[57] Kirchner H, Gutierrez JA, Solenberg PJ, Pfluger PT, Czyzyk TA, Willency JA, et al.GOAT links dietary lipids with the endocrine control of energy balance. NatMed 2009;15:741–5.
[58] Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG,et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypesof the metabolic syndrome. Proc Natl Acad Sci USA 2000;97:14478–83.
[59] Klump KL, Miller KB, Keel PK, McGue M, Iacono WG. Genetic and environ-mental influences on anorexia nervosa syndromes in a population-based twinsample. Psychol Med 2001;31:737–40.
[60] Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelinis a growth-hormone-releasing acylated peptide from stomach. Nature1999;402:656–60.
[61] Kojima M, Kangawa K. Ghrelin: Structure and function. Physiol Rev2005;85:495–522.
[62] Kojima S, Nakahara T, Nagai N, Muranaga T, Tanaka M, Yasuhara D, et al.Altered ghrelin and peptide YY responses to meals in bulimia nervosa. Clin
Endocrinol (Oxf) 2005;62:74–8.[63] Korbonits M, Bustin SA, Kojima M, Jordan S, Adams EF, Lowe DG, et al. Theexpression of the growth hormone secretagogue receptor ligand ghrelin innormal and abnormal human pituitary and other neuroendocrine tumors. JClin Endocrinol Metab 2001;86:881–7.
(2011) 2191–2207
[64] Korbonits M, Gueorguiev M, O’Grady E, Lecoeur C, Swan DC, Mein CA, et al. Avariation in the ghrelin gene increases weight and decreases insulin secretionin tall, obese children. J Clin Endocrinol Metab 2002;87:4005–8.
[65] Kuzuya M, Ando F, Iguchi A, Shimokata H. Preproghrelin Leu72Met variantcontributes to overweight in middle-aged men of a Japanese large cohort. IntJ Obes 2006;30:1609–14.
[66] Lagaud GJ, Young A, Acena A, Morton MF, Barrett TD, Shankley NP. Obestatinreduces food intake and suppresses body weight gain in rodents. BiochemBiophys Res Commun 2007;357:264–9 [Retracted article. See vol. 388, pg.619, 2009].
[67] Lall S, Balthasar N, Carmignac D, Magoulas C, Sesay A, Houston P, et al. Phys-iological studies of transgenic mice overexpressing growth hormone (GH)secretagogue receptor 1A in GH-releasing hormone neurons. Endocrinology2004;145:1602–11.
[68] Landgren S, Jerlhag E, Hallman J, Oreland L, Lissner L, Strandhagen E, et al.Genetic variation of the ghrelin signaling system in females with severe alco-hol dependence. Alcohol Clin Exp Res 2010;34:1519–24.
[69] Landgren S, Jerlhag E, Zetterberg H, Gonzalez-Quintela A, Campos J, Olofs-son U, et al. Association of pro-ghrelin and GHS-R1A gene polymorphismsand haplotypes with heavy alcohol use and body mass. Alcohol Clin Exp Res2008;32:2054–61.
[70] Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al.Hundreds of variants clustered in genomic loci and biological pathways affecthuman height. Nature 2010;467:832–8.
[71] Larsen LH, Gjesing AP, Sorensen TIA, Hamid YH, Echwald SM, Toubro S, et al.Mutation analysis of the preproghrelin gene: no association with obesity andtype 2 diabetes. Clin Biochem 2005;38:420–4.
[72] Larsson A, Edstrom L, Svensson L, Soderpalm B, Engel JA. Voluntary ethanolintake increases extracellular acetylcholine levels in the ventral tegmentalarea in the rat. Alcohol Alcohol 2005;40:349–58.
[73] Leidy HJ, Gardner JK, Frye BR, Snook ML, Schuchert MK, Richard EL, et al.Circulating ghrelin is sensitive to changes in body weight during a diet andexercise program in normal-weight young women. J Clin Endocrinol Metab2004;89:2659–64.
[74] Leite-Moreira AF, Soares JB. Physiological, pathological and potential thera-peutic roles of ghrelin. Drug Discov Today 2007;12:276–88.
[75] Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, et al. Identi-fication of ten loci associated with height highlights new biological pathwaysin human growth. Nat Genet 2008;40:584–91.
[76] Leung PK, Chow KBS, Lau PN, Chu KM, Chan CB, Cheng CHK, et al. The truncatedghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutantof the ghrelin receptor. Cell Signal 2007;19:1011–22.
[77] Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet2009;41:677–87.
[78] Lillioja S, Wilton A. Agreement among type 2 diabetes linkage studies buta poor correlation with results from genome-wide association studies. Dia-betologia 2009;52:1061–74.
