generalized eruptive histiocytosis: a possible therapeutic cure
TRANSCRIPT
CORRESPONDENCE
Influence of environmental temperature on theoccurrence of non-necrotizing cellulitis of the leg
SIR, Non-necrotizing cellulitis of the leg is a commoncutaneous bacterial infection whose risk factors includevenous insufficiency, lymphoedema and toe-web intertrigo.The role of environmental temperature remains controver-sial.1 To study the relationship between environmentaltemperature and the frequency of non-necrotizing cellulitisof the leg, we reviewed all patients hospitalized with non-necrotizing cellulitis of the leg in a university hospital in atemperate region of France during a 4-year period, andcorrelated findings with the local environmental temperatureduring the same period.
Patients included in this study were consecutive patientsreferred for non-necrotizing cellulitis of the leg from January1995 to December 1998 in the Departments of Dermatology,Internal Medicine and Infectious Diseases of the RouenUniversity Hospital. The diagnosis of cellulitis of the leg wasretrospectively identified using the database of the MedicalInformation System. The mean and maximum temperaturesof each the 8 days prior to the date of hospital admission wereobtained from the local meteorological unit for each case.Correlations between the mean and maximum daily tem-perature of each day from day )8 to the admission day (lag0–8 days) and the daily number of patients hospitalized forcellulitis of the leg were studied using a nonparametricPoisson regression model to adjust for time trends and days ofthe week (generalized additive model).2
Eight hundred and ninety-eight patients with cellulitis ofthe leg [342 men (38%) and 556 women (62%)] were
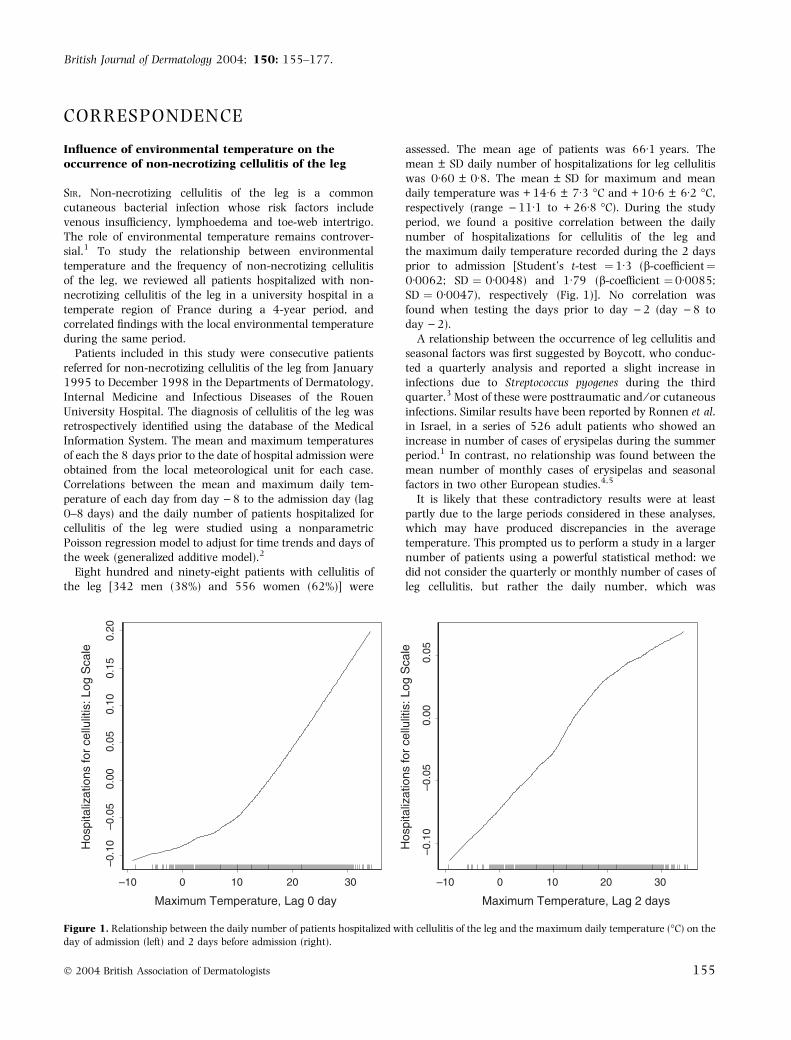
assessed. The mean age of patients was 66Æ1 years. Themean ± SD daily number of hospitalizations for leg cellulitiswas 0Æ60 ± 0Æ8. The mean ± SD for maximum and meandaily temperature was + 14Æ6 ± 7Æ3 �C and + 10Æ6 ± 6Æ2 �C,respectively (range )11Æ1 to + 26Æ8 �C). During the studyperiod, we found a positive correlation between the dailynumber of hospitalizations for cellulitis of the leg andthe maximum daily temperature recorded during the 2 daysprior to admission [Student’s t-test ¼1Æ3 (b-coefficient¼0Æ0062; SD ¼ 0Æ0048) and 1Æ79 (b-coefficient ¼0Æ0085;SD ¼ 0Æ0047), respectively (Fig. 1)]. No correlation wasfound when testing the days prior to day )2 (day )8 today )2).
A relationship between the occurrence of leg cellulitis andseasonal factors was first suggested by Boycott, who conduc-ted a quarterly analysis and reported a slight increase ininfections due to Streptococcus pyogenes during the thirdquarter.3 Most of these were posttraumatic and ⁄ or cutaneousinfections. Similar results have been reported by Ronnen et al.in Israel, in a series of 526 adult patients who showed anincrease in number of cases of erysipelas during the summerperiod.1 In contrast, no relationship was found between themean number of monthly cases of erysipelas and seasonalfactors in two other European studies.4,5
It is likely that these contradictory results were at leastpartly due to the large periods considered in these analyses,which may have produced discrepancies in the averagetemperature. This prompted us to perform a study in a largernumber of patients using a powerful statistical method: wedid not consider the quarterly or monthly number of cases ofleg cellulitis, but rather the daily number, which was
–10 0 10 20 30
–0.1
0–0
.05
0.00
0.05
0.10
0.15
0.20
Maximum Temperature, Lag 0 day Maximum Temperature, Lag 2 days
–10 100 20 30
–0.1
0–0
.05
0.00
0.05
Hos
pita
lizat
ions
for
cellu
litis
: Log
Sca
le
Hos
pita
lizat
ions
for
cellu
litis
: Log
Sca
le
Figure 1. Relationship between the daily number of patients hospitalized with cellulitis of the leg and the maximum daily temperature (�C) on the
day of admission (left) and 2 days before admission (right).
British Journal of Dermatology 2004; 150: 155–177.
� 2004 British Association of Dermatologists 155
correlated with the environmental temperature recorded foreach of the 8 days before hospital admission, using anonparametric Poisson regression model. Such a methodhas previously been used to demonstrate a correlationbetween air pollution and the number of hospital stays forrespiratory illness in the U.S.A.2 The present study showed anassociation between the number of hospital stays for legcellulitis and the maximum temperature during the 2 daysprior to hospitalization. Considering that the main risk factorsfor cellulitis—venous insufficiency and lymphoedema—areclassically aggravated by hot weather, the present studysuggests that hot weather might trigger or favour theoccurrence of non-necrotizing cellulitis of the leg.
A . M a c a r i o - B a r r e l
A . Z e g h n o u n *
P . Y o u n g
L . F r o m e n t *
H . L e v e s q u e�F . C a r o n�
P . M u s e t t e
P . J o l y
Departments ofDermatology,*Epidemiology,�Internal Medicine and �InfectiousDiseases, Centre HospitalierUniversitaire Charles Nicolle, 76031Rouen cedex, FranceCorrespondence: Pascal Joly,E-mail: [email protected]
References
1 Ronnen M, Suster S, Schewach-Millet M, Modan M. Erysipelas.
Changing faces. Int J Dermatol 1985; 24: 169–72.
2 Schwartz J. Air pollution and hospital admissions for the elderly in
Birmingham, Alabama. Am J Epidemiol 1994; 139: 589–98.
3 Boycott JA. Seasonal variations in streptococcal infections. Lancet
1966; 26: 706–7.
4 Chartier C, Grosshans E. Erysipelas. Int J Dermatol 1990; 29: 459–
67.
5 Jorup-Ronstrom C. Epidemiological, bacteriological and complica-
ting features of erysipelas. Scand J Infect Dis 1986; 18: 519–24.
Malignant proliferating trichilemmal tumour and CAV(cisplatin, adriamycin, vindesine) treatment
SIR, Malignant proliferating trichilemmal tumour (MPTT) is avery rare neoplasm originating from the hair follicle, whichoften shows high malignancy.1 Although some reportsrecommended surgical removal as the principal method oftreatment,2–4 reliable strategies for treating this type ofmalignant tumour have not yet been established. We report apatient with a highly advanced MPTT of the neck, which wastreated with chemotherapy.
A 56-year-old woman had noticed a subcutaneoustumour at her right submandibular region. Over 3 years itgradually grew in size, to become 3 cm in diameter.Pathological examination at a previous hospital had led tothe diagnosis of benign calcifying epithelioma. After surgicalremoval the tumour soon recurred, and extended up to herright subauricular region and down to the neck within4 months. A second surgical operation for removal of thetumour was performed, under general anaesthesia, at thefirst hospital. Many tumours of various sizes (1–3 cm) werefound in the neck. These formed large aggregates which
adhered to the crucial vessels and could not be removedcompletely. After the second operation, she came to theOsaka City University Hospital for treatment of the residualtumour.
Examination revealed a palpable hard subcutaneous masswith surface irregularity, which extended from the rightsubauricular region down to the supraclavicular region(Fig. 1, left). A small area of the skin on the large neoplasmhad necrosed and a curd-like content was seen there. Ourpatient complained of tenderness in the tumour. There wasno stenosis in the airway. Biopsy revealed many cysticstructures formed by the tumour cells from the superficialdermis to the deep subcutaneous tissue (Fig. 2). The tumourwas composed of variably sized lobules made of squamous
Figure 1. (Left) A palpable hard subcutaneous mass with surface
irregularity extending from the right subauricular region down to the
supraclavicular region. (Right) After 4 months many exposed
tumours could be seen.
Figure 2. The cystic structures contained keratinized material and
the tumour cells of the inner wall showed histological changes,
becoming large, clear and eosinophilic. This showed trichilemmal
keratinization, which lacks the granular cell layer during the process
of keratinizing differentiation (haematoxylin and eosin).
1 5 6 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

epithelium. The epithelium in the centre of the lobulesabruptly changed into amorphous keratin. This was trich-ilemmal keratinization, which lacks the granular cell layerduring the process of keratinizing differentiation, suggestingthat the tumour tended to follicular differentiation. Shadowcells were not seen, so the diagnosis of pilomatrixoma wasruled out. Atypia was seen in each tumour cell. Metastasisto the lymph nodes in the neck was recognized and thelesion was diagnosed as MPTT.5 Magnetic resonanceimaging examination revealed that the tumour extendedfrom the first cervical vertebral bone to the right clavicularbone. There were many tumours of various sizes withheterogeneous enhancing. The tumour invaded to thecervical and masseter muscle, and the parotid gland.Angiography showed hypervascular changes in the tumourtissue.
Our patient’s condition was estimated to be pT4N1M1and clinical stage IV. There was no indication for surgicaltreatment, and we performed chemotherapy (cisplatin,adriamycin, vindesine: CAV treatment) according to theregimen for highly advanced cases of squamous cellcarcinoma (SCC).6 After one cycle of CAV treatment, growthof the neoplasm ceased. She refused further chemotherapy,and the tumour increased in size again after a month. Inaccordance with this increase in size, levels of SCC-relatedantigen also showed a gradual increase (progressively 2Æ7,4Æ1 and 17Æ7 ng mL)1). The tumour finally extended to thechest wall and the right cheek (Fig. 1, right). Our patientdied 4 months after admission, due to stenosis of therespiratory tract. Autopsy revealed that the neoplasm hadmetastasized to the hilum lymph nodes of both lungs.
MPTT was first reported by Headington, as showingpeculiar cystic structures, aggressive invasion into the deeplayer and a poor prognosis.1 MPTT tends to metastasize at anearly stage of the clinical course.7,8 Although surgicalexcision is considered to be the first choice of curativetreatment, there are some reports on the use of radiother-apy.4,7 Injection of ethanol was recently reported as atherapeutic alternative.9 Our case showed an apparentinvasion to the carotid artery, and chemotherapy (CAVtreatment) was performed. It did not cure this neoplasm butrestricted its growth for a while, providing a life-prolongingeffect.
I . H a y a s h i
T . H a r a d a
M . M u r a o k a
M . I s h i i
Department of Plastic andReconstructive Surgery, Osaka CityUniversity Graduate School ofMedicine, Asahi 1-4-3, Abeno, Osaka545-8585, JapanE-mail: [email protected]
References
1 Headington JT. Tumors of the hair follicle. Am J Pathol 1976; 85:
479–514.
2 Amaral ALMP, Nascimento AG, Goellner JR. Proliferating pilar
(trichilemmal) cyst. Arch Pathol Lab Med 1984; 108: 808–10.
3 Mehregan AH, Lee KC. Malignant proliferating trichilemmal
tumors: report of three cases. J Dermatol Surg Oncol 1987; 13:
1339–42.
4 Weiss J, Heine M, Grimmel M et al. Malignant proliferating trich-
ilemmal cyst. J Am Acad Dermatol 1995; 32: 870–3.
5 Saida T, Oohara K, Hori Y et al. Development of a malignant
proliferating trichilemmal cyst in a patient with multiple trichi-
lemmal cysts. Dermatologica 1983; 166: 203–8.
6 Ikegawa S, Saida T, Obayashi H et al. Cisplatin combination che-
motherapy in squamous cell carcinoma and adenoid cystic carci-
noma of the skin. J Dermatol 1989; 16: 227–30.
7 Noto G, Pravata G, Arico M. Malignant proliferating trichilemmal
tumor. Am J Dermatopathol 1997; 19: 202–4.
8 Park BS, Yang SG, Cho KH. Malignant proliferating trichilemmal
tumor showing distant metastases. Am J Dermatopathol 1997; 19:
536–9.
9 Takenaka H, Kishimoto S, Shibagaki R et al. Recurrent malignant
trichilemmal tumour: local management with ethanol injection. Br
J Dermatol 1998; 139: 726–9.
Efficacy of danazol treatment in a patient with the newvariant of hereditary angio-oedema (HAE III)
SIR, Hereditary angio-oedema (HAE) is a rare autosomaldominant disease which is caused by mutations in the C1inhibitor gene or promotor, resulting in a reduced amountor reduced activity of C1 inhibitor in patients with HAEtype I or type II.1,2 Recently, a novel form of HAE has beendescribed in which C1 inhibitor function and complementcomponents are normal (HAE III).3,4 The underlyingmolecular defect is not known to date. We describe a 31-year-old woman with HAE III that responded well todanazol treatment; she has now been free of symptoms formore than 2 years.
The first swellings of the face and extremities occurred inour patient at the age of 17 years. Additionally, she sufferedfrom frequent episodes of gastrointestinal cramp-like painthat were accompanied by vomiting. Episodes usually lastedfor 1–3 days and were nonresponsive to treatment withcorticosteroids or antihistamines. No urticaria, redness oritching was present. During attacks of abdominal pain,ascites was detected by ultrasound examination. The symp-toms led to an appendectomy at the age of 18 years. In thefamily, the mother and a female cousin from the maternalside have a history of recurrent swellings of the face andextremities that were nonresponsive to steroids or antihis-tamines.
The patient presented in our hospital for diagnosticpurposes in May 1999. At that time, the novel type of HAEIII had not yet been described in the literature. Laboratorytesting revealed normal values for the amount and functionof C1 inhibitor and complement components. In the followingweeks, tests were repeated several times in different laborat-ories during attacks and during symptom-free intervals andalways showed normal values. Porphyrins, cryoglobulins,antinuclear antibodies, rheumatoid factor, angiotensin-con-verting enzyme, histamine in plasma and other routinelaboratory parameters were within the normal range. Nosensitization to preservatives, colorants or latex was detected.