[79] Luke A, Wu XD, Zhu XF, Kan DH, Su Y, Cooper R. Linkage for BMI at 3q27 regionconfirmed in an African-American population. Diabetes 2003;52:1284–7.
[80] Mackelvie KJ, Meneilly GS, Elahi D, Wong ACK, Barr SI, Chanoine JP. Regulationof appetite in lean and obese adolescents after exercise: role of acylated anddesacyl ghrelin. J Clin Endocrinol Metab 2007;92:648–54.
[81] Mager U, Degenhardt T, Pulkkinen L, Kolehmainen M, Tolppanen AM, Lind-strom J, et al. Variations in the ghrelin receptor gene associate with obesityand glucose metabolism in individuals with impaired glucose tolerance. PLoSOne 2008:3.
[82] Mager U, Kolehmainen M, Lindstrom J, Eriksson JG, Valle TT, HamalainenH, et al. Association between ghrelin gene variations and blood pressure insubjects with impaired glucose tolerance. Am J Hypertens 2006;19:920–6.
[83] Mano-Otagiri A, Nemoto T, Sekino A, Yamauchi N, Shuto Y, Sugihara H,et al. Growth hormone-releasing hormone (GHRH) neurons in the arcuatenucleus (Arc) of the hypothalamus are decreased in transgenic rats whoseexpression of ghrelin receptor is attenuated: evidence that ghrelin receptoris involved in the up-regulation of GHRH expression in the Arc. Endocrinology2006;147:4093–103.
[84] Martin GR, Loredo JC, Sun G. Lack of association of ghrelin precursor gene vari-ants and percentage body fat or serum lipid profiles. Obesity 2008;16:908–12.
[85] McKee KK, Palyha OC, Feighner SD, Hreniuk DL, Tan CP, Phillips MS, et al.Molecular analysis of rat pituitary and hypothalamic growth hormone secre-tagogue receptors. Mol Endocrinol 1997;11:415–23.
[86] Miyasaka K, Hosoya H, Sekime A, Ohta M, Amono H, Matsushita S, et al. Asso-ciation of ghrelin receptor gene polymorphism with bulimia nervosa in aJapanese population. J Neural Transm 2006;113:1279–85.
[87] Monteleone P, Martiadis V, Fabrazzo M, Serritella C, Maj M. Ghrelin and leptinresponses to food ingestion in bulimia nervosa: implications for binge-eatingand compensatory behaviours. Psychol Med 2003;33:1387–94.
[88] Monteleone P, Tortorella A, Castaldo E, Di Filippo C, Maj M. No associationof the Arg51G1n and Leu72Met polymorphisms of the ghrelin gene withanorexia nervosa or bulimia nervosa. Neurosci Lett 2006;398:325–7.
[89] Muller TD, Tschop MH, Jarick I, Ehrlich S, Scherag S, Herpertz-Dahlmann B,
et al. Genetic variation of the ghrelin activator gene ghrelin O-acyltransferase(GOAT) is associated with anorexia nervosa. J Psychiatr Res 2010.[90] Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al.Genome-wide association study identifies eight loci associated with bloodpressure. Nat Genet 2009;41:666–76.

es 32 (
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
B. Liu et al. / Peptid
[91] Nogueiras R, Pfluger P, Tovar S, Arnold M, Mitchell S, Morris A, et al. Effectsof obestatin on energy balance and growth hormone secretion in rodents.Endocrinology 2007;148:21–6.
[92] Ohlsson C, Bengtsson BA, Isaksson OGP, Andreassen TT, Slootweg MC. Growthhormone and bone. Endocr Rev 1998;19:55–79.
[93] Olszewski PK, Schioth HB, Levine AS. Ghrelin in the CNS: from hunger to arewarding and memorable meal? Brain Res Rev 2008;58:160–70.
[94] Oner-Iyidogan Y, Kocak H, Gurdol F, Oner P, Issever H, Esin D. Circulatingghrelin levels in obese women: a possible association with hypertension.Scand J Clin Lab Invest 2007;67:568–76.
[95] Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing,and other complex traits. Mol Genet Metab 2003;80:1–10.
[96] Pantel J, Legendre M, Nivot S, Morisset S, Vie-Luton MP, le Bouc Y, et al.Recessive isolated growth hormone deficiency and mutations in the ghrelinreceptor. J Clin Endocrinol Metab 2009;94:4334–41.
[97] Pedretti A, Villa M, Pallavicini M, Valoti E, Vistoli G. Construction of humanghrelin receptor (hGHS-R1a) model using a fragmental prediction approachand validation through docking analysis. J Med Chem 2006;49:3077–85.