C O R R E S P O N D E N C E 1 5 7
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

In addition, the mother of our patient showed a normalamount and function of C1 inhibitor and complementcomponents in repeated tests.
In autumn 1999, our patient had a severe attack withlaryngeal oedema and emergency admission to a peripheralears, nose and throat department. As attacks were becomingmore frequent and severe, we initiated treatment withdanazol 600 mg daily. No further attacks occurred subse-quently, and the dosage of danazol was reduced at 2-weekintervals to 100 mg daily and then over the subsequentweeks to 50 mg twice weekly. At this dose our patient hasbeen free of symptoms for more than 2 years. Regularlaboratory testing was performed and involved measurementof aminotransferases, cholestatic parameters and serum lipidsevery 6 weeks, and ultrasound examinations twice a year.Side-effects included an initial weight gain of around 8 kg, amild papulopustulous form of acne and amenorrhoea afterdiscontinuation of oral contraception. Hormone levelsshowed a slight hyperandrogenaemia, and further monitor-ing is being performed.
First reports on patients with HAE III, who have clinicalsymptoms of HAE but normal amount and function of C1inhibitor and complement components, were published in Julyand September 2000.3,4 Essential features of HAE III are: (i) along history (up to decades) of recurrent skin swelling, attacksof abdominal pain, or episodes of upper airway obstruction; (ii)familial occurrence, exclusively in female members of thefamily; (iii) no history of urticaria in the patient or other familymembers; (iv) normal C1 inhibitor and C4 concentrations inthe plasma; and (v) failure of treatment with antihistamines,corticosteroids or C1 inhibitor concentrate. All these featuresare present in our patient and her family.
Binkley and Davis3 studied one family with affectedmembers over four generations. They observed episodes ofangio-oedema only during pregnancy or following the use ofexogenous oestrogens and therefore concluded that thisfamily had an oestrogen-dependent phenotype. In ourpatient, oral contraception was not discontinued until severalmonths after danazol treatment had been started. Addition-ally, the mother of our patient reported attacks of angio-oedema even though she was neither using exogenousoestrogens, nor pregnant.
Bork et al.4 reported 36 patients from 10 independentfamilies. They also observed attacks of angio-oedema inchildhood, starting at the age of 1 year. They hypothesizedthe influence of an X-linked gene on the generation ofvasoactive peptides. One of their patients received danazol100 mg daily for 9 years and was free of symptoms for thattime. In our patient, the attacks also stopped after theintroduction of danazol treatment. Interestingly, we werethen able to reduce the dose to 50 mg twice weekly, which islower than the dose usually used for treating patients withHAE I or HAE II.
Several treatment modalities for HAE exist: these may bedivided into short-term and long-term prophylactics, andemergency intervention. Danazol is a synthetic androgen thatis indicated for the long-term prophylactic treatment of HAE I
and II. It corrects the underlying biochemical deficiencies byincreasing concentrations of C1 inhibitor.5 The exact mech-anism of action is not known but the induction of C1 inhibitorsynthesis in the liver is discussed. As the amount andfunction of C1 inhibitor are normal in patients with HAE III,the reasons for the therapeutic efficacy of danazol in ourpatient are unclear.
To date, there are no larger studies on the long-term effectsof danazol treatment. However, certain kinds of potentiallylife-threatening conditions may make the long-term use ofdanazol necessary, and initial observations in Switzerland ofwomen treated with low doses of danazol for more than15 years seem to be encouraging.1,6
Therapeutic options in an emergency situation or short-term prophylactics before dental or surgical proceduresremain unclear, as a previous report1 and our observationssuggest that application of C1 inhibitor concentrate isineffective in patients with HAE III. Further studies will benecessary to elucidate the underlying molecular defect, whichmay then lead to more specific treatment modalities.
G . H e r r m a n n
L . S c h n e i d e r *
T . K r i e g
N . H u n z e l m a n n
K . S c h a r f f e t t e r -
K o c h a n e k *
Department of Dermatology,University of Cologne, Joseph-Stelzmann-Str. 9, 50931 Cologne,and *Department of Dermatology,University of Ulm, Maienweg 12,89081 Ulm, GermanyCorrespondence: Karin Scharffetter-Kochanek,E-mail: [email protected]
References
1 Rosen FS, Charache P, Pensky J, Donaldson VH. Hereditary angio-
neurotic edema: two genetic variants. Science 1965; 148: 957–8.
2 Spath PJ, Wuthrich B. Inherited and acquired deficiencies of C1
esterase inhibitor in humans. In: The Complement System (Rother K,
Till GO, Hansch GM, eds). Berlin: Springer-Verlag, 1998: 353–
410.
3 Binkley KE, Davis A III. Clinical, biochemical, and genetic char-
acterisation of a novel and estrogen-dependent inherited form of
angioedema. J Allergy Clin Immunol 2000; 106: 546–50.
4 Bork K, Barnstedt S-E, Koch P et al. Hereditary angioedema with
normal C1-inhibitor activity in women. Lancet 2000; 356: 213–7.
5 Gelfand JA, Sherins RJ, Alling DW et al. Treatment of hereditary
angioedema with danazol: reversal of clinical and biochemical
abnormalities. N Engl J Med 1976; 295: 1444–8.
6 Zimmermann HP, Wuthrich B, Spath PJ. Hereditares Angioodem.
Ein klinischer und immunologischer Beitrag anhand von 8 eigenen
Fallen unter Langzeitbehandlung mit Androgenen. Schweiz Med
Wochenschr 1983; 113: 876–84.
Lisinopril and Kaposi’s sarcoma
SIR, I have read with interest the letter of Bilen et al.1
regarding a case of Kaposi’s sarcoma (KS) in a patient treatedwith lisinopril. This is the third case reported in the literature
1 5 8 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

in which the association between the drugs captopril–lisinopril and KS is described.1–3 These authors relate theonset of the tumour to a possible immunosuppressive activityof these drugs.4,5 Rather, I think that we are dealing with anopportunistic infective agent, namely the HHV8, the onco-genic herpetic virus now recognized as responsible for KS.
To complete these data, I would like to note that among theadverse reactions reported on the leaflet of some productsavailable in the U.S.A. of this antihypertensive class, e.g.enalapril maleate and lisinopril, cases of herpes zoster are alsoincluded, as skin reactions.
The frequency of herpes zoster reported in the leaflet is verylow (< 1%). However, this event, together with that reportedabove, could lead us to think that, given the reportedimmunosuppressive activity of these drugs (via interleukin-12 suppression), opportunistic herpetic infections can occur,clinically present as KS in the case of the oncogenic HHV8, oras herpes zoster, due to VZV.6–8
A . D i C a r l oSTD Service, S Gallicano Institute,IRCCS, Rome, Italy
References
1 Bilen N, Bayramgurler D, Aydeniz B et al. Possible causal role of
lisinopril in a case of Kaposi’s sarcoma (Letter). Br J Dermatol 2002;
147: 1042.
2 Puppin D Jr, Rybojad M, de la Chapelle C, Morel P. Kaposi’s sar-
coma associated with captopril. Lancet 1990; 336: 1251–2.
3 Larbre JP, Nicolas JF, Collet P et al. Kaposi’s sarcoma in a patient
with rheumatoid arthritis possible responsibility captopril in the
development of lesions. J Rheumatol 1991; 18: 476–7.
4 Constantinescu CS, Goodman DB, Ventura ES. Captopril and lis-
inopril suppress production of interleukin-12 by human peripheral
blood mononuclear cells. Immunol Lett 1998; 62: 25–31.
5 Boggio K, Di Carlo E, Rovero S et al. Ability of systemic interleukin-
12 to hamper progressive stages of mammary carcinogenesis in
HER2 ⁄ neu transgenic mice. Cancer Res 2000; 60: 359–64.
6 Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med 2000; 342:
1027–38.
7 Monini P, Carlini F, Sturzl M et al. Alpha interferon inhibits human
herpesvirus 8 (HHV-8) reactivation in primary effusion lymphoma
cells and reduces HHV-8 load in cultured peripheral blood mono-
nuclear cells. J Virol 1999; 73: 4029–41.
8 Giuliani M, Di Carlo A, Donati P, Mastroianni A. Onset of non-
AIDS Kaposi’s sarcoma during therapy with 2a interferon in an 82
year-old man with concomitant cutaneous T cell lymphoma. Arch
Dermatol 2002; 138: 535–7.
Lisinopril and Kaposi’s sarcoma: reply from authors
SIR, We have read the letter of Di Carlo regarding our case ofKaposi’s sarcoma (KS) in a patient treated with lisinopril.1
Indeed this is the third case reported in the literature in whichthe association between angiotensin converting enzymeinhibitors (ACEI) and KS was noted,2,3 and the first casedescribed with lisinopril. He was a 78-year-old heterosexualHIV–negative case of KS in whom the lesions appeared
6 months after the initiation of lisinopril therapy anddisappeared 4 months after its withdrawal. We thought thatthis was an induced form of KS due to possible immunosup-pressive effects of the drug. Suppression of interleukin (IL)-12production or interferon-c production,4 or the reduction oflymphocyte proliferation5 might be responsible for this effect.It was reported that IL-12 induces the enforced secretion ofantiangiogenic factors and stimulation of antitumoral immu-nity.4 The occurrence of KS in patients undergoing long-termtreatment with immunosuppressive drugs is well documen-ted, especially in renal transplant patients.6
Although the role of HHV-8, which has been calledKaposi’s sarcoma associated herpes virus (KSHV), in thecausation of KS is not completely clear, it is now recognized asresponsible for the KS.7 We could not investigate the HHV-8in the lesions or in peripheral blood cells of our patient withlisinopril-related KS, because of technical insufficiency. It iswell known that most human herpes viruses, including ofcourse varicella zoster virus, are associated with disease inimmunocompromised hosts as a result of either the reactiva-tion of latent virus or the proliferation of growth transformedcells.7 So the cases of herpes zoster related to ACEI were nosurprise for us. In the literature there are some reports ofherpes zoster due to enalapril with an incidence of < 1%.8
ACEI have been used widely in the world and thepopularity of them is increasing.9 We must keep this possibleside-effect of ACEI in mind, and in suspected cases of KS thecessation of the drug should be considered before othertreatment is planned.
N . B i l e nKocaeli Universitesi, Typ FakultesiHastanesi, Sopalyciftliði 41900Yzmit, Turkey
References
1 Bilen N, Bayramgurler D, Aydeniz B et al. Possible causal role of
lisinopril in a case of Kaposi’s sarcoma. Br J Dermatol 2002; 147:
1042 (Letter).
2 Puppin D Jr, Rybojad M, de la Chapelle C, Morel P. Kaposi’s sar-
coma associated with captopril. Lancet 1990; 336: 1251–2.
3 Larbre JP, Nicolas JF, Collet P et al. Kaposi’s sarcoma in a patient
with rheumatoid arthritis: a possible responsibility of captopril in
the development of lesions. J Rheumatol 1991; 18: 476–7.
4 Constantinescu CS, Goodman DBP, Ventura ES. Captopril and lis-
inopril suppress production of interleukin-12 by human peripheral
blood mononuclear cells. Immunol Lett 1998; 62: 25–31.
5 Johnsen SA, Aurell M. Immunosuppressive action of captopril
blocked by prostoglandin synthetase inhibitor. Lancet 1981; 1:
1005 (Letter).
6 Roszkiewicz A, Roszkiewicz J, Lange M, Tukaj C. Kaposi’s sarcoma
following longterm immunosuppressive therapy: clinical, histo-
logic, and ultrastructural study. Cutis 1998; 61: 137–41.
7 Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med 2000; 342:
1027–38.
8 Litt JZ. Drug Eruption Reference Manual. New York: Parthenon
Publishing Group, 2000: 208.
9 Sabroe RA, Black AK. Angiotensin-converting enzyme (ACE)
inhibitors and angio-oedema. Br J Dermatol 1997; 136: 153–8.
C O R R E S P O N D E N C E 1 5 9
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
Serum concentrations of vascular endothelial growthfactor-D in angiosarcoma patients
SIR, Vascular endothelial growth factor (VEGF) is a primeregulator of endothelial cell proliferation, angiogenesis, vas-culogenesis, and vascular permeability. It has been suggestedthat angiogenesis in the development and invasion oftumours is induced by VEGF secreted from tumour cells.1–4
VEGF-D, another member of the VEGF family of secretedglycoproteins, activates VEGFR-2 (KDR). In addition, VEGF-Dactivates VEGFR-3 (Flt-4), a receptor expressed on lymphaticendothelium in adult tissues, which is thought to signal forthe development of lymphatic vessels and promotion oflymphatic metastasis. Recently, VEGF-D has been reported tobe upregulated in the tumour cells of several neoplasms, andthat lymphatic vessels in the tumour expressed VEGF-Dreceptors as Flt-4.2 In the present study, we have observedthat angiosarcoma cells express VEGF-D mRNA, and thatserum levels of VEGF-D increase with advancing stages of thetumour in angiosarcoma patients.
We investigated 11 elderly patients (seven males and fourfemales; mean age 75Æ3 years, range 67–84 years) withdefinite angiosarcoma of the face and scalp (Table 1). Theserum level of VEGF was measured using a human VEGF-DQuantikine kit (R&D Systems, Minneapolis, MN, U.S.A.)according to the manufacturer’s protocol. Normal controlserum samples were obtained from 18 healthy volunteers (10males and eight females; mean age 70Æ3 years, range62–81 years). We also studied VEGF-D mRNA levels in bothISO-HAS cells and normal endothelial cells (HMvEC) (Mori-naga, Yokohama, Japan) using real-time reverse transcrip-tase-polymerase chain reaction as described by Niki et al.5
The ISO-HAS cells expressed VEGF-D mRNA, while theHMvEC did not secrete detectable levels of VEGF-D mRNA.
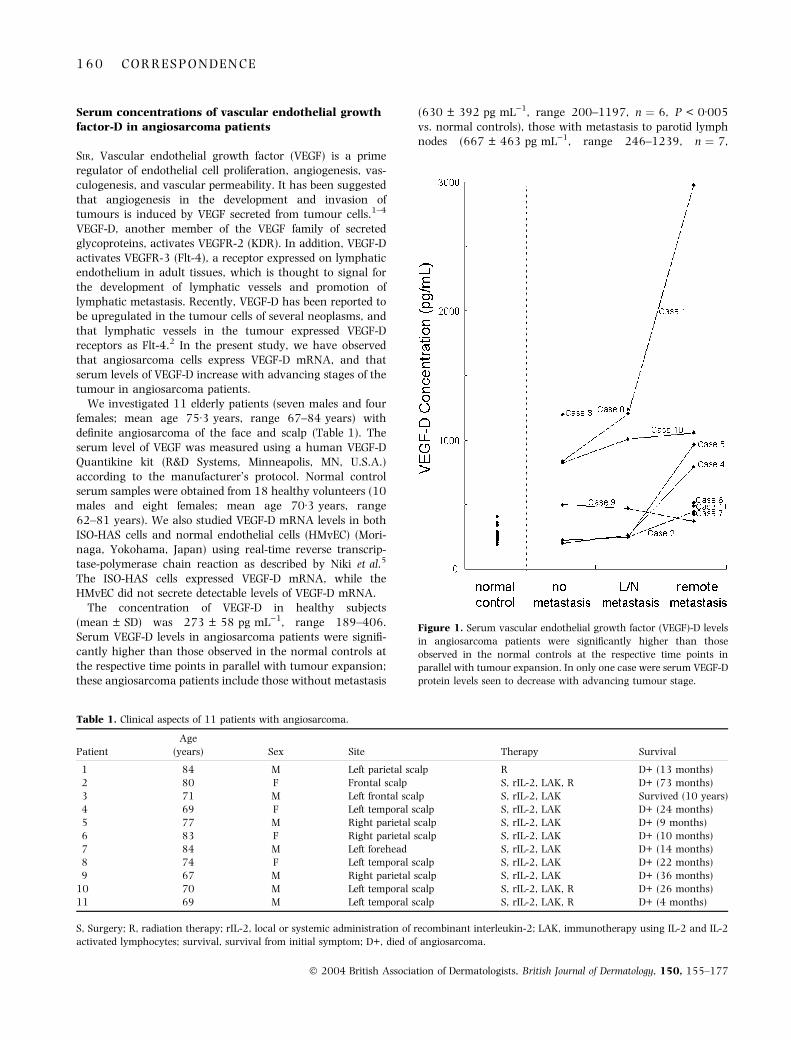
The concentration of VEGF-D in healthy subjects(mean ± SD) was 273 ± 58 pg mL)1, range 189–406.Serum VEGF-D levels in angiosarcoma patients were signifi-cantly higher than those observed in the normal controls atthe respective time points in parallel with tumour expansion;these angiosarcoma patients include those without metastasis
(630 ± 392 pg mL)1, range 200–1197, n ¼ 6, P < 0Æ005vs. normal controls), those with metastasis to parotid lymphnodes (667 ± 463 pg mL)1, range 246–1239, n ¼ 7,
Figure 1. Serum vascular endothelial growth factor (VEGF)-D levels
in angiosarcoma patients were significantly higher than those
observed in the normal controls at the respective time points in
parallel with tumour expansion. In only one case were serum VEGF-D
protein levels seen to decrease with advancing tumour stage.
Table 1. Clinical aspects of 11 patients with angiosarcoma.
Patient
Age
(years) Sex Site Therapy Survival
1 84 M Left parietal scalp R D+ (13 months)
2 80 F Frontal scalp S, rIL-2, LAK, R D+ (73 months)
3 71 M Left frontal scalp S, rIL-2, LAK Survived (10 years)
4 69 F Left temporal scalp S, rIL-2, LAK D+ (24 months)
5 77 M Right parietal scalp S, rIL-2, LAK D+ (9 months)
6 83 F Right parietal scalp S, rIL-2, LAK D+ (10 months)
7 84 M Left forehead S, rIL-2, LAK D+ (14 months)
8 74 F Left temporal scalp S, rIL-2, LAK D+ (22 months)
9 67 M Right parietal scalp S, rIL-2, LAK D+ (36 months)
10 70 M Left temporal scalp S, rIL-2, LAK, R D+ (26 months)
11 69 M Left temporal scalp S, rIL-2, LAK, R D+ (4 months)
S, Surgery; R, radiation therapy; rIL-2, local or systemic administration of recombinant interleukin-2; LAK, immunotherapy using IL-2 and IL-2
activated lymphocytes; survival, survival from initial symptom; D+, died of angiosarcoma.
1 6 0 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

P < 0Æ005 vs. normal controls), and those with remotemetastasis (892 ± 820 pg mL)1, range 370–2977, n ¼ 9,P < 0Æ005 vs. normal controls) (Fig. 1). In only one patient(case 9) were serum VEGF-D levels higher than those in thenormal controls at the respective time points; however, nocorrelation with tumour expansion was seen. This patientwas found to have a bruise-like lesion in the right parietalscalp, and was treated with surgery, local or systemicadministration of recombinant interleukin-2 (IL-2), andimmunotherapy using IL-2 and IL-2-activated lymphocytes.Three years after the first examination, the tumour invadedthe skull, and the patient died because of pyogenic meningo-encephalitis.
We recently established a human angiosarcoma cell line,ISO-HAS.6 We have previously demonstrated that tumourcells of the angiosarcoma cell line ISO-HAS secrete VEGFprotein.4 In addition, we have observed serum VEGF proteinlevels increase with advancing tumour stage in a patient fromwhom the ISO-HAS cells were derived.7,8 In the present study,we observed that ISO-HAS cells expressed VEGF-D mRNA, andthat serum VEGF-D protein levels increased as the tumourstage advanced in the angiosarcoma patients. VEGF-D caninduce both tumour angiogenesis and lymphangiogenesis,and promote the lymphatic spread of tumours.1,2 These resultssuggest that VEGF-D plays an important role in the metastatictumour spread via the lymphatic system in angiosarcoma, andthat the study of serum VEGF-D levels may be useful forpredicting the clinical course of angiosarcoma.
Acknowledgment
This work was partially supported by a grant from theParents’ Association of Kitasato University School of Medi-cine, Japan.
Y . A m o
M . M a s u z a w a
Y . H a m a d a
K . K a t s u o k a
Department of Dermatology, KitasatoUniversity School of Medicine, 1-15-1 Kitasato, Sagamihara, Kanagawa228-8555, JapanE-mail: [email protected]
References
1 Stacker SA, Caesar C, Baldwin ME et al. VEGF-D promotes the
metastatic spread of tumor cells via the lymphatic. Nature Med
2001; 7: 186–91.
2 White JD, Hewett PW, Kosuge D et al. Vascular endothelial growth
factor-D expression is an independent prognostic marker for sur-
vival in colorectal carcinoma. Cancer Res 2002; 62: 1669–75.
3 Detmar M. Tumor angiogenesis. J Invest Dermatol 2000; 5: 20–3.
4 Amo Y, Masuzawa M, Hamada Y et al. Expression of vascular
endothelial growth factor in a human hemangiosarcoma cell line
(ISO-HAS). Arch Dermatol Res 2001; 293: 296–301.
5 Niki T, Iba S, Tokunou M et al. Expression of vascular endothelial
growth factor A, B, C, and D and their relationship to lymph node
status in lung adenocarcinoma. Clin Cancer Res 2000; 6: 2431–9.
6 Masuzawa M, Fujimura T, Hamada Y et al. Establishment of a
human hemangiosarcoma cell line (ISO-HAS). Int J Cancer 1999;
81: 305–8.
7 Amo Y, Masuzawa M, Hamada Y et al. Serum levels of vascular
endothelial growth factor in a hemangiosarcoma patient with a
new-typed p53 gene point mutation. Br J Dermatol 2000; 143:
1118–19.
8 Amo Y, Masuzawa M, Hamada Y et al. Serum concentrations of
vascular endothelial growth factor with and without p53 gene
mutation in angiosarcoma. Acta Derm Venereol 2002; 82: 373–4.
Verruciform xanthoma of the scrotum in a renaltransplant patient
SIR, Verruciform xanthoma (VX) is a rare lesion usually foundin the oral cavity (buccal mucosa) and more rarely in theskin. A recent literature review found a total of 328 reportedcases, of which 46 (14%) developed on the skin,1 mainly inthe anogenital region. VXs manifest as exophytic, polypoidgrowths and display characteristic pathological featurescomprising verrucous hyperplasia of the epidermis andlipid-laden (foamy ⁄ xanthomatous) cells within the dermalpapillae. Although most VXs occur in otherwise healthypersons, five cases have been reported in the setting ofimmunosuppression (bone marrow transplantation,2–4 HIVinfection5 and common variable immunodeficiency disease).6
We report here the first case of VX developing in the setting ofsolid organ transplantation.
A 35-year-old Caucasian man received a renal allograft in1998 for end-stage renal insufficiency due to hyalinosis, andwas receiving prednisone, ciclosporin and mycophenolatemofetil. Five years later he developed on the right side of thescrotum an asymptomatic pedunculated verrucous growthmeasuring 9 · 5 mm reminiscent of condyloma. The patienthad no other mucocutaneous lesion apart from a papillomaon the elbow treated with cryotherapy. He could recall handwarts during childhood, but no genital lesions. No lipidabnormalities were found. The lesion was excised under localanaesthesia and had not recurred after 1 month.
Histologically, it consisted of an exophytic nodule with apapillomatous surface. The overlying epidermis was thick-ened, made of massive rete ridges invading the dermis(Fig. 1A). The horny layer was thin and partly parakeratotic.The stratum spinosum contained numerous neutrophils andsome dyskeratotic keratinocytes. The dermal papillae wereelongated and contained aggregates of round or polyhedralcells with a single central nucleus and a foamy cytoplasm(Fig. 1B) disclosing periodic acid–Schiff-positive granules. Themid-dermis contained several dilatated vessels and a cellinfiltrate consisting mainly of lymphocytes and plasma cells,with occasional eosinophils and neutrophils. These featureswere diagnostic of VX. Immunohistochemically, the foamycells showed strong cytoplasmic expression of vimentin and ofthe CD68 antigen (Fig. 2A), and more focally of lysozyme;one-third of them showed also cytoplasmic expression offactor XIIIa antigen (Fig. 2B). Occasional cells expressedmembrane reactivity for CD45 ⁄ LCA. Several foam cellsshowed cytoplasmic expression of epidermal keratins (AE1,AE3, K5 ⁄ 6) (Fig. 2C). K1 ⁄ 10 and K13 were expressed in theepidermis but not by foam cells; immunostaining for
C O R R E S P O N D E N C E 1 6 1
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
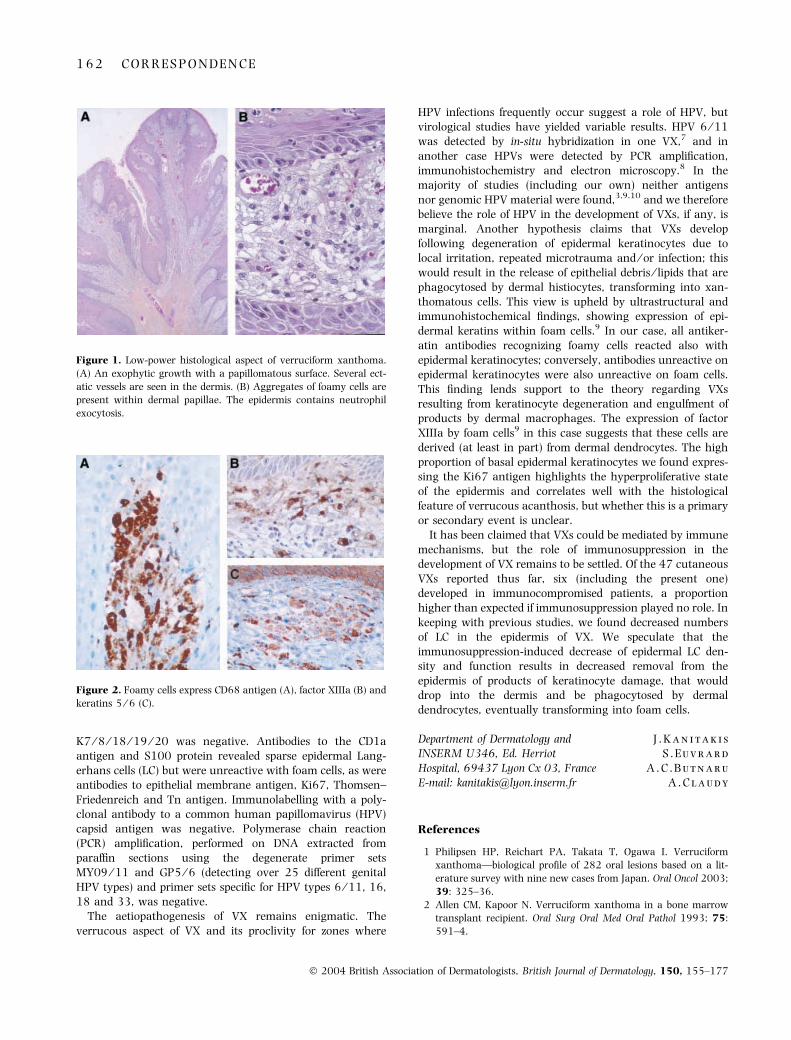
K7 ⁄ 8 ⁄ 18 ⁄ 19 ⁄ 20 was negative. Antibodies to the CD1aantigen and S100 protein revealed sparse epidermal Lang-erhans cells (LC) but were unreactive with foam cells, as wereantibodies to epithelial membrane antigen, Ki67, Thomsen–Friedenreich and Tn antigen. Immunolabelling with a poly-clonal antibody to a common human papillomavirus (HPV)capsid antigen was negative. Polymerase chain reaction(PCR) amplification, performed on DNA extracted fromparaffin sections using the degenerate primer setsMY09 ⁄ 11 and GP5 ⁄ 6 (detecting over 25 different genitalHPV types) and primer sets specific for HPV types 6 ⁄ 11, 16,18 and 33, was negative.
The aetiopathogenesis of VX remains enigmatic. Theverrucous aspect of VX and its proclivity for zones where
HPV infections frequently occur suggest a role of HPV, butvirological studies have yielded variable results. HPV 6 ⁄ 11was detected by in-situ hybridization in one VX,7 and inanother case HPVs were detected by PCR amplification,immunohistochemistry and electron microscopy.8 In themajority of studies (including our own) neither antigensnor genomic HPV material were found,3,9,10 and we thereforebelieve the role of HPV in the development of VXs, if any, ismarginal. Another hypothesis claims that VXs developfollowing degeneration of epidermal keratinocytes due tolocal irritation, repeated microtrauma and ⁄ or infection; thiswould result in the release of epithelial debris ⁄ lipids that arephagocytosed by dermal histiocytes, transforming into xan-thomatous cells. This view is upheld by ultrastructural andimmunohistochemical findings, showing expression of epi-dermal keratins within foam cells.9 In our case, all antiker-atin antibodies recognizing foamy cells reacted also withepidermal keratinocytes; conversely, antibodies unreactive onepidermal keratinocytes were also unreactive on foam cells.This finding lends support to the theory regarding VXsresulting from keratinocyte degeneration and engulfment ofproducts by dermal macrophages. The expression of factorXIIIa by foam cells9 in this case suggests that these cells arederived (at least in part) from dermal dendrocytes. The highproportion of basal epidermal keratinocytes we found expres-sing the Ki67 antigen highlights the hyperproliferative stateof the epidermis and correlates well with the histologicalfeature of verrucous acanthosis, but whether this is a primaryor secondary event is unclear.
It has been claimed that VXs could be mediated by immunemechanisms, but the role of immunosuppression in thedevelopment of VX remains to be settled. Of the 47 cutaneousVXs reported thus far, six (including the present one)developed in immunocompromised patients, a proportionhigher than expected if immunosuppression played no role. Inkeeping with previous studies, we found decreased numbersof LC in the epidermis of VX. We speculate that theimmunosuppression-induced decrease of epidermal LC den-sity and function results in decreased removal from theepidermis of products of keratinocyte damage, that woulddrop into the dermis and be phagocytosed by dermaldendrocytes, eventually transforming into foam cells.
J . K a n i t a k i s
S . E u v r a r d
A . C . B u t n a r u
A . C l a u d y
Department of Dermatology andINSERM U346, Ed. HerriotHospital, 69437 Lyon Cx 03, FranceE-mail: [email protected]
References
1 Philipsen HP, Reichart PA, Takata T, Ogawa I. Verruciform
xanthoma—biological profile of 282 oral lesions based on a lit-
erature survey with nine new cases from Japan. Oral Oncol 2003;
39: 325–36.
2 Allen CM, Kapoor N. Verruciform xanthoma in a bone marrow
transplant recipient. Oral Surg Oral Med Oral Pathol 1993; 75:
591–4.
Figure 1. Low-power histological aspect of verruciform xanthoma.
(A) An exophytic growth with a papillomatous surface. Several ect-
atic vessels are seen in the dermis. (B) Aggregates of foamy cells are
present within dermal papillae. The epidermis contains neutrophil
exocytosis.
Figure 2. Foamy cells express CD68 antigen (A), factor XIIIa (B) and
keratins 5 ⁄ 6 (C).
1 6 2 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

3 Mannes KD, Dekle CL, Requena L, Sangueza OP. Verruciform
xanthoma associated with squamous cell carcinoma. Am J Der-
matopathol 1999; 21: 66–9.
4 Helm KF, Hopfl RM, Kreider JW, Lookingbill DP. Verruciform
xanthoma in an immunocompromised patient: a case report and
immunohistochemical study. J Cutan Pathol 1993; 20: 84–6.