[98] Poykko S, Ukkola O, Kauma H, Savolainen MJ, Kesaniemi YA. Ghrelin Arg51Glnmutation is a risk factor for Type 2 diabetes and hypertension in a randomsample of middle-aged subjects. Diabetologia 2003;46:455–8.
[99] Poykko SM, Kellokoski E, Horkko S, Kauma H, Kesaniemi YA, Ukkola O. Lowplasma ghrelin is associated with insulin resistance, hypertension, and theprevalence of type 2 diabetes. Diabetes 2003;52:2546–53.
100] Pugliese-Pires PN, Fortin JP, Zhu YT, Mendonca BB, Arnhold IJ, Kopin AS, et al.Three novel mutations in the growth hormone secretagogue receptor (GHSR)gene associated with constitutional delay in growth and puberty (CDGP) orisolated growth hormone deficiency (IGHD). Horm Res 2009;72:245.
101] Purnell JQ, Weigle DS, Breen P, Cummings DE. Ghrelin levels correlate withinsulin levels, insulin resistance, and high-density lipoprotein cholesterol,but not with gender, menopausal status, or cortisol levels in humans. J ClinEndocrinol Metab 2003;88:5747–52.
102] Rice T, Rankinen T, Chagnon YC, Province MA, Perusse L, Leon AS, et al.Genomewide linkage scan of resting blood pressure – HERITAGE family study.Hypertension 2002;39:1037–43.
103] Riediger T, Bothe C, Becskei C, Lutz TA, Peptide YY. directly inhibits ghrelin-activated neurons of the arcuate nucleus and reverses fasting-induced c-Fosexpression. Neuroendocrinology 2004;79:317–26.
104] Risch N, Teng J. The relative power of family-based and case–control designsfor linkage disequilibrium studies of complex human diseases – I. DNA pool-ing. Genome Res 1998;8:1273–88.
105] Saito K, Tobe T, Minoshima S, Asakawa S, Sumiya J, Yoda M, et al. Organizationof the gene for gelatin-binding protein (GBP28). Gene 1999;229:67–73.
106] Salome N, Haage D, Perrissoud D, Moulin A, Demange L, Egecioglu E, et al.Anorexigenic and electrophysiological actions of novel ghrelin receptor (GHS-R1A) antagonists in rats. Eur J Pharmacol 2009;612:167–73.
107] Sangiao-Alvarellos S, Cordido F. Effect of ghrelin on glucose-insulin homeo-stasis: therapeutic implications. Int J Pept 2010:2010.
108] Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen WM, Bonnycastle LL, et al.Common variants in the GDF5–UQCC region are associated with variation inhuman height. Nat Genet 2008;40:198–203.
109] Saunders CL, Chiodini BD, Sham P, Lewis CM, Abkevich V, Adeyemo AA, et al.Meta-analysis of genome-wide linkage studies in BMI and obesity. Obesity2007;15:2263–75.
110] Seim I, Collet C, Herington AC, Chopin LK. Revised genomic structure of thehuman ghrelin gene and identification of novel exons, alternative splice vari-ants and natural antisense transcripts. BMC Genomics 2007;8:16.
111] Seoane LM, Al-Massadi O, Pazos Y, Pagotto U, Casanueva FF. Central obestatinadministration does not modify either spontaneous or ghrelin-induced foodintake in rats. J Endocrinol Invest 2006;29:RC13–5.
112] Sibilia V, Bresciani E, Lattuada N, Rapetti D, Locatelli V, De Luca V, et al.Intracerebroventricular acute and chronic administration of oblestatin min-imally affect food intake but not weight gain in the rat. J Endocrinol Invest2006;29:RC31–4.
113] Silverman EK, Palmer LJ. Case–control association studies for the genetics ofcomplex respiratory diseases. Am J Respir Cell Mol Biol 2000;22:645–8.
114] Steinle NI, Pollin TI, O’Connell JR, Mitchell BD, Shuldiner AR. Variants in theghrelin gene are associated with metabolic syndrome in the old order amish.J Clin Endocrinol Metab 2005;90:6672–7.
115] Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the
diabetic but not obese phenotype of ob/ob mice. Cell Metab 2006;3:379–86.116] Takezawa J, Yamada K, Morita A, Aiba N, Watanabe S. Preproghrelin genepolymorphisms in obese Japanese: Association with diabetes mellitus in menand with metabolic syndrome parameters in women. Obes Res Clin Pract2009;3:179–91.
2011) 2191–2207 2207
[117] Tholin S, Rasmussen F, Tynelius P, Karlsson J. Genetic and environmental influ-ences on eating behavior: the Swedish young male twins study. Am J Clin Nutr2005;81:564–9.