5 Smith KJ, Skelton HG, Angritt P. Changes of verruciform xan-
thoma in an HIV-1+ patient with diffuse psoriasiform skin dis-
ease. Am J Dermatopathol 1995; 17: 185–8.
6 Wu JJ, Wagner AM. Verruciform xanthoma in association with
Milroy disease and leaky capillary syndrome. Pediatr Dermatol
2003; 20: 44–7.
7 Iamaroon A, Vickers RA. Characterization of verruciform xan-
thoma by in situ hybridization and immunohistochemistry. J Oral
Pathol Med 1996; 25: 395–400.
8 Mostafa KA, Takata T, Ogawa I et al. Verruciform xanthoma of
the oral mucosa: a clinicopathological study with immunohisto-
chemical findings relating to pathogenesis. Virchows Arch A Pa-
thol Anat Histopathol 1993; 423: 243–8.
9 Mohsin SK, Lee MW, Amin MB et al. Cutaneous verruciform xan-
thoma: a report of five cases investigating the etiology and nature of
xanthomatous cells. Am J Surg Pathol 1998; 22: 479–87.
10 Takizawa H, Ohnishi T, Watanabe S. Verruciform xanthoma.
Report of a case with molecular biological analysis of HPV and
immunohistochemical analysis of cytokeratin expression. Clin
Exp Dermatol 2001; 26: 730–1.
Control of childhood pemphigus erythematosus withsteroids and azathioprine
SIR, We report the case of a 7-year-old child who presented tous in 1998 with generalized scaling and redness associatedwith pedal oedema and tightness of both upper and lowerextremities. He had developed flaccid blisters on scalp, faceand both lower extremities associated with photosensitivityover the previous 2 years. The patient had tried variousmodalities of treatment without any relief (complete details oftherapy were not available).
On examination, his vital parameters were normal. Hehad evidence of generalized erythema and scaling withcrusting in flexures. There was no evidence of oral andgenital mucosal involvement. The blood serology inclusiveof full blood count (FBC) with platelet count, serumchemistry profile, glucose levels, urine analysis, tuberculintest, triglyceride levels and chest X-ray did not reveal anyabnormality. The antinuclear antibodies (ANA) and double-stranded (ds)DNA study were normal. The bone densitom-etry showed evidence of frank osteoporosis (T ¼ )3Æ59:T < )2Æ5 is suggestive of osteoporosis). A lesional biopsy forhistopathological studies was compatible with pemphiguserythematosus. A perilesional biopsy from the buttock forimmunofluorescence studies showed deposits of IgG, C3 andfibrin antibodies in ICCS pattern (fishnet appearance) andgranular deposits of IgG and C3 at the basement membranezone confirming the diagnosis of pemphigus erythematosus.The severity of osteoporosis posed a dilemma regarding useof systemic steroids. Intravenous immunoglobulin wasconsidered but not administered because of the cost factor.Hence, he was started on prednisolone 2Æ5 mg kg)1 day)1
daily by mouth with calcium supplements, dapsone 50 mgat bedtime by mouth, as a steroid-sparing agent, andadvised to use photoprotection in the form of a broadbrimmed hat and sunscreens. The patient achieved completeremission in 10 days. As he was a native of a distant village(500 km from our clinic) he was advised to obtain regularmonitoring of blood pressure, weight, FBC with differentialwhite blood count, liver and renal function tests, glucosetest, urine analysis and ophthalmic check-up with hisresident dermatologist. His consultant dermatologist repor-ted intermittent episodes of relapses on tapering the steroiddosage and the omission of dapsone after a year because hedeveloped jaundice.
In 2001, 3 years later, the prednisolone was stoppedbecause of low-grade fever. He came back to us with newlydeveloped generalized erosions and scaling associated withphotosensitivity. There was evidence of Cushingoid facies. Onre-evaluation, his blood serology and ANA and dsDNAstudies were normal. The bone densitometry showedimprovement but was still in the osteoporotic range(T ¼ )3Æ0). The desmoglein (Dsg)1 antibody index value of301Æ95 and Dsg3 antibody index value of 2Æ36 confirmed thediagnosis of pemphigus foliaceous. Prednisolone2Æ5 mg kg)1 day)1 was reinstituted. In view of the side-effects of steroids, particularly osteoporosis, we also addedazathioprine 25 mg daily by mouth. The inclusion ofazathioprine aided the early tapering of systemic steroid andthereby we hoped to reduce the severity of the osteoporosis. Itwas also beneficial in decreasing the frequency of relapses.The patient has been in complete remission since he was11 years and 2 months old. Currently he is taking predniso-lone 5 mg daily and azathioprine 50 mg daily by mouth.There is marked improvement in his osteoporosis scores(T ¼ )2Æ76) with subsidence of the Cushingoid features. Therepeat Dsg1 antibody after 6 months was 270Æ64 showing a10% drop in the index value and indicating disease control.
Pemphigus rarely occurs in children. Our patient haderythematous scaly lesions over nose and cheeks in a butterflydistribution associated with photosensitivity. The immunoflu-orescence studies and the desmoglein results confirmed thediagnosis of pemphigus erythematosus. A judicious use ofsteroids and steroid sparing agent such as azathioprine wasmore effective than steroid alone in the control of pemphiguserythematosus in our patient. The combination of predniso-lone and azathioprine is more effective than prednisolonealone in terms of both mortality and remission.1–4 Furtherexperience and long-term outcome studies in children areneeded to determine whether some medication side-effectsmay outweigh the risks from the disease itself.5 Our patientprobably represents one of the few fully documented cases ofpemphigus erythematosus in childhood managed on a com-bination of steroids and azathioprine.
M . T . G u p t a
H . R . J e r a j a n i
Department of Dermatology, LTMGH& LTMMC, Mumbai, Maharashtra,IndiaE-mail: [email protected]
C O R R E S P O N D E N C E 1 6 3
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

References
1 Wojnarowska F, Eady RA, Burge SM. Bullous eruptions. In: Text-
book of Dermatology (Champion RH, Burton JL, Burns DA,
Breathnach SM, eds), 6th edn, Vol. 3. Oxford: Blackwell Scientific
Publications, 1998: 1861–3.
2 Carson PJ, Hameed A, Ahmed AR. Influence of treatment on the
clinical course of pemphigus vulgaris. J Am Acad Dermatol 1996;
34: 645–52.
3 Aberer W, Wolff-Schreiner EC, Stingl G, Wolff K. Azathioprine in
the treatment of pemphigus vulgaris. J Am Acad Dermatol 1987;
16: 527–33.
4 PerezBernal AM, Moreno JC, Camacho F. Ten years experience of
the treatment of 34 cases of pemphigus. J Dermatol Treat 1992; 3:
73–5.
5 Denise WM, Adelaide AH, Robert EJ. Nonendemic pemphi-
gus foliaceous in children. J Am Acad Dermatol 2002; 46: 419–
22.
Reduced therapeutic activity of warfarin duringtreatment with oral isotretinoin
SIR, Oral isotretinoin is an established effective therapy forsevere cystic acne, but it is also recommended for otherinflammatory skin disorders including Gram-negative follicu-litis, inflammatory rosacea, pyoderma faciale, acne fulminansand hidradenitis suppurativa. We report a patient undertherapy with warfarin who required an increase in coumarindosage after starting treatment with oral isotretinoin.
A 61-year-old man on oral anticoagulant therapy withwarfarin was evaluated for a sudden eruption of the face. Thelesions consisted of multiple, small follicular pustules locatedin the nasolabial line and on the upper lip and chin,associated with inflammatory papulopustular lesions of thecheeks and perioral region. His past medical history includedcardiac valve replacement for mitral stenosis 5 years previ-ously; anticoagulant therapy had been given since then. Hereported that the international normalized ratio (INR) hadbeen consistently maintained within the therapeutic rangeduring the last 2–3 years on warfarin 2Æ5 mg daily. Gram-negative organisms belonging to Klebsiella spp. were isolatedfrom the superficial follicular pustules. Our patient wasstarted on a combination treatment with oral cefpodoximeproxetil 200 mg twice daily and oral isotretinoin 30 mgdaily (0Æ4 mg kg)1). After starting the above therapy theINR decreased below 2Æ5 and returned to therapeutic levelsonly after increasing the warfarin intake to 3Æ75 mg daily.After 10 days oral cefpodoxime proxetil was discontinuedand our patient was maintained on oral isotretinoin. Hisface greatly improved and the INR subsequently remainedstable. When isotretinoin was discontinued after 40 days oftreatment, INR values progressively increased and warfarinwas reduced to the pretreatment dosage of 2Æ5 mg daily. Noabnormalities in blood tests were detected throughout thisperiod.
In our patient an increase in the warfarin dosage wasnecessary to re-establish an adequate anticoagulation afteroral treatment with cefpodoxime proxetil, and isotretinoinwas started. The evidence that warfarin had to be maintained
at the higher dosage after discontinuation of the oralantibiotic, and had to be reduced after discontinuation ofisotretinoin, suggests that the INR decrease was probably aresult of the coadministration of warfarin and isotretinoin.
A reduced therapeutic effect of warfarin has been reportedduring treatment with etretinate,1 but has never beendescribed during oral isotretinoin treatment. The cause ofthe possible interaction between isotretinoin and warfarin isnot known. Warfarin is metabolized by the liver andconsequently drugs which induce liver enzymes, e.g. barbit-urates, tend to reduce its effect. Oral isotretinoin has beenshown to induce hepatic P-450-dependent isozyme activit-ies,2 and it is possible that this caused an increased rate ofwarfarin metabolism in our patient.
Oral isotretinoin has been used in a wide variety of clinicalconditions and may occasionally be indicated in the man-agement of patients receiving anticoagulant therapy. Thus,caution should be taken when prescribing oral isotretinoin topatients on warfarin until the nature and the impact of theisotretinoin–warfarin inteaction is fully elucidated. We con-tribute this anedoctal report because the effects of isotretinoinon the anticoagulant activity of warfarin are unlikely to beinvestigated more formally.
P . F i a l l oDepartment of Health Sciences,Section of Social Dermatology,University of Genoa, Viale BenedettoXV, 7, I-16132 Genoa, ItalyE-mail: [email protected]
References
1 Ostlere LS, Langtry JAA, Jones S, Staughton RCD. Reduced
therapeutic effect of warfarin caused by etretinate. Br J Dermatol
1991; 124: 505.
2 Goerz G, Bolsen K, Kalofoutis A, Tsambaos D. Influence of oral
isotretinoin on hepatic and cutaneous P-450-dependent isozyme
activities. Arch Dermatol Res 1994; 286: 104–6.
Delayed onset of bullous reaction with severe deep skinnecrosis in association with sertraline
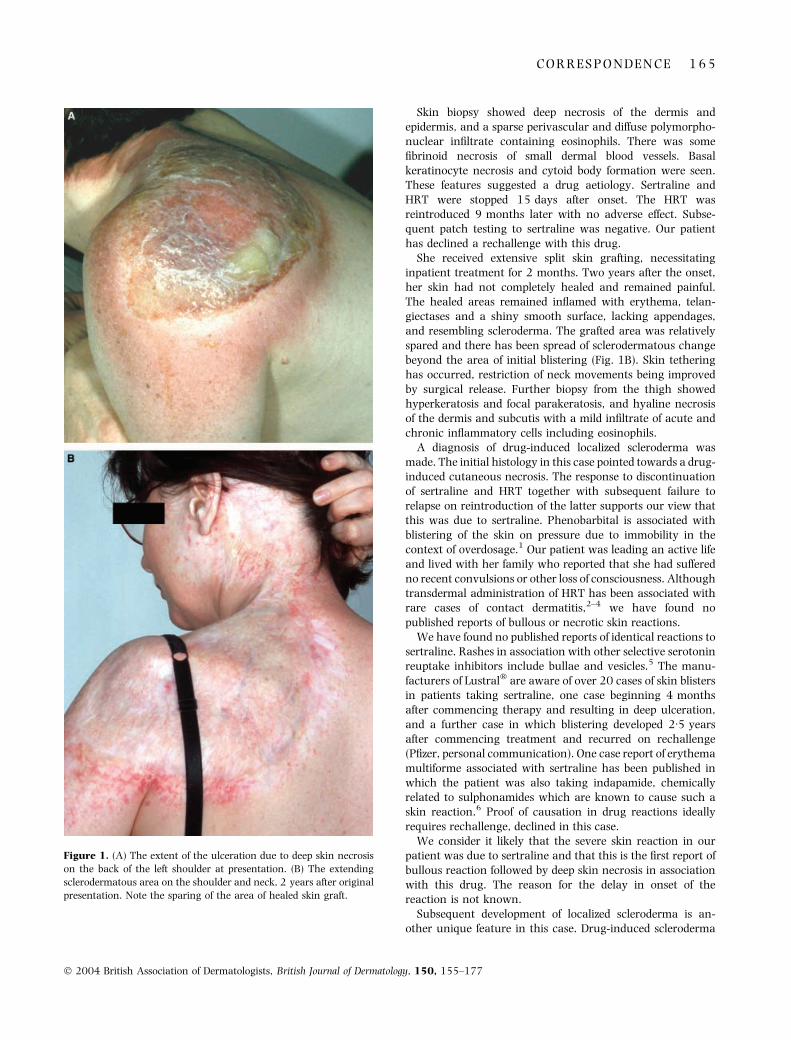
SIR, A 48-year-old woman presented 10 days after the onsetof painful erythema of the posterior and inner thighs, leftshoulder and left side of the neck, worsening despite systemicvalaciclovir and co-amoxiclav prescribed by her generalpractitioner. At presentation, blistering of the affected siteswas beginning. Ulceration of the shoulder (Fig. 1A) andthighs followed rapidly. It was apparent at the time of biopsy,3 days after admission, that full-thickness skin necrosis wasoccurring.
Our patient had a history of epilepsy, fully controlled byphenobarbital 30 mg daily. Seven months prior to presenta-tion she had commenced oral hormone replacement therapy(HRT) with combined oestradiol and levonorgestrel. Onemonth later she began taking sertraline (Lustral�; Pfizer,Tadworth, U.K.) 100 mg daily for reactive depression.
1 6 4 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
Skin biopsy showed deep necrosis of the dermis andepidermis, and a sparse perivascular and diffuse polymorpho-nuclear infiltrate containing eosinophils. There was somefibrinoid necrosis of small dermal blood vessels. Basalkeratinocyte necrosis and cytoid body formation were seen.These features suggested a drug aetiology. Sertraline andHRT were stopped 15 days after onset. The HRT wasreintroduced 9 months later with no adverse effect. Subse-quent patch testing to sertraline was negative. Our patienthas declined a rechallenge with this drug.
She received extensive split skin grafting, necessitatinginpatient treatment for 2 months. Two years after the onset,her skin had not completely healed and remained painful.The healed areas remained inflamed with erythema, telan-giectases and a shiny smooth surface, lacking appendages,and resembling scleroderma. The grafted area was relativelyspared and there has been spread of sclerodermatous changebeyond the area of initial blistering (Fig. 1B). Skin tetheringhas occurred, restriction of neck movements being improvedby surgical release. Further biopsy from the thigh showedhyperkeratosis and focal parakeratosis, and hyaline necrosisof the dermis and subcutis with a mild infiltrate of acute andchronic inflammatory cells including eosinophils.
A diagnosis of drug-induced localized scleroderma wasmade. The initial histology in this case pointed towards a drug-induced cutaneous necrosis. The response to discontinuationof sertraline and HRT together with subsequent failure torelapse on reintroduction of the latter supports our view thatthis was due to sertraline. Phenobarbital is associated withblistering of the skin on pressure due to immobility in thecontext of overdosage.1 Our patient was leading an active lifeand lived with her family who reported that she had sufferedno recent convulsions or other loss of consciousness. Althoughtransdermal administration of HRT has been associated withrare cases of contact dermatitis,2–4 we have found nopublished reports of bullous or necrotic skin reactions.
We have found no published reports of identical reactions tosertraline. Rashes in association with other selective serotoninreuptake inhibitors include bullae and vesicles.5 The manu-facturers of Lustral� are aware of over 20 cases of skin blistersin patients taking sertraline, one case beginning 4 monthsafter commencing therapy and resulting in deep ulceration,and a further case in which blistering developed 2Æ5 yearsafter commencing treatment and recurred on rechallenge(Pfizer, personal communication). One case report of erythemamultiforme associated with sertraline has been published inwhich the patient was also taking indapamide, chemicallyrelated to sulphonamides which are known to cause such askin reaction.6 Proof of causation in drug reactions ideallyrequires rechallenge, declined in this case.
We consider it likely that the severe skin reaction in ourpatient was due to sertraline and that this is the first report ofbullous reaction followed by deep skin necrosis in associationwith this drug. The reason for the delay in onset of thereaction is not known.
Subsequent development of localized scleroderma is an-other unique feature in this case. Drug-induced scleroderma
Figure 1. (A) The extent of the ulceration due to deep skin necrosis
on the back of the left shoulder at presentation. (B) The extending
sclerodermatous area on the shoulder and neck, 2 years after original
presentation. Note the sparing of the area of healed skin graft.
C O R R E S P O N D E N C E 1 6 5
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

is well recognized with penicillamine, and has also beenreported with bromocriptine.7,8 In both cases there wasrecovery on withdrawing the drug. Our patient did not beginto show sclerodermatous change until her necrotic skin hadhealed. The pathogenesis is unclear.
Acknowledgment
We thank Professor MM Black for his helpful comments onthe pathology in this case.
M . E . K i r k u p
E . A . S h e f f i e l d *
L . J . S a c k s�J . E . S a n s o m
Departments of Dermatology and*Histopathology, Bristol RoyalInfirmary, Bristol BS2 8HW, and�Department of Plastic Surgery,Frenchay Hospital, Bristol BS161ND, U.K.E-mail: [email protected]
References
1 Rall TW. Hypnotics and sedatives. In: Goodman and Gilman’s the
Pharmacological Basis of Therapeutics (Goodman Gilman AG, Rall
TW, Nies AS, Taylor P, eds), 8th edn. New York: Pergamon Press,
1990: 363.
2 Graham-Brown RAC. Dermatologic problems of the menopause.
Clin Dermatol 1997; 15: 143–5.
3 Holdiness MR. A review of contact dermatitis associated with
transdermal therapeutic systems. Contact Dermatitis 1989; 20:
3–9.
4 Torres V, Campos Lopes J, Leite L. Contact dermatitis from nitro-
glycerin and estradiol transdermal therapeutic systems. Contact
Dermatitis 1992; 26: 53–4.
5 Beauquier B, Fahs H. Effets secondaires dermatologiques des
antidepresseurs de la recapture de la serotonine: hypothese d’une
allergie croisee. A propos de deux cas. Encephale 1998; 24:
62–4.
6 Gales BJ, Gales MA. Erythema multiforme and angioedema with
indapamide and sertraline. Am J Hosp Pharm 1994; 51: 118–19
(Letter).
7 Bernstein RM, Hall MA, Gostelow BE. Morphea-like reaction to
penicillamine therapy. Ann Rheum Dis 1981; 40: 42–4.
8 Leshin B, Piette WW, Caplan RM. Morphea after bromocriptine
therapy. Int J Dermatol 1989; 28: 177–9.
Acral ulcerations and osteolysis, a severe form of thecarpal tunnel syndrome
SIR, Acral ulcerations and osteolysis are rare in carpal tunnelsyndrome. Most cases are unilateral and respond satisfactor-ily to surgery. We report two patients with unsuspectedbilateral carpal tunnel syndrome of long duration, manifest-ing with acral ulcers and acro-osteolysis.
Patient 1 was a 65-year-old man who had had a leftforearm injury 10 years previously. He consulted for markederythema and oedema in the index and middle fingers of hisright hand. He was a retired officer. He had no sensitivity inthose fingers or in the index and middle fingers of the left
hand. This anaesthesia had been present for years in the lefthand, and only for the last few months in the right.Occasionally at night he experienced tingling over the firstthree fingers of his hands which was relieved with standingand shaking his wrists.
Examination showed little bullae and small necrotic ulcerson the fingertips (Fig. 1A) with dystrophic nail changes onthe index and middle fingers of both hands. There was evidentshortening of some of these fingers. The index and middlefingers of the right hand were oedematous and erythematous,with a similar appearance to the initial manifestations ofsystemic sclerosis (Fig. 1B). Moreover, there was evidentbilateral thenar atrophy, and the patient was unable to raisehis thumbs when pressing them against a flat surface withthe palm side up. Palpation of the peripheral pulses revealedgood radial and brachial pulses.
X-ray of the hands disclosed osseous resorption of the distalphalanges in the right index and middle fingers and in the leftindex finger. There were no abnormalities in blood glucoselevels, thyroid and growth hormones, protein profile andautoantibodies. Electromyography showed bilateral severemedian nerve damage.
Figure 1. Patient 1. (A) Small necrotic ulcers on the fingertips of the
index and middle fingers. (B) The index and middle fingers of the right
hand were oedematous and erythematous, and showed acro-osteo-
lysis and dystrophic changes.
1 6 6 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

A diagnosis of bilateral carpal tunnel syndrome was made,and the patient was refered to a traumatologist for surgicaldecompression of the median nerves. Relief of cutaneouslesions was achieved a month later.
Patient 2 was a 73-year-old woman with a history of lichenplanus and high blood pressure, referred to us because ofRaynaud’s phenomenon affecting both hands for 5 years.Examination showed acral osteolysis of the third phalanges ofthe index and middle fingers of the left hand and the middlefinger of the right hand, with bilateral thenar atrophy. Theright index finger had been surgically amputated for gan-grene. The fingertips of the three affected fingers exhibitedsome small burns and ulcers. She reported that all hersymptoms, including Raynaud’s phenomenon, had beenrelieved 1 year previously by surgical decompression of bothmedian nerves. Results of routine blood tests were normal ornegative, including autoantibodies, thyroid hormones, glu-cose levels and protein profile.
Carpal tunnel syndrome is a common disorder in generalmedical practice. It is more prevalent between the fourth andthe sixth decades, and women are affected three times moreoften than men. It is due to compression of the median nervebetween the transverse carpal ligament and carpal bones.Elevated pressure in the carpal tunnel produces ischaemia ofthe median nerve, resulting in paraesthesia and pain and, ifthe condition persists, in weakness and atrophy.1 Skinsymptoms are not usually present, and they are overlookedin the English language literature. Most cases with prominentskin symptoms have been described in the French languageliterature.2–6
Skin symptoms develop only in severe forms of carpal tunnelsyndrome, and these include erythema, oedema, bullae andindolent ulcerations of both the fingertips and the subungualregions. The nails are usually affected, showing discoloration,onycholysis and other dystrophic changes. Gangrene, spon-taneous or surgical amputation and acral osteolysis of theterminal phalanges have seldom been described.2–10 Symp-toms rarely resemble those of Raynaud’s syndrome.3–5 Thefact that solely the index and middle fingers are involved maybe explained because the innervation of the thumb is mixed,coming from both the median and the radial nerves.
Acral ulcerations and osteolysis in carpal tunnel syndromeare frequently unilateral, but were bilateral in both ourpatients. We ruled out systemic causes, such as diabetesmellitus, hypothyroidism, inflammatory arthritis, amyloido-sis, acromegaly or treatment with corticosteroids or oestro-gens. The patients did not have occupations associated with ahigh incidence of this syndrome. The left forearm injuryreceived 10 years previously in patient 1 could explain themedian nerve injury of that side, but not of the right side. Notrauma could be demonstrated in patient 2.
Patient 1 was referred to us with the presumed diagnosis ofa initial form of systemic sclerosis. The distribution of thelesions and the severe anaesthesia, combined with the longhistory of the disease, ruled out that possibility. Patient 2 wasreferred to us with the diagnosis of acro-osteolysis withRaynaud’s phenomenon in spite of the fact that surgery for
carpal tunnel syndrome had been performed, and all the signshad subsequently stabilized.
Dermatologists encountering patients with fingertip ulcersassociated with sensory impairment rarely consider thepossibility of a severe form of carpal tunnel syndrome. Thisentity has also been overlooked in textbooks of dermatology,probably because it is a neurological disorder, and dermatol-ogists are consulted only for special and very rare cutaneouspresentations of the syndrome. The aim of this report is toremind dermatologists about this atypical form of carpaltunnel syndrome, because the diagnosis is easy if one takes itinto account. Diagnosis is not only of academic importance,because a good clinical response can be expected aftersurgery.2–6
C . R e u e n a
L . R e q u e n a *
S . B l a n c o
C . A l v a r e z
C . G a l a c h e
E . R o d r ı g u e z
Department of Dermatology, Hospitalde Cabuenes, 33394 Cabuenes, Gijon,and *Fundacion Jimenez Dıaz,Madrid, SpainE-mail: [email protected]
References
1 Katz JN, Simmons BP. Carpal tunnel syndrome. N Engl J Med
2002; 346: 1807–12.
2 Bouvier M, Lejeune E, Rouillat M, Marionnet J. Les formes ulcero-
mutilantes du syndrome du canal carpien. Rev Rhum 1979; 46:
169–76.
3 Geffray L, Leman J, Dehais J, Cavid-Chausse J. Deux cas de syn-
drome du canal carpien avec ulcerations digitales et acro-osteo-
lyse. Rev Rhum 1984; 51: 45–7.
4 Treves R, Arnaud JP, Benaubbou M, Desproges-Gotteron R.
Ulcerations digitales au cours d’un syndrome du canal carpien
avec syndrome de Raynaud. Rev Rhum 1980; 47: 578–9.
5 Rageot E, Maigne R, Nataf J. Guerison d’un syndrome de Ray-
naud au stade de ganfrene par une intervention portant sur le
canal carpien. Coeur Med Interne 1973; 12: 317–9.
6 Adoue D, Arlet P, Giraud P et al. Syndrome du canal carpien avec
ulcerations digitales chez un insuffisant renal en hemodialyse
periodique. Ann Dermatol Venereol 1984; 111: 1019–21.
7 Aratari E, Regesta G, Rebora A. Carpal tunnel syndrome
appearing with prominent skin symptoms. Arch Dermatol 1984;
120: 517–9.
8 Kendall D. Non penetrating injuries of the median nerve at the
wrist. Brain 1950; 73: 84–94.
9 Lagrot F, Micheau P, Costagliola M, Mansat C. A propos d’une
forme ulcerante du canal carpien. Mem Acad Chir 1966; 92: 169–
71.
10 Tosti A, Morelli R, D’Alessandro R, Bassi F. Carpal tunnel syn-
drome presenting with ischemic skin lesions, acro-osteolysis, and
nail changes. J Am Acad Dermatol 1993; 29: 287–90.
Scleromyxoedema-like eruption followinghaemodialysis or nephrogenic fibrosing dermopathy?
SIR, We were interested to read the recent article by Hubbardet al.1 The authors commented that informal communication
C O R R E S P O N D E N C E 1 6 7
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

with other renal transplant centres in the U.K. had failed toreveal any similar patient. In 2001 at our centre a 48-year-old woman, within a month of commencing haemodialysis,developed bilateral symmetrical slightly erythematous ind-urated plaques on the upper limbs including the back ofhands, and diffuse waxy thickening and hardening of thethighs and abdomen. Skin biopsies from her dorsal righthand and abdomen were consistent with scleromyxoedema,demonstrating the presence of increased mucin and fibro-blast in the dermis. She did not have paraproteinaemia, andkidney biopsies failed to show an excess of mucin deposits.This case was presented at the annual meeting of theBritish Association of Dermatologists at Edinburgh in July2002.2
We are aware of at least one other patient at our hospitalhaving similar clinicopathological cutaneous changes soonafter commencing haemodialysis. Unlike those of Hubbardet al., our patients did not have renal transplant but receivedhaemodialysis.
In September 2000, Cowper et al.3 reported a series of15 patients from four states in the U.S.A. who had received,or were in the process of receiving, renal dialysis anddeveloped a fibrotic skin condition that histopathologicallyresembled scleromyxoedema. Nine patients had receivedrenal transplants. Very recently another six similar caseswere also reported from the U.S.A.4,5 To date, a commondeterminant in all these patients was a history of renal diseaserequiring renal dialysis without associated paraproteinaemia.The sudden emergence of this condition and apparentclustering of cases raised the possibility of infectious and ⁄ ortoxic agents; however, no such agent has been demonstratedso far. In October 2001, Cowper et al.6 proposed the term�nephrogenic fibrosing dermopathy� until a specific cause canbe identified for similar pathology.
G . D a w n
S . C . H o l m e s
Department of Dermatology, GlasgowRoyal Infirmary, Glasgow G4 0SF,U.K.E-mail: [email protected]
References
1 Hubbard V, Davenport A, Jarmulowicz M, Rustin M. Scleromyx-
oedema-like changes in four renal dialysis patients. Br J Dermatol
2003; 148: 563–8.
2 Dawn G, Kavanagh D, Holmes S et al. Scleromyxoedema-like
eruption following haemodialysis. Br J Dermatol 2002; 147 (Suppl.
62): 79 (Abstract).
3 Cowper SE, Robin HS, Steinberg ST et al. Scleromyxoedema-like
cutaneous disease in renal-dialysis patients. Lancet 2000; 356:
1000–1.
4 Mackay-Wiggan JM, Cohen DJ, Hardy MA, Grossman ME. Neph-
rogenic fibrosing dermopathy (scleromyxedema-like illness of renal
disease). J Am Acad Dermatol 2003; 48: 55–60.
5 Streams BN, Liu V, Liegeois N, Moschella SM. Clinical and patho-
logic features of nephrogenic fibrosing dermopathy: a report of two
cases. J Am Acad Dermatol 2003; 48: 42–7.
6 Cowper SE, Su LD, Bhawan J et al. Nephrogenic fibrosing derm-
opathy. Am J Dermatopathol 2001; 23: 383–93.
Lichen sclerosus–lichen planus overlap in a patientwith hepatitis C virus infection
SIR, Lichen planus (LP) and lichen sclerosus (LS) share similarpathological and clinical features. There have been a fewreported cases in the literature of the coexistence of LP andLS.1–3 There is a known association between hepatitis C virus(HCV) infection and LP. However, the association of HCV andLS has never been documented. We report a case of LP–LSoverlap syndrome in a patient with HCV. To our knowledge,this is the first reported case occurring in a patient with HCVinfection.