[118] Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Hel-gadottir A, et al. Genome-wide association yields new sequence variants atseven loci that associate with measures of obesity. Nat Genet 2009;41:18–24.
[119] Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-ParikkaP, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle amongsubjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–50.
[120] Ukkola O, Ravussin E, Jacobson P, Perusse L, Rankinen T, Tschop M, et al. Roleof ghrelin polymorphisms in obesity based on three different studies. ObesRes 2002;10:782–91.
[121] Ukkola O, Ravussin E, Jacobson P, Snyder EE, Chagnon M, Sjostrom L, et al.Mutations in the preproghrelin/ghrelin gene associated with obesity inhumans. J Clin Endocrinol Metab 2001;86:3996–9.
[122] Usdan LS, Khaodhiar L, Apovian CM. The endocrinopathies of anorexia ner-vosa. Endocr Pract 2008;14:1055–63.
[123] Vartiainen J, Kesaniemi A, Ukkola O. No significant association between A-501C single nucleotide polymorphism in preproghrelin and body mass indexor waist-to-hip ratio in central European population – reply. Metabolism2008;57:1017–8.
[124] Vartiainen J, Kesaniemi YA, Ukkola O. Sequencing analysis of ghrelin gene5′ flanking region: relations between the sequence variants, fasting plasmatotal ghrelin concentrations, and body mass index. Metabolism 2006;55:1420–5.
[125] Vartiainen J, Poykko SM, Raisanen T, Kesaniemi YA, Ukkola O. Sequencinganalysis of the ghrelin receptor (growth hormone secretagogue receptor type1a) gene. Eur J Endocrinol 2004;150:457–63.
[126] Vionnet N, Hani E, Dupont S, Gallina S, Francke S, Dotte S, et al. Genomewidesearch for type 2 diabetes-susceptibility genes in French whites: Evidence fora novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome1q21–q24. Am J Hum Genet 2000;67:1470–80.
[127] Vivenza D, Rapa A, Castellino N, Bellone S, Petri A, Vacca G, et al. Ghrelin genepolymorphisms and ghrelin, insulin, IGF-I, leptin and anthropometric data inchildren and adolescents. Eur J Endocrinol 2004;151:127–33.
[128] Wang HJ, Geller F, Dempfle A, Schauble N, Friedel S, Lichtner P, et al. Ghrelinreceptor gene: identification of several sequence variants in extremelyobese children and adolescents, healthy normal-weight and underweightstudents, and children with short normal stature. J Clin Endocrinol Metab2004;89:157–62.
[129] Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M,et al. Genome-wide association analysis identifies 20 loci that influence adultheight. Nat Genet 2008;40:575–83.
[130] Willer CJ, Speliotes EK, Loos RJF, Li SX, Lindgren CM, Heid IM, et al. Six newloci associated with body mass index highlight a neuronal influence on bodyweight regulation. Nat Genet 2009;41:25–34.
[131] Wiltshire S, Frayling TM, Hattersley AT, Hitman GA, Walker M, Levy JC, et al.Evidence for linkage of stature to chromosome 3p26 in a large UK family dataset ascertained for type 2 diabetes. Am J Hum Genet 2002;70:543–6.
[132] Wortley KE, del Rincon JP, Murray JD, Garcia K, Lida K, Thorner MO,et al. Absence of ghrelin protects against early-onset obesity. J Clin Invest2005;115:3573–8.
[133] Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelinenhances appetite and increases food intake in humans. J Clin EndocrinolMetab 2001;86:5992–5.
[134] Wu XD, Cooper RS, Borecki I, Hanis C, Bray M, Lewis CE, et al. A combinedanalysis of genomewide linkage scans for body mass index, from the NationalHeart, Lung, and Blood Institute Family Blood Pressure Program. Am J HumGenet 2002;70:1247–56.
[135] Zavarella S, Petrone A, Zampetti S, Gueorguiev M, Spoletini M, Mein CA, et al.A new variation in the promoter region, the -604C>T, and the Leu72Metpolymorphism of the ghrelin gene are associated with protection to insulinresistance. Int J Obes 2008;32:663–8.
[136] Zhang JV, Klein C, Ren PG, Kass S, Donck LV, Moechars D, et al. Responseto comment on “Obestatin, a peptide encoded by the ghrelin gene, opposesghrelin’s effects on food intake”. Science 2007;315:1.
[137] Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, et al.Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effectson food intake. Science 2005;310:996–9.
[138] Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin recep-tor mRNA in the rat and the mouse brain. J Comp Neurol 2006;494:528–48.
[139] Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, et al. Micelacking ghrelin receptors resist the development of diet-induced obesity. JClin Invest 2005;115:3564–72.