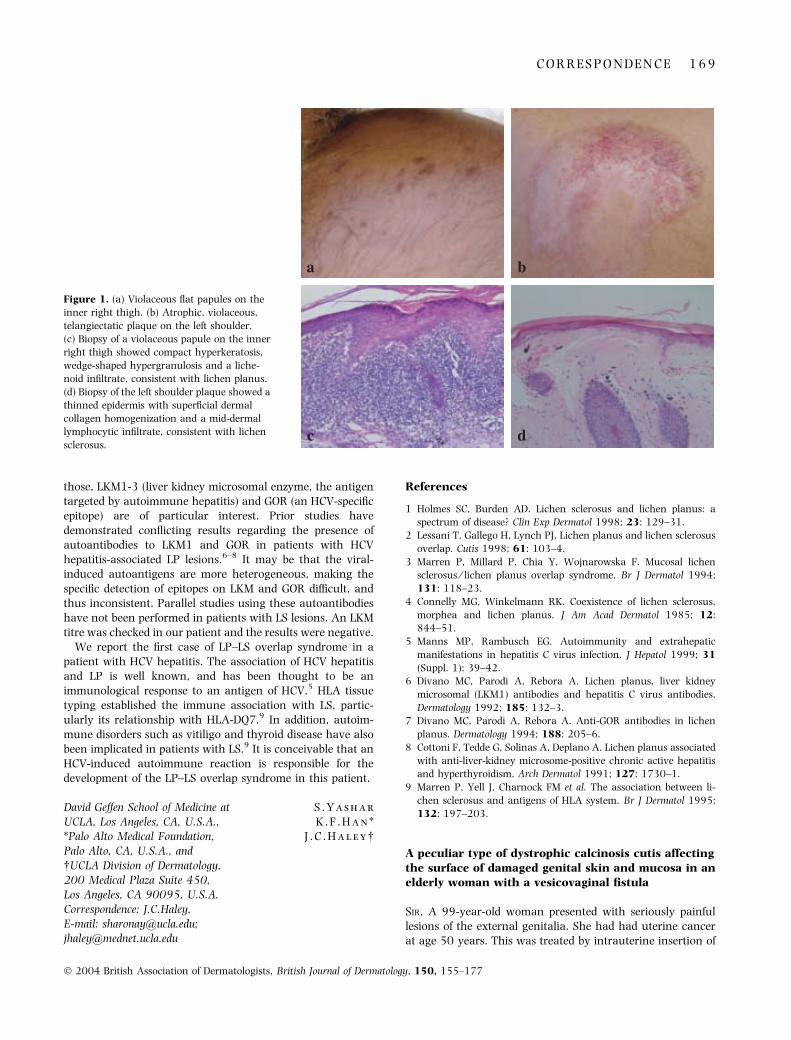
A 64-year-old man presented with a 1-year history of aseverely pruritic eruption on his scrotum, penis, left shoulderand hip. Treatments with valaciclovir and oxyconazole by hisprimary care physician were ineffective. The patient had adistant history of intravenous drug abuse. Prior laboratoryfindings were significant for a mildly elevated aspartateaminotransferase (68 U L)1) and alanine aminotransferase(55 U L)1). Hepatitis serologies showed positive HCV antibod-ies. On examination, there were multiple 1–3-mm violaceous,polygonal, flat papules on his scrotum, penile shaft, bilateralproximal lower extremities and upper left arm (Fig. 1a). Therewere also three atrophic plaques of 2 · 1 cm on the left clavicle,3 · 4 cm on the left shoulder (Fig. 1b) and 5 · 6 cm on the leftbuttock with telangiectasia and purple annular plaques withperipheral scales. There was no oral mucosal involvement. AKOH examination was negative for fungal hyphae.
Histological examination of the violaceous lesions showedorthokeratosis, wedge-shaped hypergranulosis, and a band-like lymphocytic infiltrate obscuring the dermal–epidermaljunction with vacuolar alteration of the basement membranezone. There were Civatte bodies and pigmentary incontinencein the dermis, consistent with LP (Fig. 1c). Biopsy of theatrophic plaques showed an atrophic epidermis, homogeni-zation of the upper dermis and a mid-dermal lymphocyticinfiltrate, consistent with LS (Fig. 1d). The diagnoses of LPand LS were made. The patient was treated with fluocinonideointment, with improvement.
There are only rare reports of concurrent LP and LS. Connellyand Winkelmann reported four and reviewed eight other casesof concurrent LP and LS.4 Morphoea was also present in two ofthese cases. Six of the 12 cases had other immunologicalabnormalities (eosinophilia, decreased leucocyte count) orautoimmune diseases (systemic scleroderma, vitiligo or alope-cia areata). All of these cases are suggestive of a clinicalspectrum between these two conditions. The parallel existsbetween these overlap syndromes and graft-vs.-host disease(GVHD), where in early chronic GVHD lesions appear lichen-oid, and in late chronic GVHD sclerotic lesions predominate.The pathogenesis is immunological and is considered on thespectrum of GVHD. It is therefore tempting to speculate on theimmunological relationship between LP and LS.
LP is a common cutaneous finding in association with HCVhepatitis. Autoimmunity associated with HCV hepatitis is wellknown and has been extensively investigated.5 Severalantigens are implicated in viral-induced autoimmunity. Of
1 6 8 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
those, LKM1-3 (liver kidney microsomal enzyme, the antigentargeted by autoimmune hepatitis) and GOR (an HCV-specificepitope) are of particular interest. Prior studies havedemonstrated conflicting results regarding the presence ofautoantibodies to LKM1 and GOR in patients with HCVhepatitis-associated LP lesions.6–8 It may be that the viral-induced autoantigens are more heterogeneous, making thespecific detection of epitopes on LKM and GOR difficult, andthus inconsistent. Parallel studies using these autoantibodieshave not been performed in patients with LS lesions. An LKMtitre was checked in our patient and the results were negative.
We report the first case of LP–LS overlap syndrome in apatient with HCV hepatitis. The association of HCV hepatitisand LP is well known, and has been thought to be animmunological response to an antigen of HCV.5 HLA tissuetyping established the immune association with LS, partic-ularly its relationship with HLA-DQ7.9 In addition, autoim-mune disorders such as vitiligo and thyroid disease have alsobeen implicated in patients with LS.9 It is conceivable that anHCV-induced autoimmune reaction is responsible for thedevelopment of the LP–LS overlap syndrome in this patient.
S . Y a s h a r
K . F . H a n *
J . C . H a l e y�
David Geffen School of Medicine atUCLA, Los Angeles, CA, U.S.A.,*Palo Alto Medical Foundation,Palo Alto, CA, U.S.A., and�UCLA Division of Dermatology,200 Medical Plaza Suite 450,Los Angeles, CA 90095, U.S.A.Correspondence: J.C.Haley,E-mail: [email protected];[email protected]
References
1 Holmes SC, Burden AD. Lichen sclerosus and lichen planus: a
spectrum of disease? Clin Exp Dermatol 1998; 23: 129–31.
2 Lessani T, Gallego H, Lynch PJ. Lichen planus and lichen sclerosus
overlap. Cutis 1998; 61: 103–4.
3 Marren P, Millard P, Chia Y, Wojnarowska F. Mucosal lichen
sclerosus ⁄ lichen planus overlap syndrome. Br J Dermatol 1994;
131: 118–23.
4 Connelly MG, Winkelmann RK. Coexistence of lichen sclerosus,
morphea and lichen planus. J Am Acad Dermatol 1985; 12:
844–51.
5 Manns MP, Rambusch EG. Autoimmunity and extrahepatic
manifestations in hepatitis C virus infection. J Hepatol 1999; 31
(Suppl. 1): 39–42.
6 Divano MC, Parodi A, Rebora A. Lichen planus, liver kidney
microsomal (LKM1) antibodies and hepatitis C virus antibodies.
Dermatology 1992; 185: 132–3.
7 Divano MC, Parodi A, Rebora A. Anti-GOR antibodies in lichen
planus. Dermatology 1994; 188: 205–6.
8 Cottoni F, Tedde G, Solinas A, Deplano A. Lichen planus associated
with anti-liver-kidney microsome-positive chronic active hepatitis
and hyperthyroidism. Arch Dermatol 1991; 127: 1730–1.
9 Marren P, Yell J, Charnock FM et al. The association between li-
chen sclerosus and antigens of HLA system. Br J Dermatol 1995;
132: 197–203.
A peculiar type of dystrophic calcinosis cutis affectingthe surface of damaged genital skin and mucosa in anelderly woman with a vesicovaginal fistula
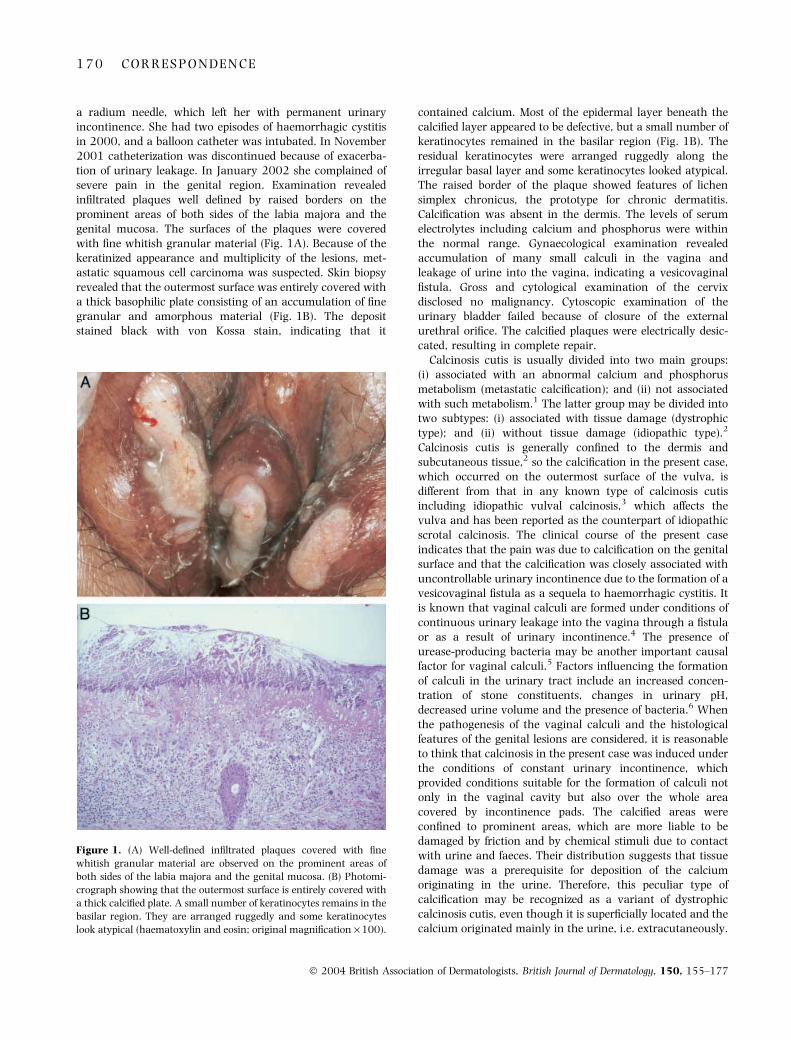
SIR, A 99-year-old woman presented with seriously painfullesions of the external genitalia. She had had uterine cancerat age 50 years. This was treated by intrauterine insertion of
a
dc
b
Figure 1. (a) Violaceous flat papules on the
inner right thigh. (b) Atrophic, violaceous,
telangiectatic plaque on the left shoulder.
(c) Biopsy of a violaceous papule on the inner
right thigh showed compact hyperkeratosis,
wedge-shaped hypergranulosis and a liche-
noid infiltrate, consistent with lichen planus.
(d) Biopsy of the left shoulder plaque showed a
thinned epidermis with superficial dermal
collagen homogenization and a mid-dermal
lymphocytic infiltrate, consistent with lichen
sclerosus.
C O R R E S P O N D E N C E 1 6 9
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
a radium needle, which left her with permanent urinaryincontinence. She had two episodes of haemorrhagic cystitisin 2000, and a balloon catheter was intubated. In November2001 catheterization was discontinued because of exacerba-tion of urinary leakage. In January 2002 she complained ofsevere pain in the genital region. Examination revealedinfiltrated plaques well defined by raised borders on theprominent areas of both sides of the labia majora and thegenital mucosa. The surfaces of the plaques were coveredwith fine whitish granular material (Fig. 1A). Because of thekeratinized appearance and multiplicity of the lesions, met-astatic squamous cell carcinoma was suspected. Skin biopsyrevealed that the outermost surface was entirely covered witha thick basophilic plate consisting of an accumulation of finegranular and amorphous material (Fig. 1B). The depositstained black with von Kossa stain, indicating that it
contained calcium. Most of the epidermal layer beneath thecalcified layer appeared to be defective, but a small number ofkeratinocytes remained in the basilar region (Fig. 1B). Theresidual keratinocytes were arranged ruggedly along theirregular basal layer and some keratinocytes looked atypical.The raised border of the plaque showed features of lichensimplex chronicus, the prototype for chronic dermatitis.Calcification was absent in the dermis. The levels of serumelectrolytes including calcium and phosphorus were withinthe normal range. Gynaecological examination revealedaccumulation of many small calculi in the vagina andleakage of urine into the vagina, indicating a vesicovaginalfistula. Gross and cytological examination of the cervixdisclosed no malignancy. Cytoscopic examination of theurinary bladder failed because of closure of the externalurethral orifice. The calcified plaques were electrically desic-cated, resulting in complete repair.
Calcinosis cutis is usually divided into two main groups:(i) associated with an abnormal calcium and phosphorusmetabolism (metastatic calcification); and (ii) not associatedwith such metabolism.1 The latter group may be divided intotwo subtypes: (i) associated with tissue damage (dystrophictype); and (ii) without tissue damage (idiopathic type).2
Calcinosis cutis is generally confined to the dermis andsubcutaneous tissue,2 so the calcification in the present case,which occurred on the outermost surface of the vulva, isdifferent from that in any known type of calcinosis cutisincluding idiopathic vulval calcinosis,3 which affects thevulva and has been reported as the counterpart of idiopathicscrotal calcinosis. The clinical course of the present caseindicates that the pain was due to calcification on the genitalsurface and that the calcification was closely associated withuncontrollable urinary incontinence due to the formation of avesicovaginal fistula as a sequela to haemorrhagic cystitis. Itis known that vaginal calculi are formed under conditions ofcontinuous urinary leakage into the vagina through a fistulaor as a result of urinary incontinence.4 The presence ofurease-producing bacteria may be another important causalfactor for vaginal calculi.5 Factors influencing the formationof calculi in the urinary tract include an increased concen-tration of stone constituents, changes in urinary pH,decreased urine volume and the presence of bacteria.6 Whenthe pathogenesis of the vaginal calculi and the histologicalfeatures of the genital lesions are considered, it is reasonableto think that calcinosis in the present case was induced underthe conditions of constant urinary incontinence, whichprovided conditions suitable for the formation of calculi notonly in the vaginal cavity but also over the whole areacovered by incontinence pads. The calcified areas wereconfined to prominent areas, which are more liable to bedamaged by friction and by chemical stimuli due to contactwith urine and faeces. Their distribution suggests that tissuedamage was a prerequisite for deposition of the calciumoriginating in the urine. Therefore, this peculiar type ofcalcification may be recognized as a variant of dystrophiccalcinosis cutis, even though it is superficially located and thecalcium originated mainly in the urine, i.e. extracutaneously.
Figure 1. (A) Well-defined infiltrated plaques covered with fine
whitish granular material are observed on the prominent areas of
both sides of the labia majora and the genital mucosa. (B) Photomi-
crograph showing that the outermost surface is entirely covered with
a thick calcified plate. A small number of keratinocytes remains in the
basilar region. They are arranged ruggedly and some keratinocytes
look atypical (haematoxylin and eosin; original magnification ·100).
1 7 0 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
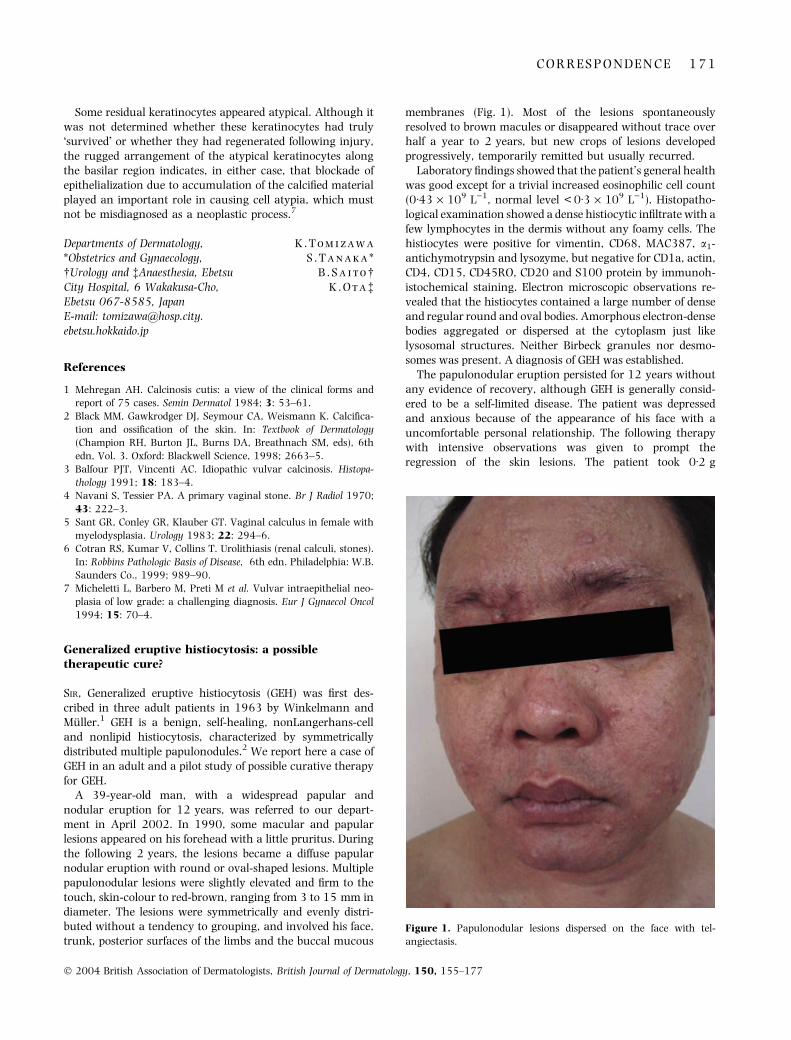
Some residual keratinocytes appeared atypical. Although itwas not determined whether these keratinocytes had truly�survived� or whether they had regenerated following injury,the rugged arrangement of the atypical keratinocytes alongthe basilar region indicates, in either case, that blockade ofepithelialization due to accumulation of the calcified materialplayed an important role in causing cell atypia, which mustnot be misdiagnosed as a neoplastic process.7
K . T o m i z a w a
S . T a n a k a *
B . S a i t o�K . O t a�
Departments of Dermatology,*Obstetrics and Gynaecology,�Urology and �Anaesthesia, EbetsuCity Hospital, 6 Wakakusa-Cho,Ebetsu 067-8585, JapanE-mail: [email protected]
References
1 Mehregan AH. Calcinosis cutis: a view of the clinical forms and
report of 75 cases. Semin Dermatol 1984; 3: 53–61.
2 Black MM, Gawkrodger DJ, Seymour CA, Weismann K. Calcifica-
tion and ossification of the skin. In: Textbook of Dermatology
(Champion RH, Burton JL, Burns DA, Breathnach SM, eds), 6th
edn, Vol. 3. Oxford: Blackwell Science, 1998; 2663–5.
3 Balfour PJT, Vincenti AC. Idiopathic vulvar calcinosis. Histopa-
thology 1991; 18: 183–4.
4 Navani S, Tessier PA. A primary vaginal stone. Br J Radiol 1970;
43: 222–3.
5 Sant GR, Conley GR, Klauber GT. Vaginal calculus in female with
myelodysplasia. Urology 1983; 22: 294–6.
6 Cotran RS, Kumar V, Collins T. Urolithiasis (renal calculi, stones).
In: Robbins Pathologic Basis of Disease, 6th edn. Philadelphia: W.B.
Saunders Co., 1999; 989–90.
7 Micheletti L, Barbero M, Preti M et al. Vulvar intraepithelial neo-
plasia of low grade: a challenging diagnosis. Eur J Gynaecol Oncol
1994; 15: 70–4.
Generalized eruptive histiocytosis: a possibletherapeutic cure?
SIR, Generalized eruptive histiocytosis (GEH) was first des-cribed in three adult patients in 1963 by Winkelmann andMuller.1 GEH is a benign, self-healing, nonLangerhans-celland nonlipid histiocytosis, characterized by symmetricallydistributed multiple papulonodules.2 We report here a case ofGEH in an adult and a pilot study of possible curative therapyfor GEH.
A 39-year-old man, with a widespread papular andnodular eruption for 12 years, was referred to our depart-ment in April 2002. In 1990, some macular and papularlesions appeared on his forehead with a little pruritus. Duringthe following 2 years, the lesions became a diffuse papularnodular eruption with round or oval-shaped lesions. Multiplepapulonodular lesions were slightly elevated and firm to thetouch, skin-colour to red-brown, ranging from 3 to 15 mm indiameter. The lesions were symmetrically and evenly distri-buted without a tendency to grouping, and involved his face,trunk, posterior surfaces of the limbs and the buccal mucous
membranes (Fig. 1). Most of the lesions spontaneouslyresolved to brown macules or disappeared without trace overhalf a year to 2 years, but new crops of lesions developedprogressively, temporarily remitted but usually recurred.
Laboratory findings showed that the patient’s general healthwas good except for a trivial increased eosinophilic cell count(0Æ43 · 109 L)1, normal level < 0Æ3 · 109 L)1). Histopatho-logical examination showed a dense histiocytic infiltrate with afew lymphocytes in the dermis without any foamy cells. Thehistiocytes were positive for vimentin, CD68, MAC387, a1-antichymotrypsin and lysozyme, but negative for CD1a, actin,CD4, CD15, CD45RO, CD20 and S100 protein by immunoh-istochemical staining. Electron microscopic observations re-vealed that the histiocytes contained a large number of denseand regular round and oval bodies. Amorphous electron-densebodies aggregated or dispersed at the cytoplasm just likelysosomal structures. Neither Birbeck granules nor desmo-somes was present. A diagnosis of GEH was established.
The papulonodular eruption persisted for 12 years withoutany evidence of recovery, although GEH is generally consid-ered to be a self-limited disease. The patient was depressedand anxious because of the appearance of his face with auncomfortable personal relationship. The following therapywith intensive observations was given to prompt theregression of the skin lesions. The patient took 0Æ2 g
Figure 1. Papulonodular lesions dispersed on the face with tel-
angiectasis.
C O R R E S P O N D E N C E 1 7 1
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177
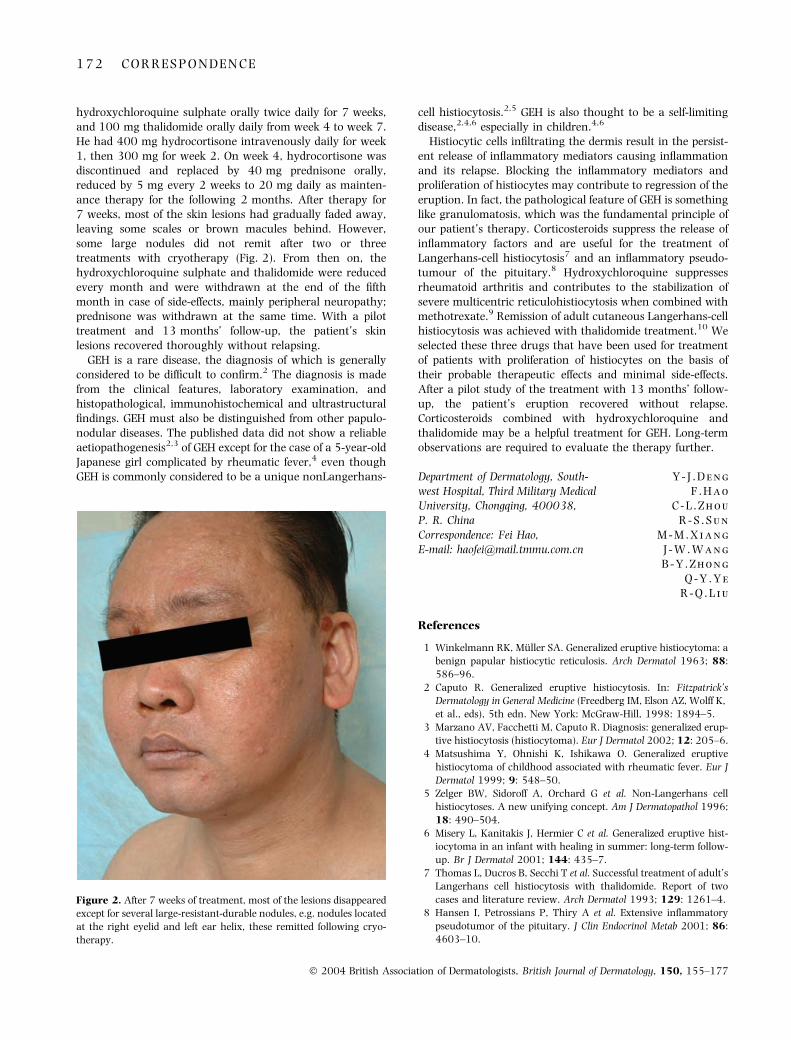
hydroxychloroquine sulphate orally twice daily for 7 weeks,and 100 mg thalidomide orally daily from week 4 to week 7.He had 400 mg hydrocortisone intravenously daily for week1, then 300 mg for week 2. On week 4, hydrocortisone wasdiscontinued and replaced by 40 mg prednisone orally,reduced by 5 mg every 2 weeks to 20 mg daily as mainten-ance therapy for the following 2 months. After therapy for7 weeks, most of the skin lesions had gradually faded away,leaving some scales or brown macules behind. However,some large nodules did not remit after two or threetreatments with cryotherapy (Fig. 2). From then on, thehydroxychloroquine sulphate and thalidomide were reducedevery month and were withdrawn at the end of the fifthmonth in case of side-effects, mainly peripheral neuropathy;prednisone was withdrawn at the same time. With a pilottreatment and 13 months’ follow-up, the patient’s skinlesions recovered thoroughly without relapsing.
GEH is a rare disease, the diagnosis of which is generallyconsidered to be difficult to confirm.2 The diagnosis is madefrom the clinical features, laboratory examination, andhistopathological, immunohistochemical and ultrastructuralfindings. GEH must also be distinguished from other papulo-nodular diseases. The published data did not show a reliableaetiopathogenesis2,3 of GEH except for the case of a 5-year-oldJapanese girl complicated by rheumatic fever,4 even thoughGEH is commonly considered to be a unique nonLangerhans-
cell histiocytosis.2,5 GEH is also thought to be a self-limitingdisease,2,4,6 especially in children.4,6
Histiocytic cells infiltrating the dermis result in the persist-ent release of inflammatory mediators causing inflammationand its relapse. Blocking the inflammatory mediators andproliferation of histiocytes may contribute to regression of theeruption. In fact, the pathological feature of GEH is somethinglike granulomatosis, which was the fundamental principle ofour patient’s therapy. Corticosteroids suppress the release ofinflammatory factors and are useful for the treatment ofLangerhans-cell histiocytosis7 and an inflammatory pseudo-tumour of the pituitary.8 Hydroxychloroquine suppressesrheumatoid arthritis and contributes to the stabilization ofsevere multicentric reticulohistiocytosis when combined withmethotrexate.9 Remission of adult cutaneous Langerhans-cellhistiocytosis was achieved with thalidomide treatment.10 Weselected these three drugs that have been used for treatmentof patients with proliferation of histiocytes on the basis oftheir probable therapeutic effects and minimal side-effects.After a pilot study of the treatment with 13 months’ follow-up, the patient’s eruption recovered without relapse.Corticosteroids combined with hydroxychloroquine andthalidomide may be a helpful treatment for GEH. Long-termobservations are required to evaluate the therapy further.
Y - J . D e n g
F . H a o
C - L . Z h o u
R - S . S u n
M - M . X i a n g
J - W . W a n g
B - Y . Z h o n g
Q - Y . Y e
R - Q . L i u
Department of Dermatology, South-west Hospital, Third Military MedicalUniversity, Chongqing, 400038,P. R. ChinaCorrespondence: Fei Hao,E-mail: [email protected]
References
1 Winkelmann RK, Muller SA. Generalized eruptive histiocytoma: a
benign papular histiocytic reticulosis. Arch Dermatol 1963; 88:
586–96.
2 Caputo R. Generalized eruptive histiocytosis. In: Fitzpatrick’s
Dermatology in General Medicine (Freedberg IM, Elson AZ, Wolff K,
et al., eds), 5th edn. New York: McGraw-Hill, 1998: 1894–5.
3 Marzano AV, Facchetti M, Caputo R. Diagnosis: generalized erup-
tive histiocytosis (histiocytoma). Eur J Dermatol 2002; 12: 205–6.
4 Matsushima Y, Ohnishi K, Ishikawa O. Generalized eruptive
histiocytoma of childhood associated with rheumatic fever. Eur J
Dermatol 1999; 9: 548–50.
5 Zelger BW, Sidoroff A, Orchard G et al. Non-Langerhans cell
histiocytoses. A new unifying concept. Am J Dermatopathol 1996;
18: 490–504.
6 Misery L, Kanitakis J, Hermier C et al. Generalized eruptive hist-
iocytoma in an infant with healing in summer: long-term follow-
up. Br J Dermatol 2001; 144: 435–7.
7 Thomas L, Ducros B, Secchi T et al. Successful treatment of adult’s
Langerhans cell histiocytosis with thalidomide. Report of two
cases and literature review. Arch Dermatol 1993; 129: 1261–4.
8 Hansen I, Petrossians P, Thiry A et al. Extensive inflammatory
pseudotumor of the pituitary. J Clin Endocrinol Metab 2001; 86:
4603–10.
Figure 2. After 7 weeks of treatment, most of the lesions disappeared
except for several large-resistant-durable nodules, e.g. nodules located
at the right eyelid and left ear helix, these remitted following cryo-
therapy.
1 7 2 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

9 Cash JM, Tyree J, Recht M. Severe multicentric reticulohistiocy-
tosis: disease stabilization achieved with methotrexate and
hydroxychloroquine. J Rheumatol 1997; 24: 2250–3.
10 Meunier L, Marck Y, Ribeyre C et al. Adult cutaneous Langerhans
cell histiocytosis: remission with thalidomide treatment. Br J
Dermatol 1995; 132: 168 (Letter).
A case of reticular erythematous mucinosis treatedwith topical tacrolimus
SIR, Reticular erythematous mucinosis (REM) is a rare diseasefirst identified in 1974 by Steigleder et al.1 Some consider itpart of the broad spectrum of �lupus erythematosus-like�diseases2,3 due to common features such as flare afterexposure to ultraviolet radiation, clinical manifestations,histology (mucin deposits) and good response to systemicantimalarials. Recently, Walker et al. described two cases ofsevere recalcitrant chronic discoid lupus erythematosus (DLE)successfully treated with topical tacrolimus.4 These reportsprompted us to try this drug in a young patient with REMwho showed initial signs of hydroxychloroquine-relatedretinopathy.
An 18-year-old man had a 4-month history of asympto-matic erythematous papules on the chest. There was nofamily history of skin manifestations of similar morphologyand site. Examination showed brownish-red reticular erythe-matous macules associated with pinkish confluent erythema-tous papules, 0Æ5–1Æ0 cm in diameter, on the chest andbilaterally in the clavicular area (Fig. 1A). Routine bloodchemistry, erythrocyte sedimentation rate and C-reactiveprotein were within normal limits. Autoantibodies (antinu-clear antibodies, extractable nuclear antigens) were notfound. Histology of a lesional biopsy showed moderateoedema and an infiltrate of lymphocytes and monocytes withperivascular and perifollicular distribution in the superficialdermis. Specific stains (Alcian blue and colloidal iron) showedmucin deposits near dermal collagen fibres. Direct immuno-fluorescence was negative. REM syndrome was diagnosed andtreatment with hydroxychloroquine sulphate 200 mg dailywas initiated.
After 3 months of therapy, no new papules had formed butthe clinical picture had not improved. The patient complainedof glare and difficulty in adjusting to changes in illumination.Ophthalmological examination showed bilateral visual acui-ties of 20 ⁄ 25, pericentral scotomata, and increased final dark-adapted rod thresholds in the two retinal areas tested. Full-fieldelectroretinograms were reduced and delayed, and focal coneelectroretinography showed abnormal foveal responses. Oph-thalmoscopy and fluorescein angiography showed bull’s-eyemaculopathy in both eyes. We therefore suspended treatmentand began therapy with tacrolimus ointment 0Æ1% (Protopic�
0Æ1%; Fujisawa, Munich, Germany) twice daily. The skinlesions showed considerable improvement, mainly in theerythematous component, after only 2 weeks, and treatmentwas maintained with tacrolimus ointment 0Æ03% (Protopic�
0Æ03%; Fujisawa) twice daily for 2 months, reduced to oncedaiy in the absence of relapse (Fig. 1B).
REM syndrome manifests with erythematous, induratedpapules and plaques associated with typical reticular macularerythema.5 Lesions are prevalent on the central chest and ⁄ orupper back, less often on the face, arms, abdomen and groin.They are frequently asymptomatic; however, in 20–30% ofcases pruritus is reported. Exposure to sunlight has beenreported to induce new lesions or to aggravate them. Amild to pronounced perivascular monocytic infiltrate andincreased dermal mucin (glycosaminoglycan) depositionare typical histological features.6 Hydroxychloroquine200–400 mg daily is the treatment of choice.5 However, insubjects not responsive to systemic antimalarials or withcontraindications for these drugs, such as the present case, noalternative therapy was hitherto available.7
Tacrolimus, a macrolide which targets T lymphocytes, isderived from Streptomyces tsukabaensis. It seems to affect thesecells by blocking calcineurin, which is required to activatecytokine genes for interleukin (IL)-2, IL-3, IL-4, granulocyte
Figure 1. (A) Typical brownish-red reticular erythematous macules
associated with pinkish confluent erythematous papules (insert) on
the chest. (B) Clinical appearance after 70 days of treatment with
topical tacrolimus.
C O R R E S P O N D E N C E 1 7 3
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

colony-stimulating factor and tumour necrosis factor-a.8 Thisimmunosuppressant activity may be why it is effective inchronic inflammatory diseases such as DLE,4 regarded bysome as pathogenetically similar to REM.2,3 Indeed, in ourpatient we observed excellent anti-inflammatory effects,whereas pigmentation remained unchanged.
In conclusion, tacrolimus was found to be a safe and validalternative to systemic antimalarials in our patient.
P . R u b e g n i
P . S b a n o
M . R i s u l o
S . P o g g i a l i
M . F i m i a n i
Department of Clinical Medicineand Immunology, Section ofDermatology, University of Siena,Policlinico �Le Scotte�,53100 Siena, ItalyE-mail: [email protected]
References
1 Steigleder GK, Gartmann H, Linker U. REM syndrome: reticular
erythematous mucinosis (round-cell erythematosis), a new entity?
Br J Dermatol 1974; 91: 191–9.
2 Bulengo-Ransby SM, Ellis CN, Griffiths CEM et al. Failure of
reticular erythematous mucinosis to respond to cyclosporin. J Am
Acad Dermatol 1992; 27: 825–8.
3 Braddock SW, Kay HD, Maennle D et al. Clinical and immuno-
logic studies in reticular erythematous mucinosis and Jessner’s
lymphocytic infiltrate of skin. J Am Acad Dermatol 1993; 28:
691–5.
4 Walker SL, Kirby B, Chalmers RJ. The effect of topical tacrolimus
on severe recalcitrant chronic discoid lupus erythematosus. Br J
Dermatol 2002; 147: 405–6.
5 Cohen PR, Rabinowitz AD, Ruszkowski AM, De Leo VA. Reticular
erythematous mucinosis syndrome: review of the world literature
and report of the syndrome in a prepubertal child. Pediatr Dermatol
1990; 7: 1–10.
6 Tominaga A, Tajima S, Ishibashi A, Kimata K. Reticular erythe-
matous mucinosis syndrome with an infiltration of factor XIIIa+
and hyaluronan synthase 2+ dermal dendrocytes. Br J Dermatol
2001; 145: 141–5.
7 Yamazaki S, Katayama I, Kurumaji Y et al. Treatment of reti-
cular erythematous mucinosis with a large dose of ultraviolet B
radiation and steroid impregnated tape. J Dermatol 1999; 26:
115–8.
8 Assmann T, Homey B, Ruzicka T. Applications of tacrolimus for
the treatment of skin disorders. Immunopharmacology 2000; 47:
203–13.
A CD56-negative case of blastic natural killer-celllymphoma (agranular CD4+ ⁄ CD56+ haematodermicneoplasm)
SIR, Four cases of blastic natural killer (NK) cell-lymphomawere reported in a recent issue of this Journal.1 This disease,also called agranular CD4+ ⁄ CD56+ haematodermic neo-plasm, represents a rare and distinct clinicopathological entitythat was described recently.2 The disease is characterized byits clinical presentation (high skin tropism, bone marrowinvolvement with or without leukaemia, very poor prognosis)and its molecular phenotype: CD4+, CD56+, CD123+ and�lineage negative�. In an editorial in the same issue, an
important role of CD56 antigen expression was highlighted inmaking the diagnosis of cutaneous lymphomas. The need forclinical and histopathological vigilance to recognize andestablish the correct diagnosis was also emphasized.3
We report the surprising case of a blastic NK cell-lymphoma which does not express the CD56 antigen. An87-year-old man presented with a 5-cm slightly reddishcutaneous nodule on the forehead (Fig. 1). The staging wasnormal and local radiotherapy was performed. A completeremission of 1 years’s duration was obtained. The patientthen relapsed with skin, cervical lymph node and bonemarrow infiltration, and a leukaemic phase. Treatment withoral etoposide (Vepesid�; Bristol-Myers Squibb, Plainsboro,N.J., U.S.A) induced a short stabilization. The patient died ofhis disease 2 years after the initial diagnosis.
Skin biopsy showed dermal infiltration by tumour cellswhich were predominantly medium sized. By immunohist-ochemistry on paraffin section these cells expressed onlyCD45, CD4 (Fig. 2a), CD43, HECA-452 and TCL1. CD43was weakly expressed on the first biopsy but stronglyexpressed on relapse biopsies. CD68 was negative on thefirst biopsy but positive on relapse biopsies. On frozensections, only CD2, CD4 and CD123 were positive. Markersof B-cell (CD20, CD79a), T-cell (CD3) myeloid-cell (CD13,CD14, CD15, CD33, CD117, MPO, lysozyme) and NK-cell(CD16, TIA-1, granzyme B) differentiation were negative.
Figure 1. A reddish nodule is evident on the forehead.
1 7 4 C O R R E S P O N D E N C E
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

Importantly, CD56 was negative on both frozen andparaffin sections of the first biopsy and subsequent relapsebiopsies (Fig. 2b).
This patient had the usual clinical presentation of blasticNK cell-lymphoma. The tumour cells showed a typicalphenotype that was CD4+, CD43+, CD123+ and lineagenegative. The case was so typical that initially we thoughtthat the CD56 negativity was due to a technical problem andseveral additional controls were performed on paraffin andfrozen sections.
As described in the new World Health Organizationclassification,4 in which this tumour is listed as a blasticNK-cell lymphoma, the precise lineage of this neoplasm isunresolved and the proposal of a NK-cell origin was linkedonly to the expression of CD56 antigen. This link was tenuousat best due to the observation that several other nonlymphoiddiseases express CD56, particularly tumours of myeloidorigin. Several very recent studies have suggested an imma-ture dendritic cell (IDC) origin for this entity.5–7 IDCs areimmune cells as originally identified. They display a peculiarphenotype: �lineage negative� with expression of CD4, CD68
and high-level CD123; they are normally CD56 negative. Aclose relationship has been established betweenCD4+ ⁄ CD56+ tumour cells and IDCs at the phenotypic5,7,8
and functional levels.6 The present case expresses CD123,HECA-452 and TCL1. These three antigens are known to beexpressed by IDCs. CD123 (interleukin 3 a-chain receptor) ishighly expressed on IDCs and at a lower level on blooddendritic cells.9 It has been shown that CD123 is regularlyexpressed and is a hallmark of agranular CD4+ ⁄ CD56+haematodermic neoplasms.5,7,8 It is not expressed by NK-celllymphomas.5 HECA-452 monoclonal antibody has also beendemonstrated to be a good marker of IDCs.10 TCL1 has beendemonstrated to be expressed by both blastic NK cell-lymphomas and IDCs.8
The present case is the first reported case of a CD56-negative blastic NK cell-lymphoma (agranular CD4+ ⁄ CD56+haematodermic neoplasm). We think that this case isinformative at two levels. First, it shows that the CD56expression is not necessary to the histopathological diagnosisof the entity, and second, it reinforces the feeling that thedisease probably originates from IDCs instead of NK cells. Inthis context the CD56 staining result must be interpretedcarefully and with a great deal of objectivity.
Acknowledgments
We thank Professor P. Souteyrand (Clermont-Ferrand), Pro-fessor M. d’Incan (Clermont-Ferrand), Dr F. Connort (Nevers)and Dr T. Lefranc (Nevers) for their help in collecting the dataand Sylvie Espin and Marie-Antoinette Lignier for theirtechnical assistance. This study was supported by grants fromthe NCI ⁄ NIH (R01CA90571, M.A.T.) and CMISE (NCC2-1364, M.A.T.). M.A.T. is a Scholar of the Leukaemia andLymphoma Society, U.S.A.
T . P e t r e l l a
M . A . T e i t e l l *
C . S p i e k e r m a n n�C . J . L . M . M e i j e r §
F . F r a n c k–
I . E n a c h e�
Centre de Pathologie, 33 Rue NicolasBornier, BP 189, and Laboratoired’Anatomie Pathologique (CentreHospitalo-Universitaire), Dijon21000, France, *Departments ofPathology and Pediatrics, Center forCell Mimetic Studies (CMISE),Jonsson Comprehensive CancerCenter, David Geffen School ofMedicine at UCLA, Los Angeles, CA90095-1732, U.S.A., Departmentsof �Pathology and �InternalMedicine, Centre HospitalierRegional, Nevers 58000, France,§Department of Pathology, VrijeUniversiteit Medical Centre,Amsterdam 1007MB, theNetherlands, and –Department ofPathology, Hotel Dieu, Clermont-Ferrand 63000, FranceE-mail:[email protected]
Figure 2. Immunostaining: (a) CD4 positive (paraffin section);
(b) CD56 negative (paraffin section).
C O R R E S P O N D E N C E 1 7 5
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

References
1 Child FJ, Mitchell TJ, Whittaker SJ et al. Blastic natural killer cell
and extranodal natural killer cell-like T-cell lymphoma presenting
in the skin: report of six cases from the U.K. Br J Dermatol 2003;
148: 507–15.
2 Petrella T, Dalac S, Maynadie M et al. CD4+ CD56+ cutaneous
neoplasms: a distinct hematological entity? Groupe Francais
d’Etude des Lymphomes Cutanes (GFELC). Am J Surg Pathol 1999;
23: 137–46.
3 Slater DN. New CD56 positive and cytotoxic T-cell cutaneous
lymphomas from the World Health Organisation. Br J Dermatol
2003; 148: 385–7.
4 Chan JKC, Jaffe ES, Ralfkiaer E. Blastic NK-cell lymphoma. In: The
World Health Organisation Classification of Tumors: Tumors of He-
matopoietic and Lymphoid Tissues (Jaffe ES, Harris NL, Stein H,
Vardiman JW, eds). Lyon: IARC Press, 2001; 214–5.
5 Petrella T, Comeau MR, Maynadie M et al. �Agranular CD4+
CD56+ hematodermic neoplasm� (blastic NK-cell lymphoma)
originates from a population of CD56+ precursor cells related to
plasmacytoid monocytes. Am J Surg Pathol 2002; 26: 852–62.
6 Chaperot L, Bendriss N, Manches O et al. Identification of a le-
ukemic counterpart of the plasmacytoid dendritic cells. Blood
2001; 97: 3210–7.
7 Feuillard J, Jacob MC, Valensi F et al. Clinical and biologic features
of CD4(+)CD56(+) malignancies. Blood 2002; 99: 1556–63.
8 Herling M, Teitell MA, Shen RR et al. TCL1 expression in plas-
macytoid dendritic cells (DC2s) and the related CD4+CD56+
blastic tumors of skin. Blood 2003; 101: 5007–9.
9 Maraskovsky E, Daro E, Roux E et al. In vivo generation of human
dendritic cell subsets by Flt3 ligand. Blood 2000; 96: 878–84.
10 Facchetti F, de Wolf-Peeters C, van den Oord JJ et al. Anti-high
endothelial venule monoclonal antibody HECA-452 recognizes
plasmacytoid T cells and delineates an �extranodular� compart-
ment in the reactive lymph node. Immunol Lett 1989; 20: 277–81.
Book Review
Dermatology – just the facts (2003). Kerdel FA, Jimenez-Acosta F, eds. New York: McGraw-Hill. ISBN: 0-07-139143-6. Price £39.99.
This multi-author text is designed to be a quick referenceguide, with easily accessible facts and an emphasis on medicaldermatology. In all of these aspects it achieves its aim. Thereare 22 chapters, each of which covers groups of disordersrather than single diseases. Inevitably, some of these are morelogical in their content than others, some chapters pullingtogether rather diverse entities (for example, the chapter ondermal infiltrates). Although medical dermatology is the mainaspect, there are four chapters on neoplasms; these discusstherapy but do not go into any detail of surgical technique.For a dermatology text from the USA, there is a refreshinglycomplete absence of cosmetic dermatology.
The style of the book is very much one of factual nuggets –the text on any disorder is broken up initially into headingssuch as epidemiology, pathophysiology etc, but is otherwiseentirely presented as a series of bullet points with a scatteringof, generally useful, tables. There are a few diagrams, and noclinical pictures, but some frustrating typos (e.g. rhynophy-ma). References are deliberately few and general, more of abackground reading list. The information presented is gener-
ally pertinent and thorough within the space constraints,although some areas are perhaps a bit too brief (HIV diseasegets less space than hand, foot and mouth disease). Thebulleted format and consistent layout makes it easy to quicklyread through individual diseases or sections.
My real problem is knowing whether this book has asignificant UK market. For countries where there is an exitexamination from dermatological training, this would be auseful revision text, quick and easy to use. It may fulfil thisfunction to some extent in the UK, for example for diplomaexams. However, it is not detailed enough to act as a formalreference text for dermatologists or dermatology trainees onthe one hand (although it would be potentially useful early intraining years), yet it is too detailed for medical students orthe average General Practitioner (even though non-specialistsand students are included in the target readership). Addi-tionally, the lack of any illustrations, whilst beneficial interms of price, means that it will not be very useful to thosewho are looking to confirm a diagnosis in clinical practice.Having said that, if viewed in the context of its subtitle – �justthe facts� – then this text achieves what it sets out to do.
N e i l H . C o x
News and Notices
II. EADV Spring Symposium, Budapest, Hungary29 April – 1 May 2004
Due to growing knowledge and complexity of dermatologyand venerology, we try to integrate international experience
regarding new techniques, research concepts and therapy inpatient management following the motto of the congress:�Tradition and Science in Clinical Practice�Major topics: Oncology, inflammatory and autoimmune
skin diseases, practical aspects of genetics, surgery andcosmetology.
1 7 6 B O O K R E V I E W
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177

European Nail Society project grant
The European Nail Society was established in 1996 and has amembership of over 70 dermatologists from 16 countriesaround the world. Our aim is to share expertise and advancesin the realm of nail biology and disease.
The Society is able to offer a grant of 2000 euros to assist inthe pursuit of a research project based on nails. Thoseinterested should apply to David de Berker for details of theapplication process. Email: [email protected]. Website: http://www.euronails.org.
IX International Congress of DermatologyBejing, 19–22 May, 2004
Organised by the International Society of Dermatology andthe Chinese Medical Association.
Email: [email protected]. Website: http://www.chinamed.com/cn/dermatology.
N E W S A N D N O T I C E S 1 7 7
� 2004 British Association of Dermatologists, British Journal of Dermatology, 150, 155–177